User login
A Double‐Edged Sword
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.

Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
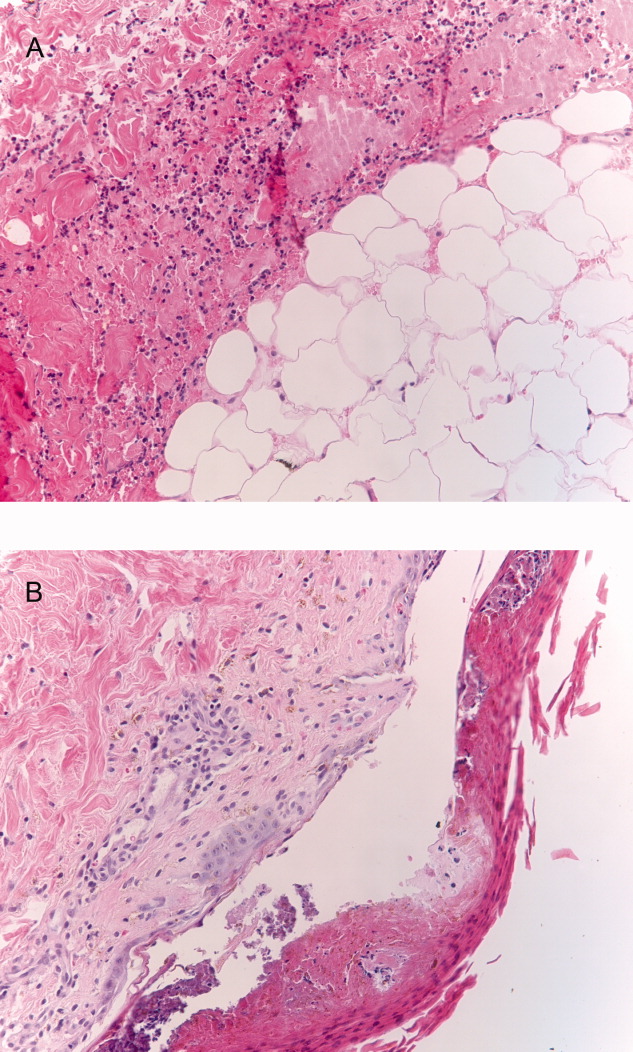
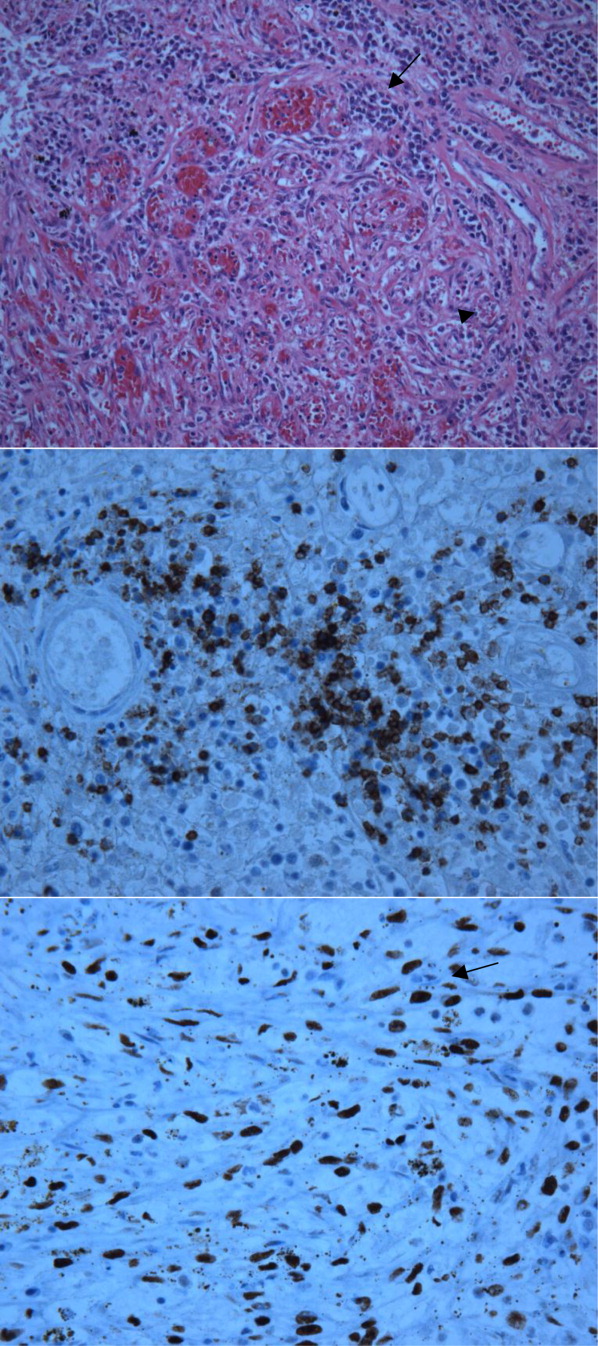
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.
Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.
Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.
Overcome by Weakness
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, <5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
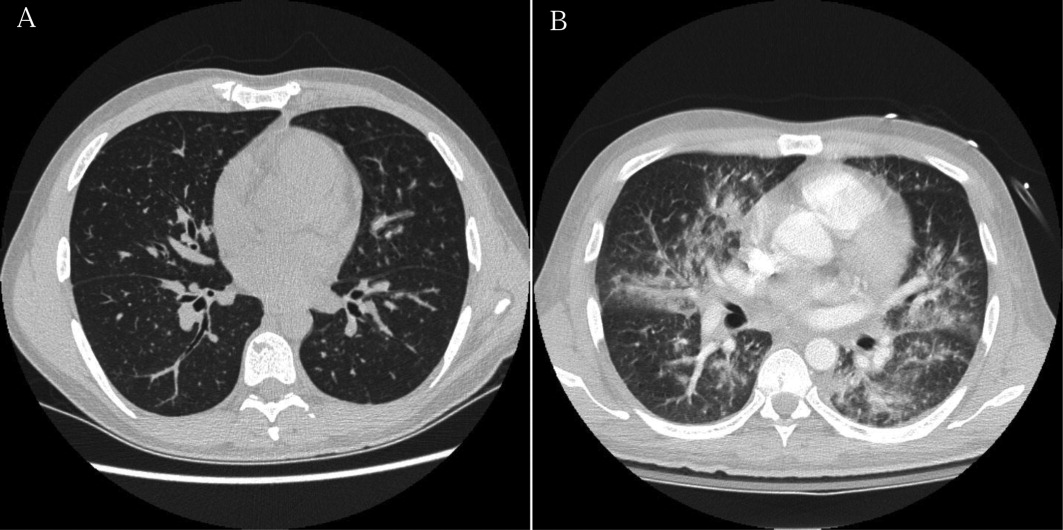
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
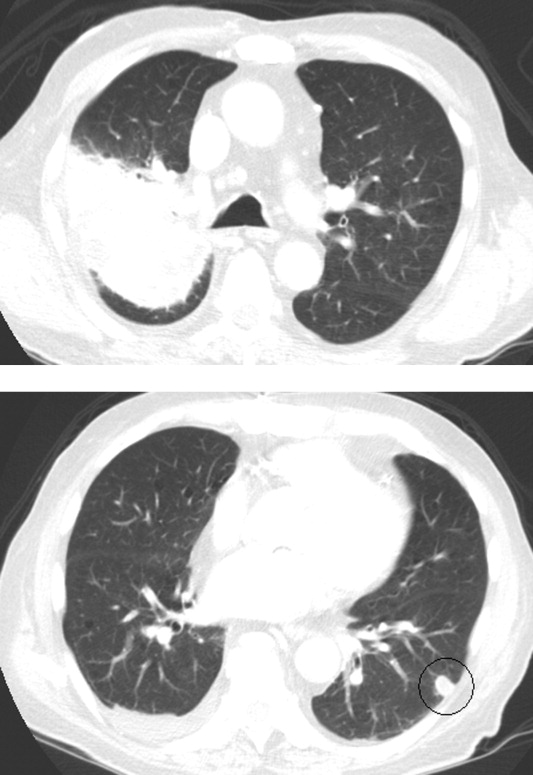
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.

Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, <5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.
Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, <5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.
Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
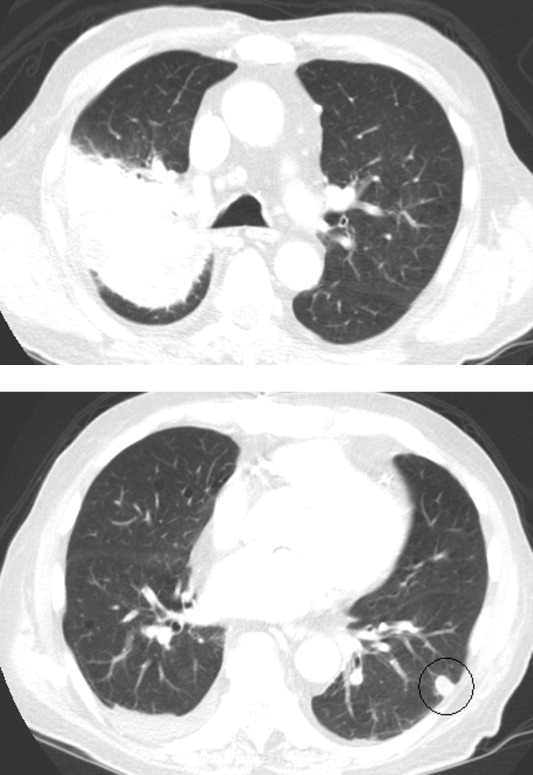
Cracking the Case
A 43‐year‐old woman presented to an outside hospital with painful plaques and patches on her bilateral lower extremities. Two weeks prior to presentation, she had noticed a single red lesion on her left ankle. Over the next two weeks, the lesion enlarged to involve the lower half of her posterior calf and subsequently turned purple and became exquisitely tender. Similar but smaller purple, tender lesions simultaneously appeared, first over her right shin and then on her bilateral thighs and hips. She also reported fatigue as well as diffuse joint pains in her hands and wrists bilaterally for the past month. She denied any swelling of these joints or functional impairment. She denied fevers, weight loss, headache, sinus symptoms, difficulty breathing, or abdominal pain.
Although we do not yet have a physical exam, the tempo, pattern of spread, and accompanying features allow some early hypotheses to be considered. Distal lower extremity lesions which darkened and spread could be erythema nodosum or erythema induratum. Malignancies rarely have such prominent skin manifestations, although leukemia cutis or an aggressive cutaneous T cell lymphoma might present with disseminated and darkened plaques, and Kaposi's sarcoma is characteristically purple and multifocal. Autoimmune disorders such as sarcoidosis, cutaneous lupus, and psoriasis may similarly present with widespread plaques. Most disseminated infections that start with patches evolve to pustules, ulcers, bullae, or other forms that reflect the invasive nature of the infection; syphilis warrants consideration for any widespread eruption of unknown etiology. Antecedent arthralgias with fatigue suggest an autoimmune condition, although infections such as hepatitis or parvovirus can do the same. Systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) would be favored initially on account of her demographics and the hand and wrist involvement, and each can be associated with vasculitis.
The significant pain as described is not compatible with most of the aforementioned diagnoses. Its presence, coupled with potential autoimmune symptoms, suggests a vasculitis such as polyarteritis nodosa (which can have prominent diffuse skin involvement), Henoch Schonlein purpura (with its predilection for the lower extremities, including extension to the hips and buttocks), cryoglobulinemia, or SLE‐ or RA‐associated vasculitis. Calciphylaxis is another ischemic vascular disorder that can cause diffuse dark painful lesions, but this only warrants consideration if advanced renal disease is present.
A skin biopsy ofher right hip was taken at the outside hospital. She was discharged on a two‐week course of prednisone for suspected vasculitis while biopsy results were pending. Over the next two weeks, none of the skin lesions improved, despite compliance with this treatment, and the skin over her left posterior calf and right shin lesions began to erode and bleed. In addition, small purple, tender lesions appeared over the pinnae of both ears. Three weeks after her initial evaluation, she presented to another emergency department for ulcerating skin lesions and worsening pain. At that point, the initial skin biopsy result was available and revealed vasculopathy of the small vessels with thrombi but no vasculitis.
The patient had no children,and denied a history of miscarriages. Her past medical history was unremarkable. She did not report any history of thrombotic events. She started a new job as a software engineer one month ago and was feeling stressed about her new responsibilities. She denied any high‐risk sexual behavior and any history of intravenous drug use. She had not traveled recently and did not own any pets. There was no family history of rheumatologic disorders, hypercoagulable states, or thrombotic events.
This picture of occluded but noninflamed vessels shifts the diagnosis away from vasculitis and focuses attention on hypercoagulable states with prominent dermal manifestations, including antiphospholipid antibody syndrome (APLS) and livedoid vasculopathy. In this young woman with arthralgias, consideration of SLE and APLS is warranted. Her recent increase in stress and widespread purpuric and ulcerative lesions could bring to mind a factitious disorder, but the histology results eliminate this possibility.
The patient's temperaturewas 36.5C, her blood pressure was 110/70 mmHg, respiratory rate was 16 breaths per minute, and her heart rate was 65 beats per minute. She was well‐appearing but in moderate pain. She did not have any oral lesions. Her cardiac, respiratory, and abdominal exams were normal. Skin exam revealed a 10‐cm by 4‐cm area of bloody granulation tissue draining serosanguinous fluid, surrounded by stellate palpable netlike purpura on her left posterior calf. There was a similar 4‐cm by 2‐cm ulcerated lesion on her right shin. Both lesions were exquisitely tender to palpation. On her bilateral thighs and hips, there were multiple stellate purpuric patches, all 4 cm in diameter or less, and only minimally tender to palpation. She also had 1‐cm purpuric bullae on the helices of both ears (Figure 1) which were slightly tender to palpation. Splinter hemorrhages were also noted on multiple nail beds bilaterally. Musculoskeletal exam did not reveal any synovitis.
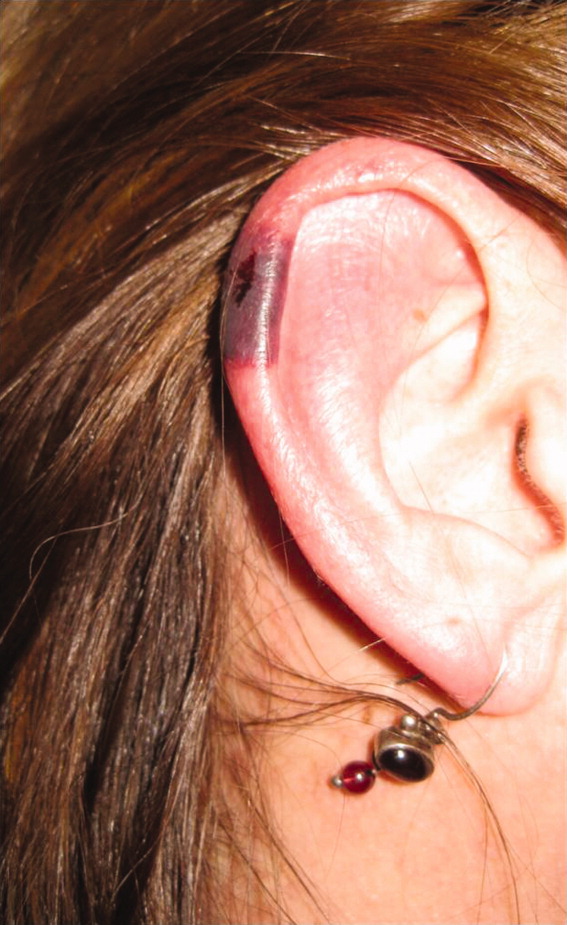
The original purpura on her calf and ear demonstrate a clear demarcation corresponding to cutaneous vascular insufficiency. The development of bullae (ear) and ulceration (calf) are compatible with ischemia. Despite the presence of multiple splinter hemorrhages, the distribution of lesions is very unusual for an embolic phenomenon (eg, endocarditis, cholesterol emboli, or atrial myxoma). The multifocal nature of the skin lesions with progression to well‐demarcated cutaneous necrosis is reminiscent of calciphylaxis or warfarin‐induced skin necrosis, although she lacks the relevant risk factors. A toxin such as cocaine or methamphetamine mediating multifocal vasoconstriction or hypercoagulability should be excluded.
The bilateral ear involvement remains decidedly unusual and makes me wonder if there is something about the ear, such as the nature of its circulation or its potentially lower temperature (as an acral organ) that might render it particularly susceptible, for instance, to cryoglobulinemia or cryofibrinogenemia‐mediated ischemia.
Laboratory studiesdemonstrated: white blood cell count of 1500/mm3 (37.3% neutrophils, 5.1% lymphocytes, 6.7% monocytes, and 1.3% eosinophils); hemoglobin 9.3 g/dl (mean corpuscular volume 91 fL); platelet count 212/mm3; erythrocyte sedimentation rate 62 mm/hr; C‐reactive protein 14.6 mg/L. Serum electrolytes, liver tests, coagulation studies, and urinalysis were normal. Fecal occult blood test was negative.
Her neutropenia and anemia suggest decreased production in the marrow by infection, malignancy, or toxin, or increased destruction, perhaps from an autoimmune process. The associated infections are usually viral, such as human immunodeficiency virus (HIV) and Epstein‐Barr virus (EBV), although their linkage with her cutaneous disease is tenuous. It is possible that malignancy could be present in the marrow with resultant dermal hypercoagulability and ischemia, but this seems unlikely. We do not know about any toxins that she has been exposed to, but these hematologic findings would mandate directed inquiry along those lines. In this young woman with cutaneous ulcers secondary to thrombotic vasculopathy, bicytopenia, antecedent arthralgias without synovitis, and elevated inflammatory markers, I favor an autoimmune process such as SLE, which I would evaluate with an antinuclear antibody (ANA) and antiphospholipid antibody studies.
She was admittedto the hospital and received hydromorphone for pain control. Corticosteroids were not administered. Peripheral blood morphology was normal. Antibodies against HIV1 and 2 were negative, as were antibodies against cytomegalovirus, EBV, parvovirus B19, mycoplasma pneumoniae, and hepatitis C virus. Bilateral lower extremity ultrasound was negative for deep vein thrombosis. Transthoracic echocardiogram was normal. Repeat skin biopsy confirmed small vessel vasculopathy without vasculitis (Figure 2). The results of the following investigations were also negative: ANA, rheumatoid factor, double‐stranded DNA (dsDNA), cyclic citrullinated peptide, ribonucleoprotein (RNP), and anti‐Smith antibodies. C3 and C4 complement levels were normal.
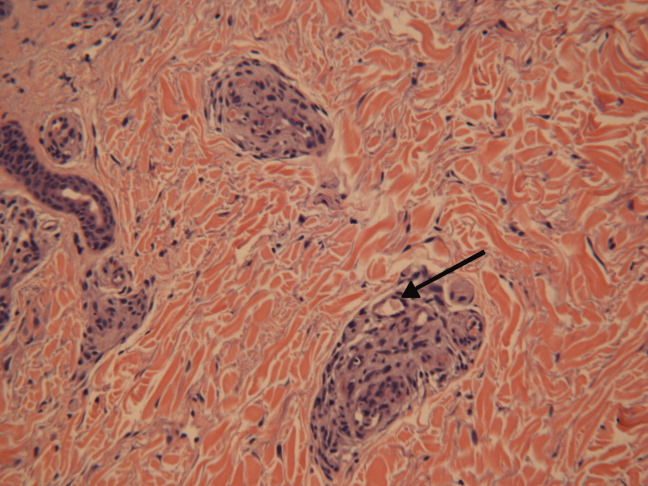
Given how much the histology is driving the clinical reasoning and focusing the differential diagnosis in this case, I agree with the decision to repeat the biopsy. In complex or undiagnosed cases, repeat histology samples allow for confirmation of the original interpretation (often with the perspective of new clinicians and pathologists) and sometimes reveal pathognomonic or additional findings that only appear after the disease has evolved over time. HIV seronegativity helps constrain the differential diagnosis, and parvovirus is another excellent consideration for arthralgias and cytopenias (with the predilection to involve cells lines other than RBCs particularly seen in HIV), although ulcers are not seen with this condition. Herpes simplex virus (HSV) is another viral infection that can cause painful skin ulcerations and cytopenias, although the duration and distribution are highly atypical. The negative ANA and dsDNA and normal complement levels make SLE unlikely. The negative lower extremity ultrasound helps frame the thromboses as a local cutaneous process rather than a systemic hypercoagulable state. Although the peripheral blood smear is normal, a bone marrow biopsy will be necessary to exclude a marrow invasive process, such as leukemia or lymphoma. A bone marrow biopsy would also provide another opportunity to examine tissue for mycobacteria or fungi which can cause ulcerations and cytopenias, although there is little reason to suspect she is susceptible to those pathogens. As this clinical picture fails to fit clearly with an infectious, autoimmune, or neoplastic disorder, I would revisit the possibility of toxinsprescription, complementary, over‐the‐counter, or illegal (eg, cocaine) at this time.
In further discussionwith the patient, she reported using cocaine intranasally for the past three months. Her urine toxicology was positive for cocaine. She was found to have positive perinuclear antineutrophil cytoplasmic antibodies (p‐ANCA), antimyeloperoxidase (MPO) antibodies, anticardiolipin (ACL) antibodies, and lupus anticoagulant (LAC). By hospital day 3, her lesions had significantly improved without any intervention, and her absolute neutrophil count increased to 1080/mm3.
The presence of widespread cutaneous ischemia (with bland thrombosis) and detectable ACL and LAC antibodies is compatible with APLS; the APLS could be deemed primary, because there is no clear associated rheumatologic or other systemic disease. However, neutropenia is not a characteristic of APLS, which has thrombocytopenia as its more frequently associated hematologic abnormality. Livedoid vasculopathy, a related disorder, is also supported by the ACL and LAC results, but also does not feature neutropenia. While the presence of diffuse thrombosis could be attributed to a widespread secondary effect of cocaine vasoconstriction, the appearance of ANCA (which can be drug‐induced, eg, propylthiouracil [PTU]) and the slowly resolving neutropenia during hospitalization without specific treatment is very suggestive of a toxin. The demographic, diffuse skin ulcers, and hematologic and serologic profile is compatible with the recently described toxidrome related to levamisole adulteration of cocaine.
A send‐out studyof a urine sample returned positive for levamisole. Based on purpuric skin lesions with a predilection for the ears, agranulocytosis, and skin biopsy revealing thrombotic vasculopathy, she was diagnosed with levamisole‐adulterated cocaine exposure. One week after discharge, her lower extremity pain and ulcerations were significantly improved. Her absolute neutrophil count increased to 2820/mm.3 Her urine toxicology screen was negative for cocaine.
DISCUSSION
Levamisole was initially developed in 1964 as an antihelminthic agent. Its incidentally discovered immunomodulatory effects led to trials for the treatment of chronic infections, inflammatory bowel disease, rheumatic diseases,1 and nephrotic syndrome in children.2 By 1990, 3 major studies supported levamisole as an adjunctive therapy in melanoma3 and colon cancer.4
Although levamisole appeared to be nontoxic at single or low doses, long‐term use in clinical trials demonstrated that 2.5%‐13% of patients developed life‐threatening agranulocytosis, and up to 10% of those instances resulted in death.5 A distinctive cutaneous pseudovasculitis was noted in children on therapeutic levamisole. They presented with purpura that had a predilection for the ears, cheeks, and thighs,6 and positive serologic markers for ANCA and antiphospholipid antibodies. Skin biopsies of the purpuric lesions revealed leukocytoclastic vasculitis, thrombotic vasculitis, and/or vascular occlusions.
Levamisole was withdrawn from the market in 2000 in the United States due to its side effects,7 but quickly found its way onto the black market. It was first detected in cocaine in 2002, and the percentage of cocaine containing levamisole has steadily been increasing since then. In July 2009, over 70% of cocaine seized by the Drug Enforcement Administration was found to contain levamisole.8 It is unclear exactly why this drug is used as an adulterant in cocaine. Theories include potentiation of the euphoric effects of cocaine, serving as a bulking agent, or functioning as a chemical signature to track distribution.9
The resurgence of levamisole has brought a new face to a problem seen over a decade ago. Current reports of levamisole toxicity describe adults presenting with purpura preferentially involving the ears, neutropenia, positive ANCA, and positive antiphospholipid antibodies.1012 Since 2002, there have been at least 20 confirmed cases of agranulocytosis and two deaths associated with levamisole‐adulterated cocaine.8, 13, 14 In September 2009, the Department of Health and Human Services issued a public health alert warning of an impending increase in levamisole‐related illness.
Levamisole is not detected on routine toxicology screens, but can be tested for using gas chromatography and mass spectrometry. Most laboratories do not offer testing for levamisole and send‐out testing is required. Given its half‐life of 5.6 hours, levamisole can only be detected in the blood within 24 hours, and in the urine within 48‐72 hours of exposure.15, 16 Urine samples are preferred over blood samples, since blood levels decline more rapidly and have lower sensitivity. Cocaine can also be sent out to local or state forensics laboratories to be tested for levamisole. The only definitive treatment for levamisole‐induced cutaneous pseudovasculitis and neutropenia is cessation of toxin exposure.
Although the discussant had familiarity with this toxidrome from local and published cases, he was only able to settle on levamisole toxicity after a series of competing hypotheses were ruled out on the basis of irreconcilable features (vasculitis and histology results; APLS and neutropenia; SLE and negative ANA with no visceral involvement) and by using analogical reasoning (eg, to infer the presence of a toxin on the basis of neutropenia [as seen with chemotherapy and other drugs] and ANCA induction [as seen with PTU]). It was a laborious process of hypothesis testing, but one that ultimately allowed him to crack the case.
Key Teaching Points
-
In patients presenting with neutropenia and purpuric skin lesionsparticularly with a predilection for the earsconsider levamisole‐adulterated cocaine exposure.
-
Tests supporting this diagnosis include positive serologies for ANCA and antiphospholipid antibodies, and skin biopsies that show leukocytoclastic vasculitis, thrombotic vasculitis, or vascular occlusion. Urine studies for levamisole are definitive if sent within 48 to 72 hours of exposure.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,.Levamisole, the story and the lessons.Int J Immunopharmocol.1992;14(3):481–486.
- British Association for Paediatric Nephrology.Levamisole for corticosteroid‐dependent nephrotic syndrome in childhood.Lancet.1991;337:1555–1557.
- ,,,,.Improved survival in patients with poor‐prognosis malignant melanoma treated with adjuvant levamisole: a phase III study by the National Cancer Institute of Canada Clinical Trials Group.J Clin Oncol.1991;9:729–735.
- ,,.Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma.N Engl J Med.1990;322:352–358.
- ,,.Studies on levamisole‐induced agranulocytosis.Blood.1980;56(3):388–396.
- ,,.Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long‐term treatment with levamisole in children.Br J Dermatol.1999;140:948–951.
- . Janssen Discontinues Ergamisol. Available at: http://findarticles.com/p/articles/mi_m3374/is_18_22/ai_68536218/. Accessed July 25,2010.
- SAMHSA. Nationwide Public Health Alert Issued Concerning Life‐Threatening Risk Posed by Cocaine Laced with Veterinary Anti‐Parasite Drug. Available at: http://www.samhsa.gov/newsroom/advisories/090921vet5101.aspx. Accessed July 20,2010.
- .Unusual adulterants in cocaine seized on Italian clandestine market.Forensic Sci Int.2007;172(2–3):e1.
- ,,.Levamisole‐induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Jun 12.
- ,,,.Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole.Ann Intern Med.2010;1;152(11):758–759.
- ,,,,.Cocaine‐associated retiform purpura and neutropenia: is levamisole the culprit?J Am Acad Dermatol.2010;Mar 19.
- ,,,.A confirmed case of agranulocytosis after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Apr 1.
- Centers for Disease Control and Prevention.Agranulocytosis associated with cocaine use—four States, March 2008–November 2009.MMWR.2009;58(49):1381–1385.
- ,,.Levamisole as a contaminant of illicit cocaine.Journal of the Clandestine Laboratory Investigating Chemists Association.2006;16:6–11. Available at: http://www.tiaft2006.org/proceedings/pdf/PT‐p‐06.pdf. Accessed July 20, 2010.
- . Cocaine Cutting Agents—A Discussion. Laboratory Medicine and Pathology, University of Alberta. Available at: http://www.vandu.org/documents/Levamisole_Cocaine.pdf. Accessed July 20,2010.
A 43‐year‐old woman presented to an outside hospital with painful plaques and patches on her bilateral lower extremities. Two weeks prior to presentation, she had noticed a single red lesion on her left ankle. Over the next two weeks, the lesion enlarged to involve the lower half of her posterior calf and subsequently turned purple and became exquisitely tender. Similar but smaller purple, tender lesions simultaneously appeared, first over her right shin and then on her bilateral thighs and hips. She also reported fatigue as well as diffuse joint pains in her hands and wrists bilaterally for the past month. She denied any swelling of these joints or functional impairment. She denied fevers, weight loss, headache, sinus symptoms, difficulty breathing, or abdominal pain.
Although we do not yet have a physical exam, the tempo, pattern of spread, and accompanying features allow some early hypotheses to be considered. Distal lower extremity lesions which darkened and spread could be erythema nodosum or erythema induratum. Malignancies rarely have such prominent skin manifestations, although leukemia cutis or an aggressive cutaneous T cell lymphoma might present with disseminated and darkened plaques, and Kaposi's sarcoma is characteristically purple and multifocal. Autoimmune disorders such as sarcoidosis, cutaneous lupus, and psoriasis may similarly present with widespread plaques. Most disseminated infections that start with patches evolve to pustules, ulcers, bullae, or other forms that reflect the invasive nature of the infection; syphilis warrants consideration for any widespread eruption of unknown etiology. Antecedent arthralgias with fatigue suggest an autoimmune condition, although infections such as hepatitis or parvovirus can do the same. Systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) would be favored initially on account of her demographics and the hand and wrist involvement, and each can be associated with vasculitis.
The significant pain as described is not compatible with most of the aforementioned diagnoses. Its presence, coupled with potential autoimmune symptoms, suggests a vasculitis such as polyarteritis nodosa (which can have prominent diffuse skin involvement), Henoch Schonlein purpura (with its predilection for the lower extremities, including extension to the hips and buttocks), cryoglobulinemia, or SLE‐ or RA‐associated vasculitis. Calciphylaxis is another ischemic vascular disorder that can cause diffuse dark painful lesions, but this only warrants consideration if advanced renal disease is present.
A skin biopsy ofher right hip was taken at the outside hospital. She was discharged on a two‐week course of prednisone for suspected vasculitis while biopsy results were pending. Over the next two weeks, none of the skin lesions improved, despite compliance with this treatment, and the skin over her left posterior calf and right shin lesions began to erode and bleed. In addition, small purple, tender lesions appeared over the pinnae of both ears. Three weeks after her initial evaluation, she presented to another emergency department for ulcerating skin lesions and worsening pain. At that point, the initial skin biopsy result was available and revealed vasculopathy of the small vessels with thrombi but no vasculitis.
The patient had no children,and denied a history of miscarriages. Her past medical history was unremarkable. She did not report any history of thrombotic events. She started a new job as a software engineer one month ago and was feeling stressed about her new responsibilities. She denied any high‐risk sexual behavior and any history of intravenous drug use. She had not traveled recently and did not own any pets. There was no family history of rheumatologic disorders, hypercoagulable states, or thrombotic events.
This picture of occluded but noninflamed vessels shifts the diagnosis away from vasculitis and focuses attention on hypercoagulable states with prominent dermal manifestations, including antiphospholipid antibody syndrome (APLS) and livedoid vasculopathy. In this young woman with arthralgias, consideration of SLE and APLS is warranted. Her recent increase in stress and widespread purpuric and ulcerative lesions could bring to mind a factitious disorder, but the histology results eliminate this possibility.
The patient's temperaturewas 36.5C, her blood pressure was 110/70 mmHg, respiratory rate was 16 breaths per minute, and her heart rate was 65 beats per minute. She was well‐appearing but in moderate pain. She did not have any oral lesions. Her cardiac, respiratory, and abdominal exams were normal. Skin exam revealed a 10‐cm by 4‐cm area of bloody granulation tissue draining serosanguinous fluid, surrounded by stellate palpable netlike purpura on her left posterior calf. There was a similar 4‐cm by 2‐cm ulcerated lesion on her right shin. Both lesions were exquisitely tender to palpation. On her bilateral thighs and hips, there were multiple stellate purpuric patches, all 4 cm in diameter or less, and only minimally tender to palpation. She also had 1‐cm purpuric bullae on the helices of both ears (Figure 1) which were slightly tender to palpation. Splinter hemorrhages were also noted on multiple nail beds bilaterally. Musculoskeletal exam did not reveal any synovitis.
The original purpura on her calf and ear demonstrate a clear demarcation corresponding to cutaneous vascular insufficiency. The development of bullae (ear) and ulceration (calf) are compatible with ischemia. Despite the presence of multiple splinter hemorrhages, the distribution of lesions is very unusual for an embolic phenomenon (eg, endocarditis, cholesterol emboli, or atrial myxoma). The multifocal nature of the skin lesions with progression to well‐demarcated cutaneous necrosis is reminiscent of calciphylaxis or warfarin‐induced skin necrosis, although she lacks the relevant risk factors. A toxin such as cocaine or methamphetamine mediating multifocal vasoconstriction or hypercoagulability should be excluded.
The bilateral ear involvement remains decidedly unusual and makes me wonder if there is something about the ear, such as the nature of its circulation or its potentially lower temperature (as an acral organ) that might render it particularly susceptible, for instance, to cryoglobulinemia or cryofibrinogenemia‐mediated ischemia.
Laboratory studiesdemonstrated: white blood cell count of 1500/mm3 (37.3% neutrophils, 5.1% lymphocytes, 6.7% monocytes, and 1.3% eosinophils); hemoglobin 9.3 g/dl (mean corpuscular volume 91 fL); platelet count 212/mm3; erythrocyte sedimentation rate 62 mm/hr; C‐reactive protein 14.6 mg/L. Serum electrolytes, liver tests, coagulation studies, and urinalysis were normal. Fecal occult blood test was negative.
Her neutropenia and anemia suggest decreased production in the marrow by infection, malignancy, or toxin, or increased destruction, perhaps from an autoimmune process. The associated infections are usually viral, such as human immunodeficiency virus (HIV) and Epstein‐Barr virus (EBV), although their linkage with her cutaneous disease is tenuous. It is possible that malignancy could be present in the marrow with resultant dermal hypercoagulability and ischemia, but this seems unlikely. We do not know about any toxins that she has been exposed to, but these hematologic findings would mandate directed inquiry along those lines. In this young woman with cutaneous ulcers secondary to thrombotic vasculopathy, bicytopenia, antecedent arthralgias without synovitis, and elevated inflammatory markers, I favor an autoimmune process such as SLE, which I would evaluate with an antinuclear antibody (ANA) and antiphospholipid antibody studies.
She was admittedto the hospital and received hydromorphone for pain control. Corticosteroids were not administered. Peripheral blood morphology was normal. Antibodies against HIV1 and 2 were negative, as were antibodies against cytomegalovirus, EBV, parvovirus B19, mycoplasma pneumoniae, and hepatitis C virus. Bilateral lower extremity ultrasound was negative for deep vein thrombosis. Transthoracic echocardiogram was normal. Repeat skin biopsy confirmed small vessel vasculopathy without vasculitis (Figure 2). The results of the following investigations were also negative: ANA, rheumatoid factor, double‐stranded DNA (dsDNA), cyclic citrullinated peptide, ribonucleoprotein (RNP), and anti‐Smith antibodies. C3 and C4 complement levels were normal.
Given how much the histology is driving the clinical reasoning and focusing the differential diagnosis in this case, I agree with the decision to repeat the biopsy. In complex or undiagnosed cases, repeat histology samples allow for confirmation of the original interpretation (often with the perspective of new clinicians and pathologists) and sometimes reveal pathognomonic or additional findings that only appear after the disease has evolved over time. HIV seronegativity helps constrain the differential diagnosis, and parvovirus is another excellent consideration for arthralgias and cytopenias (with the predilection to involve cells lines other than RBCs particularly seen in HIV), although ulcers are not seen with this condition. Herpes simplex virus (HSV) is another viral infection that can cause painful skin ulcerations and cytopenias, although the duration and distribution are highly atypical. The negative ANA and dsDNA and normal complement levels make SLE unlikely. The negative lower extremity ultrasound helps frame the thromboses as a local cutaneous process rather than a systemic hypercoagulable state. Although the peripheral blood smear is normal, a bone marrow biopsy will be necessary to exclude a marrow invasive process, such as leukemia or lymphoma. A bone marrow biopsy would also provide another opportunity to examine tissue for mycobacteria or fungi which can cause ulcerations and cytopenias, although there is little reason to suspect she is susceptible to those pathogens. As this clinical picture fails to fit clearly with an infectious, autoimmune, or neoplastic disorder, I would revisit the possibility of toxinsprescription, complementary, over‐the‐counter, or illegal (eg, cocaine) at this time.
