User login
A 7-year-old with red bumps on her nose
The finding of individual, 1- to 4-mm firm, red papules depicted in the image are consistent with facial angiofibromas, which are most commonly seen in pediatric patients as a manifestation of tuberous sclerosis (TSC). Angiofibromas, previously called adenoma sebaceum, a misnomer, are seen in TSC as smooth papules, nodules, and occasionally plaques that typically involve the malar region of the face. These lesions usually develop in childhood and adolescence and can be misdiagnosed as lesions of acne. The number of lesions tend to increase with age, though there is no significant risk of malignant transformation. Ultraviolet-induced DNA damage is thought to play a role in the development of facial angiofibromas, so sun protection is called for.1 Patients may seek treatment to minimize deformity and the stigma of angiofibromas. Recently, the mammalian target of rapamycin inhibitor sirolimus (rapamycin) topical gel received Food and Drug Administration approval for the treatment of facial angiofibromas associated with TSC in patients age at least 6 years.2
The presence of angiofibromas should prompt consideration of TSC and as such, a thorough family history, medical history, and full-body skin examination. TSC is a rare autosomal-dominant genetic disorder, caused by a pathogenic variant in either the TSC1 or TSC2 gene. This neurocutaneous disorder is characterized by the development of multiple benign hamartomas across many organ systems including the brain, eyes, heart, lung, liver, kidney, and skin. The phenotypic expression of TSC is highly variable. Besides angiofibromas, some other characteristic dermatological findings in TSC include periungual fibromas, hypopigmented macules usually elliptical in shape (known as ash-leaf spots), and irregularly shaped elevated flesh-colored fibrous tissue most often found over the lower back (known as shagreen patches).3
What is on the differential?
Agminated spitz nevi refers to multiple spitz nevi in a localized area. Spitz nevi present as a well-circumscribed, dome-shaped, pink-red or brown papules, most commonly located on the face or lower extremities.4 The finding of agminated spitz nevi is very rare and less likely for this patient given the concomitant skin findings of dental pitting, renal cysts, and cortical tubers.

Juvenile xanthogranulomas are benign,proliferations of histiocytic cells that present as reddish or yellowish-to-brown papules, plaques, or nodules that typically develop in young children around the age of 1. With time, juvenile xanthogranulomas may flatten and become more yellow.
Basal cell carcinomas present as dome-shaped lesions with centralized erosions on sun-exposed areas of the skin. They are remarkably uncommon in children but are occasionally seen in basal cell nevus syndrome (also known as nevoid basal cell carcinoma syndrome or Gorlin syndrome). Affected patients may have other findings such as developmental anomalies, bifid ribs, palmar and plantar pitting, odontogenic keratocysts, and/or medulloblastomas.5
Flat warts commonly occur in children and occur by direct skin contact with human papillomavirus. Of the various types of warts, flat warts are smaller and tend to be smooth on top. The diagnosis of cutaneous warts is based on clinical appearance, showing thrombosed capillaries underneath the overlying hyperkeratotic debris. Our patient’s history of having a common wart on her hands raises suspicion for inoculation onto her face, but the morphology, distribution, and lack of response to tretinoin makes this diagnosis much less likely.
Disease workup and course
Our patient’s physical exam revealed dental pits but no evidence of hypopigmented macules, shagreen patches, or periungual lesions. Ultrasound of the kidney displayed renal cortical cysts and brain MRI showed cortical tubers, confirming extracutaneous TSC involvement. Over time, our patient developed angiofibromas on the forehead and was ultimately started on topical sirolimus, which led to marked improvement within months.
Ms. Kleinman is a pediatric dermatology research associate, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, also in San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. They have no relevant financial disclosures.
References
1. Tyburczy ME et al. Hum Molec Genet. 2014;23(8):2023-9.
2. Food & Drug Administration. New drug application (NDA) approval for Hyftor (sirolimus topical gel). https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/213478Orig1s000ltr.pdf.
3. Webb DW et al. Br J Dermatol. 1996;135(1):1-5.
4. Ricci F et al. Eur J Dermatol. 2017;27(1):59-62.
5. Evans DG and Farndon PA. Nevoid basal cell carcinoma syndrome, in “GeneReviews®.” Seattle: University of Washington, 2002.
