User login
The plot thickens
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
After losing consciousness at a supermarket, a 70-year-old man was brought to the emergency department by paramedics. He subsequently developed chest pain.
Syncope can be difficult to evaluate, but chest pain may help narrow an otherwise broad differential diagnosis. If this patient has aortic stenosis or hypertrophic cardiomyopathy, effort syncope is the culprit. Cardiac dysrhythmia (eg, ventricular tachycardia), complete heart block, and supraventricular tachycardia each can cause syncope along with chest pain. Myocardial infarction and associated ventricular arrhythmia might also explain both chest pain and syncope. The paramedics might have noted an arrhythmia on the cardiac monitor; if possible, the rhythm strip should be reviewed. A pulmonary embolus can cause chest pain and, if large enough to cause right ventricular compromise, syncope.
According to witnesses at the supermarket, the patient dropped to the ground, lost consciousness, and convulsed for 30 seconds. He had no head trauma, tongue biting, urinary incontinence, or confusion afterward. Electrocardiogram (ECG) performed at the scene showed ST elevations in leads V1 to V3 with ST depressions in the inferior leads. On arrival in the emergency department, the patient described nonradiating substernal chest pressure exacerbated by deep inhalation. The pain did not improve with nitroglycerin. He recalled feeling light-headed before the syncope.
He had not received medical care for 20 years and had no known illnesses other than hypertension. He was not taking any medications. He previously worked as a welder and never smoked tobacco, drank alcohol, or used illicit drugs. The patient’s temperature was 36.4°C. Heart rate was 88 beats per minute, blood pressure 128/72 mm Hg, oxygen saturation 100% on room air, and respiratory rate 22 breaths per minute. The patient had conjunctival pallor. There was a grade 3/6 crescendo- decrescendo systolic murmur loudest at the right upper sternal border without radiation to the carotids. There was no jugular venous distention. Lungs were clear to auscultation bilaterally. There was no peripheral edema, rash, or lymphadenopathy.
Convulsive movements commonly occur during episodes of unconsciousness lasting more than 15 seconds—a phenomenon termed convulsive syncope and often is confused with seizures. These movements are usually clonic jerks of the extremities and trunk and slight twitching of the face, and occasionally tonic extension of the trunk and clenching of the jaw. Absence of tongue biting, urinary incontinence, and confusion in this patient’s case makes seizures less likely.
The distribution of ST segment changes on his ECG are concerning for myocardial infarction in the septal and inferior regions. Right-sided ECG should be performed to assess for right ventricular infarction. Although myocardial ischemia is the primary concern, some features warrant consideration of other etiologies of syncope. First, syncope is an unusual presentation of cardiac ischemia or infarct. The complaint of chest pressure exacerbated by deep inhalation is another atypical feature for myocardial ischemia. Although the patient’s oxygen saturation and heart rate are normal, pulmonary embolism remains a possibility.
The prominent crescendo-decrescendo systolic murmur at the right upper sternal border could indicate aortic stenosis; the carotids should be palpated to assess for pulsus parvus et tardus. A high-flow state associated with anemia could also lead to a midsystolic murmur. Conjunctival pallor typically is seen with hemoglobin levels of 6 g/dL or less. This finding may indicate severe anemia, which has the potential to cause myocardial ischemia and syncope.
Laboratory testing revealed a troponin of 0.04 ng/dL, hemoglobin 4.1 g/dL with MCV of 84.7 fL, white blood cell count 6,500/μL and platelet count 179,000/μL. Serum sodium was 130 mEq/L, urea nitrogen 16 mg/dL, creatinine 1.6 mg/dL, calcium 7.8 mg/dL, total protein 11.4 g/dL (reference range, 6.0-8.2), and albumin 2.2 g/dL. Erythrocyte sedimentation rate (ESR) was 20 mm/h. Serum iron was 48 μg/mL, total iron binding capacity 275 μg/dL, percent iron saturation 17% (reference range, 20-55), and ferritin 10 ng/mL (reference range, 30-400). The international normalized ratio (INR) was 1.5, prothrombin time 15.5 sec (reference range, 9.4-11.6), and partial thromboplastin time 24.7 sec (reference range, 22.9-30.6). Hepatitis C and HIV antibodies were negative as was the urine toxicology screen. Urine protein to creatinine ratio was 0.07. His hemoglobin rose to 7.9 g/dL with transfusion of 4 units packed red blood cells (RBC). His chest pain improved and inferior ST depressions resolved on follow-up ECG. Further history revealed multiple episodes of melena and hematochezia in the preceding weeks without nausea, vomiting, or abdominal pain.
The patient has a strikingly large gamma gap: 9.2. A gap larger than 4 is concerning for the presence of paraproteins. Given the possibility of a paraproteinemia (eg, multiple myeloma, plasmacytoma, Waldenström macroglobulinemia), the first step is to check serum and urine protein electrophoresis. The patient’s anemia is significant and reticulocyte index low. The low ferritin level combined with the inappropriately low reticulocyte count could result from iron deficiency anemia, another bone marrow process, or both. The patient’s syncope likely resulted from severe anemia and hypovolemia associated with hematochezia. The prolonged prothrombin time could be caused by a coagulation factor production problem, from vitamin K deficiency or underlying liver disease, or by a consumptive problem, from low-grade disseminated intravascular coagulation. It is controversial whether inhaling welding fumes causes cancer, but the patient’s age alone makes malignancy a definite possibility.
 and increased background staining secondary to hypergammaglobulinemia.")
Aortic stenosis is intriguing in light of the patient’s painless melena and hematochezia. Heyde syndrome is a phenomenon in which high shear stress causes a reduction in the size of von Willebrand factor predisposing to bleeding from submucosal angiodysplasia. However, Heyde syndrome has been reported to occur in the setting of severe or critical aortic stenosis and is unlikely to be the cause here. The patient needs to be examined with both upper and lower endoscopy to rule out gastrointestinal (GI) malignancy, gastric and esophageal varices, and painless peptic ulcers. High right ventricular systolic blood pressures along with echodense material in the inferior vena cava and right atrium suggest the possibility of malignancy with vascular invasion. Tricuspid regurgitation is consistent with high right-sided pressures. As left ventricular ejection fraction is reduced, some of the high right-sided pressures could also be attributable to left heart failure. CT findings of rectal wall thickening and perirectal lymph nodes could be attributable to cancer with locally metastatic disease. Blood loss caused by this cancer would explain the severe anemia on admission as well as the low ferritin level and the iron deficiency anemia. In this 70-year-old man who has not had routine health care maintenance, the leading diagnosis is colorectal cancer. However, the markedly elevated globulin gap and elevated INR strongly suggest another process (eg, multiple myeloma, other paraproteinemia) is also present.
INR remained elevated (1.5) despite vitamin K supplementation. Peripheral blood smear showed hypochromic and normocytic RBCs with moderate rouleaux formation (Figure 1). Intravenous pantoprazole and octreotide were started. Upper and lower endoscopy revealed multiple esophageal erosions and both a polypoid mass and an ulcer within the rectum with evidence of prior bleeding. The mass was resected and the ulcer biopsied.