In further discussionwith the patient, she reported using cocaine intranasally for the past three months. Her urine toxicology was positive for cocaine. She was found to have positive perinuclear antineutrophil cytoplasmic antibodies (p‐ANCA), antimyeloperoxidase (MPO) antibodies, anticardiolipin (ACL) antibodies, and lupus anticoagulant (LAC). By hospital day 3, her lesions had significantly improved without any intervention, and her absolute neutrophil count increased to 1080/mm3.
The presence of widespread cutaneous ischemia (with bland thrombosis) and detectable ACL and LAC antibodies is compatible with APLS; the APLS could be deemed primary, because there is no clear associated rheumatologic or other systemic disease. However, neutropenia is not a characteristic of APLS, which has thrombocytopenia as its more frequently associated hematologic abnormality. Livedoid vasculopathy, a related disorder, is also supported by the ACL and LAC results, but also does not feature neutropenia. While the presence of diffuse thrombosis could be attributed to a widespread secondary effect of cocaine vasoconstriction, the appearance of ANCA (which can be drug‐induced, eg, propylthiouracil [PTU]) and the slowly resolving neutropenia during hospitalization without specific treatment is very suggestive of a toxin. The demographic, diffuse skin ulcers, and hematologic and serologic profile is compatible with the recently described toxidrome related to levamisole adulteration of cocaine.
A send‐out studyof a urine sample returned positive for levamisole. Based on purpuric skin lesions with a predilection for the ears, agranulocytosis, and skin biopsy revealing thrombotic vasculopathy, she was diagnosed with levamisole‐adulterated cocaine exposure. One week after discharge, her lower extremity pain and ulcerations were significantly improved. Her absolute neutrophil count increased to 2820/mm.3 Her urine toxicology screen was negative for cocaine.
DISCUSSION
Levamisole was initially developed in 1964 as an antihelminthic agent. Its incidentally discovered immunomodulatory effects led to trials for the treatment of chronic infections, inflammatory bowel disease, rheumatic diseases,1 and nephrotic syndrome in children.2 By 1990, 3 major studies supported levamisole as an adjunctive therapy in melanoma3 and colon cancer.4
Although levamisole appeared to be nontoxic at single or low doses, long‐term use in clinical trials demonstrated that 2.5%‐13% of patients developed life‐threatening agranulocytosis, and up to 10% of those instances resulted in death.5 A distinctive cutaneous pseudovasculitis was noted in children on therapeutic levamisole. They presented with purpura that had a predilection for the ears, cheeks, and thighs,6 and positive serologic markers for ANCA and antiphospholipid antibodies. Skin biopsies of the purpuric lesions revealed leukocytoclastic vasculitis, thrombotic vasculitis, and/or vascular occlusions.
Levamisole was withdrawn from the market in 2000 in the United States due to its side effects,7 but quickly found its way onto the black market. It was first detected in cocaine in 2002, and the percentage of cocaine containing levamisole has steadily been increasing since then. In July 2009, over 70% of cocaine seized by the Drug Enforcement Administration was found to contain levamisole.8 It is unclear exactly why this drug is used as an adulterant in cocaine. Theories include potentiation of the euphoric effects of cocaine, serving as a bulking agent, or functioning as a chemical signature to track distribution.9
The resurgence of levamisole has brought a new face to a problem seen over a decade ago. Current reports of levamisole toxicity describe adults presenting with purpura preferentially involving the ears, neutropenia, positive ANCA, and positive antiphospholipid antibodies.1012 Since 2002, there have been at least 20 confirmed cases of agranulocytosis and two deaths associated with levamisole‐adulterated cocaine.8, 13, 14 In September 2009, the Department of Health and Human Services issued a public health alert warning of an impending increase in levamisole‐related illness.
Levamisole is not detected on routine toxicology screens, but can be tested for using gas chromatography and mass spectrometry. Most laboratories do not offer testing for levamisole and send‐out testing is required. Given its half‐life of 5.6 hours, levamisole can only be detected in the blood within 24 hours, and in the urine within 48‐72 hours of exposure.15, 16 Urine samples are preferred over blood samples, since blood levels decline more rapidly and have lower sensitivity. Cocaine can also be sent out to local or state forensics laboratories to be tested for levamisole. The only definitive treatment for levamisole‐induced cutaneous pseudovasculitis and neutropenia is cessation of toxin exposure.
Although the discussant had familiarity with this toxidrome from local and published cases, he was only able to settle on levamisole toxicity after a series of competing hypotheses were ruled out on the basis of irreconcilable features (vasculitis and histology results; APLS and neutropenia; SLE and negative ANA with no visceral involvement) and by using analogical reasoning (eg, to infer the presence of a toxin on the basis of neutropenia [as seen with chemotherapy and other drugs] and ANCA induction [as seen with PTU]). It was a laborious process of hypothesis testing, but one that ultimately allowed him to crack the case.
Key Teaching Points
-
In patients presenting with neutropenia and purpuric skin lesionsparticularly with a predilection for the earsconsider levamisole‐adulterated cocaine exposure.
-
Tests supporting this diagnosis include positive serologies for ANCA and antiphospholipid antibodies, and skin biopsies that show leukocytoclastic vasculitis, thrombotic vasculitis, or vascular occlusion. Urine studies for levamisole are definitive if sent within 48 to 72 hours of exposure.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 43‐year‐old woman presented to an outside hospital with painful plaques and patches on her bilateral lower extremities. Two weeks prior to presentation, she had noticed a single red lesion on her left ankle. Over the next two weeks, the lesion enlarged to involve the lower half of her posterior calf and subsequently turned purple and became exquisitely tender. Similar but smaller purple, tender lesions simultaneously appeared, first over her right shin and then on her bilateral thighs and hips. She also reported fatigue as well as diffuse joint pains in her hands and wrists bilaterally for the past month. She denied any swelling of these joints or functional impairment. She denied fevers, weight loss, headache, sinus symptoms, difficulty breathing, or abdominal pain.
Although we do not yet have a physical exam, the tempo, pattern of spread, and accompanying features allow some early hypotheses to be considered. Distal lower extremity lesions which darkened and spread could be erythema nodosum or erythema induratum. Malignancies rarely have such prominent skin manifestations, although leukemia cutis or an aggressive cutaneous T cell lymphoma might present with disseminated and darkened plaques, and Kaposi's sarcoma is characteristically purple and multifocal. Autoimmune disorders such as sarcoidosis, cutaneous lupus, and psoriasis may similarly present with widespread plaques. Most disseminated infections that start with patches evolve to pustules, ulcers, bullae, or other forms that reflect the invasive nature of the infection; syphilis warrants consideration for any widespread eruption of unknown etiology. Antecedent arthralgias with fatigue suggest an autoimmune condition, although infections such as hepatitis or parvovirus can do the same. Systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) would be favored initially on account of her demographics and the hand and wrist involvement, and each can be associated with vasculitis.
The significant pain as described is not compatible with most of the aforementioned diagnoses. Its presence, coupled with potential autoimmune symptoms, suggests a vasculitis such as polyarteritis nodosa (which can have prominent diffuse skin involvement), Henoch Schonlein purpura (with its predilection for the lower extremities, including extension to the hips and buttocks), cryoglobulinemia, or SLE‐ or RA‐associated vasculitis. Calciphylaxis is another ischemic vascular disorder that can cause diffuse dark painful lesions, but this only warrants consideration if advanced renal disease is present.
A skin biopsy ofher right hip was taken at the outside hospital. She was discharged on a two‐week course of prednisone for suspected vasculitis while biopsy results were pending. Over the next two weeks, none of the skin lesions improved, despite compliance with this treatment, and the skin over her left posterior calf and right shin lesions began to erode and bleed. In addition, small purple, tender lesions appeared over the pinnae of both ears. Three weeks after her initial evaluation, she presented to another emergency department for ulcerating skin lesions and worsening pain. At that point, the initial skin biopsy result was available and revealed vasculopathy of the small vessels with thrombi but no vasculitis.
The patient had no children,and denied a history of miscarriages. Her past medical history was unremarkable. She did not report any history of thrombotic events. She started a new job as a software engineer one month ago and was feeling stressed about her new responsibilities. She denied any high‐risk sexual behavior and any history of intravenous drug use. She had not traveled recently and did not own any pets. There was no family history of rheumatologic disorders, hypercoagulable states, or thrombotic events.
This picture of occluded but noninflamed vessels shifts the diagnosis away from vasculitis and focuses attention on hypercoagulable states with prominent dermal manifestations, including antiphospholipid antibody syndrome (APLS) and livedoid vasculopathy. In this young woman with arthralgias, consideration of SLE and APLS is warranted. Her recent increase in stress and widespread purpuric and ulcerative lesions could bring to mind a factitious disorder, but the histology results eliminate this possibility.
The patient's temperaturewas 36.5C, her blood pressure was 110/70 mmHg, respiratory rate was 16 breaths per minute, and her heart rate was 65 beats per minute. She was well‐appearing but in moderate pain. She did not have any oral lesions. Her cardiac, respiratory, and abdominal exams were normal. Skin exam revealed a 10‐cm by 4‐cm area of bloody granulation tissue draining serosanguinous fluid, surrounded by stellate palpable netlike purpura on her left posterior calf. There was a similar 4‐cm by 2‐cm ulcerated lesion on her right shin. Both lesions were exquisitely tender to palpation. On her bilateral thighs and hips, there were multiple stellate purpuric patches, all 4 cm in diameter or less, and only minimally tender to palpation. She also had 1‐cm purpuric bullae on the helices of both ears (Figure 1) which were slightly tender to palpation. Splinter hemorrhages were also noted on multiple nail beds bilaterally. Musculoskeletal exam did not reveal any synovitis.
The original purpura on her calf and ear demonstrate a clear demarcation corresponding to cutaneous vascular insufficiency. The development of bullae (ear) and ulceration (calf) are compatible with ischemia. Despite the presence of multiple splinter hemorrhages, the distribution of lesions is very unusual for an embolic phenomenon (eg, endocarditis, cholesterol emboli, or atrial myxoma). The multifocal nature of the skin lesions with progression to well‐demarcated cutaneous necrosis is reminiscent of calciphylaxis or warfarin‐induced skin necrosis, although she lacks the relevant risk factors. A toxin such as cocaine or methamphetamine mediating multifocal vasoconstriction or hypercoagulability should be excluded.
The bilateral ear involvement remains decidedly unusual and makes me wonder if there is something about the ear, such as the nature of its circulation or its potentially lower temperature (as an acral organ) that might render it particularly susceptible, for instance, to cryoglobulinemia or cryofibrinogenemia‐mediated ischemia.
Laboratory studiesdemonstrated: white blood cell count of 1500/mm3 (37.3% neutrophils, 5.1% lymphocytes, 6.7% monocytes, and 1.3% eosinophils); hemoglobin 9.3 g/dl (mean corpuscular volume 91 fL); platelet count 212/mm3; erythrocyte sedimentation rate 62 mm/hr; C‐reactive protein 14.6 mg/L. Serum electrolytes, liver tests, coagulation studies, and urinalysis were normal. Fecal occult blood test was negative.
Her neutropenia and anemia suggest decreased production in the marrow by infection, malignancy, or toxin, or increased destruction, perhaps from an autoimmune process. The associated infections are usually viral, such as human immunodeficiency virus (HIV) and Epstein‐Barr virus (EBV), although their linkage with her cutaneous disease is tenuous. It is possible that malignancy could be present in the marrow with resultant dermal hypercoagulability and ischemia, but this seems unlikely. We do not know about any toxins that she has been exposed to, but these hematologic findings would mandate directed inquiry along those lines. In this young woman with cutaneous ulcers secondary to thrombotic vasculopathy, bicytopenia, antecedent arthralgias without synovitis, and elevated inflammatory markers, I favor an autoimmune process such as SLE, which I would evaluate with an antinuclear antibody (ANA) and antiphospholipid antibody studies.
She was admittedto the hospital and received hydromorphone for pain control. Corticosteroids were not administered. Peripheral blood morphology was normal. Antibodies against HIV1 and 2 were negative, as were antibodies against cytomegalovirus, EBV, parvovirus B19, mycoplasma pneumoniae, and hepatitis C virus. Bilateral lower extremity ultrasound was negative for deep vein thrombosis. Transthoracic echocardiogram was normal. Repeat skin biopsy confirmed small vessel vasculopathy without vasculitis (Figure 2). The results of the following investigations were also negative: ANA, rheumatoid factor, double‐stranded DNA (dsDNA), cyclic citrullinated peptide, ribonucleoprotein (RNP), and anti‐Smith antibodies. C3 and C4 complement levels were normal.
Given how much the histology is driving the clinical reasoning and focusing the differential diagnosis in this case, I agree with the decision to repeat the biopsy. In complex or undiagnosed cases, repeat histology samples allow for confirmation of the original interpretation (often with the perspective of new clinicians and pathologists) and sometimes reveal pathognomonic or additional findings that only appear after the disease has evolved over time. HIV seronegativity helps constrain the differential diagnosis, and parvovirus is another excellent consideration for arthralgias and cytopenias (with the predilection to involve cells lines other than RBCs particularly seen in HIV), although ulcers are not seen with this condition. Herpes simplex virus (HSV) is another viral infection that can cause painful skin ulcerations and cytopenias, although the duration and distribution are highly atypical. The negative ANA and dsDNA and normal complement levels make SLE unlikely. The negative lower extremity ultrasound helps frame the thromboses as a local cutaneous process rather than a systemic hypercoagulable state. Although the peripheral blood smear is normal, a bone marrow biopsy will be necessary to exclude a marrow invasive process, such as leukemia or lymphoma. A bone marrow biopsy would also provide another opportunity to examine tissue for mycobacteria or fungi which can cause ulcerations and cytopenias, although there is little reason to suspect she is susceptible to those pathogens. As this clinical picture fails to fit clearly with an infectious, autoimmune, or neoplastic disorder, I would revisit the possibility of toxinsprescription, complementary, over‐the‐counter, or illegal (eg, cocaine) at this time.
In further discussionwith the patient, she reported using cocaine intranasally for the past three months. Her urine toxicology was positive for cocaine. She was found to have positive perinuclear antineutrophil cytoplasmic antibodies (p‐ANCA), antimyeloperoxidase (MPO) antibodies, anticardiolipin (ACL) antibodies, and lupus anticoagulant (LAC). By hospital day 3, her lesions had significantly improved without any intervention, and her absolute neutrophil count increased to 1080/mm3.
The presence of widespread cutaneous ischemia (with bland thrombosis) and detectable ACL and LAC antibodies is compatible with APLS; the APLS could be deemed primary, because there is no clear associated rheumatologic or other systemic disease. However, neutropenia is not a characteristic of APLS, which has thrombocytopenia as its more frequently associated hematologic abnormality. Livedoid vasculopathy, a related disorder, is also supported by the ACL and LAC results, but also does not feature neutropenia. While the presence of diffuse thrombosis could be attributed to a widespread secondary effect of cocaine vasoconstriction, the appearance of ANCA (which can be drug‐induced, eg, propylthiouracil [PTU]) and the slowly resolving neutropenia during hospitalization without specific treatment is very suggestive of a toxin. The demographic, diffuse skin ulcers, and hematologic and serologic profile is compatible with the recently described toxidrome related to levamisole adulteration of cocaine.
A send‐out studyof a urine sample returned positive for levamisole. Based on purpuric skin lesions with a predilection for the ears, agranulocytosis, and skin biopsy revealing thrombotic vasculopathy, she was diagnosed with levamisole‐adulterated cocaine exposure. One week after discharge, her lower extremity pain and ulcerations were significantly improved. Her absolute neutrophil count increased to 2820/mm.3 Her urine toxicology screen was negative for cocaine.
DISCUSSION
Levamisole was initially developed in 1964 as an antihelminthic agent. Its incidentally discovered immunomodulatory effects led to trials for the treatment of chronic infections, inflammatory bowel disease, rheumatic diseases,1 and nephrotic syndrome in children.2 By 1990, 3 major studies supported levamisole as an adjunctive therapy in melanoma3 and colon cancer.4
Although levamisole appeared to be nontoxic at single or low doses, long‐term use in clinical trials demonstrated that 2.5%‐13% of patients developed life‐threatening agranulocytosis, and up to 10% of those instances resulted in death.5 A distinctive cutaneous pseudovasculitis was noted in children on therapeutic levamisole. They presented with purpura that had a predilection for the ears, cheeks, and thighs,6 and positive serologic markers for ANCA and antiphospholipid antibodies. Skin biopsies of the purpuric lesions revealed leukocytoclastic vasculitis, thrombotic vasculitis, and/or vascular occlusions.
Levamisole was withdrawn from the market in 2000 in the United States due to its side effects,7 but quickly found its way onto the black market. It was first detected in cocaine in 2002, and the percentage of cocaine containing levamisole has steadily been increasing since then. In July 2009, over 70% of cocaine seized by the Drug Enforcement Administration was found to contain levamisole.8 It is unclear exactly why this drug is used as an adulterant in cocaine. Theories include potentiation of the euphoric effects of cocaine, serving as a bulking agent, or functioning as a chemical signature to track distribution.9
The resurgence of levamisole has brought a new face to a problem seen over a decade ago. Current reports of levamisole toxicity describe adults presenting with purpura preferentially involving the ears, neutropenia, positive ANCA, and positive antiphospholipid antibodies.1012 Since 2002, there have been at least 20 confirmed cases of agranulocytosis and two deaths associated with levamisole‐adulterated cocaine.8, 13, 14 In September 2009, the Department of Health and Human Services issued a public health alert warning of an impending increase in levamisole‐related illness.
Levamisole is not detected on routine toxicology screens, but can be tested for using gas chromatography and mass spectrometry. Most laboratories do not offer testing for levamisole and send‐out testing is required. Given its half‐life of 5.6 hours, levamisole can only be detected in the blood within 24 hours, and in the urine within 48‐72 hours of exposure.15, 16 Urine samples are preferred over blood samples, since blood levels decline more rapidly and have lower sensitivity. Cocaine can also be sent out to local or state forensics laboratories to be tested for levamisole. The only definitive treatment for levamisole‐induced cutaneous pseudovasculitis and neutropenia is cessation of toxin exposure.
Although the discussant had familiarity with this toxidrome from local and published cases, he was only able to settle on levamisole toxicity after a series of competing hypotheses were ruled out on the basis of irreconcilable features (vasculitis and histology results; APLS and neutropenia; SLE and negative ANA with no visceral involvement) and by using analogical reasoning (eg, to infer the presence of a toxin on the basis of neutropenia [as seen with chemotherapy and other drugs] and ANCA induction [as seen with PTU]). It was a laborious process of hypothesis testing, but one that ultimately allowed him to crack the case.
Key Teaching Points
-
In patients presenting with neutropenia and purpuric skin lesionsparticularly with a predilection for the earsconsider levamisole‐adulterated cocaine exposure.
-
Tests supporting this diagnosis include positive serologies for ANCA and antiphospholipid antibodies, and skin biopsies that show leukocytoclastic vasculitis, thrombotic vasculitis, or vascular occlusion. Urine studies for levamisole are definitive if sent within 48 to 72 hours of exposure.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,.Levamisole, the story and the lessons.Int J Immunopharmocol.1992;14(3):481–486.
- British Association for Paediatric Nephrology.Levamisole for corticosteroid‐dependent nephrotic syndrome in childhood.Lancet.1991;337:1555–1557.
- ,,,,.Improved survival in patients with poor‐prognosis malignant melanoma treated with adjuvant levamisole: a phase III study by the National Cancer Institute of Canada Clinical Trials Group.J Clin Oncol.1991;9:729–735.
- ,,.Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma.N Engl J Med.1990;322:352–358.
- ,,.Studies on levamisole‐induced agranulocytosis.Blood.1980;56(3):388–396.
- ,,.Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long‐term treatment with levamisole in children.Br J Dermatol.1999;140:948–951.
- . Janssen Discontinues Ergamisol. Available at: http://findarticles.com/p/articles/mi_m3374/is_18_22/ai_68536218/. Accessed July 25,2010.
- SAMHSA. Nationwide Public Health Alert Issued Concerning Life‐Threatening Risk Posed by Cocaine Laced with Veterinary Anti‐Parasite Drug. Available at: http://www.samhsa.gov/newsroom/advisories/090921vet5101.aspx. Accessed July 20,2010.
- .Unusual adulterants in cocaine seized on Italian clandestine market.Forensic Sci Int.2007;172(2–3):e1.
- ,,.Levamisole‐induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Jun 12.
- ,,,.Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole.Ann Intern Med.2010;1;152(11):758–759.
- ,,,,.Cocaine‐associated retiform purpura and neutropenia: is levamisole the culprit?J Am Acad Dermatol.2010;Mar 19.
- ,,,.A confirmed case of agranulocytosis after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Apr 1.
- Centers for Disease Control and Prevention.Agranulocytosis associated with cocaine use—four States, March 2008–November 2009.MMWR.2009;58(49):1381–1385.
- ,,.Levamisole as a contaminant of illicit cocaine.Journal of the Clandestine Laboratory Investigating Chemists Association.2006;16:6–11. Available at: http://www.tiaft2006.org/proceedings/pdf/PT‐p‐06.pdf. Accessed July 20, 2010.
- . Cocaine Cutting Agents—A Discussion. Laboratory Medicine and Pathology, University of Alberta. Available at: http://www.vandu.org/documents/Levamisole_Cocaine.pdf. Accessed July 20,2010.
- ,.Levamisole, the story and the lessons.Int J Immunopharmocol.1992;14(3):481–486.
- British Association for Paediatric Nephrology.Levamisole for corticosteroid‐dependent nephrotic syndrome in childhood.Lancet.1991;337:1555–1557.
- ,,,,.Improved survival in patients with poor‐prognosis malignant melanoma treated with adjuvant levamisole: a phase III study by the National Cancer Institute of Canada Clinical Trials Group.J Clin Oncol.1991;9:729–735.
- ,,.Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma.N Engl J Med.1990;322:352–358.
- ,,.Studies on levamisole‐induced agranulocytosis.Blood.1980;56(3):388–396.
- ,,.Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long‐term treatment with levamisole in children.Br J Dermatol.1999;140:948–951.
- . Janssen Discontinues Ergamisol. Available at: http://findarticles.com/p/articles/mi_m3374/is_18_22/ai_68536218/. Accessed July 25,2010.
- SAMHSA. Nationwide Public Health Alert Issued Concerning Life‐Threatening Risk Posed by Cocaine Laced with Veterinary Anti‐Parasite Drug. Available at: http://www.samhsa.gov/newsroom/advisories/090921vet5101.aspx. Accessed July 20,2010.
- .Unusual adulterants in cocaine seized on Italian clandestine market.Forensic Sci Int.2007;172(2–3):e1.
- ,,.Levamisole‐induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Jun 12.
- ,,,.Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole.Ann Intern Med.2010;1;152(11):758–759.
- ,,,,.Cocaine‐associated retiform purpura and neutropenia: is levamisole the culprit?J Am Acad Dermatol.2010;Mar 19.
- ,,,.A confirmed case of agranulocytosis after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Apr 1.
- Centers for Disease Control and Prevention.Agranulocytosis associated with cocaine use—four States, March 2008–November 2009.MMWR.2009;58(49):1381–1385.
- ,,.Levamisole as a contaminant of illicit cocaine.Journal of the Clandestine Laboratory Investigating Chemists Association.2006;16:6–11. Available at: http://www.tiaft2006.org/proceedings/pdf/PT‐p‐06.pdf. Accessed July 20, 2010.
- . Cocaine Cutting Agents—A Discussion. Laboratory Medicine and Pathology, University of Alberta. Available at: http://www.vandu.org/documents/Levamisole_Cocaine.pdf. Accessed July 20,2010.
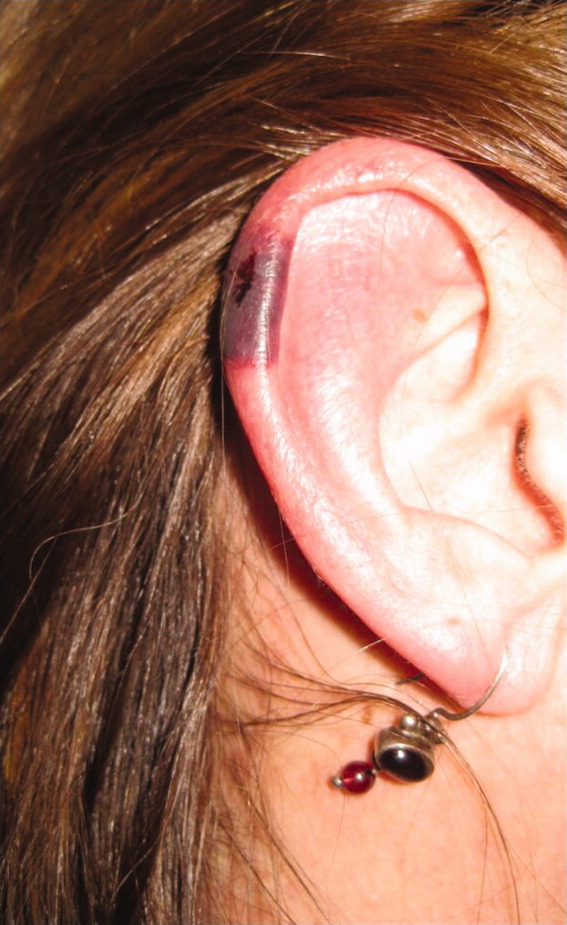
Mapping Out Diagnosis
A 19‐year‐old Japanese man was admitted to a hospital near Kyoto, Japan, because of fever and rash. Two weeks prior to admission, he developed mild headache and low‐grade fever; a rapid test for influenza was negative. His symptoms transiently improved with acetaminophen, but 8 days prior to admission, he developed fever to 38.5C and a pruritic maculopapular rash over his back that spread to his limbs. Six days prior to admission, a chest radiograph was clear; clarithromycin was prescribed for presumed upper respiratory infection. He visited the emergency department the day before admission because of continued fever of greater than 39C, fatigue, and headache. Because there was no jolt accentuation of the headache (ie, worsening with rapid horizontal rotation), or neck pain with extreme neck flexion, he was discharged on acetaminophen. He returned the next day with worsening fatigue and was admitted. He denied chills, rigor, weight loss, photosensitivity, sore throat, neck pain, cough, dyspnea, chest pain, nausea, vomiting, diarrhea, abdominal pain, back pain, and arthralgia.
Fever and diffuse rash are often due to infection, although drugs, autoimmune processes, and cancer must be considered. The presence of headache does not focus the differential diagnosis substantially, because many of the candidate diagnoses can be accompanied by meningitis or encephalitis, or even more frequently, nonspecific headaches. In one small study, jolt‐induced aggravation of headache was shown to be a sensitive indicator of cerebrospinal fluid pleocytosis. The absence of neck stiffness and the 2‐week duration makes bacterial meningitis unlikely, but a more indolent form of aseptic meningitis may need to be evaluated with a lumbar puncture.
The 2‐week illness without rapid deterioration makes some serious causes of fever and rash, such as toxic shock syndrome, disseminated meningococcal infection, or toxic epidermal necrolysis unlikely. A viral exanthema is possible, although the 2‐week duration is longer than usual. Given his youth, however, his immunization history should be queried, and acute infection with human immunodeficiency virus (HIV) should be considered. A more indolent infection, such as subacute bacterial endocarditis, disseminated gonococcal infection, or syphilis is plausible. Among autoimmune etiologies, systemic lupus erythematosus (SLE) and Behcet's disease (which is prevalent in Japan) can involve the central nervous system and cause fever. A careful inquiry directed at prescribed, complementary, and illicit drugs is required.
The patient's past medical history was notable only for mumps at the age of 10. His medications included acetaminophen, clarithromycin, and an herbal medicine, which he had been taking for the prior several days. He reported no tobacco or illicit drug use and rarely drank alcohol. He had never been sexually active. He worked in a factory and reported occasional contact with silver. He lived with his parents; there was no family history of tuberculosis or connective tissue diseases. His father was from Kyushu (the southernmost major island in Japan) and had chronic hepatitis C. The patient denied recent animal exposure or recent travel. His childhood vaccinations were said to be up to date.
Mumps at age 10 might signal general lack of immunization, in which case childhood viral exanthema‐like measles (characterized by fever, headache, and diffuse rash) would warrant consideration. The listed medications had been started after the onset of illness and therefore are unlikely to be causal. Silver causes at least 2 skin conditionscontact dermatitis and argyriabut not the systemic illness seen here. Human T lymphotropic virus‐1 (HTLV‐1) is endemic in southern Japan, but only a minority of infected humans are afflicted with associated adult T cell leukemia/lymphoma or myelopathy. Leukemia and lymphoma are the most likely cancers to cause fever, rash, and central nervous system involvement (with T cell disorders demonstrating a particular tropism for the skin). Overall, however, the differential has not changed substantially.
On physical examination, the patient was mildly overweight and appeared acutely ill. His blood pressure was 136/78 mm Hg, pulse rate was 76 and regular, temperature was 39.2C and respiratory rate was 20 with an oxygen saturation of 98% on room air. A diffuse but nonconfluent erythematous maculopapular rash was present over his chest wall, back, medial aspects of both thighs, and around the knees. There was no jolt‐induced headache. His eyes, nose, oral cavity, and throat were all clear. The neck was supple. There were palpable lymph nodes, each about 1 cm in size, which were firm and moderately tender, in his left neck and left axilla. Lungs and heart were normal. The abdomen was soft, nontender, with normal bowel sounds and no hepatosplenomegaly. His genitalia were normal. Rectal examination revealed no masses or tenderness and a scant amount of brown stool that was negative for occult blood. Neurologic examination was unremarkable.
The multifocal lymphadenopathy does not help distinguish among the categories of disease under consideration. The diffuse maculopapular rash is similarly nonspecific, occurring more frequently with infection and drug reaction than malignancy and autoimmunity. Acute HIV, Epstein‐Barr virus (EBV), syphilis, SLE, drug exposure, or a hematologic malignancy would all be suitable explanations for fever, headache, diffuse rash, and disseminated lymphadenopathy in a previously healthy young man.
Laboratory data obtained on admission was notable for a white blood cell (WBC) count of 2100/L with 72% neutrophils, 19% lymphocytes, and 9% monocytes. Hemoglobin was 13.5 mg/dL with a mean corpuscular volume of 85 fL. Platelet count was 136,000/L. Erythrocyte sedimentation rate was 26 mm/hour. Serum chemistries revealed a sodium level of 135 mEq/L, potassium level of 3.6 mEq/L, chloride level of 100 mEq/L, blood urea nitrogen of 9.8 mg/dL, creatinine level of 1.0 mg/dL, glucose level of 101 mg/dL, calcium level of 8.8 mg/dL, albumin of 4.6 mg/dL, total protein of 8.4 mg/dL, aspartate aminotransferase of 42 IU/L (normal < 35 IU/L), alanine aminotransferase of 27 IU/L, total bilirubin of 0.5 mg/dL, and lactate dehydrogenase (LDH) level of 463 IU/L (normal < 260 IU/L). Chest radiography and electrocardiogram were normal.
A mild elevation in LDH is nonspecific, but without hemolysis or infarction of the kidney, lung, or muscle, it suggests a lymphoproliferative process. Leukopenia with thrombocytopenia can be seen in a number of disorders, most commonly infections including viruses (e.g., EBV, HIV, dengue), malaria, Rocky Mountain spotted fever, or ehrlichiosis/anaplasmosis. Confirmation of his lack of travel could help prioritize those considerations. An invasive bone marrow disorder cannot be excluded, although the near‐normal hemoglobin argues against it. Autoimmune cytopenias are seen in SLE. Given his age, lymphadenopathy, LDH elevation, and absence of infectious exposures, lymphoma rises to the top of the list.
Noninvasive measures should include examination of the peripheral smear, HIV testing (including HIV RNA for acute infection), EBV serologies, and tests for syphilis and SLE. Lumbar puncture (for evaluation of aseptic meningitis) and lymph node biopsy would be informative. Skin biopsy may be helpful to evaluate for aggressive T cell lymphoproliferative disorder, but this can await the results of initial testing.
The patient was given intravenous fluids and acetaminophen as needed. Blood cultures, urine culture, cytomegalovirus and EBV serologies, hepatitis B surface antigen, hepatitis C virus antibody, HIV antibody, antinuclear antibody, complement and ferritin levels, and quantiferon‐TB were ordered. The urine was normal and a urinary antigen test for Legionella was negative. Contrast‐enhanced computed tomography scan of the chest and abdomen was normal except for mild splenomegaly and an enlarged left axillary lymph node.
The ferritin may have been ordered to help evaluate for Still's disease, which is characterized by sustained fever, lymphadenopathy, and transient rash; however, the characteristic leukocytosis and arthralgias are absent. The computed tomography findings are most notable for the absence of generalized lymphadenopathy or significant hepatosplenomegaly that is seen in lymphoma, leukemia, and lymphotropic processes such as acute EBV infection. The localization of disease to the skin (where the predominant lymphocytes are of T cell origin) with relatively modest lymphadenopathy suggests a T cell lymphoma, perhaps of an indolent variety. Vertical transmission of HTLV‐1 decades ago would make adult T cell leukemia or lymphoma a major consideration.
On the third hospital day, WBC count was 1800/L with 67% neutrophils, 22% lymphocytes, and 1% atypical lymphocytes; LDH rose to 623 IU/L. He had continued fatigue and high fever while the rash gradually faded with oral antihistamines and steroid ointment. On hospital day 4, bone marrow biopsy and skin biopsy of his left thigh were performed.
The further decline in WBC and rise in LDH are modest and therefore do not significantly modify the differential diagnosis. Likewise, 1% atypical lymphocytosis is too low to pinpoint an etiology. Because unremitting fevers start to extend into their third week without a clear source of infection, the probability of malignancy and autoimmunity rise. Improvement with oral antihistamines and topical steroids frequently suggests an underlying allergic process, but the remainder of the clinical picture is not in keeping with atopy or allergy. Cutaneous lymphomas (eg, mycosis fungoides) can have waxing and waning skin manifestations, and can be temporarily or definitively treated by topical steroids. The persistence of his fatigue is of concern given the absence of anemia, cardiopulmonary involvement, or motor weakness.
Bone marrow biopsy showed normocellular marrow with no abnormal cells and some activated macrophages with hemophagocytic activity. Skin biopsy failed to show specific pathology.
His left cervical lymph nodes gradually enlarged. Ultrasound of the neck showed multiple enlarged lymph nodes (left side dominant) with dimension of 17 mm 9 mm 31 mm. Blood and urine cultures returned negative, as did HIV antibody. cytomegalovirus and EBV serologies were consistent with previous infection and the ferritin level was 578 ng/mL (normal, 39‐340 ng/mL). Toxoplasma serology and HTLV‐1 antibody were ordered.
The absence of malignant cells on bone marrow biopsy does not exclude lymphoma, but makes a myelophthisic cause of the cytopenias less likely. The macrophage hemophagocytosis reflects immune activation, which in turn is usually caused by the same viral infections, autoimmune conditions, and lymphoproliferative disorders which constitute the current differential diagnosis.
Bone marrow and skin biopsies are both subject to sampling error, and detection of cutaneous T cell lymphoma is notoriously difficult. However, taken together, the absence of cancer on 2 specimens reduces that possibility.
Sustained unilateral cervical lymphadenopathy with fever in a young Japanese man without any histologic evidence of lymphoma points to Kikuchi's disease, ie, lymphadenitis of unknown etiology associated with varying degrees of systemic manifestations. Fever is a frequent feature, we believe, but diffuse sustained rash, cytopenias, and headache are less common or are seen in severe forms of the disease. The diagnosis of Kikuchi's requires the diligent exclusion of SLE and lymphoma. Examination of the peripheral smear and a lymph node biopsy are required.
Of note, there is also a localized form of Castleman's disease, a nonmalignant lymphoproliferative disorder, that similarly is characterized by focal lymphadenopathy. In distinction to Kikuchi's, however, localized Castleman's is largely asymptomatic and responds marvelously to excision.
