User login
A change of heart
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
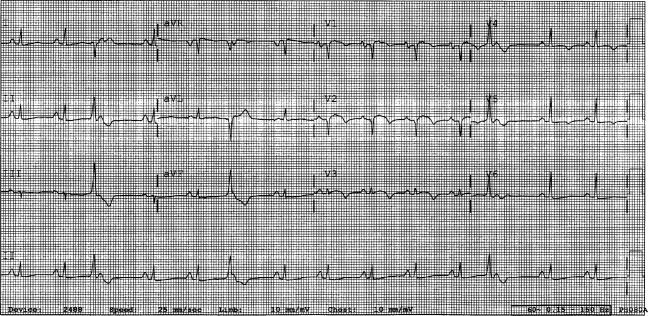
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
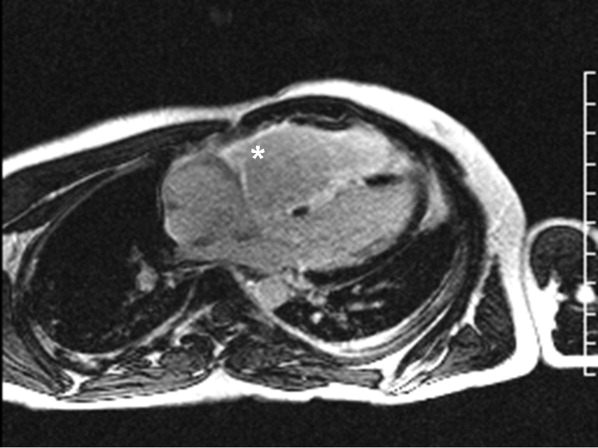
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
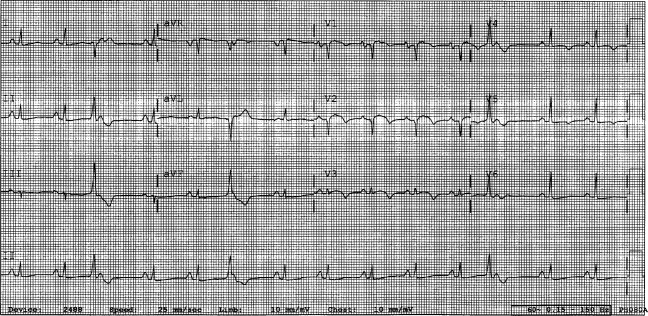
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
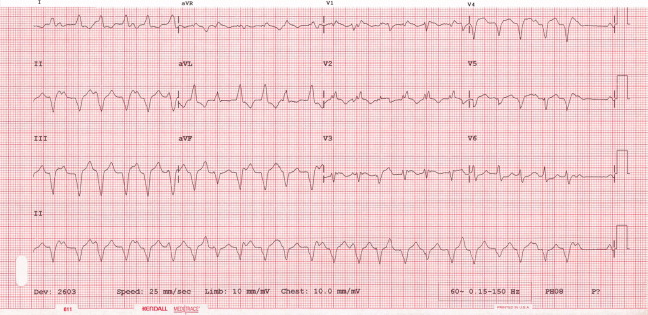
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
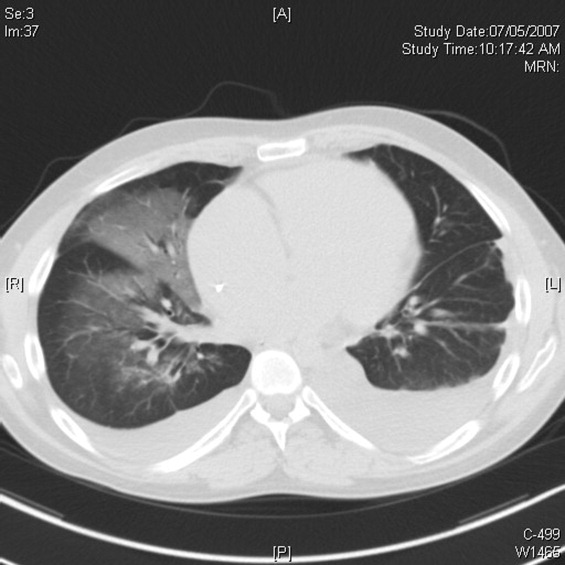
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
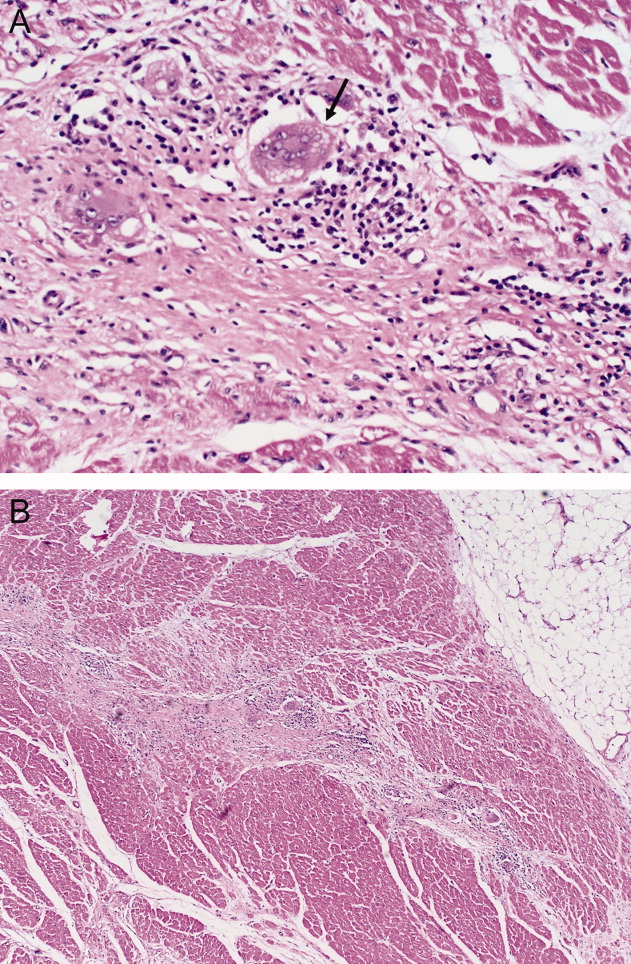
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.
Diagnosis of Exclusion
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
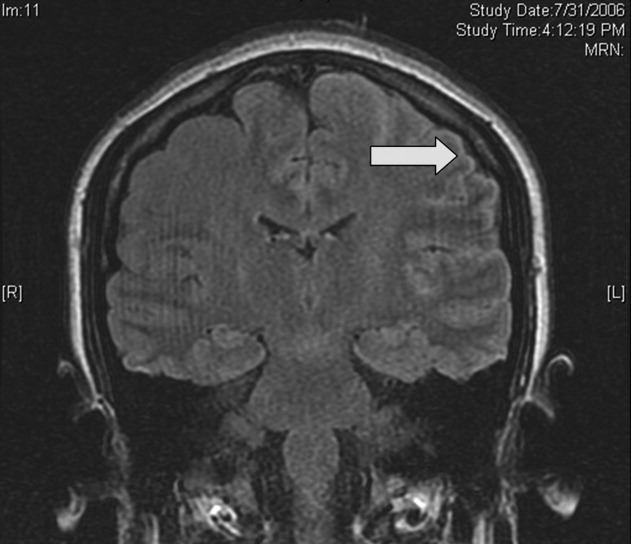
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
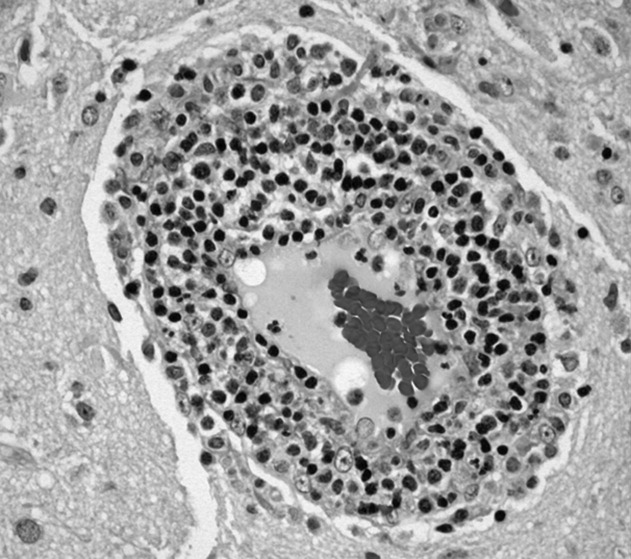
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
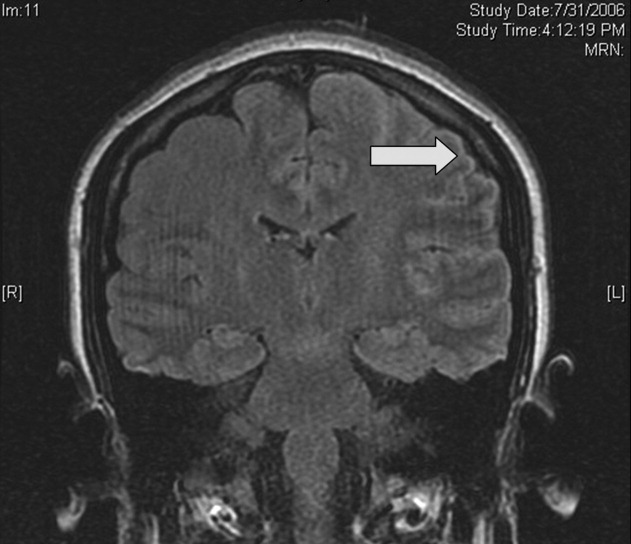
Thinking Inside the Box
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
A Rash Decision
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
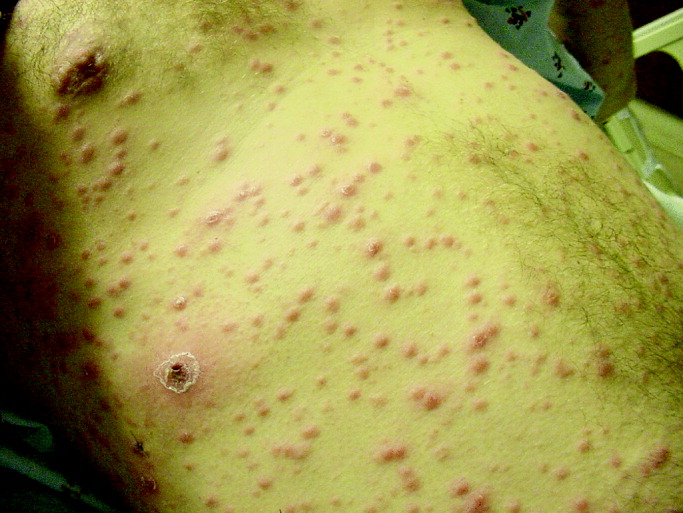
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
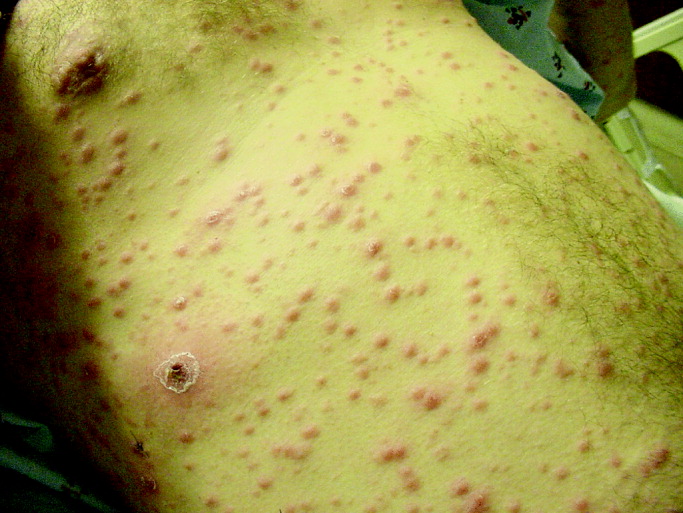
Clinical Conundrum
A 62‐year‐old man with psoriasis for more than 30 years presented to the emergency department with a scaly, pruritic rash involving his face, trunk, and extremities that he had had for the past 10 days. The rash was spreading and not responding to application of clobetasol ointment, which had helped his psoriasis in the past. He also reported mild pharyngitis, headache, and myalgias.
