User login
Continuing Medical Education Program in
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
If you wish to receive credit for this activity, which begins on the next page, please refer to the website:
Accreditation and Designation Statement
Blackwell Futura Media Services designates this educational activity for a 1 AMA PRA Category 1 Credit. Physicians should only claim credit commensurate with the extent of their participation in the activity.
Blackwell Futura Media Services is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Educational Objectives
Continuous participation in the Journal of Hospital Medicine CME program will enable learners to be better able to:
-
Interpret clinical guidelines and their applications for higher quality and more efficient care for all hospitalized patients.
-
Describe the standard of care for common illnesses and conditions treated in the hospital; such as pneumonia, COPD exacerbation, acute coronary syndrome, HF exacerbation, glycemic control, venous thromboembolic disease, stroke, etc.
-
Discuss evidence‐based recommendations involving transitions of care, including the hospital discharge process.
-
Gain insights into the roles of hospitalists as medical educators, researchers, medical ethicists, palliative care providers, and hospital‐based geriatricians.
-
Incorporate best practices for hospitalist administration, including quality improvement, patient safety, practice management, leadership, and demonstrating hospitalist value.
-
Identify evidence‐based best practices and trends for both adult and pediatric hospital medicine.
Instructions on Receiving Credit
For information on applicability and acceptance of continuing medical education credit for this activity, please consult your professional licensing board.
This activity is designed to be completed within the time designated on the title page; physicians should claim only those credits that reflect the time actually spent in the activity. To successfully earn credit, participants must complete the activity during the valid credit period that is noted on the title page.
Follow these steps to earn credit:
-
Log on to
www.blackwellpublishing.com/cme . -
Read the target audience, learning objectives, and author disclosures.
-
Read the article in print or online format.
-
Reflect on the article.
-
Access the CME Exam, and choose the best answer to each question.
-
Complete the required evaluation component of the activity.
Unscripted
A 58‐year old man was admitted with generalized weakness and acute deep venous thrombosis (DVT). His past medical history included hypertension and polymyositis/dermatomyositis (PM/DM) with anti‐synthase syndrome, which had been diagnosed 16 months prior when his creatine kinase (CK) was greater than 12,000 U/L. At that time he also was found to have bilateral lower extremity DVT, and had been treated with warfarin for 1 year. 10 days previously, he had been discharged after a 4‐day hospitalization for a polymyositis flare which was treated with methylprednisolone at 60 mg daily for 5 days. He was discharged home with daily prednisone until this follow‐up a week later, where he reported weakness and bilateral edema. Lower extremity ultrasound demonstrated acute thrombus in the right common femoral vein.
This acute extensive DVT may be a consequence of recent hospitalization and a previously damaged venous system, or may reflect ongoing hypercoagulability from an unresolved condition, such as cancer. Bilateral lower extremity edema may suggest right‐sided heart failure due to progressive interstitial lung disease, which occurs in a subset of patients with PM/DM. Edema may alternatively reflect biventricular heart failure, or liver or kidney disease.
Generalized weakness offers little in the way of focused differential diagnosis until it is characterized as motor weakness (eg, attributed to progression of the myopathy), a dyspnea‐equivalent, or an overall sense of fatigue.
His medications included weekly methotrexate, monthly intravenous immunoglobulin (IVIG) infusions, tacrolimus, hydrochlorothiazide, and aerosolized pentamidine. He had been on varying doses of prednisone for 2 years and his present dose was 40 mg daily. He was allergic to sulfa. He was married and stopped smoking 30 years previously, and did not drink alcohol or use illicit drugs.
Various medication toxicities could account for his presentation. Methotrexate causes interstitial lung disease, and IVIG and tacrolimus may cause renal failure (and fluid overload). The heavy degree of immunosuppression renders him susceptible to a wide range of infections. Aerosolized pentamidine provides incomplete protection against Pneumocystis jirovecii, especially in the lung apices.
Evaluation of the status of his myositis with motor strength assessment is important. In addition associated rashes and signs of malignancy (eg, lymphadenopathy) and infection should be sought. Proximal motor weakness would suggest a myositis flare, although care must be given to exclude competing causes of myopathy, including infections, toxins, or endocrinopathies.
His temperature was 36.2C, pulse 103 beats per minute, blood pressure 156/83 mm Hg, and respiratory rate 18 breaths per minute. He had crackles at both lung bases, and 3+ pitting edema in both lower extremities. On neurological exam his motor strength was found to be diminished at 3/5 in the lower extremities and proximal upper extremities and 4/5 in the distal upper extremities. Reflexes were uniformly at 1+/4 and his cognition was intact. Examinations of his head, skin, heart, and abdomen were normal.
The absence of elevated jugular venous pressure argues against right heart failure. He is afebrile but that is minimally reassuring given the immunosuppression. There are no clues to suggest liver or kidney dysfunction. An unrecognized occlusion of the lower abdominal venous or lymphatic system such as upward extension of the DVT into the inferior vena cava (IVC) or a pelvic obstruction of the lower extremity lymphatic vessels could be considered. It appears that his distal weakness closely mirrors his proximal weakness in distinction to most myopathies which are predominantly proximal (with some exceptions, eg, inclusion body myositis).
The white blood cell count was 26,000/L with normal differential, hemoglobin 11.2 gm/dL, and platelet count was 191,000/L (at recent discharge these values were 23,000, 11.9, and 274,000, respectively). Chemistries were normal except for creatinine of 1.4 mg/dL (baseline 1.2), blood urea nitrogen was 42 mg/dL, albumin 2.6 gm/dL (normal, 3.55.0), and CK 3,710 U/L (20220), decreased from 6,943 U/L at recent discharge. Urine dipstick testing was positive for blood and protein; the urine sediment was unremarkable. Chest radiograph revealed normal lungs and heart.
The white blood cell count is quite elevated, perhaps more so than could be attributed to chronic steroid use, and again raises the concern of an undiagnosed infection. The presence of heme (and protein) in the urine without cells is consistent with pigment nephropathy from the recent rhabdomyolysis.
He was admitted to the hospital. Unfractionated heparin and warfarin were started. No changes were made to his immunosuppressive regimen. Blood cultures were negative after 48 hours. Transthoracic echocardiogram showed an ejection fraction of 60%, normal valves, and right ventricular systolic pressure of 32 mm Hg (normal, 1525 mmHg). On hospital day 3, his platelet count was 147,000/L, and on day 5, 101,000/L. His other laboratory values remained unchanged, and there were no new clinical developments.
A declining platelet count and extensive deep vein thrombosis suggest heparin‐induced thrombocytopenia and thrombosis (HITT), especially with the greater than 50% drop in the setting of IV heparin. His platelets have continued on a downward trajectory that was evident at admission and has progressed during this hospitalization. Assuming this is not due to laboratory error or artifact such as platelet clumping, this decline could have occurred if he was sensitized to heparin during the prior hospitalization, such as for DVT prophylaxis. It is increasingly recognized that HITT can manifest even after exposure to heparin is complete, ie, posthospitalization, and there can be an immediate drop in platelet counts if an unrecognized HITT‐mediated thrombosis is treated with IV heparin. Heparin should be discontinued in favor of a direct thrombin inhibitor and tests for heparin‐induced platelet antibodies (HIPA) and serotonin‐release assay (SRA) sent.
Antiphospholipid antibody syndrome (APLS) is associated with hypercoagulability and thrombocytopenia and is more frequent in patients with autoimmune disorders. The drug list should also be examined for associations with thrombocytopenia. The peripheral smear should be scrutinized and hemoglobin and creatinine followed to exclude thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome (TTP‐HUS).
Heparin was stopped on day 5. Warfarin was continued with a therapeutic international normalized ratio (INR). Tests for antiplatelet factor 4 antibodies, HIPA, and SRA were negative. His weakness and edema improved although his CK remained between 2000 and 4000 U/L. On day 5 he developed mild hemoptysis, and a repeat chest radiograph demonstrated a new left hilar infiltrate. Computed tomography (CT) scan of the chest with contrast demonstrated a left lower lobe consolidation, scattered ground glass opacities in both lung bases, and no pulmonary embolus. He was treated with piperacillin/tazobactam and vancomycin. He remained afebrile. The same day, he erroneously received 125 mg (instead of 12.5 mg) of subcutaneous methotrexate. High‐dose leucovorin was administered on days 5 and 6.
The hemoptysis resolved after 2 days. From days 5 to 9, the platelet count dropped to 80,000/L and his hemoglobin gradually decreased to 7.3 g/dL. Anticoagulation was stopped, vitamin K administered, and an IVC filter placed. Two units of packed red blood cells (RBCs) were transfused.
In suspected HITT (which was not verified here), warfarin is typically withheld until the platelets have recovered and thrombin‐inhibitor anticoagulation has reached a steady state, to avoid the transient hypercoagulability of warfarin initiation.
The unusual time course and the 3 negative tests make HITT unlikely. The continued platelet decline after stopping heparin further supports another etiology. The excess methotrexate dosing complicates interpretation of his thrombocytopenia and anemia, which can be explained by mucosal bleeding, microangiopathic hemolytic anemia (MAHA) such as disseminated intravascular coagulation or TTP‐HUS, or autoimmunity (Evans syndrome). Bone marrow toxicity is also a major effect of methotrexate (in addition to elevation of liver enzymes and acute renal failure); however, there is typically a lag between administration and development of cytopenias. The antibiotics could also account for the ongoing (but not original) thrombocytopenia.
With the new pulmonary infiltrate, infections remain a primary concern and should be evaluated with sputum samples and perhaps bronchoscopy. Given the abnormal urine (even without cells), a pulmonary‐renal inflammatory processes should be considered also to explain the infiltrates and hemoptysis.
Haptoglobin was <20 mg/dL (normal, 37246). The direct antiglobulin test (DAT) was negative. Serum lactate dehydrogenase (LDH) was 1657 U/L (normal, 100220), with elevated LD4 and LD5 isoenzymes. Coagulation studies normalized after the administration of vitamin K. Anti‐nuclear antibody was positive at 8.7 (normal <1.5). Tests for antineutrophil cytoplasmic antibodies were negative. No sputum could be obtained. A pathologist reviewed the blood smear and reported neutrophilic leukocytosis without left shift, and thrombocytopenia with normal platelet morphology.
Low haptoglobin in the setting of an elevated LDH is highly suggestive of hemolysis, particularly the intravascular, microangiopathic varieties. Neutrophilia may reflect infection, a primary myeloproliferative process such as chronic myeloid leukemia, steroid use, or a reactive bone marrow in the setting of acute illness. The negative DAT and significant immunosuppressive regimen makes immune‐mediated hemolysis unlikely, although the history of autoimmunity and the small DAT false‐negative rate leaves Evans syndrome as an outside possibility. Medications such as tacrolimus (causing TTP) or IVIG (given the broad spectrum of antibodies it includes) are other plausible causes of the cytopenias.
At this point, I would analyze the red blood cell (RBC) morphology and check the reticulocyte count to help differentiate between hemolysis and a myelotoxin.
After transfusion, his hemoglobin remained at approximately 8.5 gm/dL and LDH remained elevated but stable. By day 12 the platelet count had fallen to 37,000/L.
With physical therapy the patient gained strength. Antibiotics were discontinued on day 12 and a follow‐up chest x‐ray demonstrated no significant disease. From days 10 to 12, his creatinine rose from 1.5 to 1.9 mg/dL, although urine output remained normal.
A hematologist observed minimal fragmentation of red cells on the blood smear. Commenting on the thrombocytopenia, anemia, and LDH isoenzymes (representative of skeletal/hepatic origin rather than hematologic), and clinical improvement after treatment of a presumed pneumonia, he felt that the continued thrombocytopenia was likely due to drug toxicity, and recommended observation, treatment of renal failure, and discontinuation of tacrolimus.
The failure to increase the hemoglobin after transfusion is consistent with (but not specific for) hemolysis. In conjunction with the progressive thrombocytopenia and persistently elevated LDH, TTP remains a consideration. While TTP can be diagnosed with minimal evidence of schistocytes, the duration of this illness, now spanning almost 2 weeks without significant end organ damagenamely more pronounced renal failure, confusion, or feveris unusual for TTP. Therefore, I think it is reasonable to withhold plasma exchange, although if the cytopenias or renal failure progress after the methotrexate, tacrolimus, and antibiotics are stopped, it may have to be undertaken empirically.
The pulmonary process remains undefined. Edema, pneumonitis (eg, aspiration), a modest pneumonia, or pulmonary hemorrhage could normalize on chest x‐ray after 1 week.
Renal ultrasound was normal. Urinalysis dipstick demonstrated 3+ blood, 3+ protein, and no nitrate or leukocyte esterase. The urine sediment showed only granular casts. Fractional excretion of sodium was 6.7%. Urine protein‐to‐creatinine ratio was 7.5, and urine myoglobin was elevated. Serum C3 and C4 complement levels and cryoglobulins were normal. Reticulocyte count was 8.5% (normal, 0.53.2).
There is significant evidence for intrinsic renal failure, starting with the elevated fractional excretion. Marked proteinuria suggests glomerular damage; nephrotic syndrome could provide an explanation for the recurrent DVT. The 3+ blood without RBCs and the markedly elevated urine myoglobin suggest pigment nephropathy from both myoglobinuria and hemoglobinuria. The elevated reticulocyte count further confirms the impression of hemolysis.
Nephrotic syndrome may result from a primary disease process, such as diabetes, systemic lupus erythematosus (SLE), or amyloidosis, for which there is no evidence to date, or as a consequence of indolent infection, malignancy, or drugs, all of which are reasonable possibilities.
The essential elements at this point include thrombocytopenia, kidney failure with proteinuria, and likely intravascular hemolysis. I would repeat the peripheral smear (looking for schistocytes) and discuss with the rheumatologist if any other medications could be discontinued.
A nephrology consultant diagnosed acute tubular necrosis (ATN) from a combination of insults (intravenous contrast, methotrexate, tacrolimus, and myoglobinuria). Over the next several days, his platelet count rose to approximately 60,000/L. The patient continued to generally feel better but the creatinine steadily increased to 4.9 mg/dL.
The hematologist's reassessment of the smear was unchanged with minimal RBC fragmentation noted. Over the next few days the hemoglobin, creatinine, and platelet count remained stable, and there were no fevers or other clinical developments. On day 21 a kidney biopsy specimen revealed evidence of thrombotic microangiopathy (TMA) and segmental glomerular necrosis, with negative immunofluorescent findings. In addition, the glomerular basement membranes were thickened and effacement of the epithelial foot processes was noted.
TTP (or other MAHA) with only a few schistocytes would be unusual at an advanced stage where organ damage has occurred, although the clinical presentation in drug‐induced variety is variable. TTP is also generally a fatal disease, so relative stability over 3 weeks without definitive therapy is atypical, unless prednisone has served as a temporizing measure. The atypical features raise the possibility of a mimic or variant of TTP such as undiagnosed cancer causing DIC or a medication (eg, tacrolimus)‐associated TTP syndrome.
At least 2 other conditions could account for the hemolysis, thrombocytopenia, and TMA. The positive ANA, glomerular disease, and cytopenias are compatible with SLE, although such progression on an intense immunosuppressive regimen would be unusual. The renal histology in a patient with an autoimmune diathesis warrants reconsideration of antiphospholipid antibody syndrome (APLS), especially in light of the earlier DVT.