 Color fundus photographs show mild dilatation of patient’s retinal veins and retinal hemorrhages. (C,D) Red-free fundus photographs show round blot hemorrhages occurring from vessels within retina (arrow) and linear, fl ame-shaped hemorrhages occurr")
On hospital day 3, the patient was transfused another unit of packed RBCs. Hemoglobin level increased from 7.4 g/dL to 8.2 g/dL, but he began to complain of headache, blurred vision, and worsened chest pain. He did not have weakness, numbness, diplopia, dysphagia, or dysarthria. External examination and extraocular movements of both eyes were normal. Visual acuity was 20/25 bilaterally. Funduscopic examination revealed mild dilation of retinal veins and retinal hemorrhages (Figure 2).
Rouleaux formation occurs as excess cathodal proteins, such as immunoglobulins or fibrinogen, adhere to RBCs and cause the cells to stack together in long chains. Classically this is associated with multiple myeloma, but can occur with Waldenström’s macroglobulinemia and other cancers or infections. It can also occur as an artifact in smear preparation but the large globulin gap in this patient supports pathologic rouleaux formation.
Venous retinopathy with hemorrhages may occur with occlusion of the arterial supply (eg, as with carotid artery obstruction) but also with hyperviscosity syndrome (HVS). Vascular disturbances throughout the body play a major role in HVS, but these changes are most easily visualized in the retina. It is interesting that the patient’s headache and blurred vision began after he received additional blood transfusions. Spuriously low hemoglobin and hematocrit levels may stem from increased plasma volume from high immunoglobulin M (IgM) concentrations in Waldenström macroglobulinemia; thus, RBC transfusions can exacerbate symptoms by elevating total RBC mass. Normocytic, normochromic anemia is characteristic of both multiple myeloma and Waldenström’s macroglobulinemia. That the patient’s chest pain recurred coincidentally with blurred vision and headache suggests the likely cause is cardiac ischemia from hyperviscosity. The serum viscosity level should be checked, and, if it is elevated, urgent serum plasmapheresis should be considered. Determining the source of excess globulin production and treating the underlying disease are crucial at this juncture.
In the general population, rectal adenocarcinoma is the most common cause of a rectal mass. In this patient, presence of a paraproteinemia may point to a different diagnosis. Extramedullary colorectal plasmacytoma can occur in the rectum but is exceedingly rare. Waldenström’s macroglobulinemia, a subtype of lymphoplasmacytic lymphoma, can be associated with a rectal lymphoma. At this point, it is not possible to confidently predict the etiology of the mass.
.")
He was started on cyclophosphamide, bortezomib and dexamethasone for IgG κ myeloma with improvement in his headache, blurred vision, chest pain, and plasma viscosity (4 to 1.8). His hemoglobin remained stable at 10 g/dL. Neoadjuvant Capecitabine and radiation therapy were initiated for his rectal cancer.
DISCUSSION
Multiple myeloma is characterized by monoclonal proliferation of plasma cells, elevated circulating monoclonal immunoglobulin, and end-organ damage.1 It accounts for approximately 0.8% of all new cancer diagnoses; average age at onset is 70 years. The patient described here had an unusual presentation, with GI bleeding and progression to HVS, and known risk factors for multiple myeloma (male sex, low socioeconomic status, welding career).2,3
An early clue in the diagnosis was the patient’s large gamma gap and concurrent anemia. Gamma gap, calculated by subtracting serum albumin from serum total protein, is so named because it often reflects an elevated gamma globulin concentration. However, it actually reflects all nonalbumin serum protein. A gamma gap larger than 3.1 g/dL is an independent risk factor for death4 and may be associated with infection, autoimmunity, and malignancy. Although there are no screening guidelines for multiple myeloma, 73% of cases are brought to attention by anemia discovered on routine laboratory investigation.5 This patient’s lack of prior medical care likely contributed to his atypical presentation. Screening colonoscopy, recommended at age 50, might have identified his rectal cancer at an earlier stage.
The patient’s anemia was likely secondary to GI hemorrhage and bone marrow suppression. His hematochezia might have been partly related to the pathophysiologic interaction of paraproteins with platelets, coagulation factors, and blood vessels.6 Amyloidosis of the GI tract is seen in 8% of AL amyloidosis7 and most frequently manifests as gastrointestinal bleeding, which is thought to be due to ischemia, vascular friability, or mucosal lesions. It less commonly presents as malabsorption or dysmotility.8 Although gastrointestinal amyloid is not typically associated with radiologic abnormalities, occasionally it may cause luminal wall thickening, adenopathy, and inflammatory stranding.9 The gold standard for diagnosis is tissue biopsy. However, presence of amyloidosis does not change the overall treatment strategy for multiple myeloma.
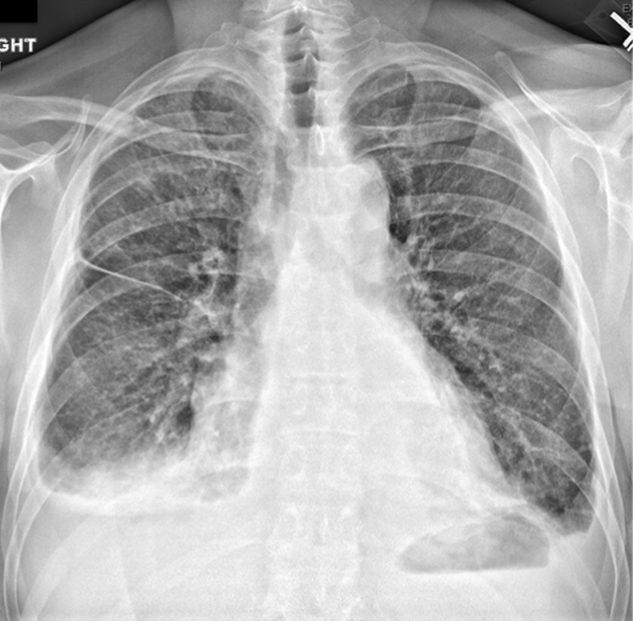
An interesting feature of this case is the development of HVS, which typically manifests with mucosal bleeding, blurred vision, and headache.10 HVS can be diagnosed on retinal examination with findings of venous tortuosity, dilatation, and intraretinal hemorrhage, as occurred in this case,11 and is confirmed with serum viscosity measurement. The first evidence of HVS in this case might have been the spontaneous echo contrast, or “smoke,” detected on echocardiogram. Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates,12 and is associated with conditions that result in left atrial stasis, such as atrial arrhythmias and mitral stenosis. This patient did not have valvular pathology or arrhythmia, and thus the “smoke” likely reflected HVS.
Of the paraproteinemias, Waldenström’s macroglobulinemia is most often associated with HVS, likely because of the pentameric structure of IgM13 and the consequential large size that predisposes to vascular occlusion. Whereas HVS can occur with IgM levels as low as 3 g/dL, it typically does not occur with IgG concentrations under 15 g/dL. This patient presented with an IgG level of 8 g/dL and developed HVS symptoms only after multiple packed RBC transfusions. Elevated IgG level likely made him susceptible to HVS, which ultimately was precipitated by blood transfusion. Therefore, this patient’s initial chest pain most likely was caused by demand cardiac ischemia secondary to anemia, whereas his subsequent, posttransfusion chest pain likely resulted from hyperviscosity angina. Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—has been described in polycythemia and connective tissue disorders.14,15 To our knowledge, however, hyperviscosity angina has not been reported in patients with multiple myeloma. Treatment of hyperviscosity with end-organ damage typically consists of plasmapheresis, but this patient was started on urgent chemotherapy, and his symptoms improved. Untreated HVS can lead to end-organ ischemia and death.