A patient with a chronic skin condition presenting with a new rash means the clinician must consider whether it is an alternative manifestation of the chronic disorder or a new illness. Psoriasis takes many forms including guttate psoriasis, which presents with small, droplike plaques and frequently follows respiratory infections (particularly those caused by Streptococcus). Well‐controlled psoriasis rarely transforms after 3 decades, so I would consider other conditions. The tempo of illness makes certain life‐threatening syndromes, including Stevens‐Johnson, toxic shock, and purpura fulminans, unlikely. An allergic reaction, atopic dermatitis, or medication reaction is possible. Infections, either systemic (eg, syphilis) or dermatologic (eg, scabies), should be considered. Photosensitivity could involve the sun‐exposed areas, such as the extremities and face. Seborrheic dermatitis can cause scaling lesions of the face and trunk but not the extremities. Vasculitis merits consideration, but dependent regions are typically affected more than the head. Mycosis fungoides or a paraneoplastic phenomenon could cause a diffuse rash in this age group.
The patient had diabetes mellitus, hypertension, diverticulosis, and depression. Three months earlier he had undergone surgical drainage of a perirectal abscess. His usual medications were lovastatin, paroxetine, insulin, hydrochlorothiazide, and lisinopril. Three weeks previously he had completed a 10‐day course of trimethoprim/sulfamethoxazole for an upper respiratory infection. Otherwise, he was taking no new medications. He was allergic to penicillin. He denied substance abuse, recent travel, or risk factors for human immunodeficiency virus (HIV) infection. He worked as an automobile painter, lived with his wife, and had a pet dog.
Physical examination revealed a well‐appearing man with normal vital signs. His skin had well‐defined circumscribed pink plaques, mostly 1‐2 cm in size, with thick, silvery scales in the ears and on the dorsal and ventral arms and legs, chest, back, face, and scalp. There were no pustules or other signs of infection (Figs. 1and 2). The nails exhibited distal onycholysis, oil spots, and rare pits. His posterior pharynx was mildly erythematous. The results of cardiovascular, pulmonary, and abdominal examinations were normal.

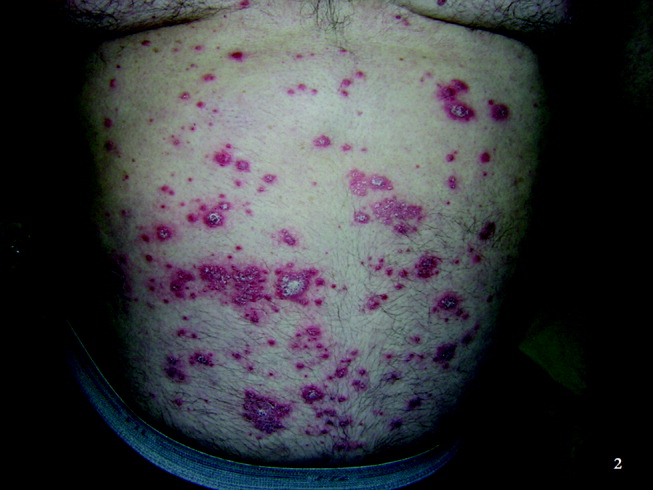
Although other scaling skin conditions such as eczema, irritant dermatitis, or malignancy remain possible, his rash is most consistent with widespread psoriasis. I would consider immunological changes that may have caused a remarkably altered and more severe expression of his chronic disease, for example, recent steroid therapy or HIV infection. The company a rash keeps helps frame the differential diagnosis. Based on the patient's well appearance, the time course, his minimal systemic symptoms, and the appearance of the rash, my leading considerations are psoriasis or an allergic dermatitis. Cutaneous T‐cell malignancy, with its indolent and sometimes protean manifestations, remains possible in a patient of his age. I would now consult a dermatologist for 3 reasons: this patient has a chronic disease that I do not manage beyond basic treatments (eg, topical steroids), he has an undiagnosed illness with substantial dermatologic manifestations, and he may need a skin biopsy for definitive diagnosis.
The dermatology team diagnosed a guttate psoriasis flare, possibly associated with streptococcal pharyngitis. The differential diagnosis included secondary syphilis, although the team believed this was less likely. The dermatology team recommended obtaining a throat culture, streptozyme assay, and rapid plasma reagin and prescribed oral erythromycin and topical steroid ointment under a sauna suit.
I would follow his response to the prescribed steroid treatments. If the patient's course deviates from the dermatologists' expectations, I would request a skin biopsy and undertake further evaluations in search of an underlying systemic disease.
The patient followed up in the dermatology clinic 3 weeks later. His rash had worsened, and he had developed patchy alopecia and progressive edema of the face, ears, and eyes. He denied mouth or tongue swelling, difficulty breathing, or hives. The streptozyme assay was positive, but the other laboratory test results were negative.
The dermatology team diagnosed a severely inflammatory psoriasis flare and prescribed an oral retinoid, acitretin, and referred him for ultraviolet light therapy. He was unable to travel for phototherapy, and acitretin was discontinued after 1 week because of elevated serum transaminase levels. The dermatologists then prescribed oral cyclosporine.
The progression of disease despite standard treatment suggests a nonpsoriatic condition. Although medications could cause the abnormal liver tests, so could another underlying illness that involves the liver. An infiltrative disorder of the skin with hair follicle destruction and local lymphedema could explain both alopecia and facial edema.
I am unable account for his clinical features with a single disease, so the differential remains broad, including severe psoriasis, an infiltrating cutaneous malignancy, or a toxic exposure. Arsenic poisoning causes hyperkeratotic skin lesions, although he lacks the associated gastrointestinal and neurological symptoms. I would not have added the potentially toxic cyclosporine.
When he returned to dermatology clinic 1 week later, his rash and facial swelling had worsened. He also reported muscle and joint aches, fatigue, lightheadedness, anorexia, nausea, abdominal pain, diarrhea, and dyspnea on exertion. He denied fever, chills, and night sweats.
He appeared ill and used a cane to arise and walk. His vital signs and oxygen saturation were normal. He had marked swelling of his face, diffuse erythema and swelling on the chest, and widespread scaly, erythematous plaques (Fig. 3). The proximal nail folds of his fingers were erythematous, with ragged cuticles. His abdomen was mildly distended, but the rest of the physical examination was normal.
He has become too systemically ill to attribute his condition to psoriasis. The nail findings suggest dermatomyositis, which could explain many of his findings. The diffuse erythema and his difficulty walking are consistent with its skin and muscle involvement. Dyspnea could be explained by dermatomyositis‐associated interstitial lung disease. A dermatomyositis‐associated hematological or solid malignancy could account for his multisystem ailments and functional decline. A point against dermatomyositis is the relatively explosive onset of his disease. He should be carefully examined for any motor weakness. With his progressive erythroderma, I am also concerned about an advancing cutaneous T‐cell lymphoma (with leukemic transformation).
Blood tests revealed the following values: white‐blood‐cell count, 8700/L; hematocrit, 46%; platelet count, 172,000/L; blood urea nitrogen, 26 mg/dL; creatinine, 1.0 mg/dL; glucose, 199 mg/dL; albumin, 3.1 g/dL; alkaline phosphatase, 172 U/L (normal range 45‐129); alanine aminotransferase, 75 U/L (normal range 0‐39 U/L); aspartate aminotransferase, 263 U/L (normal range 0‐37 U/L); total bilirubin, 1.1 mg/dL; prothrombin time, 16 seconds (normal range 11.7‐14.3 seconds), and serum creatinine, kinase, 4253 U/L (normal range 0‐194 U/L). HIV serology was negative. Urinalysis revealed trace protein. The results of chest radiographs and an electrocardiogram were normal.
The liver function tests results are consistent with medication effects or liver involvement in a systemic disease. The creatinine kinase elevation is consistent with a myopathy such as dermatomyositis. A skin biopsy would still be useful. Depending on those results, he may need a muscle biopsy, urine heavy metal testing, and computed tomography body imaging. Considering his transaminase and creatinine kinase elevations, I would discontinue lovastatin.
The patient was hospitalized. Further questioning revealed that he had typical Raynaud's phenomenon and odynophagia. A detailed neurological examination showed weakness (3/5) of the triceps and iliopsoas muscles and difficulty rising from a chair without using his arms. Dermatoscopic examination of the proximal nail folds showed dilated capillary loops and foci of hemorrhage.