Tests for antiphospholipid antibodies were negative. After multidisciplinary deliberation, a diagnosis of TMA due to tacrolimus‐associated TTP/HUS was made. Plasmapheresis was initiated and IVIG and steroids were continued. He had a complicated hospital course and required renal replacement therapy, but with pheresis, his platelet counts and hemoglobin began to recover and he was ultimately discharged in good condition. After he was discharged, testing for ADAMTS13 (a von Willebrand factor‐cleaving protease) activity was reported as 54% (normal, >66%)
Discussion
TMA in the microcirculation is the hallmark pathology of TTP‐HUS but is not specific for this disease. TMA is also seen in disseminated intravascular coagulation, sepsis, cancer, malignant hypertension, human immunodeficiency virus infection, autoimmune disorders, pregnancy‐related conditions, and in association with certain drugs.1 The first pharmacological agent to be associated with TMA was mitomycin in 1971, and since then other drug associations have been described, including antiplatelet medications such as ticlopidine and clopidogrel, antibiotics such as quinine and rifampin, interferon, and immunosuppressants such as cyclosporine and tacrolimus.2 Drug‐induced variants of TTP and TMA are challenging to diagnose because the timing of onset, clinical features, and patient factors (eg, receipt of immunosuppressants) may vary widely and mimic other conditions.2, 3 TMA is a rare complication of tacrolimus and is mostly seen in renal transplant patients at a frequency of 1%. In these patients, renal dysfunction is usually the first herald of TMA and TTP; evidence of hemolysis may be absent.3
The clinical diagnosis of TTP has historically been based on the presence of a classic pentad: MAHA, thrombocytopenia, neurological and renal abnormalities, and fever.4 Elevated levels of LDH and indirect bilirubin and the presence of fragmented RBCs and reticulocytes point toward active intravascular hemolysis. The DAT is usually negative. This textbook illness scriptthe template of a disease that is stored in a clinician's memoryis learned by physicians during training, but undergoes little modification given the limited exposure to a rare disease.
In modern practice, the pentad is rarely seen, and the characteristics of the end‐organ findings may vary substantially. For instance, while neurological symptoms including seizures, coma, and transient confusion occur in 90% of cases, renal involvement is seen in about 50% and fever in only 25% of patients.5 Although the presence of 2 or more schistocytes on the blood smear under 100 microscopy supports the diagnosis of MAHA, cases of TTP without significant schistocytosis have been reported.6
Furthermore, TTP is typically described as acute in onset, but in a quarter of patients the symptoms and signs last for weeks before diagnosis.4 This variability in disease presentation coupled with the high mortality of untreated disease has changed the diagnostic and treatment thresholds for TTP. Trials and expert opinion use MAHA, thrombocytopenia, and the exclusion of alternative causes as sufficient criteria to diagnose TTP and begin treatment.7 The measurement of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity (a von Willebrand factor‐cleaving protease) for diagnostic purposes remains controversial because assay techniques are not uniform and there is insufficient correlation between levels and clinical disease.810 For instance, the presence of severe ADAMTS 13 deficiency (ie, <5%) along with the presence of an ADAMTS13 inhibitor is considered to be very specific, but not sensitive, for the laboratory diagnosis of idiopathic TTP.11 In cohort studies, the frequency of severe deficiency among patients with idiopathic TTP ranged from 18% to 100%, and the presence of severe deficiency did not predict the development of acute episodes of TTP.9 In a registry study of 142 patients diagnosed with TTP, 81% of patients with secondary TTP (ie, not classified as idiopathic) had ADAMTS13 levels that were normal to subnormal (>25%), and patients with normal ADAMTS13 levels had a higher incidence of acute renal failure, similar to the findings in this patient.10
Untreated TTP has a mortality rate of greater than 90%, but with plasma exchange, survival has improved dramatically.4, 7 Glucocorticoids are often used in addition to plasma exchange, based on case series and reports.9 The addition of cryoprecipitate or fresh frozen plasma to plasmapheresis has not been shown to be beneficial, but rituximab, an anti CD‐20 monoclonal antibody, has shown promise in a small prospective study.12, 13
TTP is a rare disorder with a classic description but substantial variation in clinical presentation. In this case, the background autoimmune myopathy, immunosuppression, coincident acute DVT, unexplained infiltrates, complex medication regimen, and nephrotic range proteinuria (attributed to focal segmental glomerular sclerosis based on the limited evidence available from the biopsy) led the clinicians to ascribe the patient's thrombocytopenia and renal injury to more common conditions and created a challenging environment for the diagnosis of TTP. TTP is a complex disorder and the simplified understanding of the disease and its time course prevented a prompt match between the patient's clinical course and his diagnosis. The combination of a rare condition with inherent variability arising in the setting of medical complexity challenges the processes of problem representation and scripting the answer for even the most seasoned clinician.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Key Teaching Points
-
The classically described pentad of TTP is seldom seen, and the findings of otherwise unexplained MAHA and thrombocytopenia should prompt consideration of TTP.
-
TTP may be acute and idiopathic, or be secondary to drugs, infections, or other conditions. Medication‐induced TTP may present with a wide range of clinical findings.
-
Therapeutic plasma exchange may be life‐saving in cases of TTP, and when appropriate, should be initiated promptly based on clinical suspicion and without waiting to perform tissue biopsy.
- , , .Thrombotic microangiopathies. In: Tischer CC, Brenner BM, eds.Renal Pathology.2nd ed.Philadelphia, PA:JB Lippincott;1994:1154–1184.
- , , .Drug‐induced thrombotic microangiopathy: incidence, prevention and management.Drug Saf.2001;24(7):491–501.
- , , , , .FK 506‐associated thrombotic microangiopathy: report of two cases and review of the literature.Transplantation.1999;67(4):539–544.
- , .Thrombotic Thrombocytopenic purpura: report of 16 cases and review of the literature.Medicine (Baltimore).1966;45:139–159.
- , , , .Thrombotic thrombocytopenic purpura; early and late responders.Am J Hematol.1997;54:102–107.
- .Atypical presentations of thrombotic thrombocytopenic purpura: a review.J Clin Apheresis.2009;24(1)47–52.
- , , , et al.Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura.N Engl J Med.1991;325:393–397.
- , , , , , .The incidence of thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS13deficiency.J Thromb Haemost.2005;3:1432–1436.
- .Thrombotic thrombocytopenic purpura.N Engl J Med.2006;354:1927–1935.
- , , , et al.ADAMTS13 activity in thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients.Blood.2003;102:60–68.
- , , .Thrombotic thrombocytopenic purpura.J Thromb Haemost.2005;3:1663–1675.
- , , , et al.Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials.Am J Kidney Dis.2009;53:259–272.
- , , , et al.:Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13‐deficient TTP: A study of 11 cases.Blood.2005;105:1932–1937.
A 58‐year old man was admitted with generalized weakness and acute deep venous thrombosis (DVT). His past medical history included hypertension and polymyositis/dermatomyositis (PM/DM) with anti‐synthase syndrome, which had been diagnosed 16 months prior when his creatine kinase (CK) was greater than 12,000 U/L. At that time he also was found to have bilateral lower extremity DVT, and had been treated with warfarin for 1 year. 10 days previously, he had been discharged after a 4‐day hospitalization for a polymyositis flare which was treated with methylprednisolone at 60 mg daily for 5 days. He was discharged home with daily prednisone until this follow‐up a week later, where he reported weakness and bilateral edema. Lower extremity ultrasound demonstrated acute thrombus in the right common femoral vein.
This acute extensive DVT may be a consequence of recent hospitalization and a previously damaged venous system, or may reflect ongoing hypercoagulability from an unresolved condition, such as cancer. Bilateral lower extremity edema may suggest right‐sided heart failure due to progressive interstitial lung disease, which occurs in a subset of patients with PM/DM. Edema may alternatively reflect biventricular heart failure, or liver or kidney disease.
Generalized weakness offers little in the way of focused differential diagnosis until it is characterized as motor weakness (eg, attributed to progression of the myopathy), a dyspnea‐equivalent, or an overall sense of fatigue.
His medications included weekly methotrexate, monthly intravenous immunoglobulin (IVIG) infusions, tacrolimus, hydrochlorothiazide, and aerosolized pentamidine. He had been on varying doses of prednisone for 2 years and his present dose was 40 mg daily. He was allergic to sulfa. He was married and stopped smoking 30 years previously, and did not drink alcohol or use illicit drugs.
Various medication toxicities could account for his presentation. Methotrexate causes interstitial lung disease, and IVIG and tacrolimus may cause renal failure (and fluid overload). The heavy degree of immunosuppression renders him susceptible to a wide range of infections. Aerosolized pentamidine provides incomplete protection against Pneumocystis jirovecii, especially in the lung apices.
Evaluation of the status of his myositis with motor strength assessment is important. In addition associated rashes and signs of malignancy (eg, lymphadenopathy) and infection should be sought. Proximal motor weakness would suggest a myositis flare, although care must be given to exclude competing causes of myopathy, including infections, toxins, or endocrinopathies.
His temperature was 36.2C, pulse 103 beats per minute, blood pressure 156/83 mm Hg, and respiratory rate 18 breaths per minute. He had crackles at both lung bases, and 3+ pitting edema in both lower extremities. On neurological exam his motor strength was found to be diminished at 3/5 in the lower extremities and proximal upper extremities and 4/5 in the distal upper extremities. Reflexes were uniformly at 1+/4 and his cognition was intact. Examinations of his head, skin, heart, and abdomen were normal.
The absence of elevated jugular venous pressure argues against right heart failure. He is afebrile but that is minimally reassuring given the immunosuppression. There are no clues to suggest liver or kidney dysfunction. An unrecognized occlusion of the lower abdominal venous or lymphatic system such as upward extension of the DVT into the inferior vena cava (IVC) or a pelvic obstruction of the lower extremity lymphatic vessels could be considered. It appears that his distal weakness closely mirrors his proximal weakness in distinction to most myopathies which are predominantly proximal (with some exceptions, eg, inclusion body myositis).
The white blood cell count was 26,000/L with normal differential, hemoglobin 11.2 gm/dL, and platelet count was 191,000/L (at recent discharge these values were 23,000, 11.9, and 274,000, respectively). Chemistries were normal except for creatinine of 1.4 mg/dL (baseline 1.2), blood urea nitrogen was 42 mg/dL, albumin 2.6 gm/dL (normal, 3.55.0), and CK 3,710 U/L (20220), decreased from 6,943 U/L at recent discharge. Urine dipstick testing was positive for blood and protein; the urine sediment was unremarkable. Chest radiograph revealed normal lungs and heart.
The white blood cell count is quite elevated, perhaps more so than could be attributed to chronic steroid use, and again raises the concern of an undiagnosed infection. The presence of heme (and protein) in the urine without cells is consistent with pigment nephropathy from the recent rhabdomyolysis.
He was admitted to the hospital. Unfractionated heparin and warfarin were started. No changes were made to his immunosuppressive regimen. Blood cultures were negative after 48 hours. Transthoracic echocardiogram showed an ejection fraction of 60%, normal valves, and right ventricular systolic pressure of 32 mm Hg (normal, 1525 mmHg). On hospital day 3, his platelet count was 147,000/L, and on day 5, 101,000/L. His other laboratory values remained unchanged, and there were no new clinical developments.
A declining platelet count and extensive deep vein thrombosis suggest heparin‐induced thrombocytopenia and thrombosis (HITT), especially with the greater than 50% drop in the setting of IV heparin. His platelets have continued on a downward trajectory that was evident at admission and has progressed during this hospitalization. Assuming this is not due to laboratory error or artifact such as platelet clumping, this decline could have occurred if he was sensitized to heparin during the prior hospitalization, such as for DVT prophylaxis. It is increasingly recognized that HITT can manifest even after exposure to heparin is complete, ie, posthospitalization, and there can be an immediate drop in platelet counts if an unrecognized HITT‐mediated thrombosis is treated with IV heparin. Heparin should be discontinued in favor of a direct thrombin inhibitor and tests for heparin‐induced platelet antibodies (HIPA) and serotonin‐release assay (SRA) sent.
Antiphospholipid antibody syndrome (APLS) is associated with hypercoagulability and thrombocytopenia and is more frequent in patients with autoimmune disorders. The drug list should also be examined for associations with thrombocytopenia. The peripheral smear should be scrutinized and hemoglobin and creatinine followed to exclude thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome (TTP‐HUS).
Heparin was stopped on day 5. Warfarin was continued with a therapeutic international normalized ratio (INR). Tests for antiplatelet factor 4 antibodies, HIPA, and SRA were negative. His weakness and edema improved although his CK remained between 2000 and 4000 U/L. On day 5 he developed mild hemoptysis, and a repeat chest radiograph demonstrated a new left hilar infiltrate. Computed tomography (CT) scan of the chest with contrast demonstrated a left lower lobe consolidation, scattered ground glass opacities in both lung bases, and no pulmonary embolus. He was treated with piperacillin/tazobactam and vancomycin. He remained afebrile. The same day, he erroneously received 125 mg (instead of 12.5 mg) of subcutaneous methotrexate. High‐dose leucovorin was administered on days 5 and 6.
The hemoptysis resolved after 2 days. From days 5 to 9, the platelet count dropped to 80,000/L and his hemoglobin gradually decreased to 7.3 g/dL. Anticoagulation was stopped, vitamin K administered, and an IVC filter placed. Two units of packed red blood cells (RBCs) were transfused.
In suspected HITT (which was not verified here), warfarin is typically withheld until the platelets have recovered and thrombin‐inhibitor anticoagulation has reached a steady state, to avoid the transient hypercoagulability of warfarin initiation.
The unusual time course and the 3 negative tests make HITT unlikely. The continued platelet decline after stopping heparin further supports another etiology. The excess methotrexate dosing complicates interpretation of his thrombocytopenia and anemia, which can be explained by mucosal bleeding, microangiopathic hemolytic anemia (MAHA) such as disseminated intravascular coagulation or TTP‐HUS, or autoimmunity (Evans syndrome). Bone marrow toxicity is also a major effect of methotrexate (in addition to elevation of liver enzymes and acute renal failure); however, there is typically a lag between administration and development of cytopenias. The antibiotics could also account for the ongoing (but not original) thrombocytopenia.
With the new pulmonary infiltrate, infections remain a primary concern and should be evaluated with sputum samples and perhaps bronchoscopy. Given the abnormal urine (even without cells), a pulmonary‐renal inflammatory processes should be considered also to explain the infiltrates and hemoptysis.
Haptoglobin was <20 mg/dL (normal, 37246). The direct antiglobulin test (DAT) was negative. Serum lactate dehydrogenase (LDH) was 1657 U/L (normal, 100220), with elevated LD4 and LD5 isoenzymes. Coagulation studies normalized after the administration of vitamin K. Anti‐nuclear antibody was positive at 8.7 (normal <1.5). Tests for antineutrophil cytoplasmic antibodies were negative. No sputum could be obtained. A pathologist reviewed the blood smear and reported neutrophilic leukocytosis without left shift, and thrombocytopenia with normal platelet morphology.
Low haptoglobin in the setting of an elevated LDH is highly suggestive of hemolysis, particularly the intravascular, microangiopathic varieties. Neutrophilia may reflect infection, a primary myeloproliferative process such as chronic myeloid leukemia, steroid use, or a reactive bone marrow in the setting of acute illness. The negative DAT and significant immunosuppressive regimen makes immune‐mediated hemolysis unlikely, although the history of autoimmunity and the small DAT false‐negative rate leaves Evans syndrome as an outside possibility. Medications such as tacrolimus (causing TTP) or IVIG (given the broad spectrum of antibodies it includes) are other plausible causes of the cytopenias.
At this point, I would analyze the red blood cell (RBC) morphology and check the reticulocyte count to help differentiate between hemolysis and a myelotoxin.
After transfusion, his hemoglobin remained at approximately 8.5 gm/dL and LDH remained elevated but stable. By day 12 the platelet count had fallen to 37,000/L.
