User login
Sleep disorders in older adults
As humans live longer, a renewed focus on quality of life has made the prompt diagnosis and treatment of sleep-related disorders in older adults increasingly necessary.1 Normative aging results in multiple changes in sleep architecture, including decreased total sleep time, decreased sleep efficiency, decreased slow-wave sleep (SWS), and increased awakenings after sleep onset.2 Sleep disturbances in older adults are increasingly recognized as multifactorial health conditions requiring comprehensive modification of risk factors, diagnosis, and treatment.3
In this article, we discuss the effects of aging on sleep architecture and provide an overview of primary sleep disorders in older adults. We also summarize strategies for diagnosing and treating sleep disorders in these patients.
Elements of the sleep cycle
The human sleep cycle begins with light sleep (sleep stages 1 and 2), progresses into SWS (sleep stage 3), and culminates in rapid eye movement (REM) sleep. The first 3 stages are referred to as non-rapid eye movement sleep (NREM). Throughout the night, this coupling of NREM and REM cycles occurs 4 to 6 times, with each successive cycle decreasing in length until awakening.4
Two complex neurologic pathways intersect to regulate the timing of sleep and wakefulness on arousal. The first pathway, the circadian system, is located within the suprachiasmatic nucleus of the hypothalamus and is highly dependent on external stimuli (light, food, etc.) to synchronize sleep/wake cycles. The suprachiasmatic nucleus regulates melatonin secretion by the pineal gland, which signals day-night transitions. The other pathway, the homeostatic system, modifies the amount of sleep needed daily. When multiple days of poor sleep occur, homeostatic sleep pressure (colloquially described as sleep debt) compensates by increasing the amount of sleep required in the following days. Together, the circadian and homeostatic systems work in conjunction to regulate sleep quantity to approximately one-third of the total sleep-wake cycle.2,5
Age-related dysfunction of the regulatory sleep pathways leads to blunting of the ability to initiate and sustain high-quality sleep.6 Dysregulation of homeostatic sleep pressure decreases time spent in SWS, and failure of the circadian signaling apparatus results in delays in sleep/wake timing.2 While research into the underlying neurobiology of sleep reveals that some of these changes are inherent to aging (Box7-14), significant underdiagnosed pathologies may adversely affect sleep architecture, including polypharmacy, comorbid neuropathology (eg, synucleinopathies, tauopathies, etc.), and primary sleep disorders (insomnias, hypersomnias, and parasomnias).15
Box
It has long been known that sleep architecture changes significantly with age. One of the largest meta-analyses of sleep changes in healthy individuals throughout childhood into old age found that total sleep time, sleep efficiency, percentage of slow-wave sleep, percentage of rapid eye movement sleep (REM), and REM latency all decreased with normative aging.7 Other studies have also found a decreased ability to maintain sleep (increased frequency of awakenings and prolonged nocturnal awakenings).8
Based on several meta-analyses, the average total sleep time at night in the adult population decreases by approximately 10 minutes per decade in both men and women.7,9-11 However, this pattern is not observed after age 60, when the total sleep time plateaus.7 Similarly, the duration of wake after sleep onset increases by approximately 10 minutes every decade for adults age 30 to 60, and plateaus after that.7,8
Epidemiologic studies have suggested that the prevalence of daytime napping increases with age.8 This trend continues into older age without a noticeable plateau.
A study of a nationally representative sample of >7,000 Japanese participants found that a significantly higher proportion of older adults take daytime naps (27.4%) compared with middle-age adults (14.4%).12 Older adults nap more frequently because of both lifestyle and biologic changes that accompany normative aging. Polls in the United States have shown a correlation between frequent napping and an increase in excessive daytime sleepiness, depression, pain, and nocturia.13
While sleep latency steadily increases after age 50, recent studies have shown that in healthy individuals, these changes are modest at best,7,9,14 which suggests that other pathologic factors may be contributing to this problem. Although healthy older people were found to have more frequent arousals throughout the night, they retained the ability to reinitiate sleep as rapidly as younger adults.7,9
Primary sleep disorders
Obstructive sleep apnea (OSA) is one of the most common, yet frequently underdiagnosed reversible causes of sleep disturbances. It is characterized by partial or complete airway obstruction culminating in periods of involuntary cessation of respirations during sleep. The resultant fragmentation in sleep leads to significant downstream effects over time, including excessive daytime sleepiness and fatigue, poor occupational and social performance, and substantial cognitive impairment.3 While it is well known that OSA increases in prevalence throughout middle age, this relationship plateaus after age 60.16 An estimated 40% to 60% of Americans age >60 are affected by OSA.17 The hypoxemia and fragmented sleep caused by unrecognized OSA are associated with a significant decline in activities of daily living (ADL).18 Untreated OSA is strongly linked to the development and progression of several major health conditions, including cardiovascular disease, diabetes mellitus, hypertension, stroke, and depression.19 In studies of long-term care facility residents—many of whom may have comorbid cognitive decline—researchers found that unrecognized OSA often mimics the progressive cognitive decline seen in major neurocognitive disorders.20 However, classic symptoms of OSA may not always be present in these patients, and their daytime sleepiness is often attributed to old age rather than to a pathological etiology.16 Screening for OSA and prompt initiation of the appropriate treatment may reverse OSA-induced cognitive changes in these patients.21
The primary presenting symptom of OSA is snoring, which is correlated with pauses in breathing. Risk factors include increased body mass index (BMI), thick neck circumference, male sex, and advanced age. In older adults, BMI has a lower impact on the Apnea-Hypopnea Index, an indicator of the number of pauses in breathing per hour, when compared with young and middle-age adults.16 Validated screening questionnaires for OSA include the STOP-Bang Questionnaire (Table 122), OSA50, Berlin Questionnaire, and Epworth Sleepiness Scale, each of which is used in different subpopulations. The current diagnostic standard for OSA is nocturnal polysomnography in a sleep laboratory, but recent advances in home sleep apnea testing have made it a viable, low-cost alternative for patients who do not have significant medical comorbidities.23 Standard utilized cutoffs for diagnosis are ≥5 events/hour (hypopneas associated with at least 4% oxygen desaturations) in conjunction with clinical symptoms of OSA.24

Continue to: Treatment
Treatment. First-line treatment for OSA is continuous positive airway pressure therapy, but adherence rates vary widely with patient education and regular follow-up.25 Adjunctive therapy includes weight loss, oral appliances, and uvulopalatopharyngoplasty, a procedure in which tissue in the throat is remodeled or removed.
Central sleep apnea (CSA) is a pause in breathing without evidence of associated respiratory effort. In adults, the development of CSA is indicative of underlying lower brainstem dysfunction, due to intermittent failures in the pontomedullary centers responsible for regulation of rhythmic breathing.26 This can occur as a consequence of multiple diseases, including congestive heart failure, stroke, renal failure, chronic medication use (opioids), and brain tumors.
The Sleep Heart Health Study—the largest community-based cohort study to date examining CSA—estimated that the prevalence of CSA among adults age >65 was 1.1% (compared with 0.4% in those age <65).27 Subgroup analysis revealed that men had significantly higher rates of CSA compared with women (2.7% vs 0.2%, respectively).
CSA may present similarly to OSA (excessive daytime somnolence, insomnia, poor sleep quality, difficulties with attention and concentration). Symptoms may also mimic those of coexisting medical conditions in older adults, such as nocturnal angina or paroxysmal nocturnal dyspnea.27 Any older patient with daytime sleepiness and risk factors for CSA should be referred for in-laboratory nocturnal polysomnography, the gold standard diagnostic test. Unlike in OSA, ambulatory diagnostic measures (home sleep apnea testing) have not been validated for this disorder.27
Treatment. The primary treatment for CSA is to address the underlying medical problem. Positive pressure ventilation has been attempted with mixed results. Supplemental oxygen and medical management (acetazolamide or theophylline) can help stimulate breathing. Newer studies have shown favorable outcomes with transvenous neurostimulation or adaptive servoventilation.28-30
Continue to: Insomnia
Insomnia. For a primary diagnosis of insomnia, DSM-5 requires at least 3 nights per week of sleep disturbances that induce distress or functional impairment for at least 3 months.31 The International Classification of Disease, 10th Edition requires at least 1 month of symptoms (lying awake for a long time before falling asleep, sleeping for short periods, being awake for most of the night, feeling lack of sleep, waking up early) after ruling out other sleep disorders, substance use, or other medical conditions.4 Clinically, insomnia tends to present in older adults as a subjective complaint of dissatisfaction with the quality and/or quantity of their sleep. Insomnia has been consistently shown to be a significant risk factor for both the development or exacerbation of depression in older adults.32-34
While the diagnosis of insomnia is mainly clinical via a thorough sleep and medication history, assistive ancillary testing can include wrist actigraphy and screening questionnaires (the Insomnia Severity Index and the Pittsburgh Sleep Quality Index).4 Because population studies of older adults have found discrepancies between objective and subjective methods of assessing sleep quality, relying on the accuracy of self-reported symptoms alone is questionable.35
Treatment. Given that drug elimination half-life increases with age, and the risks of adverse effects are increased in older adults, the preferred treatment modalities for insomnia are nonpharmacologic.4 Sleep hygiene education (Table 2) and cognitive-behavioral therapy (CBT) for insomnia are often the first-line therapies.4,36,37 It is crucial to manage comorbidities such as heart disease and obesity, as well as sources of discomfort from conditions such as arthritic pain.38,39 If nonpharmacologic therapies are not effective, pharmacologic options can be considered.4 Before prescribing sleep medications, it may be more fruitful to treat underlying psychiatric disorders such as depression and anxiety with antidepressants.4 Although benzodiazepines are helpful for their sedative effects, they are not recommended for older adults because of an increased risk of falls, rebound insomnia, potential tolerance, and associated cognitive impairment.40 Benzodiazepine receptor agonists (eg, zolpidem, eszopiclone, zaleplon) were initially developed as a first-line treatment for insomnia to replace the reliance on benzodiazepines, but these medications have a “black-box” warning of a serious risk of complex sleep behaviors, including life-threatening parasomnias.41 As a result, guidelines suggest a shorter duration of treatment with a benzodiazepine receptor agonist may still provide benefit while limiting the risk of adverse effects.42

Doxepin is the only antidepressant FDA-approved for insomnia; it improves sleep latency (time taken to initiate sleep after lying down), duration, and quality in adults age >65.43 Melatonin receptor agonists such as ramelteon and melatonin have shown positive results in older patients with insomnia. In clinical trials of patients age ≥65, ramelteon, which is FDA-approved for insomnia, produced no rebound insomnia, withdrawal effects, memory impairment, or gait instability.44-46 Suvorexant, an orexin receptor antagonist, decreases sleep latency and increases total sleep time equally in both young and older adults.47-49Table 340-51 provides a list of medications used to treat insomnia (including off-label agents) and their common adverse effects in older adults.

Parasomnias are undesirable behaviors that occur during sleep, commonly associated with the sleep-wake transition period. These behaviors can occur during REM sleep (nightmare disorder, sleep paralysis, REM sleep behavior disorder) or NREM sleep (somnambulism [sleepwalking], confusional arousals, sleep terrors). According to a cross-sectional Norwegian study of parasomnias, the estimated lifetime prevalence of sleep walking is 22.4%; sleep talking, 66.8%; confusional arousal, 18.5%; and sleep terror, 10.4%.52
Continue to: When evaluating a patient...
When evaluating a patient with parasomnias, it is important to review their drug and substance use as well as coexisting medical conditions. Drugs and substances that can affect sleep include prescription medications (second-generation antidepressants, stimulants, dopamine agonists), excessive caffeine, alcohol, certain foods (coffee, chocolate milk, black tea, caffeinated soft drinks), environmental exposures (smoking, pesticides), and recreational drugs (amphetamines).53-56 Certain medical conditions are correlated with specific parasomnias (eg, sleep paralysis and narcolepsy, REM sleep behavior disorder and Parkinson’s disease [PD], etc.).54 Diagnosis of parasomnias is mainly clinical but supporting evidence can be obtained through in-lab polysomnography.
Treatment. For parasomnias, treatment is primarily supportive and includes creating a safe sleeping environment to reduce the risk of self-harm. Recommendations include sleeping in a room on the ground floor, minimizing furniture in the bedroom, padding any bedside furniture, child-proofing doorknobs, and locking up weapons and other dangerous household items.54
REM sleep behavior disorder (RBD). This disorder is characterized by a loss of the typical REM sleep-associated atonia and the presence of motor activity during dreaming (dream-enacted behaviors). While the estimated incidence of RBD in the general adult population is approximately 0.5%, it increases to 7.7% among those age >60.57 RBD occurs most commonly in the setting of the alpha-synucleinopathies (PD, Lewy body dementia, multisystem atrophy), but can also be found in patients with cerebral ischemia, demyelinating disorders, or alcohol misuse, or can be medication-induced (primarily antidepressants and antipsychotics).58 In patients with PD, the presence of RBD is associated with a more impaired cognitive profile, suggestive of widespread neurodegeneration.59 Recent studies revealed that RBD may also be a prodromal state of neurodegenerative diseases such as PD, which should prompt close monitoring and long-term follow up.60 Similar to other parasomnias, the diagnosis of RBD is primarily clinical, but polysomnography plays an important role in demonstrating loss of REM-related atonia.54
Treatment. Clonazepam and melatonin have been shown to be effective in treating the symptoms of RBD.54
Depression, anxiety, and sleep disturbances
Major depressive disorder (MDD) and generalized anxiety disorder (GAD) affect sleep in patients of all ages, but are underreported in older adults. According to national epidemiologic surveys, the estimated prevalence of MDD and GAD among older adults is 13% and 11.4%, respectively.61,62 Rates as high as 42% and 39% have been reported in meta-regression analyses among patients with Alzheimer’s dementia.63
Continue to: Depression and anxiety
Depression and anxiety may have additive effects and manifest as poor sleep satisfaction, increased sleep latency, insomnia, and daytime sleepiness.64 However, they may also have independent effects. Studies showed that patients with depression alone reported overall poor sleep satisfaction, whereas patients with anxiety alone reported problems with sleep latency, daytime drowsiness, and waking up at night in addition to their overall poor sleep satisfaction.65-67 Both depression and anxiety are risk factors for developing cognitive decline, and may be an early sign/prodrome of neurodegenerative diseases (dementias).68 The bidirectional relationship between depression/anxiety and sleep is complex and needs further investigation.
Treatment. Pharmacologic treatments for patients with depression/anxiety and sleep disturbances include selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, tricyclic antidepressants, and other serotonin receptor agonists.69-72 Nonpharmacologic treatments include CBT for both depression and anxiety, and problem-solving therapy for patients with mild cognitive impairment and depression.73,74 For severe depression and/or anxiety, electroconvulsive therapy is effective.75
Bottom Line
Sleep disorders in older adults are common but often underdiagnosed. Timely recognition of obstructive sleep apnea, central sleep apnea, insomnia, parasomnias, and other sleep disturbances can facilitate effective treatment and greatly improve older adults’ quality of life.
Related Resources
- American Academy of Sleep Medicine. International Classification of Sleep Disorders—Third Edition. https://aasm.org
- SleepFoundation.org. Sleep hygiene. https://www.sleepfoundation.org/articles/sleep-hygiene
Drug Brand Names
Acetazolamide • Diamox
Clonazepam • Klonopin
Doxepin • Silenor
Eszopiclone • Lunesta
Gabapentin • Neurontin
Mirtazapine • Remeron
Pramipexole • Mirapex
Quetiapine • Seroquel
Ramelteon • Rozerem
Suvorexant • Belsomra
Temazepam • Restoril
Theophylline • Elixophyllin
Tiagabine • Gabitril
Trazadone • Desyrel
Triazolam • Halcion
Zaleplon • Sonata
Zolpidem • Ambien
1. Centers for Disease Control and Prevention. The state of aging and health in America. 2013. Accessed January 27, 2021. https://www.cdc.gov/aging/pdf/state-aging-health-in-america-2013.pdf
2. Suzuki K, Miyamoto M, Hirata K. Sleep disorders in the elderly: diagnosis and management. J Gen Fam Med. 2017;18(2):61-71.
3. Inouye SK, Studenski S, Tinetti ME, et al. Geriatric syndromes: clinical, research, and policy implications of a core geriatric concept. J Am Geriatr Soc. 2007;55(5):780-791.
4. Patel D, Steinberg J, Patel P. Insomnia in the elderly: a review. J Clin Sleep Med. 2018;14(6):1017-1024.
5. Neubauer DN. A review of ramelteon in the treatment of sleep disorders. Neuropsychiatr Dis Treat. 2008;4(1):69-79.
6. Mander BA, Winer JR, Walker MP. Sleep and human aging. Neuron. 2017;94(1):19-36.
7. Ohayon MM, Carskadon MA, Guilleminault C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep. 2004;27:1255-1273.
8. Li J, Vitiello MV, Gooneratne NS. Sleep in normal aging. Sleep Med Clin. 2018;13(1):1-11.
9. Floyd JA, Medler SM, Ager JW, et al. Age-related changes in initiation and maintenance of sleep: a meta-analysis. Res Nurs Health. 2000;23(2):106-117.
10. Floyd JA, Janisse JJ, Jenuwine ES, et al. Changes in REM-sleep percentage over the adult lifespan. Sleep. 2007;30(7):829-836.
11. Dorffner G, Vitr M, Anderer P. The effects of aging on sleep architecture in healthy subjects. Adv Exp Med Biol. 2015;821:93-100.
12. Furihata R, Kaneita Y, Jike M, et al. Napping and associated factors: a Japanese nationwide general population survey. Sleep Med. 2016;20:72-79.
13. Foley DJ, Vitiello MV, Bliwise DL, et al. Frequent napping is associated with excessive daytime sleepiness, depression, pain, and nocturia in older adults: findings from the National Sleep Foundation ‘2003 Sleep in America’ Poll. Am J Geriatr Psychiatry. 2007;15(4):344-350.
14. Floyd JA, Janisse JJ, Marshall Medler S, et al. Nonlinear components of age-related change in sleep initiation. Nurs Res. 2000;49(5):290-294.
15. Miner B, Kryger MH. Sleep in the aging population. Sleep Med Clin. 2017;12(1):31-38.
16. Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;165(9):1217-1239.
17. Ancoli-Israel S, Klauber MR, Butters N, et al. Dementia in institutionalized elderly: relation to sleep apnea. J Am Geriatr Soc. 1991;39(3):258-263.
18. Spira AP, Stone KL, Rebok GW, et al. Sleep-disordered breathing and functional decline in older women. J Am Geriatr Soc. 2014;62(11):2040-2046.
19. Vijayan VK. Morbidities associated with obstructive sleep apnea. Expert Rev Respir Med. 2012;6(5):557-566.
20. Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry. 2016;24(6):496-508.
21. Dalmases M, Solé-Padullés C, Torres M, et al. Effect of CPAP on cognition, brain function, and structure among elderly patients with OSA: a randomized pilot study. Chest. 2015;148(5):1214-1223.
22. Toronto Western Hospital, University Health Network. University of Toronto. STOP-Bang Questionnaire. 2012. Accessed January 26, 2021. www.stopbang.ca
23. Freedman N. Doing it better for less: incorporating OSA management into alternative payment models. Chest. 2019;155(1):227-233.
24. Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine clinical practice guideline. J Clin Sleep Med. 2017;13(3):479-504.
25. Semelka M, Wilson J, Floyd R. Diagnosis and treatment of obstructive sleep apnea in adults. Am Fam Physician. 2016;94(5):355-360.
26. Javaheri S, Dempsey JA. Central sleep apnea. Compr Physiol. 2013;3(1):141-163.
27. Donovan LM, Kapur VK. Prevalence and characteristics of central compared to obstructive sleep apnea: analyses from the Sleep Heart Health Study cohort. Sleep. 2016;39(7):1353-1359.
28. Cao M, Cardell CY, Willes L, et al. A novel adaptive servoventilation (ASVAuto) for the treatment of central sleep apnea associated with chronic use of opioids. J Clin Sleep Med. 2014;10(8):855-861.
29. Oldenburg O, Spießhöfer J, Fox H, et al. Performance of conventional and enhanced adaptive servoventilation (ASV) in heart failure patients with central sleep apnea who have adapted to conventional ASV. Sleep Breath. 2015;19(3):795-800.
30. Costanzo MR, Ponikowski P, Javaheri S, et al. Transvenous neurostimulation for central sleep apnoea: a randomised controlled trial. Lancet. 2016;388(10048):974-982.
31. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013:362.
32. Perlis ML, Smith LJ, Lyness JM, et al. Insomnia as a risk factor for onset of depression in the elderly. Behav Sleep Med. 2006;4(2):104-113.
33. Cole MG, Dendukuri N. Risk factors for depression among elderly community subjects: a systematic review and meta-analysis. Am J Psychiatry. 2003;160(6):1147-1156.
34. Pigeon WR, Hegel M, Unützer J, et al. Is insomnia a perpetuating factor for late-life depression in the IMPACT cohort? Sleep. 2008;31(4):481-488.
35. Hughes JM, Song Y, Fung CH, et al. Measuring sleep in vulnerable older adults: a comparison of subjective and objective sleep measures. Clin Gerontol. 2018;41(2):145-157.
36. Irish LA, Kline CE, Gunn HE, et al. The role of sleep hygiene in promoting public health: a review of empirical evidence. Sleep Med Rev. 2015;22:23-36.
37. Sleep Foundation. Sleep hygiene. Accessed January 27, 2021. https://www.sleepfoundation.org/articles/sleep-hygiene
38. Foley D, Ancoli-Israel S, Britz P, et al. Sleep disturbances and chronic disease in older adults: results of the 2003 National Sleep Foundation Sleep in America Survey. J Psychosom Res. 2004;56(5):497-502.
39. Eslami V, Zimmerman ME, Grewal T, et al. Pain grade and sleep disturbance in older adults: evaluation the role of pain, and stress for depressed and non-depressed individuals. Int J Geriatr Psychiatry. 2016;31(5):450-457.
40. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
41. United States Food & Drug Administration. FDA adds Boxed Warning for risk of serious injuries caused by sleepwalking with certain prescription insomnia medicines. 2019. Accessed January 27, 2021. https://www.fda.gov/drugs/drug-safety-and-availability/fda-adds-boxed-warning-risk-serious-injuries-caused-sleepwalking-certain-prescription-insomnia
42. Schroeck JL, Ford J, Conway EL, et al. Review of safety and efficacy of sleep medicines in older adults. Clin Ther. 2016;38(11):2340-2372.
43. Krystal AD, Lankford A, Durrence HH, et al. Efficacy and safety of doxepin 3 and 6 mg in a 35-day sleep laboratory trial in adults with chronic primary insomnia. Sleep. 2011;34(10):1433-1442.
44. Roth T, Seiden D, Sainati S, et al. Effects of ramelteon on patient-reported sleep latency in older adults with chronic insomnia. Sleep Med. 2006;7(4):312-318.
45. Zammit G, Wang-Weigand S, Rosenthal M, et al. Effect of ramelteon on middle-of-the-night balance in older adults with chronic insomnia. J Clin Sleep Med. 2009;5(1):34-40.
46. Mets MAJ, de Vries JM, de Senerpont Domis LM, et al. Next-day effects of ramelteon (8 mg), zopiclone (7.5 mg), and placebo on highway driving performance, memory functioning, psychomotor performance, and mood in healthy adult subjects. Sleep. 2011;34(10):1327-1334.
47. Rhyne DN, Anderson SL. Suvorexant in insomnia: efficacy, safety and place in therapy. Ther Adv Drug Saf. 2015;6(5):189-195.
48. Norman JL, Anderson SL. Novel class of medications, orexin receptor antagonists, in the treatment of insomnia - critical appraisal of suvorexant. Nat Sci Sleep. 2016;8:239-247.
49. Owen RT. Suvorexant: efficacy and safety profile of a dual orexin receptor antagonist in treating insomnia. Drugs Today (Barc). 2016;52(1):29-40.
50. Shannon S, Lewis N, Lee H, et al. Cannabidiol in anxiety and sleep: a large case series. Perm J. 2019;23:18-041. doi: 10.7812/TPP/18-041
51. Yunusa I, Alsumali A, Garba AE, et al. Assessment of reported comparative effectiveness and safety of atypical antipsychotics in the treatment of behavioral and psychological symptoms of dementia: a network meta-analysis. JAMA Netw Open. 2019;2(3):e190828.
52. Bjorvatn B, Gronli J, Pallesen S. Prevalence of different parasomnias in the general population. Sleep Med. 2010;11(10):1031-1034.
53. Postuma RB, Montplaisir JY, Pelletier A, et al. Environmental risk factors for REM sleep behavior disorder: a multicenter case-control study. Neurology. 2012;79(5):428-434.
54. Fleetham JA, Fleming JA. Parasomnias. CMAJ. 2014;186(8):E273-E280.
55. Dinis-Oliveira RJ, Caldas I, Carvalho F, et al. Bruxism after 3,4-methylenedioxymethamphetamine (ecstasy) abuse. Clin Toxicol (Phila.) 2010;48(8):863-864.
56. Irfan MH, Howell MJ. Rapid eye movement sleep behavior disorder: overview and current perspective. Curr Sleep Medicine Rep. 2016;2:64-73.
57. Mahlknecht P, Seppi K, Frauscher B, et al. Probable RBD and association with neurodegenerative disease markers: a population-based study. Mov Disord. 2015;30(10):1417-1421.
58. Oertel WH, Depboylu C, Krenzer M, et al. [REM sleep behavior disorder as a prodromal stage of α-synucleinopathies: symptoms, epidemiology, pathophysiology, diagnosis and therapy]. Nervenarzt. 2014;85:19-25. German.
59. Jozwiak N, Postuma RB, Montplaisir J, et al. REM sleep behavior disorder and cognitive impairment in Parkinson’s disease. Sleep. 2017;40(8):zsx101. doi: 10.1093/sleep/zsx101
60. Claassen DO, Josephs KA, Ahlskog JE, et al. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology 2010;75(6):494-499.
61. Reynolds K, Pietrzak RH, El-Gabalawy R, et al. Prevalence of psychiatric disorders in U.S. older adults: findings from a nationally representative survey. World Psychiatry. 2015;14(1):74-81.
62. Lohman MC, Mezuk B, Dumenci L. Depression and frailty: concurrent risks for adverse health outcomes. Aging Ment Health. 2017;21(4):399-408.
63. Zhao QF, Tan L, Wang HF, et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: systematic review and meta-analysis. J Affect Disord. 2016;190:264-271.
64. Furihata R, Hall MH, Stone KL, et al. An aggregate measure of sleep health is associated with prevalent and incident clinically significant depression symptoms among community-dwelling older women. Sleep. 2017;40(3):zsw075. doi: 10.1093/sleep/zsw075
65. Spira AP, Stone K, Beaudreau SA, et al. Anxiety symptoms and objectively measured sleep quality in older women. Am J Geriatr Psychiatry. 2009;17(2):136-143.
66. Press Y, Punchik B, Freud T. The association between subjectively impaired sleep and symptoms of depression and anxiety in a frail elderly population. Aging Clin Exp Res. 2018;30(7):755-765.
67. Gould CE, Spira AP, Liou-Johnson V, et al. Association of anxiety symptom clusters with sleep quality and daytime sleepiness. J Gerontol B Psychol Sci Soc Sci. 2018;73(3):413-420.
68. Kassem AM, Ganguli M, Yaffe K, et al. Anxiety symptoms and risk of cognitive decline in older community-dwelling men. Int Psychogeriatr. 2017;29(7):1137-1145.
69. Frank C. Pharmacologic treatment of depression in the elderly. Can Fam Physician. 2014;60(2):121-126.
70. Subramanyam AA, Kedare J, Singh OP, et al. Clinical practice guidelines for geriatric anxiety disorders. Indian J Psychiatry. 2018;60(suppl 3):S371-S382.
71. Emsley R, Ahokas A, Suarez A, et al. Efficacy of tianeptine 25-50 mg in elderly patients with recurrent major depressive disorder: an 8-week placebo- and escitalopram-controlled study. J Clin Psychiatry. 2018;79(4):17m11741. doi: 10.4088/JCP.17m11741
72. Semel D, Murphy TK, Zlateva G, et al. Evaluation of the safety and efficacy of pregabalin in older patients with neuropathic pain: results from a pooled analysis of 11 clinical studies. BMC Fam Pract. 2010;11:85.
73. Orgeta V, Qazi A, Spector A, et al. Psychological treatments for depression and anxiety in dementia and mild cognitive impairment: systematic review and meta-analysis. Br J Psychiatry. 2015;207(4):293-298.
74. Morimoto SS, Kanellopoulos D, Manning KJ, et al. Diagnosis and treatment of depression and cognitive impairment in late life. Ann N Y Acad Sci. 2015;1345(1):36-46.
75. Casey DA. Depression in older adults: a treatable medical condition. Prim Care. 2017;44(3):499-510.
As humans live longer, a renewed focus on quality of life has made the prompt diagnosis and treatment of sleep-related disorders in older adults increasingly necessary.1 Normative aging results in multiple changes in sleep architecture, including decreased total sleep time, decreased sleep efficiency, decreased slow-wave sleep (SWS), and increased awakenings after sleep onset.2 Sleep disturbances in older adults are increasingly recognized as multifactorial health conditions requiring comprehensive modification of risk factors, diagnosis, and treatment.3
In this article, we discuss the effects of aging on sleep architecture and provide an overview of primary sleep disorders in older adults. We also summarize strategies for diagnosing and treating sleep disorders in these patients.
Elements of the sleep cycle
The human sleep cycle begins with light sleep (sleep stages 1 and 2), progresses into SWS (sleep stage 3), and culminates in rapid eye movement (REM) sleep. The first 3 stages are referred to as non-rapid eye movement sleep (NREM). Throughout the night, this coupling of NREM and REM cycles occurs 4 to 6 times, with each successive cycle decreasing in length until awakening.4
Two complex neurologic pathways intersect to regulate the timing of sleep and wakefulness on arousal. The first pathway, the circadian system, is located within the suprachiasmatic nucleus of the hypothalamus and is highly dependent on external stimuli (light, food, etc.) to synchronize sleep/wake cycles. The suprachiasmatic nucleus regulates melatonin secretion by the pineal gland, which signals day-night transitions. The other pathway, the homeostatic system, modifies the amount of sleep needed daily. When multiple days of poor sleep occur, homeostatic sleep pressure (colloquially described as sleep debt) compensates by increasing the amount of sleep required in the following days. Together, the circadian and homeostatic systems work in conjunction to regulate sleep quantity to approximately one-third of the total sleep-wake cycle.2,5
Age-related dysfunction of the regulatory sleep pathways leads to blunting of the ability to initiate and sustain high-quality sleep.6 Dysregulation of homeostatic sleep pressure decreases time spent in SWS, and failure of the circadian signaling apparatus results in delays in sleep/wake timing.2 While research into the underlying neurobiology of sleep reveals that some of these changes are inherent to aging (Box7-14), significant underdiagnosed pathologies may adversely affect sleep architecture, including polypharmacy, comorbid neuropathology (eg, synucleinopathies, tauopathies, etc.), and primary sleep disorders (insomnias, hypersomnias, and parasomnias).15
Box
It has long been known that sleep architecture changes significantly with age. One of the largest meta-analyses of sleep changes in healthy individuals throughout childhood into old age found that total sleep time, sleep efficiency, percentage of slow-wave sleep, percentage of rapid eye movement sleep (REM), and REM latency all decreased with normative aging.7 Other studies have also found a decreased ability to maintain sleep (increased frequency of awakenings and prolonged nocturnal awakenings).8
Based on several meta-analyses, the average total sleep time at night in the adult population decreases by approximately 10 minutes per decade in both men and women.7,9-11 However, this pattern is not observed after age 60, when the total sleep time plateaus.7 Similarly, the duration of wake after sleep onset increases by approximately 10 minutes every decade for adults age 30 to 60, and plateaus after that.7,8
Epidemiologic studies have suggested that the prevalence of daytime napping increases with age.8 This trend continues into older age without a noticeable plateau.
A study of a nationally representative sample of >7,000 Japanese participants found that a significantly higher proportion of older adults take daytime naps (27.4%) compared with middle-age adults (14.4%).12 Older adults nap more frequently because of both lifestyle and biologic changes that accompany normative aging. Polls in the United States have shown a correlation between frequent napping and an increase in excessive daytime sleepiness, depression, pain, and nocturia.13
While sleep latency steadily increases after age 50, recent studies have shown that in healthy individuals, these changes are modest at best,7,9,14 which suggests that other pathologic factors may be contributing to this problem. Although healthy older people were found to have more frequent arousals throughout the night, they retained the ability to reinitiate sleep as rapidly as younger adults.7,9
Primary sleep disorders
Obstructive sleep apnea (OSA) is one of the most common, yet frequently underdiagnosed reversible causes of sleep disturbances. It is characterized by partial or complete airway obstruction culminating in periods of involuntary cessation of respirations during sleep. The resultant fragmentation in sleep leads to significant downstream effects over time, including excessive daytime sleepiness and fatigue, poor occupational and social performance, and substantial cognitive impairment.3 While it is well known that OSA increases in prevalence throughout middle age, this relationship plateaus after age 60.16 An estimated 40% to 60% of Americans age >60 are affected by OSA.17 The hypoxemia and fragmented sleep caused by unrecognized OSA are associated with a significant decline in activities of daily living (ADL).18 Untreated OSA is strongly linked to the development and progression of several major health conditions, including cardiovascular disease, diabetes mellitus, hypertension, stroke, and depression.19 In studies of long-term care facility residents—many of whom may have comorbid cognitive decline—researchers found that unrecognized OSA often mimics the progressive cognitive decline seen in major neurocognitive disorders.20 However, classic symptoms of OSA may not always be present in these patients, and their daytime sleepiness is often attributed to old age rather than to a pathological etiology.16 Screening for OSA and prompt initiation of the appropriate treatment may reverse OSA-induced cognitive changes in these patients.21
The primary presenting symptom of OSA is snoring, which is correlated with pauses in breathing. Risk factors include increased body mass index (BMI), thick neck circumference, male sex, and advanced age. In older adults, BMI has a lower impact on the Apnea-Hypopnea Index, an indicator of the number of pauses in breathing per hour, when compared with young and middle-age adults.16 Validated screening questionnaires for OSA include the STOP-Bang Questionnaire (Table 122), OSA50, Berlin Questionnaire, and Epworth Sleepiness Scale, each of which is used in different subpopulations. The current diagnostic standard for OSA is nocturnal polysomnography in a sleep laboratory, but recent advances in home sleep apnea testing have made it a viable, low-cost alternative for patients who do not have significant medical comorbidities.23 Standard utilized cutoffs for diagnosis are ≥5 events/hour (hypopneas associated with at least 4% oxygen desaturations) in conjunction with clinical symptoms of OSA.24
Continue to: Treatment
Treatment. First-line treatment for OSA is continuous positive airway pressure therapy, but adherence rates vary widely with patient education and regular follow-up.25 Adjunctive therapy includes weight loss, oral appliances, and uvulopalatopharyngoplasty, a procedure in which tissue in the throat is remodeled or removed.
Central sleep apnea (CSA) is a pause in breathing without evidence of associated respiratory effort. In adults, the development of CSA is indicative of underlying lower brainstem dysfunction, due to intermittent failures in the pontomedullary centers responsible for regulation of rhythmic breathing.26 This can occur as a consequence of multiple diseases, including congestive heart failure, stroke, renal failure, chronic medication use (opioids), and brain tumors.
The Sleep Heart Health Study—the largest community-based cohort study to date examining CSA—estimated that the prevalence of CSA among adults age >65 was 1.1% (compared with 0.4% in those age <65).27 Subgroup analysis revealed that men had significantly higher rates of CSA compared with women (2.7% vs 0.2%, respectively).
CSA may present similarly to OSA (excessive daytime somnolence, insomnia, poor sleep quality, difficulties with attention and concentration). Symptoms may also mimic those of coexisting medical conditions in older adults, such as nocturnal angina or paroxysmal nocturnal dyspnea.27 Any older patient with daytime sleepiness and risk factors for CSA should be referred for in-laboratory nocturnal polysomnography, the gold standard diagnostic test. Unlike in OSA, ambulatory diagnostic measures (home sleep apnea testing) have not been validated for this disorder.27
Treatment. The primary treatment for CSA is to address the underlying medical problem. Positive pressure ventilation has been attempted with mixed results. Supplemental oxygen and medical management (acetazolamide or theophylline) can help stimulate breathing. Newer studies have shown favorable outcomes with transvenous neurostimulation or adaptive servoventilation.28-30
Continue to: Insomnia
Insomnia. For a primary diagnosis of insomnia, DSM-5 requires at least 3 nights per week of sleep disturbances that induce distress or functional impairment for at least 3 months.31 The International Classification of Disease, 10th Edition requires at least 1 month of symptoms (lying awake for a long time before falling asleep, sleeping for short periods, being awake for most of the night, feeling lack of sleep, waking up early) after ruling out other sleep disorders, substance use, or other medical conditions.4 Clinically, insomnia tends to present in older adults as a subjective complaint of dissatisfaction with the quality and/or quantity of their sleep. Insomnia has been consistently shown to be a significant risk factor for both the development or exacerbation of depression in older adults.32-34
While the diagnosis of insomnia is mainly clinical via a thorough sleep and medication history, assistive ancillary testing can include wrist actigraphy and screening questionnaires (the Insomnia Severity Index and the Pittsburgh Sleep Quality Index).4 Because population studies of older adults have found discrepancies between objective and subjective methods of assessing sleep quality, relying on the accuracy of self-reported symptoms alone is questionable.35
Treatment. Given that drug elimination half-life increases with age, and the risks of adverse effects are increased in older adults, the preferred treatment modalities for insomnia are nonpharmacologic.4 Sleep hygiene education (Table 2) and cognitive-behavioral therapy (CBT) for insomnia are often the first-line therapies.4,36,37 It is crucial to manage comorbidities such as heart disease and obesity, as well as sources of discomfort from conditions such as arthritic pain.38,39 If nonpharmacologic therapies are not effective, pharmacologic options can be considered.4 Before prescribing sleep medications, it may be more fruitful to treat underlying psychiatric disorders such as depression and anxiety with antidepressants.4 Although benzodiazepines are helpful for their sedative effects, they are not recommended for older adults because of an increased risk of falls, rebound insomnia, potential tolerance, and associated cognitive impairment.40 Benzodiazepine receptor agonists (eg, zolpidem, eszopiclone, zaleplon) were initially developed as a first-line treatment for insomnia to replace the reliance on benzodiazepines, but these medications have a “black-box” warning of a serious risk of complex sleep behaviors, including life-threatening parasomnias.41 As a result, guidelines suggest a shorter duration of treatment with a benzodiazepine receptor agonist may still provide benefit while limiting the risk of adverse effects.42
Doxepin is the only antidepressant FDA-approved for insomnia; it improves sleep latency (time taken to initiate sleep after lying down), duration, and quality in adults age >65.43 Melatonin receptor agonists such as ramelteon and melatonin have shown positive results in older patients with insomnia. In clinical trials of patients age ≥65, ramelteon, which is FDA-approved for insomnia, produced no rebound insomnia, withdrawal effects, memory impairment, or gait instability.44-46 Suvorexant, an orexin receptor antagonist, decreases sleep latency and increases total sleep time equally in both young and older adults.47-49Table 340-51 provides a list of medications used to treat insomnia (including off-label agents) and their common adverse effects in older adults.
Parasomnias are undesirable behaviors that occur during sleep, commonly associated with the sleep-wake transition period. These behaviors can occur during REM sleep (nightmare disorder, sleep paralysis, REM sleep behavior disorder) or NREM sleep (somnambulism [sleepwalking], confusional arousals, sleep terrors). According to a cross-sectional Norwegian study of parasomnias, the estimated lifetime prevalence of sleep walking is 22.4%; sleep talking, 66.8%; confusional arousal, 18.5%; and sleep terror, 10.4%.52
Continue to: When evaluating a patient...
When evaluating a patient with parasomnias, it is important to review their drug and substance use as well as coexisting medical conditions. Drugs and substances that can affect sleep include prescription medications (second-generation antidepressants, stimulants, dopamine agonists), excessive caffeine, alcohol, certain foods (coffee, chocolate milk, black tea, caffeinated soft drinks), environmental exposures (smoking, pesticides), and recreational drugs (amphetamines).53-56 Certain medical conditions are correlated with specific parasomnias (eg, sleep paralysis and narcolepsy, REM sleep behavior disorder and Parkinson’s disease [PD], etc.).54 Diagnosis of parasomnias is mainly clinical but supporting evidence can be obtained through in-lab polysomnography.
Treatment. For parasomnias, treatment is primarily supportive and includes creating a safe sleeping environment to reduce the risk of self-harm. Recommendations include sleeping in a room on the ground floor, minimizing furniture in the bedroom, padding any bedside furniture, child-proofing doorknobs, and locking up weapons and other dangerous household items.54
REM sleep behavior disorder (RBD). This disorder is characterized by a loss of the typical REM sleep-associated atonia and the presence of motor activity during dreaming (dream-enacted behaviors). While the estimated incidence of RBD in the general adult population is approximately 0.5%, it increases to 7.7% among those age >60.57 RBD occurs most commonly in the setting of the alpha-synucleinopathies (PD, Lewy body dementia, multisystem atrophy), but can also be found in patients with cerebral ischemia, demyelinating disorders, or alcohol misuse, or can be medication-induced (primarily antidepressants and antipsychotics).58 In patients with PD, the presence of RBD is associated with a more impaired cognitive profile, suggestive of widespread neurodegeneration.59 Recent studies revealed that RBD may also be a prodromal state of neurodegenerative diseases such as PD, which should prompt close monitoring and long-term follow up.60 Similar to other parasomnias, the diagnosis of RBD is primarily clinical, but polysomnography plays an important role in demonstrating loss of REM-related atonia.54
Treatment. Clonazepam and melatonin have been shown to be effective in treating the symptoms of RBD.54
Depression, anxiety, and sleep disturbances
Major depressive disorder (MDD) and generalized anxiety disorder (GAD) affect sleep in patients of all ages, but are underreported in older adults. According to national epidemiologic surveys, the estimated prevalence of MDD and GAD among older adults is 13% and 11.4%, respectively.61,62 Rates as high as 42% and 39% have been reported in meta-regression analyses among patients with Alzheimer’s dementia.63
Continue to: Depression and anxiety
Depression and anxiety may have additive effects and manifest as poor sleep satisfaction, increased sleep latency, insomnia, and daytime sleepiness.64 However, they may also have independent effects. Studies showed that patients with depression alone reported overall poor sleep satisfaction, whereas patients with anxiety alone reported problems with sleep latency, daytime drowsiness, and waking up at night in addition to their overall poor sleep satisfaction.65-67 Both depression and anxiety are risk factors for developing cognitive decline, and may be an early sign/prodrome of neurodegenerative diseases (dementias).68 The bidirectional relationship between depression/anxiety and sleep is complex and needs further investigation.
Treatment. Pharmacologic treatments for patients with depression/anxiety and sleep disturbances include selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, tricyclic antidepressants, and other serotonin receptor agonists.69-72 Nonpharmacologic treatments include CBT for both depression and anxiety, and problem-solving therapy for patients with mild cognitive impairment and depression.73,74 For severe depression and/or anxiety, electroconvulsive therapy is effective.75
Bottom Line
Sleep disorders in older adults are common but often underdiagnosed. Timely recognition of obstructive sleep apnea, central sleep apnea, insomnia, parasomnias, and other sleep disturbances can facilitate effective treatment and greatly improve older adults’ quality of life.
Related Resources
- American Academy of Sleep Medicine. International Classification of Sleep Disorders—Third Edition. https://aasm.org
- SleepFoundation.org. Sleep hygiene. https://www.sleepfoundation.org/articles/sleep-hygiene
Drug Brand Names
Acetazolamide • Diamox
Clonazepam • Klonopin
Doxepin • Silenor
Eszopiclone • Lunesta
Gabapentin • Neurontin
Mirtazapine • Remeron
Pramipexole • Mirapex
Quetiapine • Seroquel
Ramelteon • Rozerem
Suvorexant • Belsomra
Temazepam • Restoril
Theophylline • Elixophyllin
Tiagabine • Gabitril
Trazadone • Desyrel
Triazolam • Halcion
Zaleplon • Sonata
Zolpidem • Ambien
As humans live longer, a renewed focus on quality of life has made the prompt diagnosis and treatment of sleep-related disorders in older adults increasingly necessary.1 Normative aging results in multiple changes in sleep architecture, including decreased total sleep time, decreased sleep efficiency, decreased slow-wave sleep (SWS), and increased awakenings after sleep onset.2 Sleep disturbances in older adults are increasingly recognized as multifactorial health conditions requiring comprehensive modification of risk factors, diagnosis, and treatment.3
In this article, we discuss the effects of aging on sleep architecture and provide an overview of primary sleep disorders in older adults. We also summarize strategies for diagnosing and treating sleep disorders in these patients.
Elements of the sleep cycle
The human sleep cycle begins with light sleep (sleep stages 1 and 2), progresses into SWS (sleep stage 3), and culminates in rapid eye movement (REM) sleep. The first 3 stages are referred to as non-rapid eye movement sleep (NREM). Throughout the night, this coupling of NREM and REM cycles occurs 4 to 6 times, with each successive cycle decreasing in length until awakening.4
Two complex neurologic pathways intersect to regulate the timing of sleep and wakefulness on arousal. The first pathway, the circadian system, is located within the suprachiasmatic nucleus of the hypothalamus and is highly dependent on external stimuli (light, food, etc.) to synchronize sleep/wake cycles. The suprachiasmatic nucleus regulates melatonin secretion by the pineal gland, which signals day-night transitions. The other pathway, the homeostatic system, modifies the amount of sleep needed daily. When multiple days of poor sleep occur, homeostatic sleep pressure (colloquially described as sleep debt) compensates by increasing the amount of sleep required in the following days. Together, the circadian and homeostatic systems work in conjunction to regulate sleep quantity to approximately one-third of the total sleep-wake cycle.2,5
Age-related dysfunction of the regulatory sleep pathways leads to blunting of the ability to initiate and sustain high-quality sleep.6 Dysregulation of homeostatic sleep pressure decreases time spent in SWS, and failure of the circadian signaling apparatus results in delays in sleep/wake timing.2 While research into the underlying neurobiology of sleep reveals that some of these changes are inherent to aging (Box7-14), significant underdiagnosed pathologies may adversely affect sleep architecture, including polypharmacy, comorbid neuropathology (eg, synucleinopathies, tauopathies, etc.), and primary sleep disorders (insomnias, hypersomnias, and parasomnias).15
Box
It has long been known that sleep architecture changes significantly with age. One of the largest meta-analyses of sleep changes in healthy individuals throughout childhood into old age found that total sleep time, sleep efficiency, percentage of slow-wave sleep, percentage of rapid eye movement sleep (REM), and REM latency all decreased with normative aging.7 Other studies have also found a decreased ability to maintain sleep (increased frequency of awakenings and prolonged nocturnal awakenings).8
Based on several meta-analyses, the average total sleep time at night in the adult population decreases by approximately 10 minutes per decade in both men and women.7,9-11 However, this pattern is not observed after age 60, when the total sleep time plateaus.7 Similarly, the duration of wake after sleep onset increases by approximately 10 minutes every decade for adults age 30 to 60, and plateaus after that.7,8
Epidemiologic studies have suggested that the prevalence of daytime napping increases with age.8 This trend continues into older age without a noticeable plateau.
A study of a nationally representative sample of >7,000 Japanese participants found that a significantly higher proportion of older adults take daytime naps (27.4%) compared with middle-age adults (14.4%).12 Older adults nap more frequently because of both lifestyle and biologic changes that accompany normative aging. Polls in the United States have shown a correlation between frequent napping and an increase in excessive daytime sleepiness, depression, pain, and nocturia.13
While sleep latency steadily increases after age 50, recent studies have shown that in healthy individuals, these changes are modest at best,7,9,14 which suggests that other pathologic factors may be contributing to this problem. Although healthy older people were found to have more frequent arousals throughout the night, they retained the ability to reinitiate sleep as rapidly as younger adults.7,9
Primary sleep disorders
Obstructive sleep apnea (OSA) is one of the most common, yet frequently underdiagnosed reversible causes of sleep disturbances. It is characterized by partial or complete airway obstruction culminating in periods of involuntary cessation of respirations during sleep. The resultant fragmentation in sleep leads to significant downstream effects over time, including excessive daytime sleepiness and fatigue, poor occupational and social performance, and substantial cognitive impairment.3 While it is well known that OSA increases in prevalence throughout middle age, this relationship plateaus after age 60.16 An estimated 40% to 60% of Americans age >60 are affected by OSA.17 The hypoxemia and fragmented sleep caused by unrecognized OSA are associated with a significant decline in activities of daily living (ADL).18 Untreated OSA is strongly linked to the development and progression of several major health conditions, including cardiovascular disease, diabetes mellitus, hypertension, stroke, and depression.19 In studies of long-term care facility residents—many of whom may have comorbid cognitive decline—researchers found that unrecognized OSA often mimics the progressive cognitive decline seen in major neurocognitive disorders.20 However, classic symptoms of OSA may not always be present in these patients, and their daytime sleepiness is often attributed to old age rather than to a pathological etiology.16 Screening for OSA and prompt initiation of the appropriate treatment may reverse OSA-induced cognitive changes in these patients.21
The primary presenting symptom of OSA is snoring, which is correlated with pauses in breathing. Risk factors include increased body mass index (BMI), thick neck circumference, male sex, and advanced age. In older adults, BMI has a lower impact on the Apnea-Hypopnea Index, an indicator of the number of pauses in breathing per hour, when compared with young and middle-age adults.16 Validated screening questionnaires for OSA include the STOP-Bang Questionnaire (Table 122), OSA50, Berlin Questionnaire, and Epworth Sleepiness Scale, each of which is used in different subpopulations. The current diagnostic standard for OSA is nocturnal polysomnography in a sleep laboratory, but recent advances in home sleep apnea testing have made it a viable, low-cost alternative for patients who do not have significant medical comorbidities.23 Standard utilized cutoffs for diagnosis are ≥5 events/hour (hypopneas associated with at least 4% oxygen desaturations) in conjunction with clinical symptoms of OSA.24
Continue to: Treatment
Treatment. First-line treatment for OSA is continuous positive airway pressure therapy, but adherence rates vary widely with patient education and regular follow-up.25 Adjunctive therapy includes weight loss, oral appliances, and uvulopalatopharyngoplasty, a procedure in which tissue in the throat is remodeled or removed.
Central sleep apnea (CSA) is a pause in breathing without evidence of associated respiratory effort. In adults, the development of CSA is indicative of underlying lower brainstem dysfunction, due to intermittent failures in the pontomedullary centers responsible for regulation of rhythmic breathing.26 This can occur as a consequence of multiple diseases, including congestive heart failure, stroke, renal failure, chronic medication use (opioids), and brain tumors.
The Sleep Heart Health Study—the largest community-based cohort study to date examining CSA—estimated that the prevalence of CSA among adults age >65 was 1.1% (compared with 0.4% in those age <65).27 Subgroup analysis revealed that men had significantly higher rates of CSA compared with women (2.7% vs 0.2%, respectively).