The finding of individual, 1- to 4-mm firm, red papules depicted in the image are consistent with facial angiofibromas, which are most commonly seen in pediatric patients as a manifestation of tuberous sclerosis (TSC). Angiofibromas, previously called adenoma sebaceum, a misnomer, are seen in TSC as smooth papules, nodules, and occasionally plaques that typically involve the malar region of the face. These lesions usually develop in childhood and adolescence and can be misdiagnosed as lesions of acne. The number of lesions tend to increase with age, though there is no significant risk of malignant transformation. Ultraviolet-induced DNA damage is thought to play a role in the development of facial angiofibromas, so sun protection is called for.1 Patients may seek treatment to minimize deformity and the stigma of angiofibromas. Recently, the mammalian target of rapamycin inhibitor sirolimus (rapamycin) topical gel received Food and Drug Administration approval for the treatment of facial angiofibromas associated with TSC in patients age at least 6 years.2
The presence of angiofibromas should prompt consideration of TSC and as such, a thorough family history, medical history, and full-body skin examination. TSC is a rare autosomal-dominant genetic disorder, caused by a pathogenic variant in either the TSC1 or TSC2 gene. This neurocutaneous disorder is characterized by the development of multiple benign hamartomas across many organ systems including the brain, eyes, heart, lung, liver, kidney, and skin. The phenotypic expression of TSC is highly variable. Besides angiofibromas, some other characteristic dermatological findings in TSC include periungual fibromas, hypopigmented macules usually elliptical in shape (known as ash-leaf spots), and irregularly shaped elevated flesh-colored fibrous tissue most often found over the lower back (known as shagreen patches).3
What is on the differential?
Agminated spitz nevi refers to multiple spitz nevi in a localized area. Spitz nevi present as a well-circumscribed, dome-shaped, pink-red or brown papules, most commonly located on the face or lower extremities.4 The finding of agminated spitz nevi is very rare and less likely for this patient given the concomitant skin findings of dental pitting, renal cysts, and cortical tubers.
Juvenile xanthogranulomas are benign,proliferations of histiocytic cells that present as reddish or yellowish-to-brown papules, plaques, or nodules that typically develop in young children around the age of 1. With time, juvenile xanthogranulomas may flatten and become more yellow.
Basal cell carcinomas present as dome-shaped lesions with centralized erosions on sun-exposed areas of the skin. They are remarkably uncommon in children but are occasionally seen in basal cell nevus syndrome (also known as nevoid basal cell carcinoma syndrome or Gorlin syndrome). Affected patients may have other findings such as developmental anomalies, bifid ribs, palmar and plantar pitting, odontogenic keratocysts, and/or medulloblastomas.5
Flat warts commonly occur in children and occur by direct skin contact with human papillomavirus. Of the various types of warts, flat warts are smaller and tend to be smooth on top. The diagnosis of cutaneous warts is based on clinical appearance, showing thrombosed capillaries underneath the overlying hyperkeratotic debris. Our patient’s history of having a common wart on her hands raises suspicion for inoculation onto her face, but the morphology, distribution, and lack of response to tretinoin makes this diagnosis much less likely.
Disease workup and course
Our patient’s physical exam revealed dental pits but no evidence of hypopigmented macules, shagreen patches, or periungual lesions. Ultrasound of the kidney displayed renal cortical cysts and brain MRI showed cortical tubers, confirming extracutaneous TSC involvement. Over time, our patient developed angiofibromas on the forehead and was ultimately started on topical sirolimus, which led to marked improvement within months.
Ms. Kleinman is a pediatric dermatology research associate, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, also in San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. They have no relevant financial disclosures.
References
1. Tyburczy ME et al. Hum Molec Genet. 2014;23(8):2023-9.
2. Food & Drug Administration. New drug application (NDA) approval for Hyftor (sirolimus topical gel). https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/213478Orig1s000ltr.pdf.
3. Webb DW et al. Br J Dermatol. 1996;135(1):1-5.
4. Ricci F et al. Eur J Dermatol. 2017;27(1):59-62.
5. Evans DG and Farndon PA. Nevoid basal cell carcinoma syndrome, in “GeneReviews®.” Seattle: University of Washington, 2002.
The finding of individual, 1- to 4-mm firm, red papules depicted in the image are consistent with facial angiofibromas, which are most commonly seen in pediatric patients as a manifestation of tuberous sclerosis (TSC). Angiofibromas, previously called adenoma sebaceum, a misnomer, are seen in TSC as smooth papules, nodules, and occasionally plaques that typically involve the malar region of the face. These lesions usually develop in childhood and adolescence and can be misdiagnosed as lesions of acne. The number of lesions tend to increase with age, though there is no significant risk of malignant transformation. Ultraviolet-induced DNA damage is thought to play a role in the development of facial angiofibromas, so sun protection is called for.1 Patients may seek treatment to minimize deformity and the stigma of angiofibromas. Recently, the mammalian target of rapamycin inhibitor sirolimus (rapamycin) topical gel received Food and Drug Administration approval for the treatment of facial angiofibromas associated with TSC in patients age at least 6 years.2
The presence of angiofibromas should prompt consideration of TSC and as such, a thorough family history, medical history, and full-body skin examination. TSC is a rare autosomal-dominant genetic disorder, caused by a pathogenic variant in either the TSC1 or TSC2 gene. This neurocutaneous disorder is characterized by the development of multiple benign hamartomas across many organ systems including the brain, eyes, heart, lung, liver, kidney, and skin. The phenotypic expression of TSC is highly variable. Besides angiofibromas, some other characteristic dermatological findings in TSC include periungual fibromas, hypopigmented macules usually elliptical in shape (known as ash-leaf spots), and irregularly shaped elevated flesh-colored fibrous tissue most often found over the lower back (known as shagreen patches).3
What is on the differential?