With physical therapy the patient gained strength. Antibiotics were discontinued on day 12 and a follow‐up chest x‐ray demonstrated no significant disease. From days 10 to 12, his creatinine rose from 1.5 to 1.9 mg/dL, although urine output remained normal.
A hematologist observed minimal fragmentation of red cells on the blood smear. Commenting on the thrombocytopenia, anemia, and LDH isoenzymes (representative of skeletal/hepatic origin rather than hematologic), and clinical improvement after treatment of a presumed pneumonia, he felt that the continued thrombocytopenia was likely due to drug toxicity, and recommended observation, treatment of renal failure, and discontinuation of tacrolimus.
The failure to increase the hemoglobin after transfusion is consistent with (but not specific for) hemolysis. In conjunction with the progressive thrombocytopenia and persistently elevated LDH, TTP remains a consideration. While TTP can be diagnosed with minimal evidence of schistocytes, the duration of this illness, now spanning almost 2 weeks without significant end organ damagenamely more pronounced renal failure, confusion, or feveris unusual for TTP. Therefore, I think it is reasonable to withhold plasma exchange, although if the cytopenias or renal failure progress after the methotrexate, tacrolimus, and antibiotics are stopped, it may have to be undertaken empirically.
The pulmonary process remains undefined. Edema, pneumonitis (eg, aspiration), a modest pneumonia, or pulmonary hemorrhage could normalize on chest x‐ray after 1 week.
Renal ultrasound was normal. Urinalysis dipstick demonstrated 3+ blood, 3+ protein, and no nitrate or leukocyte esterase. The urine sediment showed only granular casts. Fractional excretion of sodium was 6.7%. Urine protein‐to‐creatinine ratio was 7.5, and urine myoglobin was elevated. Serum C3 and C4 complement levels and cryoglobulins were normal. Reticulocyte count was 8.5% (normal, 0.53.2).
There is significant evidence for intrinsic renal failure, starting with the elevated fractional excretion. Marked proteinuria suggests glomerular damage; nephrotic syndrome could provide an explanation for the recurrent DVT. The 3+ blood without RBCs and the markedly elevated urine myoglobin suggest pigment nephropathy from both myoglobinuria and hemoglobinuria. The elevated reticulocyte count further confirms the impression of hemolysis.
Nephrotic syndrome may result from a primary disease process, such as diabetes, systemic lupus erythematosus (SLE), or amyloidosis, for which there is no evidence to date, or as a consequence of indolent infection, malignancy, or drugs, all of which are reasonable possibilities.
The essential elements at this point include thrombocytopenia, kidney failure with proteinuria, and likely intravascular hemolysis. I would repeat the peripheral smear (looking for schistocytes) and discuss with the rheumatologist if any other medications could be discontinued.
A nephrology consultant diagnosed acute tubular necrosis (ATN) from a combination of insults (intravenous contrast, methotrexate, tacrolimus, and myoglobinuria). Over the next several days, his platelet count rose to approximately 60,000/L. The patient continued to generally feel better but the creatinine steadily increased to 4.9 mg/dL.
The hematologist's reassessment of the smear was unchanged with minimal RBC fragmentation noted. Over the next few days the hemoglobin, creatinine, and platelet count remained stable, and there were no fevers or other clinical developments. On day 21 a kidney biopsy specimen revealed evidence of thrombotic microangiopathy (TMA) and segmental glomerular necrosis, with negative immunofluorescent findings. In addition, the glomerular basement membranes were thickened and effacement of the epithelial foot processes was noted.
TTP (or other MAHA) with only a few schistocytes would be unusual at an advanced stage where organ damage has occurred, although the clinical presentation in drug‐induced variety is variable. TTP is also generally a fatal disease, so relative stability over 3 weeks without definitive therapy is atypical, unless prednisone has served as a temporizing measure. The atypical features raise the possibility of a mimic or variant of TTP such as undiagnosed cancer causing DIC or a medication (eg, tacrolimus)‐associated TTP syndrome.
At least 2 other conditions could account for the hemolysis, thrombocytopenia, and TMA. The positive ANA, glomerular disease, and cytopenias are compatible with SLE, although such progression on an intense immunosuppressive regimen would be unusual. The renal histology in a patient with an autoimmune diathesis warrants reconsideration of antiphospholipid antibody syndrome (APLS), especially in light of the earlier DVT.
Tests for antiphospholipid antibodies were negative. After multidisciplinary deliberation, a diagnosis of TMA due to tacrolimus‐associated TTP/HUS was made. Plasmapheresis was initiated and IVIG and steroids were continued. He had a complicated hospital course and required renal replacement therapy, but with pheresis, his platelet counts and hemoglobin began to recover and he was ultimately discharged in good condition. After he was discharged, testing for ADAMTS13 (a von Willebrand factor‐cleaving protease) activity was reported as 54% (normal, >66%)
Discussion
TMA in the microcirculation is the hallmark pathology of TTP‐HUS but is not specific for this disease. TMA is also seen in disseminated intravascular coagulation, sepsis, cancer, malignant hypertension, human immunodeficiency virus infection, autoimmune disorders, pregnancy‐related conditions, and in association with certain drugs.1 The first pharmacological agent to be associated with TMA was mitomycin in 1971, and since then other drug associations have been described, including antiplatelet medications such as ticlopidine and clopidogrel, antibiotics such as quinine and rifampin, interferon, and immunosuppressants such as cyclosporine and tacrolimus.2 Drug‐induced variants of TTP and TMA are challenging to diagnose because the timing of onset, clinical features, and patient factors (eg, receipt of immunosuppressants) may vary widely and mimic other conditions.2, 3 TMA is a rare complication of tacrolimus and is mostly seen in renal transplant patients at a frequency of 1%. In these patients, renal dysfunction is usually the first herald of TMA and TTP; evidence of hemolysis may be absent.3
The clinical diagnosis of TTP has historically been based on the presence of a classic pentad: MAHA, thrombocytopenia, neurological and renal abnormalities, and fever.4 Elevated levels of LDH and indirect bilirubin and the presence of fragmented RBCs and reticulocytes point toward active intravascular hemolysis. The DAT is usually negative. This textbook illness scriptthe template of a disease that is stored in a clinician's memoryis learned by physicians during training, but undergoes little modification given the limited exposure to a rare disease.
In modern practice, the pentad is rarely seen, and the characteristics of the end‐organ findings may vary substantially. For instance, while neurological symptoms including seizures, coma, and transient confusion occur in 90% of cases, renal involvement is seen in about 50% and fever in only 25% of patients.5 Although the presence of 2 or more schistocytes on the blood smear under 100 microscopy supports the diagnosis of MAHA, cases of TTP without significant schistocytosis have been reported.6
Furthermore, TTP is typically described as acute in onset, but in a quarter of patients the symptoms and signs last for weeks before diagnosis.4 This variability in disease presentation coupled with the high mortality of untreated disease has changed the diagnostic and treatment thresholds for TTP. Trials and expert opinion use MAHA, thrombocytopenia, and the exclusion of alternative causes as sufficient criteria to diagnose TTP and begin treatment.7 The measurement of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity (a von Willebrand factor‐cleaving protease) for diagnostic purposes remains controversial because assay techniques are not uniform and there is insufficient correlation between levels and clinical disease.810 For instance, the presence of severe ADAMTS 13 deficiency (ie, <5%) along with the presence of an ADAMTS13 inhibitor is considered to be very specific, but not sensitive, for the laboratory diagnosis of idiopathic TTP.11 In cohort studies, the frequency of severe deficiency among patients with idiopathic TTP ranged from 18% to 100%, and the presence of severe deficiency did not predict the development of acute episodes of TTP.9 In a registry study of 142 patients diagnosed with TTP, 81% of patients with secondary TTP (ie, not classified as idiopathic) had ADAMTS13 levels that were normal to subnormal (>25%), and patients with normal ADAMTS13 levels had a higher incidence of acute renal failure, similar to the findings in this patient.10
Untreated TTP has a mortality rate of greater than 90%, but with plasma exchange, survival has improved dramatically.4, 7 Glucocorticoids are often used in addition to plasma exchange, based on case series and reports.9 The addition of cryoprecipitate or fresh frozen plasma to plasmapheresis has not been shown to be beneficial, but rituximab, an anti CD‐20 monoclonal antibody, has shown promise in a small prospective study.12, 13
TTP is a rare disorder with a classic description but substantial variation in clinical presentation. In this case, the background autoimmune myopathy, immunosuppression, coincident acute DVT, unexplained infiltrates, complex medication regimen, and nephrotic range proteinuria (attributed to focal segmental glomerular sclerosis based on the limited evidence available from the biopsy) led the clinicians to ascribe the patient's thrombocytopenia and renal injury to more common conditions and created a challenging environment for the diagnosis of TTP. TTP is a complex disorder and the simplified understanding of the disease and its time course prevented a prompt match between the patient's clinical course and his diagnosis. The combination of a rare condition with inherent variability arising in the setting of medical complexity challenges the processes of problem representation and scripting the answer for even the most seasoned clinician.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Key Teaching Points
-
The classically described pentad of TTP is seldom seen, and the findings of otherwise unexplained MAHA and thrombocytopenia should prompt consideration of TTP.
-
TTP may be acute and idiopathic, or be secondary to drugs, infections, or other conditions. Medication‐induced TTP may present with a wide range of clinical findings.
-
Therapeutic plasma exchange may be life‐saving in cases of TTP, and when appropriate, should be initiated promptly based on clinical suspicion and without waiting to perform tissue biopsy.
A 58‐year old man was admitted with generalized weakness and acute deep venous thrombosis (DVT). His past medical history included hypertension and polymyositis/dermatomyositis (PM/DM) with anti‐synthase syndrome, which had been diagnosed 16 months prior when his creatine kinase (CK) was greater than 12,000 U/L. At that time he also was found to have bilateral lower extremity DVT, and had been treated with warfarin for 1 year. 10 days previously, he had been discharged after a 4‐day hospitalization for a polymyositis flare which was treated with methylprednisolone at 60 mg daily for 5 days. He was discharged home with daily prednisone until this follow‐up a week later, where he reported weakness and bilateral edema. Lower extremity ultrasound demonstrated acute thrombus in the right common femoral vein.
This acute extensive DVT may be a consequence of recent hospitalization and a previously damaged venous system, or may reflect ongoing hypercoagulability from an unresolved condition, such as cancer. Bilateral lower extremity edema may suggest right‐sided heart failure due to progressive interstitial lung disease, which occurs in a subset of patients with PM/DM. Edema may alternatively reflect biventricular heart failure, or liver or kidney disease.
Generalized weakness offers little in the way of focused differential diagnosis until it is characterized as motor weakness (eg, attributed to progression of the myopathy), a dyspnea‐equivalent, or an overall sense of fatigue.
His medications included weekly methotrexate, monthly intravenous immunoglobulin (IVIG) infusions, tacrolimus, hydrochlorothiazide, and aerosolized pentamidine. He had been on varying doses of prednisone for 2 years and his present dose was 40 mg daily. He was allergic to sulfa. He was married and stopped smoking 30 years previously, and did not drink alcohol or use illicit drugs.
Various medication toxicities could account for his presentation. Methotrexate causes interstitial lung disease, and IVIG and tacrolimus may cause renal failure (and fluid overload). The heavy degree of immunosuppression renders him susceptible to a wide range of infections. Aerosolized pentamidine provides incomplete protection against Pneumocystis jirovecii, especially in the lung apices.
Evaluation of the status of his myositis with motor strength assessment is important. In addition associated rashes and signs of malignancy (eg, lymphadenopathy) and infection should be sought. Proximal motor weakness would suggest a myositis flare, although care must be given to exclude competing causes of myopathy, including infections, toxins, or endocrinopathies.
His temperature was 36.2C, pulse 103 beats per minute, blood pressure 156/83 mm Hg, and respiratory rate 18 breaths per minute. He had crackles at both lung bases, and 3+ pitting edema in both lower extremities. On neurological exam his motor strength was found to be diminished at 3/5 in the lower extremities and proximal upper extremities and 4/5 in the distal upper extremities. Reflexes were uniformly at 1+/4 and his cognition was intact. Examinations of his head, skin, heart, and abdomen were normal.
The absence of elevated jugular venous pressure argues against right heart failure. He is afebrile but that is minimally reassuring given the immunosuppression. There are no clues to suggest liver or kidney dysfunction. An unrecognized occlusion of the lower abdominal venous or lymphatic system such as upward extension of the DVT into the inferior vena cava (IVC) or a pelvic obstruction of the lower extremity lymphatic vessels could be considered. It appears that his distal weakness closely mirrors his proximal weakness in distinction to most myopathies which are predominantly proximal (with some exceptions, eg, inclusion body myositis).
The white blood cell count was 26,000/L with normal differential, hemoglobin 11.2 gm/dL, and platelet count was 191,000/L (at recent discharge these values were 23,000, 11.9, and 274,000, respectively). Chemistries were normal except for creatinine of 1.4 mg/dL (baseline 1.2), blood urea nitrogen was 42 mg/dL, albumin 2.6 gm/dL (normal, 3.55.0), and CK 3,710 U/L (20220), decreased from 6,943 U/L at recent discharge. Urine dipstick testing was positive for blood and protein; the urine sediment was unremarkable. Chest radiograph revealed normal lungs and heart.
The white blood cell count is quite elevated, perhaps more so than could be attributed to chronic steroid use, and again raises the concern of an undiagnosed infection. The presence of heme (and protein) in the urine without cells is consistent with pigment nephropathy from the recent rhabdomyolysis.
He was admitted to the hospital. Unfractionated heparin and warfarin were started. No changes were made to his immunosuppressive regimen. Blood cultures were negative after 48 hours. Transthoracic echocardiogram showed an ejection fraction of 60%, normal valves, and right ventricular systolic pressure of 32 mm Hg (normal, 1525 mmHg). On hospital day 3, his platelet count was 147,000/L, and on day 5, 101,000/L. His other laboratory values remained unchanged, and there were no new clinical developments.
A declining platelet count and extensive deep vein thrombosis suggest heparin‐induced thrombocytopenia and thrombosis (HITT), especially with the greater than 50% drop in the setting of IV heparin. His platelets have continued on a downward trajectory that was evident at admission and has progressed during this hospitalization. Assuming this is not due to laboratory error or artifact such as platelet clumping, this decline could have occurred if he was sensitized to heparin during the prior hospitalization, such as for DVT prophylaxis. It is increasingly recognized that HITT can manifest even after exposure to heparin is complete, ie, posthospitalization, and there can be an immediate drop in platelet counts if an unrecognized HITT‐mediated thrombosis is treated with IV heparin. Heparin should be discontinued in favor of a direct thrombin inhibitor and tests for heparin‐induced platelet antibodies (HIPA) and serotonin‐release assay (SRA) sent.
Antiphospholipid antibody syndrome (APLS) is associated with hypercoagulability and thrombocytopenia and is more frequent in patients with autoimmune disorders. The drug list should also be examined for associations with thrombocytopenia. The peripheral smear should be scrutinized and hemoglobin and creatinine followed to exclude thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome (TTP‐HUS).
Heparin was stopped on day 5. Warfarin was continued with a therapeutic international normalized ratio (INR). Tests for antiplatelet factor 4 antibodies, HIPA, and SRA were negative. His weakness and edema improved although his CK remained between 2000 and 4000 U/L. On day 5 he developed mild hemoptysis, and a repeat chest radiograph demonstrated a new left hilar infiltrate. Computed tomography (CT) scan of the chest with contrast demonstrated a left lower lobe consolidation, scattered ground glass opacities in both lung bases, and no pulmonary embolus. He was treated with piperacillin/tazobactam and vancomycin. He remained afebrile. The same day, he erroneously received 125 mg (instead of 12.5 mg) of subcutaneous methotrexate. High‐dose leucovorin was administered on days 5 and 6.