This patient had a multitude of seemingly disparate symptoms and abnormalities that ultimately were united in a diagnosis of IgG κ multiple myeloma. Subsequently diagnosed rectal adenocarcinoma may have led to ongoing blood loss, which worsened the anemia, but had no evident relation to the primary diagnosis of multiple myeloma. This case exemplifies the fact that HVS is a rare but important iatrogenic complication of multiple myeloma treated with blood transfusion. As this patient’s hospital course progressed, the plot, and his blood, thickened.
KEY TEACHING POINTS
- Multiple myeloma is occasionally associated with HVS, which manifests with mucosal bleeding, blurred vision, and headache.
- Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—should be considered in patients with paraproteinemias and chest pain.
- Plasmapheresis reverses the clinical manifestations of HVS but not the underlying disease process (eg, Waldenström’s macroglobulinemia, multiple myeloma, leukemia, polycythemia).
- Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates, and is associated with left atrial stasis, commonly from atrial fibrillation or mitral stenosis, but might be present in HVS.
Acknowledgment
The authors thank Peter Campochiaro, MD, and Whitney Green, MD, for their contributions to the images used in this article.
Disclosure
Dr. Sedighi Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The other authors have nothing to report.
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046-1060. PubMed
2. Koessel SL, Theis MK, Vaughan TL, et al. Socioeconomic status and the incidence of multiple myeloma. Epidemiology. 1996;7(1):4-8. PubMed
3. Fritschi L, Siemiatycki J. Lymphoma, myeloma and occupation: results of a case-control study. Int J Cancer. 1996;67(4):498-503. PubMed
4. Juraschek SP, Moliterno AR, Checkley W, Miller ER 3rd. The gamma gap and all-cause mortality. PLoS One. 2015;10(12):e0143494. PubMed
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. PubMed
6. Eby CS. Bleeding and thrombosis risks in plasma cell dyscrasias. Hematology Am Soc Hematol Educ Program. 2007:158-164. PubMed
7. Menke DM, Kyle RA, Fleming CR, Wolfe JT 3rd, Kurtin PJ, Oldenburg WA. Symptomatic gastric amyloidosis in patients with primary systemic amyloidosis. Mayo Clin Proc. 1993;68(8):763-767. PubMed
8. Levy DJ, Franklin GO, Rosenthal WS. Gastrointestinal bleeding and amyloidosis. Am J Gastroenterol. 1982;77(6):422-426. PubMed
9. Araoz PA, Batts KP, MacCarty RL. Amyloidosis of the alimentary canal: radiologic-pathologic correlation of CT findings. Abdom Imaging. 2000;25(1):38-44. PubMed
10. Stone MJ, Bogen SA. Evidence-based focused review of management of hyperviscosity syndrome. Blood. 2012;119(10):2205-2208. PubMed
11. Rajagopal R, Apte RS. Seeing through thick and through thin: retinal manifestations of thrombophilic and hyperviscosity syndromes. Surv Ophthalmol. 2016;61(2):236-247. PubMed
12. Black IW. Spontaneous echo contrast: where there’s smoke there’s fire. Echocardiography. 2000;17(4):373-382. PubMed
13. Kwaan HC. Hyperviscosity in plasma cell dyscrasias. Clin Hemorheol Microcirc. 2013;55(1):75-83. PubMed
14. Piccirillo G, Fimognari FL, Valdivia JL, Marigliano V. Effects of phlebotomy on a patient with secondary polycythemia and angina pectoris. Int J Cardiol. 1994;44(2):175-177. PubMed
15. Ovadia S, Lysyy L, Floru S. Emergency plasmapheresis for unstable angina in a patient with hyperviscosity syndrome. Am J Emerg Med. 2005;23(6):811-812. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
After losing consciousness at a supermarket, a 70-year-old man was brought to the emergency department by paramedics. He subsequently developed chest pain.
Syncope can be difficult to evaluate, but chest pain may help narrow an otherwise broad differential diagnosis. If this patient has aortic stenosis or hypertrophic cardiomyopathy, effort syncope is the culprit. Cardiac dysrhythmia (eg, ventricular tachycardia), complete heart block, and supraventricular tachycardia each can cause syncope along with chest pain. Myocardial infarction and associated ventricular arrhythmia might also explain both chest pain and syncope. The paramedics might have noted an arrhythmia on the cardiac monitor; if possible, the rhythm strip should be reviewed. A pulmonary embolus can cause chest pain and, if large enough to cause right ventricular compromise, syncope.
According to witnesses at the supermarket, the patient dropped to the ground, lost consciousness, and convulsed for 30 seconds. He had no head trauma, tongue biting, urinary incontinence, or confusion afterward. Electrocardiogram (ECG) performed at the scene showed ST elevations in leads V1 to V3 with ST depressions in the inferior leads. On arrival in the emergency department, the patient described nonradiating substernal chest pressure exacerbated by deep inhalation. The pain did not improve with nitroglycerin. He recalled feeling light-headed before the syncope.
He had not received medical care for 20 years and had no known illnesses other than hypertension. He was not taking any medications. He previously worked as a welder and never smoked tobacco, drank alcohol, or used illicit drugs. The patient’s temperature was 36.4°C. Heart rate was 88 beats per minute, blood pressure 128/72 mm Hg, oxygen saturation 100% on room air, and respiratory rate 22 breaths per minute. The patient had conjunctival pallor. There was a grade 3/6 crescendo- decrescendo systolic murmur loudest at the right upper sternal border without radiation to the carotids. There was no jugular venous distention. Lungs were clear to auscultation bilaterally. There was no peripheral edema, rash, or lymphadenopathy.
Convulsive movements commonly occur during episodes of unconsciousness lasting more than 15 seconds—a phenomenon termed convulsive syncope and often is confused with seizures. These movements are usually clonic jerks of the extremities and trunk and slight twitching of the face, and occasionally tonic extension of the trunk and clenching of the jaw. Absence of tongue biting, urinary incontinence, and confusion in this patient’s case makes seizures less likely.
The distribution of ST segment changes on his ECG are concerning for myocardial infarction in the septal and inferior regions. Right-sided ECG should be performed to assess for right ventricular infarction. Although myocardial ischemia is the primary concern, some features warrant consideration of other etiologies of syncope. First, syncope is an unusual presentation of cardiac ischemia or infarct. The complaint of chest pressure exacerbated by deep inhalation is another atypical feature for myocardial ischemia. Although the patient’s oxygen saturation and heart rate are normal, pulmonary embolism remains a possibility.