Agminated spitz nevi refers to multiple spitz nevi in a localized area. Spitz nevi present as a well-circumscribed, dome-shaped, pink-red or brown papules, most commonly located on the face or lower extremities.4 The finding of agminated spitz nevi is very rare and less likely for this patient given the concomitant skin findings of dental pitting, renal cysts, and cortical tubers.
Juvenile xanthogranulomas are benign,proliferations of histiocytic cells that present as reddish or yellowish-to-brown papules, plaques, or nodules that typically develop in young children around the age of 1. With time, juvenile xanthogranulomas may flatten and become more yellow.
Basal cell carcinomas present as dome-shaped lesions with centralized erosions on sun-exposed areas of the skin. They are remarkably uncommon in children but are occasionally seen in basal cell nevus syndrome (also known as nevoid basal cell carcinoma syndrome or Gorlin syndrome). Affected patients may have other findings such as developmental anomalies, bifid ribs, palmar and plantar pitting, odontogenic keratocysts, and/or medulloblastomas.5
Flat warts commonly occur in children and occur by direct skin contact with human papillomavirus. Of the various types of warts, flat warts are smaller and tend to be smooth on top. The diagnosis of cutaneous warts is based on clinical appearance, showing thrombosed capillaries underneath the overlying hyperkeratotic debris. Our patient’s history of having a common wart on her hands raises suspicion for inoculation onto her face, but the morphology, distribution, and lack of response to tretinoin makes this diagnosis much less likely.
Disease workup and course
Our patient’s physical exam revealed dental pits but no evidence of hypopigmented macules, shagreen patches, or periungual lesions. Ultrasound of the kidney displayed renal cortical cysts and brain MRI showed cortical tubers, confirming extracutaneous TSC involvement. Over time, our patient developed angiofibromas on the forehead and was ultimately started on topical sirolimus, which led to marked improvement within months.
Ms. Kleinman is a pediatric dermatology research associate, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, also in San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. They have no relevant financial disclosures.
References
1. Tyburczy ME et al. Hum Molec Genet. 2014;23(8):2023-9.
2. Food & Drug Administration. New drug application (NDA) approval for Hyftor (sirolimus topical gel). https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/213478Orig1s000ltr.pdf.
3. Webb DW et al. Br J Dermatol. 1996;135(1):1-5.
4. Ricci F et al. Eur J Dermatol. 2017;27(1):59-62.
5. Evans DG and Farndon PA. Nevoid basal cell carcinoma syndrome, in “GeneReviews®.” Seattle: University of Washington, 2002.
A 7-year-old female presented with a bump on the bridge of her nose that was present for 10 months, with subsequent development of multiple papules on the nose and cheeks.
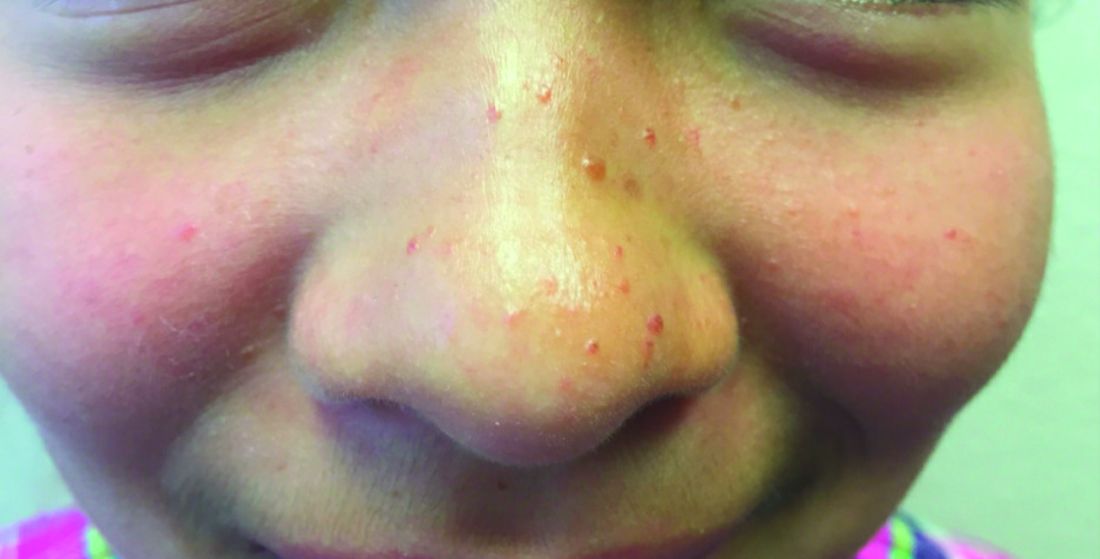
A 7-year-old, previously healthy female presented with a bump on the bridge of her nose that was present for 10 months, with subsequent development of multiple papules on the nose and cheeks. She has no significant medical history aside from a wart on her hand that was recently frozen with liquid nitrogen and resolved. She denied pruritus, bumps, or skin changes elsewhere on the body. The patient was prescribed tretinoin 0.1% cream applied nightly for several months without response.