CSA may present similarly to OSA (excessive daytime somnolence, insomnia, poor sleep quality, difficulties with attention and concentration). Symptoms may also mimic those of coexisting medical conditions in older adults, such as nocturnal angina or paroxysmal nocturnal dyspnea.27 Any older patient with daytime sleepiness and risk factors for CSA should be referred for in-laboratory nocturnal polysomnography, the gold standard diagnostic test. Unlike in OSA, ambulatory diagnostic measures (home sleep apnea testing) have not been validated for this disorder.27
Treatment. The primary treatment for CSA is to address the underlying medical problem. Positive pressure ventilation has been attempted with mixed results. Supplemental oxygen and medical management (acetazolamide or theophylline) can help stimulate breathing. Newer studies have shown favorable outcomes with transvenous neurostimulation or adaptive servoventilation.28-30
Continue to: Insomnia
Insomnia. For a primary diagnosis of insomnia, DSM-5 requires at least 3 nights per week of sleep disturbances that induce distress or functional impairment for at least 3 months.31 The International Classification of Disease, 10th Edition requires at least 1 month of symptoms (lying awake for a long time before falling asleep, sleeping for short periods, being awake for most of the night, feeling lack of sleep, waking up early) after ruling out other sleep disorders, substance use, or other medical conditions.4 Clinically, insomnia tends to present in older adults as a subjective complaint of dissatisfaction with the quality and/or quantity of their sleep. Insomnia has been consistently shown to be a significant risk factor for both the development or exacerbation of depression in older adults.32-34
While the diagnosis of insomnia is mainly clinical via a thorough sleep and medication history, assistive ancillary testing can include wrist actigraphy and screening questionnaires (the Insomnia Severity Index and the Pittsburgh Sleep Quality Index).4 Because population studies of older adults have found discrepancies between objective and subjective methods of assessing sleep quality, relying on the accuracy of self-reported symptoms alone is questionable.35
Treatment. Given that drug elimination half-life increases with age, and the risks of adverse effects are increased in older adults, the preferred treatment modalities for insomnia are nonpharmacologic.4 Sleep hygiene education (Table 2) and cognitive-behavioral therapy (CBT) for insomnia are often the first-line therapies.4,36,37 It is crucial to manage comorbidities such as heart disease and obesity, as well as sources of discomfort from conditions such as arthritic pain.38,39 If nonpharmacologic therapies are not effective, pharmacologic options can be considered.4 Before prescribing sleep medications, it may be more fruitful to treat underlying psychiatric disorders such as depression and anxiety with antidepressants.4 Although benzodiazepines are helpful for their sedative effects, they are not recommended for older adults because of an increased risk of falls, rebound insomnia, potential tolerance, and associated cognitive impairment.40 Benzodiazepine receptor agonists (eg, zolpidem, eszopiclone, zaleplon) were initially developed as a first-line treatment for insomnia to replace the reliance on benzodiazepines, but these medications have a “black-box” warning of a serious risk of complex sleep behaviors, including life-threatening parasomnias.41 As a result, guidelines suggest a shorter duration of treatment with a benzodiazepine receptor agonist may still provide benefit while limiting the risk of adverse effects.42
Doxepin is the only antidepressant FDA-approved for insomnia; it improves sleep latency (time taken to initiate sleep after lying down), duration, and quality in adults age >65.43 Melatonin receptor agonists such as ramelteon and melatonin have shown positive results in older patients with insomnia. In clinical trials of patients age ≥65, ramelteon, which is FDA-approved for insomnia, produced no rebound insomnia, withdrawal effects, memory impairment, or gait instability.44-46 Suvorexant, an orexin receptor antagonist, decreases sleep latency and increases total sleep time equally in both young and older adults.47-49Table 340-51 provides a list of medications used to treat insomnia (including off-label agents) and their common adverse effects in older adults.
Parasomnias are undesirable behaviors that occur during sleep, commonly associated with the sleep-wake transition period. These behaviors can occur during REM sleep (nightmare disorder, sleep paralysis, REM sleep behavior disorder) or NREM sleep (somnambulism [sleepwalking], confusional arousals, sleep terrors). According to a cross-sectional Norwegian study of parasomnias, the estimated lifetime prevalence of sleep walking is 22.4%; sleep talking, 66.8%; confusional arousal, 18.5%; and sleep terror, 10.4%.52
Continue to: When evaluating a patient...
When evaluating a patient with parasomnias, it is important to review their drug and substance use as well as coexisting medical conditions. Drugs and substances that can affect sleep include prescription medications (second-generation antidepressants, stimulants, dopamine agonists), excessive caffeine, alcohol, certain foods (coffee, chocolate milk, black tea, caffeinated soft drinks), environmental exposures (smoking, pesticides), and recreational drugs (amphetamines).53-56 Certain medical conditions are correlated with specific parasomnias (eg, sleep paralysis and narcolepsy, REM sleep behavior disorder and Parkinson’s disease [PD], etc.).54 Diagnosis of parasomnias is mainly clinical but supporting evidence can be obtained through in-lab polysomnography.
Treatment. For parasomnias, treatment is primarily supportive and includes creating a safe sleeping environment to reduce the risk of self-harm. Recommendations include sleeping in a room on the ground floor, minimizing furniture in the bedroom, padding any bedside furniture, child-proofing doorknobs, and locking up weapons and other dangerous household items.54
REM sleep behavior disorder (RBD). This disorder is characterized by a loss of the typical REM sleep-associated atonia and the presence of motor activity during dreaming (dream-enacted behaviors). While the estimated incidence of RBD in the general adult population is approximately 0.5%, it increases to 7.7% among those age >60.57 RBD occurs most commonly in the setting of the alpha-synucleinopathies (PD, Lewy body dementia, multisystem atrophy), but can also be found in patients with cerebral ischemia, demyelinating disorders, or alcohol misuse, or can be medication-induced (primarily antidepressants and antipsychotics).58 In patients with PD, the presence of RBD is associated with a more impaired cognitive profile, suggestive of widespread neurodegeneration.59 Recent studies revealed that RBD may also be a prodromal state of neurodegenerative diseases such as PD, which should prompt close monitoring and long-term follow up.60 Similar to other parasomnias, the diagnosis of RBD is primarily clinical, but polysomnography plays an important role in demonstrating loss of REM-related atonia.54
Treatment. Clonazepam and melatonin have been shown to be effective in treating the symptoms of RBD.54
Depression, anxiety, and sleep disturbances
Major depressive disorder (MDD) and generalized anxiety disorder (GAD) affect sleep in patients of all ages, but are underreported in older adults. According to national epidemiologic surveys, the estimated prevalence of MDD and GAD among older adults is 13% and 11.4%, respectively.61,62 Rates as high as 42% and 39% have been reported in meta-regression analyses among patients with Alzheimer’s dementia.63
Continue to: Depression and anxiety
Depression and anxiety may have additive effects and manifest as poor sleep satisfaction, increased sleep latency, insomnia, and daytime sleepiness.64 However, they may also have independent effects. Studies showed that patients with depression alone reported overall poor sleep satisfaction, whereas patients with anxiety alone reported problems with sleep latency, daytime drowsiness, and waking up at night in addition to their overall poor sleep satisfaction.65-67 Both depression and anxiety are risk factors for developing cognitive decline, and may be an early sign/prodrome of neurodegenerative diseases (dementias).68 The bidirectional relationship between depression/anxiety and sleep is complex and needs further investigation.
Treatment. Pharmacologic treatments for patients with depression/anxiety and sleep disturbances include selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, tricyclic antidepressants, and other serotonin receptor agonists.69-72 Nonpharmacologic treatments include CBT for both depression and anxiety, and problem-solving therapy for patients with mild cognitive impairment and depression.73,74 For severe depression and/or anxiety, electroconvulsive therapy is effective.75
Bottom Line
Sleep disorders in older adults are common but often underdiagnosed. Timely recognition of obstructive sleep apnea, central sleep apnea, insomnia, parasomnias, and other sleep disturbances can facilitate effective treatment and greatly improve older adults’ quality of life.
Related Resources
- American Academy of Sleep Medicine. International Classification of Sleep Disorders—Third Edition. https://aasm.org
- SleepFoundation.org. Sleep hygiene. https://www.sleepfoundation.org/articles/sleep-hygiene
Drug Brand Names
Acetazolamide • Diamox
Clonazepam • Klonopin
Doxepin • Silenor
Eszopiclone • Lunesta
Gabapentin • Neurontin
Mirtazapine • Remeron
Pramipexole • Mirapex
Quetiapine • Seroquel
Ramelteon • Rozerem
Suvorexant • Belsomra
Temazepam • Restoril
Theophylline • Elixophyllin
Tiagabine • Gabitril
Trazadone • Desyrel
Triazolam • Halcion
Zaleplon • Sonata
Zolpidem • Ambien
1. Centers for Disease Control and Prevention. The state of aging and health in America. 2013. Accessed January 27, 2021. https://www.cdc.gov/aging/pdf/state-aging-health-in-america-2013.pdf
2. Suzuki K, Miyamoto M, Hirata K. Sleep disorders in the elderly: diagnosis and management. J Gen Fam Med. 2017;18(2):61-71.
3. Inouye SK, Studenski S, Tinetti ME, et al. Geriatric syndromes: clinical, research, and policy implications of a core geriatric concept. J Am Geriatr Soc. 2007;55(5):780-791.
4. Patel D, Steinberg J, Patel P. Insomnia in the elderly: a review. J Clin Sleep Med. 2018;14(6):1017-1024.
5. Neubauer DN. A review of ramelteon in the treatment of sleep disorders. Neuropsychiatr Dis Treat. 2008;4(1):69-79.
6. Mander BA, Winer JR, Walker MP. Sleep and human aging. Neuron. 2017;94(1):19-36.
7. Ohayon MM, Carskadon MA, Guilleminault C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep. 2004;27:1255-1273.
8. Li J, Vitiello MV, Gooneratne NS. Sleep in normal aging. Sleep Med Clin. 2018;13(1):1-11.
9. Floyd JA, Medler SM, Ager JW, et al. Age-related changes in initiation and maintenance of sleep: a meta-analysis. Res Nurs Health. 2000;23(2):106-117.
10. Floyd JA, Janisse JJ, Jenuwine ES, et al. Changes in REM-sleep percentage over the adult lifespan. Sleep. 2007;30(7):829-836.
11. Dorffner G, Vitr M, Anderer P. The effects of aging on sleep architecture in healthy subjects. Adv Exp Med Biol. 2015;821:93-100.
12. Furihata R, Kaneita Y, Jike M, et al. Napping and associated factors: a Japanese nationwide general population survey. Sleep Med. 2016;20:72-79.
13. Foley DJ, Vitiello MV, Bliwise DL, et al. Frequent napping is associated with excessive daytime sleepiness, depression, pain, and nocturia in older adults: findings from the National Sleep Foundation ‘2003 Sleep in America’ Poll. Am J Geriatr Psychiatry. 2007;15(4):344-350.
14. Floyd JA, Janisse JJ, Marshall Medler S, et al. Nonlinear components of age-related change in sleep initiation. Nurs Res. 2000;49(5):290-294.
15. Miner B, Kryger MH. Sleep in the aging population. Sleep Med Clin. 2017;12(1):31-38.
16. Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;165(9):1217-1239.
17. Ancoli-Israel S, Klauber MR, Butters N, et al. Dementia in institutionalized elderly: relation to sleep apnea. J Am Geriatr Soc. 1991;39(3):258-263.
18. Spira AP, Stone KL, Rebok GW, et al. Sleep-disordered breathing and functional decline in older women. J Am Geriatr Soc. 2014;62(11):2040-2046.
19. Vijayan VK. Morbidities associated with obstructive sleep apnea. Expert Rev Respir Med. 2012;6(5):557-566.
20. Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry. 2016;24(6):496-508.
21. Dalmases M, Solé-Padullés C, Torres M, et al. Effect of CPAP on cognition, brain function, and structure among elderly patients with OSA: a randomized pilot study. Chest. 2015;148(5):1214-1223.
22. Toronto Western Hospital, University Health Network. University of Toronto. STOP-Bang Questionnaire. 2012. Accessed January 26, 2021. www.stopbang.ca
23. Freedman N. Doing it better for less: incorporating OSA management into alternative payment models. Chest. 2019;155(1):227-233.
24. Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine clinical practice guideline. J Clin Sleep Med. 2017;13(3):479-504.
25. Semelka M, Wilson J, Floyd R. Diagnosis and treatment of obstructive sleep apnea in adults. Am Fam Physician. 2016;94(5):355-360.
26. Javaheri S, Dempsey JA. Central sleep apnea. Compr Physiol. 2013;3(1):141-163.
27. Donovan LM, Kapur VK. Prevalence and characteristics of central compared to obstructive sleep apnea: analyses from the Sleep Heart Health Study cohort. Sleep. 2016;39(7):1353-1359.
28. Cao M, Cardell CY, Willes L, et al. A novel adaptive servoventilation (ASVAuto) for the treatment of central sleep apnea associated with chronic use of opioids. J Clin Sleep Med. 2014;10(8):855-861.
29. Oldenburg O, Spießhöfer J, Fox H, et al. Performance of conventional and enhanced adaptive servoventilation (ASV) in heart failure patients with central sleep apnea who have adapted to conventional ASV. Sleep Breath. 2015;19(3):795-800.
30. Costanzo MR, Ponikowski P, Javaheri S, et al. Transvenous neurostimulation for central sleep apnoea: a randomised controlled trial. Lancet. 2016;388(10048):974-982.
31. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013:362.
32. Perlis ML, Smith LJ, Lyness JM, et al. Insomnia as a risk factor for onset of depression in the elderly. Behav Sleep Med. 2006;4(2):104-113.
33. Cole MG, Dendukuri N. Risk factors for depression among elderly community subjects: a systematic review and meta-analysis. Am J Psychiatry. 2003;160(6):1147-1156.
34. Pigeon WR, Hegel M, Unützer J, et al. Is insomnia a perpetuating factor for late-life depression in the IMPACT cohort? Sleep. 2008;31(4):481-488.
35. Hughes JM, Song Y, Fung CH, et al. Measuring sleep in vulnerable older adults: a comparison of subjective and objective sleep measures. Clin Gerontol. 2018;41(2):145-157.
36. Irish LA, Kline CE, Gunn HE, et al. The role of sleep hygiene in promoting public health: a review of empirical evidence. Sleep Med Rev. 2015;22:23-36.
37. Sleep Foundation. Sleep hygiene. Accessed January 27, 2021. https://www.sleepfoundation.org/articles/sleep-hygiene
38. Foley D, Ancoli-Israel S, Britz P, et al. Sleep disturbances and chronic disease in older adults: results of the 2003 National Sleep Foundation Sleep in America Survey. J Psychosom Res. 2004;56(5):497-502.
39. Eslami V, Zimmerman ME, Grewal T, et al. Pain grade and sleep disturbance in older adults: evaluation the role of pain, and stress for depressed and non-depressed individuals. Int J Geriatr Psychiatry. 2016;31(5):450-457.
40. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
41. United States Food & Drug Administration. FDA adds Boxed Warning for risk of serious injuries caused by sleepwalking with certain prescription insomnia medicines. 2019. Accessed January 27, 2021. https://www.fda.gov/drugs/drug-safety-and-availability/fda-adds-boxed-warning-risk-serious-injuries-caused-sleepwalking-certain-prescription-insomnia
42. Schroeck JL, Ford J, Conway EL, et al. Review of safety and efficacy of sleep medicines in older adults. Clin Ther. 2016;38(11):2340-2372.
43. Krystal AD, Lankford A, Durrence HH, et al. Efficacy and safety of doxepin 3 and 6 mg in a 35-day sleep laboratory trial in adults with chronic primary insomnia. Sleep. 2011;34(10):1433-1442.
44. Roth T, Seiden D, Sainati S, et al. Effects of ramelteon on patient-reported sleep latency in older adults with chronic insomnia. Sleep Med. 2006;7(4):312-318.
45. Zammit G, Wang-Weigand S, Rosenthal M, et al. Effect of ramelteon on middle-of-the-night balance in older adults with chronic insomnia. J Clin Sleep Med. 2009;5(1):34-40.
46. Mets MAJ, de Vries JM, de Senerpont Domis LM, et al. Next-day effects of ramelteon (8 mg), zopiclone (7.5 mg), and placebo on highway driving performance, memory functioning, psychomotor performance, and mood in healthy adult subjects. Sleep. 2011;34(10):1327-1334.
47. Rhyne DN, Anderson SL. Suvorexant in insomnia: efficacy, safety and place in therapy. Ther Adv Drug Saf. 2015;6(5):189-195.
48. Norman JL, Anderson SL. Novel class of medications, orexin receptor antagonists, in the treatment of insomnia - critical appraisal of suvorexant. Nat Sci Sleep. 2016;8:239-247.
49. Owen RT. Suvorexant: efficacy and safety profile of a dual orexin receptor antagonist in treating insomnia. Drugs Today (Barc). 2016;52(1):29-40.
50. Shannon S, Lewis N, Lee H, et al. Cannabidiol in anxiety and sleep: a large case series. Perm J. 2019;23:18-041. doi: 10.7812/TPP/18-041
51. Yunusa I, Alsumali A, Garba AE, et al. Assessment of reported comparative effectiveness and safety of atypical antipsychotics in the treatment of behavioral and psychological symptoms of dementia: a network meta-analysis. JAMA Netw Open. 2019;2(3):e190828.
52. Bjorvatn B, Gronli J, Pallesen S. Prevalence of different parasomnias in the general population. Sleep Med. 2010;11(10):1031-1034.
53. Postuma RB, Montplaisir JY, Pelletier A, et al. Environmental risk factors for REM sleep behavior disorder: a multicenter case-control study. Neurology. 2012;79(5):428-434.
54. Fleetham JA, Fleming JA. Parasomnias. CMAJ. 2014;186(8):E273-E280.
55. Dinis-Oliveira RJ, Caldas I, Carvalho F, et al. Bruxism after 3,4-methylenedioxymethamphetamine (ecstasy) abuse. Clin Toxicol (Phila.) 2010;48(8):863-864.
56. Irfan MH, Howell MJ. Rapid eye movement sleep behavior disorder: overview and current perspective. Curr Sleep Medicine Rep. 2016;2:64-73.
57. Mahlknecht P, Seppi K, Frauscher B, et al. Probable RBD and association with neurodegenerative disease markers: a population-based study. Mov Disord. 2015;30(10):1417-1421.
58. Oertel WH, Depboylu C, Krenzer M, et al. [REM sleep behavior disorder as a prodromal stage of α-synucleinopathies: symptoms, epidemiology, pathophysiology, diagnosis and therapy]. Nervenarzt. 2014;85:19-25. German.
59. Jozwiak N, Postuma RB, Montplaisir J, et al. REM sleep behavior disorder and cognitive impairment in Parkinson’s disease. Sleep. 2017;40(8):zsx101. doi: 10.1093/sleep/zsx101
60. Claassen DO, Josephs KA, Ahlskog JE, et al. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology 2010;75(6):494-499.
61. Reynolds K, Pietrzak RH, El-Gabalawy R, et al. Prevalence of psychiatric disorders in U.S. older adults: findings from a nationally representative survey. World Psychiatry. 2015;14(1):74-81.
62. Lohman MC, Mezuk B, Dumenci L. Depression and frailty: concurrent risks for adverse health outcomes. Aging Ment Health. 2017;21(4):399-408.
63. Zhao QF, Tan L, Wang HF, et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: systematic review and meta-analysis. J Affect Disord. 2016;190:264-271.
64. Furihata R, Hall MH, Stone KL, et al. An aggregate measure of sleep health is associated with prevalent and incident clinically significant depression symptoms among community-dwelling older women. Sleep. 2017;40(3):zsw075. doi: 10.1093/sleep/zsw075
65. Spira AP, Stone K, Beaudreau SA, et al. Anxiety symptoms and objectively measured sleep quality in older women. Am J Geriatr Psychiatry. 2009;17(2):136-143.
66. Press Y, Punchik B, Freud T. The association between subjectively impaired sleep and symptoms of depression and anxiety in a frail elderly population. Aging Clin Exp Res. 2018;30(7):755-765.
67. Gould CE, Spira AP, Liou-Johnson V, et al. Association of anxiety symptom clusters with sleep quality and daytime sleepiness. J Gerontol B Psychol Sci Soc Sci. 2018;73(3):413-420.
68. Kassem AM, Ganguli M, Yaffe K, et al. Anxiety symptoms and risk of cognitive decline in older community-dwelling men. Int Psychogeriatr. 2017;29(7):1137-1145.
69. Frank C. Pharmacologic treatment of depression in the elderly. Can Fam Physician. 2014;60(2):121-126.
70. Subramanyam AA, Kedare J, Singh OP, et al. Clinical practice guidelines for geriatric anxiety disorders. Indian J Psychiatry. 2018;60(suppl 3):S371-S382.
71. Emsley R, Ahokas A, Suarez A, et al. Efficacy of tianeptine 25-50 mg in elderly patients with recurrent major depressive disorder: an 8-week placebo- and escitalopram-controlled study. J Clin Psychiatry. 2018;79(4):17m11741. doi: 10.4088/JCP.17m11741
72. Semel D, Murphy TK, Zlateva G, et al. Evaluation of the safety and efficacy of pregabalin in older patients with neuropathic pain: results from a pooled analysis of 11 clinical studies. BMC Fam Pract. 2010;11:85.
73. Orgeta V, Qazi A, Spector A, et al. Psychological treatments for depression and anxiety in dementia and mild cognitive impairment: systematic review and meta-analysis. Br J Psychiatry. 2015;207(4):293-298.
74. Morimoto SS, Kanellopoulos D, Manning KJ, et al. Diagnosis and treatment of depression and cognitive impairment in late life. Ann N Y Acad Sci. 2015;1345(1):36-46.
75. Casey DA. Depression in older adults: a treatable medical condition. Prim Care. 2017;44(3):499-510.
1. Centers for Disease Control and Prevention. The state of aging and health in America. 2013. Accessed January 27, 2021. https://www.cdc.gov/aging/pdf/state-aging-health-in-america-2013.pdf
2. Suzuki K, Miyamoto M, Hirata K. Sleep disorders in the elderly: diagnosis and management. J Gen Fam Med. 2017;18(2):61-71.
3. Inouye SK, Studenski S, Tinetti ME, et al. Geriatric syndromes: clinical, research, and policy implications of a core geriatric concept. J Am Geriatr Soc. 2007;55(5):780-791.
4. Patel D, Steinberg J, Patel P. Insomnia in the elderly: a review. J Clin Sleep Med. 2018;14(6):1017-1024.
5. Neubauer DN. A review of ramelteon in the treatment of sleep disorders. Neuropsychiatr Dis Treat. 2008;4(1):69-79.
6. Mander BA, Winer JR, Walker MP. Sleep and human aging. Neuron. 2017;94(1):19-36.
7. Ohayon MM, Carskadon MA, Guilleminault C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep. 2004;27:1255-1273.
8. Li J, Vitiello MV, Gooneratne NS. Sleep in normal aging. Sleep Med Clin. 2018;13(1):1-11.
9. Floyd JA, Medler SM, Ager JW, et al. Age-related changes in initiation and maintenance of sleep: a meta-analysis. Res Nurs Health. 2000;23(2):106-117.
10. Floyd JA, Janisse JJ, Jenuwine ES, et al. Changes in REM-sleep percentage over the adult lifespan. Sleep. 2007;30(7):829-836.
11. Dorffner G, Vitr M, Anderer P. The effects of aging on sleep architecture in healthy subjects. Adv Exp Med Biol. 2015;821:93-100.
12. Furihata R, Kaneita Y, Jike M, et al. Napping and associated factors: a Japanese nationwide general population survey. Sleep Med. 2016;20:72-79.
13. Foley DJ, Vitiello MV, Bliwise DL, et al. Frequent napping is associated with excessive daytime sleepiness, depression, pain, and nocturia in older adults: findings from the National Sleep Foundation ‘2003 Sleep in America’ Poll. Am J Geriatr Psychiatry. 2007;15(4):344-350.
14. Floyd JA, Janisse JJ, Marshall Medler S, et al. Nonlinear components of age-related change in sleep initiation. Nurs Res. 2000;49(5):290-294.
15. Miner B, Kryger MH. Sleep in the aging population. Sleep Med Clin. 2017;12(1):31-38.
16. Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;165(9):1217-1239.
17. Ancoli-Israel S, Klauber MR, Butters N, et al. Dementia in institutionalized elderly: relation to sleep apnea. J Am Geriatr Soc. 1991;39(3):258-263.
18. Spira AP, Stone KL, Rebok GW, et al. Sleep-disordered breathing and functional decline in older women. J Am Geriatr Soc. 2014;62(11):2040-2046.
19. Vijayan VK. Morbidities associated with obstructive sleep apnea. Expert Rev Respir Med. 2012;6(5):557-566.
20. Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry. 2016;24(6):496-508.
21. Dalmases M, Solé-Padullés C, Torres M, et al. Effect of CPAP on cognition, brain function, and structure among elderly patients with OSA: a randomized pilot study. Chest. 2015;148(5):1214-1223.
22. Toronto Western Hospital, University Health Network. University of Toronto. STOP-Bang Questionnaire. 2012. Accessed January 26, 2021. www.stopbang.ca
23. Freedman N. Doing it better for less: incorporating OSA management into alternative payment models. Chest. 2019;155(1):227-233.
24. Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine clinical practice guideline. J Clin Sleep Med. 2017;13(3):479-504.
25. Semelka M, Wilson J, Floyd R. Diagnosis and treatment of obstructive sleep apnea in adults. Am Fam Physician. 2016;94(5):355-360.
26. Javaheri S, Dempsey JA. Central sleep apnea. Compr Physiol. 2013;3(1):141-163.
27. Donovan LM, Kapur VK. Prevalence and characteristics of central compared to obstructive sleep apnea: analyses from the Sleep Heart Health Study cohort. Sleep. 2016;39(7):1353-1359.
28. Cao M, Cardell CY, Willes L, et al. A novel adaptive servoventilation (ASVAuto) for the treatment of central sleep apnea associated with chronic use of opioids. J Clin Sleep Med. 2014;10(8):855-861.
29. Oldenburg O, Spießhöfer J, Fox H, et al. Performance of conventional and enhanced adaptive servoventilation (ASV) in heart failure patients with central sleep apnea who have adapted to conventional ASV. Sleep Breath. 2015;19(3):795-800.
30. Costanzo MR, Ponikowski P, Javaheri S, et al. Transvenous neurostimulation for central sleep apnoea: a randomised controlled trial. Lancet. 2016;388(10048):974-982.
31. Diagnostic and statistical manual of mental disorders, 5th ed. American Psychiatric Association; 2013:362.
32. Perlis ML, Smith LJ, Lyness JM, et al. Insomnia as a risk factor for onset of depression in the elderly. Behav Sleep Med. 2006;4(2):104-113.
33. Cole MG, Dendukuri N. Risk factors for depression among elderly community subjects: a systematic review and meta-analysis. Am J Psychiatry. 2003;160(6):1147-1156.
34. Pigeon WR, Hegel M, Unützer J, et al. Is insomnia a perpetuating factor for late-life depression in the IMPACT cohort? Sleep. 2008;31(4):481-488.
35. Hughes JM, Song Y, Fung CH, et al. Measuring sleep in vulnerable older adults: a comparison of subjective and objective sleep measures. Clin Gerontol. 2018;41(2):145-157.
36. Irish LA, Kline CE, Gunn HE, et al. The role of sleep hygiene in promoting public health: a review of empirical evidence. Sleep Med Rev. 2015;22:23-36.
37. Sleep Foundation. Sleep hygiene. Accessed January 27, 2021. https://www.sleepfoundation.org/articles/sleep-hygiene
38. Foley D, Ancoli-Israel S, Britz P, et al. Sleep disturbances and chronic disease in older adults: results of the 2003 National Sleep Foundation Sleep in America Survey. J Psychosom Res. 2004;56(5):497-502.
39. Eslami V, Zimmerman ME, Grewal T, et al. Pain grade and sleep disturbance in older adults: evaluation the role of pain, and stress for depressed and non-depressed individuals. Int J Geriatr Psychiatry. 2016;31(5):450-457.
40. American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
41. United States Food & Drug Administration. FDA adds Boxed Warning for risk of serious injuries caused by sleepwalking with certain prescription insomnia medicines. 2019. Accessed January 27, 2021. https://www.fda.gov/drugs/drug-safety-and-availability/fda-adds-boxed-warning-risk-serious-injuries-caused-sleepwalking-certain-prescription-insomnia
42. Schroeck JL, Ford J, Conway EL, et al. Review of safety and efficacy of sleep medicines in older adults. Clin Ther. 2016;38(11):2340-2372.
43. Krystal AD, Lankford A, Durrence HH, et al. Efficacy and safety of doxepin 3 and 6 mg in a 35-day sleep laboratory trial in adults with chronic primary insomnia. Sleep. 2011;34(10):1433-1442.
44. Roth T, Seiden D, Sainati S, et al. Effects of ramelteon on patient-reported sleep latency in older adults with chronic insomnia. Sleep Med. 2006;7(4):312-318.
45. Zammit G, Wang-Weigand S, Rosenthal M, et al. Effect of ramelteon on middle-of-the-night balance in older adults with chronic insomnia. J Clin Sleep Med. 2009;5(1):34-40.
46. Mets MAJ, de Vries JM, de Senerpont Domis LM, et al. Next-day effects of ramelteon (8 mg), zopiclone (7.5 mg), and placebo on highway driving performance, memory functioning, psychomotor performance, and mood in healthy adult subjects. Sleep. 2011;34(10):1327-1334.
47. Rhyne DN, Anderson SL. Suvorexant in insomnia: efficacy, safety and place in therapy. Ther Adv Drug Saf. 2015;6(5):189-195.
48. Norman JL, Anderson SL. Novel class of medications, orexin receptor antagonists, in the treatment of insomnia - critical appraisal of suvorexant. Nat Sci Sleep. 2016;8:239-247.
49. Owen RT. Suvorexant: efficacy and safety profile of a dual orexin receptor antagonist in treating insomnia. Drugs Today (Barc). 2016;52(1):29-40.
50. Shannon S, Lewis N, Lee H, et al. Cannabidiol in anxiety and sleep: a large case series. Perm J. 2019;23:18-041. doi: 10.7812/TPP/18-041
51. Yunusa I, Alsumali A, Garba AE, et al. Assessment of reported comparative effectiveness and safety of atypical antipsychotics in the treatment of behavioral and psychological symptoms of dementia: a network meta-analysis. JAMA Netw Open. 2019;2(3):e190828.
52. Bjorvatn B, Gronli J, Pallesen S. Prevalence of different parasomnias in the general population. Sleep Med. 2010;11(10):1031-1034.
53. Postuma RB, Montplaisir JY, Pelletier A, et al. Environmental risk factors for REM sleep behavior disorder: a multicenter case-control study. Neurology. 2012;79(5):428-434.
54. Fleetham JA, Fleming JA. Parasomnias. CMAJ. 2014;186(8):E273-E280.
55. Dinis-Oliveira RJ, Caldas I, Carvalho F, et al. Bruxism after 3,4-methylenedioxymethamphetamine (ecstasy) abuse. Clin Toxicol (Phila.) 2010;48(8):863-864.
56. Irfan MH, Howell MJ. Rapid eye movement sleep behavior disorder: overview and current perspective. Curr Sleep Medicine Rep. 2016;2:64-73.
57. Mahlknecht P, Seppi K, Frauscher B, et al. Probable RBD and association with neurodegenerative disease markers: a population-based study. Mov Disord. 2015;30(10):1417-1421.
58. Oertel WH, Depboylu C, Krenzer M, et al. [REM sleep behavior disorder as a prodromal stage of α-synucleinopathies: symptoms, epidemiology, pathophysiology, diagnosis and therapy]. Nervenarzt. 2014;85:19-25. German.
59. Jozwiak N, Postuma RB, Montplaisir J, et al. REM sleep behavior disorder and cognitive impairment in Parkinson’s disease. Sleep. 2017;40(8):zsx101. doi: 10.1093/sleep/zsx101
60. Claassen DO, Josephs KA, Ahlskog JE, et al. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology 2010;75(6):494-499.
61. Reynolds K, Pietrzak RH, El-Gabalawy R, et al. Prevalence of psychiatric disorders in U.S. older adults: findings from a nationally representative survey. World Psychiatry. 2015;14(1):74-81.
62. Lohman MC, Mezuk B, Dumenci L. Depression and frailty: concurrent risks for adverse health outcomes. Aging Ment Health. 2017;21(4):399-408.
63. Zhao QF, Tan L, Wang HF, et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: systematic review and meta-analysis. J Affect Disord. 2016;190:264-271.
64. Furihata R, Hall MH, Stone KL, et al. An aggregate measure of sleep health is associated with prevalent and incident clinically significant depression symptoms among community-dwelling older women. Sleep. 2017;40(3):zsw075. doi: 10.1093/sleep/zsw075
65. Spira AP, Stone K, Beaudreau SA, et al. Anxiety symptoms and objectively measured sleep quality in older women. Am J Geriatr Psychiatry. 2009;17(2):136-143.
66. Press Y, Punchik B, Freud T. The association between subjectively impaired sleep and symptoms of depression and anxiety in a frail elderly population. Aging Clin Exp Res. 2018;30(7):755-765.
67. Gould CE, Spira AP, Liou-Johnson V, et al. Association of anxiety symptom clusters with sleep quality and daytime sleepiness. J Gerontol B Psychol Sci Soc Sci. 2018;73(3):413-420.
68. Kassem AM, Ganguli M, Yaffe K, et al. Anxiety symptoms and risk of cognitive decline in older community-dwelling men. Int Psychogeriatr. 2017;29(7):1137-1145.
69. Frank C. Pharmacologic treatment of depression in the elderly. Can Fam Physician. 2014;60(2):121-126.
70. Subramanyam AA, Kedare J, Singh OP, et al. Clinical practice guidelines for geriatric anxiety disorders. Indian J Psychiatry. 2018;60(suppl 3):S371-S382.
71. Emsley R, Ahokas A, Suarez A, et al. Efficacy of tianeptine 25-50 mg in elderly patients with recurrent major depressive disorder: an 8-week placebo- and escitalopram-controlled study. J Clin Psychiatry. 2018;79(4):17m11741. doi: 10.4088/JCP.17m11741
72. Semel D, Murphy TK, Zlateva G, et al. Evaluation of the safety and efficacy of pregabalin in older patients with neuropathic pain: results from a pooled analysis of 11 clinical studies. BMC Fam Pract. 2010;11:85.
73. Orgeta V, Qazi A, Spector A, et al. Psychological treatments for depression and anxiety in dementia and mild cognitive impairment: systematic review and meta-analysis. Br J Psychiatry. 2015;207(4):293-298.
74. Morimoto SS, Kanellopoulos D, Manning KJ, et al. Diagnosis and treatment of depression and cognitive impairment in late life. Ann N Y Acad Sci. 2015;1345(1):36-46.
75. Casey DA. Depression in older adults: a treatable medical condition. Prim Care. 2017;44(3):499-510.

Can lifestyle modifications delay or prevent Alzheimer’s disease?
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4

Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75

On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4
Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75
On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
Clinicians have devoted strenuous efforts to secondary prevention of Alzheimer’s disease (AD) by diagnosing and treating patients as early as possible. Unfortunately, there is no cure for AD, and the field has witnessed recurrent failures of several pharmacotherapy candidates with either symptomatic or disease-modifying properties.1 An estimated one-third of AD cases can be attributed to modifiable risk factors.2 Thus, implementing primary prevention measures by addressing modifiable risk factors thought to contribute to the disease, with the goal of reducing the risk of developing AD, or at least delaying its onset, is a crucial public health strategy.
Cardiovascular risk factors, such as hypertension, hyperlipidemia, diabetes, hyperhomocysteinemia, obesity, and smoking, have emerged as substantive risk factors for AD.3 Optimal management of these major risk factors, especially in mid-life, may be a preventive approach against AD. Although detailing the evidence on the impact of managing cardiovascular risk factors to delay or prevent AD is beyond the scope of this article, it is becoming clear that “what is good for the heart is good for the brain.”
Additional modifiable risk factors are related to lifestyle habits, such as physical exercise, mental and social activity, meditation/spiritual activity, and diet. This article reviews the importance of pursuing a healthy lifestyle in delaying AD, with the corresponding levels of evidence that support each specific lifestyle modification. The levels of evidence are defined in Table 1.4
Physical exercise
Twenty-one percent of AD cases in the United States are attributable to physical inactivity.5 In addition to its beneficial effect on metabolic syndrome, in animal and human research, regular exercise has been shown to have direct neuroprotective effects. High levels of physical activity increase hippocampal neurogenesis and neuroplasticity, increase vascular circulation in the brain regions implicated in AD, and modulate inflammatory mediators as well as brain growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF-1).6
The definition of regular physical exercise varies across the literature, but usually implies aerobic exercise—an ongoing activity sufficient to increase the heart rate and the need for oxygen, sustained for 20 to 30 minutes per session.7 Modalities include household activities and leisure-time activities. In a large prospective cohort study, Scarmeas et al8 categorized leisure-time activities into 3 types:
- light (walking, dancing, calisthenics, golfing, bowling, gardening, horseback riding)
- moderate (bicycling, swimming, hiking, playing tennis)
- vigorous (aerobic dancing, jogging, playing handball).
These types of physical exercise were weighed by the frequency of participation per week. Compared with being physically inactive, low levels of weekly physical activity (0.1 hours of vigorous, 0.8 hours of moderate, or 1.3 hours of light exercise) were associated with a 29% to 41% lower risk of developing AD, while higher weekly physical activity (1.3 hours of vigorous, 2.3 hours of moderate, or 3.8 hours of light exercise) were associated with a 37% to 50% lower risk (level III).8
In another 20-year cohort study, engaging in leisure-time physical activity at least twice a week in mid-life was significantly associated with a reduced risk of AD, after adjusting for age, sex, education, follow-up time, locomotor disorders, apolipoprotein E (ApoE) genotype, vascular disorders, smoking, and alcohol intake (level III).9 Moreover, a systematic review of 29 randomized controlled trials (RCTs) showed that aerobic exercise training, such as brisk walking, jogging, and biking, was associated with improvements in attention, processing speed, executive function, and memory among healthy older adults and those with mild cognitive impairment (MCI; level IA).10
Continue to: From a pathophysiological standpoint...
From a pathophysiological standpoint, higher levels of physical exercise in cognitively intact older adults have been associated with reduced brain amyloid beta deposits, especially in ApoE4 carriers.11 This inverse relationship also has been demonstrated in patients who are presymptomatic who carry 1 of the 3 known autosomal dominant mutations for the familial forms of AD.12
Overall, physicians should recommend that patients—especially those with cardiovascular risk factors that increase their risk for AD—exercise regularly by following the guidelines of the American Heart Association or the American College of Sports Medicine.13 These include muscle-strengthening activities (legs, hips, back, abdomen, shoulders, and arms) at least 2 days/week, in addition to either 30 minutes/day of moderate-intensity aerobic activity such as brisk walking, 5 days/week; or 25 minutes of vigorous aerobic activity such as jogging and running, 3 days/week14 (level IA evidence for overall improvement in cognitive function; level III evidence for AD delay/risk reduction). Neuromotor exercise, such as yoga and tai chi, and flexibility exercise such as muscle stretching, especially after a hot bath, 2 to 3 days/week are also recommended (level III).15
Mental activity
Nineteen percent of AD cases worldwide and 7% in the United States. can be attributed to low educational attainment, which is associated with low brain cognitive reserve.5 Cognitive resilience in later life may be enhanced by building brain reserves through intellectual stimulation, which affects neuronal branching and plasticity.16 Higher levels of complex mental activities measured across the lifespan, such as education, occupation, reading, and writing, are correlated with significantly less hippocampal volume shrinkage over time.17 Frequent participation in mentally stimulating activities—such as listening to the radio; reading newspapers, magazines, or books; playing games (cards, checkers, crosswords or other puzzles); and visiting museums—was associated with an up to 64% reduction in the odds of developing AD in a cohort of cognitively intact older adults followed for 4 years.18 The correlation between mental activity and AD was found to be independent of physical activity, social activity, or baseline cognitive function.19
In a large cohort of cognitively intact older adults (mean age 70), engaging in a mentally stimulating activity (craft activities, computer use, or going to the theater/movies) once to twice a week was significantly associated with a reduced incidence of amnestic MCI.20 Another prospective 21-year study demonstrated a significant reduction in AD risk in community-dwelling cognitively intact older adults (age 75 to 85) who participated in cognitively stimulating activities, such as reading books or newspapers, writing for pleasure, doing crossword puzzles, playing board games or cards, or playing musical instruments, several times/week.21
Growing scientific evidence also suggests that lifelong multilingualism can delay AD onset by 4 to 5 years.22 Multilingualism is associated with greater cognitive reserve, gray matter volume, functional connectivity and white matter density.23
Continue to: Physicians should encourage their patients...
Physicians should encourage their patients to engage in intellectually stimulating activities and creative leisure-time activities several times/week to enhance their cognitive reserves and delay AD onset (level III evidence with respect to AD risk reduction/delay).
Social activity
Social engagement may be an additional protective factor against AD. In a large 4-year prospective study, increased loneliness in cognitively intact older adults doubled the risk of AD.24 Data from the large French cohort PAQUID (Personnes Agées QUID) emphasized the importance of a patient’s social network as a protective factor against AD. In this cohort, the perception of reciprocity in relationships with others (the perception that a person had received more than he or she had given) was associated with a 53% reduction in AD risk (level III).25 In another longitudinal cohort study, social activity was found to decrease the incidence of subjective cognitive decline, which is a prodromal syndrome for MCI and AD (level III).26
A major confounder in studies assessing for social activity is the uncertainty if social withdrawal is a modifiable risk factor or an early manifestation of AD, since apathetic patients with AD tend to be socially withdrawn.27 Another limitation of measuring the impact of social activity relative to AD risk is the difficulty in isolating social activities from activities that have physical and mental activity components, such as leisure-time activities.28
Meditation/spiritual activity
Chronic psychological stress is believed to compromise limbic structures that regulate stress-related behaviors and the memory network, which might explain how being prone to psychological distress may be associated with MCI or AD.29 Cognitive stress may increase the oxidative stress and telomere shortening implicated in the neurodegenerative processes of AD.30 In one study, participants who were highly prone to psychological distress were found to be at 3 times increased risk for developing AD, after adjusting for depression symptoms and physical and mental activities (level III).31 By reducing chronic psychological stress, meditation techniques offer a promising preventive option against AD.
Mindfulness-based interventions (MBI) have gained increased attention in the past decade. They entail directing one’s attention towards the present moment, thereby decreasing ruminative thoughts and stress arousal.32 Recent RCTs have shown that MBI may promote brain health in older adults not only by improving psychological well-being but also by improving attentional control33 and functional connectivity in brain regions implicated in executive functioning,34 as well as by modulating inflammatory processes implicated in AD.35 Furthermore, an RCT of patients diagnosed with MCI found that compared with memory enhancement training, a weekly 60-minute yoga session improved memory and executive functioning.36
Continue to: Kirtan Kriya is a medication technique...
Kirtan Kriya is a meditation technique that is easy to learn and practice by older adults and can improve memory in patients at risk for developing AD.37 However, more rigorous RCTs conducted in larger samples of older adults are needed to better evaluate the effect of all meditation techniques for delaying or preventing AD (level IB with respect to improvement in cognitive functioning/level III for AD delay/risk reduction).38
Spiritual activities, such as going to places of worship or religious meditation, have been associated with a lower prevalence of AD. Attending religious services, gatherings, or retreats involves a social component because these activities often are practiced in groups. They also confer a method of dealing with psychological distress and depression. Additionally, frequent readings of religious texts represents a mentally stimulating activity that may also contribute to delaying/preventing AD (level III).39
Diet
In the past decade, a growing body of evidence has linked diet to cognition. Individuals with a higher intake of calories and fat are at higher risk for developing AD.40 The incidence of AD rose in Japan after the country transitioned to a more Westernized diet.41 A modern Western diet rich in saturated fatty acids and simple carbohydrates may negatively impact hippocampus-mediated functions such as memory and learning, and is associated with an increased risk of AD.42 In contrast with high-glycemic and fatty diets, a “healthy diet” is associated with a decrease in beta-amyloid burden, inflammation, and oxidative stress.43,44
Studies focusing on dietary patterns rather than a single nutrient for delaying or preventing AD have yielded more robust and consistent results.45 In a recent meta-analysis, adhering to a Mediterranean diet—which is rich in fruits and vegetables, whole grains, olive oil, and fish; moderate in some dairy products and wine; and low in red meat—was associated with a decreased risk of AD; this evidence was derived mostly from epidemiologic studies.46 Scarmeas et al8 found that high adherence to the Mediterranean diet was associated with 32% to 40% reduced risk of AD. Combining this diet with physical exercise was associated with an up to 67% reduced risk (level III). The Dietary Approaches to Stop Hypertension (DASH) diet, which is rich in total grains, fruits, vegetables, and dairy products, but low in sodium and sweets, correlated with neurocognitive improvement in patients with hypertension.47 Both the Mediterranean and DASH diets have been associated with better cognitive function48 and slower cognitive decline.49 Thus, an attempt to combine the neuroprotective components from both diets led to the creation of the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet, which also has been associated with a lower incidence of AD.50
Besides specific diets, some food groups have also been found to promote brain health and may help delay or prevent AD. Berries have the highest amount of antioxidants of all fruit. Among vegetables, tomatoes and green leafy vegetables have the highest amount of nutrients for the brain. Nuts, such as walnuts, which are rich in omega-3 fatty acids, are also considered “power foods” for the brain; however, they should be consumed in moderation because they are also rich in fat. Monounsaturated fatty acids, which are found in olives and olive oil, are also beneficial for the brain. Among the 3 types of omega-3 fatty acids, the most important for cognition is docosahexaenoic acid (DHA) because it constitutes 40% of all fatty acids in the brain. Mainly found in oily fish, DHA has antioxidant and anti-inflammatory properties that may delay or prevent AD. Low levels of DHA have been found in patients with AD.51
Continue to: Curcumin, which is derived from...
Curcumin, which is derived from the curry spice turmeric, is a polyphenol with anti-inflammatory, antioxidant, and anti-amyloid properties that may have a promising role in preventing AD in cognitively intact individuals. Initial trials with curcumin have yielded mixed results on cognition, which was partly related to the low solubility and bioavailability of its formulation.52 However, a recent 18-month double-blind randomized placebo-controlled trial found positive effects on memory and attention, as well as reduction of amyloid plaques and tau tangles deposition in the brain, in non-demented older adults age 51 to 84 who took Theracumin, a highly absorptive oral form of curcumin dispersed with colloidal nanoparticles.53 A longer follow-up is required to determine if curcumin can delay or prevent AD.
Alcohol
The role of alcohol in AD prevention is controversial. Overall, data from prospective studies has shown that low to moderate alcohol consumption may be associated with a reduced risk of AD (level III).54 Alcohol drinking in mid-life showed a U-shaped relationship with cognitive impairment; both abstainers and heavy drinkers had an increased risk of cognitive decline compared with light to moderate drinkers (level III).55 Binge drinking significantly increased the odds of cognitive decline, even after controlling for total alcohol consumption per week.55
The definition of low-to-moderate drinking varies substantially among countries. In addition, the size and amount of alcohol contained in a standard drink may differ.56 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA),57 moderate drinking is defined as up to 1 drink daily for women and 2 drinks daily for men. Binge drinking involves drinking >4 drinks for women and >5 drinks for men, in approximately 2 hours, at least monthly. In the United States, one standard drink contains 14 grams of pure alcohol, which is usually found in 12 ounces of regular beer, 5 ounces of wine, and 1.5 ounces of distilled spirits (vodka or whiskey).58
In a 5-year prospective Canadian study, having 1 drink weekly (especially wine) was associated with an up to 50% reduced risk of AD (level III).59 In the French cohort PAQUID, mild drinkers (<1 to 2 drinks/day) and moderate drinkers (3 to 4 drinks daily) had a reduced incidence of AD compared with non-drinkers. Wine was the most frequently consumed beverage in this study.60 Other studies have found cognitive benefits from mild to moderate drinking regardless of beverage type.54 However, a recent study that included a 30-year follow-up failed to find a significant protective effect of light drinking over abstinence in terms of hippocampal atrophy.61 Atrophy of the hippocampus was correlated with increasing alcohol amounts in a dose-dependent manner, starting at 7 to 14 drinks/week (level III).61
Research has shown that moderate and heavy alcohol use or misuse can directly induce microglial activation and inflammatory mediators’ release, which induce amyloid beta pathology and leads to brain atrophy.62 Hence, non-drinkers should not be advised to begin drinking, because of the lack of RCTs and the concern that beginning to drink may lead to heavy drinking. All drinkers should be advised to adhere to the NIAAA recommendations.13
Continue to: Coffee/tea
Coffee/tea
Although studies of caffeinated coffee have been heterogeneous and yielded mixed results (beneficial effect vs no effect on delaying cognitive decline), systematic reviews and meta-analyses of cross-sectional, case-control, and longitudinal cohort studies have found a general trend towards a favorable preventive role (level III).63-65 Caffeine exhibits its neuroprotective effect by increasing brain serotonin and acetylcholine, and by stabilizing blood-brain-barrier integrity.66 Moreover, in an animal study, mice given caffeine in their drinking water from young adulthood into older age had lower amyloid beta plasma levels compared with those given decaffeinated water.67 These findings suggest that in humans, 5 cups of regular caffeinated coffee daily, equivalent to 500 mg of caffeine,
An Italian study showed that older adults who don’t or rarely drink coffee (<1 cup daily) and those who recently increased their consumption pattern to >1 cup daily had a higher incidence of MCI than those who habitually consumed 1 to 2 cups daily.69 Therefore, it is not recommended to advise a change in coffee drinking pattern in old age. Older adults who are coffee drinkers should, however, be educated about the association between heavier caffeine intake and anxiety, insomnia, and cardiac arrhythmias.70
Despite its more modest caffeine levels, green tea is rich in polyphenols, which belong to the family of catechins and are characterized by antioxidant and anti-inflammatory properties.71 In a Japanese cohort, higher green tea consumption (up to 1 cup daily) was associated with a decreased incidence of MCI in older adults.72 More studies are needed to confirm its potential preventative role in AD.
Which lifestyle change is the most important?
Focusing on a single lifestyle change may be insufficient, especially because the bulk of evidence for individual interventions comes from population-based cohort studies (level III), rather than strong RCTs with a long follow-up. There is increasing evidence that combining multiple lifestyle modifications may yield better outcomes in maintaining or improving cognition.73
The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), a large, 2-year RCT that included community-dwelling older adults (age 60 to 77) with no diagnosis of major neurocognitive disorder, found that compared with regular health advice, multi-domain interventions reduced cognitive decline and improved overall cognition, executive functioning, and processing speed. The interventions evaluated in this study combined the following 4 modalities74:
- a healthy diet according to the Finnish nutrition recommendations (eating vegetables, fruits, and berries [minimum: 500 g/d], whole grain cereals [several times a day], and fish [2 to 3 times/week]; using low-salt products; consuming fat-free or low-fat milk products; and limiting red meat consumption to <500 g/week
- regular physical exercise tailored for improving muscle strength (1 to 3 times/week) coupled with aerobic exercise (2 to 5 times/week)
- cognitive training, including group sessions that have a social activity component and computer-based individual sessions 3 times/week that target episodic and working memory and executive functioning
- optimal management of cardiovascular risk factors.
Continue to: This multi-domain approach...
This multi-domain approach for lifestyle modification should be strongly recommended to cognitively intact older patients (level IB).
Modeled after the FINGER study, the Alzheimer’s Association U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER) is a 2-year, multicenter, controlled clinical trial aimed at testing the ability of a multidimensional lifestyle intervention to prevent AD in at-risk older adults (age 60 to 79, with established metabolic and cardiovascular risk factors). Interventions include a combination of physical exercise, nutritional counseling and management, cognitive and social stimulation, and improved management of cardiovascular risk factors. Recruitment for this large-scale trial was estimated to begin in January 2019 (NCT03688126).75
On a practical basis, Desai et al13 have proposed a checklist (Table 213) that physicians can use in their routine consultations to improve primary prevention of AD among their older patients.
Bottom Line
Advise patients that pursuing a healthy lifestyle is a key to delaying or preventing Alzheimer’s disease. This involves managing cardiovascular risk factors and a combination of staying physically, mentally, socially, and spiritually active, in addition to adhering to a healthy diet such as the Mediterranean diet.
Related Resources
- Anderson K, Grossberg GT. Brain games to slow cognitive decline in Alzheimer’s disease. J Am Med Dir Assoc. 2014;15(8):536-537.
- Small G, Vorgan G. The memory prescription: Dr. Garry Small’s 14-day plan to keep your brain and body young. New York, NY: Hyperion; 2004.
- Small G, Vorgan G. The Alzheimer’s prevention program; keep your brain healthy for the rest of your life. New York, NY: Workman Publishing Company, Inc.; 2012.
Drug Brand Name
Curcumin • Theracurmin
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.
1. Mehta D, Jackson R, Paul G, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs. 2017;26(6):735-739.
2. Norton S, Matthews FE, Barnes DE, et al. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788-794.
3. Meng XF, Yu JT, Wang HF, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42(4):1295-1310.
4. Shekelle PG, Woolf SH, Eccles M, et al. Developing clinical guidelines. West J Med. 1999;170(6):348-351.
5. Barnes DE, Yaffe Y. The projected impact of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819-828.
6. Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30(9):464-472.
7. Ahlskog JE, Geda YE, Graff-Radford NR, et al. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin Proc. 2011;86(9):876-884.
8. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer Disease. JAMA. 2009;302(6):627-637.
9. Rovio S, Kåreholt I, Helkala EL, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005;4(11):705-711.
10. Smith PJ et al. Aerobic exercise and neurocognitive performance: a meta-analytic review of randomized controlled trials. Psychosom Med. 2010;72(3):239-252.
11. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18(8):875-881.
12. Brown BM, Sohrabi HR, Taddei K, et al. Habitual exercise levels are associated with cerebral amyloid load in presymptomatic autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2017;13(11):1197-1206.
13. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26(1):1-16.
14. Centers for Disease Control and Prevention. Physical activity: how much physical activity do older adults need?
15. Garber CE, Blissmer B, Deschenes MR, et al; American College of Sports Medicine. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334-1359.
16. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113);2673-2734.
17. Valenzuela MJ, Sachdev P, Wen W, et al. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS One. 2008;3(7):e2598. doi.org/10.1371/journal.pone.0002598.
18. Wilson RS, Bennett DA, Bienias JL, et al. Cognitive activity and incident AD in a population-based sample of older persons. Neurology. 2002;59(12):1910-1914.
19. Wilson RS, Scherr PA, Schneider JA, et al. Relation of cognitive activity to risk of developing Alzheimer disease. Neurology. 2007;69(20):1911-1920.
20. Krell-Roesch J, Vemuri P, Pink A, et al. Association between mentally stimulating activities in late life and the outcome of incident mild cognitive impairment, with an analysis of the apoe ε4 genotype. JAMA Neurol. 2017;74(3):332-338.
21. Verghese J, Lipton RB, Katz MJ, et al. Leisure activities and the risk of dementia in the elderly. N Engl J Med. 2003;348(25):2508-2516.
22. Klein RM, Christie J, Parkvall M. Does multilingualism affect the incidence of Alzheimer’s disease?: a worldwide analysis by country. SSM Popul Health. 2016;2:463-467.
23. Grundy JG, Anderson JAE, Bialystok E. Neural correlates of cognitive processing in monolinguals and bilinguals. Ann N Y Acad Sci. 2017;1396(1):183-201.
24. Wilson RS, Krueger KR, Arnold SE, et al. Loneliness and risk of Alzheimer disease. Arch Gen Psychiatry. 2007;64(2):234-240.
25. Amieva H, Stoykova R, Matharan F, et al. What aspects of social network are protective for dementia? Not the quantity but the quality of social interactions is protective up to 15 years later. Psychosom Med. 2010;72(9):905-911.
26. Kuiper JS, Oude Voshaar RC, Zuidema SU, et al. The relationship between social functioning and subjective memory complaints in older persons: a population-based longitudinal cohort study. Int J Geriatr Psychiatry. 2017;32(10):1059-1071.
27. Robert P, Onyike CU, Leentjens AF, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98-104.
28. Marioni RE, Proust-Lima C, Amieva H, et al. Social activity, cognitive decline and dementia risk: a 20-year prospective cohort study. BMC Public Health. 2015;15:1089.
29. Wilson RS, Schneider JA, Boyle PA, et al. Chronic distress and incidence of mild cognitive impairment. Neurology. 2007;68(24):2085-2092.
30. Cai Z, Yan LJ, Ratka A. Telomere shortening and Alzheimer’s disease. Neuromolecular Med. 2013;15(1):25-48.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
32. Epel E, Daubenmier J, Moskowitz JT, et al. Can meditation slow rate of cellular aging? Cognitive stress, mindfulness, and telomeres. Ann N Y Acad Sci. 2009;1172:34-53.
33. Malinowski P, Moore AW, Mead Br, et al. Mindful aging: the effects of regular brief mindfulness practice on electrophysiological markers of cognitive and affective processing in older adults. Mindfulness (N Y). 2017;8(1):78-94.
34. Taren AA, Gianaros PJ, Greco CM, et al. Mindfulness meditation training and executive control network resting state functional connectivity: a randomized controlled trial. Psychosom Med. 2017;79(6):674-683.
35. Fountain-Zaragoza S, Prakash RS. Mindfulness training for healthy aging: impact on attention, well-being, and inflammation. Front in Aging Neurosci. 2017;9:11.
36. Eyre HA, Siddarth P, Acevedo B, et al. A randomized controlled trial of Kundalini yoga in mild cognitive impairment. Int Psychogeriatr. 2017;29(4):557-567.
37. Khalsa DS. Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis. 2015;48(1):1-12.
38. Berk L, van Boxtel M, van Os J. Can mindfulness-based interventions influence cognitive functioning in older adults? A review and considerations for future research. Aging Ment Health. 2017;21(11):1113-1120.
39. Hosseini S, Chaurasia A, Oremus M. The effect of religion and spirituality on cognitive function: a systematic review. Gerontologist. 2017. doi: 10.1093/geront/gnx024.
40. Luchsinger JA, Tang MX, Shea S, et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59(8):1258-1263.
41. Grant WB. Trends in diet and Alzheimer’s disease during the nutrition transition in Japan and developing countries. J Alzheimers Dis. 2014;38(3):611-620.
42. Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103(1):59-68.
43. Hu N, Yu JT, Tan L, et al. Nutrition and the risk of Alzheimer’s disease. Biomed Res Int. 2013;2013:524820. doi: 10.1155/2013/524820.
44. Taylor MK, Sullivan DK, Swerdlow RH, et al. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am J Clin Nutr. 2017;106(6):1463-1470.
45. van de Rest O, Berendsen AM, Haveman-Nies A, et al. Dietary patterns, cognitive decline, and dementia: a systematic review. Adv Nutr. 2015;6(2):154-168.
46. Petersson SD, Philippou E. Mediterranean diet, cognitive function, and dementia: a systematic review of the evidence. Adv Nutr. 2016;7(5):889-904.
47. Smith PJ, Blumenthal JA, Babyak MA, et al. Effects of the dietary approaches to stop hypertension diet, exercise, and caloric restriction on neurocognition in overweight adults with high blood pressure. Hypertension. 2010;55(6):1331-1338.
48. Wengreen H, Munger RG, Cutler A, et al. Prospective study of dietary approaches to stop hypertension- and Mediterranean-style dietary patterns and age-related cognitive change: the Cache County study on memory, health and aging. Am J Clin Nutr. 2013;98(5):1263-1271.
49. Tangney CC, Li H, Wang Y, et al. Relation of DASH- and Mediterranean-like dietary patterns to cognitive decline in older persons. Neurology. 2014;83(16):1410-1416.
50. Morris MC, Tangney CC, Wang Y, et al. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007-1014.
51. Desai AK, Rush J, Naveen L, et al. Nutrition and nutritional supplements to promote brain health. In: Hartman-Stein PE, Rue AL, eds. Enhancing cognitive fitness in adults: a guide to the use and development of community-based programs. New York, NY: Springer; 2011:249-269.
52. Goozee KG, Shah TM, Sohrabi HR, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449-465.
53. Small GW, Siddarth P, Li Z, et al. Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: a double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry. 2018;26(3):266-277.
54. Kim JW, Lee DY, Lee BC, et al. Alcohol and cognition in the elderly: a review. Psychiatry Investig. 2012;9(1):8-16.
55. Virtaa JJ, Järvenpää T, Heikkilä K, et al. Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up study. J Alzheimers Dis. 2010;22(3):939-948.
56. Kerr WC, Stockwell T. Understanding standard drinks and drinking guidelines. Drug and Alcohol Rev. 2012;31(2):200-205.
57. National Institute on Alcohol Abuse and Alcoholism. Drinking levels defined. https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/moderate-binge-drinking. Accessed December 9, 2017.
58. National Institute on Alcohol Abuse and Alcoholism. What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed November 9, 2017.
59. Lindsay J, Laurin D, Verreault R, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian study of health and aging. Am J Epidemiol. 2002;156(5):445-453.
60. Orgogozo JM, Dartigues JF, Lafont S, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris). 1997;153(3):185-192.
61. Topiwala A, Allan CL, Valkanova V, et al. Moderate alcohol consumption as risk factor for adverse brain outcomes and cognitive decline: longitudinal cohort study. BMJ. 2017;357.
62. Venkataraman A, Kalk N, Sewell G, et al. Alcohol and Alzheimer’s disease-does alcohol dependence contribute to beta-amyloid deposition, neuroinflammation and neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017;52(2):151-158.
63. Ma QP, Huang C, Cui QY, et al. Meta-analysis of the association between tea intake and the risk of cognitive disorders. PLoS One. 2016;11(11):e0165861. doi: 10.1371/journal.pone.0165861.
64. Santos C, Costa J, Santos J, et al. Caffeine intake and dementia: systematic review and meta-analysis. J Alzheimers Dis. 2010;20(Suppl 1):S187-204.
65. Panza F, Solfrizzi V, Barulli MR, et al. Coffee, tea, and caffeine consumption and prevention of late-life cognitive decline and dementia: a systematic review. J Nutr Health Aging. 2015;19(3):313-328.
66. Wierzejska R. Can coffee consumption lower the risk of Alzheimer’s disease and Parkinson’s disease? A literature review. Arch Med Sci. 2017;13(3):507-514.
67. Arendash GW, Cao C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis. 2010;20 (Suppl 1):S117-S126.
68. Eskelinen MH, Ngandu T, Tuomilehto J, et al. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16(1):85-91.
69. Solfrizzi V, Panza F, Imbimbo BP, et al. Coffee consumption habits and the risk of mild cognitive impairment: the Italian longitudinal study on aging. J Alzheimers Dis. 2015;47(4):889-899.
70. Vittoria Mattioli. Beverages of daily life: impact of caffeine on atrial fibrillation. J Atr Fibrillation. 2014;7(2):1133.
71. Chacko SM, Thambi PT, Kuttan R, et al. Beneficial effects of green tea: a literature review. Chin Med. 2010;5:13.
72. Noguchi-Shinohara M, Yuki S, Dohmoto C, et al. Consumption of green tea, but not black tea or coffee, is associated with reduced risk of cognitive decline. PLoS One. 2014;9(5):e96013. doi: 10.1371/journal.pone.0096013.
73. Schneider N, Yvon C. A review of multidomain interventions to support healthy cognitive ageing. J Nutr Health Aging. 2013;17(3):252-257.
74. Ngandu T, Lehitsalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
75. U.S. National Library of Medicing. ClinicalTrials.gov. U.S. study to protect brain health through lifestyle intervention to reduce risk (POINTER). https://clinicaltrials.gov/ct2/show/NCT03688126?term=pointer&cond=Alzheimer+Disease&rank=1. Published September 28, 2018. Accessed November 3, 2018.

Botulinum toxin: Emerging psychiatric indications
Botulinum toxin, a potent neurotoxic protein produced by the bacterium Clostridium botulinum, has been used as treatment for a variety of medical indications for more than 25 years (Box1-12). Recently, researchers have been exploring the role of botulinum toxin in psychiatry, primarily as an adjunctive treatment for depression, but also for several other possible indications. Several studies, including randomized controlled trials (RCTs), have provided evidence that glabellar botulinum toxin injections may be a safe and effective treatment for depression. In this article, we provide an update on the latest clinical trials that evaluated botulinum toxin for depression, and also summarize the evidence regarding other potential clinical psychiatric applications of botulinum toxin.
Several RCTs suggest efficacy for depression
The use of botulinum toxin to treat depression is based on the facial feedback hypothesis, which was first proposed by Charles Darwin in 187213 and further elaborated by William James,14,15 who emphasized the importance of the sensation of bodily changes in emotion. Contrary to the popular belief that emotions trigger physiological changes in the body, James postulated that peripheral bodily changes secondary to stimuli perception would exert a sensory feedback, generating emotions. The manipulation of human facial expression with an expression that is associated with a particular emotion (eg, holding a pen with teeth, leading to risorius/zygomaticus muscles contraction and a smile simulation) was found to influence participants’ affective responses in the presence of emotional stimuli (eg, rating cartoons as funnier), reinforcing the facial-feedback hypothesis.16,17
From a neurobiologic standpoint, facial botulinum toxin A (BTA) injections in rats were associated with increased serotonin and norepinephrine concentrations in the hypothalamus and striatum, respectively.18 Moreover, amygdala activity in response to angry vs happy faces, measured via functional magnetic resonance imaging (fMRI), was found to be attenuated after BTA applications to muscles involved in angry facial expressions.19,20 Both the neurotransmitters as well as the aforementioned brain regions have been implicated in the pathophysiology of depression.21,22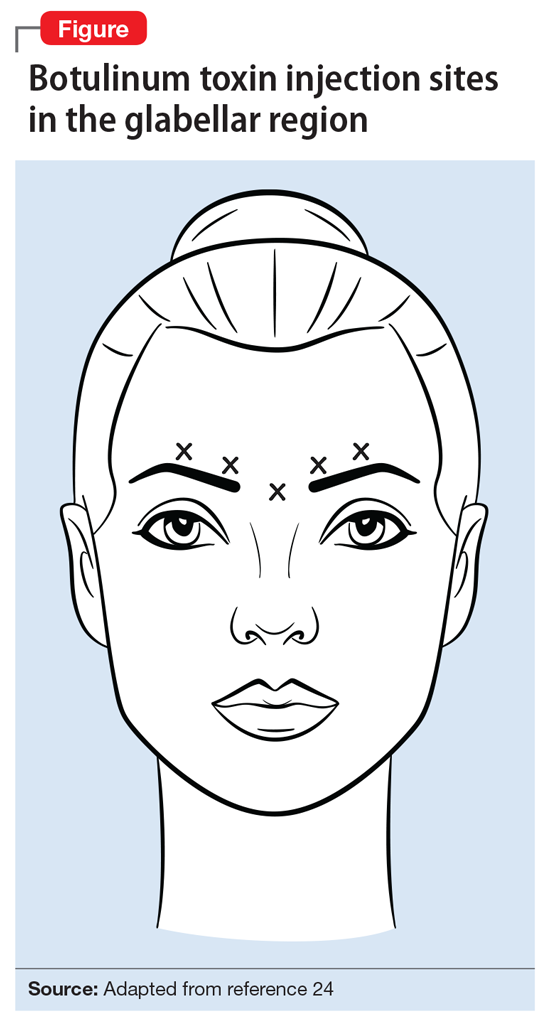
Compared with those in the placebo group, participants in the BTA group had a higher response rate as measured by the HAM-D17 at 6 weeks after treatment (P = .02), especially female patients (P = .002). Response to BTA, defined as ≥50% reduction on the HAM-D17, occurred within 2 weeks, and lasted another 6 weeks before slightly wearing off. Assessment of the CSS-GFL showed a statistically significant change at 6 weeks (P < .001). This small study failed, however, to show significant remission rates (HAM-D17 ≤7) in the BTA group compared with placebo.
Box
Botulinum toxin is a potent neurotoxin from Clostridium botulinum. Its potential for therapeutic use was first noticed in 1817 by physician Justinus Kerner, who coined the term botulism.1 In 1897, bacteriologist Emile van Ermengem isolated the causative bacterium C. botulinum.2 It was later discovered that the toxin induces muscle paralysis by inhibiting acetylcholine release from presynaptic motor neurons at the neuromuscular junction3 and was then mainly investigated as a treatment for medical conditions involving excessive or abnormal muscular contraction.
In 1989, the FDA approved botulinum toxin A (BTA) for the treatment of strabismus, blepharospasm, and other facial nerve disorders. In 2000, both BTA and botulinum toxin B (BTB) were FDA-approved for the treatment of cervical dystonia, and BTA was approved for the cosmetic treatment of frown lines (glabellar, canthal, and forehead lines).4 Other approved clinical indications for BTA include urinary incontinence due to detrusor overactivity associated with a neurologic condition such as spinal cord injury or multiple sclerosis; prophylaxis of headaches in chronic migraine patients; treatment of both upper and lower limb spasticity; severe axillary hyperhidrosis inadequately managed by topical agents; and the reduction of the severity of abnormal head position and neck pain.5 Its anticholinergic effects have been also investigated for treatment of hyperhidrosis as well as sialorrhea caused by neurodegenerative disorders such as amyotrophic lateral sclerosis.6-8 Multiple studies have shown that botulinum toxin can alleviate spasms of the gastrointestinal tract, aiding patients with dysphagia and achalasia.9-11 There is also growing evidence supporting the use of botulinum toxin in the treatment of chronic pain, including non-migraine types of headaches such as tension headaches; myofascial syndrome; and neuropathic pain.12
Continue to: In a second RCT involving 74 patients with depression...
In a second RCT involving 74 patients with depression, Finzi and Rosenthal25 observed statistically significant response and remission rates in participants who received BTA injections, as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS). Participants were given either BTA or saline injections and assessed at 3 visits across 6 weeks using the MADRS, CGI, and Beck Depression Inventory-II (BDI-II). Photographs of participants’ facial expressions were assessed using frown scores to see whether changes in facial expression were associated with improvement of depression.
This study was able to reproduce on a larger scale the results observed by Wollmer et al.23 It found a statistically significant increase in the rate of remission (MADRS ≤10) at 6 weeks following BTA injections (27%, P < .02), and that even patients who were not resistant to antidepressants could benefit from BTA. However, although there was an observable trend in improvement of frown scores associated with improved depression scores, the correlation between these 2 variables was not statistically significant.
In a crossover RCT, Magid et al26 observed the response to BTA vs placebo saline injections in 30 patients with moderate to severe frown lines. The study lasted 24 weeks; participants switched treatments at Week 12. Mood improvement was assessed using the 21-item Hamilton Depression Rating Scale (HDRS-21), BDI, and Patient Health Questionnaire-9 (PHQ-9). Compared with patients who received placebo injections, those treated with BTA injections showed statistically significant response rates, but not remission rates. This study demonstrated continued improvement throughout the 24 weeks in participants who initially received BTA injections, despite having received placebo for the last 12 weeks, by which time the cosmetic effects of the initial injection had worn off. This suggests that the antidepressant effects of botulinum toxin may not depend entirely on its paralytic effects, but also on its impact on the neurotransmitters involved in the pathophysiology of depression.18 By demonstrating improvement in the placebo group once they were started on botulinum toxin, this study also was able to exclude the possibility that other variables may be responsible for the difference in the clinical course between the 2 groups. However, this study was limited by a small sample size, and it only included participants who had moderate to severe frown lines at baseline.
Zamanian et al27 examined the therapeutic effects of BTA injections in 28 Iranian patients with major depressive disorder (MDD) diagnosed according to DSM-5 criteria. At 6 weeks, there were significant improvements in BDI scores in patients who received BTA vs those receiving placebo. However, these changes were demonstrated at 6 weeks (not as early as 2 weeks), and patients didn’t achieve remission.
A large-scale, multicenter U.S. phase II RCT investigated the safety, tolerability, and efficacy of a single administration of 2 different doses of BTA (30 units or 50 units) as monotherapy for the treatment of moderate to severe depression in 258 women.28 Effects on depression were measured at 3, 6, and 9 weeks using the MADRS. Participants who received the 30-unit injection showed statistically significant improvement at 3 weeks (
More recently, in a case series, Chugh et al30 examined the effect of BTA in 42 patients (55% men) with severe treatment-resistant depression. Participants were given BTA injections in the glabellar region as an adjunctive treatment to antidepressants and observed for at least 6 weeks. Depression severity was measured using HAM-D17, MADRS, and BDI at baseline and at 3 weeks. Changes in glabellar frown lines also were assessed using the CSS-GFL. The authors reported statistically significant improvements in HAM-D17 (
A summary of the RCTs of BTA for treating depression appears in Table 1.23,25-28
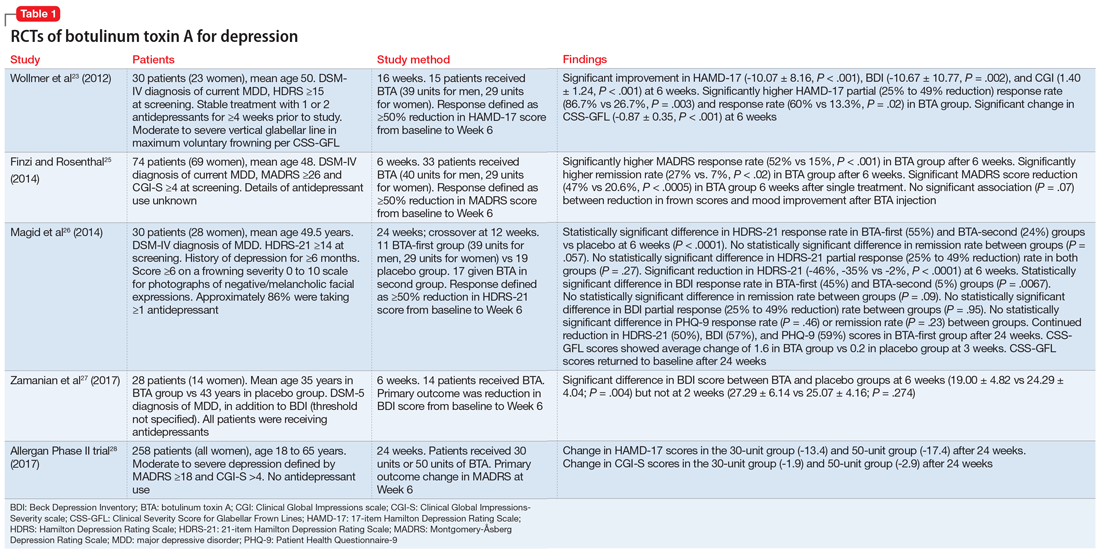
Continue to: Benefits for other psychiatric indications
Benefits for other psychiatric indications
Borderline personality disorder. In a case series of 6 women, BTA injections in the glabellar region were reported to be particularly effective for the treatment of borderline personality disorder symptoms that were resistant to psychotherapy and pharmacotherapy.31 Two to 6 weeks after a 29-unit injection, borderline personality disorder symptoms as measured by the Zanarini Rating Scale for Borderline Personality Disorder and/or the Borderline Symptom List were shown to significantly improve by 49% to 94% from baseline (P ≤ .05). These findings emphasize the promising therapeutic role of BTA on depressive symptoms concomitant with the emotional lability, impulsivity, and negative emotions that usually characterize this personality disorder.31,32 A small sample size and lack of a placebo comparator are limitations of this research.
Neuroleptic-induced sialorrhea. Botulinum toxin injections in the salivary glands have been investigated for treating clozapine-induced sialorrhea because they are thought to directly inhibit the release of acetylcholine from salivary glands. One small RCT that used botulinum toxin B (BTB)33 and 1 case report that used BTA34 reported successful reduction in hypersalivation, with doses ranging from 150 to 500 units injected in each of the parotid and/or submandibular glands bilaterally. Although the treatment was well tolerated and lasted up to 16 weeks, larger studies are needed to replicate these findings.33-35
Orofacial tardive dyskinesia. Several case reports of orofacial tardive dyskinesia, including lingual dyskinesia and lingual protrusion dystonia, have found improvements in hyperkinetic movements following muscular BTA injections, such as in the genioglossus muscle in the case of tongue involvement.36-39 These cases were, however, described in the literature before the recent FDA approval of the vesicular monoamine transporter 2 inhibitors valbenazine and deutetrabenazine for the treatment of tardive dyskinesia.40,41
Studies examining botulinum toxin’s application in areas of psychiatry other than depression are summarized in Table 2.31,33,36-38
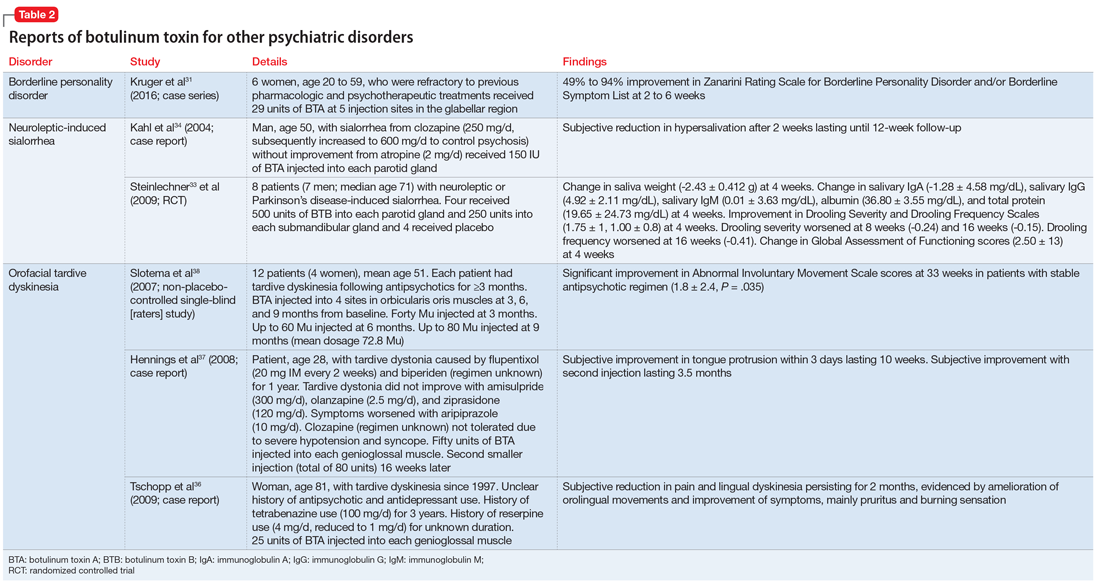
Continue to: Promising initial findings but multiple limitations
Promising initial findings but multiple limitations
Although BTA injections have been explored as a potential treatment for several psychiatric conditions, the bulk of recent evidence is derived from studies in patients with depressive disorders. BTA injections in the glabellar regions have been shown in small RCTs to be well-tolerated with overall promising improvement of depressive symptoms, optimally 6 weeks after a single injection. Moreover, BTA has been shown to be safe and long-lasting, which would be convenient for patients and might improve adherence to therapy.42-44 BTA’s antidepressant effects were shown to be independent of frown line severity or patient satisfaction with cosmetic effects.45 The trials by Wollmer et al,23 Finzi and Rosenthal,25 and Magid et al26 mainly studied BTA as an adjunctive treatment to antidepressants in patients with ongoing unipolar depression. However, Finzi and Rosenthal25 included patients who were not medicated at the time of the study.
Pooled analysis of these 3 RCTs found that patients who received BTA monotherapy improved equally to those who received it as an adjunctive treatment to antidepressants. Overall, on primary endpoint measures, a response rate of 54.2% was obtained in the BTA group compared with 10.7% among patients who received placebo saline injections (odds ratio [OR] 11.1, 95% confidence interval [CI], 4.3 to 28.8, number needed to treat [NNT] = 2.3) and a remission rate of 30.5% with BTA compared with 6.7% with placebo (OR 7.3, 95 % CI, 2.4 to 22.5, NNT = 4.2).46 However, remission rates tend to be higher in the augmentation groups, and so further studies are needed to compare both treatment strategies.
Nevertheless, these positive findings have been recently challenged by the results of the largest U.S. multicenter phase II RCT,28 which failed to find a significant antidepressant effect at 6 weeks with the 30-unit BTA injection, and also failed to prove a dose-effect relationship, as the 50-unit injection wasn’t superior to the lower dose and didn’t significantly differ from placebo. One hypothesis to explain this discrepancy may be the difference in injection sites between the treatment and placebo groups.47 Future studies need to address the various limitations of earlier clinical trials that mainly yielded promising results with BTA.
A major concern is the high rate of unblinding of participants and researchers in BTA trials, as the cosmetic effects of botulinum toxin injections make them easy to distinguish from saline injections. Ninety percent of participants in the Wollmer et al study23 were able to correctly guess their group allocation, while 60% of evaluators guessed correctly. Finzi and Rozenthal25 reported 52% of participants in the BTA group, 46% in the placebo group, and 73% of evaluators correctly guessed their allocation. Magid et al26 reported 75% of participants were able to guess the order of intervention they received. The high unblinding rates in these trials remains a significant limitation. There is a concern that this may lead to an underestimation of the placebo effect relative to clinical improvement, thus causing inflation of outcome differences between groups. Although various methods have been tried to minimize evaluator unblinding, such as placing surgical caps on participants’ faces during visits to hide the glabellar region, better methods need to be implemented to prevent unblinding of both raters and participants.
Furthermore, except for the multicenter phase II trial, most studies have been conducted in small samples, which limits their statistical power. Larger controlled trials will be needed to replicate the positive findings obtained in smaller RCTs.
Another limitation is that the majority of the well-designed RCTs were conducted in populations that were predominantly female, which makes it difficult to reliably assess treatment efficacy in men. This may be because cosmetic treatment with botulinum toxin injection is more favorably received by women than by men. A recent comparison48 of the studies by Wollmer et al23 and Finzi and Rosenthal25 discussed an interesting observation. Wollmer et al did not explicitly mention botulinum toxin when recruiting for the study, while Finzi and Rosenthal did. While approximately a quarter of the participants in the Wollmer et al study were male, Finzi and Rosenthal attracted an almost entirely female population. Perhaps there is a potential bias for females to be more attracted to these studies due to the secondary gain of receiving a cosmetic procedure.
In an attempt to understand predictors of positive response to botulinum toxin in patients with depression, Wollmer et al49 conducted a follow-up study in which they reassessed the data obtained from their initial RCT using the HAM-D agitation item scores to separate the 15 participants who received BTA into low-agitation (≤1 score on agitation item of the HAM-D scale) and high-agitation (≥2 score on agitation item of the HAM-D scale) groups. They found that the 9 participants who responded to BTA treatment had significantly higher baseline agitation scores than participants who did not respond (1.56 ± 0.88 vs 0.33 ± 0.52, P = .01). All of the participants who presented with higher agitation levels experienced response, compared with 40% of those with lower agitation levels (P = .04), although there was no significant difference in magnitude of improvement (14.2 ± 1.92 vs 8.0 ± 9.37, P = .07). The study added additional support to the facial feedback hypothesis, as it links the improvement of depression to facial muscle activation targeted by the injections. It also introduced a potential predictor of response to botulinum toxin treatment, highlighting potential factors to consider when enrolling patients in future investigations.
The case series of patients with borderline personality disorder31 also shed light on the potential positive effect of BTA treatment for a particular subtype of patients with depression—those with comorbid emotional instability—to consider as a therapeutic target for the future. Hence, inclusion criteria for future trials might potentially include patient age, gender, existence/quantification of prominent frown lines at baseline, severity of MDD, duration of depression, and personality characteristics of enrolled participants.
In conclusion, BTA injections appear promising as a treatment for depression as well as for other psychiatric disorders. Future studies should focus on identifying optimal candidates for this innovative treatment modality. Furthermore, BTA dosing and administration strategies (monotherapy vs adjunctive treatment to antidepressants) need to be further explored. As retrograde axonal transport of botulinum toxin has been demonstrated in animal studies, it would be interesting to further examine its effects in the human CNS to enhance our knowledge of the pathophysiology of botulinum and its potential applications in psychiatry.50
Bottom Line
Botulinum toxin shows promising antidepressant effects and may have a role in the treatment of several other psychiatric disorders. More research is needed to address limitations of previous studies and to establish an adequate treatment regimen.
Related Resources
- Wollmer MA, Magid M, Kruger TH. Botulinum toxin treatment in depression. In: Bewley A, Taylor RE, Reichenberg JS, et al (eds). Practical psychodermatology. Oxford, UK: Wiley; 2014.
- Wollmer MA, Neumann I, Magid M. et al. Shrink that frown! Botulinum toxin therapy is lifting the face of psychiatry. G Ital Dermatol Venereol. 2018;153(4):540-548.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Biperiden • Akineton
Botulinum toxin A • Botox
Botulinum toxin B • Myobloc
Clozapine • Clozaril
Deutetrabenazine • Austedo
Flupentixol • Prolixin
Imipramine • Tofranil
Olanzapine • Zyprexa
Reserpine • Serpalan, Serpasil
Tetrabenazine • Xenazine
Valbenazine • Ingrezza
Ziprasidone • Geodon
1. Erbguth FJ, Naumann M. Historical aspects of botulinum toxin. Justinus Kerner (1786-1862) and the “sausage” poison. Neurology. 1999;53(8):1850-1853.
2. Devriese PP. On the discovery of Clostridium botulinum. J History Neurosci. 1999;8(1):43-50.
3. Burgen ASV, Dickens F, Zatman LJ. The action of botulinum toxin on the neuro-muscular junction. J Physiol. 1949;109(1-2):10-24.
4. Jankovic J. Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry. 2004;75(7):951-957.
5. BOTOX (OnabotulinumtoxinA) [package insert]. Allergan, Inc., Irvine, CA; 2015.
6. Saadia D, Voustianiouk A, Wang AK, et al. Botulinum toxin type A in primary palmar hyperhidrosis. Randomized, single-blind, two-dose study. Neurology. 2001;57(11):2095-2099.
7. Naumann MK, Lowe NJ. Effect of botulinum toxin type A on quality of life measures in patients with excessive axillary sweating: a randomized controlled trial. Br J Dermatol. 2002;147(6):1218-1226.
8. Giess R, Naumann M, Werner E, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhea in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2000;69(1):121-123.
9. Restivo DA, Palmeri A, Marchese-Ragona R. Botulinum toxin for cricopharyngeal dysfunction in Parkinson’s disease. N Engl J Med. 2002;346(15):1174-1175.
10. Pasricha PJ, Ravich WJ, Hendrix T, et al. Intrasphincteric botulinum toxin for the treatment of achalasia. N Engl J Med. 1995(12);322:774-778.
11. Schiano TD, Parkman HP, Miller LS, et al. Use of botulinum toxin in the treatment of achalasia. Dig Dis. 1998;16(1):14-22.
12. Sim WS. Application of botulinum toxin in pain management. Korean J Pain. 2011;24(1):1-6.
13. Darwin C. The expression of the emotions in man and animals. London, UK: John Murray; 1872:366.
14. James W. The principles of psychology, vol. 2. New York, NY: Henry Holt and Company; 1890.
15. James W. II. —What is an emotion? Mind. 1884;os-IX(34):188-205.
16. Strack R, Martin LL, Stepper S. Inhibiting and facilitating conditions of facial expressions: a nonobtrusive test of the facial feedback hypothesis. J Pers Soc Psychol. 1988;54(5):768-777.
17. Larsen RJ, Kasimatis M, Frey K. Facilitating the furrowed brow: an unobtrusive test of the facial feedback hypothesis applied to unpleasant affect. Cogn Emot. 1992;6(5):321-338.
18. Ibragic S, Matak I, Dracic A, et al. Effects of botulinum toxin type A facial injection on monoamines and their metabolites in sensory, limbic, and motor brain regions in rats. Neurosci Lett. 2016;617:213-217.
19. Hennenlotter A, Dresel C, Castrop F, et al. The link between facial feedback and neural activity within central circuitries of emotion—new insights from botulinum toxin-induced denervation of frown muscles. Cereb Cortex. 2009;19(3):537-42
20. Kim MJ, Neta M, Davis FC, et al. Botulinum toxin-induced facial muscle paralysis affects amygdala responses to the perception emotional expressions: preliminary findings from an A-B-A design. Biol Mood Anxiety Disord. 2014;4:11.
21. Nestler EJ, Barrot M, DiLeone RJ, et al. Neurobiology of depression. Neuron. 2002;34(1):13-25.
22. Pandya M, Altinay M, Malone DA Jr, et al. Where in the brain is depression? Curr Psychiatry Rep. 2012;14(6):634-642.
23. Wollmer MA, de Boer C, Kalak N, et al. Facing depression with botulinum toxin: a randomized controlled trial. J Psychiatr Res. 2012;46:574-581.
24. BOTOX Cosmetic [prescribing information]. Allergan, Inc., Irvine, CA; 2017.
25. Finzi E, Rosenthal NE. Treatment of depression with onabotulinumtoxinA; a randomized, double-blind, placebo controlled trial. J Psychiatr Res. 2014;52:1-6.
26. Magid M, Reichenberg JS, Poth PE, et al. The treatment of major depressive disorder using botulinum toxin A: a 24 week randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2014;75(8):837-844.
27. Zamanian A, Ghanbari Jolfaei A, Mehran G, et al. Efficacy of botox versus placebo for treatment of patients with major depression. Iran J Public Health. 2017;46(7):982-984.
28. Allergan. OnabotulinumtoxinA as treatment for major depressive disorder in adult females. 2017. https://clinicaltrials.gov/ct2/show/NCT02116361. Accessed October 26, 2018.
29. Allergan. Allergan reports topline phase II data supporting advancement of BOTOX® (onabotulinumtoxinA) for the treatment of major depressive disorder (MDD). April 5, 2017. https://www.allergan.com/news/news/thomson-reuters/allergan-reports-topline-phase-ii-data-supporting. Accessed October 26, 2018.
30. Chugh S, Chhabria A, Jung S, et al. Botulinum toxin as a treatment for depression in a real-world setting. J Psychiatr Pract. 2018;24(1):15-20.
31. Kruger TH, Magid M, Wollmer MA. Can botulinum toxin help patients with borderline personality disorder? Am J Psychiatry. 2016;173(9):940-941.
32. Baumeister JC, Papa G, Foroni F. Deeper than skin deep – the effect of botulinum toxin-A on emotion processing. Toxicon. 2016;119:86-90.
33. Steinlechner S, Klein C, Moser A, et al. Botulinum toxin B as an effective and safe treatment for neuroleptic-induced sialorrhea. Psychopharmacology (Berl). 2010;207(4):593-597.
34. Kahl KG, Hagenah J, Zapf S, et al. Botulinum toxin as an effective treatment of clozapine-induced hypersalivation. Psychopharmacology (Berl). 2004;173(1-2):229-230.
35. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
36. Tschopp L, Salazar Z, Micheli F. Botulinum toxin in painful tardive dyskinesia. Clin Neuropharmacol. 2009;32(3):165-166.
37. Hennings JM, Krause E, Bötzel K, et al. Successful treatment of tardive lingual dystonia with botulinum toxin: case report and review of the literature. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1167-1171.
38. Slotema CW, van Harten PN, Bruggeman R, et al. Botulinum toxin in the treatment of orofacial tardive dyskinesia: a single blind study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):507-509.
39. Esper CD, Freeman A, Factor SA. Lingual protrusion dystonia: frequency, etiology and botulinum toxin therapy. Parkinsonism Relat Disord. 2010;16(7):438-441.
40. Seeberger LC, Hauser RA. Valbenazine for the treatment of tardive dyskinesia. Expert Opin Pharmacother. 2017;18(12):1279-1287.
41. Citrome L. Deutetrabenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication—What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(11):e13030.
42. Brin MF, Boodhoo TI, Pogoda JM, et al. Safety and tolerability of onabotulinumtoxinA in the tretment of facial lines: a meta-analysis of individual patient data from global clinical registration studies in 1678 participants. J Am Acad Dermatol. 2009;61:961-970.
43. Beer K. Cost effectiveness of botulinum toxins for the treatment of depression: preliminary observations. J Drugs Dermatol. 2010;9(1):27-30.
44. Serna MC, Cruz I, Real J, et al. Duration and adherence of antidepressant treatment (2003-2007) based on prescription database. Eur Psychiatry. 2010;25(4):206-213.
45. Rechenberg JS, Hauptman AJ, Robertson HT, et al. Botulinum toxin for depression: Does patient appearance matter? J Am Acad Dermatol. 2016;74(1):171-173.
46. Magid M, Finzi E, Kruger THC, et al. Treating depression with botulinum toxin: a pooled analysis of randomized controlled trials. Pharmacopsychiatry. 2015;48(6):205-210.
47. Court, E. Allergan is still hopeful about using Botox to treat depression. April 8, 2017. https://www.marketwatch.com/story/allergan-is-still-hopeful-about-using-botox-to-treat-depression-2017-04-07. Accessed October 26, 2018.
48. Rudorfer MV. Botulinum toxin: does it have a place in the management of depression? CNS Drugs. 2018;32(2):97-100.
49. Wollmer MA, Kalak N, Jung S, et al. Agitation predicts response of depression to botulinum toxin treatment in a randomized controlled trial. Front Psychiatry. 2014;5:36
50. Antonucci F, Rossi C, Gianfranceschi L, et al. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28(14):3689-3696.
Botulinum toxin, a potent neurotoxic protein produced by the bacterium Clostridium botulinum, has been used as treatment for a variety of medical indications for more than 25 years (Box1-12). Recently, researchers have been exploring the role of botulinum toxin in psychiatry, primarily as an adjunctive treatment for depression, but also for several other possible indications. Several studies, including randomized controlled trials (RCTs), have provided evidence that glabellar botulinum toxin injections may be a safe and effective treatment for depression. In this article, we provide an update on the latest clinical trials that evaluated botulinum toxin for depression, and also summarize the evidence regarding other potential clinical psychiatric applications of botulinum toxin.
Several RCTs suggest efficacy for depression
The use of botulinum toxin to treat depression is based on the facial feedback hypothesis, which was first proposed by Charles Darwin in 187213 and further elaborated by William James,14,15 who emphasized the importance of the sensation of bodily changes in emotion. Contrary to the popular belief that emotions trigger physiological changes in the body, James postulated that peripheral bodily changes secondary to stimuli perception would exert a sensory feedback, generating emotions. The manipulation of human facial expression with an expression that is associated with a particular emotion (eg, holding a pen with teeth, leading to risorius/zygomaticus muscles contraction and a smile simulation) was found to influence participants’ affective responses in the presence of emotional stimuli (eg, rating cartoons as funnier), reinforcing the facial-feedback hypothesis.16,17
From a neurobiologic standpoint, facial botulinum toxin A (BTA) injections in rats were associated with increased serotonin and norepinephrine concentrations in the hypothalamus and striatum, respectively.18 Moreover, amygdala activity in response to angry vs happy faces, measured via functional magnetic resonance imaging (fMRI), was found to be attenuated after BTA applications to muscles involved in angry facial expressions.19,20 Both the neurotransmitters as well as the aforementioned brain regions have been implicated in the pathophysiology of depression.21,22
Compared with those in the placebo group, participants in the BTA group had a higher response rate as measured by the HAM-D17 at 6 weeks after treatment (P = .02), especially female patients (P = .002). Response to BTA, defined as ≥50% reduction on the HAM-D17, occurred within 2 weeks, and lasted another 6 weeks before slightly wearing off. Assessment of the CSS-GFL showed a statistically significant change at 6 weeks (P < .001). This small study failed, however, to show significant remission rates (HAM-D17 ≤7) in the BTA group compared with placebo.
Box
Botulinum toxin is a potent neurotoxin from Clostridium botulinum. Its potential for therapeutic use was first noticed in 1817 by physician Justinus Kerner, who coined the term botulism.1 In 1897, bacteriologist Emile van Ermengem isolated the causative bacterium C. botulinum.2 It was later discovered that the toxin induces muscle paralysis by inhibiting acetylcholine release from presynaptic motor neurons at the neuromuscular junction3 and was then mainly investigated as a treatment for medical conditions involving excessive or abnormal muscular contraction.
In 1989, the FDA approved botulinum toxin A (BTA) for the treatment of strabismus, blepharospasm, and other facial nerve disorders. In 2000, both BTA and botulinum toxin B (BTB) were FDA-approved for the treatment of cervical dystonia, and BTA was approved for the cosmetic treatment of frown lines (glabellar, canthal, and forehead lines).4 Other approved clinical indications for BTA include urinary incontinence due to detrusor overactivity associated with a neurologic condition such as spinal cord injury or multiple sclerosis; prophylaxis of headaches in chronic migraine patients; treatment of both upper and lower limb spasticity; severe axillary hyperhidrosis inadequately managed by topical agents; and the reduction of the severity of abnormal head position and neck pain.5 Its anticholinergic effects have been also investigated for treatment of hyperhidrosis as well as sialorrhea caused by neurodegenerative disorders such as amyotrophic lateral sclerosis.6-8 Multiple studies have shown that botulinum toxin can alleviate spasms of the gastrointestinal tract, aiding patients with dysphagia and achalasia.9-11 There is also growing evidence supporting the use of botulinum toxin in the treatment of chronic pain, including non-migraine types of headaches such as tension headaches; myofascial syndrome; and neuropathic pain.12
Continue to: In a second RCT involving 74 patients with depression...
In a second RCT involving 74 patients with depression, Finzi and Rosenthal25 observed statistically significant response and remission rates in participants who received BTA injections, as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS). Participants were given either BTA or saline injections and assessed at 3 visits across 6 weeks using the MADRS, CGI, and Beck Depression Inventory-II (BDI-II). Photographs of participants’ facial expressions were assessed using frown scores to see whether changes in facial expression were associated with improvement of depression.
This study was able to reproduce on a larger scale the results observed by Wollmer et al.23 It found a statistically significant increase in the rate of remission (MADRS ≤10) at 6 weeks following BTA injections (27%, P < .02), and that even patients who were not resistant to antidepressants could benefit from BTA. However, although there was an observable trend in improvement of frown scores associated with improved depression scores, the correlation between these 2 variables was not statistically significant.
In a crossover RCT, Magid et al26 observed the response to BTA vs placebo saline injections in 30 patients with moderate to severe frown lines. The study lasted 24 weeks; participants switched treatments at Week 12. Mood improvement was assessed using the 21-item Hamilton Depression Rating Scale (HDRS-21), BDI, and Patient Health Questionnaire-9 (PHQ-9). Compared with patients who received placebo injections, those treated with BTA injections showed statistically significant response rates, but not remission rates. This study demonstrated continued improvement throughout the 24 weeks in participants who initially received BTA injections, despite having received placebo for the last 12 weeks, by which time the cosmetic effects of the initial injection had worn off. This suggests that the antidepressant effects of botulinum toxin may not depend entirely on its paralytic effects, but also on its impact on the neurotransmitters involved in the pathophysiology of depression.18 By demonstrating improvement in the placebo group once they were started on botulinum toxin, this study also was able to exclude the possibility that other variables may be responsible for the difference in the clinical course between the 2 groups. However, this study was limited by a small sample size, and it only included participants who had moderate to severe frown lines at baseline.
Zamanian et al27 examined the therapeutic effects of BTA injections in 28 Iranian patients with major depressive disorder (MDD) diagnosed according to DSM-5 criteria. At 6 weeks, there were significant improvements in BDI scores in patients who received BTA vs those receiving placebo. However, these changes were demonstrated at 6 weeks (not as early as 2 weeks), and patients didn’t achieve remission.
A large-scale, multicenter U.S. phase II RCT investigated the safety, tolerability, and efficacy of a single administration of 2 different doses of BTA (30 units or 50 units) as monotherapy for the treatment of moderate to severe depression in 258 women.28 Effects on depression were measured at 3, 6, and 9 weeks using the MADRS. Participants who received the 30-unit injection showed statistically significant improvement at 3 weeks (
More recently, in a case series, Chugh et al30 examined the effect of BTA in 42 patients (55% men) with severe treatment-resistant depression. Participants were given BTA injections in the glabellar region as an adjunctive treatment to antidepressants and observed for at least 6 weeks. Depression severity was measured using HAM-D17, MADRS, and BDI at baseline and at 3 weeks. Changes in glabellar frown lines also were assessed using the CSS-GFL. The authors reported statistically significant improvements in HAM-D17 (
A summary of the RCTs of BTA for treating depression appears in Table 1.23,25-28
Continue to: Benefits for other psychiatric indications
Benefits for other psychiatric indications
Borderline personality disorder. In a case series of 6 women, BTA injections in the glabellar region were reported to be particularly effective for the treatment of borderline personality disorder symptoms that were resistant to psychotherapy and pharmacotherapy.31 Two to 6 weeks after a 29-unit injection, borderline personality disorder symptoms as measured by the Zanarini Rating Scale for Borderline Personality Disorder and/or the Borderline Symptom List were shown to significantly improve by 49% to 94% from baseline (P ≤ .05). These findings emphasize the promising therapeutic role of BTA on depressive symptoms concomitant with the emotional lability, impulsivity, and negative emotions that usually characterize this personality disorder.31,32 A small sample size and lack of a placebo comparator are limitations of this research.
Neuroleptic-induced sialorrhea. Botulinum toxin injections in the salivary glands have been investigated for treating clozapine-induced sialorrhea because they are thought to directly inhibit the release of acetylcholine from salivary glands. One small RCT that used botulinum toxin B (BTB)33 and 1 case report that used BTA34 reported successful reduction in hypersalivation, with doses ranging from 150 to 500 units injected in each of the parotid and/or submandibular glands bilaterally. Although the treatment was well tolerated and lasted up to 16 weeks, larger studies are needed to replicate these findings.33-35
Orofacial tardive dyskinesia. Several case reports of orofacial tardive dyskinesia, including lingual dyskinesia and lingual protrusion dystonia, have found improvements in hyperkinetic movements following muscular BTA injections, such as in the genioglossus muscle in the case of tongue involvement.36-39 These cases were, however, described in the literature before the recent FDA approval of the vesicular monoamine transporter 2 inhibitors valbenazine and deutetrabenazine for the treatment of tardive dyskinesia.40,41
Studies examining botulinum toxin’s application in areas of psychiatry other than depression are summarized in Table 2.31,33,36-38
Continue to: Promising initial findings but multiple limitations
Promising initial findings but multiple limitations
Although BTA injections have been explored as a potential treatment for several psychiatric conditions, the bulk of recent evidence is derived from studies in patients with depressive disorders. BTA injections in the glabellar regions have been shown in small RCTs to be well-tolerated with overall promising improvement of depressive symptoms, optimally 6 weeks after a single injection. Moreover, BTA has been shown to be safe and long-lasting, which would be convenient for patients and might improve adherence to therapy.42-44 BTA’s antidepressant effects were shown to be independent of frown line severity or patient satisfaction with cosmetic effects.45 The trials by Wollmer et al,23 Finzi and Rosenthal,25 and Magid et al26 mainly studied BTA as an adjunctive treatment to antidepressants in patients with ongoing unipolar depression. However, Finzi and Rosenthal25 included patients who were not medicated at the time of the study.
Pooled analysis of these 3 RCTs found that patients who received BTA monotherapy improved equally to those who received it as an adjunctive treatment to antidepressants. Overall, on primary endpoint measures, a response rate of 54.2% was obtained in the BTA group compared with 10.7% among patients who received placebo saline injections (odds ratio [OR] 11.1, 95% confidence interval [CI], 4.3 to 28.8, number needed to treat [NNT] = 2.3) and a remission rate of 30.5% with BTA compared with 6.7% with placebo (OR 7.3, 95 % CI, 2.4 to 22.5, NNT = 4.2).46 However, remission rates tend to be higher in the augmentation groups, and so further studies are needed to compare both treatment strategies.
Nevertheless, these positive findings have been recently challenged by the results of the largest U.S. multicenter phase II RCT,28 which failed to find a significant antidepressant effect at 6 weeks with the 30-unit BTA injection, and also failed to prove a dose-effect relationship, as the 50-unit injection wasn’t superior to the lower dose and didn’t significantly differ from placebo. One hypothesis to explain this discrepancy may be the difference in injection sites between the treatment and placebo groups.47 Future studies need to address the various limitations of earlier clinical trials that mainly yielded promising results with BTA.
A major concern is the high rate of unblinding of participants and researchers in BTA trials, as the cosmetic effects of botulinum toxin injections make them easy to distinguish from saline injections. Ninety percent of participants in the Wollmer et al study23 were able to correctly guess their group allocation, while 60% of evaluators guessed correctly. Finzi and Rozenthal25 reported 52% of participants in the BTA group, 46% in the placebo group, and 73% of evaluators correctly guessed their allocation. Magid et al26 reported 75% of participants were able to guess the order of intervention they received. The high unblinding rates in these trials remains a significant limitation. There is a concern that this may lead to an underestimation of the placebo effect relative to clinical improvement, thus causing inflation of outcome differences between groups. Although various methods have been tried to minimize evaluator unblinding, such as placing surgical caps on participants’ faces during visits to hide the glabellar region, better methods need to be implemented to prevent unblinding of both raters and participants.
Furthermore, except for the multicenter phase II trial, most studies have been conducted in small samples, which limits their statistical power. Larger controlled trials will be needed to replicate the positive findings obtained in smaller RCTs.
Another limitation is that the majority of the well-designed RCTs were conducted in populations that were predominantly female, which makes it difficult to reliably assess treatment efficacy in men. This may be because cosmetic treatment with botulinum toxin injection is more favorably received by women than by men. A recent comparison48 of the studies by Wollmer et al23 and Finzi and Rosenthal25 discussed an interesting observation. Wollmer et al did not explicitly mention botulinum toxin when recruiting for the study, while Finzi and Rosenthal did. While approximately a quarter of the participants in the Wollmer et al study were male, Finzi and Rosenthal attracted an almost entirely female population. Perhaps there is a potential bias for females to be more attracted to these studies due to the secondary gain of receiving a cosmetic procedure.
In an attempt to understand predictors of positive response to botulinum toxin in patients with depression, Wollmer et al49 conducted a follow-up study in which they reassessed the data obtained from their initial RCT using the HAM-D agitation item scores to separate the 15 participants who received BTA into low-agitation (≤1 score on agitation item of the HAM-D scale) and high-agitation (≥2 score on agitation item of the HAM-D scale) groups. They found that the 9 participants who responded to BTA treatment had significantly higher baseline agitation scores than participants who did not respond (1.56 ± 0.88 vs 0.33 ± 0.52, P = .01). All of the participants who presented with higher agitation levels experienced response, compared with 40% of those with lower agitation levels (P = .04), although there was no significant difference in magnitude of improvement (14.2 ± 1.92 vs 8.0 ± 9.37, P = .07). The study added additional support to the facial feedback hypothesis, as it links the improvement of depression to facial muscle activation targeted by the injections. It also introduced a potential predictor of response to botulinum toxin treatment, highlighting potential factors to consider when enrolling patients in future investigations.