The prominent crescendo-decrescendo systolic murmur at the right upper sternal border could indicate aortic stenosis; the carotids should be palpated to assess for pulsus parvus et tardus. A high-flow state associated with anemia could also lead to a midsystolic murmur. Conjunctival pallor typically is seen with hemoglobin levels of 6 g/dL or less. This finding may indicate severe anemia, which has the potential to cause myocardial ischemia and syncope.
Laboratory testing revealed a troponin of 0.04 ng/dL, hemoglobin 4.1 g/dL with MCV of 84.7 fL, white blood cell count 6,500/μL and platelet count 179,000/μL. Serum sodium was 130 mEq/L, urea nitrogen 16 mg/dL, creatinine 1.6 mg/dL, calcium 7.8 mg/dL, total protein 11.4 g/dL (reference range, 6.0-8.2), and albumin 2.2 g/dL. Erythrocyte sedimentation rate (ESR) was 20 mm/h. Serum iron was 48 μg/mL, total iron binding capacity 275 μg/dL, percent iron saturation 17% (reference range, 20-55), and ferritin 10 ng/mL (reference range, 30-400). The international normalized ratio (INR) was 1.5, prothrombin time 15.5 sec (reference range, 9.4-11.6), and partial thromboplastin time 24.7 sec (reference range, 22.9-30.6). Hepatitis C and HIV antibodies were negative as was the urine toxicology screen. Urine protein to creatinine ratio was 0.07. His hemoglobin rose to 7.9 g/dL with transfusion of 4 units packed red blood cells (RBC). His chest pain improved and inferior ST depressions resolved on follow-up ECG. Further history revealed multiple episodes of melena and hematochezia in the preceding weeks without nausea, vomiting, or abdominal pain.
The patient has a strikingly large gamma gap: 9.2. A gap larger than 4 is concerning for the presence of paraproteins. Given the possibility of a paraproteinemia (eg, multiple myeloma, plasmacytoma, Waldenström macroglobulinemia), the first step is to check serum and urine protein electrophoresis. The patient’s anemia is significant and reticulocyte index low. The low ferritin level combined with the inappropriately low reticulocyte count could result from iron deficiency anemia, another bone marrow process, or both. The patient’s syncope likely resulted from severe anemia and hypovolemia associated with hematochezia. The prolonged prothrombin time could be caused by a coagulation factor production problem, from vitamin K deficiency or underlying liver disease, or by a consumptive problem, from low-grade disseminated intravascular coagulation. It is controversial whether inhaling welding fumes causes cancer, but the patient’s age alone makes malignancy a definite possibility.
Aortic stenosis is intriguing in light of the patient’s painless melena and hematochezia. Heyde syndrome is a phenomenon in which high shear stress causes a reduction in the size of von Willebrand factor predisposing to bleeding from submucosal angiodysplasia. However, Heyde syndrome has been reported to occur in the setting of severe or critical aortic stenosis and is unlikely to be the cause here. The patient needs to be examined with both upper and lower endoscopy to rule out gastrointestinal (GI) malignancy, gastric and esophageal varices, and painless peptic ulcers. High right ventricular systolic blood pressures along with echodense material in the inferior vena cava and right atrium suggest the possibility of malignancy with vascular invasion. Tricuspid regurgitation is consistent with high right-sided pressures. As left ventricular ejection fraction is reduced, some of the high right-sided pressures could also be attributable to left heart failure. CT findings of rectal wall thickening and perirectal lymph nodes could be attributable to cancer with locally metastatic disease. Blood loss caused by this cancer would explain the severe anemia on admission as well as the low ferritin level and the iron deficiency anemia. In this 70-year-old man who has not had routine health care maintenance, the leading diagnosis is colorectal cancer. However, the markedly elevated globulin gap and elevated INR strongly suggest another process (eg, multiple myeloma, other paraproteinemia) is also present.
INR remained elevated (1.5) despite vitamin K supplementation. Peripheral blood smear showed hypochromic and normocytic RBCs with moderate rouleaux formation (Figure 1). Intravenous pantoprazole and octreotide were started. Upper and lower endoscopy revealed multiple esophageal erosions and both a polypoid mass and an ulcer within the rectum with evidence of prior bleeding. The mass was resected and the ulcer biopsied.
On hospital day 3, the patient was transfused another unit of packed RBCs. Hemoglobin level increased from 7.4 g/dL to 8.2 g/dL, but he began to complain of headache, blurred vision, and worsened chest pain. He did not have weakness, numbness, diplopia, dysphagia, or dysarthria. External examination and extraocular movements of both eyes were normal. Visual acuity was 20/25 bilaterally. Funduscopic examination revealed mild dilation of retinal veins and retinal hemorrhages (Figure 2).
Rouleaux formation occurs as excess cathodal proteins, such as immunoglobulins or fibrinogen, adhere to RBCs and cause the cells to stack together in long chains. Classically this is associated with multiple myeloma, but can occur with Waldenström’s macroglobulinemia and other cancers or infections. It can also occur as an artifact in smear preparation but the large globulin gap in this patient supports pathologic rouleaux formation.
Venous retinopathy with hemorrhages may occur with occlusion of the arterial supply (eg, as with carotid artery obstruction) but also with hyperviscosity syndrome (HVS). Vascular disturbances throughout the body play a major role in HVS, but these changes are most easily visualized in the retina. It is interesting that the patient’s headache and blurred vision began after he received additional blood transfusions. Spuriously low hemoglobin and hematocrit levels may stem from increased plasma volume from high immunoglobulin M (IgM) concentrations in Waldenström macroglobulinemia; thus, RBC transfusions can exacerbate symptoms by elevating total RBC mass. Normocytic, normochromic anemia is characteristic of both multiple myeloma and Waldenström’s macroglobulinemia. That the patient’s chest pain recurred coincidentally with blurred vision and headache suggests the likely cause is cardiac ischemia from hyperviscosity. The serum viscosity level should be checked, and, if it is elevated, urgent serum plasmapheresis should be considered. Determining the source of excess globulin production and treating the underlying disease are crucial at this juncture.
In the general population, rectal adenocarcinoma is the most common cause of a rectal mass. In this patient, presence of a paraproteinemia may point to a different diagnosis. Extramedullary colorectal plasmacytoma can occur in the rectum but is exceedingly rare. Waldenström’s macroglobulinemia, a subtype of lymphoplasmacytic lymphoma, can be associated with a rectal lymphoma. At this point, it is not possible to confidently predict the etiology of the mass.
He was started on cyclophosphamide, bortezomib and dexamethasone for IgG κ myeloma with improvement in his headache, blurred vision, chest pain, and plasma viscosity (4 to 1.8). His hemoglobin remained stable at 10 g/dL. Neoadjuvant Capecitabine and radiation therapy were initiated for his rectal cancer.