On hospital day 9, an excisional biopsy of his left anterior cervical lymph nodes was performed, which revealed paracortical foci with necrosis and a histiocytic cellular infiltrate consistent with subacute necrotizing lymphadenitis (Kikuchi‐Fujimoto disease). Antinuclear antibody, Toxoplasma, and HTLV‐1 antibodies returned negative.
There is no treatment for Kikuchi's. It is usually self‐limited, but steroids are sometimes given for symptomatic control.
His condition began to improve after hospital day 9 without specific treatment, including his WBC count and LDH level. He was discharged home on hospital day 15. In the outpatient clinic 1 and 3 months later, he was well and active without recurrences of any symptoms or laboratory abnormalities. His WBC count was 6600/L and LDH was 268 IU/L.
Commentary
Kikuchi‐Fujimoto disease (KFD), also called Kikuchi's disease, is a benign histiocytic necrotizing lymphadenitis described by both Kikuchi and Fujimoto in 1972.1, 2 It is rare in the United States, but seems more common in Asia, especially Japan, where at least 143 cases have been reported since 1972. The etiology has not been determined, but a viral causeincluding EBV, and human herpesvirus 6 and 8has been suggested.3 An autoimmune etiology is also implicated because of infrequent association with SLE. In general, young women are most likely to be affected. In a review of 244 cases by Kucukardali and colleagues, 77% of patients were female and the mean age was 25; 70% were younger than 30 years of age.4
The common presentation is low‐grade fever with unilateral cervical lymphadenopathy.4 Although generalized lymphadenopathy can occur, it is rare. Other common clinical manifestations include malaise, joint pain, rash, arthritis, and hepatosplenomegaly. No specific laboratory tests for diagnosis are available, but leukopenia (seen in 43% of patients), increased erythrocyte sedimentation rate (40%), and anemia (23%) may be observed.4 In this case, atypical lymphocytes were seen, and are reported in one‐third of patients.5 KFD is generally diagnosed by lymph node biopsy, which typically shows irregular paracortical areas of coagulation necrosis that can distort the nodal architecture, while different types of histiocytes are observed at the margin of necrotic areas.
Other diseases in the differential diagnosisseveral of which were considered by the discussantinclude lymphoma, tuberculosis, SLE, and even metastatic adenocarcinoma. KFD is self‐limited; symptoms typically resolve within 1 to 4 months. Patients with severe manifestations have been treated with anti‐inflammatory drugs and glucocorticosteroids. A recurrence rate of 3% to 4% has been reported.6
The clinicians taking care of this patient initially focused on ruling out those infections occasionally resulting in prolonged fever in a previously healthy young man, such as viruses from the herpes family, HIV, viral hepatitis, tuberculosis, syphilis, infective endocarditis, and intra‐abdominal abscess. Physical examination, specifically lymphadenopathy and mild splenomegaly, made Herpesviridae infections, tuberculosis, syphilis, and lymphoma difficult to exclude. Once the initial evaluation ruled out common infections, attention focused on malignancy and histiocytic necrotizing lymphadenitis, given his ethnicity and geographic location.
The discussant was similarly concerned about infection, malignancy, and noninfectious inflammatory diseases, such as SLE, as possible causes. As evidence of these treatable diseases failed to accumulate, the discussant, an American physician with teaching and clinical experience in Japan, considered endemic diseases such as Behcet's, HTLV‐1, and KFD because they fit the unfolding pattern. Given our global society, clinicians will increasingly benefit from becoming familiar with the less common diseases that afflict the various populations around the world.
Teaching Points
-
The combination of fever, lymphadenopathy, and leukopenia in young adults suggests SLE, lymphoma, and HIV. Clinicians should also consider KFD in patients from Japan and neighboring countries.
-
Lymph node biopsy is usually diagnostic of KFD, although interpretation of histopathology can be difficult and sometimes leads to confusion with SLE and lymphoma.
-
KFD typically resolves without specific treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- .Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study.Acta Hematol Jpn.1972;35:379–380.
- ,,.Cervical subacute necrotizing lymphadenitis: a new clinicopathological agent.Naika.1972;20:920–927.
- ,,,.Enigmatic Kikuchi‐Fujimoto disease: a comprehensive review.Am J Clin Pathol.2004;122:141–152.
- ,,,,,.Kikuchi‐Fujimoto Disease: analysis of 244 cases.Clin Rheumatol.2007;26:50–54.
- ,,,,.Kikuchi's disease: A review and analysis of 61 cases.Otolaryngol Head Neck Surg.2003;128:650–653.
- .Histiocytic necrotizing lymphadenitis of Kikuchi and Fujimoto.Arch Pathol Lab Med.1987;11:1026–1029.
A 19‐year‐old Japanese man was admitted to a hospital near Kyoto, Japan, because of fever and rash. Two weeks prior to admission, he developed mild headache and low‐grade fever; a rapid test for influenza was negative. His symptoms transiently improved with acetaminophen, but 8 days prior to admission, he developed fever to 38.5C and a pruritic maculopapular rash over his back that spread to his limbs. Six days prior to admission, a chest radiograph was clear; clarithromycin was prescribed for presumed upper respiratory infection. He visited the emergency department the day before admission because of continued fever of greater than 39C, fatigue, and headache. Because there was no jolt accentuation of the headache (ie, worsening with rapid horizontal rotation), or neck pain with extreme neck flexion, he was discharged on acetaminophen. He returned the next day with worsening fatigue and was admitted. He denied chills, rigor, weight loss, photosensitivity, sore throat, neck pain, cough, dyspnea, chest pain, nausea, vomiting, diarrhea, abdominal pain, back pain, and arthralgia.
Fever and diffuse rash are often due to infection, although drugs, autoimmune processes, and cancer must be considered. The presence of headache does not focus the differential diagnosis substantially, because many of the candidate diagnoses can be accompanied by meningitis or encephalitis, or even more frequently, nonspecific headaches. In one small study, jolt‐induced aggravation of headache was shown to be a sensitive indicator of cerebrospinal fluid pleocytosis. The absence of neck stiffness and the 2‐week duration makes bacterial meningitis unlikely, but a more indolent form of aseptic meningitis may need to be evaluated with a lumbar puncture.
The 2‐week illness without rapid deterioration makes some serious causes of fever and rash, such as toxic shock syndrome, disseminated meningococcal infection, or toxic epidermal necrolysis unlikely. A viral exanthema is possible, although the 2‐week duration is longer than usual. Given his youth, however, his immunization history should be queried, and acute infection with human immunodeficiency virus (HIV) should be considered. A more indolent infection, such as subacute bacterial endocarditis, disseminated gonococcal infection, or syphilis is plausible. Among autoimmune etiologies, systemic lupus erythematosus (SLE) and Behcet's disease (which is prevalent in Japan) can involve the central nervous system and cause fever. A careful inquiry directed at prescribed, complementary, and illicit drugs is required.
The patient's past medical history was notable only for mumps at the age of 10. His medications included acetaminophen, clarithromycin, and an herbal medicine, which he had been taking for the prior several days. He reported no tobacco or illicit drug use and rarely drank alcohol. He had never been sexually active. He worked in a factory and reported occasional contact with silver. He lived with his parents; there was no family history of tuberculosis or connective tissue diseases. His father was from Kyushu (the southernmost major island in Japan) and had chronic hepatitis C. The patient denied recent animal exposure or recent travel. His childhood vaccinations were said to be up to date.
Mumps at age 10 might signal general lack of immunization, in which case childhood viral exanthema‐like measles (characterized by fever, headache, and diffuse rash) would warrant consideration. The listed medications had been started after the onset of illness and therefore are unlikely to be causal. Silver causes at least 2 skin conditionscontact dermatitis and argyriabut not the systemic illness seen here. Human T lymphotropic virus‐1 (HTLV‐1) is endemic in southern Japan, but only a minority of infected humans are afflicted with associated adult T cell leukemia/lymphoma or myelopathy. Leukemia and lymphoma are the most likely cancers to cause fever, rash, and central nervous system involvement (with T cell disorders demonstrating a particular tropism for the skin). Overall, however, the differential has not changed substantially.
On physical examination, the patient was mildly overweight and appeared acutely ill. His blood pressure was 136/78 mm Hg, pulse rate was 76 and regular, temperature was 39.2C and respiratory rate was 20 with an oxygen saturation of 98% on room air. A diffuse but nonconfluent erythematous maculopapular rash was present over his chest wall, back, medial aspects of both thighs, and around the knees. There was no jolt‐induced headache. His eyes, nose, oral cavity, and throat were all clear. The neck was supple. There were palpable lymph nodes, each about 1 cm in size, which were firm and moderately tender, in his left neck and left axilla. Lungs and heart were normal. The abdomen was soft, nontender, with normal bowel sounds and no hepatosplenomegaly. His genitalia were normal. Rectal examination revealed no masses or tenderness and a scant amount of brown stool that was negative for occult blood. Neurologic examination was unremarkable.
The multifocal lymphadenopathy does not help distinguish among the categories of disease under consideration. The diffuse maculopapular rash is similarly nonspecific, occurring more frequently with infection and drug reaction than malignancy and autoimmunity. Acute HIV, Epstein‐Barr virus (EBV), syphilis, SLE, drug exposure, or a hematologic malignancy would all be suitable explanations for fever, headache, diffuse rash, and disseminated lymphadenopathy in a previously healthy young man.
Laboratory data obtained on admission was notable for a white blood cell (WBC) count of 2100/L with 72% neutrophils, 19% lymphocytes, and 9% monocytes. Hemoglobin was 13.5 mg/dL with a mean corpuscular volume of 85 fL. Platelet count was 136,000/L. Erythrocyte sedimentation rate was 26 mm/hour. Serum chemistries revealed a sodium level of 135 mEq/L, potassium level of 3.6 mEq/L, chloride level of 100 mEq/L, blood urea nitrogen of 9.8 mg/dL, creatinine level of 1.0 mg/dL, glucose level of 101 mg/dL, calcium level of 8.8 mg/dL, albumin of 4.6 mg/dL, total protein of 8.4 mg/dL, aspartate aminotransferase of 42 IU/L (normal < 35 IU/L), alanine aminotransferase of 27 IU/L, total bilirubin of 0.5 mg/dL, and lactate dehydrogenase (LDH) level of 463 IU/L (normal < 260 IU/L). Chest radiography and electrocardiogram were normal.
A mild elevation in LDH is nonspecific, but without hemolysis or infarction of the kidney, lung, or muscle, it suggests a lymphoproliferative process. Leukopenia with thrombocytopenia can be seen in a number of disorders, most commonly infections including viruses (e.g., EBV, HIV, dengue), malaria, Rocky Mountain spotted fever, or ehrlichiosis/anaplasmosis. Confirmation of his lack of travel could help prioritize those considerations. An invasive bone marrow disorder cannot be excluded, although the near‐normal hemoglobin argues against it. Autoimmune cytopenias are seen in SLE. Given his age, lymphadenopathy, LDH elevation, and absence of infectious exposures, lymphoma rises to the top of the list.
Noninvasive measures should include examination of the peripheral smear, HIV testing (including HIV RNA for acute infection), EBV serologies, and tests for syphilis and SLE. Lumbar puncture (for evaluation of aseptic meningitis) and lymph node biopsy would be informative. Skin biopsy may be helpful to evaluate for aggressive T cell lymphoproliferative disorder, but this can await the results of initial testing.
The patient was given intravenous fluids and acetaminophen as needed. Blood cultures, urine culture, cytomegalovirus and EBV serologies, hepatitis B surface antigen, hepatitis C virus antibody, HIV antibody, antinuclear antibody, complement and ferritin levels, and quantiferon‐TB were ordered. The urine was normal and a urinary antigen test for Legionella was negative. Contrast‐enhanced computed tomography scan of the chest and abdomen was normal except for mild splenomegaly and an enlarged left axillary lymph node.
The ferritin may have been ordered to help evaluate for Still's disease, which is characterized by sustained fever, lymphadenopathy, and transient rash; however, the characteristic leukocytosis and arthralgias are absent. The computed tomography findings are most notable for the absence of generalized lymphadenopathy or significant hepatosplenomegaly that is seen in lymphoma, leukemia, and lymphotropic processes such as acute EBV infection. The localization of disease to the skin (where the predominant lymphocytes are of T cell origin) with relatively modest lymphadenopathy suggests a T cell lymphoma, perhaps of an indolent variety. Vertical transmission of HTLV‐1 decades ago would make adult T cell leukemia or lymphoma a major consideration.
On the third hospital day, WBC count was 1800/L with 67% neutrophils, 22% lymphocytes, and 1% atypical lymphocytes; LDH rose to 623 IU/L. He had continued fatigue and high fever while the rash gradually faded with oral antihistamines and steroid ointment. On hospital day 4, bone marrow biopsy and skin biopsy of his left thigh were performed.
The further decline in WBC and rise in LDH are modest and therefore do not significantly modify the differential diagnosis. Likewise, 1% atypical lymphocytosis is too low to pinpoint an etiology. Because unremitting fevers start to extend into their third week without a clear source of infection, the probability of malignancy and autoimmunity rise. Improvement with oral antihistamines and topical steroids frequently suggests an underlying allergic process, but the remainder of the clinical picture is not in keeping with atopy or allergy. Cutaneous lymphomas (eg, mycosis fungoides) can have waxing and waning skin manifestations, and can be temporarily or definitively treated by topical steroids. The persistence of his fatigue is of concern given the absence of anemia, cardiopulmonary involvement, or motor weakness.
Bone marrow biopsy showed normocellular marrow with no abnormal cells and some activated macrophages with hemophagocytic activity. Skin biopsy failed to show specific pathology.
His left cervical lymph nodes gradually enlarged. Ultrasound of the neck showed multiple enlarged lymph nodes (left side dominant) with dimension of 17 mm 9 mm 31 mm. Blood and urine cultures returned negative, as did HIV antibody. cytomegalovirus and EBV serologies were consistent with previous infection and the ferritin level was 578 ng/mL (normal, 39‐340 ng/mL). Toxoplasma serology and HTLV‐1 antibody were ordered.
The absence of malignant cells on bone marrow biopsy does not exclude lymphoma, but makes a myelophthisic cause of the cytopenias less likely. The macrophage hemophagocytosis reflects immune activation, which in turn is usually caused by the same viral infections, autoimmune conditions, and lymphoproliferative disorders which constitute the current differential diagnosis.
Bone marrow and skin biopsies are both subject to sampling error, and detection of cutaneous T cell lymphoma is notoriously difficult. However, taken together, the absence of cancer on 2 specimens reduces that possibility.
Sustained unilateral cervical lymphadenopathy with fever in a young Japanese man without any histologic evidence of lymphoma points to Kikuchi's disease, ie, lymphadenitis of unknown etiology associated with varying degrees of systemic manifestations. Fever is a frequent feature, we believe, but diffuse sustained rash, cytopenias, and headache are less common or are seen in severe forms of the disease. The diagnosis of Kikuchi's requires the diligent exclusion of SLE and lymphoma. Examination of the peripheral smear and a lymph node biopsy are required.
Of note, there is also a localized form of Castleman's disease, a nonmalignant lymphoproliferative disorder, that similarly is characterized by focal lymphadenopathy. In distinction to Kikuchi's, however, localized Castleman's is largely asymptomatic and responds marvelously to excision.
On hospital day 9, an excisional biopsy of his left anterior cervical lymph nodes was performed, which revealed paracortical foci with necrosis and a histiocytic cellular infiltrate consistent with subacute necrotizing lymphadenitis (Kikuchi‐Fujimoto disease). Antinuclear antibody, Toxoplasma, and HTLV‐1 antibodies returned negative.
There is no treatment for Kikuchi's. It is usually self‐limited, but steroids are sometimes given for symptomatic control.
His condition began to improve after hospital day 9 without specific treatment, including his WBC count and LDH level. He was discharged home on hospital day 15. In the outpatient clinic 1 and 3 months later, he was well and active without recurrences of any symptoms or laboratory abnormalities. His WBC count was 6600/L and LDH was 268 IU/L.
Commentary
Kikuchi‐Fujimoto disease (KFD), also called Kikuchi's disease, is a benign histiocytic necrotizing lymphadenitis described by both Kikuchi and Fujimoto in 1972.1, 2 It is rare in the United States, but seems more common in Asia, especially Japan, where at least 143 cases have been reported since 1972. The etiology has not been determined, but a viral causeincluding EBV, and human herpesvirus 6 and 8has been suggested.3 An autoimmune etiology is also implicated because of infrequent association with SLE. In general, young women are most likely to be affected. In a review of 244 cases by Kucukardali and colleagues, 77% of patients were female and the mean age was 25; 70% were younger than 30 years of age.4
The common presentation is low‐grade fever with unilateral cervical lymphadenopathy.4 Although generalized lymphadenopathy can occur, it is rare. Other common clinical manifestations include malaise, joint pain, rash, arthritis, and hepatosplenomegaly. No specific laboratory tests for diagnosis are available, but leukopenia (seen in 43% of patients), increased erythrocyte sedimentation rate (40%), and anemia (23%) may be observed.4 In this case, atypical lymphocytes were seen, and are reported in one‐third of patients.5 KFD is generally diagnosed by lymph node biopsy, which typically shows irregular paracortical areas of coagulation necrosis that can distort the nodal architecture, while different types of histiocytes are observed at the margin of necrotic areas.
Other diseases in the differential diagnosisseveral of which were considered by the discussantinclude lymphoma, tuberculosis, SLE, and even metastatic adenocarcinoma. KFD is self‐limited; symptoms typically resolve within 1 to 4 months. Patients with severe manifestations have been treated with anti‐inflammatory drugs and glucocorticosteroids. A recurrence rate of 3% to 4% has been reported.6
The clinicians taking care of this patient initially focused on ruling out those infections occasionally resulting in prolonged fever in a previously healthy young man, such as viruses from the herpes family, HIV, viral hepatitis, tuberculosis, syphilis, infective endocarditis, and intra‐abdominal abscess. Physical examination, specifically lymphadenopathy and mild splenomegaly, made Herpesviridae infections, tuberculosis, syphilis, and lymphoma difficult to exclude. Once the initial evaluation ruled out common infections, attention focused on malignancy and histiocytic necrotizing lymphadenitis, given his ethnicity and geographic location.
The discussant was similarly concerned about infection, malignancy, and noninfectious inflammatory diseases, such as SLE, as possible causes. As evidence of these treatable diseases failed to accumulate, the discussant, an American physician with teaching and clinical experience in Japan, considered endemic diseases such as Behcet's, HTLV‐1, and KFD because they fit the unfolding pattern. Given our global society, clinicians will increasingly benefit from becoming familiar with the less common diseases that afflict the various populations around the world.
Teaching Points
-
The combination of fever, lymphadenopathy, and leukopenia in young adults suggests SLE, lymphoma, and HIV. Clinicians should also consider KFD in patients from Japan and neighboring countries.
-
Lymph node biopsy is usually diagnostic of KFD, although interpretation of histopathology can be difficult and sometimes leads to confusion with SLE and lymphoma.
-
KFD typically resolves without specific treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 19‐year‐old Japanese man was admitted to a hospital near Kyoto, Japan, because of fever and rash. Two weeks prior to admission, he developed mild headache and low‐grade fever; a rapid test for influenza was negative. His symptoms transiently improved with acetaminophen, but 8 days prior to admission, he developed fever to 38.5C and a pruritic maculopapular rash over his back that spread to his limbs. Six days prior to admission, a chest radiograph was clear; clarithromycin was prescribed for presumed upper respiratory infection. He visited the emergency department the day before admission because of continued fever of greater than 39C, fatigue, and headache. Because there was no jolt accentuation of the headache (ie, worsening with rapid horizontal rotation), or neck pain with extreme neck flexion, he was discharged on acetaminophen. He returned the next day with worsening fatigue and was admitted. He denied chills, rigor, weight loss, photosensitivity, sore throat, neck pain, cough, dyspnea, chest pain, nausea, vomiting, diarrhea, abdominal pain, back pain, and arthralgia.
Fever and diffuse rash are often due to infection, although drugs, autoimmune processes, and cancer must be considered. The presence of headache does not focus the differential diagnosis substantially, because many of the candidate diagnoses can be accompanied by meningitis or encephalitis, or even more frequently, nonspecific headaches. In one small study, jolt‐induced aggravation of headache was shown to be a sensitive indicator of cerebrospinal fluid pleocytosis. The absence of neck stiffness and the 2‐week duration makes bacterial meningitis unlikely, but a more indolent form of aseptic meningitis may need to be evaluated with a lumbar puncture.
The 2‐week illness without rapid deterioration makes some serious causes of fever and rash, such as toxic shock syndrome, disseminated meningococcal infection, or toxic epidermal necrolysis unlikely. A viral exanthema is possible, although the 2‐week duration is longer than usual. Given his youth, however, his immunization history should be queried, and acute infection with human immunodeficiency virus (HIV) should be considered. A more indolent infection, such as subacute bacterial endocarditis, disseminated gonococcal infection, or syphilis is plausible. Among autoimmune etiologies, systemic lupus erythematosus (SLE) and Behcet's disease (which is prevalent in Japan) can involve the central nervous system and cause fever. A careful inquiry directed at prescribed, complementary, and illicit drugs is required.
The patient's past medical history was notable only for mumps at the age of 10. His medications included acetaminophen, clarithromycin, and an herbal medicine, which he had been taking for the prior several days. He reported no tobacco or illicit drug use and rarely drank alcohol. He had never been sexually active. He worked in a factory and reported occasional contact with silver. He lived with his parents; there was no family history of tuberculosis or connective tissue diseases. His father was from Kyushu (the southernmost major island in Japan) and had chronic hepatitis C. The patient denied recent animal exposure or recent travel. His childhood vaccinations were said to be up to date.
Mumps at age 10 might signal general lack of immunization, in which case childhood viral exanthema‐like measles (characterized by fever, headache, and diffuse rash) would warrant consideration. The listed medications had been started after the onset of illness and therefore are unlikely to be causal. Silver causes at least 2 skin conditionscontact dermatitis and argyriabut not the systemic illness seen here. Human T lymphotropic virus‐1 (HTLV‐1) is endemic in southern Japan, but only a minority of infected humans are afflicted with associated adult T cell leukemia/lymphoma or myelopathy. Leukemia and lymphoma are the most likely cancers to cause fever, rash, and central nervous system involvement (with T cell disorders demonstrating a particular tropism for the skin). Overall, however, the differential has not changed substantially.
On physical examination, the patient was mildly overweight and appeared acutely ill. His blood pressure was 136/78 mm Hg, pulse rate was 76 and regular, temperature was 39.2C and respiratory rate was 20 with an oxygen saturation of 98% on room air. A diffuse but nonconfluent erythematous maculopapular rash was present over his chest wall, back, medial aspects of both thighs, and around the knees. There was no jolt‐induced headache. His eyes, nose, oral cavity, and throat were all clear. The neck was supple. There were palpable lymph nodes, each about 1 cm in size, which were firm and moderately tender, in his left neck and left axilla. Lungs and heart were normal. The abdomen was soft, nontender, with normal bowel sounds and no hepatosplenomegaly. His genitalia were normal. Rectal examination revealed no masses or tenderness and a scant amount of brown stool that was negative for occult blood. Neurologic examination was unremarkable.
The multifocal lymphadenopathy does not help distinguish among the categories of disease under consideration. The diffuse maculopapular rash is similarly nonspecific, occurring more frequently with infection and drug reaction than malignancy and autoimmunity. Acute HIV, Epstein‐Barr virus (EBV), syphilis, SLE, drug exposure, or a hematologic malignancy would all be suitable explanations for fever, headache, diffuse rash, and disseminated lymphadenopathy in a previously healthy young man.
Laboratory data obtained on admission was notable for a white blood cell (WBC) count of 2100/L with 72% neutrophils, 19% lymphocytes, and 9% monocytes. Hemoglobin was 13.5 mg/dL with a mean corpuscular volume of 85 fL. Platelet count was 136,000/L. Erythrocyte sedimentation rate was 26 mm/hour. Serum chemistries revealed a sodium level of 135 mEq/L, potassium level of 3.6 mEq/L, chloride level of 100 mEq/L, blood urea nitrogen of 9.8 mg/dL, creatinine level of 1.0 mg/dL, glucose level of 101 mg/dL, calcium level of 8.8 mg/dL, albumin of 4.6 mg/dL, total protein of 8.4 mg/dL, aspartate aminotransferase of 42 IU/L (normal < 35 IU/L), alanine aminotransferase of 27 IU/L, total bilirubin of 0.5 mg/dL, and lactate dehydrogenase (LDH) level of 463 IU/L (normal < 260 IU/L). Chest radiography and electrocardiogram were normal.
A mild elevation in LDH is nonspecific, but without hemolysis or infarction of the kidney, lung, or muscle, it suggests a lymphoproliferative process. Leukopenia with thrombocytopenia can be seen in a number of disorders, most commonly infections including viruses (e.g., EBV, HIV, dengue), malaria, Rocky Mountain spotted fever, or ehrlichiosis/anaplasmosis. Confirmation of his lack of travel could help prioritize those considerations. An invasive bone marrow disorder cannot be excluded, although the near‐normal hemoglobin argues against it. Autoimmune cytopenias are seen in SLE. Given his age, lymphadenopathy, LDH elevation, and absence of infectious exposures, lymphoma rises to the top of the list.
Noninvasive measures should include examination of the peripheral smear, HIV testing (including HIV RNA for acute infection), EBV serologies, and tests for syphilis and SLE. Lumbar puncture (for evaluation of aseptic meningitis) and lymph node biopsy would be informative. Skin biopsy may be helpful to evaluate for aggressive T cell lymphoproliferative disorder, but this can await the results of initial testing.
The patient was given intravenous fluids and acetaminophen as needed. Blood cultures, urine culture, cytomegalovirus and EBV serologies, hepatitis B surface antigen, hepatitis C virus antibody, HIV antibody, antinuclear antibody, complement and ferritin levels, and quantiferon‐TB were ordered. The urine was normal and a urinary antigen test for Legionella was negative. Contrast‐enhanced computed tomography scan of the chest and abdomen was normal except for mild splenomegaly and an enlarged left axillary lymph node.
The ferritin may have been ordered to help evaluate for Still's disease, which is characterized by sustained fever, lymphadenopathy, and transient rash; however, the characteristic leukocytosis and arthralgias are absent. The computed tomography findings are most notable for the absence of generalized lymphadenopathy or significant hepatosplenomegaly that is seen in lymphoma, leukemia, and lymphotropic processes such as acute EBV infection. The localization of disease to the skin (where the predominant lymphocytes are of T cell origin) with relatively modest lymphadenopathy suggests a T cell lymphoma, perhaps of an indolent variety. Vertical transmission of HTLV‐1 decades ago would make adult T cell leukemia or lymphoma a major consideration.
On the third hospital day, WBC count was 1800/L with 67% neutrophils, 22% lymphocytes, and 1% atypical lymphocytes; LDH rose to 623 IU/L. He had continued fatigue and high fever while the rash gradually faded with oral antihistamines and steroid ointment. On hospital day 4, bone marrow biopsy and skin biopsy of his left thigh were performed.
The further decline in WBC and rise in LDH are modest and therefore do not significantly modify the differential diagnosis. Likewise, 1% atypical lymphocytosis is too low to pinpoint an etiology. Because unremitting fevers start to extend into their third week without a clear source of infection, the probability of malignancy and autoimmunity rise. Improvement with oral antihistamines and topical steroids frequently suggests an underlying allergic process, but the remainder of the clinical picture is not in keeping with atopy or allergy. Cutaneous lymphomas (eg, mycosis fungoides) can have waxing and waning skin manifestations, and can be temporarily or definitively treated by topical steroids. The persistence of his fatigue is of concern given the absence of anemia, cardiopulmonary involvement, or motor weakness.
Bone marrow biopsy showed normocellular marrow with no abnormal cells and some activated macrophages with hemophagocytic activity. Skin biopsy failed to show specific pathology.
His left cervical lymph nodes gradually enlarged. Ultrasound of the neck showed multiple enlarged lymph nodes (left side dominant) with dimension of 17 mm 9 mm 31 mm. Blood and urine cultures returned negative, as did HIV antibody. cytomegalovirus and EBV serologies were consistent with previous infection and the ferritin level was 578 ng/mL (normal, 39‐340 ng/mL). Toxoplasma serology and HTLV‐1 antibody were ordered.
The absence of malignant cells on bone marrow biopsy does not exclude lymphoma, but makes a myelophthisic cause of the cytopenias less likely. The macrophage hemophagocytosis reflects immune activation, which in turn is usually caused by the same viral infections, autoimmune conditions, and lymphoproliferative disorders which constitute the current differential diagnosis.
Bone marrow and skin biopsies are both subject to sampling error, and detection of cutaneous T cell lymphoma is notoriously difficult. However, taken together, the absence of cancer on 2 specimens reduces that possibility.
Sustained unilateral cervical lymphadenopathy with fever in a young Japanese man without any histologic evidence of lymphoma points to Kikuchi's disease, ie, lymphadenitis of unknown etiology associated with varying degrees of systemic manifestations. Fever is a frequent feature, we believe, but diffuse sustained rash, cytopenias, and headache are less common or are seen in severe forms of the disease. The diagnosis of Kikuchi's requires the diligent exclusion of SLE and lymphoma. Examination of the peripheral smear and a lymph node biopsy are required.
Of note, there is also a localized form of Castleman's disease, a nonmalignant lymphoproliferative disorder, that similarly is characterized by focal lymphadenopathy. In distinction to Kikuchi's, however, localized Castleman's is largely asymptomatic and responds marvelously to excision.
On hospital day 9, an excisional biopsy of his left anterior cervical lymph nodes was performed, which revealed paracortical foci with necrosis and a histiocytic cellular infiltrate consistent with subacute necrotizing lymphadenitis (Kikuchi‐Fujimoto disease). Antinuclear antibody, Toxoplasma, and HTLV‐1 antibodies returned negative.
There is no treatment for Kikuchi's. It is usually self‐limited, but steroids are sometimes given for symptomatic control.
His condition began to improve after hospital day 9 without specific treatment, including his WBC count and LDH level. He was discharged home on hospital day 15. In the outpatient clinic 1 and 3 months later, he was well and active without recurrences of any symptoms or laboratory abnormalities. His WBC count was 6600/L and LDH was 268 IU/L.
Commentary
Kikuchi‐Fujimoto disease (KFD), also called Kikuchi's disease, is a benign histiocytic necrotizing lymphadenitis described by both Kikuchi and Fujimoto in 1972.1, 2 It is rare in the United States, but seems more common in Asia, especially Japan, where at least 143 cases have been reported since 1972. The etiology has not been determined, but a viral causeincluding EBV, and human herpesvirus 6 and 8has been suggested.3 An autoimmune etiology is also implicated because of infrequent association with SLE. In general, young women are most likely to be affected. In a review of 244 cases by Kucukardali and colleagues, 77% of patients were female and the mean age was 25; 70% were younger than 30 years of age.4
The common presentation is low‐grade fever with unilateral cervical lymphadenopathy.4 Although generalized lymphadenopathy can occur, it is rare. Other common clinical manifestations include malaise, joint pain, rash, arthritis, and hepatosplenomegaly. No specific laboratory tests for diagnosis are available, but leukopenia (seen in 43% of patients), increased erythrocyte sedimentation rate (40%), and anemia (23%) may be observed.4 In this case, atypical lymphocytes were seen, and are reported in one‐third of patients.5 KFD is generally diagnosed by lymph node biopsy, which typically shows irregular paracortical areas of coagulation necrosis that can distort the nodal architecture, while different types of histiocytes are observed at the margin of necrotic areas.
Other diseases in the differential diagnosisseveral of which were considered by the discussantinclude lymphoma, tuberculosis, SLE, and even metastatic adenocarcinoma. KFD is self‐limited; symptoms typically resolve within 1 to 4 months. Patients with severe manifestations have been treated with anti‐inflammatory drugs and glucocorticosteroids. A recurrence rate of 3% to 4% has been reported.6
The clinicians taking care of this patient initially focused on ruling out those infections occasionally resulting in prolonged fever in a previously healthy young man, such as viruses from the herpes family, HIV, viral hepatitis, tuberculosis, syphilis, infective endocarditis, and intra‐abdominal abscess. Physical examination, specifically lymphadenopathy and mild splenomegaly, made Herpesviridae infections, tuberculosis, syphilis, and lymphoma difficult to exclude. Once the initial evaluation ruled out common infections, attention focused on malignancy and histiocytic necrotizing lymphadenitis, given his ethnicity and geographic location.
The discussant was similarly concerned about infection, malignancy, and noninfectious inflammatory diseases, such as SLE, as possible causes. As evidence of these treatable diseases failed to accumulate, the discussant, an American physician with teaching and clinical experience in Japan, considered endemic diseases such as Behcet's, HTLV‐1, and KFD because they fit the unfolding pattern. Given our global society, clinicians will increasingly benefit from becoming familiar with the less common diseases that afflict the various populations around the world.
Teaching Points
-
The combination of fever, lymphadenopathy, and leukopenia in young adults suggests SLE, lymphoma, and HIV. Clinicians should also consider KFD in patients from Japan and neighboring countries.
-
Lymph node biopsy is usually diagnostic of KFD, although interpretation of histopathology can be difficult and sometimes leads to confusion with SLE and lymphoma.
-
KFD typically resolves without specific treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- .Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study.Acta Hematol Jpn.1972;35:379–380.
- ,,.Cervical subacute necrotizing lymphadenitis: a new clinicopathological agent.Naika.1972;20:920–927.
- ,,,.Enigmatic Kikuchi‐Fujimoto disease: a comprehensive review.Am J Clin Pathol.2004;122:141–152.
- ,,,,,.Kikuchi‐Fujimoto Disease: analysis of 244 cases.Clin Rheumatol.2007;26:50–54.
- ,,,,.Kikuchi's disease: A review and analysis of 61 cases.Otolaryngol Head Neck Surg.2003;128:650–653.
- .Histiocytic necrotizing lymphadenitis of Kikuchi and Fujimoto.Arch Pathol Lab Med.1987;11:1026–1029.
- .Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes: a clinicopathological study.Acta Hematol Jpn.1972;35:379–380.
- ,,.Cervical subacute necrotizing lymphadenitis: a new clinicopathological agent.Naika.1972;20:920–927.
- ,,,.Enigmatic Kikuchi‐Fujimoto disease: a comprehensive review.Am J Clin Pathol.2004;122:141–152.
- ,,,,,.Kikuchi‐Fujimoto Disease: analysis of 244 cases.Clin Rheumatol.2007;26:50–54.
- ,,,,.Kikuchi's disease: A review and analysis of 61 cases.Otolaryngol Head Neck Surg.2003;128:650–653.
- .Histiocytic necrotizing lymphadenitis of Kikuchi and Fujimoto.Arch Pathol Lab Med.1987;11:1026–1029.
In the Face of It All
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, <57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, <2), and lambda was 4.25 mg/dL (normal range, <2).
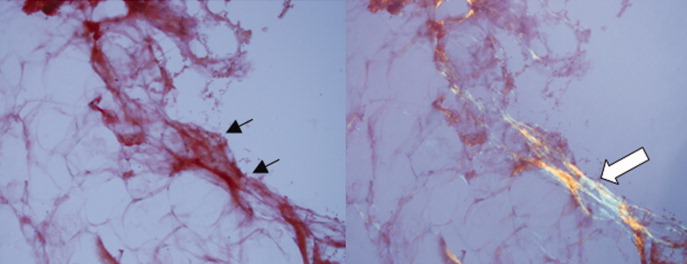
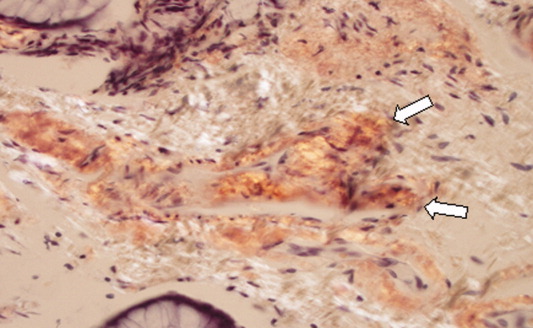
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, <57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, <2), and lambda was 4.25 mg/dL (normal range, <2).
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, <57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, <2), and lambda was 4.25 mg/dL (normal range, <2).