The hemoptysis resolved after 2 days. From days 5 to 9, the platelet count dropped to 80,000/L and his hemoglobin gradually decreased to 7.3 g/dL. Anticoagulation was stopped, vitamin K administered, and an IVC filter placed. Two units of packed red blood cells (RBCs) were transfused.
In suspected HITT (which was not verified here), warfarin is typically withheld until the platelets have recovered and thrombin‐inhibitor anticoagulation has reached a steady state, to avoid the transient hypercoagulability of warfarin initiation.
The unusual time course and the 3 negative tests make HITT unlikely. The continued platelet decline after stopping heparin further supports another etiology. The excess methotrexate dosing complicates interpretation of his thrombocytopenia and anemia, which can be explained by mucosal bleeding, microangiopathic hemolytic anemia (MAHA) such as disseminated intravascular coagulation or TTP‐HUS, or autoimmunity (Evans syndrome). Bone marrow toxicity is also a major effect of methotrexate (in addition to elevation of liver enzymes and acute renal failure); however, there is typically a lag between administration and development of cytopenias. The antibiotics could also account for the ongoing (but not original) thrombocytopenia.
With the new pulmonary infiltrate, infections remain a primary concern and should be evaluated with sputum samples and perhaps bronchoscopy. Given the abnormal urine (even without cells), a pulmonary‐renal inflammatory processes should be considered also to explain the infiltrates and hemoptysis.
Haptoglobin was <20 mg/dL (normal, 37246). The direct antiglobulin test (DAT) was negative. Serum lactate dehydrogenase (LDH) was 1657 U/L (normal, 100220), with elevated LD4 and LD5 isoenzymes. Coagulation studies normalized after the administration of vitamin K. Anti‐nuclear antibody was positive at 8.7 (normal <1.5). Tests for antineutrophil cytoplasmic antibodies were negative. No sputum could be obtained. A pathologist reviewed the blood smear and reported neutrophilic leukocytosis without left shift, and thrombocytopenia with normal platelet morphology.
Low haptoglobin in the setting of an elevated LDH is highly suggestive of hemolysis, particularly the intravascular, microangiopathic varieties. Neutrophilia may reflect infection, a primary myeloproliferative process such as chronic myeloid leukemia, steroid use, or a reactive bone marrow in the setting of acute illness. The negative DAT and significant immunosuppressive regimen makes immune‐mediated hemolysis unlikely, although the history of autoimmunity and the small DAT false‐negative rate leaves Evans syndrome as an outside possibility. Medications such as tacrolimus (causing TTP) or IVIG (given the broad spectrum of antibodies it includes) are other plausible causes of the cytopenias.
At this point, I would analyze the red blood cell (RBC) morphology and check the reticulocyte count to help differentiate between hemolysis and a myelotoxin.
After transfusion, his hemoglobin remained at approximately 8.5 gm/dL and LDH remained elevated but stable. By day 12 the platelet count had fallen to 37,000/L.
With physical therapy the patient gained strength. Antibiotics were discontinued on day 12 and a follow‐up chest x‐ray demonstrated no significant disease. From days 10 to 12, his creatinine rose from 1.5 to 1.9 mg/dL, although urine output remained normal.
A hematologist observed minimal fragmentation of red cells on the blood smear. Commenting on the thrombocytopenia, anemia, and LDH isoenzymes (representative of skeletal/hepatic origin rather than hematologic), and clinical improvement after treatment of a presumed pneumonia, he felt that the continued thrombocytopenia was likely due to drug toxicity, and recommended observation, treatment of renal failure, and discontinuation of tacrolimus.
The failure to increase the hemoglobin after transfusion is consistent with (but not specific for) hemolysis. In conjunction with the progressive thrombocytopenia and persistently elevated LDH, TTP remains a consideration. While TTP can be diagnosed with minimal evidence of schistocytes, the duration of this illness, now spanning almost 2 weeks without significant end organ damagenamely more pronounced renal failure, confusion, or feveris unusual for TTP. Therefore, I think it is reasonable to withhold plasma exchange, although if the cytopenias or renal failure progress after the methotrexate, tacrolimus, and antibiotics are stopped, it may have to be undertaken empirically.
The pulmonary process remains undefined. Edema, pneumonitis (eg, aspiration), a modest pneumonia, or pulmonary hemorrhage could normalize on chest x‐ray after 1 week.
Renal ultrasound was normal. Urinalysis dipstick demonstrated 3+ blood, 3+ protein, and no nitrate or leukocyte esterase. The urine sediment showed only granular casts. Fractional excretion of sodium was 6.7%. Urine protein‐to‐creatinine ratio was 7.5, and urine myoglobin was elevated. Serum C3 and C4 complement levels and cryoglobulins were normal. Reticulocyte count was 8.5% (normal, 0.53.2).
There is significant evidence for intrinsic renal failure, starting with the elevated fractional excretion. Marked proteinuria suggests glomerular damage; nephrotic syndrome could provide an explanation for the recurrent DVT. The 3+ blood without RBCs and the markedly elevated urine myoglobin suggest pigment nephropathy from both myoglobinuria and hemoglobinuria. The elevated reticulocyte count further confirms the impression of hemolysis.
Nephrotic syndrome may result from a primary disease process, such as diabetes, systemic lupus erythematosus (SLE), or amyloidosis, for which there is no evidence to date, or as a consequence of indolent infection, malignancy, or drugs, all of which are reasonable possibilities.
The essential elements at this point include thrombocytopenia, kidney failure with proteinuria, and likely intravascular hemolysis. I would repeat the peripheral smear (looking for schistocytes) and discuss with the rheumatologist if any other medications could be discontinued.
A nephrology consultant diagnosed acute tubular necrosis (ATN) from a combination of insults (intravenous contrast, methotrexate, tacrolimus, and myoglobinuria). Over the next several days, his platelet count rose to approximately 60,000/L. The patient continued to generally feel better but the creatinine steadily increased to 4.9 mg/dL.
The hematologist's reassessment of the smear was unchanged with minimal RBC fragmentation noted. Over the next few days the hemoglobin, creatinine, and platelet count remained stable, and there were no fevers or other clinical developments. On day 21 a kidney biopsy specimen revealed evidence of thrombotic microangiopathy (TMA) and segmental glomerular necrosis, with negative immunofluorescent findings. In addition, the glomerular basement membranes were thickened and effacement of the epithelial foot processes was noted.
TTP (or other MAHA) with only a few schistocytes would be unusual at an advanced stage where organ damage has occurred, although the clinical presentation in drug‐induced variety is variable. TTP is also generally a fatal disease, so relative stability over 3 weeks without definitive therapy is atypical, unless prednisone has served as a temporizing measure. The atypical features raise the possibility of a mimic or variant of TTP such as undiagnosed cancer causing DIC or a medication (eg, tacrolimus)‐associated TTP syndrome.
At least 2 other conditions could account for the hemolysis, thrombocytopenia, and TMA. The positive ANA, glomerular disease, and cytopenias are compatible with SLE, although such progression on an intense immunosuppressive regimen would be unusual. The renal histology in a patient with an autoimmune diathesis warrants reconsideration of antiphospholipid antibody syndrome (APLS), especially in light of the earlier DVT.
Tests for antiphospholipid antibodies were negative. After multidisciplinary deliberation, a diagnosis of TMA due to tacrolimus‐associated TTP/HUS was made. Plasmapheresis was initiated and IVIG and steroids were continued. He had a complicated hospital course and required renal replacement therapy, but with pheresis, his platelet counts and hemoglobin began to recover and he was ultimately discharged in good condition. After he was discharged, testing for ADAMTS13 (a von Willebrand factor‐cleaving protease) activity was reported as 54% (normal, >66%)
Discussion
TMA in the microcirculation is the hallmark pathology of TTP‐HUS but is not specific for this disease. TMA is also seen in disseminated intravascular coagulation, sepsis, cancer, malignant hypertension, human immunodeficiency virus infection, autoimmune disorders, pregnancy‐related conditions, and in association with certain drugs.1 The first pharmacological agent to be associated with TMA was mitomycin in 1971, and since then other drug associations have been described, including antiplatelet medications such as ticlopidine and clopidogrel, antibiotics such as quinine and rifampin, interferon, and immunosuppressants such as cyclosporine and tacrolimus.2 Drug‐induced variants of TTP and TMA are challenging to diagnose because the timing of onset, clinical features, and patient factors (eg, receipt of immunosuppressants) may vary widely and mimic other conditions.2, 3 TMA is a rare complication of tacrolimus and is mostly seen in renal transplant patients at a frequency of 1%. In these patients, renal dysfunction is usually the first herald of TMA and TTP; evidence of hemolysis may be absent.3
The clinical diagnosis of TTP has historically been based on the presence of a classic pentad: MAHA, thrombocytopenia, neurological and renal abnormalities, and fever.4 Elevated levels of LDH and indirect bilirubin and the presence of fragmented RBCs and reticulocytes point toward active intravascular hemolysis. The DAT is usually negative. This textbook illness scriptthe template of a disease that is stored in a clinician's memoryis learned by physicians during training, but undergoes little modification given the limited exposure to a rare disease.
In modern practice, the pentad is rarely seen, and the characteristics of the end‐organ findings may vary substantially. For instance, while neurological symptoms including seizures, coma, and transient confusion occur in 90% of cases, renal involvement is seen in about 50% and fever in only 25% of patients.5 Although the presence of 2 or more schistocytes on the blood smear under 100 microscopy supports the diagnosis of MAHA, cases of TTP without significant schistocytosis have been reported.6
Furthermore, TTP is typically described as acute in onset, but in a quarter of patients the symptoms and signs last for weeks before diagnosis.4 This variability in disease presentation coupled with the high mortality of untreated disease has changed the diagnostic and treatment thresholds for TTP. Trials and expert opinion use MAHA, thrombocytopenia, and the exclusion of alternative causes as sufficient criteria to diagnose TTP and begin treatment.7 The measurement of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity (a von Willebrand factor‐cleaving protease) for diagnostic purposes remains controversial because assay techniques are not uniform and there is insufficient correlation between levels and clinical disease.810 For instance, the presence of severe ADAMTS 13 deficiency (ie, <5%) along with the presence of an ADAMTS13 inhibitor is considered to be very specific, but not sensitive, for the laboratory diagnosis of idiopathic TTP.11 In cohort studies, the frequency of severe deficiency among patients with idiopathic TTP ranged from 18% to 100%, and the presence of severe deficiency did not predict the development of acute episodes of TTP.9 In a registry study of 142 patients diagnosed with TTP, 81% of patients with secondary TTP (ie, not classified as idiopathic) had ADAMTS13 levels that were normal to subnormal (>25%), and patients with normal ADAMTS13 levels had a higher incidence of acute renal failure, similar to the findings in this patient.10
Untreated TTP has a mortality rate of greater than 90%, but with plasma exchange, survival has improved dramatically.4, 7 Glucocorticoids are often used in addition to plasma exchange, based on case series and reports.9 The addition of cryoprecipitate or fresh frozen plasma to plasmapheresis has not been shown to be beneficial, but rituximab, an anti CD‐20 monoclonal antibody, has shown promise in a small prospective study.12, 13
TTP is a rare disorder with a classic description but substantial variation in clinical presentation. In this case, the background autoimmune myopathy, immunosuppression, coincident acute DVT, unexplained infiltrates, complex medication regimen, and nephrotic range proteinuria (attributed to focal segmental glomerular sclerosis based on the limited evidence available from the biopsy) led the clinicians to ascribe the patient's thrombocytopenia and renal injury to more common conditions and created a challenging environment for the diagnosis of TTP. TTP is a complex disorder and the simplified understanding of the disease and its time course prevented a prompt match between the patient's clinical course and his diagnosis. The combination of a rare condition with inherent variability arising in the setting of medical complexity challenges the processes of problem representation and scripting the answer for even the most seasoned clinician.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Key Teaching Points
-
The classically described pentad of TTP is seldom seen, and the findings of otherwise unexplained MAHA and thrombocytopenia should prompt consideration of TTP.
-
TTP may be acute and idiopathic, or be secondary to drugs, infections, or other conditions. Medication‐induced TTP may present with a wide range of clinical findings.
-
Therapeutic plasma exchange may be life‐saving in cases of TTP, and when appropriate, should be initiated promptly based on clinical suspicion and without waiting to perform tissue biopsy.
- , , .Thrombotic microangiopathies. In: Tischer CC, Brenner BM, eds.Renal Pathology.2nd ed.Philadelphia, PA:JB Lippincott;1994:1154–1184.
- , , .Drug‐induced thrombotic microangiopathy: incidence, prevention and management.Drug Saf.2001;24(7):491–501.
- , , , , .FK 506‐associated thrombotic microangiopathy: report of two cases and review of the literature.Transplantation.1999;67(4):539–544.
- , .Thrombotic Thrombocytopenic purpura: report of 16 cases and review of the literature.Medicine (Baltimore).1966;45:139–159.
- , , , .Thrombotic thrombocytopenic purpura; early and late responders.Am J Hematol.1997;54:102–107.
- .Atypical presentations of thrombotic thrombocytopenic purpura: a review.J Clin Apheresis.2009;24(1)47–52.
- , , , et al.Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura.N Engl J Med.1991;325:393–397.
- , , , , , .The incidence of thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS13deficiency.J Thromb Haemost.2005;3:1432–1436.
- .Thrombotic thrombocytopenic purpura.N Engl J Med.2006;354:1927–1935.
- , , , et al.ADAMTS13 activity in thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients.Blood.2003;102:60–68.
- , , .Thrombotic thrombocytopenic purpura.J Thromb Haemost.2005;3:1663–1675.
- , , , et al.Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials.Am J Kidney Dis.2009;53:259–272.
- , , , et al.:Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13‐deficient TTP: A study of 11 cases.Blood.2005;105:1932–1937.
- , , .Thrombotic microangiopathies. In: Tischer CC, Brenner BM, eds.Renal Pathology.2nd ed.Philadelphia, PA:JB Lippincott;1994:1154–1184.
- , , .Drug‐induced thrombotic microangiopathy: incidence, prevention and management.Drug Saf.2001;24(7):491–501.
- , , , , .FK 506‐associated thrombotic microangiopathy: report of two cases and review of the literature.Transplantation.1999;67(4):539–544.
- , .Thrombotic Thrombocytopenic purpura: report of 16 cases and review of the literature.Medicine (Baltimore).1966;45:139–159.
- , , , .Thrombotic thrombocytopenic purpura; early and late responders.Am J Hematol.1997;54:102–107.
- .Atypical presentations of thrombotic thrombocytopenic purpura: a review.J Clin Apheresis.2009;24(1)47–52.
- , , , et al.Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura.N Engl J Med.1991;325:393–397.
- , , , , , .The incidence of thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS13deficiency.J Thromb Haemost.2005;3:1432–1436.
- .Thrombotic thrombocytopenic purpura.N Engl J Med.2006;354:1927–1935.
- , , , et al.ADAMTS13 activity in thrombotic thrombocytopenic purpura‐hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients.Blood.2003;102:60–68.
- , , .Thrombotic thrombocytopenic purpura.J Thromb Haemost.2005;3:1663–1675.
- , , , et al.Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials.Am J Kidney Dis.2009;53:259–272.
- , , , et al.:Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13‐deficient TTP: A study of 11 cases.Blood.2005;105:1932–1937.
Positional Atrial Flutter?