DISCUSSION
Multiple myeloma is characterized by monoclonal proliferation of plasma cells, elevated circulating monoclonal immunoglobulin, and end-organ damage.1 It accounts for approximately 0.8% of all new cancer diagnoses; average age at onset is 70 years. The patient described here had an unusual presentation, with GI bleeding and progression to HVS, and known risk factors for multiple myeloma (male sex, low socioeconomic status, welding career).2,3
An early clue in the diagnosis was the patient’s large gamma gap and concurrent anemia. Gamma gap, calculated by subtracting serum albumin from serum total protein, is so named because it often reflects an elevated gamma globulin concentration. However, it actually reflects all nonalbumin serum protein. A gamma gap larger than 3.1 g/dL is an independent risk factor for death4 and may be associated with infection, autoimmunity, and malignancy. Although there are no screening guidelines for multiple myeloma, 73% of cases are brought to attention by anemia discovered on routine laboratory investigation.5 This patient’s lack of prior medical care likely contributed to his atypical presentation. Screening colonoscopy, recommended at age 50, might have identified his rectal cancer at an earlier stage.
The patient’s anemia was likely secondary to GI hemorrhage and bone marrow suppression. His hematochezia might have been partly related to the pathophysiologic interaction of paraproteins with platelets, coagulation factors, and blood vessels.6 Amyloidosis of the GI tract is seen in 8% of AL amyloidosis7 and most frequently manifests as gastrointestinal bleeding, which is thought to be due to ischemia, vascular friability, or mucosal lesions. It less commonly presents as malabsorption or dysmotility.8 Although gastrointestinal amyloid is not typically associated with radiologic abnormalities, occasionally it may cause luminal wall thickening, adenopathy, and inflammatory stranding.9 The gold standard for diagnosis is tissue biopsy. However, presence of amyloidosis does not change the overall treatment strategy for multiple myeloma.
An interesting feature of this case is the development of HVS, which typically manifests with mucosal bleeding, blurred vision, and headache.10 HVS can be diagnosed on retinal examination with findings of venous tortuosity, dilatation, and intraretinal hemorrhage, as occurred in this case,11 and is confirmed with serum viscosity measurement. The first evidence of HVS in this case might have been the spontaneous echo contrast, or “smoke,” detected on echocardiogram. Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates,12 and is associated with conditions that result in left atrial stasis, such as atrial arrhythmias and mitral stenosis. This patient did not have valvular pathology or arrhythmia, and thus the “smoke” likely reflected HVS.
Of the paraproteinemias, Waldenström’s macroglobulinemia is most often associated with HVS, likely because of the pentameric structure of IgM13 and the consequential large size that predisposes to vascular occlusion. Whereas HVS can occur with IgM levels as low as 3 g/dL, it typically does not occur with IgG concentrations under 15 g/dL. This patient presented with an IgG level of 8 g/dL and developed HVS symptoms only after multiple packed RBC transfusions. Elevated IgG level likely made him susceptible to HVS, which ultimately was precipitated by blood transfusion. Therefore, this patient’s initial chest pain most likely was caused by demand cardiac ischemia secondary to anemia, whereas his subsequent, posttransfusion chest pain likely resulted from hyperviscosity angina. Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—has been described in polycythemia and connective tissue disorders.14,15 To our knowledge, however, hyperviscosity angina has not been reported in patients with multiple myeloma. Treatment of hyperviscosity with end-organ damage typically consists of plasmapheresis, but this patient was started on urgent chemotherapy, and his symptoms improved. Untreated HVS can lead to end-organ ischemia and death.
This patient had a multitude of seemingly disparate symptoms and abnormalities that ultimately were united in a diagnosis of IgG κ multiple myeloma. Subsequently diagnosed rectal adenocarcinoma may have led to ongoing blood loss, which worsened the anemia, but had no evident relation to the primary diagnosis of multiple myeloma. This case exemplifies the fact that HVS is a rare but important iatrogenic complication of multiple myeloma treated with blood transfusion. As this patient’s hospital course progressed, the plot, and his blood, thickened.
KEY TEACHING POINTS
- Multiple myeloma is occasionally associated with HVS, which manifests with mucosal bleeding, blurred vision, and headache.
- Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—should be considered in patients with paraproteinemias and chest pain.
- Plasmapheresis reverses the clinical manifestations of HVS but not the underlying disease process (eg, Waldenström’s macroglobulinemia, multiple myeloma, leukemia, polycythemia).
- Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates, and is associated with left atrial stasis, commonly from atrial fibrillation or mitral stenosis, but might be present in HVS.
Acknowledgment
The authors thank Peter Campochiaro, MD, and Whitney Green, MD, for their contributions to the images used in this article.
Disclosure
Dr. Sedighi Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The other authors have nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
After losing consciousness at a supermarket, a 70-year-old man was brought to the emergency department by paramedics. He subsequently developed chest pain.
Syncope can be difficult to evaluate, but chest pain may help narrow an otherwise broad differential diagnosis. If this patient has aortic stenosis or hypertrophic cardiomyopathy, effort syncope is the culprit. Cardiac dysrhythmia (eg, ventricular tachycardia), complete heart block, and supraventricular tachycardia each can cause syncope along with chest pain. Myocardial infarction and associated ventricular arrhythmia might also explain both chest pain and syncope. The paramedics might have noted an arrhythmia on the cardiac monitor; if possible, the rhythm strip should be reviewed. A pulmonary embolus can cause chest pain and, if large enough to cause right ventricular compromise, syncope.
According to witnesses at the supermarket, the patient dropped to the ground, lost consciousness, and convulsed for 30 seconds. He had no head trauma, tongue biting, urinary incontinence, or confusion afterward. Electrocardiogram (ECG) performed at the scene showed ST elevations in leads V1 to V3 with ST depressions in the inferior leads. On arrival in the emergency department, the patient described nonradiating substernal chest pressure exacerbated by deep inhalation. The pain did not improve with nitroglycerin. He recalled feeling light-headed before the syncope.
He had not received medical care for 20 years and had no known illnesses other than hypertension. He was not taking any medications. He previously worked as a welder and never smoked tobacco, drank alcohol, or used illicit drugs. The patient’s temperature was 36.4°C. Heart rate was 88 beats per minute, blood pressure 128/72 mm Hg, oxygen saturation 100% on room air, and respiratory rate 22 breaths per minute. The patient had conjunctival pallor. There was a grade 3/6 crescendo- decrescendo systolic murmur loudest at the right upper sternal border without radiation to the carotids. There was no jugular venous distention. Lungs were clear to auscultation bilaterally. There was no peripheral edema, rash, or lymphadenopathy.
Convulsive movements commonly occur during episodes of unconsciousness lasting more than 15 seconds—a phenomenon termed convulsive syncope and often is confused with seizures. These movements are usually clonic jerks of the extremities and trunk and slight twitching of the face, and occasionally tonic extension of the trunk and clenching of the jaw. Absence of tongue biting, urinary incontinence, and confusion in this patient’s case makes seizures less likely.
The distribution of ST segment changes on his ECG are concerning for myocardial infarction in the septal and inferior regions. Right-sided ECG should be performed to assess for right ventricular infarction. Although myocardial ischemia is the primary concern, some features warrant consideration of other etiologies of syncope. First, syncope is an unusual presentation of cardiac ischemia or infarct. The complaint of chest pressure exacerbated by deep inhalation is another atypical feature for myocardial ischemia. Although the patient’s oxygen saturation and heart rate are normal, pulmonary embolism remains a possibility.