Blood tests showed a lactate dehydrogenase level of 456 U/L (normal range 0‐249 U/L) and an aldolase of 38 U/L (normal range 1.2‐7.6 U/L). Tests for antinuclear antibodies, anti‐Jo antibody, and antimyeloperoxidase antibodies were negative. Two skin biopsies were interpreted by general pathology as consistent with partially treated psoriasis, whereas another showed nonspecific changes with minimal superficial perivascular lymphohistiocytic inflammation (Fig. 4). Lisinopril was discontinued because of its possible contribution to the facial edema.
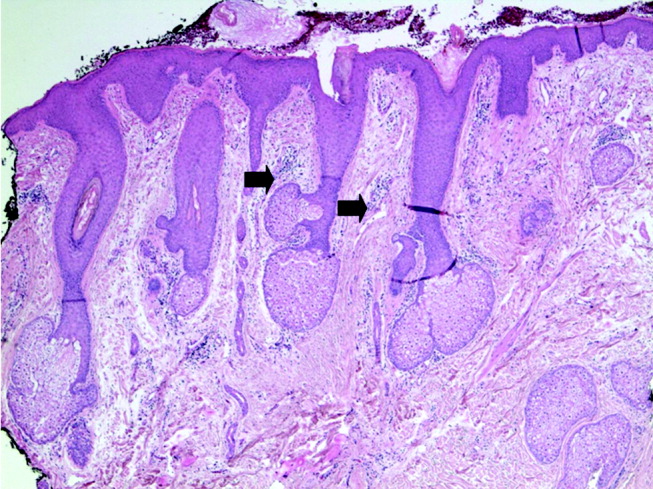
Dermatomyositis is now the leading diagnosis. Characteristic features include his proximal muscle weakness, Raynaud's phenomenon, and dilated nailfold capillary loops. I am not overly dissuaded by the negative antinuclear antibodies, but because of additional atypical features (ie, extensive cutaneous edema, rapid onset, illness severity, prominent gastrointestinal symptoms), a confirmatory muscle biopsy is needed. Endoscopy of the proximal aerodigestive tract would help evaluate the odynophagia. There is little to suggest infection, malignancy, or metabolic derangement.
The inpatient medical team considered myositis related to retinoid or cyclosporine therapy. They discontinued cyclosporine and began systemic corticosteroid therapy. Within a few days, the patient's rash, muscle pain, and weakness improved, and the elevated transaminase and creatinine kinase levels decreased.
Dermatology recommended an evaluation for dermatomyositis‐associated malignancy, but the medicine team and rheumatology consultants, noting the lack of classic skin findings (heliotrope rash and Gottron's papules) and the uncharacteristically rapid onset and improvement of myositis, suggested delaying the evaluation until dermatomyositis was proven.
An immediate improvement in symptoms with steroids is nonspecific, often occurring in autoimmune, infectious, and neoplastic diseases. This juncture in the case is common in complex multisystem illnesses, where various consultants may arrive at differing conclusions. With both typical and atypical features of dermatomyositis, where should one set the therapeutic threshold, that is, the point where one ends testing, accepts a diagnosis, and initiates treatment? Several factors raise the level of certainty I would require. First, dermatomyositis is quite rare. Adding atypical features further increases the burden of proof for that illness. Second, the existence of alternative possibilities (admittedly of equal uncertainty) gives me some pause. Finally, the toxicity of the proposed treatments raises the therapeutic threshold. Acknowledging that empiric treatment may be indicated for a severely ill patient at a lower level of certainty, I would hesitate to commit a patient to long‐term steroids without being confident of the diagnosis. I would therefore require a muscle biopsy, or at least electromyography to support or exclude dermatomyositis.
The patient was discharged from the hospital on high‐dose prednisone. He underwent electromyography, which revealed inflammatory myopathic changes more apparent in the proximal than distal muscles. These findings were thought to be compatible with dermatomyositis, although the fibrillations and positive sharp waves characteristic of acute inflammation were absent, perhaps because of corticosteroid therapy.
The patient mistakenly stopped taking his prednisone. Within days, his weakness and skin rash worsened, and he developed nausea with vomiting. He returned to clinic, where his creatinine kinase level was again found to be elevated, and he was rehospitalized. Oral corticosteroid therapy was restarted with prompt improvement. On review of the original skin biopsies, a dermatopathologist observed areas of thickened dermal collagen and a superficial and deep perivascular lymphocytic infiltrate, both consistent with connective tissue disease.
These 3 additional findings (ie, electromyography results, temporally established steroid responsiveness, and the new skin biopsy interpretation) in aggregate support the diagnosis of dermatomyositis, but the nausea and vomiting are unusual. I would discuss these results with a rheumatologist and still request a confirmatory muscle biopsy. Because diagnosing dermatomyositis should prompt consideration of seeking an underlying malignancy in a patient of this age group, I would repeat a targeted history and physical examination along with age‐ and risk‐factor‐appropriate screening. If muscle biopsy results are not definitive, finding an underlying malignancy would lend support to dermatomyositis.
While hospitalized, the patient complained of continued odynophagia and was noted to have oral candidiasis. Upper endoscopy, undertaken to evaluate for esophageal candidiasis, revealed a mass at the gastroesophageal junction. Biopsy revealed gastric‐type adenocarcinoma. An abdominal computed tomography scan demonstrated 3 hypodense hepatic lesions, evidence of cirrhosis, and ascites. Cytology of paracentesis fluid revealed cells compatible with adenocarcinoma. The patient died in hospice care 2 weeks later.
At autopsy, he had metastatic gastric‐type adenocarcinoma. A muscle biopsy (Fig. 5) revealed muscle atrophy with small foci of lymphocytic infiltrates, most compatible with dermatomyositis. Another dermatopathologist reviewed the skin biopsies and noted interface dermatitis, which is typical of connective tissue diseases like dermatomyositis (Fig. 6A,B).
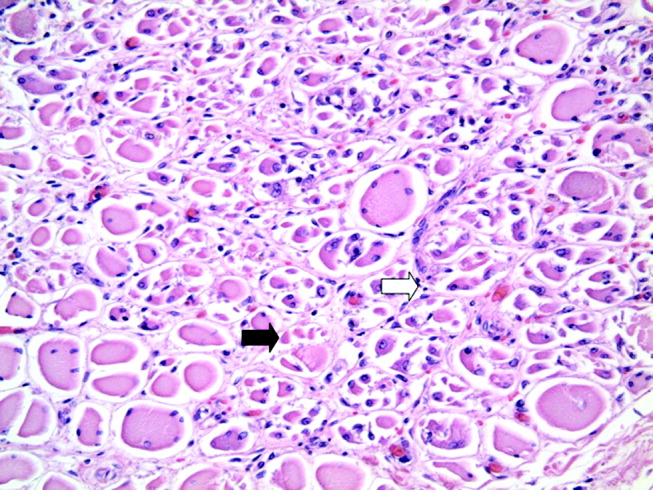
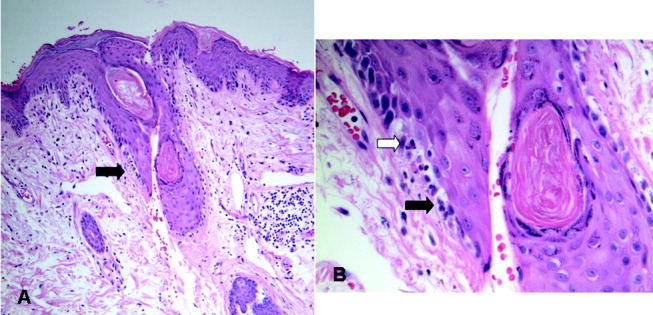
COMMENTARY
Dermatomyositis is an idiopathic inflammatory myopathy characterized by endomysial inflammation and muscle weakness and differentiated from other myopathies by the presence of a rash.1 Muscle disease may manifest with or precede the rash, but up to 40% of patients present with skin manifestations alone, an entity called amyopathic dermatomyositis.2 When present, the myositis generally develops over months, but the onset can be acute.1 The weakness is typically symmetrical and proximal,1 and many patients have oropharyngeal dysphagia.3
The characteristic rash is erythematous, symmetrical, and photodistributed.4 Classic cutaneous findings are the heliotrope rash (violaceous eyelid erythema), which is pathognomonic but uncommon, and the more common Gottron's papules (violaceous, slightly elevated papules and plaques on bony prominences and extensor surfaces, especially the knuckles).4 Other findings include periorbital edema, scalp dermatitis, poikiloderma (ie, hyperpigmentation, hypopigmentation, atrophy, and telangiectasia), periungual erythema, and dystrophic cuticles.2 The cutaneous manifestations of dermatomyositis may be similar to those of psoriasis, systemic lupus erythematosus, lichen planus, rosacea, polymorphous light eruption, drug eruption, atopic dermatitis, seborrheic dermatitis, or allergic contact dermatitis.4
Diagnosing dermatomyositis requires considering clinical, laboratory, electromyographical, and histological evidence, as there are no widely accepted, validated diagnostic criteria.1, 5 The diagnosis is usually suspected if there is a characteristic rash and symptoms of myositis (eg, proximal muscle weakness, myalgias, fatigue, or an inability to swallow). When the patient has an atypical rash, skin biopsy can differentiate dermatomyositis from other conditions, except lupus, which shares the key finding of interface dermatitis.2 The histological findings can be variable and subtle,6 so consultation with a dermatopathologist may be helpful.