A 68‐year‐old man with a history of congestive heart failure and hypertension presented to the emergency department with fatigue and dyspnea of 3 weeks duration. Physical examination was consistent with heart failure. In addition, a right upper extremity resting tremor was noticed. An electrocardiogram (ECG) revealed an atrial flutter with a conduction ratio of 4:1 (Figure 1A). He denied palpitations or a previous history of atrial flutter/fibrillation. Unlike typical atrial flutter, these flutter like waves were distinctly absent in lead III, the only limb lead not connected to the right arm.

While holding the patient's right arm to control the tremor, a second ECG tracing was obtained. As expected the flutter like waves disappeared (Figure 1B). These ECG findings were attributed to the patient's tremor. A neurological consultation established a clinical diagnosis of Parkinson's disease. His congestive heart failure (CHF) was treated with increasing diuretics and appropriate treatment for Parkinson's disease was initiated.
A 68‐year‐old man with a history of congestive heart failure and hypertension presented to the emergency department with fatigue and dyspnea of 3 weeks duration. Physical examination was consistent with heart failure. In addition, a right upper extremity resting tremor was noticed. An electrocardiogram (ECG) revealed an atrial flutter with a conduction ratio of 4:1 (Figure 1A). He denied palpitations or a previous history of atrial flutter/fibrillation. Unlike typical atrial flutter, these flutter like waves were distinctly absent in lead III, the only limb lead not connected to the right arm.
While holding the patient's right arm to control the tremor, a second ECG tracing was obtained. As expected the flutter like waves disappeared (Figure 1B). These ECG findings were attributed to the patient's tremor. A neurological consultation established a clinical diagnosis of Parkinson's disease. His congestive heart failure (CHF) was treated with increasing diuretics and appropriate treatment for Parkinson's disease was initiated.
A 68‐year‐old man with a history of congestive heart failure and hypertension presented to the emergency department with fatigue and dyspnea of 3 weeks duration. Physical examination was consistent with heart failure. In addition, a right upper extremity resting tremor was noticed. An electrocardiogram (ECG) revealed an atrial flutter with a conduction ratio of 4:1 (Figure 1A). He denied palpitations or a previous history of atrial flutter/fibrillation. Unlike typical atrial flutter, these flutter like waves were distinctly absent in lead III, the only limb lead not connected to the right arm.
While holding the patient's right arm to control the tremor, a second ECG tracing was obtained. As expected the flutter like waves disappeared (Figure 1B). These ECG findings were attributed to the patient's tremor. A neurological consultation established a clinical diagnosis of Parkinson's disease. His congestive heart failure (CHF) was treated with increasing diuretics and appropriate treatment for Parkinson's disease was initiated.
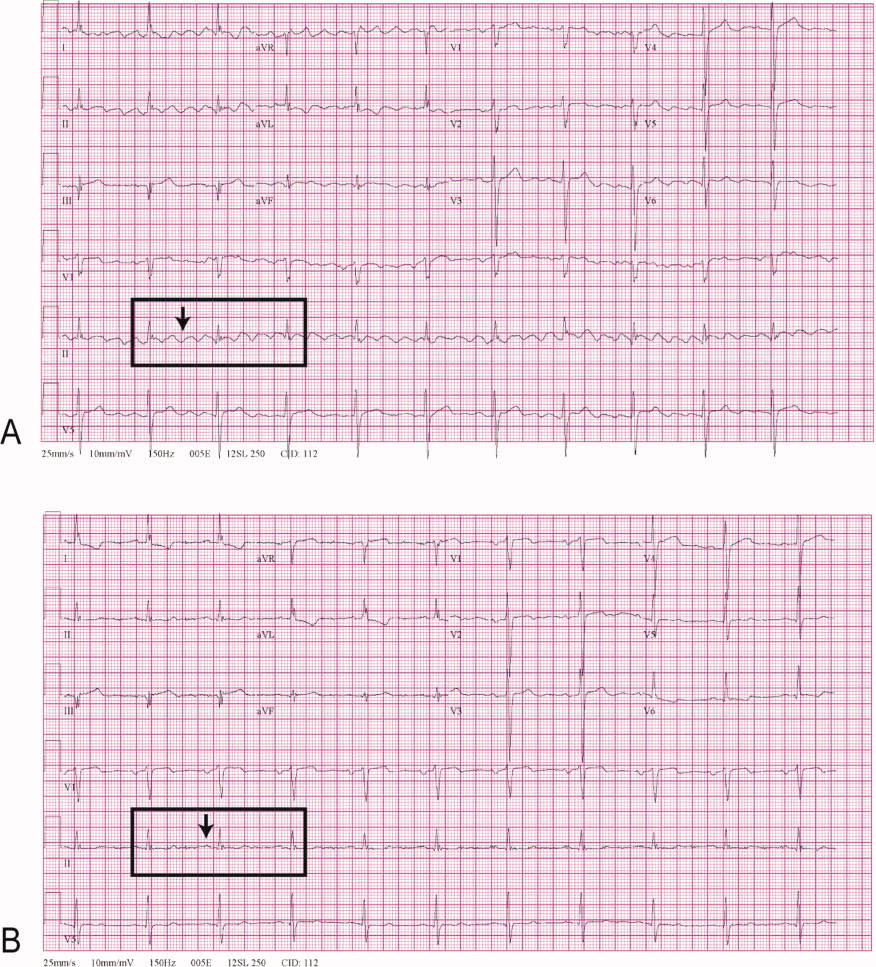
Postoperative myocardial infarction and in-hospital mortality predictors in patients undergoing elective noncardiac surgery
Incidence and predictors of postoperative heart failure in patients undergoing elective noncardiac surgery
Bilateral Adrenal Hemorrhage Complication
A 52‐year‐old man presented to the emergency department (ED) from a skilled nursing facility with a complaint of bilateral upper‐quadrant abdominal pain of 48 hours' duration. The pain was sharp, nonradiating, constant, and was associated with nausea, vomiting, and constipation. The patient denied any fever, back pain, dysuria, melena, or hematochezia. In the rehabilitation facility the patient had been initially evaluated for this pain. He was given laxatives and stool softeners for presumed constipation but these measures had not been effective. A computed tomography (CT) scan of the abdomen had only showed stool in the colon and he was sent to the ED for further evaluation.
Apart from severe degenerative joint disease in both his knees he was in good health. He was in the skilled nursing facility (SNF) for rehabilitation for bilateral knee replacement surgery done 9 days prior to this presentation. His postoperative course was unremarkable. He had been maintained on prophylaxis for venous thromboembolism with enoxaparin since postoperative day 1 at a daily dose of 40 mg subcutaneously, and was transferred to the SNF on postoperative day 6 on the same dose. His was receiving oxycodone and Tylenol for pain. He was on no other medications.
Vital signs on presentation revealed a temperature of 97.5F, a heart rate of 100 beats per minute, a respiratory rate of 16 breaths per minute, and a blood pressure of 136/69 mmHg. He was alert and oriented and in mild distress from the abdominal pain. Examination was normal except for tenderness in the upper quadrants of the abdomen though no rigidity or rebound tenderness were noted. Routine chemistries were normal except for sodium of 134 mg/dL. His white count, hemoglobin, hematocrit, and platelet levels were noted to be at 17.5K/L, 10 g/dL, 30%, and 345K/L, respectively, and were stable with regard to his discharge laboratory values. His serum eosinophil level was normal. A complete workup for hypercoagulable state and bleeding disorders including assays for antibodies associated with heparin‐induced thrombocytopenia were negative. He was admitted for further evaluation and treatment.
The patient had another CT scan of the abdomen (Figure 1), which when compared to the one done at the SNF 2 days prior showed markedly enlarged bilateral adrenal glands suggestive of bilateral acute adrenal hemorrhage. The enoxaparin was discontinued and empiric steroid replacement therapy was begun. A random cortisol level was normal but a cosyntropin stimulation test showed an absolute increase in cortisol level of only 0.8 g/dL at both 30 and 60 minutes after administration of 250 g of cosyntropin. An investigation was undertaken to determine if the patient had any prior risk factors for bleeding. There was no evidence of infection and a comprehensive evaluation for bleeding, and coagulation disorders was normal. The bilateral adrenal hemorrhage was attributed to the use of enoxaparin in the postoperative setting. Unfortunately, the patient subsequently developed a deep venous thrombosis in his lower extremity and an inferior vena cava (IVC) filter was placed before discharge. He was doing well 6 months later, and is still continued on glucocorticoid and mineralocorticoid replacement therapy and follows up with endocrinology as an outpatient.
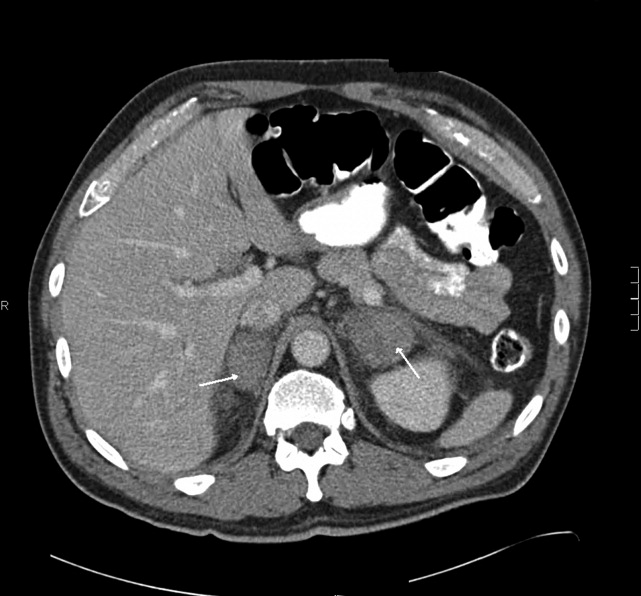
Discussion
Bilateral adrenal hemorrhage is usually associated with massive sepsis from Gram‐negative organisms such as Neisseria meningitides, Pseudomonas aeroginosa, Escherichia coli, and Bacteroides fragilis. Rupert Waterhouse, in 1911, was the first person to describe a patient with severe meningococcal sepsis resulting in acute adrenal hemorrhage and collapse. This was also later described independently by Carl Friderichsen in 1918, and is now referred to as the Waterhouse‐Friderichsen syndrome. Other causes include antiphospholipid antibody syndrome, heparin‐associated thrombocytopenia (HIT), and severe physical stress. Bilateral adrenal hemorrhage can also spontaneously occur in the postoperative period, especially after cardiothoracic or orthopedic surgery. This phenomenon may be related to the frequent use of prophylactic anticoagulants after these types of procedures.
The first case report of bilateral adrenal hemorrhage secondary to use of anticoagulants was described in 1947, and the first case report of successful resuscitation after corticosteroid administration in a patient with bilateral adrenal hemorrhage secondary to anticoagulant use was described by Thorn in 1956.1 A review of the literature demonstrates multiple case reports of adrenal hemorrhage reported in the postoperative period, particularly after joint arthroplasty, and especially after knee replacement surgeries. Most of the recent cases have been associated with use of prophylactic low‐dose heparin or low‐molecular‐weight heparin at the time of adrenal hemorrhage. In a study of 157 case reports of individuals with bilateral hemorrhage (including 22 autopsies), 48 cases were associated with administration of anticoagulants, although the dose and effect were not specified.2 Amador et al.1 showed that out of 4325 autopsies performed from 1949 to 1962 in their institution, 30 cases were found of bilateral hemorrhage, of which 10 were receiving heparin at presumably prophylactic doses; 5 of these patients were also receiving dicumarol.
Mayo Clinic investigators performed a retrospective review of all cases of adrenal hemorrhage over a period of 25 years at their hospital, and found 141 cases of adrenal hemorrhage, of which 78 were bilateral and 63 were unilateral,3 and in 67 patients the condition was diagnosed at autopsy. In this study 14 patients had adrenal hemorrhage in the postoperative period in the absence of lupus anticoagulant or HIT; there was no specific mention in this study of the use of postoperative anticoagulants. Finally, a multicenter case control study was undertaken by Kovacs et al.4 to assess putative risk factors for development of bilateral massive adrenal hemorrhage. In the multivariate analysis, thrombocytopenia, exposure to heparin, and sepsis were found to be strongly associated with risk of hemorrhage. Of 23 patients with bilateral, massive adrenal hemorrhage, 16 had been exposed to heparin, and at least 6 were on exclusively subcutaneous heparin. The authors concluded that heparin exposure was a much bigger risk factor than other coagulopathies, and those exposed to heparin of any route or type for 4 to 6 days and those exposed for more than 6 days were about 17 and 34 times, respectively, more likely to develop bilateral hemorrhage than those who had less than 4 days or no exposure.
The clinical presentation of adrenal insufficiency due to bilateral adrenal hemorrhage is often nonspecific. Symptoms may include abdominal pain, back pain, fever, nausea, vomiting, weakness, obtundation, confusion, and hypotensionall of which are also common postoperative symptoms and can be missed or ignored.5 Rao et al.6 profiled the clinical presentation of 64 cases of bilateral hemorrhage and found the following: abdominal, flank, back, or chest pain (86%); anorexia, nausea, or vomiting (47%); psychiatric symptoms (42%); fever (66%); hypotension recognized before shock episode (19%); and abdominal rigidity or rebound (22%). Adrenal insufficiency becomes clinically evident once 90% of the gland is destroyed. About 50% of patients do not manifest typical laboratory abnormalities, so a high degree of suspicion is necessary to diagnose the condition.3 Also, the laboratory diagnosis of adrenal insufficiency using random cortisol levels is unreliable, as reference ranges in patients experiencing stress (as in the postoperative period) have not been well studied or established. In patients with bilateral hemorrhage postoperatively on prophylactic anticoagulants, the coagulation profile is usually within normal limits and there is typically no evidence of spontaneous bleeding elsewhere. In later stages, the typical laboratory findings of abnormal adrenal function such as hypokalemia, hyponatremia, declining cortisol levels, and an inappropriate response to adrenocorticotropic hormone stimulation test may be seen. A significant drop in hemoglobin secondary to hemorrhage may also be encountered in some patients secondary to the bleed.
CT is the most reliable and extensively used imaging modality for making the diagnosis, although magnetic resonance imaging (MRI) or ultrasound may also be utilized. Early in the course of adrenal hemorrhage, CT findings may be negative, and repeated imaging is appropriate when clinical suspicion is high. The presence of bilateral adrenal enlargement with increased signal attenuation suggests bilateral adrenal hemorrhage. MRI can both characterize adrenal hematomas, and estimate their age.7, 8
Postoperative adrenal hemorrhage and insufficiency is easily treatable and has excellent outcomes; survivors will need lifelong corticosteroid replacement (and usually mineralocorticoid replacement as well). In the Mayo Clinic study, survival was 100% with treatment vs. 17% without treatment. In comparison, sepsis‐induced or stress‐induced adrenal insufficiency has poor outcomes despite adequate treatment (9% survival with treatment vs. 6% survival without treatment).3 Death can occur within hours to days of symptoms if untreated. Treatment includes timely initiation of adrenal hormone replacement and reversal of coagulopathies.
Postoperative venous thromboembolism (VTE) prophylaxis with anticoagulants is the appropriate care in many cases, but, along with the postoperative state itself, also appears to be a risk factor for this unusual condition. Postoperative bilateral adrenal hemorrhage is rare and potentially fatal. Early identification and prompt initiation of steroid replacement therapy and reversal of coagulopathies can prove to be lifesaving. Making this diagnosis can be very challenging, as the clinical presentation and laboratory findings of adrenal hemorrhage are vague and nonspecific and mimic many nonlife threatening postoperative complications. Radiological diagnosis by CT may initially be normal and thus further confound the diagnosis. Hence, providers should remain vigilant for associated complications even with low‐dose prophylactic heparin or low‐molecular‐weight heparin in postoperative patients, and prompt, presumptive treatment with corticosteroids should be started while awaiting confirmation by imaging and laboratory testing.