The prominent crescendo-decrescendo systolic murmur at the right upper sternal border could indicate aortic stenosis; the carotids should be palpated to assess for pulsus parvus et tardus. A high-flow state associated with anemia could also lead to a midsystolic murmur. Conjunctival pallor typically is seen with hemoglobin levels of 6 g/dL or less. This finding may indicate severe anemia, which has the potential to cause myocardial ischemia and syncope.
Laboratory testing revealed a troponin of 0.04 ng/dL, hemoglobin 4.1 g/dL with MCV of 84.7 fL, white blood cell count 6,500/μL and platelet count 179,000/μL. Serum sodium was 130 mEq/L, urea nitrogen 16 mg/dL, creatinine 1.6 mg/dL, calcium 7.8 mg/dL, total protein 11.4 g/dL (reference range, 6.0-8.2), and albumin 2.2 g/dL. Erythrocyte sedimentation rate (ESR) was 20 mm/h. Serum iron was 48 μg/mL, total iron binding capacity 275 μg/dL, percent iron saturation 17% (reference range, 20-55), and ferritin 10 ng/mL (reference range, 30-400). The international normalized ratio (INR) was 1.5, prothrombin time 15.5 sec (reference range, 9.4-11.6), and partial thromboplastin time 24.7 sec (reference range, 22.9-30.6). Hepatitis C and HIV antibodies were negative as was the urine toxicology screen. Urine protein to creatinine ratio was 0.07. His hemoglobin rose to 7.9 g/dL with transfusion of 4 units packed red blood cells (RBC). His chest pain improved and inferior ST depressions resolved on follow-up ECG. Further history revealed multiple episodes of melena and hematochezia in the preceding weeks without nausea, vomiting, or abdominal pain.
The patient has a strikingly large gamma gap: 9.2. A gap larger than 4 is concerning for the presence of paraproteins. Given the possibility of a paraproteinemia (eg, multiple myeloma, plasmacytoma, Waldenström macroglobulinemia), the first step is to check serum and urine protein electrophoresis. The patient’s anemia is significant and reticulocyte index low. The low ferritin level combined with the inappropriately low reticulocyte count could result from iron deficiency anemia, another bone marrow process, or both. The patient’s syncope likely resulted from severe anemia and hypovolemia associated with hematochezia. The prolonged prothrombin time could be caused by a coagulation factor production problem, from vitamin K deficiency or underlying liver disease, or by a consumptive problem, from low-grade disseminated intravascular coagulation. It is controversial whether inhaling welding fumes causes cancer, but the patient’s age alone makes malignancy a definite possibility.
Aortic stenosis is intriguing in light of the patient’s painless melena and hematochezia. Heyde syndrome is a phenomenon in which high shear stress causes a reduction in the size of von Willebrand factor predisposing to bleeding from submucosal angiodysplasia. However, Heyde syndrome has been reported to occur in the setting of severe or critical aortic stenosis and is unlikely to be the cause here. The patient needs to be examined with both upper and lower endoscopy to rule out gastrointestinal (GI) malignancy, gastric and esophageal varices, and painless peptic ulcers. High right ventricular systolic blood pressures along with echodense material in the inferior vena cava and right atrium suggest the possibility of malignancy with vascular invasion. Tricuspid regurgitation is consistent with high right-sided pressures. As left ventricular ejection fraction is reduced, some of the high right-sided pressures could also be attributable to left heart failure. CT findings of rectal wall thickening and perirectal lymph nodes could be attributable to cancer with locally metastatic disease. Blood loss caused by this cancer would explain the severe anemia on admission as well as the low ferritin level and the iron deficiency anemia. In this 70-year-old man who has not had routine health care maintenance, the leading diagnosis is colorectal cancer. However, the markedly elevated globulin gap and elevated INR strongly suggest another process (eg, multiple myeloma, other paraproteinemia) is also present.
INR remained elevated (1.5) despite vitamin K supplementation. Peripheral blood smear showed hypochromic and normocytic RBCs with moderate rouleaux formation (Figure 1). Intravenous pantoprazole and octreotide were started. Upper and lower endoscopy revealed multiple esophageal erosions and both a polypoid mass and an ulcer within the rectum with evidence of prior bleeding. The mass was resected and the ulcer biopsied.
On hospital day 3, the patient was transfused another unit of packed RBCs. Hemoglobin level increased from 7.4 g/dL to 8.2 g/dL, but he began to complain of headache, blurred vision, and worsened chest pain. He did not have weakness, numbness, diplopia, dysphagia, or dysarthria. External examination and extraocular movements of both eyes were normal. Visual acuity was 20/25 bilaterally. Funduscopic examination revealed mild dilation of retinal veins and retinal hemorrhages (Figure 2).
Rouleaux formation occurs as excess cathodal proteins, such as immunoglobulins or fibrinogen, adhere to RBCs and cause the cells to stack together in long chains. Classically this is associated with multiple myeloma, but can occur with Waldenström’s macroglobulinemia and other cancers or infections. It can also occur as an artifact in smear preparation but the large globulin gap in this patient supports pathologic rouleaux formation.
Venous retinopathy with hemorrhages may occur with occlusion of the arterial supply (eg, as with carotid artery obstruction) but also with hyperviscosity syndrome (HVS). Vascular disturbances throughout the body play a major role in HVS, but these changes are most easily visualized in the retina. It is interesting that the patient’s headache and blurred vision began after he received additional blood transfusions. Spuriously low hemoglobin and hematocrit levels may stem from increased plasma volume from high immunoglobulin M (IgM) concentrations in Waldenström macroglobulinemia; thus, RBC transfusions can exacerbate symptoms by elevating total RBC mass. Normocytic, normochromic anemia is characteristic of both multiple myeloma and Waldenström’s macroglobulinemia. That the patient’s chest pain recurred coincidentally with blurred vision and headache suggests the likely cause is cardiac ischemia from hyperviscosity. The serum viscosity level should be checked, and, if it is elevated, urgent serum plasmapheresis should be considered. Determining the source of excess globulin production and treating the underlying disease are crucial at this juncture.
In the general population, rectal adenocarcinoma is the most common cause of a rectal mass. In this patient, presence of a paraproteinemia may point to a different diagnosis. Extramedullary colorectal plasmacytoma can occur in the rectum but is exceedingly rare. Waldenström’s macroglobulinemia, a subtype of lymphoplasmacytic lymphoma, can be associated with a rectal lymphoma. At this point, it is not possible to confidently predict the etiology of the mass.
He was started on cyclophosphamide, bortezomib and dexamethasone for IgG κ myeloma with improvement in his headache, blurred vision, chest pain, and plasma viscosity (4 to 1.8). His hemoglobin remained stable at 10 g/dL. Neoadjuvant Capecitabine and radiation therapy were initiated for his rectal cancer.