Myositis may be confirmed by various studies. Most patients have elevated muscle enzymes (ie, creatinine kinase, aldolase, lactate dehydrogenase, or transaminases)1; for those who do not, magnetic resonance imaging can be helpful in detecting muscle involvement and locating the best site for muscle biopsy.7 Electromyography reveals nonspecific muscle membrane instability.8 Muscle biopsy shows muscle fiber necrosis, perifascicular atrophy, and perivascular and perifascicular lymphocytic infiltrates. These can be patchy, diminished by steroid use, and occasionally seen in noninflammatory muscular dystrophies.8 For a patient with typical myositis and a characteristic rash, muscle biopsy may be unnecessary.1
The clinical utility of serologic testing for diagnosing dermatomyositis is controversial.2 Myositis‐specific antibody testing is insensitive but specific; these antibodies include Jo‐1, an antisynthetase antibody that predicts incomplete response to therapy and lung involvement, and Mi‐2, which is associated with better response to therapy.2, 9, 10 The sensitivity and specificity of antinuclear antibodies are both approximately 60%.10
Patients with dermatomyositis have higher rates of cancers than age‐matched controls, and nearly 25% of patients are diagnosed with a malignancy at some point during the course of the disease.11 Malignancies are typically solid tumors that manifest within 3 years of the diagnosis,1214 although the increased risk may exist for at least 5 years.14 There is a 10‐fold higher risk of ovarian cancer in women with dermatomyositis.12, 15 Other associated malignancies include lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma.14
Recommendations for screening affected patients for cancer have changed over the years, with increasing evidence of an association between dermatomyositis and malignancy and evolving improvements in diagnostic techniques.16 Many authorities recommend that all adult patients with dermatomyositis be evaluated for cancer, including a complete physical examination, basic hematological tests, age‐ and sex‐appropriate screening (eg, mammography, pap smear, and colonoscopy), and chest x‐ray.16 Some would add upper endoscopy; imaging of the chest, abdomen, and pelvis; gynecological examination; and serum CA‐125 level to better evaluate for the most common malignancies (ie, ovarian, gastric, lung, and pancreatic carcinomas and non‐Hodgkins lymphoma).12, 1720
In 19% of adults, dermatomyositis overlaps with other autoimmune disorders, usually systemic lupus erythematosus and systemic sclerosis.21 These manifest as Raynaud's phenomenon, arthritis, esophageal dysmotility, renal disease, or neuropathy.21 Other potentially serious systemic manifestations of dermatomyositis include proximal dysphagia from pharyngeal myopathy; distal dysphagia from esophageal dysmotility in systemic sclerosis overlap; pulmonary disease from autoimmune interstitial lung disease or aspiration; cardiac disease from conduction abnormalities, myocarditis, pericarditis, and valvular disease; and rhabdomyolysis.2
Treatment of dermatomyositis requires systemic immunosuppression with 1 or more agents. The prognosis of dermatomyositis is variable. Mortality at 5 years ranges from 23% to 73%. At least a third of patients are left with mild to severe disability.1 In addition to older age, predictors of poor outcome include male sex, dysphagia, longstanding symptoms before treatment, pulmonary or cardiac involvement, and presence of antisynthetase antibodies.22
Dermatomyositis is often treated in the outpatient setting, but there are many reasons for hospitalization. Complications of treatment, like infection or adverse effects of medications, could result in hospitalization. Treatment with intravenous pulse corticosteroids or IVIG may require inpatient administration if no infusion center is available. Other indications for inpatient evaluation include the consequences of various malignancies and the more severe expression of systemic complications of dermatomyositis (eg, dysphagia and pulmonary, cardiac, or renal disease).
Every parent knows the plaintive backseat whine, Are we there, yet? Clinicians may also experience this feeling when attempting to diagnose a perplexing illness, especially one that lacks a definitive diagnostic test. It was easy for this patient's doctors to assume initially that his new rash was a manifestation of his long‐standing psoriasis. Having done so, they could understandably attribute the subsequent findings to either evolution of this disease or to consequences of the prescribed treatments, rather than considering a novel diagnosis. Only when faced with new (or newly appreciated) findings suggesting myopathy did the clinicians (and our discussant) consider the diagnosis of dermatomyositis. Even then, the primary inpatient medical team and their consultants were unsure when they had sufficient evidence to be certain.
Several factors compounded the difficulty of making a diagnosis in this case: the clinicians were dealing with a rare disease; they were considering alternative diagnoses (ie, psoriasis or a toxic effect of medication); and the disease presented somewhat atypically. The clinicians initially failed to consider and then accept the correct diagnosis because the patient's rash was not classic, his biopsy was interpreted as nonspecific, and he lacked myositis at presentation. Furthermore, when the generalists sought expert assistance, they encountered a difference of opinion among the consultants. These complex situations should goad the clinician into carefully considering the therapeutic threshold, that is, the transition point from diagnostic testing to therapeutic intervention.23 With complex cases like this, it may be difficult to know when one has reached a strongly supported diagnosis, and frequently asking whether we are there yet may be appropriate.
Take‐Home Points for the Hospitalist
-
A skin rash, which may have typical or atypical features, distinguishes dermatomyositis from other acquired myopathies.
-
Consider consultation with pathology specialists for skin and muscle biopsies.
-
Ovarian, lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma are the most common cancers associated with dermatomyositis.
-
In addition to age‐appropriate cancer screening, consider obtaining upper endoscopy, imaging of the chest/abdomen/pelvis, and CA‐125.
-
Patients with dermatomyositis and no obvious concurrent malignancy need long‐term outpatient follow‐up for repeated malignancy screening.
- ,.Polymyositis and dermatomyositis.Lancet.2003;362:971–982.
- .Dermatomyositis.Lancet.2000;355:53–47.
- ,,,.Oropharyngeal dysphagia in polymyositis/dermatomyositis.Clin Neurol Neurosurg.2004;107(1):32–37.
- ,.Skin involvement in dermatomyositis.Curr Opin Rheumatol.2003;15:714–22.
- ,,,,,.Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients.Medicine (Baltimore).2005;84:231–249.
- .Skin Pathology.2nd ed.New York:Churchill Livingstone;2002.
- ,.Utility of magnetic resonance imaging in the evaluation of patients with inflammatory myopathies.Curr Rheumatol Rep.2001;3:334–245.
- ,,.Is it really myositis? A consideration of the differential diagnosis.Curr Opin Rheumatol2004;16:684–691.
- .Idiopathic inflammatory myopathy: autoantibody update.Curr Rheumatol Rep.2002;4:434–441.
- ,,.Laboratory assessment in musculoskeletal disorders.Best Pract Res Clin Rheumatol.2003;17:475–494.
- ,.Dermatomyositis.Clin Dermatol.2006;24:363–373.
- ,,, et al.Frequency of specific cancer types in dermatomyositis and polymyositis: a population‐based study.Lancet.2001;357:96–100.
- ,,, et al.Cancer‐associated myositis: clinical features and prognostic signs.Ann N Y Acad Sci.2005;1051:64–71.
- ,,,,.Incidence of malignant disease in biopsy‐proven inflammatory myopathy. A population‐based cohort study.Ann Intern Med.2001;134:1087–1095.
- ,,.Risk of cancer in patients with dermatomyositis or polymyositis, and follow‐up implications: a Scottish population‐based cohort study.Br J Cancer.2001;85 (1):41–45.
- .When and how should the patient with dermatomyositis or amyopathic dermatomyositis be assessed for possible cancer?Arch Dermatol.2002;138:969–971.
- ,,.Ovarian cancer in patients with dermatomyositis.Medicine (Baltimore).1994;73(3):153–160.
- ,,,.Dermatomyositis sine myositis: association with malignancy.J Rheumatol.1996;23 (1):101–105.
- ,,, et al.Tumor antigen markers for the detection of solid cancers in inflammatory myopathies.Cancer Epidemiol Biomarkers Prev.2005;14:1279–1282.
- ,,, et al.Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients.Arch Dermatol.2002;138:885–890.
- ,,,,,.Dermatomyositis: a dermatology‐based case series.J Am Acad Dermatol.1998;38:397–404.
- ,,, et al.Long‐term outcome in polymyositis and dermatomyositis.Ann Rheum Dis.2006;65:1456–1461.
- .Our stubborn quest for diagnostic certainty. A cause of excessive testing.N Engl J Med.1989;320:1489–1491.
A 62‐year‐old man with psoriasis for more than 30 years presented to the emergency department with a scaly, pruritic rash involving his face, trunk, and extremities that he had had for the past 10 days. The rash was spreading and not responding to application of clobetasol ointment, which had helped his psoriasis in the past. He also reported mild pharyngitis, headache, and myalgias.
A patient with a chronic skin condition presenting with a new rash means the clinician must consider whether it is an alternative manifestation of the chronic disorder or a new illness. Psoriasis takes many forms including guttate psoriasis, which presents with small, droplike plaques and frequently follows respiratory infections (particularly those caused by Streptococcus). Well‐controlled psoriasis rarely transforms after 3 decades, so I would consider other conditions. The tempo of illness makes certain life‐threatening syndromes, including Stevens‐Johnson, toxic shock, and purpura fulminans, unlikely. An allergic reaction, atopic dermatitis, or medication reaction is possible. Infections, either systemic (eg, syphilis) or dermatologic (eg, scabies), should be considered. Photosensitivity could involve the sun‐exposed areas, such as the extremities and face. Seborrheic dermatitis can cause scaling lesions of the face and trunk but not the extremities. Vasculitis merits consideration, but dependent regions are typically affected more than the head. Mycosis fungoides or a paraneoplastic phenomenon could cause a diffuse rash in this age group.
The patient had diabetes mellitus, hypertension, diverticulosis, and depression. Three months earlier he had undergone surgical drainage of a perirectal abscess. His usual medications were lovastatin, paroxetine, insulin, hydrochlorothiazide, and lisinopril. Three weeks previously he had completed a 10‐day course of trimethoprim/sulfamethoxazole for an upper respiratory infection. Otherwise, he was taking no new medications. He was allergic to penicillin. He denied substance abuse, recent travel, or risk factors for human immunodeficiency virus (HIV) infection. He worked as an automobile painter, lived with his wife, and had a pet dog.
Physical examination revealed a well‐appearing man with normal vital signs. His skin had well‐defined circumscribed pink plaques, mostly 1‐2 cm in size, with thick, silvery scales in the ears and on the dorsal and ventral arms and legs, chest, back, face, and scalp. There were no pustules or other signs of infection (Figs. 1and 2). The nails exhibited distal onycholysis, oil spots, and rare pits. His posterior pharynx was mildly erythematous. The results of cardiovascular, pulmonary, and abdominal examinations were normal.
Although other scaling skin conditions such as eczema, irritant dermatitis, or malignancy remain possible, his rash is most consistent with widespread psoriasis. I would consider immunological changes that may have caused a remarkably altered and more severe expression of his chronic disease, for example, recent steroid therapy or HIV infection. The company a rash keeps helps frame the differential diagnosis. Based on the patient's well appearance, the time course, his minimal systemic symptoms, and the appearance of the rash, my leading considerations are psoriasis or an allergic dermatitis. Cutaneous T‐cell malignancy, with its indolent and sometimes protean manifestations, remains possible in a patient of his age. I would now consult a dermatologist for 3 reasons: this patient has a chronic disease that I do not manage beyond basic treatments (eg, topical steroids), he has an undiagnosed illness with substantial dermatologic manifestations, and he may need a skin biopsy for definitive diagnosis.
