User login
Hot in the tropics
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 42-year-old Malaysian construction worker with subjective fevers of 4 days’ duration presented to an emergency department in Singapore. He reported nonproductive cough, chills without rigors, sore throat, and body aches. He denied sick contacts. Past medical history included chronic hepatitis B virus (HBV) infection. The patient was not taking any medications.
For this patient presenting acutely with subjective fevers, nonproductive cough, chills, aches, and lethargy, initial considerations include infection with a common virus (influenza virus, adenovirus, Epstein-Barr virus [EBV]), acute human immunodeficiency virus (HIV) infection, emerging infection (severe acute respiratory syndrome [SARS], Middle Eastern respiratory syndrome coronavirus [MERS-CoV] infection, avian influenza), and tropical infection (dengue, chikungunya). Also possible are bacterial infections (eg, with Salmonella typhi or Rickettsia or Mycoplasma species), parasitic infections (eg, malaria), and noninfectious illnesses (eg, autoimmune diseases, thyroiditis, acute leukemia, environmental exposures).
The patient’s temperature was 38.5°C; blood pressure, 133/73 mm Hg; heart rate, 95 beats per minute; respiratory rate, 18 breaths per minute; and oxygen saturation, 100% on ambient air. On physical examination, he appeared comfortable, and heart, lung, abdomen, skin, and extremities were normal. Laboratory test results included white blood cell (WBC) count, 4400/μL (with normal differential); hemoglobin, 16.1 g/dL; and platelet count, 207,000/μL. Serum chemistries were normal. C-reactive protein (CRP) level was 44.6 mg/L (reference range, 0.2-9.1 mg/L), and procalcitonin level was 0.13 ng/mL (reference range, <0.50 ng/mL). Chest radiograph was normal. Dengue antibodies (immunoglobulin M, immunoglobulin G [IgG]) and dengue NS1 antigen were negative. The patient was discharged with a presumptive diagnosis of viral upper respiratory tract infection.
There is no left shift characteristic of bacterial infection or lymphopenia characteristic of rickettsial disease or acute HIV infection. The serologic testing and the patient’s overall appearance make dengue unlikely. The low procalcitonin supports a nonbacterial cause of illness. CRP elevation may indicate an inflammatory process and is relatively nonspecific.
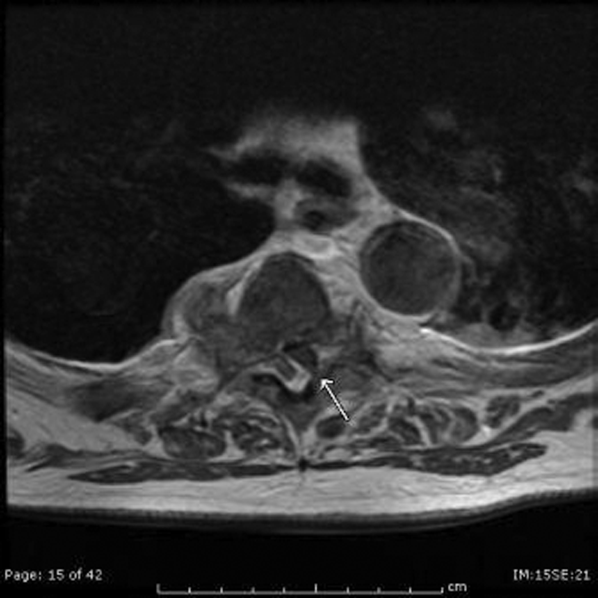
Myalgias, pharyngitis, and cough improved over several days, but fevers persisted, and a rash developed over the lower abdomen. The patient returned to the emergency department and was admitted. He denied weight loss and night sweats. He had multiple female sexual partners, including commercial sex workers, within the previous 6 months. Temperature was 38.5°C. The posterior oropharynx was slightly erythematous. There was no lymphadenopathy. Firm, mildly erythematous macules were present on the anterior abdominal wall (Figure 1). The rest of the physical examination was normal.

Laboratory testing revealed WBC count, 5800/μL (75% neutrophils, 19% lymphocytes, 3% monocytes, 2% atypical mononuclear cells); hemoglobin, 16.3 g/dL; platelet count, 185,000/μL; sodium, 131 mmol/L; potassium, 3.4 mmol/L; creatinine, 0.9 mg/dL; albumin, 3.2 g/dL; alanine aminotransferase (ALT), 99 U/L; aspartate aminotransferase (AST), 137 U/L; alkaline phosphatase (ALP), 63 U/L; and total bilirubin, 1.9 mg/dL. Prothrombin time was 11.1 seconds; partial thromboplastin time, 36.1 seconds; erythrocyte sedimentation rate, 14 mm/h; and CRP, 62.2 mg/L.
EBV, acute HIV, and cytomegalovirus infections often present with adenopathy, which is absent here. Disseminated gonococcal infection can manifest with fever, body aches, and rash, but his rash and the absence of penile discharge, migratory arthritis, and enthesitis are not characteristic. Mycoplasma infection can present with macules, urticaria, or erythema multiforme. Rickettsia illnesses typically cause vasculitis with progression to petechiae or purpura resulting from endothelial damage. Patients with secondary syphilis may have widespread macular lesions, and the accompanying syphilitic hepatitis often manifests with elevations in ALP instead of ALT and AST. The mild elevation in ALT and AST can occur with many systemic viral infections. Sweet syndrome may manifest with febrile illness and rash, but the acuity of this patient’s illness and the rapid evolution favor infection.
The patient’s fevers (35°-40°C) continued without pattern over the next 3 days. Blood and urine cultures were negative. Polymerase chain reaction (PCR) test of the nasal mucosa was negative for respiratory viruses. PCR blood tests for EBV, HIV-1, and cytomegalovirus were also negative. Antistreptolysin O (ASO) titer was 400 IU/mm (reference range, <200 IU/mm). Antinuclear antibodies were negative, and rheumatoid factor was 12.4 U/mL (reference range, <10.3 U/mL). Computed tomography (CT) of the thorax, abdomen, and pelvis was normal. Results of a biopsy of an anterior abdominal wall skin lesion showed perivascular and periadnexal lymphocytic inflammation. Amoxicillin was started for the treatment of possible group A streptococcal infection.
PCR for HIV would be positive at a high level in acute HIV. The skin biopsy is not characteristic of Sweet syndrome, which typically shows neutrophilic infiltrate without leukocytoclastic vasculitis, or of syphilis, which typically shows a plasma cell infiltrate.
The patient’s erythematous oropharynx may indicate recent streptococcal pharyngitis. The fevers, elevated ASO titer, and CRP level are consistent with acute rheumatic fever, but arthritis, carditis, and neurologic manifestations are lacking. Erythema marginatum manifests on the trunk and limbs as macules or papules with central clearing as the lesions spread outward—and differs from the patient’s rash, which is firm and restricted to the abdominal wall.
Fevers persisted through hospital day 7. The WBC count was 1100/μL (75.7% neutrophils, 22.5% lymphocytes), hemoglobin was 10.3 g/dL, and platelet count was 52,000/μL. Additional laboratory test results included ALP, 234 U/L; ALT, 250 U/L; AST, 459 U/L; lactate dehydrogenase, 2303 U/L (reference range, 222-454 U/L); and ferritin, 14,964 ng/mL (reference range, 47-452 ng/mL).
The duration of illness and negative diagnostic tests for infections increases suspicion for a noninfectious illness. Conditions commonly associated with marked hyperferritinemia include adult-onset Still disease (AOSD) and hemophagocytic lymphohistiocytosis (HLH). Of the 9 AOSD diagnostic (Yamaguchi) criteria, 5 are met in this case: fever, rash, sore throat, abnormal liver function tests, and negative rheumatologic tests. However, the patient lacks arthritis, leukocytosis, lymphadenopathy, and hepatosplenomegaly. Except for the elevated ferritin, the AOSD criteria overlap substantially with the criteria for acute rheumatic fever, and still require that infections be adequately excluded. HLH, a state of abnormal immune activation with resultant organ dysfunction, can be a primary disorder, but in adults more often is secondary to underlying infectious, autoimmune, or malignant (often lymphoma) conditions. Elevated ferritin, cytopenias, elevated ALT and AST, elevated CRP and erythrocyte sedimentation rate, and elevated lactate dehydrogenase are consistent with HLH. The HLH diagnosis can be more firmly established with the more specific findings of hypertriglyceridemia, hypofibrinogenemia, and elevated soluble CD25 level. The histopathologic finding of hemophagocytosis in the bone marrow, lymph nodes, or liver may further support the diagnosis of HLH.
Rash and fevers persisted. Hepatitis A, hepatitis C, Rickettsia IgG, Burkholderia pseudomallei (the causative organism of melioidosis), and Leptospira serologies, as well as PCR for herpes simplex virus and parvovirus, were all negative. Hepatitis B viral load was 962 IU/mL (2.98 log), hepatitis B envelope antigen was negative, and hepatitis B envelope antibody was positive. Orientia tsutsugamushi (organism responsible for scrub typhus) IgG titer was elevated at 1:128. Antiliver kidney microsomal antibodies and antineutrophil cytoplasmic antibodies were negative. Fibrinogen level was 0.69 g/L (reference range, 1.8-4.8 g/L), and beta-2 microglobulin level was 5078 ng/mL (reference range, 878-2000 ng/mL). Bone marrow biopsy results showed hypocellular marrow with suppressed myelopoiesis, few atypical lymphoid cells, and few hemophagocytes. Flow cytometry was negative for clonal B lymphocytes and aberrant expression of T lymphocytes. Bone marrow myobacterial PCR and fungal cultures were negative.
The patient’s chronic HBV infection is unlikely to be related to his presentation given his low viral load and absence of signs of hepatic dysfunction. Excluding rickettsial disease requires paired acute and convalescent serologies. O tsutsugamushi, the causative agent of the rickettsial disease scrub typhus, is endemic in Malaysia; thus, his positive O tsutsugamushi IgG may indicate past exposure. His fevers, myalgias, truncal rash, and hepatitis are consistent with scrub typhus, but he lacks the characteristic severe headache and generalized lymphadenopathy. Although eschar formation with evolution of a papular rash is common in scrub typhus, it is often absent in the variant found in Southeast Asia. Although elevated β2 microglobulin level is used as a prognostic marker in multiple myeloma and Waldenström macroglobulinemia, it can be elevated in many immune-active states. The patient likely has HLH, which is supported by the hemophagocytosis seen on bone marrow biopsy, and the hypofibrinogenemia. Potential HLH triggers include O tsutsugamushi infection or recent streptococcal pharyngitis.
A deep-punch skin biopsy of the anterior abdominal wall skin lesion was performed because of the absence of subcutaneous fat in the first biopsy specimen. The latest biopsy results showed irregular interstitial expansion of medium-size lymphocytes in a lobular panniculated pattern. The lymphocytes contained enlarged, irregularly contoured nucleoli and were positive for T-cell markers CD2 and CD3 with reduction in CD5 expression. The lymphomatous cells were of CD8+ with uniform expression of activated cytotoxic granule protein granzyme B and were positive for T-cell hemireceptor β.
Positron emission tomography (PET) CT, obtained for staging purposes, showed multiple hypermetabolic subcutaneous and cutaneous lesions over the torso and upper and lower limbs—compatible with lymphomatous infiltrates (Figure 2). Examination, pathology, and imaging findings suggested a rare neoplasm: subcutaneous panniculitis-like T-cell lymphoma (SPTCL). SPTCL was confirmed by T-cell receptor gene rearrangements studies. with surrounding patchy foci of subcutaneous fat stranding (blue-grey) in anterior abdominal wall and upper left arm, compatible with areas o")
HLH was diagnosed on the basis of the fevers, cytopenias, hypofibrinogenemia, elevated ferritin level, and evidence of hemophagocytosis. SPTCL was suspected as the HLH trigger.
The patient was treated with cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone. While on this regimen, he developed new skin lesions, and his ferritin level was persistently elevated. He was switched to romidepsin, a histone deacetylase inhibitor that specifically targets cutaneous T-cell lymphoma, but the lesions continued to progress. The patient then was treated with gemcitabine, dexamethasone, and cisplatin, and the rashes resolved. The most recent PET-CT showed nearly complete resolution of the subcutaneous lesions.
DISCUSSION
When residents or visitors to tropical or sub-tropical regions, those located near or between the Tropics of Cancer and Capricorn, present with fever, physicians usually first think of infectious diseases. This patient’s case is a reminder that these important first considerations should not be the last.
Generating a differential diagnosis for tropical illnesses begins with the patient’s history. Factors to be considered include location (regional disease prevalence), exposures (food/water ingestion, outdoor work/recreation, sexual contact, animal contact), and timing (temporal relationship of symptom development to possible exposure). Common tropical infections are malaria, dengue, typhoid, and emerging infections such as chikungunya, avian influenza, and Zika virus infection.1This case underscores the need to analyze diagnostic tests critically. Interpreting tests as simply positive or negative, irrespective of disease features, epidemiology, and test characteristics, can contribute to diagnostic error. For example, the patient’s positive ASO titer requires an understanding of disease features and a nuanced interpretation based on the clinical presentation. The erythematous posterior oropharynx prompted concern for postinfectious sequelae of streptococcal pharyngitis, but his illness was more severe and more prolonged than is typical of that condition. The isolated elevated O tsutsugamushi IgG titer provides an example of the role of epidemiology in test interpretation. Although a single positive value might indicate a new exposure for a visitor to an endemic region, IgG seropositivity in Singapore, where scrub typhus is endemic, likely reflects prior exposure to the organism. Diagnosing an acute scrub typhus infection in a patient in an endemic region requires PCR testing. The skin biopsy results highlight the importance of understanding test characteristics. A skin biopsy specimen must be adequate in order to draw valid and accurate conclusions. In this case, the initial skin biopsy was superficial, and the specimen inadequate, but the test was not “negative.” In the diagnostic skin biopsy, deeper tissue was sampled, and panniculitis (inflammation of subcutaneous fat), which arises in inflammatory, infectious, traumatic, enzymatic, and malignant conditions, was identified. An adequate biopsy specimen that contains subcutaneous fat is essential in making this diagnosis.2This patient eventually manifested several elements of hemophagocytic lymphohistiocytosis (HLH), a syndrome of excessive inflammation and resultant organ injury relating to abnormal immune activation and excessive inflammation. HLH results from deficient down-regulation of activated macrophages and lymphocytes.3 It was initially described in pediatric patients but is now recognized in adults, and associated with mortality as high as 50%.3 A high ferritin level (>2000 ng/mL) has 70% sensitivity and 68% specificity for pediatric HLH and should trigger consideration of HLH in any age group.4 The diagnostic criteria for HLH initially proposed in 2004 by the Histiocyte Society to identify patients for recruitment into a clinical trial included molecular testing consistent with HLH and/or 5 of 8 clinical, laboratory, or histopathologic features (Table 1).5 HScore is a more recent validated scoring system that predicts the probability of HLH (Table 2). A score above 169 signifies diagnostic sensitivity of 93% and specificity of 86%.6
The diagnosis of HLH warrants a search for its underlying cause. Common triggers are viral infections (eg, EBV), autoimmune diseases (eg, systemic lupus erythematosus), and hematologic malignancies. These triggers typically stimulate or suppress the immune system. Initial management involves treatment of the underlying trigger and, potentially, immunosuppression with high
In this case, SPTCL triggered HLH. SPTCL is a rare non-Hodgkin lymphoma characterized by painless subcutaneous nodules or indurated plaques (panniculitis-like) on the trunk or extremities, constitutional symptoms, and, in some cases, HLH.7-10 SPTCL is diagnosed by deep skin biopsy, with immunohistochemistry showing CD8-positive pathologic T cells expressing cytotoxic proteins (eg, granzyme B).9,11 SPTCL can either have an alpha/beta T-cell phenotype (SPTCL-AB) or gamma/delta T-cell phenotype (SPTCL-GD). Seventeen percent of patients with SPTCL-AB and 45% of patients with SPTCL-GD have HLH on diagnosis. Concomitant HLH is associated with decreased 5-year survival.12This patient presented with fevers and was ultimately diagnosed with HLH secondary to SPLTCL. His case is a reminder that not all diseases in the tropics are tropical diseases. In the diagnosis of a febrile illness, a broad evaluative framework and rigorous test results evaluation are essential—no matter where a patient lives or visits.
")
KEY TEACHING POINTS
- A febrile illness acquired in the tropics is not always attributable to a tropical infection.
- To avoid diagnostic error, weigh positive or negative test results against disease features, patient epidemiology, and test characteristics.
- HLH is characterized by fevers, cytopenias, hepatosplenomegaly, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia. In tissue specimens, hemophagocytosis may help differentiate HLH from competing conditions.
- After HLH is diagnosed, try to determine its underlying cause, which may be an infection, autoimmunity, or a malignancy (commonly, a lymphoma).
Disclosure
Nothing to report.
1. Centers for Disease Control and Prevention. Destinations [list]. http://wwwnc.cdc.gov/travel/destinations/list/. Accessed April 22, 2016.
2. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22(6):530-549. PubMed
3. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503-1516. PubMed
4. Lehmberg K, McClain KL, Janka GE, Allen CE. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2014;61(11):2101-2103. PubMed
5. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. PubMed
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. PubMed
7. Aronson IK, Worobed CM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23(4):389-402. PubMed
8. Willemze R, Jansen PM, Cerroni L, et al; EORTC Cutaneous Lymphoma Group. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111(2):838-845. PubMed
9. Kumar S, Krenacs L, Medeiros J, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol. 1998;29(4):397-403. PubMed
10. Salhany KE, Macon WR, Choi JK, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol. 1998;22(7):881-893. PubMed
11. Jaffe ES, Nicolae A, Pittaluga S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: pearls and pitfalls. Mod Pathol. 2013;26(suppl 1):S71-S87. PubMed
12. Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M; ESMO Guidelines Working Group. Primary cutaneous lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl 6):vi149-vi154. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 42-year-old Malaysian construction worker with subjective fevers of 4 days’ duration presented to an emergency department in Singapore. He reported nonproductive cough, chills without rigors, sore throat, and body aches. He denied sick contacts. Past medical history included chronic hepatitis B virus (HBV) infection. The patient was not taking any medications.
For this patient presenting acutely with subjective fevers, nonproductive cough, chills, aches, and lethargy, initial considerations include infection with a common virus (influenza virus, adenovirus, Epstein-Barr virus [EBV]), acute human immunodeficiency virus (HIV) infection, emerging infection (severe acute respiratory syndrome [SARS], Middle Eastern respiratory syndrome coronavirus [MERS-CoV] infection, avian influenza), and tropical infection (dengue, chikungunya). Also possible are bacterial infections (eg, with Salmonella typhi or Rickettsia or Mycoplasma species), parasitic infections (eg, malaria), and noninfectious illnesses (eg, autoimmune diseases, thyroiditis, acute leukemia, environmental exposures).
The patient’s temperature was 38.5°C; blood pressure, 133/73 mm Hg; heart rate, 95 beats per minute; respiratory rate, 18 breaths per minute; and oxygen saturation, 100% on ambient air. On physical examination, he appeared comfortable, and heart, lung, abdomen, skin, and extremities were normal. Laboratory test results included white blood cell (WBC) count, 4400/μL (with normal differential); hemoglobin, 16.1 g/dL; and platelet count, 207,000/μL. Serum chemistries were normal. C-reactive protein (CRP) level was 44.6 mg/L (reference range, 0.2-9.1 mg/L), and procalcitonin level was 0.13 ng/mL (reference range, <0.50 ng/mL). Chest radiograph was normal. Dengue antibodies (immunoglobulin M, immunoglobulin G [IgG]) and dengue NS1 antigen were negative. The patient was discharged with a presumptive diagnosis of viral upper respiratory tract infection.
There is no left shift characteristic of bacterial infection or lymphopenia characteristic of rickettsial disease or acute HIV infection. The serologic testing and the patient’s overall appearance make dengue unlikely. The low procalcitonin supports a nonbacterial cause of illness. CRP elevation may indicate an inflammatory process and is relatively nonspecific.
Myalgias, pharyngitis, and cough improved over several days, but fevers persisted, and a rash developed over the lower abdomen. The patient returned to the emergency department and was admitted. He denied weight loss and night sweats. He had multiple female sexual partners, including commercial sex workers, within the previous 6 months. Temperature was 38.5°C. The posterior oropharynx was slightly erythematous. There was no lymphadenopathy. Firm, mildly erythematous macules were present on the anterior abdominal wall (Figure 1). The rest of the physical examination was normal.
Laboratory testing revealed WBC count, 5800/μL (75% neutrophils, 19% lymphocytes, 3% monocytes, 2% atypical mononuclear cells); hemoglobin, 16.3 g/dL; platelet count, 185,000/μL; sodium, 131 mmol/L; potassium, 3.4 mmol/L; creatinine, 0.9 mg/dL; albumin, 3.2 g/dL; alanine aminotransferase (ALT), 99 U/L; aspartate aminotransferase (AST), 137 U/L; alkaline phosphatase (ALP), 63 U/L; and total bilirubin, 1.9 mg/dL. Prothrombin time was 11.1 seconds; partial thromboplastin time, 36.1 seconds; erythrocyte sedimentation rate, 14 mm/h; and CRP, 62.2 mg/L.
EBV, acute HIV, and cytomegalovirus infections often present with adenopathy, which is absent here. Disseminated gonococcal infection can manifest with fever, body aches, and rash, but his rash and the absence of penile discharge, migratory arthritis, and enthesitis are not characteristic. Mycoplasma infection can present with macules, urticaria, or erythema multiforme. Rickettsia illnesses typically cause vasculitis with progression to petechiae or purpura resulting from endothelial damage. Patients with secondary syphilis may have widespread macular lesions, and the accompanying syphilitic hepatitis often manifests with elevations in ALP instead of ALT and AST. The mild elevation in ALT and AST can occur with many systemic viral infections. Sweet syndrome may manifest with febrile illness and rash, but the acuity of this patient’s illness and the rapid evolution favor infection.
The patient’s fevers (35°-40°C) continued without pattern over the next 3 days. Blood and urine cultures were negative. Polymerase chain reaction (PCR) test of the nasal mucosa was negative for respiratory viruses. PCR blood tests for EBV, HIV-1, and cytomegalovirus were also negative. Antistreptolysin O (ASO) titer was 400 IU/mm (reference range, <200 IU/mm). Antinuclear antibodies were negative, and rheumatoid factor was 12.4 U/mL (reference range, <10.3 U/mL). Computed tomography (CT) of the thorax, abdomen, and pelvis was normal. Results of a biopsy of an anterior abdominal wall skin lesion showed perivascular and periadnexal lymphocytic inflammation. Amoxicillin was started for the treatment of possible group A streptococcal infection.
PCR for HIV would be positive at a high level in acute HIV. The skin biopsy is not characteristic of Sweet syndrome, which typically shows neutrophilic infiltrate without leukocytoclastic vasculitis, or of syphilis, which typically shows a plasma cell infiltrate.
The patient’s erythematous oropharynx may indicate recent streptococcal pharyngitis. The fevers, elevated ASO titer, and CRP level are consistent with acute rheumatic fever, but arthritis, carditis, and neurologic manifestations are lacking. Erythema marginatum manifests on the trunk and limbs as macules or papules with central clearing as the lesions spread outward—and differs from the patient’s rash, which is firm and restricted to the abdominal wall.
Fevers persisted through hospital day 7. The WBC count was 1100/μL (75.7% neutrophils, 22.5% lymphocytes), hemoglobin was 10.3 g/dL, and platelet count was 52,000/μL. Additional laboratory test results included ALP, 234 U/L; ALT, 250 U/L; AST, 459 U/L; lactate dehydrogenase, 2303 U/L (reference range, 222-454 U/L); and ferritin, 14,964 ng/mL (reference range, 47-452 ng/mL).
The duration of illness and negative diagnostic tests for infections increases suspicion for a noninfectious illness. Conditions commonly associated with marked hyperferritinemia include adult-onset Still disease (AOSD) and hemophagocytic lymphohistiocytosis (HLH). Of the 9 AOSD diagnostic (Yamaguchi) criteria, 5 are met in this case: fever, rash, sore throat, abnormal liver function tests, and negative rheumatologic tests. However, the patient lacks arthritis, leukocytosis, lymphadenopathy, and hepatosplenomegaly. Except for the elevated ferritin, the AOSD criteria overlap substantially with the criteria for acute rheumatic fever, and still require that infections be adequately excluded. HLH, a state of abnormal immune activation with resultant organ dysfunction, can be a primary disorder, but in adults more often is secondary to underlying infectious, autoimmune, or malignant (often lymphoma) conditions. Elevated ferritin, cytopenias, elevated ALT and AST, elevated CRP and erythrocyte sedimentation rate, and elevated lactate dehydrogenase are consistent with HLH. The HLH diagnosis can be more firmly established with the more specific findings of hypertriglyceridemia, hypofibrinogenemia, and elevated soluble CD25 level. The histopathologic finding of hemophagocytosis in the bone marrow, lymph nodes, or liver may further support the diagnosis of HLH.
Rash and fevers persisted. Hepatitis A, hepatitis C, Rickettsia IgG, Burkholderia pseudomallei (the causative organism of melioidosis), and Leptospira serologies, as well as PCR for herpes simplex virus and parvovirus, were all negative. Hepatitis B viral load was 962 IU/mL (2.98 log), hepatitis B envelope antigen was negative, and hepatitis B envelope antibody was positive. Orientia tsutsugamushi (organism responsible for scrub typhus) IgG titer was elevated at 1:128. Antiliver kidney microsomal antibodies and antineutrophil cytoplasmic antibodies were negative. Fibrinogen level was 0.69 g/L (reference range, 1.8-4.8 g/L), and beta-2 microglobulin level was 5078 ng/mL (reference range, 878-2000 ng/mL). Bone marrow biopsy results showed hypocellular marrow with suppressed myelopoiesis, few atypical lymphoid cells, and few hemophagocytes. Flow cytometry was negative for clonal B lymphocytes and aberrant expression of T lymphocytes. Bone marrow myobacterial PCR and fungal cultures were negative.
The patient’s chronic HBV infection is unlikely to be related to his presentation given his low viral load and absence of signs of hepatic dysfunction. Excluding rickettsial disease requires paired acute and convalescent serologies. O tsutsugamushi, the causative agent of the rickettsial disease scrub typhus, is endemic in Malaysia; thus, his positive O tsutsugamushi IgG may indicate past exposure. His fevers, myalgias, truncal rash, and hepatitis are consistent with scrub typhus, but he lacks the characteristic severe headache and generalized lymphadenopathy. Although eschar formation with evolution of a papular rash is common in scrub typhus, it is often absent in the variant found in Southeast Asia. Although elevated β2 microglobulin level is used as a prognostic marker in multiple myeloma and Waldenström macroglobulinemia, it can be elevated in many immune-active states. The patient likely has HLH, which is supported by the hemophagocytosis seen on bone marrow biopsy, and the hypofibrinogenemia. Potential HLH triggers include O tsutsugamushi infection or recent streptococcal pharyngitis.
A deep-punch skin biopsy of the anterior abdominal wall skin lesion was performed because of the absence of subcutaneous fat in the first biopsy specimen. The latest biopsy results showed irregular interstitial expansion of medium-size lymphocytes in a lobular panniculated pattern. The lymphocytes contained enlarged, irregularly contoured nucleoli and were positive for T-cell markers CD2 and CD3 with reduction in CD5 expression. The lymphomatous cells were of CD8+ with uniform expression of activated cytotoxic granule protein granzyme B and were positive for T-cell hemireceptor β.
Positron emission tomography (PET) CT, obtained for staging purposes, showed multiple hypermetabolic subcutaneous and cutaneous lesions over the torso and upper and lower limbs—compatible with lymphomatous infiltrates (Figure 2). Examination, pathology, and imaging findings suggested a rare neoplasm: subcutaneous panniculitis-like T-cell lymphoma (SPTCL). SPTCL was confirmed by T-cell receptor gene rearrangements studies.
HLH was diagnosed on the basis of the fevers, cytopenias, hypofibrinogenemia, elevated ferritin level, and evidence of hemophagocytosis. SPTCL was suspected as the HLH trigger.
The patient was treated with cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone. While on this regimen, he developed new skin lesions, and his ferritin level was persistently elevated. He was switched to romidepsin, a histone deacetylase inhibitor that specifically targets cutaneous T-cell lymphoma, but the lesions continued to progress. The patient then was treated with gemcitabine, dexamethasone, and cisplatin, and the rashes resolved. The most recent PET-CT showed nearly complete resolution of the subcutaneous lesions.
DISCUSSION
When residents or visitors to tropical or sub-tropical regions, those located near or between the Tropics of Cancer and Capricorn, present with fever, physicians usually first think of infectious diseases. This patient’s case is a reminder that these important first considerations should not be the last.
Generating a differential diagnosis for tropical illnesses begins with the patient’s history. Factors to be considered include location (regional disease prevalence), exposures (food/water ingestion, outdoor work/recreation, sexual contact, animal contact), and timing (temporal relationship of symptom development to possible exposure). Common tropical infections are malaria, dengue, typhoid, and emerging infections such as chikungunya, avian influenza, and Zika virus infection.1This case underscores the need to analyze diagnostic tests critically. Interpreting tests as simply positive or negative, irrespective of disease features, epidemiology, and test characteristics, can contribute to diagnostic error. For example, the patient’s positive ASO titer requires an understanding of disease features and a nuanced interpretation based on the clinical presentation. The erythematous posterior oropharynx prompted concern for postinfectious sequelae of streptococcal pharyngitis, but his illness was more severe and more prolonged than is typical of that condition. The isolated elevated O tsutsugamushi IgG titer provides an example of the role of epidemiology in test interpretation. Although a single positive value might indicate a new exposure for a visitor to an endemic region, IgG seropositivity in Singapore, where scrub typhus is endemic, likely reflects prior exposure to the organism. Diagnosing an acute scrub typhus infection in a patient in an endemic region requires PCR testing. The skin biopsy results highlight the importance of understanding test characteristics. A skin biopsy specimen must be adequate in order to draw valid and accurate conclusions. In this case, the initial skin biopsy was superficial, and the specimen inadequate, but the test was not “negative.” In the diagnostic skin biopsy, deeper tissue was sampled, and panniculitis (inflammation of subcutaneous fat), which arises in inflammatory, infectious, traumatic, enzymatic, and malignant conditions, was identified. An adequate biopsy specimen that contains subcutaneous fat is essential in making this diagnosis.2This patient eventually manifested several elements of hemophagocytic lymphohistiocytosis (HLH), a syndrome of excessive inflammation and resultant organ injury relating to abnormal immune activation and excessive inflammation. HLH results from deficient down-regulation of activated macrophages and lymphocytes.3 It was initially described in pediatric patients but is now recognized in adults, and associated with mortality as high as 50%.3 A high ferritin level (>2000 ng/mL) has 70% sensitivity and 68% specificity for pediatric HLH and should trigger consideration of HLH in any age group.4 The diagnostic criteria for HLH initially proposed in 2004 by the Histiocyte Society to identify patients for recruitment into a clinical trial included molecular testing consistent with HLH and/or 5 of 8 clinical, laboratory, or histopathologic features (Table 1).5 HScore is a more recent validated scoring system that predicts the probability of HLH (Table 2). A score above 169 signifies diagnostic sensitivity of 93% and specificity of 86%.6
The diagnosis of HLH warrants a search for its underlying cause. Common triggers are viral infections (eg, EBV), autoimmune diseases (eg, systemic lupus erythematosus), and hematologic malignancies. These triggers typically stimulate or suppress the immune system. Initial management involves treatment of the underlying trigger and, potentially, immunosuppression with high
In this case, SPTCL triggered HLH. SPTCL is a rare non-Hodgkin lymphoma characterized by painless subcutaneous nodules or indurated plaques (panniculitis-like) on the trunk or extremities, constitutional symptoms, and, in some cases, HLH.7-10 SPTCL is diagnosed by deep skin biopsy, with immunohistochemistry showing CD8-positive pathologic T cells expressing cytotoxic proteins (eg, granzyme B).9,11 SPTCL can either have an alpha/beta T-cell phenotype (SPTCL-AB) or gamma/delta T-cell phenotype (SPTCL-GD). Seventeen percent of patients with SPTCL-AB and 45% of patients with SPTCL-GD have HLH on diagnosis. Concomitant HLH is associated with decreased 5-year survival.12This patient presented with fevers and was ultimately diagnosed with HLH secondary to SPLTCL. His case is a reminder that not all diseases in the tropics are tropical diseases. In the diagnosis of a febrile illness, a broad evaluative framework and rigorous test results evaluation are essential—no matter where a patient lives or visits.
KEY TEACHING POINTS
- A febrile illness acquired in the tropics is not always attributable to a tropical infection.
- To avoid diagnostic error, weigh positive or negative test results against disease features, patient epidemiology, and test characteristics.
- HLH is characterized by fevers, cytopenias, hepatosplenomegaly, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia. In tissue specimens, hemophagocytosis may help differentiate HLH from competing conditions.
- After HLH is diagnosed, try to determine its underlying cause, which may be an infection, autoimmunity, or a malignancy (commonly, a lymphoma).
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 42-year-old Malaysian construction worker with subjective fevers of 4 days’ duration presented to an emergency department in Singapore. He reported nonproductive cough, chills without rigors, sore throat, and body aches. He denied sick contacts. Past medical history included chronic hepatitis B virus (HBV) infection. The patient was not taking any medications.
For this patient presenting acutely with subjective fevers, nonproductive cough, chills, aches, and lethargy, initial considerations include infection with a common virus (influenza virus, adenovirus, Epstein-Barr virus [EBV]), acute human immunodeficiency virus (HIV) infection, emerging infection (severe acute respiratory syndrome [SARS], Middle Eastern respiratory syndrome coronavirus [MERS-CoV] infection, avian influenza), and tropical infection (dengue, chikungunya). Also possible are bacterial infections (eg, with Salmonella typhi or Rickettsia or Mycoplasma species), parasitic infections (eg, malaria), and noninfectious illnesses (eg, autoimmune diseases, thyroiditis, acute leukemia, environmental exposures).
The patient’s temperature was 38.5°C; blood pressure, 133/73 mm Hg; heart rate, 95 beats per minute; respiratory rate, 18 breaths per minute; and oxygen saturation, 100% on ambient air. On physical examination, he appeared comfortable, and heart, lung, abdomen, skin, and extremities were normal. Laboratory test results included white blood cell (WBC) count, 4400/μL (with normal differential); hemoglobin, 16.1 g/dL; and platelet count, 207,000/μL. Serum chemistries were normal. C-reactive protein (CRP) level was 44.6 mg/L (reference range, 0.2-9.1 mg/L), and procalcitonin level was 0.13 ng/mL (reference range, <0.50 ng/mL). Chest radiograph was normal. Dengue antibodies (immunoglobulin M, immunoglobulin G [IgG]) and dengue NS1 antigen were negative. The patient was discharged with a presumptive diagnosis of viral upper respiratory tract infection.
There is no left shift characteristic of bacterial infection or lymphopenia characteristic of rickettsial disease or acute HIV infection. The serologic testing and the patient’s overall appearance make dengue unlikely. The low procalcitonin supports a nonbacterial cause of illness. CRP elevation may indicate an inflammatory process and is relatively nonspecific.
Myalgias, pharyngitis, and cough improved over several days, but fevers persisted, and a rash developed over the lower abdomen. The patient returned to the emergency department and was admitted. He denied weight loss and night sweats. He had multiple female sexual partners, including commercial sex workers, within the previous 6 months. Temperature was 38.5°C. The posterior oropharynx was slightly erythematous. There was no lymphadenopathy. Firm, mildly erythematous macules were present on the anterior abdominal wall (Figure 1). The rest of the physical examination was normal.
Laboratory testing revealed WBC count, 5800/μL (75% neutrophils, 19% lymphocytes, 3% monocytes, 2% atypical mononuclear cells); hemoglobin, 16.3 g/dL; platelet count, 185,000/μL; sodium, 131 mmol/L; potassium, 3.4 mmol/L; creatinine, 0.9 mg/dL; albumin, 3.2 g/dL; alanine aminotransferase (ALT), 99 U/L; aspartate aminotransferase (AST), 137 U/L; alkaline phosphatase (ALP), 63 U/L; and total bilirubin, 1.9 mg/dL. Prothrombin time was 11.1 seconds; partial thromboplastin time, 36.1 seconds; erythrocyte sedimentation rate, 14 mm/h; and CRP, 62.2 mg/L.
EBV, acute HIV, and cytomegalovirus infections often present with adenopathy, which is absent here. Disseminated gonococcal infection can manifest with fever, body aches, and rash, but his rash and the absence of penile discharge, migratory arthritis, and enthesitis are not characteristic. Mycoplasma infection can present with macules, urticaria, or erythema multiforme. Rickettsia illnesses typically cause vasculitis with progression to petechiae or purpura resulting from endothelial damage. Patients with secondary syphilis may have widespread macular lesions, and the accompanying syphilitic hepatitis often manifests with elevations in ALP instead of ALT and AST. The mild elevation in ALT and AST can occur with many systemic viral infections. Sweet syndrome may manifest with febrile illness and rash, but the acuity of this patient’s illness and the rapid evolution favor infection.
The patient’s fevers (35°-40°C) continued without pattern over the next 3 days. Blood and urine cultures were negative. Polymerase chain reaction (PCR) test of the nasal mucosa was negative for respiratory viruses. PCR blood tests for EBV, HIV-1, and cytomegalovirus were also negative. Antistreptolysin O (ASO) titer was 400 IU/mm (reference range, <200 IU/mm). Antinuclear antibodies were negative, and rheumatoid factor was 12.4 U/mL (reference range, <10.3 U/mL). Computed tomography (CT) of the thorax, abdomen, and pelvis was normal. Results of a biopsy of an anterior abdominal wall skin lesion showed perivascular and periadnexal lymphocytic inflammation. Amoxicillin was started for the treatment of possible group A streptococcal infection.
PCR for HIV would be positive at a high level in acute HIV. The skin biopsy is not characteristic of Sweet syndrome, which typically shows neutrophilic infiltrate without leukocytoclastic vasculitis, or of syphilis, which typically shows a plasma cell infiltrate.
The patient’s erythematous oropharynx may indicate recent streptococcal pharyngitis. The fevers, elevated ASO titer, and CRP level are consistent with acute rheumatic fever, but arthritis, carditis, and neurologic manifestations are lacking. Erythema marginatum manifests on the trunk and limbs as macules or papules with central clearing as the lesions spread outward—and differs from the patient’s rash, which is firm and restricted to the abdominal wall.
Fevers persisted through hospital day 7. The WBC count was 1100/μL (75.7% neutrophils, 22.5% lymphocytes), hemoglobin was 10.3 g/dL, and platelet count was 52,000/μL. Additional laboratory test results included ALP, 234 U/L; ALT, 250 U/L; AST, 459 U/L; lactate dehydrogenase, 2303 U/L (reference range, 222-454 U/L); and ferritin, 14,964 ng/mL (reference range, 47-452 ng/mL).
The duration of illness and negative diagnostic tests for infections increases suspicion for a noninfectious illness. Conditions commonly associated with marked hyperferritinemia include adult-onset Still disease (AOSD) and hemophagocytic lymphohistiocytosis (HLH). Of the 9 AOSD diagnostic (Yamaguchi) criteria, 5 are met in this case: fever, rash, sore throat, abnormal liver function tests, and negative rheumatologic tests. However, the patient lacks arthritis, leukocytosis, lymphadenopathy, and hepatosplenomegaly. Except for the elevated ferritin, the AOSD criteria overlap substantially with the criteria for acute rheumatic fever, and still require that infections be adequately excluded. HLH, a state of abnormal immune activation with resultant organ dysfunction, can be a primary disorder, but in adults more often is secondary to underlying infectious, autoimmune, or malignant (often lymphoma) conditions. Elevated ferritin, cytopenias, elevated ALT and AST, elevated CRP and erythrocyte sedimentation rate, and elevated lactate dehydrogenase are consistent with HLH. The HLH diagnosis can be more firmly established with the more specific findings of hypertriglyceridemia, hypofibrinogenemia, and elevated soluble CD25 level. The histopathologic finding of hemophagocytosis in the bone marrow, lymph nodes, or liver may further support the diagnosis of HLH.
Rash and fevers persisted. Hepatitis A, hepatitis C, Rickettsia IgG, Burkholderia pseudomallei (the causative organism of melioidosis), and Leptospira serologies, as well as PCR for herpes simplex virus and parvovirus, were all negative. Hepatitis B viral load was 962 IU/mL (2.98 log), hepatitis B envelope antigen was negative, and hepatitis B envelope antibody was positive. Orientia tsutsugamushi (organism responsible for scrub typhus) IgG titer was elevated at 1:128. Antiliver kidney microsomal antibodies and antineutrophil cytoplasmic antibodies were negative. Fibrinogen level was 0.69 g/L (reference range, 1.8-4.8 g/L), and beta-2 microglobulin level was 5078 ng/mL (reference range, 878-2000 ng/mL). Bone marrow biopsy results showed hypocellular marrow with suppressed myelopoiesis, few atypical lymphoid cells, and few hemophagocytes. Flow cytometry was negative for clonal B lymphocytes and aberrant expression of T lymphocytes. Bone marrow myobacterial PCR and fungal cultures were negative.
The patient’s chronic HBV infection is unlikely to be related to his presentation given his low viral load and absence of signs of hepatic dysfunction. Excluding rickettsial disease requires paired acute and convalescent serologies. O tsutsugamushi, the causative agent of the rickettsial disease scrub typhus, is endemic in Malaysia; thus, his positive O tsutsugamushi IgG may indicate past exposure. His fevers, myalgias, truncal rash, and hepatitis are consistent with scrub typhus, but he lacks the characteristic severe headache and generalized lymphadenopathy. Although eschar formation with evolution of a papular rash is common in scrub typhus, it is often absent in the variant found in Southeast Asia. Although elevated β2 microglobulin level is used as a prognostic marker in multiple myeloma and Waldenström macroglobulinemia, it can be elevated in many immune-active states. The patient likely has HLH, which is supported by the hemophagocytosis seen on bone marrow biopsy, and the hypofibrinogenemia. Potential HLH triggers include O tsutsugamushi infection or recent streptococcal pharyngitis.
A deep-punch skin biopsy of the anterior abdominal wall skin lesion was performed because of the absence of subcutaneous fat in the first biopsy specimen. The latest biopsy results showed irregular interstitial expansion of medium-size lymphocytes in a lobular panniculated pattern. The lymphocytes contained enlarged, irregularly contoured nucleoli and were positive for T-cell markers CD2 and CD3 with reduction in CD5 expression. The lymphomatous cells were of CD8+ with uniform expression of activated cytotoxic granule protein granzyme B and were positive for T-cell hemireceptor β.
Positron emission tomography (PET) CT, obtained for staging purposes, showed multiple hypermetabolic subcutaneous and cutaneous lesions over the torso and upper and lower limbs—compatible with lymphomatous infiltrates (Figure 2). Examination, pathology, and imaging findings suggested a rare neoplasm: subcutaneous panniculitis-like T-cell lymphoma (SPTCL). SPTCL was confirmed by T-cell receptor gene rearrangements studies.
HLH was diagnosed on the basis of the fevers, cytopenias, hypofibrinogenemia, elevated ferritin level, and evidence of hemophagocytosis. SPTCL was suspected as the HLH trigger.
The patient was treated with cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone. While on this regimen, he developed new skin lesions, and his ferritin level was persistently elevated. He was switched to romidepsin, a histone deacetylase inhibitor that specifically targets cutaneous T-cell lymphoma, but the lesions continued to progress. The patient then was treated with gemcitabine, dexamethasone, and cisplatin, and the rashes resolved. The most recent PET-CT showed nearly complete resolution of the subcutaneous lesions.
DISCUSSION
When residents or visitors to tropical or sub-tropical regions, those located near or between the Tropics of Cancer and Capricorn, present with fever, physicians usually first think of infectious diseases. This patient’s case is a reminder that these important first considerations should not be the last.
Generating a differential diagnosis for tropical illnesses begins with the patient’s history. Factors to be considered include location (regional disease prevalence), exposures (food/water ingestion, outdoor work/recreation, sexual contact, animal contact), and timing (temporal relationship of symptom development to possible exposure). Common tropical infections are malaria, dengue, typhoid, and emerging infections such as chikungunya, avian influenza, and Zika virus infection.1This case underscores the need to analyze diagnostic tests critically. Interpreting tests as simply positive or negative, irrespective of disease features, epidemiology, and test characteristics, can contribute to diagnostic error. For example, the patient’s positive ASO titer requires an understanding of disease features and a nuanced interpretation based on the clinical presentation. The erythematous posterior oropharynx prompted concern for postinfectious sequelae of streptococcal pharyngitis, but his illness was more severe and more prolonged than is typical of that condition. The isolated elevated O tsutsugamushi IgG titer provides an example of the role of epidemiology in test interpretation. Although a single positive value might indicate a new exposure for a visitor to an endemic region, IgG seropositivity in Singapore, where scrub typhus is endemic, likely reflects prior exposure to the organism. Diagnosing an acute scrub typhus infection in a patient in an endemic region requires PCR testing. The skin biopsy results highlight the importance of understanding test characteristics. A skin biopsy specimen must be adequate in order to draw valid and accurate conclusions. In this case, the initial skin biopsy was superficial, and the specimen inadequate, but the test was not “negative.” In the diagnostic skin biopsy, deeper tissue was sampled, and panniculitis (inflammation of subcutaneous fat), which arises in inflammatory, infectious, traumatic, enzymatic, and malignant conditions, was identified. An adequate biopsy specimen that contains subcutaneous fat is essential in making this diagnosis.2This patient eventually manifested several elements of hemophagocytic lymphohistiocytosis (HLH), a syndrome of excessive inflammation and resultant organ injury relating to abnormal immune activation and excessive inflammation. HLH results from deficient down-regulation of activated macrophages and lymphocytes.3 It was initially described in pediatric patients but is now recognized in adults, and associated with mortality as high as 50%.3 A high ferritin level (>2000 ng/mL) has 70% sensitivity and 68% specificity for pediatric HLH and should trigger consideration of HLH in any age group.4 The diagnostic criteria for HLH initially proposed in 2004 by the Histiocyte Society to identify patients for recruitment into a clinical trial included molecular testing consistent with HLH and/or 5 of 8 clinical, laboratory, or histopathologic features (Table 1).5 HScore is a more recent validated scoring system that predicts the probability of HLH (Table 2). A score above 169 signifies diagnostic sensitivity of 93% and specificity of 86%.6
The diagnosis of HLH warrants a search for its underlying cause. Common triggers are viral infections (eg, EBV), autoimmune diseases (eg, systemic lupus erythematosus), and hematologic malignancies. These triggers typically stimulate or suppress the immune system. Initial management involves treatment of the underlying trigger and, potentially, immunosuppression with high
In this case, SPTCL triggered HLH. SPTCL is a rare non-Hodgkin lymphoma characterized by painless subcutaneous nodules or indurated plaques (panniculitis-like) on the trunk or extremities, constitutional symptoms, and, in some cases, HLH.7-10 SPTCL is diagnosed by deep skin biopsy, with immunohistochemistry showing CD8-positive pathologic T cells expressing cytotoxic proteins (eg, granzyme B).9,11 SPTCL can either have an alpha/beta T-cell phenotype (SPTCL-AB) or gamma/delta T-cell phenotype (SPTCL-GD). Seventeen percent of patients with SPTCL-AB and 45% of patients with SPTCL-GD have HLH on diagnosis. Concomitant HLH is associated with decreased 5-year survival.12This patient presented with fevers and was ultimately diagnosed with HLH secondary to SPLTCL. His case is a reminder that not all diseases in the tropics are tropical diseases. In the diagnosis of a febrile illness, a broad evaluative framework and rigorous test results evaluation are essential—no matter where a patient lives or visits.