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
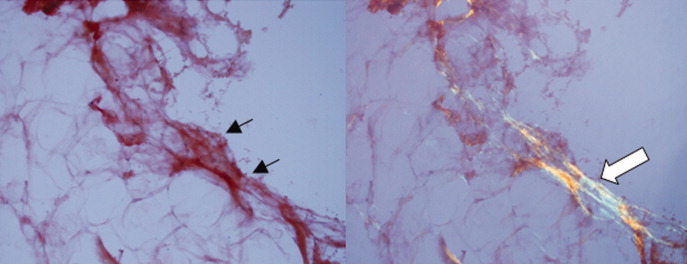
Continuing Medical Education Program in
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
Unscripted
A 58‐year old man was admitted with generalized weakness and acute deep venous thrombosis (DVT). His past medical history included hypertension and polymyositis/dermatomyositis (PM/DM) with anti‐synthase syndrome, which had been diagnosed 16 months prior when his creatine kinase (CK) was greater than 12,000 U/L. At that time he also was found to have bilateral lower extremity DVT, and had been treated with warfarin for 1 year. 10 days previously, he had been discharged after a 4‐day hospitalization for a polymyositis flare which was treated with methylprednisolone at 60 mg daily for 5 days. He was discharged home with daily prednisone until this follow‐up a week later, where he reported weakness and bilateral edema. Lower extremity ultrasound demonstrated acute thrombus in the right common femoral vein.
This acute extensive DVT may be a consequence of recent hospitalization and a previously damaged venous system, or may reflect ongoing hypercoagulability from an unresolved condition, such as cancer. Bilateral lower extremity edema may suggest right‐sided heart failure due to progressive interstitial lung disease, which occurs in a subset of patients with PM/DM. Edema may alternatively reflect biventricular heart failure, or liver or kidney disease.
Generalized weakness offers little in the way of focused differential diagnosis until it is characterized as motor weakness (eg, attributed to progression of the myopathy), a dyspnea‐equivalent, or an overall sense of fatigue.
His medications included weekly methotrexate, monthly intravenous immunoglobulin (IVIG) infusions, tacrolimus, hydrochlorothiazide, and aerosolized pentamidine. He had been on varying doses of prednisone for 2 years and his present dose was 40 mg daily. He was allergic to sulfa. He was married and stopped smoking 30 years previously, and did not drink alcohol or use illicit drugs.
Various medication toxicities could account for his presentation. Methotrexate causes interstitial lung disease, and IVIG and tacrolimus may cause renal failure (and fluid overload). The heavy degree of immunosuppression renders him susceptible to a wide range of infections. Aerosolized pentamidine provides incomplete protection against Pneumocystis jirovecii, especially in the lung apices.
Evaluation of the status of his myositis with motor strength assessment is important. In addition associated rashes and signs of malignancy (eg, lymphadenopathy) and infection should be sought. Proximal motor weakness would suggest a myositis flare, although care must be given to exclude competing causes of myopathy, including infections, toxins, or endocrinopathies.
His temperature was 36.2C, pulse 103 beats per minute, blood pressure 156/83 mm Hg, and respiratory rate 18 breaths per minute. He had crackles at both lung bases, and 3+ pitting edema in both lower extremities. On neurological exam his motor strength was found to be diminished at 3/5 in the lower extremities and proximal upper extremities and 4/5 in the distal upper extremities. Reflexes were uniformly at 1+/4 and his cognition was intact. Examinations of his head, skin, heart, and abdomen were normal.
The absence of elevated jugular venous pressure argues against right heart failure. He is afebrile but that is minimally reassuring given the immunosuppression. There are no clues to suggest liver or kidney dysfunction. An unrecognized occlusion of the lower abdominal venous or lymphatic system such as upward extension of the DVT into the inferior vena cava (IVC) or a pelvic obstruction of the lower extremity lymphatic vessels could be considered. It appears that his distal weakness closely mirrors his proximal weakness in distinction to most myopathies which are predominantly proximal (with some exceptions, eg, inclusion body myositis).
The white blood cell count was 26,000/L with normal differential, hemoglobin 11.2 gm/dL, and platelet count was 191,000/L (at recent discharge these values were 23,000, 11.9, and 274,000, respectively). Chemistries were normal except for creatinine of 1.4 mg/dL (baseline 1.2), blood urea nitrogen was 42 mg/dL, albumin 2.6 gm/dL (normal, 3.55.0), and CK 3,710 U/L (20220), decreased from 6,943 U/L at recent discharge. Urine dipstick testing was positive for blood and protein; the urine sediment was unremarkable. Chest radiograph revealed normal lungs and heart.
The white blood cell count is quite elevated, perhaps more so than could be attributed to chronic steroid use, and again raises the concern of an undiagnosed infection. The presence of heme (and protein) in the urine without cells is consistent with pigment nephropathy from the recent rhabdomyolysis.
He was admitted to the hospital. Unfractionated heparin and warfarin were started. No changes were made to his immunosuppressive regimen. Blood cultures were negative after 48 hours. Transthoracic echocardiogram showed an ejection fraction of 60%, normal valves, and right ventricular systolic pressure of 32 mm Hg (normal, 1525 mmHg). On hospital day 3, his platelet count was 147,000/L, and on day 5, 101,000/L. His other laboratory values remained unchanged, and there were no new clinical developments.
A declining platelet count and extensive deep vein thrombosis suggest heparin‐induced thrombocytopenia and thrombosis (HITT), especially with the greater than 50% drop in the setting of IV heparin. His platelets have continued on a downward trajectory that was evident at admission and has progressed during this hospitalization. Assuming this is not due to laboratory error or artifact such as platelet clumping, this decline could have occurred if he was sensitized to heparin during the prior hospitalization, such as for DVT prophylaxis. It is increasingly recognized that HITT can manifest even after exposure to heparin is complete, ie, posthospitalization, and there can be an immediate drop in platelet counts if an unrecognized HITT‐mediated thrombosis is treated with IV heparin. Heparin should be discontinued in favor of a direct thrombin inhibitor and tests for heparin‐induced platelet antibodies (HIPA) and serotonin‐release assay (SRA) sent.
Antiphospholipid antibody syndrome (APLS) is associated with hypercoagulability and thrombocytopenia and is more frequent in patients with autoimmune disorders. The drug list should also be examined for associations with thrombocytopenia. The peripheral smear should be scrutinized and hemoglobin and creatinine followed to exclude thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome (TTP‐HUS).
Heparin was stopped on day 5. Warfarin was continued with a therapeutic international normalized ratio (INR). Tests for antiplatelet factor 4 antibodies, HIPA, and SRA were negative. His weakness and edema improved although his CK remained between 2000 and 4000 U/L. On day 5 he developed mild hemoptysis, and a repeat chest radiograph demonstrated a new left hilar infiltrate. Computed tomography (CT) scan of the chest with contrast demonstrated a left lower lobe consolidation, scattered ground glass opacities in both lung bases, and no pulmonary embolus. He was treated with piperacillin/tazobactam and vancomycin. He remained afebrile. The same day, he erroneously received 125 mg (instead of 12.5 mg) of subcutaneous methotrexate. High‐dose leucovorin was administered on days 5 and 6.
The hemoptysis resolved after 2 days. From days 5 to 9, the platelet count dropped to 80,000/L and his hemoglobin gradually decreased to 7.3 g/dL. Anticoagulation was stopped, vitamin K administered, and an IVC filter placed. Two units of packed red blood cells (RBCs) were transfused.
In suspected HITT (which was not verified here), warfarin is typically withheld until the platelets have recovered and thrombin‐inhibitor anticoagulation has reached a steady state, to avoid the transient hypercoagulability of warfarin initiation.
The unusual time course and the 3 negative tests make HITT unlikely. The continued platelet decline after stopping heparin further supports another etiology. The excess methotrexate dosing complicates interpretation of his thrombocytopenia and anemia, which can be explained by mucosal bleeding, microangiopathic hemolytic anemia (MAHA) such as disseminated intravascular coagulation or TTP‐HUS, or autoimmunity (Evans syndrome). Bone marrow toxicity is also a major effect of methotrexate (in addition to elevation of liver enzymes and acute renal failure); however, there is typically a lag between administration and development of cytopenias. The antibiotics could also account for the ongoing (but not original) thrombocytopenia.
With the new pulmonary infiltrate, infections remain a primary concern and should be evaluated with sputum samples and perhaps bronchoscopy. Given the abnormal urine (even without cells), a pulmonary‐renal inflammatory processes should be considered also to explain the infiltrates and hemoptysis.
Haptoglobin was <20 mg/dL (normal, 37246). The direct antiglobulin test (DAT) was negative. Serum lactate dehydrogenase (LDH) was 1657 U/L (normal, 100220), with elevated LD4 and LD5 isoenzymes. Coagulation studies normalized after the administration of vitamin K. Anti‐nuclear antibody was positive at 8.7 (normal <1.5). Tests for antineutrophil cytoplasmic antibodies were negative. No sputum could be obtained. A pathologist reviewed the blood smear and reported neutrophilic leukocytosis without left shift, and thrombocytopenia with normal platelet morphology.
Low haptoglobin in the setting of an elevated LDH is highly suggestive of hemolysis, particularly the intravascular, microangiopathic varieties. Neutrophilia may reflect infection, a primary myeloproliferative process such as chronic myeloid leukemia, steroid use, or a reactive bone marrow in the setting of acute illness. The negative DAT and significant immunosuppressive regimen makes immune‐mediated hemolysis unlikely, although the history of autoimmunity and the small DAT false‐negative rate leaves Evans syndrome as an outside possibility. Medications such as tacrolimus (causing TTP) or IVIG (given the broad spectrum of antibodies it includes) are other plausible causes of the cytopenias.
At this point, I would analyze the red blood cell (RBC) morphology and check the reticulocyte count to help differentiate between hemolysis and a myelotoxin.
After transfusion, his hemoglobin remained at approximately 8.5 gm/dL and LDH remained elevated but stable. By day 12 the platelet count had fallen to 37,000/L.
With physical therapy the patient gained strength. Antibiotics were discontinued on day 12 and a follow‐up chest x‐ray demonstrated no significant disease. From days 10 to 12, his creatinine rose from 1.5 to 1.9 mg/dL, although urine output remained normal.
A hematologist observed minimal fragmentation of red cells on the blood smear. Commenting on the thrombocytopenia, anemia, and LDH isoenzymes (representative of skeletal/hepatic origin rather than hematologic), and clinical improvement after treatment of a presumed pneumonia, he felt that the continued thrombocytopenia was likely due to drug toxicity, and recommended observation, treatment of renal failure, and discontinuation of tacrolimus.
The failure to increase the hemoglobin after transfusion is consistent with (but not specific for) hemolysis. In conjunction with the progressive thrombocytopenia and persistently elevated LDH, TTP remains a consideration. While TTP can be diagnosed with minimal evidence of schistocytes, the duration of this illness, now spanning almost 2 weeks without significant end organ damagenamely more pronounced renal failure, confusion, or feveris unusual for TTP. Therefore, I think it is reasonable to withhold plasma exchange, although if the cytopenias or renal failure progress after the methotrexate, tacrolimus, and antibiotics are stopped, it may have to be undertaken empirically.
The pulmonary process remains undefined. Edema, pneumonitis (eg, aspiration), a modest pneumonia, or pulmonary hemorrhage could normalize on chest x‐ray after 1 week.
Renal ultrasound was normal. Urinalysis dipstick demonstrated 3+ blood, 3+ protein, and no nitrate or leukocyte esterase. The urine sediment showed only granular casts. Fractional excretion of sodium was 6.7%. Urine protein‐to‐creatinine ratio was 7.5, and urine myoglobin was elevated. Serum C3 and C4 complement levels and cryoglobulins were normal. Reticulocyte count was 8.5% (normal, 0.53.2).
There is significant evidence for intrinsic renal failure, starting with the elevated fractional excretion. Marked proteinuria suggests glomerular damage; nephrotic syndrome could provide an explanation for the recurrent DVT. The 3+ blood without RBCs and the markedly elevated urine myoglobin suggest pigment nephropathy from both myoglobinuria and hemoglobinuria. The elevated reticulocyte count further confirms the impression of hemolysis.
Nephrotic syndrome may result from a primary disease process, such as diabetes, systemic lupus erythematosus (SLE), or amyloidosis, for which there is no evidence to date, or as a consequence of indolent infection, malignancy, or drugs, all of which are reasonable possibilities.
The essential elements at this point include thrombocytopenia, kidney failure with proteinuria, and likely intravascular hemolysis. I would repeat the peripheral smear (looking for schistocytes) and discuss with the rheumatologist if any other medications could be discontinued.
A nephrology consultant diagnosed acute tubular necrosis (ATN) from a combination of insults (intravenous contrast, methotrexate, tacrolimus, and myoglobinuria). Over the next several days, his platelet count rose to approximately 60,000/L. The patient continued to generally feel better but the creatinine steadily increased to 4.9 mg/dL.
The hematologist's reassessment of the smear was unchanged with minimal RBC fragmentation noted. Over the next few days the hemoglobin, creatinine, and platelet count remained stable, and there were no fevers or other clinical developments. On day 21 a kidney biopsy specimen revealed evidence of thrombotic microangiopathy (TMA) and segmental glomerular necrosis, with negative immunofluorescent findings. In addition, the glomerular basement membranes were thickened and effacement of the epithelial foot processes was noted.
TTP (or other MAHA) with only a few schistocytes would be unusual at an advanced stage where organ damage has occurred, although the clinical presentation in drug‐induced variety is variable. TTP is also generally a fatal disease, so relative stability over 3 weeks without definitive therapy is atypical, unless prednisone has served as a temporizing measure. The atypical features raise the possibility of a mimic or variant of TTP such as undiagnosed cancer causing DIC or a medication (eg, tacrolimus)‐associated TTP syndrome.
At least 2 other conditions could account for the hemolysis, thrombocytopenia, and TMA. The positive ANA, glomerular disease, and cytopenias are compatible with SLE, although such progression on an intense immunosuppressive regimen would be unusual. The renal histology in a patient with an autoimmune diathesis warrants reconsideration of antiphospholipid antibody syndrome (APLS), especially in light of the earlier DVT.
Tests for antiphospholipid antibodies were negative. After multidisciplinary deliberation, a diagnosis of TMA due to tacrolimus‐associated TTP/HUS was made. Plasmapheresis was initiated and IVIG and steroids were continued. He had a complicated hospital course and required renal replacement therapy, but with pheresis, his platelet counts and hemoglobin began to recover and he was ultimately discharged in good condition. After he was discharged, testing for ADAMTS13 (a von Willebrand factor‐cleaving protease) activity was reported as 54% (normal, >66%)
Discussion
TMA in the microcirculation is the hallmark pathology of TTP‐HUS but is not specific for this disease. TMA is also seen in disseminated intravascular coagulation, sepsis, cancer, malignant hypertension, human immunodeficiency virus infection, autoimmune disorders, pregnancy‐related conditions, and in association with certain drugs.1 The first pharmacological agent to be associated with TMA was mitomycin in 1971, and since then other drug associations have been described, including antiplatelet medications such as ticlopidine and clopidogrel, antibiotics such as quinine and rifampin, interferon, and immunosuppressants such as cyclosporine and tacrolimus.2 Drug‐induced variants of TTP and TMA are challenging to diagnose because the timing of onset, clinical features, and patient factors (eg, receipt of immunosuppressants) may vary widely and mimic other conditions.2, 3 TMA is a rare complication of tacrolimus and is mostly seen in renal transplant patients at a frequency of 1%. In these patients, renal dysfunction is usually the first herald of TMA and TTP; evidence of hemolysis may be absent.3
The clinical diagnosis of TTP has historically been based on the presence of a classic pentad: MAHA, thrombocytopenia, neurological and renal abnormalities, and fever.4 Elevated levels of LDH and indirect bilirubin and the presence of fragmented RBCs and reticulocytes point toward active intravascular hemolysis. The DAT is usually negative. This textbook illness scriptthe template of a disease that is stored in a clinician's memoryis learned by physicians during training, but undergoes little modification given the limited exposure to a rare disease.
In modern practice, the pentad is rarely seen, and the characteristics of the end‐organ findings may vary substantially. For instance, while neurological symptoms including seizures, coma, and transient confusion occur in 90% of cases, renal involvement is seen in about 50% and fever in only 25% of patients.5 Although the presence of 2 or more schistocytes on the blood smear under 100 microscopy supports the diagnosis of MAHA, cases of TTP without significant schistocytosis have been reported.6
Furthermore, TTP is typically described as acute in onset, but in a quarter of patients the symptoms and signs last for weeks before diagnosis.4 This variability in disease presentation coupled with the high mortality of untreated disease has changed the diagnostic and treatment thresholds for TTP. Trials and expert opinion use MAHA, thrombocytopenia, and the exclusion of alternative causes as sufficient criteria to diagnose TTP and begin treatment.7 The measurement of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity (a von Willebrand factor‐cleaving protease) for diagnostic purposes remains controversial because assay techniques are not uniform and there is insufficient correlation between levels and clinical disease.810 For instance, the presence of severe ADAMTS 13 deficiency (ie, <5%) along with the presence of an ADAMTS13 inhibitor is considered to be very specific, but not sensitive, for the laboratory diagnosis of idiopathic TTP.11 In cohort studies, the frequency of severe deficiency among patients with idiopathic TTP ranged from 18% to 100%, and the presence of severe deficiency did not predict the development of acute episodes of TTP.9 In a registry study of 142 patients diagnosed with TTP, 81% of patients with secondary TTP (ie, not classified as idiopathic) had ADAMTS13 levels that were normal to subnormal (>25%), and patients with normal ADAMTS13 levels had a higher incidence of acute renal failure, similar to the findings in this patient.10
Untreated TTP has a mortality rate of greater than 90%, but with plasma exchange, survival has improved dramatically.4, 7 Glucocorticoids are often used in addition to plasma exchange, based on case series and reports.9 The addition of cryoprecipitate or fresh frozen plasma to plasmapheresis has not been shown to be beneficial, but rituximab, an anti CD‐20 monoclonal antibody, has shown promise in a small prospective study.12, 13
TTP is a rare disorder with a classic description but substantial variation in clinical presentation. In this case, the background autoimmune myopathy, immunosuppression, coincident acute DVT, unexplained infiltrates, complex medication regimen, and nephrotic range proteinuria (attributed to focal segmental glomerular sclerosis based on the limited evidence available from the biopsy) led the clinicians to ascribe the patient's thrombocytopenia and renal injury to more common conditions and created a challenging environment for the diagnosis of TTP. TTP is a complex disorder and the simplified understanding of the disease and its time course prevented a prompt match between the patient's clinical course and his diagnosis. The combination of a rare condition with inherent variability arising in the setting of medical complexity challenges the processes of problem representation and scripting the answer for even the most seasoned clinician.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Key Teaching Points
-
The classically described pentad of TTP is seldom seen, and the findings of otherwise unexplained MAHA and thrombocytopenia should prompt consideration of TTP.
-
TTP may be acute and idiopathic, or be secondary to drugs, infections, or other conditions. Medication‐induced TTP may present with a wide range of clinical findings.
-
Therapeutic plasma exchange may be life‐saving in cases of TTP, and when appropriate, should be initiated promptly based on clinical suspicion and without waiting to perform tissue biopsy.
- , , .Thrombotic microangiopathies. In: Tischer CC, Brenner BM, eds.Renal Pathology.2nd ed.Philadelphia, PA:JB Lippincott;1994:1154–1184.
- , , .Drug‐induced thrombotic microangiopathy: incidence, prevention and management.Drug Saf.2001;24(7):491–501.
- , , , , .FK 506‐associated thrombotic microangiopathy: report of two cases and review of the literature.Transplantation.1999;67(4):539–544.
- , .Thrombotic Thrombocytopenic purpura: report of 16 cases and review of the literature.Medicine (Baltimore).1966;45:139–159.
- , , , .Thrombotic thrombocytopenic purpura; early and late responders.Am J Hematol.1997;54:102–107.
- .Atypical presentations of thrombotic thrombocytopenic purpura: a review.J Clin Apheresis.2009;24(1)47–52.
- , , , et al.Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura.N Engl J Med.1991;325:393–397.
- , , , , , .The incidence of thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS13deficiency.J Thromb Haemost.2005;3:1432–1436.
- .Thrombotic thrombocytopenic purpura.N Engl J Med.2006;354:1927–1935.
- , , , et al.ADAMTS13 activity in thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients.Blood.2003;102:60–68.
- , , .Thrombotic thrombocytopenic purpura.J Thromb Haemost.2005;3:1663–1675.
- , , , et al.Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials.Am J Kidney Dis.2009;53:259–272.
- , , , et al.:Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13‐deficient TTP: A study of 11 cases.Blood.2005;105:1932–1937.
A 58‐year old man was admitted with generalized weakness and acute deep venous thrombosis (DVT). His past medical history included hypertension and polymyositis/dermatomyositis (PM/DM) with anti‐synthase syndrome, which had been diagnosed 16 months prior when his creatine kinase (CK) was greater than 12,000 U/L. At that time he also was found to have bilateral lower extremity DVT, and had been treated with warfarin for 1 year. 10 days previously, he had been discharged after a 4‐day hospitalization for a polymyositis flare which was treated with methylprednisolone at 60 mg daily for 5 days. He was discharged home with daily prednisone until this follow‐up a week later, where he reported weakness and bilateral edema. Lower extremity ultrasound demonstrated acute thrombus in the right common femoral vein.
This acute extensive DVT may be a consequence of recent hospitalization and a previously damaged venous system, or may reflect ongoing hypercoagulability from an unresolved condition, such as cancer. Bilateral lower extremity edema may suggest right‐sided heart failure due to progressive interstitial lung disease, which occurs in a subset of patients with PM/DM. Edema may alternatively reflect biventricular heart failure, or liver or kidney disease.
Generalized weakness offers little in the way of focused differential diagnosis until it is characterized as motor weakness (eg, attributed to progression of the myopathy), a dyspnea‐equivalent, or an overall sense of fatigue.
His medications included weekly methotrexate, monthly intravenous immunoglobulin (IVIG) infusions, tacrolimus, hydrochlorothiazide, and aerosolized pentamidine. He had been on varying doses of prednisone for 2 years and his present dose was 40 mg daily. He was allergic to sulfa. He was married and stopped smoking 30 years previously, and did not drink alcohol or use illicit drugs.
Various medication toxicities could account for his presentation. Methotrexate causes interstitial lung disease, and IVIG and tacrolimus may cause renal failure (and fluid overload). The heavy degree of immunosuppression renders him susceptible to a wide range of infections. Aerosolized pentamidine provides incomplete protection against Pneumocystis jirovecii, especially in the lung apices.
Evaluation of the status of his myositis with motor strength assessment is important. In addition associated rashes and signs of malignancy (eg, lymphadenopathy) and infection should be sought. Proximal motor weakness would suggest a myositis flare, although care must be given to exclude competing causes of myopathy, including infections, toxins, or endocrinopathies.
His temperature was 36.2C, pulse 103 beats per minute, blood pressure 156/83 mm Hg, and respiratory rate 18 breaths per minute. He had crackles at both lung bases, and 3+ pitting edema in both lower extremities. On neurological exam his motor strength was found to be diminished at 3/5 in the lower extremities and proximal upper extremities and 4/5 in the distal upper extremities. Reflexes were uniformly at 1+/4 and his cognition was intact. Examinations of his head, skin, heart, and abdomen were normal.
The absence of elevated jugular venous pressure argues against right heart failure. He is afebrile but that is minimally reassuring given the immunosuppression. There are no clues to suggest liver or kidney dysfunction. An unrecognized occlusion of the lower abdominal venous or lymphatic system such as upward extension of the DVT into the inferior vena cava (IVC) or a pelvic obstruction of the lower extremity lymphatic vessels could be considered. It appears that his distal weakness closely mirrors his proximal weakness in distinction to most myopathies which are predominantly proximal (with some exceptions, eg, inclusion body myositis).
The white blood cell count was 26,000/L with normal differential, hemoglobin 11.2 gm/dL, and platelet count was 191,000/L (at recent discharge these values were 23,000, 11.9, and 274,000, respectively). Chemistries were normal except for creatinine of 1.4 mg/dL (baseline 1.2), blood urea nitrogen was 42 mg/dL, albumin 2.6 gm/dL (normal, 3.55.0), and CK 3,710 U/L (20220), decreased from 6,943 U/L at recent discharge. Urine dipstick testing was positive for blood and protein; the urine sediment was unremarkable. Chest radiograph revealed normal lungs and heart.
The white blood cell count is quite elevated, perhaps more so than could be attributed to chronic steroid use, and again raises the concern of an undiagnosed infection. The presence of heme (and protein) in the urine without cells is consistent with pigment nephropathy from the recent rhabdomyolysis.
He was admitted to the hospital. Unfractionated heparin and warfarin were started. No changes were made to his immunosuppressive regimen. Blood cultures were negative after 48 hours. Transthoracic echocardiogram showed an ejection fraction of 60%, normal valves, and right ventricular systolic pressure of 32 mm Hg (normal, 1525 mmHg). On hospital day 3, his platelet count was 147,000/L, and on day 5, 101,000/L. His other laboratory values remained unchanged, and there were no new clinical developments.
A declining platelet count and extensive deep vein thrombosis suggest heparin‐induced thrombocytopenia and thrombosis (HITT), especially with the greater than 50% drop in the setting of IV heparin. His platelets have continued on a downward trajectory that was evident at admission and has progressed during this hospitalization. Assuming this is not due to laboratory error or artifact such as platelet clumping, this decline could have occurred if he was sensitized to heparin during the prior hospitalization, such as for DVT prophylaxis. It is increasingly recognized that HITT can manifest even after exposure to heparin is complete, ie, posthospitalization, and there can be an immediate drop in platelet counts if an unrecognized HITT‐mediated thrombosis is treated with IV heparin. Heparin should be discontinued in favor of a direct thrombin inhibitor and tests for heparin‐induced platelet antibodies (HIPA) and serotonin‐release assay (SRA) sent.
Antiphospholipid antibody syndrome (APLS) is associated with hypercoagulability and thrombocytopenia and is more frequent in patients with autoimmune disorders. The drug list should also be examined for associations with thrombocytopenia. The peripheral smear should be scrutinized and hemoglobin and creatinine followed to exclude thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome (TTP‐HUS).
Heparin was stopped on day 5. Warfarin was continued with a therapeutic international normalized ratio (INR). Tests for antiplatelet factor 4 antibodies, HIPA, and SRA were negative. His weakness and edema improved although his CK remained between 2000 and 4000 U/L. On day 5 he developed mild hemoptysis, and a repeat chest radiograph demonstrated a new left hilar infiltrate. Computed tomography (CT) scan of the chest with contrast demonstrated a left lower lobe consolidation, scattered ground glass opacities in both lung bases, and no pulmonary embolus. He was treated with piperacillin/tazobactam and vancomycin. He remained afebrile. The same day, he erroneously received 125 mg (instead of 12.5 mg) of subcutaneous methotrexate. High‐dose leucovorin was administered on days 5 and 6.
The hemoptysis resolved after 2 days. From days 5 to 9, the platelet count dropped to 80,000/L and his hemoglobin gradually decreased to 7.3 g/dL. Anticoagulation was stopped, vitamin K administered, and an IVC filter placed. Two units of packed red blood cells (RBCs) were transfused.
In suspected HITT (which was not verified here), warfarin is typically withheld until the platelets have recovered and thrombin‐inhibitor anticoagulation has reached a steady state, to avoid the transient hypercoagulability of warfarin initiation.
The unusual time course and the 3 negative tests make HITT unlikely. The continued platelet decline after stopping heparin further supports another etiology. The excess methotrexate dosing complicates interpretation of his thrombocytopenia and anemia, which can be explained by mucosal bleeding, microangiopathic hemolytic anemia (MAHA) such as disseminated intravascular coagulation or TTP‐HUS, or autoimmunity (Evans syndrome). Bone marrow toxicity is also a major effect of methotrexate (in addition to elevation of liver enzymes and acute renal failure); however, there is typically a lag between administration and development of cytopenias. The antibiotics could also account for the ongoing (but not original) thrombocytopenia.
With the new pulmonary infiltrate, infections remain a primary concern and should be evaluated with sputum samples and perhaps bronchoscopy. Given the abnormal urine (even without cells), a pulmonary‐renal inflammatory processes should be considered also to explain the infiltrates and hemoptysis.
Haptoglobin was <20 mg/dL (normal, 37246). The direct antiglobulin test (DAT) was negative. Serum lactate dehydrogenase (LDH) was 1657 U/L (normal, 100220), with elevated LD4 and LD5 isoenzymes. Coagulation studies normalized after the administration of vitamin K. Anti‐nuclear antibody was positive at 8.7 (normal <1.5). Tests for antineutrophil cytoplasmic antibodies were negative. No sputum could be obtained. A pathologist reviewed the blood smear and reported neutrophilic leukocytosis without left shift, and thrombocytopenia with normal platelet morphology.
Low haptoglobin in the setting of an elevated LDH is highly suggestive of hemolysis, particularly the intravascular, microangiopathic varieties. Neutrophilia may reflect infection, a primary myeloproliferative process such as chronic myeloid leukemia, steroid use, or a reactive bone marrow in the setting of acute illness. The negative DAT and significant immunosuppressive regimen makes immune‐mediated hemolysis unlikely, although the history of autoimmunity and the small DAT false‐negative rate leaves Evans syndrome as an outside possibility. Medications such as tacrolimus (causing TTP) or IVIG (given the broad spectrum of antibodies it includes) are other plausible causes of the cytopenias.
At this point, I would analyze the red blood cell (RBC) morphology and check the reticulocyte count to help differentiate between hemolysis and a myelotoxin.
After transfusion, his hemoglobin remained at approximately 8.5 gm/dL and LDH remained elevated but stable. By day 12 the platelet count had fallen to 37,000/L.
With physical therapy the patient gained strength. Antibiotics were discontinued on day 12 and a follow‐up chest x‐ray demonstrated no significant disease. From days 10 to 12, his creatinine rose from 1.5 to 1.9 mg/dL, although urine output remained normal.
A hematologist observed minimal fragmentation of red cells on the blood smear. Commenting on the thrombocytopenia, anemia, and LDH isoenzymes (representative of skeletal/hepatic origin rather than hematologic), and clinical improvement after treatment of a presumed pneumonia, he felt that the continued thrombocytopenia was likely due to drug toxicity, and recommended observation, treatment of renal failure, and discontinuation of tacrolimus.
The failure to increase the hemoglobin after transfusion is consistent with (but not specific for) hemolysis. In conjunction with the progressive thrombocytopenia and persistently elevated LDH, TTP remains a consideration. While TTP can be diagnosed with minimal evidence of schistocytes, the duration of this illness, now spanning almost 2 weeks without significant end organ damagenamely more pronounced renal failure, confusion, or feveris unusual for TTP. Therefore, I think it is reasonable to withhold plasma exchange, although if the cytopenias or renal failure progress after the methotrexate, tacrolimus, and antibiotics are stopped, it may have to be undertaken empirically.
The pulmonary process remains undefined. Edema, pneumonitis (eg, aspiration), a modest pneumonia, or pulmonary hemorrhage could normalize on chest x‐ray after 1 week.
Renal ultrasound was normal. Urinalysis dipstick demonstrated 3+ blood, 3+ protein, and no nitrate or leukocyte esterase. The urine sediment showed only granular casts. Fractional excretion of sodium was 6.7%. Urine protein‐to‐creatinine ratio was 7.5, and urine myoglobin was elevated. Serum C3 and C4 complement levels and cryoglobulins were normal. Reticulocyte count was 8.5% (normal, 0.53.2).
There is significant evidence for intrinsic renal failure, starting with the elevated fractional excretion. Marked proteinuria suggests glomerular damage; nephrotic syndrome could provide an explanation for the recurrent DVT. The 3+ blood without RBCs and the markedly elevated urine myoglobin suggest pigment nephropathy from both myoglobinuria and hemoglobinuria. The elevated reticulocyte count further confirms the impression of hemolysis.
Nephrotic syndrome may result from a primary disease process, such as diabetes, systemic lupus erythematosus (SLE), or amyloidosis, for which there is no evidence to date, or as a consequence of indolent infection, malignancy, or drugs, all of which are reasonable possibilities.
The essential elements at this point include thrombocytopenia, kidney failure with proteinuria, and likely intravascular hemolysis. I would repeat the peripheral smear (looking for schistocytes) and discuss with the rheumatologist if any other medications could be discontinued.
A nephrology consultant diagnosed acute tubular necrosis (ATN) from a combination of insults (intravenous contrast, methotrexate, tacrolimus, and myoglobinuria). Over the next several days, his platelet count rose to approximately 60,000/L. The patient continued to generally feel better but the creatinine steadily increased to 4.9 mg/dL.
The hematologist's reassessment of the smear was unchanged with minimal RBC fragmentation noted. Over the next few days the hemoglobin, creatinine, and platelet count remained stable, and there were no fevers or other clinical developments. On day 21 a kidney biopsy specimen revealed evidence of thrombotic microangiopathy (TMA) and segmental glomerular necrosis, with negative immunofluorescent findings. In addition, the glomerular basement membranes were thickened and effacement of the epithelial foot processes was noted.
TTP (or other MAHA) with only a few schistocytes would be unusual at an advanced stage where organ damage has occurred, although the clinical presentation in drug‐induced variety is variable. TTP is also generally a fatal disease, so relative stability over 3 weeks without definitive therapy is atypical, unless prednisone has served as a temporizing measure. The atypical features raise the possibility of a mimic or variant of TTP such as undiagnosed cancer causing DIC or a medication (eg, tacrolimus)‐associated TTP syndrome.
At least 2 other conditions could account for the hemolysis, thrombocytopenia, and TMA. The positive ANA, glomerular disease, and cytopenias are compatible with SLE, although such progression on an intense immunosuppressive regimen would be unusual. The renal histology in a patient with an autoimmune diathesis warrants reconsideration of antiphospholipid antibody syndrome (APLS), especially in light of the earlier DVT.
Tests for antiphospholipid antibodies were negative. After multidisciplinary deliberation, a diagnosis of TMA due to tacrolimus‐associated TTP/HUS was made. Plasmapheresis was initiated and IVIG and steroids were continued. He had a complicated hospital course and required renal replacement therapy, but with pheresis, his platelet counts and hemoglobin began to recover and he was ultimately discharged in good condition. After he was discharged, testing for ADAMTS13 (a von Willebrand factor‐cleaving protease) activity was reported as 54% (normal, >66%)
Discussion
TMA in the microcirculation is the hallmark pathology of TTP‐HUS but is not specific for this disease. TMA is also seen in disseminated intravascular coagulation, sepsis, cancer, malignant hypertension, human immunodeficiency virus infection, autoimmune disorders, pregnancy‐related conditions, and in association with certain drugs.1 The first pharmacological agent to be associated with TMA was mitomycin in 1971, and since then other drug associations have been described, including antiplatelet medications such as ticlopidine and clopidogrel, antibiotics such as quinine and rifampin, interferon, and immunosuppressants such as cyclosporine and tacrolimus.2 Drug‐induced variants of TTP and TMA are challenging to diagnose because the timing of onset, clinical features, and patient factors (eg, receipt of immunosuppressants) may vary widely and mimic other conditions.2, 3 TMA is a rare complication of tacrolimus and is mostly seen in renal transplant patients at a frequency of 1%. In these patients, renal dysfunction is usually the first herald of TMA and TTP; evidence of hemolysis may be absent.3
The clinical diagnosis of TTP has historically been based on the presence of a classic pentad: MAHA, thrombocytopenia, neurological and renal abnormalities, and fever.4 Elevated levels of LDH and indirect bilirubin and the presence of fragmented RBCs and reticulocytes point toward active intravascular hemolysis. The DAT is usually negative. This textbook illness scriptthe template of a disease that is stored in a clinician's memoryis learned by physicians during training, but undergoes little modification given the limited exposure to a rare disease.