- .Adrenal hemorrhage during anticoagulant therapy. A clinical and pathological study of ten cases.Ann Intern Med.1965;63(4):559–571.
- ,,,,,.Adrenal hemorrhage in the adult.Medicine.1978;57(3):211–221.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76(2):161–168.
- ,,.Bilateral massive adrenal hemorrhage. Assessment of putative risk factors by the case‐control method.Medicine.2001;80(1):45–53.
- .Bilateral massive adrenal hemorrhage.Med Clin North Am.1995;79(1):107–129.
- ,,.Bilateral massive adrenal hemorrhage: early recognition and treatment.Ann Intern Med.1989;110(3):227–235.
- ,,, et al.Imaging of nontraumatic hemorrhage of the adrenal gland.Radiographics.1999;19(4):949–963.
- ,,,,.Spontaneous unilateral adrenal hemorrhage: computerized tomography and magnetic resonance imaging findings in 8 cases.J Urol.1995;154(5):1647–1651.
A 52‐year‐old man presented to the emergency department (ED) from a skilled nursing facility with a complaint of bilateral upper‐quadrant abdominal pain of 48 hours' duration. The pain was sharp, nonradiating, constant, and was associated with nausea, vomiting, and constipation. The patient denied any fever, back pain, dysuria, melena, or hematochezia. In the rehabilitation facility the patient had been initially evaluated for this pain. He was given laxatives and stool softeners for presumed constipation but these measures had not been effective. A computed tomography (CT) scan of the abdomen had only showed stool in the colon and he was sent to the ED for further evaluation.
Apart from severe degenerative joint disease in both his knees he was in good health. He was in the skilled nursing facility (SNF) for rehabilitation for bilateral knee replacement surgery done 9 days prior to this presentation. His postoperative course was unremarkable. He had been maintained on prophylaxis for venous thromboembolism with enoxaparin since postoperative day 1 at a daily dose of 40 mg subcutaneously, and was transferred to the SNF on postoperative day 6 on the same dose. His was receiving oxycodone and Tylenol for pain. He was on no other medications.
Vital signs on presentation revealed a temperature of 97.5F, a heart rate of 100 beats per minute, a respiratory rate of 16 breaths per minute, and a blood pressure of 136/69 mmHg. He was alert and oriented and in mild distress from the abdominal pain. Examination was normal except for tenderness in the upper quadrants of the abdomen though no rigidity or rebound tenderness were noted. Routine chemistries were normal except for sodium of 134 mg/dL. His white count, hemoglobin, hematocrit, and platelet levels were noted to be at 17.5K/L, 10 g/dL, 30%, and 345K/L, respectively, and were stable with regard to his discharge laboratory values. His serum eosinophil level was normal. A complete workup for hypercoagulable state and bleeding disorders including assays for antibodies associated with heparin‐induced thrombocytopenia were negative. He was admitted for further evaluation and treatment.
The patient had another CT scan of the abdomen (Figure 1), which when compared to the one done at the SNF 2 days prior showed markedly enlarged bilateral adrenal glands suggestive of bilateral acute adrenal hemorrhage. The enoxaparin was discontinued and empiric steroid replacement therapy was begun. A random cortisol level was normal but a cosyntropin stimulation test showed an absolute increase in cortisol level of only 0.8 g/dL at both 30 and 60 minutes after administration of 250 g of cosyntropin. An investigation was undertaken to determine if the patient had any prior risk factors for bleeding. There was no evidence of infection and a comprehensive evaluation for bleeding, and coagulation disorders was normal. The bilateral adrenal hemorrhage was attributed to the use of enoxaparin in the postoperative setting. Unfortunately, the patient subsequently developed a deep venous thrombosis in his lower extremity and an inferior vena cava (IVC) filter was placed before discharge. He was doing well 6 months later, and is still continued on glucocorticoid and mineralocorticoid replacement therapy and follows up with endocrinology as an outpatient.
Discussion
Bilateral adrenal hemorrhage is usually associated with massive sepsis from Gram‐negative organisms such as Neisseria meningitides, Pseudomonas aeroginosa, Escherichia coli, and Bacteroides fragilis. Rupert Waterhouse, in 1911, was the first person to describe a patient with severe meningococcal sepsis resulting in acute adrenal hemorrhage and collapse. This was also later described independently by Carl Friderichsen in 1918, and is now referred to as the Waterhouse‐Friderichsen syndrome. Other causes include antiphospholipid antibody syndrome, heparin‐associated thrombocytopenia (HIT), and severe physical stress. Bilateral adrenal hemorrhage can also spontaneously occur in the postoperative period, especially after cardiothoracic or orthopedic surgery. This phenomenon may be related to the frequent use of prophylactic anticoagulants after these types of procedures.
The first case report of bilateral adrenal hemorrhage secondary to use of anticoagulants was described in 1947, and the first case report of successful resuscitation after corticosteroid administration in a patient with bilateral adrenal hemorrhage secondary to anticoagulant use was described by Thorn in 1956.1 A review of the literature demonstrates multiple case reports of adrenal hemorrhage reported in the postoperative period, particularly after joint arthroplasty, and especially after knee replacement surgeries. Most of the recent cases have been associated with use of prophylactic low‐dose heparin or low‐molecular‐weight heparin at the time of adrenal hemorrhage. In a study of 157 case reports of individuals with bilateral hemorrhage (including 22 autopsies), 48 cases were associated with administration of anticoagulants, although the dose and effect were not specified.2 Amador et al.1 showed that out of 4325 autopsies performed from 1949 to 1962 in their institution, 30 cases were found of bilateral hemorrhage, of which 10 were receiving heparin at presumably prophylactic doses; 5 of these patients were also receiving dicumarol.
Mayo Clinic investigators performed a retrospective review of all cases of adrenal hemorrhage over a period of 25 years at their hospital, and found 141 cases of adrenal hemorrhage, of which 78 were bilateral and 63 were unilateral,3 and in 67 patients the condition was diagnosed at autopsy. In this study 14 patients had adrenal hemorrhage in the postoperative period in the absence of lupus anticoagulant or HIT; there was no specific mention in this study of the use of postoperative anticoagulants. Finally, a multicenter case control study was undertaken by Kovacs et al.4 to assess putative risk factors for development of bilateral massive adrenal hemorrhage. In the multivariate analysis, thrombocytopenia, exposure to heparin, and sepsis were found to be strongly associated with risk of hemorrhage. Of 23 patients with bilateral, massive adrenal hemorrhage, 16 had been exposed to heparin, and at least 6 were on exclusively subcutaneous heparin. The authors concluded that heparin exposure was a much bigger risk factor than other coagulopathies, and those exposed to heparin of any route or type for 4 to 6 days and those exposed for more than 6 days were about 17 and 34 times, respectively, more likely to develop bilateral hemorrhage than those who had less than 4 days or no exposure.
The clinical presentation of adrenal insufficiency due to bilateral adrenal hemorrhage is often nonspecific. Symptoms may include abdominal pain, back pain, fever, nausea, vomiting, weakness, obtundation, confusion, and hypotensionall of which are also common postoperative symptoms and can be missed or ignored.5 Rao et al.6 profiled the clinical presentation of 64 cases of bilateral hemorrhage and found the following: abdominal, flank, back, or chest pain (86%); anorexia, nausea, or vomiting (47%); psychiatric symptoms (42%); fever (66%); hypotension recognized before shock episode (19%); and abdominal rigidity or rebound (22%). Adrenal insufficiency becomes clinically evident once 90% of the gland is destroyed. About 50% of patients do not manifest typical laboratory abnormalities, so a high degree of suspicion is necessary to diagnose the condition.3 Also, the laboratory diagnosis of adrenal insufficiency using random cortisol levels is unreliable, as reference ranges in patients experiencing stress (as in the postoperative period) have not been well studied or established. In patients with bilateral hemorrhage postoperatively on prophylactic anticoagulants, the coagulation profile is usually within normal limits and there is typically no evidence of spontaneous bleeding elsewhere. In later stages, the typical laboratory findings of abnormal adrenal function such as hypokalemia, hyponatremia, declining cortisol levels, and an inappropriate response to adrenocorticotropic hormone stimulation test may be seen. A significant drop in hemoglobin secondary to hemorrhage may also be encountered in some patients secondary to the bleed.
CT is the most reliable and extensively used imaging modality for making the diagnosis, although magnetic resonance imaging (MRI) or ultrasound may also be utilized. Early in the course of adrenal hemorrhage, CT findings may be negative, and repeated imaging is appropriate when clinical suspicion is high. The presence of bilateral adrenal enlargement with increased signal attenuation suggests bilateral adrenal hemorrhage. MRI can both characterize adrenal hematomas, and estimate their age.7, 8
Postoperative adrenal hemorrhage and insufficiency is easily treatable and has excellent outcomes; survivors will need lifelong corticosteroid replacement (and usually mineralocorticoid replacement as well). In the Mayo Clinic study, survival was 100% with treatment vs. 17% without treatment. In comparison, sepsis‐induced or stress‐induced adrenal insufficiency has poor outcomes despite adequate treatment (9% survival with treatment vs. 6% survival without treatment).3 Death can occur within hours to days of symptoms if untreated. Treatment includes timely initiation of adrenal hormone replacement and reversal of coagulopathies.
Postoperative venous thromboembolism (VTE) prophylaxis with anticoagulants is the appropriate care in many cases, but, along with the postoperative state itself, also appears to be a risk factor for this unusual condition. Postoperative bilateral adrenal hemorrhage is rare and potentially fatal. Early identification and prompt initiation of steroid replacement therapy and reversal of coagulopathies can prove to be lifesaving. Making this diagnosis can be very challenging, as the clinical presentation and laboratory findings of adrenal hemorrhage are vague and nonspecific and mimic many nonlife threatening postoperative complications. Radiological diagnosis by CT may initially be normal and thus further confound the diagnosis. Hence, providers should remain vigilant for associated complications even with low‐dose prophylactic heparin or low‐molecular‐weight heparin in postoperative patients, and prompt, presumptive treatment with corticosteroids should be started while awaiting confirmation by imaging and laboratory testing.
A 52‐year‐old man presented to the emergency department (ED) from a skilled nursing facility with a complaint of bilateral upper‐quadrant abdominal pain of 48 hours' duration. The pain was sharp, nonradiating, constant, and was associated with nausea, vomiting, and constipation. The patient denied any fever, back pain, dysuria, melena, or hematochezia. In the rehabilitation facility the patient had been initially evaluated for this pain. He was given laxatives and stool softeners for presumed constipation but these measures had not been effective. A computed tomography (CT) scan of the abdomen had only showed stool in the colon and he was sent to the ED for further evaluation.
Apart from severe degenerative joint disease in both his knees he was in good health. He was in the skilled nursing facility (SNF) for rehabilitation for bilateral knee replacement surgery done 9 days prior to this presentation. His postoperative course was unremarkable. He had been maintained on prophylaxis for venous thromboembolism with enoxaparin since postoperative day 1 at a daily dose of 40 mg subcutaneously, and was transferred to the SNF on postoperative day 6 on the same dose. His was receiving oxycodone and Tylenol for pain. He was on no other medications.
Vital signs on presentation revealed a temperature of 97.5F, a heart rate of 100 beats per minute, a respiratory rate of 16 breaths per minute, and a blood pressure of 136/69 mmHg. He was alert and oriented and in mild distress from the abdominal pain. Examination was normal except for tenderness in the upper quadrants of the abdomen though no rigidity or rebound tenderness were noted. Routine chemistries were normal except for sodium of 134 mg/dL. His white count, hemoglobin, hematocrit, and platelet levels were noted to be at 17.5K/L, 10 g/dL, 30%, and 345K/L, respectively, and were stable with regard to his discharge laboratory values. His serum eosinophil level was normal. A complete workup for hypercoagulable state and bleeding disorders including assays for antibodies associated with heparin‐induced thrombocytopenia were negative. He was admitted for further evaluation and treatment.
The patient had another CT scan of the abdomen (Figure 1), which when compared to the one done at the SNF 2 days prior showed markedly enlarged bilateral adrenal glands suggestive of bilateral acute adrenal hemorrhage. The enoxaparin was discontinued and empiric steroid replacement therapy was begun. A random cortisol level was normal but a cosyntropin stimulation test showed an absolute increase in cortisol level of only 0.8 g/dL at both 30 and 60 minutes after administration of 250 g of cosyntropin. An investigation was undertaken to determine if the patient had any prior risk factors for bleeding. There was no evidence of infection and a comprehensive evaluation for bleeding, and coagulation disorders was normal. The bilateral adrenal hemorrhage was attributed to the use of enoxaparin in the postoperative setting. Unfortunately, the patient subsequently developed a deep venous thrombosis in his lower extremity and an inferior vena cava (IVC) filter was placed before discharge. He was doing well 6 months later, and is still continued on glucocorticoid and mineralocorticoid replacement therapy and follows up with endocrinology as an outpatient.
Discussion
Bilateral adrenal hemorrhage is usually associated with massive sepsis from Gram‐negative organisms such as Neisseria meningitides, Pseudomonas aeroginosa, Escherichia coli, and Bacteroides fragilis. Rupert Waterhouse, in 1911, was the first person to describe a patient with severe meningococcal sepsis resulting in acute adrenal hemorrhage and collapse. This was also later described independently by Carl Friderichsen in 1918, and is now referred to as the Waterhouse‐Friderichsen syndrome. Other causes include antiphospholipid antibody syndrome, heparin‐associated thrombocytopenia (HIT), and severe physical stress. Bilateral adrenal hemorrhage can also spontaneously occur in the postoperative period, especially after cardiothoracic or orthopedic surgery. This phenomenon may be related to the frequent use of prophylactic anticoagulants after these types of procedures.
The first case report of bilateral adrenal hemorrhage secondary to use of anticoagulants was described in 1947, and the first case report of successful resuscitation after corticosteroid administration in a patient with bilateral adrenal hemorrhage secondary to anticoagulant use was described by Thorn in 1956.1 A review of the literature demonstrates multiple case reports of adrenal hemorrhage reported in the postoperative period, particularly after joint arthroplasty, and especially after knee replacement surgeries. Most of the recent cases have been associated with use of prophylactic low‐dose heparin or low‐molecular‐weight heparin at the time of adrenal hemorrhage. In a study of 157 case reports of individuals with bilateral hemorrhage (including 22 autopsies), 48 cases were associated with administration of anticoagulants, although the dose and effect were not specified.2 Amador et al.1 showed that out of 4325 autopsies performed from 1949 to 1962 in their institution, 30 cases were found of bilateral hemorrhage, of which 10 were receiving heparin at presumably prophylactic doses; 5 of these patients were also receiving dicumarol.
Mayo Clinic investigators performed a retrospective review of all cases of adrenal hemorrhage over a period of 25 years at their hospital, and found 141 cases of adrenal hemorrhage, of which 78 were bilateral and 63 were unilateral,3 and in 67 patients the condition was diagnosed at autopsy. In this study 14 patients had adrenal hemorrhage in the postoperative period in the absence of lupus anticoagulant or HIT; there was no specific mention in this study of the use of postoperative anticoagulants. Finally, a multicenter case control study was undertaken by Kovacs et al.4 to assess putative risk factors for development of bilateral massive adrenal hemorrhage. In the multivariate analysis, thrombocytopenia, exposure to heparin, and sepsis were found to be strongly associated with risk of hemorrhage. Of 23 patients with bilateral, massive adrenal hemorrhage, 16 had been exposed to heparin, and at least 6 were on exclusively subcutaneous heparin. The authors concluded that heparin exposure was a much bigger risk factor than other coagulopathies, and those exposed to heparin of any route or type for 4 to 6 days and those exposed for more than 6 days were about 17 and 34 times, respectively, more likely to develop bilateral hemorrhage than those who had less than 4 days or no exposure.