KEY TEACHING POINTS
- A febrile illness acquired in the tropics is not always attributable to a tropical infection.
- To avoid diagnostic error, weigh positive or negative test results against disease features, patient epidemiology, and test characteristics.
- HLH is characterized by fevers, cytopenias, hepatosplenomegaly, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia. In tissue specimens, hemophagocytosis may help differentiate HLH from competing conditions.
- After HLH is diagnosed, try to determine its underlying cause, which may be an infection, autoimmunity, or a malignancy (commonly, a lymphoma).
Disclosure
Nothing to report.
1. Centers for Disease Control and Prevention. Destinations [list]. http://wwwnc.cdc.gov/travel/destinations/list/. Accessed April 22, 2016.
2. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22(6):530-549. PubMed
3. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503-1516. PubMed
4. Lehmberg K, McClain KL, Janka GE, Allen CE. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2014;61(11):2101-2103. PubMed
5. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. PubMed
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. PubMed
7. Aronson IK, Worobed CM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23(4):389-402. PubMed
8. Willemze R, Jansen PM, Cerroni L, et al; EORTC Cutaneous Lymphoma Group. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111(2):838-845. PubMed
9. Kumar S, Krenacs L, Medeiros J, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol. 1998;29(4):397-403. PubMed
10. Salhany KE, Macon WR, Choi JK, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol. 1998;22(7):881-893. PubMed
11. Jaffe ES, Nicolae A, Pittaluga S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: pearls and pitfalls. Mod Pathol. 2013;26(suppl 1):S71-S87. PubMed
12. Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M; ESMO Guidelines Working Group. Primary cutaneous lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl 6):vi149-vi154. PubMed
1. Centers for Disease Control and Prevention. Destinations [list]. http://wwwnc.cdc.gov/travel/destinations/list/. Accessed April 22, 2016.
2. Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol. 2000;22(6):530-549. PubMed
3. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503-1516. PubMed
4. Lehmberg K, McClain KL, Janka GE, Allen CE. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2014;61(11):2101-2103. PubMed
5. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. PubMed
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. PubMed
7. Aronson IK, Worobed CM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23(4):389-402. PubMed
8. Willemze R, Jansen PM, Cerroni L, et al; EORTC Cutaneous Lymphoma Group. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008;111(2):838-845. PubMed
9. Kumar S, Krenacs L, Medeiros J, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol. 1998;29(4):397-403. PubMed
10. Salhany KE, Macon WR, Choi JK, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol. 1998;22(7):881-893. PubMed
11. Jaffe ES, Nicolae A, Pittaluga S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: pearls and pitfalls. Mod Pathol. 2013;26(suppl 1):S71-S87. PubMed
12. Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M; ESMO Guidelines Working Group. Primary cutaneous lymphomas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl 6):vi149-vi154. PubMed
© 2017 Society of Hospital Medicine
A shocking diagnosis
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 75-year-old man was brought by ambulance to the emergency department (ED) after the acute onset of palpitations, lightheadedness, and confusion. His medical history, provided by his wife, included osteoarthritis and remote cholecystectomy. He was not a smoker but drank 2 to 4 cans of beer daily. His medications were aspirin 162 mg daily and naproxen as needed. There was no history of bruising, diarrhea, melena, or bleeding.
Palpitations may represent an arrhythmia arising from an ischemic or alcoholic cardiomyopathy. Mental status changes usually have metabolic, infectious, structural (eg, hemorrhage, tumor), or toxic causes. Lightheadedness and confusion could occur with arrhythmia-associated cerebral hypoperfusion or a seizure. Daily alcohol use could cause confusion through acute intoxication, thiamine or B12 deficiency, repeated head trauma, or liver failure.
The patient’s systolic blood pressure (BP) was 60 mm Hg, heart rate (HR) was 120 beats per minute (bpm), and oral temperature was 98.4°F. Rousing him was difficult. There were no localizing neurologic abnormalities, and the rest of the physical examination findings were normal. Point-of-care blood glucose level was 155 mg/dL. Blood cultures were obtained and broad-spectrum antibiotics initiated. After fluid resuscitation, BP improved to 116/87 mm Hg, HR fell to 105 bpm, and the patient became alert and oriented. He denied chest pain, fever, or diaphoresis.
The patient’s improvement with intravenous (IV) fluids makes cardiogenic shock unlikely but does not exclude an underlying compensated cardiomyopathy that may be predisposing to arrhythmia. Hypotension, tachycardia, and somnolence may represent sepsis, but the near normalization of vital signs and mental status shortly after administration of IV fluids, the normal temperature, and the absence of localizing signs of infection favor withholding additional antibiotics. Other causes of hypotension are hypovolemia, medication effects, adrenal insufficiency, anaphylaxis, and autonomic insufficiency. There was no reported nausea, vomiting, diarrhea, bleeding, polyuria, or impaired oral intake to support hypovolemia, though the response to IV fluids suggests hypovolemia may still be playing a role.
White blood cell (WBC) count was 15,450/µL with a normal differential; hemoglobin level was 15.8 g/dL; and platelet count was 176,000/µL. Electrolytes, liver function tests, cardiac enzymes, and urinalysis were normal. Electrocardiogram showed sinus tachycardia with premature atrial complexes and no ST-segment abnormalities. Radiograph of the chest and computed tomography scan of the head were normal. Echocardiogram showed moderate left ventricular hypertrophy with a normal ejection fraction and no valvular abnormalities. Exercise nuclear cardiac stress test was negative for ischemia. Blood cultures were sterile. The patient quickly became asymptomatic and remained so during his 3-day hospitalization. There were no arrhythmias on telemetry. The patient was discharged with follow-up scheduled with his primary care physician.
The nonlocalizing history and physical examination findings, normal chest radiograph and urinalysis, absence of fevers, negative blood cultures, and quick recovery make infection unlikely, despite the moderate leukocytosis. Conditions that present with acute and transient hypotension and altered mental status include arrhythmias, seizures, and reactions to drugs or toxins. Given the cardiac test results, a chronic cardiomyopathy seems unlikely, but arrhythmia is still possible. Continuous outpatient monitoring is required to assess the palpitations and the frequency of the premature atrial complexes.
Two days after discharge, the patient suddenly became diaphoretic and lost consciousness while walking to the bathroom. He was taken to the ED, where his BP was 90/60 mm Hg and HR was 108 bpm. Family members reported that he had appeared flushed during the syncopal episode, showed no seizure activity, and been unconscious for 15 to 20 minutes. The patient denied chest pain, dyspnea, fever, bowel or bladder incontinence, focal weakness, slurred speech, visual changes, nausea or vomiting either before or after the episode. Physical examination revealed a tongue laceration and facial erythema; all other findings were normal. In the ED, there was an asymptomatic 7-beat run of nonsustained ventricular tachycardia, and the hypotension resolved after fluid resuscitation. The patient now reported 2 similar syncopal episodes in the past. The first occurred in a restaurant 6 years earlier, and the second occurred 3 years later, at which time he was hospitalized and no etiology was found.
The loss of consciousness is attributable to cerebral hypoperfusion. Hypotension has 3 principal categories: hypovolemic, cardiogenic, and distributive. With syncopal episodes recurring over several years, hypovolemia seems unlikely. Given the palpitations and ventricular tachycardia, it is reasonable to suspect a cardiogenic cause. Although his heart appears to be structurally normal on echocardiogram, genetic, electrophysiologic, or magnetic resonance imaging (MRI) testing will occasionally reveal an unsuspected substrate for arrhythmia.
The recurring yet self-limited nature, diaphoresis, flushing, and facial erythema suggest a non-sepsis distributive cause of hypotension. It is possible the patient is recurrently exposed to a toxin (eg, alcohol) that causes both flushing and dehydration. Flushing disorders include carcinoid syndrome, pheochromocytoma, drug reaction with eosinophilia and systemic symptoms (DRESS), and mastocytosis. Carcinoid syndrome is characterized by bronchospasm and diarrhea and, in some cases, right-sided valvulopathy, all of which are absent in this patient. Pheochromocytoma is associated with orthostasis, but patients typically are hypertensive at baseline. DRESS, which may arise from nonsteroidal anti-inflammatory drug (NSAID) or aspirin use, can cause facial erythema and swelling but is also characterized by liver, renal, and hematologic abnormalities, none of which was demonstrated. Furthermore, DRESS typically does not cause hypotension. Mastocytosis can manifest as isolated or recurrent anaphylaxis.
It is important to investigate antecedents of these syncopal episodes. If the earlier episodes were food-related—one occurred at a restaurant—then deglutition syncope (syncope precipitated by swallowing) should be considered. If an NSAID or aspirin was ingested before each episode, then medication hypersensitivity or mast cell degranulation (which can be triggered by these medications) should be further examined. Loss of consciousness lasting 20 minutes without causing any neurologic sequelae is unusual for most causes of recurrent syncope. This feature raises the possibility that a toxin or mediator might still be present in the patient’s system.
Serial cardiac enzymes and electrocardiogram were normal. A tilt-table study was negative. The cortisol response to ACTH (cosyntropin) stimulation was normal. The level of serum tryptase, drawn 2 days after syncope, was 18.4 ng/dL (normal, <11.5 ng/dL). Computed tomography scan of chest and abdomen was negative for pulmonary embolism but showed a 1.4×1.3-cm hypervascular lesion in the tail of pancreas. The following neuroendocrine tests were within normal limits: serum and urine catecholamines; urine 5-hydroxyindoleacetic acid (5-HIAA); and serum chromogranin A, insulin, serotonin, vasoactive intestinal polypeptide (VIP), and somatostatin (Table 1). The patient remained asymptomatic during his hospital stay and was discharged home with appointments for cardiology follow-up and endoscopic ultrasound-guided biopsy of the pancreatic mass.

Pheochromocytoma is unlikely with normal serum and urine catecholamine levels and normal adrenal images. The differential diagnosis for a pancreatic mass includes pancreatic carcinoma, lymphoma, cystic neoplasm, and neuroendocrine tumor. All markers of neuroendocrine excess are normal, though elevations can be episodic. The normal 5-HIAA level makes carcinoid syndrome unlikely. VIPomas are associated with flushing, but the absence of profound and protracted diarrhea makes a VIPoma unlikely.
As hypoglycemia from a pancreatic insulinoma is plausible as a cause of episodic loss of consciousness lasting 15 minutes or more, it is important to inquire if giving food or drink helped resolve previous episodes. The normal insulin level reported here is of limited value, because it is the combination of insulin and C-peptide levels at time of hypoglycemia that is diagnostic. The normal glucose level recorded during one of the earlier episodes and the hypotension argue against hypoglycemia.
The elevated tryptase level is an indicator of mast cell degranulation. Tryptase levels are transiently elevated during the initial 2 to 4 hours after an anaphylactic episode and then normalize. An elevated level many hours or days later is considered a sign of mast cell excess. Although there is no evidence of the multi-organ disease (eg, cytopenia, bone disease, hepatosplenomegaly) seen in patients with a high systemic burden of mast cells, mast cell disorders exist on a spectrum. There may be a focal excess of mast cells confined to one organ or an isolated mass.
The same day as discharge, the patient’s wife drove them to the grocery store. He remained in the car while she shopped. When she returned, she found him confused and minimally responsive with subsequent brief loss of consciousness. He was taken to an ED, where he was flushed and hypotensive (systolic BP, 60 mm Hg) and tachycardic. Other examination findings were normal. After fluid resuscitation he became alert and oriented. WBC count was 20,850/μL with 89% neutrophils, hemoglobin level was 14.6 g/dL, and platelet count was 168,000/μL. Serum lactate level was 3.7 mmol/L (normal, <2.3 mmol/L). Chest radiograph was normal. He was treated with broad-spectrum antibiotic therapy and admitted to the hospital. Blood and urine cultures were sterile. Fine-needle aspiration of the pancreatic mass demonstrated nonspecific inflammation. Four days after admission (3 days after pancreatic mass biopsy) the patient developed palpitations, felt unwell, and had marked flushing of the face and trunk, with concomitant BP of 90/50 mm Hg and HR of 140 bpm.
The salient features of this case are recurrent hypotension, tachycardia, and flushing. Autonomic insufficiency, to which elderly patients are prone, causes hemodynamic perturbations but rarely flushing. The patient does not have diabetes mellitus, Parkinson disease, or another condition that puts him at risk for dysautonomia. Pancreatic neuroendocrine tumors secrete mediators that lead to vasodilation and hypotension but are unlikely given the clinical and biochemical data.
The patient’s symptoms are consistent with anaphylaxis, though prototypical immunoglobulin E (IgE)–mediated anaphylaxis is usually accompanied by urticaria, angioedema, and wheezing, which have been absent during his presentations. There are no clear food, pharmacologic, or environmental precipitants.
Recurrent anaphylaxis can be a manifestation of mast cell excess (eg, cutaneous or systemic mastocytosis). A markedly elevated tryptase level during an anaphylactic episode is consistent with mastocytosis or IgE-mediated anaphylaxis. An elevated baseline tryptase level days after an anaphylactic episode signals increased mast cell burden. There may be a reservoir of mast cells in the bone marrow. Alternatively, the hypervascular pancreatic mass may be a mastocytoma or a mast cell sarcoma (missed because of inadequate sampling or staining).
The lactic acidosis likely reflects global tissue hypoperfusion from vasodilatory hypotension. The leukocytosis may reflect WBC mobilization secondary to endogenous corticosteroids and catecholamines in response to hypotension or may be a direct response to the release of mast cell–derived mediators of inflammation.
The patient was treated with diphenhydramine and ranitidine. Serum tryptase level was 46.8 ng/mL (normal, <11.5 ng/mL), and 24-hour urine histamine level was 95 µ g/dL (normal, <60 µ g/dL). Bone marrow biopsy results showed multifocal dense infiltrative aggregates of mast cells (>15 cells/aggregate), which were confirmed by CD117 (Kit) and tryptase positivity (Figure). Mutation analysis for Kit Asp816Val, which is present in 80% to 90% of patients with mastocytosis, was positive. He fulfilled the 2008 World Health Organization criteria for systemic mastocytosis (Table 2). Prednisone, histamine inhibitors, and montelukast were prescribed. Six months later, magnetic resonance imaging of the abdomen showed no change in the pancreatic mass, which was now characterized as a possible splenule. The patient had no additional episodes of flushing or syncope over 2 years.


DISCUSSION
Cardiovascular collapse (hypotension, tachycardia, syncope) in an elderly patient prompts clinicians to focus on life-threatening conditions, such as acute coronary syndrome, pulmonary embolus, arrhythmia, and sepsis. Each of these diagnoses was considered early in the course of this patient’s presentations, but each was deemed unlikely as it became apparent that the episodes were self-limited and recurrent over years. Incorporating flushing into the diagnostic problem representation allowed the clinicians to focus on a subset of causes of hypotension.
Flushing disorders may be classified by whether they are mediated by the autonomic nervous system (wet flushes, because they are usually accompanied by diaphoresis) or by exogenous or endogenous vasoactive substances (dry flushes).1 Autonomic nervous system flushing is triggered by emotions, fever, exercise, perimenopause (hot flashes), and neurologic conditions (eg, Parkinson disease, spinal cord injury, multiple sclerosis). Vasoactive flushing precipitants include drugs (eg, niacin); alcohol (secondary to cutaneous vasodilation, or acetaldehyde particularly in people with insufficient acetaldehyde dehydrogenase activity)2; foods that contain capsaicin, tyramine, sulfites, or histamine (eg, eating improperly handled fish can cause scombroid poisoning); and anaphylaxis. Rare causes of vasoactive flushing include carcinoid syndrome, pheochromocytoma, medullary thyroid carcinoma, VIPoma, and mastocytosis.2
Mastocytosis is a rare clonal disorder characterized by the accumulation of abnormal mast cells in the skin (cutaneous mastocytosis), in multiple organs (systemic mastocytosis), or in a solid tumor (mastocytoma). Urticaria pigmentosa is the most common form of cutaneous mastocytosis; it is seen more often in children than in adults and typically is associated with a maculopapular rash and dermatographism. Systemic mastocytosis is the most common form of the disorder in adults.3 Symptoms are related to mast cell infiltration or mast cell mediator–related effects, which range from itching, flushing, and diarrhea to hypotension and anaphylaxis. Other manifestations are fatigue, urticaria pigmentosa, osteoporosis, hepatosplenomegaly, bone pain, cytopenias, and lymphadenopathy.4
Systemic mastocytosis can occur at any age and should be considered in patients with recurrent unexplained flushing, syncope, or hypotension. Eighty percent to 90% of patients with systemic mastocytosis have a mutation in Kit,5 a transmembrane tyrosine kinase that is the receptor for stem cell factor. The Asp816Val mutation leads to increased proliferation and reduced apoptosis of mast cells.3,6,7 Proposed diagnostic algorithms8-11 involve measurement of serum tryptase levels and examination of bone marrow. Bone marrow biopsy and testing for the Asp816Val
The primary goals of treatment are managing mast cell–mediated symptoms and, in advanced cases, achieving cytoreduction. Alcohol can trigger mast cell degranulation in indolent systemic mastocytosis and should be avoided. Mast cell–mediated symptoms are managed with histamine blockers, leukotriene antagonists, and mast cell stabilizers.12 Targeted therapy with tyrosine kinase inhibitors (eg, imatinib) in patients with transmembrane Kit mutation (eg, Phe522Cys, Lys509Ile) associated with systemic mastocytosis has had promising results.13,14 However, this patient’s Asp816Val mutation is in the Kit catalytic domain, not the transmembrane region, and therefore would not be expected to respond to imatinib. A recent open-label trial of the multikinase inhibitor midostaurin demonstrated resolution of organ damage, reduced bone marrow burden, and lowered serum tryptase levels in patients with advanced systemic mastocytosis.15 Interferon, cladribine, and high-dose corticosteroids are prescribed in patients for whom other therapies have been ineffective.8
The differential diagnosis is broad for both hypotension and for flushing, but the differential diagnosis for recurrent hypotension and flushing is limited. Recognizing that flushing was an essential feature of this patient’s hypotensive condition, and not an epiphenomenon of syncope, allowed the clinicians to focus on the overlap and make a shocking diagnosis.
Acknowledgment
The authors thank David Bosler, MD (Cleveland Clinic) for interpreting the pathology image.
Disclosure
Nothing to report.
1. Wilkin JK. The red face: flushing disorders. Clin Dermatol. 1993;11(2):211-223. PubMed
2. Izikson L, English JC 3rd, Zirwas MJ. The flushing patient: differential diagnosis, workup, and treatment. J Am Acad Dermatol. 2006;55(2):193-208. PubMed
3. Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435-453. PubMed
4. Hermans MA, Rietveld MJ, van Laar JA, et al. Systemic mastocytosis: a cohort study on clinical characteristics of 136 patients in a large tertiary centre. Eur J Intern Med. 2016;30:25-30. PubMed
5. Kristensen T, Vestergaard H, Bindslev-Jensen C, Møller MB, Broesby-Olsen S; Mastocytosis Centre, Odense University Hospital (MastOUH). Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493-498. PubMed
6. Verstovsek S. Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. Eur J Haematol. 2013;90(2):89-98. PubMed
7. Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7):2366-2372. PubMed
8. Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90(3):250-262. PubMed
9. Valent P, Aberer E, Beham-Schmid C, et al. Guidelines and diagnostic algorithm for patients with suspected systemic mastocytosis: a proposal of the Austrian Competence Network (AUCNM). Am J Blood Res. 2013;3(2):174-180. PubMed
10. Valent P, Escribano L, Broesby-Olsen S, et al; European Competence Network on Mastocytosis. Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. Allergy. 2014;69(10):1267-1274. PubMed
11. Akin C, Soto D, Brittain E, et al. Tryptase haplotype in mastocytosis: relationship to disease variant and diagnostic utility of total tryptase levels. Clin Immunol. 2007;123(3):268-271. PubMed
12. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(19):1885-1886. PubMed
13. Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222-3225. PubMed
14. Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30(4):373-378. PubMed
15. Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530-2541. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 75-year-old man was brought by ambulance to the emergency department (ED) after the acute onset of palpitations, lightheadedness, and confusion. His medical history, provided by his wife, included osteoarthritis and remote cholecystectomy. He was not a smoker but drank 2 to 4 cans of beer daily. His medications were aspirin 162 mg daily and naproxen as needed. There was no history of bruising, diarrhea, melena, or bleeding.
Palpitations may represent an arrhythmia arising from an ischemic or alcoholic cardiomyopathy. Mental status changes usually have metabolic, infectious, structural (eg, hemorrhage, tumor), or toxic causes. Lightheadedness and confusion could occur with arrhythmia-associated cerebral hypoperfusion or a seizure. Daily alcohol use could cause confusion through acute intoxication, thiamine or B12 deficiency, repeated head trauma, or liver failure.
The patient’s systolic blood pressure (BP) was 60 mm Hg, heart rate (HR) was 120 beats per minute (bpm), and oral temperature was 98.4°F. Rousing him was difficult. There were no localizing neurologic abnormalities, and the rest of the physical examination findings were normal. Point-of-care blood glucose level was 155 mg/dL. Blood cultures were obtained and broad-spectrum antibiotics initiated. After fluid resuscitation, BP improved to 116/87 mm Hg, HR fell to 105 bpm, and the patient became alert and oriented. He denied chest pain, fever, or diaphoresis.
The patient’s improvement with intravenous (IV) fluids makes cardiogenic shock unlikely but does not exclude an underlying compensated cardiomyopathy that may be predisposing to arrhythmia. Hypotension, tachycardia, and somnolence may represent sepsis, but the near normalization of vital signs and mental status shortly after administration of IV fluids, the normal temperature, and the absence of localizing signs of infection favor withholding additional antibiotics. Other causes of hypotension are hypovolemia, medication effects, adrenal insufficiency, anaphylaxis, and autonomic insufficiency. There was no reported nausea, vomiting, diarrhea, bleeding, polyuria, or impaired oral intake to support hypovolemia, though the response to IV fluids suggests hypovolemia may still be playing a role.
White blood cell (WBC) count was 15,450/µL with a normal differential; hemoglobin level was 15.8 g/dL; and platelet count was 176,000/µL. Electrolytes, liver function tests, cardiac enzymes, and urinalysis were normal. Electrocardiogram showed sinus tachycardia with premature atrial complexes and no ST-segment abnormalities. Radiograph of the chest and computed tomography scan of the head were normal. Echocardiogram showed moderate left ventricular hypertrophy with a normal ejection fraction and no valvular abnormalities. Exercise nuclear cardiac stress test was negative for ischemia. Blood cultures were sterile. The patient quickly became asymptomatic and remained so during his 3-day hospitalization. There were no arrhythmias on telemetry. The patient was discharged with follow-up scheduled with his primary care physician.
The nonlocalizing history and physical examination findings, normal chest radiograph and urinalysis, absence of fevers, negative blood cultures, and quick recovery make infection unlikely, despite the moderate leukocytosis. Conditions that present with acute and transient hypotension and altered mental status include arrhythmias, seizures, and reactions to drugs or toxins. Given the cardiac test results, a chronic cardiomyopathy seems unlikely, but arrhythmia is still possible. Continuous outpatient monitoring is required to assess the palpitations and the frequency of the premature atrial complexes.
Two days after discharge, the patient suddenly became diaphoretic and lost consciousness while walking to the bathroom. He was taken to the ED, where his BP was 90/60 mm Hg and HR was 108 bpm. Family members reported that he had appeared flushed during the syncopal episode, showed no seizure activity, and been unconscious for 15 to 20 minutes. The patient denied chest pain, dyspnea, fever, bowel or bladder incontinence, focal weakness, slurred speech, visual changes, nausea or vomiting either before or after the episode. Physical examination revealed a tongue laceration and facial erythema; all other findings were normal. In the ED, there was an asymptomatic 7-beat run of nonsustained ventricular tachycardia, and the hypotension resolved after fluid resuscitation. The patient now reported 2 similar syncopal episodes in the past. The first occurred in a restaurant 6 years earlier, and the second occurred 3 years later, at which time he was hospitalized and no etiology was found.
The loss of consciousness is attributable to cerebral hypoperfusion. Hypotension has 3 principal categories: hypovolemic, cardiogenic, and distributive. With syncopal episodes recurring over several years, hypovolemia seems unlikely. Given the palpitations and ventricular tachycardia, it is reasonable to suspect a cardiogenic cause. Although his heart appears to be structurally normal on echocardiogram, genetic, electrophysiologic, or magnetic resonance imaging (MRI) testing will occasionally reveal an unsuspected substrate for arrhythmia.
The recurring yet self-limited nature, diaphoresis, flushing, and facial erythema suggest a non-sepsis distributive cause of hypotension. It is possible the patient is recurrently exposed to a toxin (eg, alcohol) that causes both flushing and dehydration. Flushing disorders include carcinoid syndrome, pheochromocytoma, drug reaction with eosinophilia and systemic symptoms (DRESS), and mastocytosis. Carcinoid syndrome is characterized by bronchospasm and diarrhea and, in some cases, right-sided valvulopathy, all of which are absent in this patient. Pheochromocytoma is associated with orthostasis, but patients typically are hypertensive at baseline. DRESS, which may arise from nonsteroidal anti-inflammatory drug (NSAID) or aspirin use, can cause facial erythema and swelling but is also characterized by liver, renal, and hematologic abnormalities, none of which was demonstrated. Furthermore, DRESS typically does not cause hypotension. Mastocytosis can manifest as isolated or recurrent anaphylaxis.
It is important to investigate antecedents of these syncopal episodes. If the earlier episodes were food-related—one occurred at a restaurant—then deglutition syncope (syncope precipitated by swallowing) should be considered. If an NSAID or aspirin was ingested before each episode, then medication hypersensitivity or mast cell degranulation (which can be triggered by these medications) should be further examined. Loss of consciousness lasting 20 minutes without causing any neurologic sequelae is unusual for most causes of recurrent syncope. This feature raises the possibility that a toxin or mediator might still be present in the patient’s system.
Serial cardiac enzymes and electrocardiogram were normal. A tilt-table study was negative. The cortisol response to ACTH (cosyntropin) stimulation was normal. The level of serum tryptase, drawn 2 days after syncope, was 18.4 ng/dL (normal, <11.5 ng/dL). Computed tomography scan of chest and abdomen was negative for pulmonary embolism but showed a 1.4×1.3-cm hypervascular lesion in the tail of pancreas. The following neuroendocrine tests were within normal limits: serum and urine catecholamines; urine 5-hydroxyindoleacetic acid (5-HIAA); and serum chromogranin A, insulin, serotonin, vasoactive intestinal polypeptide (VIP), and somatostatin (Table 1). The patient remained asymptomatic during his hospital stay and was discharged home with appointments for cardiology follow-up and endoscopic ultrasound-guided biopsy of the pancreatic mass.
Pheochromocytoma is unlikely with normal serum and urine catecholamine levels and normal adrenal images. The differential diagnosis for a pancreatic mass includes pancreatic carcinoma, lymphoma, cystic neoplasm, and neuroendocrine tumor. All markers of neuroendocrine excess are normal, though elevations can be episodic. The normal 5-HIAA level makes carcinoid syndrome unlikely. VIPomas are associated with flushing, but the absence of profound and protracted diarrhea makes a VIPoma unlikely.
As hypoglycemia from a pancreatic insulinoma is plausible as a cause of episodic loss of consciousness lasting 15 minutes or more, it is important to inquire if giving food or drink helped resolve previous episodes. The normal insulin level reported here is of limited value, because it is the combination of insulin and C-peptide levels at time of hypoglycemia that is diagnostic. The normal glucose level recorded during one of the earlier episodes and the hypotension argue against hypoglycemia.
The elevated tryptase level is an indicator of mast cell degranulation. Tryptase levels are transiently elevated during the initial 2 to 4 hours after an anaphylactic episode and then normalize. An elevated level many hours or days later is considered a sign of mast cell excess. Although there is no evidence of the multi-organ disease (eg, cytopenia, bone disease, hepatosplenomegaly) seen in patients with a high systemic burden of mast cells, mast cell disorders exist on a spectrum. There may be a focal excess of mast cells confined to one organ or an isolated mass.
The same day as discharge, the patient’s wife drove them to the grocery store. He remained in the car while she shopped. When she returned, she found him confused and minimally responsive with subsequent brief loss of consciousness. He was taken to an ED, where he was flushed and hypotensive (systolic BP, 60 mm Hg) and tachycardic. Other examination findings were normal. After fluid resuscitation he became alert and oriented. WBC count was 20,850/μL with 89% neutrophils, hemoglobin level was 14.6 g/dL, and platelet count was 168,000/μL. Serum lactate level was 3.7 mmol/L (normal, <2.3 mmol/L). Chest radiograph was normal. He was treated with broad-spectrum antibiotic therapy and admitted to the hospital. Blood and urine cultures were sterile. Fine-needle aspiration of the pancreatic mass demonstrated nonspecific inflammation. Four days after admission (3 days after pancreatic mass biopsy) the patient developed palpitations, felt unwell, and had marked flushing of the face and trunk, with concomitant BP of 90/50 mm Hg and HR of 140 bpm.
The salient features of this case are recurrent hypotension, tachycardia, and flushing. Autonomic insufficiency, to which elderly patients are prone, causes hemodynamic perturbations but rarely flushing. The patient does not have diabetes mellitus, Parkinson disease, or another condition that puts him at risk for dysautonomia. Pancreatic neuroendocrine tumors secrete mediators that lead to vasodilation and hypotension but are unlikely given the clinical and biochemical data.
The patient’s symptoms are consistent with anaphylaxis, though prototypical immunoglobulin E (IgE)–mediated anaphylaxis is usually accompanied by urticaria, angioedema, and wheezing, which have been absent during his presentations. There are no clear food, pharmacologic, or environmental precipitants.
Recurrent anaphylaxis can be a manifestation of mast cell excess (eg, cutaneous or systemic mastocytosis). A markedly elevated tryptase level during an anaphylactic episode is consistent with mastocytosis or IgE-mediated anaphylaxis. An elevated baseline tryptase level days after an anaphylactic episode signals increased mast cell burden. There may be a reservoir of mast cells in the bone marrow. Alternatively, the hypervascular pancreatic mass may be a mastocytoma or a mast cell sarcoma (missed because of inadequate sampling or staining).
The lactic acidosis likely reflects global tissue hypoperfusion from vasodilatory hypotension. The leukocytosis may reflect WBC mobilization secondary to endogenous corticosteroids and catecholamines in response to hypotension or may be a direct response to the release of mast cell–derived mediators of inflammation.
The patient was treated with diphenhydramine and ranitidine. Serum tryptase level was 46.8 ng/mL (normal, <11.5 ng/mL), and 24-hour urine histamine level was 95 µ g/dL (normal, <60 µ g/dL). Bone marrow biopsy results showed multifocal dense infiltrative aggregates of mast cells (>15 cells/aggregate), which were confirmed by CD117 (Kit) and tryptase positivity (Figure). Mutation analysis for Kit Asp816Val, which is present in 80% to 90% of patients with mastocytosis, was positive. He fulfilled the 2008 World Health Organization criteria for systemic mastocytosis (Table 2). Prednisone, histamine inhibitors, and montelukast were prescribed. Six months later, magnetic resonance imaging of the abdomen showed no change in the pancreatic mass, which was now characterized as a possible splenule. The patient had no additional episodes of flushing or syncope over 2 years.
DISCUSSION
Cardiovascular collapse (hypotension, tachycardia, syncope) in an elderly patient prompts clinicians to focus on life-threatening conditions, such as acute coronary syndrome, pulmonary embolus, arrhythmia, and sepsis. Each of these diagnoses was considered early in the course of this patient’s presentations, but each was deemed unlikely as it became apparent that the episodes were self-limited and recurrent over years. Incorporating flushing into the diagnostic problem representation allowed the clinicians to focus on a subset of causes of hypotension.
Flushing disorders may be classified by whether they are mediated by the autonomic nervous system (wet flushes, because they are usually accompanied by diaphoresis) or by exogenous or endogenous vasoactive substances (dry flushes).1 Autonomic nervous system flushing is triggered by emotions, fever, exercise, perimenopause (hot flashes), and neurologic conditions (eg, Parkinson disease, spinal cord injury, multiple sclerosis). Vasoactive flushing precipitants include drugs (eg, niacin); alcohol (secondary to cutaneous vasodilation, or acetaldehyde particularly in people with insufficient acetaldehyde dehydrogenase activity)2; foods that contain capsaicin, tyramine, sulfites, or histamine (eg, eating improperly handled fish can cause scombroid poisoning); and anaphylaxis. Rare causes of vasoactive flushing include carcinoid syndrome, pheochromocytoma, medullary thyroid carcinoma, VIPoma, and mastocytosis.2
Mastocytosis is a rare clonal disorder characterized by the accumulation of abnormal mast cells in the skin (cutaneous mastocytosis), in multiple organs (systemic mastocytosis), or in a solid tumor (mastocytoma). Urticaria pigmentosa is the most common form of cutaneous mastocytosis; it is seen more often in children than in adults and typically is associated with a maculopapular rash and dermatographism. Systemic mastocytosis is the most common form of the disorder in adults.3 Symptoms are related to mast cell infiltration or mast cell mediator–related effects, which range from itching, flushing, and diarrhea to hypotension and anaphylaxis. Other manifestations are fatigue, urticaria pigmentosa, osteoporosis, hepatosplenomegaly, bone pain, cytopenias, and lymphadenopathy.4
Systemic mastocytosis can occur at any age and should be considered in patients with recurrent unexplained flushing, syncope, or hypotension. Eighty percent to 90% of patients with systemic mastocytosis have a mutation in Kit,5 a transmembrane tyrosine kinase that is the receptor for stem cell factor. The Asp816Val mutation leads to increased proliferation and reduced apoptosis of mast cells.3,6,7 Proposed diagnostic algorithms8-11 involve measurement of serum tryptase levels and examination of bone marrow. Bone marrow biopsy and testing for the Asp816Val
The primary goals of treatment are managing mast cell–mediated symptoms and, in advanced cases, achieving cytoreduction. Alcohol can trigger mast cell degranulation in indolent systemic mastocytosis and should be avoided. Mast cell–mediated symptoms are managed with histamine blockers, leukotriene antagonists, and mast cell stabilizers.12 Targeted therapy with tyrosine kinase inhibitors (eg, imatinib) in patients with transmembrane Kit mutation (eg, Phe522Cys, Lys509Ile) associated with systemic mastocytosis has had promising results.13,14 However, this patient’s Asp816Val mutation is in the Kit catalytic domain, not the transmembrane region, and therefore would not be expected to respond to imatinib. A recent open-label trial of the multikinase inhibitor midostaurin demonstrated resolution of organ damage, reduced bone marrow burden, and lowered serum tryptase levels in patients with advanced systemic mastocytosis.15 Interferon, cladribine, and high-dose corticosteroids are prescribed in patients for whom other therapies have been ineffective.8
The differential diagnosis is broad for both hypotension and for flushing, but the differential diagnosis for recurrent hypotension and flushing is limited. Recognizing that flushing was an essential feature of this patient’s hypotensive condition, and not an epiphenomenon of syncope, allowed the clinicians to focus on the overlap and make a shocking diagnosis.
Acknowledgment
The authors thank David Bosler, MD (Cleveland Clinic) for interpreting the pathology image.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 75-year-old man was brought by ambulance to the emergency department (ED) after the acute onset of palpitations, lightheadedness, and confusion. His medical history, provided by his wife, included osteoarthritis and remote cholecystectomy. He was not a smoker but drank 2 to 4 cans of beer daily. His medications were aspirin 162 mg daily and naproxen as needed. There was no history of bruising, diarrhea, melena, or bleeding.
Palpitations may represent an arrhythmia arising from an ischemic or alcoholic cardiomyopathy. Mental status changes usually have metabolic, infectious, structural (eg, hemorrhage, tumor), or toxic causes. Lightheadedness and confusion could occur with arrhythmia-associated cerebral hypoperfusion or a seizure. Daily alcohol use could cause confusion through acute intoxication, thiamine or B12 deficiency, repeated head trauma, or liver failure.
The patient’s systolic blood pressure (BP) was 60 mm Hg, heart rate (HR) was 120 beats per minute (bpm), and oral temperature was 98.4°F. Rousing him was difficult. There were no localizing neurologic abnormalities, and the rest of the physical examination findings were normal. Point-of-care blood glucose level was 155 mg/dL. Blood cultures were obtained and broad-spectrum antibiotics initiated. After fluid resuscitation, BP improved to 116/87 mm Hg, HR fell to 105 bpm, and the patient became alert and oriented. He denied chest pain, fever, or diaphoresis.
The patient’s improvement with intravenous (IV) fluids makes cardiogenic shock unlikely but does not exclude an underlying compensated cardiomyopathy that may be predisposing to arrhythmia. Hypotension, tachycardia, and somnolence may represent sepsis, but the near normalization of vital signs and mental status shortly after administration of IV fluids, the normal temperature, and the absence of localizing signs of infection favor withholding additional antibiotics. Other causes of hypotension are hypovolemia, medication effects, adrenal insufficiency, anaphylaxis, and autonomic insufficiency. There was no reported nausea, vomiting, diarrhea, bleeding, polyuria, or impaired oral intake to support hypovolemia, though the response to IV fluids suggests hypovolemia may still be playing a role.
White blood cell (WBC) count was 15,450/µL with a normal differential; hemoglobin level was 15.8 g/dL; and platelet count was 176,000/µL. Electrolytes, liver function tests, cardiac enzymes, and urinalysis were normal. Electrocardiogram showed sinus tachycardia with premature atrial complexes and no ST-segment abnormalities. Radiograph of the chest and computed tomography scan of the head were normal. Echocardiogram showed moderate left ventricular hypertrophy with a normal ejection fraction and no valvular abnormalities. Exercise nuclear cardiac stress test was negative for ischemia. Blood cultures were sterile. The patient quickly became asymptomatic and remained so during his 3-day hospitalization. There were no arrhythmias on telemetry. The patient was discharged with follow-up scheduled with his primary care physician.
The nonlocalizing history and physical examination findings, normal chest radiograph and urinalysis, absence of fevers, negative blood cultures, and quick recovery make infection unlikely, despite the moderate leukocytosis. Conditions that present with acute and transient hypotension and altered mental status include arrhythmias, seizures, and reactions to drugs or toxins. Given the cardiac test results, a chronic cardiomyopathy seems unlikely, but arrhythmia is still possible. Continuous outpatient monitoring is required to assess the palpitations and the frequency of the premature atrial complexes.
Two days after discharge, the patient suddenly became diaphoretic and lost consciousness while walking to the bathroom. He was taken to the ED, where his BP was 90/60 mm Hg and HR was 108 bpm. Family members reported that he had appeared flushed during the syncopal episode, showed no seizure activity, and been unconscious for 15 to 20 minutes. The patient denied chest pain, dyspnea, fever, bowel or bladder incontinence, focal weakness, slurred speech, visual changes, nausea or vomiting either before or after the episode. Physical examination revealed a tongue laceration and facial erythema; all other findings were normal. In the ED, there was an asymptomatic 7-beat run of nonsustained ventricular tachycardia, and the hypotension resolved after fluid resuscitation. The patient now reported 2 similar syncopal episodes in the past. The first occurred in a restaurant 6 years earlier, and the second occurred 3 years later, at which time he was hospitalized and no etiology was found.
The loss of consciousness is attributable to cerebral hypoperfusion. Hypotension has 3 principal categories: hypovolemic, cardiogenic, and distributive. With syncopal episodes recurring over several years, hypovolemia seems unlikely. Given the palpitations and ventricular tachycardia, it is reasonable to suspect a cardiogenic cause. Although his heart appears to be structurally normal on echocardiogram, genetic, electrophysiologic, or magnetic resonance imaging (MRI) testing will occasionally reveal an unsuspected substrate for arrhythmia.
The recurring yet self-limited nature, diaphoresis, flushing, and facial erythema suggest a non-sepsis distributive cause of hypotension. It is possible the patient is recurrently exposed to a toxin (eg, alcohol) that causes both flushing and dehydration. Flushing disorders include carcinoid syndrome, pheochromocytoma, drug reaction with eosinophilia and systemic symptoms (DRESS), and mastocytosis. Carcinoid syndrome is characterized by bronchospasm and diarrhea and, in some cases, right-sided valvulopathy, all of which are absent in this patient. Pheochromocytoma is associated with orthostasis, but patients typically are hypertensive at baseline. DRESS, which may arise from nonsteroidal anti-inflammatory drug (NSAID) or aspirin use, can cause facial erythema and swelling but is also characterized by liver, renal, and hematologic abnormalities, none of which was demonstrated. Furthermore, DRESS typically does not cause hypotension. Mastocytosis can manifest as isolated or recurrent anaphylaxis.
It is important to investigate antecedents of these syncopal episodes. If the earlier episodes were food-related—one occurred at a restaurant—then deglutition syncope (syncope precipitated by swallowing) should be considered. If an NSAID or aspirin was ingested before each episode, then medication hypersensitivity or mast cell degranulation (which can be triggered by these medications) should be further examined. Loss of consciousness lasting 20 minutes without causing any neurologic sequelae is unusual for most causes of recurrent syncope. This feature raises the possibility that a toxin or mediator might still be present in the patient’s system.
Serial cardiac enzymes and electrocardiogram were normal. A tilt-table study was negative. The cortisol response to ACTH (cosyntropin) stimulation was normal. The level of serum tryptase, drawn 2 days after syncope, was 18.4 ng/dL (normal, <11.5 ng/dL). Computed tomography scan of chest and abdomen was negative for pulmonary embolism but showed a 1.4×1.3-cm hypervascular lesion in the tail of pancreas. The following neuroendocrine tests were within normal limits: serum and urine catecholamines; urine 5-hydroxyindoleacetic acid (5-HIAA); and serum chromogranin A, insulin, serotonin, vasoactive intestinal polypeptide (VIP), and somatostatin (Table 1). The patient remained asymptomatic during his hospital stay and was discharged home with appointments for cardiology follow-up and endoscopic ultrasound-guided biopsy of the pancreatic mass.
Pheochromocytoma is unlikely with normal serum and urine catecholamine levels and normal adrenal images. The differential diagnosis for a pancreatic mass includes pancreatic carcinoma, lymphoma, cystic neoplasm, and neuroendocrine tumor. All markers of neuroendocrine excess are normal, though elevations can be episodic. The normal 5-HIAA level makes carcinoid syndrome unlikely. VIPomas are associated with flushing, but the absence of profound and protracted diarrhea makes a VIPoma unlikely.
As hypoglycemia from a pancreatic insulinoma is plausible as a cause of episodic loss of consciousness lasting 15 minutes or more, it is important to inquire if giving food or drink helped resolve previous episodes. The normal insulin level reported here is of limited value, because it is the combination of insulin and C-peptide levels at time of hypoglycemia that is diagnostic. The normal glucose level recorded during one of the earlier episodes and the hypotension argue against hypoglycemia.
The elevated tryptase level is an indicator of mast cell degranulation. Tryptase levels are transiently elevated during the initial 2 to 4 hours after an anaphylactic episode and then normalize. An elevated level many hours or days later is considered a sign of mast cell excess. Although there is no evidence of the multi-organ disease (eg, cytopenia, bone disease, hepatosplenomegaly) seen in patients with a high systemic burden of mast cells, mast cell disorders exist on a spectrum. There may be a focal excess of mast cells confined to one organ or an isolated mass.
The same day as discharge, the patient’s wife drove them to the grocery store. He remained in the car while she shopped. When she returned, she found him confused and minimally responsive with subsequent brief loss of consciousness. He was taken to an ED, where he was flushed and hypotensive (systolic BP, 60 mm Hg) and tachycardic. Other examination findings were normal. After fluid resuscitation he became alert and oriented. WBC count was 20,850/μL with 89% neutrophils, hemoglobin level was 14.6 g/dL, and platelet count was 168,000/μL. Serum lactate level was 3.7 mmol/L (normal, <2.3 mmol/L). Chest radiograph was normal. He was treated with broad-spectrum antibiotic therapy and admitted to the hospital. Blood and urine cultures were sterile. Fine-needle aspiration of the pancreatic mass demonstrated nonspecific inflammation. Four days after admission (3 days after pancreatic mass biopsy) the patient developed palpitations, felt unwell, and had marked flushing of the face and trunk, with concomitant BP of 90/50 mm Hg and HR of 140 bpm.