The case series of patients with borderline personality disorder31 also shed light on the potential positive effect of BTA treatment for a particular subtype of patients with depression—those with comorbid emotional instability—to consider as a therapeutic target for the future. Hence, inclusion criteria for future trials might potentially include patient age, gender, existence/quantification of prominent frown lines at baseline, severity of MDD, duration of depression, and personality characteristics of enrolled participants.
In conclusion, BTA injections appear promising as a treatment for depression as well as for other psychiatric disorders. Future studies should focus on identifying optimal candidates for this innovative treatment modality. Furthermore, BTA dosing and administration strategies (monotherapy vs adjunctive treatment to antidepressants) need to be further explored. As retrograde axonal transport of botulinum toxin has been demonstrated in animal studies, it would be interesting to further examine its effects in the human CNS to enhance our knowledge of the pathophysiology of botulinum and its potential applications in psychiatry.50
Bottom Line
Botulinum toxin shows promising antidepressant effects and may have a role in the treatment of several other psychiatric disorders. More research is needed to address limitations of previous studies and to establish an adequate treatment regimen.
Related Resources
- Wollmer MA, Magid M, Kruger TH. Botulinum toxin treatment in depression. In: Bewley A, Taylor RE, Reichenberg JS, et al (eds). Practical psychodermatology. Oxford, UK: Wiley; 2014.
- Wollmer MA, Neumann I, Magid M. et al. Shrink that frown! Botulinum toxin therapy is lifting the face of psychiatry. G Ital Dermatol Venereol. 2018;153(4):540-548.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Biperiden • Akineton
Botulinum toxin A • Botox
Botulinum toxin B • Myobloc
Clozapine • Clozaril
Deutetrabenazine • Austedo
Flupentixol • Prolixin
Imipramine • Tofranil
Olanzapine • Zyprexa
Reserpine • Serpalan, Serpasil
Tetrabenazine • Xenazine
Valbenazine • Ingrezza
Ziprasidone • Geodon
Botulinum toxin, a potent neurotoxic protein produced by the bacterium Clostridium botulinum, has been used as treatment for a variety of medical indications for more than 25 years (Box1-12). Recently, researchers have been exploring the role of botulinum toxin in psychiatry, primarily as an adjunctive treatment for depression, but also for several other possible indications. Several studies, including randomized controlled trials (RCTs), have provided evidence that glabellar botulinum toxin injections may be a safe and effective treatment for depression. In this article, we provide an update on the latest clinical trials that evaluated botulinum toxin for depression, and also summarize the evidence regarding other potential clinical psychiatric applications of botulinum toxin.
Several RCTs suggest efficacy for depression
The use of botulinum toxin to treat depression is based on the facial feedback hypothesis, which was first proposed by Charles Darwin in 187213 and further elaborated by William James,14,15 who emphasized the importance of the sensation of bodily changes in emotion. Contrary to the popular belief that emotions trigger physiological changes in the body, James postulated that peripheral bodily changes secondary to stimuli perception would exert a sensory feedback, generating emotions. The manipulation of human facial expression with an expression that is associated with a particular emotion (eg, holding a pen with teeth, leading to risorius/zygomaticus muscles contraction and a smile simulation) was found to influence participants’ affective responses in the presence of emotional stimuli (eg, rating cartoons as funnier), reinforcing the facial-feedback hypothesis.16,17
From a neurobiologic standpoint, facial botulinum toxin A (BTA) injections in rats were associated with increased serotonin and norepinephrine concentrations in the hypothalamus and striatum, respectively.18 Moreover, amygdala activity in response to angry vs happy faces, measured via functional magnetic resonance imaging (fMRI), was found to be attenuated after BTA applications to muscles involved in angry facial expressions.19,20 Both the neurotransmitters as well as the aforementioned brain regions have been implicated in the pathophysiology of depression.21,22
Compared with those in the placebo group, participants in the BTA group had a higher response rate as measured by the HAM-D17 at 6 weeks after treatment (P = .02), especially female patients (P = .002). Response to BTA, defined as ≥50% reduction on the HAM-D17, occurred within 2 weeks, and lasted another 6 weeks before slightly wearing off. Assessment of the CSS-GFL showed a statistically significant change at 6 weeks (P < .001). This small study failed, however, to show significant remission rates (HAM-D17 ≤7) in the BTA group compared with placebo.
Box
Botulinum toxin is a potent neurotoxin from Clostridium botulinum. Its potential for therapeutic use was first noticed in 1817 by physician Justinus Kerner, who coined the term botulism.1 In 1897, bacteriologist Emile van Ermengem isolated the causative bacterium C. botulinum.2 It was later discovered that the toxin induces muscle paralysis by inhibiting acetylcholine release from presynaptic motor neurons at the neuromuscular junction3 and was then mainly investigated as a treatment for medical conditions involving excessive or abnormal muscular contraction.
In 1989, the FDA approved botulinum toxin A (BTA) for the treatment of strabismus, blepharospasm, and other facial nerve disorders. In 2000, both BTA and botulinum toxin B (BTB) were FDA-approved for the treatment of cervical dystonia, and BTA was approved for the cosmetic treatment of frown lines (glabellar, canthal, and forehead lines).4 Other approved clinical indications for BTA include urinary incontinence due to detrusor overactivity associated with a neurologic condition such as spinal cord injury or multiple sclerosis; prophylaxis of headaches in chronic migraine patients; treatment of both upper and lower limb spasticity; severe axillary hyperhidrosis inadequately managed by topical agents; and the reduction of the severity of abnormal head position and neck pain.5 Its anticholinergic effects have been also investigated for treatment of hyperhidrosis as well as sialorrhea caused by neurodegenerative disorders such as amyotrophic lateral sclerosis.6-8 Multiple studies have shown that botulinum toxin can alleviate spasms of the gastrointestinal tract, aiding patients with dysphagia and achalasia.9-11 There is also growing evidence supporting the use of botulinum toxin in the treatment of chronic pain, including non-migraine types of headaches such as tension headaches; myofascial syndrome; and neuropathic pain.12
Continue to: In a second RCT involving 74 patients with depression...
In a second RCT involving 74 patients with depression, Finzi and Rosenthal25 observed statistically significant response and remission rates in participants who received BTA injections, as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS). Participants were given either BTA or saline injections and assessed at 3 visits across 6 weeks using the MADRS, CGI, and Beck Depression Inventory-II (BDI-II). Photographs of participants’ facial expressions were assessed using frown scores to see whether changes in facial expression were associated with improvement of depression.
This study was able to reproduce on a larger scale the results observed by Wollmer et al.23 It found a statistically significant increase in the rate of remission (MADRS ≤10) at 6 weeks following BTA injections (27%, P < .02), and that even patients who were not resistant to antidepressants could benefit from BTA. However, although there was an observable trend in improvement of frown scores associated with improved depression scores, the correlation between these 2 variables was not statistically significant.
In a crossover RCT, Magid et al26 observed the response to BTA vs placebo saline injections in 30 patients with moderate to severe frown lines. The study lasted 24 weeks; participants switched treatments at Week 12. Mood improvement was assessed using the 21-item Hamilton Depression Rating Scale (HDRS-21), BDI, and Patient Health Questionnaire-9 (PHQ-9). Compared with patients who received placebo injections, those treated with BTA injections showed statistically significant response rates, but not remission rates. This study demonstrated continued improvement throughout the 24 weeks in participants who initially received BTA injections, despite having received placebo for the last 12 weeks, by which time the cosmetic effects of the initial injection had worn off. This suggests that the antidepressant effects of botulinum toxin may not depend entirely on its paralytic effects, but also on its impact on the neurotransmitters involved in the pathophysiology of depression.18 By demonstrating improvement in the placebo group once they were started on botulinum toxin, this study also was able to exclude the possibility that other variables may be responsible for the difference in the clinical course between the 2 groups. However, this study was limited by a small sample size, and it only included participants who had moderate to severe frown lines at baseline.
Zamanian et al27 examined the therapeutic effects of BTA injections in 28 Iranian patients with major depressive disorder (MDD) diagnosed according to DSM-5 criteria. At 6 weeks, there were significant improvements in BDI scores in patients who received BTA vs those receiving placebo. However, these changes were demonstrated at 6 weeks (not as early as 2 weeks), and patients didn’t achieve remission.
A large-scale, multicenter U.S. phase II RCT investigated the safety, tolerability, and efficacy of a single administration of 2 different doses of BTA (30 units or 50 units) as monotherapy for the treatment of moderate to severe depression in 258 women.28 Effects on depression were measured at 3, 6, and 9 weeks using the MADRS. Participants who received the 30-unit injection showed statistically significant improvement at 3 weeks (
More recently, in a case series, Chugh et al30 examined the effect of BTA in 42 patients (55% men) with severe treatment-resistant depression. Participants were given BTA injections in the glabellar region as an adjunctive treatment to antidepressants and observed for at least 6 weeks. Depression severity was measured using HAM-D17, MADRS, and BDI at baseline and at 3 weeks. Changes in glabellar frown lines also were assessed using the CSS-GFL. The authors reported statistically significant improvements in HAM-D17 (
A summary of the RCTs of BTA for treating depression appears in Table 1.23,25-28
Continue to: Benefits for other psychiatric indications
Benefits for other psychiatric indications
Borderline personality disorder. In a case series of 6 women, BTA injections in the glabellar region were reported to be particularly effective for the treatment of borderline personality disorder symptoms that were resistant to psychotherapy and pharmacotherapy.31 Two to 6 weeks after a 29-unit injection, borderline personality disorder symptoms as measured by the Zanarini Rating Scale for Borderline Personality Disorder and/or the Borderline Symptom List were shown to significantly improve by 49% to 94% from baseline (P ≤ .05). These findings emphasize the promising therapeutic role of BTA on depressive symptoms concomitant with the emotional lability, impulsivity, and negative emotions that usually characterize this personality disorder.31,32 A small sample size and lack of a placebo comparator are limitations of this research.
Neuroleptic-induced sialorrhea. Botulinum toxin injections in the salivary glands have been investigated for treating clozapine-induced sialorrhea because they are thought to directly inhibit the release of acetylcholine from salivary glands. One small RCT that used botulinum toxin B (BTB)33 and 1 case report that used BTA34 reported successful reduction in hypersalivation, with doses ranging from 150 to 500 units injected in each of the parotid and/or submandibular glands bilaterally. Although the treatment was well tolerated and lasted up to 16 weeks, larger studies are needed to replicate these findings.33-35
Orofacial tardive dyskinesia. Several case reports of orofacial tardive dyskinesia, including lingual dyskinesia and lingual protrusion dystonia, have found improvements in hyperkinetic movements following muscular BTA injections, such as in the genioglossus muscle in the case of tongue involvement.36-39 These cases were, however, described in the literature before the recent FDA approval of the vesicular monoamine transporter 2 inhibitors valbenazine and deutetrabenazine for the treatment of tardive dyskinesia.40,41
Studies examining botulinum toxin’s application in areas of psychiatry other than depression are summarized in Table 2.31,33,36-38
Continue to: Promising initial findings but multiple limitations
Promising initial findings but multiple limitations
Although BTA injections have been explored as a potential treatment for several psychiatric conditions, the bulk of recent evidence is derived from studies in patients with depressive disorders. BTA injections in the glabellar regions have been shown in small RCTs to be well-tolerated with overall promising improvement of depressive symptoms, optimally 6 weeks after a single injection. Moreover, BTA has been shown to be safe and long-lasting, which would be convenient for patients and might improve adherence to therapy.42-44 BTA’s antidepressant effects were shown to be independent of frown line severity or patient satisfaction with cosmetic effects.45 The trials by Wollmer et al,23 Finzi and Rosenthal,25 and Magid et al26 mainly studied BTA as an adjunctive treatment to antidepressants in patients with ongoing unipolar depression. However, Finzi and Rosenthal25 included patients who were not medicated at the time of the study.
Pooled analysis of these 3 RCTs found that patients who received BTA monotherapy improved equally to those who received it as an adjunctive treatment to antidepressants. Overall, on primary endpoint measures, a response rate of 54.2% was obtained in the BTA group compared with 10.7% among patients who received placebo saline injections (odds ratio [OR] 11.1, 95% confidence interval [CI], 4.3 to 28.8, number needed to treat [NNT] = 2.3) and a remission rate of 30.5% with BTA compared with 6.7% with placebo (OR 7.3, 95 % CI, 2.4 to 22.5, NNT = 4.2).46 However, remission rates tend to be higher in the augmentation groups, and so further studies are needed to compare both treatment strategies.
Nevertheless, these positive findings have been recently challenged by the results of the largest U.S. multicenter phase II RCT,28 which failed to find a significant antidepressant effect at 6 weeks with the 30-unit BTA injection, and also failed to prove a dose-effect relationship, as the 50-unit injection wasn’t superior to the lower dose and didn’t significantly differ from placebo. One hypothesis to explain this discrepancy may be the difference in injection sites between the treatment and placebo groups.47 Future studies need to address the various limitations of earlier clinical trials that mainly yielded promising results with BTA.
A major concern is the high rate of unblinding of participants and researchers in BTA trials, as the cosmetic effects of botulinum toxin injections make them easy to distinguish from saline injections. Ninety percent of participants in the Wollmer et al study23 were able to correctly guess their group allocation, while 60% of evaluators guessed correctly. Finzi and Rozenthal25 reported 52% of participants in the BTA group, 46% in the placebo group, and 73% of evaluators correctly guessed their allocation. Magid et al26 reported 75% of participants were able to guess the order of intervention they received. The high unblinding rates in these trials remains a significant limitation. There is a concern that this may lead to an underestimation of the placebo effect relative to clinical improvement, thus causing inflation of outcome differences between groups. Although various methods have been tried to minimize evaluator unblinding, such as placing surgical caps on participants’ faces during visits to hide the glabellar region, better methods need to be implemented to prevent unblinding of both raters and participants.
Furthermore, except for the multicenter phase II trial, most studies have been conducted in small samples, which limits their statistical power. Larger controlled trials will be needed to replicate the positive findings obtained in smaller RCTs.
Another limitation is that the majority of the well-designed RCTs were conducted in populations that were predominantly female, which makes it difficult to reliably assess treatment efficacy in men. This may be because cosmetic treatment with botulinum toxin injection is more favorably received by women than by men. A recent comparison48 of the studies by Wollmer et al23 and Finzi and Rosenthal25 discussed an interesting observation. Wollmer et al did not explicitly mention botulinum toxin when recruiting for the study, while Finzi and Rosenthal did. While approximately a quarter of the participants in the Wollmer et al study were male, Finzi and Rosenthal attracted an almost entirely female population. Perhaps there is a potential bias for females to be more attracted to these studies due to the secondary gain of receiving a cosmetic procedure.
In an attempt to understand predictors of positive response to botulinum toxin in patients with depression, Wollmer et al49 conducted a follow-up study in which they reassessed the data obtained from their initial RCT using the HAM-D agitation item scores to separate the 15 participants who received BTA into low-agitation (≤1 score on agitation item of the HAM-D scale) and high-agitation (≥2 score on agitation item of the HAM-D scale) groups. They found that the 9 participants who responded to BTA treatment had significantly higher baseline agitation scores than participants who did not respond (1.56 ± 0.88 vs 0.33 ± 0.52, P = .01). All of the participants who presented with higher agitation levels experienced response, compared with 40% of those with lower agitation levels (P = .04), although there was no significant difference in magnitude of improvement (14.2 ± 1.92 vs 8.0 ± 9.37, P = .07). The study added additional support to the facial feedback hypothesis, as it links the improvement of depression to facial muscle activation targeted by the injections. It also introduced a potential predictor of response to botulinum toxin treatment, highlighting potential factors to consider when enrolling patients in future investigations.
The case series of patients with borderline personality disorder31 also shed light on the potential positive effect of BTA treatment for a particular subtype of patients with depression—those with comorbid emotional instability—to consider as a therapeutic target for the future. Hence, inclusion criteria for future trials might potentially include patient age, gender, existence/quantification of prominent frown lines at baseline, severity of MDD, duration of depression, and personality characteristics of enrolled participants.
In conclusion, BTA injections appear promising as a treatment for depression as well as for other psychiatric disorders. Future studies should focus on identifying optimal candidates for this innovative treatment modality. Furthermore, BTA dosing and administration strategies (monotherapy vs adjunctive treatment to antidepressants) need to be further explored. As retrograde axonal transport of botulinum toxin has been demonstrated in animal studies, it would be interesting to further examine its effects in the human CNS to enhance our knowledge of the pathophysiology of botulinum and its potential applications in psychiatry.50
Bottom Line
Botulinum toxin shows promising antidepressant effects and may have a role in the treatment of several other psychiatric disorders. More research is needed to address limitations of previous studies and to establish an adequate treatment regimen.
Related Resources
- Wollmer MA, Magid M, Kruger TH. Botulinum toxin treatment in depression. In: Bewley A, Taylor RE, Reichenberg JS, et al (eds). Practical psychodermatology. Oxford, UK: Wiley; 2014.
- Wollmer MA, Neumann I, Magid M. et al. Shrink that frown! Botulinum toxin therapy is lifting the face of psychiatry. G Ital Dermatol Venereol. 2018;153(4):540-548.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Biperiden • Akineton
Botulinum toxin A • Botox
Botulinum toxin B • Myobloc
Clozapine • Clozaril
Deutetrabenazine • Austedo
Flupentixol • Prolixin
Imipramine • Tofranil
Olanzapine • Zyprexa
Reserpine • Serpalan, Serpasil
Tetrabenazine • Xenazine
Valbenazine • Ingrezza
Ziprasidone • Geodon
1. Erbguth FJ, Naumann M. Historical aspects of botulinum toxin. Justinus Kerner (1786-1862) and the “sausage” poison. Neurology. 1999;53(8):1850-1853.
2. Devriese PP. On the discovery of Clostridium botulinum. J History Neurosci. 1999;8(1):43-50.
3. Burgen ASV, Dickens F, Zatman LJ. The action of botulinum toxin on the neuro-muscular junction. J Physiol. 1949;109(1-2):10-24.
4. Jankovic J. Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry. 2004;75(7):951-957.
5. BOTOX (OnabotulinumtoxinA) [package insert]. Allergan, Inc., Irvine, CA; 2015.
6. Saadia D, Voustianiouk A, Wang AK, et al. Botulinum toxin type A in primary palmar hyperhidrosis. Randomized, single-blind, two-dose study. Neurology. 2001;57(11):2095-2099.
7. Naumann MK, Lowe NJ. Effect of botulinum toxin type A on quality of life measures in patients with excessive axillary sweating: a randomized controlled trial. Br J Dermatol. 2002;147(6):1218-1226.
8. Giess R, Naumann M, Werner E, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhea in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2000;69(1):121-123.
9. Restivo DA, Palmeri A, Marchese-Ragona R. Botulinum toxin for cricopharyngeal dysfunction in Parkinson’s disease. N Engl J Med. 2002;346(15):1174-1175.
10. Pasricha PJ, Ravich WJ, Hendrix T, et al. Intrasphincteric botulinum toxin for the treatment of achalasia. N Engl J Med. 1995(12);322:774-778.
11. Schiano TD, Parkman HP, Miller LS, et al. Use of botulinum toxin in the treatment of achalasia. Dig Dis. 1998;16(1):14-22.
12. Sim WS. Application of botulinum toxin in pain management. Korean J Pain. 2011;24(1):1-6.
13. Darwin C. The expression of the emotions in man and animals. London, UK: John Murray; 1872:366.
14. James W. The principles of psychology, vol. 2. New York, NY: Henry Holt and Company; 1890.
15. James W. II. —What is an emotion? Mind. 1884;os-IX(34):188-205.
16. Strack R, Martin LL, Stepper S. Inhibiting and facilitating conditions of facial expressions: a nonobtrusive test of the facial feedback hypothesis. J Pers Soc Psychol. 1988;54(5):768-777.
17. Larsen RJ, Kasimatis M, Frey K. Facilitating the furrowed brow: an unobtrusive test of the facial feedback hypothesis applied to unpleasant affect. Cogn Emot. 1992;6(5):321-338.
18. Ibragic S, Matak I, Dracic A, et al. Effects of botulinum toxin type A facial injection on monoamines and their metabolites in sensory, limbic, and motor brain regions in rats. Neurosci Lett. 2016;617:213-217.
19. Hennenlotter A, Dresel C, Castrop F, et al. The link between facial feedback and neural activity within central circuitries of emotion—new insights from botulinum toxin-induced denervation of frown muscles. Cereb Cortex. 2009;19(3):537-42
20. Kim MJ, Neta M, Davis FC, et al. Botulinum toxin-induced facial muscle paralysis affects amygdala responses to the perception emotional expressions: preliminary findings from an A-B-A design. Biol Mood Anxiety Disord. 2014;4:11.
21. Nestler EJ, Barrot M, DiLeone RJ, et al. Neurobiology of depression. Neuron. 2002;34(1):13-25.
22. Pandya M, Altinay M, Malone DA Jr, et al. Where in the brain is depression? Curr Psychiatry Rep. 2012;14(6):634-642.
23. Wollmer MA, de Boer C, Kalak N, et al. Facing depression with botulinum toxin: a randomized controlled trial. J Psychiatr Res. 2012;46:574-581.
24. BOTOX Cosmetic [prescribing information]. Allergan, Inc., Irvine, CA; 2017.
25. Finzi E, Rosenthal NE. Treatment of depression with onabotulinumtoxinA; a randomized, double-blind, placebo controlled trial. J Psychiatr Res. 2014;52:1-6.
26. Magid M, Reichenberg JS, Poth PE, et al. The treatment of major depressive disorder using botulinum toxin A: a 24 week randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2014;75(8):837-844.
27. Zamanian A, Ghanbari Jolfaei A, Mehran G, et al. Efficacy of botox versus placebo for treatment of patients with major depression. Iran J Public Health. 2017;46(7):982-984.
28. Allergan. OnabotulinumtoxinA as treatment for major depressive disorder in adult females. 2017. https://clinicaltrials.gov/ct2/show/NCT02116361. Accessed October 26, 2018.
29. Allergan. Allergan reports topline phase II data supporting advancement of BOTOX® (onabotulinumtoxinA) for the treatment of major depressive disorder (MDD). April 5, 2017. https://www.allergan.com/news/news/thomson-reuters/allergan-reports-topline-phase-ii-data-supporting. Accessed October 26, 2018.
30. Chugh S, Chhabria A, Jung S, et al. Botulinum toxin as a treatment for depression in a real-world setting. J Psychiatr Pract. 2018;24(1):15-20.
31. Kruger TH, Magid M, Wollmer MA. Can botulinum toxin help patients with borderline personality disorder? Am J Psychiatry. 2016;173(9):940-941.
32. Baumeister JC, Papa G, Foroni F. Deeper than skin deep – the effect of botulinum toxin-A on emotion processing. Toxicon. 2016;119:86-90.
33. Steinlechner S, Klein C, Moser A, et al. Botulinum toxin B as an effective and safe treatment for neuroleptic-induced sialorrhea. Psychopharmacology (Berl). 2010;207(4):593-597.
34. Kahl KG, Hagenah J, Zapf S, et al. Botulinum toxin as an effective treatment of clozapine-induced hypersalivation. Psychopharmacology (Berl). 2004;173(1-2):229-230.
35. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
36. Tschopp L, Salazar Z, Micheli F. Botulinum toxin in painful tardive dyskinesia. Clin Neuropharmacol. 2009;32(3):165-166.
37. Hennings JM, Krause E, Bötzel K, et al. Successful treatment of tardive lingual dystonia with botulinum toxin: case report and review of the literature. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1167-1171.
38. Slotema CW, van Harten PN, Bruggeman R, et al. Botulinum toxin in the treatment of orofacial tardive dyskinesia: a single blind study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):507-509.
39. Esper CD, Freeman A, Factor SA. Lingual protrusion dystonia: frequency, etiology and botulinum toxin therapy. Parkinsonism Relat Disord. 2010;16(7):438-441.
40. Seeberger LC, Hauser RA. Valbenazine for the treatment of tardive dyskinesia. Expert Opin Pharmacother. 2017;18(12):1279-1287.
41. Citrome L. Deutetrabenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication—What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(11):e13030.
42. Brin MF, Boodhoo TI, Pogoda JM, et al. Safety and tolerability of onabotulinumtoxinA in the tretment of facial lines: a meta-analysis of individual patient data from global clinical registration studies in 1678 participants. J Am Acad Dermatol. 2009;61:961-970.
43. Beer K. Cost effectiveness of botulinum toxins for the treatment of depression: preliminary observations. J Drugs Dermatol. 2010;9(1):27-30.
44. Serna MC, Cruz I, Real J, et al. Duration and adherence of antidepressant treatment (2003-2007) based on prescription database. Eur Psychiatry. 2010;25(4):206-213.
45. Rechenberg JS, Hauptman AJ, Robertson HT, et al. Botulinum toxin for depression: Does patient appearance matter? J Am Acad Dermatol. 2016;74(1):171-173.
46. Magid M, Finzi E, Kruger THC, et al. Treating depression with botulinum toxin: a pooled analysis of randomized controlled trials. Pharmacopsychiatry. 2015;48(6):205-210.
47. Court, E. Allergan is still hopeful about using Botox to treat depression. April 8, 2017. https://www.marketwatch.com/story/allergan-is-still-hopeful-about-using-botox-to-treat-depression-2017-04-07. Accessed October 26, 2018.
48. Rudorfer MV. Botulinum toxin: does it have a place in the management of depression? CNS Drugs. 2018;32(2):97-100.
49. Wollmer MA, Kalak N, Jung S, et al. Agitation predicts response of depression to botulinum toxin treatment in a randomized controlled trial. Front Psychiatry. 2014;5:36
50. Antonucci F, Rossi C, Gianfranceschi L, et al. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28(14):3689-3696.
1. Erbguth FJ, Naumann M. Historical aspects of botulinum toxin. Justinus Kerner (1786-1862) and the “sausage” poison. Neurology. 1999;53(8):1850-1853.
2. Devriese PP. On the discovery of Clostridium botulinum. J History Neurosci. 1999;8(1):43-50.
3. Burgen ASV, Dickens F, Zatman LJ. The action of botulinum toxin on the neuro-muscular junction. J Physiol. 1949;109(1-2):10-24.
4. Jankovic J. Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry. 2004;75(7):951-957.
5. BOTOX (OnabotulinumtoxinA) [package insert]. Allergan, Inc., Irvine, CA; 2015.
6. Saadia D, Voustianiouk A, Wang AK, et al. Botulinum toxin type A in primary palmar hyperhidrosis. Randomized, single-blind, two-dose study. Neurology. 2001;57(11):2095-2099.
7. Naumann MK, Lowe NJ. Effect of botulinum toxin type A on quality of life measures in patients with excessive axillary sweating: a randomized controlled trial. Br J Dermatol. 2002;147(6):1218-1226.
8. Giess R, Naumann M, Werner E, et al. Injections of botulinum toxin A into the salivary glands improve sialorrhea in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2000;69(1):121-123.
9. Restivo DA, Palmeri A, Marchese-Ragona R. Botulinum toxin for cricopharyngeal dysfunction in Parkinson’s disease. N Engl J Med. 2002;346(15):1174-1175.
10. Pasricha PJ, Ravich WJ, Hendrix T, et al. Intrasphincteric botulinum toxin for the treatment of achalasia. N Engl J Med. 1995(12);322:774-778.
11. Schiano TD, Parkman HP, Miller LS, et al. Use of botulinum toxin in the treatment of achalasia. Dig Dis. 1998;16(1):14-22.
12. Sim WS. Application of botulinum toxin in pain management. Korean J Pain. 2011;24(1):1-6.
13. Darwin C. The expression of the emotions in man and animals. London, UK: John Murray; 1872:366.
14. James W. The principles of psychology, vol. 2. New York, NY: Henry Holt and Company; 1890.
15. James W. II. —What is an emotion? Mind. 1884;os-IX(34):188-205.
16. Strack R, Martin LL, Stepper S. Inhibiting and facilitating conditions of facial expressions: a nonobtrusive test of the facial feedback hypothesis. J Pers Soc Psychol. 1988;54(5):768-777.
17. Larsen RJ, Kasimatis M, Frey K. Facilitating the furrowed brow: an unobtrusive test of the facial feedback hypothesis applied to unpleasant affect. Cogn Emot. 1992;6(5):321-338.
18. Ibragic S, Matak I, Dracic A, et al. Effects of botulinum toxin type A facial injection on monoamines and their metabolites in sensory, limbic, and motor brain regions in rats. Neurosci Lett. 2016;617:213-217.
19. Hennenlotter A, Dresel C, Castrop F, et al. The link between facial feedback and neural activity within central circuitries of emotion—new insights from botulinum toxin-induced denervation of frown muscles. Cereb Cortex. 2009;19(3):537-42
20. Kim MJ, Neta M, Davis FC, et al. Botulinum toxin-induced facial muscle paralysis affects amygdala responses to the perception emotional expressions: preliminary findings from an A-B-A design. Biol Mood Anxiety Disord. 2014;4:11.
21. Nestler EJ, Barrot M, DiLeone RJ, et al. Neurobiology of depression. Neuron. 2002;34(1):13-25.
22. Pandya M, Altinay M, Malone DA Jr, et al. Where in the brain is depression? Curr Psychiatry Rep. 2012;14(6):634-642.
23. Wollmer MA, de Boer C, Kalak N, et al. Facing depression with botulinum toxin: a randomized controlled trial. J Psychiatr Res. 2012;46:574-581.
24. BOTOX Cosmetic [prescribing information]. Allergan, Inc., Irvine, CA; 2017.
25. Finzi E, Rosenthal NE. Treatment of depression with onabotulinumtoxinA; a randomized, double-blind, placebo controlled trial. J Psychiatr Res. 2014;52:1-6.
26. Magid M, Reichenberg JS, Poth PE, et al. The treatment of major depressive disorder using botulinum toxin A: a 24 week randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2014;75(8):837-844.
27. Zamanian A, Ghanbari Jolfaei A, Mehran G, et al. Efficacy of botox versus placebo for treatment of patients with major depression. Iran J Public Health. 2017;46(7):982-984.
28. Allergan. OnabotulinumtoxinA as treatment for major depressive disorder in adult females. 2017. https://clinicaltrials.gov/ct2/show/NCT02116361. Accessed October 26, 2018.
29. Allergan. Allergan reports topline phase II data supporting advancement of BOTOX® (onabotulinumtoxinA) for the treatment of major depressive disorder (MDD). April 5, 2017. https://www.allergan.com/news/news/thomson-reuters/allergan-reports-topline-phase-ii-data-supporting. Accessed October 26, 2018.
30. Chugh S, Chhabria A, Jung S, et al. Botulinum toxin as a treatment for depression in a real-world setting. J Psychiatr Pract. 2018;24(1):15-20.
31. Kruger TH, Magid M, Wollmer MA. Can botulinum toxin help patients with borderline personality disorder? Am J Psychiatry. 2016;173(9):940-941.
32. Baumeister JC, Papa G, Foroni F. Deeper than skin deep – the effect of botulinum toxin-A on emotion processing. Toxicon. 2016;119:86-90.
33. Steinlechner S, Klein C, Moser A, et al. Botulinum toxin B as an effective and safe treatment for neuroleptic-induced sialorrhea. Psychopharmacology (Berl). 2010;207(4):593-597.
34. Kahl KG, Hagenah J, Zapf S, et al. Botulinum toxin as an effective treatment of clozapine-induced hypersalivation. Psychopharmacology (Berl). 2004;173(1-2):229-230.
35. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
36. Tschopp L, Salazar Z, Micheli F. Botulinum toxin in painful tardive dyskinesia. Clin Neuropharmacol. 2009;32(3):165-166.
37. Hennings JM, Krause E, Bötzel K, et al. Successful treatment of tardive lingual dystonia with botulinum toxin: case report and review of the literature. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1167-1171.
38. Slotema CW, van Harten PN, Bruggeman R, et al. Botulinum toxin in the treatment of orofacial tardive dyskinesia: a single blind study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):507-509.
39. Esper CD, Freeman A, Factor SA. Lingual protrusion dystonia: frequency, etiology and botulinum toxin therapy. Parkinsonism Relat Disord. 2010;16(7):438-441.
40. Seeberger LC, Hauser RA. Valbenazine for the treatment of tardive dyskinesia. Expert Opin Pharmacother. 2017;18(12):1279-1287.
41. Citrome L. Deutetrabenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication—What is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(11):e13030.
42. Brin MF, Boodhoo TI, Pogoda JM, et al. Safety and tolerability of onabotulinumtoxinA in the tretment of facial lines: a meta-analysis of individual patient data from global clinical registration studies in 1678 participants. J Am Acad Dermatol. 2009;61:961-970.
43. Beer K. Cost effectiveness of botulinum toxins for the treatment of depression: preliminary observations. J Drugs Dermatol. 2010;9(1):27-30.
44. Serna MC, Cruz I, Real J, et al. Duration and adherence of antidepressant treatment (2003-2007) based on prescription database. Eur Psychiatry. 2010;25(4):206-213.
45. Rechenberg JS, Hauptman AJ, Robertson HT, et al. Botulinum toxin for depression: Does patient appearance matter? J Am Acad Dermatol. 2016;74(1):171-173.
46. Magid M, Finzi E, Kruger THC, et al. Treating depression with botulinum toxin: a pooled analysis of randomized controlled trials. Pharmacopsychiatry. 2015;48(6):205-210.
47. Court, E. Allergan is still hopeful about using Botox to treat depression. April 8, 2017. https://www.marketwatch.com/story/allergan-is-still-hopeful-about-using-botox-to-treat-depression-2017-04-07. Accessed October 26, 2018.
48. Rudorfer MV. Botulinum toxin: does it have a place in the management of depression? CNS Drugs. 2018;32(2):97-100.
49. Wollmer MA, Kalak N, Jung S, et al. Agitation predicts response of depression to botulinum toxin treatment in a randomized controlled trial. Front Psychiatry. 2014;5:36
50. Antonucci F, Rossi C, Gianfranceschi L, et al. Long-distance retrograde effects of botulinum neurotoxin A. J Neurosci. 2008;28(14):3689-3696.
Psychiatric consultations in long-term care: An evidence-based practical guide
Long-term care (LTC) services provide health care to >8 million people in approximately 30,000 nursing homes and assisted living/residential care communities in the United States.1 One-half of older adults in LTC have neurocognitive disorders (NCDs), and one-third have depressive syndromes.2 Common reasons for psychiatric consultation include these 2 major diagnoses, as well as delirium, behavioral and psychological symptoms of dementia (BPSD), bipolar disorder, anxiety, sleep disorders, and pain management.
Psychiatric assessment of individuals in LTC can be challenging because of atypical presentations, cognitive impairment, and multiple comorbidities. Establishing a management plan involves eliciting a careful history from both the patient and caretakers, examining previous records and medications, and selecting appropriate screening tools and laboratory tests (Table 1 and Table 2).
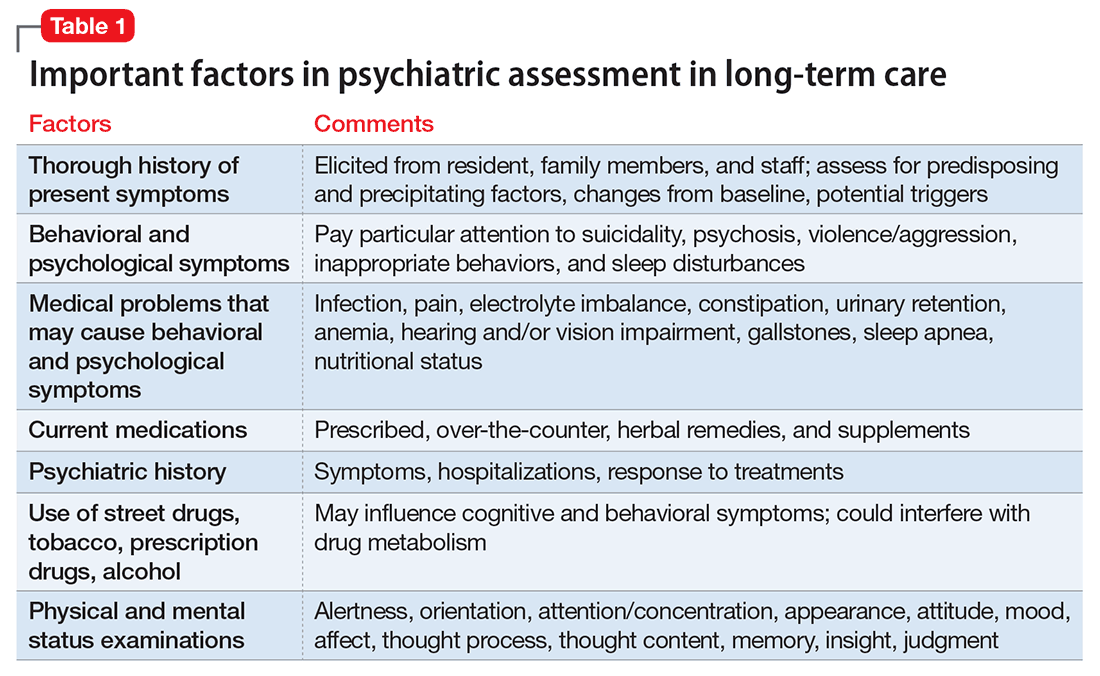
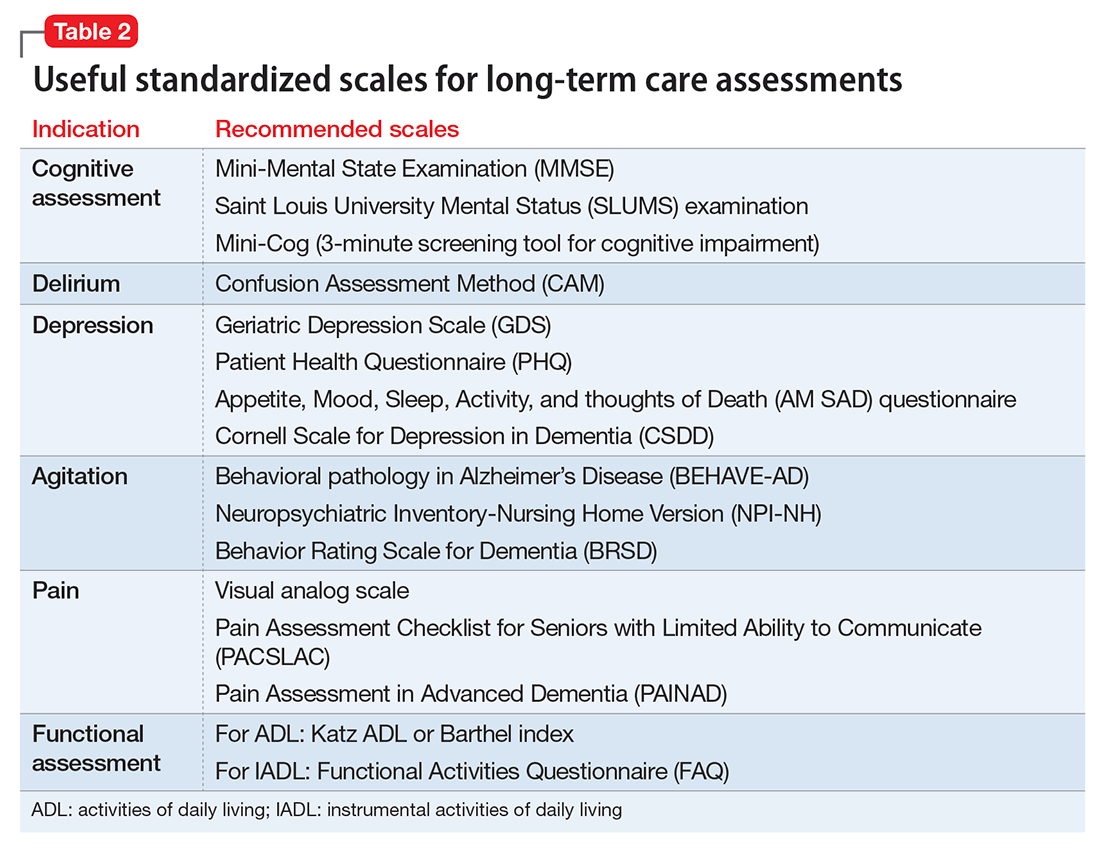
This article offers a practical approach to assess and manage common psychiatric conditions in LTC. We include new evidence about:
- assessment tools for psychiatric symptoms in LTC
- potentially inappropriate medication use in older adults
- antipsychotic use for agitation and psychosis with dementia
- nonpharmacologic interventions to help prevent cognitive decline
- antipsychotic review in reducing antipsychotic use and mortality.
Delirium
Delirium is an important topic in LTC because it is highly prevalent, poorly recognized, and can be difficult to manage. Common causes of delirium in LTC include infection (often urinary), dehydration, medications, long-standing constipation, and urinary retention (Table 3).3 Early recognition is key because delirium has been associated with cognitive decline, decreased functional status, increased caregiver burden, and increased mortality.4,5
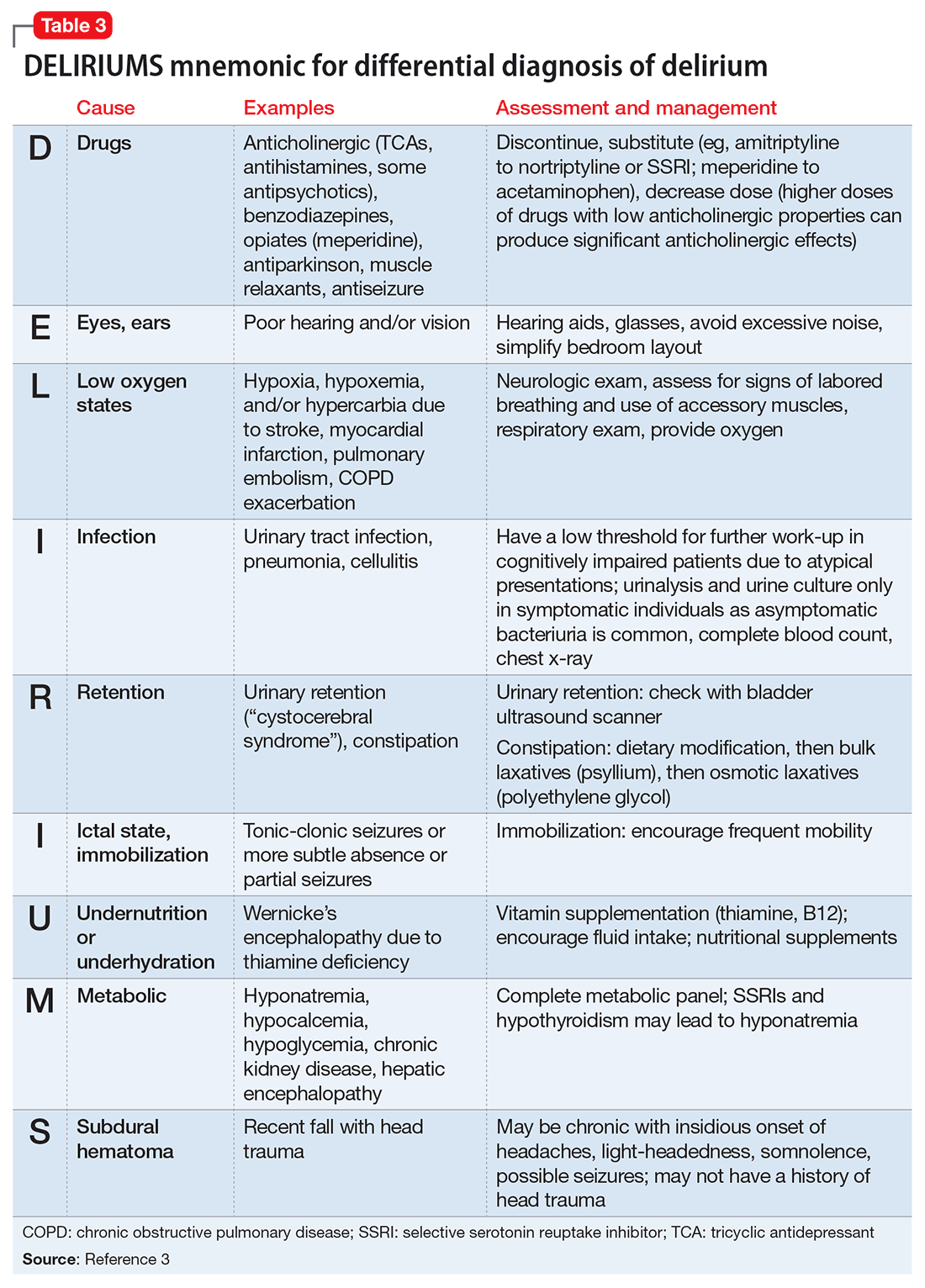
The Confusion Assessment Method (CAM) is a quick tool with 4 features to differentiate delirium from other forms of cognitive impairment.6 The 2 core features are an acute change or fluctuating course of mental status and inattention. Family members or caregivers can provide information about an acute change. To assess inattention, ask the patient to say the days of the week backward or spell the word “world” backward. The 2 other features of delirium—one of which must be present when using the CAM—are disorganized thinking and altered level of consciousness.
Individuals with delirium may present with hyperactive or hypoactive psychomotor activity. Hypoactive delirium’s features, such as sluggishness and lethargy, could be confused with depression.7 A careful history to determine symptom onset and fluctuation in course can help differentiate between the 2.
Management. Delirium management always should begin by addressing underlying causes and implementing psychosocial and environmental interventions. Pharmacologic interventions have not demonstrated consistent benefit for delirium in well-designed trials and are not recommended as first-line treatment.8 The American Geriatrics Society (AGS) Beers Criteria for Potentially Inappropriate Medication Use in Older Adults recommends avoiding benzodiazepines in this population.9 Antipsychotics could be used in patients with severe agitation who pose harm to themselves or others. Nonpharmacologic approaches to delirium in LTC include:
- frequent reorientation (clocks, daily schedule)
- one-on-one monitoring by staff or family members
- use of hearing aids and eye-glasses, if needed
- maintaining an appropriate sleep-wake cycle by encouraging exposure to bright light during the day and avoiding night-time interruptions.
Restraints should not be used; they appear to worsen delirium severity, and their removal does not increase the rate of falls or fall-related injury.10
Various methods for managing a patient with delirium have been proposed, such as the TADA approach (tolerate, anticipate, and don’t agitate).5,11,12 For example, if a patient’s agitation worsens with attempted reorientation, distraction or playing along with the disorientation could be more beneficial.12
Keep in mind delirium’s overlapping presentation with Lewy body dementia (LBD). Patients with LBD demonstrate a progressive decline in cognitive functioning associated with fluctuating cognition, visual hallucinations, and parkinsonism features. Consider LBD when no cause for delirium-like symptoms is found. These patients may show increased sensitivity to neuroleptics and extrapyramidal side effects.
Neurocognitive disorders
Reversible causes. Although most individuals with major NCDs are diagnosed before entering LTC, the consulting psychiatrist’s review of potentially reversible causes of neurocognitive symptoms can lead to dramatically different treatment regimens (Table 43). For example, anticholinergic medications can harm the aging brain and have been linked to delirium, increased brain atrophy, and lower scores on tests of cognitive functioning.13 Given the prevalence of polypharmacy in older adults, be aware of unexpected anticholinergic properties of many common drugs, as rated by the Aging Brain Care initiative.14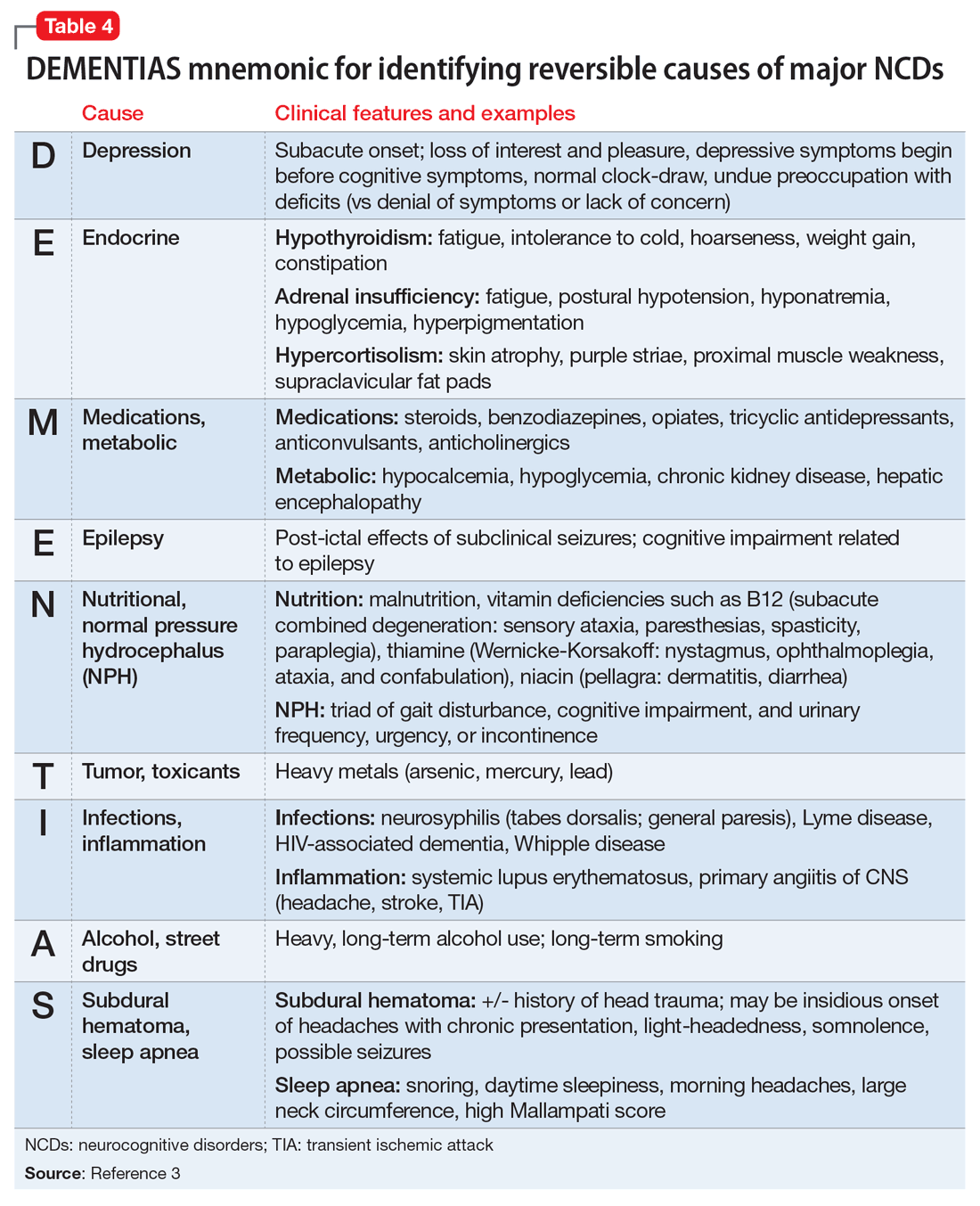
Mild cognitive impairment. Should patients showing signs of cognitive impairment or those at risk for major NCDs begin pharmacotherapy? The FDA has approved no medications for this indication, and clinical trials with agents such as cholinesterase inhibitors (ChEIs) have shown inconsistent results.
The randomized, double-blind Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability provides convincing data that a nonpharmacologic approach could benefit older adults at risk for a major NCD. A 2-year intervention of nutritional advice, aerobic and strength training, cognitive training, social activities, and blood pressure and weight monitoring was more effective in improving or maintaining cognitive function in individuals age 60 to 77, compared with general health advice given to a control group.15
Behavioral and psychological symptoms. Psychiatrists are likely to be consulted in LTC when a person with a major NCD presents with an acute episode of increased confusion and cognitive worsening, often accompanied by behavioral symptoms. BPSD may include agitation, aggression, apathy, depression, sleep problems, socially inappropriate behaviors, and psychosis. One study of patients with Alzheimer’s disease (AD) reported a cumulative 51% incidence of new-onset hallucinations and delusions at 4 years.16
Increased vulnerability to stressors, unmet needs, over- or under-stimulation, or lack of routines may predispose individuals with major NCDs to developing BPSD.17 Nonpharmacologic approaches usually are tried first, although supporting evidence is not substantial.18 Changes in environment, behavioral redirection, sensory interventions, or music therapy may reduce disruptive behaviors.19 Patients with increased confusion and agitation in late afternoon and evening (“sundowning”) may benefit from short naps after lunch, light therapy, calming activities in late afternoon, and reduced noise (such as from dishes, loud speakers, staff conversations).20
Antipsychotics. The drugs most commonly used to manage BPSD are antipsychotics, antidepressants, mood stabilizers/anticonvulsants, ChEIs, and the N-methyl-
When nonpharmacotherapeutic interventions are not successful, most guidelines agree that using an atypical antipsychotic is warranted in AD patients with severe agitation and/or psychosis that pose a risk to the patient or others or severely impair their quality of life.9,22,23
Antipsychotic review. Recent guidelines from the American Psychiatric Association (APA) recommend that attempts to taper and withdraw antipsychotic drugs be made within 4 months of initiating treatment in patients with dementia who display an adequate response.23 In a recent nursing home study, antipsychotic review was found to reduce antipsychotic use by 50% and, when combined with a social intervention, to reduce mortality compared with a group receiving neither intervention.24
Interestingly, patients receiving antipsychotic review alone showed an increase in overall neuropsychiatric symptoms.24 A previous study of patients with AD whose psychosis or agitation responded to risperidone also found an increased risk of relapse when risperidone was discontinued.25 These results highlight the importance of making patient-centered decisions, frequent re-assessments, and adding non-pharmacologic interventions (eg, positive social interactions or exercise) when attempting to discontinue antipsychotics.