DISCUSSION
Multiple myeloma is characterized by monoclonal proliferation of plasma cells, elevated circulating monoclonal immunoglobulin, and end-organ damage.1 It accounts for approximately 0.8% of all new cancer diagnoses; average age at onset is 70 years. The patient described here had an unusual presentation, with GI bleeding and progression to HVS, and known risk factors for multiple myeloma (male sex, low socioeconomic status, welding career).2,3
An early clue in the diagnosis was the patient’s large gamma gap and concurrent anemia. Gamma gap, calculated by subtracting serum albumin from serum total protein, is so named because it often reflects an elevated gamma globulin concentration. However, it actually reflects all nonalbumin serum protein. A gamma gap larger than 3.1 g/dL is an independent risk factor for death4 and may be associated with infection, autoimmunity, and malignancy. Although there are no screening guidelines for multiple myeloma, 73% of cases are brought to attention by anemia discovered on routine laboratory investigation.5 This patient’s lack of prior medical care likely contributed to his atypical presentation. Screening colonoscopy, recommended at age 50, might have identified his rectal cancer at an earlier stage.
The patient’s anemia was likely secondary to GI hemorrhage and bone marrow suppression. His hematochezia might have been partly related to the pathophysiologic interaction of paraproteins with platelets, coagulation factors, and blood vessels.6 Amyloidosis of the GI tract is seen in 8% of AL amyloidosis7 and most frequently manifests as gastrointestinal bleeding, which is thought to be due to ischemia, vascular friability, or mucosal lesions. It less commonly presents as malabsorption or dysmotility.8 Although gastrointestinal amyloid is not typically associated with radiologic abnormalities, occasionally it may cause luminal wall thickening, adenopathy, and inflammatory stranding.9 The gold standard for diagnosis is tissue biopsy. However, presence of amyloidosis does not change the overall treatment strategy for multiple myeloma.
An interesting feature of this case is the development of HVS, which typically manifests with mucosal bleeding, blurred vision, and headache.10 HVS can be diagnosed on retinal examination with findings of venous tortuosity, dilatation, and intraretinal hemorrhage, as occurred in this case,11 and is confirmed with serum viscosity measurement. The first evidence of HVS in this case might have been the spontaneous echo contrast, or “smoke,” detected on echocardiogram. Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates,12 and is associated with conditions that result in left atrial stasis, such as atrial arrhythmias and mitral stenosis. This patient did not have valvular pathology or arrhythmia, and thus the “smoke” likely reflected HVS.
Of the paraproteinemias, Waldenström’s macroglobulinemia is most often associated with HVS, likely because of the pentameric structure of IgM13 and the consequential large size that predisposes to vascular occlusion. Whereas HVS can occur with IgM levels as low as 3 g/dL, it typically does not occur with IgG concentrations under 15 g/dL. This patient presented with an IgG level of 8 g/dL and developed HVS symptoms only after multiple packed RBC transfusions. Elevated IgG level likely made him susceptible to HVS, which ultimately was precipitated by blood transfusion. Therefore, this patient’s initial chest pain most likely was caused by demand cardiac ischemia secondary to anemia, whereas his subsequent, posttransfusion chest pain likely resulted from hyperviscosity angina. Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—has been described in polycythemia and connective tissue disorders.14,15 To our knowledge, however, hyperviscosity angina has not been reported in patients with multiple myeloma. Treatment of hyperviscosity with end-organ damage typically consists of plasmapheresis, but this patient was started on urgent chemotherapy, and his symptoms improved. Untreated HVS can lead to end-organ ischemia and death.
This patient had a multitude of seemingly disparate symptoms and abnormalities that ultimately were united in a diagnosis of IgG κ multiple myeloma. Subsequently diagnosed rectal adenocarcinoma may have led to ongoing blood loss, which worsened the anemia, but had no evident relation to the primary diagnosis of multiple myeloma. This case exemplifies the fact that HVS is a rare but important iatrogenic complication of multiple myeloma treated with blood transfusion. As this patient’s hospital course progressed, the plot, and his blood, thickened.
KEY TEACHING POINTS
- Multiple myeloma is occasionally associated with HVS, which manifests with mucosal bleeding, blurred vision, and headache.
- Hyperviscosity angina—cardiac ischemia resulting from poor coronary perfusion caused by hyperviscous blood—should be considered in patients with paraproteinemias and chest pain.
- Plasmapheresis reverses the clinical manifestations of HVS but not the underlying disease process (eg, Waldenström’s macroglobulinemia, multiple myeloma, leukemia, polycythemia).
- Spontaneous echo contrast represents increased RBC aggregation, from interaction of RBCs and plasma proteins, at low shear rates, and is associated with left atrial stasis, commonly from atrial fibrillation or mitral stenosis, but might be present in HVS.