The salient features of this case are recurrent hypotension, tachycardia, and flushing. Autonomic insufficiency, to which elderly patients are prone, causes hemodynamic perturbations but rarely flushing. The patient does not have diabetes mellitus, Parkinson disease, or another condition that puts him at risk for dysautonomia. Pancreatic neuroendocrine tumors secrete mediators that lead to vasodilation and hypotension but are unlikely given the clinical and biochemical data.
The patient’s symptoms are consistent with anaphylaxis, though prototypical immunoglobulin E (IgE)–mediated anaphylaxis is usually accompanied by urticaria, angioedema, and wheezing, which have been absent during his presentations. There are no clear food, pharmacologic, or environmental precipitants.
Recurrent anaphylaxis can be a manifestation of mast cell excess (eg, cutaneous or systemic mastocytosis). A markedly elevated tryptase level during an anaphylactic episode is consistent with mastocytosis or IgE-mediated anaphylaxis. An elevated baseline tryptase level days after an anaphylactic episode signals increased mast cell burden. There may be a reservoir of mast cells in the bone marrow. Alternatively, the hypervascular pancreatic mass may be a mastocytoma or a mast cell sarcoma (missed because of inadequate sampling or staining).
The lactic acidosis likely reflects global tissue hypoperfusion from vasodilatory hypotension. The leukocytosis may reflect WBC mobilization secondary to endogenous corticosteroids and catecholamines in response to hypotension or may be a direct response to the release of mast cell–derived mediators of inflammation.
The patient was treated with diphenhydramine and ranitidine. Serum tryptase level was 46.8 ng/mL (normal, <11.5 ng/mL), and 24-hour urine histamine level was 95 µ g/dL (normal, <60 µ g/dL). Bone marrow biopsy results showed multifocal dense infiltrative aggregates of mast cells (>15 cells/aggregate), which were confirmed by CD117 (Kit) and tryptase positivity (Figure). Mutation analysis for Kit Asp816Val, which is present in 80% to 90% of patients with mastocytosis, was positive. He fulfilled the 2008 World Health Organization criteria for systemic mastocytosis (Table 2). Prednisone, histamine inhibitors, and montelukast were prescribed. Six months later, magnetic resonance imaging of the abdomen showed no change in the pancreatic mass, which was now characterized as a possible splenule. The patient had no additional episodes of flushing or syncope over 2 years.
DISCUSSION
Cardiovascular collapse (hypotension, tachycardia, syncope) in an elderly patient prompts clinicians to focus on life-threatening conditions, such as acute coronary syndrome, pulmonary embolus, arrhythmia, and sepsis. Each of these diagnoses was considered early in the course of this patient’s presentations, but each was deemed unlikely as it became apparent that the episodes were self-limited and recurrent over years. Incorporating flushing into the diagnostic problem representation allowed the clinicians to focus on a subset of causes of hypotension.
Flushing disorders may be classified by whether they are mediated by the autonomic nervous system (wet flushes, because they are usually accompanied by diaphoresis) or by exogenous or endogenous vasoactive substances (dry flushes).1 Autonomic nervous system flushing is triggered by emotions, fever, exercise, perimenopause (hot flashes), and neurologic conditions (eg, Parkinson disease, spinal cord injury, multiple sclerosis). Vasoactive flushing precipitants include drugs (eg, niacin); alcohol (secondary to cutaneous vasodilation, or acetaldehyde particularly in people with insufficient acetaldehyde dehydrogenase activity)2; foods that contain capsaicin, tyramine, sulfites, or histamine (eg, eating improperly handled fish can cause scombroid poisoning); and anaphylaxis. Rare causes of vasoactive flushing include carcinoid syndrome, pheochromocytoma, medullary thyroid carcinoma, VIPoma, and mastocytosis.2
Mastocytosis is a rare clonal disorder characterized by the accumulation of abnormal mast cells in the skin (cutaneous mastocytosis), in multiple organs (systemic mastocytosis), or in a solid tumor (mastocytoma). Urticaria pigmentosa is the most common form of cutaneous mastocytosis; it is seen more often in children than in adults and typically is associated with a maculopapular rash and dermatographism. Systemic mastocytosis is the most common form of the disorder in adults.3 Symptoms are related to mast cell infiltration or mast cell mediator–related effects, which range from itching, flushing, and diarrhea to hypotension and anaphylaxis. Other manifestations are fatigue, urticaria pigmentosa, osteoporosis, hepatosplenomegaly, bone pain, cytopenias, and lymphadenopathy.4
Systemic mastocytosis can occur at any age and should be considered in patients with recurrent unexplained flushing, syncope, or hypotension. Eighty percent to 90% of patients with systemic mastocytosis have a mutation in Kit,5 a transmembrane tyrosine kinase that is the receptor for stem cell factor. The Asp816Val mutation leads to increased proliferation and reduced apoptosis of mast cells.3,6,7 Proposed diagnostic algorithms8-11 involve measurement of serum tryptase levels and examination of bone marrow. Bone marrow biopsy and testing for the Asp816Val
The primary goals of treatment are managing mast cell–mediated symptoms and, in advanced cases, achieving cytoreduction. Alcohol can trigger mast cell degranulation in indolent systemic mastocytosis and should be avoided. Mast cell–mediated symptoms are managed with histamine blockers, leukotriene antagonists, and mast cell stabilizers.12 Targeted therapy with tyrosine kinase inhibitors (eg, imatinib) in patients with transmembrane Kit mutation (eg, Phe522Cys, Lys509Ile) associated with systemic mastocytosis has had promising results.13,14 However, this patient’s Asp816Val mutation is in the Kit catalytic domain, not the transmembrane region, and therefore would not be expected to respond to imatinib. A recent open-label trial of the multikinase inhibitor midostaurin demonstrated resolution of organ damage, reduced bone marrow burden, and lowered serum tryptase levels in patients with advanced systemic mastocytosis.15 Interferon, cladribine, and high-dose corticosteroids are prescribed in patients for whom other therapies have been ineffective.8
The differential diagnosis is broad for both hypotension and for flushing, but the differential diagnosis for recurrent hypotension and flushing is limited. Recognizing that flushing was an essential feature of this patient’s hypotensive condition, and not an epiphenomenon of syncope, allowed the clinicians to focus on the overlap and make a shocking diagnosis.
Acknowledgment
The authors thank David Bosler, MD (Cleveland Clinic) for interpreting the pathology image.
Disclosure
Nothing to report.
1. Wilkin JK. The red face: flushing disorders. Clin Dermatol. 1993;11(2):211-223. PubMed
2. Izikson L, English JC 3rd, Zirwas MJ. The flushing patient: differential diagnosis, workup, and treatment. J Am Acad Dermatol. 2006;55(2):193-208. PubMed
3. Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435-453. PubMed
4. Hermans MA, Rietveld MJ, van Laar JA, et al. Systemic mastocytosis: a cohort study on clinical characteristics of 136 patients in a large tertiary centre. Eur J Intern Med. 2016;30:25-30. PubMed
5. Kristensen T, Vestergaard H, Bindslev-Jensen C, Møller MB, Broesby-Olsen S; Mastocytosis Centre, Odense University Hospital (MastOUH). Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493-498. PubMed
6. Verstovsek S. Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. Eur J Haematol. 2013;90(2):89-98. PubMed
7. Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7):2366-2372. PubMed
8. Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90(3):250-262. PubMed
9. Valent P, Aberer E, Beham-Schmid C, et al. Guidelines and diagnostic algorithm for patients with suspected systemic mastocytosis: a proposal of the Austrian Competence Network (AUCNM). Am J Blood Res. 2013;3(2):174-180. PubMed
10. Valent P, Escribano L, Broesby-Olsen S, et al; European Competence Network on Mastocytosis. Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. Allergy. 2014;69(10):1267-1274. PubMed
11. Akin C, Soto D, Brittain E, et al. Tryptase haplotype in mastocytosis: relationship to disease variant and diagnostic utility of total tryptase levels. Clin Immunol. 2007;123(3):268-271. PubMed
12. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(19):1885-1886. PubMed
13. Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222-3225. PubMed
14. Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30(4):373-378. PubMed
15. Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530-2541. PubMed
1. Wilkin JK. The red face: flushing disorders. Clin Dermatol. 1993;11(2):211-223. PubMed
2. Izikson L, English JC 3rd, Zirwas MJ. The flushing patient: differential diagnosis, workup, and treatment. J Am Acad Dermatol. 2006;55(2):193-208. PubMed
3. Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37(6):435-453. PubMed
4. Hermans MA, Rietveld MJ, van Laar JA, et al. Systemic mastocytosis: a cohort study on clinical characteristics of 136 patients in a large tertiary centre. Eur J Intern Med. 2016;30:25-30. PubMed
5. Kristensen T, Vestergaard H, Bindslev-Jensen C, Møller MB, Broesby-Olsen S; Mastocytosis Centre, Odense University Hospital (MastOUH). Sensitive KIT D816V mutation analysis of blood as a diagnostic test in mastocytosis. Am J Hematol. 2014;89(5):493-498. PubMed
6. Verstovsek S. Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. Eur J Haematol. 2013;90(2):89-98. PubMed
7. Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7):2366-2372. PubMed
8. Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90(3):250-262. PubMed
9. Valent P, Aberer E, Beham-Schmid C, et al. Guidelines and diagnostic algorithm for patients with suspected systemic mastocytosis: a proposal of the Austrian Competence Network (AUCNM). Am J Blood Res. 2013;3(2):174-180. PubMed
10. Valent P, Escribano L, Broesby-Olsen S, et al; European Competence Network on Mastocytosis. Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. Allergy. 2014;69(10):1267-1274. PubMed
11. Akin C, Soto D, Brittain E, et al. Tryptase haplotype in mastocytosis: relationship to disease variant and diagnostic utility of total tryptase levels. Clin Immunol. 2007;123(3):268-271. PubMed
12. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(19):1885-1886. PubMed
13. Akin C, Fumo G, Yavuz AS, Lipsky PE, Neckers L, Metcalfe DD. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103(8):3222-3225. PubMed
14. Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—in vitro and in vivo responsiveness to imatinib therapy. Leuk Res. 2006;30(4):373-378. PubMed
15. Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374(26):2530-2541. PubMed
© 2017 Society of Hospital Medicine
A Physician With Thigh Pain
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
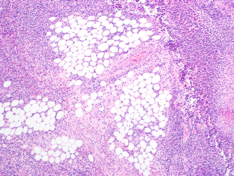
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
Necrotizing soft-tissue infection (NSTI) often is difficult to distinguish from a superficial soft-tissue infection like cellulitis. Both conditions present with pain, edema, and erythema and can be accompanied by fever and malaise. The diagnosis of NSTI must be made quickly because successful treatment requires early surgical debridement and broad-spectrum antibiotics. The following case demonstrates the challenge of diagnosing NSTI.
Case Presentation
A 50-year-old physician developed a sore throat with subjective fevers, night sweats, and chills. After 2 days, his symptoms resolved. The next day he developed right thigh pain while playing tennis and limped off the court. That night he had fevers, chills, and sweats. For the next 3 days, his right thigh pain persisted with waxing and waning fevers.
The patient’s medical history included gastroesophageal reflux disease, vitamin D deficiency, and a positive purified protein derivative test for which he had completed 1 year of isoniazid therapy. The patient was married and in a monogamous relationship with his wife. He had traveled to the Sierra National Forest and Yosemite Park during the preceding winter. He did not swim in a lake or recall a tick bite. He had not consumed raw food, imported meats, or dairy products. He recently started oral fluconazole for tinea corporis.
The patient’s temperature was 39.5°C, heart rate was 115 beats per minute, blood pressure (BP) was 142/88 mm Hg, and respiratory rate was 18 breaths per minute with an oxygen saturation of 95% while breathing ambient air. He was drenched in sweat yet remained comfortable throughout the interview. The oropharyngeal mucosa was moist without lesions or erythema. There was no rash or lymphadenopathy. The lungs were clear to auscultation. The cardiac exam revealed tachycardia. There was point tenderness to deep palpation of the mid-anterior right thigh without crepitus, erythema, or edema.
The patient’s sodium level was 129 mmol/L (normal range 135-145 mmol/L), bicarbonate was 20 mmol/L (normal range 22-32 mmol/L), creatinine was 1.1 mg/dL (normal range 0.7-1.2 mg/dL), and glucose was 194 mg/dL. The white blood cell count (WBC) was 12,900 cells/mm3 (normal range 3,400-10,000 cells/mm3) with 96% neutrophils. The hematocrit was 41% (normal range 41-53%), and the platelet count was 347,000 cells/mm3 (normal range 140,000-450,000 cells/mm3). The lactate level was 2.2 mmol/L (normal range 0-2 mmol/L). The creatine kinase level was 347 U/L (normal range 50-388 U/L), and the lactate dehydrogenase level was 254 U/L (normal range 102-199 U/L). A rapid group A streptococcal (GAS) antigen test was negative. A radiograph of the right femur revealed mildly edematous soft tissue. On ultrasound the right quadriceps appeared mildly edematous, but there was no evidence of abscess or discrete fluid collection (eFigure 1).
eFigure 1. Ultrasound of the Right Anterior Thigh Ultrasound revealed heterogeneous, mildly edematous quadriceps muscle. There was no abscess or discrete fluid collection. There was trace fluid along the fascia of the quadriceps muscle.
Four liters of normal saline, acetaminophen, ceftriaxone, and doxycycline were administered to the patient. Overnight he was afebrile, tachycardic, and normotensive. The following morning his BP decreased to 81/53 mm Hg. His WBC count was 33,000 cells/mm3 with 96% neutrophils. A peripheral blood smear showed immature granulocytes. The sodium and creatinine increased to 135 mmol/L and 1.3 mg/dL, respectively. The erythrocyte sedimentation rate was 20 mm/h (normal range 0-10 mm/h), and the C-reactive protein level was 174 mg/L (normal range < 6.3 mg/L).The right thigh became erythematous and edematous.
Given concern for necrotizing fasciitis, antibiotics were changed to vancomycin, piperacillin-tazobactam, and clindamycin. The patient was taken to the operating room (OR). The right quadriceps muscle was markedly edematous with overlying necrotic fibrofatty tissue with easy separation of the fascia from the anterolateral rectus femoris and rectus lateralis muscles. Necrotizing fasciitis was diagnosed.
The tissue was debrided, and surgical pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation (eFigure 2). After the anterolateral space between the fascia and underlying thigh muscle was drained, a Penrose drain was placed, and the wound was left open with plans for a second-look operation within 24 hours.
eFigure 2. Surgical Pathology of Debrided Right Thigh
Pathology revealed fibroadipose tissue with extensive necrosis and dense acute inflammation.
eFigure 3. Right Anterior Thigh
Two Penrose drains inserted after second operation.
In the ensuing hours erythema extended proximal to the operative site. The patient was emergently taken to the OR. The focus of necrotizing fasciitis along the anterolateral aspect of the thigh had extended posteriorly and superiorly. This area was irrigated, all loculations were disrupted, and a second Penrose drain was placed.
The wound was left open for 6 more days. On hospital day 9, operative exploration revealed no necrotizing fasciitis. The fascia and skin wound were then closed (eFigure 3).
Cultures from the fascia grew the GAS bacteria Streptococcus pyogenes (S pyogenes), which was sensitive to penicillin. The blood cultures from admission were sterile. A test for Epstein-Barr virus immunoglobulin M antibody was negative. The patient was discharged after 10 days in the hospital to complete a 2-week course of IV penicillin. Two months later he resumed playing tennis and returned to his clinical duties.
Discussion
In the U.S., there are approximately 3.5 cases of invasive GAS infection per 100,000 persons.1 Type I NSTI is polymicrobial (aerobic and anaerobic organisms). Risk factors include recent surgery, immunocompromised states, drug use, diabetes mellitus, and traumatic wounds.2 Type II NSTI is caused by GAS or other β-hemolytic streptococci either alone or in association with another organism, most commonly Staphylococcus aureus. Type II NSTI is classically found on the extremities and occurs in young, healthy, immunocompetent patients—such as this patient.3
The portal of entry in nearly half of type II NSTI is unknown; minor local trauma is often suspected.4 However, cases have been reported in which the only identifiable source was a preceding sore throat.4 The origin of this patient’s GAS remains unknown, but perhaps his pharyngitis led to transient bacteremia, which then seeded his injured thigh muscle. An in vitro model demonstrated that injured muscles increase surface expression of the cytoskeletal protein vimentin, which binds GAS.5 Exotoxins and endotoxins produced by S pyogenes may lead to microvascular thrombosis, tissue ischemia, liquefactive necrosis, and systemic release of cytokines followed by systemic illness, multiorgan dysfunction, and death.6
The Laboratory Risk Indicator for Necrotizing Fasciitis (LRINEC) score was developed to aid in early diagnosis of NSTI.7 It was derived from a series of 2,555 patients admitted with cellulitis or abscesses at a single institution. Scores > 8 have a positive predictive value of 93% for NSTI. This patient had a LRINEC score of 9. Radiographs or computed tomography scans may demonstrate soft-tissue air collections but lack sensitivity and are often nondiagnostic.8,9 T1-weighted magnetic resonance imaging can delineate the anatomic extent of soft-tissue infections but is time consuming and may delay treatment.10 When the pretest probability is high, proceeding directly to the OR for direct visualization and possible debridement is advisable. Histologic features of necrotizing fasciitis include inflammation with polymorphonuclear cells and necrosis of the subcutaneous fat and fascia with relative sparing of the muscle.11Necrotizing soft-tissue infection requires early surgical debridement and broad-spectrum antibiotic coverage. Without surgical debridement, the mortality rate approaches 100%.2 Antibiotics should include activity against Gram-positive, Gram-negative, and anaerobic organisms. The duration of antibiotic therapy has not been defined and is dependent on the patient’s clinical status. Adjunctive treatment options may include IV immunoglobulin and hyperbaric oxygen therapy, although the data supporting their utility are limited.12,13
Conclusion
Despite the LRINEC scoring systems and advanced imaging, necrotizing fasciitis remains challenging to diagnose in a timely manner. In this case, close monitoring of the patient facilitated timely evaluation and treatment of a fatal disease.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
1. O'Loughlin RE, Roberson A, Cieslak PR, et al; Active Bacterial Core Surveillance Team. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853-857.
2. Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis. 2007;44(5):705-710.
3. Naqvi GA, Malik SA, Jan W. Necrotizing fasciitis of the lower extremity: a case report and current concept of diagnosis and management. Scand J Trauma Resusc Emerg Med. 2009;17:28.
4. Stevens DL. Streptococcal toxic-shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1195;1(3):69-78.
5. Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis. 2006;193(12):1685-1692.
6. Cainzos M, Gonzalez-Rodriguez FJ. Necrotizing soft tissue infections. Curr Opin Crit Care. 2007;13(4):433-439.
7. Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med. 2004;32(7):1535-1541.
8. Goh T, Goh LG, Ang CH, Wong CH. Early diagnosis of necrotizing fasciitis. Br J Surg. 2014;101(1):119-125.
9. Lancerotto L, Tocco I, Salmaso R, Vindigni V, Basetto F. Necrotizing fasciitis: classification, diagnosis and management. J Trauma Acute Care Surg. 2012;72(3):560-566.
10. Brothers TE, Tagge DU, Stutley JE, Conway WF, Del Schutte H Jr, Byrne TK. Magnetic resonance imaging differentiates between necrotizing and non-necrotizing fasciitis of the lower extremity. J Am Coll Surg. 1998;187(4):416-421.
11. Bakleh M, Wold LE, Mandrekar JN, Harmsen WS, Dimashkieh HH, Baddour LM. Correlation of histopathologic findings with clinical outcome in necrotizing fasciitis. Clin Infect Dis. 2005;40(3):410-414.
12. Barry W, Hudgins L, Donta ST, Pesanti EL. Intravenous immunoglobulin therapy for toxic shock syndrome. JAMA. 1992;267(24):3315-3316.
13. Wilkinson D, Doolette D. Hyperbaric oxygen treatment and survival from necrotizing soft tissue infection. Arch Surg. 2004;139(12):1339-1345.
Another Spin
A previously healthy 11‐year‐old boy presented to the emergency department after referral from his pediatrician for 1 week of fevers. Seven days prior to admission he developed a fever to 40.8C, vomiting, and mild left knee pain. The vomiting resolved within 2 days. Five days prior to admission he developed a pruritic, pinpoint rash over his abdomen that resolved within 24 hours. He also developed red, cracked lips, redness of his tongue, redness surrounding his eyes, and slight swelling of his hands. Three days prior to admission his pediatrician noted a 1‐cm anterior cervical lymph node. His fevers occurred throughout each of the prior 7 days without a discernible pattern, and his mild knee pain persisted at the time of presentation.
This preteen has had high fevers for 1 week associated with arthralgia, pruritic rash, emesis, and oral mucosal erythema. His rash, lip and tongue erythema, and swollen hands are classic features of Kawasaki disease (KD), but he lacks the other characteristic physical examination findings. The diagnosis of KD requires fever for at least 5 days accompanied by 4 of the following 5 signs: polymorphous rash, oral mucous membrane changes, peripheral extremity changes such as swelling or skin desquamation, bilateral bulbar conjunctival injection, and cervical lymphadenopathy >1.5 cm in diameter. Children meeting fewer than 4 of these criteria may have an incomplete form of KD.
Because most patients with KD (80%) are under 5 years old, alternative diagnoses such as autoimmune illnesses or a hypersensitivity reaction should be considered. Travel, medication, and animal exposure histories may reveal clues to an infectious or drug‐induced etiology of his fever. Immunization status should be assessed, as measles is also associated with fever, rash, and mucosal changes. Arthralgia or arthritis may occur in KD, but these findings suggest the need to entertain other possibilities, including bone or joint infection, infective endocarditis, inflammatory bowel disease, juvenile idiopathic arthritis (JIA), or systemic lupus erythematosus (SLE).
The child's only past medical history was an episode of croup as an infant. There was no family history of autoimmune diseases. He was not taking any medications and had no known allergies. His immunizations were up to date, including measles, mumps, rubella, and varicella. He lived with his parents and his dog. He swam in fresh water during a trip to Maine 2 months earlier. Neither he nor his family recalled a tick bite. He had no exposure to raw meat or unpasteurized dairy products.
The travel to New England raises the possibility of Lyme disease, although a 2‐month interval between exposure and a high, prolonged fever would be very unusual. Knee arthralgia or arthritis is common in children with late‐stage Lyme disease, but can also be seen in early‐disseminated disease. The prior description of the rash is not suggestive of erythema chronicum migrans, which is seen in early‐stage Lyme disease.
C‐reactive protein (CRP) was 189 mg/L (normal <6.3 mg/L). An echocardiogram was normal. Intravenous immunoglobulin (IVIG) was administered for presumed KD, with immediate improvement of the periorbital erythema, tongue redness, and hand swelling. He was discharged the next day on aspirin with cardiology clinic follow‐up.
Improvement after IVIG supports the diagnosis of KD. It is typical to discharge KD patients from the hospital when they have been afebrile for 24 hours or when the CRP level has declined by approximately 50%.
Over the next 48 hours he felt unwell with high‐grade fevers, continued left knee pain, and new left hip pain. He was readmitted to the hospital. His temperature was 39.4C, respiratory rate was 22 breaths per minute, heart rate was 122 beats per minute, blood pressure was 103/50 mm Hg, and oxygen saturation was 100% while breathing ambient air. He appeared mildly uncomfortable. His conjunctivae were normal. His lips were dry, red, and cracked, and his tongue was red with prominent papillae. His neck was supple without lymphadenopathy. His lungs were clear to auscultation. His heart exam was without murmurs. His abdomen was soft, and the liver and spleen were not enlarged. He had no swelling or erythema of his joints; however, he experienced pain with range of motion of his left knee, and tenderness and restricted range of motion of his left hip. His neurologic exam was normal. There were no rashes.
He has persistent fever, tachycardia, and tachypnea, now without features of KD except oral mucosal changes including prominent tongue papillae consistent with a strawberry tongue. Continued or recurrent fever may suggest persistent KD with ongoing inflammation or the need to search for an alternative diagnoses. An echocardiogram should be repeated, as the coronary artery abnormalities in KD can evolve rapidly, particularly when inflammation persists. Additional findings may include decreased left ventricular function, mitral regurgitation, or pericardial effusion. A second dose of IVIG is necessary to control fever and inflammation in about 15% of patients with KD, although in this case IVIG should be withheld pending further evaluation.
Arthralgia occurs commonly in KD, whereas frank arthritis is less typical. Polyarticular or oligoarticular arthritis involving small or large joints (especially knee or ankle) affects 5% to 10% of patients. The severity of findings in his left hip warrants consideration of septic arthritis with pain referred to the knee; pelvic or femoral osteomyelitis; psoas abscess; or pyomyositis. Following basic lab tests, imaging of the left hip region is indicated.
Laboratory evaluation revealed: white blood cell (WBC) count 10,000/L (absolute neutrophil count 8,460/L, absolute lymphocyte count 530/L), hemoglobin 10.6 g/dL, platelet count 208,000/L, serum sodium 130 mmol/L, serum potassium 3.3 mmol/L, serum urea nitrogen 11 mg/dL, serum creatinine 0.54 mg/dL, aspartate transaminase (AST) 26 U/L, alanine transaminase (ALT) 31 U/L, albumin 1.7 g/dL, erythrocyte sedimentation rate (ESR) > 100 mm/h, and CRP 263 mg/L. No blast cells were seen on peripheral blood smear.
Hypoalbuminemia and markedly elevated inflammatory markers indicate an inflammatory condition that has been active for more than a week. Assessing ESR after IVIG therapy is not useful because exogenous globulins increase the ESR; however, CRP is useful to monitor inflammation and remains elevated here.
Incomplete KD is still possible. Hyponatremia, hypoalbuminemia, and anemia are all features of persistent KD, and have been utilized in several clinical scoring systems in Japan to identify KD patients at increased risk for developing coronary complications. A neoplastic process cannot be excluded, but does not appear likely based on the acuity of his presentation and peripheral blood smear review.
Upon readmission he received a second dose of 2 g/kg IVIG. He remained on aspirin and continued to have fevers. A repeat echocardiogram was normal. He had worsening pain in his left knee and hip with difficulty straightening his left leg. Physical examination was notable for tenderness to palpation over his left hip joint, refusal to bear weight, and resistance to passive range of motion. On hospital day 2, an ultrasound of his left hip and knee revealed a complex left hip effusion and small left knee effusion.
KD becomes less likely in the presence of persistent fevers after IVIG and a repeatedly normal echocardiogram. Worsening left leg symptoms including impaired hip extension with a complex hip effusion suggests an infectious process in or adjacent to the left hip, such as septic arthritis, myositis, or osteomyelitis of the pelvis or proximal femur. A complex hip effusion is less likely to be present with arthritis related to JIA or SLE. The patient needs an emergent hip aspiration and possibly magnetic resonance imaging (MRI) to evaluate adjacent structures.
Arthrotomy and open drainage of his left hip revealed purulent fluid with a WBC count of 49,000/L with 89% neutrophils and 2% lymphocytes. Gram stain was negative. A left knee aspirate demonstrated straw‐colored synovial fluid (which was not sent for cell counts). Bacterial, fungal, and acid‐fast bacilli cultures were requested from hip and knee aspirates. Intravenous ceftriaxone and vancomycin were administered.
The most likely organism in pediatric pyogenic arthritis is Staphylococcus aureus, but there is a long list of other potential pathogens, including Streptococcus pyogenes (group A streptococcus) and Streptococcus pneumoniae. Most pediatric patients with acute pyogenic arthritis have synovial fluid WBC counts in excess of 75,000 to 100,000/L. The protracted course and the initial lack of hip symptoms raise the possibility of a primary osteomyelitis of the femur (particularly the intracapsular portion of the femoral neck or head) or of the acetabulum, with subsequent extension into the hip joint. Pyogenic myositis involving muscle groups adjacent to the hip would be unlikely to spread into the hip space, but can lead to synovial irritation, characterized by sterile joint fluid and WBC counts that fall short of the usual numbers seen in septic arthritis. The blood supply to the femoral head can become compromised with prolonged inflammation and increased intracapsular pressure, resulting in aseptic necrosis.
All cultures from his hip and knee aspirations were sterile. He continued to have daily fevers and persistent tachycardia while receiving intravenous ceftriaxone and vancomycin. Additional testing was notable for: antinuclear antibody (ANA) 1:80, anti‐streptolysin O (ASO) titer 344 IU (normal <150 IU), AST and ALT within normal limits, ferritin 568 ng/mL (normal <322 ng/mL), and lactate dehydrogenase (LDH) 212 units/L (normal <257 units/L). Abdominal ultrasound revealed borderline hepatosplenomegaly. An ophthalmologic examination was normal.
On postoperative day 4 he developed left upper thigh swelling. An MRI showed rim‐enhancing juxta‐articular complex fluid collections surrounding the left femur with decreased marrow enhancement of the left proximal femur (Figure 1).
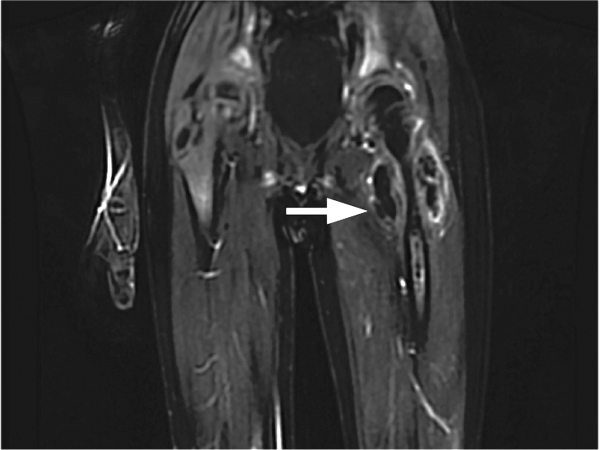
The limited rheumatologic evaluation is unrevealing; the ANA result is nondiagnostic and the ASO titer is normal for age. Laboratories generally report adult normal values for streptococcal antibodies regardless of the patient's age; children from ages 7 to 12 years are at their life peak frequency of group A streptococcal pharyngitis and typically have higher normal values of streptococcal antibodies, including ASO (up to about 480640 IU). The moderately elevated ferritin level is most likely an acute phase reactant and not high enough to suggest macrophage activation syndrome, which is unlikely with the normal AST, ALT, and LDH levels, the absence of significant splenomegaly, and the lack of cytopenias. Continued fever with progressive left upper thigh swelling point to osteomyelitis of the proximal femur, which may have initially ruptured into the hip and then infiltrated the femoral cortex and spread infection into the adjacent soft tissues. Surgical debridement is indicated.
The relative prevalence of methicillin‐sensitive S aureus (MSSA) and methicillin‐resistant S aureus vary widely with geography. MSSA strains are more likely to be highly toxigenic. The elaboration of 1 or more extracellular toxins could account for the patient's initial symptoms.
The patient was brought back to the operating room for drainage of the juxta‐articular fluid collections and a biopsy of his femur. The fluid collections were grossly purulent. His intraoperative cultures were positive for MSSA. The bone biopsy revealed necrotic tissue, acute inflammation, and bacterial colonies, consistent with acute osteomyelitis. Further testing of his S aureus isolate was positive for staphylococcal enterotoxin B. He completed a 4‐week course of oral clindamycin with subsequent normalization of his hip exam and inflammatory markers. At a follow‐up visit the patient was feeling better, but had developed skin peeling on the lateral aspects of his feet consistent with late sequelae of toxin‐mediated disease (Figure 2). Three months after discharge the patient had returned to his baseline activity level and remained asymptomatic.
COMMENTARY
The patient presented with a constellation of symptoms that was initially mistaken for incomplete KD until focal progression of his symptoms exposed an underlying femoral osteomyelitis with periarticular abscess formation. Bacterial cultures and subsequent toxin assay revealed an enterotoxin B‐producing strain of S aureus.
Certain staphylococcal strains secrete superantigens that may lead to the development of a systemic toxin‐mediated syndrome. Toxins elaborated by S aureus include toxic shock syndrome toxin‐1 (TSST‐1) and enterotoxins, which have been implicated in menstrual and nonmenstrual toxic shock syndromes.[1, 2] Enterotoxin B is a staphylococcal superantigen found in most strains of the USA400 clonal group, and has been frequently associated with skin and soft tissue infections.[3] Enterotoxin B production has been reported in nearly half of S aureus isolates from skin, soft tissue, and bone infections.[4]
Staphylococcal and streptococcal toxin‐mediated diseases can mimic vasculitis, systemic juvenile idiopathic arthritis, viral infections, and Stevens‐Johnson syndrome. Glossitis in toxin‐mediated syndromes manifests with a swollen, red tongue with overlying enlarged papillae, giving the appearance of a strawberry. Although pediatric providers often equate strawberry tongue, conjunctival injection, rash, and erythematous lips with KD, these findings are also seen in toxin‐mediated diseases, such as scarlet fever or staphylococcal toxic shock syndrome. Enterotoxin B mediated staphylococcal disease masquerading as KD has been reported in 2 cases: a 7‐month‐old boy with multifocal S aureus osteomyelitis and a 5‐year‐old boy with S aureus bacteremia. Both staphylococcal isolates produced enterotoxin B but were negative for other staphylococcus‐related toxins including TSST‐1.[5]
The Institute of Medicine (IOM) recently released its report Improving Diagnosis in Health Care, highlighting the under‐recognized quality and safety issue of diagnostic error.[6] The report uses the following broad and patient‐centered definition of diagnostic error: the failure to (a) establish an accurate and timely explanation of the patient's health problem(s) or (b) communicate that explanation to the patient. The IOM's conceptual model of diagnosis emphasizes the iterative nature of the diagnostic process, including the importance of generating a working diagnosis, gathering and incorporating new information in the reassessment of that diagnosis, and integrating treatment response into the formulation of the final diagnosis (Figure 3).

Even though the patient initially had several features consistent with KD, the increasing number of atypical features could have prompted the clinical team to reconsider their working diagnosis. The patient's age was atypical for KD, he had progressive knee and hip arthritis, and his fevers persisted after IVIG. An expanded differential should have included toxic shock syndrome; the resolution of conjunctival and mucosal injection and edema after IVIG may have been the result of antibodies in the IVIG preparation with neutralizing activity against superantigens. This antitoxin activity has established a role for IVIG in the management of staphylococcal toxic shock syndrome.[7] Ultimately, his imaging and surgical drainage revealed a focal staphylococcal toxin‐producing infectious source from which his fevers, rash, and mucosal and extremity changes emanated. This case reminds us that the more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should take it for another spin around the diagnostic wheel in search of a more suitable alternative.
KEY LEARNING POINTS
- Staphylococcal toxin‐mediated disease may mimic KD, with common features including strawberry tongue, oral and conjunctival injection, and skin desquamation.
- Improvement after treatment with IVIG is characteristic but not diagnostic of KD, and may be seen in toxin‐mediated disease.
- KD may present with arthralgia or arthritis, but severe joint abnormalities warrant consideration of infectious and other autoimmune conditions.
- The more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should gather, interpret, and integrate new information in search of a more suitable alternative.
Disclosure: Nothing to report.
- . Staphylococcal enterotoxin B and toxic shock syndrome toxin‐1 are significantly associated with non‐menstrual TSS. Lancet. 1986;1:1149–1150.
- , , , , . Toxic shock syndrome caused by a strain of staphylococcus aureus that produces enterotoxin C but not toxic shock syndrome toxin‐1. Am J Dis Child. 1989;143 (7):848–849.
- , , , , , . Staphylococcus aureus isolates encode variant staphylococcal enterotoxin B proteins that are diverse in superantigenicity and lethality. PLoS One. 2012;7(7):e41157.
- , , , et al. Variability of antibiotic susceptibility and toxin production of Staphylococcal aureus stains isolated from skin, soft tissue, and bone related infections. BMC Microbiol. 2013;13:188.
- , , , , . Kawasaki syndrome‐like illness associated with infection caused by enterotoxin B‐secreting Staphylococcus aureus. Clin Microbiol Rev. 2013;26:422–447.
- National Academies of Sciences, Engineering, and Medicine. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press; 2015.
- , , , et al. Intravenous immunoglobulin G therapy in streptococcal toxic shock syndrome: a European randomized, double‐blind, placebo‐controlled trial. Clin Infect Dis. 2003;37(3):333–340.
A previously healthy 11‐year‐old boy presented to the emergency department after referral from his pediatrician for 1 week of fevers. Seven days prior to admission he developed a fever to 40.8C, vomiting, and mild left knee pain. The vomiting resolved within 2 days. Five days prior to admission he developed a pruritic, pinpoint rash over his abdomen that resolved within 24 hours. He also developed red, cracked lips, redness of his tongue, redness surrounding his eyes, and slight swelling of his hands. Three days prior to admission his pediatrician noted a 1‐cm anterior cervical lymph node. His fevers occurred throughout each of the prior 7 days without a discernible pattern, and his mild knee pain persisted at the time of presentation.
This preteen has had high fevers for 1 week associated with arthralgia, pruritic rash, emesis, and oral mucosal erythema. His rash, lip and tongue erythema, and swollen hands are classic features of Kawasaki disease (KD), but he lacks the other characteristic physical examination findings. The diagnosis of KD requires fever for at least 5 days accompanied by 4 of the following 5 signs: polymorphous rash, oral mucous membrane changes, peripheral extremity changes such as swelling or skin desquamation, bilateral bulbar conjunctival injection, and cervical lymphadenopathy >1.5 cm in diameter. Children meeting fewer than 4 of these criteria may have an incomplete form of KD.
Because most patients with KD (80%) are under 5 years old, alternative diagnoses such as autoimmune illnesses or a hypersensitivity reaction should be considered. Travel, medication, and animal exposure histories may reveal clues to an infectious or drug‐induced etiology of his fever. Immunization status should be assessed, as measles is also associated with fever, rash, and mucosal changes. Arthralgia or arthritis may occur in KD, but these findings suggest the need to entertain other possibilities, including bone or joint infection, infective endocarditis, inflammatory bowel disease, juvenile idiopathic arthritis (JIA), or systemic lupus erythematosus (SLE).
The child's only past medical history was an episode of croup as an infant. There was no family history of autoimmune diseases. He was not taking any medications and had no known allergies. His immunizations were up to date, including measles, mumps, rubella, and varicella. He lived with his parents and his dog. He swam in fresh water during a trip to Maine 2 months earlier. Neither he nor his family recalled a tick bite. He had no exposure to raw meat or unpasteurized dairy products.
The travel to New England raises the possibility of Lyme disease, although a 2‐month interval between exposure and a high, prolonged fever would be very unusual. Knee arthralgia or arthritis is common in children with late‐stage Lyme disease, but can also be seen in early‐disseminated disease. The prior description of the rash is not suggestive of erythema chronicum migrans, which is seen in early‐stage Lyme disease.
C‐reactive protein (CRP) was 189 mg/L (normal <6.3 mg/L). An echocardiogram was normal. Intravenous immunoglobulin (IVIG) was administered for presumed KD, with immediate improvement of the periorbital erythema, tongue redness, and hand swelling. He was discharged the next day on aspirin with cardiology clinic follow‐up.
Improvement after IVIG supports the diagnosis of KD. It is typical to discharge KD patients from the hospital when they have been afebrile for 24 hours or when the CRP level has declined by approximately 50%.
Over the next 48 hours he felt unwell with high‐grade fevers, continued left knee pain, and new left hip pain. He was readmitted to the hospital. His temperature was 39.4C, respiratory rate was 22 breaths per minute, heart rate was 122 beats per minute, blood pressure was 103/50 mm Hg, and oxygen saturation was 100% while breathing ambient air. He appeared mildly uncomfortable. His conjunctivae were normal. His lips were dry, red, and cracked, and his tongue was red with prominent papillae. His neck was supple without lymphadenopathy. His lungs were clear to auscultation. His heart exam was without murmurs. His abdomen was soft, and the liver and spleen were not enlarged. He had no swelling or erythema of his joints; however, he experienced pain with range of motion of his left knee, and tenderness and restricted range of motion of his left hip. His neurologic exam was normal. There were no rashes.
He has persistent fever, tachycardia, and tachypnea, now without features of KD except oral mucosal changes including prominent tongue papillae consistent with a strawberry tongue. Continued or recurrent fever may suggest persistent KD with ongoing inflammation or the need to search for an alternative diagnoses. An echocardiogram should be repeated, as the coronary artery abnormalities in KD can evolve rapidly, particularly when inflammation persists. Additional findings may include decreased left ventricular function, mitral regurgitation, or pericardial effusion. A second dose of IVIG is necessary to control fever and inflammation in about 15% of patients with KD, although in this case IVIG should be withheld pending further evaluation.
Arthralgia occurs commonly in KD, whereas frank arthritis is less typical. Polyarticular or oligoarticular arthritis involving small or large joints (especially knee or ankle) affects 5% to 10% of patients. The severity of findings in his left hip warrants consideration of septic arthritis with pain referred to the knee; pelvic or femoral osteomyelitis; psoas abscess; or pyomyositis. Following basic lab tests, imaging of the left hip region is indicated.
Laboratory evaluation revealed: white blood cell (WBC) count 10,000/L (absolute neutrophil count 8,460/L, absolute lymphocyte count 530/L), hemoglobin 10.6 g/dL, platelet count 208,000/L, serum sodium 130 mmol/L, serum potassium 3.3 mmol/L, serum urea nitrogen 11 mg/dL, serum creatinine 0.54 mg/dL, aspartate transaminase (AST) 26 U/L, alanine transaminase (ALT) 31 U/L, albumin 1.7 g/dL, erythrocyte sedimentation rate (ESR) > 100 mm/h, and CRP 263 mg/L. No blast cells were seen on peripheral blood smear.
Hypoalbuminemia and markedly elevated inflammatory markers indicate an inflammatory condition that has been active for more than a week. Assessing ESR after IVIG therapy is not useful because exogenous globulins increase the ESR; however, CRP is useful to monitor inflammation and remains elevated here.
Incomplete KD is still possible. Hyponatremia, hypoalbuminemia, and anemia are all features of persistent KD, and have been utilized in several clinical scoring systems in Japan to identify KD patients at increased risk for developing coronary complications. A neoplastic process cannot be excluded, but does not appear likely based on the acuity of his presentation and peripheral blood smear review.
Upon readmission he received a second dose of 2 g/kg IVIG. He remained on aspirin and continued to have fevers. A repeat echocardiogram was normal. He had worsening pain in his left knee and hip with difficulty straightening his left leg. Physical examination was notable for tenderness to palpation over his left hip joint, refusal to bear weight, and resistance to passive range of motion. On hospital day 2, an ultrasound of his left hip and knee revealed a complex left hip effusion and small left knee effusion.
KD becomes less likely in the presence of persistent fevers after IVIG and a repeatedly normal echocardiogram. Worsening left leg symptoms including impaired hip extension with a complex hip effusion suggests an infectious process in or adjacent to the left hip, such as septic arthritis, myositis, or osteomyelitis of the pelvis or proximal femur. A complex hip effusion is less likely to be present with arthritis related to JIA or SLE. The patient needs an emergent hip aspiration and possibly magnetic resonance imaging (MRI) to evaluate adjacent structures.
Arthrotomy and open drainage of his left hip revealed purulent fluid with a WBC count of 49,000/L with 89% neutrophils and 2% lymphocytes. Gram stain was negative. A left knee aspirate demonstrated straw‐colored synovial fluid (which was not sent for cell counts). Bacterial, fungal, and acid‐fast bacilli cultures were requested from hip and knee aspirates. Intravenous ceftriaxone and vancomycin were administered.
The most likely organism in pediatric pyogenic arthritis is Staphylococcus aureus, but there is a long list of other potential pathogens, including Streptococcus pyogenes (group A streptococcus) and Streptococcus pneumoniae. Most pediatric patients with acute pyogenic arthritis have synovial fluid WBC counts in excess of 75,000 to 100,000/L. The protracted course and the initial lack of hip symptoms raise the possibility of a primary osteomyelitis of the femur (particularly the intracapsular portion of the femoral neck or head) or of the acetabulum, with subsequent extension into the hip joint. Pyogenic myositis involving muscle groups adjacent to the hip would be unlikely to spread into the hip space, but can lead to synovial irritation, characterized by sterile joint fluid and WBC counts that fall short of the usual numbers seen in septic arthritis. The blood supply to the femoral head can become compromised with prolonged inflammation and increased intracapsular pressure, resulting in aseptic necrosis.
All cultures from his hip and knee aspirations were sterile. He continued to have daily fevers and persistent tachycardia while receiving intravenous ceftriaxone and vancomycin. Additional testing was notable for: antinuclear antibody (ANA) 1:80, anti‐streptolysin O (ASO) titer 344 IU (normal <150 IU), AST and ALT within normal limits, ferritin 568 ng/mL (normal <322 ng/mL), and lactate dehydrogenase (LDH) 212 units/L (normal <257 units/L). Abdominal ultrasound revealed borderline hepatosplenomegaly. An ophthalmologic examination was normal.
On postoperative day 4 he developed left upper thigh swelling. An MRI showed rim‐enhancing juxta‐articular complex fluid collections surrounding the left femur with decreased marrow enhancement of the left proximal femur (Figure 1).
The limited rheumatologic evaluation is unrevealing; the ANA result is nondiagnostic and the ASO titer is normal for age. Laboratories generally report adult normal values for streptococcal antibodies regardless of the patient's age; children from ages 7 to 12 years are at their life peak frequency of group A streptococcal pharyngitis and typically have higher normal values of streptococcal antibodies, including ASO (up to about 480640 IU). The moderately elevated ferritin level is most likely an acute phase reactant and not high enough to suggest macrophage activation syndrome, which is unlikely with the normal AST, ALT, and LDH levels, the absence of significant splenomegaly, and the lack of cytopenias. Continued fever with progressive left upper thigh swelling point to osteomyelitis of the proximal femur, which may have initially ruptured into the hip and then infiltrated the femoral cortex and spread infection into the adjacent soft tissues. Surgical debridement is indicated.