The clinical presentation of adrenal insufficiency due to bilateral adrenal hemorrhage is often nonspecific. Symptoms may include abdominal pain, back pain, fever, nausea, vomiting, weakness, obtundation, confusion, and hypotensionall of which are also common postoperative symptoms and can be missed or ignored.5 Rao et al.6 profiled the clinical presentation of 64 cases of bilateral hemorrhage and found the following: abdominal, flank, back, or chest pain (86%); anorexia, nausea, or vomiting (47%); psychiatric symptoms (42%); fever (66%); hypotension recognized before shock episode (19%); and abdominal rigidity or rebound (22%). Adrenal insufficiency becomes clinically evident once 90% of the gland is destroyed. About 50% of patients do not manifest typical laboratory abnormalities, so a high degree of suspicion is necessary to diagnose the condition.3 Also, the laboratory diagnosis of adrenal insufficiency using random cortisol levels is unreliable, as reference ranges in patients experiencing stress (as in the postoperative period) have not been well studied or established. In patients with bilateral hemorrhage postoperatively on prophylactic anticoagulants, the coagulation profile is usually within normal limits and there is typically no evidence of spontaneous bleeding elsewhere. In later stages, the typical laboratory findings of abnormal adrenal function such as hypokalemia, hyponatremia, declining cortisol levels, and an inappropriate response to adrenocorticotropic hormone stimulation test may be seen. A significant drop in hemoglobin secondary to hemorrhage may also be encountered in some patients secondary to the bleed.
CT is the most reliable and extensively used imaging modality for making the diagnosis, although magnetic resonance imaging (MRI) or ultrasound may also be utilized. Early in the course of adrenal hemorrhage, CT findings may be negative, and repeated imaging is appropriate when clinical suspicion is high. The presence of bilateral adrenal enlargement with increased signal attenuation suggests bilateral adrenal hemorrhage. MRI can both characterize adrenal hematomas, and estimate their age.7, 8
Postoperative adrenal hemorrhage and insufficiency is easily treatable and has excellent outcomes; survivors will need lifelong corticosteroid replacement (and usually mineralocorticoid replacement as well). In the Mayo Clinic study, survival was 100% with treatment vs. 17% without treatment. In comparison, sepsis‐induced or stress‐induced adrenal insufficiency has poor outcomes despite adequate treatment (9% survival with treatment vs. 6% survival without treatment).3 Death can occur within hours to days of symptoms if untreated. Treatment includes timely initiation of adrenal hormone replacement and reversal of coagulopathies.
Postoperative venous thromboembolism (VTE) prophylaxis with anticoagulants is the appropriate care in many cases, but, along with the postoperative state itself, also appears to be a risk factor for this unusual condition. Postoperative bilateral adrenal hemorrhage is rare and potentially fatal. Early identification and prompt initiation of steroid replacement therapy and reversal of coagulopathies can prove to be lifesaving. Making this diagnosis can be very challenging, as the clinical presentation and laboratory findings of adrenal hemorrhage are vague and nonspecific and mimic many nonlife threatening postoperative complications. Radiological diagnosis by CT may initially be normal and thus further confound the diagnosis. Hence, providers should remain vigilant for associated complications even with low‐dose prophylactic heparin or low‐molecular‐weight heparin in postoperative patients, and prompt, presumptive treatment with corticosteroids should be started while awaiting confirmation by imaging and laboratory testing.
- .Adrenal hemorrhage during anticoagulant therapy. A clinical and pathological study of ten cases.Ann Intern Med.1965;63(4):559–571.
- ,,,,,.Adrenal hemorrhage in the adult.Medicine.1978;57(3):211–221.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76(2):161–168.
- ,,.Bilateral massive adrenal hemorrhage. Assessment of putative risk factors by the case‐control method.Medicine.2001;80(1):45–53.
- .Bilateral massive adrenal hemorrhage.Med Clin North Am.1995;79(1):107–129.
- ,,.Bilateral massive adrenal hemorrhage: early recognition and treatment.Ann Intern Med.1989;110(3):227–235.
- ,,, et al.Imaging of nontraumatic hemorrhage of the adrenal gland.Radiographics.1999;19(4):949–963.
- ,,,,.Spontaneous unilateral adrenal hemorrhage: computerized tomography and magnetic resonance imaging findings in 8 cases.J Urol.1995;154(5):1647–1651.
- .Adrenal hemorrhage during anticoagulant therapy. A clinical and pathological study of ten cases.Ann Intern Med.1965;63(4):559–571.
- ,,,,,.Adrenal hemorrhage in the adult.Medicine.1978;57(3):211–221.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76(2):161–168.
- ,,.Bilateral massive adrenal hemorrhage. Assessment of putative risk factors by the case‐control method.Medicine.2001;80(1):45–53.
- .Bilateral massive adrenal hemorrhage.Med Clin North Am.1995;79(1):107–129.
- ,,.Bilateral massive adrenal hemorrhage: early recognition and treatment.Ann Intern Med.1989;110(3):227–235.
- ,,, et al.Imaging of nontraumatic hemorrhage of the adrenal gland.Radiographics.1999;19(4):949–963.
- ,,,,.Spontaneous unilateral adrenal hemorrhage: computerized tomography and magnetic resonance imaging findings in 8 cases.J Urol.1995;154(5):1647–1651.
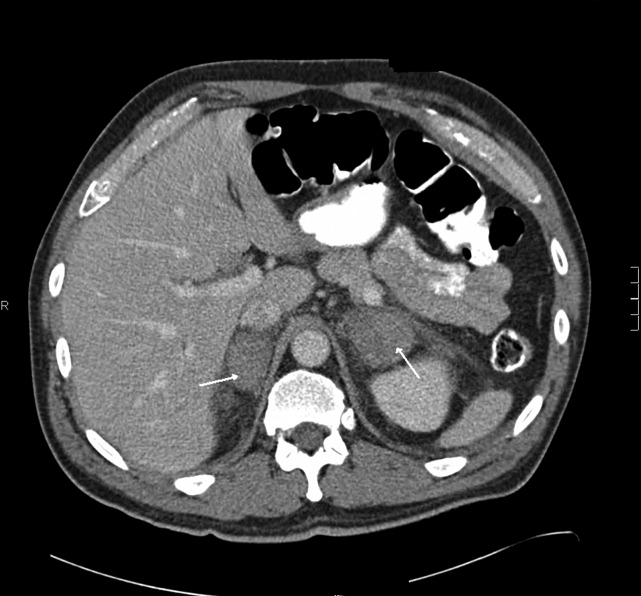
Pin the Pinworm
An 84‐year‐old female patient with hypertension, osteoarthritis, hypothyroidism, and remote breast cancer was admitted with complaints of generalized abdominal pain of 2 months' duration. Pain was described as noncolicky in nature and was associated with diarrhea. She reported 78 daily episodes of watery, non‐foul‐smelling diarrhea. She denied any nausea, vomiting, fever, joint pains, oral ulcers, eye redness, stool incontinence, melena, hematochezia, or weight loss. There was no history of recent travel, antibiotic use, or exposure to sick contacts. She had no risk factors for HIV infection or other sexually transmitted infections. Her social history was significant for dining out on a regular basis and living in an assisted living facility. However, she denied any relationship between her abdominal symptoms and any particular food intake or with bowel movements. She denied any anal pruritis but reported seeing white squiggly things on tissue paper after bowel movements. She denied use of over‐the‐counter laxatives or herbal supplements. None of her prescription medications had diarrhea as a major side effect. Her social history was unremarkable for smoking, alcohol use, or illicit drug use. There were no prior abdominal surgeries. The patient's physical exam showed normal vitals on presentation and was unremarkable except for vague, generalized abdominal tenderness with no involuntary guarding or rebound pain. Her initial laboratory evaluation showed normal complete blood counts with no eosinophilia and normal serum electrolytes and liver and thyroid panel. Acute‐phase reactants, erythrocyte sedimentation rate, and C‐reactive protein were not elevated. Stool evaluation was unremarkable for Clostridium difficile toxin, fat droplets, leukocytes, erythrocytes, ova, parasites, or any bacterial growth on cultures. Computed tomography scans of the abdomen and pelvis were nonrevealing. Her colonoscopic examination 1 year prior was significant only for diverticulosis.1, 2
The patient was treated with loperamide as an outpatient with no relief. She was then admitted to the hospital for further diagnostic workup. Hospital workup included a Scotch tape test, which showed adult pinworms. She was treated with a single dose of 400 mg of albendazole with complete resolution of her symptoms within 2 days. No further workup was done. Patient was discharged with advice to contact her primary care doctor for reevaluation if symptoms recurred. However, the patient remained symptom free 1 year after discharge.
DISCUSSION
Enterobius vermicularis is a parasite that infects 2040 million people annually in the United States and about 200 million people worldwide. Equal infection rates are seen in all races, socioeconomic classes, and cultures.1 It is more prevalent among those in crowded living conditions. Humans are the primary natural host for the parasite, although it has been documented in cockroaches and primates. Transmission occurs via the feco‐oral route or via airborne eggs that are dislodged from contaminated clothing or bed linen. Its life cycle begins with parasite eggs hatching in the duodenum, usually within 6 hours of ingestion. They mature into adults in as little as 2 weeks and have a life span of approximately 2 months. Enterobius vermicularis normally inhabits distal small bowel including the terminal ileum, cecum, and vermiform appendix, as well as the proximal ascending colon. After copulation, an adult female will migrate to the perineum, often at night, and lay an average of 10,00015,000 eggs. These eggs mature in about 6 hours and are then transmitted to a new host by the feco‐oral route. The worms live mainly in the intestinal lumen and do not invade tissue. Hence, pinworm infections, unlike many other parasitic infections, are rarely associated with serum eosinophilia or elevated serum IgE levels.
E. vermicularis is generally considered to be an innocuous parasite. Perianal pruritis, especially during the nighttime, is the most common symptom. Patients may develop secondary bacterial infection of the irritated anal skin. Rarely, E. vermicularis infection may result in a life‐threatening illness. A literature review showed pinworm infection to be an infrequent cause of eosinophilic enterocolitis, appendicitis, intestinal obstruction, intestinal perforation, hepatic infection, urinary tract infection, sialoadenitis, salpingitis, enterocolitis, eosinophilic ileocolitis, vulvovaginitis, pelvic inflammatory disease, conditions mimicking inflammatory bowel diseases, perianal abscesses, and perianal granulomas. In a retrospective review of 180 colonoscopies done on patients with rectal bleeding or suspected inflammatory bowel disease, E. vermicularis was identified macroscopically in 31 cases (17.2%). Data collected on 23 of these cases showed that symptoms were present for a mean of 17 months; the symptoms with the highest frequency were abdominal pain (73%), rectal bleeding (62%), chronic diarrhea (50%), and weight loss (42%). None of these patients experienced perianal pruritis or developed inflammatory bowel disease during the follow‐up period of up to 5 years, although 21 patients demonstrated histopathological evidence of nonspecific colitis.6
The gold standard for diagnosing E. vermicularis infection is by visualizing the worms directly or by examination of the parasitic eggs under a microscope. The Scotch tape test is a simple, inexpensive, and quick way for confirming the infection. It is performed by doubling clear cellophane Scotch tape onto a wooden stick so that the sticky side points outward and pressing it against the perianal skin. The kidney‐bean‐shaped eggs (50 25 m) will stick to the tape and can then be directly visualized under a microscope. Pinworms are most active during the night, and eggs are deposited around the perianal region and are best recovered before defecation, early in the morning. The sensitivity of this test is 90% if done on 3 consecutive mornings and goes up to 99% when performed on 5 consecutive mornings.2, 3 Female adult worms are pin‐shaped, about 813 mm long, and white in color. They may be seen by direct visualization in the perianal region or more invasively by an anoscopic or colonoscopic examination. However, endoscopic examination may sometimes give false‐negative results as the worms are small, (ie, only a few millimeters in length) and may be missed if the endoscopist is not actively looking for them.
A single oral dose of benzimidazoles (100 mg of mebendazole or 400 mg of albendazole) results in a cure of rate of 95% and 100%, respectively. Despite the high initial cure rates, reinfection remains common; hence, a second dose 12 weeks after the initial treatment is often given to prevent it.4, 5 Pyrantel pamoate and piperazine are alternate treatments. However, they have lower efficacy and are more toxic than benzimidazoles.
Close contacts such as household members are often concurrently infected, and treatment of the remaining household members or of the group institution is also indicated. All bedding and clothes should be laundered. Personal hygiene such as fingernail clipping, frequent hand washing, and bathing should also be encouraged.
Although the pinworm's entire life cycle is in the human intestinal tract, gastrointestinal symptoms have seldom been reported. However, this may be because of underreporting. Given the increasing number of patients living in institutionalized environments such as nursing homes and assisted living, it is important to consider the possibility of E. vermicularis infection early on in a diagnostic workup of patients presenting with symptoms of colitis, even when not accompanied by anal pruritis. In a patient presenting with symptoms of inflammatory bowel disease with histopathological evaluation of nonspecific colitis should prompt clinicians to consider E. vermicularis infection.6 On the other hand, in patients who fail to respond to antiparasitic therapy or those who present with weight loss, change in bowel habits, or melena, colonscopic examination is warranted. Considering pinworm infection early during evaluation of nonspecific abdominal complaints may avoid an unnecessary and expensive diagnostic workup.
KEY POINTS
-
Recognize early on that Enterobius vermicularis infection is an important differential diagnosis for patients presenting with symptoms of colitis, thus avoiding unnecessary, expensive, and potentially harmful invasive testing.
-
Recognize that a simple and inexpensive Scotch tape test and/or direct visualization is an easy and effective way of confirming diagnosis and that stool examination may be unhelpful.
-
Recognize that reinfection may be prevented using a second dose of the antiparasitic drug.
- .The pinworm, Enterobius vermicularis.Prim Care.1991;18:13–24.
- ,,,,.Prevalence of intestinal parasites in three socioeconomically‐different regions of Sivas, Turkey.J Health Popul Nutr.2005;23:184–191.
- ,,,.Pinworm infection.Gastrointest Endosc.2001;53:210.
- ,,.Mebendazole (R 17635) in enterobiasis. A clinical trial in mental retardates.Chemotherapy.1975;21:255–260.
- ,,, et al.Field trials on the efficacy of albendazole composite against intestinal nematodiasis.Chung Kuo Chi Sheng Chung Hsueh Yu Chi Sheng Chung Ping Tsa Chih.1998;16:1–5.
- ,,.Enterobius vermicularis and colitis in children.J Pediatr Gastroenterol Nutr.2006;43:610–612.