Acknowledgment
The authors thank Peter Campochiaro, MD, and Whitney Green, MD, for their contributions to the images used in this article.
Disclosure
Dr. Sedighi Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The other authors have nothing to report.
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046-1060. PubMed
2. Koessel SL, Theis MK, Vaughan TL, et al. Socioeconomic status and the incidence of multiple myeloma. Epidemiology. 1996;7(1):4-8. PubMed
3. Fritschi L, Siemiatycki J. Lymphoma, myeloma and occupation: results of a case-control study. Int J Cancer. 1996;67(4):498-503. PubMed
4. Juraschek SP, Moliterno AR, Checkley W, Miller ER 3rd. The gamma gap and all-cause mortality. PLoS One. 2015;10(12):e0143494. PubMed
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. PubMed
6. Eby CS. Bleeding and thrombosis risks in plasma cell dyscrasias. Hematology Am Soc Hematol Educ Program. 2007:158-164. PubMed
7. Menke DM, Kyle RA, Fleming CR, Wolfe JT 3rd, Kurtin PJ, Oldenburg WA. Symptomatic gastric amyloidosis in patients with primary systemic amyloidosis. Mayo Clin Proc. 1993;68(8):763-767. PubMed
8. Levy DJ, Franklin GO, Rosenthal WS. Gastrointestinal bleeding and amyloidosis. Am J Gastroenterol. 1982;77(6):422-426. PubMed
9. Araoz PA, Batts KP, MacCarty RL. Amyloidosis of the alimentary canal: radiologic-pathologic correlation of CT findings. Abdom Imaging. 2000;25(1):38-44. PubMed
10. Stone MJ, Bogen SA. Evidence-based focused review of management of hyperviscosity syndrome. Blood. 2012;119(10):2205-2208. PubMed
11. Rajagopal R, Apte RS. Seeing through thick and through thin: retinal manifestations of thrombophilic and hyperviscosity syndromes. Surv Ophthalmol. 2016;61(2):236-247. PubMed
12. Black IW. Spontaneous echo contrast: where there’s smoke there’s fire. Echocardiography. 2000;17(4):373-382. PubMed
13. Kwaan HC. Hyperviscosity in plasma cell dyscrasias. Clin Hemorheol Microcirc. 2013;55(1):75-83. PubMed
14. Piccirillo G, Fimognari FL, Valdivia JL, Marigliano V. Effects of phlebotomy on a patient with secondary polycythemia and angina pectoris. Int J Cardiol. 1994;44(2):175-177. PubMed
15. Ovadia S, Lysyy L, Floru S. Emergency plasmapheresis for unstable angina in a patient with hyperviscosity syndrome. Am J Emerg Med. 2005;23(6):811-812. PubMed
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046-1060. PubMed
2. Koessel SL, Theis MK, Vaughan TL, et al. Socioeconomic status and the incidence of multiple myeloma. Epidemiology. 1996;7(1):4-8. PubMed
3. Fritschi L, Siemiatycki J. Lymphoma, myeloma and occupation: results of a case-control study. Int J Cancer. 1996;67(4):498-503. PubMed
4. Juraschek SP, Moliterno AR, Checkley W, Miller ER 3rd. The gamma gap and all-cause mortality. PLoS One. 2015;10(12):e0143494. PubMed
5. Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21-33. PubMed
6. Eby CS. Bleeding and thrombosis risks in plasma cell dyscrasias. Hematology Am Soc Hematol Educ Program. 2007:158-164. PubMed
7. Menke DM, Kyle RA, Fleming CR, Wolfe JT 3rd, Kurtin PJ, Oldenburg WA. Symptomatic gastric amyloidosis in patients with primary systemic amyloidosis. Mayo Clin Proc. 1993;68(8):763-767. PubMed
8. Levy DJ, Franklin GO, Rosenthal WS. Gastrointestinal bleeding and amyloidosis. Am J Gastroenterol. 1982;77(6):422-426. PubMed
9. Araoz PA, Batts KP, MacCarty RL. Amyloidosis of the alimentary canal: radiologic-pathologic correlation of CT findings. Abdom Imaging. 2000;25(1):38-44. PubMed
10. Stone MJ, Bogen SA. Evidence-based focused review of management of hyperviscosity syndrome. Blood. 2012;119(10):2205-2208. PubMed
11. Rajagopal R, Apte RS. Seeing through thick and through thin: retinal manifestations of thrombophilic and hyperviscosity syndromes. Surv Ophthalmol. 2016;61(2):236-247. PubMed
12. Black IW. Spontaneous echo contrast: where there’s smoke there’s fire. Echocardiography. 2000;17(4):373-382. PubMed
13. Kwaan HC. Hyperviscosity in plasma cell dyscrasias. Clin Hemorheol Microcirc. 2013;55(1):75-83. PubMed
14. Piccirillo G, Fimognari FL, Valdivia JL, Marigliano V. Effects of phlebotomy on a patient with secondary polycythemia and angina pectoris. Int J Cardiol. 1994;44(2):175-177. PubMed
15. Ovadia S, Lysyy L, Floru S. Emergency plasmapheresis for unstable angina in a patient with hyperviscosity syndrome. Am J Emerg Med. 2005;23(6):811-812. PubMed
© 2017 Society of Hospital Medicine
The Missing Element
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.

Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
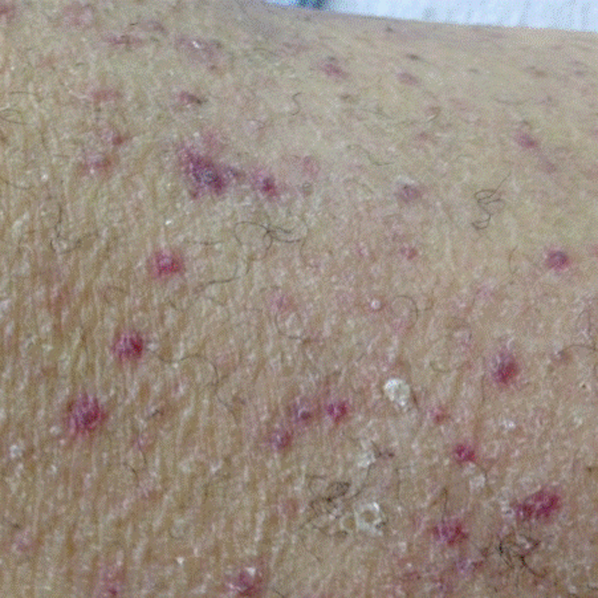
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.
Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.
Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
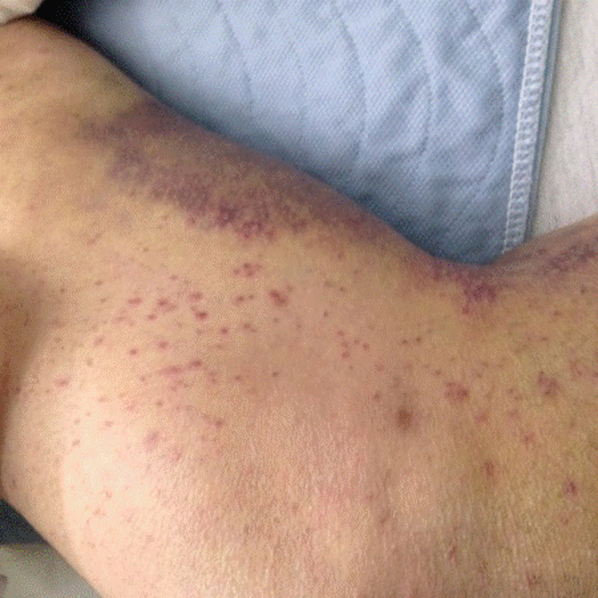
A Physician With Thigh Pain
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
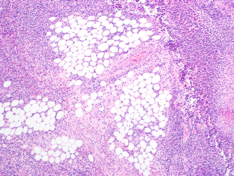
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
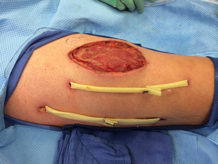
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
A coat with a clue
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
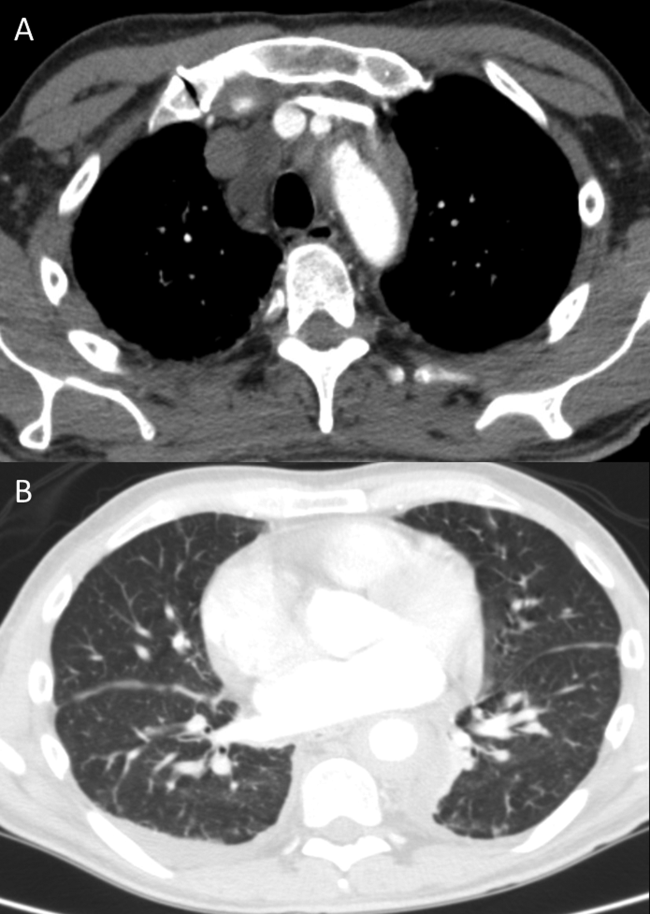
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.