The relative prevalence of methicillin‐sensitive S aureus (MSSA) and methicillin‐resistant S aureus vary widely with geography. MSSA strains are more likely to be highly toxigenic. The elaboration of 1 or more extracellular toxins could account for the patient's initial symptoms.
The patient was brought back to the operating room for drainage of the juxta‐articular fluid collections and a biopsy of his femur. The fluid collections were grossly purulent. His intraoperative cultures were positive for MSSA. The bone biopsy revealed necrotic tissue, acute inflammation, and bacterial colonies, consistent with acute osteomyelitis. Further testing of his S aureus isolate was positive for staphylococcal enterotoxin B. He completed a 4‐week course of oral clindamycin with subsequent normalization of his hip exam and inflammatory markers. At a follow‐up visit the patient was feeling better, but had developed skin peeling on the lateral aspects of his feet consistent with late sequelae of toxin‐mediated disease (Figure 2). Three months after discharge the patient had returned to his baseline activity level and remained asymptomatic.
COMMENTARY
The patient presented with a constellation of symptoms that was initially mistaken for incomplete KD until focal progression of his symptoms exposed an underlying femoral osteomyelitis with periarticular abscess formation. Bacterial cultures and subsequent toxin assay revealed an enterotoxin B‐producing strain of S aureus.
Certain staphylococcal strains secrete superantigens that may lead to the development of a systemic toxin‐mediated syndrome. Toxins elaborated by S aureus include toxic shock syndrome toxin‐1 (TSST‐1) and enterotoxins, which have been implicated in menstrual and nonmenstrual toxic shock syndromes.[1, 2] Enterotoxin B is a staphylococcal superantigen found in most strains of the USA400 clonal group, and has been frequently associated with skin and soft tissue infections.[3] Enterotoxin B production has been reported in nearly half of S aureus isolates from skin, soft tissue, and bone infections.[4]
Staphylococcal and streptococcal toxin‐mediated diseases can mimic vasculitis, systemic juvenile idiopathic arthritis, viral infections, and Stevens‐Johnson syndrome. Glossitis in toxin‐mediated syndromes manifests with a swollen, red tongue with overlying enlarged papillae, giving the appearance of a strawberry. Although pediatric providers often equate strawberry tongue, conjunctival injection, rash, and erythematous lips with KD, these findings are also seen in toxin‐mediated diseases, such as scarlet fever or staphylococcal toxic shock syndrome. Enterotoxin B mediated staphylococcal disease masquerading as KD has been reported in 2 cases: a 7‐month‐old boy with multifocal S aureus osteomyelitis and a 5‐year‐old boy with S aureus bacteremia. Both staphylococcal isolates produced enterotoxin B but were negative for other staphylococcus‐related toxins including TSST‐1.[5]
The Institute of Medicine (IOM) recently released its report Improving Diagnosis in Health Care, highlighting the under‐recognized quality and safety issue of diagnostic error.[6] The report uses the following broad and patient‐centered definition of diagnostic error: the failure to (a) establish an accurate and timely explanation of the patient's health problem(s) or (b) communicate that explanation to the patient. The IOM's conceptual model of diagnosis emphasizes the iterative nature of the diagnostic process, including the importance of generating a working diagnosis, gathering and incorporating new information in the reassessment of that diagnosis, and integrating treatment response into the formulation of the final diagnosis (Figure 3).
Even though the patient initially had several features consistent with KD, the increasing number of atypical features could have prompted the clinical team to reconsider their working diagnosis. The patient's age was atypical for KD, he had progressive knee and hip arthritis, and his fevers persisted after IVIG. An expanded differential should have included toxic shock syndrome; the resolution of conjunctival and mucosal injection and edema after IVIG may have been the result of antibodies in the IVIG preparation with neutralizing activity against superantigens. This antitoxin activity has established a role for IVIG in the management of staphylococcal toxic shock syndrome.[7] Ultimately, his imaging and surgical drainage revealed a focal staphylococcal toxin‐producing infectious source from which his fevers, rash, and mucosal and extremity changes emanated. This case reminds us that the more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should take it for another spin around the diagnostic wheel in search of a more suitable alternative.
KEY LEARNING POINTS
- Staphylococcal toxin‐mediated disease may mimic KD, with common features including strawberry tongue, oral and conjunctival injection, and skin desquamation.
- Improvement after treatment with IVIG is characteristic but not diagnostic of KD, and may be seen in toxin‐mediated disease.
- KD may present with arthralgia or arthritis, but severe joint abnormalities warrant consideration of infectious and other autoimmune conditions.
- The more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should gather, interpret, and integrate new information in search of a more suitable alternative.
Disclosure: Nothing to report.
A previously healthy 11‐year‐old boy presented to the emergency department after referral from his pediatrician for 1 week of fevers. Seven days prior to admission he developed a fever to 40.8C, vomiting, and mild left knee pain. The vomiting resolved within 2 days. Five days prior to admission he developed a pruritic, pinpoint rash over his abdomen that resolved within 24 hours. He also developed red, cracked lips, redness of his tongue, redness surrounding his eyes, and slight swelling of his hands. Three days prior to admission his pediatrician noted a 1‐cm anterior cervical lymph node. His fevers occurred throughout each of the prior 7 days without a discernible pattern, and his mild knee pain persisted at the time of presentation.
This preteen has had high fevers for 1 week associated with arthralgia, pruritic rash, emesis, and oral mucosal erythema. His rash, lip and tongue erythema, and swollen hands are classic features of Kawasaki disease (KD), but he lacks the other characteristic physical examination findings. The diagnosis of KD requires fever for at least 5 days accompanied by 4 of the following 5 signs: polymorphous rash, oral mucous membrane changes, peripheral extremity changes such as swelling or skin desquamation, bilateral bulbar conjunctival injection, and cervical lymphadenopathy >1.5 cm in diameter. Children meeting fewer than 4 of these criteria may have an incomplete form of KD.
Because most patients with KD (80%) are under 5 years old, alternative diagnoses such as autoimmune illnesses or a hypersensitivity reaction should be considered. Travel, medication, and animal exposure histories may reveal clues to an infectious or drug‐induced etiology of his fever. Immunization status should be assessed, as measles is also associated with fever, rash, and mucosal changes. Arthralgia or arthritis may occur in KD, but these findings suggest the need to entertain other possibilities, including bone or joint infection, infective endocarditis, inflammatory bowel disease, juvenile idiopathic arthritis (JIA), or systemic lupus erythematosus (SLE).
The child's only past medical history was an episode of croup as an infant. There was no family history of autoimmune diseases. He was not taking any medications and had no known allergies. His immunizations were up to date, including measles, mumps, rubella, and varicella. He lived with his parents and his dog. He swam in fresh water during a trip to Maine 2 months earlier. Neither he nor his family recalled a tick bite. He had no exposure to raw meat or unpasteurized dairy products.
The travel to New England raises the possibility of Lyme disease, although a 2‐month interval between exposure and a high, prolonged fever would be very unusual. Knee arthralgia or arthritis is common in children with late‐stage Lyme disease, but can also be seen in early‐disseminated disease. The prior description of the rash is not suggestive of erythema chronicum migrans, which is seen in early‐stage Lyme disease.
C‐reactive protein (CRP) was 189 mg/L (normal <6.3 mg/L). An echocardiogram was normal. Intravenous immunoglobulin (IVIG) was administered for presumed KD, with immediate improvement of the periorbital erythema, tongue redness, and hand swelling. He was discharged the next day on aspirin with cardiology clinic follow‐up.
Improvement after IVIG supports the diagnosis of KD. It is typical to discharge KD patients from the hospital when they have been afebrile for 24 hours or when the CRP level has declined by approximately 50%.
Over the next 48 hours he felt unwell with high‐grade fevers, continued left knee pain, and new left hip pain. He was readmitted to the hospital. His temperature was 39.4C, respiratory rate was 22 breaths per minute, heart rate was 122 beats per minute, blood pressure was 103/50 mm Hg, and oxygen saturation was 100% while breathing ambient air. He appeared mildly uncomfortable. His conjunctivae were normal. His lips were dry, red, and cracked, and his tongue was red with prominent papillae. His neck was supple without lymphadenopathy. His lungs were clear to auscultation. His heart exam was without murmurs. His abdomen was soft, and the liver and spleen were not enlarged. He had no swelling or erythema of his joints; however, he experienced pain with range of motion of his left knee, and tenderness and restricted range of motion of his left hip. His neurologic exam was normal. There were no rashes.
He has persistent fever, tachycardia, and tachypnea, now without features of KD except oral mucosal changes including prominent tongue papillae consistent with a strawberry tongue. Continued or recurrent fever may suggest persistent KD with ongoing inflammation or the need to search for an alternative diagnoses. An echocardiogram should be repeated, as the coronary artery abnormalities in KD can evolve rapidly, particularly when inflammation persists. Additional findings may include decreased left ventricular function, mitral regurgitation, or pericardial effusion. A second dose of IVIG is necessary to control fever and inflammation in about 15% of patients with KD, although in this case IVIG should be withheld pending further evaluation.
Arthralgia occurs commonly in KD, whereas frank arthritis is less typical. Polyarticular or oligoarticular arthritis involving small or large joints (especially knee or ankle) affects 5% to 10% of patients. The severity of findings in his left hip warrants consideration of septic arthritis with pain referred to the knee; pelvic or femoral osteomyelitis; psoas abscess; or pyomyositis. Following basic lab tests, imaging of the left hip region is indicated.
Laboratory evaluation revealed: white blood cell (WBC) count 10,000/L (absolute neutrophil count 8,460/L, absolute lymphocyte count 530/L), hemoglobin 10.6 g/dL, platelet count 208,000/L, serum sodium 130 mmol/L, serum potassium 3.3 mmol/L, serum urea nitrogen 11 mg/dL, serum creatinine 0.54 mg/dL, aspartate transaminase (AST) 26 U/L, alanine transaminase (ALT) 31 U/L, albumin 1.7 g/dL, erythrocyte sedimentation rate (ESR) > 100 mm/h, and CRP 263 mg/L. No blast cells were seen on peripheral blood smear.
Hypoalbuminemia and markedly elevated inflammatory markers indicate an inflammatory condition that has been active for more than a week. Assessing ESR after IVIG therapy is not useful because exogenous globulins increase the ESR; however, CRP is useful to monitor inflammation and remains elevated here.
Incomplete KD is still possible. Hyponatremia, hypoalbuminemia, and anemia are all features of persistent KD, and have been utilized in several clinical scoring systems in Japan to identify KD patients at increased risk for developing coronary complications. A neoplastic process cannot be excluded, but does not appear likely based on the acuity of his presentation and peripheral blood smear review.
Upon readmission he received a second dose of 2 g/kg IVIG. He remained on aspirin and continued to have fevers. A repeat echocardiogram was normal. He had worsening pain in his left knee and hip with difficulty straightening his left leg. Physical examination was notable for tenderness to palpation over his left hip joint, refusal to bear weight, and resistance to passive range of motion. On hospital day 2, an ultrasound of his left hip and knee revealed a complex left hip effusion and small left knee effusion.
KD becomes less likely in the presence of persistent fevers after IVIG and a repeatedly normal echocardiogram. Worsening left leg symptoms including impaired hip extension with a complex hip effusion suggests an infectious process in or adjacent to the left hip, such as septic arthritis, myositis, or osteomyelitis of the pelvis or proximal femur. A complex hip effusion is less likely to be present with arthritis related to JIA or SLE. The patient needs an emergent hip aspiration and possibly magnetic resonance imaging (MRI) to evaluate adjacent structures.
Arthrotomy and open drainage of his left hip revealed purulent fluid with a WBC count of 49,000/L with 89% neutrophils and 2% lymphocytes. Gram stain was negative. A left knee aspirate demonstrated straw‐colored synovial fluid (which was not sent for cell counts). Bacterial, fungal, and acid‐fast bacilli cultures were requested from hip and knee aspirates. Intravenous ceftriaxone and vancomycin were administered.
The most likely organism in pediatric pyogenic arthritis is Staphylococcus aureus, but there is a long list of other potential pathogens, including Streptococcus pyogenes (group A streptococcus) and Streptococcus pneumoniae. Most pediatric patients with acute pyogenic arthritis have synovial fluid WBC counts in excess of 75,000 to 100,000/L. The protracted course and the initial lack of hip symptoms raise the possibility of a primary osteomyelitis of the femur (particularly the intracapsular portion of the femoral neck or head) or of the acetabulum, with subsequent extension into the hip joint. Pyogenic myositis involving muscle groups adjacent to the hip would be unlikely to spread into the hip space, but can lead to synovial irritation, characterized by sterile joint fluid and WBC counts that fall short of the usual numbers seen in septic arthritis. The blood supply to the femoral head can become compromised with prolonged inflammation and increased intracapsular pressure, resulting in aseptic necrosis.
All cultures from his hip and knee aspirations were sterile. He continued to have daily fevers and persistent tachycardia while receiving intravenous ceftriaxone and vancomycin. Additional testing was notable for: antinuclear antibody (ANA) 1:80, anti‐streptolysin O (ASO) titer 344 IU (normal <150 IU), AST and ALT within normal limits, ferritin 568 ng/mL (normal <322 ng/mL), and lactate dehydrogenase (LDH) 212 units/L (normal <257 units/L). Abdominal ultrasound revealed borderline hepatosplenomegaly. An ophthalmologic examination was normal.
On postoperative day 4 he developed left upper thigh swelling. An MRI showed rim‐enhancing juxta‐articular complex fluid collections surrounding the left femur with decreased marrow enhancement of the left proximal femur (Figure 1).
The limited rheumatologic evaluation is unrevealing; the ANA result is nondiagnostic and the ASO titer is normal for age. Laboratories generally report adult normal values for streptococcal antibodies regardless of the patient's age; children from ages 7 to 12 years are at their life peak frequency of group A streptococcal pharyngitis and typically have higher normal values of streptococcal antibodies, including ASO (up to about 480640 IU). The moderately elevated ferritin level is most likely an acute phase reactant and not high enough to suggest macrophage activation syndrome, which is unlikely with the normal AST, ALT, and LDH levels, the absence of significant splenomegaly, and the lack of cytopenias. Continued fever with progressive left upper thigh swelling point to osteomyelitis of the proximal femur, which may have initially ruptured into the hip and then infiltrated the femoral cortex and spread infection into the adjacent soft tissues. Surgical debridement is indicated.
The relative prevalence of methicillin‐sensitive S aureus (MSSA) and methicillin‐resistant S aureus vary widely with geography. MSSA strains are more likely to be highly toxigenic. The elaboration of 1 or more extracellular toxins could account for the patient's initial symptoms.
The patient was brought back to the operating room for drainage of the juxta‐articular fluid collections and a biopsy of his femur. The fluid collections were grossly purulent. His intraoperative cultures were positive for MSSA. The bone biopsy revealed necrotic tissue, acute inflammation, and bacterial colonies, consistent with acute osteomyelitis. Further testing of his S aureus isolate was positive for staphylococcal enterotoxin B. He completed a 4‐week course of oral clindamycin with subsequent normalization of his hip exam and inflammatory markers. At a follow‐up visit the patient was feeling better, but had developed skin peeling on the lateral aspects of his feet consistent with late sequelae of toxin‐mediated disease (Figure 2). Three months after discharge the patient had returned to his baseline activity level and remained asymptomatic.
COMMENTARY
The patient presented with a constellation of symptoms that was initially mistaken for incomplete KD until focal progression of his symptoms exposed an underlying femoral osteomyelitis with periarticular abscess formation. Bacterial cultures and subsequent toxin assay revealed an enterotoxin B‐producing strain of S aureus.
Certain staphylococcal strains secrete superantigens that may lead to the development of a systemic toxin‐mediated syndrome. Toxins elaborated by S aureus include toxic shock syndrome toxin‐1 (TSST‐1) and enterotoxins, which have been implicated in menstrual and nonmenstrual toxic shock syndromes.[1, 2] Enterotoxin B is a staphylococcal superantigen found in most strains of the USA400 clonal group, and has been frequently associated with skin and soft tissue infections.[3] Enterotoxin B production has been reported in nearly half of S aureus isolates from skin, soft tissue, and bone infections.[4]
Staphylococcal and streptococcal toxin‐mediated diseases can mimic vasculitis, systemic juvenile idiopathic arthritis, viral infections, and Stevens‐Johnson syndrome. Glossitis in toxin‐mediated syndromes manifests with a swollen, red tongue with overlying enlarged papillae, giving the appearance of a strawberry. Although pediatric providers often equate strawberry tongue, conjunctival injection, rash, and erythematous lips with KD, these findings are also seen in toxin‐mediated diseases, such as scarlet fever or staphylococcal toxic shock syndrome. Enterotoxin B mediated staphylococcal disease masquerading as KD has been reported in 2 cases: a 7‐month‐old boy with multifocal S aureus osteomyelitis and a 5‐year‐old boy with S aureus bacteremia. Both staphylococcal isolates produced enterotoxin B but were negative for other staphylococcus‐related toxins including TSST‐1.[5]
The Institute of Medicine (IOM) recently released its report Improving Diagnosis in Health Care, highlighting the under‐recognized quality and safety issue of diagnostic error.[6] The report uses the following broad and patient‐centered definition of diagnostic error: the failure to (a) establish an accurate and timely explanation of the patient's health problem(s) or (b) communicate that explanation to the patient. The IOM's conceptual model of diagnosis emphasizes the iterative nature of the diagnostic process, including the importance of generating a working diagnosis, gathering and incorporating new information in the reassessment of that diagnosis, and integrating treatment response into the formulation of the final diagnosis (Figure 3).
Even though the patient initially had several features consistent with KD, the increasing number of atypical features could have prompted the clinical team to reconsider their working diagnosis. The patient's age was atypical for KD, he had progressive knee and hip arthritis, and his fevers persisted after IVIG. An expanded differential should have included toxic shock syndrome; the resolution of conjunctival and mucosal injection and edema after IVIG may have been the result of antibodies in the IVIG preparation with neutralizing activity against superantigens. This antitoxin activity has established a role for IVIG in the management of staphylococcal toxic shock syndrome.[7] Ultimately, his imaging and surgical drainage revealed a focal staphylococcal toxin‐producing infectious source from which his fevers, rash, and mucosal and extremity changes emanated. This case reminds us that the more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should take it for another spin around the diagnostic wheel in search of a more suitable alternative.
KEY LEARNING POINTS
- Staphylococcal toxin‐mediated disease may mimic KD, with common features including strawberry tongue, oral and conjunctival injection, and skin desquamation.
- Improvement after treatment with IVIG is characteristic but not diagnostic of KD, and may be seen in toxin‐mediated disease.
- KD may present with arthralgia or arthritis, but severe joint abnormalities warrant consideration of infectious and other autoimmune conditions.
- The more atypical a working diagnosis iseither in its presentation or treatment responsethe more readily clinicians should gather, interpret, and integrate new information in search of a more suitable alternative.
Disclosure: Nothing to report.
- . Staphylococcal enterotoxin B and toxic shock syndrome toxin‐1 are significantly associated with non‐menstrual TSS. Lancet. 1986;1:1149–1150.
- , , , , . Toxic shock syndrome caused by a strain of staphylococcus aureus that produces enterotoxin C but not toxic shock syndrome toxin‐1. Am J Dis Child. 1989;143 (7):848–849.
- , , , , , . Staphylococcus aureus isolates encode variant staphylococcal enterotoxin B proteins that are diverse in superantigenicity and lethality. PLoS One. 2012;7(7):e41157.
- , , , et al. Variability of antibiotic susceptibility and toxin production of Staphylococcal aureus stains isolated from skin, soft tissue, and bone related infections. BMC Microbiol. 2013;13:188.
- , , , , . Kawasaki syndrome‐like illness associated with infection caused by enterotoxin B‐secreting Staphylococcus aureus. Clin Microbiol Rev. 2013;26:422–447.
- National Academies of Sciences, Engineering, and Medicine. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press; 2015.
- , , , et al. Intravenous immunoglobulin G therapy in streptococcal toxic shock syndrome: a European randomized, double‐blind, placebo‐controlled trial. Clin Infect Dis. 2003;37(3):333–340.
- . Staphylococcal enterotoxin B and toxic shock syndrome toxin‐1 are significantly associated with non‐menstrual TSS. Lancet. 1986;1:1149–1150.
- , , , , . Toxic shock syndrome caused by a strain of staphylococcus aureus that produces enterotoxin C but not toxic shock syndrome toxin‐1. Am J Dis Child. 1989;143 (7):848–849.
- , , , , , . Staphylococcus aureus isolates encode variant staphylococcal enterotoxin B proteins that are diverse in superantigenicity and lethality. PLoS One. 2012;7(7):e41157.
- , , , et al. Variability of antibiotic susceptibility and toxin production of Staphylococcal aureus stains isolated from skin, soft tissue, and bone related infections. BMC Microbiol. 2013;13:188.
- , , , , . Kawasaki syndrome‐like illness associated with infection caused by enterotoxin B‐secreting Staphylococcus aureus. Clin Microbiol Rev. 2013;26:422–447.
- National Academies of Sciences, Engineering, and Medicine. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press; 2015.
- , , , et al. Intravenous immunoglobulin G therapy in streptococcal toxic shock syndrome: a European randomized, double‐blind, placebo‐controlled trial. Clin Infect Dis. 2003;37(3):333–340.
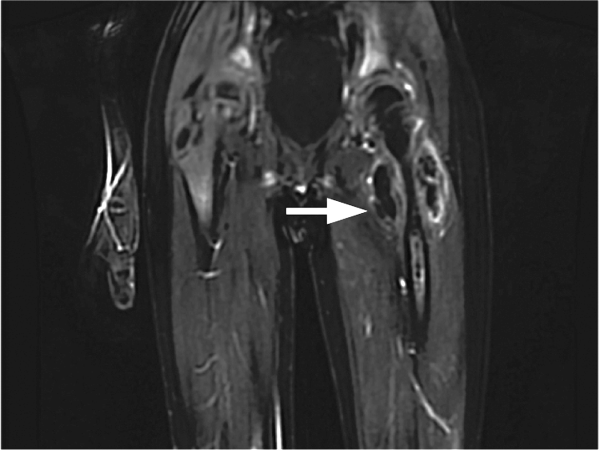
Breakdown
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
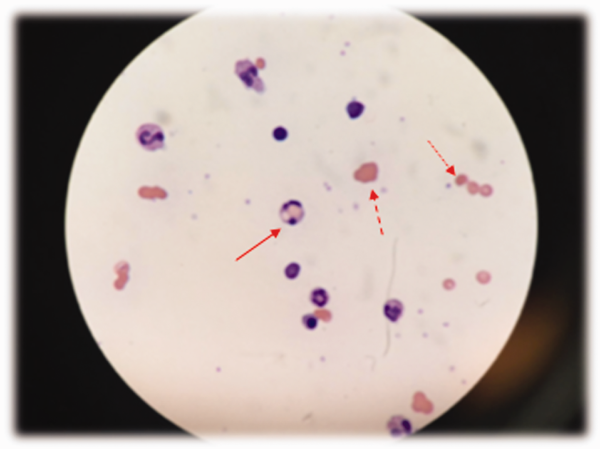
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
The patient's tachycardia and leukocytosis suggest sepsis. Potential sources include soft tissue infection or osteomyelitis from his sacral ulcers, Clostridium difficile, or a urinary tract infection. Impaired visceral sensation from his spinal cord injury may dampen his response to an intra‐abdominal process, such as mesenteric ischemia or toxic megacolon. Records from other hospitals should be reviewed to assess the acuity of change in his WBC count, hemoglobin, and creatinine. His anemia may be from chronic inflammation (eg, osteomyelitis), renal insufficiency, hemolysis, or occult blood loss, including retroperitoneal and gastrointestinal sources. His kidney injury may be from tubular necrosis in the setting of sepsis or obstructive uropathy related to a neurogenic bladder.
Potential contributors to his PEA and cardiovascular collapse are drug use (cocaine), alcohol withdrawal, infection, hypovolemia, myocardial ischemia, or heart failure. Severe hemorrhage, hyperkalemia, or acidosis from acute kidney injury and sepsis could also account for his cardiac arrest. His paraplegia and hospitalization raise the risk of venous thromboembolism, which can lead to PEA from pulmonary embolus and prolonged hypoxia.
His profound anemia is the likely cause of his PEA arrest and severe lactic acidosis. Massive hemolysis is most likely given no overt evidence of bleeding to account for the precipitous fall in hematocrit. Hemolysis can result from disorders intrinsic or extrinsic to the red blood cell (RBC). Intrinsic defects are usually congenital and involve the membrane, hemoglobin, or metabolic enzymes within the RBC. Extrinsic hemolysis arises from processes that injure the RBC from the outside: antibodies, infections, and mechanical shearing.
A rapidly declining platelet count is seen in microangiopathic hemolytic conditions such as disseminated intravascular coagulation (DIC) or thrombotic thrombocytopenic purpura (TTP), where platelets are consumed along with RBCs; sepsis makes DIC more likely. Autoimmune hemolytic anemia (AIHA) is sometimes accompanied by immune thrombocytopenia. AIHA arises from antibodies that are idiopathic or produced in response to infection, autoimmune conditions (eg, systemic lupus erythematosus), lymphoproliferative disease, or drugs (eg, ‐lactam antibiotics). The antiphospholipid syndrome can lead to thrombocytopenia, hemolysis, and kidney injury. Devitalized tissue in his sacral ulcers may predispose the patient to infection with Clostridium perfringens, which can elaborate enzymes that trigger massive hemolysis.
Because automated hemoglobin measurement is performed by spectrophotometry (light absorption and scatter), high concentrations of poorly soluble autoantibodies can increase the turbidity of the sample and preclude the measurement of hemoglobin concentration. This could lead to the report of interfering substances.
Low haptoglobin, elevated LDH, and hyperbilirubinemia confirm hemolysis. A more robust reticulocytosis is expected in the face of profound anemia, but the patient may also suffer from a concomitant hypoproliferative state (eg, nutritional deficiency). More likely, the rapidity of his decline outpaced the marrow's response, which can be delayed by days.
The most common cause of a combined elevation of the INR/PT and aPTT in a critically ill patient is DIC. Although no schistocytes were detected on the peripheral smear, they can be absent in up to 50% of DIC cases. TTP is associated with hemolytic anemia, kidney injury, and thrombocytopenia, but it generally does not cause coagulopathy.
The combination of red cell agglutination and hemophagocytosis suggests that the RBCs are coated with autoantibodies that cross‐link the cells and make them targets for phagocytosis by neutrophils in the circulation. This is distinct from the hemophagocytic syndrome, a rare immune activation syndrome characterized by macrophage phagocytosis of RBCs in the reticuloendothelial system. The blood smear also shows microspherocytes, which are seen in AIHA and hereditary spherocytosis.
Acute tubular necrosis could result from sepsis, ischemic injury from DIC, hypotension during cardiac arrest, or heme pigment toxicity. Urine sediment should be reviewed for dysmorphic RBCs or RBC casts that would indicate glomerulonephritis (eg, from an underlying autoimmune process associated with AIHA).
Urine hemoglobin that is disproportionate to the degree of hematuria suggests hemoglobinuria, which in turn defines the hemolysis as intravascular. Processes that directly lyse RBCs in circulation via mechanical shearing, activation of complement, infection of the RBC, or enzymatic or oxidative destruction of the membrane cause intravascular hemolysis. Leading considerations include microangiopathy (eg, DIC, TTP), clostridial sepsis, and AIHA.
AIHA can be broadly classified as warm or cold. Warm AIHA is caused by immunoglobulin IgG antibodies that bind most avidly at body temperature. Because warm AIHA does not activate complement, patients present with evidence of extravascular hemolysis that is typically chronic and mild to moderate in severity. It does not typically cause the acute, fulminant, intravascular hemolytic condition seen here.
Cold AIHA is characterized by autoantibodies that bind at lower temperatures and comes in 2 forms: cold agglutinin disease and (rarely) paroxysmal cold hemoglobinuria (PCH). Cold agglutinins are most often IgM antibodies produced in response to infection (Mycoplasma pneumoniae, infectious mononucleosis), drugs, or a hematologic malignancy. These IgM antibodies bind RBCs, causing them to agglutinate, and fix complement (including C3) to the surface of RBCs when blood circulates to cooler parts of the body. This results in complement activation, formation of the membrane attack complex, and intravascular hemolysis when bound and activated complement is present in large numbers. Acute infection can increase the complement available for binding to the surface of RBCs. Through a slightly different mechanism, PCH causes intravascular hemolysis through direct IgG activation of complement fixed to the surface of RBCs. During a hemolytic episode the direct antibody test (DAT) is positive using anti‐C3 and negative for IgG.
Based on the patient's clinical evidence of intravascular hemolysis and a suspected autoimmune etiology, the leading diagnosis at this time is cold AIHA.
The DAT detects IgG or complement adherent to RBCs. This patient has tested positive for both IgG and C3, though much more strongly for IgG, suggesting an unusual ability of the patient's IgG to activate complement. The phenomenon of mixed AIHA, in which the patient has both warm‐ and cold‐reacting antibodies, is rare.
Regarding infections associated with AIHA, there is no cough or rash to suggest M pneumoniae, and there is no sore throat, fever, lymphadenopathy, splenomegaly, or atypical lymphocytosis to suggest infectious mononucleosis. He should be tested for human immunodeficiency virus, which is also associated with AIHA. His leukocytosis may raise suspicion for an underlying hematologic malignancy, but he does not have blasts, dysplastic leukocytes, or lymphocytosis on his peripheral blood smear. Systemic lupus erythematosus can be associated with AIHA, thrombocytopenia, and renal failure, but he lacks the more common clinical manifestations of rash, arthralgias, and fever.
Drug‐induced immune hemolytic anemia (DIIHA) can cause both the clinical and serologic profile of an AIHA, as seen here. DIIHA can be distinguished from mixed AIHA if hemolysis abates with discontinuation of an offending drug. His deterioration is temporally associated with drug administration at the time of admission. Cephalosporins and ‐lactams (e.g., piperacillin) are the most common causes of DIIHA, and ‐lactamases such as tazobactam have also been implicated. By exclusion of other causes, DIIHA secondary to piperacillin is most likely responsible for his massive intravascular hemolysis.
COMMENTARY
This case illustrates a dramatic presentation of fulminant intravascular hemolysis secondary to piperacillin. The incidence of DIIHA is estimated to be 1 in 1 million.[1] Historically, methyldopa and high‐dose penicillin have been responsible for the majority of cases,[2] but in recent years complex penicillins, including piperacillin, and second‐ and third‐generation cephalosporins have been implicated.[3, 4] Cases of DIIHA are often underdiagnosed or misdiagnosed, as smoldering or less severe cases may not be recognized or are attributed to other causes.
A positive DAT, suggesting immunoglobulin and/or complement binding to RBCs, is the most reliable laboratory finding in DIIHA.[5] However, a positive DAT does not identify the source of the antigen and may result in misattribution of the immune hemolysis to autoimmunity rather than to a drug. Repeated or continued administration of the offending drug (as in this case) may perpetuate or worsen the hemolysis. Drug‐specific antibody tests may help to confirm the diagnosis, but these tests are complex and take significant time for specialized laboratories to run.
Severe hemolysis should be considered when a patient has a sudden and dramatic drop in his hemoglobin level in the absence of bleeding. Because DIIHA can be rapidly progressive, discontinuing a suspected culprit drug is the most important diagnostic and therapeutic measure. Typically, when an offending drug is stopped, the hemolysis stops as well. The time course over which this occurs depends on the rapidity of drug clearance.[4] Hemodialysis or plasmapheresis may be required in cases where the medication is renally excreted, particularly in cases of concomitant kidney injury. Evidence supporting corticosteroid use in DIIHA is limited, as the offending agent is usually discontinued by the time corticosteroids are initiated.[4]
This patient's DAT confirmed both IgG and complement activation, consistent with DIIHA caused by an immune complexlike reaction. This mechanism involves the antibody binding to a mixed epitope of the drug and a RBC membrane glycoprotein.[6] The offending drug was stopped only when review of his medical records established a clear temporal association between antibiotic administration and prior hemolysis.
The 2009 Health Information Technology for Economic and Clinical Health Act created an electronic health record (EHR) incentive program (meaningful use criteria).[7] By 2012, only 6% of hospitals met all of the stage 2 criteria, which include EHR interoperability across health systems.[8] The patient's preceding hemolytic event was described in records faxed by the outside hospitals, but without EHR interoperability, the treating clinicians did not have timely access to this information. Instead, the familiar manual process of obtaining outside records involving signed forms, phone calls, fax machines, and reams of paper progressed at its usual pace. Real‐time access to health records might have guided providers to select an alternative antibiotic regimen. Instead, a communication breakdown contributed to a catastrophic drug reaction and to this tragic patient outcome.
KEY TEACHING POINTS
- In a patient presenting with acute hemolysis and a positive DAT, consider DIIHA.
- Both piperacillin and tazobactam can cause a severe, complement‐mediated immune hemolytic anemia (DIIHA).
- Drug‐induced antibodies are detected by direct antiglobulin testing, but a complete medication history is the key to diagnosis.
- Management of drug‐induced hemolytic anemia involves immediate discontinuation of the culprit medication, supportive care, and potentially corticosteroids, plasmapheresis, and/or hemodialysis to expedite removal of the offending agent.
- EHR interoperability may provide timely access to important health information across different hospitals, expedite health information exchange, and reduce adverse patient outcomes that stem from communication delays.
This case was submitted anonymously to AHRQ WebM&M on July 18, 2014, and was accepted on August 7, 2014. The case and WebM&M commentary were published online on October 26, 2015.[9] This separate commentary on the same case was later submitted to the Journal of Hospital Medicine on September 2, 2015, accepted on November 24, 2015, and published on January 22, 2016. The 2 publications are written by different authors, and although they reference the same case, they make different but valuable points.
Disclosure
Nothing to report.
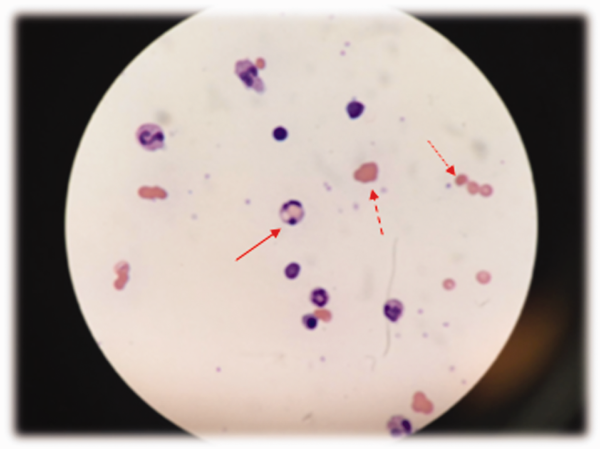
A coat with a clue
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
Not a Textbook Case
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
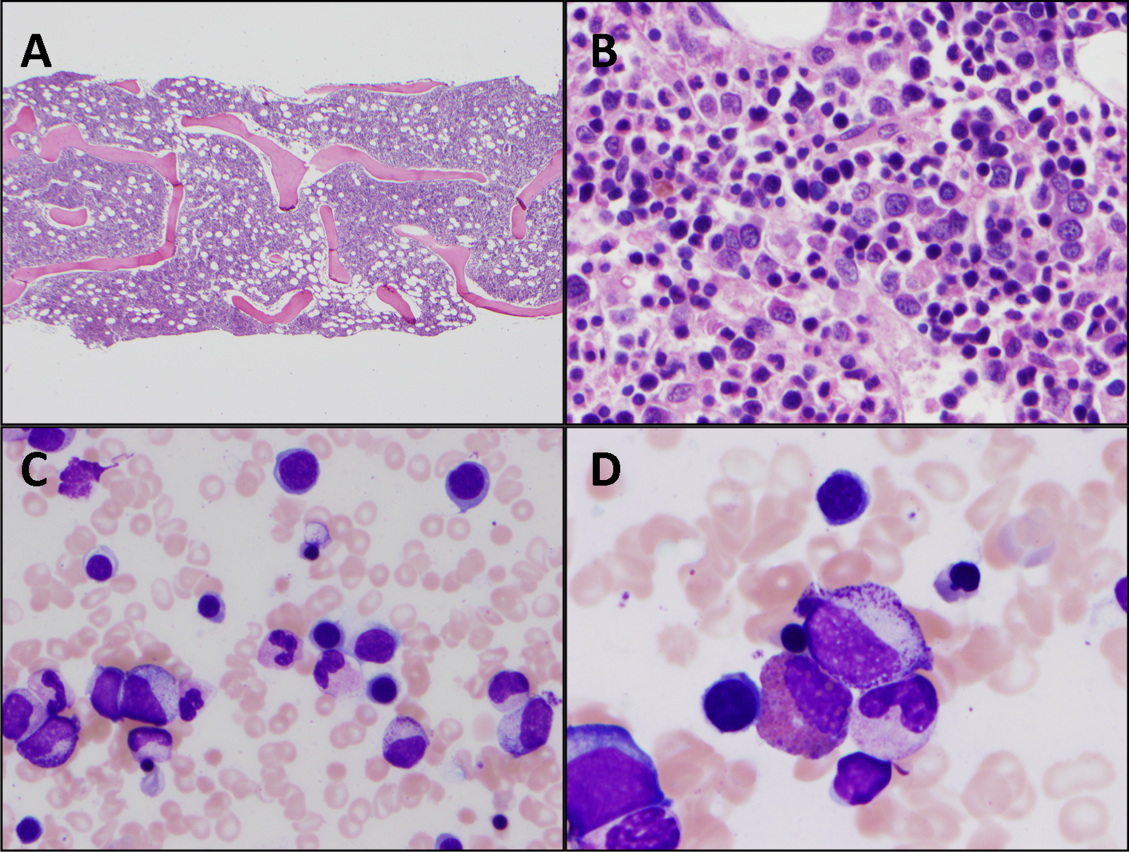
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.
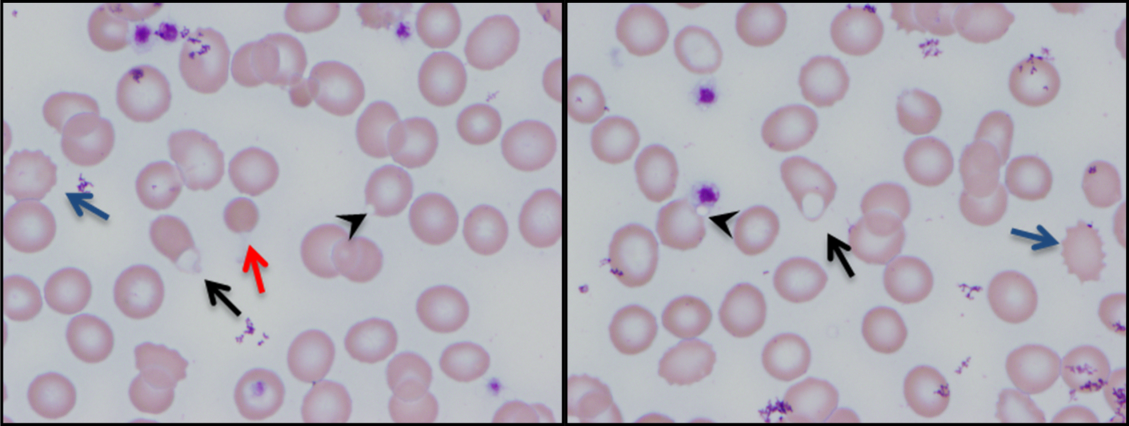
A Validated Delirium Prediction Rule
Delirium is characterized by fluctuating disturbances in cognition and consciousness and is a common complication of hospitalization in medical and surgical patients. Studies estimate the prevalence of delirium in hospitalized patients[1] to be 14% to 56%, and up to 70% in critically ill elderly patients.[2] Estimates of total healthcare costs associated with delirium range from $38 to $152 billion per year in the United States.[3] Delirious patients are more likely to be discharged to a nursing home and have increased hospital mortality and longer lengths of stay.[4, 5, 6] Recent data suggest long‐term effects of delirium including cognitive impairments up to 1 year following the illness[7] and an increased likelihood of developing[8] or worsening dementia.[9]
It is estimated that one‐third of hospital‐acquired delirium cases could be prevented with appropriate interventions.[10] A prediction rule that easily and accurately identifies high‐risk patients upon admission could therefore have a substantial clinical impact. In addition, a prediction rule could be used to identify patients in whom new targeted interventions for delirium prevention could be investigated. A number of risk factors for delirium have been identified, including older age, preexisting cognitive dysfunction, vision and hearing impairment, severe illness, dehydration, electrolyte abnormalities, overmedication, and alcohol abuse.[11, 12, 13, 14, 15, 16] Existing prediction rules using various combinations of these measures have been limited by their complexity,[17] do not predict incident delirium,[18, 19] or are restricted to surgical[20, 21, 22] or intensive care[23] patients and therefore are not broadly applicable to the general medical population, which is particularly susceptible to developing delirium.
We conducted this study to develop a simple, efficient, and accurate prediction rule for hospital‐acquired delirium in adult medical inpatients assessed at the time of admission. Our a priori hypothesis was that a delirium prediction rule would consist of a combination of known risk factors and most likely incorporate old age, illness severity, and preexisting cognitive dysfunction.
METHODS
Design and Setting
This was a prospective cohort study with a derivation phase from May 2010 to November 2010 at 2 hospitals at the University of California, San Francisco (UCSF) (Moffitt‐Long and Mount Zion Hospitals) and a validation phase from October 2011 to March 2012 at the San Francisco Veterans Affairs Medical Center (SFVAMC).
Participants and Measurements
Subject identification, recruitment, and inclusion and exclusion criteria were identical for the derivation and validation cohorts. Subjects were identified by reviewing daily admission logs. All non‐intensive care unit patients aged 50 years or older admitted through the emergency department to the medicine, cardiology, or neurology services were screened for eligibility through chart review or in person within 24 hours of admission by a trained research assistant. One research assistant, a college graduate, conducted all screening for the derivation cohort, and 2 research assistants, 1 a fourth‐year medical student and the other a third‐year psychology graduate student, conducted screening for the validation cohort. In‐person screening included an assessment for delirium using the long version of the Confusion Assessment Method (CAM).[24] To minimize the possibility of enrolling delirious subjects, research assistants were instructed to notify the study supervisor (V.C.D.), a board‐certified neurologist, to discuss every case in which any yes checkbox was marked on the CAM score sheet. Subjects delirious upon initial evaluation, admitted for alcohol withdrawal, admitted for comfort care, who were aphasic or who could not speak English were excluded. For all patients, or if they were unable to provide consent, their surrogates provided written informed consent, and the study was approved by the institutional review boards at UCSF and SFVAMC.
In the derivation cohort, 1241 patients were screened, and 439 were eligible for enrollment. Of these, 180 declined, 50 were discharged prior to the first follow‐up visit, and 209 were included. In the validation cohort, 420 patients were screened, and 368 were eligible for enrollment. Of these, 144 declined, 59 were discharged prior to the first follow‐up visit, and 165 were included.
Baseline data regarding known delirium risk factors[11, 12, 13, 14, 15, 16] were collected from subjects in the derivation cohort. Cognitive performance was assessed with the Mini Mental Status Examination (MMSE),[25] forward digit span,[26] and clock draw.[27] Permission for administration of the MMSE was granted by Psychological Assessment Resources, Inc., and each administration was paid for. A structured interview was conducted with validated questions regarding visual and hearing impairment, pain, mobility, place of residence, and alcohol, tobacco, and drug use.[28, 29, 30, 31] A whisper test for hearing loss was performed.[32] Subjects' charts were reviewed for demographic, clinical, and laboratory data. Illness severity was assessed by asking each subject's nurse to rate their patient on a scale from not ill to mildly ill, moderately ill, severely ill, or moribund.[33] Each nurse was shown these 5 choices, but more specific definitions of what each level of illness severity meant were not provided. We chose this method to assess illness severity because this rating scale was incorporated into a previous validated and widely cited delirium prediction rule.[17] This illness severity scale has been validated as a predictor of outcomes and correlates with other measures of illness severity and comorbidity when graded by physicians.[33, 34] Nurse and physician ratings of illness severity have been shown to be comparable,[35] and therefore if the scale were incorporated into the prediction rule it would allow nurses to perform it independently. In the validation cohort, only data required to complete the baseline CAM and apply the prediction rule were collected.
Assessment of Outcomes
All subjects were assessed for delirium daily for 6 days after enrollment or until discharge, whichever came first. Follow‐up was limited to 6 days, based on the assumption that delirium occurring beyond 1 week is more likely due to events during the hospitalization as opposed to factors measurable at admission. Delirium was assessed using the short CAM, an internationally recognized and validated tool.[24] To complete the CAM during follow‐up visits, subjects and their nurses were interviewed using a written script, and an MMSE and forward digit span were performed.
Daily follow‐up assessments were performed by research assistants who were not blinded to the initial assessment but who, in the validation phase, were blinded to the prediction rule score. Some weekend follow‐ups were performed by postgraduate year 2, 3, or 4 neurology residents, or internal medicine faculty experienced in the assessment of delirium and blinded to both the initial assessment and prediction rule score. Neurology residents and internists read the CAM training manual and were educated in the administration and scoring of the CAM by 1 of the senior investigators (V.C.D.) prior to their first shift; these nonstudy personnel covered 17 of 189 days of follow‐up in the derivation cohort and 21 of 169 days of follow‐up in the validation cohort. To maximize sensitivity of delirium detection, for any change in cognition, MMSE score, or forward digit span compared to baseline, a board‐certified neurologist blinded to the initial assessment was notified to discuss the case and validate the diagnosis of delirium in person (derivation cohort) or over the phone (validation cohort). All research assistants were trained by a board‐certified neurologist (V.C.D.) in the administration and interpretation of the CAM using published methods prior to enrollment of any subjects.[36] Training included the performance of independent long‐version CAMs by the trainer and the trainee on a series of delirious and nondelirious patients until there was consistent agreement for each item on the CAM in 5 consecutive patients. In addition, a board‐certified neurologist supervised the first 5 administrations of the CAM performed by each research assistant.