In modern practice, the pentad is rarely seen, and the characteristics of the end‐organ findings may vary substantially. For instance, while neurological symptoms including seizures, coma, and transient confusion occur in 90% of cases, renal involvement is seen in about 50% and fever in only 25% of patients.5 Although the presence of 2 or more schistocytes on the blood smear under 100 microscopy supports the diagnosis of MAHA, cases of TTP without significant schistocytosis have been reported.6
Furthermore, TTP is typically described as acute in onset, but in a quarter of patients the symptoms and signs last for weeks before diagnosis.4 This variability in disease presentation coupled with the high mortality of untreated disease has changed the diagnostic and treatment thresholds for TTP. Trials and expert opinion use MAHA, thrombocytopenia, and the exclusion of alternative causes as sufficient criteria to diagnose TTP and begin treatment.7 The measurement of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity (a von Willebrand factor‐cleaving protease) for diagnostic purposes remains controversial because assay techniques are not uniform and there is insufficient correlation between levels and clinical disease.810 For instance, the presence of severe ADAMTS 13 deficiency (ie, <5%) along with the presence of an ADAMTS13 inhibitor is considered to be very specific, but not sensitive, for the laboratory diagnosis of idiopathic TTP.11 In cohort studies, the frequency of severe deficiency among patients with idiopathic TTP ranged from 18% to 100%, and the presence of severe deficiency did not predict the development of acute episodes of TTP.9 In a registry study of 142 patients diagnosed with TTP, 81% of patients with secondary TTP (ie, not classified as idiopathic) had ADAMTS13 levels that were normal to subnormal (>25%), and patients with normal ADAMTS13 levels had a higher incidence of acute renal failure, similar to the findings in this patient.10
Untreated TTP has a mortality rate of greater than 90%, but with plasma exchange, survival has improved dramatically.4, 7 Glucocorticoids are often used in addition to plasma exchange, based on case series and reports.9 The addition of cryoprecipitate or fresh frozen plasma to plasmapheresis has not been shown to be beneficial, but rituximab, an anti CD‐20 monoclonal antibody, has shown promise in a small prospective study.12, 13
TTP is a rare disorder with a classic description but substantial variation in clinical presentation. In this case, the background autoimmune myopathy, immunosuppression, coincident acute DVT, unexplained infiltrates, complex medication regimen, and nephrotic range proteinuria (attributed to focal segmental glomerular sclerosis based on the limited evidence available from the biopsy) led the clinicians to ascribe the patient's thrombocytopenia and renal injury to more common conditions and created a challenging environment for the diagnosis of TTP. TTP is a complex disorder and the simplified understanding of the disease and its time course prevented a prompt match between the patient's clinical course and his diagnosis. The combination of a rare condition with inherent variability arising in the setting of medical complexity challenges the processes of problem representation and scripting the answer for even the most seasoned clinician.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Key Teaching Points
-
The classically described pentad of TTP is seldom seen, and the findings of otherwise unexplained MAHA and thrombocytopenia should prompt consideration of TTP.
-
TTP may be acute and idiopathic, or be secondary to drugs, infections, or other conditions. Medication‐induced TTP may present with a wide range of clinical findings.
-
Therapeutic plasma exchange may be life‐saving in cases of TTP, and when appropriate, should be initiated promptly based on clinical suspicion and without waiting to perform tissue biopsy.
A 58‐year old man was admitted with generalized weakness and acute deep venous thrombosis (DVT). His past medical history included hypertension and polymyositis/dermatomyositis (PM/DM) with anti‐synthase syndrome, which had been diagnosed 16 months prior when his creatine kinase (CK) was greater than 12,000 U/L. At that time he also was found to have bilateral lower extremity DVT, and had been treated with warfarin for 1 year. 10 days previously, he had been discharged after a 4‐day hospitalization for a polymyositis flare which was treated with methylprednisolone at 60 mg daily for 5 days. He was discharged home with daily prednisone until this follow‐up a week later, where he reported weakness and bilateral edema. Lower extremity ultrasound demonstrated acute thrombus in the right common femoral vein.
This acute extensive DVT may be a consequence of recent hospitalization and a previously damaged venous system, or may reflect ongoing hypercoagulability from an unresolved condition, such as cancer. Bilateral lower extremity edema may suggest right‐sided heart failure due to progressive interstitial lung disease, which occurs in a subset of patients with PM/DM. Edema may alternatively reflect biventricular heart failure, or liver or kidney disease.
Generalized weakness offers little in the way of focused differential diagnosis until it is characterized as motor weakness (eg, attributed to progression of the myopathy), a dyspnea‐equivalent, or an overall sense of fatigue.
His medications included weekly methotrexate, monthly intravenous immunoglobulin (IVIG) infusions, tacrolimus, hydrochlorothiazide, and aerosolized pentamidine. He had been on varying doses of prednisone for 2 years and his present dose was 40 mg daily. He was allergic to sulfa. He was married and stopped smoking 30 years previously, and did not drink alcohol or use illicit drugs.
Various medication toxicities could account for his presentation. Methotrexate causes interstitial lung disease, and IVIG and tacrolimus may cause renal failure (and fluid overload). The heavy degree of immunosuppression renders him susceptible to a wide range of infections. Aerosolized pentamidine provides incomplete protection against Pneumocystis jirovecii, especially in the lung apices.
Evaluation of the status of his myositis with motor strength assessment is important. In addition associated rashes and signs of malignancy (eg, lymphadenopathy) and infection should be sought. Proximal motor weakness would suggest a myositis flare, although care must be given to exclude competing causes of myopathy, including infections, toxins, or endocrinopathies.
His temperature was 36.2C, pulse 103 beats per minute, blood pressure 156/83 mm Hg, and respiratory rate 18 breaths per minute. He had crackles at both lung bases, and 3+ pitting edema in both lower extremities. On neurological exam his motor strength was found to be diminished at 3/5 in the lower extremities and proximal upper extremities and 4/5 in the distal upper extremities. Reflexes were uniformly at 1+/4 and his cognition was intact. Examinations of his head, skin, heart, and abdomen were normal.
The absence of elevated jugular venous pressure argues against right heart failure. He is afebrile but that is minimally reassuring given the immunosuppression. There are no clues to suggest liver or kidney dysfunction. An unrecognized occlusion of the lower abdominal venous or lymphatic system such as upward extension of the DVT into the inferior vena cava (IVC) or a pelvic obstruction of the lower extremity lymphatic vessels could be considered. It appears that his distal weakness closely mirrors his proximal weakness in distinction to most myopathies which are predominantly proximal (with some exceptions, eg, inclusion body myositis).
The white blood cell count was 26,000/L with normal differential, hemoglobin 11.2 gm/dL, and platelet count was 191,000/L (at recent discharge these values were 23,000, 11.9, and 274,000, respectively). Chemistries were normal except for creatinine of 1.4 mg/dL (baseline 1.2), blood urea nitrogen was 42 mg/dL, albumin 2.6 gm/dL (normal, 3.55.0), and CK 3,710 U/L (20220), decreased from 6,943 U/L at recent discharge. Urine dipstick testing was positive for blood and protein; the urine sediment was unremarkable. Chest radiograph revealed normal lungs and heart.
The white blood cell count is quite elevated, perhaps more so than could be attributed to chronic steroid use, and again raises the concern of an undiagnosed infection. The presence of heme (and protein) in the urine without cells is consistent with pigment nephropathy from the recent rhabdomyolysis.
He was admitted to the hospital. Unfractionated heparin and warfarin were started. No changes were made to his immunosuppressive regimen. Blood cultures were negative after 48 hours. Transthoracic echocardiogram showed an ejection fraction of 60%, normal valves, and right ventricular systolic pressure of 32 mm Hg (normal, 1525 mmHg). On hospital day 3, his platelet count was 147,000/L, and on day 5, 101,000/L. His other laboratory values remained unchanged, and there were no new clinical developments.
A declining platelet count and extensive deep vein thrombosis suggest heparin‐induced thrombocytopenia and thrombosis (HITT), especially with the greater than 50% drop in the setting of IV heparin. His platelets have continued on a downward trajectory that was evident at admission and has progressed during this hospitalization. Assuming this is not due to laboratory error or artifact such as platelet clumping, this decline could have occurred if he was sensitized to heparin during the prior hospitalization, such as for DVT prophylaxis. It is increasingly recognized that HITT can manifest even after exposure to heparin is complete, ie, posthospitalization, and there can be an immediate drop in platelet counts if an unrecognized HITT‐mediated thrombosis is treated with IV heparin. Heparin should be discontinued in favor of a direct thrombin inhibitor and tests for heparin‐induced platelet antibodies (HIPA) and serotonin‐release assay (SRA) sent.
Antiphospholipid antibody syndrome (APLS) is associated with hypercoagulability and thrombocytopenia and is more frequent in patients with autoimmune disorders. The drug list should also be examined for associations with thrombocytopenia. The peripheral smear should be scrutinized and hemoglobin and creatinine followed to exclude thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome (TTP‐HUS).
Heparin was stopped on day 5. Warfarin was continued with a therapeutic international normalized ratio (INR). Tests for antiplatelet factor 4 antibodies, HIPA, and SRA were negative. His weakness and edema improved although his CK remained between 2000 and 4000 U/L. On day 5 he developed mild hemoptysis, and a repeat chest radiograph demonstrated a new left hilar infiltrate. Computed tomography (CT) scan of the chest with contrast demonstrated a left lower lobe consolidation, scattered ground glass opacities in both lung bases, and no pulmonary embolus. He was treated with piperacillin/tazobactam and vancomycin. He remained afebrile. The same day, he erroneously received 125 mg (instead of 12.5 mg) of subcutaneous methotrexate. High‐dose leucovorin was administered on days 5 and 6.
The hemoptysis resolved after 2 days. From days 5 to 9, the platelet count dropped to 80,000/L and his hemoglobin gradually decreased to 7.3 g/dL. Anticoagulation was stopped, vitamin K administered, and an IVC filter placed. Two units of packed red blood cells (RBCs) were transfused.
In suspected HITT (which was not verified here), warfarin is typically withheld until the platelets have recovered and thrombin‐inhibitor anticoagulation has reached a steady state, to avoid the transient hypercoagulability of warfarin initiation.
The unusual time course and the 3 negative tests make HITT unlikely. The continued platelet decline after stopping heparin further supports another etiology. The excess methotrexate dosing complicates interpretation of his thrombocytopenia and anemia, which can be explained by mucosal bleeding, microangiopathic hemolytic anemia (MAHA) such as disseminated intravascular coagulation or TTP‐HUS, or autoimmunity (Evans syndrome). Bone marrow toxicity is also a major effect of methotrexate (in addition to elevation of liver enzymes and acute renal failure); however, there is typically a lag between administration and development of cytopenias. The antibiotics could also account for the ongoing (but not original) thrombocytopenia.
With the new pulmonary infiltrate, infections remain a primary concern and should be evaluated with sputum samples and perhaps bronchoscopy. Given the abnormal urine (even without cells), a pulmonary‐renal inflammatory processes should be considered also to explain the infiltrates and hemoptysis.
Haptoglobin was <20 mg/dL (normal, 37246). The direct antiglobulin test (DAT) was negative. Serum lactate dehydrogenase (LDH) was 1657 U/L (normal, 100220), with elevated LD4 and LD5 isoenzymes. Coagulation studies normalized after the administration of vitamin K. Anti‐nuclear antibody was positive at 8.7 (normal <1.5). Tests for antineutrophil cytoplasmic antibodies were negative. No sputum could be obtained. A pathologist reviewed the blood smear and reported neutrophilic leukocytosis without left shift, and thrombocytopenia with normal platelet morphology.
Low haptoglobin in the setting of an elevated LDH is highly suggestive of hemolysis, particularly the intravascular, microangiopathic varieties. Neutrophilia may reflect infection, a primary myeloproliferative process such as chronic myeloid leukemia, steroid use, or a reactive bone marrow in the setting of acute illness. The negative DAT and significant immunosuppressive regimen makes immune‐mediated hemolysis unlikely, although the history of autoimmunity and the small DAT false‐negative rate leaves Evans syndrome as an outside possibility. Medications such as tacrolimus (causing TTP) or IVIG (given the broad spectrum of antibodies it includes) are other plausible causes of the cytopenias.
At this point, I would analyze the red blood cell (RBC) morphology and check the reticulocyte count to help differentiate between hemolysis and a myelotoxin.
After transfusion, his hemoglobin remained at approximately 8.5 gm/dL and LDH remained elevated but stable. By day 12 the platelet count had fallen to 37,000/L.
With physical therapy the patient gained strength. Antibiotics were discontinued on day 12 and a follow‐up chest x‐ray demonstrated no significant disease. From days 10 to 12, his creatinine rose from 1.5 to 1.9 mg/dL, although urine output remained normal.
A hematologist observed minimal fragmentation of red cells on the blood smear. Commenting on the thrombocytopenia, anemia, and LDH isoenzymes (representative of skeletal/hepatic origin rather than hematologic), and clinical improvement after treatment of a presumed pneumonia, he felt that the continued thrombocytopenia was likely due to drug toxicity, and recommended observation, treatment of renal failure, and discontinuation of tacrolimus.
The failure to increase the hemoglobin after transfusion is consistent with (but not specific for) hemolysis. In conjunction with the progressive thrombocytopenia and persistently elevated LDH, TTP remains a consideration. While TTP can be diagnosed with minimal evidence of schistocytes, the duration of this illness, now spanning almost 2 weeks without significant end organ damagenamely more pronounced renal failure, confusion, or feveris unusual for TTP. Therefore, I think it is reasonable to withhold plasma exchange, although if the cytopenias or renal failure progress after the methotrexate, tacrolimus, and antibiotics are stopped, it may have to be undertaken empirically.
The pulmonary process remains undefined. Edema, pneumonitis (eg, aspiration), a modest pneumonia, or pulmonary hemorrhage could normalize on chest x‐ray after 1 week.
Renal ultrasound was normal. Urinalysis dipstick demonstrated 3+ blood, 3+ protein, and no nitrate or leukocyte esterase. The urine sediment showed only granular casts. Fractional excretion of sodium was 6.7%. Urine protein‐to‐creatinine ratio was 7.5, and urine myoglobin was elevated. Serum C3 and C4 complement levels and cryoglobulins were normal. Reticulocyte count was 8.5% (normal, 0.53.2).
There is significant evidence for intrinsic renal failure, starting with the elevated fractional excretion. Marked proteinuria suggests glomerular damage; nephrotic syndrome could provide an explanation for the recurrent DVT. The 3+ blood without RBCs and the markedly elevated urine myoglobin suggest pigment nephropathy from both myoglobinuria and hemoglobinuria. The elevated reticulocyte count further confirms the impression of hemolysis.
Nephrotic syndrome may result from a primary disease process, such as diabetes, systemic lupus erythematosus (SLE), or amyloidosis, for which there is no evidence to date, or as a consequence of indolent infection, malignancy, or drugs, all of which are reasonable possibilities.
The essential elements at this point include thrombocytopenia, kidney failure with proteinuria, and likely intravascular hemolysis. I would repeat the peripheral smear (looking for schistocytes) and discuss with the rheumatologist if any other medications could be discontinued.
A nephrology consultant diagnosed acute tubular necrosis (ATN) from a combination of insults (intravenous contrast, methotrexate, tacrolimus, and myoglobinuria). Over the next several days, his platelet count rose to approximately 60,000/L. The patient continued to generally feel better but the creatinine steadily increased to 4.9 mg/dL.
The hematologist's reassessment of the smear was unchanged with minimal RBC fragmentation noted. Over the next few days the hemoglobin, creatinine, and platelet count remained stable, and there were no fevers or other clinical developments. On day 21 a kidney biopsy specimen revealed evidence of thrombotic microangiopathy (TMA) and segmental glomerular necrosis, with negative immunofluorescent findings. In addition, the glomerular basement membranes were thickened and effacement of the epithelial foot processes was noted.
TTP (or other MAHA) with only a few schistocytes would be unusual at an advanced stage where organ damage has occurred, although the clinical presentation in drug‐induced variety is variable. TTP is also generally a fatal disease, so relative stability over 3 weeks without definitive therapy is atypical, unless prednisone has served as a temporizing measure. The atypical features raise the possibility of a mimic or variant of TTP such as undiagnosed cancer causing DIC or a medication (eg, tacrolimus)‐associated TTP syndrome.
At least 2 other conditions could account for the hemolysis, thrombocytopenia, and TMA. The positive ANA, glomerular disease, and cytopenias are compatible with SLE, although such progression on an intense immunosuppressive regimen would be unusual. The renal histology in a patient with an autoimmune diathesis warrants reconsideration of antiphospholipid antibody syndrome (APLS), especially in light of the earlier DVT.
Tests for antiphospholipid antibodies were negative. After multidisciplinary deliberation, a diagnosis of TMA due to tacrolimus‐associated TTP/HUS was made. Plasmapheresis was initiated and IVIG and steroids were continued. He had a complicated hospital course and required renal replacement therapy, but with pheresis, his platelet counts and hemoglobin began to recover and he was ultimately discharged in good condition. After he was discharged, testing for ADAMTS13 (a von Willebrand factor‐cleaving protease) activity was reported as 54% (normal, >66%)
Discussion
TMA in the microcirculation is the hallmark pathology of TTP‐HUS but is not specific for this disease. TMA is also seen in disseminated intravascular coagulation, sepsis, cancer, malignant hypertension, human immunodeficiency virus infection, autoimmune disorders, pregnancy‐related conditions, and in association with certain drugs.1 The first pharmacological agent to be associated with TMA was mitomycin in 1971, and since then other drug associations have been described, including antiplatelet medications such as ticlopidine and clopidogrel, antibiotics such as quinine and rifampin, interferon, and immunosuppressants such as cyclosporine and tacrolimus.2 Drug‐induced variants of TTP and TMA are challenging to diagnose because the timing of onset, clinical features, and patient factors (eg, receipt of immunosuppressants) may vary widely and mimic other conditions.2, 3 TMA is a rare complication of tacrolimus and is mostly seen in renal transplant patients at a frequency of 1%. In these patients, renal dysfunction is usually the first herald of TMA and TTP; evidence of hemolysis may be absent.3
The clinical diagnosis of TTP has historically been based on the presence of a classic pentad: MAHA, thrombocytopenia, neurological and renal abnormalities, and fever.4 Elevated levels of LDH and indirect bilirubin and the presence of fragmented RBCs and reticulocytes point toward active intravascular hemolysis. The DAT is usually negative. This textbook illness scriptthe template of a disease that is stored in a clinician's memoryis learned by physicians during training, but undergoes little modification given the limited exposure to a rare disease.
In modern practice, the pentad is rarely seen, and the characteristics of the end‐organ findings may vary substantially. For instance, while neurological symptoms including seizures, coma, and transient confusion occur in 90% of cases, renal involvement is seen in about 50% and fever in only 25% of patients.5 Although the presence of 2 or more schistocytes on the blood smear under 100 microscopy supports the diagnosis of MAHA, cases of TTP without significant schistocytosis have been reported.6
Furthermore, TTP is typically described as acute in onset, but in a quarter of patients the symptoms and signs last for weeks before diagnosis.4 This variability in disease presentation coupled with the high mortality of untreated disease has changed the diagnostic and treatment thresholds for TTP. Trials and expert opinion use MAHA, thrombocytopenia, and the exclusion of alternative causes as sufficient criteria to diagnose TTP and begin treatment.7 The measurement of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity (a von Willebrand factor‐cleaving protease) for diagnostic purposes remains controversial because assay techniques are not uniform and there is insufficient correlation between levels and clinical disease.810 For instance, the presence of severe ADAMTS 13 deficiency (ie, <5%) along with the presence of an ADAMTS13 inhibitor is considered to be very specific, but not sensitive, for the laboratory diagnosis of idiopathic TTP.11 In cohort studies, the frequency of severe deficiency among patients with idiopathic TTP ranged from 18% to 100%, and the presence of severe deficiency did not predict the development of acute episodes of TTP.9 In a registry study of 142 patients diagnosed with TTP, 81% of patients with secondary TTP (ie, not classified as idiopathic) had ADAMTS13 levels that were normal to subnormal (>25%), and patients with normal ADAMTS13 levels had a higher incidence of acute renal failure, similar to the findings in this patient.10
Untreated TTP has a mortality rate of greater than 90%, but with plasma exchange, survival has improved dramatically.4, 7 Glucocorticoids are often used in addition to plasma exchange, based on case series and reports.9 The addition of cryoprecipitate or fresh frozen plasma to plasmapheresis has not been shown to be beneficial, but rituximab, an anti CD‐20 monoclonal antibody, has shown promise in a small prospective study.12, 13
TTP is a rare disorder with a classic description but substantial variation in clinical presentation. In this case, the background autoimmune myopathy, immunosuppression, coincident acute DVT, unexplained infiltrates, complex medication regimen, and nephrotic range proteinuria (attributed to focal segmental glomerular sclerosis based on the limited evidence available from the biopsy) led the clinicians to ascribe the patient's thrombocytopenia and renal injury to more common conditions and created a challenging environment for the diagnosis of TTP. TTP is a complex disorder and the simplified understanding of the disease and its time course prevented a prompt match between the patient's clinical course and his diagnosis. The combination of a rare condition with inherent variability arising in the setting of medical complexity challenges the processes of problem representation and scripting the answer for even the most seasoned clinician.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Key Teaching Points
-
The classically described pentad of TTP is seldom seen, and the findings of otherwise unexplained MAHA and thrombocytopenia should prompt consideration of TTP.
-
TTP may be acute and idiopathic, or be secondary to drugs, infections, or other conditions. Medication‐induced TTP may present with a wide range of clinical findings.
-
Therapeutic plasma exchange may be life‐saving in cases of TTP, and when appropriate, should be initiated promptly based on clinical suspicion and without waiting to perform tissue biopsy.
- , , .Thrombotic microangiopathies. In: Tischer CC, Brenner BM, eds.Renal Pathology.2nd ed.Philadelphia, PA:JB Lippincott;1994:1154–1184.
- , , .Drug‐induced thrombotic microangiopathy: incidence, prevention and management.Drug Saf.2001;24(7):491–501.
- , , , , .FK 506‐associated thrombotic microangiopathy: report of two cases and review of the literature.Transplantation.1999;67(4):539–544.
- , .Thrombotic Thrombocytopenic purpura: report of 16 cases and review of the literature.Medicine (Baltimore).1966;45:139–159.
- , , , .Thrombotic thrombocytopenic purpura; early and late responders.Am J Hematol.1997;54:102–107.
- .Atypical presentations of thrombotic thrombocytopenic purpura: a review.J Clin Apheresis.2009;24(1)47–52.
- , , , et al.Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura.N Engl J Med.1991;325:393–397.
- , , , , , .The incidence of thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS13deficiency.J Thromb Haemost.2005;3:1432–1436.
- .Thrombotic thrombocytopenic purpura.N Engl J Med.2006;354:1927–1935.
- , , , et al.ADAMTS13 activity in thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients.Blood.2003;102:60–68.
- , , .Thrombotic thrombocytopenic purpura.J Thromb Haemost.2005;3:1663–1675.
- , , , et al.Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials.Am J Kidney Dis.2009;53:259–272.
- , , , et al.:Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13‐deficient TTP: A study of 11 cases.Blood.2005;105:1932–1937.
- , , .Thrombotic microangiopathies. In: Tischer CC, Brenner BM, eds.Renal Pathology.2nd ed.Philadelphia, PA:JB Lippincott;1994:1154–1184.
- , , .Drug‐induced thrombotic microangiopathy: incidence, prevention and management.Drug Saf.2001;24(7):491–501.
- , , , , .FK 506‐associated thrombotic microangiopathy: report of two cases and review of the literature.Transplantation.1999;67(4):539–544.
- , .Thrombotic Thrombocytopenic purpura: report of 16 cases and review of the literature.Medicine (Baltimore).1966;45:139–159.
- , , , .Thrombotic thrombocytopenic purpura; early and late responders.Am J Hematol.1997;54:102–107.
- .Atypical presentations of thrombotic thrombocytopenic purpura: a review.J Clin Apheresis.2009;24(1)47–52.
- , , , et al.Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura.N Engl J Med.1991;325:393–397.
- , , , , , .The incidence of thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS13deficiency.J Thromb Haemost.2005;3:1432–1436.
- .Thrombotic thrombocytopenic purpura.N Engl J Med.2006;354:1927–1935.
- , , , et al.ADAMTS13 activity in thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients.Blood.2003;102:60–68.
- , , .Thrombotic thrombocytopenic purpura.J Thromb Haemost.2005;3:1663–1675.
- , , , et al.Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials.Am J Kidney Dis.2009;53:259–272.
- , , , et al.:Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13‐deficient TTP: A study of 11 cases.Blood.2005;105:1932–1937.
Evaluation of Hemostasis
A previously healthy 25‐year‐old Guatemalan man presented to the emergency department with 1 day of fever, nausea, vomiting, and right lower quadrant abdominal pain. A computed tomography (CT) scan revealed acute appendicitis. The patient underwent an uncomplicated laparoscopic appendectomy and was discharged in stable condition after 48 hours.
Five days after the operation he returned to the emergency department with abdominal pain, nausea, vomiting, and lightheadedness. He was tachycardic, and his hemoglobin was 9.5 g/dL (normal, 13.3‐17.7 g/dL), decreased from 14.4 g/dL prior to his appendectomy. A CT scan showed intraperitoneal blood with active extravasation of contrast at the site of the appendectomy.
Additional laboratory testing revealed an activated partial thromboplastin time (aPTT) of 52 seconds (normal, <37 seconds) and protime (also prothrombin time [PT]) of 14 seconds (normal, <14.1 seconds). The platelet count was 449,000/L (normal, 150‐400,000/L) and the fibrinogen level was 337 mg/dL (normal, 170‐440 mg/dL). Crystalloid and packed red blood cells were administered. Since further laboratory evaluation of the prolonged aPTT was not immediately available, the patient was empirically treated with fresh frozen plasma (FFP), cryoprecipitate, and Factor VIII/von Willebrand factor concentrate. At laparotomy, bleeding was observed at the previous operative site, and 2 L of intraperitoneal blood was evacuated.
The next morning, Factor VIII and Factor IX (FIX) activities and the ristocetin cofactor study performed on specimens obtained immediately prior to the second operation were normal, but the FIX activity was 5% of normal. The diagnosis of FXI deficiency was made and 2 to 3 units of FFP (the amount necessary to maintain the patient's measured FXI activity near 20% of normal) were transfused daily. Nine days of FFP infusions were required to achieve complete wound hemostasis. The patient had no further bleeding episodes after discharge.
Upon further interviewing, the patient revealed that 2 months prior he sustained a small laceration on his arm that bled for a long time and that his brother had experienced prolonged bleeding after a dental extraction.
Commentary
Routine performance of preprocedural laboratory testing, and complete reliance on the results as a means of excluding a propensity to bleeding, may not only lead to excessive testing and delayed procedures, but also provides false reassurance because normal routine laboratory studies cannot be used to exclude some bleeding disorders (Table 1).
| Von Willebrand disease |
| Mild hemophilia A (Factor VIII deficiency) |
| Mild hemophilia B (Factor IX deficiency) |
| Mild hemophilia C (Factor XI deficiency) |
| Qualitative platelet disorders (congenital or acquired) |
| Factor XIII deficiency |
| Disorders of fibrinolysis (eg, antiplasmin deficiency, plasminogen activator inhibitor type 1 deficiency) |
| Disorders of the vasculature or integument (hereditary hemorrhagic telangiectasia, Ehlers‐Danlos syndrome) |
Most studies evaluating routine laboratory testing of hemostatic variables prior to invasive procedures come from patients undergoing elective general surgery. A 1988 study concluded that there is no benefit in the routine preoperative use of the PT, aPTT, platelet count, and bleeding time in the absence of clinical evidence of a hemostatic defect, as assessed by a patient questionnaire and a thorough physical examination.1 A subsequent European, prospective, multicenter study confirmed that abnormalities of preoperative laboratory screening in the absence of a history of bleeding or clinical abnormality were not associated with worse surgical morbidity or mortality, compared to patients with normal screening laboratory studies.2 A recent systematic review has also confirmed the poor positive predictive value of screening tests when used in isolation, and recommended a history‐based and physical exam‐based approach.3 Questionnaires have been shown to be particularly important tools for eliciting clinically significant bleeding disorders that may require revision of the surgical plan.1,4
FXI is a serine protease whose activity is crucial for robust fibrin clot formation and inhibition of fibrinolysis at sites of vascular injury.5 FXI deficiency is an autosomal recessive disorder with an incidence of 1 per 1,000,000 in the general population, with a significantly higher incidence in the Ashkenazi Jewish population. While the risk of spontaneous hemorrhage is typically low, life‐threatening bleeding may occur after surgery or trauma. The severity of the measured FXI level deficiency does not always correlate with risk of bleeding. Periprocedural prophylaxis and treatment of bleeding aim to replace FXI to the low‐normal range by administering FXI concentrate, (not available in the United States) or FFP. Antifibrinolytic agents such as tranexamic acid or ϵ‐aminocaproic acid may be used adjunctively in cases of mucosal bleeding.5
In this case, preoperative screening, either using a questionnaire or careful history‐taking, would have identified the patient's personal and family history of bleeding and prompted appropriate preoperative coagulation testing, which could have exposed the hemostatic defect, allowing for modification of the perioperative medical plan.
In summary, preoperative bleeding evaluations should be performed routinely and should begin with a careful history (use of a questionnaire may be considered) and physical examination. Excessive bleeding after prior surgery, trauma, dental extractions, parturition, or circumcision; bleeding tendency in family members; current use of medications that may increase bleeding risk (such as anticoagulants or aspirin); and physical signs associated with bleeding should be assessed. If clinical details fail to expose a potential bleeding disorder, it is safe and cost‐effective1 to proceed with surgery without performing additional laboratory testing. In contrast, any abnormality on the clinical assessment should trigger preoperative laboratory analysis of basic hemostatic parameters, which may prompt further testing or hematology consultation.
- ,,.A prospective evaluation of the efficacy of preoperative coagulation testing.Ann Surg.1988;208(5):554–557.
- ,,,,.A prospective multicenter evaluation of preoperative hemostatic screening tests. The French Associations for Surgical Research.Am J Surg.1995;170(1):19–23.
- ,,,.Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures.Br J Haematol.2008;140:496–504.
- ,,, et al.A practical concept for preoperative identification of patients with impaired primary hemostasis.Clin Appl Thrombosis Haemost.2004;10(3):195–204.
- ,.Factor XI deficiency.Haemophilia.2008;14(6):1183–1189.
A previously healthy 25‐year‐old Guatemalan man presented to the emergency department with 1 day of fever, nausea, vomiting, and right lower quadrant abdominal pain. A computed tomography (CT) scan revealed acute appendicitis. The patient underwent an uncomplicated laparoscopic appendectomy and was discharged in stable condition after 48 hours.
Five days after the operation he returned to the emergency department with abdominal pain, nausea, vomiting, and lightheadedness. He was tachycardic, and his hemoglobin was 9.5 g/dL (normal, 13.3‐17.7 g/dL), decreased from 14.4 g/dL prior to his appendectomy. A CT scan showed intraperitoneal blood with active extravasation of contrast at the site of the appendectomy.
Additional laboratory testing revealed an activated partial thromboplastin time (aPTT) of 52 seconds (normal, <37 seconds) and protime (also prothrombin time [PT]) of 14 seconds (normal, <14.1 seconds). The platelet count was 449,000/L (normal, 150‐400,000/L) and the fibrinogen level was 337 mg/dL (normal, 170‐440 mg/dL). Crystalloid and packed red blood cells were administered. Since further laboratory evaluation of the prolonged aPTT was not immediately available, the patient was empirically treated with fresh frozen plasma (FFP), cryoprecipitate, and Factor VIII/von Willebrand factor concentrate. At laparotomy, bleeding was observed at the previous operative site, and 2 L of intraperitoneal blood was evacuated.
The next morning, Factor VIII and Factor IX (FIX) activities and the ristocetin cofactor study performed on specimens obtained immediately prior to the second operation were normal, but the FIX activity was 5% of normal. The diagnosis of FXI deficiency was made and 2 to 3 units of FFP (the amount necessary to maintain the patient's measured FXI activity near 20% of normal) were transfused daily. Nine days of FFP infusions were required to achieve complete wound hemostasis. The patient had no further bleeding episodes after discharge.
Upon further interviewing, the patient revealed that 2 months prior he sustained a small laceration on his arm that bled for a long time and that his brother had experienced prolonged bleeding after a dental extraction.
Commentary
Routine performance of preprocedural laboratory testing, and complete reliance on the results as a means of excluding a propensity to bleeding, may not only lead to excessive testing and delayed procedures, but also provides false reassurance because normal routine laboratory studies cannot be used to exclude some bleeding disorders (Table 1).
| Von Willebrand disease |
| Mild hemophilia A (Factor VIII deficiency) |
| Mild hemophilia B (Factor IX deficiency) |
| Mild hemophilia C (Factor XI deficiency) |
| Qualitative platelet disorders (congenital or acquired) |
| Factor XIII deficiency |
| Disorders of fibrinolysis (eg, antiplasmin deficiency, plasminogen activator inhibitor type 1 deficiency) |
| Disorders of the vasculature or integument (hereditary hemorrhagic telangiectasia, Ehlers‐Danlos syndrome) |
Most studies evaluating routine laboratory testing of hemostatic variables prior to invasive procedures come from patients undergoing elective general surgery. A 1988 study concluded that there is no benefit in the routine preoperative use of the PT, aPTT, platelet count, and bleeding time in the absence of clinical evidence of a hemostatic defect, as assessed by a patient questionnaire and a thorough physical examination.1 A subsequent European, prospective, multicenter study confirmed that abnormalities of preoperative laboratory screening in the absence of a history of bleeding or clinical abnormality were not associated with worse surgical morbidity or mortality, compared to patients with normal screening laboratory studies.2 A recent systematic review has also confirmed the poor positive predictive value of screening tests when used in isolation, and recommended a history‐based and physical exam‐based approach.3 Questionnaires have been shown to be particularly important tools for eliciting clinically significant bleeding disorders that may require revision of the surgical plan.1,4
FXI is a serine protease whose activity is crucial for robust fibrin clot formation and inhibition of fibrinolysis at sites of vascular injury.5 FXI deficiency is an autosomal recessive disorder with an incidence of 1 per 1,000,000 in the general population, with a significantly higher incidence in the Ashkenazi Jewish population. While the risk of spontaneous hemorrhage is typically low, life‐threatening bleeding may occur after surgery or trauma. The severity of the measured FXI level deficiency does not always correlate with risk of bleeding. Periprocedural prophylaxis and treatment of bleeding aim to replace FXI to the low‐normal range by administering FXI concentrate, (not available in the United States) or FFP. Antifibrinolytic agents such as tranexamic acid or ϵ‐aminocaproic acid may be used adjunctively in cases of mucosal bleeding.5
In this case, preoperative screening, either using a questionnaire or careful history‐taking, would have identified the patient's personal and family history of bleeding and prompted appropriate preoperative coagulation testing, which could have exposed the hemostatic defect, allowing for modification of the perioperative medical plan.
In summary, preoperative bleeding evaluations should be performed routinely and should begin with a careful history (use of a questionnaire may be considered) and physical examination. Excessive bleeding after prior surgery, trauma, dental extractions, parturition, or circumcision; bleeding tendency in family members; current use of medications that may increase bleeding risk (such as anticoagulants or aspirin); and physical signs associated with bleeding should be assessed. If clinical details fail to expose a potential bleeding disorder, it is safe and cost‐effective1 to proceed with surgery without performing additional laboratory testing. In contrast, any abnormality on the clinical assessment should trigger preoperative laboratory analysis of basic hemostatic parameters, which may prompt further testing or hematology consultation.
A previously healthy 25‐year‐old Guatemalan man presented to the emergency department with 1 day of fever, nausea, vomiting, and right lower quadrant abdominal pain. A computed tomography (CT) scan revealed acute appendicitis. The patient underwent an uncomplicated laparoscopic appendectomy and was discharged in stable condition after 48 hours.
Five days after the operation he returned to the emergency department with abdominal pain, nausea, vomiting, and lightheadedness. He was tachycardic, and his hemoglobin was 9.5 g/dL (normal, 13.3‐17.7 g/dL), decreased from 14.4 g/dL prior to his appendectomy. A CT scan showed intraperitoneal blood with active extravasation of contrast at the site of the appendectomy.
Additional laboratory testing revealed an activated partial thromboplastin time (aPTT) of 52 seconds (normal, <37 seconds) and protime (also prothrombin time [PT]) of 14 seconds (normal, <14.1 seconds). The platelet count was 449,000/L (normal, 150‐400,000/L) and the fibrinogen level was 337 mg/dL (normal, 170‐440 mg/dL). Crystalloid and packed red blood cells were administered. Since further laboratory evaluation of the prolonged aPTT was not immediately available, the patient was empirically treated with fresh frozen plasma (FFP), cryoprecipitate, and Factor VIII/von Willebrand factor concentrate. At laparotomy, bleeding was observed at the previous operative site, and 2 L of intraperitoneal blood was evacuated.
The next morning, Factor VIII and Factor IX (FIX) activities and the ristocetin cofactor study performed on specimens obtained immediately prior to the second operation were normal, but the FIX activity was 5% of normal. The diagnosis of FXI deficiency was made and 2 to 3 units of FFP (the amount necessary to maintain the patient's measured FXI activity near 20% of normal) were transfused daily. Nine days of FFP infusions were required to achieve complete wound hemostasis. The patient had no further bleeding episodes after discharge.
Upon further interviewing, the patient revealed that 2 months prior he sustained a small laceration on his arm that bled for a long time and that his brother had experienced prolonged bleeding after a dental extraction.