The dermatology team diagnosed a guttate psoriasis flare, possibly associated with streptococcal pharyngitis. The differential diagnosis included secondary syphilis, although the team believed this was less likely. The dermatology team recommended obtaining a throat culture, streptozyme assay, and rapid plasma reagin and prescribed oral erythromycin and topical steroid ointment under a sauna suit.
I would follow his response to the prescribed steroid treatments. If the patient's course deviates from the dermatologists' expectations, I would request a skin biopsy and undertake further evaluations in search of an underlying systemic disease.
The patient followed up in the dermatology clinic 3 weeks later. His rash had worsened, and he had developed patchy alopecia and progressive edema of the face, ears, and eyes. He denied mouth or tongue swelling, difficulty breathing, or hives. The streptozyme assay was positive, but the other laboratory test results were negative.
The dermatology team diagnosed a severely inflammatory psoriasis flare and prescribed an oral retinoid, acitretin, and referred him for ultraviolet light therapy. He was unable to travel for phototherapy, and acitretin was discontinued after 1 week because of elevated serum transaminase levels. The dermatologists then prescribed oral cyclosporine.
The progression of disease despite standard treatment suggests a nonpsoriatic condition. Although medications could cause the abnormal liver tests, so could another underlying illness that involves the liver. An infiltrative disorder of the skin with hair follicle destruction and local lymphedema could explain both alopecia and facial edema.
I am unable account for his clinical features with a single disease, so the differential remains broad, including severe psoriasis, an infiltrating cutaneous malignancy, or a toxic exposure. Arsenic poisoning causes hyperkeratotic skin lesions, although he lacks the associated gastrointestinal and neurological symptoms. I would not have added the potentially toxic cyclosporine.
When he returned to dermatology clinic 1 week later, his rash and facial swelling had worsened. He also reported muscle and joint aches, fatigue, lightheadedness, anorexia, nausea, abdominal pain, diarrhea, and dyspnea on exertion. He denied fever, chills, and night sweats.
He appeared ill and used a cane to arise and walk. His vital signs and oxygen saturation were normal. He had marked swelling of his face, diffuse erythema and swelling on the chest, and widespread scaly, erythematous plaques (Fig. 3). The proximal nail folds of his fingers were erythematous, with ragged cuticles. His abdomen was mildly distended, but the rest of the physical examination was normal.
He has become too systemically ill to attribute his condition to psoriasis. The nail findings suggest dermatomyositis, which could explain many of his findings. The diffuse erythema and his difficulty walking are consistent with its skin and muscle involvement. Dyspnea could be explained by dermatomyositis‐associated interstitial lung disease. A dermatomyositis‐associated hematological or solid malignancy could account for his multisystem ailments and functional decline. A point against dermatomyositis is the relatively explosive onset of his disease. He should be carefully examined for any motor weakness. With his progressive erythroderma, I am also concerned about an advancing cutaneous T‐cell lymphoma (with leukemic transformation).
Blood tests revealed the following values: white‐blood‐cell count, 8700/L; hematocrit, 46%; platelet count, 172,000/L; blood urea nitrogen, 26 mg/dL; creatinine, 1.0 mg/dL; glucose, 199 mg/dL; albumin, 3.1 g/dL; alkaline phosphatase, 172 U/L (normal range 45‐129); alanine aminotransferase, 75 U/L (normal range 0‐39 U/L); aspartate aminotransferase, 263 U/L (normal range 0‐37 U/L); total bilirubin, 1.1 mg/dL; prothrombin time, 16 seconds (normal range 11.7‐14.3 seconds), and serum creatinine, kinase, 4253 U/L (normal range 0‐194 U/L). HIV serology was negative. Urinalysis revealed trace protein. The results of chest radiographs and an electrocardiogram were normal.
The liver function tests results are consistent with medication effects or liver involvement in a systemic disease. The creatinine kinase elevation is consistent with a myopathy such as dermatomyositis. A skin biopsy would still be useful. Depending on those results, he may need a muscle biopsy, urine heavy metal testing, and computed tomography body imaging. Considering his transaminase and creatinine kinase elevations, I would discontinue lovastatin.
The patient was hospitalized. Further questioning revealed that he had typical Raynaud's phenomenon and odynophagia. A detailed neurological examination showed weakness (3/5) of the triceps and iliopsoas muscles and difficulty rising from a chair without using his arms. Dermatoscopic examination of the proximal nail folds showed dilated capillary loops and foci of hemorrhage.
Blood tests showed a lactate dehydrogenase level of 456 U/L (normal range 0‐249 U/L) and an aldolase of 38 U/L (normal range 1.2‐7.6 U/L). Tests for antinuclear antibodies, anti‐Jo antibody, and antimyeloperoxidase antibodies were negative. Two skin biopsies were interpreted by general pathology as consistent with partially treated psoriasis, whereas another showed nonspecific changes with minimal superficial perivascular lymphohistiocytic inflammation (Fig. 4). Lisinopril was discontinued because of its possible contribution to the facial edema.
Dermatomyositis is now the leading diagnosis. Characteristic features include his proximal muscle weakness, Raynaud's phenomenon, and dilated nailfold capillary loops. I am not overly dissuaded by the negative antinuclear antibodies, but because of additional atypical features (ie, extensive cutaneous edema, rapid onset, illness severity, prominent gastrointestinal symptoms), a confirmatory muscle biopsy is needed. Endoscopy of the proximal aerodigestive tract would help evaluate the odynophagia. There is little to suggest infection, malignancy, or metabolic derangement.
The inpatient medical team considered myositis related to retinoid or cyclosporine therapy. They discontinued cyclosporine and began systemic corticosteroid therapy. Within a few days, the patient's rash, muscle pain, and weakness improved, and the elevated transaminase and creatinine kinase levels decreased.
Dermatology recommended an evaluation for dermatomyositis‐associated malignancy, but the medicine team and rheumatology consultants, noting the lack of classic skin findings (heliotrope rash and Gottron's papules) and the uncharacteristically rapid onset and improvement of myositis, suggested delaying the evaluation until dermatomyositis was proven.
An immediate improvement in symptoms with steroids is nonspecific, often occurring in autoimmune, infectious, and neoplastic diseases. This juncture in the case is common in complex multisystem illnesses, where various consultants may arrive at differing conclusions. With both typical and atypical features of dermatomyositis, where should one set the therapeutic threshold, that is, the point where one ends testing, accepts a diagnosis, and initiates treatment? Several factors raise the level of certainty I would require. First, dermatomyositis is quite rare. Adding atypical features further increases the burden of proof for that illness. Second, the existence of alternative possibilities (admittedly of equal uncertainty) gives me some pause. Finally, the toxicity of the proposed treatments raises the therapeutic threshold. Acknowledging that empiric treatment may be indicated for a severely ill patient at a lower level of certainty, I would hesitate to commit a patient to long‐term steroids without being confident of the diagnosis. I would therefore require a muscle biopsy, or at least electromyography to support or exclude dermatomyositis.
The patient was discharged from the hospital on high‐dose prednisone. He underwent electromyography, which revealed inflammatory myopathic changes more apparent in the proximal than distal muscles. These findings were thought to be compatible with dermatomyositis, although the fibrillations and positive sharp waves characteristic of acute inflammation were absent, perhaps because of corticosteroid therapy.
The patient mistakenly stopped taking his prednisone. Within days, his weakness and skin rash worsened, and he developed nausea with vomiting. He returned to clinic, where his creatinine kinase level was again found to be elevated, and he was rehospitalized. Oral corticosteroid therapy was restarted with prompt improvement. On review of the original skin biopsies, a dermatopathologist observed areas of thickened dermal collagen and a superficial and deep perivascular lymphocytic infiltrate, both consistent with connective tissue disease.
These 3 additional findings (ie, electromyography results, temporally established steroid responsiveness, and the new skin biopsy interpretation) in aggregate support the diagnosis of dermatomyositis, but the nausea and vomiting are unusual. I would discuss these results with a rheumatologist and still request a confirmatory muscle biopsy. Because diagnosing dermatomyositis should prompt consideration of seeking an underlying malignancy in a patient of this age group, I would repeat a targeted history and physical examination along with age‐ and risk‐factor‐appropriate screening. If muscle biopsy results are not definitive, finding an underlying malignancy would lend support to dermatomyositis.
While hospitalized, the patient complained of continued odynophagia and was noted to have oral candidiasis. Upper endoscopy, undertaken to evaluate for esophageal candidiasis, revealed a mass at the gastroesophageal junction. Biopsy revealed gastric‐type adenocarcinoma. An abdominal computed tomography scan demonstrated 3 hypodense hepatic lesions, evidence of cirrhosis, and ascites. Cytology of paracentesis fluid revealed cells compatible with adenocarcinoma. The patient died in hospice care 2 weeks later.
At autopsy, he had metastatic gastric‐type adenocarcinoma. A muscle biopsy (Fig. 5) revealed muscle atrophy with small foci of lymphocytic infiltrates, most compatible with dermatomyositis. Another dermatopathologist reviewed the skin biopsies and noted interface dermatitis, which is typical of connective tissue diseases like dermatomyositis (Fig. 6A,B).
COMMENTARY
Dermatomyositis is an idiopathic inflammatory myopathy characterized by endomysial inflammation and muscle weakness and differentiated from other myopathies by the presence of a rash.1 Muscle disease may manifest with or precede the rash, but up to 40% of patients present with skin manifestations alone, an entity called amyopathic dermatomyositis.2 When present, the myositis generally develops over months, but the onset can be acute.1 The weakness is typically symmetrical and proximal,1 and many patients have oropharyngeal dysphagia.3
The characteristic rash is erythematous, symmetrical, and photodistributed.4 Classic cutaneous findings are the heliotrope rash (violaceous eyelid erythema), which is pathognomonic but uncommon, and the more common Gottron's papules (violaceous, slightly elevated papules and plaques on bony prominences and extensor surfaces, especially the knuckles).4 Other findings include periorbital edema, scalp dermatitis, poikiloderma (ie, hyperpigmentation, hypopigmentation, atrophy, and telangiectasia), periungual erythema, and dystrophic cuticles.2 The cutaneous manifestations of dermatomyositis may be similar to those of psoriasis, systemic lupus erythematosus, lichen planus, rosacea, polymorphous light eruption, drug eruption, atopic dermatitis, seborrheic dermatitis, or allergic contact dermatitis.4
Diagnosing dermatomyositis requires considering clinical, laboratory, electromyographical, and histological evidence, as there are no widely accepted, validated diagnostic criteria.1, 5 The diagnosis is usually suspected if there is a characteristic rash and symptoms of myositis (eg, proximal muscle weakness, myalgias, fatigue, or an inability to swallow). When the patient has an atypical rash, skin biopsy can differentiate dermatomyositis from other conditions, except lupus, which shares the key finding of interface dermatitis.2 The histological findings can be variable and subtle,6 so consultation with a dermatopathologist may be helpful.