Statistical Analysis
Sample size for the derivation cohort was based on the predicted ability to detect a difference in rates of delirium among those with and without cognitive impairment, the strongest risk factor for delirium. Using a [2] test with an of 0.05 and of 0.80, we estimated we would need to enroll 260 subjects, assuming a prevalence of cognitive dysfunction in our cohort of 10% and an estimated rate of delirium of 24% and 6% among those with and without cognitive dysfunction respectively.[14, 16, 17, 20] We were unable to reach enrollment targets because of a short funding period and slower than expected recruitment.
To construct the prediction rule in the derivation cohort, all variables were dichotomized. Age was dichotomized at 80 years because old age is a known risk factor for delirium, and only 1 of 46 subjects between the ages of 70 and 80 years became delirious in the derivation cohort. Components of the MMSE were dichotomized as correct/emncorrect, with a correct response requiring perfect performance based on expert consensus. For 3 subjects who would not attempt to spell world backward (2 in the derivation and 1 in the validation cohort), their score on serial 7s was used instead. The total MMSE score was not used because our objective was to develop a prediction rule using elements that could be assessed quickly in the fast‐paced environment of the hospital. Illness severity was dichotomized at moderate or worse/mild or better because there were only 15 subjects in the severe illness category, and the majority of delirium (22 outcomes) occurred in the moderate illness category. High blood urea nitrogen:creatinine ratio was defined as >18.[37]
The association between predictor variables and occurrence of delirium was analyzed using univariate logistic regression. A forward stepwise logistic regression was then performed using the variables associated with the outcome at a significance level of P<0.05 in univariate analysis. Variables were eligible for addition to the multivariable model if they were associated with the outcome at a significance level of <0.05. The 4 independent predictors thus identified were combined into a prediction rule by assigning each predictor 1 point if present. The performance of the prediction rule was assessed by using Cuzick's nonparametric test for a trend across groups ordered by score.[38]
The prediction rule was tested in the validation cohort using the nonparametric test for trend. Receiver operating characteristic (ROC) curves were compared between the derivation and validation cohorts. All statistical analysis was performed using Stata software (StataCorp, College Station, TX).
RESULTS
The derivation cohort consisted of elderly patients (mean age, 68.0811.96 years; interquartile range, 5096 years), and included more males than females (54.1% vs 45.9%). Subjects were predominantly white (73.7%) and lived at home (90%) (Table 1). The mean admission MMSE score was 27.0 (standard deviation [SD], 3.4; range, 730). Median follow‐up was 2 days (interquartile range, 13). Delirium developed in 12% (n=25) of the cohort.
| Derivation Cohort, N=209 | Validation Cohort, N=165 | |
|---|---|---|
| ||
| Gender, No. (%) | ||
| Male | 113 (54) | 157 (95) |
| Female | 96 (46) | 8 (4.8) |
| Race, No. (%) | ||
| White | 154 (74) | 125 (76) |
| African American | 34 (16) | 25 (15) |
| Asian | 21 (10.0) | 13 (7.9) |
| Native American | 0 | 2 (1.2) |
| Illness severity, No. (%) | ||
| Not ill | 1 (0.5) | 0 |
| Mildly ill | 49 (23) | 62 (38) |
| Moderately ill | 129 (62) | 86 (52) |
| Severely ill | 15 (7.2) | 17 (10) |
| Moribund | 0 | 0 |
| Living situation, No. (%) | ||
| Home | 188 (90) | 147 (89) |
| Assisted living | 11 (5.3) | 6 (3.6) |
| Hotel | 4 (1.9) | 5 (3.0) |
| SNF | 1 (0.5) | 3 (1.8) |
| Homeless | 4 (1.9) | 4 (2.4) |
| Developed delirium | 25 (12) | 14 (8.5) |
Univariate analysis of the derivation study identified 10 variables significantly associated (P<0.05) with delirium (Table 2). Predictors of delirium included abnormal scores on 4 subtests of the MMSE, low score on the Mini‐Cog, living in an assisted living or skilled nursing facility, moderate to severe illness, old age, a past history of dementia, and hearing loss as assessed by the whisper test. These predictors were then entered into a stepwise logistic regression analysis that identified 4 independent predictors of delirium (Table 3).
| Variable | No. (%) Without Delirium | No. (%) With Delirium | Odds Ratio | P Value | 95% Confidence Interval |
|---|---|---|---|---|---|
| |||||
| Age 80 years | 30 (16) | 13 (52) | 5.6 | <0.001 | 2.313.4 |
| Male sex | 99 (54) | 14 (56) | 1.1 | 0.84 | 0.52.5 |
| White race | 135 (73) | 19 (76) | 1.2 | 0.78 | 0.433.1 |
| Score <5 on date questions of MMSE | 37 (20) | 12 (48) | 3.7 | 0.003 | 1.68.7 |
| Score <5 on place questions of MMSE | 50 (27) | 14 (56) | 3.4 | 0.005 | 1.58.0 |
| Score <3 on MMSE recall | 89 (48) | 18 (72) | 2.7 | 0.03 | 1.16.9 |
| Score <5 on MMSE W‐O‐R‐L‐D backward | 37 (20) | 13 (52) | 4.3 | 0.001 | 1.810.2 |
| Score 0 on MMSE pentagon copy, n=203 | 53 (30) | 12 (48) | 2.2 | 0.07 | 0.935.1 |
| Score 0 on clock draw, n=203 | 70 (39) | 15 (60) | 2.3 | 0.05 | 0.985.4 |
| MiniCog score 02, n=203[27] | 46 (26) | 12 (48) | 2.7 | 0.03 | 1.16.2 |
| Self‐rated vision fair, poor, or very poor | 55 (30) | 8 (32) | 1.1 | 0.83 | 0.452.7 |
| Endorses hearing loss | 89 (48) | 12 (48) | 0.99 | 0.97 | 0.432.3 |
| Uses hearing aid | 19 (10) | 2 (8) | 0.76 | 0.72 | 0.173.5 |
| Fails whisper test in either ear | 39 (21) | 10 (40) | 2.5 | 0.04 | 1.05.9 |
| Prior episode of delirium per patient or informant | 70 (38) | 13 (52) | 1.8 | 0.19 | 0.764.1 |
| Dementia in past medical history | 3 (2) | 3 (12) | 8.2 | 0.01 | 1.643.3 |
| Depression in past medical history | 16 (9) | 1 (4) | 0.44 | 0.43 | 0.063.5 |
| Lives in assisted living or SNF | 8 (4) | 4 (16) | 4.2 | 0.03 | 1.215.1 |
| Endorses pain | 82 (45) | 7 (28) | 0.48 | 0.12 | 0.191.2 |
| Less than independent for transfers | 11 (6) | 3 (12) | 2.1 | 0.27 | 0.568.3 |
| Less than independent for mobility on a level surface | 36 (20) | 7 (28) | 1.6 | 0.33 | 0.624.1 |
| Score of 24 on CAGE questionnaire[29] | 5 (3) | 0 (0) | No outcomes | ||
| Drinks any alcohol | 84 (46) | 10 (40) | 0.79 | 0.60 | 0.341.9 |
| Current smoker | 20 (11) | 2 (8) | 0.71 | 0.66 | 0.164.1 |
| Uses illicit drugs | 13 (7) | 2 (8) | 1.2 | 0.83 | 0.255.6 |
| Moderately or severely ill on nursing assessment, n=194 | 121 (71) | 23 (96) | 9.3 | 0.031 | 1.270.9 |
| Fever | 8 (4) | 0 (0) | No outcomes | ||
| Serum sodium <134mmol/L | 38 (21) | 3 (12) | 0.52 | 0.31 | 0.151.8 |
| WBC count>10109/L, n=208 | 57 (31) | 6 (24) | 0.70 | 0.47 | 0.261.8 |
| AST>41 U/L, n=131 | 27 (23) | 2 (15) | 0.61 | 0.54 | 0.132.9 |
| BUN:Cr>18, n=208 | 66 (36) | 13 (52) | 1.9 | 0.13 | 0.834.5 |
| Infection as admission diagnosis | 28 (15) | 4 (16) | 1.1 | 0.92 | 0.343.3 |
| Variable | Odds Ratio | 95% Confidence Interval | P Value | Points Toward AWOL Score |
|---|---|---|---|---|
| Age 80 years | 5.7 | 2.115.6 | 0.001 | 1 |
| Unable to correctly spell world backward | 3.5 | 1.39.6 | 0.01 | 1 |
| Not oriented to city, state, county, hospital name, and floor | 2.9 | 1.17.9 | 0.03 | 1 |
| Nursing illness severity assessment of moderately ill, severely ill, or moribund (as opposed to not ill or mildly ill) | 10.5 | 1.386.9 | 0.03 | 1 |
These 4 independent predictors were assigned 1 point each if present to create a prediction rule with a range of possible scores from 0 to 4. There was a significant trend predicting higher rates of delirium with higher scores, with no subjects who scored 0 becoming delirious, compared to 40% of those subjects scoring 3 or 4 (P for trend<0.001) (Table 4).
| Derivation Cohorta | Validation Cohort | Combined Cohorts | ||||
|---|---|---|---|---|---|---|
| AWOL Score | Not Delirious | Delirious | Not Delirious | Delirious | Not Delirious | Delirious |
| ||||||
| 0 | 26 (100%) | 0 (0%) | 24 (96%) | 1 (4%) | 49 (98%) | 1 (2%) |
| 1 | 86 (95%) | 5 (5%) | 57 (97%) | 2 (3%) | 136 (96%) | 5 (4%) |
| 2 | 41 (85%) | 7 (15%) | 44 (90%) | 5 (10%) | 92 (86%) | 15 (14%) |
| 3 | 17 (74%) | 6 (26%) | 22 (79%) | 6 (21%) | 40 (80%) | 10 (20%) |
| 4 | 0 (0%) | 6 (100%) | 4 (100%) | 0 (0%) | 4 (36%) | 7 (64%) |
| Total | 170 | 24 | 151 | 14 | 321 | 38 |
| P<0.001 | P=0.025 | P<0.001 | ||||
The validation cohort consisted of adults with a mean age of 70.7210.6 years, (interquartile range, 5194 years), who were predominantly white (75.8%) and overwhelmingly male (95.2%) (Table 1). The mean admission MMSE score was 26.75 (SD, 2.8; range, 1730). Median follow‐up was 2 days (interquartile range, 15). Delirium developed in 8.5% (n=14) of the cohort. In the validation cohort, 4% of subjects with a score of 0 became delirious, whereas 19% of those scoring 3 or 4 became delirious (P for trend 0.025) (Table 4).
ROC curves were compared for the derivation and validation cohorts. The area under the ROC curve for the derivation cohort (0.81, 95% confidence interval [CI]: 0.720.90) was slightly better than that in the validation cohort (0.69, 95% CI: 0.540.83), but the difference did not reach statistical significance (P=0.14) (Figure 1).

DISCUSSION
We derived and validated a prediction rule to assess the risk of developing delirium in hospitalized adult medical patients. Four variables easily assessed on admission in a screen lasting less than 2 minutes were independently associated with the development of delirium. The prediction rule can be remembered with the following mnemonic: AWOL (Age80 years; unable to spell World backward; not fully Oriented to place; and moderate or severe iLlness severity).
It is estimated up to a third of hospital acquired delirium cases can be prevented.[10] Recent guidelines recommend the use of a multicomponent intervention to prevent delirium and provide evidence that such a strategy would be cost‐effective.[39] Nevertheless, such interventions are resource intense, requiring specialized nurse training and staffing[40] and have not been widely implemented. Acute care for the elderly units, where interventions to prevent delirium might logically be implemented, also require physical remodeling to provide carpeted hallways, handrails, and elevated toilet seats and door levers.[41] A method of risk stratification to identify the patients who would benefit most from resource‐intensive prevention strategies would be valuable.
The AWOL tool may provide a practical alternative to existing delirium prediction rules for adult medical inpatients. Because it can be completed by a nurse in <2 minutes, the AWOL tool may be easier to apply and disseminate than a previously described score relying on the MMSE, Acute Physiology and Chronic Health Evaluation scores, and measured visual acuity.[17] Two other tools, 1 based on chart abstraction[18] and the other based on clinical variables measured at admission,[19] are similarly easy to apply but only predict prevalent and not incident delirium, making them less clinically useful.
This study's strengths include its prospective cohort design and the derivation and validation being performed in different hospitals. The derivation cohort consisted of patients admitted to a tertiary care academic medical center or an affiliated hospital where routine mixed gender general medical patients are treated, whereas validation was performed at the SFVAMC, where patients are predominantly older men with a high incidence of vascular risk factors. The outcome was assessed on a daily basis, and the likelihood any cases were missed was low. Although there is some potential for bias because the outcome was assessed by a research assistant not blinded to baseline characteristics, this was mitigated by having each outcome validated by a blinded neurologist and in the validation cohort having the research assistant blinded to the AWOL score. Other strengths are the broad inclusion criteria, with both middle‐aged and elderly patients having a wide range of medical and neurological conditions, allowing for wide application of the results. Although many studies of delirium focus on patients over age 70 years, we chose to include patients aged 50 years or older because hospital‐acquired delirium still occurs in this age group (17 of 195 [8%] patients aged 5069 years became delirious in this study), and risk factors such as severe illness and cognitive dysfunction are likely to be predictors of delirium even at younger ages. Additionally, the inclusion of nurses' clinical judgment to assess illness severity using a straightforward rating scale allows bedside nurses to readily administer the prediction rule in practice.[34]
This study has several potential limitations. The number of outcomes in the derivation cohort was small compared to the number of predictors chosen for the prediction rule. This could potentially have led to overfitting the model in the derivation cohort and thus an overly optimistic estimation of the model's performance. In the validation cohort, the area under the ROC curve was lower than in the derivation cohort, and although the difference did not reach statistical significance, this may have been due to the small sample size. In addition, none of the 4 subjects with an AWOL score of 4 became delirious, potentially reflecting poor calibration of the prediction rule. However, the trend of higher rates of delirium among subjects with higher AWOL scores still reached statistical significance, and the prediction rule demonstrated good discrimination between patients at high and low risk for developing delirium.
To test whether a better prediction tool could be derived from our data, we combined the derivation and validation cohorts and repeated a stepwise multivariable logistic regression with the same variables used for derivation of the AWOL tool (with the exception of the whisper test of hearing and a past medical history of dementia, because these data were not collected in the validation cohort). This model produced the same 4 independent predictors of delirium used in the AWOL tool. We then used bootstrapping to internally validate the prediction rule, suggesting that the predictors in the AWOL tool were the best fit for the available data. However, given the small number of outcomes in our study, the AWOL tool may benefit from further validation in a larger independent cohort to more precisely calibrate the number of expected outcomes with each score.
Although the majority of medical inpatients were eligible for enrollment in our study, some populations were excluded, and our results may not generalize to these populations. Non‐English speaking patients were excluded to preserve the validity of our study instruments. In addition, patients with profound aphasia or an admission diagnosis of alcohol withdrawal were excluded. Patients discharged on the first day of their hospitalization were excluded either because they were discharged prior to screening or prior to their first follow‐up visit. Therefore, our results may only be valid in patients who remained in the hospital for over 24 hours. In addition, because we only included medical patients, our results cannot necessarily be generalized to the surgical population.
Finally, parts of the prediction rule (orientation and spelling world backward) are also components of the CAM and were used in the assessment of the outcome, and this may introduce a potential tautology: if patients are disoriented or have poor attention because they cannot spell world backward at admission, they already have fulfilled part of the criteria for delirium. However, a diagnosis of delirium using the CAM involves a comprehensive patient and caregiver interview, and in addition to poor attention, requires the presence of an acute change in mental status and disorganized thinking or altered level of consciousness. Therefore, it is possible, and common, for patients to be disoriented to place and/or unable to spell world backward, yet not be delirious, and predicting a subsequent change in cognition during the hospitalization is still clinically important. It is possible the AWOL tool works by identifying patients with impaired attention and subclinical delirium, but one could argue this makes a strong case for its validity because these patients especially should be triaged to an inpatient unit that specializes in delirium prevention. It is also possible the cognitive tasks that are part of the AWOL tool detect preexisting cognitive impairment, which is in turn a major risk factor for delirium.
Recognizing and classifying the risk of delirium during hospitalization is imperative, considering the illness' significant contribution to healthcare costs, morbidity, and mortality. The cost‐effectiveness of proven interventions to detect and prevent delirium could be magnified with focused implementation in those patients at highest risk.[39, 40, 41] Further research is required to determine whether the combination of delirium prediction rules such as those developed here and prevention strategies will result in decreased rates of delirium and economic savings for the healthcare system.
Acknowledgments
The following University of California, San Francisco neurology residents provided follow‐up of study subjects on weekends and were financially compensated: Amar Dhand, MD, DPhil; Tim West, MD; Sarah Shalev, MD; Karen DaSilva, MD; Mark Burish, MD, PhD; Maggie Waung, MD, PhD; Raquel Gardner, MD; Molly Burnett, MD; Adam Ziemann, MD, PhD; Kathryn Kvam, MD; Neel Singhal, MD, PhD; James Orengo, MD, PhD; Kelly Mills, MD; and Joanna Hellmuth, MD, MHS. The authors are grateful to Dr. Douglas Bauer for assisting with the study design.
Disclosures
Drs. Douglas, Hessler, Dhaliwal, Betjemann, Lucatorto, Johnston, Josephson, and Ms. Fukuda and Ms. Alameddine have no conflicts of interest or financial disclosures. This research was made possible by the Ruth E. Raskin Fund and a UCSF Dean's Research Scholarship. These funding agencies had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
- , , . Occurrence and outcome of delirium in medical in‐patients: a systematic literature review. Age Ageing. 2006;35(4):350–364.
- , , , , , . Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591–598.
- , , , , . One‐year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27–32.
- , , , , . Does delirium contribute to poor hospital outcomes? A three‐site epidemiologic study. J Gen Intern Med. 1998;13(4):234–242.
- , , , , , . Delirium duration and mortality in lightly sedated, mechanically ventilated intensive care patients. Crit Care Med. 2010;38(12):2311–2318.
- , , , et al. Delirium epidemiology in critical care (DECCA): an international study. Crit Care. 2010;14(6):R210.
- , , , et al. Delirium as a predictor of long‐term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513–1520.
- , , , , , . Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta‐analysis. JAMA. 2010;304(4):443–451.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156(12):848–856.
- , , , et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669–676.
- , , , et al. Simple cognitive testing (Mini‐Cog) predicts in‐hospital delirium in the elderly. J Am Geriatr Soc. 2007;55(2):314–316.
- , , . A prospective study of delirium in hospitalized elderly. JAMA. 1990;263(8):1097–1101.
- , . Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275(11):852–857.
- , , , , , . Risk factors for delirium at discharge: development and validation of a predictive model. Arch Intern Med. 2007;167(13):1406–1413.
- , . Delirium in vascular surgery. Eur J Vasc Endovasc Surg. 2007;34(2):131–134.
- , , , , , . Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42(8):809–815.
- , , , , . A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119(6):474–481.
- , , , , , . Validation of a medical record‐based delirium risk assessment. J Am Geriatr Soc. 2011;59(suppl 2):S289–S294.
- , , , et al. Derivation and validation of a clinical prediction rule for delirium in patients admitted to a medical ward: an observational study. BMJ Open. 2012;2(5) pii: e001599.
- , , , et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134–139.
- , , , , , . Prediction of postoperative delirium after abdominal surgery in the elderly. J Anesth. 2009;23(1):51–56.
- , , , et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229–236.
- , , , et al. Development and validation of PRE‐DELIRIC (PREdiction of DELIRium in ICu patients) delirium prediction model for intensive care patients: observational multicentre study. BMJ. 2012;344:e420.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , . “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198.
- . Wechsler Memory Scale‐III. New York, NY: Psychological Corp.; 1997.
- , , , , . The mini‐cog: a cognitive 'vital signs' measure for dementia screening in multi‐lingual elderly. Int J Geriatr Psychiatry. 2000;15(11):1021–1027.
- , . Functional evaluation: the Barthel index. Md State Med J. 1965;14:61–65.
- , , . The CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry. 1974;131(10):1121–1123.
- , , , , , . Is the NEI‐VFQ‐25 a useful tool in identifying visual impairment in an elderly population? BMC Ophthalmol. 2006;6:24.
- , , , et al. Validation of self‐reported hearing loss. The Blue Mountains Hearing Study. Int J Epidemiol. 2001;30(6):1371–1378.
- , , . Does this patient have hearing impairment? JAMA. 2006;295(4):416–428.
- , , , , , . Realizing the potential of clinical judgment: a real‐time strategy for predicting outcomes and cost for medical inpatients. Am J Med. 2000;109(3):189–195.
- , , , , , . Assessing illness severity: does clinical judgment work? J Chronic Dis. 1986;39(6):439–452.
- , , , , , . Prognostication in acutely admitted older patients by nurses and physicians. J Gen Intern Med. 2008;23(11):1883–1889.
- . The Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, CT: Yale University School of Medicine; 2003.
- , , , . Acute confusional states and dementia in the elderly: the role of dehydration/volume depletion, physical illness and age. Age Ageing. 1980;9(3):137–146.
- . A Wilcoxon‐type test for trend. Stat Med. 1985;4(1):87–90.
- , , , . Synopsis of the National Institute for Health and Clinical Excellence guideline for prevention of delirium. Ann Intern Med. 2011;154(11):746–751.
- , , , , . The Hospital Elder Life Program: a model of care to prevent cognitive and functional decline in older hospitalized patients. Hospital Elder Life Program. J Am Geriatr Soc. 2000;48(12):1697–1706.
- , , , , . A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338–1344.
Delirium is characterized by fluctuating disturbances in cognition and consciousness and is a common complication of hospitalization in medical and surgical patients. Studies estimate the prevalence of delirium in hospitalized patients[1] to be 14% to 56%, and up to 70% in critically ill elderly patients.[2] Estimates of total healthcare costs associated with delirium range from $38 to $152 billion per year in the United States.[3] Delirious patients are more likely to be discharged to a nursing home and have increased hospital mortality and longer lengths of stay.[4, 5, 6] Recent data suggest long‐term effects of delirium including cognitive impairments up to 1 year following the illness[7] and an increased likelihood of developing[8] or worsening dementia.[9]
It is estimated that one‐third of hospital‐acquired delirium cases could be prevented with appropriate interventions.[10] A prediction rule that easily and accurately identifies high‐risk patients upon admission could therefore have a substantial clinical impact. In addition, a prediction rule could be used to identify patients in whom new targeted interventions for delirium prevention could be investigated. A number of risk factors for delirium have been identified, including older age, preexisting cognitive dysfunction, vision and hearing impairment, severe illness, dehydration, electrolyte abnormalities, overmedication, and alcohol abuse.[11, 12, 13, 14, 15, 16] Existing prediction rules using various combinations of these measures have been limited by their complexity,[17] do not predict incident delirium,[18, 19] or are restricted to surgical[20, 21, 22] or intensive care[23] patients and therefore are not broadly applicable to the general medical population, which is particularly susceptible to developing delirium.
We conducted this study to develop a simple, efficient, and accurate prediction rule for hospital‐acquired delirium in adult medical inpatients assessed at the time of admission. Our a priori hypothesis was that a delirium prediction rule would consist of a combination of known risk factors and most likely incorporate old age, illness severity, and preexisting cognitive dysfunction.
METHODS
Design and Setting
This was a prospective cohort study with a derivation phase from May 2010 to November 2010 at 2 hospitals at the University of California, San Francisco (UCSF) (Moffitt‐Long and Mount Zion Hospitals) and a validation phase from October 2011 to March 2012 at the San Francisco Veterans Affairs Medical Center (SFVAMC).
Participants and Measurements
Subject identification, recruitment, and inclusion and exclusion criteria were identical for the derivation and validation cohorts. Subjects were identified by reviewing daily admission logs. All non‐intensive care unit patients aged 50 years or older admitted through the emergency department to the medicine, cardiology, or neurology services were screened for eligibility through chart review or in person within 24 hours of admission by a trained research assistant. One research assistant, a college graduate, conducted all screening for the derivation cohort, and 2 research assistants, 1 a fourth‐year medical student and the other a third‐year psychology graduate student, conducted screening for the validation cohort. In‐person screening included an assessment for delirium using the long version of the Confusion Assessment Method (CAM).[24] To minimize the possibility of enrolling delirious subjects, research assistants were instructed to notify the study supervisor (V.C.D.), a board‐certified neurologist, to discuss every case in which any yes checkbox was marked on the CAM score sheet. Subjects delirious upon initial evaluation, admitted for alcohol withdrawal, admitted for comfort care, who were aphasic or who could not speak English were excluded. For all patients, or if they were unable to provide consent, their surrogates provided written informed consent, and the study was approved by the institutional review boards at UCSF and SFVAMC.
In the derivation cohort, 1241 patients were screened, and 439 were eligible for enrollment. Of these, 180 declined, 50 were discharged prior to the first follow‐up visit, and 209 were included. In the validation cohort, 420 patients were screened, and 368 were eligible for enrollment. Of these, 144 declined, 59 were discharged prior to the first follow‐up visit, and 165 were included.
Baseline data regarding known delirium risk factors[11, 12, 13, 14, 15, 16] were collected from subjects in the derivation cohort. Cognitive performance was assessed with the Mini Mental Status Examination (MMSE),[25] forward digit span,[26] and clock draw.[27] Permission for administration of the MMSE was granted by Psychological Assessment Resources, Inc., and each administration was paid for. A structured interview was conducted with validated questions regarding visual and hearing impairment, pain, mobility, place of residence, and alcohol, tobacco, and drug use.[28, 29, 30, 31] A whisper test for hearing loss was performed.[32] Subjects' charts were reviewed for demographic, clinical, and laboratory data. Illness severity was assessed by asking each subject's nurse to rate their patient on a scale from not ill to mildly ill, moderately ill, severely ill, or moribund.[33] Each nurse was shown these 5 choices, but more specific definitions of what each level of illness severity meant were not provided. We chose this method to assess illness severity because this rating scale was incorporated into a previous validated and widely cited delirium prediction rule.[17] This illness severity scale has been validated as a predictor of outcomes and correlates with other measures of illness severity and comorbidity when graded by physicians.[33, 34] Nurse and physician ratings of illness severity have been shown to be comparable,[35] and therefore if the scale were incorporated into the prediction rule it would allow nurses to perform it independently. In the validation cohort, only data required to complete the baseline CAM and apply the prediction rule were collected.
Assessment of Outcomes
All subjects were assessed for delirium daily for 6 days after enrollment or until discharge, whichever came first. Follow‐up was limited to 6 days, based on the assumption that delirium occurring beyond 1 week is more likely due to events during the hospitalization as opposed to factors measurable at admission. Delirium was assessed using the short CAM, an internationally recognized and validated tool.[24] To complete the CAM during follow‐up visits, subjects and their nurses were interviewed using a written script, and an MMSE and forward digit span were performed.
Daily follow‐up assessments were performed by research assistants who were not blinded to the initial assessment but who, in the validation phase, were blinded to the prediction rule score. Some weekend follow‐ups were performed by postgraduate year 2, 3, or 4 neurology residents, or internal medicine faculty experienced in the assessment of delirium and blinded to both the initial assessment and prediction rule score. Neurology residents and internists read the CAM training manual and were educated in the administration and scoring of the CAM by 1 of the senior investigators (V.C.D.) prior to their first shift; these nonstudy personnel covered 17 of 189 days of follow‐up in the derivation cohort and 21 of 169 days of follow‐up in the validation cohort. To maximize sensitivity of delirium detection, for any change in cognition, MMSE score, or forward digit span compared to baseline, a board‐certified neurologist blinded to the initial assessment was notified to discuss the case and validate the diagnosis of delirium in person (derivation cohort) or over the phone (validation cohort). All research assistants were trained by a board‐certified neurologist (V.C.D.) in the administration and interpretation of the CAM using published methods prior to enrollment of any subjects.[36] Training included the performance of independent long‐version CAMs by the trainer and the trainee on a series of delirious and nondelirious patients until there was consistent agreement for each item on the CAM in 5 consecutive patients. In addition, a board‐certified neurologist supervised the first 5 administrations of the CAM performed by each research assistant.
Statistical Analysis
Sample size for the derivation cohort was based on the predicted ability to detect a difference in rates of delirium among those with and without cognitive impairment, the strongest risk factor for delirium. Using a [2] test with an of 0.05 and of 0.80, we estimated we would need to enroll 260 subjects, assuming a prevalence of cognitive dysfunction in our cohort of 10% and an estimated rate of delirium of 24% and 6% among those with and without cognitive dysfunction respectively.[14, 16, 17, 20] We were unable to reach enrollment targets because of a short funding period and slower than expected recruitment.
To construct the prediction rule in the derivation cohort, all variables were dichotomized. Age was dichotomized at 80 years because old age is a known risk factor for delirium, and only 1 of 46 subjects between the ages of 70 and 80 years became delirious in the derivation cohort. Components of the MMSE were dichotomized as correct/emncorrect, with a correct response requiring perfect performance based on expert consensus. For 3 subjects who would not attempt to spell world backward (2 in the derivation and 1 in the validation cohort), their score on serial 7s was used instead. The total MMSE score was not used because our objective was to develop a prediction rule using elements that could be assessed quickly in the fast‐paced environment of the hospital. Illness severity was dichotomized at moderate or worse/mild or better because there were only 15 subjects in the severe illness category, and the majority of delirium (22 outcomes) occurred in the moderate illness category. High blood urea nitrogen:creatinine ratio was defined as >18.[37]
The association between predictor variables and occurrence of delirium was analyzed using univariate logistic regression. A forward stepwise logistic regression was then performed using the variables associated with the outcome at a significance level of P<0.05 in univariate analysis. Variables were eligible for addition to the multivariable model if they were associated with the outcome at a significance level of <0.05. The 4 independent predictors thus identified were combined into a prediction rule by assigning each predictor 1 point if present. The performance of the prediction rule was assessed by using Cuzick's nonparametric test for a trend across groups ordered by score.[38]
The prediction rule was tested in the validation cohort using the nonparametric test for trend. Receiver operating characteristic (ROC) curves were compared between the derivation and validation cohorts. All statistical analysis was performed using Stata software (StataCorp, College Station, TX).
RESULTS
The derivation cohort consisted of elderly patients (mean age, 68.0811.96 years; interquartile range, 5096 years), and included more males than females (54.1% vs 45.9%). Subjects were predominantly white (73.7%) and lived at home (90%) (Table 1). The mean admission MMSE score was 27.0 (standard deviation [SD], 3.4; range, 730). Median follow‐up was 2 days (interquartile range, 13). Delirium developed in 12% (n=25) of the cohort.
| Derivation Cohort, N=209 | Validation Cohort, N=165 | |
|---|---|---|
| ||
| Gender, No. (%) | ||
| Male | 113 (54) | 157 (95) |
| Female | 96 (46) | 8 (4.8) |
| Race, No. (%) | ||
| White | 154 (74) | 125 (76) |
| African American | 34 (16) | 25 (15) |
| Asian | 21 (10.0) | 13 (7.9) |
| Native American | 0 | 2 (1.2) |
| Illness severity, No. (%) | ||
| Not ill | 1 (0.5) | 0 |
| Mildly ill | 49 (23) | 62 (38) |
| Moderately ill | 129 (62) | 86 (52) |
| Severely ill | 15 (7.2) | 17 (10) |
| Moribund | 0 | 0 |
| Living situation, No. (%) | ||
| Home | 188 (90) | 147 (89) |
| Assisted living | 11 (5.3) | 6 (3.6) |
| Hotel | 4 (1.9) | 5 (3.0) |
| SNF | 1 (0.5) | 3 (1.8) |
| Homeless | 4 (1.9) | 4 (2.4) |
| Developed delirium | 25 (12) | 14 (8.5) |
Univariate analysis of the derivation study identified 10 variables significantly associated (P<0.05) with delirium (Table 2). Predictors of delirium included abnormal scores on 4 subtests of the MMSE, low score on the Mini‐Cog, living in an assisted living or skilled nursing facility, moderate to severe illness, old age, a past history of dementia, and hearing loss as assessed by the whisper test. These predictors were then entered into a stepwise logistic regression analysis that identified 4 independent predictors of delirium (Table 3).
| Variable | No. (%) Without Delirium | No. (%) With Delirium | Odds Ratio | P Value | 95% Confidence Interval |
|---|---|---|---|---|---|
| |||||
| Age 80 years | 30 (16) | 13 (52) | 5.6 | <0.001 | 2.313.4 |
| Male sex | 99 (54) | 14 (56) | 1.1 | 0.84 | 0.52.5 |
| White race | 135 (73) | 19 (76) | 1.2 | 0.78 | 0.433.1 |
| Score <5 on date questions of MMSE | 37 (20) | 12 (48) | 3.7 | 0.003 | 1.68.7 |
| Score <5 on place questions of MMSE | 50 (27) | 14 (56) | 3.4 | 0.005 | 1.58.0 |
| Score <3 on MMSE recall | 89 (48) | 18 (72) | 2.7 | 0.03 | 1.16.9 |
| Score <5 on MMSE W‐O‐R‐L‐D backward | 37 (20) | 13 (52) | 4.3 | 0.001 | 1.810.2 |
| Score 0 on MMSE pentagon copy, n=203 | 53 (30) | 12 (48) | 2.2 | 0.07 | 0.935.1 |
| Score 0 on clock draw, n=203 | 70 (39) | 15 (60) | 2.3 | 0.05 | 0.985.4 |
| MiniCog score 02, n=203[27] | 46 (26) | 12 (48) | 2.7 | 0.03 | 1.16.2 |
| Self‐rated vision fair, poor, or very poor | 55 (30) | 8 (32) | 1.1 | 0.83 | 0.452.7 |
| Endorses hearing loss | 89 (48) | 12 (48) | 0.99 | 0.97 | 0.432.3 |
| Uses hearing aid | 19 (10) | 2 (8) | 0.76 | 0.72 | 0.173.5 |
| Fails whisper test in either ear | 39 (21) | 10 (40) | 2.5 | 0.04 | 1.05.9 |
| Prior episode of delirium per patient or informant | 70 (38) | 13 (52) | 1.8 | 0.19 | 0.764.1 |
| Dementia in past medical history | 3 (2) | 3 (12) | 8.2 | 0.01 | 1.643.3 |
| Depression in past medical history | 16 (9) | 1 (4) | 0.44 | 0.43 | 0.063.5 |
| Lives in assisted living or SNF | 8 (4) | 4 (16) | 4.2 | 0.03 | 1.215.1 |
| Endorses pain | 82 (45) | 7 (28) | 0.48 | 0.12 | 0.191.2 |
| Less than independent for transfers | 11 (6) | 3 (12) | 2.1 | 0.27 | 0.568.3 |
| Less than independent for mobility on a level surface | 36 (20) | 7 (28) | 1.6 | 0.33 | 0.624.1 |
| Score of 24 on CAGE questionnaire[29] | 5 (3) | 0 (0) | No outcomes | ||
| Drinks any alcohol | 84 (46) | 10 (40) | 0.79 | 0.60 | 0.341.9 |
| Current smoker | 20 (11) | 2 (8) | 0.71 | 0.66 | 0.164.1 |
| Uses illicit drugs | 13 (7) | 2 (8) | 1.2 | 0.83 | 0.255.6 |
| Moderately or severely ill on nursing assessment, n=194 | 121 (71) | 23 (96) | 9.3 | 0.031 | 1.270.9 |
| Fever | 8 (4) | 0 (0) | No outcomes | ||
| Serum sodium <134mmol/L | 38 (21) | 3 (12) | 0.52 | 0.31 | 0.151.8 |
| WBC count>10109/L, n=208 | 57 (31) | 6 (24) | 0.70 | 0.47 | 0.261.8 |
| AST>41 U/L, n=131 | 27 (23) | 2 (15) | 0.61 | 0.54 | 0.132.9 |
| BUN:Cr>18, n=208 | 66 (36) | 13 (52) | 1.9 | 0.13 | 0.834.5 |
| Infection as admission diagnosis | 28 (15) | 4 (16) | 1.1 | 0.92 | 0.343.3 |
| Variable | Odds Ratio | 95% Confidence Interval | P Value | Points Toward AWOL Score |
|---|---|---|---|---|
| Age 80 years | 5.7 | 2.115.6 | 0.001 | 1 |
| Unable to correctly spell world backward | 3.5 | 1.39.6 | 0.01 | 1 |
| Not oriented to city, state, county, hospital name, and floor | 2.9 | 1.17.9 | 0.03 | 1 |
| Nursing illness severity assessment of moderately ill, severely ill, or moribund (as opposed to not ill or mildly ill) | 10.5 | 1.386.9 | 0.03 | 1 |
These 4 independent predictors were assigned 1 point each if present to create a prediction rule with a range of possible scores from 0 to 4. There was a significant trend predicting higher rates of delirium with higher scores, with no subjects who scored 0 becoming delirious, compared to 40% of those subjects scoring 3 or 4 (P for trend<0.001) (Table 4).
| Derivation Cohorta | Validation Cohort | Combined Cohorts | ||||
|---|---|---|---|---|---|---|
| AWOL Score | Not Delirious | Delirious | Not Delirious | Delirious | Not Delirious | Delirious |
| ||||||
| 0 | 26 (100%) | 0 (0%) | 24 (96%) | 1 (4%) | 49 (98%) | 1 (2%) |
| 1 | 86 (95%) | 5 (5%) | 57 (97%) | 2 (3%) | 136 (96%) | 5 (4%) |
| 2 | 41 (85%) | 7 (15%) | 44 (90%) | 5 (10%) | 92 (86%) | 15 (14%) |
| 3 | 17 (74%) | 6 (26%) | 22 (79%) | 6 (21%) | 40 (80%) | 10 (20%) |
| 4 | 0 (0%) | 6 (100%) | 4 (100%) | 0 (0%) | 4 (36%) | 7 (64%) |
| Total | 170 | 24 | 151 | 14 | 321 | 38 |
| P<0.001 | P=0.025 | P<0.001 | ||||
The validation cohort consisted of adults with a mean age of 70.7210.6 years, (interquartile range, 5194 years), who were predominantly white (75.8%) and overwhelmingly male (95.2%) (Table 1). The mean admission MMSE score was 26.75 (SD, 2.8; range, 1730). Median follow‐up was 2 days (interquartile range, 15). Delirium developed in 8.5% (n=14) of the cohort. In the validation cohort, 4% of subjects with a score of 0 became delirious, whereas 19% of those scoring 3 or 4 became delirious (P for trend 0.025) (Table 4).
ROC curves were compared for the derivation and validation cohorts. The area under the ROC curve for the derivation cohort (0.81, 95% confidence interval [CI]: 0.720.90) was slightly better than that in the validation cohort (0.69, 95% CI: 0.540.83), but the difference did not reach statistical significance (P=0.14) (Figure 1).
DISCUSSION
We derived and validated a prediction rule to assess the risk of developing delirium in hospitalized adult medical patients. Four variables easily assessed on admission in a screen lasting less than 2 minutes were independently associated with the development of delirium. The prediction rule can be remembered with the following mnemonic: AWOL (Age80 years; unable to spell World backward; not fully Oriented to place; and moderate or severe iLlness severity).
It is estimated up to a third of hospital acquired delirium cases can be prevented.[10] Recent guidelines recommend the use of a multicomponent intervention to prevent delirium and provide evidence that such a strategy would be cost‐effective.[39] Nevertheless, such interventions are resource intense, requiring specialized nurse training and staffing[40] and have not been widely implemented. Acute care for the elderly units, where interventions to prevent delirium might logically be implemented, also require physical remodeling to provide carpeted hallways, handrails, and elevated toilet seats and door levers.[41] A method of risk stratification to identify the patients who would benefit most from resource‐intensive prevention strategies would be valuable.
The AWOL tool may provide a practical alternative to existing delirium prediction rules for adult medical inpatients. Because it can be completed by a nurse in <2 minutes, the AWOL tool may be easier to apply and disseminate than a previously described score relying on the MMSE, Acute Physiology and Chronic Health Evaluation scores, and measured visual acuity.[17] Two other tools, 1 based on chart abstraction[18] and the other based on clinical variables measured at admission,[19] are similarly easy to apply but only predict prevalent and not incident delirium, making them less clinically useful.
This study's strengths include its prospective cohort design and the derivation and validation being performed in different hospitals. The derivation cohort consisted of patients admitted to a tertiary care academic medical center or an affiliated hospital where routine mixed gender general medical patients are treated, whereas validation was performed at the SFVAMC, where patients are predominantly older men with a high incidence of vascular risk factors. The outcome was assessed on a daily basis, and the likelihood any cases were missed was low. Although there is some potential for bias because the outcome was assessed by a research assistant not blinded to baseline characteristics, this was mitigated by having each outcome validated by a blinded neurologist and in the validation cohort having the research assistant blinded to the AWOL score. Other strengths are the broad inclusion criteria, with both middle‐aged and elderly patients having a wide range of medical and neurological conditions, allowing for wide application of the results. Although many studies of delirium focus on patients over age 70 years, we chose to include patients aged 50 years or older because hospital‐acquired delirium still occurs in this age group (17 of 195 [8%] patients aged 5069 years became delirious in this study), and risk factors such as severe illness and cognitive dysfunction are likely to be predictors of delirium even at younger ages. Additionally, the inclusion of nurses' clinical judgment to assess illness severity using a straightforward rating scale allows bedside nurses to readily administer the prediction rule in practice.[34]
This study has several potential limitations. The number of outcomes in the derivation cohort was small compared to the number of predictors chosen for the prediction rule. This could potentially have led to overfitting the model in the derivation cohort and thus an overly optimistic estimation of the model's performance. In the validation cohort, the area under the ROC curve was lower than in the derivation cohort, and although the difference did not reach statistical significance, this may have been due to the small sample size. In addition, none of the 4 subjects with an AWOL score of 4 became delirious, potentially reflecting poor calibration of the prediction rule. However, the trend of higher rates of delirium among subjects with higher AWOL scores still reached statistical significance, and the prediction rule demonstrated good discrimination between patients at high and low risk for developing delirium.
To test whether a better prediction tool could be derived from our data, we combined the derivation and validation cohorts and repeated a stepwise multivariable logistic regression with the same variables used for derivation of the AWOL tool (with the exception of the whisper test of hearing and a past medical history of dementia, because these data were not collected in the validation cohort). This model produced the same 4 independent predictors of delirium used in the AWOL tool. We then used bootstrapping to internally validate the prediction rule, suggesting that the predictors in the AWOL tool were the best fit for the available data. However, given the small number of outcomes in our study, the AWOL tool may benefit from further validation in a larger independent cohort to more precisely calibrate the number of expected outcomes with each score.
Although the majority of medical inpatients were eligible for enrollment in our study, some populations were excluded, and our results may not generalize to these populations. Non‐English speaking patients were excluded to preserve the validity of our study instruments. In addition, patients with profound aphasia or an admission diagnosis of alcohol withdrawal were excluded. Patients discharged on the first day of their hospitalization were excluded either because they were discharged prior to screening or prior to their first follow‐up visit. Therefore, our results may only be valid in patients who remained in the hospital for over 24 hours. In addition, because we only included medical patients, our results cannot necessarily be generalized to the surgical population.
Finally, parts of the prediction rule (orientation and spelling world backward) are also components of the CAM and were used in the assessment of the outcome, and this may introduce a potential tautology: if patients are disoriented or have poor attention because they cannot spell world backward at admission, they already have fulfilled part of the criteria for delirium. However, a diagnosis of delirium using the CAM involves a comprehensive patient and caregiver interview, and in addition to poor attention, requires the presence of an acute change in mental status and disorganized thinking or altered level of consciousness. Therefore, it is possible, and common, for patients to be disoriented to place and/or unable to spell world backward, yet not be delirious, and predicting a subsequent change in cognition during the hospitalization is still clinically important. It is possible the AWOL tool works by identifying patients with impaired attention and subclinical delirium, but one could argue this makes a strong case for its validity because these patients especially should be triaged to an inpatient unit that specializes in delirium prevention. It is also possible the cognitive tasks that are part of the AWOL tool detect preexisting cognitive impairment, which is in turn a major risk factor for delirium.
Recognizing and classifying the risk of delirium during hospitalization is imperative, considering the illness' significant contribution to healthcare costs, morbidity, and mortality. The cost‐effectiveness of proven interventions to detect and prevent delirium could be magnified with focused implementation in those patients at highest risk.[39, 40, 41] Further research is required to determine whether the combination of delirium prediction rules such as those developed here and prevention strategies will result in decreased rates of delirium and economic savings for the healthcare system.
Acknowledgments
The following University of California, San Francisco neurology residents provided follow‐up of study subjects on weekends and were financially compensated: Amar Dhand, MD, DPhil; Tim West, MD; Sarah Shalev, MD; Karen DaSilva, MD; Mark Burish, MD, PhD; Maggie Waung, MD, PhD; Raquel Gardner, MD; Molly Burnett, MD; Adam Ziemann, MD, PhD; Kathryn Kvam, MD; Neel Singhal, MD, PhD; James Orengo, MD, PhD; Kelly Mills, MD; and Joanna Hellmuth, MD, MHS. The authors are grateful to Dr. Douglas Bauer for assisting with the study design.
Disclosures
Drs. Douglas, Hessler, Dhaliwal, Betjemann, Lucatorto, Johnston, Josephson, and Ms. Fukuda and Ms. Alameddine have no conflicts of interest or financial disclosures. This research was made possible by the Ruth E. Raskin Fund and a UCSF Dean's Research Scholarship. These funding agencies had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Delirium is characterized by fluctuating disturbances in cognition and consciousness and is a common complication of hospitalization in medical and surgical patients. Studies estimate the prevalence of delirium in hospitalized patients[1] to be 14% to 56%, and up to 70% in critically ill elderly patients.[2] Estimates of total healthcare costs associated with delirium range from $38 to $152 billion per year in the United States.[3] Delirious patients are more likely to be discharged to a nursing home and have increased hospital mortality and longer lengths of stay.[4, 5, 6] Recent data suggest long‐term effects of delirium including cognitive impairments up to 1 year following the illness[7] and an increased likelihood of developing[8] or worsening dementia.[9]
It is estimated that one‐third of hospital‐acquired delirium cases could be prevented with appropriate interventions.[10] A prediction rule that easily and accurately identifies high‐risk patients upon admission could therefore have a substantial clinical impact. In addition, a prediction rule could be used to identify patients in whom new targeted interventions for delirium prevention could be investigated. A number of risk factors for delirium have been identified, including older age, preexisting cognitive dysfunction, vision and hearing impairment, severe illness, dehydration, electrolyte abnormalities, overmedication, and alcohol abuse.[11, 12, 13, 14, 15, 16] Existing prediction rules using various combinations of these measures have been limited by their complexity,[17] do not predict incident delirium,[18, 19] or are restricted to surgical[20, 21, 22] or intensive care[23] patients and therefore are not broadly applicable to the general medical population, which is particularly susceptible to developing delirium.
