User login
Quotes to live by: Paving the way to personal and professional success
In the first 2 years of medical school, the most common reasons for unsuccessful performance are a deficiency in cognitive knowledge, inefficient time management, and poor study skills. Thereafter, however, the principal reasons for poor performance in training or practice are personality issues and/or unprofessional behavior.
In this article, I review the attributes expected of a physician and the factors that undermine professionalism. I then offer suggestions for smoothing the pathway for personal and professional success. I crafted these suggestions with the “help” of some unlikely medical philosophers. (Note: Some variations of the cited quotations may exist.) I have tempered their guidance with my own personal experiences as a spouse, parent, and grandparent and my professional experiences over almost 50 years, during which I served as a career military officer, student clerkship director, residency program director, fellowship program director, and associate dean for student affairs. I readily acknowledge that, as major league baseball player Yogi Berra reputedly said, “I made too many wrong mistakes,” and that bad experiences are a tough way to ultimately learn good judgment. I hope these suggestions will help you avoid many of my “wrong mistakes.”
High expectations for the medical professional
“To whom much is given, much shall be required.”
—Luke 12:48
Medicine is a higher calling. It is not the usual type of business, and our patients certainly are not just customers or clients. In the unique moment of personal contact, we are asked to put the interest and well-being of our patient above all else. Our patients rightly have high expectations for what type of person their physician should be. The personal strengths expected of a physician include:
- humility
- honesty—personal and fiscal
- integrity
- strong moral compass
- fairness
- responsible
- diligent
- accountable
- insightful
- wise
- technically competent
- perseverant
- sympathetic
- empathetic
- inspiring.
To exhibit all these characteristics consistently is a herculean task and one that is impossible to fulfill. Many factors conspire to undermine our ability to steadfastly be all that we can be. Among these factors are:
- time constraints
- financial pressures
- physical illness
- emotional illness
- the explosion of information technology and scientific knowledge
- bureaucratic inefficiencies.
Therefore, we need to acknowledge with the philosopher Voltaire that “Perfect is the enemy of good.” We need to set our performance bar at excellence, not perfection. If we expect perfection of ourselves, we are destined to be consistently disappointed.
What follows is a series of well-intentioned and good-natured suggestions for keeping ourselves on an even keel, personally and professionally, and maintaining our compass setting on true north.
Continue to: Practical suggestions...
Practical suggestions
“It may not be that the race always goes to the swift nor the battle to the strong, but that is the way to bet.”
—Damon Runyon, journalist
The message is to study hard, work hard, practice our technical skills, and stay on top of our game. We must commit ourselves to a lifetime of learning.
“Chance favors the prepared mind.”
—Louis Pasteur, scientist
One of the best examples of this adage is Alexander Fleming’s “chance” discovery of the bactericidal effect of a mold growing on a culture plate in his laboratory. This observation led to the development of penicillin, an amazing antibiotic that, over the course of the past century, has saved the lives of literally hundreds of thousands of patients. We need to sustain our scientific curiosity throughout our careers and always remain open to new discoveries. Moreover, we need to maintain our capacity for awe and wonder as we consider the exquisite beauty of the scientific world.
“I have a dream.”
—Martin Luther King Jr, civil rights leader
Like Reverend King, we must aspire to a world where civility, peace, and social justice prevail, a world where we embrace diversity and inclusiveness and eschew prejudice, mean-spiritedness, and narrow-mindedness. We must acknowledge that some truths and moral principles are absolute, not relative.
“Once you learn to quit, it becomes a habit.”
—Vince Lombardi, professional football coach
Our lesson: Never quit. We must be fiercely determined to do the right thing, even in troubled and confusing times.
“A pessimist sees the difficulty in every opportunity; an optimist sees the opportunity in every difficulty.”
—Winston Churchill, British prime minister
Until proven wrong, always think the best of everyone. The bright side is far superior to the dark side. We must strive to consistently have a positive attitude and to be part of the solution to a problem, not the problem itself.
“It’s all such a delicate balance.”
—From “It’s a Delicate Balance” by Tom Dundee, folk singer and songwriter
Our top 3 priorities should always be our own emotional and physical well-being, the well-being and security of our loved ones, and the well-being of our patients. The order of these priorities may change, depending upon circumstances. When urgent patient care demands our presence and we miss a birthday celebration, anniversary dinner, soccer game, or dance recital, we need to make certain that, the next time a conflict arises, we arrange to have a colleague cover our clinical or administrative responsibilities.