Commentary
Routine performance of preprocedural laboratory testing, and complete reliance on the results as a means of excluding a propensity to bleeding, may not only lead to excessive testing and delayed procedures, but also provides false reassurance because normal routine laboratory studies cannot be used to exclude some bleeding disorders (Table 1).
| Von Willebrand disease |
| Mild hemophilia A (Factor VIII deficiency) |
| Mild hemophilia B (Factor IX deficiency) |
| Mild hemophilia C (Factor XI deficiency) |
| Qualitative platelet disorders (congenital or acquired) |
| Factor XIII deficiency |
| Disorders of fibrinolysis (eg, antiplasmin deficiency, plasminogen activator inhibitor type 1 deficiency) |
| Disorders of the vasculature or integument (hereditary hemorrhagic telangiectasia, Ehlers‐Danlos syndrome) |
Most studies evaluating routine laboratory testing of hemostatic variables prior to invasive procedures come from patients undergoing elective general surgery. A 1988 study concluded that there is no benefit in the routine preoperative use of the PT, aPTT, platelet count, and bleeding time in the absence of clinical evidence of a hemostatic defect, as assessed by a patient questionnaire and a thorough physical examination.1 A subsequent European, prospective, multicenter study confirmed that abnormalities of preoperative laboratory screening in the absence of a history of bleeding or clinical abnormality were not associated with worse surgical morbidity or mortality, compared to patients with normal screening laboratory studies.2 A recent systematic review has also confirmed the poor positive predictive value of screening tests when used in isolation, and recommended a history‐based and physical exam‐based approach.3 Questionnaires have been shown to be particularly important tools for eliciting clinically significant bleeding disorders that may require revision of the surgical plan.1,4
FXI is a serine protease whose activity is crucial for robust fibrin clot formation and inhibition of fibrinolysis at sites of vascular injury.5 FXI deficiency is an autosomal recessive disorder with an incidence of 1 per 1,000,000 in the general population, with a significantly higher incidence in the Ashkenazi Jewish population. While the risk of spontaneous hemorrhage is typically low, life‐threatening bleeding may occur after surgery or trauma. The severity of the measured FXI level deficiency does not always correlate with risk of bleeding. Periprocedural prophylaxis and treatment of bleeding aim to replace FXI to the low‐normal range by administering FXI concentrate, (not available in the United States) or FFP. Antifibrinolytic agents such as tranexamic acid or ϵ‐aminocaproic acid may be used adjunctively in cases of mucosal bleeding.5
In this case, preoperative screening, either using a questionnaire or careful history‐taking, would have identified the patient's personal and family history of bleeding and prompted appropriate preoperative coagulation testing, which could have exposed the hemostatic defect, allowing for modification of the perioperative medical plan.
In summary, preoperative bleeding evaluations should be performed routinely and should begin with a careful history (use of a questionnaire may be considered) and physical examination. Excessive bleeding after prior surgery, trauma, dental extractions, parturition, or circumcision; bleeding tendency in family members; current use of medications that may increase bleeding risk (such as anticoagulants or aspirin); and physical signs associated with bleeding should be assessed. If clinical details fail to expose a potential bleeding disorder, it is safe and cost‐effective1 to proceed with surgery without performing additional laboratory testing. In contrast, any abnormality on the clinical assessment should trigger preoperative laboratory analysis of basic hemostatic parameters, which may prompt further testing or hematology consultation.
- ,,.A prospective evaluation of the efficacy of preoperative coagulation testing.Ann Surg.1988;208(5):554–557.
- ,,,,.A prospective multicenter evaluation of preoperative hemostatic screening tests. The French Associations for Surgical Research.Am J Surg.1995;170(1):19–23.
- ,,,.Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures.Br J Haematol.2008;140:496–504.
- ,,, et al.A practical concept for preoperative identification of patients with impaired primary hemostasis.Clin Appl Thrombosis Haemost.2004;10(3):195–204.
- ,.Factor XI deficiency.Haemophilia.2008;14(6):1183–1189.
- ,,.A prospective evaluation of the efficacy of preoperative coagulation testing.Ann Surg.1988;208(5):554–557.
- ,,,,.A prospective multicenter evaluation of preoperative hemostatic screening tests. The French Associations for Surgical Research.Am J Surg.1995;170(1):19–23.
- ,,,.Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures.Br J Haematol.2008;140:496–504.
- ,,, et al.A practical concept for preoperative identification of patients with impaired primary hemostasis.Clin Appl Thrombosis Haemost.2004;10(3):195–204.
- ,.Factor XI deficiency.Haemophilia.2008;14(6):1183–1189.
Flushing Out the Diagnosis
A 42‐year‐old woman with a history of mild asthma presented to the emergency department (ED) following 1 week of headache. She had been in her usual state of good health until 1 week prior to her presentation, when she noticed intermittent frontal headaches without neck stiffness or other neurologic symptoms. She then developed diffuse myalgias, fatigue, subjective fevers, and rigors for the 24 hours prior to presentation. On the morning of presentation, chest tightness, palpitations, and shortness of breath occurred. She used her albuterol metered‐dose inhaler without relief and went to the hospital.
Many of these features can be explained by a viral syndrome exacerbating underlying asthma or by a psychiatric condition such as anxiety or depression, but they may also be a harbinger of a systemic process, including infection, malignancy, or autoimmunity. Because the onset of headache is temporally distant from the other symptoms, I am more inclined to believe that it represents a primary intracranial process than I would if it were coincident with the onset of the other acute symptoms. If the fevers and rigors are verified, infection would be the initial concern. Failure to respond to her inhalers may either signify a severe asthma exacerbation or a nonbronchospastic cause of dyspnea.
She reported mild nausea, but denied photophobia, vomiting, abdominal pain, diarrhea, melena, or hematochezia. She did not have recent ill contacts, animal bites, or travel. Her medical history included asthma, diverticulitis, chronic right ankle pain, and obesity. She reported an allergic rash to amoxicillin. Her medications were sulindac and fluticasone/salmeterol, and albuterol metered‐dose inhalers. She worked as a preschool teacher and was married with 2 children. She denied any tobacco use and seldom drank alcoholic beverages. On exam, temperature was 36.7C, pulse was 107 beats per minute, blood pressure was 129/91 mm Hg, respiratory rate was 19 breaths per minute, and oxygen saturation was 98% while breathing ambient air. Her face and anterior neck were flushed and diaphoretic, and her sclerae were icteric. There was no nuchal rigidity. Her cardiac rhythm was regular without murmurs, lungs were clear to auscultation, and the abdomen was mildly tender to palpation in the epigastrium and right upper quadrant. The white blood cell (WBC) count was 9200/L, with 84% neutrophils, 3% lymphocytes, 6% monocytes, and 7% eosinophils. The hemoglobin was 14.8 g/dL and the platelet count was 166,000/L. Total serum bilirubin was 4.6 mg/dL, aspartate aminotransferase (AST) was 459 U/L (normal range, 8‐31), alanine aminotransferase (ALT) was 667 U/L (normal range, 7‐31), and alkaline phosphatase was 146 U/L (normal range, 39‐117). Serum electrolytes, creatinine, lactate, lipase, thyrotropin, coagulation studies, and cardiac enzymes were all normal. Urinalysis showed trace leukocyte esterase and bilirubin, as well as 3 WBCs and 2 red cells per high‐power field. Chest radiography and an electrocardiogram demonstrated no abnormalities.
The major findingwhich is critical to focusing problem‐solving in the face of a broad range of symptomsis her hepatitis. The common etiologies for hepatitis of this degree include viruses (hepatitis A and cytomegalovirus [CMV] should be considered given her work in preschool), toxins, autoimmunity, and vascular events. Liver disease in association with flushing raises the possibility of carcinoid syndrome with liver metastases. The lack of wheezing makes the bronchospasm of asthma or carcinoid less suitable explanations for her shortness of breath. Her eosinophilia is mild but probably is not accounted for alone by well‐controlled asthma in a person with no history of atopic disease. I would also ask her about any alternative and over‐the‐counter remedies. The paucity of lymphocytes raises the possibility of human immunodeficiency virus (HIV), Hodgkin's disease, or systemic lupus erythematosus. Although she does not have a documented fever or leukocytosis, she reported fevers and chills and is diaphoretic and tachycardic, so exclusion of biliary obstruction and cholangitis is the highest priority.
An abdominal ultrasound demonstrated hepatomegaly with moderate fatty infiltration and a normal gallbladder without pericholecystic fluid. The intrahepatic and extrahepatic biliary ducts were normal and the hepatic and portal veins were patent. Computed tomography of the abdomen showed slight thickening of the sigmoid colon wall. Ciprofloxacin and metronidazole were administered for possible diverticulitis. Over the first 48 hours of hospitalization her symptoms improved markedly. Her flushing resolved and she had no recorded fevers in the hospital. Serologies were negative for hepatitis A immunoglobulin M (IgM), hepatitis B surface antibody, hepatitis B surface antigen, and hepatitis C antibody. A monospot test was negative and the erythrocyte sedimentation rate was 11 mm/hour. Blood and urine cultures were negative. On the second hospital day the absolute eosinophil count rose to 855/L (15% of 5700 WBCs). On the fourth hospital day, the absolute eosinophil count was 1092/L, the total bilirubin was 1.9 mg/dL, and the AST and ALT were 174 U/L and 476 U/L, respectively. Antibiotics were stopped and she was discharged home.
Her prompt improvement suggests either a self‐limited condition or a response to the antibiotics. The rapid but incomplete resolution of her hepatitis is in keeping with a withdrawal of a toxin, relief of biliary obstruction, or a transient vascular event, and is less consistent with a viral hepatitis or an infiltrative process. With normal biliary system imaging, sterile blood cultures, and the absence of fever or leukocytosis, cholangitis is unlikely. Likewise, there is no suggestion of a vascular event, either obstructive or hemodynamic, that is impairing the liver.
A common cause of eosinophilia in hospitalized patients is medications, so it would be useful to monitor that count after the new antibiotics. At this point, I also wonder if the eosinophils are a feature of the underlying illness, as they were present to a modest degree on admission before any new medications were administered. The overlap of eosinophilia and hepatitis brings to mind a medication reaction (eg, to sulindac) or a hepatobiliary parasite, such as ascaris or clonorchis, for which she lacks a known exposure. Many patients experience flushing in the setting of fever or stress, but sustained flushing may suggest a systemic illness characterized by the release of vasoactive mediators such as carcinoid syndrome or mastocytosis. The latter might be considered more strongly if the eosinophilia is deemed to be primary (rather than reactive) after a thorough evaluation.
After 2 days at home, the patient had recurrence of subjective fevers, with chest, back, and abdominal pain, fatigue, loose stools, and rigors. She returned to the ED, where she was noted to have facial erythema and injected sclerae, but the remainder of her physical exam was normal. The total serum bilirubin was 1.1 mg/dL, AST was 156 U/L, ALT was 214 U/L, and alkaline phosphatase was 240 U/L. Serum lipase was normal. WBC count was 14,000/L, with 94% neutrophils, 3% lymphocytes, 2% monocytes, and 1% eosinophils. She was again treated empirically with ciprofloxacin and metronidazole. Endoscopic ultrasound was normal, with no evidence of gallbladder sludge or microlithiasis. Stool cultures, assay for Clostridium difficile, and examination for ova and parasites were negative. The 24‐hour urine demonstrated no elevation in 5‐hydroxyindoleacetic acid. An adrenocorticotropic hormone (ACTH) stimulation test was normal. HIV antibody was negative. Her symptoms improved within 2 days. The eosinophil count rose and peaked at 1541/L by the third hospital day, while the transaminase elevations resolved. Antibiotics were discontinued. A liver biopsy showed mixed macrovesicular and microvesicular fatty metamorphosis and steatohepatitis with eosinophils (Figures 1 and 2). She was discharged home on the sixth hospital day.

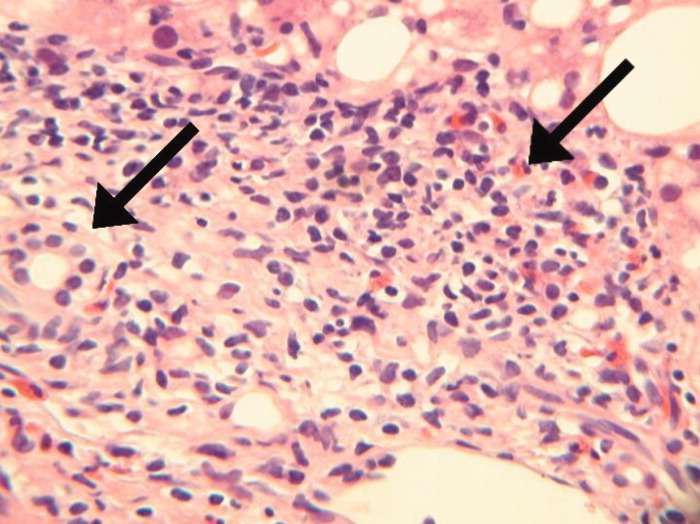
Her illness can now be characterized as relapsing inflammation, which given the frequency (over days) suggests either an indolent infectious focus that periodically causes systemic inflammation or reexposure to a toxic substance. The 2 most notable laboratory abnormalities, the hepatitis and the eosinophilia, persist but have differing trajectories. While the liver function tests have progressively normalized despite clinical relapses, the eosinophils have had a more fluctuating course characterized by increases during the hospitalization and higher levels during the second hospitalization. The absence of an infection, recurrent systemic inflammation, and eosinophilic hepatitis suggest a hypersensitivity reaction to a medication or other substance. She is most likely being reexposed at home, where her symptoms occur, and not in the hospital, where her symptoms resolve. Sulindac is a leading candidate, because nonsteroidal antiinflammatory drugs (NSAIDs) cause a number of hypersensitivity reactions and are frequently stopped when sick patients enter the hospital.
Seven days after discharge she developed acute onset of subjective fever, nausea, diffuse myalgias, and flushing, identical to the 2 prior episodes, and she again returned to the ED. Her temperature was 39.1C, heart rate was 120 beats per minute, blood pressure was 87/50 mm Hg, respiratory rate was 18 breaths per minute, and the oxygen saturation was 96% while breathing room air. She had diffuse flushing from her neck over her torso and was diaphoretic with injected sclera and conjunctiva. The WBC count was 11,400/L with 97% neutrophils and 3% lymphocytes. Total bilirubin was 0.8 mg/dL, AST was 134 U/L, ALT was 140 U/L, and alkaline phosphatase was 144 U/L. She was readmitted to the hospital. Following admission, she had no fevers, the flushing resolved, and AST and ALT levels decreased. The only treatment the patient received in the ED and during her hospital stay was acetaminophen as needed for pain or fever. The eosinophil count peaked at 1404/L by hospital day 4. Blood and urine cultures were negative. IgM antibodies to Epstein‐Barr virus were not detected, CMV DNA was not detected, and a rapid plasma reagin (RPR) test was nonreactive. Ferritin, ceruloplasmin, alpha‐1‐antitrypsin, and tryptase levels were normal. Antimitochondrial, antismooth muscle, antineutrophil cytoplasmic, and antinuclear antibodies were negative. There was no monoclonal band on serum protein electrophoresis. A blood smear for Borrelia detected no spirochetes.
A complete picture of the uncommon but classic flushing disorders, namely carcinoid, mastocytosis, and pheochromocytoma, has not emerged. The constellation of inflammation, mucosal and hepatic involvement, and eosinophilia are most consistent with a drug hypersensitivity reaction. Additionally, the recurrent inflammation is becoming more severe, as manifest by the fever and hemodynamic derangements, which suggests an increasing sensitization to the offending agent. I would review every drug she has received in the hospital, but given the recurrences after discharge her home medications are the most likely explanation. Of these, sulindac is the most likely culprit.
On further questioning, it was learned that the patient began taking sulindac 200 mg twice daily to treat her chronic ankle pain 6 weeks before the first admission. The medication had been stopped on each admission. She was instructed to discontinue sulindac. She has had no recurrences of symptoms and her hepatitis and eosinophilia have resolved.
DISCUSSION
This patient presented with recurrent skin findings, eosinophilia, hepatitis, and constitutional symptoms caused by hypersensitivity to sulindac. This drug‐induced hypersensitivity syndrome was originally described with anticonvulsant drugs (carbamazepine, phenytoin, and phenobarbitone) and named anticonvulsant hypersensitivity syndrome,1, 2 but has been observed with many other medications, including allopurinol, dapsone, minocycline, and nevirapine. The term drug rash with eosinophilia and systemic symptoms (DRESS) syndrome has been recently adopted to convey the cardinal features that characterize this disorder.3
DRESS syndrome is defined by rash, fever, and internal organ involvement.4 Also included in the diagnostic criteria are hematologic abnormalities (eosinophilia 1.5 109/mm3 or the presence of atypical lymphocytes) and lymphadenopathy.3 The multiorgan involvement distinguishes DRESS from other cutaneous drug eruptions. In a review of the French Pharmacovigilance Database for all cases of DRESS over a 15‐year period, 73% to 100% of patients were reported to have dermatologic abnormalities, most frequently a maculopapular rash or erythroderma. Less common skin findings include vesicles, bullae, pustules, erythroderma, and purpuric lesions. Liver abnormalities were observed in more than 60% of patients and were the most frequent systemic finding.5 Eosinophilia was the most common hematologic abnormality, present in more than 50% of cases. As this case underscores, DRESS syndrome typically begins 3 to 8 weeks after initiation of the drug because it is a delayed type IV hypersensitivity reaction.6 Fever can occur within hours on rechallenge because of the presence of memory T cells.
The main treatment for DRESS syndrome is withdrawal of the offending drug. Systemic corticosteroids have been recommended in cases with life‐threatening pulmonary or cardiac involvement, but have not been shown to be helpful in reversing renal or hepatic disease. Mortality, usually from end‐organ damage, occurs in about 10% of cases. The most common drugs are phenobarbital, carbamazepine, and phenytoin, with incidences of 1 in 5000 to 1 in 10,000. NSAIDs and antibiotics also have been implicated frequently. Human herpesvirus type 6 coinfection and genetically inherited slow acetylation have been associated with DRESS, although causal links have yet to be established.7, 8
The initial challenge in caring for a patient with multiple symptoms, exam findings, and test abnormalities is the coherent framing of the key clinical features that require explanation. This process, called problem representation, allows clinicians to search among a bounded list of possible diagnoses (or solutions) rather than invoking a differential diagnosis for every single abnormality. In searching for the proper diagnosis, this patient's clinical course required frequent reframing as more data became available.
Initially, the problem was framed as a 42‐year‐old woman with hepatitis. As the flushing and eosinophilia, which initially appeared to be transient and possibly nonspecific, became more prominent, the problem representation was revised to a 42‐year‐old woman with hepatitis, eosinophilia, and flushing. Since this triad did not immediately invoke a single diagnosis for the treating clinicians or the discussant, the differential diagnosis of hepatitis and eosinophilia and the differential diagnosis of flushing were considered in parallel.
Hepatitis and eosinophilia can occur coincidentally in the setting of parasitic infections, particularly helminths (ascaris, strongyloidiasis, and toxocaris) and liver flukes (opisthorchis and clonorchis), which invade the hepatobiliary system and induce a reactive eosinophilia. Some neoplasms, such as lymphomas and leukemias, and myeloproliferative disorders, including hypereosinophilic syndrome and mastocytosis, may have neoplastic cellular invasion of the liver and induce eosinophilia. Systemic drug hypersensitivity reactions are typically characterized by eosinophilia, transaminase elevations,9 and hepatitis on histology.10 As with this patient, liver biopsy in drug‐induced hepatitis shows a mixed microvesicular and macrovesicular steatosis, often with eosinophils.10
The flushing prompted a thorough but negative workup for the classic flushing disorders. The discussant's attempts to unify the flushing with the other clinical features illustrate how framing can affect our reasoning. Hepatitis, eosinophilia, and flushing defied an obvious single explanation and led the discussant down parallel diagnostic reasoning pathways. Although flushing is 1 of the well‐described dermatologic manifestations in drug‐hypersensitivity reactions, framing the central features as hepatitis, eosinophilia, and rash would have more readily suggested a drug reaction as a unifying diagnosis.11
The tempo and periodicity of this patient's illness provided the final formulation of a 42‐year‐old woman with hepatitis, eosinophilia, and flushing (or rash) that occurs every few days at home and resolves in the hospital. This formulation and the increasingly severe presentation, suggesting sensitization, were highly suggestive of an exogenous cause of her illness.
This case highlights how easily medication side effects can be overlooked during an extensive evaluation and how vigilant medication reconciliation coupled with an increased understanding of the spectrum of drug reactions can lead to early detection and prevention of potentially serious effects. In the case of DRESS, recognizing an association between a rash and organ involvement is central to making the diagnosis. Eosinophilia that accompanies a rash can further aid in narrowing the differential diagnosis.
The case also serves as a reminder of how framing with the slightest imprecision (eg, flushing instead of rash) can derail or delay the diagnostic process, yet is indispensable in tackling a complicated case. Finally, a time‐honored lesson in diagnosis is highlighted yet again: the diagnosis can usually be flushed out from the history.1214
Key Teaching Points
-
The constellation of skin findings, eosinophilia, organ involvement (particularly hepatitis), and constitutional symptoms should prompt consideration of DRESS syndrome and a hunt for a culprit drug.
-
Symptoms that resolve during hospitalization and repeatedly recur after discharge should prompt consideration of an exposure unique to the home, which may be environmental or pharmacologic.
-
Problem representation is critical in solving a complicated case, but adopting an inaccurate frame (representation) can derail or delay the diagnostic process.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,.Anticonvulsant hypersensitivity syndrome. In vitro assessment of risk.J Clin Invest.1988;82:1826–1832.
- ,,.Anticonvulsant hypersensitivity syndrome: incidence, prevention, and management.Drug Saf.1999;21:489–501.
- ,,,.Drug rash with eosinophilia and systemic symptoms vs toxic epidermal necrolysis: the dilemma of classification.Clin Dermatol.2005;23:311–314.
- ,.Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update.Dermatology.2003;206:353–356.
- ,,,,,Blayac J‐P; Network of the French Pharmacovigilance Centers. Variability in the clinical pattern of cutaneous side‐effects of drugs with systemic symptoms: does a DRESS syndrome really exist?Br J Dermatol.2006;155:422–428.
- ,.The immunological and clinical spectrum of delayed drug‐induced exanthems.Curr Opin Allergy Clin Immunol.2004;4:411–419.
- ,,.Association between anticonvulsant hypersensitivity syndrome and human herpesvirus 6 reactivation and hypogammaglobulinemia.Arch Dermatol.2004;140:183–188.
- ,.Treatment of severe drug reactions: Stevens‐Johnson Syndrome, toxic epidermal necrolysis and hypersensitivity syndrome.Dermatol Online J.2002;8:5.
- ,.Drug reactions. In: Bolognia JL, Jorizzo JL, Rapini RP, eds.Dermatology.2nd ed.Spain:Mosby Elsevier;2008:310–311.
- Pathology Outlines. Liver and intrahepatic bile ducts—non tumor. Hepatitis (non‐infectious). Drug/toxin‐induced hepatitis. Available at: http://www.pathologyoutlines.com/liver.html#drugtoxin. Accessed August2009.
- ,,.The flushing patient: differential diagnosis, workup and treatment.J Am Acad Dermatol.2006;55(2):193–208.
- ,,,,.Relative contributions of history‐taking, physical examination, and laboratory investigation to diagnosis and management of medical outpatients.Br Med J.1975;2(5969):486–489.
- ,,,,.Contributions to the history, physical examination, and laboratory investigation in making medical diagnosis.West J Med.1992;156(2):163–165.
- ,.A study on relative contributions of the history, physical examination and investigations in making medical diagnosis.J Assoc Physicians India.2000;48(8):771–775.
A 42‐year‐old woman with a history of mild asthma presented to the emergency department (ED) following 1 week of headache. She had been in her usual state of good health until 1 week prior to her presentation, when she noticed intermittent frontal headaches without neck stiffness or other neurologic symptoms. She then developed diffuse myalgias, fatigue, subjective fevers, and rigors for the 24 hours prior to presentation. On the morning of presentation, chest tightness, palpitations, and shortness of breath occurred. She used her albuterol metered‐dose inhaler without relief and went to the hospital.
Many of these features can be explained by a viral syndrome exacerbating underlying asthma or by a psychiatric condition such as anxiety or depression, but they may also be a harbinger of a systemic process, including infection, malignancy, or autoimmunity. Because the onset of headache is temporally distant from the other symptoms, I am more inclined to believe that it represents a primary intracranial process than I would if it were coincident with the onset of the other acute symptoms. If the fevers and rigors are verified, infection would be the initial concern. Failure to respond to her inhalers may either signify a severe asthma exacerbation or a nonbronchospastic cause of dyspnea.
She reported mild nausea, but denied photophobia, vomiting, abdominal pain, diarrhea, melena, or hematochezia. She did not have recent ill contacts, animal bites, or travel. Her medical history included asthma, diverticulitis, chronic right ankle pain, and obesity. She reported an allergic rash to amoxicillin. Her medications were sulindac and fluticasone/salmeterol, and albuterol metered‐dose inhalers. She worked as a preschool teacher and was married with 2 children. She denied any tobacco use and seldom drank alcoholic beverages. On exam, temperature was 36.7C, pulse was 107 beats per minute, blood pressure was 129/91 mm Hg, respiratory rate was 19 breaths per minute, and oxygen saturation was 98% while breathing ambient air. Her face and anterior neck were flushed and diaphoretic, and her sclerae were icteric. There was no nuchal rigidity. Her cardiac rhythm was regular without murmurs, lungs were clear to auscultation, and the abdomen was mildly tender to palpation in the epigastrium and right upper quadrant. The white blood cell (WBC) count was 9200/L, with 84% neutrophils, 3% lymphocytes, 6% monocytes, and 7% eosinophils. The hemoglobin was 14.8 g/dL and the platelet count was 166,000/L. Total serum bilirubin was 4.6 mg/dL, aspartate aminotransferase (AST) was 459 U/L (normal range, 8‐31), alanine aminotransferase (ALT) was 667 U/L (normal range, 7‐31), and alkaline phosphatase was 146 U/L (normal range, 39‐117). Serum electrolytes, creatinine, lactate, lipase, thyrotropin, coagulation studies, and cardiac enzymes were all normal. Urinalysis showed trace leukocyte esterase and bilirubin, as well as 3 WBCs and 2 red cells per high‐power field. Chest radiography and an electrocardiogram demonstrated no abnormalities.
The major findingwhich is critical to focusing problem‐solving in the face of a broad range of symptomsis her hepatitis. The common etiologies for hepatitis of this degree include viruses (hepatitis A and cytomegalovirus [CMV] should be considered given her work in preschool), toxins, autoimmunity, and vascular events. Liver disease in association with flushing raises the possibility of carcinoid syndrome with liver metastases. The lack of wheezing makes the bronchospasm of asthma or carcinoid less suitable explanations for her shortness of breath. Her eosinophilia is mild but probably is not accounted for alone by well‐controlled asthma in a person with no history of atopic disease. I would also ask her about any alternative and over‐the‐counter remedies. The paucity of lymphocytes raises the possibility of human immunodeficiency virus (HIV), Hodgkin's disease, or systemic lupus erythematosus. Although she does not have a documented fever or leukocytosis, she reported fevers and chills and is diaphoretic and tachycardic, so exclusion of biliary obstruction and cholangitis is the highest priority.
An abdominal ultrasound demonstrated hepatomegaly with moderate fatty infiltration and a normal gallbladder without pericholecystic fluid. The intrahepatic and extrahepatic biliary ducts were normal and the hepatic and portal veins were patent. Computed tomography of the abdomen showed slight thickening of the sigmoid colon wall. Ciprofloxacin and metronidazole were administered for possible diverticulitis. Over the first 48 hours of hospitalization her symptoms improved markedly. Her flushing resolved and she had no recorded fevers in the hospital. Serologies were negative for hepatitis A immunoglobulin M (IgM), hepatitis B surface antibody, hepatitis B surface antigen, and hepatitis C antibody. A monospot test was negative and the erythrocyte sedimentation rate was 11 mm/hour. Blood and urine cultures were negative. On the second hospital day the absolute eosinophil count rose to 855/L (15% of 5700 WBCs). On the fourth hospital day, the absolute eosinophil count was 1092/L, the total bilirubin was 1.9 mg/dL, and the AST and ALT were 174 U/L and 476 U/L, respectively. Antibiotics were stopped and she was discharged home.
Her prompt improvement suggests either a self‐limited condition or a response to the antibiotics. The rapid but incomplete resolution of her hepatitis is in keeping with a withdrawal of a toxin, relief of biliary obstruction, or a transient vascular event, and is less consistent with a viral hepatitis or an infiltrative process. With normal biliary system imaging, sterile blood cultures, and the absence of fever or leukocytosis, cholangitis is unlikely. Likewise, there is no suggestion of a vascular event, either obstructive or hemodynamic, that is impairing the liver.
A common cause of eosinophilia in hospitalized patients is medications, so it would be useful to monitor that count after the new antibiotics. At this point, I also wonder if the eosinophils are a feature of the underlying illness, as they were present to a modest degree on admission before any new medications were administered. The overlap of eosinophilia and hepatitis brings to mind a medication reaction (eg, to sulindac) or a hepatobiliary parasite, such as ascaris or clonorchis, for which she lacks a known exposure. Many patients experience flushing in the setting of fever or stress, but sustained flushing may suggest a systemic illness characterized by the release of vasoactive mediators such as carcinoid syndrome or mastocytosis. The latter might be considered more strongly if the eosinophilia is deemed to be primary (rather than reactive) after a thorough evaluation.
After 2 days at home, the patient had recurrence of subjective fevers, with chest, back, and abdominal pain, fatigue, loose stools, and rigors. She returned to the ED, where she was noted to have facial erythema and injected sclerae, but the remainder of her physical exam was normal. The total serum bilirubin was 1.1 mg/dL, AST was 156 U/L, ALT was 214 U/L, and alkaline phosphatase was 240 U/L. Serum lipase was normal. WBC count was 14,000/L, with 94% neutrophils, 3% lymphocytes, 2% monocytes, and 1% eosinophils. She was again treated empirically with ciprofloxacin and metronidazole. Endoscopic ultrasound was normal, with no evidence of gallbladder sludge or microlithiasis. Stool cultures, assay for Clostridium difficile, and examination for ova and parasites were negative. The 24‐hour urine demonstrated no elevation in 5‐hydroxyindoleacetic acid. An adrenocorticotropic hormone (ACTH) stimulation test was normal. HIV antibody was negative. Her symptoms improved within 2 days. The eosinophil count rose and peaked at 1541/L by the third hospital day, while the transaminase elevations resolved. Antibiotics were discontinued. A liver biopsy showed mixed macrovesicular and microvesicular fatty metamorphosis and steatohepatitis with eosinophils (Figures 1 and 2). She was discharged home on the sixth hospital day.
Her illness can now be characterized as relapsing inflammation, which given the frequency (over days) suggests either an indolent infectious focus that periodically causes systemic inflammation or reexposure to a toxic substance. The 2 most notable laboratory abnormalities, the hepatitis and the eosinophilia, persist but have differing trajectories. While the liver function tests have progressively normalized despite clinical relapses, the eosinophils have had a more fluctuating course characterized by increases during the hospitalization and higher levels during the second hospitalization. The absence of an infection, recurrent systemic inflammation, and eosinophilic hepatitis suggest a hypersensitivity reaction to a medication or other substance. She is most likely being reexposed at home, where her symptoms occur, and not in the hospital, where her symptoms resolve. Sulindac is a leading candidate, because nonsteroidal antiinflammatory drugs (NSAIDs) cause a number of hypersensitivity reactions and are frequently stopped when sick patients enter the hospital.
Seven days after discharge she developed acute onset of subjective fever, nausea, diffuse myalgias, and flushing, identical to the 2 prior episodes, and she again returned to the ED. Her temperature was 39.1C, heart rate was 120 beats per minute, blood pressure was 87/50 mm Hg, respiratory rate was 18 breaths per minute, and the oxygen saturation was 96% while breathing room air. She had diffuse flushing from her neck over her torso and was diaphoretic with injected sclera and conjunctiva. The WBC count was 11,400/L with 97% neutrophils and 3% lymphocytes. Total bilirubin was 0.8 mg/dL, AST was 134 U/L, ALT was 140 U/L, and alkaline phosphatase was 144 U/L. She was readmitted to the hospital. Following admission, she had no fevers, the flushing resolved, and AST and ALT levels decreased. The only treatment the patient received in the ED and during her hospital stay was acetaminophen as needed for pain or fever. The eosinophil count peaked at 1404/L by hospital day 4. Blood and urine cultures were negative. IgM antibodies to Epstein‐Barr virus were not detected, CMV DNA was not detected, and a rapid plasma reagin (RPR) test was nonreactive. Ferritin, ceruloplasmin, alpha‐1‐antitrypsin, and tryptase levels were normal. Antimitochondrial, antismooth muscle, antineutrophil cytoplasmic, and antinuclear antibodies were negative. There was no monoclonal band on serum protein electrophoresis. A blood smear for Borrelia detected no spirochetes.
A complete picture of the uncommon but classic flushing disorders, namely carcinoid, mastocytosis, and pheochromocytoma, has not emerged. The constellation of inflammation, mucosal and hepatic involvement, and eosinophilia are most consistent with a drug hypersensitivity reaction. Additionally, the recurrent inflammation is becoming more severe, as manifest by the fever and hemodynamic derangements, which suggests an increasing sensitization to the offending agent. I would review every drug she has received in the hospital, but given the recurrences after discharge her home medications are the most likely explanation. Of these, sulindac is the most likely culprit.
On further questioning, it was learned that the patient began taking sulindac 200 mg twice daily to treat her chronic ankle pain 6 weeks before the first admission. The medication had been stopped on each admission. She was instructed to discontinue sulindac. She has had no recurrences of symptoms and her hepatitis and eosinophilia have resolved.
DISCUSSION
This patient presented with recurrent skin findings, eosinophilia, hepatitis, and constitutional symptoms caused by hypersensitivity to sulindac. This drug‐induced hypersensitivity syndrome was originally described with anticonvulsant drugs (carbamazepine, phenytoin, and phenobarbitone) and named anticonvulsant hypersensitivity syndrome,1, 2 but has been observed with many other medications, including allopurinol, dapsone, minocycline, and nevirapine. The term drug rash with eosinophilia and systemic symptoms (DRESS) syndrome has been recently adopted to convey the cardinal features that characterize this disorder.3
DRESS syndrome is defined by rash, fever, and internal organ involvement.4 Also included in the diagnostic criteria are hematologic abnormalities (eosinophilia 1.5 109/mm3 or the presence of atypical lymphocytes) and lymphadenopathy.3 The multiorgan involvement distinguishes DRESS from other cutaneous drug eruptions. In a review of the French Pharmacovigilance Database for all cases of DRESS over a 15‐year period, 73% to 100% of patients were reported to have dermatologic abnormalities, most frequently a maculopapular rash or erythroderma. Less common skin findings include vesicles, bullae, pustules, erythroderma, and purpuric lesions. Liver abnormalities were observed in more than 60% of patients and were the most frequent systemic finding.5 Eosinophilia was the most common hematologic abnormality, present in more than 50% of cases. As this case underscores, DRESS syndrome typically begins 3 to 8 weeks after initiation of the drug because it is a delayed type IV hypersensitivity reaction.6 Fever can occur within hours on rechallenge because of the presence of memory T cells.
The main treatment for DRESS syndrome is withdrawal of the offending drug. Systemic corticosteroids have been recommended in cases with life‐threatening pulmonary or cardiac involvement, but have not been shown to be helpful in reversing renal or hepatic disease. Mortality, usually from end‐organ damage, occurs in about 10% of cases. The most common drugs are phenobarbital, carbamazepine, and phenytoin, with incidences of 1 in 5000 to 1 in 10,000. NSAIDs and antibiotics also have been implicated frequently. Human herpesvirus type 6 coinfection and genetically inherited slow acetylation have been associated with DRESS, although causal links have yet to be established.7, 8
The initial challenge in caring for a patient with multiple symptoms, exam findings, and test abnormalities is the coherent framing of the key clinical features that require explanation. This process, called problem representation, allows clinicians to search among a bounded list of possible diagnoses (or solutions) rather than invoking a differential diagnosis for every single abnormality. In searching for the proper diagnosis, this patient's clinical course required frequent reframing as more data became available.