We conducted this study to develop a simple, efficient, and accurate prediction rule for hospital‐acquired delirium in adult medical inpatients assessed at the time of admission. Our a priori hypothesis was that a delirium prediction rule would consist of a combination of known risk factors and most likely incorporate old age, illness severity, and preexisting cognitive dysfunction.
METHODS
Design and Setting
This was a prospective cohort study with a derivation phase from May 2010 to November 2010 at 2 hospitals at the University of California, San Francisco (UCSF) (Moffitt‐Long and Mount Zion Hospitals) and a validation phase from October 2011 to March 2012 at the San Francisco Veterans Affairs Medical Center (SFVAMC).
Participants and Measurements
Subject identification, recruitment, and inclusion and exclusion criteria were identical for the derivation and validation cohorts. Subjects were identified by reviewing daily admission logs. All non‐intensive care unit patients aged 50 years or older admitted through the emergency department to the medicine, cardiology, or neurology services were screened for eligibility through chart review or in person within 24 hours of admission by a trained research assistant. One research assistant, a college graduate, conducted all screening for the derivation cohort, and 2 research assistants, 1 a fourth‐year medical student and the other a third‐year psychology graduate student, conducted screening for the validation cohort. In‐person screening included an assessment for delirium using the long version of the Confusion Assessment Method (CAM).[24] To minimize the possibility of enrolling delirious subjects, research assistants were instructed to notify the study supervisor (V.C.D.), a board‐certified neurologist, to discuss every case in which any yes checkbox was marked on the CAM score sheet. Subjects delirious upon initial evaluation, admitted for alcohol withdrawal, admitted for comfort care, who were aphasic or who could not speak English were excluded. For all patients, or if they were unable to provide consent, their surrogates provided written informed consent, and the study was approved by the institutional review boards at UCSF and SFVAMC.
In the derivation cohort, 1241 patients were screened, and 439 were eligible for enrollment. Of these, 180 declined, 50 were discharged prior to the first follow‐up visit, and 209 were included. In the validation cohort, 420 patients were screened, and 368 were eligible for enrollment. Of these, 144 declined, 59 were discharged prior to the first follow‐up visit, and 165 were included.
Baseline data regarding known delirium risk factors[11, 12, 13, 14, 15, 16] were collected from subjects in the derivation cohort. Cognitive performance was assessed with the Mini Mental Status Examination (MMSE),[25] forward digit span,[26] and clock draw.[27] Permission for administration of the MMSE was granted by Psychological Assessment Resources, Inc., and each administration was paid for. A structured interview was conducted with validated questions regarding visual and hearing impairment, pain, mobility, place of residence, and alcohol, tobacco, and drug use.[28, 29, 30, 31] A whisper test for hearing loss was performed.[32] Subjects' charts were reviewed for demographic, clinical, and laboratory data. Illness severity was assessed by asking each subject's nurse to rate their patient on a scale from not ill to mildly ill, moderately ill, severely ill, or moribund.[33] Each nurse was shown these 5 choices, but more specific definitions of what each level of illness severity meant were not provided. We chose this method to assess illness severity because this rating scale was incorporated into a previous validated and widely cited delirium prediction rule.[17] This illness severity scale has been validated as a predictor of outcomes and correlates with other measures of illness severity and comorbidity when graded by physicians.[33, 34] Nurse and physician ratings of illness severity have been shown to be comparable,[35] and therefore if the scale were incorporated into the prediction rule it would allow nurses to perform it independently. In the validation cohort, only data required to complete the baseline CAM and apply the prediction rule were collected.
Assessment of Outcomes
All subjects were assessed for delirium daily for 6 days after enrollment or until discharge, whichever came first. Follow‐up was limited to 6 days, based on the assumption that delirium occurring beyond 1 week is more likely due to events during the hospitalization as opposed to factors measurable at admission. Delirium was assessed using the short CAM, an internationally recognized and validated tool.[24] To complete the CAM during follow‐up visits, subjects and their nurses were interviewed using a written script, and an MMSE and forward digit span were performed.
Daily follow‐up assessments were performed by research assistants who were not blinded to the initial assessment but who, in the validation phase, were blinded to the prediction rule score. Some weekend follow‐ups were performed by postgraduate year 2, 3, or 4 neurology residents, or internal medicine faculty experienced in the assessment of delirium and blinded to both the initial assessment and prediction rule score. Neurology residents and internists read the CAM training manual and were educated in the administration and scoring of the CAM by 1 of the senior investigators (V.C.D.) prior to their first shift; these nonstudy personnel covered 17 of 189 days of follow‐up in the derivation cohort and 21 of 169 days of follow‐up in the validation cohort. To maximize sensitivity of delirium detection, for any change in cognition, MMSE score, or forward digit span compared to baseline, a board‐certified neurologist blinded to the initial assessment was notified to discuss the case and validate the diagnosis of delirium in person (derivation cohort) or over the phone (validation cohort). All research assistants were trained by a board‐certified neurologist (V.C.D.) in the administration and interpretation of the CAM using published methods prior to enrollment of any subjects.[36] Training included the performance of independent long‐version CAMs by the trainer and the trainee on a series of delirious and nondelirious patients until there was consistent agreement for each item on the CAM in 5 consecutive patients. In addition, a board‐certified neurologist supervised the first 5 administrations of the CAM performed by each research assistant.
Statistical Analysis
Sample size for the derivation cohort was based on the predicted ability to detect a difference in rates of delirium among those with and without cognitive impairment, the strongest risk factor for delirium. Using a [2] test with an of 0.05 and of 0.80, we estimated we would need to enroll 260 subjects, assuming a prevalence of cognitive dysfunction in our cohort of 10% and an estimated rate of delirium of 24% and 6% among those with and without cognitive dysfunction respectively.[14, 16, 17, 20] We were unable to reach enrollment targets because of a short funding period and slower than expected recruitment.
To construct the prediction rule in the derivation cohort, all variables were dichotomized. Age was dichotomized at 80 years because old age is a known risk factor for delirium, and only 1 of 46 subjects between the ages of 70 and 80 years became delirious in the derivation cohort. Components of the MMSE were dichotomized as correct/emncorrect, with a correct response requiring perfect performance based on expert consensus. For 3 subjects who would not attempt to spell world backward (2 in the derivation and 1 in the validation cohort), their score on serial 7s was used instead. The total MMSE score was not used because our objective was to develop a prediction rule using elements that could be assessed quickly in the fast‐paced environment of the hospital. Illness severity was dichotomized at moderate or worse/mild or better because there were only 15 subjects in the severe illness category, and the majority of delirium (22 outcomes) occurred in the moderate illness category. High blood urea nitrogen:creatinine ratio was defined as >18.[37]
The association between predictor variables and occurrence of delirium was analyzed using univariate logistic regression. A forward stepwise logistic regression was then performed using the variables associated with the outcome at a significance level of P<0.05 in univariate analysis. Variables were eligible for addition to the multivariable model if they were associated with the outcome at a significance level of <0.05. The 4 independent predictors thus identified were combined into a prediction rule by assigning each predictor 1 point if present. The performance of the prediction rule was assessed by using Cuzick's nonparametric test for a trend across groups ordered by score.[38]
The prediction rule was tested in the validation cohort using the nonparametric test for trend. Receiver operating characteristic (ROC) curves were compared between the derivation and validation cohorts. All statistical analysis was performed using Stata software (StataCorp, College Station, TX).
RESULTS
The derivation cohort consisted of elderly patients (mean age, 68.0811.96 years; interquartile range, 5096 years), and included more males than females (54.1% vs 45.9%). Subjects were predominantly white (73.7%) and lived at home (90%) (Table 1). The mean admission MMSE score was 27.0 (standard deviation [SD], 3.4; range, 730). Median follow‐up was 2 days (interquartile range, 13). Delirium developed in 12% (n=25) of the cohort.
| Derivation Cohort, N=209 | Validation Cohort, N=165 | |
|---|---|---|
| ||
| Gender, No. (%) | ||
| Male | 113 (54) | 157 (95) |
| Female | 96 (46) | 8 (4.8) |
| Race, No. (%) | ||
| White | 154 (74) | 125 (76) |
| African American | 34 (16) | 25 (15) |
| Asian | 21 (10.0) | 13 (7.9) |
| Native American | 0 | 2 (1.2) |
| Illness severity, No. (%) | ||
| Not ill | 1 (0.5) | 0 |
| Mildly ill | 49 (23) | 62 (38) |
| Moderately ill | 129 (62) | 86 (52) |
| Severely ill | 15 (7.2) | 17 (10) |
| Moribund | 0 | 0 |
| Living situation, No. (%) | ||
| Home | 188 (90) | 147 (89) |
| Assisted living | 11 (5.3) | 6 (3.6) |
| Hotel | 4 (1.9) | 5 (3.0) |
| SNF | 1 (0.5) | 3 (1.8) |
| Homeless | 4 (1.9) | 4 (2.4) |
| Developed delirium | 25 (12) | 14 (8.5) |
Univariate analysis of the derivation study identified 10 variables significantly associated (P<0.05) with delirium (Table 2). Predictors of delirium included abnormal scores on 4 subtests of the MMSE, low score on the Mini‐Cog, living in an assisted living or skilled nursing facility, moderate to severe illness, old age, a past history of dementia, and hearing loss as assessed by the whisper test. These predictors were then entered into a stepwise logistic regression analysis that identified 4 independent predictors of delirium (Table 3).
| Variable | No. (%) Without Delirium | No. (%) With Delirium | Odds Ratio | P Value | 95% Confidence Interval |
|---|---|---|---|---|---|
| |||||
| Age 80 years | 30 (16) | 13 (52) | 5.6 | <0.001 | 2.313.4 |
| Male sex | 99 (54) | 14 (56) | 1.1 | 0.84 | 0.52.5 |
| White race | 135 (73) | 19 (76) | 1.2 | 0.78 | 0.433.1 |
| Score <5 on date questions of MMSE | 37 (20) | 12 (48) | 3.7 | 0.003 | 1.68.7 |
| Score <5 on place questions of MMSE | 50 (27) | 14 (56) | 3.4 | 0.005 | 1.58.0 |
| Score <3 on MMSE recall | 89 (48) | 18 (72) | 2.7 | 0.03 | 1.16.9 |
| Score <5 on MMSE W‐O‐R‐L‐D backward | 37 (20) | 13 (52) | 4.3 | 0.001 | 1.810.2 |
| Score 0 on MMSE pentagon copy, n=203 | 53 (30) | 12 (48) | 2.2 | 0.07 | 0.935.1 |
| Score 0 on clock draw, n=203 | 70 (39) | 15 (60) | 2.3 | 0.05 | 0.985.4 |
| MiniCog score 02, n=203[27] | 46 (26) | 12 (48) | 2.7 | 0.03 | 1.16.2 |
| Self‐rated vision fair, poor, or very poor | 55 (30) | 8 (32) | 1.1 | 0.83 | 0.452.7 |
| Endorses hearing loss | 89 (48) | 12 (48) | 0.99 | 0.97 | 0.432.3 |
| Uses hearing aid | 19 (10) | 2 (8) | 0.76 | 0.72 | 0.173.5 |
| Fails whisper test in either ear | 39 (21) | 10 (40) | 2.5 | 0.04 | 1.05.9 |
| Prior episode of delirium per patient or informant | 70 (38) | 13 (52) | 1.8 | 0.19 | 0.764.1 |
| Dementia in past medical history | 3 (2) | 3 (12) | 8.2 | 0.01 | 1.643.3 |
| Depression in past medical history | 16 (9) | 1 (4) | 0.44 | 0.43 | 0.063.5 |
| Lives in assisted living or SNF | 8 (4) | 4 (16) | 4.2 | 0.03 | 1.215.1 |
| Endorses pain | 82 (45) | 7 (28) | 0.48 | 0.12 | 0.191.2 |
| Less than independent for transfers | 11 (6) | 3 (12) | 2.1 | 0.27 | 0.568.3 |
| Less than independent for mobility on a level surface | 36 (20) | 7 (28) | 1.6 | 0.33 | 0.624.1 |
| Score of 24 on CAGE questionnaire[29] | 5 (3) | 0 (0) | No outcomes | ||
| Drinks any alcohol | 84 (46) | 10 (40) | 0.79 | 0.60 | 0.341.9 |
| Current smoker | 20 (11) | 2 (8) | 0.71 | 0.66 | 0.164.1 |
| Uses illicit drugs | 13 (7) | 2 (8) | 1.2 | 0.83 | 0.255.6 |
| Moderately or severely ill on nursing assessment, n=194 | 121 (71) | 23 (96) | 9.3 | 0.031 | 1.270.9 |
| Fever | 8 (4) | 0 (0) | No outcomes | ||
| Serum sodium <134mmol/L | 38 (21) | 3 (12) | 0.52 | 0.31 | 0.151.8 |
| WBC count>10109/L, n=208 | 57 (31) | 6 (24) | 0.70 | 0.47 | 0.261.8 |
| AST>41 U/L, n=131 | 27 (23) | 2 (15) | 0.61 | 0.54 | 0.132.9 |
| BUN:Cr>18, n=208 | 66 (36) | 13 (52) | 1.9 | 0.13 | 0.834.5 |
| Infection as admission diagnosis | 28 (15) | 4 (16) | 1.1 | 0.92 | 0.343.3 |
| Variable | Odds Ratio | 95% Confidence Interval | P Value | Points Toward AWOL Score |
|---|---|---|---|---|
| Age 80 years | 5.7 | 2.115.6 | 0.001 | 1 |
| Unable to correctly spell world backward | 3.5 | 1.39.6 | 0.01 | 1 |
| Not oriented to city, state, county, hospital name, and floor | 2.9 | 1.17.9 | 0.03 | 1 |
| Nursing illness severity assessment of moderately ill, severely ill, or moribund (as opposed to not ill or mildly ill) | 10.5 | 1.386.9 | 0.03 | 1 |
These 4 independent predictors were assigned 1 point each if present to create a prediction rule with a range of possible scores from 0 to 4. There was a significant trend predicting higher rates of delirium with higher scores, with no subjects who scored 0 becoming delirious, compared to 40% of those subjects scoring 3 or 4 (P for trend<0.001) (Table 4).
| Derivation Cohorta | Validation Cohort | Combined Cohorts | ||||
|---|---|---|---|---|---|---|
| AWOL Score | Not Delirious | Delirious | Not Delirious | Delirious | Not Delirious | Delirious |
| ||||||
| 0 | 26 (100%) | 0 (0%) | 24 (96%) | 1 (4%) | 49 (98%) | 1 (2%) |
| 1 | 86 (95%) | 5 (5%) | 57 (97%) | 2 (3%) | 136 (96%) | 5 (4%) |
| 2 | 41 (85%) | 7 (15%) | 44 (90%) | 5 (10%) | 92 (86%) | 15 (14%) |
| 3 | 17 (74%) | 6 (26%) | 22 (79%) | 6 (21%) | 40 (80%) | 10 (20%) |
| 4 | 0 (0%) | 6 (100%) | 4 (100%) | 0 (0%) | 4 (36%) | 7 (64%) |
| Total | 170 | 24 | 151 | 14 | 321 | 38 |
| P<0.001 | P=0.025 | P<0.001 | ||||
The validation cohort consisted of adults with a mean age of 70.7210.6 years, (interquartile range, 5194 years), who were predominantly white (75.8%) and overwhelmingly male (95.2%) (Table 1). The mean admission MMSE score was 26.75 (SD, 2.8; range, 1730). Median follow‐up was 2 days (interquartile range, 15). Delirium developed in 8.5% (n=14) of the cohort. In the validation cohort, 4% of subjects with a score of 0 became delirious, whereas 19% of those scoring 3 or 4 became delirious (P for trend 0.025) (Table 4).
ROC curves were compared for the derivation and validation cohorts. The area under the ROC curve for the derivation cohort (0.81, 95% confidence interval [CI]: 0.720.90) was slightly better than that in the validation cohort (0.69, 95% CI: 0.540.83), but the difference did not reach statistical significance (P=0.14) (Figure 1).
DISCUSSION
We derived and validated a prediction rule to assess the risk of developing delirium in hospitalized adult medical patients. Four variables easily assessed on admission in a screen lasting less than 2 minutes were independently associated with the development of delirium. The prediction rule can be remembered with the following mnemonic: AWOL (Age80 years; unable to spell World backward; not fully Oriented to place; and moderate or severe iLlness severity).
It is estimated up to a third of hospital acquired delirium cases can be prevented.[10] Recent guidelines recommend the use of a multicomponent intervention to prevent delirium and provide evidence that such a strategy would be cost‐effective.[39] Nevertheless, such interventions are resource intense, requiring specialized nurse training and staffing[40] and have not been widely implemented. Acute care for the elderly units, where interventions to prevent delirium might logically be implemented, also require physical remodeling to provide carpeted hallways, handrails, and elevated toilet seats and door levers.[41] A method of risk stratification to identify the patients who would benefit most from resource‐intensive prevention strategies would be valuable.
The AWOL tool may provide a practical alternative to existing delirium prediction rules for adult medical inpatients. Because it can be completed by a nurse in <2 minutes, the AWOL tool may be easier to apply and disseminate than a previously described score relying on the MMSE, Acute Physiology and Chronic Health Evaluation scores, and measured visual acuity.[17] Two other tools, 1 based on chart abstraction[18] and the other based on clinical variables measured at admission,[19] are similarly easy to apply but only predict prevalent and not incident delirium, making them less clinically useful.
This study's strengths include its prospective cohort design and the derivation and validation being performed in different hospitals. The derivation cohort consisted of patients admitted to a tertiary care academic medical center or an affiliated hospital where routine mixed gender general medical patients are treated, whereas validation was performed at the SFVAMC, where patients are predominantly older men with a high incidence of vascular risk factors. The outcome was assessed on a daily basis, and the likelihood any cases were missed was low. Although there is some potential for bias because the outcome was assessed by a research assistant not blinded to baseline characteristics, this was mitigated by having each outcome validated by a blinded neurologist and in the validation cohort having the research assistant blinded to the AWOL score. Other strengths are the broad inclusion criteria, with both middle‐aged and elderly patients having a wide range of medical and neurological conditions, allowing for wide application of the results. Although many studies of delirium focus on patients over age 70 years, we chose to include patients aged 50 years or older because hospital‐acquired delirium still occurs in this age group (17 of 195 [8%] patients aged 5069 years became delirious in this study), and risk factors such as severe illness and cognitive dysfunction are likely to be predictors of delirium even at younger ages. Additionally, the inclusion of nurses' clinical judgment to assess illness severity using a straightforward rating scale allows bedside nurses to readily administer the prediction rule in practice.[34]
This study has several potential limitations. The number of outcomes in the derivation cohort was small compared to the number of predictors chosen for the prediction rule. This could potentially have led to overfitting the model in the derivation cohort and thus an overly optimistic estimation of the model's performance. In the validation cohort, the area under the ROC curve was lower than in the derivation cohort, and although the difference did not reach statistical significance, this may have been due to the small sample size. In addition, none of the 4 subjects with an AWOL score of 4 became delirious, potentially reflecting poor calibration of the prediction rule. However, the trend of higher rates of delirium among subjects with higher AWOL scores still reached statistical significance, and the prediction rule demonstrated good discrimination between patients at high and low risk for developing delirium.
To test whether a better prediction tool could be derived from our data, we combined the derivation and validation cohorts and repeated a stepwise multivariable logistic regression with the same variables used for derivation of the AWOL tool (with the exception of the whisper test of hearing and a past medical history of dementia, because these data were not collected in the validation cohort). This model produced the same 4 independent predictors of delirium used in the AWOL tool. We then used bootstrapping to internally validate the prediction rule, suggesting that the predictors in the AWOL tool were the best fit for the available data. However, given the small number of outcomes in our study, the AWOL tool may benefit from further validation in a larger independent cohort to more precisely calibrate the number of expected outcomes with each score.
Although the majority of medical inpatients were eligible for enrollment in our study, some populations were excluded, and our results may not generalize to these populations. Non‐English speaking patients were excluded to preserve the validity of our study instruments. In addition, patients with profound aphasia or an admission diagnosis of alcohol withdrawal were excluded. Patients discharged on the first day of their hospitalization were excluded either because they were discharged prior to screening or prior to their first follow‐up visit. Therefore, our results may only be valid in patients who remained in the hospital for over 24 hours. In addition, because we only included medical patients, our results cannot necessarily be generalized to the surgical population.
Finally, parts of the prediction rule (orientation and spelling world backward) are also components of the CAM and were used in the assessment of the outcome, and this may introduce a potential tautology: if patients are disoriented or have poor attention because they cannot spell world backward at admission, they already have fulfilled part of the criteria for delirium. However, a diagnosis of delirium using the CAM involves a comprehensive patient and caregiver interview, and in addition to poor attention, requires the presence of an acute change in mental status and disorganized thinking or altered level of consciousness. Therefore, it is possible, and common, for patients to be disoriented to place and/or unable to spell world backward, yet not be delirious, and predicting a subsequent change in cognition during the hospitalization is still clinically important. It is possible the AWOL tool works by identifying patients with impaired attention and subclinical delirium, but one could argue this makes a strong case for its validity because these patients especially should be triaged to an inpatient unit that specializes in delirium prevention. It is also possible the cognitive tasks that are part of the AWOL tool detect preexisting cognitive impairment, which is in turn a major risk factor for delirium.
Recognizing and classifying the risk of delirium during hospitalization is imperative, considering the illness' significant contribution to healthcare costs, morbidity, and mortality. The cost‐effectiveness of proven interventions to detect and prevent delirium could be magnified with focused implementation in those patients at highest risk.[39, 40, 41] Further research is required to determine whether the combination of delirium prediction rules such as those developed here and prevention strategies will result in decreased rates of delirium and economic savings for the healthcare system.
Acknowledgments
The following University of California, San Francisco neurology residents provided follow‐up of study subjects on weekends and were financially compensated: Amar Dhand, MD, DPhil; Tim West, MD; Sarah Shalev, MD; Karen DaSilva, MD; Mark Burish, MD, PhD; Maggie Waung, MD, PhD; Raquel Gardner, MD; Molly Burnett, MD; Adam Ziemann, MD, PhD; Kathryn Kvam, MD; Neel Singhal, MD, PhD; James Orengo, MD, PhD; Kelly Mills, MD; and Joanna Hellmuth, MD, MHS. The authors are grateful to Dr. Douglas Bauer for assisting with the study design.
Disclosures
Drs. Douglas, Hessler, Dhaliwal, Betjemann, Lucatorto, Johnston, Josephson, and Ms. Fukuda and Ms. Alameddine have no conflicts of interest or financial disclosures. This research was made possible by the Ruth E. Raskin Fund and a UCSF Dean's Research Scholarship. These funding agencies had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
- , , . Occurrence and outcome of delirium in medical in‐patients: a systematic literature review. Age Ageing. 2006;35(4):350–364.
- , , , , , . Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591–598.
- , , , , . One‐year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27–32.
- , , , , . Does delirium contribute to poor hospital outcomes? A three‐site epidemiologic study. J Gen Intern Med. 1998;13(4):234–242.
- , , , , , . Delirium duration and mortality in lightly sedated, mechanically ventilated intensive care patients. Crit Care Med. 2010;38(12):2311–2318.
- , , , et al. Delirium epidemiology in critical care (DECCA): an international study. Crit Care. 2010;14(6):R210.
- , , , et al. Delirium as a predictor of long‐term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513–1520.
- , , , , , . Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta‐analysis. JAMA. 2010;304(4):443–451.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156(12):848–856.
- , , , et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669–676.
- , , , et al. Simple cognitive testing (Mini‐Cog) predicts in‐hospital delirium in the elderly. J Am Geriatr Soc. 2007;55(2):314–316.
- , , . A prospective study of delirium in hospitalized elderly. JAMA. 1990;263(8):1097–1101.
- , . Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275(11):852–857.
- , , , , , . Risk factors for delirium at discharge: development and validation of a predictive model. Arch Intern Med. 2007;167(13):1406–1413.
- , . Delirium in vascular surgery. Eur J Vasc Endovasc Surg. 2007;34(2):131–134.
- , , , , , . Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42(8):809–815.
- , , , , . A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119(6):474–481.
- , , , , , . Validation of a medical record‐based delirium risk assessment. J Am Geriatr Soc. 2011;59(suppl 2):S289–S294.
- , , , et al. Derivation and validation of a clinical prediction rule for delirium in patients admitted to a medical ward: an observational study. BMJ Open. 2012;2(5) pii: e001599.
- , , , et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134–139.
- , , , , , . Prediction of postoperative delirium after abdominal surgery in the elderly. J Anesth. 2009;23(1):51–56.
- , , , et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229–236.
- , , , et al. Development and validation of PRE‐DELIRIC (PREdiction of DELIRium in ICu patients) delirium prediction model for intensive care patients: observational multicentre study. BMJ. 2012;344:e420.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , . “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198.
- . Wechsler Memory Scale‐III. New York, NY: Psychological Corp.; 1997.
- , , , , . The mini‐cog: a cognitive 'vital signs' measure for dementia screening in multi‐lingual elderly. Int J Geriatr Psychiatry. 2000;15(11):1021–1027.
- , . Functional evaluation: the Barthel index. Md State Med J. 1965;14:61–65.
- , , . The CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry. 1974;131(10):1121–1123.
- , , , , , . Is the NEI‐VFQ‐25 a useful tool in identifying visual impairment in an elderly population? BMC Ophthalmol. 2006;6:24.
- , , , et al. Validation of self‐reported hearing loss. The Blue Mountains Hearing Study. Int J Epidemiol. 2001;30(6):1371–1378.
- , , . Does this patient have hearing impairment? JAMA. 2006;295(4):416–428.
- , , , , , . Realizing the potential of clinical judgment: a real‐time strategy for predicting outcomes and cost for medical inpatients. Am J Med. 2000;109(3):189–195.
- , , , , , . Assessing illness severity: does clinical judgment work? J Chronic Dis. 1986;39(6):439–452.
- , , , , , . Prognostication in acutely admitted older patients by nurses and physicians. J Gen Intern Med. 2008;23(11):1883–1889.
- . The Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, CT: Yale University School of Medicine; 2003.
- , , , . Acute confusional states and dementia in the elderly: the role of dehydration/volume depletion, physical illness and age. Age Ageing. 1980;9(3):137–146.
- . A Wilcoxon‐type test for trend. Stat Med. 1985;4(1):87–90.
- , , , . Synopsis of the National Institute for Health and Clinical Excellence guideline for prevention of delirium. Ann Intern Med. 2011;154(11):746–751.
- , , , , . The Hospital Elder Life Program: a model of care to prevent cognitive and functional decline in older hospitalized patients. Hospital Elder Life Program. J Am Geriatr Soc. 2000;48(12):1697–1706.
- , , , , . A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338–1344.
- , , . Occurrence and outcome of delirium in medical in‐patients: a systematic literature review. Age Ageing. 2006;35(4):350–364.
- , , , , , . Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591–598.
- , , , , . One‐year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27–32.
- , , , , . Does delirium contribute to poor hospital outcomes? A three‐site epidemiologic study. J Gen Intern Med. 1998;13(4):234–242.
- , , , , , . Delirium duration and mortality in lightly sedated, mechanically ventilated intensive care patients. Crit Care Med. 2010;38(12):2311–2318.
- , , , et al. Delirium epidemiology in critical care (DECCA): an international study. Crit Care. 2010;14(6):R210.
- , , , et al. Delirium as a predictor of long‐term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513–1520.
- , , , , , . Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta‐analysis. JAMA. 2010;304(4):443–451.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156(12):848–856.
- , , , et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669–676.
- , , , et al. Simple cognitive testing (Mini‐Cog) predicts in‐hospital delirium in the elderly. J Am Geriatr Soc. 2007;55(2):314–316.
- , , . A prospective study of delirium in hospitalized elderly. JAMA. 1990;263(8):1097–1101.
- , . Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275(11):852–857.
- , , , , , . Risk factors for delirium at discharge: development and validation of a predictive model. Arch Intern Med. 2007;167(13):1406–1413.
- , . Delirium in vascular surgery. Eur J Vasc Endovasc Surg. 2007;34(2):131–134.
- , , , , , . Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42(8):809–815.
- , , , , . A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119(6):474–481.
- , , , , , . Validation of a medical record‐based delirium risk assessment. J Am Geriatr Soc. 2011;59(suppl 2):S289–S294.
- , , , et al. Derivation and validation of a clinical prediction rule for delirium in patients admitted to a medical ward: an observational study. BMJ Open. 2012;2(5) pii: e001599.
- , , , et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134–139.
- , , , , , . Prediction of postoperative delirium after abdominal surgery in the elderly. J Anesth. 2009;23(1):51–56.
- , , , et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229–236.
- , , , et al. Development and validation of PRE‐DELIRIC (PREdiction of DELIRium in ICu patients) delirium prediction model for intensive care patients: observational multicentre study. BMJ. 2012;344:e420.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , . “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198.
- . Wechsler Memory Scale‐III. New York, NY: Psychological Corp.; 1997.
- , , , , . The mini‐cog: a cognitive 'vital signs' measure for dementia screening in multi‐lingual elderly. Int J Geriatr Psychiatry. 2000;15(11):1021–1027.
- , . Functional evaluation: the Barthel index. Md State Med J. 1965;14:61–65.
- , , . The CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry. 1974;131(10):1121–1123.
- , , , , , . Is the NEI‐VFQ‐25 a useful tool in identifying visual impairment in an elderly population? BMC Ophthalmol. 2006;6:24.
- , , , et al. Validation of self‐reported hearing loss. The Blue Mountains Hearing Study. Int J Epidemiol. 2001;30(6):1371–1378.
- , , . Does this patient have hearing impairment? JAMA. 2006;295(4):416–428.
- , , , , , . Realizing the potential of clinical judgment: a real‐time strategy for predicting outcomes and cost for medical inpatients. Am J Med. 2000;109(3):189–195.
- , , , , , . Assessing illness severity: does clinical judgment work? J Chronic Dis. 1986;39(6):439–452.
- , , , , , . Prognostication in acutely admitted older patients by nurses and physicians. J Gen Intern Med. 2008;23(11):1883–1889.
- . The Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, CT: Yale University School of Medicine; 2003.
- , , , . Acute confusional states and dementia in the elderly: the role of dehydration/volume depletion, physical illness and age. Age Ageing. 1980;9(3):137–146.
- . A Wilcoxon‐type test for trend. Stat Med. 1985;4(1):87–90.
- , , , . Synopsis of the National Institute for Health and Clinical Excellence guideline for prevention of delirium. Ann Intern Med. 2011;154(11):746–751.
- , , , , . The Hospital Elder Life Program: a model of care to prevent cognitive and functional decline in older hospitalized patients. Hospital Elder Life Program. J Am Geriatr Soc. 2000;48(12):1697–1706.
- , , , , . A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338–1344.
Copyright © 2013 Society of Hospital Medicine
A Raw Deal
A 39‐year‐old woman presented to the emergency department (ED) with fever and headache. One to two weeks prior to presentation, she developed nightly fevers that gradually increased to as high as 39.4C. She subsequently developed generalized throbbing headaches, malaise, and diffuse body pain. The headache gradually worsened. The day prior to presentation, she developed photophobia, nausea, and vomiting. She also reported right scalp pain while combing her hair, difficulty emptying her bladder, and left buttock pain radiating down the leg. She denied rash, joint pain, visual changes, dysarthria, cough, chest pain, abdominal pain, or diarrhea.
Fever and headache can be explained by meningitis, encephalitis, or brain abscess. The combination is seen far more frequently, however, in patients with common systemic infections such as influenza. For either bacterial meningitis or influenza, a 2‐week course is prolonged and atypical. The progressive nature of the symptoms and photophobia suggest a chronic meningitis, and the development of nausea and vomiting, although nonspecific, is also consistent with elevated intracranial pressure. In a young woman, subacute fever and aches should prompt consideration of an autoimmune disorder such as systemic lupus erythematosus (SLE), although early central nervous system (CNS) involvement is atypical. Migraine headaches are characterized by light sensitivity, nausea, and vomiting and can be precipitated by a viral syndrome, but in this case, the headaches were present at the outset, and 2 weeks is too long for a migraine attack.
Pain while combing hair is not characteristic of the aforementioned syndromes. The scalp should be examined to confirm that there are no skin lesions associated with herpes zoster and no arterial prominence associated with temporal arteritis. She is young for the latter, which would otherwise be a suitable explanation for fever, headache, scalp tenderness, and visual complaints (usually impairment not photophobia).
Incomplete bladder emptying and left buttock pain suggest that there might be a concomitant lumbosacral myelopathy or radiculopathy. Some nonbacterial causes of meningitis such as cytomegalovirus (CMV), syphilis, and cancer simultaneously involve the CNS and peripheral nerve roots. It is also possible that the scalp tenderness associated with combing reflects a cervical sensory radiculopathy.
She had presented to the ED 2 and 4 days before the current (third) ED visit. Both times her main complaint was left buttock pain and left leg paresthesias. Although she had no skin lesions, she was diagnosed with prodromal herpes zoster in the S2 dermatomal distribution and was prescribed valacyclovir (to be started should eruptions develop, which never occurred).
She reported intermittent self‐limited fevers at 3‐ to 4‐week intervals during the prior 6 months; two fever episodes were accompanied by an influenza‐like illness, and one was associated with gastrointestinal symptoms. Her last fever prior to this evaluation was 6 weeks earlier when she was treated with azithromycin for suspected pneumonia at an outside facility.
Her past medical history included hypothyroidism, gastroesophageal reflux disease, diverticulitis, and gluten intolerance. Her medications included porcine (natural) thyroid, fish oil, ibuprofen, and acetaminophen. She lived in Michigan and traveled to the northeast United States (Maine, Cape Cod, New Hampshire, Connecticut, and Vermont) 7 months prior to this evaluation. She was married and had no pets at home. She denied any tobacco, alcohol, or illicit drug use.
Her illness now appears to be chronic, associated with fever, and multisystem (potentially involving the pulmonary and gastrointestinal tract). None of her medical problems would predispose her to subacute meningitis, myelopathy, or radiculopathy. Hypothyroidism raises the possibility of a concomitant autoimmune disorder which causes meningitis, such as SLE or Behet's disease. Sarcoidosis can cause chronic meningitis and neuropathy with concomitant lung and gastrointestinal involvement and rarely fever.
Residency in the upper Midwest increases exposure to chronic infections that rarely cause subacute meningitis such as histoplasmosis, blastomycosis, or human granulocytic anaplasmosis. Travel to the northeast United States 1 month before the onset of her symptoms raises the possibility of other endemic infections like Lyme disease, babesiosis, and tularemia, which may account for her recurrent fevers. Of these, Lyme is most likely to present as chronic meningitis with cranial neuropathy and radiculoneuropathy.
Although the diagnosis of pneumonia was made late in her 6‐month illness, its etiology and treatment may be relevant. If the recent pneumonia was viral, a subsequent viral meningitis may be manifesting now or may have triggered an autoimmune process, such as acute disseminated encephalomyelitis. Bacterial pneumonia is a common precursor to bacterial meningitis, and treatment with azithromycin for the pneumonia may have delayed the meningitis onset or muted its course; this should be taken into account when interpreting cerebrospinal fluid (CSF) culture results.
On physical examination, her temperature was 39.1C, blood pressure was 135/91 mm Hg, with pulse of 87 beats per minute, respiratory rate of 16 breaths per minute, and oxygenation saturation of 97% on room air. She appeared in distress and was covering her eyes. She was alert and oriented. She had photophobia and mild nuchal rigidity. Pupils were equal and reactive to light, but she could not tolerate the eye exam for papilledema. Lung, heart, and abdominal exam were normal. No cranial nerve abnormalities were noted, and muscle strength was 5/5 in all 4 extremities. She had decreased sensation to light touch with allodynia throughout her lower extremities in addition to the lateral portion of the right scalp, which was also tender to palpation. Deep tendon reflexes were 2+ and symmetric in her bilateral upper and lower extremities. She did not have joint swelling, edema, lymphadenopathy, or a rash.
Her fever, headache, nuchal rigidity and photophobia collectively suggest meningitis, which requires evaluation by a lumbar puncture. There is no rash that supports herpes zoster or SLE. She does not have signs of myelopathy that would explain the urinary complaints, but lower motor neuron involvement has not been excluded. The sensory abnormalities in the scalp and leg are consistent with a polyneuroradiculopathy. Anterior lateral scalp tenderness may signal trigeminal nerve involvement, whereas posterior scalp tenderness would localize to the upper cervical cord nerve roots. The contralateral distribution of the scalp and leg sensory deficits suggests a multifocal peripheral nervous system process rather than a single CNS lesion.
Initial laboratory data showed serum white blood cell count (WBC) of 12,000/mm3 (79% polymorphonuclear leukocytes). Hemoglobin was 14.2 g/dL, and platelets were 251,000/mm3. Electrolytes, renal function, and liver function were normal. Thyroid‐stimulating hormone, erythrocyte sedimentation rate, and C‐reactive protein were normal. Urinalysis was negative. Chest x‐ray was normal. Noncontrast head computed tomography (CT) was normal. The patient was unable to void; 500 mL of urine returned when catheterization was performed.
CSF WBC count was 1,280/mm3 (39% neutrophils and 49% lymphocytes). CSF total protein was 175 mg/dL, and glucose was 48 mg/dL; serum glucose was 104 mg/dL. Opening pressure was not recorded. Gram stain was negative. Ceftriaxone, vancomycin, ampicillin, and acyclovir were administered for presumed bacterial or viral meningitis. Magnetic resonance imaging (MRI) of the brain and spine showed diffuse leptomeningeal enhancement (Figure 1).
The urinary retention in the absence of myelopathic findings on exam or MRI suggests a sacral polyradiculoneuropathy. Diffuse leptomeningeal enhancement is consistent with many, if not all, causes of meningitis. The high WBC count, elevated protein, and low glucosecollectively signaling active inflammation in the CNSare highly compatible with bacterial meningitis, although the lymphocytic predominance and other clinical data point to nonbacterial etiologies. The negative Gram stain further lowers the probability of bacterial meningitis, but it has limited sensitivity, may be affected by recent antibiotics, and is typically negative with Listeria. Enterovirus, acute human immunodeficiency virus (HIV), and herpes viruses (eg, CMV or herpes simplex virus [HSV]) are important considerations, with the latter 2 causing associated polyneuroradiculopathy. Patients with genital HSV (not detected here) can have a concomitant sacral radiculitis leading to urinary retention.
Fungal and mycobacterial meningitis is a possibility (especially with the high protein), but the patient does not have the typical multisystem disease or immunosuppression that frequently accompanies those conditions when CNS disease is present. Autoimmune conditions like SLE, Behet's disease, and sarcoidosis remain important conditions, especially with the polyneuroradiculopathy or mononeuritis multiplex, which may reflect multifocal nerve infarction or invasion. Similarly, lymphomatous or carcinomatous meningitis should be considered, although an isolated manifestation in the CNS is unusual. Based on the multifocal neurologic deficits, I favor a viral, spirochete, or malignant etiology of her meningoencephalitis.
Despite ongoing broad spectrum antibiotics and supportive care, she became confused on hospital day 3 and developed anomia, agitation, and worsening headache. A repeat CT of the brain did not show any new abnormalities, but repeat lumbar puncture demonstrated elevated intracranial pressure (opening pressure of 47 cm water) with 427 WBC/mm3. Blood and CSF cultures remained negative.
Detailed questioning of the family revealed that she had been horseback riding 3 weeks prior to admission; there were no other livestock where she rode horses. In addition, the family reported that she and other family members routinely drank raw milk from a cow share program.
HIV antibody test was negative. Herpes simplex, varicella zoster, enteroviruses, and adenovirus CSF polymerase chain reaction (PCR) were negative. Cytomegalovirus and Epstein‐Barr virus PCR were negative in serum and CSF. Arbovirus, lymphocytic choriomeningitis, Coccidioides, Blastomyces, Histoplasma, Brucella, and Lyme serologies were negative. Cryptococcus neoformans antigen was negative in CSF. Serum QuantiFERON‐TB test was negative. Blood and CSF acid‐fast bacilli smears (and eventually mycobacterial cultures) were also negative. Her CSF flow cytometry and cytology were negative for lymphoma.
Unpasteurized milk conveys multiple infectious risks. Listeriosis is a food‐borne illness that can cause meningoencephalitis, but peripheral neuropathies are not characteristic. Brucellosis is usually characterized by severe bone pain, pancytopenia, and hepatosplenomegaly, which are absent. Infection with Mycobacterium bovis mimics Mycobacterium tuberculosis and can cause multisystem disease, typically involving the lung. Campylobacter infection is characterized by gastroenteritis, which has not been prominent.
Rhodococcus equi is a horse‐related pathogen which leads to pulmonary infections in immunocompromised hosts but not meningitis. Rather than focusing on horse exposure alone, however, it may be useful to consider her at risk for vector‐borne pathogens based on her time outdoors, such as Lyme disease (which can cause radiculopathy and encephalopathy), West Nile virus (although motor weakness rather than sensory symptoms is typical), or eastern equine encephalitis.
The absence of weight loss, cytopenias, lymphadenopathy, and organomegaly with the negative CSF cytology and flow cytometry makes lymphomatous meningitis unlikely. The case for an autoimmune disorder is not strong in the absence of joint pains, rash, or autoimmune serologies. In a young woman with unexplained encephalitis, antibodies to the N‐methyl‐D‐aspartate receptor should be assayed.
Although the CSF leukocytosis is declining, the elevated pressure and clinical deterioration signal that the disease process is not controlled. At this point I am uncertain as to the cause of her progressive meningoencephalitis with polyneuroradiculopathy. The latter feature makes me favor a viral or spirochete etiology.
On hospital day 4, Coxiella burnetii serologies were reported as positive (phase II immunoglobulin [Ig] G 1:256; phase II IgM <1:16; phase I IgG <1:16; phase I IgM <1:16) suggesting acute Q fever. Antibiotics were changed to intravenous doxycycline and ciprofloxacin. Her increased intracranial pressure was managed with serial lumbar punctures. The patient was discharged after 12 days of hospitalization taking oral doxycycline and ciprofloxacin. Her symptoms resolved over 10 weeks. No vegetations were seen on transesophageal echocardiogram. She had no evidence of chronic Q fever on repeat serologies.
I was not aware that Q fever causes meningitis or meningoencephalitis. However, I should have considered it in light of her indirect exposure to cows. It is possible that her pneumonia 6 weeks earlier represented acute Q fever, as pneumonia and hepatitis are among the most typical acute manifestations of this infection.
COMMENTARY
Hospitalists are commonly confronted by the combination of fever, headache, and confusion and are familiar with the diagnostic and therapeutic dilemmas related to prompt discrimination between CNS and non‐CNS processes, particularly infections. At the time of this patient's final ED presentation, her illness unambiguously localized to the CNS. As common and emergent conditions such as acute bacterial meningitis were excluded, the greatest challenge was finding the clue that could direct investigations into less common causes of meningoencephalitis.
The Infectious Disease Society of America has developed clinical practice guidelines for the diagnosis and management of encephalitis which highlight the importance of epidemiology and risk factor assessment.[1] This approach requires the clinician to examine potential clues and to go beyond initial associationsfor instance, not simply linking horseback riding to horse‐associated pathogens, but interpreting horseback riding as a proxy for outdoor exposure, which places her at risk for contact with mosquitos, which transmit West Nile virus or eastern equine encephalitis. Similarly, ingestion of raw milk, which is typically linked to Listeria monocytogenes, Brucella, and other pathogens prompted the infectious disease consultant to think more broadly and include livestock (cow)‐associated pathogens including C. burnetii.
Although involvement of the CNS is common in chronic Q fever endocarditis due to septic embolism, neurologic involvement in acute Q fever varies in prevalence (range of 1.7%22%).[2, 3, 4] The 3 major neurological syndromes of acute Q fever are (1) meningoencephalitis or encephalitis, (2) lymphocytic meningitis, and (3) peripheral neuropathy (myelitis, polyradiculoneuritis, or peripheral neuritis). CSF analysis usually shows mild pleocytosis with a predominance of lymphocytic cells; CSF protein elevation is variable, and glucose is usually normal. Neuroradiologic examination is usually normal, and there are no pathognomonic imaging abnormalities for Q fever meningoencephalitis.[2, 3] The mechanism by which C. burnetii causes neurologic injury and dysfunction is unknown.
The diagnosis of Q fever is usually established by serologic testing. In acute Q fever, antibodies to phase II antigen are higher than the phase I antibody titer. Phase II IgM antibodies are the first to appear, but then decline on average after week 8, often reaching undetectable levels 10 to 12 weeks after disease onset.[5] If this patient's pneumonia 6 weeks prior to this presentation was acute Q fever pneumonia, her IgM titers may have been declining by the time her neurologic illness developed. A false negative test result is also possible; immunofluorescence assays are more specific than sensitive in acute Q fever.[5]
Evaluating this case in isolation may raise some doubt as to the accuracy of the diagnosis as she did not have a 4‐fold rise in the phase II IgG titer and did not have a detectable phase II IgM. However, she was part of a cluster of individuals who regularly consumed raw milk from the same dairy and had evidence of C. burnetii infection. This group included her spouse, who had a robust serologic evidence of C. burnetii, characterized by a >4‐fold rise in phase II IgM and IgG titers.[6]
C. burnetii is found primarily in cattle, sheep, and goats and is shed in large quantities by infected periparturient animals in their urine, feces, and milk.[7] Inhalation of contaminated aerosols is the principal route of transmission.[7, 8] Acute Q fever is underdiagnosed because the majority of acute infections are asymptomatic (60%) or present as a nonspecific flu‐like illness.[7] This case represents a rare manifestation of a rare infection acquired through a rare route of transmission, but highlights the importance of epidemiology and risk factor assessment when clinicians are faced with a diagnostic challenge.