Other treatment options. Because patients with LBD often display increased sensitivity to neuroleptics, agents such as quetiapine or aripiprazole (with a lower risk of EPS) are preferred when managing severe psychosis/aggression. ChEIs may show some benefit for behavioral disturbances in patients with LBD.26
In patients with AD, ChEIs have shown inconsistent results in benefiting neuropsychiatric symptoms. Preliminary data suggest some benefit with citalopram (also associated with prolonged QTc)27 and the dextromethorphan/quinidine combination FDA-approved for pseudobulbar affect, but more studies are needed.28 Pimavanserin, a 5-HT2A receptor inverse agonist, recently was approved for treating hallucinations and delusions associated with Parkinson’s disease psychosis and currently is in clinical trials for Alzheimer’s disease psychosis.
Electroconvulsive therapy (ECT) may be a therapeutic option for agitation and aggression in people with dementia.29 ECT has no absolute contraindications and can be safely performed in individuals with pacemakers or implantable cardioverter defibrillators. Common adverse effects include transient changes in blood pressure or heart rate, headache, and nausea. Cognitive adverse effects from ECT may include:
- anterograde amnesia, which typically resolves after a few weeks
- retrograde amnesia, which typically manifests as loss of impersonal memories occurring in the past few months.
Depression
The prevalence of depression in nursing home residents is an estimated 3 to 4 times that of community-dwelling older adults.30 Assessing for depression is particularly important in people with mild cognitive impairment, as depressive symptoms have been associated with progression to AD.31 Quick screening tools (Table 2) include short forms of the Patient Health Questionnaire (PHQ-2 or PHQ-9)32 or the Saint Louis University Appetite, Mood, Sleep, Activity, and thoughts of Death (SLU “AM SAD”) scale.33 The Cornell Scale for Depression in Dementia is useful for individuals with major NCDs because it relies on interviews with the patient and nursing staff or family.34
To test for other causes of depression, order a complete blood count for anemia, serum glucose, thyroid-stimulating hormone for hypothyroidism or hyperthyroidism, B12 and folate levels, and a cognitive screen such as the Saint Louis University Mental Status examination.35
Treatment. Antidepressants are generally considered effective in older patients with depression. Selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs) are first-line treatments because of safety concerns with tricyclic antidepressants. All 3 classes have shown similar efficacy in comparison trials in geriatric populations.
When initiating these agents, take care in the first few days and weeks to monitor for potential serious adverse effects, such as nausea and vomiting, which may be associated with substantial morbidity in patients with comorbidities. For monitoring treatment response, the PHQ-9 can effectively distinguish patients with persistent major depression, partial remission, or full remission.36
The optimal duration of a short-term antidepressant trial before switching to a different agent is unclear, although a good therapeutic trial typically is 4 to 12 weeks. In one study of older adults with depression, 4 weeks was enough to reliably identify those likely to benefit from a change in treatment plan.37
Cognitive-behavioral therapy (CBT) can be used in older adults not wishing to pursue pharmacotherapy or as an adjunct to antidepressants. Randomized controlled trials have shown some benefit for those with depression, anxiety, and insomnia.38 Individuals with significant cognitive deficits or those not motivated to apply CBT strategies might not benefit.
ECT may be appropriate for treating depression in older adults with:
- urgent need of a therapeutic response (eg, suicidal ideation or nutritional compromise)
- lack of response to antidepressant medication
- major depressive disorder with psychotic or catatonic features.
Evidence regarding ECT’s efficacy for late-life depression is derived primarily from clinical experience and open-label trials.39
Bipolar disorder
Most individuals with bipolar disorder present before age 50, although 9% of first manic episodes occur after age 60.40 Earlier age of onset appears to predict poor outcomes, and early-onset bipolar disorder may worsen with advanced age related to increased comorbidities and difficulty in medical management.41 Compared with younger patients, features of bipolar disorder in older adults include increased prominence of rapid cycling, more time spent in a depressed state than in manic state, and less severe manic and psychotic symptoms.42
When older patients present with depression, always evaluate for clinical features more consistent with late-onset bipolar disorder than with major depressive disorder: hypomania, family history of bipolar disorder, higher number of prior depressive episodes, and higher levels of fear and inner tension.43 The differential diagnosis for new-onset manic symptoms in older adults includes:
- general medical conditions (stroke, brain tumors, hyperthyroidism, neurosyphilis)
- medications (corticosteroids, dopaminergic drugs, St. John’s wort)
- substance use.
Hyperthyroidism deserves special attention because it can present in older adults with either manic-like symptoms and hyperkinesis or features of apathy, depression, and somnolence. Given that older age and bipolar disorder both are associated with increased suicide risk, monitor these individuals for signs of hopelessness and statements of suicide.44
Treatment. Managing bipolar disorder in older adults often requires complex medication regimens. Acute treatment options for geriatric mania and hypomania with the most supporting evidence include lithium, valproate, quetiapine, and olanzapine.45-47 The therapeutic index of lithium is small, and older individuals are more vulnerable to adverse effects related to physiologic changes (eg, decreased glomerular filtration rate or low volume of distribution) that impair lithium clearance. Lithium also interacts with many drugs commonly used by older patients, such as nonsteroidal anti-inflammatory drugs (NSAIDs) and diuretics. Common adverse events associated with lithium include memory impairment, diarrhea, falls, and tremors.
Maintenance treatment for bipolar disorder is generally the same medication used to induce remission. The evidence for maintenance treatment of bipolar disorder in older adults is limited mostly to subgroup analyses. In one retrospective analysis of patients age ≥55 in 2 randomized trials, lamotrigine and lithium were effective and well-tolerated in delaying time to intervention.48
Anxiety disorders
Anxiety among LTC residents may manifest as irritability, insomnia, restlessness, and verbal and/or physical agitation/aggression.49 Typical causes include:
- primary anxiety disorders
- anxiety symptoms during depressive episodes or bereavement
- adverse effects of medications
- complications of major NCDs or delirium.
Anxiety disorders and subsyndromal anxiety have been associated with poorer quality of life, decreased sleep, and increased distress and impairment.50
Assessment begins with a self-report of symptoms, although this may be difficult in people with major NCDs. Factors that may differentiate true anxiety from major NCDs include restlessness, irritability, muscle tension, fears, and respiratory symptoms in addition to excessive anxiety and worry.51 The Geriatric Anxiety Inventory is a useful screening tool.52 The newer Brief Anxiety and Depression Scale may identify and differentiate patients with major depressive episodes and generalized anxiety disorder (GAD).53 Potential instruments for patients with comorbid anxiety and major NCDs include the Neuropsychiatric Inventory, Rating Anxiety in Dementia scale,54 and the Anxiety in Cognitive Impairment and Dementia scale.55 Because medications can cause akathisia that may mimic anxiety symptoms, screen for the recent addition of antidepressants, antipsychotics, sympathomimetics, thyroid supplements, and corticosteroids.
Treatment of anxiety disorders—such as panic disorder, social phobia, or GAD—generally starts with SSRIs or SNRIs. Although benzodiazepines are commonly used for anxiety in older adults,56 these drugs are associated with a high rate of adverse effects: increased risk of agitation, falls, impaired cognition, and possibly dementia.57 In general, reserve benzodiazepines for treating acute episodes of severe anxiety in this population.
A particularly prevalent source of anxiety in LTC is fear of falling, which may affect up to 50% of residents and cause them to restrict their activities.58 Interventions such as CBT, exercise, or tai chi may be beneficial, although supporting evidence is lacking.
Pain and sleep management
Addressing pain. Age-related changes in pain perception and difficulty in reporting pain likely contribute to under-recognition of pain in LTC residents. Two useful methods to recognize their pain are to:
- observe for pain behaviors, such as facial expressions (grimacing and brow lowering), vocalizations, and body movements (clenched fists)
- solicit reports from nurses and other caregivers.59
Self-report may be a reliable indicator of pain for individuals with mild-to-moderate NCDs. Observational pain scales, such as the Pain Assessment Checklist for Seniors with Limited Ability to Communicate, may be useful in severe NCDs.60
The AGS recommends acetaminophen as initial pharmacotherapy to manage persistent pain.61 NSAIDs may be another option, but caution is warranted for patients with acid-peptic disease or chronic kidney disease. Opioids may be considered for severe pain, but otherwise avoid using them.
Sleep disturbances are common in LTC because of physiologic changes associated with aging (altered circadian rhythm), comorbidities (depression), and environmental factors.62 A strong association appears to exist between insomnia and use of sedative-hypnotic drugs, and the AGS Beers Criteria recommend avoiding non-benzodiazepine receptor agonists and benzodiazepines when treating insomnia in older adults.9
Assess factors that may contribute to sleep disturbances, including medications and use of caffeine or alcohol. Have the resident or caregiver document sleep patterns in a sleep diary.
Consider administrating medications at different times (eg, switch donepezil from bedtime to morning) or replace with alternatives (switch from the more anticholinergic amitriptyline to nortriptyline). Ensure that residents engage in physical activity and have at least 30 minutes daily exposure to sunlight.
In addition to behavioral interventions and CBT, treatment in older adults can involve melatonin—which has mixed evidence—or sedating antidepressants, such as mirtazapine or trazodone, in patients with comorbid depression.
1. Harris-Kojetin L, Sengupta M, Park-Lee E, et al. Long-term care services in the United States: 2013 overview. Vital Health Stat 3. 2013(37):1-107.
2. Seitz D, Purandare N, Conn D. Prevalence of psychiatric disorders among older adults in long-term care homes: a systematic review. Int Psychogeriatr. 2010;22(7):1025-1039.
3. Flaherty J, Tumosa N. Saint Louis University Geriatric Evaluation Mnemonics and Screening Tools. http://aging.slu.edu/uploads/pdf/Saint-Louis-University-Geriatric-Evaluation_2013.pdf. Accessed October 5, 2016.
4. Boockvar K, Signor D, Ramaswamy R, et al. Delirium during acute illness in nursing home residents. J Am Med Dir Assoc. 2013;14(9):656-660.
5. Inouye SK, Westendorp RG, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
6. Wei LA, Fearing MA, Sternberg EJ, et al. The Confusion Assessment Method: a systematic review of current usage. J Am Geriatr Soc. 2008;56(5):823-830.
7. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
8. Flaherty JH, Gonzales JP, Dong B. Antipsychotics in the treatment of delirium in older hospitalized adults: a systematic review. J Am Geriatr Soc. 2011;59(suppl 2):S269-S276.
9. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
10. Capezuti E, Strumpf NE, Evans LK, et al. The relationship between physical restraint removal and falls and injuries among nursing home residents. J Gerontol A Biol Sci Med Sci. 1998;53(1):M47-M52.
11. Flaherty JH, Morley JE. Delirium in the nursing home. J Am Med Dir Assoc. 2013;14(9):632-634.
12. Flaherty JH. The evaluation and management of delirium among older persons. Med Clin North Am. 2011;95(3):555-577, xi.
13. Risacher SL, McDonald BC, Tallman EF, et al. Association between anticholinergic medication use and cognition, brain metabolism, and brain atrophy in cognitively normal older adults. JAMA Neurol. 2016;73(6):721-732.
14. Anticholinergic Cognitive Burden Scale. Aging Brain Care. http://agingbraincare.org/uploads/products/ACB_scale_-_legal_size.pdf. Published 2012. Accessed October 5, 2016.
15. Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
16. Paulsen JS, Salmon DP, Thal LJ, et al. Incidence of and risk factors for hallucinations and delusions in patients with probable AD. Neurology. 2000;54(10):1965-1971.
17. Gitlin LN, Kales HC, Lyketsos CG. Nonpharmacologic management of behavioral symptoms in dementia. JAMA. 2012;308(19):2020-2029.
18. Livingston G, Kelly L, Lewis-Holmes E, et al. A systematic review of the clinical effectiveness and cost-effectiveness of sensory, psychological and behavioural interventions for managing agitation in older adults with dementia. Health Technol Assess. 2014;18(39):1-226, v-vi.
19. Kong EH, Evans LK, Guevara JP. Nonpharmacological intervention for agitation in dementia: a systematic review and meta-analysis. Aging Ment Health. 2009;13(4):512-520.
20. Khachiyants N, Trinkle D, Son SJ, et al. Sundown syndrome in persons with dementia: an update. Psychiatry Investig. 2011;8(4):275-287.
21. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525-1538.
22. Jennings L, Grossberg GT. Antipsychotics continue to have a place in the management of difficult behavior problems in patients with dementia. J Am Med Dir Assoc. 2013;14(6):447-449.
23. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
24. Ballard C, Orrell M, YongZhong S, et al. Impact of antipsychotic review and nonpharmacological intervention on antipsychotic use, neuropsychiatric symptoms, and mortality in people with dementia living in nursing homes: a factorial cluster-randomized controlled trial by the Well-Being and Health for People With Dementia (WHELD) program. Am J Psychiatry. 2015;173(3):252-262.
25. Devanand DP, Mintzer J, Schultz SK, et al. Relapse risk after discontinuation of risperidone in Alzheimer’s disease. N Engl J Med. 2012;367(16):1497-1507.
26. Matsunaga S, Kishi T, Yasue I, et al. Cholinesterase inhibitors for Lewy body disorders: a meta-analysis. Int J Neuropsychopharmacol. 2015;19(2). doi: 10.1093/ijnp/pyv086.
27. Porsteinsson AP, Drye LT, Pollock BG, et al; CitAD Research Group. Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. JAMA. 2014;311(7):682-691.
28. Cummings JL, Lyketsos CG, Peskind ER, et al. Effect of dextromethorphan-quinidine on agitation in patients with Alzheimer disease dementia: a randomized clinical trial. JAMA. 2015;314(12):1242-1254.
29. Ujkaj M, Davidoff DA, Seiner SJ, et al. Safety and efficacy of electroconvulsive therapy for the treatment of agitation and aggression in patients with dementia. Am J Geriatr Psychiatry. 2012;20(1):61-72.
30. Jongenelis K, Pot AM, Eisses AM, et al. Prevalence and risk indicators of depression in elderly nursing home patients: the AGED study. J Affect Disord. 2004;83(2-3):135-142.
31. Van der Mussele S, Fransen E, Struyfs H, et al. Depression in mild cognitive impairment is associated with progression to Alzheimer’s disease: a longitudinal study. J Alzheimers Dis. 2014;42(4):1239-1250.
32. Li C, Friedman B, Conwell Y, et al. Validity of the Patient Health Questionnaire 2 (PHQ-2) in identifying major depression in older people. J Am Geriatr Soc. 2007;55(4):596-602.
33. Chakkamparambil B, Chibnall JT, Graypel EA, et al. Development of a brief validated geriatric depression screening tool: the SLU “AM SAD”. Am J Geriatr Psychiatry. 2015;23(8):780-783.
34. Korner A, Lauritzen L, Abelskov K, et al. The Geriatric Depression Scale and the Cornell Scale for Depression in Dementia. A validity study. Nord J Psychiatry. 2006;60(5):360-364.
35. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the Mini-Mental State Examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
36. Löwe B, Unützer J, Callahan CM, et al. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care. 2004;42(12):1194-1201.
37. Mulsant BH, Houck PR, Gildengers AG, et al. What is the optimal duration of a short-term antidepressant trial when treating geriatric depression? J Clin Psychopharmacol. 2006;26(2):113-120.
38. Chand SP, Grossberg GT. How to adapt cognitive-behavioral therapy for older adults. Current Psychiatry. 2013;12(3):10-15.
39. Van der Wurff FB, Stek ML, Hoogendijk WL, et al. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev. 2003;(2):CD003593.
40. Kennedy N, Everitt B, Boydell J, et al. Incidence and distribution of first-episode mania by age: results from a 35-year study. Psychol Med. 2005;35(6):855-863.
41. Carter TD, Mundo E, Parikh SV, et al. Early age at onset as a risk factor for poor outcome of bipolar disorder. J Psychiatr Res. 2003;37(4):297-303.
42. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
43. Perlis RH, Brown E, Baker RW, et al. Clinical features of bipolar depression versus major depressive disorder in large multicenter trials. Am J Psychiatry. 2006;163(2):225-231.
44. Aizenberg D, Olmer A, Barak Y. Suicide attempts amongst elderly bipolar patients. J Affect Disord. 2006;91(1):91-94.
45. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
46. Young RC, Gyulai L, Mulsant BH, et al. Pharmacotherapy of bipolar disorder in old age: review and recommendations. Am J Geriatr Psychiatry. 2004;12(4):342-357.
47. Sajatovic M, Calabrese JR, Mullen J. Quetiapine for the treatment of bipolar mania in older adults. Bipolar Disord. 2008;10(6):662-671.
48. Sajatovic M, Gyulai L, Calabrese JR, et al. Maintenance treatment outcomes in older patients with bipolar I disorder. Am J Geriatr Psychiatry. 2005;13(4):305-311.
49. Gum AM, King-Kallimanis B, Kohn R. Prevalence of mood, anxiety, and substance-abuse disorders for older Americans in the National Comorbidity Survey-Replication. Am J Geriatr Psychiatry. 2009;17(9):769-781.
50. Wetherell JL, Le Roux H, Gatz M. DSM-IV criteria for generalized anxiety disorder in older adults: distinguishing the worried from the well. Psychol Aging. 2003;18(3):622-627.
51. Starkstein SE, Jorge R, Petracca G, et al. The construct of generalized anxiety disorder in Alzheimer disease. Am J Geriatr Psychiatry. 2007;15(1):42-49.
52. Pachana NA, Byrne GJ, Siddle H, et al. Development and validation of the Geriatric Anxiety Inventory. Int Psychogeriatr. 2007;19(1):103-114.
53. Mansbach WE, Mace RA, Clark KM. The Brief Anxiety and Depression Scale (BADS): a new instrument for detecting anxiety and depression in long-term care residents. Int Psychogeriatr. 2015;27(4):673-681.
54. Seignourel PJ, Kunik ME, Snow L, et al. Anxiety in dementia: a critical review. Clin Psychol Rev. 2008;28(7):1071-1082.
55. Gerolimatos LA, Ciliberti CM, Gregg JJ, et al. Development and preliminary evaluation of the Anxiety in Cognitive Impairment and Dementia (ACID) scales. Int Psychogeriatr. 2015;27(11):1825-1838.
56. Benitez CI, Smith K, Vasile RG, et al. Use of benzodiazepines and selective serotonin reuptake inhibitors in middle-aged and older adults with anxiety disorders: a longitudinal and prospective study. Am J Geriatr Psychiatry. 2008;16(1):5-13.
57. Billioti de Gage S, Moride Y, Ducruet T, et al. Benzodiazepine use and risk of Alzheimer’s disease: case control study. BMJ. 2014;349:g5205.
58. Lach HW, Parsons JL. Impact of fear of falling in long term care: an integrative review. J Am Med Dir Assoc. 2013;14(8):573-577.
59. Hadjistavropoulos T, Herr K, Prkachin KM, et al. Pain assessment in elderly adults with dementia. Lancet Neurol. 2014;13(12):1216-1227.
60. Zwakhalen SM, Hamers JP, Abu-Saad HH, et al. Pain in elderly people with severe dementia: a systematic review of behavioural pain assessment tools [published online January 27, 2006]. BMC Geriatr. doi: 10.1186/1471-2318-6-3.
61. American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Adults. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc. 2009;57(8):1331-1346.
62. Gindin J, Shochat T, Chetrit A, et al; SHELTER project. Insomnia in long-term care facilities: a comparison of seven European countries and Israel: the Services and Health for Elderly in Long TERm care study. J Am Geriatr Soc. 2014;62(11):2033-2039.
Long-term care (LTC) services provide health care to >8 million people in approximately 30,000 nursing homes and assisted living/residential care communities in the United States.1 One-half of older adults in LTC have neurocognitive disorders (NCDs), and one-third have depressive syndromes.2 Common reasons for psychiatric consultation include these 2 major diagnoses, as well as delirium, behavioral and psychological symptoms of dementia (BPSD), bipolar disorder, anxiety, sleep disorders, and pain management.
Psychiatric assessment of individuals in LTC can be challenging because of atypical presentations, cognitive impairment, and multiple comorbidities. Establishing a management plan involves eliciting a careful history from both the patient and caretakers, examining previous records and medications, and selecting appropriate screening tools and laboratory tests (Table 1 and Table 2).
This article offers a practical approach to assess and manage common psychiatric conditions in LTC. We include new evidence about:
- assessment tools for psychiatric symptoms in LTC
- potentially inappropriate medication use in older adults
- antipsychotic use for agitation and psychosis with dementia
- nonpharmacologic interventions to help prevent cognitive decline
- antipsychotic review in reducing antipsychotic use and mortality.
Delirium
Delirium is an important topic in LTC because it is highly prevalent, poorly recognized, and can be difficult to manage. Common causes of delirium in LTC include infection (often urinary), dehydration, medications, long-standing constipation, and urinary retention (Table 3).3 Early recognition is key because delirium has been associated with cognitive decline, decreased functional status, increased caregiver burden, and increased mortality.4,5
The Confusion Assessment Method (CAM) is a quick tool with 4 features to differentiate delirium from other forms of cognitive impairment.6 The 2 core features are an acute change or fluctuating course of mental status and inattention. Family members or caregivers can provide information about an acute change. To assess inattention, ask the patient to say the days of the week backward or spell the word “world” backward. The 2 other features of delirium—one of which must be present when using the CAM—are disorganized thinking and altered level of consciousness.
Individuals with delirium may present with hyperactive or hypoactive psychomotor activity. Hypoactive delirium’s features, such as sluggishness and lethargy, could be confused with depression.7 A careful history to determine symptom onset and fluctuation in course can help differentiate between the 2.
Management. Delirium management always should begin by addressing underlying causes and implementing psychosocial and environmental interventions. Pharmacologic interventions have not demonstrated consistent benefit for delirium in well-designed trials and are not recommended as first-line treatment.8 The American Geriatrics Society (AGS) Beers Criteria for Potentially Inappropriate Medication Use in Older Adults recommends avoiding benzodiazepines in this population.9 Antipsychotics could be used in patients with severe agitation who pose harm to themselves or others. Nonpharmacologic approaches to delirium in LTC include:
- frequent reorientation (clocks, daily schedule)
- one-on-one monitoring by staff or family members
- use of hearing aids and eye-glasses, if needed
- maintaining an appropriate sleep-wake cycle by encouraging exposure to bright light during the day and avoiding night-time interruptions.
Restraints should not be used; they appear to worsen delirium severity, and their removal does not increase the rate of falls or fall-related injury.10
Various methods for managing a patient with delirium have been proposed, such as the TADA approach (tolerate, anticipate, and don’t agitate).5,11,12 For example, if a patient’s agitation worsens with attempted reorientation, distraction or playing along with the disorientation could be more beneficial.12
Keep in mind delirium’s overlapping presentation with Lewy body dementia (LBD). Patients with LBD demonstrate a progressive decline in cognitive functioning associated with fluctuating cognition, visual hallucinations, and parkinsonism features. Consider LBD when no cause for delirium-like symptoms is found. These patients may show increased sensitivity to neuroleptics and extrapyramidal side effects.
Neurocognitive disorders
Reversible causes. Although most individuals with major NCDs are diagnosed before entering LTC, the consulting psychiatrist’s review of potentially reversible causes of neurocognitive symptoms can lead to dramatically different treatment regimens (Table 43). For example, anticholinergic medications can harm the aging brain and have been linked to delirium, increased brain atrophy, and lower scores on tests of cognitive functioning.13 Given the prevalence of polypharmacy in older adults, be aware of unexpected anticholinergic properties of many common drugs, as rated by the Aging Brain Care initiative.14
Mild cognitive impairment. Should patients showing signs of cognitive impairment or those at risk for major NCDs begin pharmacotherapy? The FDA has approved no medications for this indication, and clinical trials with agents such as cholinesterase inhibitors (ChEIs) have shown inconsistent results.
The randomized, double-blind Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability provides convincing data that a nonpharmacologic approach could benefit older adults at risk for a major NCD. A 2-year intervention of nutritional advice, aerobic and strength training, cognitive training, social activities, and blood pressure and weight monitoring was more effective in improving or maintaining cognitive function in individuals age 60 to 77, compared with general health advice given to a control group.15
Behavioral and psychological symptoms. Psychiatrists are likely to be consulted in LTC when a person with a major NCD presents with an acute episode of increased confusion and cognitive worsening, often accompanied by behavioral symptoms. BPSD may include agitation, aggression, apathy, depression, sleep problems, socially inappropriate behaviors, and psychosis. One study of patients with Alzheimer’s disease (AD) reported a cumulative 51% incidence of new-onset hallucinations and delusions at 4 years.16
Increased vulnerability to stressors, unmet needs, over- or under-stimulation, or lack of routines may predispose individuals with major NCDs to developing BPSD.17 Nonpharmacologic approaches usually are tried first, although supporting evidence is not substantial.18 Changes in environment, behavioral redirection, sensory interventions, or music therapy may reduce disruptive behaviors.19 Patients with increased confusion and agitation in late afternoon and evening (“sundowning”) may benefit from short naps after lunch, light therapy, calming activities in late afternoon, and reduced noise (such as from dishes, loud speakers, staff conversations).20
Antipsychotics. The drugs most commonly used to manage BPSD are antipsychotics, antidepressants, mood stabilizers/anticonvulsants, ChEIs, and the N-methyl-
When nonpharmacotherapeutic interventions are not successful, most guidelines agree that using an atypical antipsychotic is warranted in AD patients with severe agitation and/or psychosis that pose a risk to the patient or others or severely impair their quality of life.9,22,23
Antipsychotic review. Recent guidelines from the American Psychiatric Association (APA) recommend that attempts to taper and withdraw antipsychotic drugs be made within 4 months of initiating treatment in patients with dementia who display an adequate response.23 In a recent nursing home study, antipsychotic review was found to reduce antipsychotic use by 50% and, when combined with a social intervention, to reduce mortality compared with a group receiving neither intervention.24
Interestingly, patients receiving antipsychotic review alone showed an increase in overall neuropsychiatric symptoms.24 A previous study of patients with AD whose psychosis or agitation responded to risperidone also found an increased risk of relapse when risperidone was discontinued.25 These results highlight the importance of making patient-centered decisions, frequent re-assessments, and adding non-pharmacologic interventions (eg, positive social interactions or exercise) when attempting to discontinue antipsychotics.
Other treatment options. Because patients with LBD often display increased sensitivity to neuroleptics, agents such as quetiapine or aripiprazole (with a lower risk of EPS) are preferred when managing severe psychosis/aggression. ChEIs may show some benefit for behavioral disturbances in patients with LBD.26
In patients with AD, ChEIs have shown inconsistent results in benefiting neuropsychiatric symptoms. Preliminary data suggest some benefit with citalopram (also associated with prolonged QTc)27 and the dextromethorphan/quinidine combination FDA-approved for pseudobulbar affect, but more studies are needed.28 Pimavanserin, a 5-HT2A receptor inverse agonist, recently was approved for treating hallucinations and delusions associated with Parkinson’s disease psychosis and currently is in clinical trials for Alzheimer’s disease psychosis.
Electroconvulsive therapy (ECT) may be a therapeutic option for agitation and aggression in people with dementia.29 ECT has no absolute contraindications and can be safely performed in individuals with pacemakers or implantable cardioverter defibrillators. Common adverse effects include transient changes in blood pressure or heart rate, headache, and nausea. Cognitive adverse effects from ECT may include:
- anterograde amnesia, which typically resolves after a few weeks
- retrograde amnesia, which typically manifests as loss of impersonal memories occurring in the past few months.
Depression
The prevalence of depression in nursing home residents is an estimated 3 to 4 times that of community-dwelling older adults.30 Assessing for depression is particularly important in people with mild cognitive impairment, as depressive symptoms have been associated with progression to AD.31 Quick screening tools (Table 2) include short forms of the Patient Health Questionnaire (PHQ-2 or PHQ-9)32 or the Saint Louis University Appetite, Mood, Sleep, Activity, and thoughts of Death (SLU “AM SAD”) scale.33 The Cornell Scale for Depression in Dementia is useful for individuals with major NCDs because it relies on interviews with the patient and nursing staff or family.34
To test for other causes of depression, order a complete blood count for anemia, serum glucose, thyroid-stimulating hormone for hypothyroidism or hyperthyroidism, B12 and folate levels, and a cognitive screen such as the Saint Louis University Mental Status examination.35
Treatment. Antidepressants are generally considered effective in older patients with depression. Selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs) are first-line treatments because of safety concerns with tricyclic antidepressants. All 3 classes have shown similar efficacy in comparison trials in geriatric populations.
When initiating these agents, take care in the first few days and weeks to monitor for potential serious adverse effects, such as nausea and vomiting, which may be associated with substantial morbidity in patients with comorbidities. For monitoring treatment response, the PHQ-9 can effectively distinguish patients with persistent major depression, partial remission, or full remission.36
The optimal duration of a short-term antidepressant trial before switching to a different agent is unclear, although a good therapeutic trial typically is 4 to 12 weeks. In one study of older adults with depression, 4 weeks was enough to reliably identify those likely to benefit from a change in treatment plan.37
Cognitive-behavioral therapy (CBT) can be used in older adults not wishing to pursue pharmacotherapy or as an adjunct to antidepressants. Randomized controlled trials have shown some benefit for those with depression, anxiety, and insomnia.38 Individuals with significant cognitive deficits or those not motivated to apply CBT strategies might not benefit.
ECT may be appropriate for treating depression in older adults with:
- urgent need of a therapeutic response (eg, suicidal ideation or nutritional compromise)
- lack of response to antidepressant medication
- major depressive disorder with psychotic or catatonic features.
Evidence regarding ECT’s efficacy for late-life depression is derived primarily from clinical experience and open-label trials.39
Bipolar disorder
Most individuals with bipolar disorder present before age 50, although 9% of first manic episodes occur after age 60.40 Earlier age of onset appears to predict poor outcomes, and early-onset bipolar disorder may worsen with advanced age related to increased comorbidities and difficulty in medical management.41 Compared with younger patients, features of bipolar disorder in older adults include increased prominence of rapid cycling, more time spent in a depressed state than in manic state, and less severe manic and psychotic symptoms.42
When older patients present with depression, always evaluate for clinical features more consistent with late-onset bipolar disorder than with major depressive disorder: hypomania, family history of bipolar disorder, higher number of prior depressive episodes, and higher levels of fear and inner tension.43 The differential diagnosis for new-onset manic symptoms in older adults includes:
- general medical conditions (stroke, brain tumors, hyperthyroidism, neurosyphilis)
- medications (corticosteroids, dopaminergic drugs, St. John’s wort)
- substance use.
Hyperthyroidism deserves special attention because it can present in older adults with either manic-like symptoms and hyperkinesis or features of apathy, depression, and somnolence. Given that older age and bipolar disorder both are associated with increased suicide risk, monitor these individuals for signs of hopelessness and statements of suicide.44
Treatment. Managing bipolar disorder in older adults often requires complex medication regimens. Acute treatment options for geriatric mania and hypomania with the most supporting evidence include lithium, valproate, quetiapine, and olanzapine.45-47 The therapeutic index of lithium is small, and older individuals are more vulnerable to adverse effects related to physiologic changes (eg, decreased glomerular filtration rate or low volume of distribution) that impair lithium clearance. Lithium also interacts with many drugs commonly used by older patients, such as nonsteroidal anti-inflammatory drugs (NSAIDs) and diuretics. Common adverse events associated with lithium include memory impairment, diarrhea, falls, and tremors.
Maintenance treatment for bipolar disorder is generally the same medication used to induce remission. The evidence for maintenance treatment of bipolar disorder in older adults is limited mostly to subgroup analyses. In one retrospective analysis of patients age ≥55 in 2 randomized trials, lamotrigine and lithium were effective and well-tolerated in delaying time to intervention.48
Anxiety disorders
Anxiety among LTC residents may manifest as irritability, insomnia, restlessness, and verbal and/or physical agitation/aggression.49 Typical causes include:
- primary anxiety disorders
- anxiety symptoms during depressive episodes or bereavement
- adverse effects of medications
- complications of major NCDs or delirium.
Anxiety disorders and subsyndromal anxiety have been associated with poorer quality of life, decreased sleep, and increased distress and impairment.50
Assessment begins with a self-report of symptoms, although this may be difficult in people with major NCDs. Factors that may differentiate true anxiety from major NCDs include restlessness, irritability, muscle tension, fears, and respiratory symptoms in addition to excessive anxiety and worry.51 The Geriatric Anxiety Inventory is a useful screening tool.52 The newer Brief Anxiety and Depression Scale may identify and differentiate patients with major depressive episodes and generalized anxiety disorder (GAD).53 Potential instruments for patients with comorbid anxiety and major NCDs include the Neuropsychiatric Inventory, Rating Anxiety in Dementia scale,54 and the Anxiety in Cognitive Impairment and Dementia scale.55 Because medications can cause akathisia that may mimic anxiety symptoms, screen for the recent addition of antidepressants, antipsychotics, sympathomimetics, thyroid supplements, and corticosteroids.
Treatment of anxiety disorders—such as panic disorder, social phobia, or GAD—generally starts with SSRIs or SNRIs. Although benzodiazepines are commonly used for anxiety in older adults,56 these drugs are associated with a high rate of adverse effects: increased risk of agitation, falls, impaired cognition, and possibly dementia.57 In general, reserve benzodiazepines for treating acute episodes of severe anxiety in this population.
A particularly prevalent source of anxiety in LTC is fear of falling, which may affect up to 50% of residents and cause them to restrict their activities.58 Interventions such as CBT, exercise, or tai chi may be beneficial, although supporting evidence is lacking.
Pain and sleep management
Addressing pain. Age-related changes in pain perception and difficulty in reporting pain likely contribute to under-recognition of pain in LTC residents. Two useful methods to recognize their pain are to:
- observe for pain behaviors, such as facial expressions (grimacing and brow lowering), vocalizations, and body movements (clenched fists)
- solicit reports from nurses and other caregivers.59
Self-report may be a reliable indicator of pain for individuals with mild-to-moderate NCDs. Observational pain scales, such as the Pain Assessment Checklist for Seniors with Limited Ability to Communicate, may be useful in severe NCDs.60
The AGS recommends acetaminophen as initial pharmacotherapy to manage persistent pain.61 NSAIDs may be another option, but caution is warranted for patients with acid-peptic disease or chronic kidney disease. Opioids may be considered for severe pain, but otherwise avoid using them.
Sleep disturbances are common in LTC because of physiologic changes associated with aging (altered circadian rhythm), comorbidities (depression), and environmental factors.62 A strong association appears to exist between insomnia and use of sedative-hypnotic drugs, and the AGS Beers Criteria recommend avoiding non-benzodiazepine receptor agonists and benzodiazepines when treating insomnia in older adults.9
Assess factors that may contribute to sleep disturbances, including medications and use of caffeine or alcohol. Have the resident or caregiver document sleep patterns in a sleep diary.
Consider administrating medications at different times (eg, switch donepezil from bedtime to morning) or replace with alternatives (switch from the more anticholinergic amitriptyline to nortriptyline). Ensure that residents engage in physical activity and have at least 30 minutes daily exposure to sunlight.
In addition to behavioral interventions and CBT, treatment in older adults can involve melatonin—which has mixed evidence—or sedating antidepressants, such as mirtazapine or trazodone, in patients with comorbid depression.
Long-term care (LTC) services provide health care to >8 million people in approximately 30,000 nursing homes and assisted living/residential care communities in the United States.1 One-half of older adults in LTC have neurocognitive disorders (NCDs), and one-third have depressive syndromes.2 Common reasons for psychiatric consultation include these 2 major diagnoses, as well as delirium, behavioral and psychological symptoms of dementia (BPSD), bipolar disorder, anxiety, sleep disorders, and pain management.
Psychiatric assessment of individuals in LTC can be challenging because of atypical presentations, cognitive impairment, and multiple comorbidities. Establishing a management plan involves eliciting a careful history from both the patient and caretakers, examining previous records and medications, and selecting appropriate screening tools and laboratory tests (Table 1 and Table 2).
This article offers a practical approach to assess and manage common psychiatric conditions in LTC. We include new evidence about:
- assessment tools for psychiatric symptoms in LTC
- potentially inappropriate medication use in older adults
- antipsychotic use for agitation and psychosis with dementia
- nonpharmacologic interventions to help prevent cognitive decline
- antipsychotic review in reducing antipsychotic use and mortality.
Delirium
Delirium is an important topic in LTC because it is highly prevalent, poorly recognized, and can be difficult to manage. Common causes of delirium in LTC include infection (often urinary), dehydration, medications, long-standing constipation, and urinary retention (Table 3).3 Early recognition is key because delirium has been associated with cognitive decline, decreased functional status, increased caregiver burden, and increased mortality.4,5
The Confusion Assessment Method (CAM) is a quick tool with 4 features to differentiate delirium from other forms of cognitive impairment.6 The 2 core features are an acute change or fluctuating course of mental status and inattention. Family members or caregivers can provide information about an acute change. To assess inattention, ask the patient to say the days of the week backward or spell the word “world” backward. The 2 other features of delirium—one of which must be present when using the CAM—are disorganized thinking and altered level of consciousness.
Individuals with delirium may present with hyperactive or hypoactive psychomotor activity. Hypoactive delirium’s features, such as sluggishness and lethargy, could be confused with depression.7 A careful history to determine symptom onset and fluctuation in course can help differentiate between the 2.
Management. Delirium management always should begin by addressing underlying causes and implementing psychosocial and environmental interventions. Pharmacologic interventions have not demonstrated consistent benefit for delirium in well-designed trials and are not recommended as first-line treatment.8 The American Geriatrics Society (AGS) Beers Criteria for Potentially Inappropriate Medication Use in Older Adults recommends avoiding benzodiazepines in this population.9 Antipsychotics could be used in patients with severe agitation who pose harm to themselves or others. Nonpharmacologic approaches to delirium in LTC include:
- frequent reorientation (clocks, daily schedule)
- one-on-one monitoring by staff or family members
- use of hearing aids and eye-glasses, if needed
- maintaining an appropriate sleep-wake cycle by encouraging exposure to bright light during the day and avoiding night-time interruptions.
Restraints should not be used; they appear to worsen delirium severity, and their removal does not increase the rate of falls or fall-related injury.10
Various methods for managing a patient with delirium have been proposed, such as the TADA approach (tolerate, anticipate, and don’t agitate).5,11,12 For example, if a patient’s agitation worsens with attempted reorientation, distraction or playing along with the disorientation could be more beneficial.12
Keep in mind delirium’s overlapping presentation with Lewy body dementia (LBD). Patients with LBD demonstrate a progressive decline in cognitive functioning associated with fluctuating cognition, visual hallucinations, and parkinsonism features. Consider LBD when no cause for delirium-like symptoms is found. These patients may show increased sensitivity to neuroleptics and extrapyramidal side effects.
Neurocognitive disorders
Reversible causes. Although most individuals with major NCDs are diagnosed before entering LTC, the consulting psychiatrist’s review of potentially reversible causes of neurocognitive symptoms can lead to dramatically different treatment regimens (Table 43). For example, anticholinergic medications can harm the aging brain and have been linked to delirium, increased brain atrophy, and lower scores on tests of cognitive functioning.13 Given the prevalence of polypharmacy in older adults, be aware of unexpected anticholinergic properties of many common drugs, as rated by the Aging Brain Care initiative.14
Mild cognitive impairment. Should patients showing signs of cognitive impairment or those at risk for major NCDs begin pharmacotherapy? The FDA has approved no medications for this indication, and clinical trials with agents such as cholinesterase inhibitors (ChEIs) have shown inconsistent results.
The randomized, double-blind Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability provides convincing data that a nonpharmacologic approach could benefit older adults at risk for a major NCD. A 2-year intervention of nutritional advice, aerobic and strength training, cognitive training, social activities, and blood pressure and weight monitoring was more effective in improving or maintaining cognitive function in individuals age 60 to 77, compared with general health advice given to a control group.15
Behavioral and psychological symptoms. Psychiatrists are likely to be consulted in LTC when a person with a major NCD presents with an acute episode of increased confusion and cognitive worsening, often accompanied by behavioral symptoms. BPSD may include agitation, aggression, apathy, depression, sleep problems, socially inappropriate behaviors, and psychosis. One study of patients with Alzheimer’s disease (AD) reported a cumulative 51% incidence of new-onset hallucinations and delusions at 4 years.16
Increased vulnerability to stressors, unmet needs, over- or under-stimulation, or lack of routines may predispose individuals with major NCDs to developing BPSD.17 Nonpharmacologic approaches usually are tried first, although supporting evidence is not substantial.18 Changes in environment, behavioral redirection, sensory interventions, or music therapy may reduce disruptive behaviors.19 Patients with increased confusion and agitation in late afternoon and evening (“sundowning”) may benefit from short naps after lunch, light therapy, calming activities in late afternoon, and reduced noise (such as from dishes, loud speakers, staff conversations).20
Antipsychotics. The drugs most commonly used to manage BPSD are antipsychotics, antidepressants, mood stabilizers/anticonvulsants, ChEIs, and the N-methyl-
When nonpharmacotherapeutic interventions are not successful, most guidelines agree that using an atypical antipsychotic is warranted in AD patients with severe agitation and/or psychosis that pose a risk to the patient or others or severely impair their quality of life.9,22,23
Antipsychotic review. Recent guidelines from the American Psychiatric Association (APA) recommend that attempts to taper and withdraw antipsychotic drugs be made within 4 months of initiating treatment in patients with dementia who display an adequate response.23 In a recent nursing home study, antipsychotic review was found to reduce antipsychotic use by 50% and, when combined with a social intervention, to reduce mortality compared with a group receiving neither intervention.24
Interestingly, patients receiving antipsychotic review alone showed an increase in overall neuropsychiatric symptoms.24 A previous study of patients with AD whose psychosis or agitation responded to risperidone also found an increased risk of relapse when risperidone was discontinued.25 These results highlight the importance of making patient-centered decisions, frequent re-assessments, and adding non-pharmacologic interventions (eg, positive social interactions or exercise) when attempting to discontinue antipsychotics.
Other treatment options. Because patients with LBD often display increased sensitivity to neuroleptics, agents such as quetiapine or aripiprazole (with a lower risk of EPS) are preferred when managing severe psychosis/aggression. ChEIs may show some benefit for behavioral disturbances in patients with LBD.26
In patients with AD, ChEIs have shown inconsistent results in benefiting neuropsychiatric symptoms. Preliminary data suggest some benefit with citalopram (also associated with prolonged QTc)27 and the dextromethorphan/quinidine combination FDA-approved for pseudobulbar affect, but more studies are needed.28 Pimavanserin, a 5-HT2A receptor inverse agonist, recently was approved for treating hallucinations and delusions associated with Parkinson’s disease psychosis and currently is in clinical trials for Alzheimer’s disease psychosis.
Electroconvulsive therapy (ECT) may be a therapeutic option for agitation and aggression in people with dementia.29 ECT has no absolute contraindications and can be safely performed in individuals with pacemakers or implantable cardioverter defibrillators. Common adverse effects include transient changes in blood pressure or heart rate, headache, and nausea. Cognitive adverse effects from ECT may include:
- anterograde amnesia, which typically resolves after a few weeks
- retrograde amnesia, which typically manifests as loss of impersonal memories occurring in the past few months.
Depression
The prevalence of depression in nursing home residents is an estimated 3 to 4 times that of community-dwelling older adults.30 Assessing for depression is particularly important in people with mild cognitive impairment, as depressive symptoms have been associated with progression to AD.31 Quick screening tools (Table 2) include short forms of the Patient Health Questionnaire (PHQ-2 or PHQ-9)32 or the Saint Louis University Appetite, Mood, Sleep, Activity, and thoughts of Death (SLU “AM SAD”) scale.33 The Cornell Scale for Depression in Dementia is useful for individuals with major NCDs because it relies on interviews with the patient and nursing staff or family.34
To test for other causes of depression, order a complete blood count for anemia, serum glucose, thyroid-stimulating hormone for hypothyroidism or hyperthyroidism, B12 and folate levels, and a cognitive screen such as the Saint Louis University Mental Status examination.35
Treatment. Antidepressants are generally considered effective in older patients with depression. Selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs) are first-line treatments because of safety concerns with tricyclic antidepressants. All 3 classes have shown similar efficacy in comparison trials in geriatric populations.
When initiating these agents, take care in the first few days and weeks to monitor for potential serious adverse effects, such as nausea and vomiting, which may be associated with substantial morbidity in patients with comorbidities. For monitoring treatment response, the PHQ-9 can effectively distinguish patients with persistent major depression, partial remission, or full remission.36
The optimal duration of a short-term antidepressant trial before switching to a different agent is unclear, although a good therapeutic trial typically is 4 to 12 weeks. In one study of older adults with depression, 4 weeks was enough to reliably identify those likely to benefit from a change in treatment plan.37
Cognitive-behavioral therapy (CBT) can be used in older adults not wishing to pursue pharmacotherapy or as an adjunct to antidepressants. Randomized controlled trials have shown some benefit for those with depression, anxiety, and insomnia.38 Individuals with significant cognitive deficits or those not motivated to apply CBT strategies might not benefit.
ECT may be appropriate for treating depression in older adults with:
- urgent need of a therapeutic response (eg, suicidal ideation or nutritional compromise)
- lack of response to antidepressant medication
- major depressive disorder with psychotic or catatonic features.
Evidence regarding ECT’s efficacy for late-life depression is derived primarily from clinical experience and open-label trials.39
Bipolar disorder
Most individuals with bipolar disorder present before age 50, although 9% of first manic episodes occur after age 60.40 Earlier age of onset appears to predict poor outcomes, and early-onset bipolar disorder may worsen with advanced age related to increased comorbidities and difficulty in medical management.41 Compared with younger patients, features of bipolar disorder in older adults include increased prominence of rapid cycling, more time spent in a depressed state than in manic state, and less severe manic and psychotic symptoms.42
When older patients present with depression, always evaluate for clinical features more consistent with late-onset bipolar disorder than with major depressive disorder: hypomania, family history of bipolar disorder, higher number of prior depressive episodes, and higher levels of fear and inner tension.43 The differential diagnosis for new-onset manic symptoms in older adults includes:
- general medical conditions (stroke, brain tumors, hyperthyroidism, neurosyphilis)
- medications (corticosteroids, dopaminergic drugs, St. John’s wort)
- substance use.
Hyperthyroidism deserves special attention because it can present in older adults with either manic-like symptoms and hyperkinesis or features of apathy, depression, and somnolence. Given that older age and bipolar disorder both are associated with increased suicide risk, monitor these individuals for signs of hopelessness and statements of suicide.44
Treatment. Managing bipolar disorder in older adults often requires complex medication regimens. Acute treatment options for geriatric mania and hypomania with the most supporting evidence include lithium, valproate, quetiapine, and olanzapine.45-47 The therapeutic index of lithium is small, and older individuals are more vulnerable to adverse effects related to physiologic changes (eg, decreased glomerular filtration rate or low volume of distribution) that impair lithium clearance. Lithium also interacts with many drugs commonly used by older patients, such as nonsteroidal anti-inflammatory drugs (NSAIDs) and diuretics. Common adverse events associated with lithium include memory impairment, diarrhea, falls, and tremors.
Maintenance treatment for bipolar disorder is generally the same medication used to induce remission. The evidence for maintenance treatment of bipolar disorder in older adults is limited mostly to subgroup analyses. In one retrospective analysis of patients age ≥55 in 2 randomized trials, lamotrigine and lithium were effective and well-tolerated in delaying time to intervention.48
Anxiety disorders
Anxiety among LTC residents may manifest as irritability, insomnia, restlessness, and verbal and/or physical agitation/aggression.49 Typical causes include:
- primary anxiety disorders
- anxiety symptoms during depressive episodes or bereavement
- adverse effects of medications
- complications of major NCDs or delirium.
Anxiety disorders and subsyndromal anxiety have been associated with poorer quality of life, decreased sleep, and increased distress and impairment.50
Assessment begins with a self-report of symptoms, although this may be difficult in people with major NCDs. Factors that may differentiate true anxiety from major NCDs include restlessness, irritability, muscle tension, fears, and respiratory symptoms in addition to excessive anxiety and worry.51 The Geriatric Anxiety Inventory is a useful screening tool.52 The newer Brief Anxiety and Depression Scale may identify and differentiate patients with major depressive episodes and generalized anxiety disorder (GAD).53 Potential instruments for patients with comorbid anxiety and major NCDs include the Neuropsychiatric Inventory, Rating Anxiety in Dementia scale,54 and the Anxiety in Cognitive Impairment and Dementia scale.55 Because medications can cause akathisia that may mimic anxiety symptoms, screen for the recent addition of antidepressants, antipsychotics, sympathomimetics, thyroid supplements, and corticosteroids.
Treatment of anxiety disorders—such as panic disorder, social phobia, or GAD—generally starts with SSRIs or SNRIs. Although benzodiazepines are commonly used for anxiety in older adults,56 these drugs are associated with a high rate of adverse effects: increased risk of agitation, falls, impaired cognition, and possibly dementia.57 In general, reserve benzodiazepines for treating acute episodes of severe anxiety in this population.
A particularly prevalent source of anxiety in LTC is fear of falling, which may affect up to 50% of residents and cause them to restrict their activities.58 Interventions such as CBT, exercise, or tai chi may be beneficial, although supporting evidence is lacking.
Pain and sleep management
Addressing pain. Age-related changes in pain perception and difficulty in reporting pain likely contribute to under-recognition of pain in LTC residents. Two useful methods to recognize their pain are to:
- observe for pain behaviors, such as facial expressions (grimacing and brow lowering), vocalizations, and body movements (clenched fists)
- solicit reports from nurses and other caregivers.59
Self-report may be a reliable indicator of pain for individuals with mild-to-moderate NCDs. Observational pain scales, such as the Pain Assessment Checklist for Seniors with Limited Ability to Communicate, may be useful in severe NCDs.60
The AGS recommends acetaminophen as initial pharmacotherapy to manage persistent pain.61 NSAIDs may be another option, but caution is warranted for patients with acid-peptic disease or chronic kidney disease. Opioids may be considered for severe pain, but otherwise avoid using them.
Sleep disturbances are common in LTC because of physiologic changes associated with aging (altered circadian rhythm), comorbidities (depression), and environmental factors.62 A strong association appears to exist between insomnia and use of sedative-hypnotic drugs, and the AGS Beers Criteria recommend avoiding non-benzodiazepine receptor agonists and benzodiazepines when treating insomnia in older adults.9
Assess factors that may contribute to sleep disturbances, including medications and use of caffeine or alcohol. Have the resident or caregiver document sleep patterns in a sleep diary.
Consider administrating medications at different times (eg, switch donepezil from bedtime to morning) or replace with alternatives (switch from the more anticholinergic amitriptyline to nortriptyline). Ensure that residents engage in physical activity and have at least 30 minutes daily exposure to sunlight.
In addition to behavioral interventions and CBT, treatment in older adults can involve melatonin—which has mixed evidence—or sedating antidepressants, such as mirtazapine or trazodone, in patients with comorbid depression.
1. Harris-Kojetin L, Sengupta M, Park-Lee E, et al. Long-term care services in the United States: 2013 overview. Vital Health Stat 3. 2013(37):1-107.
2. Seitz D, Purandare N, Conn D. Prevalence of psychiatric disorders among older adults in long-term care homes: a systematic review. Int Psychogeriatr. 2010;22(7):1025-1039.
3. Flaherty J, Tumosa N. Saint Louis University Geriatric Evaluation Mnemonics and Screening Tools. http://aging.slu.edu/uploads/pdf/Saint-Louis-University-Geriatric-Evaluation_2013.pdf. Accessed October 5, 2016.
4. Boockvar K, Signor D, Ramaswamy R, et al. Delirium during acute illness in nursing home residents. J Am Med Dir Assoc. 2013;14(9):656-660.