A 19-month-old vaccinated female with 2 days of rash
Acute hemorrhagic edema of infancy (AHEI) is a leukocytoclastic vasculitis that typically affects children between 4 months and 2 years of age.1 Etiology is unknown but the majority of cases are preceded by infections, vaccinations, or certain medications.2
AHEI is a self-limited disease that runs a benign course with spontaneous resolution within days to 3 weeks.3 Classic presentation involves acute onset of fever, purpura, ecchymosis, and inflammatory edema. Edema is often the first sign, and may involve the face, ears, scrotum, or extremities. Hemorrhagic lesions may vary in size but often coalesce and present in a distinctive “cockade” or rosette pattern with scalloped borders. Systemic manifestations are rare, but renal and joint involvement may occur.4 Despite the dramatic and sometimes extensive appearance of the dermatologic manifestations, patients with AHEI are usually not in significant distress.
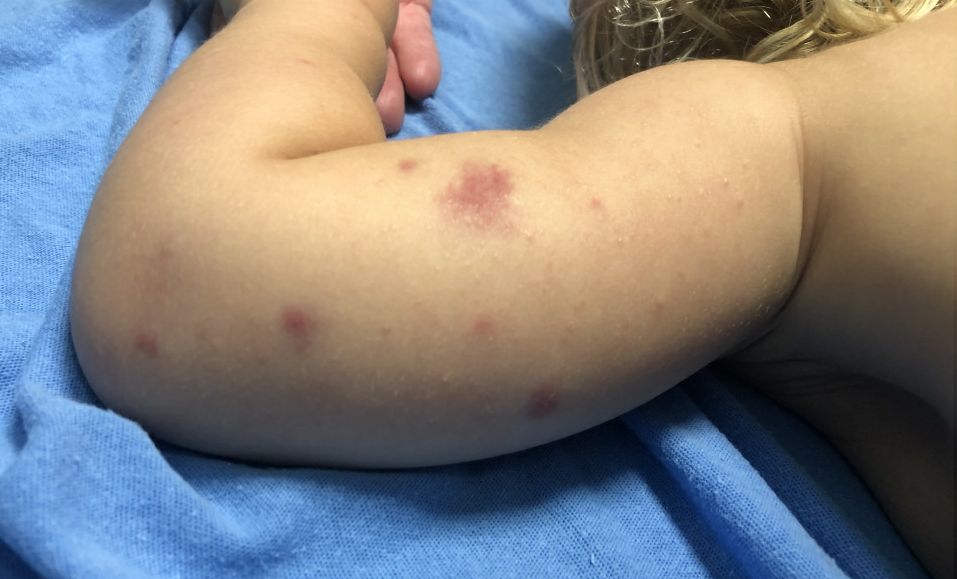
Diagnosis is clinical, but skin biopsy may show leukocytoclastic vasculitis of the superficial small vessels with infiltrations of neutrophils, extravasation of red blood cells, and fibrinoid necrosis.5 In most cases, immunofluorescence is negative for perivascular IgA deposition. Treatment is symptomatic as the disease resolves spontaneously. Recurrence is uncommon but may occur, and usually occurs early.

What is on the differential?
Kawasaki disease. Similar to AHEI, patients with Kawasaki disease also may present with facial and extremity edema. However, patients with Kawasaki disease appear sicker, have associated lymphadenopathy, conjunctivitis, and fever longer than 5 days. The lack of elevated inflammatory markers, acute-onset, classic dermatologic lesions, and nontoxic appearance in our patient rule out Kawasaki disease and make AHEI more likely.
IgA vasculitis/Henoch-Schönlein purpura. The distinction between AHEI and Henoch-Schönlein purpura is among the most challenging. AHEI commonly afflicts younger children ranging from 4 months to 2 years, whereas Henoch-Schönlein purpura occurs in older children from 3 to 6 years of age. Visceral involvement is rare in AHEI, but frequently presents in Henoch-Schönlein purpura with gastrointestinal and renal complications. Although our patient had both mild renal involvement and a distribution primarily on the buttocks and lower limbs, similar to the classic distribution of Henoch-Schönlein purpura, the younger age and lack of gastrointestinal and arthritic manifestations make AHEI more likely.