TEACHING POINTS
- Exploration of epidemiology and exposure history is central to diagnosing meningoencephalitis with negative bacterial cultures and undetectable HSV PCR, although the etiology of meningoencephalitis can elude identification even after exhaustive investigation.
- Inhalation of contaminated aerosols is the principal route of transmission for C. burnetii, but it can also be transmitted via infected unpasteurized milk.[7, 9]
- Acute presentations of Q fever, which may warrant admission, include pneumonia, hepatitis, or meningoencephalitis.
- Q fever is diagnosed by serologic testing, and doxycycline is the antibiotic of choice.
Disclosures
This case was presented at the 2012 Annual Meeting of the Society of Hospital Medicine. It was subsequently reported in the epidemiologic report of the outbreak.[6] The authors report no conflicts of interest.
- , , , et al. The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2008;47:303–327.
- , , , et al. Q fever 1985–1998 clinical and epidemiologic features of 1,383 Infections. Medicine. 2000;79:109–123.
- , , , et al. Neurological involvement in acute Q fever: a report of 29 cases and review of the literature. Arch Intern Med. 2002;162:693–700.
- , , . Q fever in Plymouth 1972–88, a review with particular reference to neurological manifestations. Epidemiol Infect. 1990;105:391–408.
- , , . Diagnosis of Q fever. J Clin Microbiol. 1998;36:1823–1834.
- , , . Q fever cluster among raw milk drinkers in Michigan, (2011). Clin Infect Dis. 2012;55:1387–1389.
- , . Q fever. Clin Micorbial Rev. 1999;12:18–53.
- , , , et al. A large outbreak of Q fever in the West Midlands: windborne spread into a metropolitan area? Commun Dis Public Health. 1998;1:180–187.
- , . A cluster of Coxiella burnetii infections associated with exposure to vaccinated goats and their unpasteurized dairy products. Am J Trop Med Hyg. 1992;47:35–40.
A 39‐year‐old woman presented to the emergency department (ED) with fever and headache. One to two weeks prior to presentation, she developed nightly fevers that gradually increased to as high as 39.4C. She subsequently developed generalized throbbing headaches, malaise, and diffuse body pain. The headache gradually worsened. The day prior to presentation, she developed photophobia, nausea, and vomiting. She also reported right scalp pain while combing her hair, difficulty emptying her bladder, and left buttock pain radiating down the leg. She denied rash, joint pain, visual changes, dysarthria, cough, chest pain, abdominal pain, or diarrhea.
Fever and headache can be explained by meningitis, encephalitis, or brain abscess. The combination is seen far more frequently, however, in patients with common systemic infections such as influenza. For either bacterial meningitis or influenza, a 2‐week course is prolonged and atypical. The progressive nature of the symptoms and photophobia suggest a chronic meningitis, and the development of nausea and vomiting, although nonspecific, is also consistent with elevated intracranial pressure. In a young woman, subacute fever and aches should prompt consideration of an autoimmune disorder such as systemic lupus erythematosus (SLE), although early central nervous system (CNS) involvement is atypical. Migraine headaches are characterized by light sensitivity, nausea, and vomiting and can be precipitated by a viral syndrome, but in this case, the headaches were present at the outset, and 2 weeks is too long for a migraine attack.
Pain while combing hair is not characteristic of the aforementioned syndromes. The scalp should be examined to confirm that there are no skin lesions associated with herpes zoster and no arterial prominence associated with temporal arteritis. She is young for the latter, which would otherwise be a suitable explanation for fever, headache, scalp tenderness, and visual complaints (usually impairment not photophobia).
Incomplete bladder emptying and left buttock pain suggest that there might be a concomitant lumbosacral myelopathy or radiculopathy. Some nonbacterial causes of meningitis such as cytomegalovirus (CMV), syphilis, and cancer simultaneously involve the CNS and peripheral nerve roots. It is also possible that the scalp tenderness associated with combing reflects a cervical sensory radiculopathy.
She had presented to the ED 2 and 4 days before the current (third) ED visit. Both times her main complaint was left buttock pain and left leg paresthesias. Although she had no skin lesions, she was diagnosed with prodromal herpes zoster in the S2 dermatomal distribution and was prescribed valacyclovir (to be started should eruptions develop, which never occurred).
She reported intermittent self‐limited fevers at 3‐ to 4‐week intervals during the prior 6 months; two fever episodes were accompanied by an influenza‐like illness, and one was associated with gastrointestinal symptoms. Her last fever prior to this evaluation was 6 weeks earlier when she was treated with azithromycin for suspected pneumonia at an outside facility.
Her past medical history included hypothyroidism, gastroesophageal reflux disease, diverticulitis, and gluten intolerance. Her medications included porcine (natural) thyroid, fish oil, ibuprofen, and acetaminophen. She lived in Michigan and traveled to the northeast United States (Maine, Cape Cod, New Hampshire, Connecticut, and Vermont) 7 months prior to this evaluation. She was married and had no pets at home. She denied any tobacco, alcohol, or illicit drug use.
Her illness now appears to be chronic, associated with fever, and multisystem (potentially involving the pulmonary and gastrointestinal tract). None of her medical problems would predispose her to subacute meningitis, myelopathy, or radiculopathy. Hypothyroidism raises the possibility of a concomitant autoimmune disorder which causes meningitis, such as SLE or Behet's disease. Sarcoidosis can cause chronic meningitis and neuropathy with concomitant lung and gastrointestinal involvement and rarely fever.
Residency in the upper Midwest increases exposure to chronic infections that rarely cause subacute meningitis such as histoplasmosis, blastomycosis, or human granulocytic anaplasmosis. Travel to the northeast United States 1 month before the onset of her symptoms raises the possibility of other endemic infections like Lyme disease, babesiosis, and tularemia, which may account for her recurrent fevers. Of these, Lyme is most likely to present as chronic meningitis with cranial neuropathy and radiculoneuropathy.
Although the diagnosis of pneumonia was made late in her 6‐month illness, its etiology and treatment may be relevant. If the recent pneumonia was viral, a subsequent viral meningitis may be manifesting now or may have triggered an autoimmune process, such as acute disseminated encephalomyelitis. Bacterial pneumonia is a common precursor to bacterial meningitis, and treatment with azithromycin for the pneumonia may have delayed the meningitis onset or muted its course; this should be taken into account when interpreting cerebrospinal fluid (CSF) culture results.
On physical examination, her temperature was 39.1C, blood pressure was 135/91 mm Hg, with pulse of 87 beats per minute, respiratory rate of 16 breaths per minute, and oxygenation saturation of 97% on room air. She appeared in distress and was covering her eyes. She was alert and oriented. She had photophobia and mild nuchal rigidity. Pupils were equal and reactive to light, but she could not tolerate the eye exam for papilledema. Lung, heart, and abdominal exam were normal. No cranial nerve abnormalities were noted, and muscle strength was 5/5 in all 4 extremities. She had decreased sensation to light touch with allodynia throughout her lower extremities in addition to the lateral portion of the right scalp, which was also tender to palpation. Deep tendon reflexes were 2+ and symmetric in her bilateral upper and lower extremities. She did not have joint swelling, edema, lymphadenopathy, or a rash.
Her fever, headache, nuchal rigidity and photophobia collectively suggest meningitis, which requires evaluation by a lumbar puncture. There is no rash that supports herpes zoster or SLE. She does not have signs of myelopathy that would explain the urinary complaints, but lower motor neuron involvement has not been excluded. The sensory abnormalities in the scalp and leg are consistent with a polyneuroradiculopathy. Anterior lateral scalp tenderness may signal trigeminal nerve involvement, whereas posterior scalp tenderness would localize to the upper cervical cord nerve roots. The contralateral distribution of the scalp and leg sensory deficits suggests a multifocal peripheral nervous system process rather than a single CNS lesion.
Initial laboratory data showed serum white blood cell count (WBC) of 12,000/mm3 (79% polymorphonuclear leukocytes). Hemoglobin was 14.2 g/dL, and platelets were 251,000/mm3. Electrolytes, renal function, and liver function were normal. Thyroid‐stimulating hormone, erythrocyte sedimentation rate, and C‐reactive protein were normal. Urinalysis was negative. Chest x‐ray was normal. Noncontrast head computed tomography (CT) was normal. The patient was unable to void; 500 mL of urine returned when catheterization was performed.
CSF WBC count was 1,280/mm3 (39% neutrophils and 49% lymphocytes). CSF total protein was 175 mg/dL, and glucose was 48 mg/dL; serum glucose was 104 mg/dL. Opening pressure was not recorded. Gram stain was negative. Ceftriaxone, vancomycin, ampicillin, and acyclovir were administered for presumed bacterial or viral meningitis. Magnetic resonance imaging (MRI) of the brain and spine showed diffuse leptomeningeal enhancement (Figure 1).
The urinary retention in the absence of myelopathic findings on exam or MRI suggests a sacral polyradiculoneuropathy. Diffuse leptomeningeal enhancement is consistent with many, if not all, causes of meningitis. The high WBC count, elevated protein, and low glucosecollectively signaling active inflammation in the CNSare highly compatible with bacterial meningitis, although the lymphocytic predominance and other clinical data point to nonbacterial etiologies. The negative Gram stain further lowers the probability of bacterial meningitis, but it has limited sensitivity, may be affected by recent antibiotics, and is typically negative with Listeria. Enterovirus, acute human immunodeficiency virus (HIV), and herpes viruses (eg, CMV or herpes simplex virus [HSV]) are important considerations, with the latter 2 causing associated polyneuroradiculopathy. Patients with genital HSV (not detected here) can have a concomitant sacral radiculitis leading to urinary retention.
Fungal and mycobacterial meningitis is a possibility (especially with the high protein), but the patient does not have the typical multisystem disease or immunosuppression that frequently accompanies those conditions when CNS disease is present. Autoimmune conditions like SLE, Behet's disease, and sarcoidosis remain important conditions, especially with the polyneuroradiculopathy or mononeuritis multiplex, which may reflect multifocal nerve infarction or invasion. Similarly, lymphomatous or carcinomatous meningitis should be considered, although an isolated manifestation in the CNS is unusual. Based on the multifocal neurologic deficits, I favor a viral, spirochete, or malignant etiology of her meningoencephalitis.
Despite ongoing broad spectrum antibiotics and supportive care, she became confused on hospital day 3 and developed anomia, agitation, and worsening headache. A repeat CT of the brain did not show any new abnormalities, but repeat lumbar puncture demonstrated elevated intracranial pressure (opening pressure of 47 cm water) with 427 WBC/mm3. Blood and CSF cultures remained negative.
Detailed questioning of the family revealed that she had been horseback riding 3 weeks prior to admission; there were no other livestock where she rode horses. In addition, the family reported that she and other family members routinely drank raw milk from a cow share program.
HIV antibody test was negative. Herpes simplex, varicella zoster, enteroviruses, and adenovirus CSF polymerase chain reaction (PCR) were negative. Cytomegalovirus and Epstein‐Barr virus PCR were negative in serum and CSF. Arbovirus, lymphocytic choriomeningitis, Coccidioides, Blastomyces, Histoplasma, Brucella, and Lyme serologies were negative. Cryptococcus neoformans antigen was negative in CSF. Serum QuantiFERON‐TB test was negative. Blood and CSF acid‐fast bacilli smears (and eventually mycobacterial cultures) were also negative. Her CSF flow cytometry and cytology were negative for lymphoma.
Unpasteurized milk conveys multiple infectious risks. Listeriosis is a food‐borne illness that can cause meningoencephalitis, but peripheral neuropathies are not characteristic. Brucellosis is usually characterized by severe bone pain, pancytopenia, and hepatosplenomegaly, which are absent. Infection with Mycobacterium bovis mimics Mycobacterium tuberculosis and can cause multisystem disease, typically involving the lung. Campylobacter infection is characterized by gastroenteritis, which has not been prominent.
Rhodococcus equi is a horse‐related pathogen which leads to pulmonary infections in immunocompromised hosts but not meningitis. Rather than focusing on horse exposure alone, however, it may be useful to consider her at risk for vector‐borne pathogens based on her time outdoors, such as Lyme disease (which can cause radiculopathy and encephalopathy), West Nile virus (although motor weakness rather than sensory symptoms is typical), or eastern equine encephalitis.
The absence of weight loss, cytopenias, lymphadenopathy, and organomegaly with the negative CSF cytology and flow cytometry makes lymphomatous meningitis unlikely. The case for an autoimmune disorder is not strong in the absence of joint pains, rash, or autoimmune serologies. In a young woman with unexplained encephalitis, antibodies to the N‐methyl‐D‐aspartate receptor should be assayed.
Although the CSF leukocytosis is declining, the elevated pressure and clinical deterioration signal that the disease process is not controlled. At this point I am uncertain as to the cause of her progressive meningoencephalitis with polyneuroradiculopathy. The latter feature makes me favor a viral or spirochete etiology.
On hospital day 4, Coxiella burnetii serologies were reported as positive (phase II immunoglobulin [Ig] G 1:256; phase II IgM <1:16; phase I IgG <1:16; phase I IgM <1:16) suggesting acute Q fever. Antibiotics were changed to intravenous doxycycline and ciprofloxacin. Her increased intracranial pressure was managed with serial lumbar punctures. The patient was discharged after 12 days of hospitalization taking oral doxycycline and ciprofloxacin. Her symptoms resolved over 10 weeks. No vegetations were seen on transesophageal echocardiogram. She had no evidence of chronic Q fever on repeat serologies.
I was not aware that Q fever causes meningitis or meningoencephalitis. However, I should have considered it in light of her indirect exposure to cows. It is possible that her pneumonia 6 weeks earlier represented acute Q fever, as pneumonia and hepatitis are among the most typical acute manifestations of this infection.
COMMENTARY
Hospitalists are commonly confronted by the combination of fever, headache, and confusion and are familiar with the diagnostic and therapeutic dilemmas related to prompt discrimination between CNS and non‐CNS processes, particularly infections. At the time of this patient's final ED presentation, her illness unambiguously localized to the CNS. As common and emergent conditions such as acute bacterial meningitis were excluded, the greatest challenge was finding the clue that could direct investigations into less common causes of meningoencephalitis.
The Infectious Disease Society of America has developed clinical practice guidelines for the diagnosis and management of encephalitis which highlight the importance of epidemiology and risk factor assessment.[1] This approach requires the clinician to examine potential clues and to go beyond initial associationsfor instance, not simply linking horseback riding to horse‐associated pathogens, but interpreting horseback riding as a proxy for outdoor exposure, which places her at risk for contact with mosquitos, which transmit West Nile virus or eastern equine encephalitis. Similarly, ingestion of raw milk, which is typically linked to Listeria monocytogenes, Brucella, and other pathogens prompted the infectious disease consultant to think more broadly and include livestock (cow)‐associated pathogens including C. burnetii.
Although involvement of the CNS is common in chronic Q fever endocarditis due to septic embolism, neurologic involvement in acute Q fever varies in prevalence (range of 1.7%22%).[2, 3, 4] The 3 major neurological syndromes of acute Q fever are (1) meningoencephalitis or encephalitis, (2) lymphocytic meningitis, and (3) peripheral neuropathy (myelitis, polyradiculoneuritis, or peripheral neuritis). CSF analysis usually shows mild pleocytosis with a predominance of lymphocytic cells; CSF protein elevation is variable, and glucose is usually normal. Neuroradiologic examination is usually normal, and there are no pathognomonic imaging abnormalities for Q fever meningoencephalitis.[2, 3] The mechanism by which C. burnetii causes neurologic injury and dysfunction is unknown.
The diagnosis of Q fever is usually established by serologic testing. In acute Q fever, antibodies to phase II antigen are higher than the phase I antibody titer. Phase II IgM antibodies are the first to appear, but then decline on average after week 8, often reaching undetectable levels 10 to 12 weeks after disease onset.[5] If this patient's pneumonia 6 weeks prior to this presentation was acute Q fever pneumonia, her IgM titers may have been declining by the time her neurologic illness developed. A false negative test result is also possible; immunofluorescence assays are more specific than sensitive in acute Q fever.[5]
Evaluating this case in isolation may raise some doubt as to the accuracy of the diagnosis as she did not have a 4‐fold rise in the phase II IgG titer and did not have a detectable phase II IgM. However, she was part of a cluster of individuals who regularly consumed raw milk from the same dairy and had evidence of C. burnetii infection. This group included her spouse, who had a robust serologic evidence of C. burnetii, characterized by a >4‐fold rise in phase II IgM and IgG titers.[6]
C. burnetii is found primarily in cattle, sheep, and goats and is shed in large quantities by infected periparturient animals in their urine, feces, and milk.[7] Inhalation of contaminated aerosols is the principal route of transmission.[7, 8] Acute Q fever is underdiagnosed because the majority of acute infections are asymptomatic (60%) or present as a nonspecific flu‐like illness.[7] This case represents a rare manifestation of a rare infection acquired through a rare route of transmission, but highlights the importance of epidemiology and risk factor assessment when clinicians are faced with a diagnostic challenge.
TEACHING POINTS
- Exploration of epidemiology and exposure history is central to diagnosing meningoencephalitis with negative bacterial cultures and undetectable HSV PCR, although the etiology of meningoencephalitis can elude identification even after exhaustive investigation.
- Inhalation of contaminated aerosols is the principal route of transmission for C. burnetii, but it can also be transmitted via infected unpasteurized milk.[7, 9]
- Acute presentations of Q fever, which may warrant admission, include pneumonia, hepatitis, or meningoencephalitis.
- Q fever is diagnosed by serologic testing, and doxycycline is the antibiotic of choice.
Disclosures
This case was presented at the 2012 Annual Meeting of the Society of Hospital Medicine. It was subsequently reported in the epidemiologic report of the outbreak.[6] The authors report no conflicts of interest.
A 39‐year‐old woman presented to the emergency department (ED) with fever and headache. One to two weeks prior to presentation, she developed nightly fevers that gradually increased to as high as 39.4C. She subsequently developed generalized throbbing headaches, malaise, and diffuse body pain. The headache gradually worsened. The day prior to presentation, she developed photophobia, nausea, and vomiting. She also reported right scalp pain while combing her hair, difficulty emptying her bladder, and left buttock pain radiating down the leg. She denied rash, joint pain, visual changes, dysarthria, cough, chest pain, abdominal pain, or diarrhea.
Fever and headache can be explained by meningitis, encephalitis, or brain abscess. The combination is seen far more frequently, however, in patients with common systemic infections such as influenza. For either bacterial meningitis or influenza, a 2‐week course is prolonged and atypical. The progressive nature of the symptoms and photophobia suggest a chronic meningitis, and the development of nausea and vomiting, although nonspecific, is also consistent with elevated intracranial pressure. In a young woman, subacute fever and aches should prompt consideration of an autoimmune disorder such as systemic lupus erythematosus (SLE), although early central nervous system (CNS) involvement is atypical. Migraine headaches are characterized by light sensitivity, nausea, and vomiting and can be precipitated by a viral syndrome, but in this case, the headaches were present at the outset, and 2 weeks is too long for a migraine attack.
Pain while combing hair is not characteristic of the aforementioned syndromes. The scalp should be examined to confirm that there are no skin lesions associated with herpes zoster and no arterial prominence associated with temporal arteritis. She is young for the latter, which would otherwise be a suitable explanation for fever, headache, scalp tenderness, and visual complaints (usually impairment not photophobia).
Incomplete bladder emptying and left buttock pain suggest that there might be a concomitant lumbosacral myelopathy or radiculopathy. Some nonbacterial causes of meningitis such as cytomegalovirus (CMV), syphilis, and cancer simultaneously involve the CNS and peripheral nerve roots. It is also possible that the scalp tenderness associated with combing reflects a cervical sensory radiculopathy.
She had presented to the ED 2 and 4 days before the current (third) ED visit. Both times her main complaint was left buttock pain and left leg paresthesias. Although she had no skin lesions, she was diagnosed with prodromal herpes zoster in the S2 dermatomal distribution and was prescribed valacyclovir (to be started should eruptions develop, which never occurred).
She reported intermittent self‐limited fevers at 3‐ to 4‐week intervals during the prior 6 months; two fever episodes were accompanied by an influenza‐like illness, and one was associated with gastrointestinal symptoms. Her last fever prior to this evaluation was 6 weeks earlier when she was treated with azithromycin for suspected pneumonia at an outside facility.
Her past medical history included hypothyroidism, gastroesophageal reflux disease, diverticulitis, and gluten intolerance. Her medications included porcine (natural) thyroid, fish oil, ibuprofen, and acetaminophen. She lived in Michigan and traveled to the northeast United States (Maine, Cape Cod, New Hampshire, Connecticut, and Vermont) 7 months prior to this evaluation. She was married and had no pets at home. She denied any tobacco, alcohol, or illicit drug use.
Her illness now appears to be chronic, associated with fever, and multisystem (potentially involving the pulmonary and gastrointestinal tract). None of her medical problems would predispose her to subacute meningitis, myelopathy, or radiculopathy. Hypothyroidism raises the possibility of a concomitant autoimmune disorder which causes meningitis, such as SLE or Behet's disease. Sarcoidosis can cause chronic meningitis and neuropathy with concomitant lung and gastrointestinal involvement and rarely fever.
Residency in the upper Midwest increases exposure to chronic infections that rarely cause subacute meningitis such as histoplasmosis, blastomycosis, or human granulocytic anaplasmosis. Travel to the northeast United States 1 month before the onset of her symptoms raises the possibility of other endemic infections like Lyme disease, babesiosis, and tularemia, which may account for her recurrent fevers. Of these, Lyme is most likely to present as chronic meningitis with cranial neuropathy and radiculoneuropathy.
Although the diagnosis of pneumonia was made late in her 6‐month illness, its etiology and treatment may be relevant. If the recent pneumonia was viral, a subsequent viral meningitis may be manifesting now or may have triggered an autoimmune process, such as acute disseminated encephalomyelitis. Bacterial pneumonia is a common precursor to bacterial meningitis, and treatment with azithromycin for the pneumonia may have delayed the meningitis onset or muted its course; this should be taken into account when interpreting cerebrospinal fluid (CSF) culture results.
On physical examination, her temperature was 39.1C, blood pressure was 135/91 mm Hg, with pulse of 87 beats per minute, respiratory rate of 16 breaths per minute, and oxygenation saturation of 97% on room air. She appeared in distress and was covering her eyes. She was alert and oriented. She had photophobia and mild nuchal rigidity. Pupils were equal and reactive to light, but she could not tolerate the eye exam for papilledema. Lung, heart, and abdominal exam were normal. No cranial nerve abnormalities were noted, and muscle strength was 5/5 in all 4 extremities. She had decreased sensation to light touch with allodynia throughout her lower extremities in addition to the lateral portion of the right scalp, which was also tender to palpation. Deep tendon reflexes were 2+ and symmetric in her bilateral upper and lower extremities. She did not have joint swelling, edema, lymphadenopathy, or a rash.
Her fever, headache, nuchal rigidity and photophobia collectively suggest meningitis, which requires evaluation by a lumbar puncture. There is no rash that supports herpes zoster or SLE. She does not have signs of myelopathy that would explain the urinary complaints, but lower motor neuron involvement has not been excluded. The sensory abnormalities in the scalp and leg are consistent with a polyneuroradiculopathy. Anterior lateral scalp tenderness may signal trigeminal nerve involvement, whereas posterior scalp tenderness would localize to the upper cervical cord nerve roots. The contralateral distribution of the scalp and leg sensory deficits suggests a multifocal peripheral nervous system process rather than a single CNS lesion.
Initial laboratory data showed serum white blood cell count (WBC) of 12,000/mm3 (79% polymorphonuclear leukocytes). Hemoglobin was 14.2 g/dL, and platelets were 251,000/mm3. Electrolytes, renal function, and liver function were normal. Thyroid‐stimulating hormone, erythrocyte sedimentation rate, and C‐reactive protein were normal. Urinalysis was negative. Chest x‐ray was normal. Noncontrast head computed tomography (CT) was normal. The patient was unable to void; 500 mL of urine returned when catheterization was performed.
CSF WBC count was 1,280/mm3 (39% neutrophils and 49% lymphocytes). CSF total protein was 175 mg/dL, and glucose was 48 mg/dL; serum glucose was 104 mg/dL. Opening pressure was not recorded. Gram stain was negative. Ceftriaxone, vancomycin, ampicillin, and acyclovir were administered for presumed bacterial or viral meningitis. Magnetic resonance imaging (MRI) of the brain and spine showed diffuse leptomeningeal enhancement (Figure 1).
The urinary retention in the absence of myelopathic findings on exam or MRI suggests a sacral polyradiculoneuropathy. Diffuse leptomeningeal enhancement is consistent with many, if not all, causes of meningitis. The high WBC count, elevated protein, and low glucosecollectively signaling active inflammation in the CNSare highly compatible with bacterial meningitis, although the lymphocytic predominance and other clinical data point to nonbacterial etiologies. The negative Gram stain further lowers the probability of bacterial meningitis, but it has limited sensitivity, may be affected by recent antibiotics, and is typically negative with Listeria. Enterovirus, acute human immunodeficiency virus (HIV), and herpes viruses (eg, CMV or herpes simplex virus [HSV]) are important considerations, with the latter 2 causing associated polyneuroradiculopathy. Patients with genital HSV (not detected here) can have a concomitant sacral radiculitis leading to urinary retention.
Fungal and mycobacterial meningitis is a possibility (especially with the high protein), but the patient does not have the typical multisystem disease or immunosuppression that frequently accompanies those conditions when CNS disease is present. Autoimmune conditions like SLE, Behet's disease, and sarcoidosis remain important conditions, especially with the polyneuroradiculopathy or mononeuritis multiplex, which may reflect multifocal nerve infarction or invasion. Similarly, lymphomatous or carcinomatous meningitis should be considered, although an isolated manifestation in the CNS is unusual. Based on the multifocal neurologic deficits, I favor a viral, spirochete, or malignant etiology of her meningoencephalitis.
Despite ongoing broad spectrum antibiotics and supportive care, she became confused on hospital day 3 and developed anomia, agitation, and worsening headache. A repeat CT of the brain did not show any new abnormalities, but repeat lumbar puncture demonstrated elevated intracranial pressure (opening pressure of 47 cm water) with 427 WBC/mm3. Blood and CSF cultures remained negative.
Detailed questioning of the family revealed that she had been horseback riding 3 weeks prior to admission; there were no other livestock where she rode horses. In addition, the family reported that she and other family members routinely drank raw milk from a cow share program.
HIV antibody test was negative. Herpes simplex, varicella zoster, enteroviruses, and adenovirus CSF polymerase chain reaction (PCR) were negative. Cytomegalovirus and Epstein‐Barr virus PCR were negative in serum and CSF. Arbovirus, lymphocytic choriomeningitis, Coccidioides, Blastomyces, Histoplasma, Brucella, and Lyme serologies were negative. Cryptococcus neoformans antigen was negative in CSF. Serum QuantiFERON‐TB test was negative. Blood and CSF acid‐fast bacilli smears (and eventually mycobacterial cultures) were also negative. Her CSF flow cytometry and cytology were negative for lymphoma.
Unpasteurized milk conveys multiple infectious risks. Listeriosis is a food‐borne illness that can cause meningoencephalitis, but peripheral neuropathies are not characteristic. Brucellosis is usually characterized by severe bone pain, pancytopenia, and hepatosplenomegaly, which are absent. Infection with Mycobacterium bovis mimics Mycobacterium tuberculosis and can cause multisystem disease, typically involving the lung. Campylobacter infection is characterized by gastroenteritis, which has not been prominent.
Rhodococcus equi is a horse‐related pathogen which leads to pulmonary infections in immunocompromised hosts but not meningitis. Rather than focusing on horse exposure alone, however, it may be useful to consider her at risk for vector‐borne pathogens based on her time outdoors, such as Lyme disease (which can cause radiculopathy and encephalopathy), West Nile virus (although motor weakness rather than sensory symptoms is typical), or eastern equine encephalitis.
The absence of weight loss, cytopenias, lymphadenopathy, and organomegaly with the negative CSF cytology and flow cytometry makes lymphomatous meningitis unlikely. The case for an autoimmune disorder is not strong in the absence of joint pains, rash, or autoimmune serologies. In a young woman with unexplained encephalitis, antibodies to the N‐methyl‐D‐aspartate receptor should be assayed.
Although the CSF leukocytosis is declining, the elevated pressure and clinical deterioration signal that the disease process is not controlled. At this point I am uncertain as to the cause of her progressive meningoencephalitis with polyneuroradiculopathy. The latter feature makes me favor a viral or spirochete etiology.
On hospital day 4, Coxiella burnetii serologies were reported as positive (phase II immunoglobulin [Ig] G 1:256; phase II IgM <1:16; phase I IgG <1:16; phase I IgM <1:16) suggesting acute Q fever. Antibiotics were changed to intravenous doxycycline and ciprofloxacin. Her increased intracranial pressure was managed with serial lumbar punctures. The patient was discharged after 12 days of hospitalization taking oral doxycycline and ciprofloxacin. Her symptoms resolved over 10 weeks. No vegetations were seen on transesophageal echocardiogram. She had no evidence of chronic Q fever on repeat serologies.
I was not aware that Q fever causes meningitis or meningoencephalitis. However, I should have considered it in light of her indirect exposure to cows. It is possible that her pneumonia 6 weeks earlier represented acute Q fever, as pneumonia and hepatitis are among the most typical acute manifestations of this infection.
COMMENTARY
Hospitalists are commonly confronted by the combination of fever, headache, and confusion and are familiar with the diagnostic and therapeutic dilemmas related to prompt discrimination between CNS and non‐CNS processes, particularly infections. At the time of this patient's final ED presentation, her illness unambiguously localized to the CNS. As common and emergent conditions such as acute bacterial meningitis were excluded, the greatest challenge was finding the clue that could direct investigations into less common causes of meningoencephalitis.
The Infectious Disease Society of America has developed clinical practice guidelines for the diagnosis and management of encephalitis which highlight the importance of epidemiology and risk factor assessment.[1] This approach requires the clinician to examine potential clues and to go beyond initial associationsfor instance, not simply linking horseback riding to horse‐associated pathogens, but interpreting horseback riding as a proxy for outdoor exposure, which places her at risk for contact with mosquitos, which transmit West Nile virus or eastern equine encephalitis. Similarly, ingestion of raw milk, which is typically linked to Listeria monocytogenes, Brucella, and other pathogens prompted the infectious disease consultant to think more broadly and include livestock (cow)‐associated pathogens including C. burnetii.
Although involvement of the CNS is common in chronic Q fever endocarditis due to septic embolism, neurologic involvement in acute Q fever varies in prevalence (range of 1.7%22%).[2, 3, 4] The 3 major neurological syndromes of acute Q fever are (1) meningoencephalitis or encephalitis, (2) lymphocytic meningitis, and (3) peripheral neuropathy (myelitis, polyradiculoneuritis, or peripheral neuritis). CSF analysis usually shows mild pleocytosis with a predominance of lymphocytic cells; CSF protein elevation is variable, and glucose is usually normal. Neuroradiologic examination is usually normal, and there are no pathognomonic imaging abnormalities for Q fever meningoencephalitis.[2, 3] The mechanism by which C. burnetii causes neurologic injury and dysfunction is unknown.
The diagnosis of Q fever is usually established by serologic testing. In acute Q fever, antibodies to phase II antigen are higher than the phase I antibody titer. Phase II IgM antibodies are the first to appear, but then decline on average after week 8, often reaching undetectable levels 10 to 12 weeks after disease onset.[5] If this patient's pneumonia 6 weeks prior to this presentation was acute Q fever pneumonia, her IgM titers may have been declining by the time her neurologic illness developed. A false negative test result is also possible; immunofluorescence assays are more specific than sensitive in acute Q fever.[5]
Evaluating this case in isolation may raise some doubt as to the accuracy of the diagnosis as she did not have a 4‐fold rise in the phase II IgG titer and did not have a detectable phase II IgM. However, she was part of a cluster of individuals who regularly consumed raw milk from the same dairy and had evidence of C. burnetii infection. This group included her spouse, who had a robust serologic evidence of C. burnetii, characterized by a >4‐fold rise in phase II IgM and IgG titers.[6]
C. burnetii is found primarily in cattle, sheep, and goats and is shed in large quantities by infected periparturient animals in their urine, feces, and milk.[7] Inhalation of contaminated aerosols is the principal route of transmission.[7, 8] Acute Q fever is underdiagnosed because the majority of acute infections are asymptomatic (60%) or present as a nonspecific flu‐like illness.[7] This case represents a rare manifestation of a rare infection acquired through a rare route of transmission, but highlights the importance of epidemiology and risk factor assessment when clinicians are faced with a diagnostic challenge.
TEACHING POINTS
- Exploration of epidemiology and exposure history is central to diagnosing meningoencephalitis with negative bacterial cultures and undetectable HSV PCR, although the etiology of meningoencephalitis can elude identification even after exhaustive investigation.
- Inhalation of contaminated aerosols is the principal route of transmission for C. burnetii, but it can also be transmitted via infected unpasteurized milk.[7, 9]
- Acute presentations of Q fever, which may warrant admission, include pneumonia, hepatitis, or meningoencephalitis.
- Q fever is diagnosed by serologic testing, and doxycycline is the antibiotic of choice.
Disclosures
This case was presented at the 2012 Annual Meeting of the Society of Hospital Medicine. It was subsequently reported in the epidemiologic report of the outbreak.[6] The authors report no conflicts of interest.
- , , , et al. The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2008;47:303–327.
- , , , et al. Q fever 1985–1998 clinical and epidemiologic features of 1,383 Infections. Medicine. 2000;79:109–123.
- , , , et al. Neurological involvement in acute Q fever: a report of 29 cases and review of the literature. Arch Intern Med. 2002;162:693–700.
- , , . Q fever in Plymouth 1972–88, a review with particular reference to neurological manifestations. Epidemiol Infect. 1990;105:391–408.
- , , . Diagnosis of Q fever. J Clin Microbiol. 1998;36:1823–1834.
- , , . Q fever cluster among raw milk drinkers in Michigan, (2011). Clin Infect Dis. 2012;55:1387–1389.
- , . Q fever. Clin Micorbial Rev. 1999;12:18–53.
- , , , et al. A large outbreak of Q fever in the West Midlands: windborne spread into a metropolitan area? Commun Dis Public Health. 1998;1:180–187.
- , . A cluster of Coxiella burnetii infections associated with exposure to vaccinated goats and their unpasteurized dairy products. Am J Trop Med Hyg. 1992;47:35–40.
- , , , et al. The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2008;47:303–327.
- , , , et al. Q fever 1985–1998 clinical and epidemiologic features of 1,383 Infections. Medicine. 2000;79:109–123.
- , , , et al. Neurological involvement in acute Q fever: a report of 29 cases and review of the literature. Arch Intern Med. 2002;162:693–700.
- , , . Q fever in Plymouth 1972–88, a review with particular reference to neurological manifestations. Epidemiol Infect. 1990;105:391–408.
- , , . Diagnosis of Q fever. J Clin Microbiol. 1998;36:1823–1834.
- , , . Q fever cluster among raw milk drinkers in Michigan, (2011). Clin Infect Dis. 2012;55:1387–1389.
- , . Q fever. Clin Micorbial Rev. 1999;12:18–53.
- , , , et al. A large outbreak of Q fever in the West Midlands: windborne spread into a metropolitan area? Commun Dis Public Health. 1998;1:180–187.
- , . A cluster of Coxiella burnetii infections associated with exposure to vaccinated goats and their unpasteurized dairy products. Am J Trop Med Hyg. 1992;47:35–40.
A Multifaceted Case
Box
1
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Box
2
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
A 67‐year‐old male presented to an outside hospital with a 1‐day history of fevers up to 39.4C, bilateral upper extremity weakness, and confusion. Forty‐eight hours prior to his presentation he had undergone uncomplicated bilateral carpal tunnel release surgery for the complaint of bilateral upper extremity paresthesias.
Bilateral carpal tunnel syndrome should prompt consideration of systemic diseases that infiltrate or impinge both canals (eg, rheumatoid arthritis, acromegaly, hypothyroidism, amyloidosis), although it is most frequently explained by a bilateral repetitive stress (eg, workplace typing). The development of upper extremity weakness suggests that an alternative condition such as cervical myelopathy, bilateral radiculopathy, or a rapidly progressive peripheral neuropathy may be responsible for his paresthesias. It would be unusual for a central nervous system process to selectively cause bilateral upper extremity weakness. Occasionally, patients emerge from surgery with limb weakness caused by peripheral nerve injury sustained from malpositioning of the extremity, but this would have been evident immediately following the operation.
Postoperative fevers are frequently unexplained, but require a search for common healthcare‐associated infections, such as pneumonia, urinary tract infection, intravenous catheter thrombophlebitis, wound infection, or Clostridium difficile colitis. However, such complications are unlikely following an ambulatory procedure. Confusion and fever together point to a central nervous system infection (meningoencephalitis or brain abscess) or a systemic infection that has impaired cognition. Malignancies can cause fever and altered mental status, but these are typically asynchronous events.
His past medical history was notable for hypertension, dyslipidemia, gout, actinic keratosis, and gastroesophageal reflux. His surgical history included bilateral knee replacements, repair of a left rotator cuff injury, and a herniorrhaphy. He was a nonsmoker who consumed 4 to 6 beers daily. His medications included clonidine, colchicine, atorvastatin, extended release metoprolol, triamterene‐hydrochlorothiazide, probenecid, and as‐needed ibuprofen and omeprazole.
Upon presentation he was cooperative and in no distress. Temperature was 38.9C, pulse 119 beats per minute, blood pressure 140/90 mm Hg, and oxygen saturation 94% on room air. He was noted to have logical thinking but impaired concentration. His upper extremity movement was restricted because of postoperative discomfort and swelling rather than true weakness. The rest of the exam was normal.
Metabolic, infectious, structural (intracranial), and toxic disorders can cause altered mental status. His heavy alcohol use puts him at risk for alcohol withdrawal and infections (such as Listeria meningitis), both of which may explain his fever and altered mental status. Signs and symptoms of meningitis are absent at this time. His knee prostheses could have harbored an infection preoperatively and therefore warrant close examination. Patients sometimes have adverse reactions to medications they have been prescribed but are not exposed to until hospitalization, although his surgical procedure was likely done on an outpatient basis. Empiric thiamine should be administered early given his confusion and alcohol habits.
Basic laboratories revealed a hemoglobin of 11.2 g/dL, white blood cell (WBC) count of 6,900/mm3 with 75% neutrophils, platelets of 206,000/mm3. Mean corpuscular volume was 97 mm3. Serum albumin was 2.4 g/dl, sodium 134 mmol/L, potassium 3.9 mmol/L, blood urea nitrogen 12 mg/dL, and creatinine 0.9 mg/dL. The aspartate aminotransferase was 93 U/L, alanine aminotransferase 73 U/L, alkaline phosphatase 254 U/L, and total bilirubin 1.0 mg/dL. Urinalysis was normal. Over the next 16 days fevers and waxing and waning mentation continued. The following studies were normal or negative: blood and urine cultures; transthoracic echocardiogram, antinuclear antibodies, hepatitis B surface antigen, hepatitis C antibody, and human immunodeficiency virus antibody; magnetic resonance imaging of the brain, electroencephalogram, and lower extremity venous ultrasound.
Hypoalbuminemia may signal chronic illness, hypoproduction from liver disease (caused by his heavy alcohol use), or losses from the kidney or gastrointestinal tract. His anemia may reflect chronic disease or point toward a specific underlying disorder. For example, fever and anemia could arise from hemolytic processes such as thrombotic thrombocytopenic purpura or clostridial infections.
An extensive workup has not revealed a cause for his prolonged fever (eg, infection, malignancy, autoimmune condition, or toxin). Likewise, an explanation for confusion is lacking. Because systemic illness and structural brain disease have not been uncovered, a lumbar puncture is indicated.
A lumbar puncture under fluoroscopic guidance revealed a cerebrospinal fluid (CSF) WBC count of 6/mm3, red blood cell count (RBC) 2255/mm3, protein 49 mg/dL, and glucose 54 mg/dL. The WBC differential was not reported. No growth was reported on bacterial cultures. Polymerase chain reactions for enterovirus and herpes simplex viruses 1 and 2 were negative. Cryptococcal antigen and Venereal Disease Research Laboratory serologies were also negative.
A CSF WBC count of 6 is out of the normal range, but could be explained by a traumatic tap given the elevated RBC; the protein and glucose are likewise at the border of normal. Collectively, these are nonspecific findings that could point to an infectious or noninfectious cause of intrathecal or paraspinous inflammation, but are not suggestive of bacterial meningitis.
The patient developed pneumonia, for which he received ertapenem. On hospital day 17 he was intubated for hypoxia and respiratory distress and was extubated after 4 days of mechanical ventilation. Increasing weakness in all extremities prompted magnetic resonance imaging of the spine, which revealed fluid and enhancement involving the soft tissues around C3‐C4 and C5‐C6, raising concerns for discitis and osteomyelitis. Possible septic arthritis at the C3‐C4 and C4‐C5 facets was noted. Ring enhancing fluid collections from T2‐T8 compatible with an epidural abscess with cord compression at T4‐T5 and T6‐T7 were seen. Enhancement and fluid involving the facet joints between T2‐T7 was also consistent with septic arthritis (Figure 1).
His pneumonia appears to have developed many days into his hospitalization, and therefore is unlikely to account for his initial fever and confusion. Blood cultures and echocardiogram have not suggested an endovascular infection that could account for such widespread vertebral and epidural deposition. A wide number of bacteria can cause epidural abscesses and septic arthritis, most commonly Staphylococcus aureus. Less common pathogens with a predilection for osteoarticular involvement, such as Brucella species, warrant consideration when there is appropriate epidemiologic risk.
Systemic bacterial infection remains a concern with his alcoholism rendering him partially immunosuppressed. However, a large number of adjacent spinal joints harboring a bacterial infection is unusual, and a working diagnosis of multilevel spinal infection, therefore, should prompt consideration of noninfectious processes. When a patient develops a swollen peripheral joint and fever in the postoperative setting, gout or pseudogout is a leading consideration. That same thinking should be applied to the vertebrae, where spinal gout can manifest. Surgery itself or associated changes in alcohol consumption patterns or changes in medications (at least 4 of which are relevant to goutcolchicine, hydrochlorothiazide, probenecid, and ibuprofen) could predispose him to a flare.
Aspiration of the epidural collection yielded a negative Gram stain and culture. He developed swelling in the bilateral proximal interphalangeal joints and was treated with steroids and colchicine for suspected gout flare. Vancomycin and piperacillin‐tazobactam were initiated, and on hospital day 22 the patient was transferred to another hospital for further evaluation by neurosurgery.
The negative Gram stain and culture argues against septic arthritis, but these are imperfect tests and will not detect atypical pathogens (eg, spinal tuberculosis). Reexamination of the aspirate for urate and calcium pyrophosphate crystals would be useful. Initiation of steroids in the setting of potentially undiagnosed infection requires a careful risk/benefit analysis. It may be reasonable to treat the patient with colchicine alone while withholding steroids and avoiding nonsteroidal agents in case invasive procedures are planned.
On exam his temperature was 36C, blood pressure 156/92 mm Hg, pulse 100 beats per minute, respirations 21 per minute, and oxygenation 97% on room air. He was not in acute distress and was only oriented to self. Bilateral 2+ lower extremity pitting edema up to the knees was noted. Examination of the heart and lungs was unremarkable. Gouty tophi were noted over both elbows. His joints were normal.
Cranial nerves IIXII were normal. Motor exam revealed normal muscle tone and bulk. Muscle strength was approximately 3/5 in the right upper extremity and 4+/5 in the left upper extremity. Bilateral lower extremity strength was 3/5 in hip flexion, knee flexion, and knee extension. Dorsiflexion and plantar flexion were approximately 2/5 bilaterally. Sensation was intact to light touch and pinprick, and proprioception was normal. Gait was not tested. A Foley catheter was in place.
This examination confirms ongoing encephalopathy and incomplete quadriplegia. The lower extremity weakness is nearly equal proximally and distally, which can be seen with an advanced peripheral neuropathy but is more characteristic of myelopathy. The expected concomitant sensory deficit of myelopathy is not present, although this may be difficult to detect in a confused patient. Reflex testing would help in distinguishing myelopathy (favored because of the imaging findings) from a rapid progressive peripheral motor neuropathy (eg, acute inflammatory demyelinating polyneuropathy or acute intermittent porphyria).
The pitting edema likely represents fluid overload, which can be nonspecific after prolonged immobility during hospitalization; hypoalbuminemia is oftentimes speculated to play a role when this develops. His alcohol use puts him at risk for heart failure (although there is no evidence of this on exam) and liver disease (which his liver function tests suggest). The tophi speak to the extent and chronicity of his hyperuricemia.
On arrival he reported recent onset diarrhea. Medications at transfer included metoprolol, omeprazole, prednisone, piperacillin/tazobactam, vancomycin, and colchicine; acetaminophen, bisacodyl, diphenhydramine, fentanyl, subcutaneous insulin, and labetalol were administered as needed. Laboratory studies included a hemoglobin of 9.5 g/dL, WBC count of 7,300/mm3 with 95% neutrophils, platelets 301,000/mm3, sodium 151 mmol/L, potassium 2.9 mmol/L, blood urea nitrogen 76 mg/dL, creatinine 2.0 mg/dL, aspartate aminotransferase 171 U/L, and alanine aminotransferase 127 U/L. Serum albumin was 1.7 g/dL.