5. Inouye SK, Westendorp RG, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
6. Wei LA, Fearing MA, Sternberg EJ, et al. The Confusion Assessment Method: a systematic review of current usage. J Am Geriatr Soc. 2008;56(5):823-830.
7. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
8. Flaherty JH, Gonzales JP, Dong B. Antipsychotics in the treatment of delirium in older hospitalized adults: a systematic review. J Am Geriatr Soc. 2011;59(suppl 2):S269-S276.
9. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
10. Capezuti E, Strumpf NE, Evans LK, et al. The relationship between physical restraint removal and falls and injuries among nursing home residents. J Gerontol A Biol Sci Med Sci. 1998;53(1):M47-M52.
11. Flaherty JH, Morley JE. Delirium in the nursing home. J Am Med Dir Assoc. 2013;14(9):632-634.
12. Flaherty JH. The evaluation and management of delirium among older persons. Med Clin North Am. 2011;95(3):555-577, xi.
13. Risacher SL, McDonald BC, Tallman EF, et al. Association between anticholinergic medication use and cognition, brain metabolism, and brain atrophy in cognitively normal older adults. JAMA Neurol. 2016;73(6):721-732.
14. Anticholinergic Cognitive Burden Scale. Aging Brain Care. http://agingbraincare.org/uploads/products/ACB_scale_-_legal_size.pdf. Published 2012. Accessed October 5, 2016.
15. Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
16. Paulsen JS, Salmon DP, Thal LJ, et al. Incidence of and risk factors for hallucinations and delusions in patients with probable AD. Neurology. 2000;54(10):1965-1971.
17. Gitlin LN, Kales HC, Lyketsos CG. Nonpharmacologic management of behavioral symptoms in dementia. JAMA. 2012;308(19):2020-2029.
18. Livingston G, Kelly L, Lewis-Holmes E, et al. A systematic review of the clinical effectiveness and cost-effectiveness of sensory, psychological and behavioural interventions for managing agitation in older adults with dementia. Health Technol Assess. 2014;18(39):1-226, v-vi.
19. Kong EH, Evans LK, Guevara JP. Nonpharmacological intervention for agitation in dementia: a systematic review and meta-analysis. Aging Ment Health. 2009;13(4):512-520.
20. Khachiyants N, Trinkle D, Son SJ, et al. Sundown syndrome in persons with dementia: an update. Psychiatry Investig. 2011;8(4):275-287.
21. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525-1538.
22. Jennings L, Grossberg GT. Antipsychotics continue to have a place in the management of difficult behavior problems in patients with dementia. J Am Med Dir Assoc. 2013;14(6):447-449.
23. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
24. Ballard C, Orrell M, YongZhong S, et al. Impact of antipsychotic review and nonpharmacological intervention on antipsychotic use, neuropsychiatric symptoms, and mortality in people with dementia living in nursing homes: a factorial cluster-randomized controlled trial by the Well-Being and Health for People With Dementia (WHELD) program. Am J Psychiatry. 2015;173(3):252-262.
25. Devanand DP, Mintzer J, Schultz SK, et al. Relapse risk after discontinuation of risperidone in Alzheimer’s disease. N Engl J Med. 2012;367(16):1497-1507.
26. Matsunaga S, Kishi T, Yasue I, et al. Cholinesterase inhibitors for Lewy body disorders: a meta-analysis. Int J Neuropsychopharmacol. 2015;19(2). doi: 10.1093/ijnp/pyv086.
27. Porsteinsson AP, Drye LT, Pollock BG, et al; CitAD Research Group. Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. JAMA. 2014;311(7):682-691.
28. Cummings JL, Lyketsos CG, Peskind ER, et al. Effect of dextromethorphan-quinidine on agitation in patients with Alzheimer disease dementia: a randomized clinical trial. JAMA. 2015;314(12):1242-1254.
29. Ujkaj M, Davidoff DA, Seiner SJ, et al. Safety and efficacy of electroconvulsive therapy for the treatment of agitation and aggression in patients with dementia. Am J Geriatr Psychiatry. 2012;20(1):61-72.
30. Jongenelis K, Pot AM, Eisses AM, et al. Prevalence and risk indicators of depression in elderly nursing home patients: the AGED study. J Affect Disord. 2004;83(2-3):135-142.
31. Van der Mussele S, Fransen E, Struyfs H, et al. Depression in mild cognitive impairment is associated with progression to Alzheimer’s disease: a longitudinal study. J Alzheimers Dis. 2014;42(4):1239-1250.
32. Li C, Friedman B, Conwell Y, et al. Validity of the Patient Health Questionnaire 2 (PHQ-2) in identifying major depression in older people. J Am Geriatr Soc. 2007;55(4):596-602.
33. Chakkamparambil B, Chibnall JT, Graypel EA, et al. Development of a brief validated geriatric depression screening tool: the SLU “AM SAD”. Am J Geriatr Psychiatry. 2015;23(8):780-783.
34. Korner A, Lauritzen L, Abelskov K, et al. The Geriatric Depression Scale and the Cornell Scale for Depression in Dementia. A validity study. Nord J Psychiatry. 2006;60(5):360-364.
35. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the Mini-Mental State Examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
36. Löwe B, Unützer J, Callahan CM, et al. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care. 2004;42(12):1194-1201.
37. Mulsant BH, Houck PR, Gildengers AG, et al. What is the optimal duration of a short-term antidepressant trial when treating geriatric depression? J Clin Psychopharmacol. 2006;26(2):113-120.
38. Chand SP, Grossberg GT. How to adapt cognitive-behavioral therapy for older adults. Current Psychiatry. 2013;12(3):10-15.
39. Van der Wurff FB, Stek ML, Hoogendijk WL, et al. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev. 2003;(2):CD003593.
40. Kennedy N, Everitt B, Boydell J, et al. Incidence and distribution of first-episode mania by age: results from a 35-year study. Psychol Med. 2005;35(6):855-863.
41. Carter TD, Mundo E, Parikh SV, et al. Early age at onset as a risk factor for poor outcome of bipolar disorder. J Psychiatr Res. 2003;37(4):297-303.
42. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
43. Perlis RH, Brown E, Baker RW, et al. Clinical features of bipolar depression versus major depressive disorder in large multicenter trials. Am J Psychiatry. 2006;163(2):225-231.
44. Aizenberg D, Olmer A, Barak Y. Suicide attempts amongst elderly bipolar patients. J Affect Disord. 2006;91(1):91-94.
45. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
46. Young RC, Gyulai L, Mulsant BH, et al. Pharmacotherapy of bipolar disorder in old age: review and recommendations. Am J Geriatr Psychiatry. 2004;12(4):342-357.
47. Sajatovic M, Calabrese JR, Mullen J. Quetiapine for the treatment of bipolar mania in older adults. Bipolar Disord. 2008;10(6):662-671.
48. Sajatovic M, Gyulai L, Calabrese JR, et al. Maintenance treatment outcomes in older patients with bipolar I disorder. Am J Geriatr Psychiatry. 2005;13(4):305-311.
49. Gum AM, King-Kallimanis B, Kohn R. Prevalence of mood, anxiety, and substance-abuse disorders for older Americans in the National Comorbidity Survey-Replication. Am J Geriatr Psychiatry. 2009;17(9):769-781.
50. Wetherell JL, Le Roux H, Gatz M. DSM-IV criteria for generalized anxiety disorder in older adults: distinguishing the worried from the well. Psychol Aging. 2003;18(3):622-627.
51. Starkstein SE, Jorge R, Petracca G, et al. The construct of generalized anxiety disorder in Alzheimer disease. Am J Geriatr Psychiatry. 2007;15(1):42-49.
52. Pachana NA, Byrne GJ, Siddle H, et al. Development and validation of the Geriatric Anxiety Inventory. Int Psychogeriatr. 2007;19(1):103-114.
53. Mansbach WE, Mace RA, Clark KM. The Brief Anxiety and Depression Scale (BADS): a new instrument for detecting anxiety and depression in long-term care residents. Int Psychogeriatr. 2015;27(4):673-681.
54. Seignourel PJ, Kunik ME, Snow L, et al. Anxiety in dementia: a critical review. Clin Psychol Rev. 2008;28(7):1071-1082.
55. Gerolimatos LA, Ciliberti CM, Gregg JJ, et al. Development and preliminary evaluation of the Anxiety in Cognitive Impairment and Dementia (ACID) scales. Int Psychogeriatr. 2015;27(11):1825-1838.
56. Benitez CI, Smith K, Vasile RG, et al. Use of benzodiazepines and selective serotonin reuptake inhibitors in middle-aged and older adults with anxiety disorders: a longitudinal and prospective study. Am J Geriatr Psychiatry. 2008;16(1):5-13.
57. Billioti de Gage S, Moride Y, Ducruet T, et al. Benzodiazepine use and risk of Alzheimer’s disease: case control study. BMJ. 2014;349:g5205.
58. Lach HW, Parsons JL. Impact of fear of falling in long term care: an integrative review. J Am Med Dir Assoc. 2013;14(8):573-577.
59. Hadjistavropoulos T, Herr K, Prkachin KM, et al. Pain assessment in elderly adults with dementia. Lancet Neurol. 2014;13(12):1216-1227.
60. Zwakhalen SM, Hamers JP, Abu-Saad HH, et al. Pain in elderly people with severe dementia: a systematic review of behavioural pain assessment tools [published online January 27, 2006]. BMC Geriatr. doi: 10.1186/1471-2318-6-3.
61. American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Adults. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc. 2009;57(8):1331-1346.
62. Gindin J, Shochat T, Chetrit A, et al; SHELTER project. Insomnia in long-term care facilities: a comparison of seven European countries and Israel: the Services and Health for Elderly in Long TERm care study. J Am Geriatr Soc. 2014;62(11):2033-2039.
1. Harris-Kojetin L, Sengupta M, Park-Lee E, et al. Long-term care services in the United States: 2013 overview. Vital Health Stat 3. 2013(37):1-107.
2. Seitz D, Purandare N, Conn D. Prevalence of psychiatric disorders among older adults in long-term care homes: a systematic review. Int Psychogeriatr. 2010;22(7):1025-1039.
3. Flaherty J, Tumosa N. Saint Louis University Geriatric Evaluation Mnemonics and Screening Tools. http://aging.slu.edu/uploads/pdf/Saint-Louis-University-Geriatric-Evaluation_2013.pdf. Accessed October 5, 2016.
4. Boockvar K, Signor D, Ramaswamy R, et al. Delirium during acute illness in nursing home residents. J Am Med Dir Assoc. 2013;14(9):656-660.
5. Inouye SK, Westendorp RG, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
6. Wei LA, Fearing MA, Sternberg EJ, et al. The Confusion Assessment Method: a systematic review of current usage. J Am Geriatr Soc. 2008;56(5):823-830.
7. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
8. Flaherty JH, Gonzales JP, Dong B. Antipsychotics in the treatment of delirium in older hospitalized adults: a systematic review. J Am Geriatr Soc. 2011;59(suppl 2):S269-S276.
9. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
10. Capezuti E, Strumpf NE, Evans LK, et al. The relationship between physical restraint removal and falls and injuries among nursing home residents. J Gerontol A Biol Sci Med Sci. 1998;53(1):M47-M52.
11. Flaherty JH, Morley JE. Delirium in the nursing home. J Am Med Dir Assoc. 2013;14(9):632-634.
12. Flaherty JH. The evaluation and management of delirium among older persons. Med Clin North Am. 2011;95(3):555-577, xi.
13. Risacher SL, McDonald BC, Tallman EF, et al. Association between anticholinergic medication use and cognition, brain metabolism, and brain atrophy in cognitively normal older adults. JAMA Neurol. 2016;73(6):721-732.
14. Anticholinergic Cognitive Burden Scale. Aging Brain Care. http://agingbraincare.org/uploads/products/ACB_scale_-_legal_size.pdf. Published 2012. Accessed October 5, 2016.
15. Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263.
16. Paulsen JS, Salmon DP, Thal LJ, et al. Incidence of and risk factors for hallucinations and delusions in patients with probable AD. Neurology. 2000;54(10):1965-1971.
17. Gitlin LN, Kales HC, Lyketsos CG. Nonpharmacologic management of behavioral symptoms in dementia. JAMA. 2012;308(19):2020-2029.
18. Livingston G, Kelly L, Lewis-Holmes E, et al. A systematic review of the clinical effectiveness and cost-effectiveness of sensory, psychological and behavioural interventions for managing agitation in older adults with dementia. Health Technol Assess. 2014;18(39):1-226, v-vi.
19. Kong EH, Evans LK, Guevara JP. Nonpharmacological intervention for agitation in dementia: a systematic review and meta-analysis. Aging Ment Health. 2009;13(4):512-520.
20. Khachiyants N, Trinkle D, Son SJ, et al. Sundown syndrome in persons with dementia: an update. Psychiatry Investig. 2011;8(4):275-287.
21. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355(15):1525-1538.
22. Jennings L, Grossberg GT. Antipsychotics continue to have a place in the management of difficult behavior problems in patients with dementia. J Am Med Dir Assoc. 2013;14(6):447-449.
23. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
24. Ballard C, Orrell M, YongZhong S, et al. Impact of antipsychotic review and nonpharmacological intervention on antipsychotic use, neuropsychiatric symptoms, and mortality in people with dementia living in nursing homes: a factorial cluster-randomized controlled trial by the Well-Being and Health for People With Dementia (WHELD) program. Am J Psychiatry. 2015;173(3):252-262.
25. Devanand DP, Mintzer J, Schultz SK, et al. Relapse risk after discontinuation of risperidone in Alzheimer’s disease. N Engl J Med. 2012;367(16):1497-1507.
26. Matsunaga S, Kishi T, Yasue I, et al. Cholinesterase inhibitors for Lewy body disorders: a meta-analysis. Int J Neuropsychopharmacol. 2015;19(2). doi: 10.1093/ijnp/pyv086.
27. Porsteinsson AP, Drye LT, Pollock BG, et al; CitAD Research Group. Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. JAMA. 2014;311(7):682-691.
28. Cummings JL, Lyketsos CG, Peskind ER, et al. Effect of dextromethorphan-quinidine on agitation in patients with Alzheimer disease dementia: a randomized clinical trial. JAMA. 2015;314(12):1242-1254.
29. Ujkaj M, Davidoff DA, Seiner SJ, et al. Safety and efficacy of electroconvulsive therapy for the treatment of agitation and aggression in patients with dementia. Am J Geriatr Psychiatry. 2012;20(1):61-72.
30. Jongenelis K, Pot AM, Eisses AM, et al. Prevalence and risk indicators of depression in elderly nursing home patients: the AGED study. J Affect Disord. 2004;83(2-3):135-142.
31. Van der Mussele S, Fransen E, Struyfs H, et al. Depression in mild cognitive impairment is associated with progression to Alzheimer’s disease: a longitudinal study. J Alzheimers Dis. 2014;42(4):1239-1250.
32. Li C, Friedman B, Conwell Y, et al. Validity of the Patient Health Questionnaire 2 (PHQ-2) in identifying major depression in older people. J Am Geriatr Soc. 2007;55(4):596-602.
33. Chakkamparambil B, Chibnall JT, Graypel EA, et al. Development of a brief validated geriatric depression screening tool: the SLU “AM SAD”. Am J Geriatr Psychiatry. 2015;23(8):780-783.
34. Korner A, Lauritzen L, Abelskov K, et al. The Geriatric Depression Scale and the Cornell Scale for Depression in Dementia. A validity study. Nord J Psychiatry. 2006;60(5):360-364.
35. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the Mini-Mental State Examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
36. Löwe B, Unützer J, Callahan CM, et al. Monitoring depression treatment outcomes with the Patient Health Questionnaire-9. Med Care. 2004;42(12):1194-1201.
37. Mulsant BH, Houck PR, Gildengers AG, et al. What is the optimal duration of a short-term antidepressant trial when treating geriatric depression? J Clin Psychopharmacol. 2006;26(2):113-120.
38. Chand SP, Grossberg GT. How to adapt cognitive-behavioral therapy for older adults. Current Psychiatry. 2013;12(3):10-15.
39. Van der Wurff FB, Stek ML, Hoogendijk WL, et al. Electroconvulsive therapy for the depressed elderly. Cochrane Database Syst Rev. 2003;(2):CD003593.
40. Kennedy N, Everitt B, Boydell J, et al. Incidence and distribution of first-episode mania by age: results from a 35-year study. Psychol Med. 2005;35(6):855-863.
41. Carter TD, Mundo E, Parikh SV, et al. Early age at onset as a risk factor for poor outcome of bipolar disorder. J Psychiatr Res. 2003;37(4):297-303.
42. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
43. Perlis RH, Brown E, Baker RW, et al. Clinical features of bipolar depression versus major depressive disorder in large multicenter trials. Am J Psychiatry. 2006;163(2):225-231.
44. Aizenberg D, Olmer A, Barak Y. Suicide attempts amongst elderly bipolar patients. J Affect Disord. 2006;91(1):91-94.
45. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
46. Young RC, Gyulai L, Mulsant BH, et al. Pharmacotherapy of bipolar disorder in old age: review and recommendations. Am J Geriatr Psychiatry. 2004;12(4):342-357.
47. Sajatovic M, Calabrese JR, Mullen J. Quetiapine for the treatment of bipolar mania in older adults. Bipolar Disord. 2008;10(6):662-671.
48. Sajatovic M, Gyulai L, Calabrese JR, et al. Maintenance treatment outcomes in older patients with bipolar I disorder. Am J Geriatr Psychiatry. 2005;13(4):305-311.
49. Gum AM, King-Kallimanis B, Kohn R. Prevalence of mood, anxiety, and substance-abuse disorders for older Americans in the National Comorbidity Survey-Replication. Am J Geriatr Psychiatry. 2009;17(9):769-781.
50. Wetherell JL, Le Roux H, Gatz M. DSM-IV criteria for generalized anxiety disorder in older adults: distinguishing the worried from the well. Psychol Aging. 2003;18(3):622-627.
51. Starkstein SE, Jorge R, Petracca G, et al. The construct of generalized anxiety disorder in Alzheimer disease. Am J Geriatr Psychiatry. 2007;15(1):42-49.
52. Pachana NA, Byrne GJ, Siddle H, et al. Development and validation of the Geriatric Anxiety Inventory. Int Psychogeriatr. 2007;19(1):103-114.
53. Mansbach WE, Mace RA, Clark KM. The Brief Anxiety and Depression Scale (BADS): a new instrument for detecting anxiety and depression in long-term care residents. Int Psychogeriatr. 2015;27(4):673-681.
54. Seignourel PJ, Kunik ME, Snow L, et al. Anxiety in dementia: a critical review. Clin Psychol Rev. 2008;28(7):1071-1082.
55. Gerolimatos LA, Ciliberti CM, Gregg JJ, et al. Development and preliminary evaluation of the Anxiety in Cognitive Impairment and Dementia (ACID) scales. Int Psychogeriatr. 2015;27(11):1825-1838.
56. Benitez CI, Smith K, Vasile RG, et al. Use of benzodiazepines and selective serotonin reuptake inhibitors in middle-aged and older adults with anxiety disorders: a longitudinal and prospective study. Am J Geriatr Psychiatry. 2008;16(1):5-13.
57. Billioti de Gage S, Moride Y, Ducruet T, et al. Benzodiazepine use and risk of Alzheimer’s disease: case control study. BMJ. 2014;349:g5205.
58. Lach HW, Parsons JL. Impact of fear of falling in long term care: an integrative review. J Am Med Dir Assoc. 2013;14(8):573-577.
59. Hadjistavropoulos T, Herr K, Prkachin KM, et al. Pain assessment in elderly adults with dementia. Lancet Neurol. 2014;13(12):1216-1227.
60. Zwakhalen SM, Hamers JP, Abu-Saad HH, et al. Pain in elderly people with severe dementia: a systematic review of behavioural pain assessment tools [published online January 27, 2006]. BMC Geriatr. doi: 10.1186/1471-2318-6-3.
61. American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Adults. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc. 2009;57(8):1331-1346.
62. Gindin J, Shochat T, Chetrit A, et al; SHELTER project. Insomnia in long-term care facilities: a comparison of seven European countries and Israel: the Services and Health for Elderly in Long TERm care study. J Am Geriatr Soc. 2014;62(11):2033-2039.

Promoting your older patient’s healthy ‘brain aging’

‘We need to protect the brain’ Addressing the growing problem of chronic traumatic encephalopathy
The National Football League (NFL) had its highest concussion tally last year: 182 such injuries reported1 in the 2014-2015 regular season. The true rate of concussion in the NFL is likely higher, as a result of multiple factors (fear of “letting the team [or the coach] down,” fear of retaliation from team owners,2 etc.).
To simply call a head injury a “concussion” is a disservice to players and their family: Any blow to the head, severe or otherwise, has the potential to cause microvascular disruption in the brain; repeated blows to the head undoubtedly cause further damage.
In reality, a “concussion” is a mild traumatic brain injury (mTBI). With repeated blows, an mTBI can lead to chronic traumatic encephalopathy (CTE). In 2015, eighty-seven of 91 brains from autopsied former NFL players displayed some stage of CTE.3
Pathophysiology and presentation
CTE comprises 4 histological stages; Stage 4 is the most advanced. Alzheimer’s disease (AD) and CTE display similarities, which suggests a separate classification of CTE-AD; the presence of amyloid β plaques correlates with (1) more severe hyperphosphorylated tau (pTau) pathology and (2) advanced stages of the disease and clinical presentations. Death tends to occur 10 years earlier in CTE-AD than in AD, suggesting that repetitive mTBI might change the deposition and accumulation of amyloid β plaques, and even accelerate the aging process in the brain.4
Symptoms. The case series by Omalu et al4 (which inspired the 2015 motion picture Concussion) and the case series presented by McKee et al5 described severe psychiatric symptoms associated with CTE:
- decreased speed of information processing
- increase in religiosity
- lack of insight
- poor judgment
- involvement in illegal activities
- substance abuse
- indiscretion
- verbal and physical abuse
- problems with interpersonal relationships
- isolation
- restlessness and hyperactivity
- somatic complaints.
The 2 groups of researchers also noted hopelessness, social phobia, anxiety, agitation, mania, labile mood, insomnia, explosivity, and suicidal ideation, attempt, and completion.4,5
By Stage 4, all affected patients are symptomatic. Cognitive impairment is severe; many are described as having “severe memory loss with dementia,”5 “profound” inattention and loss of concentration,5 and dysarthria. Paranoia may develop. Mood symptoms can be severe: Approximately 31% of subjects studied have contemplated suicide; of those, 26% had “suicidal tendencies” and 14% completed suicide.5
Two distinct types of CTE progression are apparent:
- patients who display cognitive deficits first; they progress to dementia but live longer
- patients who display mood and behavioral symptoms first; they tend to be younger, more violent, depressed, and explosive.6
CTE cannot be diagnosed with imaging. There are, however, a few positron emission tomography (PET) ligands for pTau that show promise:
- [F-18]FDDNP, which consistently identifies pTau deposits in brains in which CTE is clinically suspected, in the same distribution of pTau neurofibrillary tangles on autopsy.
- [11C]DPA-713, which detected TBI-related inflammation of neurons in 9 former NFL players in whom CTE was suspected based on the clinical presentation.
- PiB amyloid ligand, under investigation for use in PET neuroimaging.7
Casualties
In January 2016 alone, at least 3 former NFL players were found to have CTE posthumously.
Earl Morrall. Former quarterback who had a 21-year NFL career. Official cause of death in 2014 at age 79 was recorded as “complications of Parkinson’s disease.” In 2016, Stage-4 CTE was discovered on autopsy.8
Ken Stabler. Former quarterback for several NFL teams over 15 seasons. Died of colon cancer at age 69 in 2015. On autopsy, was found to have Stage-3 CTE.9
Tyler Sash. Former University of Iowa and New York Giants football player. Died in September 2015 at age 27 of an apparent drug overdose; posthumously, determined to have Stage-2 CTE. His family reported memory loss, minor fits of rage, confusion, inattention, lack of focus, and chronic pain.
Sash’s mother said, “My son knew something was wrong, but he couldn’t express it. He was such a good person, and it’s sad that he struggled so with this—not knowing where to go with it. Now it makes sense.”10 Sash played 16 years of football in all, sustaining at least 5 concussions. (“If you’ve played football, you know there are often other incidents [of head trauma],” Sash’s father said.10)
Cultural and medical mindsets about contact sports
In the United States, children as young as age 5, with a low weight limit of 35 pounds, routinely are introduced to football.11 Reports of 5 high school players dying from football-related injury in the 2014 season, and 3 deaths in the 2015 season, led a St. Louis, Missouri, area school district to defund their football program entirely. The district’s 2015 homecoming game was a soccer match; students and parents seemed to embrace the change.12
On its face, soccer seems a good alternative to football. When children are instructed to “head” the ball, however, concern arises about CTE: Mild CTE changes have been reported in 2 young soccer players, and late-stage CTE changes were seen in a retired soccer player with dementia.13
Perhaps most disturbing is that players who develop symptoms of CTE, or are at risk, are unlikely to seek psychiatric help. We, as psychiatric clinicians, must be diligent about questioning young patients about their extracurricular activities. It is not enough to simply ask about a history of head trauma: Ask patients about any blow to the head, and don’t limit your questioning to whether they sustained a “concussion” during practice or play.
When speaking with adult and geriatric patients, ask about a history of playing interscholastic or collegiate contact sports, such as football, hockey, and soccer.
Is the solution to better shield the head?
That is not a solution: Helmets and other protective headgear appear to be insufficient to protect the brain from traumatic injury. Perhaps keeping children from engaging in violent sports that put them at high risk of CTE later is the preventive approach that merits the most attention.
1. Blackstone J. NFL tackles alarming increase in concussions. CBS News. http://www.cbsnews.com/news/nfl-studying-how-to-tackle-alarming-increase-in-concussions. Published February 2, 2016. Accessed February 3, 2016.
2. McNamee M, Partridge B, Anderson L. Concussion ethics and sports medicine. Clin Sports Med. 2015;35(2):257-267.
3. Abreu MA, Cromartie FJ, Spradley BD; United States Sports Academy. Chronic traumatic encephalopathy (CTE) and former National Football League player suicides. The Sport Journal. http://thesportjournal.org/article/chronic-traumatic-encephalopathy-cte-and-former-national-football-league-player-suicides. Published January 29, 2016. Accessed January 29, 2016.
4. Omalu B, Bailes J, Hamilton RL, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in american athletes. Neurosurgery. 2011;69(1):173-183; discussion 183.
5. McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(pt 1):43-64.
6. Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81(13):1122-1129.
7. Eisenmenger LB, Huo EJ, Hoffman JM, et al. Advances in PET imaging of degenerative, cerebrovascular, and traumatic causes of dementia. Semin Nucl Med. 2016;46(1):57-87.
8. Jackson B. Report: former Miami Dolphins QB Earl Morrall had brain disease CTE. Miami Herald. http://www.miamiherald.com/sports/nfl/miami-dolphins/article58794523.html. Published February 5, 2016. Accessed February 6, 2016.
9. Fantz A. Ex-NFL player Ken Stabler had concussion disease CTE, doctor says. CNN. http://www.cnn.com/2016/02/03/health/ken-stabler-cte. Updated February 4, 2016. Accessed February 9, 2016.
10. Pennington B. C.T.E. is found in an Ex-Giant Tyler Sash, who died at 27. The New York Times. http://www.nytimes.com/2016/01/27/sports/football/former-giants-safety-tyler-sash-found-to-have-cte.html?_r=0. Published January 26, 2016. Accessed January 27, 2016.
11. Pop Warner Little Scholars, Inc. Ages and weights for tackle football programs. http://www.popwarner.com/football/footballstructure.htm. Accessed February 5, 2016.
12. Fowler L. No football for homecoming? No problem at Maplewood-Richmond Heights High. St. Louis Post Dispatch. http://www.stltoday.com/news/local/education/no-football-for-homecoming-no-problem-at-maplewood-richmond-heights/article_cc8dc31b-5097-5114-ba9b-9b3584f478b9.html. Published October 9, 2015. Accessed February 3, 2016.
13. Hales C, Neill S, Gearing M, et al. Late-stage CTE pathology in a retired soccer player with dementia. Neurology. 2014;83(24):2307-2309. doi: 10.1212/WNL.0000000000001081.
The National Football League (NFL) had its highest concussion tally last year: 182 such injuries reported1 in the 2014-2015 regular season. The true rate of concussion in the NFL is likely higher, as a result of multiple factors (fear of “letting the team [or the coach] down,” fear of retaliation from team owners,2 etc.).
To simply call a head injury a “concussion” is a disservice to players and their family: Any blow to the head, severe or otherwise, has the potential to cause microvascular disruption in the brain; repeated blows to the head undoubtedly cause further damage.
In reality, a “concussion” is a mild traumatic brain injury (mTBI). With repeated blows, an mTBI can lead to chronic traumatic encephalopathy (CTE). In 2015, eighty-seven of 91 brains from autopsied former NFL players displayed some stage of CTE.3
Pathophysiology and presentation
CTE comprises 4 histological stages; Stage 4 is the most advanced. Alzheimer’s disease (AD) and CTE display similarities, which suggests a separate classification of CTE-AD; the presence of amyloid β plaques correlates with (1) more severe hyperphosphorylated tau (pTau) pathology and (2) advanced stages of the disease and clinical presentations. Death tends to occur 10 years earlier in CTE-AD than in AD, suggesting that repetitive mTBI might change the deposition and accumulation of amyloid β plaques, and even accelerate the aging process in the brain.4
Symptoms. The case series by Omalu et al4 (which inspired the 2015 motion picture Concussion) and the case series presented by McKee et al5 described severe psychiatric symptoms associated with CTE:
- decreased speed of information processing
- increase in religiosity
- lack of insight
- poor judgment
- involvement in illegal activities
- substance abuse
- indiscretion
- verbal and physical abuse
- problems with interpersonal relationships
- isolation
- restlessness and hyperactivity
- somatic complaints.
The 2 groups of researchers also noted hopelessness, social phobia, anxiety, agitation, mania, labile mood, insomnia, explosivity, and suicidal ideation, attempt, and completion.4,5
By Stage 4, all affected patients are symptomatic. Cognitive impairment is severe; many are described as having “severe memory loss with dementia,”5 “profound” inattention and loss of concentration,5 and dysarthria. Paranoia may develop. Mood symptoms can be severe: Approximately 31% of subjects studied have contemplated suicide; of those, 26% had “suicidal tendencies” and 14% completed suicide.5
Two distinct types of CTE progression are apparent:
- patients who display cognitive deficits first; they progress to dementia but live longer
- patients who display mood and behavioral symptoms first; they tend to be younger, more violent, depressed, and explosive.6
CTE cannot be diagnosed with imaging. There are, however, a few positron emission tomography (PET) ligands for pTau that show promise:
- [F-18]FDDNP, which consistently identifies pTau deposits in brains in which CTE is clinically suspected, in the same distribution of pTau neurofibrillary tangles on autopsy.
- [11C]DPA-713, which detected TBI-related inflammation of neurons in 9 former NFL players in whom CTE was suspected based on the clinical presentation.
- PiB amyloid ligand, under investigation for use in PET neuroimaging.7
Casualties
In January 2016 alone, at least 3 former NFL players were found to have CTE posthumously.
Earl Morrall. Former quarterback who had a 21-year NFL career. Official cause of death in 2014 at age 79 was recorded as “complications of Parkinson’s disease.” In 2016, Stage-4 CTE was discovered on autopsy.8
Ken Stabler. Former quarterback for several NFL teams over 15 seasons. Died of colon cancer at age 69 in 2015. On autopsy, was found to have Stage-3 CTE.9
Tyler Sash. Former University of Iowa and New York Giants football player. Died in September 2015 at age 27 of an apparent drug overdose; posthumously, determined to have Stage-2 CTE. His family reported memory loss, minor fits of rage, confusion, inattention, lack of focus, and chronic pain.
Sash’s mother said, “My son knew something was wrong, but he couldn’t express it. He was such a good person, and it’s sad that he struggled so with this—not knowing where to go with it. Now it makes sense.”10 Sash played 16 years of football in all, sustaining at least 5 concussions. (“If you’ve played football, you know there are often other incidents [of head trauma],” Sash’s father said.10)
Cultural and medical mindsets about contact sports
In the United States, children as young as age 5, with a low weight limit of 35 pounds, routinely are introduced to football.11 Reports of 5 high school players dying from football-related injury in the 2014 season, and 3 deaths in the 2015 season, led a St. Louis, Missouri, area school district to defund their football program entirely. The district’s 2015 homecoming game was a soccer match; students and parents seemed to embrace the change.12
On its face, soccer seems a good alternative to football. When children are instructed to “head” the ball, however, concern arises about CTE: Mild CTE changes have been reported in 2 young soccer players, and late-stage CTE changes were seen in a retired soccer player with dementia.13
Perhaps most disturbing is that players who develop symptoms of CTE, or are at risk, are unlikely to seek psychiatric help. We, as psychiatric clinicians, must be diligent about questioning young patients about their extracurricular activities. It is not enough to simply ask about a history of head trauma: Ask patients about any blow to the head, and don’t limit your questioning to whether they sustained a “concussion” during practice or play.
When speaking with adult and geriatric patients, ask about a history of playing interscholastic or collegiate contact sports, such as football, hockey, and soccer.
Is the solution to better shield the head?
That is not a solution: Helmets and other protective headgear appear to be insufficient to protect the brain from traumatic injury. Perhaps keeping children from engaging in violent sports that put them at high risk of CTE later is the preventive approach that merits the most attention.
The National Football League (NFL) had its highest concussion tally last year: 182 such injuries reported1 in the 2014-2015 regular season. The true rate of concussion in the NFL is likely higher, as a result of multiple factors (fear of “letting the team [or the coach] down,” fear of retaliation from team owners,2 etc.).
To simply call a head injury a “concussion” is a disservice to players and their family: Any blow to the head, severe or otherwise, has the potential to cause microvascular disruption in the brain; repeated blows to the head undoubtedly cause further damage.
In reality, a “concussion” is a mild traumatic brain injury (mTBI). With repeated blows, an mTBI can lead to chronic traumatic encephalopathy (CTE). In 2015, eighty-seven of 91 brains from autopsied former NFL players displayed some stage of CTE.3
Pathophysiology and presentation
CTE comprises 4 histological stages; Stage 4 is the most advanced. Alzheimer’s disease (AD) and CTE display similarities, which suggests a separate classification of CTE-AD; the presence of amyloid β plaques correlates with (1) more severe hyperphosphorylated tau (pTau) pathology and (2) advanced stages of the disease and clinical presentations. Death tends to occur 10 years earlier in CTE-AD than in AD, suggesting that repetitive mTBI might change the deposition and accumulation of amyloid β plaques, and even accelerate the aging process in the brain.4
Symptoms. The case series by Omalu et al4 (which inspired the 2015 motion picture Concussion) and the case series presented by McKee et al5 described severe psychiatric symptoms associated with CTE:
- decreased speed of information processing
- increase in religiosity
- lack of insight
- poor judgment
- involvement in illegal activities
- substance abuse
- indiscretion
- verbal and physical abuse
- problems with interpersonal relationships
- isolation
- restlessness and hyperactivity
- somatic complaints.
The 2 groups of researchers also noted hopelessness, social phobia, anxiety, agitation, mania, labile mood, insomnia, explosivity, and suicidal ideation, attempt, and completion.4,5
By Stage 4, all affected patients are symptomatic. Cognitive impairment is severe; many are described as having “severe memory loss with dementia,”5 “profound” inattention and loss of concentration,5 and dysarthria. Paranoia may develop. Mood symptoms can be severe: Approximately 31% of subjects studied have contemplated suicide; of those, 26% had “suicidal tendencies” and 14% completed suicide.5
Two distinct types of CTE progression are apparent:
- patients who display cognitive deficits first; they progress to dementia but live longer
- patients who display mood and behavioral symptoms first; they tend to be younger, more violent, depressed, and explosive.6
CTE cannot be diagnosed with imaging. There are, however, a few positron emission tomography (PET) ligands for pTau that show promise:
- [F-18]FDDNP, which consistently identifies pTau deposits in brains in which CTE is clinically suspected, in the same distribution of pTau neurofibrillary tangles on autopsy.
- [11C]DPA-713, which detected TBI-related inflammation of neurons in 9 former NFL players in whom CTE was suspected based on the clinical presentation.
- PiB amyloid ligand, under investigation for use in PET neuroimaging.7
Casualties
In January 2016 alone, at least 3 former NFL players were found to have CTE posthumously.
Earl Morrall. Former quarterback who had a 21-year NFL career. Official cause of death in 2014 at age 79 was recorded as “complications of Parkinson’s disease.” In 2016, Stage-4 CTE was discovered on autopsy.8
Ken Stabler. Former quarterback for several NFL teams over 15 seasons. Died of colon cancer at age 69 in 2015. On autopsy, was found to have Stage-3 CTE.9
Tyler Sash. Former University of Iowa and New York Giants football player. Died in September 2015 at age 27 of an apparent drug overdose; posthumously, determined to have Stage-2 CTE. His family reported memory loss, minor fits of rage, confusion, inattention, lack of focus, and chronic pain.
Sash’s mother said, “My son knew something was wrong, but he couldn’t express it. He was such a good person, and it’s sad that he struggled so with this—not knowing where to go with it. Now it makes sense.”10 Sash played 16 years of football in all, sustaining at least 5 concussions. (“If you’ve played football, you know there are often other incidents [of head trauma],” Sash’s father said.10)
Cultural and medical mindsets about contact sports
In the United States, children as young as age 5, with a low weight limit of 35 pounds, routinely are introduced to football.11 Reports of 5 high school players dying from football-related injury in the 2014 season, and 3 deaths in the 2015 season, led a St. Louis, Missouri, area school district to defund their football program entirely. The district’s 2015 homecoming game was a soccer match; students and parents seemed to embrace the change.12
On its face, soccer seems a good alternative to football. When children are instructed to “head” the ball, however, concern arises about CTE: Mild CTE changes have been reported in 2 young soccer players, and late-stage CTE changes were seen in a retired soccer player with dementia.13
Perhaps most disturbing is that players who develop symptoms of CTE, or are at risk, are unlikely to seek psychiatric help. We, as psychiatric clinicians, must be diligent about questioning young patients about their extracurricular activities. It is not enough to simply ask about a history of head trauma: Ask patients about any blow to the head, and don’t limit your questioning to whether they sustained a “concussion” during practice or play.
When speaking with adult and geriatric patients, ask about a history of playing interscholastic or collegiate contact sports, such as football, hockey, and soccer.
Is the solution to better shield the head?
That is not a solution: Helmets and other protective headgear appear to be insufficient to protect the brain from traumatic injury. Perhaps keeping children from engaging in violent sports that put them at high risk of CTE later is the preventive approach that merits the most attention.
1. Blackstone J. NFL tackles alarming increase in concussions. CBS News. http://www.cbsnews.com/news/nfl-studying-how-to-tackle-alarming-increase-in-concussions. Published February 2, 2016. Accessed February 3, 2016.
2. McNamee M, Partridge B, Anderson L. Concussion ethics and sports medicine. Clin Sports Med. 2015;35(2):257-267.
3. Abreu MA, Cromartie FJ, Spradley BD; United States Sports Academy. Chronic traumatic encephalopathy (CTE) and former National Football League player suicides. The Sport Journal. http://thesportjournal.org/article/chronic-traumatic-encephalopathy-cte-and-former-national-football-league-player-suicides. Published January 29, 2016. Accessed January 29, 2016.
4. Omalu B, Bailes J, Hamilton RL, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in american athletes. Neurosurgery. 2011;69(1):173-183; discussion 183.
5. McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(pt 1):43-64.
6. Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81(13):1122-1129.
7. Eisenmenger LB, Huo EJ, Hoffman JM, et al. Advances in PET imaging of degenerative, cerebrovascular, and traumatic causes of dementia. Semin Nucl Med. 2016;46(1):57-87.
8. Jackson B. Report: former Miami Dolphins QB Earl Morrall had brain disease CTE. Miami Herald. http://www.miamiherald.com/sports/nfl/miami-dolphins/article58794523.html. Published February 5, 2016. Accessed February 6, 2016.
9. Fantz A. Ex-NFL player Ken Stabler had concussion disease CTE, doctor says. CNN. http://www.cnn.com/2016/02/03/health/ken-stabler-cte. Updated February 4, 2016. Accessed February 9, 2016.
10. Pennington B. C.T.E. is found in an Ex-Giant Tyler Sash, who died at 27. The New York Times. http://www.nytimes.com/2016/01/27/sports/football/former-giants-safety-tyler-sash-found-to-have-cte.html?_r=0. Published January 26, 2016. Accessed January 27, 2016.
11. Pop Warner Little Scholars, Inc. Ages and weights for tackle football programs. http://www.popwarner.com/football/footballstructure.htm. Accessed February 5, 2016.
12. Fowler L. No football for homecoming? No problem at Maplewood-Richmond Heights High. St. Louis Post Dispatch. http://www.stltoday.com/news/local/education/no-football-for-homecoming-no-problem-at-maplewood-richmond-heights/article_cc8dc31b-5097-5114-ba9b-9b3584f478b9.html. Published October 9, 2015. Accessed February 3, 2016.
13. Hales C, Neill S, Gearing M, et al. Late-stage CTE pathology in a retired soccer player with dementia. Neurology. 2014;83(24):2307-2309. doi: 10.1212/WNL.0000000000001081.
1. Blackstone J. NFL tackles alarming increase in concussions. CBS News. http://www.cbsnews.com/news/nfl-studying-how-to-tackle-alarming-increase-in-concussions. Published February 2, 2016. Accessed February 3, 2016.
2. McNamee M, Partridge B, Anderson L. Concussion ethics and sports medicine. Clin Sports Med. 2015;35(2):257-267.
3. Abreu MA, Cromartie FJ, Spradley BD; United States Sports Academy. Chronic traumatic encephalopathy (CTE) and former National Football League player suicides. The Sport Journal. http://thesportjournal.org/article/chronic-traumatic-encephalopathy-cte-and-former-national-football-league-player-suicides. Published January 29, 2016. Accessed January 29, 2016.
4. Omalu B, Bailes J, Hamilton RL, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in american athletes. Neurosurgery. 2011;69(1):173-183; discussion 183.
5. McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(pt 1):43-64.
6. Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. 2013;81(13):1122-1129.
7. Eisenmenger LB, Huo EJ, Hoffman JM, et al. Advances in PET imaging of degenerative, cerebrovascular, and traumatic causes of dementia. Semin Nucl Med. 2016;46(1):57-87.
8. Jackson B. Report: former Miami Dolphins QB Earl Morrall had brain disease CTE. Miami Herald. http://www.miamiherald.com/sports/nfl/miami-dolphins/article58794523.html. Published February 5, 2016. Accessed February 6, 2016.
9. Fantz A. Ex-NFL player Ken Stabler had concussion disease CTE, doctor says. CNN. http://www.cnn.com/2016/02/03/health/ken-stabler-cte. Updated February 4, 2016. Accessed February 9, 2016.
10. Pennington B. C.T.E. is found in an Ex-Giant Tyler Sash, who died at 27. The New York Times. http://www.nytimes.com/2016/01/27/sports/football/former-giants-safety-tyler-sash-found-to-have-cte.html?_r=0. Published January 26, 2016. Accessed January 27, 2016.
11. Pop Warner Little Scholars, Inc. Ages and weights for tackle football programs. http://www.popwarner.com/football/footballstructure.htm. Accessed February 5, 2016.
12. Fowler L. No football for homecoming? No problem at Maplewood-Richmond Heights High. St. Louis Post Dispatch. http://www.stltoday.com/news/local/education/no-football-for-homecoming-no-problem-at-maplewood-richmond-heights/article_cc8dc31b-5097-5114-ba9b-9b3584f478b9.html. Published October 9, 2015. Accessed February 3, 2016.
13. Hales C, Neill S, Gearing M, et al. Late-stage CTE pathology in a retired soccer player with dementia. Neurology. 2014;83(24):2307-2309. doi: 10.1212/WNL.0000000000001081.

Is it Alzheimer’s? How to pare down the possibilities
Accurate and early diagnosis of Alzheimer’s disease (AD) is evolving, and—although not yet definitive—is no longer one of exclusion. With a careful in-office work-up and routine assessment tools, you can accurately identify >90% of patients with late-onset AD.1
AD is by far the most common cause of dementia in older patients. To help you make the diagnosis, this state-of-the-art article discusses:
- AD’s clinical presentation and course
- the role of neuropsychological tests for assessing cognitive and functional status
- neuropsychiatric and medical findings that differentiate AD from other dementia causes
- indications for structural neuroimaging with CT or MRI.
Presentation and course
Variability. AD’s gradual onset and progression are characterized by prominent memory loss, anomia, constructional apraxia, anosognosia, and personality changes with affect deregulation, behavioral disturbance, and distorted perception.1 Amnesia—particularly deficits in anterograde episodic memory—is the most common presentation, but the disease course is heterogeneous and may be affected by:
- patient age at onset
- illness severity at diagnosis
- comorbid medical and neuropsychiatric illnesses
- premorbid cerebral reserves (amount of brain damage a person can sustain before reaching a threshold for the clinical expression of dementia).1-3
Researchers are investigating surrogates for detecting Alzheimer’s disease (AD) and monitoring disease progression.5
Serum and CSF markers. AD is viewed as a series of sequential events, beginning with beta-amyloid (β-amyloid) accumulation and progressing through a pathophysiologic cascade to cell death, transmitter deficit, and dementia. A unique biomarker may be associated with each event, either in the primary disease process of β-amyloid production and accumulation or intermediate processes such as tau hyperphosphorylation, oxidation, and inflammation.5,6
These biochemical markers are found more consistently in cerebrospinal fluid (CSF) than peripherally. Lower CSF β-amyloid (especially β-amyloid 42) and higher CSF tau and tau-phosphorylated (p-tau) have been found in AD patients compared with normal and disease controls.7 Some overlap exists, however, among AD and other dementias. Other possible serum, CSF, and urine markers include isoprostanes, sulfatides, oxysterols, homocysteine, apolipoprotein E, alpha 1-antichymotrypsin, 3-nitrotyrosine, and more.8 No biomarkers are available or recommended for clinical use at this time.
Neuroimaging. Amyloid imaging tracers may increase the capacity of single photon emission computed tomography (SPECT) and positron emission tomography (PET) to detect AD pathology. These tracers have high binding affinity for amyloid and may enable PET/SPECT to detect amyloid deposits in vivo.
Amyloid radioligands are being developed and tested as potential clinical diagnostic tools and surrogate biomarkers of antiamyloid therapies. A radioligand that targets amyloid and neurofibrillary tangles in AD has been developed recently for use as a research tool.
Mild AD. An individual or close companion may notice increased forgetfulness and word-finding difficulties, a tendency to lose or misplace things, repeated questioning, and some disorientation. Motor skills are intact.
Severe AD. An individual with late-stage disease has severe impairment and can be bedridden, incontinent, and unable to under-stand or speak. Full-time care is required.
Staging informs treatment. In clinical trials, patients with mild-to-moderate AD consistently show small improvements in cognitive and global function when treated with acetylcholinesterase inhibitors (AChEIs) such as donepezil, rivastigmine, and galantamine.4 Donepezil also is approved for use in severe AD.
Memantine is indicated for symptomatic treatment of moderate-to-severe AD. It differs in mechanism of action from the AChEIs and is thought to inhibit cytotoxic overstimulation of glutamatergic neurons.4 For moderately advanced AD, memantine appears to be beneficial alone or in combination with AChEIs.
Dementia assessment
Clinical assessment has low sensitivity for early-phase AD and compromised specificity in advanced stages, where all dementia subtypes are similar and comorbidities may confuse the picture. Promising surrogate biomarkers and other diagnostic tools are being developed (Box 1),5-8 but definitive AD diagnosis still requires post-mortem histopathologic examination of the cerebral cortex.
Neuropsychological tests disclose a degree of intellectual impairment that correlates with functional impairment and may be particularly useful for assessing:
- mild cognitive impairment when diagnosis is doubtful
- cases where major lifestyle changes may be required, such as driving cessation or assisted-living placement.
These tests can examine performance across different domains of cognitive function, including orientation, memory, attention, naming, comprehension, and praxis.
Limitations. Neuropsychological tests have limitations, including cost and administration time. Some older patients find the tests distressing or tiring, and those with severe dementia are incapable of participating. Patients’ anxiety about taking tests, poor test-taking skills, low motivation/effort, and language, cultural, and educational variables limit these tests’ usefulness and may influence results.
Interpret a neuropsychological evaluation in the context of other clinical data, such as informant-based history of cognitive decline, evidence of impairment in independent activities of daily living, educational background, depression assessment, sensory impairment, or factors other than dementia that may account for impaired performance.
History and physical exam. Depending on the AD stage at presentation, patients might not be a reliable source of information. For a realistic and unbiased history and evaluation, assess the patient separately and obtain collateral information from reliable informants.
In typical cases, the history guides the physical/neurologic examination. Advancing age and family history are confirmed risk factors for AD; others may include:
- female gender (after age 80)
- cardiovascular disease (such as cerebral infarcts, hypertension, elevated cholesterol/homocysteine, smoking, and diabetes mellitus)
- history of head trauma, especially with loss of consciousness.
Early and accurate diagnosis of AD is challenging in patients with mixed dementias, comorbid neurologic diseases, or atypical features. Patients with these presentations may require referral to an expert clinician, extensive workup, or longitudinal follow-up before the diagnosis becomes clear.
Neuropsychological testing. Most mental status tests examine orientation, attention/concentration, learning, memory, language, and constructional praxis. The Folstein Mini-Mental State Examination (MMSE)9 is the most widely used and well-validated mental status test. A score of 10 to 20 on the MMSE is generally considered as moderate AD, and 10 Other mental status testing options include:
- Blessed Information-Memory-Concentration (BIMC)
- Blessed Orientation-Memory-Concentration (BOMC)
- Short Test of Mental Status (STMS)
- Saint Louis University Mental Status (SLUMS).11,12
Reversible causes. If the patient is generally healthy, a core of laboratory tests is recommended in the diagnostic workup (Table 1).6,15 Other options include:
- CSF examination for atypical presentations, such as unusually rapid symptom progression, altered consciousness, or other neurologic manifestations
- EEG to differentiate delirium, seizure disorders, encephalopathies, or a rapidly progressing dementia such as CreutzfeldtJakob disease.
Because delirium may be the initial presentation of AD or reversible causes, re-evaluate patients for dementia after delirium clears.
Neuroimaging. Structural neuroimaging with a noncontrast CT or MRI is appropriate in the initial evaluation of patients with dementia.17 More routinely, it is used to exclude rare but potentially correctable dementia causes, such as space-occupying lesions.18 Hippocampal and entorhinal volume are measured most often in discriminating AD from non-demented aging and other dementias.19
Positron emission tomography (PET) using fluorine-18-labeled deoxyglucose (FDG) may help differentiate characteristic patterns of cerebral hypometabolism in the temporoparietal lobes in AD from fronto-temporal dementia (FTD) and other less common dementias, particularly during the earliest stages of the disease.19 Medi-care reimbursement for brain PET is limited to differentiating FTD from AD.
Table 1
Recommended lab tests for Alzheimer’s disease workup
| Test | Rationale |
|---|---|
| CBC | Anemia and signs of infection |
| Vitamin B12 | Related to reversible dementia, anemia |
| Folate | Related to reversible dementia, anemia |
| Homocysteine | More accurate than individual B12/folate tests |
| C-reactive protein | Ongoing inflamatory reaction |
| Thyroid function | Hypothyroidism (reversible dementia) |
| Liver function | Metabolic causes of cognitive impairment |
| Renal function | Uremia, metabolic causes of cognitive impairment |
| Electrolytes | Hypo/hypernatremia as a cognitive impairment cause |
| Glucose | Recurrent hypoglycemia, diabetes mellitus |
| Lipid panel | Vascular dementia risk factor |
| Baseline ECG | Cardiac abnormalities as vascular risk factors |
| STS (optional) | Neurosyphilis |
| CBC: complete blood count; ECG: electrocardiogram; | |
| STS: serologic test for syphilis | |
| Source: Adapted from references 6,15 | |
Detecting causes of potentially reversible cognitive impairment
| Cause | Examples | Suggested tests |
|---|---|---|
| Space-occupying lesions | Subdural hematoma, benign tumors, hydrocephalus | CT/MRI without contrast |
| Infectious diseases | AIDS dementia complex, syphilis, Lyme disease | Serologic tests |
| Endocrinopathies/ metabolic/autoimmune disorders | Hypothyroidism, Cushing’s disease, uremia, hepatic encephalopathy, Wilson’s disease, recurrent hypoglycemia, chronic hypocalcemia, multiple sclerosis, disseminated SLE, sarcoidosis | Thyroid panel, renal and liver function tests, electrolytes, slit lamp test, serum ceruloplasmin |
| Psychiatric | Depression, alcohol dependence | Geriatric Depression Scale, assess vitamin deficiency states |
| Nutritional deficiencies | Vitamin B12, thiamine (Wernicke-Korsakoff syndrome), pyridoxine, niacin (pellagra) | Vitamin B12, homocysteine |
| Medication effects | Benzodiazepines, barbiturates, anticholinergics, opioid analgesics, antihypertensives, antiarrhythmics, antidepressants, anticonvulsants, cardiac drugs such as digitalis and derivatives (among others) | Review patients’ medications for drugs that can cause cognitive changes |
| Others | Autoimmune diseases, heavy metals, illicit drugs, obstructive sleep apnea | Drug screens and specific tests |
Diagnostic criteria
NINCDS-ADRDA. Neuropsychological AD assessment criteria developed by the National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) classify AD as probable, possible, or definite:
Possible AD is considered when a patient has an atypical onset, presentation, or course and other secondary illnesses capable of producing dementia are not believed to be the cause.