Myositis may be confirmed by various studies. Most patients have elevated muscle enzymes (ie, creatinine kinase, aldolase, lactate dehydrogenase, or transaminases)1; for those who do not, magnetic resonance imaging can be helpful in detecting muscle involvement and locating the best site for muscle biopsy.7 Electromyography reveals nonspecific muscle membrane instability.8 Muscle biopsy shows muscle fiber necrosis, perifascicular atrophy, and perivascular and perifascicular lymphocytic infiltrates. These can be patchy, diminished by steroid use, and occasionally seen in noninflammatory muscular dystrophies.8 For a patient with typical myositis and a characteristic rash, muscle biopsy may be unnecessary.1
The clinical utility of serologic testing for diagnosing dermatomyositis is controversial.2 Myositis‐specific antibody testing is insensitive but specific; these antibodies include Jo‐1, an antisynthetase antibody that predicts incomplete response to therapy and lung involvement, and Mi‐2, which is associated with better response to therapy.2, 9, 10 The sensitivity and specificity of antinuclear antibodies are both approximately 60%.10
Patients with dermatomyositis have higher rates of cancers than age‐matched controls, and nearly 25% of patients are diagnosed with a malignancy at some point during the course of the disease.11 Malignancies are typically solid tumors that manifest within 3 years of the diagnosis,1214 although the increased risk may exist for at least 5 years.14 There is a 10‐fold higher risk of ovarian cancer in women with dermatomyositis.12, 15 Other associated malignancies include lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma.14
Recommendations for screening affected patients for cancer have changed over the years, with increasing evidence of an association between dermatomyositis and malignancy and evolving improvements in diagnostic techniques.16 Many authorities recommend that all adult patients with dermatomyositis be evaluated for cancer, including a complete physical examination, basic hematological tests, age‐ and sex‐appropriate screening (eg, mammography, pap smear, and colonoscopy), and chest x‐ray.16 Some would add upper endoscopy; imaging of the chest, abdomen, and pelvis; gynecological examination; and serum CA‐125 level to better evaluate for the most common malignancies (ie, ovarian, gastric, lung, and pancreatic carcinomas and non‐Hodgkins lymphoma).12, 1720
In 19% of adults, dermatomyositis overlaps with other autoimmune disorders, usually systemic lupus erythematosus and systemic sclerosis.21 These manifest as Raynaud's phenomenon, arthritis, esophageal dysmotility, renal disease, or neuropathy.21 Other potentially serious systemic manifestations of dermatomyositis include proximal dysphagia from pharyngeal myopathy; distal dysphagia from esophageal dysmotility in systemic sclerosis overlap; pulmonary disease from autoimmune interstitial lung disease or aspiration; cardiac disease from conduction abnormalities, myocarditis, pericarditis, and valvular disease; and rhabdomyolysis.2
Treatment of dermatomyositis requires systemic immunosuppression with 1 or more agents. The prognosis of dermatomyositis is variable. Mortality at 5 years ranges from 23% to 73%. At least a third of patients are left with mild to severe disability.1 In addition to older age, predictors of poor outcome include male sex, dysphagia, longstanding symptoms before treatment, pulmonary or cardiac involvement, and presence of antisynthetase antibodies.22
Dermatomyositis is often treated in the outpatient setting, but there are many reasons for hospitalization. Complications of treatment, like infection or adverse effects of medications, could result in hospitalization. Treatment with intravenous pulse corticosteroids or IVIG may require inpatient administration if no infusion center is available. Other indications for inpatient evaluation include the consequences of various malignancies and the more severe expression of systemic complications of dermatomyositis (eg, dysphagia and pulmonary, cardiac, or renal disease).
Every parent knows the plaintive backseat whine, Are we there, yet? Clinicians may also experience this feeling when attempting to diagnose a perplexing illness, especially one that lacks a definitive diagnostic test. It was easy for this patient's doctors to assume initially that his new rash was a manifestation of his long‐standing psoriasis. Having done so, they could understandably attribute the subsequent findings to either evolution of this disease or to consequences of the prescribed treatments, rather than considering a novel diagnosis. Only when faced with new (or newly appreciated) findings suggesting myopathy did the clinicians (and our discussant) consider the diagnosis of dermatomyositis. Even then, the primary inpatient medical team and their consultants were unsure when they had sufficient evidence to be certain.
Several factors compounded the difficulty of making a diagnosis in this case: the clinicians were dealing with a rare disease; they were considering alternative diagnoses (ie, psoriasis or a toxic effect of medication); and the disease presented somewhat atypically. The clinicians initially failed to consider and then accept the correct diagnosis because the patient's rash was not classic, his biopsy was interpreted as nonspecific, and he lacked myositis at presentation. Furthermore, when the generalists sought expert assistance, they encountered a difference of opinion among the consultants. These complex situations should goad the clinician into carefully considering the therapeutic threshold, that is, the transition point from diagnostic testing to therapeutic intervention.23 With complex cases like this, it may be difficult to know when one has reached a strongly supported diagnosis, and frequently asking whether we are there yet may be appropriate.
Take‐Home Points for the Hospitalist
-
A skin rash, which may have typical or atypical features, distinguishes dermatomyositis from other acquired myopathies.
-
Consider consultation with pathology specialists for skin and muscle biopsies.
-
Ovarian, lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma are the most common cancers associated with dermatomyositis.
-
In addition to age‐appropriate cancer screening, consider obtaining upper endoscopy, imaging of the chest/abdomen/pelvis, and CA‐125.
-
Patients with dermatomyositis and no obvious concurrent malignancy need long‐term outpatient follow‐up for repeated malignancy screening.
A 62‐year‐old man with psoriasis for more than 30 years presented to the emergency department with a scaly, pruritic rash involving his face, trunk, and extremities that he had had for the past 10 days. The rash was spreading and not responding to application of clobetasol ointment, which had helped his psoriasis in the past. He also reported mild pharyngitis, headache, and myalgias.
A patient with a chronic skin condition presenting with a new rash means the clinician must consider whether it is an alternative manifestation of the chronic disorder or a new illness. Psoriasis takes many forms including guttate psoriasis, which presents with small, droplike plaques and frequently follows respiratory infections (particularly those caused by Streptococcus). Well‐controlled psoriasis rarely transforms after 3 decades, so I would consider other conditions. The tempo of illness makes certain life‐threatening syndromes, including Stevens‐Johnson, toxic shock, and purpura fulminans, unlikely. An allergic reaction, atopic dermatitis, or medication reaction is possible. Infections, either systemic (eg, syphilis) or dermatologic (eg, scabies), should be considered. Photosensitivity could involve the sun‐exposed areas, such as the extremities and face. Seborrheic dermatitis can cause scaling lesions of the face and trunk but not the extremities. Vasculitis merits consideration, but dependent regions are typically affected more than the head. Mycosis fungoides or a paraneoplastic phenomenon could cause a diffuse rash in this age group.
The patient had diabetes mellitus, hypertension, diverticulosis, and depression. Three months earlier he had undergone surgical drainage of a perirectal abscess. His usual medications were lovastatin, paroxetine, insulin, hydrochlorothiazide, and lisinopril. Three weeks previously he had completed a 10‐day course of trimethoprim/sulfamethoxazole for an upper respiratory infection. Otherwise, he was taking no new medications. He was allergic to penicillin. He denied substance abuse, recent travel, or risk factors for human immunodeficiency virus (HIV) infection. He worked as an automobile painter, lived with his wife, and had a pet dog.
Physical examination revealed a well‐appearing man with normal vital signs. His skin had well‐defined circumscribed pink plaques, mostly 1‐2 cm in size, with thick, silvery scales in the ears and on the dorsal and ventral arms and legs, chest, back, face, and scalp. There were no pustules or other signs of infection (Figs. 1and 2). The nails exhibited distal onycholysis, oil spots, and rare pits. His posterior pharynx was mildly erythematous. The results of cardiovascular, pulmonary, and abdominal examinations were normal.
Although other scaling skin conditions such as eczema, irritant dermatitis, or malignancy remain possible, his rash is most consistent with widespread psoriasis. I would consider immunological changes that may have caused a remarkably altered and more severe expression of his chronic disease, for example, recent steroid therapy or HIV infection. The company a rash keeps helps frame the differential diagnosis. Based on the patient's well appearance, the time course, his minimal systemic symptoms, and the appearance of the rash, my leading considerations are psoriasis or an allergic dermatitis. Cutaneous T‐cell malignancy, with its indolent and sometimes protean manifestations, remains possible in a patient of his age. I would now consult a dermatologist for 3 reasons: this patient has a chronic disease that I do not manage beyond basic treatments (eg, topical steroids), he has an undiagnosed illness with substantial dermatologic manifestations, and he may need a skin biopsy for definitive diagnosis.
The dermatology team diagnosed a guttate psoriasis flare, possibly associated with streptococcal pharyngitis. The differential diagnosis included secondary syphilis, although the team believed this was less likely. The dermatology team recommended obtaining a throat culture, streptozyme assay, and rapid plasma reagin and prescribed oral erythromycin and topical steroid ointment under a sauna suit.
I would follow his response to the prescribed steroid treatments. If the patient's course deviates from the dermatologists' expectations, I would request a skin biopsy and undertake further evaluations in search of an underlying systemic disease.
The patient followed up in the dermatology clinic 3 weeks later. His rash had worsened, and he had developed patchy alopecia and progressive edema of the face, ears, and eyes. He denied mouth or tongue swelling, difficulty breathing, or hives. The streptozyme assay was positive, but the other laboratory test results were negative.
The dermatology team diagnosed a severely inflammatory psoriasis flare and prescribed an oral retinoid, acitretin, and referred him for ultraviolet light therapy. He was unable to travel for phototherapy, and acitretin was discontinued after 1 week because of elevated serum transaminase levels. The dermatologists then prescribed oral cyclosporine.