Initially, the problem was framed as a 42‐year‐old woman with hepatitis. As the flushing and eosinophilia, which initially appeared to be transient and possibly nonspecific, became more prominent, the problem representation was revised to a 42‐year‐old woman with hepatitis, eosinophilia, and flushing. Since this triad did not immediately invoke a single diagnosis for the treating clinicians or the discussant, the differential diagnosis of hepatitis and eosinophilia and the differential diagnosis of flushing were considered in parallel.
Hepatitis and eosinophilia can occur coincidentally in the setting of parasitic infections, particularly helminths (ascaris, strongyloidiasis, and toxocaris) and liver flukes (opisthorchis and clonorchis), which invade the hepatobiliary system and induce a reactive eosinophilia. Some neoplasms, such as lymphomas and leukemias, and myeloproliferative disorders, including hypereosinophilic syndrome and mastocytosis, may have neoplastic cellular invasion of the liver and induce eosinophilia. Systemic drug hypersensitivity reactions are typically characterized by eosinophilia, transaminase elevations,9 and hepatitis on histology.10 As with this patient, liver biopsy in drug‐induced hepatitis shows a mixed microvesicular and macrovesicular steatosis, often with eosinophils.10
The flushing prompted a thorough but negative workup for the classic flushing disorders. The discussant's attempts to unify the flushing with the other clinical features illustrate how framing can affect our reasoning. Hepatitis, eosinophilia, and flushing defied an obvious single explanation and led the discussant down parallel diagnostic reasoning pathways. Although flushing is 1 of the well‐described dermatologic manifestations in drug‐hypersensitivity reactions, framing the central features as hepatitis, eosinophilia, and rash would have more readily suggested a drug reaction as a unifying diagnosis.11
The tempo and periodicity of this patient's illness provided the final formulation of a 42‐year‐old woman with hepatitis, eosinophilia, and flushing (or rash) that occurs every few days at home and resolves in the hospital. This formulation and the increasingly severe presentation, suggesting sensitization, were highly suggestive of an exogenous cause of her illness.
This case highlights how easily medication side effects can be overlooked during an extensive evaluation and how vigilant medication reconciliation coupled with an increased understanding of the spectrum of drug reactions can lead to early detection and prevention of potentially serious effects. In the case of DRESS, recognizing an association between a rash and organ involvement is central to making the diagnosis. Eosinophilia that accompanies a rash can further aid in narrowing the differential diagnosis.
The case also serves as a reminder of how framing with the slightest imprecision (eg, flushing instead of rash) can derail or delay the diagnostic process, yet is indispensable in tackling a complicated case. Finally, a time‐honored lesson in diagnosis is highlighted yet again: the diagnosis can usually be flushed out from the history.1214
Key Teaching Points
-
The constellation of skin findings, eosinophilia, organ involvement (particularly hepatitis), and constitutional symptoms should prompt consideration of DRESS syndrome and a hunt for a culprit drug.
-
Symptoms that resolve during hospitalization and repeatedly recur after discharge should prompt consideration of an exposure unique to the home, which may be environmental or pharmacologic.
-
Problem representation is critical in solving a complicated case, but adopting an inaccurate frame (representation) can derail or delay the diagnostic process.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 42‐year‐old woman with a history of mild asthma presented to the emergency department (ED) following 1 week of headache. She had been in her usual state of good health until 1 week prior to her presentation, when she noticed intermittent frontal headaches without neck stiffness or other neurologic symptoms. She then developed diffuse myalgias, fatigue, subjective fevers, and rigors for the 24 hours prior to presentation. On the morning of presentation, chest tightness, palpitations, and shortness of breath occurred. She used her albuterol metered‐dose inhaler without relief and went to the hospital.
Many of these features can be explained by a viral syndrome exacerbating underlying asthma or by a psychiatric condition such as anxiety or depression, but they may also be a harbinger of a systemic process, including infection, malignancy, or autoimmunity. Because the onset of headache is temporally distant from the other symptoms, I am more inclined to believe that it represents a primary intracranial process than I would if it were coincident with the onset of the other acute symptoms. If the fevers and rigors are verified, infection would be the initial concern. Failure to respond to her inhalers may either signify a severe asthma exacerbation or a nonbronchospastic cause of dyspnea.
She reported mild nausea, but denied photophobia, vomiting, abdominal pain, diarrhea, melena, or hematochezia. She did not have recent ill contacts, animal bites, or travel. Her medical history included asthma, diverticulitis, chronic right ankle pain, and obesity. She reported an allergic rash to amoxicillin. Her medications were sulindac and fluticasone/salmeterol, and albuterol metered‐dose inhalers. She worked as a preschool teacher and was married with 2 children. She denied any tobacco use and seldom drank alcoholic beverages. On exam, temperature was 36.7C, pulse was 107 beats per minute, blood pressure was 129/91 mm Hg, respiratory rate was 19 breaths per minute, and oxygen saturation was 98% while breathing ambient air. Her face and anterior neck were flushed and diaphoretic, and her sclerae were icteric. There was no nuchal rigidity. Her cardiac rhythm was regular without murmurs, lungs were clear to auscultation, and the abdomen was mildly tender to palpation in the epigastrium and right upper quadrant. The white blood cell (WBC) count was 9200/L, with 84% neutrophils, 3% lymphocytes, 6% monocytes, and 7% eosinophils. The hemoglobin was 14.8 g/dL and the platelet count was 166,000/L. Total serum bilirubin was 4.6 mg/dL, aspartate aminotransferase (AST) was 459 U/L (normal range, 8‐31), alanine aminotransferase (ALT) was 667 U/L (normal range, 7‐31), and alkaline phosphatase was 146 U/L (normal range, 39‐117). Serum electrolytes, creatinine, lactate, lipase, thyrotropin, coagulation studies, and cardiac enzymes were all normal. Urinalysis showed trace leukocyte esterase and bilirubin, as well as 3 WBCs and 2 red cells per high‐power field. Chest radiography and an electrocardiogram demonstrated no abnormalities.
The major findingwhich is critical to focusing problem‐solving in the face of a broad range of symptomsis her hepatitis. The common etiologies for hepatitis of this degree include viruses (hepatitis A and cytomegalovirus [CMV] should be considered given her work in preschool), toxins, autoimmunity, and vascular events. Liver disease in association with flushing raises the possibility of carcinoid syndrome with liver metastases. The lack of wheezing makes the bronchospasm of asthma or carcinoid less suitable explanations for her shortness of breath. Her eosinophilia is mild but probably is not accounted for alone by well‐controlled asthma in a person with no history of atopic disease. I would also ask her about any alternative and over‐the‐counter remedies. The paucity of lymphocytes raises the possibility of human immunodeficiency virus (HIV), Hodgkin's disease, or systemic lupus erythematosus. Although she does not have a documented fever or leukocytosis, she reported fevers and chills and is diaphoretic and tachycardic, so exclusion of biliary obstruction and cholangitis is the highest priority.
An abdominal ultrasound demonstrated hepatomegaly with moderate fatty infiltration and a normal gallbladder without pericholecystic fluid. The intrahepatic and extrahepatic biliary ducts were normal and the hepatic and portal veins were patent. Computed tomography of the abdomen showed slight thickening of the sigmoid colon wall. Ciprofloxacin and metronidazole were administered for possible diverticulitis. Over the first 48 hours of hospitalization her symptoms improved markedly. Her flushing resolved and she had no recorded fevers in the hospital. Serologies were negative for hepatitis A immunoglobulin M (IgM), hepatitis B surface antibody, hepatitis B surface antigen, and hepatitis C antibody. A monospot test was negative and the erythrocyte sedimentation rate was 11 mm/hour. Blood and urine cultures were negative. On the second hospital day the absolute eosinophil count rose to 855/L (15% of 5700 WBCs). On the fourth hospital day, the absolute eosinophil count was 1092/L, the total bilirubin was 1.9 mg/dL, and the AST and ALT were 174 U/L and 476 U/L, respectively. Antibiotics were stopped and she was discharged home.
Her prompt improvement suggests either a self‐limited condition or a response to the antibiotics. The rapid but incomplete resolution of her hepatitis is in keeping with a withdrawal of a toxin, relief of biliary obstruction, or a transient vascular event, and is less consistent with a viral hepatitis or an infiltrative process. With normal biliary system imaging, sterile blood cultures, and the absence of fever or leukocytosis, cholangitis is unlikely. Likewise, there is no suggestion of a vascular event, either obstructive or hemodynamic, that is impairing the liver.
A common cause of eosinophilia in hospitalized patients is medications, so it would be useful to monitor that count after the new antibiotics. At this point, I also wonder if the eosinophils are a feature of the underlying illness, as they were present to a modest degree on admission before any new medications were administered. The overlap of eosinophilia and hepatitis brings to mind a medication reaction (eg, to sulindac) or a hepatobiliary parasite, such as ascaris or clonorchis, for which she lacks a known exposure. Many patients experience flushing in the setting of fever or stress, but sustained flushing may suggest a systemic illness characterized by the release of vasoactive mediators such as carcinoid syndrome or mastocytosis. The latter might be considered more strongly if the eosinophilia is deemed to be primary (rather than reactive) after a thorough evaluation.
After 2 days at home, the patient had recurrence of subjective fevers, with chest, back, and abdominal pain, fatigue, loose stools, and rigors. She returned to the ED, where she was noted to have facial erythema and injected sclerae, but the remainder of her physical exam was normal. The total serum bilirubin was 1.1 mg/dL, AST was 156 U/L, ALT was 214 U/L, and alkaline phosphatase was 240 U/L. Serum lipase was normal. WBC count was 14,000/L, with 94% neutrophils, 3% lymphocytes, 2% monocytes, and 1% eosinophils. She was again treated empirically with ciprofloxacin and metronidazole. Endoscopic ultrasound was normal, with no evidence of gallbladder sludge or microlithiasis. Stool cultures, assay for Clostridium difficile, and examination for ova and parasites were negative. The 24‐hour urine demonstrated no elevation in 5‐hydroxyindoleacetic acid. An adrenocorticotropic hormone (ACTH) stimulation test was normal. HIV antibody was negative. Her symptoms improved within 2 days. The eosinophil count rose and peaked at 1541/L by the third hospital day, while the transaminase elevations resolved. Antibiotics were discontinued. A liver biopsy showed mixed macrovesicular and microvesicular fatty metamorphosis and steatohepatitis with eosinophils (Figures 1 and 2). She was discharged home on the sixth hospital day.
Her illness can now be characterized as relapsing inflammation, which given the frequency (over days) suggests either an indolent infectious focus that periodically causes systemic inflammation or reexposure to a toxic substance. The 2 most notable laboratory abnormalities, the hepatitis and the eosinophilia, persist but have differing trajectories. While the liver function tests have progressively normalized despite clinical relapses, the eosinophils have had a more fluctuating course characterized by increases during the hospitalization and higher levels during the second hospitalization. The absence of an infection, recurrent systemic inflammation, and eosinophilic hepatitis suggest a hypersensitivity reaction to a medication or other substance. She is most likely being reexposed at home, where her symptoms occur, and not in the hospital, where her symptoms resolve. Sulindac is a leading candidate, because nonsteroidal antiinflammatory drugs (NSAIDs) cause a number of hypersensitivity reactions and are frequently stopped when sick patients enter the hospital.
Seven days after discharge she developed acute onset of subjective fever, nausea, diffuse myalgias, and flushing, identical to the 2 prior episodes, and she again returned to the ED. Her temperature was 39.1C, heart rate was 120 beats per minute, blood pressure was 87/50 mm Hg, respiratory rate was 18 breaths per minute, and the oxygen saturation was 96% while breathing room air. She had diffuse flushing from her neck over her torso and was diaphoretic with injected sclera and conjunctiva. The WBC count was 11,400/L with 97% neutrophils and 3% lymphocytes. Total bilirubin was 0.8 mg/dL, AST was 134 U/L, ALT was 140 U/L, and alkaline phosphatase was 144 U/L. She was readmitted to the hospital. Following admission, she had no fevers, the flushing resolved, and AST and ALT levels decreased. The only treatment the patient received in the ED and during her hospital stay was acetaminophen as needed for pain or fever. The eosinophil count peaked at 1404/L by hospital day 4. Blood and urine cultures were negative. IgM antibodies to Epstein‐Barr virus were not detected, CMV DNA was not detected, and a rapid plasma reagin (RPR) test was nonreactive. Ferritin, ceruloplasmin, alpha‐1‐antitrypsin, and tryptase levels were normal. Antimitochondrial, antismooth muscle, antineutrophil cytoplasmic, and antinuclear antibodies were negative. There was no monoclonal band on serum protein electrophoresis. A blood smear for Borrelia detected no spirochetes.
A complete picture of the uncommon but classic flushing disorders, namely carcinoid, mastocytosis, and pheochromocytoma, has not emerged. The constellation of inflammation, mucosal and hepatic involvement, and eosinophilia are most consistent with a drug hypersensitivity reaction. Additionally, the recurrent inflammation is becoming more severe, as manifest by the fever and hemodynamic derangements, which suggests an increasing sensitization to the offending agent. I would review every drug she has received in the hospital, but given the recurrences after discharge her home medications are the most likely explanation. Of these, sulindac is the most likely culprit.
On further questioning, it was learned that the patient began taking sulindac 200 mg twice daily to treat her chronic ankle pain 6 weeks before the first admission. The medication had been stopped on each admission. She was instructed to discontinue sulindac. She has had no recurrences of symptoms and her hepatitis and eosinophilia have resolved.
DISCUSSION
This patient presented with recurrent skin findings, eosinophilia, hepatitis, and constitutional symptoms caused by hypersensitivity to sulindac. This drug‐induced hypersensitivity syndrome was originally described with anticonvulsant drugs (carbamazepine, phenytoin, and phenobarbitone) and named anticonvulsant hypersensitivity syndrome,1, 2 but has been observed with many other medications, including allopurinol, dapsone, minocycline, and nevirapine. The term drug rash with eosinophilia and systemic symptoms (DRESS) syndrome has been recently adopted to convey the cardinal features that characterize this disorder.3
DRESS syndrome is defined by rash, fever, and internal organ involvement.4 Also included in the diagnostic criteria are hematologic abnormalities (eosinophilia 1.5 109/mm3 or the presence of atypical lymphocytes) and lymphadenopathy.3 The multiorgan involvement distinguishes DRESS from other cutaneous drug eruptions. In a review of the French Pharmacovigilance Database for all cases of DRESS over a 15‐year period, 73% to 100% of patients were reported to have dermatologic abnormalities, most frequently a maculopapular rash or erythroderma. Less common skin findings include vesicles, bullae, pustules, erythroderma, and purpuric lesions. Liver abnormalities were observed in more than 60% of patients and were the most frequent systemic finding.5 Eosinophilia was the most common hematologic abnormality, present in more than 50% of cases. As this case underscores, DRESS syndrome typically begins 3 to 8 weeks after initiation of the drug because it is a delayed type IV hypersensitivity reaction.6 Fever can occur within hours on rechallenge because of the presence of memory T cells.
The main treatment for DRESS syndrome is withdrawal of the offending drug. Systemic corticosteroids have been recommended in cases with life‐threatening pulmonary or cardiac involvement, but have not been shown to be helpful in reversing renal or hepatic disease. Mortality, usually from end‐organ damage, occurs in about 10% of cases. The most common drugs are phenobarbital, carbamazepine, and phenytoin, with incidences of 1 in 5000 to 1 in 10,000. NSAIDs and antibiotics also have been implicated frequently. Human herpesvirus type 6 coinfection and genetically inherited slow acetylation have been associated with DRESS, although causal links have yet to be established.7, 8
The initial challenge in caring for a patient with multiple symptoms, exam findings, and test abnormalities is the coherent framing of the key clinical features that require explanation. This process, called problem representation, allows clinicians to search among a bounded list of possible diagnoses (or solutions) rather than invoking a differential diagnosis for every single abnormality. In searching for the proper diagnosis, this patient's clinical course required frequent reframing as more data became available.
Initially, the problem was framed as a 42‐year‐old woman with hepatitis. As the flushing and eosinophilia, which initially appeared to be transient and possibly nonspecific, became more prominent, the problem representation was revised to a 42‐year‐old woman with hepatitis, eosinophilia, and flushing. Since this triad did not immediately invoke a single diagnosis for the treating clinicians or the discussant, the differential diagnosis of hepatitis and eosinophilia and the differential diagnosis of flushing were considered in parallel.
Hepatitis and eosinophilia can occur coincidentally in the setting of parasitic infections, particularly helminths (ascaris, strongyloidiasis, and toxocaris) and liver flukes (opisthorchis and clonorchis), which invade the hepatobiliary system and induce a reactive eosinophilia. Some neoplasms, such as lymphomas and leukemias, and myeloproliferative disorders, including hypereosinophilic syndrome and mastocytosis, may have neoplastic cellular invasion of the liver and induce eosinophilia. Systemic drug hypersensitivity reactions are typically characterized by eosinophilia, transaminase elevations,9 and hepatitis on histology.10 As with this patient, liver biopsy in drug‐induced hepatitis shows a mixed microvesicular and macrovesicular steatosis, often with eosinophils.10
The flushing prompted a thorough but negative workup for the classic flushing disorders. The discussant's attempts to unify the flushing with the other clinical features illustrate how framing can affect our reasoning. Hepatitis, eosinophilia, and flushing defied an obvious single explanation and led the discussant down parallel diagnostic reasoning pathways. Although flushing is 1 of the well‐described dermatologic manifestations in drug‐hypersensitivity reactions, framing the central features as hepatitis, eosinophilia, and rash would have more readily suggested a drug reaction as a unifying diagnosis.11
The tempo and periodicity of this patient's illness provided the final formulation of a 42‐year‐old woman with hepatitis, eosinophilia, and flushing (or rash) that occurs every few days at home and resolves in the hospital. This formulation and the increasingly severe presentation, suggesting sensitization, were highly suggestive of an exogenous cause of her illness.
This case highlights how easily medication side effects can be overlooked during an extensive evaluation and how vigilant medication reconciliation coupled with an increased understanding of the spectrum of drug reactions can lead to early detection and prevention of potentially serious effects. In the case of DRESS, recognizing an association between a rash and organ involvement is central to making the diagnosis. Eosinophilia that accompanies a rash can further aid in narrowing the differential diagnosis.
The case also serves as a reminder of how framing with the slightest imprecision (eg, flushing instead of rash) can derail or delay the diagnostic process, yet is indispensable in tackling a complicated case. Finally, a time‐honored lesson in diagnosis is highlighted yet again: the diagnosis can usually be flushed out from the history.1214
Key Teaching Points
-
The constellation of skin findings, eosinophilia, organ involvement (particularly hepatitis), and constitutional symptoms should prompt consideration of DRESS syndrome and a hunt for a culprit drug.
-
Symptoms that resolve during hospitalization and repeatedly recur after discharge should prompt consideration of an exposure unique to the home, which may be environmental or pharmacologic.
-
Problem representation is critical in solving a complicated case, but adopting an inaccurate frame (representation) can derail or delay the diagnostic process.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,.Anticonvulsant hypersensitivity syndrome. In vitro assessment of risk.J Clin Invest.1988;82:1826–1832.
- ,,.Anticonvulsant hypersensitivity syndrome: incidence, prevention, and management.Drug Saf.1999;21:489–501.
- ,,,.Drug rash with eosinophilia and systemic symptoms vs toxic epidermal necrolysis: the dilemma of classification.Clin Dermatol.2005;23:311–314.
- ,.Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update.Dermatology.2003;206:353–356.
- ,,,,,Blayac J‐P; Network of the French Pharmacovigilance Centers. Variability in the clinical pattern of cutaneous side‐effects of drugs with systemic symptoms: does a DRESS syndrome really exist?Br J Dermatol.2006;155:422–428.
- ,.The immunological and clinical spectrum of delayed drug‐induced exanthems.Curr Opin Allergy Clin Immunol.2004;4:411–419.
- ,,.Association between anticonvulsant hypersensitivity syndrome and human herpesvirus 6 reactivation and hypogammaglobulinemia.Arch Dermatol.2004;140:183–188.
- ,.Treatment of severe drug reactions: Stevens‐Johnson Syndrome, toxic epidermal necrolysis and hypersensitivity syndrome.Dermatol Online J.2002;8:5.
- ,.Drug reactions. In: Bolognia JL, Jorizzo JL, Rapini RP, eds.Dermatology.2nd ed.Spain:Mosby Elsevier;2008:310–311.
- Pathology Outlines. Liver and intrahepatic bile ducts—non tumor. Hepatitis (non‐infectious). Drug/toxin‐induced hepatitis. Available at: http://www.pathologyoutlines.com/liver.html#drugtoxin. Accessed August2009.
- ,,.The flushing patient: differential diagnosis, workup and treatment.J Am Acad Dermatol.2006;55(2):193–208.
- ,,,,.Relative contributions of history‐taking, physical examination, and laboratory investigation to diagnosis and management of medical outpatients.Br Med J.1975;2(5969):486–489.
- ,,,,.Contributions to the history, physical examination, and laboratory investigation in making medical diagnosis.West J Med.1992;156(2):163–165.
- ,.A study on relative contributions of the history, physical examination and investigations in making medical diagnosis.J Assoc Physicians India.2000;48(8):771–775.
- ,.Anticonvulsant hypersensitivity syndrome. In vitro assessment of risk.J Clin Invest.1988;82:1826–1832.
- ,,.Anticonvulsant hypersensitivity syndrome: incidence, prevention, and management.Drug Saf.1999;21:489–501.
- ,,,.Drug rash with eosinophilia and systemic symptoms vs toxic epidermal necrolysis: the dilemma of classification.Clin Dermatol.2005;23:311–314.
- ,.Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update.Dermatology.2003;206:353–356.
- ,,,,,Blayac J‐P; Network of the French Pharmacovigilance Centers. Variability in the clinical pattern of cutaneous side‐effects of drugs with systemic symptoms: does a DRESS syndrome really exist?Br J Dermatol.2006;155:422–428.
- ,.The immunological and clinical spectrum of delayed drug‐induced exanthems.Curr Opin Allergy Clin Immunol.2004;4:411–419.
- ,,.Association between anticonvulsant hypersensitivity syndrome and human herpesvirus 6 reactivation and hypogammaglobulinemia.Arch Dermatol.2004;140:183–188.
- ,.Treatment of severe drug reactions: Stevens‐Johnson Syndrome, toxic epidermal necrolysis and hypersensitivity syndrome.Dermatol Online J.2002;8:5.
- ,.Drug reactions. In: Bolognia JL, Jorizzo JL, Rapini RP, eds.Dermatology.2nd ed.Spain:Mosby Elsevier;2008:310–311.
- Pathology Outlines. Liver and intrahepatic bile ducts—non tumor. Hepatitis (non‐infectious). Drug/toxin‐induced hepatitis. Available at: http://www.pathologyoutlines.com/liver.html#drugtoxin. Accessed August2009.
- ,,.The flushing patient: differential diagnosis, workup and treatment.J Am Acad Dermatol.2006;55(2):193–208.
- ,,,,.Relative contributions of history‐taking, physical examination, and laboratory investigation to diagnosis and management of medical outpatients.Br Med J.1975;2(5969):486–489.
- ,,,,.Contributions to the history, physical examination, and laboratory investigation in making medical diagnosis.West J Med.1992;156(2):163–165.
- ,.A study on relative contributions of the history, physical examination and investigations in making medical diagnosis.J Assoc Physicians India.2000;48(8):771–775.
The Tip of the Iceberg
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 33‐year‐old African American man was seen in the emergency department for bilateral wrist pain and forearm swelling. He was a professional mover and had transported a piano 3 days before. Several hours after the move, he noticed wrist pain and a few small red, slightly pruritic bumps on the palmar aspect of both wrists. The following day, he developed nausea, vomiting, and watery diarrhea. His wrists and forearms became more swollen and painful.
The most distinctive aspect of the patient's symptom complex is forearm swelling. I am not certain if the primary pathology lies in the wrist or in the forearm. Examination should focus on the presence or absence of arthritis and whether the forearm swelling is simply adjacent to the wrist and rash or extends the entire length from the wrist to the elbow.
Strenuous lifting can lead to rhabdomyolysis and forearm compartment syndrome. However, transporting a piano is not an unusual task for a professional mover. More common causes of arm swelling such as fracture, cellulitis, deep vein thrombosis, and lymphatic obstruction are possible but are unexpected in this patient because of the bilateral findings. An inflammatory myopathy would not present in the distal extremities, but an infectious myositis, such as trichinosis (which can have early gastrointestinal symptoms), could.
Given the patient's age and the temporal correlation, I am inclined to pursue a unifying diagnosis between his gastrointestinal and upper extremity symptoms. The gastrointestinal symptoms could reflect vasculitis (eg, polyarteritis nodosa) or a nonspecific manifestation of systemic illness (eg, sepsis), whereas the rash could be due to infection with petechiae (eg, meningococcemia, gonococcemia, or endocarditis) or an infection with a predilection for peripheral skin lesions that progress centripetally as the illness progresses, such as Rocky Mountain spotted fever.
Although the patient had not seen spiders, he was concerned that his skin lesions might have resulted from spider bites. He had a history of atopic dermatitis but took no medications, did not smoke, and drank alcohol rarely. He lived in Denver, CO, had not traveled outside of the region, and had not visited rural areas. He was monogamous with a female partner and had no known exposures to human immunodeficiency virus. He had no family history of rheumatological disorders.
Patients and physicians frequently attribute papules, ulcers, or necrotic skin lesions to spider bites far out of proportion to their true prevalence. Bites by more innocuous arthropods such as ticks, fleas, bedbugs, or mites are much more common. The symmetry of the lesions and extensive swelling, however, make such bites unlikely.
Although the patient hails from the southwestern United States, there is no compelling evidence for endemic illness. Hantavirus infection causes a viral prodrome, but a severe pulmonary syndrome is its primary manifestation. Plague presents in bubonic, pulmonary, or septicemic forms, but other than the gastrointestinal symptoms, there is nothing to suggest such a systemic illness. The initial pulmonary infection of coccidioidomycosis is often unnoticed, and patients may present with extrapulmonary manifestations, including skin lesions and skeletal disease.
More common ailments remain on the differential. Disseminated gonococcemia must be considered in a sexually active adult with skin lesions and what may be tenosynovitis or arthritis of the distal extremities. The history of atopic dermatitis supports a diagnosis of allergic or contact dermatitis on the wrists (perhaps inoculated during the move), explaining the rash and adjacent swelling (but not the gastrointestinal symptoms).
The patient's temperature was 36.5C with a pulse of 103 beats per minute, a blood pressure of 103/67 mm Hg, and a respiratory rate of 24 breaths per minute. He appeared uncomfortable and was in moderate distress. His sclerae were injected, and his mucous membranes were dry. He had diffusely swollen fingers and firm nonpitting edema in both hands and forearms. His wrists and hands were tender and warm but had no appreciable redness. Two 1‐mm ulcerated lesions were present on his right wrist, and one 2‐mm ulcerated lesion was present on his left wrist. No lymphadenopathy was present.
The patient meets the criteria for systemic inflammatory response syndrome (SIRS), and considering his ill appearance, I am concerned that he may have an infectious process that has evolved into sepsis. The absence of cutaneous erythema rules out cellulitis, and although skin findings in necrotizing fasciitis can be modest in comparison with the underlying infection, an examination with nothing more than punctate ulcerations would be atypical. Plague and tularemia cause ulcerative skin lesions and systemic illness but usually have prominent lymphadenopathy. Cutaneous anthrax can cause intense local edema but is usually accompanied by some degree of necrosis.
I am struck by the degree of local edema. It would be a remarkable coincidence for spider bites to occur on both wrists; however, given the size of the lesions, this remains a consideration. Spider bites can cause severe local swelling and occasionally even SIRS.
The white blood cellcount was 6000/mm3 with a normal differential. The hemoglobin level was 16.6 g/dL, and the platelet count was 227,000/mm3. The serum sodium level was measured to be 135 mmol/L, the potassium level was 3.6 mmol/L, the chloride level was 94 mmol/L, and the bicarbonate level was 9 mmol/L. The urea nitrogen level was measured to be 48 mg/dL (normal, 622), and the serum creatinine level was 5.6 mg/dL (normal, 0.41.2). An arterial blood gas test on room air revealed a pH of 7.22, a partial pressure of carbon dioxide of 24 mm Hg, and a partial pressure of oxygen of 128 mm Hg. Liver enzymes were normal. The serum creatine kinase level was measured to be 194 U/L (normal, 0250). A chest radiograph revealed clear lung fields.
The anion gap is elevated and could be explained in part by renal failure, but it is quite pronounced, and I suspect that there is lactic acidosis either from SIRS and systemic hypoperfusion or from local underperfusion of the distal upper extremities (eg, compartment syndrome). Rhabdomyolysis can cause an anion gap acidosis and acute renal failure but is ruled out by a normal creatine kinase level.
Rheumatological diseases (such as polyarteritis nodosa and systemic scleroderma) can cause renal failure, gastrointestinal symptoms, and cutaneous lesions with skin ulceration but are often accompanied by hypertension. The combination of SIRS and volume depletion from nausea, vomiting, and diarrhea seems more likely, although an etiology for SIRS remains elusive. I suspect infectionmost likely of the upper extremitiesis the underlying cause in this patient despite his normal body temperature and white cell count. I would therefore start with plain films of the arms and hands. If these are unrevealing, I would proceed with an ultrasound of the arms to evaluate the soft tissues and particularly the vessels. Although imaging studies are unlikely to provide a diagnosis, they will provide guidance in choosing the next step, such as biopsy or culture.
The patient wasvolume‐resuscitated with saline and treated with clindamycin, pipercillin‐tazobactam, and vancomycin. Radiographs of the wrists and hands demonstrated edema but no subcutaneous gas and no abnormalities of the bones or joints. An ultrasound of the upper extremities was negative for superficial or deep venous thrombosis. Renal ultrasonography revealed normal‐sized kidneys and no hydronephrosis.
Short of superior vena cava thrombosis, which the ultrasound could not visualize, deep venous thrombosis can be ruled out with confidence. Superior vena cava syndrome could account for the bilateral upper extremity symptoms, but complete sparing of the face would be unusual, and the chest radiograph was normal.
Despite the bilateral nature, I doubt that this patient has an arthritis of the wrists and hands (eg, rheumatoid or psoriatic arthritis). The plain films did not show evidence of joint destruction, although that would not be expected in the first few days of most noninfectious arthropathies. Many rheumatological diseases and vasculitides have renal manifestations, but I do not find convincing evidence of such diseases yet.
Despite intravenous fluidsover the next 4 hours, the patient remained anuric. His upper extremity edema worsened, and he became increasingly tachycardic and hypotensive. Vasopressor support was required. Repeat laboratory studies demonstrated a serum creatinine level of 7.0 mg/dL, a bicarbonate level of 6 mmol/L, and a creatine kinase level of 1183 U/L. The serum lactate level was measured to be 6.5. Blood cultures obtained on admission were negative. An echocardiogram demonstrated marked impairment of left ventricular systolic dysfunction with an estimated ejection fraction of 35%. Repeat chest radiography revealed only mild pulmonary vascular congestion.
The patient has become progressively ill despite initial resuscitation. Any infection (cellulitis, fasciitis, or myositis) may progress to such a point (although to do so without fever or leukocytosis with this degree of illness is unusual) and would prompt me to ask a surgeon to explore the upper extremities for diagnostic and possibly therapeutic purposes. Among the spectrum of possible soft tissue infections, this clinical presentation is most consistent with pyomyositis (caused by Staphylococcus aureus) because of the relatively modest creatine kinase elevations that accompany it and the overall absence of cutaneous findings (save the punctuate lesions), which I would expect to be present with cellulitis or necrotizing fasciitis. A deep forearm infection and perhaps compartment syndrome leading to sepsis could explain lactic acidosis, decreased cardiac function, hypotension, and acute renal failure.
Because of the unusual characteristics noted so farparticularly the bilateral diseaseI continue to also consider systemic diseases that can cause skin lesions, cardiomyopathy, renal disease, ocular involvement (eg, keratoconjunctivitis or uveitis), and myositis. Sarcoidosis is possible, although in most of the aforementioned organs, histological disease is far more common than clinical disease, and sarcoidosis typically does not cause this degree of illness. Furthermore, over 90% of patients with sarcoidosis have pulmonary involvement. Polyarteritis nodosa could explain the multiorgan involvement and the brisk pace. If no infection is present on exploration, I would ask the surgeon to biopsy the muscle, particularly looking for granulomas or vasculitis. A progressive soft tissue infection leading to sepsis remains my leading consideration at this point.
Surgical consultation was obtained. A muscle biopsy of the left biceps and left forearm revealed group A streptococcus within the muscle and evidence of necrosis (Figure 1). Debridement of both arms and wrists was performed. The patient subsequently developed erythema of the palms and soles followed by diffuse sloughing of the skin. Streptococcal toxic shock syndrome with necrotizing myositis was diagnosed, and the antimicrobial regimen was changed to intravenous penicillin, clindamycin, and intravenous immunoglobulin G. Repeat debridement of the arms was required.
Even when a soft tissue infection is suspected, it can be challenging to preoperatively localize or characterize with precision. In retrospect, the overall severity of his illness and his previously good health status perhaps favor necrotizing myositis (and fasciitis) over pyomyositis.
I may have put undue emphasis on the absence of skin findings. Although the symmetric nature of the disease is unusual, the small skin lesions may have been portals of entry, and in retrospect, they represent the tip of the iceberg. The absence of fever and leukocytosisor hypothermia and leukopeniain a young, previously healthy patient along with the bilateral and symmetric findings made me hesitant to definitely label his illness as a deep soft tissue infection early on, but the gravity of the illness, the lack of a plausible alternative explanation, and his precipitous decline all made surgical exploration imperative.
The patient's skin sloughing progressed, and he was transferred to a regional burn unit. Three additional operations were required for debridement of both upper extremities. Despite apparent control of the initial infection, the patient continued to require significant hemodynamic and ventilator support. He subsequently developed neutropenia, thrombocytopenia, and Escherichia coli urosepsis. Necrosis of the lower extremities developed, and additional surgical debridement was recommended. After extensive discussion regarding prognosis, the family decided against further surgery and withdrew life support. The patient died shortly after extubation. The family refused an autopsy.
Commentary
Expert clinicians employ a variety of approaches to solve complex clinical problems. One of the most effective strategies is pattern recognition, in which the clinician divides the case into recognizable portions and compares these to previous cases that he or she has encountered.1, 2 If patterns from the new case appear similar or identical to those of previous cases, a diagnosis can be made quickly and without the need for unnecessary testing. However, when features of the case are unusual or atypical, pattern recognition may be disrupted. For example, although the discussant suspected a soft tissue infection, a number of features (including a normal white blood cell count, minimal skin findings, and a bilateral and symmetric distribution of swelling) did not match his illness script (a mental representation of a disease) of necrotizing fasciitis.
When pattern recognition fails, other strategies are available.3 Hypothetico‐deductive reasoning is a data‐to‐diagnosis method whereby the clinician uses the presenting information to construct a list of diagnostic possibilities.4 Additional testing and gathering of information are then used to continuously revise the diagnostic possibilities until confirmatory information is obtained and the diagnosis is established. After a pattern failed to materialize, the discussant employed this analytical strategy by noting the unusual characteristics of the case and incorporating laboratory and physiological data to revise his differential diagnosis. As a result, he requested the appropriate diagnostic test: surgical exploration of the forearms.
Necrotizing soft tissue infections are characterized by fulminant tissue destruction, rapid spread along tissue planes, and local vascular thrombosis. Mixed aerobic and anaerobic infections typically occur after penetrating skin injury or following surgery in patients with diabetes mellitus or vascular disease. In contrast, monomicrobial infections with S. aureus or group A streptococcus generally occur in healthy individuals. The prevalence of necrotizing group A streptococcal infections has increased dramatically in the last 15 to 20 years.5 Over one‐third of these cases are complicated by toxic shock syndrome.57 Mortality rates for necrotizing fasciitis with toxic shock exceed 30%, and early surgical consultation is directly associated with a reduction in morbidity and mortality.5, 8
Toxic shock is an inflammatory response syndrome caused by release of exotoxins from group A streptococcus and S. aureus.9, 10 In streptococcal toxic shock, Streptococcus pyogenes exotoxin A and Streptococcus pyogenes exotoxin B are the major toxins produced.10 These toxins activate the systemic production of inflammatory cytokines such as interleukin‐1, gamma‐interferon, and tumor necrosis factor, resulting in capillary leak, systemic hypotension, tissue hypoperfusion, and organ failure. The most common initial symptom is diffuse or localized pain that is severe and abrupt in onset and often precedes or is out of proportion to other physical findings of soft tissue infection.8 Up to 20% of individuals may also develop a viral‐like syndrome with myalgias, fever, nausea, vomiting, and diarrhea.11 Erythroderma of the skin and mucous membranes can be another early finding. The rash is diffuse, erythematous, and macular, resembling a sunburn. It involves the palms and soles but can be subtle and fleeting. Erythroderma may be particularly difficult to detect in dark‐skinned individuals.12 It is also important to consider that the absence of fever, erythroderma, or leukocytosis does not necessarily rule out the possibility of serious infection in necrotizing fasciitis patients, as the development of these signs and symptoms may occur later in the disease process.