Definite AD requires histopathologic evidence of AD in addition to fulfilling criteria for probable AD.20
DSM-IV-TR. Similar but broader DSM-IVTR criteria describe an insidious progressive cognitive decline that affects recent memory and ≥1 other cognitive domain (apraxia, aphasia, agnosia, or executive functioning). This cognitive decline impairs social and occupational function, represents a change from a higher level, and is not due to other causes such as delirium.21
NINCDS-ADRDA and DSM-IV-TR criteria have comparable sensitivity and specificity for clinical AD diagnosis. Neither requires neuropathologic or genetic assessment (Box 3).15,17,22-24 Neuroimaging and other tests may be required to rule out other brain diseases that may cause dementia.
Other causes of dementia
Mild cognitive impairment (MCI) may represent a prodromal state for the earliest clinical manifestations of dementia. Symptoms include memory complaints but generally preserved activities of daily living.
Originally introduced to define a progressive, single-symptom amnestic syndrome, MCI has evolved into a classification of amnestic and non-amnestic MCI with single or multiple domains.25 Amnestic MCI is the most specifically correlated with AD.26 Neurobiologic similarities between amnestic MCI and clinically diagnosed AD include:
- neuropsychiatric symptoms, such as apathy, mood disturbance, irritability and anxiety
- over-representation of the APOE ε4 allele
- volumetric loss in the entorhinal cortex and hippocampus as measured by MRI
- Glucose hypometabolism in AD-typical regions as measured by FDG-PET
- neuronal loss in vulnerable brain regions.26
Dementia with Lewy bodies (DLB) is the second most common dementing disorder in late life—after Alzheimer’s dementia— and two-thirds of DLB cases overlap with AD. Core DLB clinical features include early recurrent visual hallucinations, fluctuating cognition, spontaneous parkinsonism, and sensitivity to conventional antipsychotics.15,28
Parkinson’s disease (PD) and DLB may represent a clinicopathologic continuum, and substantial overlap exists among AD, DLB, and PD in underlying disease process and clinical presentation.15,29 Hallucinations, depression, delusions, and delusional misidentification are seen more often in patients with DLB than AD.15
Vascular dementia (VaD) was once thought to account for 15% to 20% of dementing illnesses, but discrete VaD is now viewed as much less common. Whatever the underlying vasculopathy, vascular lesions often co-exist with other causes of dementia—usually AD (in 77% of presumed VaD cases).30
Compared with AD, patients with VaD have a more subcortical dementia with difficulty retrieving words, organizing and solving complex problems, “absent-mindedness,” and psychomotor slowing with relatively preserved language skills. VaD is thought to have a more abrupt onset than AD and “stepladder” deterioration.
Frontotemporal dementia (FTD)—such as Pick’s disease—is associated with focal atrophy of the frontal and/or temporal lobes. Mean onset is age 52 to 56, and FTD is less common than AD, VaD, or DLB.
FTD often presents with gradual personality changes (with inappropriate responses or activities) or language changes (with severe naming difficulty and problems with word meaning).31 Features that may help differentiate FTD from AD include:
- disinhibition/apathy with personality change
- affect disregulation
- behavioral disturbance (frontal type) and expressive/receptive language changes (semantic or primary progressive aphasia) with relatively mild memory loss.32,33
Other neurodegenerative diseases that might present with dementia include PD, Huntington’s disease, progressive supra-nuclear palsy, corticobasal degeneration, and Creutzfeldt-Jakob disease.33
Genetic testing may become important for high-risk patients or early-stage Alzheimer’s disease (AD) when preventive/ disease-modifying therapy becomes available. At this time, however, the clinical value and implications of genetic tests remain controversial.17,22
Apolipoprotein E (APOE). The APOE ε4 allele is an established risk factor for AD,23,24 but limitations of APOE testing include:
- inability to predict with sufficient certainty whether or when a person might develop AD
- risk of false alarm or false reassurance
- no established treatment exists for a person with this genetic risk.
Amyloid precursor protein (APP), presenilin 1 (PS1), presenilin 2 (PS2). Age 15
- Mutations are rare (~1% of AD cases).
- Increased APP transcriptional activity is an AD risk factor; onset age correlates inversely with levels of APP expression.
- PS1 mutation testing may benefit patients with early-onset familial AD. If this mutation is found, other presymptomatic at-risk family members may wish to be tested so they can make important life decisions based on the results.17,22 Careful pre- and post-test counseling is critical.
Related resources
- Morris JC. Dementia update 2005. Alzheimer Dis Assoc Disord 2005;19:100-17.
- Boeve BF. A review of the non-Alzheimer dementias. J Clin Psychiatry 2006;67(12)1985-2001.
- Medscape. Alzheimer’s disease resource center. www.medscape.com/resource/alzheimers.
- Donepezil • Aricept
- Memantine • Namenda
- Galantamine • Razadyne
- Rivastigmine • Exelon
Dr. Gebretsadik reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Bristol-Myers Squibb, Forest Laboratories, Eli Lilly and Company, Novartis, Pfizer Inc., Wyeth, Elan, Myriad, Ono Pharmaceutical, and the Alzheimer’s Disease Cooperative Study Consortium. He is a consultant to Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, Janssen Pharmaceutica, Novartis, AstraZeneca, Wyeth, Pfizer Inc., Takeda, and Sepracor.
1. Cummings JL. Clinical evaluation as a biomarker for Alzheimer’s disease. J Alzheimer’s Dis 2005;8:327-37.
2. Hodges JR. Alzheimer’s centennial legacy: origins, landmarks and the current status of knowledge concerning cognitive aspects. Brain 2006;129:2811-22.
3. Stern Y. Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord 2006;20:112-7.
4. Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer’s disease. Annu Rev Med 2006;57:513-33.
5. Kennedy GJ, Golde TE, Tarriot PN, Cummings JL. Amyloid-based interventions in Alzheimer’s disease. CNS Spectr 2007;12: 1(suppl 1):1-14.
6. Van der Flier WM, Scheltens P. Use of laboratory and imaging investigations in dementia. J Neurol Neurosurg Psychiatry 2005;76:45-52.
7. Galasko D. Biomarkers for Alzheimer’s disease—clinical needs and application. J Alzheimer’s Dis 2005;8:339-46.
8. Sunderland T, Hampel H, Takeda M, et al. Biomarkers in the diagnosis of Alzheimer’s disease: are we ready? J Geriatr Psychiatry Neurol 2006;19:172-9.
9. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189-98.
10. Perneczky R, Wagenpfeil S, Komossa K, et al. Mapping scores onto stages: Mini-Mental State Examination and Clinical Dementia Rating. Am J Geriatr Psychiatry 2006;14:139-44.
11. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the Mini-Mental State Examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry 2006;14(11):897-9.
12. Agency for Health Care Policy and Research Recognition and initial assessment of Alzheimer’s disease and related dementias. Comparison of mental and functional status tests according to three phases of discrimination difficulty. Available at:http://ncbi.nlm.nih.gov/books/bv.fcgi?rid=hstat6.table.31677. Accesssed November 6, 2007.
13. Sano M. Neuropsychological testing in the diagnosis of dementia. J Geriatr Psychiatry Neurol 2006;19:155-9.
14. Mohs RC. Neuropsychological assessment of patients with Alzheimer’s disease. In: Psychopharmacology—the fourth generation of progress American College of Neuropsychopharmacology. Available at: http://www.acnp.org/ g4/GN401000133/Default.htm. Accessed November 6, 2007.
15. Morris JC. Dementia update 2005. Alzheimer Dis Assoc Disord 2005;19:100-17.
16. Clarfield AM. Reversible dementia—the implications of a fall in prevalence. Age Ageing 2005;34:544-5.
17. Roberts JS, Cupples LA, Relkin NR, et al. Genetic risk assessment for adult children of people with Alzheimer’s disease: the Risk Evaluation and Education for AD (REVEAL) study. J Geriatr Psychiatry Neurol 2005;18:250-5.
18. Frisoni GB. Structural imaging in the clinical diagnosis of Alzheimer’s disease: problems and tools. J Neurol Neurosurg Psychiatry 2001;70:711-18.
19. Ramani A, Jensen JH, Helpern JA. Quantitative MR imaging in Alzheimer disease. Radiology 2006;241(1):26-44.
20. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34(7):939-44.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington, DC: American Psychiatric Association; 2000.
22. Roberts JS, Barber M, Brown T, et al. Who seeks genetic susceptibility testing for Alzheimer’s disease? Findings from a multi-site, randomized clinical trial. Genet Med 2004;6(4):197-203.
23. Van der Flier WM, Scheltens P. Epidemiology and risk factors of dementia. J Neurol Neurosurg Psychiatry 2005;76:2-7.
24. Blacker D, Lovestone S. Genetics and dementia nosology. J Geriatr Psychiatry Neurol 2006;19:186-91.
25. Busse A, Bischkopf J, Reidel-Heller SG, Angermeyer MS. Subclassifications for mild cognitive impairment: prevalence and predictive validity. Psychol Med 2003;33(6):1029-38.
26. Rasquin SM, Lodder J, Visser PJ, et al. Predictive accuracy of MCI subtypes for Alzheimer’s disease and vascular dementia in subjects with mild cognitive impairment: a 2-year followup study. Dement Geriatr Cogn Disord 2005;19(2-3):113-19.
27. Boyle PA, Wilson RS, Aggarwal NT, et al. Mild cognitive impairment: risk of Alzheimer disease and rate of cognitive decline. Neurology 2006;67:441-5.
28. Geser F, Wenning GK, Poewe W, McKeith I. How to diagnose dementia with Lewy bodies: state of the art. Mov Disord 2005;20(suppl 12):S11-S20.
29. Hardy J. The relationship between Lewy body disease, Parkinson’s disease, and Alzheimer’s disease. Ann NY Acad Sci 2003;991:167-70.
30. Jellinger KA. Vascular-ischemic dementia: an update. J Neural Transm 2002;62(suppl):1-23.
31. McKhann GM, Albert MS, Grossman M, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 2001;58:1803-9.
32. Boxer AL, Miller BL. Clinical features of frontotemporal dementia. Alzheimer Dis Assoc Disord 2005;19(suppl):S3-S6.
33. Boeve BF. A review of the non-Alzheimer dementias. J Clin Psychiatry 2006;67(12):1985-2001.
Accurate and early diagnosis of Alzheimer’s disease (AD) is evolving, and—although not yet definitive—is no longer one of exclusion. With a careful in-office work-up and routine assessment tools, you can accurately identify >90% of patients with late-onset AD.1
AD is by far the most common cause of dementia in older patients. To help you make the diagnosis, this state-of-the-art article discusses:
- AD’s clinical presentation and course
- the role of neuropsychological tests for assessing cognitive and functional status
- neuropsychiatric and medical findings that differentiate AD from other dementia causes
- indications for structural neuroimaging with CT or MRI.
Presentation and course
Variability. AD’s gradual onset and progression are characterized by prominent memory loss, anomia, constructional apraxia, anosognosia, and personality changes with affect deregulation, behavioral disturbance, and distorted perception.1 Amnesia—particularly deficits in anterograde episodic memory—is the most common presentation, but the disease course is heterogeneous and may be affected by:
- patient age at onset
- illness severity at diagnosis
- comorbid medical and neuropsychiatric illnesses
- premorbid cerebral reserves (amount of brain damage a person can sustain before reaching a threshold for the clinical expression of dementia).1-3
Researchers are investigating surrogates for detecting Alzheimer’s disease (AD) and monitoring disease progression.5
Serum and CSF markers. AD is viewed as a series of sequential events, beginning with beta-amyloid (β-amyloid) accumulation and progressing through a pathophysiologic cascade to cell death, transmitter deficit, and dementia. A unique biomarker may be associated with each event, either in the primary disease process of β-amyloid production and accumulation or intermediate processes such as tau hyperphosphorylation, oxidation, and inflammation.5,6
These biochemical markers are found more consistently in cerebrospinal fluid (CSF) than peripherally. Lower CSF β-amyloid (especially β-amyloid 42) and higher CSF tau and tau-phosphorylated (p-tau) have been found in AD patients compared with normal and disease controls.7 Some overlap exists, however, among AD and other dementias. Other possible serum, CSF, and urine markers include isoprostanes, sulfatides, oxysterols, homocysteine, apolipoprotein E, alpha 1-antichymotrypsin, 3-nitrotyrosine, and more.8 No biomarkers are available or recommended for clinical use at this time.
Neuroimaging. Amyloid imaging tracers may increase the capacity of single photon emission computed tomography (SPECT) and positron emission tomography (PET) to detect AD pathology. These tracers have high binding affinity for amyloid and may enable PET/SPECT to detect amyloid deposits in vivo.
Amyloid radioligands are being developed and tested as potential clinical diagnostic tools and surrogate biomarkers of antiamyloid therapies. A radioligand that targets amyloid and neurofibrillary tangles in AD has been developed recently for use as a research tool.
Mild AD. An individual or close companion may notice increased forgetfulness and word-finding difficulties, a tendency to lose or misplace things, repeated questioning, and some disorientation. Motor skills are intact.
Severe AD. An individual with late-stage disease has severe impairment and can be bedridden, incontinent, and unable to under-stand or speak. Full-time care is required.
Staging informs treatment. In clinical trials, patients with mild-to-moderate AD consistently show small improvements in cognitive and global function when treated with acetylcholinesterase inhibitors (AChEIs) such as donepezil, rivastigmine, and galantamine.4 Donepezil also is approved for use in severe AD.
Memantine is indicated for symptomatic treatment of moderate-to-severe AD. It differs in mechanism of action from the AChEIs and is thought to inhibit cytotoxic overstimulation of glutamatergic neurons.4 For moderately advanced AD, memantine appears to be beneficial alone or in combination with AChEIs.
Dementia assessment
Clinical assessment has low sensitivity for early-phase AD and compromised specificity in advanced stages, where all dementia subtypes are similar and comorbidities may confuse the picture. Promising surrogate biomarkers and other diagnostic tools are being developed (Box 1),5-8 but definitive AD diagnosis still requires post-mortem histopathologic examination of the cerebral cortex.
Neuropsychological tests disclose a degree of intellectual impairment that correlates with functional impairment and may be particularly useful for assessing:
- mild cognitive impairment when diagnosis is doubtful
- cases where major lifestyle changes may be required, such as driving cessation or assisted-living placement.
These tests can examine performance across different domains of cognitive function, including orientation, memory, attention, naming, comprehension, and praxis.
Limitations. Neuropsychological tests have limitations, including cost and administration time. Some older patients find the tests distressing or tiring, and those with severe dementia are incapable of participating. Patients’ anxiety about taking tests, poor test-taking skills, low motivation/effort, and language, cultural, and educational variables limit these tests’ usefulness and may influence results.
Interpret a neuropsychological evaluation in the context of other clinical data, such as informant-based history of cognitive decline, evidence of impairment in independent activities of daily living, educational background, depression assessment, sensory impairment, or factors other than dementia that may account for impaired performance.
History and physical exam. Depending on the AD stage at presentation, patients might not be a reliable source of information. For a realistic and unbiased history and evaluation, assess the patient separately and obtain collateral information from reliable informants.
In typical cases, the history guides the physical/neurologic examination. Advancing age and family history are confirmed risk factors for AD; others may include:
- female gender (after age 80)
- cardiovascular disease (such as cerebral infarcts, hypertension, elevated cholesterol/homocysteine, smoking, and diabetes mellitus)
- history of head trauma, especially with loss of consciousness.
Early and accurate diagnosis of AD is challenging in patients with mixed dementias, comorbid neurologic diseases, or atypical features. Patients with these presentations may require referral to an expert clinician, extensive workup, or longitudinal follow-up before the diagnosis becomes clear.
Neuropsychological testing. Most mental status tests examine orientation, attention/concentration, learning, memory, language, and constructional praxis. The Folstein Mini-Mental State Examination (MMSE)9 is the most widely used and well-validated mental status test. A score of 10 to 20 on the MMSE is generally considered as moderate AD, and 10 Other mental status testing options include:
- Blessed Information-Memory-Concentration (BIMC)
- Blessed Orientation-Memory-Concentration (BOMC)
- Short Test of Mental Status (STMS)
- Saint Louis University Mental Status (SLUMS).11,12
Reversible causes. If the patient is generally healthy, a core of laboratory tests is recommended in the diagnostic workup (Table 1).6,15 Other options include:
- CSF examination for atypical presentations, such as unusually rapid symptom progression, altered consciousness, or other neurologic manifestations
- EEG to differentiate delirium, seizure disorders, encephalopathies, or a rapidly progressing dementia such as CreutzfeldtJakob disease.
Because delirium may be the initial presentation of AD or reversible causes, re-evaluate patients for dementia after delirium clears.
Neuroimaging. Structural neuroimaging with a noncontrast CT or MRI is appropriate in the initial evaluation of patients with dementia.17 More routinely, it is used to exclude rare but potentially correctable dementia causes, such as space-occupying lesions.18 Hippocampal and entorhinal volume are measured most often in discriminating AD from non-demented aging and other dementias.19
Positron emission tomography (PET) using fluorine-18-labeled deoxyglucose (FDG) may help differentiate characteristic patterns of cerebral hypometabolism in the temporoparietal lobes in AD from fronto-temporal dementia (FTD) and other less common dementias, particularly during the earliest stages of the disease.19 Medi-care reimbursement for brain PET is limited to differentiating FTD from AD.
Table 1
Recommended lab tests for Alzheimer’s disease workup
| Test | Rationale |
|---|---|
| CBC | Anemia and signs of infection |
| Vitamin B12 | Related to reversible dementia, anemia |
| Folate | Related to reversible dementia, anemia |
| Homocysteine | More accurate than individual B12/folate tests |
| C-reactive protein | Ongoing inflamatory reaction |
| Thyroid function | Hypothyroidism (reversible dementia) |
| Liver function | Metabolic causes of cognitive impairment |
| Renal function | Uremia, metabolic causes of cognitive impairment |
| Electrolytes | Hypo/hypernatremia as a cognitive impairment cause |
| Glucose | Recurrent hypoglycemia, diabetes mellitus |
| Lipid panel | Vascular dementia risk factor |
| Baseline ECG | Cardiac abnormalities as vascular risk factors |
| STS (optional) | Neurosyphilis |
| CBC: complete blood count; ECG: electrocardiogram; | |
| STS: serologic test for syphilis | |
| Source: Adapted from references 6,15 | |
Detecting causes of potentially reversible cognitive impairment
| Cause | Examples | Suggested tests |
|---|---|---|
| Space-occupying lesions | Subdural hematoma, benign tumors, hydrocephalus | CT/MRI without contrast |
| Infectious diseases | AIDS dementia complex, syphilis, Lyme disease | Serologic tests |
| Endocrinopathies/ metabolic/autoimmune disorders | Hypothyroidism, Cushing’s disease, uremia, hepatic encephalopathy, Wilson’s disease, recurrent hypoglycemia, chronic hypocalcemia, multiple sclerosis, disseminated SLE, sarcoidosis | Thyroid panel, renal and liver function tests, electrolytes, slit lamp test, serum ceruloplasmin |
| Psychiatric | Depression, alcohol dependence | Geriatric Depression Scale, assess vitamin deficiency states |
| Nutritional deficiencies | Vitamin B12, thiamine (Wernicke-Korsakoff syndrome), pyridoxine, niacin (pellagra) | Vitamin B12, homocysteine |
| Medication effects | Benzodiazepines, barbiturates, anticholinergics, opioid analgesics, antihypertensives, antiarrhythmics, antidepressants, anticonvulsants, cardiac drugs such as digitalis and derivatives (among others) | Review patients’ medications for drugs that can cause cognitive changes |
| Others | Autoimmune diseases, heavy metals, illicit drugs, obstructive sleep apnea | Drug screens and specific tests |
Diagnostic criteria
NINCDS-ADRDA. Neuropsychological AD assessment criteria developed by the National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) classify AD as probable, possible, or definite:
Possible AD is considered when a patient has an atypical onset, presentation, or course and other secondary illnesses capable of producing dementia are not believed to be the cause.
Definite AD requires histopathologic evidence of AD in addition to fulfilling criteria for probable AD.20
DSM-IV-TR. Similar but broader DSM-IVTR criteria describe an insidious progressive cognitive decline that affects recent memory and ≥1 other cognitive domain (apraxia, aphasia, agnosia, or executive functioning). This cognitive decline impairs social and occupational function, represents a change from a higher level, and is not due to other causes such as delirium.21
NINCDS-ADRDA and DSM-IV-TR criteria have comparable sensitivity and specificity for clinical AD diagnosis. Neither requires neuropathologic or genetic assessment (Box 3).15,17,22-24 Neuroimaging and other tests may be required to rule out other brain diseases that may cause dementia.
Other causes of dementia
Mild cognitive impairment (MCI) may represent a prodromal state for the earliest clinical manifestations of dementia. Symptoms include memory complaints but generally preserved activities of daily living.
Originally introduced to define a progressive, single-symptom amnestic syndrome, MCI has evolved into a classification of amnestic and non-amnestic MCI with single or multiple domains.25 Amnestic MCI is the most specifically correlated with AD.26 Neurobiologic similarities between amnestic MCI and clinically diagnosed AD include:
- neuropsychiatric symptoms, such as apathy, mood disturbance, irritability and anxiety
- over-representation of the APOE ε4 allele
- volumetric loss in the entorhinal cortex and hippocampus as measured by MRI
- Glucose hypometabolism in AD-typical regions as measured by FDG-PET
- neuronal loss in vulnerable brain regions.26
Dementia with Lewy bodies (DLB) is the second most common dementing disorder in late life—after Alzheimer’s dementia— and two-thirds of DLB cases overlap with AD. Core DLB clinical features include early recurrent visual hallucinations, fluctuating cognition, spontaneous parkinsonism, and sensitivity to conventional antipsychotics.15,28
Parkinson’s disease (PD) and DLB may represent a clinicopathologic continuum, and substantial overlap exists among AD, DLB, and PD in underlying disease process and clinical presentation.15,29 Hallucinations, depression, delusions, and delusional misidentification are seen more often in patients with DLB than AD.15
Vascular dementia (VaD) was once thought to account for 15% to 20% of dementing illnesses, but discrete VaD is now viewed as much less common. Whatever the underlying vasculopathy, vascular lesions often co-exist with other causes of dementia—usually AD (in 77% of presumed VaD cases).30
Compared with AD, patients with VaD have a more subcortical dementia with difficulty retrieving words, organizing and solving complex problems, “absent-mindedness,” and psychomotor slowing with relatively preserved language skills. VaD is thought to have a more abrupt onset than AD and “stepladder” deterioration.
Frontotemporal dementia (FTD)—such as Pick’s disease—is associated with focal atrophy of the frontal and/or temporal lobes. Mean onset is age 52 to 56, and FTD is less common than AD, VaD, or DLB.
FTD often presents with gradual personality changes (with inappropriate responses or activities) or language changes (with severe naming difficulty and problems with word meaning).31 Features that may help differentiate FTD from AD include:
- disinhibition/apathy with personality change
- affect disregulation
- behavioral disturbance (frontal type) and expressive/receptive language changes (semantic or primary progressive aphasia) with relatively mild memory loss.32,33
Other neurodegenerative diseases that might present with dementia include PD, Huntington’s disease, progressive supra-nuclear palsy, corticobasal degeneration, and Creutzfeldt-Jakob disease.33
Genetic testing may become important for high-risk patients or early-stage Alzheimer’s disease (AD) when preventive/ disease-modifying therapy becomes available. At this time, however, the clinical value and implications of genetic tests remain controversial.17,22
Apolipoprotein E (APOE). The APOE ε4 allele is an established risk factor for AD,23,24 but limitations of APOE testing include:
- inability to predict with sufficient certainty whether or when a person might develop AD
- risk of false alarm or false reassurance
- no established treatment exists for a person with this genetic risk.
Amyloid precursor protein (APP), presenilin 1 (PS1), presenilin 2 (PS2). Age 15
- Mutations are rare (~1% of AD cases).
- Increased APP transcriptional activity is an AD risk factor; onset age correlates inversely with levels of APP expression.
- PS1 mutation testing may benefit patients with early-onset familial AD. If this mutation is found, other presymptomatic at-risk family members may wish to be tested so they can make important life decisions based on the results.17,22 Careful pre- and post-test counseling is critical.
Related resources
- Morris JC. Dementia update 2005. Alzheimer Dis Assoc Disord 2005;19:100-17.
- Boeve BF. A review of the non-Alzheimer dementias. J Clin Psychiatry 2006;67(12)1985-2001.
- Medscape. Alzheimer’s disease resource center. www.medscape.com/resource/alzheimers.
- Donepezil • Aricept
- Memantine • Namenda
- Galantamine • Razadyne
- Rivastigmine • Exelon
Dr. Gebretsadik reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Bristol-Myers Squibb, Forest Laboratories, Eli Lilly and Company, Novartis, Pfizer Inc., Wyeth, Elan, Myriad, Ono Pharmaceutical, and the Alzheimer’s Disease Cooperative Study Consortium. He is a consultant to Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, Janssen Pharmaceutica, Novartis, AstraZeneca, Wyeth, Pfizer Inc., Takeda, and Sepracor.
Accurate and early diagnosis of Alzheimer’s disease (AD) is evolving, and—although not yet definitive—is no longer one of exclusion. With a careful in-office work-up and routine assessment tools, you can accurately identify >90% of patients with late-onset AD.1
AD is by far the most common cause of dementia in older patients. To help you make the diagnosis, this state-of-the-art article discusses:
- AD’s clinical presentation and course
- the role of neuropsychological tests for assessing cognitive and functional status
- neuropsychiatric and medical findings that differentiate AD from other dementia causes
- indications for structural neuroimaging with CT or MRI.
Presentation and course
Variability. AD’s gradual onset and progression are characterized by prominent memory loss, anomia, constructional apraxia, anosognosia, and personality changes with affect deregulation, behavioral disturbance, and distorted perception.1 Amnesia—particularly deficits in anterograde episodic memory—is the most common presentation, but the disease course is heterogeneous and may be affected by:
- patient age at onset
- illness severity at diagnosis
- comorbid medical and neuropsychiatric illnesses
- premorbid cerebral reserves (amount of brain damage a person can sustain before reaching a threshold for the clinical expression of dementia).1-3
Researchers are investigating surrogates for detecting Alzheimer’s disease (AD) and monitoring disease progression.5
Serum and CSF markers. AD is viewed as a series of sequential events, beginning with beta-amyloid (β-amyloid) accumulation and progressing through a pathophysiologic cascade to cell death, transmitter deficit, and dementia. A unique biomarker may be associated with each event, either in the primary disease process of β-amyloid production and accumulation or intermediate processes such as tau hyperphosphorylation, oxidation, and inflammation.5,6
These biochemical markers are found more consistently in cerebrospinal fluid (CSF) than peripherally. Lower CSF β-amyloid (especially β-amyloid 42) and higher CSF tau and tau-phosphorylated (p-tau) have been found in AD patients compared with normal and disease controls.7 Some overlap exists, however, among AD and other dementias. Other possible serum, CSF, and urine markers include isoprostanes, sulfatides, oxysterols, homocysteine, apolipoprotein E, alpha 1-antichymotrypsin, 3-nitrotyrosine, and more.8 No biomarkers are available or recommended for clinical use at this time.
Neuroimaging. Amyloid imaging tracers may increase the capacity of single photon emission computed tomography (SPECT) and positron emission tomography (PET) to detect AD pathology. These tracers have high binding affinity for amyloid and may enable PET/SPECT to detect amyloid deposits in vivo.
Amyloid radioligands are being developed and tested as potential clinical diagnostic tools and surrogate biomarkers of antiamyloid therapies. A radioligand that targets amyloid and neurofibrillary tangles in AD has been developed recently for use as a research tool.
Mild AD. An individual or close companion may notice increased forgetfulness and word-finding difficulties, a tendency to lose or misplace things, repeated questioning, and some disorientation. Motor skills are intact.
Severe AD. An individual with late-stage disease has severe impairment and can be bedridden, incontinent, and unable to under-stand or speak. Full-time care is required.
Staging informs treatment. In clinical trials, patients with mild-to-moderate AD consistently show small improvements in cognitive and global function when treated with acetylcholinesterase inhibitors (AChEIs) such as donepezil, rivastigmine, and galantamine.4 Donepezil also is approved for use in severe AD.
Memantine is indicated for symptomatic treatment of moderate-to-severe AD. It differs in mechanism of action from the AChEIs and is thought to inhibit cytotoxic overstimulation of glutamatergic neurons.4 For moderately advanced AD, memantine appears to be beneficial alone or in combination with AChEIs.
Dementia assessment
Clinical assessment has low sensitivity for early-phase AD and compromised specificity in advanced stages, where all dementia subtypes are similar and comorbidities may confuse the picture. Promising surrogate biomarkers and other diagnostic tools are being developed (Box 1),5-8 but definitive AD diagnosis still requires post-mortem histopathologic examination of the cerebral cortex.
Neuropsychological tests disclose a degree of intellectual impairment that correlates with functional impairment and may be particularly useful for assessing:
- mild cognitive impairment when diagnosis is doubtful
- cases where major lifestyle changes may be required, such as driving cessation or assisted-living placement.
These tests can examine performance across different domains of cognitive function, including orientation, memory, attention, naming, comprehension, and praxis.
Limitations. Neuropsychological tests have limitations, including cost and administration time. Some older patients find the tests distressing or tiring, and those with severe dementia are incapable of participating. Patients’ anxiety about taking tests, poor test-taking skills, low motivation/effort, and language, cultural, and educational variables limit these tests’ usefulness and may influence results.
Interpret a neuropsychological evaluation in the context of other clinical data, such as informant-based history of cognitive decline, evidence of impairment in independent activities of daily living, educational background, depression assessment, sensory impairment, or factors other than dementia that may account for impaired performance.
History and physical exam. Depending on the AD stage at presentation, patients might not be a reliable source of information. For a realistic and unbiased history and evaluation, assess the patient separately and obtain collateral information from reliable informants.
In typical cases, the history guides the physical/neurologic examination. Advancing age and family history are confirmed risk factors for AD; others may include:
- female gender (after age 80)
- cardiovascular disease (such as cerebral infarcts, hypertension, elevated cholesterol/homocysteine, smoking, and diabetes mellitus)
- history of head trauma, especially with loss of consciousness.
Early and accurate diagnosis of AD is challenging in patients with mixed dementias, comorbid neurologic diseases, or atypical features. Patients with these presentations may require referral to an expert clinician, extensive workup, or longitudinal follow-up before the diagnosis becomes clear.
Neuropsychological testing. Most mental status tests examine orientation, attention/concentration, learning, memory, language, and constructional praxis. The Folstein Mini-Mental State Examination (MMSE)9 is the most widely used and well-validated mental status test. A score of 10 to 20 on the MMSE is generally considered as moderate AD, and 10 Other mental status testing options include:
- Blessed Information-Memory-Concentration (BIMC)
- Blessed Orientation-Memory-Concentration (BOMC)
- Short Test of Mental Status (STMS)
- Saint Louis University Mental Status (SLUMS).11,12
Reversible causes. If the patient is generally healthy, a core of laboratory tests is recommended in the diagnostic workup (Table 1).6,15 Other options include:
- CSF examination for atypical presentations, such as unusually rapid symptom progression, altered consciousness, or other neurologic manifestations
- EEG to differentiate delirium, seizure disorders, encephalopathies, or a rapidly progressing dementia such as CreutzfeldtJakob disease.
Because delirium may be the initial presentation of AD or reversible causes, re-evaluate patients for dementia after delirium clears.
Neuroimaging. Structural neuroimaging with a noncontrast CT or MRI is appropriate in the initial evaluation of patients with dementia.17 More routinely, it is used to exclude rare but potentially correctable dementia causes, such as space-occupying lesions.18 Hippocampal and entorhinal volume are measured most often in discriminating AD from non-demented aging and other dementias.19
Positron emission tomography (PET) using fluorine-18-labeled deoxyglucose (FDG) may help differentiate characteristic patterns of cerebral hypometabolism in the temporoparietal lobes in AD from fronto-temporal dementia (FTD) and other less common dementias, particularly during the earliest stages of the disease.19 Medi-care reimbursement for brain PET is limited to differentiating FTD from AD.
Table 1
Recommended lab tests for Alzheimer’s disease workup
| Test | Rationale |
|---|---|
| CBC | Anemia and signs of infection |
| Vitamin B12 | Related to reversible dementia, anemia |
| Folate | Related to reversible dementia, anemia |
| Homocysteine | More accurate than individual B12/folate tests |
| C-reactive protein | Ongoing inflamatory reaction |
| Thyroid function | Hypothyroidism (reversible dementia) |
| Liver function | Metabolic causes of cognitive impairment |
| Renal function | Uremia, metabolic causes of cognitive impairment |
| Electrolytes | Hypo/hypernatremia as a cognitive impairment cause |
| Glucose | Recurrent hypoglycemia, diabetes mellitus |
| Lipid panel | Vascular dementia risk factor |
| Baseline ECG | Cardiac abnormalities as vascular risk factors |
| STS (optional) | Neurosyphilis |
| CBC: complete blood count; ECG: electrocardiogram; | |
| STS: serologic test for syphilis | |
| Source: Adapted from references 6,15 | |
Detecting causes of potentially reversible cognitive impairment
| Cause | Examples | Suggested tests |
|---|---|---|
| Space-occupying lesions | Subdural hematoma, benign tumors, hydrocephalus | CT/MRI without contrast |
| Infectious diseases | AIDS dementia complex, syphilis, Lyme disease | Serologic tests |
| Endocrinopathies/ metabolic/autoimmune disorders | Hypothyroidism, Cushing’s disease, uremia, hepatic encephalopathy, Wilson’s disease, recurrent hypoglycemia, chronic hypocalcemia, multiple sclerosis, disseminated SLE, sarcoidosis | Thyroid panel, renal and liver function tests, electrolytes, slit lamp test, serum ceruloplasmin |
| Psychiatric | Depression, alcohol dependence | Geriatric Depression Scale, assess vitamin deficiency states |
| Nutritional deficiencies | Vitamin B12, thiamine (Wernicke-Korsakoff syndrome), pyridoxine, niacin (pellagra) | Vitamin B12, homocysteine |
| Medication effects | Benzodiazepines, barbiturates, anticholinergics, opioid analgesics, antihypertensives, antiarrhythmics, antidepressants, anticonvulsants, cardiac drugs such as digitalis and derivatives (among others) | Review patients’ medications for drugs that can cause cognitive changes |
| Others | Autoimmune diseases, heavy metals, illicit drugs, obstructive sleep apnea | Drug screens and specific tests |
Diagnostic criteria
NINCDS-ADRDA. Neuropsychological AD assessment criteria developed by the National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) classify AD as probable, possible, or definite:
Possible AD is considered when a patient has an atypical onset, presentation, or course and other secondary illnesses capable of producing dementia are not believed to be the cause.
Definite AD requires histopathologic evidence of AD in addition to fulfilling criteria for probable AD.20
DSM-IV-TR. Similar but broader DSM-IVTR criteria describe an insidious progressive cognitive decline that affects recent memory and ≥1 other cognitive domain (apraxia, aphasia, agnosia, or executive functioning). This cognitive decline impairs social and occupational function, represents a change from a higher level, and is not due to other causes such as delirium.21
NINCDS-ADRDA and DSM-IV-TR criteria have comparable sensitivity and specificity for clinical AD diagnosis. Neither requires neuropathologic or genetic assessment (Box 3).15,17,22-24 Neuroimaging and other tests may be required to rule out other brain diseases that may cause dementia.
Other causes of dementia
Mild cognitive impairment (MCI) may represent a prodromal state for the earliest clinical manifestations of dementia. Symptoms include memory complaints but generally preserved activities of daily living.
Originally introduced to define a progressive, single-symptom amnestic syndrome, MCI has evolved into a classification of amnestic and non-amnestic MCI with single or multiple domains.25 Amnestic MCI is the most specifically correlated with AD.26 Neurobiologic similarities between amnestic MCI and clinically diagnosed AD include:
- neuropsychiatric symptoms, such as apathy, mood disturbance, irritability and anxiety
- over-representation of the APOE ε4 allele
- volumetric loss in the entorhinal cortex and hippocampus as measured by MRI
- Glucose hypometabolism in AD-typical regions as measured by FDG-PET
- neuronal loss in vulnerable brain regions.26
Dementia with Lewy bodies (DLB) is the second most common dementing disorder in late life—after Alzheimer’s dementia— and two-thirds of DLB cases overlap with AD. Core DLB clinical features include early recurrent visual hallucinations, fluctuating cognition, spontaneous parkinsonism, and sensitivity to conventional antipsychotics.15,28
Parkinson’s disease (PD) and DLB may represent a clinicopathologic continuum, and substantial overlap exists among AD, DLB, and PD in underlying disease process and clinical presentation.15,29 Hallucinations, depression, delusions, and delusional misidentification are seen more often in patients with DLB than AD.15
Vascular dementia (VaD) was once thought to account for 15% to 20% of dementing illnesses, but discrete VaD is now viewed as much less common. Whatever the underlying vasculopathy, vascular lesions often co-exist with other causes of dementia—usually AD (in 77% of presumed VaD cases).30
Compared with AD, patients with VaD have a more subcortical dementia with difficulty retrieving words, organizing and solving complex problems, “absent-mindedness,” and psychomotor slowing with relatively preserved language skills. VaD is thought to have a more abrupt onset than AD and “stepladder” deterioration.
Frontotemporal dementia (FTD)—such as Pick’s disease—is associated with focal atrophy of the frontal and/or temporal lobes. Mean onset is age 52 to 56, and FTD is less common than AD, VaD, or DLB.
FTD often presents with gradual personality changes (with inappropriate responses or activities) or language changes (with severe naming difficulty and problems with word meaning).31 Features that may help differentiate FTD from AD include:
- disinhibition/apathy with personality change
- affect disregulation
- behavioral disturbance (frontal type) and expressive/receptive language changes (semantic or primary progressive aphasia) with relatively mild memory loss.32,33
Other neurodegenerative diseases that might present with dementia include PD, Huntington’s disease, progressive supra-nuclear palsy, corticobasal degeneration, and Creutzfeldt-Jakob disease.33
Genetic testing may become important for high-risk patients or early-stage Alzheimer’s disease (AD) when preventive/ disease-modifying therapy becomes available. At this time, however, the clinical value and implications of genetic tests remain controversial.17,22
Apolipoprotein E (APOE). The APOE ε4 allele is an established risk factor for AD,23,24 but limitations of APOE testing include:
- inability to predict with sufficient certainty whether or when a person might develop AD
- risk of false alarm or false reassurance
- no established treatment exists for a person with this genetic risk.
Amyloid precursor protein (APP), presenilin 1 (PS1), presenilin 2 (PS2). Age 15
- Mutations are rare (~1% of AD cases).
- Increased APP transcriptional activity is an AD risk factor; onset age correlates inversely with levels of APP expression.
- PS1 mutation testing may benefit patients with early-onset familial AD. If this mutation is found, other presymptomatic at-risk family members may wish to be tested so they can make important life decisions based on the results.17,22 Careful pre- and post-test counseling is critical.
Related resources
- Morris JC. Dementia update 2005. Alzheimer Dis Assoc Disord 2005;19:100-17.
- Boeve BF. A review of the non-Alzheimer dementias. J Clin Psychiatry 2006;67(12)1985-2001.
- Medscape. Alzheimer’s disease resource center. www.medscape.com/resource/alzheimers.
- Donepezil • Aricept
- Memantine • Namenda
- Galantamine • Razadyne
- Rivastigmine • Exelon
Dr. Gebretsadik reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Bristol-Myers Squibb, Forest Laboratories, Eli Lilly and Company, Novartis, Pfizer Inc., Wyeth, Elan, Myriad, Ono Pharmaceutical, and the Alzheimer’s Disease Cooperative Study Consortium. He is a consultant to Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, Janssen Pharmaceutica, Novartis, AstraZeneca, Wyeth, Pfizer Inc., Takeda, and Sepracor.
1. Cummings JL. Clinical evaluation as a biomarker for Alzheimer’s disease. J Alzheimer’s Dis 2005;8:327-37.
2. Hodges JR. Alzheimer’s centennial legacy: origins, landmarks and the current status of knowledge concerning cognitive aspects. Brain 2006;129:2811-22.
3. Stern Y. Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord 2006;20:112-7.
4. Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer’s disease. Annu Rev Med 2006;57:513-33.
5. Kennedy GJ, Golde TE, Tarriot PN, Cummings JL. Amyloid-based interventions in Alzheimer’s disease. CNS Spectr 2007;12: 1(suppl 1):1-14.
6. Van der Flier WM, Scheltens P. Use of laboratory and imaging investigations in dementia. J Neurol Neurosurg Psychiatry 2005;76:45-52.
7. Galasko D. Biomarkers for Alzheimer’s disease—clinical needs and application. J Alzheimer’s Dis 2005;8:339-46.
8. Sunderland T, Hampel H, Takeda M, et al. Biomarkers in the diagnosis of Alzheimer’s disease: are we ready? J Geriatr Psychiatry Neurol 2006;19:172-9.
9. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189-98.
10. Perneczky R, Wagenpfeil S, Komossa K, et al. Mapping scores onto stages: Mini-Mental State Examination and Clinical Dementia Rating. Am J Geriatr Psychiatry 2006;14:139-44.
11. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the Mini-Mental State Examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry 2006;14(11):897-9.
12. Agency for Health Care Policy and Research Recognition and initial assessment of Alzheimer’s disease and related dementias. Comparison of mental and functional status tests according to three phases of discrimination difficulty. Available at:http://ncbi.nlm.nih.gov/books/bv.fcgi?rid=hstat6.table.31677. Accesssed November 6, 2007.
13. Sano M. Neuropsychological testing in the diagnosis of dementia. J Geriatr Psychiatry Neurol 2006;19:155-9.
14. Mohs RC. Neuropsychological assessment of patients with Alzheimer’s disease. In: Psychopharmacology—the fourth generation of progress American College of Neuropsychopharmacology. Available at: http://www.acnp.org/ g4/GN401000133/Default.htm. Accessed November 6, 2007.
15. Morris JC. Dementia update 2005. Alzheimer Dis Assoc Disord 2005;19:100-17.
16. Clarfield AM. Reversible dementia—the implications of a fall in prevalence. Age Ageing 2005;34:544-5.
17. Roberts JS, Cupples LA, Relkin NR, et al. Genetic risk assessment for adult children of people with Alzheimer’s disease: the Risk Evaluation and Education for AD (REVEAL) study. J Geriatr Psychiatry Neurol 2005;18:250-5.
18. Frisoni GB. Structural imaging in the clinical diagnosis of Alzheimer’s disease: problems and tools. J Neurol Neurosurg Psychiatry 2001;70:711-18.
19. Ramani A, Jensen JH, Helpern JA. Quantitative MR imaging in Alzheimer disease. Radiology 2006;241(1):26-44.
20. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34(7):939-44.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington, DC: American Psychiatric Association; 2000.
22. Roberts JS, Barber M, Brown T, et al. Who seeks genetic susceptibility testing for Alzheimer’s disease? Findings from a multi-site, randomized clinical trial. Genet Med 2004;6(4):197-203.
23. Van der Flier WM, Scheltens P. Epidemiology and risk factors of dementia. J Neurol Neurosurg Psychiatry 2005;76:2-7.
24. Blacker D, Lovestone S. Genetics and dementia nosology. J Geriatr Psychiatry Neurol 2006;19:186-91.
25. Busse A, Bischkopf J, Reidel-Heller SG, Angermeyer MS. Subclassifications for mild cognitive impairment: prevalence and predictive validity. Psychol Med 2003;33(6):1029-38.
26. Rasquin SM, Lodder J, Visser PJ, et al. Predictive accuracy of MCI subtypes for Alzheimer’s disease and vascular dementia in subjects with mild cognitive impairment: a 2-year followup study. Dement Geriatr Cogn Disord 2005;19(2-3):113-19.
27. Boyle PA, Wilson RS, Aggarwal NT, et al. Mild cognitive impairment: risk of Alzheimer disease and rate of cognitive decline. Neurology 2006;67:441-5.
28. Geser F, Wenning GK, Poewe W, McKeith I. How to diagnose dementia with Lewy bodies: state of the art. Mov Disord 2005;20(suppl 12):S11-S20.
29. Hardy J. The relationship between Lewy body disease, Parkinson’s disease, and Alzheimer’s disease. Ann NY Acad Sci 2003;991:167-70.
30. Jellinger KA. Vascular-ischemic dementia: an update. J Neural Transm 2002;62(suppl):1-23.
31. McKhann GM, Albert MS, Grossman M, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 2001;58:1803-9.
32. Boxer AL, Miller BL. Clinical features of frontotemporal dementia. Alzheimer Dis Assoc Disord 2005;19(suppl):S3-S6.
33. Boeve BF. A review of the non-Alzheimer dementias. J Clin Psychiatry 2006;67(12):1985-2001.
1. Cummings JL. Clinical evaluation as a biomarker for Alzheimer’s disease. J Alzheimer’s Dis 2005;8:327-37.
2. Hodges JR. Alzheimer’s centennial legacy: origins, landmarks and the current status of knowledge concerning cognitive aspects. Brain 2006;129:2811-22.
3. Stern Y. Cognitive reserve and Alzheimer disease. Alzheimer Dis Assoc Disord 2006;20:112-7.
4. Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer’s disease. Annu Rev Med 2006;57:513-33.
5. Kennedy GJ, Golde TE, Tarriot PN, Cummings JL. Amyloid-based interventions in Alzheimer’s disease. CNS Spectr 2007;12: 1(suppl 1):1-14.
6. Van der Flier WM, Scheltens P. Use of laboratory and imaging investigations in dementia. J Neurol Neurosurg Psychiatry 2005;76:45-52.
7. Galasko D. Biomarkers for Alzheimer’s disease—clinical needs and application. J Alzheimer’s Dis 2005;8:339-46.
8. Sunderland T, Hampel H, Takeda M, et al. Biomarkers in the diagnosis of Alzheimer’s disease: are we ready? J Geriatr Psychiatry Neurol 2006;19:172-9.
9. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189-98.
10. Perneczky R, Wagenpfeil S, Komossa K, et al. Mapping scores onto stages: Mini-Mental State Examination and Clinical Dementia Rating. Am J Geriatr Psychiatry 2006;14:139-44.
11. Tariq SH, Tumosa N, Chibnall JT, et al. Comparison of the Saint Louis University mental status examination and the Mini-Mental State Examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry 2006;14(11):897-9.
12. Agency for Health Care Policy and Research Recognition and initial assessment of Alzheimer’s disease and related dementias. Comparison of mental and functional status tests according to three phases of discrimination difficulty. Available at:http://ncbi.nlm.nih.gov/books/bv.fcgi?rid=hstat6.table.31677. Accesssed November 6, 2007.
13. Sano M. Neuropsychological testing in the diagnosis of dementia. J Geriatr Psychiatry Neurol 2006;19:155-9.
14. Mohs RC. Neuropsychological assessment of patients with Alzheimer’s disease. In: Psychopharmacology—the fourth generation of progress American College of Neuropsychopharmacology. Available at: http://www.acnp.org/ g4/GN401000133/Default.htm. Accessed November 6, 2007.
15. Morris JC. Dementia update 2005. Alzheimer Dis Assoc Disord 2005;19:100-17.
16. Clarfield AM. Reversible dementia—the implications of a fall in prevalence. Age Ageing 2005;34:544-5.
17. Roberts JS, Cupples LA, Relkin NR, et al. Genetic risk assessment for adult children of people with Alzheimer’s disease: the Risk Evaluation and Education for AD (REVEAL) study. J Geriatr Psychiatry Neurol 2005;18:250-5.
18. Frisoni GB. Structural imaging in the clinical diagnosis of Alzheimer’s disease: problems and tools. J Neurol Neurosurg Psychiatry 2001;70:711-18.
19. Ramani A, Jensen JH, Helpern JA. Quantitative MR imaging in Alzheimer disease. Radiology 2006;241(1):26-44.
20. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34(7):939-44.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington, DC: American Psychiatric Association; 2000.
22. Roberts JS, Barber M, Brown T, et al. Who seeks genetic susceptibility testing for Alzheimer’s disease? Findings from a multi-site, randomized clinical trial. Genet Med 2004;6(4):197-203.
23. Van der Flier WM, Scheltens P. Epidemiology and risk factors of dementia. J Neurol Neurosurg Psychiatry 2005;76:2-7.
24. Blacker D, Lovestone S. Genetics and dementia nosology. J Geriatr Psychiatry Neurol 2006;19:186-91.
25. Busse A, Bischkopf J, Reidel-Heller SG, Angermeyer MS. Subclassifications for mild cognitive impairment: prevalence and predictive validity. Psychol Med 2003;33(6):1029-38.
26. Rasquin SM, Lodder J, Visser PJ, et al. Predictive accuracy of MCI subtypes for Alzheimer’s disease and vascular dementia in subjects with mild cognitive impairment: a 2-year followup study. Dement Geriatr Cogn Disord 2005;19(2-3):113-19.
27. Boyle PA, Wilson RS, Aggarwal NT, et al. Mild cognitive impairment: risk of Alzheimer disease and rate of cognitive decline. Neurology 2006;67:441-5.
28. Geser F, Wenning GK, Poewe W, McKeith I. How to diagnose dementia with Lewy bodies: state of the art. Mov Disord 2005;20(suppl 12):S11-S20.
29. Hardy J. The relationship between Lewy body disease, Parkinson’s disease, and Alzheimer’s disease. Ann NY Acad Sci 2003;991:167-70.
30. Jellinger KA. Vascular-ischemic dementia: an update. J Neural Transm 2002;62(suppl):1-23.
31. McKhann GM, Albert MS, Grossman M, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 2001;58:1803-9.
32. Boxer AL, Miller BL. Clinical features of frontotemporal dementia. Alzheimer Dis Assoc Disord 2005;19(suppl):S3-S6.
33. Boeve BF. A review of the non-Alzheimer dementias. J Clin Psychiatry 2006;67(12):1985-2001.
Which cholinesterase inhibitor for early dementia?
Using a cholinesterase inhibitor (ChEI) makes sense for any disorder with a significant cholinergic deficit, such as Alzheimer’s disease (AD) and other forms of mild-to-moderate dementia (Box 1).1-3 Yet the ChEIs tacrine, donepezil, rivastigmine, and galantamine have pharmacologic differences, and individual patients respond differently to them.
To help you choose the safest, most effective treatment for each patient, we discuss:
- three cases that show how ChEIs differ in mechanism of action, administration, and side effects
- evidence of ChEIs’ efficacy in AD—for which they are approved—and in other dementias for which they have been tried
- when to switch agents, and how long to continue treatment.
Probable Alzheimer’s disease (AD) accounts for 64% of all dementias in the United States. Less-common causes include:
- vascular dementia (5%)
- combined vascular dementia and AD (10%)
- probable dementia with Lewy bodies, Parkinson’s dementia, or diffuse Lewy body disease (9%)
- Lewy body variant of AD, or AD and dementia with Lewy bodies (6%)
- frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, or Creutzfeldt-Jakob disease (6%).2,3
In our experience, many primary care physicians choose to follow their patients with dementia, even when clinical features are atypical or suggest unusual causes. Psychiatrists are asked most often to assist in diagnosis and management of patients with:
- uncommon dementias, including frontotemporal dementia or dementia with Lewy bodies
- rapidly progressive dementia
- dementia in a patient age
- dementia with psychiatric comorbidities or severe behavior disturbances.4
How Cheis Differ
Although dementia remains incurable, recognizing cognitive decline early allows you to start ChEI therapy before substantial neuronal loss occurs (Box 2).3,4 The goal of early treatment is to improve or stabilize cognition, behavior, and activities of daily living for as long as possible.