Gianotti-Crosti syndrome. Gianotti-Crosti syndrome, also known as papulovesicular acrodermatitis of childhood, mainly affects children between the ages of 6 months and 12 years. Like AHEI, Gianotti-Crosti is a self-limiting condition likely triggered by viral infection or immunization. However, Gianotti-Crosti is characterized by a papular rash that may last for several weeks. Neither AHEI nor Gianotti-Crosti are pruritic, but patients with Gianotti-Crosti tend to have either inguinal or axillary lymphadenopathy. Our patient’s large, coalescing dusky red patches and edematous plaques without lymphadenopathy are more consistent with AHEI.
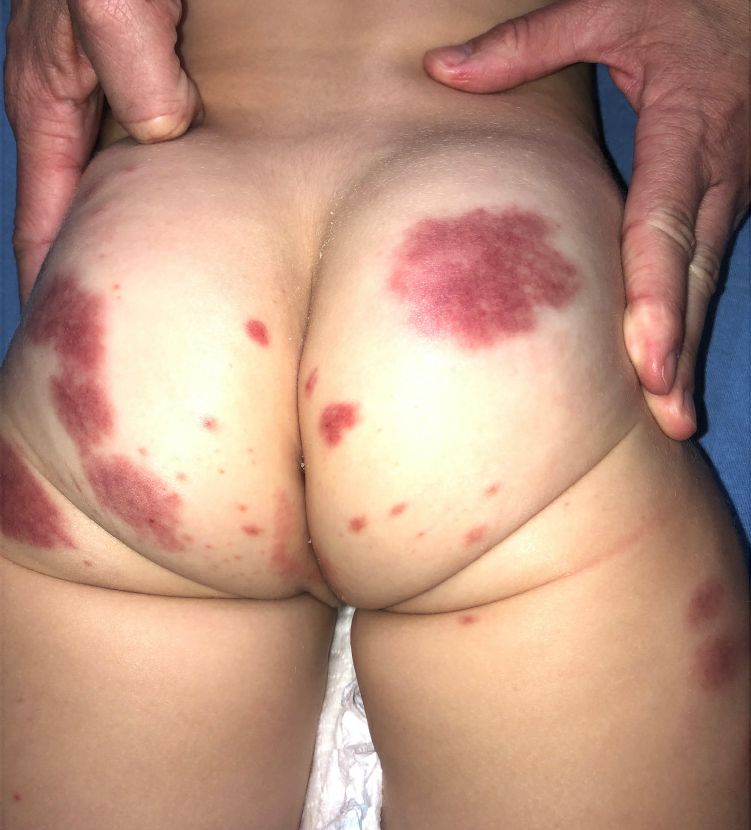
Erythema multiforme. Erythema multiforme is an acute, immune-mediated condition characterized by distinctive target-like lesions on the skin often accompanied by erosions or bullae. Unlike AHEI, erythema multiforme can involve the oral, genital, and/or ocular mucosae. Erythema multiforme is rare before the age of 4 years. Although the targetoid or annular purpuric configuration of erythema multiforme may present similarly to AHEI in some cases, the young age of our patient and the lack of mucosal involvement make AHEI more likely.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Kleinman is a pediatric dermatology research associate at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Dr. Matiz nor Ms. Kleinman has any relevant financial disclosures.
References
1. Savino F et al. Pediatr Dermatol. 2013;30(6):e149-e152.
2. Carboni E et al. F1000Res. 2019;8:1771. 2019 Oct 17.
3. Fiore E et al. J Am Acad Dermatol. 2008;59(4):684-95.
4. Watanabe T and Sato Y. Pediatr Nephrol. 2007;22(11):1979-81.
5. Cunha DF et al. Autops Case Rep. 2015;5(3):37-41.
Acute hemorrhagic edema of infancy (AHEI) is a leukocytoclastic vasculitis that typically affects children between 4 months and 2 years of age.1 Etiology is unknown but the majority of cases are preceded by infections, vaccinations, or certain medications.2
AHEI is a self-limited disease that runs a benign course with spontaneous resolution within days to 3 weeks.3 Classic presentation involves acute onset of fever, purpura, ecchymosis, and inflammatory edema. Edema is often the first sign, and may involve the face, ears, scrotum, or extremities. Hemorrhagic lesions may vary in size but often coalesce and present in a distinctive “cockade” or rosette pattern with scalloped borders. Systemic manifestations are rare, but renal and joint involvement may occur.4 Despite the dramatic and sometimes extensive appearance of the dermatologic manifestations, patients with AHEI are usually not in significant distress.
Diagnosis is clinical, but skin biopsy may show leukocytoclastic vasculitis of the superficial small vessels with infiltrations of neutrophils, extravasation of red blood cells, and fibrinoid necrosis.5 In most cases, immunofluorescence is negative for perivascular IgA deposition. Treatment is symptomatic as the disease resolves spontaneously. Recurrence is uncommon but may occur, and usually occurs early.
What is on the differential?
Kawasaki disease. Similar to AHEI, patients with Kawasaki disease also may present with facial and extremity edema. However, patients with Kawasaki disease appear sicker, have associated lymphadenopathy, conjunctivitis, and fever longer than 5 days. The lack of elevated inflammatory markers, acute-onset, classic dermatologic lesions, and nontoxic appearance in our patient rule out Kawasaki disease and make AHEI more likely.