At least 3 of his medicationsdiphenhydramine, fentanyl, and prednisonemay be contributing to his ongoing altered mental status, which may be further compounded by hypernatremia. Although his liver disease remains uncharacterized, hepatic encephalopathy may be contributing to his confusion as well.
Colchicine is likely responsible for his diarrhea, which would be the most readily available explanation for his hypernatremia, hypokalemia, and acute kidney injury (AKI). Acute kidney injury could result from progressive liver disease (hepatorenal syndrome), decreased arterial perfusion (suggested by third spacing or his diarrhea), acute tubular necrosis (from infection or medication), or urinary retention secondary to catheter obstruction. Acute hyperuricemia can also cause AKI (urate nephropathy).
Anemia has progressed and requires evaluation for blood loss as well as hemolysis. Hepatotoxicity from any of his medications (eg, acetaminophen) must be considered. Coagulation studies and review of the previous abdominal computed tomography would help determine the extent of his liver disease.
Neurosurgical consultation was obtained and the patient and his family elected to proceed with a thoracic laminectomy. Cheesy fluid was identified at the facet joints at T6‐T7, which was found to contain rare deposits of monosodium urate crystals. Surgical specimen cultures were sterile. His mental status and strength slowly improved to baseline following the surgery. He was discharged on postoperative day 7 to a rehabilitation facility. On the telephone follow‐up he reported that he has regained his strength completely.
The fluid analysis and clinical course confirms spinal gout. The presenting encephalopathy remains unexplained; I am unaware of gout leading to altered mental status.
COMMENTARY
Gout is an inflammatory condition triggered by the deposition of monosodium urate crystals in tissues in association with hyperuricemia.[1] Based on the 20072008 National Health and Nutrition Examination Survey, the prevalence of gout among US adults was 3.9% (8.3 million individuals).[2] These rates are increasing and are thought to be spurred by the aging population, increasing rates of obesity, and changing dietary habits including increases in the consumption of soft drinks and red meat.[3, 4, 5] The development of gout during hospitalization can prolong length of stay, and the implementation of a management protocol appears to help decrease treatment delays and the inappropriate discontinuation of gout prophylaxis.[6, 7] Surgery, with its associated physiologic stressors, can trigger gout, which is often polyarticular and presents with fever leading to testing and consultations for the febrile episode.[8]
Gout is an ancient disease that is familiar to most clinicians. In 1666, Daniel Sennert, a German physician, described gout as the physician's shame because of its infrequent recognition.[9] Clinical gout spans 3 stages: asymptomatic hyperuricemia, acute and intercritical gout, and chronic gouty arthritis. The typical acute presentation is monoarticular with the abrupt onset of pain, swelling, warmth, and erythema in a peripheral joint. It manifests most characteristically in the first metatarsophalangeal joint (podagra), but also frequently involves the midfoot, ankle, knee, and wrist and sometimes affects multiple joints simultaneously (polyarticular gout).[1, 10] The visualization of monosodium urate crystals either in synovial fluid or from a tophus is diagnostic of gout; however, guidelines recognize that a classic presentation of gout may be diagnosed based on clinical criteria alone.[11] Dual energy computerized tomography and ultrasonography are emerging as techniques for the visualization of monosodium urate crystals; however, they are not currently routinely recommended.[12]
There are many unusual presentations of gout, with an increase in such reports paralleling both the overall increase in the prevalence of gout and improvements in available imaging techniques.[13] Atypical presentations present diagnostic challenges and are often caused by tophaceous deposits in unusual locations. Reports of atypical gout have described entrapment neuropathies (eg, gouty deposits inducing carpal tunnel syndrome), ocular gout manifested as conjunctival deposits and uveitis, pancreatic gout presenting as a mass, and dermatologic manifestations including panniculitis.[13, 14]
Spinal gout (also known as axial gout) manifests when crystal‐induced inflammation, erosive arthritis, and tophaceous deposits occur along the spinal column. A cross‐sectional study of patients with poorly controlled gout reported the prevalence of spinal gout diagnosed by computerized tomography to be 35%. These radiographic findings were not consistently correlated with back pain.[15] Imaging features that are suggestive of spinal gout include intra‐articular and juxta‐articular erosions with sclerotic margins and density greater than the surrounding muscle. Periosteal new bone formation adjacent to bony destruction can form overhanging edges.[16] When retrospectively presented with the final diagnosis, the radiologist at our institution noted that the appearance was typical gout in an atypical location.
Spinal gout can be confused with spinal metastasis, infection, and stenosis. It can remain asymptomatic or present with back pain, radiculopathy, or cord compression. The lumbar spine is the most frequently affected site.[17, 18] Many patients with spinal gout have had chronic tophaceous gout with radiologic evidence of erosions in the peripheral joints.[15] Patients with spinal gout also have elevated urate levels and markers of inflammation.[18] Surgical decompression and stabilization is recommended when there is frank cord compression, progressive neurologic compromise, or lack of improvement with gout therapy alone.[18]
This patient's male gender, history of gout, hypertension, alcohol consumption, and thiazide diuretic use placed him at an increased risk of a gout attack.[19, 20] The possible interruption of urate‐lowering therapy for the surgical procedure and surgery itself further heightened his risk of suffering acute gouty arthritis in the perioperative period.[21] The patient's encephalopathy may have masked back pain and precluded an accurate neurologic exam. There is one case report to our knowledge describing encephalopathy that improved with colchicine and was possibly related to gout.[22] This patient's encephalopathy was deemed multifactorial and attributed to alcohol withdrawal, medications (including opioids and steroids), and infection (pneumonia).
Gout is best known for its peripheral arthritis and is rarely invoked in the consideration of spinal and myelopathic processes where more pressing competing diagnoses, such as infection and malignancy, are typically considered. In addition, when surgical specimens are submitted for examination for pathology in formaldehyde (rather than alcohol), monosodium urate crystals are dissolved and are thus difficult to identify in the specimen.
This case reminds us that gout remains a diagnostic challenge and should be considered in the differential of an inflammatory process. Recognition of the multifaceted nature of gout can allow for the earlier recognition and treatment of the less typical presentations of this ancient malady.
KEY TEACHING POINTS
- Crystalline disease is a common cause of postoperative arthritis.
- Gout (and pseudogout) should be considered in cases of focal inflammation (detected by examination or imaging) when the evidence or predisposition for infection is limited or nonexistent.
- Spinal gout presents with back pain, radiculopathy, or cord compression and may be confused with spinal metastasis, infection, and stenosis.
Acknowledgements
The authors thank Dr. Kari Waddell and Elaine Bammerlin for their assistance in the preparation of this manuscript.
Disclosure: Nothing to report.
- , . Clinical features and treatment of gout. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O'Dell JR, eds. Kelley's Textbook of Rheumatology. Vol 2. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013:1544–1575.
- , , . Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141.
- , , , . Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31(8):1582–1587.
- , , , , . Purine‐rich foods, dairy and protein intake, and the risk of gout in men. New Engl J Med. 2004;350(11):1093–1103.
- , , . Fructose‐rich beverages and risk of gout in women. JAMA. 2010;304(20):2270–2278.
- , . Healthcare burden of in‐hospital gout. Intern Med J. 2012;42(11):1261–1263.
- , , , , , . Improved management of acute gout during hospitalization following introduction of a protocol. Int J Rheum Dis. 2012;15(6):512–520.
- , , . Postsurgical gout. Am Surg. 1995;61(1):56–59.
- , . Evolution of modern medicine. Arch Intern Med. 1960;105(4):640–644.
- . Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452.
- . Management of gout: a 57‐year‐old man with a history of podagra, hyperuricemia, and mild renal insufficiency. JAMA. 2012;308(20):2133–2141.
- , , , et al. Diagnostic imaging of gout: comparison of high‐resolution US versus conventional X‐ray. Eur Radiol. 2008;18(3):621–630.
- , . The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42(2):146–154.
- , . Unusual clinical presentations of gout. Curr Opin Rheumatol. 2010;22(2):181–187.
- , , , , , . Correlates of axial gout: a cross‐sectional study. J Rheumatol. 2012;39(7):1445–1449.
- , , , , , . Axial gouty arthropathy. Am J Med Sci. 2009;338(2):140–146.
- , , . Axial (spinal) gout. Curr Rheumatol Rep. 2012;14(2):161–164.
- , , , . Spinal gout in a renal transplant patient: a case report and literature review. Surg Neurol. 2007;67(1):65–73.
- , , , et al. Alcohol consumption as a trigger of recurrent gout attacks. Am J Med. 2006;119(9):800.e11–800.e16.
- , , , , , . Recent diuretic use and the risk of recurrent gout attacks: the online case‐crossover gout study. J Rheumatol. 2006;33(7):1341–1345.
- , , , , . Clinical features and risk factors of postsurgical gout. Ann Rheum Dis. 2008;67(9):1271–1275.
- , , , . Gouty encephalopathy: myth or reality [in French]? Rev Med Interne. 1997;18(6):474–476.
Box
1
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Box
2
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
A 67‐year‐old male presented to an outside hospital with a 1‐day history of fevers up to 39.4C, bilateral upper extremity weakness, and confusion. Forty‐eight hours prior to his presentation he had undergone uncomplicated bilateral carpal tunnel release surgery for the complaint of bilateral upper extremity paresthesias.
Bilateral carpal tunnel syndrome should prompt consideration of systemic diseases that infiltrate or impinge both canals (eg, rheumatoid arthritis, acromegaly, hypothyroidism, amyloidosis), although it is most frequently explained by a bilateral repetitive stress (eg, workplace typing). The development of upper extremity weakness suggests that an alternative condition such as cervical myelopathy, bilateral radiculopathy, or a rapidly progressive peripheral neuropathy may be responsible for his paresthesias. It would be unusual for a central nervous system process to selectively cause bilateral upper extremity weakness. Occasionally, patients emerge from surgery with limb weakness caused by peripheral nerve injury sustained from malpositioning of the extremity, but this would have been evident immediately following the operation.
Postoperative fevers are frequently unexplained, but require a search for common healthcare‐associated infections, such as pneumonia, urinary tract infection, intravenous catheter thrombophlebitis, wound infection, or Clostridium difficile colitis. However, such complications are unlikely following an ambulatory procedure. Confusion and fever together point to a central nervous system infection (meningoencephalitis or brain abscess) or a systemic infection that has impaired cognition. Malignancies can cause fever and altered mental status, but these are typically asynchronous events.
His past medical history was notable for hypertension, dyslipidemia, gout, actinic keratosis, and gastroesophageal reflux. His surgical history included bilateral knee replacements, repair of a left rotator cuff injury, and a herniorrhaphy. He was a nonsmoker who consumed 4 to 6 beers daily. His medications included clonidine, colchicine, atorvastatin, extended release metoprolol, triamterene‐hydrochlorothiazide, probenecid, and as‐needed ibuprofen and omeprazole.
Upon presentation he was cooperative and in no distress. Temperature was 38.9C, pulse 119 beats per minute, blood pressure 140/90 mm Hg, and oxygen saturation 94% on room air. He was noted to have logical thinking but impaired concentration. His upper extremity movement was restricted because of postoperative discomfort and swelling rather than true weakness. The rest of the exam was normal.
Metabolic, infectious, structural (intracranial), and toxic disorders can cause altered mental status. His heavy alcohol use puts him at risk for alcohol withdrawal and infections (such as Listeria meningitis), both of which may explain his fever and altered mental status. Signs and symptoms of meningitis are absent at this time. His knee prostheses could have harbored an infection preoperatively and therefore warrant close examination. Patients sometimes have adverse reactions to medications they have been prescribed but are not exposed to until hospitalization, although his surgical procedure was likely done on an outpatient basis. Empiric thiamine should be administered early given his confusion and alcohol habits.
Basic laboratories revealed a hemoglobin of 11.2 g/dL, white blood cell (WBC) count of 6,900/mm3 with 75% neutrophils, platelets of 206,000/mm3. Mean corpuscular volume was 97 mm3. Serum albumin was 2.4 g/dl, sodium 134 mmol/L, potassium 3.9 mmol/L, blood urea nitrogen 12 mg/dL, and creatinine 0.9 mg/dL. The aspartate aminotransferase was 93 U/L, alanine aminotransferase 73 U/L, alkaline phosphatase 254 U/L, and total bilirubin 1.0 mg/dL. Urinalysis was normal. Over the next 16 days fevers and waxing and waning mentation continued. The following studies were normal or negative: blood and urine cultures; transthoracic echocardiogram, antinuclear antibodies, hepatitis B surface antigen, hepatitis C antibody, and human immunodeficiency virus antibody; magnetic resonance imaging of the brain, electroencephalogram, and lower extremity venous ultrasound.
Hypoalbuminemia may signal chronic illness, hypoproduction from liver disease (caused by his heavy alcohol use), or losses from the kidney or gastrointestinal tract. His anemia may reflect chronic disease or point toward a specific underlying disorder. For example, fever and anemia could arise from hemolytic processes such as thrombotic thrombocytopenic purpura or clostridial infections.
An extensive workup has not revealed a cause for his prolonged fever (eg, infection, malignancy, autoimmune condition, or toxin). Likewise, an explanation for confusion is lacking. Because systemic illness and structural brain disease have not been uncovered, a lumbar puncture is indicated.
A lumbar puncture under fluoroscopic guidance revealed a cerebrospinal fluid (CSF) WBC count of 6/mm3, red blood cell count (RBC) 2255/mm3, protein 49 mg/dL, and glucose 54 mg/dL. The WBC differential was not reported. No growth was reported on bacterial cultures. Polymerase chain reactions for enterovirus and herpes simplex viruses 1 and 2 were negative. Cryptococcal antigen and Venereal Disease Research Laboratory serologies were also negative.
A CSF WBC count of 6 is out of the normal range, but could be explained by a traumatic tap given the elevated RBC; the protein and glucose are likewise at the border of normal. Collectively, these are nonspecific findings that could point to an infectious or noninfectious cause of intrathecal or paraspinous inflammation, but are not suggestive of bacterial meningitis.
The patient developed pneumonia, for which he received ertapenem. On hospital day 17 he was intubated for hypoxia and respiratory distress and was extubated after 4 days of mechanical ventilation. Increasing weakness in all extremities prompted magnetic resonance imaging of the spine, which revealed fluid and enhancement involving the soft tissues around C3‐C4 and C5‐C6, raising concerns for discitis and osteomyelitis. Possible septic arthritis at the C3‐C4 and C4‐C5 facets was noted. Ring enhancing fluid collections from T2‐T8 compatible with an epidural abscess with cord compression at T4‐T5 and T6‐T7 were seen. Enhancement and fluid involving the facet joints between T2‐T7 was also consistent with septic arthritis (Figure 1).
His pneumonia appears to have developed many days into his hospitalization, and therefore is unlikely to account for his initial fever and confusion. Blood cultures and echocardiogram have not suggested an endovascular infection that could account for such widespread vertebral and epidural deposition. A wide number of bacteria can cause epidural abscesses and septic arthritis, most commonly Staphylococcus aureus. Less common pathogens with a predilection for osteoarticular involvement, such as Brucella species, warrant consideration when there is appropriate epidemiologic risk.
Systemic bacterial infection remains a concern with his alcoholism rendering him partially immunosuppressed. However, a large number of adjacent spinal joints harboring a bacterial infection is unusual, and a working diagnosis of multilevel spinal infection, therefore, should prompt consideration of noninfectious processes. When a patient develops a swollen peripheral joint and fever in the postoperative setting, gout or pseudogout is a leading consideration. That same thinking should be applied to the vertebrae, where spinal gout can manifest. Surgery itself or associated changes in alcohol consumption patterns or changes in medications (at least 4 of which are relevant to goutcolchicine, hydrochlorothiazide, probenecid, and ibuprofen) could predispose him to a flare.
Aspiration of the epidural collection yielded a negative Gram stain and culture. He developed swelling in the bilateral proximal interphalangeal joints and was treated with steroids and colchicine for suspected gout flare. Vancomycin and piperacillin‐tazobactam were initiated, and on hospital day 22 the patient was transferred to another hospital for further evaluation by neurosurgery.
The negative Gram stain and culture argues against septic arthritis, but these are imperfect tests and will not detect atypical pathogens (eg, spinal tuberculosis). Reexamination of the aspirate for urate and calcium pyrophosphate crystals would be useful. Initiation of steroids in the setting of potentially undiagnosed infection requires a careful risk/benefit analysis. It may be reasonable to treat the patient with colchicine alone while withholding steroids and avoiding nonsteroidal agents in case invasive procedures are planned.
On exam his temperature was 36C, blood pressure 156/92 mm Hg, pulse 100 beats per minute, respirations 21 per minute, and oxygenation 97% on room air. He was not in acute distress and was only oriented to self. Bilateral 2+ lower extremity pitting edema up to the knees was noted. Examination of the heart and lungs was unremarkable. Gouty tophi were noted over both elbows. His joints were normal.
Cranial nerves IIXII were normal. Motor exam revealed normal muscle tone and bulk. Muscle strength was approximately 3/5 in the right upper extremity and 4+/5 in the left upper extremity. Bilateral lower extremity strength was 3/5 in hip flexion, knee flexion, and knee extension. Dorsiflexion and plantar flexion were approximately 2/5 bilaterally. Sensation was intact to light touch and pinprick, and proprioception was normal. Gait was not tested. A Foley catheter was in place.
This examination confirms ongoing encephalopathy and incomplete quadriplegia. The lower extremity weakness is nearly equal proximally and distally, which can be seen with an advanced peripheral neuropathy but is more characteristic of myelopathy. The expected concomitant sensory deficit of myelopathy is not present, although this may be difficult to detect in a confused patient. Reflex testing would help in distinguishing myelopathy (favored because of the imaging findings) from a rapid progressive peripheral motor neuropathy (eg, acute inflammatory demyelinating polyneuropathy or acute intermittent porphyria).
The pitting edema likely represents fluid overload, which can be nonspecific after prolonged immobility during hospitalization; hypoalbuminemia is oftentimes speculated to play a role when this develops. His alcohol use puts him at risk for heart failure (although there is no evidence of this on exam) and liver disease (which his liver function tests suggest). The tophi speak to the extent and chronicity of his hyperuricemia.
On arrival he reported recent onset diarrhea. Medications at transfer included metoprolol, omeprazole, prednisone, piperacillin/tazobactam, vancomycin, and colchicine; acetaminophen, bisacodyl, diphenhydramine, fentanyl, subcutaneous insulin, and labetalol were administered as needed. Laboratory studies included a hemoglobin of 9.5 g/dL, WBC count of 7,300/mm3 with 95% neutrophils, platelets 301,000/mm3, sodium 151 mmol/L, potassium 2.9 mmol/L, blood urea nitrogen 76 mg/dL, creatinine 2.0 mg/dL, aspartate aminotransferase 171 U/L, and alanine aminotransferase 127 U/L. Serum albumin was 1.7 g/dL.
At least 3 of his medicationsdiphenhydramine, fentanyl, and prednisonemay be contributing to his ongoing altered mental status, which may be further compounded by hypernatremia. Although his liver disease remains uncharacterized, hepatic encephalopathy may be contributing to his confusion as well.
Colchicine is likely responsible for his diarrhea, which would be the most readily available explanation for his hypernatremia, hypokalemia, and acute kidney injury (AKI). Acute kidney injury could result from progressive liver disease (hepatorenal syndrome), decreased arterial perfusion (suggested by third spacing or his diarrhea), acute tubular necrosis (from infection or medication), or urinary retention secondary to catheter obstruction. Acute hyperuricemia can also cause AKI (urate nephropathy).
Anemia has progressed and requires evaluation for blood loss as well as hemolysis. Hepatotoxicity from any of his medications (eg, acetaminophen) must be considered. Coagulation studies and review of the previous abdominal computed tomography would help determine the extent of his liver disease.
Neurosurgical consultation was obtained and the patient and his family elected to proceed with a thoracic laminectomy. Cheesy fluid was identified at the facet joints at T6‐T7, which was found to contain rare deposits of monosodium urate crystals. Surgical specimen cultures were sterile. His mental status and strength slowly improved to baseline following the surgery. He was discharged on postoperative day 7 to a rehabilitation facility. On the telephone follow‐up he reported that he has regained his strength completely.
The fluid analysis and clinical course confirms spinal gout. The presenting encephalopathy remains unexplained; I am unaware of gout leading to altered mental status.
COMMENTARY
Gout is an inflammatory condition triggered by the deposition of monosodium urate crystals in tissues in association with hyperuricemia.[1] Based on the 20072008 National Health and Nutrition Examination Survey, the prevalence of gout among US adults was 3.9% (8.3 million individuals).[2] These rates are increasing and are thought to be spurred by the aging population, increasing rates of obesity, and changing dietary habits including increases in the consumption of soft drinks and red meat.[3, 4, 5] The development of gout during hospitalization can prolong length of stay, and the implementation of a management protocol appears to help decrease treatment delays and the inappropriate discontinuation of gout prophylaxis.[6, 7] Surgery, with its associated physiologic stressors, can trigger gout, which is often polyarticular and presents with fever leading to testing and consultations for the febrile episode.[8]
Gout is an ancient disease that is familiar to most clinicians. In 1666, Daniel Sennert, a German physician, described gout as the physician's shame because of its infrequent recognition.[9] Clinical gout spans 3 stages: asymptomatic hyperuricemia, acute and intercritical gout, and chronic gouty arthritis. The typical acute presentation is monoarticular with the abrupt onset of pain, swelling, warmth, and erythema in a peripheral joint. It manifests most characteristically in the first metatarsophalangeal joint (podagra), but also frequently involves the midfoot, ankle, knee, and wrist and sometimes affects multiple joints simultaneously (polyarticular gout).[1, 10] The visualization of monosodium urate crystals either in synovial fluid or from a tophus is diagnostic of gout; however, guidelines recognize that a classic presentation of gout may be diagnosed based on clinical criteria alone.[11] Dual energy computerized tomography and ultrasonography are emerging as techniques for the visualization of monosodium urate crystals; however, they are not currently routinely recommended.[12]
There are many unusual presentations of gout, with an increase in such reports paralleling both the overall increase in the prevalence of gout and improvements in available imaging techniques.[13] Atypical presentations present diagnostic challenges and are often caused by tophaceous deposits in unusual locations. Reports of atypical gout have described entrapment neuropathies (eg, gouty deposits inducing carpal tunnel syndrome), ocular gout manifested as conjunctival deposits and uveitis, pancreatic gout presenting as a mass, and dermatologic manifestations including panniculitis.[13, 14]
Spinal gout (also known as axial gout) manifests when crystal‐induced inflammation, erosive arthritis, and tophaceous deposits occur along the spinal column. A cross‐sectional study of patients with poorly controlled gout reported the prevalence of spinal gout diagnosed by computerized tomography to be 35%. These radiographic findings were not consistently correlated with back pain.[15] Imaging features that are suggestive of spinal gout include intra‐articular and juxta‐articular erosions with sclerotic margins and density greater than the surrounding muscle. Periosteal new bone formation adjacent to bony destruction can form overhanging edges.[16] When retrospectively presented with the final diagnosis, the radiologist at our institution noted that the appearance was typical gout in an atypical location.
Spinal gout can be confused with spinal metastasis, infection, and stenosis. It can remain asymptomatic or present with back pain, radiculopathy, or cord compression. The lumbar spine is the most frequently affected site.[17, 18] Many patients with spinal gout have had chronic tophaceous gout with radiologic evidence of erosions in the peripheral joints.[15] Patients with spinal gout also have elevated urate levels and markers of inflammation.[18] Surgical decompression and stabilization is recommended when there is frank cord compression, progressive neurologic compromise, or lack of improvement with gout therapy alone.[18]
This patient's male gender, history of gout, hypertension, alcohol consumption, and thiazide diuretic use placed him at an increased risk of a gout attack.[19, 20] The possible interruption of urate‐lowering therapy for the surgical procedure and surgery itself further heightened his risk of suffering acute gouty arthritis in the perioperative period.[21] The patient's encephalopathy may have masked back pain and precluded an accurate neurologic exam. There is one case report to our knowledge describing encephalopathy that improved with colchicine and was possibly related to gout.[22] This patient's encephalopathy was deemed multifactorial and attributed to alcohol withdrawal, medications (including opioids and steroids), and infection (pneumonia).
Gout is best known for its peripheral arthritis and is rarely invoked in the consideration of spinal and myelopathic processes where more pressing competing diagnoses, such as infection and malignancy, are typically considered. In addition, when surgical specimens are submitted for examination for pathology in formaldehyde (rather than alcohol), monosodium urate crystals are dissolved and are thus difficult to identify in the specimen.
This case reminds us that gout remains a diagnostic challenge and should be considered in the differential of an inflammatory process. Recognition of the multifaceted nature of gout can allow for the earlier recognition and treatment of the less typical presentations of this ancient malady.
KEY TEACHING POINTS
- Crystalline disease is a common cause of postoperative arthritis.
- Gout (and pseudogout) should be considered in cases of focal inflammation (detected by examination or imaging) when the evidence or predisposition for infection is limited or nonexistent.
- Spinal gout presents with back pain, radiculopathy, or cord compression and may be confused with spinal metastasis, infection, and stenosis.
Acknowledgements
The authors thank Dr. Kari Waddell and Elaine Bammerlin for their assistance in the preparation of this manuscript.
Disclosure: Nothing to report.
Box
1
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Box
2
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
A 67‐year‐old male presented to an outside hospital with a 1‐day history of fevers up to 39.4C, bilateral upper extremity weakness, and confusion. Forty‐eight hours prior to his presentation he had undergone uncomplicated bilateral carpal tunnel release surgery for the complaint of bilateral upper extremity paresthesias.
Bilateral carpal tunnel syndrome should prompt consideration of systemic diseases that infiltrate or impinge both canals (eg, rheumatoid arthritis, acromegaly, hypothyroidism, amyloidosis), although it is most frequently explained by a bilateral repetitive stress (eg, workplace typing). The development of upper extremity weakness suggests that an alternative condition such as cervical myelopathy, bilateral radiculopathy, or a rapidly progressive peripheral neuropathy may be responsible for his paresthesias. It would be unusual for a central nervous system process to selectively cause bilateral upper extremity weakness. Occasionally, patients emerge from surgery with limb weakness caused by peripheral nerve injury sustained from malpositioning of the extremity, but this would have been evident immediately following the operation.
Postoperative fevers are frequently unexplained, but require a search for common healthcare‐associated infections, such as pneumonia, urinary tract infection, intravenous catheter thrombophlebitis, wound infection, or Clostridium difficile colitis. However, such complications are unlikely following an ambulatory procedure. Confusion and fever together point to a central nervous system infection (meningoencephalitis or brain abscess) or a systemic infection that has impaired cognition. Malignancies can cause fever and altered mental status, but these are typically asynchronous events.
His past medical history was notable for hypertension, dyslipidemia, gout, actinic keratosis, and gastroesophageal reflux. His surgical history included bilateral knee replacements, repair of a left rotator cuff injury, and a herniorrhaphy. He was a nonsmoker who consumed 4 to 6 beers daily. His medications included clonidine, colchicine, atorvastatin, extended release metoprolol, triamterene‐hydrochlorothiazide, probenecid, and as‐needed ibuprofen and omeprazole.
Upon presentation he was cooperative and in no distress. Temperature was 38.9C, pulse 119 beats per minute, blood pressure 140/90 mm Hg, and oxygen saturation 94% on room air. He was noted to have logical thinking but impaired concentration. His upper extremity movement was restricted because of postoperative discomfort and swelling rather than true weakness. The rest of the exam was normal.
Metabolic, infectious, structural (intracranial), and toxic disorders can cause altered mental status. His heavy alcohol use puts him at risk for alcohol withdrawal and infections (such as Listeria meningitis), both of which may explain his fever and altered mental status. Signs and symptoms of meningitis are absent at this time. His knee prostheses could have harbored an infection preoperatively and therefore warrant close examination. Patients sometimes have adverse reactions to medications they have been prescribed but are not exposed to until hospitalization, although his surgical procedure was likely done on an outpatient basis. Empiric thiamine should be administered early given his confusion and alcohol habits.
Basic laboratories revealed a hemoglobin of 11.2 g/dL, white blood cell (WBC) count of 6,900/mm3 with 75% neutrophils, platelets of 206,000/mm3. Mean corpuscular volume was 97 mm3. Serum albumin was 2.4 g/dl, sodium 134 mmol/L, potassium 3.9 mmol/L, blood urea nitrogen 12 mg/dL, and creatinine 0.9 mg/dL. The aspartate aminotransferase was 93 U/L, alanine aminotransferase 73 U/L, alkaline phosphatase 254 U/L, and total bilirubin 1.0 mg/dL. Urinalysis was normal. Over the next 16 days fevers and waxing and waning mentation continued. The following studies were normal or negative: blood and urine cultures; transthoracic echocardiogram, antinuclear antibodies, hepatitis B surface antigen, hepatitis C antibody, and human immunodeficiency virus antibody; magnetic resonance imaging of the brain, electroencephalogram, and lower extremity venous ultrasound.
Hypoalbuminemia may signal chronic illness, hypoproduction from liver disease (caused by his heavy alcohol use), or losses from the kidney or gastrointestinal tract. His anemia may reflect chronic disease or point toward a specific underlying disorder. For example, fever and anemia could arise from hemolytic processes such as thrombotic thrombocytopenic purpura or clostridial infections.
An extensive workup has not revealed a cause for his prolonged fever (eg, infection, malignancy, autoimmune condition, or toxin). Likewise, an explanation for confusion is lacking. Because systemic illness and structural brain disease have not been uncovered, a lumbar puncture is indicated.
A lumbar puncture under fluoroscopic guidance revealed a cerebrospinal fluid (CSF) WBC count of 6/mm3, red blood cell count (RBC) 2255/mm3, protein 49 mg/dL, and glucose 54 mg/dL. The WBC differential was not reported. No growth was reported on bacterial cultures. Polymerase chain reactions for enterovirus and herpes simplex viruses 1 and 2 were negative. Cryptococcal antigen and Venereal Disease Research Laboratory serologies were also negative.
A CSF WBC count of 6 is out of the normal range, but could be explained by a traumatic tap given the elevated RBC; the protein and glucose are likewise at the border of normal. Collectively, these are nonspecific findings that could point to an infectious or noninfectious cause of intrathecal or paraspinous inflammation, but are not suggestive of bacterial meningitis.
The patient developed pneumonia, for which he received ertapenem. On hospital day 17 he was intubated for hypoxia and respiratory distress and was extubated after 4 days of mechanical ventilation. Increasing weakness in all extremities prompted magnetic resonance imaging of the spine, which revealed fluid and enhancement involving the soft tissues around C3‐C4 and C5‐C6, raising concerns for discitis and osteomyelitis. Possible septic arthritis at the C3‐C4 and C4‐C5 facets was noted. Ring enhancing fluid collections from T2‐T8 compatible with an epidural abscess with cord compression at T4‐T5 and T6‐T7 were seen. Enhancement and fluid involving the facet joints between T2‐T7 was also consistent with septic arthritis (Figure 1).
His pneumonia appears to have developed many days into his hospitalization, and therefore is unlikely to account for his initial fever and confusion. Blood cultures and echocardiogram have not suggested an endovascular infection that could account for such widespread vertebral and epidural deposition. A wide number of bacteria can cause epidural abscesses and septic arthritis, most commonly Staphylococcus aureus. Less common pathogens with a predilection for osteoarticular involvement, such as Brucella species, warrant consideration when there is appropriate epidemiologic risk.
Systemic bacterial infection remains a concern with his alcoholism rendering him partially immunosuppressed. However, a large number of adjacent spinal joints harboring a bacterial infection is unusual, and a working diagnosis of multilevel spinal infection, therefore, should prompt consideration of noninfectious processes. When a patient develops a swollen peripheral joint and fever in the postoperative setting, gout or pseudogout is a leading consideration. That same thinking should be applied to the vertebrae, where spinal gout can manifest. Surgery itself or associated changes in alcohol consumption patterns or changes in medications (at least 4 of which are relevant to goutcolchicine, hydrochlorothiazide, probenecid, and ibuprofen) could predispose him to a flare.
Aspiration of the epidural collection yielded a negative Gram stain and culture. He developed swelling in the bilateral proximal interphalangeal joints and was treated with steroids and colchicine for suspected gout flare. Vancomycin and piperacillin‐tazobactam were initiated, and on hospital day 22 the patient was transferred to another hospital for further evaluation by neurosurgery.
The negative Gram stain and culture argues against septic arthritis, but these are imperfect tests and will not detect atypical pathogens (eg, spinal tuberculosis). Reexamination of the aspirate for urate and calcium pyrophosphate crystals would be useful. Initiation of steroids in the setting of potentially undiagnosed infection requires a careful risk/benefit analysis. It may be reasonable to treat the patient with colchicine alone while withholding steroids and avoiding nonsteroidal agents in case invasive procedures are planned.
On exam his temperature was 36C, blood pressure 156/92 mm Hg, pulse 100 beats per minute, respirations 21 per minute, and oxygenation 97% on room air. He was not in acute distress and was only oriented to self. Bilateral 2+ lower extremity pitting edema up to the knees was noted. Examination of the heart and lungs was unremarkable. Gouty tophi were noted over both elbows. His joints were normal.
Cranial nerves IIXII were normal. Motor exam revealed normal muscle tone and bulk. Muscle strength was approximately 3/5 in the right upper extremity and 4+/5 in the left upper extremity. Bilateral lower extremity strength was 3/5 in hip flexion, knee flexion, and knee extension. Dorsiflexion and plantar flexion were approximately 2/5 bilaterally. Sensation was intact to light touch and pinprick, and proprioception was normal. Gait was not tested. A Foley catheter was in place.
This examination confirms ongoing encephalopathy and incomplete quadriplegia. The lower extremity weakness is nearly equal proximally and distally, which can be seen with an advanced peripheral neuropathy but is more characteristic of myelopathy. The expected concomitant sensory deficit of myelopathy is not present, although this may be difficult to detect in a confused patient. Reflex testing would help in distinguishing myelopathy (favored because of the imaging findings) from a rapid progressive peripheral motor neuropathy (eg, acute inflammatory demyelinating polyneuropathy or acute intermittent porphyria).
The pitting edema likely represents fluid overload, which can be nonspecific after prolonged immobility during hospitalization; hypoalbuminemia is oftentimes speculated to play a role when this develops. His alcohol use puts him at risk for heart failure (although there is no evidence of this on exam) and liver disease (which his liver function tests suggest). The tophi speak to the extent and chronicity of his hyperuricemia.
On arrival he reported recent onset diarrhea. Medications at transfer included metoprolol, omeprazole, prednisone, piperacillin/tazobactam, vancomycin, and colchicine; acetaminophen, bisacodyl, diphenhydramine, fentanyl, subcutaneous insulin, and labetalol were administered as needed. Laboratory studies included a hemoglobin of 9.5 g/dL, WBC count of 7,300/mm3 with 95% neutrophils, platelets 301,000/mm3, sodium 151 mmol/L, potassium 2.9 mmol/L, blood urea nitrogen 76 mg/dL, creatinine 2.0 mg/dL, aspartate aminotransferase 171 U/L, and alanine aminotransferase 127 U/L. Serum albumin was 1.7 g/dL.
At least 3 of his medicationsdiphenhydramine, fentanyl, and prednisonemay be contributing to his ongoing altered mental status, which may be further compounded by hypernatremia. Although his liver disease remains uncharacterized, hepatic encephalopathy may be contributing to his confusion as well.
Colchicine is likely responsible for his diarrhea, which would be the most readily available explanation for his hypernatremia, hypokalemia, and acute kidney injury (AKI). Acute kidney injury could result from progressive liver disease (hepatorenal syndrome), decreased arterial perfusion (suggested by third spacing or his diarrhea), acute tubular necrosis (from infection or medication), or urinary retention secondary to catheter obstruction. Acute hyperuricemia can also cause AKI (urate nephropathy).
Anemia has progressed and requires evaluation for blood loss as well as hemolysis. Hepatotoxicity from any of his medications (eg, acetaminophen) must be considered. Coagulation studies and review of the previous abdominal computed tomography would help determine the extent of his liver disease.
Neurosurgical consultation was obtained and the patient and his family elected to proceed with a thoracic laminectomy. Cheesy fluid was identified at the facet joints at T6‐T7, which was found to contain rare deposits of monosodium urate crystals. Surgical specimen cultures were sterile. His mental status and strength slowly improved to baseline following the surgery. He was discharged on postoperative day 7 to a rehabilitation facility. On the telephone follow‐up he reported that he has regained his strength completely.
The fluid analysis and clinical course confirms spinal gout. The presenting encephalopathy remains unexplained; I am unaware of gout leading to altered mental status.
COMMENTARY
Gout is an inflammatory condition triggered by the deposition of monosodium urate crystals in tissues in association with hyperuricemia.[1] Based on the 20072008 National Health and Nutrition Examination Survey, the prevalence of gout among US adults was 3.9% (8.3 million individuals).[2] These rates are increasing and are thought to be spurred by the aging population, increasing rates of obesity, and changing dietary habits including increases in the consumption of soft drinks and red meat.[3, 4, 5] The development of gout during hospitalization can prolong length of stay, and the implementation of a management protocol appears to help decrease treatment delays and the inappropriate discontinuation of gout prophylaxis.[6, 7] Surgery, with its associated physiologic stressors, can trigger gout, which is often polyarticular and presents with fever leading to testing and consultations for the febrile episode.[8]
Gout is an ancient disease that is familiar to most clinicians. In 1666, Daniel Sennert, a German physician, described gout as the physician's shame because of its infrequent recognition.[9] Clinical gout spans 3 stages: asymptomatic hyperuricemia, acute and intercritical gout, and chronic gouty arthritis. The typical acute presentation is monoarticular with the abrupt onset of pain, swelling, warmth, and erythema in a peripheral joint. It manifests most characteristically in the first metatarsophalangeal joint (podagra), but also frequently involves the midfoot, ankle, knee, and wrist and sometimes affects multiple joints simultaneously (polyarticular gout).[1, 10] The visualization of monosodium urate crystals either in synovial fluid or from a tophus is diagnostic of gout; however, guidelines recognize that a classic presentation of gout may be diagnosed based on clinical criteria alone.[11] Dual energy computerized tomography and ultrasonography are emerging as techniques for the visualization of monosodium urate crystals; however, they are not currently routinely recommended.[12]
There are many unusual presentations of gout, with an increase in such reports paralleling both the overall increase in the prevalence of gout and improvements in available imaging techniques.[13] Atypical presentations present diagnostic challenges and are often caused by tophaceous deposits in unusual locations. Reports of atypical gout have described entrapment neuropathies (eg, gouty deposits inducing carpal tunnel syndrome), ocular gout manifested as conjunctival deposits and uveitis, pancreatic gout presenting as a mass, and dermatologic manifestations including panniculitis.[13, 14]
Spinal gout (also known as axial gout) manifests when crystal‐induced inflammation, erosive arthritis, and tophaceous deposits occur along the spinal column. A cross‐sectional study of patients with poorly controlled gout reported the prevalence of spinal gout diagnosed by computerized tomography to be 35%. These radiographic findings were not consistently correlated with back pain.[15] Imaging features that are suggestive of spinal gout include intra‐articular and juxta‐articular erosions with sclerotic margins and density greater than the surrounding muscle. Periosteal new bone formation adjacent to bony destruction can form overhanging edges.[16] When retrospectively presented with the final diagnosis, the radiologist at our institution noted that the appearance was typical gout in an atypical location.
Spinal gout can be confused with spinal metastasis, infection, and stenosis. It can remain asymptomatic or present with back pain, radiculopathy, or cord compression. The lumbar spine is the most frequently affected site.[17, 18] Many patients with spinal gout have had chronic tophaceous gout with radiologic evidence of erosions in the peripheral joints.[15] Patients with spinal gout also have elevated urate levels and markers of inflammation.[18] Surgical decompression and stabilization is recommended when there is frank cord compression, progressive neurologic compromise, or lack of improvement with gout therapy alone.[18]
This patient's male gender, history of gout, hypertension, alcohol consumption, and thiazide diuretic use placed him at an increased risk of a gout attack.[19, 20] The possible interruption of urate‐lowering therapy for the surgical procedure and surgery itself further heightened his risk of suffering acute gouty arthritis in the perioperative period.[21] The patient's encephalopathy may have masked back pain and precluded an accurate neurologic exam. There is one case report to our knowledge describing encephalopathy that improved with colchicine and was possibly related to gout.[22] This patient's encephalopathy was deemed multifactorial and attributed to alcohol withdrawal, medications (including opioids and steroids), and infection (pneumonia).
Gout is best known for its peripheral arthritis and is rarely invoked in the consideration of spinal and myelopathic processes where more pressing competing diagnoses, such as infection and malignancy, are typically considered. In addition, when surgical specimens are submitted for examination for pathology in formaldehyde (rather than alcohol), monosodium urate crystals are dissolved and are thus difficult to identify in the specimen.
This case reminds us that gout remains a diagnostic challenge and should be considered in the differential of an inflammatory process. Recognition of the multifaceted nature of gout can allow for the earlier recognition and treatment of the less typical presentations of this ancient malady.
KEY TEACHING POINTS
- Crystalline disease is a common cause of postoperative arthritis.
- Gout (and pseudogout) should be considered in cases of focal inflammation (detected by examination or imaging) when the evidence or predisposition for infection is limited or nonexistent.
- Spinal gout presents with back pain, radiculopathy, or cord compression and may be confused with spinal metastasis, infection, and stenosis.
Acknowledgements
The authors thank Dr. Kari Waddell and Elaine Bammerlin for their assistance in the preparation of this manuscript.
Disclosure: Nothing to report.
- , . Clinical features and treatment of gout. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O'Dell JR, eds. Kelley's Textbook of Rheumatology. Vol 2. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013:1544–1575.
- , , . Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141.
- , , , . Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31(8):1582–1587.
- , , , , . Purine‐rich foods, dairy and protein intake, and the risk of gout in men. New Engl J Med. 2004;350(11):1093–1103.
- , , . Fructose‐rich beverages and risk of gout in women. JAMA. 2010;304(20):2270–2278.
- , . Healthcare burden of in‐hospital gout. Intern Med J. 2012;42(11):1261–1263.
- , , , , , . Improved management of acute gout during hospitalization following introduction of a protocol. Int J Rheum Dis. 2012;15(6):512–520.
- , , . Postsurgical gout. Am Surg. 1995;61(1):56–59.
- , . Evolution of modern medicine. Arch Intern Med. 1960;105(4):640–644.
- . Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452.
- . Management of gout: a 57‐year‐old man with a history of podagra, hyperuricemia, and mild renal insufficiency. JAMA. 2012;308(20):2133–2141.
- , , , et al. Diagnostic imaging of gout: comparison of high‐resolution US versus conventional X‐ray. Eur Radiol. 2008;18(3):621–630.
- , . The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42(2):146–154.
- , . Unusual clinical presentations of gout. Curr Opin Rheumatol. 2010;22(2):181–187.
- , , , , , . Correlates of axial gout: a cross‐sectional study. J Rheumatol. 2012;39(7):1445–1449.
- , , , , , . Axial gouty arthropathy. Am J Med Sci. 2009;338(2):140–146.
- , , . Axial (spinal) gout. Curr Rheumatol Rep. 2012;14(2):161–164.
- , , , . Spinal gout in a renal transplant patient: a case report and literature review. Surg Neurol. 2007;67(1):65–73.
- , , , et al. Alcohol consumption as a trigger of recurrent gout attacks. Am J Med. 2006;119(9):800.e11–800.e16.
- , , , , , . Recent diuretic use and the risk of recurrent gout attacks: the online case‐crossover gout study. J Rheumatol. 2006;33(7):1341–1345.
- , , , , . Clinical features and risk factors of postsurgical gout. Ann Rheum Dis. 2008;67(9):1271–1275.
- , , , . Gouty encephalopathy: myth or reality [in French]? Rev Med Interne. 1997;18(6):474–476.
- , . Clinical features and treatment of gout. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O'Dell JR, eds. Kelley's Textbook of Rheumatology. Vol 2. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013:1544–1575.
- , , . Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141.
- , , , . Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31(8):1582–1587.
- , , , , . Purine‐rich foods, dairy and protein intake, and the risk of gout in men. New Engl J Med. 2004;350(11):1093–1103.
- , , . Fructose‐rich beverages and risk of gout in women. JAMA. 2010;304(20):2270–2278.
- , . Healthcare burden of in‐hospital gout. Intern Med J. 2012;42(11):1261–1263.
- , , , , , . Improved management of acute gout during hospitalization following introduction of a protocol. Int J Rheum Dis. 2012;15(6):512–520.
- , , . Postsurgical gout. Am Surg. 1995;61(1):56–59.
- , . Evolution of modern medicine. Arch Intern Med. 1960;105(4):640–644.
- . Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452.
- . Management of gout: a 57‐year‐old man with a history of podagra, hyperuricemia, and mild renal insufficiency. JAMA. 2012;308(20):2133–2141.
- , , , et al. Diagnostic imaging of gout: comparison of high‐resolution US versus conventional X‐ray. Eur Radiol. 2008;18(3):621–630.
- , . The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42(2):146–154.
- , . Unusual clinical presentations of gout. Curr Opin Rheumatol. 2010;22(2):181–187.
- , , , , , . Correlates of axial gout: a cross‐sectional study. J Rheumatol. 2012;39(7):1445–1449.
- , , , , , . Axial gouty arthropathy. Am J Med Sci. 2009;338(2):140–146.
- , , . Axial (spinal) gout. Curr Rheumatol Rep. 2012;14(2):161–164.
- , , , . Spinal gout in a renal transplant patient: a case report and literature review. Surg Neurol. 2007;67(1):65–73.
- , , , et al. Alcohol consumption as a trigger of recurrent gout attacks. Am J Med. 2006;119(9):800.e11–800.e16.
- , , , , , . Recent diuretic use and the risk of recurrent gout attacks: the online case‐crossover gout study. J Rheumatol. 2006;33(7):1341–1345.
- , , , , . Clinical features and risk factors of postsurgical gout. Ann Rheum Dis. 2008;67(9):1271–1275.
- , , , . Gouty encephalopathy: myth or reality [in French]? Rev Med Interne. 1997;18(6):474–476.