The progression of disease despite standard treatment suggests a nonpsoriatic condition. Although medications could cause the abnormal liver tests, so could another underlying illness that involves the liver. An infiltrative disorder of the skin with hair follicle destruction and local lymphedema could explain both alopecia and facial edema.
I am unable account for his clinical features with a single disease, so the differential remains broad, including severe psoriasis, an infiltrating cutaneous malignancy, or a toxic exposure. Arsenic poisoning causes hyperkeratotic skin lesions, although he lacks the associated gastrointestinal and neurological symptoms. I would not have added the potentially toxic cyclosporine.
When he returned to dermatology clinic 1 week later, his rash and facial swelling had worsened. He also reported muscle and joint aches, fatigue, lightheadedness, anorexia, nausea, abdominal pain, diarrhea, and dyspnea on exertion. He denied fever, chills, and night sweats.
He appeared ill and used a cane to arise and walk. His vital signs and oxygen saturation were normal. He had marked swelling of his face, diffuse erythema and swelling on the chest, and widespread scaly, erythematous plaques (Fig. 3). The proximal nail folds of his fingers were erythematous, with ragged cuticles. His abdomen was mildly distended, but the rest of the physical examination was normal.
He has become too systemically ill to attribute his condition to psoriasis. The nail findings suggest dermatomyositis, which could explain many of his findings. The diffuse erythema and his difficulty walking are consistent with its skin and muscle involvement. Dyspnea could be explained by dermatomyositis‐associated interstitial lung disease. A dermatomyositis‐associated hematological or solid malignancy could account for his multisystem ailments and functional decline. A point against dermatomyositis is the relatively explosive onset of his disease. He should be carefully examined for any motor weakness. With his progressive erythroderma, I am also concerned about an advancing cutaneous T‐cell lymphoma (with leukemic transformation).
Blood tests revealed the following values: white‐blood‐cell count, 8700/L; hematocrit, 46%; platelet count, 172,000/L; blood urea nitrogen, 26 mg/dL; creatinine, 1.0 mg/dL; glucose, 199 mg/dL; albumin, 3.1 g/dL; alkaline phosphatase, 172 U/L (normal range 45‐129); alanine aminotransferase, 75 U/L (normal range 0‐39 U/L); aspartate aminotransferase, 263 U/L (normal range 0‐37 U/L); total bilirubin, 1.1 mg/dL; prothrombin time, 16 seconds (normal range 11.7‐14.3 seconds), and serum creatinine, kinase, 4253 U/L (normal range 0‐194 U/L). HIV serology was negative. Urinalysis revealed trace protein. The results of chest radiographs and an electrocardiogram were normal.
The liver function tests results are consistent with medication effects or liver involvement in a systemic disease. The creatinine kinase elevation is consistent with a myopathy such as dermatomyositis. A skin biopsy would still be useful. Depending on those results, he may need a muscle biopsy, urine heavy metal testing, and computed tomography body imaging. Considering his transaminase and creatinine kinase elevations, I would discontinue lovastatin.
The patient was hospitalized. Further questioning revealed that he had typical Raynaud's phenomenon and odynophagia. A detailed neurological examination showed weakness (3/5) of the triceps and iliopsoas muscles and difficulty rising from a chair without using his arms. Dermatoscopic examination of the proximal nail folds showed dilated capillary loops and foci of hemorrhage.
Blood tests showed a lactate dehydrogenase level of 456 U/L (normal range 0‐249 U/L) and an aldolase of 38 U/L (normal range 1.2‐7.6 U/L). Tests for antinuclear antibodies, anti‐Jo antibody, and antimyeloperoxidase antibodies were negative. Two skin biopsies were interpreted by general pathology as consistent with partially treated psoriasis, whereas another showed nonspecific changes with minimal superficial perivascular lymphohistiocytic inflammation (Fig. 4). Lisinopril was discontinued because of its possible contribution to the facial edema.
Dermatomyositis is now the leading diagnosis. Characteristic features include his proximal muscle weakness, Raynaud's phenomenon, and dilated nailfold capillary loops. I am not overly dissuaded by the negative antinuclear antibodies, but because of additional atypical features (ie, extensive cutaneous edema, rapid onset, illness severity, prominent gastrointestinal symptoms), a confirmatory muscle biopsy is needed. Endoscopy of the proximal aerodigestive tract would help evaluate the odynophagia. There is little to suggest infection, malignancy, or metabolic derangement.
The inpatient medical team considered myositis related to retinoid or cyclosporine therapy. They discontinued cyclosporine and began systemic corticosteroid therapy. Within a few days, the patient's rash, muscle pain, and weakness improved, and the elevated transaminase and creatinine kinase levels decreased.
Dermatology recommended an evaluation for dermatomyositis‐associated malignancy, but the medicine team and rheumatology consultants, noting the lack of classic skin findings (heliotrope rash and Gottron's papules) and the uncharacteristically rapid onset and improvement of myositis, suggested delaying the evaluation until dermatomyositis was proven.
An immediate improvement in symptoms with steroids is nonspecific, often occurring in autoimmune, infectious, and neoplastic diseases. This juncture in the case is common in complex multisystem illnesses, where various consultants may arrive at differing conclusions. With both typical and atypical features of dermatomyositis, where should one set the therapeutic threshold, that is, the point where one ends testing, accepts a diagnosis, and initiates treatment? Several factors raise the level of certainty I would require. First, dermatomyositis is quite rare. Adding atypical features further increases the burden of proof for that illness. Second, the existence of alternative possibilities (admittedly of equal uncertainty) gives me some pause. Finally, the toxicity of the proposed treatments raises the therapeutic threshold. Acknowledging that empiric treatment may be indicated for a severely ill patient at a lower level of certainty, I would hesitate to commit a patient to long‐term steroids without being confident of the diagnosis. I would therefore require a muscle biopsy, or at least electromyography to support or exclude dermatomyositis.
The patient was discharged from the hospital on high‐dose prednisone. He underwent electromyography, which revealed inflammatory myopathic changes more apparent in the proximal than distal muscles. These findings were thought to be compatible with dermatomyositis, although the fibrillations and positive sharp waves characteristic of acute inflammation were absent, perhaps because of corticosteroid therapy.
The patient mistakenly stopped taking his prednisone. Within days, his weakness and skin rash worsened, and he developed nausea with vomiting. He returned to clinic, where his creatinine kinase level was again found to be elevated, and he was rehospitalized. Oral corticosteroid therapy was restarted with prompt improvement. On review of the original skin biopsies, a dermatopathologist observed areas of thickened dermal collagen and a superficial and deep perivascular lymphocytic infiltrate, both consistent with connective tissue disease.
These 3 additional findings (ie, electromyography results, temporally established steroid responsiveness, and the new skin biopsy interpretation) in aggregate support the diagnosis of dermatomyositis, but the nausea and vomiting are unusual. I would discuss these results with a rheumatologist and still request a confirmatory muscle biopsy. Because diagnosing dermatomyositis should prompt consideration of seeking an underlying malignancy in a patient of this age group, I would repeat a targeted history and physical examination along with age‐ and risk‐factor‐appropriate screening. If muscle biopsy results are not definitive, finding an underlying malignancy would lend support to dermatomyositis.
While hospitalized, the patient complained of continued odynophagia and was noted to have oral candidiasis. Upper endoscopy, undertaken to evaluate for esophageal candidiasis, revealed a mass at the gastroesophageal junction. Biopsy revealed gastric‐type adenocarcinoma. An abdominal computed tomography scan demonstrated 3 hypodense hepatic lesions, evidence of cirrhosis, and ascites. Cytology of paracentesis fluid revealed cells compatible with adenocarcinoma. The patient died in hospice care 2 weeks later.
At autopsy, he had metastatic gastric‐type adenocarcinoma. A muscle biopsy (Fig. 5) revealed muscle atrophy with small foci of lymphocytic infiltrates, most compatible with dermatomyositis. Another dermatopathologist reviewed the skin biopsies and noted interface dermatitis, which is typical of connective tissue diseases like dermatomyositis (Fig. 6A,B).
COMMENTARY
Dermatomyositis is an idiopathic inflammatory myopathy characterized by endomysial inflammation and muscle weakness and differentiated from other myopathies by the presence of a rash.1 Muscle disease may manifest with or precede the rash, but up to 40% of patients present with skin manifestations alone, an entity called amyopathic dermatomyositis.2 When present, the myositis generally develops over months, but the onset can be acute.1 The weakness is typically symmetrical and proximal,1 and many patients have oropharyngeal dysphagia.3
The characteristic rash is erythematous, symmetrical, and photodistributed.4 Classic cutaneous findings are the heliotrope rash (violaceous eyelid erythema), which is pathognomonic but uncommon, and the more common Gottron's papules (violaceous, slightly elevated papules and plaques on bony prominences and extensor surfaces, especially the knuckles).4 Other findings include periorbital edema, scalp dermatitis, poikiloderma (ie, hyperpigmentation, hypopigmentation, atrophy, and telangiectasia), periungual erythema, and dystrophic cuticles.2 The cutaneous manifestations of dermatomyositis may be similar to those of psoriasis, systemic lupus erythematosus, lichen planus, rosacea, polymorphous light eruption, drug eruption, atopic dermatitis, seborrheic dermatitis, or allergic contact dermatitis.4
Diagnosing dermatomyositis requires considering clinical, laboratory, electromyographical, and histological evidence, as there are no widely accepted, validated diagnostic criteria.1, 5 The diagnosis is usually suspected if there is a characteristic rash and symptoms of myositis (eg, proximal muscle weakness, myalgias, fatigue, or an inability to swallow). When the patient has an atypical rash, skin biopsy can differentiate dermatomyositis from other conditions, except lupus, which shares the key finding of interface dermatitis.2 The histological findings can be variable and subtle,6 so consultation with a dermatopathologist may be helpful.