IgA vasculitis/Henoch-Schönlein purpura. The distinction between AHEI and Henoch-Schönlein purpura is among the most challenging. AHEI commonly afflicts younger children ranging from 4 months to 2 years, whereas Henoch-Schönlein purpura occurs in older children from 3 to 6 years of age. Visceral involvement is rare in AHEI, but frequently presents in Henoch-Schönlein purpura with gastrointestinal and renal complications. Although our patient had both mild renal involvement and a distribution primarily on the buttocks and lower limbs, similar to the classic distribution of Henoch-Schönlein purpura, the younger age and lack of gastrointestinal and arthritic manifestations make AHEI more likely.
Gianotti-Crosti syndrome. Gianotti-Crosti syndrome, also known as papulovesicular acrodermatitis of childhood, mainly affects children between the ages of 6 months and 12 years. Like AHEI, Gianotti-Crosti is a self-limiting condition likely triggered by viral infection or immunization. However, Gianotti-Crosti is characterized by a papular rash that may last for several weeks. Neither AHEI nor Gianotti-Crosti are pruritic, but patients with Gianotti-Crosti tend to have either inguinal or axillary lymphadenopathy. Our patient’s large, coalescing dusky red patches and edematous plaques without lymphadenopathy are more consistent with AHEI.
Erythema multiforme. Erythema multiforme is an acute, immune-mediated condition characterized by distinctive target-like lesions on the skin often accompanied by erosions or bullae. Unlike AHEI, erythema multiforme can involve the oral, genital, and/or ocular mucosae. Erythema multiforme is rare before the age of 4 years. Although the targetoid or annular purpuric configuration of erythema multiforme may present similarly to AHEI in some cases, the young age of our patient and the lack of mucosal involvement make AHEI more likely.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Kleinman is a pediatric dermatology research associate at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Dr. Matiz nor Ms. Kleinman has any relevant financial disclosures.
References
1. Savino F et al. Pediatr Dermatol. 2013;30(6):e149-e152.
2. Carboni E et al. F1000Res. 2019;8:1771. 2019 Oct 17.
3. Fiore E et al. J Am Acad Dermatol. 2008;59(4):684-95.
4. Watanabe T and Sato Y. Pediatr Nephrol. 2007;22(11):1979-81.
5. Cunha DF et al. Autops Case Rep. 2015;5(3):37-41.
Acute hemorrhagic edema of infancy (AHEI) is a leukocytoclastic vasculitis that typically affects children between 4 months and 2 years of age.1 Etiology is unknown but the majority of cases are preceded by infections, vaccinations, or certain medications.2
AHEI is a self-limited disease that runs a benign course with spontaneous resolution within days to 3 weeks.3 Classic presentation involves acute onset of fever, purpura, ecchymosis, and inflammatory edema. Edema is often the first sign, and may involve the face, ears, scrotum, or extremities. Hemorrhagic lesions may vary in size but often coalesce and present in a distinctive “cockade” or rosette pattern with scalloped borders. Systemic manifestations are rare, but renal and joint involvement may occur.4 Despite the dramatic and sometimes extensive appearance of the dermatologic manifestations, patients with AHEI are usually not in significant distress.
Diagnosis is clinical, but skin biopsy may show leukocytoclastic vasculitis of the superficial small vessels with infiltrations of neutrophils, extravasation of red blood cells, and fibrinoid necrosis.5 In most cases, immunofluorescence is negative for perivascular IgA deposition. Treatment is symptomatic as the disease resolves spontaneously. Recurrence is uncommon but may occur, and usually occurs early.
What is on the differential?
Kawasaki disease. Similar to AHEI, patients with Kawasaki disease also may present with facial and extremity edema. However, patients with Kawasaki disease appear sicker, have associated lymphadenopathy, conjunctivitis, and fever longer than 5 days. The lack of elevated inflammatory markers, acute-onset, classic dermatologic lesions, and nontoxic appearance in our patient rule out Kawasaki disease and make AHEI more likely.
IgA vasculitis/Henoch-Schönlein purpura. The distinction between AHEI and Henoch-Schönlein purpura is among the most challenging. AHEI commonly afflicts younger children ranging from 4 months to 2 years, whereas Henoch-Schönlein purpura occurs in older children from 3 to 6 years of age. Visceral involvement is rare in AHEI, but frequently presents in Henoch-Schönlein purpura with gastrointestinal and renal complications. Although our patient had both mild renal involvement and a distribution primarily on the buttocks and lower limbs, similar to the classic distribution of Henoch-Schönlein purpura, the younger age and lack of gastrointestinal and arthritic manifestations make AHEI more likely.