An 84‐year‐old female patient with hypertension, osteoarthritis, hypothyroidism, and remote breast cancer was admitted with complaints of generalized abdominal pain of 2 months' duration. Pain was described as noncolicky in nature and was associated with diarrhea. She reported 78 daily episodes of watery, non‐foul‐smelling diarrhea. She denied any nausea, vomiting, fever, joint pains, oral ulcers, eye redness, stool incontinence, melena, hematochezia, or weight loss. There was no history of recent travel, antibiotic use, or exposure to sick contacts. She had no risk factors for HIV infection or other sexually transmitted infections. Her social history was significant for dining out on a regular basis and living in an assisted living facility. However, she denied any relationship between her abdominal symptoms and any particular food intake or with bowel movements. She denied any anal pruritis but reported seeing white squiggly things on tissue paper after bowel movements. She denied use of over‐the‐counter laxatives or herbal supplements. None of her prescription medications had diarrhea as a major side effect. Her social history was unremarkable for smoking, alcohol use, or illicit drug use. There were no prior abdominal surgeries. The patient's physical exam showed normal vitals on presentation and was unremarkable except for vague, generalized abdominal tenderness with no involuntary guarding or rebound pain. Her initial laboratory evaluation showed normal complete blood counts with no eosinophilia and normal serum electrolytes and liver and thyroid panel. Acute‐phase reactants, erythrocyte sedimentation rate, and C‐reactive protein were not elevated. Stool evaluation was unremarkable for Clostridium difficile toxin, fat droplets, leukocytes, erythrocytes, ova, parasites, or any bacterial growth on cultures. Computed tomography scans of the abdomen and pelvis were nonrevealing. Her colonoscopic examination 1 year prior was significant only for diverticulosis.1, 2
The patient was treated with loperamide as an outpatient with no relief. She was then admitted to the hospital for further diagnostic workup. Hospital workup included a Scotch tape test, which showed adult pinworms. She was treated with a single dose of 400 mg of albendazole with complete resolution of her symptoms within 2 days. No further workup was done. Patient was discharged with advice to contact her primary care doctor for reevaluation if symptoms recurred. However, the patient remained symptom free 1 year after discharge.
DISCUSSION
Enterobius vermicularis is a parasite that infects 2040 million people annually in the United States and about 200 million people worldwide. Equal infection rates are seen in all races, socioeconomic classes, and cultures.1 It is more prevalent among those in crowded living conditions. Humans are the primary natural host for the parasite, although it has been documented in cockroaches and primates. Transmission occurs via the feco‐oral route or via airborne eggs that are dislodged from contaminated clothing or bed linen. Its life cycle begins with parasite eggs hatching in the duodenum, usually within 6 hours of ingestion. They mature into adults in as little as 2 weeks and have a life span of approximately 2 months. Enterobius vermicularis normally inhabits distal small bowel including the terminal ileum, cecum, and vermiform appendix, as well as the proximal ascending colon. After copulation, an adult female will migrate to the perineum, often at night, and lay an average of 10,00015,000 eggs. These eggs mature in about 6 hours and are then transmitted to a new host by the feco‐oral route. The worms live mainly in the intestinal lumen and do not invade tissue. Hence, pinworm infections, unlike many other parasitic infections, are rarely associated with serum eosinophilia or elevated serum IgE levels.
E. vermicularis is generally considered to be an innocuous parasite. Perianal pruritis, especially during the nighttime, is the most common symptom. Patients may develop secondary bacterial infection of the irritated anal skin. Rarely, E. vermicularis infection may result in a life‐threatening illness. A literature review showed pinworm infection to be an infrequent cause of eosinophilic enterocolitis, appendicitis, intestinal obstruction, intestinal perforation, hepatic infection, urinary tract infection, sialoadenitis, salpingitis, enterocolitis, eosinophilic ileocolitis, vulvovaginitis, pelvic inflammatory disease, conditions mimicking inflammatory bowel diseases, perianal abscesses, and perianal granulomas. In a retrospective review of 180 colonoscopies done on patients with rectal bleeding or suspected inflammatory bowel disease, E. vermicularis was identified macroscopically in 31 cases (17.2%). Data collected on 23 of these cases showed that symptoms were present for a mean of 17 months; the symptoms with the highest frequency were abdominal pain (73%), rectal bleeding (62%), chronic diarrhea (50%), and weight loss (42%). None of these patients experienced perianal pruritis or developed inflammatory bowel disease during the follow‐up period of up to 5 years, although 21 patients demonstrated histopathological evidence of nonspecific colitis.6
The gold standard for diagnosing E. vermicularis infection is by visualizing the worms directly or by examination of the parasitic eggs under a microscope. The Scotch tape test is a simple, inexpensive, and quick way for confirming the infection. It is performed by doubling clear cellophane Scotch tape onto a wooden stick so that the sticky side points outward and pressing it against the perianal skin. The kidney‐bean‐shaped eggs (50 25 m) will stick to the tape and can then be directly visualized under a microscope. Pinworms are most active during the night, and eggs are deposited around the perianal region and are best recovered before defecation, early in the morning. The sensitivity of this test is 90% if done on 3 consecutive mornings and goes up to 99% when performed on 5 consecutive mornings.2, 3 Female adult worms are pin‐shaped, about 813 mm long, and white in color. They may be seen by direct visualization in the perianal region or more invasively by an anoscopic or colonoscopic examination. However, endoscopic examination may sometimes give false‐negative results as the worms are small, (ie, only a few millimeters in length) and may be missed if the endoscopist is not actively looking for them.
A single oral dose of benzimidazoles (100 mg of mebendazole or 400 mg of albendazole) results in a cure of rate of 95% and 100%, respectively. Despite the high initial cure rates, reinfection remains common; hence, a second dose 12 weeks after the initial treatment is often given to prevent it.4, 5 Pyrantel pamoate and piperazine are alternate treatments. However, they have lower efficacy and are more toxic than benzimidazoles.
Close contacts such as household members are often concurrently infected, and treatment of the remaining household members or of the group institution is also indicated. All bedding and clothes should be laundered. Personal hygiene such as fingernail clipping, frequent hand washing, and bathing should also be encouraged.
Although the pinworm's entire life cycle is in the human intestinal tract, gastrointestinal symptoms have seldom been reported. However, this may be because of underreporting. Given the increasing number of patients living in institutionalized environments such as nursing homes and assisted living, it is important to consider the possibility of E. vermicularis infection early on in a diagnostic workup of patients presenting with symptoms of colitis, even when not accompanied by anal pruritis. In a patient presenting with symptoms of inflammatory bowel disease with histopathological evaluation of nonspecific colitis should prompt clinicians to consider E. vermicularis infection.6 On the other hand, in patients who fail to respond to antiparasitic therapy or those who present with weight loss, change in bowel habits, or melena, colonscopic examination is warranted. Considering pinworm infection early during evaluation of nonspecific abdominal complaints may avoid an unnecessary and expensive diagnostic workup.
KEY POINTS
-
Recognize early on that Enterobius vermicularis infection is an important differential diagnosis for patients presenting with symptoms of colitis, thus avoiding unnecessary, expensive, and potentially harmful invasive testing.
-
Recognize that a simple and inexpensive Scotch tape test and/or direct visualization is an easy and effective way of confirming diagnosis and that stool examination may be unhelpful.
-
Recognize that reinfection may be prevented using a second dose of the antiparasitic drug.
An 84‐year‐old female patient with hypertension, osteoarthritis, hypothyroidism, and remote breast cancer was admitted with complaints of generalized abdominal pain of 2 months' duration. Pain was described as noncolicky in nature and was associated with diarrhea. She reported 78 daily episodes of watery, non‐foul‐smelling diarrhea. She denied any nausea, vomiting, fever, joint pains, oral ulcers, eye redness, stool incontinence, melena, hematochezia, or weight loss. There was no history of recent travel, antibiotic use, or exposure to sick contacts. She had no risk factors for HIV infection or other sexually transmitted infections. Her social history was significant for dining out on a regular basis and living in an assisted living facility. However, she denied any relationship between her abdominal symptoms and any particular food intake or with bowel movements. She denied any anal pruritis but reported seeing white squiggly things on tissue paper after bowel movements. She denied use of over‐the‐counter laxatives or herbal supplements. None of her prescription medications had diarrhea as a major side effect. Her social history was unremarkable for smoking, alcohol use, or illicit drug use. There were no prior abdominal surgeries. The patient's physical exam showed normal vitals on presentation and was unremarkable except for vague, generalized abdominal tenderness with no involuntary guarding or rebound pain. Her initial laboratory evaluation showed normal complete blood counts with no eosinophilia and normal serum electrolytes and liver and thyroid panel. Acute‐phase reactants, erythrocyte sedimentation rate, and C‐reactive protein were not elevated. Stool evaluation was unremarkable for Clostridium difficile toxin, fat droplets, leukocytes, erythrocytes, ova, parasites, or any bacterial growth on cultures. Computed tomography scans of the abdomen and pelvis were nonrevealing. Her colonoscopic examination 1 year prior was significant only for diverticulosis.1, 2
The patient was treated with loperamide as an outpatient with no relief. She was then admitted to the hospital for further diagnostic workup. Hospital workup included a Scotch tape test, which showed adult pinworms. She was treated with a single dose of 400 mg of albendazole with complete resolution of her symptoms within 2 days. No further workup was done. Patient was discharged with advice to contact her primary care doctor for reevaluation if symptoms recurred. However, the patient remained symptom free 1 year after discharge.
DISCUSSION
Enterobius vermicularis is a parasite that infects 2040 million people annually in the United States and about 200 million people worldwide. Equal infection rates are seen in all races, socioeconomic classes, and cultures.1 It is more prevalent among those in crowded living conditions. Humans are the primary natural host for the parasite, although it has been documented in cockroaches and primates. Transmission occurs via the feco‐oral route or via airborne eggs that are dislodged from contaminated clothing or bed linen. Its life cycle begins with parasite eggs hatching in the duodenum, usually within 6 hours of ingestion. They mature into adults in as little as 2 weeks and have a life span of approximately 2 months. Enterobius vermicularis normally inhabits distal small bowel including the terminal ileum, cecum, and vermiform appendix, as well as the proximal ascending colon. After copulation, an adult female will migrate to the perineum, often at night, and lay an average of 10,00015,000 eggs. These eggs mature in about 6 hours and are then transmitted to a new host by the feco‐oral route. The worms live mainly in the intestinal lumen and do not invade tissue. Hence, pinworm infections, unlike many other parasitic infections, are rarely associated with serum eosinophilia or elevated serum IgE levels.
E. vermicularis is generally considered to be an innocuous parasite. Perianal pruritis, especially during the nighttime, is the most common symptom. Patients may develop secondary bacterial infection of the irritated anal skin. Rarely, E. vermicularis infection may result in a life‐threatening illness. A literature review showed pinworm infection to be an infrequent cause of eosinophilic enterocolitis, appendicitis, intestinal obstruction, intestinal perforation, hepatic infection, urinary tract infection, sialoadenitis, salpingitis, enterocolitis, eosinophilic ileocolitis, vulvovaginitis, pelvic inflammatory disease, conditions mimicking inflammatory bowel diseases, perianal abscesses, and perianal granulomas. In a retrospective review of 180 colonoscopies done on patients with rectal bleeding or suspected inflammatory bowel disease, E. vermicularis was identified macroscopically in 31 cases (17.2%). Data collected on 23 of these cases showed that symptoms were present for a mean of 17 months; the symptoms with the highest frequency were abdominal pain (73%), rectal bleeding (62%), chronic diarrhea (50%), and weight loss (42%). None of these patients experienced perianal pruritis or developed inflammatory bowel disease during the follow‐up period of up to 5 years, although 21 patients demonstrated histopathological evidence of nonspecific colitis.6
The gold standard for diagnosing E. vermicularis infection is by visualizing the worms directly or by examination of the parasitic eggs under a microscope. The Scotch tape test is a simple, inexpensive, and quick way for confirming the infection. It is performed by doubling clear cellophane Scotch tape onto a wooden stick so that the sticky side points outward and pressing it against the perianal skin. The kidney‐bean‐shaped eggs (50 25 m) will stick to the tape and can then be directly visualized under a microscope. Pinworms are most active during the night, and eggs are deposited around the perianal region and are best recovered before defecation, early in the morning. The sensitivity of this test is 90% if done on 3 consecutive mornings and goes up to 99% when performed on 5 consecutive mornings.2, 3 Female adult worms are pin‐shaped, about 813 mm long, and white in color. They may be seen by direct visualization in the perianal region or more invasively by an anoscopic or colonoscopic examination. However, endoscopic examination may sometimes give false‐negative results as the worms are small, (ie, only a few millimeters in length) and may be missed if the endoscopist is not actively looking for them.
A single oral dose of benzimidazoles (100 mg of mebendazole or 400 mg of albendazole) results in a cure of rate of 95% and 100%, respectively. Despite the high initial cure rates, reinfection remains common; hence, a second dose 12 weeks after the initial treatment is often given to prevent it.4, 5 Pyrantel pamoate and piperazine are alternate treatments. However, they have lower efficacy and are more toxic than benzimidazoles.
Close contacts such as household members are often concurrently infected, and treatment of the remaining household members or of the group institution is also indicated. All bedding and clothes should be laundered. Personal hygiene such as fingernail clipping, frequent hand washing, and bathing should also be encouraged.
Although the pinworm's entire life cycle is in the human intestinal tract, gastrointestinal symptoms have seldom been reported. However, this may be because of underreporting. Given the increasing number of patients living in institutionalized environments such as nursing homes and assisted living, it is important to consider the possibility of E. vermicularis infection early on in a diagnostic workup of patients presenting with symptoms of colitis, even when not accompanied by anal pruritis. In a patient presenting with symptoms of inflammatory bowel disease with histopathological evaluation of nonspecific colitis should prompt clinicians to consider E. vermicularis infection.6 On the other hand, in patients who fail to respond to antiparasitic therapy or those who present with weight loss, change in bowel habits, or melena, colonscopic examination is warranted. Considering pinworm infection early during evaluation of nonspecific abdominal complaints may avoid an unnecessary and expensive diagnostic workup.
KEY POINTS
-
Recognize early on that Enterobius vermicularis infection is an important differential diagnosis for patients presenting with symptoms of colitis, thus avoiding unnecessary, expensive, and potentially harmful invasive testing.
-
Recognize that a simple and inexpensive Scotch tape test and/or direct visualization is an easy and effective way of confirming diagnosis and that stool examination may be unhelpful.
-
Recognize that reinfection may be prevented using a second dose of the antiparasitic drug.
- .The pinworm, Enterobius vermicularis.Prim Care.1991;18:13–24.
- ,,,,.Prevalence of intestinal parasites in three socioeconomically‐different regions of Sivas, Turkey.J Health Popul Nutr.2005;23:184–191.
- ,,,.Pinworm infection.Gastrointest Endosc.2001;53:210.
- ,,.Mebendazole (R 17635) in enterobiasis. A clinical trial in mental retardates.Chemotherapy.1975;21:255–260.
- ,,, et al.Field trials on the efficacy of albendazole composite against intestinal nematodiasis.Chung Kuo Chi Sheng Chung Hsueh Yu Chi Sheng Chung Ping Tsa Chih.1998;16:1–5.
- ,,.Enterobius vermicularis and colitis in children.J Pediatr Gastroenterol Nutr.2006;43:610–612.
- .The pinworm, Enterobius vermicularis.Prim Care.1991;18:13–24.
- ,,,,.Prevalence of intestinal parasites in three socioeconomically‐different regions of Sivas, Turkey.J Health Popul Nutr.2005;23:184–191.
- ,,,.Pinworm infection.Gastrointest Endosc.2001;53:210.
- ,,.Mebendazole (R 17635) in enterobiasis. A clinical trial in mental retardates.Chemotherapy.1975;21:255–260.
- ,,, et al.Field trials on the efficacy of albendazole composite against intestinal nematodiasis.Chung Kuo Chi Sheng Chung Hsueh Yu Chi Sheng Chung Ping Tsa Chih.1998;16:1–5.
- ,,.Enterobius vermicularis and colitis in children.J Pediatr Gastroenterol Nutr.2006;43:610–612.