Myositis may be confirmed by various studies. Most patients have elevated muscle enzymes (ie, creatinine kinase, aldolase, lactate dehydrogenase, or transaminases)1; for those who do not, magnetic resonance imaging can be helpful in detecting muscle involvement and locating the best site for muscle biopsy.7 Electromyography reveals nonspecific muscle membrane instability.8 Muscle biopsy shows muscle fiber necrosis, perifascicular atrophy, and perivascular and perifascicular lymphocytic infiltrates. These can be patchy, diminished by steroid use, and occasionally seen in noninflammatory muscular dystrophies.8 For a patient with typical myositis and a characteristic rash, muscle biopsy may be unnecessary.1
The clinical utility of serologic testing for diagnosing dermatomyositis is controversial.2 Myositis‐specific antibody testing is insensitive but specific; these antibodies include Jo‐1, an antisynthetase antibody that predicts incomplete response to therapy and lung involvement, and Mi‐2, which is associated with better response to therapy.2, 9, 10 The sensitivity and specificity of antinuclear antibodies are both approximately 60%.10
Patients with dermatomyositis have higher rates of cancers than age‐matched controls, and nearly 25% of patients are diagnosed with a malignancy at some point during the course of the disease.11 Malignancies are typically solid tumors that manifest within 3 years of the diagnosis,1214 although the increased risk may exist for at least 5 years.14 There is a 10‐fold higher risk of ovarian cancer in women with dermatomyositis.12, 15 Other associated malignancies include lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma.14
Recommendations for screening affected patients for cancer have changed over the years, with increasing evidence of an association between dermatomyositis and malignancy and evolving improvements in diagnostic techniques.16 Many authorities recommend that all adult patients with dermatomyositis be evaluated for cancer, including a complete physical examination, basic hematological tests, age‐ and sex‐appropriate screening (eg, mammography, pap smear, and colonoscopy), and chest x‐ray.16 Some would add upper endoscopy; imaging of the chest, abdomen, and pelvis; gynecological examination; and serum CA‐125 level to better evaluate for the most common malignancies (ie, ovarian, gastric, lung, and pancreatic carcinomas and non‐Hodgkins lymphoma).12, 1720
In 19% of adults, dermatomyositis overlaps with other autoimmune disorders, usually systemic lupus erythematosus and systemic sclerosis.21 These manifest as Raynaud's phenomenon, arthritis, esophageal dysmotility, renal disease, or neuropathy.21 Other potentially serious systemic manifestations of dermatomyositis include proximal dysphagia from pharyngeal myopathy; distal dysphagia from esophageal dysmotility in systemic sclerosis overlap; pulmonary disease from autoimmune interstitial lung disease or aspiration; cardiac disease from conduction abnormalities, myocarditis, pericarditis, and valvular disease; and rhabdomyolysis.2
Treatment of dermatomyositis requires systemic immunosuppression with 1 or more agents. The prognosis of dermatomyositis is variable. Mortality at 5 years ranges from 23% to 73%. At least a third of patients are left with mild to severe disability.1 In addition to older age, predictors of poor outcome include male sex, dysphagia, longstanding symptoms before treatment, pulmonary or cardiac involvement, and presence of antisynthetase antibodies.22
Dermatomyositis is often treated in the outpatient setting, but there are many reasons for hospitalization. Complications of treatment, like infection or adverse effects of medications, could result in hospitalization. Treatment with intravenous pulse corticosteroids or IVIG may require inpatient administration if no infusion center is available. Other indications for inpatient evaluation include the consequences of various malignancies and the more severe expression of systemic complications of dermatomyositis (eg, dysphagia and pulmonary, cardiac, or renal disease).
Every parent knows the plaintive backseat whine, Are we there, yet? Clinicians may also experience this feeling when attempting to diagnose a perplexing illness, especially one that lacks a definitive diagnostic test. It was easy for this patient's doctors to assume initially that his new rash was a manifestation of his long‐standing psoriasis. Having done so, they could understandably attribute the subsequent findings to either evolution of this disease or to consequences of the prescribed treatments, rather than considering a novel diagnosis. Only when faced with new (or newly appreciated) findings suggesting myopathy did the clinicians (and our discussant) consider the diagnosis of dermatomyositis. Even then, the primary inpatient medical team and their consultants were unsure when they had sufficient evidence to be certain.
Several factors compounded the difficulty of making a diagnosis in this case: the clinicians were dealing with a rare disease; they were considering alternative diagnoses (ie, psoriasis or a toxic effect of medication); and the disease presented somewhat atypically. The clinicians initially failed to consider and then accept the correct diagnosis because the patient's rash was not classic, his biopsy was interpreted as nonspecific, and he lacked myositis at presentation. Furthermore, when the generalists sought expert assistance, they encountered a difference of opinion among the consultants. These complex situations should goad the clinician into carefully considering the therapeutic threshold, that is, the transition point from diagnostic testing to therapeutic intervention.23 With complex cases like this, it may be difficult to know when one has reached a strongly supported diagnosis, and frequently asking whether we are there yet may be appropriate.
Take‐Home Points for the Hospitalist
-
A skin rash, which may have typical or atypical features, distinguishes dermatomyositis from other acquired myopathies.
-
Consider consultation with pathology specialists for skin and muscle biopsies.
-
Ovarian, lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma are the most common cancers associated with dermatomyositis.
-
In addition to age‐appropriate cancer screening, consider obtaining upper endoscopy, imaging of the chest/abdomen/pelvis, and CA‐125.
-
Patients with dermatomyositis and no obvious concurrent malignancy need long‐term outpatient follow‐up for repeated malignancy screening.
- ,.Polymyositis and dermatomyositis.Lancet.2003;362:971–982.
- .Dermatomyositis.Lancet.2000;355:53–47.
- ,,,.Oropharyngeal dysphagia in polymyositis/dermatomyositis.Clin Neurol Neurosurg.2004;107(1):32–37.
- ,.Skin involvement in dermatomyositis.Curr Opin Rheumatol.2003;15:714–22.
- ,,,,,.Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients.Medicine (Baltimore).2005;84:231–249.
- .Skin Pathology.2nd ed.New York:Churchill Livingstone;2002.
- ,.Utility of magnetic resonance imaging in the evaluation of patients with inflammatory myopathies.Curr Rheumatol Rep.2001;3:334–245.
- ,,.Is it really myositis? A consideration of the differential diagnosis.Curr Opin Rheumatol2004;16:684–691.
- .Idiopathic inflammatory myopathy: autoantibody update.Curr Rheumatol Rep.2002;4:434–441.
- ,,.Laboratory assessment in musculoskeletal disorders.Best Pract Res Clin Rheumatol.2003;17:475–494.
- ,.Dermatomyositis.Clin Dermatol.2006;24:363–373.
- ,,, et al.Frequency of specific cancer types in dermatomyositis and polymyositis: a population‐based study.Lancet.2001;357:96–100.
- ,,, et al.Cancer‐associated myositis: clinical features and prognostic signs.Ann N Y Acad Sci.2005;1051:64–71.
- ,,,,.Incidence of malignant disease in biopsy‐proven inflammatory myopathy. A population‐based cohort study.Ann Intern Med.2001;134:1087–1095.
- ,,.Risk of cancer in patients with dermatomyositis or polymyositis, and follow‐up implications: a Scottish population‐based cohort study.Br J Cancer.2001;85 (1):41–45.
- .When and how should the patient with dermatomyositis or amyopathic dermatomyositis be assessed for possible cancer?Arch Dermatol.2002;138:969–971.
- ,,.Ovarian cancer in patients with dermatomyositis.Medicine (Baltimore).1994;73(3):153–160.
- ,,,.Dermatomyositis sine myositis: association with malignancy.J Rheumatol.1996;23 (1):101–105.
- ,,, et al.Tumor antigen markers for the detection of solid cancers in inflammatory myopathies.Cancer Epidemiol Biomarkers Prev.2005;14:1279–1282.
- ,,, et al.Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients.Arch Dermatol.2002;138:885–890.
- ,,,,,.Dermatomyositis: a dermatology‐based case series.J Am Acad Dermatol.1998;38:397–404.
- ,,, et al.Long‐term outcome in polymyositis and dermatomyositis.Ann Rheum Dis.2006;65:1456–1461.
- .Our stubborn quest for diagnostic certainty. A cause of excessive testing.N Engl J Med.1989;320:1489–1491.
- ,.Polymyositis and dermatomyositis.Lancet.2003;362:971–982.
- .Dermatomyositis.Lancet.2000;355:53–47.
- ,,,.Oropharyngeal dysphagia in polymyositis/dermatomyositis.Clin Neurol Neurosurg.2004;107(1):32–37.
- ,.Skin involvement in dermatomyositis.Curr Opin Rheumatol.2003;15:714–22.
- ,,,,,.Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients.Medicine (Baltimore).2005;84:231–249.
- .Skin Pathology.2nd ed.New York:Churchill Livingstone;2002.
- ,.Utility of magnetic resonance imaging in the evaluation of patients with inflammatory myopathies.Curr Rheumatol Rep.2001;3:334–245.
- ,,.Is it really myositis? A consideration of the differential diagnosis.Curr Opin Rheumatol2004;16:684–691.
- .Idiopathic inflammatory myopathy: autoantibody update.Curr Rheumatol Rep.2002;4:434–441.
- ,,.Laboratory assessment in musculoskeletal disorders.Best Pract Res Clin Rheumatol.2003;17:475–494.
- ,.Dermatomyositis.Clin Dermatol.2006;24:363–373.
- ,,, et al.Frequency of specific cancer types in dermatomyositis and polymyositis: a population‐based study.Lancet.2001;357:96–100.
- ,,, et al.Cancer‐associated myositis: clinical features and prognostic signs.Ann N Y Acad Sci.2005;1051:64–71.
- ,,,,.Incidence of malignant disease in biopsy‐proven inflammatory myopathy. A population‐based cohort study.Ann Intern Med.2001;134:1087–1095.
- ,,.Risk of cancer in patients with dermatomyositis or polymyositis, and follow‐up implications: a Scottish population‐based cohort study.Br J Cancer.2001;85 (1):41–45.
- .When and how should the patient with dermatomyositis or amyopathic dermatomyositis be assessed for possible cancer?Arch Dermatol.2002;138:969–971.
- ,,.Ovarian cancer in patients with dermatomyositis.Medicine (Baltimore).1994;73(3):153–160.
- ,,,.Dermatomyositis sine myositis: association with malignancy.J Rheumatol.1996;23 (1):101–105.
- ,,, et al.Tumor antigen markers for the detection of solid cancers in inflammatory myopathies.Cancer Epidemiol Biomarkers Prev.2005;14:1279–1282.
- ,,, et al.Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients.Arch Dermatol.2002;138:885–890.
- ,,,,,.Dermatomyositis: a dermatology‐based case series.J Am Acad Dermatol.1998;38:397–404.
- ,,, et al.Long‐term outcome in polymyositis and dermatomyositis.Ann Rheum Dis.2006;65:1456–1461.
- .Our stubborn quest for diagnostic certainty. A cause of excessive testing.N Engl J Med.1989;320:1489–1491.