The most common portals of entry for group A streptococcus are the skin, vagina, and pharynx. Predisposing factors include varicella infection, penetrating injuries, minor cuts, burns, splinters, and surgery. Interestingly, a portal of entry cannot be identified in 45% of cases.8 These patients in particular are at risk for developing severe necrotizing myositis or fasciitis at the site of a minor injury such as a strained muscle. Hematogenous translocation from the pharynx to the site of injury is the probable mechanism13 and would provide one scenario by which our piano mover developed bilateral and symmetric disease. An alternative explanation would be direct extension of bacteria from small breaks in the skin to adjacent areas of muscle strain. Regardless of the portal of entry, as the small skin lesions demonstrate in this patient, the smallest physical finding can represent the tip of the iceberg.
- ,,,,,.Fast and frugal models of clinical judgment in novice and expert physicians.Med Decis Making.2003;23(4):293–300.
- .What is it to be an expert? In: Chi M, Farr MJ, Glaser R, eds.The Nature of Expertise.Hillsdale, NJ:Lawrence Erlbaum;1988.
- .Clinical decision‐making: understanding how clinicians make a diagnosis. In: Saint S, Drazen JM, Solomon CG, eds.Clinical Problem‐Solving.New York, NY:McGraw‐Hill;2006.
- ,,.Medical Problem Solving: An Analysis of Clinical Reasoning.Cambridge, MA:Harvard University Press;1978.
- ,,,,.Population‐based surveillance for group A streptococcal necrotizing fasciitis: clinical features, prognostic indicators, and microbiologic analysis of seventy‐seven cases. Ontario Group A Streptococcal Study.Am J Med.1997;103(1):18–24.
- ,,, et al.Invasive group A streptococcal infections in Sweden in 1994 and 1995: epidemiology and clinical spectrum.Scand J Infect Dis.2000;32(6):609–614.
- ,,,.Reemergence of emm1 and a changed superantigen profile for group A streptococci causing invasive infections: results from a nationwide study.J Clin Microbiol.2005;43(4):1789–1796.
- ,,, et al.Severe group A streptococcal infections associated with a toxic shock‐like syndrome and scarlet fever toxin A.N Engl J Med.1989;321(1):1–7.
- ,,,.Rapidly progressive necrotizing fasciitis caused by Staphylococcus aureus.J Microbiol Immunol Infect.2005;38(5):361–364.
- ,.Streptococcal infections of skin and soft tissues.N Engl J Med.1996;334(4):240–245.
- ,.Streptococcal toxic shock syndrome after breast reconstruction.Ann Plast Surg.2005;54(5):553–556.
- ,.The influence of pigmentation and illumination on the perception of erythema.Photodermatol Photoimmunol Photomed.1992;9(2):45–47.
- .Streptococcal toxic‐shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment.Emerg Infect Dis.1995;1(3):69–78.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 33‐year‐old African American man was seen in the emergency department for bilateral wrist pain and forearm swelling. He was a professional mover and had transported a piano 3 days before. Several hours after the move, he noticed wrist pain and a few small red, slightly pruritic bumps on the palmar aspect of both wrists. The following day, he developed nausea, vomiting, and watery diarrhea. His wrists and forearms became more swollen and painful.
The most distinctive aspect of the patient's symptom complex is forearm swelling. I am not certain if the primary pathology lies in the wrist or in the forearm. Examination should focus on the presence or absence of arthritis and whether the forearm swelling is simply adjacent to the wrist and rash or extends the entire length from the wrist to the elbow.
Strenuous lifting can lead to rhabdomyolysis and forearm compartment syndrome. However, transporting a piano is not an unusual task for a professional mover. More common causes of arm swelling such as fracture, cellulitis, deep vein thrombosis, and lymphatic obstruction are possible but are unexpected in this patient because of the bilateral findings. An inflammatory myopathy would not present in the distal extremities, but an infectious myositis, such as trichinosis (which can have early gastrointestinal symptoms), could.
Given the patient's age and the temporal correlation, I am inclined to pursue a unifying diagnosis between his gastrointestinal and upper extremity symptoms. The gastrointestinal symptoms could reflect vasculitis (eg, polyarteritis nodosa) or a nonspecific manifestation of systemic illness (eg, sepsis), whereas the rash could be due to infection with petechiae (eg, meningococcemia, gonococcemia, or endocarditis) or an infection with a predilection for peripheral skin lesions that progress centripetally as the illness progresses, such as Rocky Mountain spotted fever.
Although the patient had not seen spiders, he was concerned that his skin lesions might have resulted from spider bites. He had a history of atopic dermatitis but took no medications, did not smoke, and drank alcohol rarely. He lived in Denver, CO, had not traveled outside of the region, and had not visited rural areas. He was monogamous with a female partner and had no known exposures to human immunodeficiency virus. He had no family history of rheumatological disorders.
Patients and physicians frequently attribute papules, ulcers, or necrotic skin lesions to spider bites far out of proportion to their true prevalence. Bites by more innocuous arthropods such as ticks, fleas, bedbugs, or mites are much more common. The symmetry of the lesions and extensive swelling, however, make such bites unlikely.
Although the patient hails from the southwestern United States, there is no compelling evidence for endemic illness. Hantavirus infection causes a viral prodrome, but a severe pulmonary syndrome is its primary manifestation. Plague presents in bubonic, pulmonary, or septicemic forms, but other than the gastrointestinal symptoms, there is nothing to suggest such a systemic illness. The initial pulmonary infection of coccidioidomycosis is often unnoticed, and patients may present with extrapulmonary manifestations, including skin lesions and skeletal disease.
More common ailments remain on the differential. Disseminated gonococcemia must be considered in a sexually active adult with skin lesions and what may be tenosynovitis or arthritis of the distal extremities. The history of atopic dermatitis supports a diagnosis of allergic or contact dermatitis on the wrists (perhaps inoculated during the move), explaining the rash and adjacent swelling (but not the gastrointestinal symptoms).
The patient's temperature was 36.5C with a pulse of 103 beats per minute, a blood pressure of 103/67 mm Hg, and a respiratory rate of 24 breaths per minute. He appeared uncomfortable and was in moderate distress. His sclerae were injected, and his mucous membranes were dry. He had diffusely swollen fingers and firm nonpitting edema in both hands and forearms. His wrists and hands were tender and warm but had no appreciable redness. Two 1‐mm ulcerated lesions were present on his right wrist, and one 2‐mm ulcerated lesion was present on his left wrist. No lymphadenopathy was present.
The patient meets the criteria for systemic inflammatory response syndrome (SIRS), and considering his ill appearance, I am concerned that he may have an infectious process that has evolved into sepsis. The absence of cutaneous erythema rules out cellulitis, and although skin findings in necrotizing fasciitis can be modest in comparison with the underlying infection, an examination with nothing more than punctate ulcerations would be atypical. Plague and tularemia cause ulcerative skin lesions and systemic illness but usually have prominent lymphadenopathy. Cutaneous anthrax can cause intense local edema but is usually accompanied by some degree of necrosis.
I am struck by the degree of local edema. It would be a remarkable coincidence for spider bites to occur on both wrists; however, given the size of the lesions, this remains a consideration. Spider bites can cause severe local swelling and occasionally even SIRS.
The white blood cellcount was 6000/mm3 with a normal differential. The hemoglobin level was 16.6 g/dL, and the platelet count was 227,000/mm3. The serum sodium level was measured to be 135 mmol/L, the potassium level was 3.6 mmol/L, the chloride level was 94 mmol/L, and the bicarbonate level was 9 mmol/L. The urea nitrogen level was measured to be 48 mg/dL (normal, 622), and the serum creatinine level was 5.6 mg/dL (normal, 0.41.2). An arterial blood gas test on room air revealed a pH of 7.22, a partial pressure of carbon dioxide of 24 mm Hg, and a partial pressure of oxygen of 128 mm Hg. Liver enzymes were normal. The serum creatine kinase level was measured to be 194 U/L (normal, 0250). A chest radiograph revealed clear lung fields.
The anion gap is elevated and could be explained in part by renal failure, but it is quite pronounced, and I suspect that there is lactic acidosis either from SIRS and systemic hypoperfusion or from local underperfusion of the distal upper extremities (eg, compartment syndrome). Rhabdomyolysis can cause an anion gap acidosis and acute renal failure but is ruled out by a normal creatine kinase level.
Rheumatological diseases (such as polyarteritis nodosa and systemic scleroderma) can cause renal failure, gastrointestinal symptoms, and cutaneous lesions with skin ulceration but are often accompanied by hypertension. The combination of SIRS and volume depletion from nausea, vomiting, and diarrhea seems more likely, although an etiology for SIRS remains elusive. I suspect infectionmost likely of the upper extremitiesis the underlying cause in this patient despite his normal body temperature and white cell count. I would therefore start with plain films of the arms and hands. If these are unrevealing, I would proceed with an ultrasound of the arms to evaluate the soft tissues and particularly the vessels. Although imaging studies are unlikely to provide a diagnosis, they will provide guidance in choosing the next step, such as biopsy or culture.
The patient wasvolume‐resuscitated with saline and treated with clindamycin, pipercillin‐tazobactam, and vancomycin. Radiographs of the wrists and hands demonstrated edema but no subcutaneous gas and no abnormalities of the bones or joints. An ultrasound of the upper extremities was negative for superficial or deep venous thrombosis. Renal ultrasonography revealed normal‐sized kidneys and no hydronephrosis.
Short of superior vena cava thrombosis, which the ultrasound could not visualize, deep venous thrombosis can be ruled out with confidence. Superior vena cava syndrome could account for the bilateral upper extremity symptoms, but complete sparing of the face would be unusual, and the chest radiograph was normal.
Despite the bilateral nature, I doubt that this patient has an arthritis of the wrists and hands (eg, rheumatoid or psoriatic arthritis). The plain films did not show evidence of joint destruction, although that would not be expected in the first few days of most noninfectious arthropathies. Many rheumatological diseases and vasculitides have renal manifestations, but I do not find convincing evidence of such diseases yet.
Despite intravenous fluidsover the next 4 hours, the patient remained anuric. His upper extremity edema worsened, and he became increasingly tachycardic and hypotensive. Vasopressor support was required. Repeat laboratory studies demonstrated a serum creatinine level of 7.0 mg/dL, a bicarbonate level of 6 mmol/L, and a creatine kinase level of 1183 U/L. The serum lactate level was measured to be 6.5. Blood cultures obtained on admission were negative. An echocardiogram demonstrated marked impairment of left ventricular systolic dysfunction with an estimated ejection fraction of 35%. Repeat chest radiography revealed only mild pulmonary vascular congestion.
The patient has become progressively ill despite initial resuscitation. Any infection (cellulitis, fasciitis, or myositis) may progress to such a point (although to do so without fever or leukocytosis with this degree of illness is unusual) and would prompt me to ask a surgeon to explore the upper extremities for diagnostic and possibly therapeutic purposes. Among the spectrum of possible soft tissue infections, this clinical presentation is most consistent with pyomyositis (caused by Staphylococcus aureus) because of the relatively modest creatine kinase elevations that accompany it and the overall absence of cutaneous findings (save the punctuate lesions), which I would expect to be present with cellulitis or necrotizing fasciitis. A deep forearm infection and perhaps compartment syndrome leading to sepsis could explain lactic acidosis, decreased cardiac function, hypotension, and acute renal failure.
Because of the unusual characteristics noted so farparticularly the bilateral diseaseI continue to also consider systemic diseases that can cause skin lesions, cardiomyopathy, renal disease, ocular involvement (eg, keratoconjunctivitis or uveitis), and myositis. Sarcoidosis is possible, although in most of the aforementioned organs, histological disease is far more common than clinical disease, and sarcoidosis typically does not cause this degree of illness. Furthermore, over 90% of patients with sarcoidosis have pulmonary involvement. Polyarteritis nodosa could explain the multiorgan involvement and the brisk pace. If no infection is present on exploration, I would ask the surgeon to biopsy the muscle, particularly looking for granulomas or vasculitis. A progressive soft tissue infection leading to sepsis remains my leading consideration at this point.
Surgical consultation was obtained. A muscle biopsy of the left biceps and left forearm revealed group A streptococcus within the muscle and evidence of necrosis (Figure 1). Debridement of both arms and wrists was performed. The patient subsequently developed erythema of the palms and soles followed by diffuse sloughing of the skin. Streptococcal toxic shock syndrome with necrotizing myositis was diagnosed, and the antimicrobial regimen was changed to intravenous penicillin, clindamycin, and intravenous immunoglobulin G. Repeat debridement of the arms was required.
Even when a soft tissue infection is suspected, it can be challenging to preoperatively localize or characterize with precision. In retrospect, the overall severity of his illness and his previously good health status perhaps favor necrotizing myositis (and fasciitis) over pyomyositis.
I may have put undue emphasis on the absence of skin findings. Although the symmetric nature of the disease is unusual, the small skin lesions may have been portals of entry, and in retrospect, they represent the tip of the iceberg. The absence of fever and leukocytosisor hypothermia and leukopeniain a young, previously healthy patient along with the bilateral and symmetric findings made me hesitant to definitely label his illness as a deep soft tissue infection early on, but the gravity of the illness, the lack of a plausible alternative explanation, and his precipitous decline all made surgical exploration imperative.
The patient's skin sloughing progressed, and he was transferred to a regional burn unit. Three additional operations were required for debridement of both upper extremities. Despite apparent control of the initial infection, the patient continued to require significant hemodynamic and ventilator support. He subsequently developed neutropenia, thrombocytopenia, and Escherichia coli urosepsis. Necrosis of the lower extremities developed, and additional surgical debridement was recommended. After extensive discussion regarding prognosis, the family decided against further surgery and withdrew life support. The patient died shortly after extubation. The family refused an autopsy.
Commentary
Expert clinicians employ a variety of approaches to solve complex clinical problems. One of the most effective strategies is pattern recognition, in which the clinician divides the case into recognizable portions and compares these to previous cases that he or she has encountered.1, 2 If patterns from the new case appear similar or identical to those of previous cases, a diagnosis can be made quickly and without the need for unnecessary testing. However, when features of the case are unusual or atypical, pattern recognition may be disrupted. For example, although the discussant suspected a soft tissue infection, a number of features (including a normal white blood cell count, minimal skin findings, and a bilateral and symmetric distribution of swelling) did not match his illness script (a mental representation of a disease) of necrotizing fasciitis.
When pattern recognition fails, other strategies are available.3 Hypothetico‐deductive reasoning is a data‐to‐diagnosis method whereby the clinician uses the presenting information to construct a list of diagnostic possibilities.4 Additional testing and gathering of information are then used to continuously revise the diagnostic possibilities until confirmatory information is obtained and the diagnosis is established. After a pattern failed to materialize, the discussant employed this analytical strategy by noting the unusual characteristics of the case and incorporating laboratory and physiological data to revise his differential diagnosis. As a result, he requested the appropriate diagnostic test: surgical exploration of the forearms.
Necrotizing soft tissue infections are characterized by fulminant tissue destruction, rapid spread along tissue planes, and local vascular thrombosis. Mixed aerobic and anaerobic infections typically occur after penetrating skin injury or following surgery in patients with diabetes mellitus or vascular disease. In contrast, monomicrobial infections with S. aureus or group A streptococcus generally occur in healthy individuals. The prevalence of necrotizing group A streptococcal infections has increased dramatically in the last 15 to 20 years.5 Over one‐third of these cases are complicated by toxic shock syndrome.57 Mortality rates for necrotizing fasciitis with toxic shock exceed 30%, and early surgical consultation is directly associated with a reduction in morbidity and mortality.5, 8
Toxic shock is an inflammatory response syndrome caused by release of exotoxins from group A streptococcus and S. aureus.9, 10 In streptococcal toxic shock, Streptococcus pyogenes exotoxin A and Streptococcus pyogenes exotoxin B are the major toxins produced.10 These toxins activate the systemic production of inflammatory cytokines such as interleukin‐1, gamma‐interferon, and tumor necrosis factor, resulting in capillary leak, systemic hypotension, tissue hypoperfusion, and organ failure. The most common initial symptom is diffuse or localized pain that is severe and abrupt in onset and often precedes or is out of proportion to other physical findings of soft tissue infection.8 Up to 20% of individuals may also develop a viral‐like syndrome with myalgias, fever, nausea, vomiting, and diarrhea.11 Erythroderma of the skin and mucous membranes can be another early finding. The rash is diffuse, erythematous, and macular, resembling a sunburn. It involves the palms and soles but can be subtle and fleeting. Erythroderma may be particularly difficult to detect in dark‐skinned individuals.12 It is also important to consider that the absence of fever, erythroderma, or leukocytosis does not necessarily rule out the possibility of serious infection in necrotizing fasciitis patients, as the development of these signs and symptoms may occur later in the disease process.
The most common portals of entry for group A streptococcus are the skin, vagina, and pharynx. Predisposing factors include varicella infection, penetrating injuries, minor cuts, burns, splinters, and surgery. Interestingly, a portal of entry cannot be identified in 45% of cases.8 These patients in particular are at risk for developing severe necrotizing myositis or fasciitis at the site of a minor injury such as a strained muscle. Hematogenous translocation from the pharynx to the site of injury is the probable mechanism13 and would provide one scenario by which our piano mover developed bilateral and symmetric disease. An alternative explanation would be direct extension of bacteria from small breaks in the skin to adjacent areas of muscle strain. Regardless of the portal of entry, as the small skin lesions demonstrate in this patient, the smallest physical finding can represent the tip of the iceberg.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 33‐year‐old African American man was seen in the emergency department for bilateral wrist pain and forearm swelling. He was a professional mover and had transported a piano 3 days before. Several hours after the move, he noticed wrist pain and a few small red, slightly pruritic bumps on the palmar aspect of both wrists. The following day, he developed nausea, vomiting, and watery diarrhea. His wrists and forearms became more swollen and painful.
The most distinctive aspect of the patient's symptom complex is forearm swelling. I am not certain if the primary pathology lies in the wrist or in the forearm. Examination should focus on the presence or absence of arthritis and whether the forearm swelling is simply adjacent to the wrist and rash or extends the entire length from the wrist to the elbow.
Strenuous lifting can lead to rhabdomyolysis and forearm compartment syndrome. However, transporting a piano is not an unusual task for a professional mover. More common causes of arm swelling such as fracture, cellulitis, deep vein thrombosis, and lymphatic obstruction are possible but are unexpected in this patient because of the bilateral findings. An inflammatory myopathy would not present in the distal extremities, but an infectious myositis, such as trichinosis (which can have early gastrointestinal symptoms), could.
Given the patient's age and the temporal correlation, I am inclined to pursue a unifying diagnosis between his gastrointestinal and upper extremity symptoms. The gastrointestinal symptoms could reflect vasculitis (eg, polyarteritis nodosa) or a nonspecific manifestation of systemic illness (eg, sepsis), whereas the rash could be due to infection with petechiae (eg, meningococcemia, gonococcemia, or endocarditis) or an infection with a predilection for peripheral skin lesions that progress centripetally as the illness progresses, such as Rocky Mountain spotted fever.
Although the patient had not seen spiders, he was concerned that his skin lesions might have resulted from spider bites. He had a history of atopic dermatitis but took no medications, did not smoke, and drank alcohol rarely. He lived in Denver, CO, had not traveled outside of the region, and had not visited rural areas. He was monogamous with a female partner and had no known exposures to human immunodeficiency virus. He had no family history of rheumatological disorders.
Patients and physicians frequently attribute papules, ulcers, or necrotic skin lesions to spider bites far out of proportion to their true prevalence. Bites by more innocuous arthropods such as ticks, fleas, bedbugs, or mites are much more common. The symmetry of the lesions and extensive swelling, however, make such bites unlikely.
Although the patient hails from the southwestern United States, there is no compelling evidence for endemic illness. Hantavirus infection causes a viral prodrome, but a severe pulmonary syndrome is its primary manifestation. Plague presents in bubonic, pulmonary, or septicemic forms, but other than the gastrointestinal symptoms, there is nothing to suggest such a systemic illness. The initial pulmonary infection of coccidioidomycosis is often unnoticed, and patients may present with extrapulmonary manifestations, including skin lesions and skeletal disease.
More common ailments remain on the differential. Disseminated gonococcemia must be considered in a sexually active adult with skin lesions and what may be tenosynovitis or arthritis of the distal extremities. The history of atopic dermatitis supports a diagnosis of allergic or contact dermatitis on the wrists (perhaps inoculated during the move), explaining the rash and adjacent swelling (but not the gastrointestinal symptoms).
The patient's temperature was 36.5C with a pulse of 103 beats per minute, a blood pressure of 103/67 mm Hg, and a respiratory rate of 24 breaths per minute. He appeared uncomfortable and was in moderate distress. His sclerae were injected, and his mucous membranes were dry. He had diffusely swollen fingers and firm nonpitting edema in both hands and forearms. His wrists and hands were tender and warm but had no appreciable redness. Two 1‐mm ulcerated lesions were present on his right wrist, and one 2‐mm ulcerated lesion was present on his left wrist. No lymphadenopathy was present.
The patient meets the criteria for systemic inflammatory response syndrome (SIRS), and considering his ill appearance, I am concerned that he may have an infectious process that has evolved into sepsis. The absence of cutaneous erythema rules out cellulitis, and although skin findings in necrotizing fasciitis can be modest in comparison with the underlying infection, an examination with nothing more than punctate ulcerations would be atypical. Plague and tularemia cause ulcerative skin lesions and systemic illness but usually have prominent lymphadenopathy. Cutaneous anthrax can cause intense local edema but is usually accompanied by some degree of necrosis.
I am struck by the degree of local edema. It would be a remarkable coincidence for spider bites to occur on both wrists; however, given the size of the lesions, this remains a consideration. Spider bites can cause severe local swelling and occasionally even SIRS.
The white blood cellcount was 6000/mm3 with a normal differential. The hemoglobin level was 16.6 g/dL, and the platelet count was 227,000/mm3. The serum sodium level was measured to be 135 mmol/L, the potassium level was 3.6 mmol/L, the chloride level was 94 mmol/L, and the bicarbonate level was 9 mmol/L. The urea nitrogen level was measured to be 48 mg/dL (normal, 622), and the serum creatinine level was 5.6 mg/dL (normal, 0.41.2). An arterial blood gas test on room air revealed a pH of 7.22, a partial pressure of carbon dioxide of 24 mm Hg, and a partial pressure of oxygen of 128 mm Hg. Liver enzymes were normal. The serum creatine kinase level was measured to be 194 U/L (normal, 0250). A chest radiograph revealed clear lung fields.
The anion gap is elevated and could be explained in part by renal failure, but it is quite pronounced, and I suspect that there is lactic acidosis either from SIRS and systemic hypoperfusion or from local underperfusion of the distal upper extremities (eg, compartment syndrome). Rhabdomyolysis can cause an anion gap acidosis and acute renal failure but is ruled out by a normal creatine kinase level.
Rheumatological diseases (such as polyarteritis nodosa and systemic scleroderma) can cause renal failure, gastrointestinal symptoms, and cutaneous lesions with skin ulceration but are often accompanied by hypertension. The combination of SIRS and volume depletion from nausea, vomiting, and diarrhea seems more likely, although an etiology for SIRS remains elusive. I suspect infectionmost likely of the upper extremitiesis the underlying cause in this patient despite his normal body temperature and white cell count. I would therefore start with plain films of the arms and hands. If these are unrevealing, I would proceed with an ultrasound of the arms to evaluate the soft tissues and particularly the vessels. Although imaging studies are unlikely to provide a diagnosis, they will provide guidance in choosing the next step, such as biopsy or culture.
The patient wasvolume‐resuscitated with saline and treated with clindamycin, pipercillin‐tazobactam, and vancomycin. Radiographs of the wrists and hands demonstrated edema but no subcutaneous gas and no abnormalities of the bones or joints. An ultrasound of the upper extremities was negative for superficial or deep venous thrombosis. Renal ultrasonography revealed normal‐sized kidneys and no hydronephrosis.
Short of superior vena cava thrombosis, which the ultrasound could not visualize, deep venous thrombosis can be ruled out with confidence. Superior vena cava syndrome could account for the bilateral upper extremity symptoms, but complete sparing of the face would be unusual, and the chest radiograph was normal.
Despite the bilateral nature, I doubt that this patient has an arthritis of the wrists and hands (eg, rheumatoid or psoriatic arthritis). The plain films did not show evidence of joint destruction, although that would not be expected in the first few days of most noninfectious arthropathies. Many rheumatological diseases and vasculitides have renal manifestations, but I do not find convincing evidence of such diseases yet.
Despite intravenous fluidsover the next 4 hours, the patient remained anuric. His upper extremity edema worsened, and he became increasingly tachycardic and hypotensive. Vasopressor support was required. Repeat laboratory studies demonstrated a serum creatinine level of 7.0 mg/dL, a bicarbonate level of 6 mmol/L, and a creatine kinase level of 1183 U/L. The serum lactate level was measured to be 6.5. Blood cultures obtained on admission were negative. An echocardiogram demonstrated marked impairment of left ventricular systolic dysfunction with an estimated ejection fraction of 35%. Repeat chest radiography revealed only mild pulmonary vascular congestion.
The patient has become progressively ill despite initial resuscitation. Any infection (cellulitis, fasciitis, or myositis) may progress to such a point (although to do so without fever or leukocytosis with this degree of illness is unusual) and would prompt me to ask a surgeon to explore the upper extremities for diagnostic and possibly therapeutic purposes. Among the spectrum of possible soft tissue infections, this clinical presentation is most consistent with pyomyositis (caused by Staphylococcus aureus) because of the relatively modest creatine kinase elevations that accompany it and the overall absence of cutaneous findings (save the punctuate lesions), which I would expect to be present with cellulitis or necrotizing fasciitis. A deep forearm infection and perhaps compartment syndrome leading to sepsis could explain lactic acidosis, decreased cardiac function, hypotension, and acute renal failure.
Because of the unusual characteristics noted so farparticularly the bilateral diseaseI continue to also consider systemic diseases that can cause skin lesions, cardiomyopathy, renal disease, ocular involvement (eg, keratoconjunctivitis or uveitis), and myositis. Sarcoidosis is possible, although in most of the aforementioned organs, histological disease is far more common than clinical disease, and sarcoidosis typically does not cause this degree of illness. Furthermore, over 90% of patients with sarcoidosis have pulmonary involvement. Polyarteritis nodosa could explain the multiorgan involvement and the brisk pace. If no infection is present on exploration, I would ask the surgeon to biopsy the muscle, particularly looking for granulomas or vasculitis. A progressive soft tissue infection leading to sepsis remains my leading consideration at this point.
Surgical consultation was obtained. A muscle biopsy of the left biceps and left forearm revealed group A streptococcus within the muscle and evidence of necrosis (Figure 1). Debridement of both arms and wrists was performed. The patient subsequently developed erythema of the palms and soles followed by diffuse sloughing of the skin. Streptococcal toxic shock syndrome with necrotizing myositis was diagnosed, and the antimicrobial regimen was changed to intravenous penicillin, clindamycin, and intravenous immunoglobulin G. Repeat debridement of the arms was required.
Even when a soft tissue infection is suspected, it can be challenging to preoperatively localize or characterize with precision. In retrospect, the overall severity of his illness and his previously good health status perhaps favor necrotizing myositis (and fasciitis) over pyomyositis.
I may have put undue emphasis on the absence of skin findings. Although the symmetric nature of the disease is unusual, the small skin lesions may have been portals of entry, and in retrospect, they represent the tip of the iceberg. The absence of fever and leukocytosisor hypothermia and leukopeniain a young, previously healthy patient along with the bilateral and symmetric findings made me hesitant to definitely label his illness as a deep soft tissue infection early on, but the gravity of the illness, the lack of a plausible alternative explanation, and his precipitous decline all made surgical exploration imperative.
The patient's skin sloughing progressed, and he was transferred to a regional burn unit. Three additional operations were required for debridement of both upper extremities. Despite apparent control of the initial infection, the patient continued to require significant hemodynamic and ventilator support. He subsequently developed neutropenia, thrombocytopenia, and Escherichia coli urosepsis. Necrosis of the lower extremities developed, and additional surgical debridement was recommended. After extensive discussion regarding prognosis, the family decided against further surgery and withdrew life support. The patient died shortly after extubation. The family refused an autopsy.
Commentary
Expert clinicians employ a variety of approaches to solve complex clinical problems. One of the most effective strategies is pattern recognition, in which the clinician divides the case into recognizable portions and compares these to previous cases that he or she has encountered.1, 2 If patterns from the new case appear similar or identical to those of previous cases, a diagnosis can be made quickly and without the need for unnecessary testing. However, when features of the case are unusual or atypical, pattern recognition may be disrupted. For example, although the discussant suspected a soft tissue infection, a number of features (including a normal white blood cell count, minimal skin findings, and a bilateral and symmetric distribution of swelling) did not match his illness script (a mental representation of a disease) of necrotizing fasciitis.
When pattern recognition fails, other strategies are available.3 Hypothetico‐deductive reasoning is a data‐to‐diagnosis method whereby the clinician uses the presenting information to construct a list of diagnostic possibilities.4 Additional testing and gathering of information are then used to continuously revise the diagnostic possibilities until confirmatory information is obtained and the diagnosis is established. After a pattern failed to materialize, the discussant employed this analytical strategy by noting the unusual characteristics of the case and incorporating laboratory and physiological data to revise his differential diagnosis. As a result, he requested the appropriate diagnostic test: surgical exploration of the forearms.
Necrotizing soft tissue infections are characterized by fulminant tissue destruction, rapid spread along tissue planes, and local vascular thrombosis. Mixed aerobic and anaerobic infections typically occur after penetrating skin injury or following surgery in patients with diabetes mellitus or vascular disease. In contrast, monomicrobial infections with S. aureus or group A streptococcus generally occur in healthy individuals. The prevalence of necrotizing group A streptococcal infections has increased dramatically in the last 15 to 20 years.5 Over one‐third of these cases are complicated by toxic shock syndrome.57 Mortality rates for necrotizing fasciitis with toxic shock exceed 30%, and early surgical consultation is directly associated with a reduction in morbidity and mortality.5, 8
Toxic shock is an inflammatory response syndrome caused by release of exotoxins from group A streptococcus and S. aureus.9, 10 In streptococcal toxic shock, Streptococcus pyogenes exotoxin A and Streptococcus pyogenes exotoxin B are the major toxins produced.10 These toxins activate the systemic production of inflammatory cytokines such as interleukin‐1, gamma‐interferon, and tumor necrosis factor, resulting in capillary leak, systemic hypotension, tissue hypoperfusion, and organ failure. The most common initial symptom is diffuse or localized pain that is severe and abrupt in onset and often precedes or is out of proportion to other physical findings of soft tissue infection.8 Up to 20% of individuals may also develop a viral‐like syndrome with myalgias, fever, nausea, vomiting, and diarrhea.11 Erythroderma of the skin and mucous membranes can be another early finding. The rash is diffuse, erythematous, and macular, resembling a sunburn. It involves the palms and soles but can be subtle and fleeting. Erythroderma may be particularly difficult to detect in dark‐skinned individuals.12 It is also important to consider that the absence of fever, erythroderma, or leukocytosis does not necessarily rule out the possibility of serious infection in necrotizing fasciitis patients, as the development of these signs and symptoms may occur later in the disease process.
The most common portals of entry for group A streptococcus are the skin, vagina, and pharynx. Predisposing factors include varicella infection, penetrating injuries, minor cuts, burns, splinters, and surgery. Interestingly, a portal of entry cannot be identified in 45% of cases.8 These patients in particular are at risk for developing severe necrotizing myositis or fasciitis at the site of a minor injury such as a strained muscle. Hematogenous translocation from the pharynx to the site of injury is the probable mechanism13 and would provide one scenario by which our piano mover developed bilateral and symmetric disease. An alternative explanation would be direct extension of bacteria from small breaks in the skin to adjacent areas of muscle strain. Regardless of the portal of entry, as the small skin lesions demonstrate in this patient, the smallest physical finding can represent the tip of the iceberg.
- ,,,,,.Fast and frugal models of clinical judgment in novice and expert physicians.Med Decis Making.2003;23(4):293–300.
- .What is it to be an expert? In: Chi M, Farr MJ, Glaser R, eds.The Nature of Expertise.Hillsdale, NJ:Lawrence Erlbaum;1988.
- .Clinical decision‐making: understanding how clinicians make a diagnosis. In: Saint S, Drazen JM, Solomon CG, eds.Clinical Problem‐Solving.New York, NY:McGraw‐Hill;2006.
- ,,.Medical Problem Solving: An Analysis of Clinical Reasoning.Cambridge, MA:Harvard University Press;1978.
- ,,,,.Population‐based surveillance for group A streptococcal necrotizing fasciitis: clinical features, prognostic indicators, and microbiologic analysis of seventy‐seven cases. Ontario Group A Streptococcal Study.Am J Med.1997;103(1):18–24.
- ,,, et al.Invasive group A streptococcal infections in Sweden in 1994 and 1995: epidemiology and clinical spectrum.Scand J Infect Dis.2000;32(6):609–614.
- ,,,.Reemergence of emm1 and a changed superantigen profile for group A streptococci causing invasive infections: results from a nationwide study.J Clin Microbiol.2005;43(4):1789–1796.
- ,,, et al.Severe group A streptococcal infections associated with a toxic shock‐like syndrome and scarlet fever toxin A.N Engl J Med.1989;321(1):1–7.
- ,,,.Rapidly progressive necrotizing fasciitis caused by Staphylococcus aureus.J Microbiol Immunol Infect.2005;38(5):361–364.
- ,.Streptococcal infections of skin and soft tissues.N Engl J Med.1996;334(4):240–245.
- ,.Streptococcal toxic shock syndrome after breast reconstruction.Ann Plast Surg.2005;54(5):553–556.
- ,.The influence of pigmentation and illumination on the perception of erythema.Photodermatol Photoimmunol Photomed.1992;9(2):45–47.
- .Streptococcal toxic‐shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment.Emerg Infect Dis.1995;1(3):69–78.
- ,,,,,.Fast and frugal models of clinical judgment in novice and expert physicians.Med Decis Making.2003;23(4):293–300.
- .What is it to be an expert? In: Chi M, Farr MJ, Glaser R, eds.The Nature of Expertise.Hillsdale, NJ:Lawrence Erlbaum;1988.
- .Clinical decision‐making: understanding how clinicians make a diagnosis. In: Saint S, Drazen JM, Solomon CG, eds.Clinical Problem‐Solving.New York, NY:McGraw‐Hill;2006.
- ,,.Medical Problem Solving: An Analysis of Clinical Reasoning.Cambridge, MA:Harvard University Press;1978.
- ,,,,.Population‐based surveillance for group A streptococcal necrotizing fasciitis: clinical features, prognostic indicators, and microbiologic analysis of seventy‐seven cases. Ontario Group A Streptococcal Study.Am J Med.1997;103(1):18–24.
- ,,, et al.Invasive group A streptococcal infections in Sweden in 1994 and 1995: epidemiology and clinical spectrum.Scand J Infect Dis.2000;32(6):609–614.
- ,,,.Reemergence of emm1 and a changed superantigen profile for group A streptococci causing invasive infections: results from a nationwide study.J Clin Microbiol.2005;43(4):1789–1796.
- ,,, et al.Severe group A streptococcal infections associated with a toxic shock‐like syndrome and scarlet fever toxin A.N Engl J Med.1989;321(1):1–7.
- ,,,.Rapidly progressive necrotizing fasciitis caused by Staphylococcus aureus.J Microbiol Immunol Infect.2005;38(5):361–364.
- ,.Streptococcal infections of skin and soft tissues.N Engl J Med.1996;334(4):240–245.
- ,.Streptococcal toxic shock syndrome after breast reconstruction.Ann Plast Surg.2005;54(5):553–556.
- ,.The influence of pigmentation and illumination on the perception of erythema.Photodermatol Photoimmunol Photomed.1992;9(2):45–47.
- .Streptococcal toxic‐shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment.Emerg Infect Dis.1995;1(3):69–78.