Gianotti-Crosti syndrome. Gianotti-Crosti syndrome, also known as papulovesicular acrodermatitis of childhood, mainly affects children between the ages of 6 months and 12 years. Like AHEI, Gianotti-Crosti is a self-limiting condition likely triggered by viral infection or immunization. However, Gianotti-Crosti is characterized by a papular rash that may last for several weeks. Neither AHEI nor Gianotti-Crosti are pruritic, but patients with Gianotti-Crosti tend to have either inguinal or axillary lymphadenopathy. Our patient’s large, coalescing dusky red patches and edematous plaques without lymphadenopathy are more consistent with AHEI.
Erythema multiforme. Erythema multiforme is an acute, immune-mediated condition characterized by distinctive target-like lesions on the skin often accompanied by erosions or bullae. Unlike AHEI, erythema multiforme can involve the oral, genital, and/or ocular mucosae. Erythema multiforme is rare before the age of 4 years. Although the targetoid or annular purpuric configuration of erythema multiforme may present similarly to AHEI in some cases, the young age of our patient and the lack of mucosal involvement make AHEI more likely.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Kleinman is a pediatric dermatology research associate at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Dr. Matiz nor Ms. Kleinman has any relevant financial disclosures.
References
1. Savino F et al. Pediatr Dermatol. 2013;30(6):e149-e152.
2. Carboni E et al. F1000Res. 2019;8:1771. 2019 Oct 17.
3. Fiore E et al. J Am Acad Dermatol. 2008;59(4):684-95.
4. Watanabe T and Sato Y. Pediatr Nephrol. 2007;22(11):1979-81.
5. Cunha DF et al. Autops Case Rep. 2015;5(3):37-41.
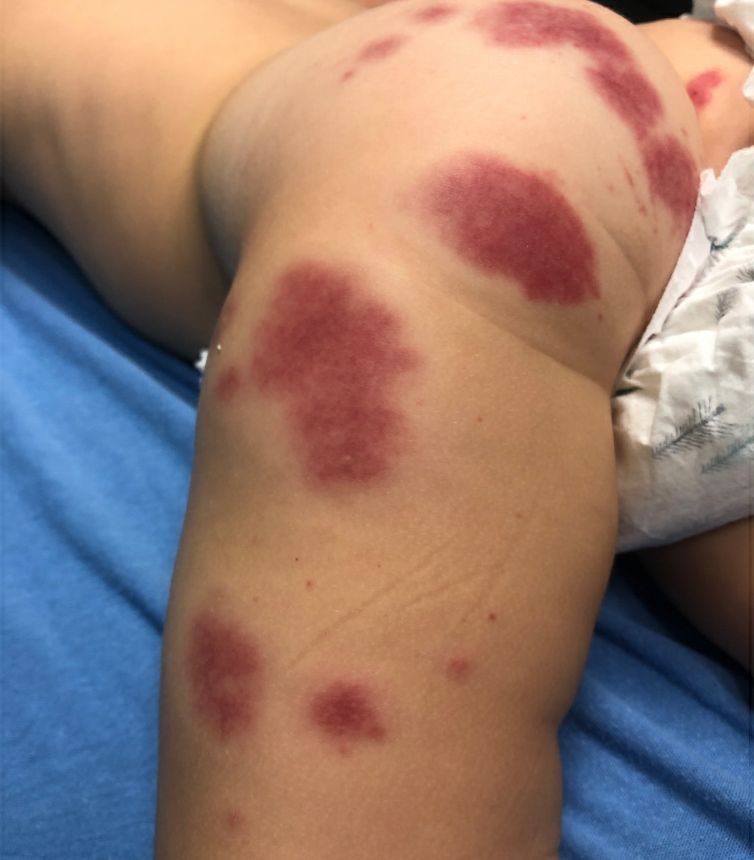
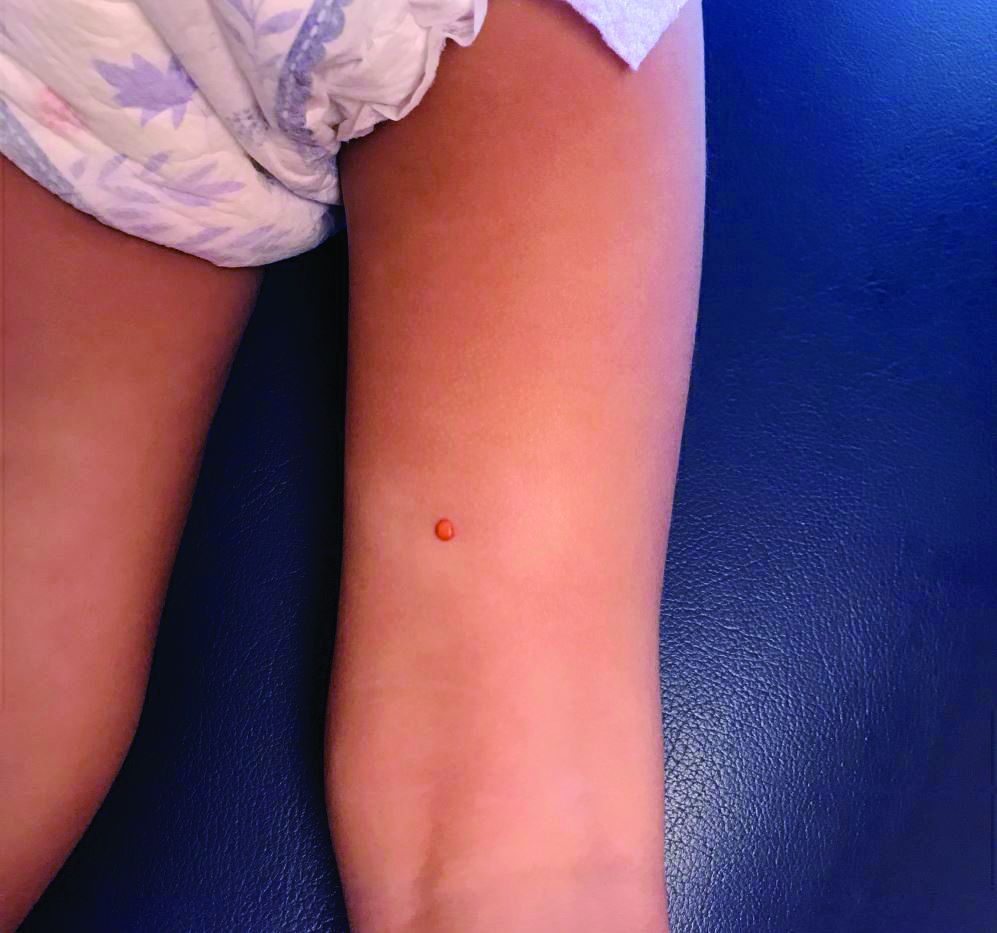
A female toddler presents with an itchy yellow nodule
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
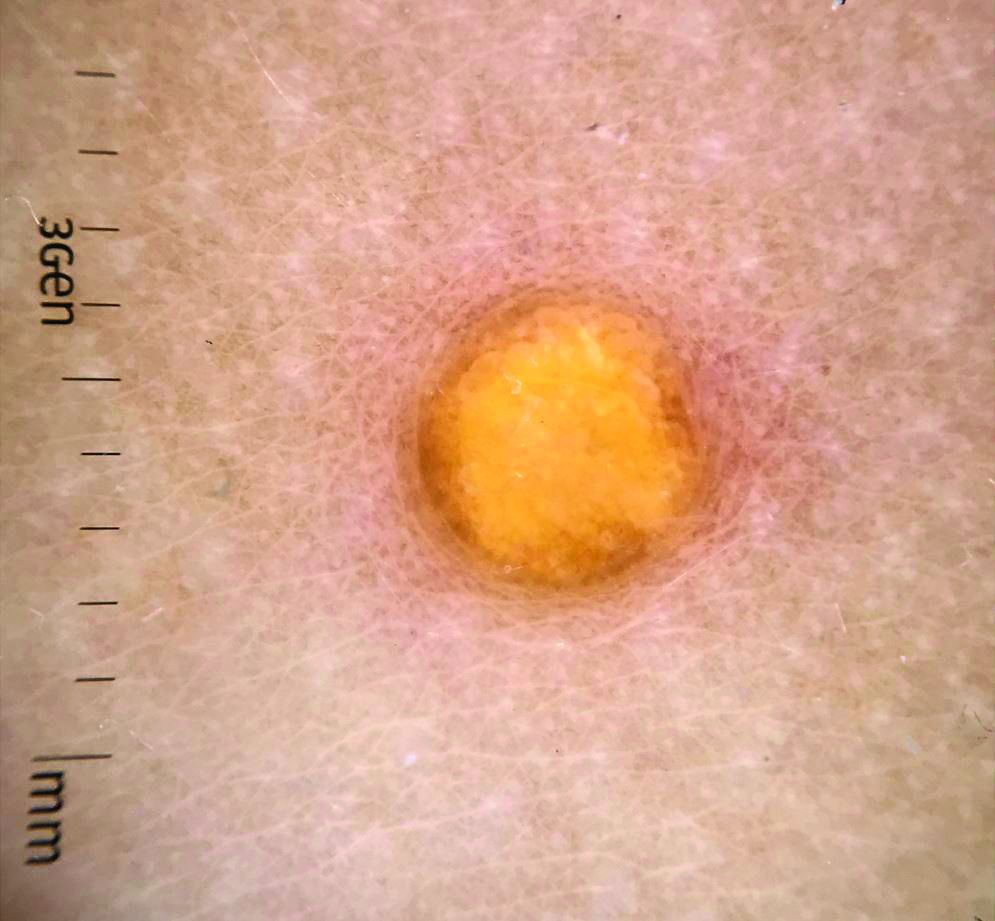
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.
Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.
Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.
Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.