In comparison studies,5,6 ChEIs have shown differences in tolerability but not consistent differences in efficacy for mild to moderate AD—though these studies had methodologic limitations. Because the agents appear similarly effective, the initial ChEI choice often depends on how their differences might benefit your patient (Table 1). Consider the following cases:
An early dementia diagnosis enables you educate the patient and family (Box 3) and begin the most effective treatment for the person with cognitive decline. Although dementia remains incurable, early recognition presents the opportunity to start cholinesterase inhibitors before substantial neuronal loss occurs.3,4
Patient workup. The Alzheimer’s Association offers online information for health care professionals on AD diagnosis and treatment protocols (see Related resources). A detailed history, physical examination, and Mini-Mental State Examination (MMSE) are necessary if you suspect Alzheimer’s or a related dementia.
Also recommended are a comprehensive metabolic screen, complete blood counts with differential, urine analysis, serum B12 and folate studies, homocysteine levels, thyroid studies, chest radiography, ECG, lipid profile, and brain scan (MRI or CT). Perform studies such as the rapid plasma reagin test for syphilis and HIV testing as appropriate.
Similarities and differences among cholinesterase inhibitors
| Tacrine | Donepezil | Rivastigmine | Galantamine | |
|---|---|---|---|---|
| Administration | Four times daily | Once daily | Twice daily with full meals | Once daily (extended-release formulation) |
| AChE inhibitor | Yes | Yes | Yes | Yes |
| BuChE inhibitor | Yes | No | Yes | No |
| Allosteric modulation of nicotinic receptor | No | Yes | No | Yes |
| Pharmacodynamic nicotinic/muscarinic effect | Yes | Yes | Yes | Yes |
| GI side effects | Present | Present | Present | Present |
| Hepatotoxicity | Present | Absent | Absent | Absent |
| Metabolism | CYP-450 | CYP-450 | Autohydrolysis | CYP-450 |
| Drug–drug interactions | Yes | Yes | None reported | Yes |
| AChE: acetylcholinesterase | ||||
| BuChE: butyrylcholinesterase | ||||
| CYP-450: cytochrome P-450 hepatic isoenzymes | ||||
Case 1: Gradual Memory Loss
Mrs. J, age 76, has experienced a slow, insidious memory decline across 5 years. She has become socially withdrawn and shows some language difficulties. She has had peptic ulcer disease and often does not take medications as prescribed.
Her psychiatrist diagnoses probable AD and chooses donepezil with its easy dosing schedule because of Mrs. J’s history of nonadherence. Donepezil’s GI tolerability is also a factor in this choice because of the patient’s peptic ulcer disease.
Case 2: Dementia And Motor Deficits
Mr. L, age 82, has gradually developed memory loss and parkinsonian symptoms, including slowness of movement and shuffling gait. He has visual hallucinations of people and episodic confusion. His medications include warfarin and digoxin for atrial fibrillation and congestive heart failure.
Mr. L is diagnosed with probable dementia with Lewy bodies. His psychiatrist chooses rivastigmine because it has shown efficacy in this type of dementia and is not known to interact significantly with cardiovascular medications.
Case 3: Stroke, Then Rapid Decline
Mrs. D, age 68, has a history of hypertension and suffered a stroke in the past. Her family says her memory and behavior—anger outbursts and excessive irritability—have worsened rapidly across 2 years. Examination reveals some focal neurologic deficits.
Her psychiatrist diagnoses probable vascular dementia and chooses galantamine for its efficacy in patients with this dementia type. Mrs. D has no history of GI illness and will likely tolerate the drug’s GI side effects. Follow-up care will include monitoring for tolerability.
Mechanism. Donepezil inhibits the enzyme acetylcholinesterase, and rivastigmine inhibits acetylcholinesterase and butyrylcholinesterase. Galantamine inhibits acetylcholinesterase and shows allosteric modulation of the presynaptic nicotinic receptor.
Data indicating that rivastigmine is particularly effective in patients with rapidly progressive illness is consistent with the possible advantage of inhibiting both butyrylcholinesterase and acetylcholinesterase. It has been argued that galantamine’s binding to nicotinic receptors modulates their function, which may enhance acetylcholine release.
Among the three agents, only rivastigmine shows a consistent, linear dose-response relationship. It is rapidly and extensively metabolized, primarily via cholinesterase-mediated hydrolysis to the decarbamylated metabolite (autohydrolysis). Minimal metabolism occurs via the major cytochrome P (CYP)-450 isoenzymes. Donepezil and galantamine are metabolized by isoenzymes 2D6 and 3A4 and undergo glucuronidation.7
Drug interactions. Because rivastigmine avoids hepatic metabolism, interactions with drugs metabolized by CYP-450 isoenzymes have not been reported.8
Donepezil interacts with ketoconazole and quinidine, which inhibit donepezil metabolism and increase mean donepezil concentrations. Galantamine interacts with ketoconazole, paroxetine, and erythromycin, which increase mean galantamine concentrations.9
Administration. Donepezil and extended-release galantamine are given once daily because of their long half-lives, whereas regular galantamine and rivastigmine are taken twice daily with meals to minimize GI effects (Table 2). Nausea and vomiting can occur with any of the ChEIs but are more common and troublesome with rivastigmine and galantamine.
Table 2
How to use cholinesterase inhibitors for patients with dementia
| Drug | Recommended dosing | Possible side effects | Titration | Administration |
|---|---|---|---|---|
| Tacrine | Initial: 40 mg/d Maximum: 160 mg/d | Liver damage causing increase in ALT levels, GI effects (nausea, indigestion, vomiting, diarrhea, abdominal pain), skin rash | Dosage can be increased every 4 weeks | Divide into four doses; take on empty stomach |
| Donepezil | Initial: 5 mg/d Maximum: 10 mg/d | GI effects (nausea, diarrhea, vomiting, loss of appetite), insomnia, muscle cramps, fatigue | Increase dosage after 4 weeks | Once daily in morning or at bedtime |
| Rivastigmine | Initial: 3 mg/d Maximum: 12 mg/d | GI effects (nausea, vomiting, loss of appetite, weight loss, diarrhea, heartburn) | Increase dosage every 4 weeks | Twice daily after meals |
| Galantamine (regular, ER) | Initial: 8 mg/d Maximum: 24 mg/d | GI effects (nausea, vomiting, diarrhea, weight loss), possible increased mortality risk in patients with MCI | Increase dosage every 4 weeks | Regular: Twice daily after meals ER: Once daily after a meal |
| ALT: alanine transferase | ||||
| ER: extended-release formulation | ||||
| MCI: mild cognitive impairment | ||||
Efficacy In Early AD
In controlled clinical trials, all four ChEIs have significantly improved cognition, behavior, and activities of daily living in patients with mild-to-moderate AD.10-12 Tacrine—the first FDA-approved ChEI—is rarely used because its associated hepatoxicity requires ongoing liver enzyme monitoring.13 Among the other three:
Donepezil. A review of 16 trials involving 4,365 participants10 found significant benefits in cognitive functioning, activities of daily living, and behavior in persons with mild, moderate, or severe AD who were treated with donepezil for 12, 24, or 52 weeks.
Rivastigmine improved or maintained cognitive function, activities of daily living, and behavior for up to 52 weeks in patients with mild to moderate AD, according to a review of studies from 1995 to 2002.11 GI irritation was the most common adverse effect. Giving rivastigmine for up to 2 years may reduce the cost of caring for patients with AD, mostly by delaying nursing home placement.
Galantamine has beneficial effects on cognition, global function, activities of daily living, and behavior in patients with AD, vascular dementia, and AD with cerebrovascular components, according to a review of clinical studies.12 Adverse events are generally mild to moderate, transient, and gastrointestinal.
Efficacy In Other Dementias
In addition to their FDA-approved use for mildto-moderate AD, ChEIs also have been studied in persons with other types of dementia and mild cognitive impairment (MCI).
Dementia with Lewy bodies. Rivastigmine given with flexible titration from 6 to 12 mg/d improved behavior in 120 patients with Lewy body dementia.14 In the double-blind, multicenter study, patients taking rivastigmine, mean 9.7 mg/d for 20 weeks, were less apathetic and anxious and had fewer delusions and hallucinations than did those taking placebo. The drug was judged to be safe and well tolerated.
Vascular dementia. Patients with vascular dementia showed improved cognition and global function when treated with donepezil, 5 or 10 mg/d, for up to 24 weeks. Donepezil was well tolerated in this combined analysis of two randomized, placebo-controlled trials.15
Kumar et al16 compared two rivastigmine dosages in patients with mild-to-moderate AD, some of whom also had vascular dementia risk factors. Patients were randomly assigned to placebo, low-dose rivastigmine (1 to 4 mg/d), or high-dose rivastigmine (6 to 12 mg/d) for 26 weeks. Cognition, activities of daily living, and disease severity improved with rivastigmine in patients with or without vascular risk factors. Greater benefit was seen with high-dose than low-dose rivastigmine and in patients with AD plus vascular risk factors than in those with AD alone.
In a multicenter, double-blind trial,17 patients with vascular dementia or AD with vascular risk factors received galantamine, up to 24 mg/d, or placebo for 6 months. Compared with controls, those taking galantamine showed improved cognition, behavior, and function. The drug overall was well tolerated, with nausea and vomiting the most common side effects.
Parkinson’s dementia. Emre et al18 evaluated rivastigmine’s efficacy and safety in patients whose mild-to-moderate dementia developed at least 2 years after a clinical diagnosis of Parkinson’s disease (PD). Patients were randomly assigned to placebo or rivastigmine, 3 to 12 mg/d, for 24 weeks, and 410 of 541 enrollees completed the study. Compared with placebo, rivastigmine was associated with statistically significant improvements in cognition and global measures in dementia associated with PD but also with higher rates of nausea, vomiting, and tremor. PD’s motor symptoms did not change significantly in either group.
Mixed dementia states. As mentioned, galantamine improved cognitive and noncognitive abilities in patients with vascular dementia or AD with vascular risk factors in a 6-month, double-blind trial.17 Patients who received galantamine or placebo could then continue open-label galantamine, 24 mg/d, for another 6 months. In patients treated the full 12 months, galantamine continued to improve or maintain:
- cognition, based on Alzheimer’s Disease Assessment Scale-cognitive subscale scores
- functional ability, measured by the 40-item Disability Assessment for Dementia
- behavior, measured by the Neuro-psychiatric Inventory.19
After 12 months, the rivastigmine-treated patients were less behaviorally impaired than the matched patients, and their caregivers reported reduced stress. Rivastigmine did not prevent cognitive deterioration, as assessed with the Mini-Mental State Examination (MMSE).
Mild cognitive impairment. Persons with MCI have objective psychometric evidence of memory loss compared with their peers, but they are not significantly impaired in activities of daily living or other cognitive functions (language, abstract thinking, or problem-solving).
At this time, we do not recommend using ChEIs to treat MCI. These agents have shown little benefit and potential risk in patients who do not meet diagnostic criteria for dementia:
- Salloway et al21 tested donepezil’s efficacy and safety in 270 patients with MCI in a 24-week, double-blind, placebo-controlled trial. Donepezil was started at 5 mg/d for 42 days, then escalated to 10 mg/d. Compared with placebo, donepezil showed no significant effects on recall, but some improvements were seen in attention and psychomotor speed.
- In two unpublished placebo-controlled trials, galantamine did not improve memory when given for 2 years to elderly patients with MCI. A precaution was added to the drug’s prescribing information because 13 of the 1,026 patients taking galantamine died, compared with 1 of 1,022 taking placebo. Vascular disease caused one-half of the galantamine group deaths. No evidence of increased mortality risk has been seen in studies of galantamine in patients with mild-to-moderate AD, for which it is indicated.
Getting The Greatest Response
To gauge response to ChEI therapy, family reports about the patient are helpful—such as that cognition has improved or cognitive decline has not progressed as rapidly as before. Assessment tools such as the MMSE can document improvement or stabilization.
We recommend trying an initial ChEI for at least 6 months to determine its efficacy. If your patient cannot tolerate one ChEI or fails to respond to initial treatment, two consensus panels22,23 recommend that you consider changing ChEIs:
- If switching because of intolerable side effects, wait at least 2 to 3 days after stopping the first ChEI before starting another.
- If switching because of poor response, you can start a different ChEI immediately after the first one is stopped.
- Cholinesterase inhibitors may help improve or stabilize cognition, behavior, and/or activities of daily living
- Persons receiving these agents may decline more slowly than those who have not been treated
- Common side effects include nausea, vomiting, diarrhea, and loss of appetite
- Other less-common side effects are muscle cramps, slowed heart rate, dizziness, and fainting
- Because of differences in these agents, it may make sense to switch to another cholinesterase inhibitor if the patient has intolerable side effects or does not improve with the first one tried
Related resources
- Alzheimer’s Association. Information for health care professionals:
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- • Treating cognitive symptoms www.alz.org/Health/Treating/symptoms.asp
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- American Association for Geriatric Psychiatry. Information for older patients and their families on Alzheimer’s disease, other dementias. www.gmhfonline.org/gmhf/consumer/alzheimers.html.
- Tacrine • Cognex
- Donepezil • Aricept
- Rivastigmine • Exelon
- Galantamine • Razadyne (was Reminyl)
Drs. Kamat and LeFevre report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Boehringer Ingelheim Pharmaceuticals, Cyberonics, Eli Lilly and Co., Eunoe, Forest Pharmaceuticals, Novartis Pharmaceuticals Corp., Pfizer, and Wyeth Pharmaceuticals. He is a consultant to AstraZeneca Pharmaceuticals, Forest Pharmaceuticals, Janssen Pharmaceutica, KV Pharma, Novartis Pharmaceuticals Corp., Organon International, and Sanofi-Synthelabo.
Acknowledgment
The authors thank Anjali Baliga, MD, for her contribution and help in preparing this article
1. Small GW, Rabins PV, Barry PP, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA 1997;278(16):1363-71.
2. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 suppl 5):S4-S9.
3. Grossberg GT, Lake JT. The role of the psychiatrist in Alzheimer’s disease. J Clin Psychiatry 1998;59(suppl 9):3-6.
4. Doraiswamy PM, Steffens DC, Pitchumoni S, Tabrizi S. Early recognition of Alzheimer’s disease: what is consensual? What is controversial? What is practical? J Clin Psychiatry 1998;59(suppl 13):6-18.
5. Wilkinson DG, Passmore AP, Bullock R, et al. A multinational, randomised, 12-week, comparative study of donepezil and rivastigmine in patients with mild to moderate Alzheimer’s disease. Int J Clin Pract 2002;56(6):441-6
6. Jones RW, Soininen H, Hager K, et al. A multinational, randomised, 12-week study comparing the effects of donepezil and galantamine in patients with mild to moderate Alzheimer’s disease. Int J Geriatr Psychiatry 2004;19(1):58-67.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twentytwo classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. U. S. Bureau of the Census. 2004 International database: Midyear population, by age and sex. Table 094. U.S. Bureau of the Census; 2004.
9. Reminyl (galantamine HBr). Physicians’ desk reference (59th ed). Montvale, NJ: Thomson PDR; 2005:1739.
10. Birks JS, Harvey R. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev 2003;(3):CD001190.-
11. Williams BR, Nazarians A, Gill MA. A review of rivastigmine: a reversible cholinesterase inhibitor. Clin Ther 2003;25(6):1634-53.
12. Corey-Bloom J. Galantamine: a review of its use in Alzheimer’s disease and vascular dementia. Int J Clin Pract 2003;57(3):219-23.
13. Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994;271(13):992-8.
14. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000;356(9247):2031-6.
15. Passmore AP, Bayer AJ, Steinhagen-Thiessen E. Cognitive, global, and functional benefits of donepezil in Alzheimer’s disease and vascular dementia: results from large-scale clinical trials. J Neurol Sci 2005;229-30:141-6.
16. Kumar V, Anand R, Messina J, et al. An efficacy and safety analysis of rivastigmine in Alzheimer’s disease patients with concurrent vascular risk factors. Eur J Neurol 2000;7(2):159-69.
17. Kurz AF, Erkinjuntti T, Gauthier S, et al. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002;359(9314):1283-90.
18. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351(24):2509-18.
19. Erkinjuntti T, Kurz A, Small GW, et al. An open-label extension trial of galantamine in patients with probable vascular dementia and mixed dementia. Clin Ther 2003;25(6):1765-82.
20. Moretti R, Torre P, Antonello RM, et al. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004;21(14):931-7.
21. Salloway S, Ferris S, Kluger A, et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology 2004;63(4):651-7.
22. Emre M. Switching cholinesterase inhibitors in patients with Alzheimer’s disease. Int J Clin Pract Suppl 2002;(127):64-72.
23. Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int J Clin Pract Suppl 2002;(127):45-63.
Using a cholinesterase inhibitor (ChEI) makes sense for any disorder with a significant cholinergic deficit, such as Alzheimer’s disease (AD) and other forms of mild-to-moderate dementia (Box 1).1-3 Yet the ChEIs tacrine, donepezil, rivastigmine, and galantamine have pharmacologic differences, and individual patients respond differently to them.
To help you choose the safest, most effective treatment for each patient, we discuss:
- three cases that show how ChEIs differ in mechanism of action, administration, and side effects
- evidence of ChEIs’ efficacy in AD—for which they are approved—and in other dementias for which they have been tried
- when to switch agents, and how long to continue treatment.
Probable Alzheimer’s disease (AD) accounts for 64% of all dementias in the United States. Less-common causes include:
- vascular dementia (5%)
- combined vascular dementia and AD (10%)
- probable dementia with Lewy bodies, Parkinson’s dementia, or diffuse Lewy body disease (9%)
- Lewy body variant of AD, or AD and dementia with Lewy bodies (6%)
- frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, or Creutzfeldt-Jakob disease (6%).2,3
In our experience, many primary care physicians choose to follow their patients with dementia, even when clinical features are atypical or suggest unusual causes. Psychiatrists are asked most often to assist in diagnosis and management of patients with:
- uncommon dementias, including frontotemporal dementia or dementia with Lewy bodies
- rapidly progressive dementia
- dementia in a patient age
- dementia with psychiatric comorbidities or severe behavior disturbances.4
How Cheis Differ
Although dementia remains incurable, recognizing cognitive decline early allows you to start ChEI therapy before substantial neuronal loss occurs (Box 2).3,4 The goal of early treatment is to improve or stabilize cognition, behavior, and activities of daily living for as long as possible.
In comparison studies,5,6 ChEIs have shown differences in tolerability but not consistent differences in efficacy for mild to moderate AD—though these studies had methodologic limitations. Because the agents appear similarly effective, the initial ChEI choice often depends on how their differences might benefit your patient (Table 1). Consider the following cases:
An early dementia diagnosis enables you educate the patient and family (Box 3) and begin the most effective treatment for the person with cognitive decline. Although dementia remains incurable, early recognition presents the opportunity to start cholinesterase inhibitors before substantial neuronal loss occurs.3,4
Patient workup. The Alzheimer’s Association offers online information for health care professionals on AD diagnosis and treatment protocols (see Related resources). A detailed history, physical examination, and Mini-Mental State Examination (MMSE) are necessary if you suspect Alzheimer’s or a related dementia.
Also recommended are a comprehensive metabolic screen, complete blood counts with differential, urine analysis, serum B12 and folate studies, homocysteine levels, thyroid studies, chest radiography, ECG, lipid profile, and brain scan (MRI or CT). Perform studies such as the rapid plasma reagin test for syphilis and HIV testing as appropriate.
Similarities and differences among cholinesterase inhibitors
| Tacrine | Donepezil | Rivastigmine | Galantamine | |
|---|---|---|---|---|
| Administration | Four times daily | Once daily | Twice daily with full meals | Once daily (extended-release formulation) |
| AChE inhibitor | Yes | Yes | Yes | Yes |
| BuChE inhibitor | Yes | No | Yes | No |
| Allosteric modulation of nicotinic receptor | No | Yes | No | Yes |
| Pharmacodynamic nicotinic/muscarinic effect | Yes | Yes | Yes | Yes |
| GI side effects | Present | Present | Present | Present |
| Hepatotoxicity | Present | Absent | Absent | Absent |
| Metabolism | CYP-450 | CYP-450 | Autohydrolysis | CYP-450 |
| Drug–drug interactions | Yes | Yes | None reported | Yes |
| AChE: acetylcholinesterase | ||||
| BuChE: butyrylcholinesterase | ||||
| CYP-450: cytochrome P-450 hepatic isoenzymes | ||||
Case 1: Gradual Memory Loss
Mrs. J, age 76, has experienced a slow, insidious memory decline across 5 years. She has become socially withdrawn and shows some language difficulties. She has had peptic ulcer disease and often does not take medications as prescribed.
Her psychiatrist diagnoses probable AD and chooses donepezil with its easy dosing schedule because of Mrs. J’s history of nonadherence. Donepezil’s GI tolerability is also a factor in this choice because of the patient’s peptic ulcer disease.
Case 2: Dementia And Motor Deficits
Mr. L, age 82, has gradually developed memory loss and parkinsonian symptoms, including slowness of movement and shuffling gait. He has visual hallucinations of people and episodic confusion. His medications include warfarin and digoxin for atrial fibrillation and congestive heart failure.
Mr. L is diagnosed with probable dementia with Lewy bodies. His psychiatrist chooses rivastigmine because it has shown efficacy in this type of dementia and is not known to interact significantly with cardiovascular medications.
Case 3: Stroke, Then Rapid Decline
Mrs. D, age 68, has a history of hypertension and suffered a stroke in the past. Her family says her memory and behavior—anger outbursts and excessive irritability—have worsened rapidly across 2 years. Examination reveals some focal neurologic deficits.
Her psychiatrist diagnoses probable vascular dementia and chooses galantamine for its efficacy in patients with this dementia type. Mrs. D has no history of GI illness and will likely tolerate the drug’s GI side effects. Follow-up care will include monitoring for tolerability.
Mechanism. Donepezil inhibits the enzyme acetylcholinesterase, and rivastigmine inhibits acetylcholinesterase and butyrylcholinesterase. Galantamine inhibits acetylcholinesterase and shows allosteric modulation of the presynaptic nicotinic receptor.
Data indicating that rivastigmine is particularly effective in patients with rapidly progressive illness is consistent with the possible advantage of inhibiting both butyrylcholinesterase and acetylcholinesterase. It has been argued that galantamine’s binding to nicotinic receptors modulates their function, which may enhance acetylcholine release.
Among the three agents, only rivastigmine shows a consistent, linear dose-response relationship. It is rapidly and extensively metabolized, primarily via cholinesterase-mediated hydrolysis to the decarbamylated metabolite (autohydrolysis). Minimal metabolism occurs via the major cytochrome P (CYP)-450 isoenzymes. Donepezil and galantamine are metabolized by isoenzymes 2D6 and 3A4 and undergo glucuronidation.7
Drug interactions. Because rivastigmine avoids hepatic metabolism, interactions with drugs metabolized by CYP-450 isoenzymes have not been reported.8
Donepezil interacts with ketoconazole and quinidine, which inhibit donepezil metabolism and increase mean donepezil concentrations. Galantamine interacts with ketoconazole, paroxetine, and erythromycin, which increase mean galantamine concentrations.9
Administration. Donepezil and extended-release galantamine are given once daily because of their long half-lives, whereas regular galantamine and rivastigmine are taken twice daily with meals to minimize GI effects (Table 2). Nausea and vomiting can occur with any of the ChEIs but are more common and troublesome with rivastigmine and galantamine.
Table 2
How to use cholinesterase inhibitors for patients with dementia
| Drug | Recommended dosing | Possible side effects | Titration | Administration |
|---|---|---|---|---|
| Tacrine | Initial: 40 mg/d Maximum: 160 mg/d | Liver damage causing increase in ALT levels, GI effects (nausea, indigestion, vomiting, diarrhea, abdominal pain), skin rash | Dosage can be increased every 4 weeks | Divide into four doses; take on empty stomach |
| Donepezil | Initial: 5 mg/d Maximum: 10 mg/d | GI effects (nausea, diarrhea, vomiting, loss of appetite), insomnia, muscle cramps, fatigue | Increase dosage after 4 weeks | Once daily in morning or at bedtime |
| Rivastigmine | Initial: 3 mg/d Maximum: 12 mg/d | GI effects (nausea, vomiting, loss of appetite, weight loss, diarrhea, heartburn) | Increase dosage every 4 weeks | Twice daily after meals |
| Galantamine (regular, ER) | Initial: 8 mg/d Maximum: 24 mg/d | GI effects (nausea, vomiting, diarrhea, weight loss), possible increased mortality risk in patients with MCI | Increase dosage every 4 weeks | Regular: Twice daily after meals ER: Once daily after a meal |
| ALT: alanine transferase | ||||
| ER: extended-release formulation | ||||
| MCI: mild cognitive impairment | ||||
Efficacy In Early AD
In controlled clinical trials, all four ChEIs have significantly improved cognition, behavior, and activities of daily living in patients with mild-to-moderate AD.10-12 Tacrine—the first FDA-approved ChEI—is rarely used because its associated hepatoxicity requires ongoing liver enzyme monitoring.13 Among the other three:
Donepezil. A review of 16 trials involving 4,365 participants10 found significant benefits in cognitive functioning, activities of daily living, and behavior in persons with mild, moderate, or severe AD who were treated with donepezil for 12, 24, or 52 weeks.
Rivastigmine improved or maintained cognitive function, activities of daily living, and behavior for up to 52 weeks in patients with mild to moderate AD, according to a review of studies from 1995 to 2002.11 GI irritation was the most common adverse effect. Giving rivastigmine for up to 2 years may reduce the cost of caring for patients with AD, mostly by delaying nursing home placement.
Galantamine has beneficial effects on cognition, global function, activities of daily living, and behavior in patients with AD, vascular dementia, and AD with cerebrovascular components, according to a review of clinical studies.12 Adverse events are generally mild to moderate, transient, and gastrointestinal.
Efficacy In Other Dementias
In addition to their FDA-approved use for mildto-moderate AD, ChEIs also have been studied in persons with other types of dementia and mild cognitive impairment (MCI).
Dementia with Lewy bodies. Rivastigmine given with flexible titration from 6 to 12 mg/d improved behavior in 120 patients with Lewy body dementia.14 In the double-blind, multicenter study, patients taking rivastigmine, mean 9.7 mg/d for 20 weeks, were less apathetic and anxious and had fewer delusions and hallucinations than did those taking placebo. The drug was judged to be safe and well tolerated.
Vascular dementia. Patients with vascular dementia showed improved cognition and global function when treated with donepezil, 5 or 10 mg/d, for up to 24 weeks. Donepezil was well tolerated in this combined analysis of two randomized, placebo-controlled trials.15
Kumar et al16 compared two rivastigmine dosages in patients with mild-to-moderate AD, some of whom also had vascular dementia risk factors. Patients were randomly assigned to placebo, low-dose rivastigmine (1 to 4 mg/d), or high-dose rivastigmine (6 to 12 mg/d) for 26 weeks. Cognition, activities of daily living, and disease severity improved with rivastigmine in patients with or without vascular risk factors. Greater benefit was seen with high-dose than low-dose rivastigmine and in patients with AD plus vascular risk factors than in those with AD alone.
In a multicenter, double-blind trial,17 patients with vascular dementia or AD with vascular risk factors received galantamine, up to 24 mg/d, or placebo for 6 months. Compared with controls, those taking galantamine showed improved cognition, behavior, and function. The drug overall was well tolerated, with nausea and vomiting the most common side effects.
Parkinson’s dementia. Emre et al18 evaluated rivastigmine’s efficacy and safety in patients whose mild-to-moderate dementia developed at least 2 years after a clinical diagnosis of Parkinson’s disease (PD). Patients were randomly assigned to placebo or rivastigmine, 3 to 12 mg/d, for 24 weeks, and 410 of 541 enrollees completed the study. Compared with placebo, rivastigmine was associated with statistically significant improvements in cognition and global measures in dementia associated with PD but also with higher rates of nausea, vomiting, and tremor. PD’s motor symptoms did not change significantly in either group.
Mixed dementia states. As mentioned, galantamine improved cognitive and noncognitive abilities in patients with vascular dementia or AD with vascular risk factors in a 6-month, double-blind trial.17 Patients who received galantamine or placebo could then continue open-label galantamine, 24 mg/d, for another 6 months. In patients treated the full 12 months, galantamine continued to improve or maintain:
- cognition, based on Alzheimer’s Disease Assessment Scale-cognitive subscale scores
- functional ability, measured by the 40-item Disability Assessment for Dementia
- behavior, measured by the Neuro-psychiatric Inventory.19
After 12 months, the rivastigmine-treated patients were less behaviorally impaired than the matched patients, and their caregivers reported reduced stress. Rivastigmine did not prevent cognitive deterioration, as assessed with the Mini-Mental State Examination (MMSE).
Mild cognitive impairment. Persons with MCI have objective psychometric evidence of memory loss compared with their peers, but they are not significantly impaired in activities of daily living or other cognitive functions (language, abstract thinking, or problem-solving).
At this time, we do not recommend using ChEIs to treat MCI. These agents have shown little benefit and potential risk in patients who do not meet diagnostic criteria for dementia:
- Salloway et al21 tested donepezil’s efficacy and safety in 270 patients with MCI in a 24-week, double-blind, placebo-controlled trial. Donepezil was started at 5 mg/d for 42 days, then escalated to 10 mg/d. Compared with placebo, donepezil showed no significant effects on recall, but some improvements were seen in attention and psychomotor speed.
- In two unpublished placebo-controlled trials, galantamine did not improve memory when given for 2 years to elderly patients with MCI. A precaution was added to the drug’s prescribing information because 13 of the 1,026 patients taking galantamine died, compared with 1 of 1,022 taking placebo. Vascular disease caused one-half of the galantamine group deaths. No evidence of increased mortality risk has been seen in studies of galantamine in patients with mild-to-moderate AD, for which it is indicated.
Getting The Greatest Response
To gauge response to ChEI therapy, family reports about the patient are helpful—such as that cognition has improved or cognitive decline has not progressed as rapidly as before. Assessment tools such as the MMSE can document improvement or stabilization.
We recommend trying an initial ChEI for at least 6 months to determine its efficacy. If your patient cannot tolerate one ChEI or fails to respond to initial treatment, two consensus panels22,23 recommend that you consider changing ChEIs:
- If switching because of intolerable side effects, wait at least 2 to 3 days after stopping the first ChEI before starting another.
- If switching because of poor response, you can start a different ChEI immediately after the first one is stopped.
- Cholinesterase inhibitors may help improve or stabilize cognition, behavior, and/or activities of daily living
- Persons receiving these agents may decline more slowly than those who have not been treated
- Common side effects include nausea, vomiting, diarrhea, and loss of appetite
- Other less-common side effects are muscle cramps, slowed heart rate, dizziness, and fainting
- Because of differences in these agents, it may make sense to switch to another cholinesterase inhibitor if the patient has intolerable side effects or does not improve with the first one tried
Related resources
- Alzheimer’s Association. Information for health care professionals:
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- • Treating cognitive symptoms www.alz.org/Health/Treating/symptoms.asp
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- American Association for Geriatric Psychiatry. Information for older patients and their families on Alzheimer’s disease, other dementias. www.gmhfonline.org/gmhf/consumer/alzheimers.html.
- Tacrine • Cognex
- Donepezil • Aricept
- Rivastigmine • Exelon
- Galantamine • Razadyne (was Reminyl)
Drs. Kamat and LeFevre report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Boehringer Ingelheim Pharmaceuticals, Cyberonics, Eli Lilly and Co., Eunoe, Forest Pharmaceuticals, Novartis Pharmaceuticals Corp., Pfizer, and Wyeth Pharmaceuticals. He is a consultant to AstraZeneca Pharmaceuticals, Forest Pharmaceuticals, Janssen Pharmaceutica, KV Pharma, Novartis Pharmaceuticals Corp., Organon International, and Sanofi-Synthelabo.
Acknowledgment
The authors thank Anjali Baliga, MD, for her contribution and help in preparing this article
Using a cholinesterase inhibitor (ChEI) makes sense for any disorder with a significant cholinergic deficit, such as Alzheimer’s disease (AD) and other forms of mild-to-moderate dementia (Box 1).1-3 Yet the ChEIs tacrine, donepezil, rivastigmine, and galantamine have pharmacologic differences, and individual patients respond differently to them.
To help you choose the safest, most effective treatment for each patient, we discuss:
- three cases that show how ChEIs differ in mechanism of action, administration, and side effects
- evidence of ChEIs’ efficacy in AD—for which they are approved—and in other dementias for which they have been tried
- when to switch agents, and how long to continue treatment.
Probable Alzheimer’s disease (AD) accounts for 64% of all dementias in the United States. Less-common causes include:
- vascular dementia (5%)
- combined vascular dementia and AD (10%)
- probable dementia with Lewy bodies, Parkinson’s dementia, or diffuse Lewy body disease (9%)
- Lewy body variant of AD, or AD and dementia with Lewy bodies (6%)
- frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, or Creutzfeldt-Jakob disease (6%).2,3
In our experience, many primary care physicians choose to follow their patients with dementia, even when clinical features are atypical or suggest unusual causes. Psychiatrists are asked most often to assist in diagnosis and management of patients with:
- uncommon dementias, including frontotemporal dementia or dementia with Lewy bodies
- rapidly progressive dementia
- dementia in a patient age
- dementia with psychiatric comorbidities or severe behavior disturbances.4
How Cheis Differ
Although dementia remains incurable, recognizing cognitive decline early allows you to start ChEI therapy before substantial neuronal loss occurs (Box 2).3,4 The goal of early treatment is to improve or stabilize cognition, behavior, and activities of daily living for as long as possible.
In comparison studies,5,6 ChEIs have shown differences in tolerability but not consistent differences in efficacy for mild to moderate AD—though these studies had methodologic limitations. Because the agents appear similarly effective, the initial ChEI choice often depends on how their differences might benefit your patient (Table 1). Consider the following cases:
An early dementia diagnosis enables you educate the patient and family (Box 3) and begin the most effective treatment for the person with cognitive decline. Although dementia remains incurable, early recognition presents the opportunity to start cholinesterase inhibitors before substantial neuronal loss occurs.3,4
Patient workup. The Alzheimer’s Association offers online information for health care professionals on AD diagnosis and treatment protocols (see Related resources). A detailed history, physical examination, and Mini-Mental State Examination (MMSE) are necessary if you suspect Alzheimer’s or a related dementia.
Also recommended are a comprehensive metabolic screen, complete blood counts with differential, urine analysis, serum B12 and folate studies, homocysteine levels, thyroid studies, chest radiography, ECG, lipid profile, and brain scan (MRI or CT). Perform studies such as the rapid plasma reagin test for syphilis and HIV testing as appropriate.
Similarities and differences among cholinesterase inhibitors
| Tacrine | Donepezil | Rivastigmine | Galantamine | |
|---|---|---|---|---|
| Administration | Four times daily | Once daily | Twice daily with full meals | Once daily (extended-release formulation) |
| AChE inhibitor | Yes | Yes | Yes | Yes |
| BuChE inhibitor | Yes | No | Yes | No |
| Allosteric modulation of nicotinic receptor | No | Yes | No | Yes |
| Pharmacodynamic nicotinic/muscarinic effect | Yes | Yes | Yes | Yes |
| GI side effects | Present | Present | Present | Present |
| Hepatotoxicity | Present | Absent | Absent | Absent |
| Metabolism | CYP-450 | CYP-450 | Autohydrolysis | CYP-450 |
| Drug–drug interactions | Yes | Yes | None reported | Yes |
| AChE: acetylcholinesterase | ||||
| BuChE: butyrylcholinesterase | ||||
| CYP-450: cytochrome P-450 hepatic isoenzymes | ||||
Case 1: Gradual Memory Loss
Mrs. J, age 76, has experienced a slow, insidious memory decline across 5 years. She has become socially withdrawn and shows some language difficulties. She has had peptic ulcer disease and often does not take medications as prescribed.
Her psychiatrist diagnoses probable AD and chooses donepezil with its easy dosing schedule because of Mrs. J’s history of nonadherence. Donepezil’s GI tolerability is also a factor in this choice because of the patient’s peptic ulcer disease.
Case 2: Dementia And Motor Deficits
Mr. L, age 82, has gradually developed memory loss and parkinsonian symptoms, including slowness of movement and shuffling gait. He has visual hallucinations of people and episodic confusion. His medications include warfarin and digoxin for atrial fibrillation and congestive heart failure.
Mr. L is diagnosed with probable dementia with Lewy bodies. His psychiatrist chooses rivastigmine because it has shown efficacy in this type of dementia and is not known to interact significantly with cardiovascular medications.
Case 3: Stroke, Then Rapid Decline
Mrs. D, age 68, has a history of hypertension and suffered a stroke in the past. Her family says her memory and behavior—anger outbursts and excessive irritability—have worsened rapidly across 2 years. Examination reveals some focal neurologic deficits.
Her psychiatrist diagnoses probable vascular dementia and chooses galantamine for its efficacy in patients with this dementia type. Mrs. D has no history of GI illness and will likely tolerate the drug’s GI side effects. Follow-up care will include monitoring for tolerability.
Mechanism. Donepezil inhibits the enzyme acetylcholinesterase, and rivastigmine inhibits acetylcholinesterase and butyrylcholinesterase. Galantamine inhibits acetylcholinesterase and shows allosteric modulation of the presynaptic nicotinic receptor.
Data indicating that rivastigmine is particularly effective in patients with rapidly progressive illness is consistent with the possible advantage of inhibiting both butyrylcholinesterase and acetylcholinesterase. It has been argued that galantamine’s binding to nicotinic receptors modulates their function, which may enhance acetylcholine release.
Among the three agents, only rivastigmine shows a consistent, linear dose-response relationship. It is rapidly and extensively metabolized, primarily via cholinesterase-mediated hydrolysis to the decarbamylated metabolite (autohydrolysis). Minimal metabolism occurs via the major cytochrome P (CYP)-450 isoenzymes. Donepezil and galantamine are metabolized by isoenzymes 2D6 and 3A4 and undergo glucuronidation.7
Drug interactions. Because rivastigmine avoids hepatic metabolism, interactions with drugs metabolized by CYP-450 isoenzymes have not been reported.8
Donepezil interacts with ketoconazole and quinidine, which inhibit donepezil metabolism and increase mean donepezil concentrations. Galantamine interacts with ketoconazole, paroxetine, and erythromycin, which increase mean galantamine concentrations.9
Administration. Donepezil and extended-release galantamine are given once daily because of their long half-lives, whereas regular galantamine and rivastigmine are taken twice daily with meals to minimize GI effects (Table 2). Nausea and vomiting can occur with any of the ChEIs but are more common and troublesome with rivastigmine and galantamine.
Table 2
How to use cholinesterase inhibitors for patients with dementia
| Drug | Recommended dosing | Possible side effects | Titration | Administration |
|---|---|---|---|---|
| Tacrine | Initial: 40 mg/d Maximum: 160 mg/d | Liver damage causing increase in ALT levels, GI effects (nausea, indigestion, vomiting, diarrhea, abdominal pain), skin rash | Dosage can be increased every 4 weeks | Divide into four doses; take on empty stomach |
| Donepezil | Initial: 5 mg/d Maximum: 10 mg/d | GI effects (nausea, diarrhea, vomiting, loss of appetite), insomnia, muscle cramps, fatigue | Increase dosage after 4 weeks | Once daily in morning or at bedtime |
| Rivastigmine | Initial: 3 mg/d Maximum: 12 mg/d | GI effects (nausea, vomiting, loss of appetite, weight loss, diarrhea, heartburn) | Increase dosage every 4 weeks | Twice daily after meals |
| Galantamine (regular, ER) | Initial: 8 mg/d Maximum: 24 mg/d | GI effects (nausea, vomiting, diarrhea, weight loss), possible increased mortality risk in patients with MCI | Increase dosage every 4 weeks | Regular: Twice daily after meals ER: Once daily after a meal |
| ALT: alanine transferase | ||||
| ER: extended-release formulation | ||||
| MCI: mild cognitive impairment | ||||
Efficacy In Early AD
In controlled clinical trials, all four ChEIs have significantly improved cognition, behavior, and activities of daily living in patients with mild-to-moderate AD.10-12 Tacrine—the first FDA-approved ChEI—is rarely used because its associated hepatoxicity requires ongoing liver enzyme monitoring.13 Among the other three:
Donepezil. A review of 16 trials involving 4,365 participants10 found significant benefits in cognitive functioning, activities of daily living, and behavior in persons with mild, moderate, or severe AD who were treated with donepezil for 12, 24, or 52 weeks.
Rivastigmine improved or maintained cognitive function, activities of daily living, and behavior for up to 52 weeks in patients with mild to moderate AD, according to a review of studies from 1995 to 2002.11 GI irritation was the most common adverse effect. Giving rivastigmine for up to 2 years may reduce the cost of caring for patients with AD, mostly by delaying nursing home placement.
Galantamine has beneficial effects on cognition, global function, activities of daily living, and behavior in patients with AD, vascular dementia, and AD with cerebrovascular components, according to a review of clinical studies.12 Adverse events are generally mild to moderate, transient, and gastrointestinal.
Efficacy In Other Dementias
In addition to their FDA-approved use for mildto-moderate AD, ChEIs also have been studied in persons with other types of dementia and mild cognitive impairment (MCI).
Dementia with Lewy bodies. Rivastigmine given with flexible titration from 6 to 12 mg/d improved behavior in 120 patients with Lewy body dementia.14 In the double-blind, multicenter study, patients taking rivastigmine, mean 9.7 mg/d for 20 weeks, were less apathetic and anxious and had fewer delusions and hallucinations than did those taking placebo. The drug was judged to be safe and well tolerated.
Vascular dementia. Patients with vascular dementia showed improved cognition and global function when treated with donepezil, 5 or 10 mg/d, for up to 24 weeks. Donepezil was well tolerated in this combined analysis of two randomized, placebo-controlled trials.15
Kumar et al16 compared two rivastigmine dosages in patients with mild-to-moderate AD, some of whom also had vascular dementia risk factors. Patients were randomly assigned to placebo, low-dose rivastigmine (1 to 4 mg/d), or high-dose rivastigmine (6 to 12 mg/d) for 26 weeks. Cognition, activities of daily living, and disease severity improved with rivastigmine in patients with or without vascular risk factors. Greater benefit was seen with high-dose than low-dose rivastigmine and in patients with AD plus vascular risk factors than in those with AD alone.
In a multicenter, double-blind trial,17 patients with vascular dementia or AD with vascular risk factors received galantamine, up to 24 mg/d, or placebo for 6 months. Compared with controls, those taking galantamine showed improved cognition, behavior, and function. The drug overall was well tolerated, with nausea and vomiting the most common side effects.
Parkinson’s dementia. Emre et al18 evaluated rivastigmine’s efficacy and safety in patients whose mild-to-moderate dementia developed at least 2 years after a clinical diagnosis of Parkinson’s disease (PD). Patients were randomly assigned to placebo or rivastigmine, 3 to 12 mg/d, for 24 weeks, and 410 of 541 enrollees completed the study. Compared with placebo, rivastigmine was associated with statistically significant improvements in cognition and global measures in dementia associated with PD but also with higher rates of nausea, vomiting, and tremor. PD’s motor symptoms did not change significantly in either group.
Mixed dementia states. As mentioned, galantamine improved cognitive and noncognitive abilities in patients with vascular dementia or AD with vascular risk factors in a 6-month, double-blind trial.17 Patients who received galantamine or placebo could then continue open-label galantamine, 24 mg/d, for another 6 months. In patients treated the full 12 months, galantamine continued to improve or maintain:
- cognition, based on Alzheimer’s Disease Assessment Scale-cognitive subscale scores
- functional ability, measured by the 40-item Disability Assessment for Dementia
- behavior, measured by the Neuro-psychiatric Inventory.19
After 12 months, the rivastigmine-treated patients were less behaviorally impaired than the matched patients, and their caregivers reported reduced stress. Rivastigmine did not prevent cognitive deterioration, as assessed with the Mini-Mental State Examination (MMSE).
Mild cognitive impairment. Persons with MCI have objective psychometric evidence of memory loss compared with their peers, but they are not significantly impaired in activities of daily living or other cognitive functions (language, abstract thinking, or problem-solving).
At this time, we do not recommend using ChEIs to treat MCI. These agents have shown little benefit and potential risk in patients who do not meet diagnostic criteria for dementia:
- Salloway et al21 tested donepezil’s efficacy and safety in 270 patients with MCI in a 24-week, double-blind, placebo-controlled trial. Donepezil was started at 5 mg/d for 42 days, then escalated to 10 mg/d. Compared with placebo, donepezil showed no significant effects on recall, but some improvements were seen in attention and psychomotor speed.
- In two unpublished placebo-controlled trials, galantamine did not improve memory when given for 2 years to elderly patients with MCI. A precaution was added to the drug’s prescribing information because 13 of the 1,026 patients taking galantamine died, compared with 1 of 1,022 taking placebo. Vascular disease caused one-half of the galantamine group deaths. No evidence of increased mortality risk has been seen in studies of galantamine in patients with mild-to-moderate AD, for which it is indicated.
Getting The Greatest Response
To gauge response to ChEI therapy, family reports about the patient are helpful—such as that cognition has improved or cognitive decline has not progressed as rapidly as before. Assessment tools such as the MMSE can document improvement or stabilization.
We recommend trying an initial ChEI for at least 6 months to determine its efficacy. If your patient cannot tolerate one ChEI or fails to respond to initial treatment, two consensus panels22,23 recommend that you consider changing ChEIs:
- If switching because of intolerable side effects, wait at least 2 to 3 days after stopping the first ChEI before starting another.
- If switching because of poor response, you can start a different ChEI immediately after the first one is stopped.
- Cholinesterase inhibitors may help improve or stabilize cognition, behavior, and/or activities of daily living
- Persons receiving these agents may decline more slowly than those who have not been treated
- Common side effects include nausea, vomiting, diarrhea, and loss of appetite
- Other less-common side effects are muscle cramps, slowed heart rate, dizziness, and fainting
- Because of differences in these agents, it may make sense to switch to another cholinesterase inhibitor if the patient has intolerable side effects or does not improve with the first one tried
Related resources
- Alzheimer’s Association. Information for health care professionals:
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- • Treating cognitive symptoms www.alz.org/Health/Treating/symptoms.asp
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- American Association for Geriatric Psychiatry. Information for older patients and their families on Alzheimer’s disease, other dementias. www.gmhfonline.org/gmhf/consumer/alzheimers.html.
- Tacrine • Cognex
- Donepezil • Aricept
- Rivastigmine • Exelon
- Galantamine • Razadyne (was Reminyl)
Drs. Kamat and LeFevre report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Boehringer Ingelheim Pharmaceuticals, Cyberonics, Eli Lilly and Co., Eunoe, Forest Pharmaceuticals, Novartis Pharmaceuticals Corp., Pfizer, and Wyeth Pharmaceuticals. He is a consultant to AstraZeneca Pharmaceuticals, Forest Pharmaceuticals, Janssen Pharmaceutica, KV Pharma, Novartis Pharmaceuticals Corp., Organon International, and Sanofi-Synthelabo.
Acknowledgment
The authors thank Anjali Baliga, MD, for her contribution and help in preparing this article
1. Small GW, Rabins PV, Barry PP, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA 1997;278(16):1363-71.
2. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 suppl 5):S4-S9.
3. Grossberg GT, Lake JT. The role of the psychiatrist in Alzheimer’s disease. J Clin Psychiatry 1998;59(suppl 9):3-6.
4. Doraiswamy PM, Steffens DC, Pitchumoni S, Tabrizi S. Early recognition of Alzheimer’s disease: what is consensual? What is controversial? What is practical? J Clin Psychiatry 1998;59(suppl 13):6-18.
5. Wilkinson DG, Passmore AP, Bullock R, et al. A multinational, randomised, 12-week, comparative study of donepezil and rivastigmine in patients with mild to moderate Alzheimer’s disease. Int J Clin Pract 2002;56(6):441-6
6. Jones RW, Soininen H, Hager K, et al. A multinational, randomised, 12-week study comparing the effects of donepezil and galantamine in patients with mild to moderate Alzheimer’s disease. Int J Geriatr Psychiatry 2004;19(1):58-67.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twentytwo classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. U. S. Bureau of the Census. 2004 International database: Midyear population, by age and sex. Table 094. U.S. Bureau of the Census; 2004.
9. Reminyl (galantamine HBr). Physicians’ desk reference (59th ed). Montvale, NJ: Thomson PDR; 2005:1739.
10. Birks JS, Harvey R. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev 2003;(3):CD001190.-
11. Williams BR, Nazarians A, Gill MA. A review of rivastigmine: a reversible cholinesterase inhibitor. Clin Ther 2003;25(6):1634-53.
12. Corey-Bloom J. Galantamine: a review of its use in Alzheimer’s disease and vascular dementia. Int J Clin Pract 2003;57(3):219-23.
13. Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994;271(13):992-8.
14. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000;356(9247):2031-6.
15. Passmore AP, Bayer AJ, Steinhagen-Thiessen E. Cognitive, global, and functional benefits of donepezil in Alzheimer’s disease and vascular dementia: results from large-scale clinical trials. J Neurol Sci 2005;229-30:141-6.
16. Kumar V, Anand R, Messina J, et al. An efficacy and safety analysis of rivastigmine in Alzheimer’s disease patients with concurrent vascular risk factors. Eur J Neurol 2000;7(2):159-69.
17. Kurz AF, Erkinjuntti T, Gauthier S, et al. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002;359(9314):1283-90.
18. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351(24):2509-18.
19. Erkinjuntti T, Kurz A, Small GW, et al. An open-label extension trial of galantamine in patients with probable vascular dementia and mixed dementia. Clin Ther 2003;25(6):1765-82.
20. Moretti R, Torre P, Antonello RM, et al. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004;21(14):931-7.
21. Salloway S, Ferris S, Kluger A, et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology 2004;63(4):651-7.
22. Emre M. Switching cholinesterase inhibitors in patients with Alzheimer’s disease. Int J Clin Pract Suppl 2002;(127):64-72.
23. Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int J Clin Pract Suppl 2002;(127):45-63.
1. Small GW, Rabins PV, Barry PP, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA 1997;278(16):1363-71.
2. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 suppl 5):S4-S9.
3. Grossberg GT, Lake JT. The role of the psychiatrist in Alzheimer’s disease. J Clin Psychiatry 1998;59(suppl 9):3-6.
4. Doraiswamy PM, Steffens DC, Pitchumoni S, Tabrizi S. Early recognition of Alzheimer’s disease: what is consensual? What is controversial? What is practical? J Clin Psychiatry 1998;59(suppl 13):6-18.
5. Wilkinson DG, Passmore AP, Bullock R, et al. A multinational, randomised, 12-week, comparative study of donepezil and rivastigmine in patients with mild to moderate Alzheimer’s disease. Int J Clin Pract 2002;56(6):441-6
6. Jones RW, Soininen H, Hager K, et al. A multinational, randomised, 12-week study comparing the effects of donepezil and galantamine in patients with mild to moderate Alzheimer’s disease. Int J Geriatr Psychiatry 2004;19(1):58-67.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twentytwo classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. U. S. Bureau of the Census. 2004 International database: Midyear population, by age and sex. Table 094. U.S. Bureau of the Census; 2004.
9. Reminyl (galantamine HBr). Physicians’ desk reference (59th ed). Montvale, NJ: Thomson PDR; 2005:1739.
10. Birks JS, Harvey R. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev 2003;(3):CD001190.-
11. Williams BR, Nazarians A, Gill MA. A review of rivastigmine: a reversible cholinesterase inhibitor. Clin Ther 2003;25(6):1634-53.
12. Corey-Bloom J. Galantamine: a review of its use in Alzheimer’s disease and vascular dementia. Int J Clin Pract 2003;57(3):219-23.
13. Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994;271(13):992-8.
14. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000;356(9247):2031-6.
15. Passmore AP, Bayer AJ, Steinhagen-Thiessen E. Cognitive, global, and functional benefits of donepezil in Alzheimer’s disease and vascular dementia: results from large-scale clinical trials. J Neurol Sci 2005;229-30:141-6.
16. Kumar V, Anand R, Messina J, et al. An efficacy and safety analysis of rivastigmine in Alzheimer’s disease patients with concurrent vascular risk factors. Eur J Neurol 2000;7(2):159-69.
17. Kurz AF, Erkinjuntti T, Gauthier S, et al. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002;359(9314):1283-90.
18. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351(24):2509-18.
19. Erkinjuntti T, Kurz A, Small GW, et al. An open-label extension trial of galantamine in patients with probable vascular dementia and mixed dementia. Clin Ther 2003;25(6):1765-82.
20. Moretti R, Torre P, Antonello RM, et al. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004;21(14):931-7.
21. Salloway S, Ferris S, Kluger A, et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology 2004;63(4):651-7.
22. Emre M. Switching cholinesterase inhibitors in patients with Alzheimer’s disease. Int J Clin Pract Suppl 2002;(127):64-72.
23. Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int J Clin Pract Suppl 2002;(127):45-63.