We must learn to say no when our plate is too full. Failure to say no inevitably leads to life-work imbalance. It is always flattering to be asked to make a presentation, serve on a committee, or prepare a textbook chapter, and it is natural to be concerned that, if we decline, we will not be invited again. However, that concern is unwarranted. Rather, others will respect us for acknowledging when we are too busy and will be grateful that we did not accept an invitation and then miss important deadlines. Conversely, when we do say yes, we need to honor that commitment in a timely manner.
Continue to: The importance of time...
The importance of time
Perhaps the most common complaints that patients have with respect to their interactions with physicians are that they were forced to wait too long and then felt rushed through their appointment. Therefore:
- We must respect our patients’ time and recognize that their time is as valuable as ours.
- We must schedule our patient appointments appropriately and allow different amounts of time depending upon the complexity of a patient’s condition. We should not consistently overschedule. We need to offer a genuine apology when we keep a patient waiting for more than 15 minutes in the absence of an outright emergency that requires our attention elsewhere.
- When we interact with patients, we should sit down, establish eye-to-eye contact, and never appear hurried.
“You don’t make your character in a crisis; you exhibit it.”
—Oren Arnold, journalist and novelist
In the often-chaotic environment of the operating room or the labor and delivery suite, we must be the calm voice of reason at the center of the storm. We should not yell and make demands of others. We must strive to be unflappable. The other members of the team will be appreciative if they recognize that we have a steady hand on the tiller.
“To do good is noble. To teach others to do good is nobler—and less trouble.”
—Mark Twain, humorist
We need to teach our patients about their condition(s) so that they can assume more responsibility for their own care. We also need to teach our students and colleagues so that they can help us provide the best possible care for our patients. Being a good teacher is inherent in being a good physician. As the famous scientist Albert Einstein said, “If you cannot explain it simply, you do not understand it well enough.”
“It ain’t the things you don’t know that get you. It’s the things you think you know that ain’t so.”
—Artemus Ward, humorist
We must constantly strive to practice evidence-based medicine. We should not be the first to embrace the new or the last to give up the old. In medicine, as opposed to the highway, the best place to be is usually in the middle of the road. However, our commitment to evidence-based medicine cannot be absolute. In fact, no more than half of all our present treatment guidelines are based on level 1 evidence. At times, good old-fashioned common sense tempered by years of sobering experience should carry the day.
“We may be lost, but we’re making good time.”
—Yogi Berra, major league baseball player
In my experience, only the minority of mistakes in medicine result from lack of fundamental knowledge or a deficiency in technical skill. Rather, most result from imprudent haste and/or attempts to multitask. Therefore, our lesson is to slow down, concentrate on one task at a time, complete that task, and then refocus on the next challenge.
“The single greatest problem in communication is the illusion that it has taken place.”
—George Bernard Shaw, playwright
We must be sure that we always “close the loop” in our written and verbal communication so that we can avoid misunderstandings that threaten personal relationships and/or patient safety.
“You raise me up so I can stand on mountains.”
—From “You Raise Me Up” as sung by Josh Groban
All of us need a mentor to raise us up. We must choose our mentors carefully and recognize that we may need different mentors at different stages of our career. As we benefit from effective mentoring, we must pay it forward and be a good mentor to others.
“Worrying is a total waste of time. It accomplishes nothing, changes nothing, and robs you of joy. It is like paying a debt that you don’t owe.”
—Mark Twain, humorist
We have to assiduously cultivate the strength of resilience. We must accept that mistakes inevitably will occur and that perfection in practice is simply not possible, despite our best intentions. We then have to learn from these errors and ensure that they never occur again. We need to apologize for our mistakes and move on. If we carry our last strikeout into our next at bat, we are likely doomed to more misfortune.
“Feeling gratitude and not expressing it is like wrapping a present and not giving it.”
—William Arthur Ward, motivational writer
Our lesson is to be keenly aware of the importance of showing gratitude to those around us. The height of our success will depend directly on the depth of our gratitude. The higher we rise in the hierarchy of the medical profession, the more gracious and kind we need to be.
“Kindness is the language which the deaf can hear and the blind can see.”
—Mark Twain, humorist
“Kindness is the only service that will stand the storm of life and not wash out.”
—Abraham Lincoln, American president
There is never an excuse for rudeness or hubris. We should never teach or conduct business by intimidation. The words please, thank you, and I’m sorry should be front and center in our vocabulary. We must learn not to take ourselves too seriously, to remember that the best part of life is the laughter, and to always strive for grace and humility.
“The secret of the care of the patient is in caring for the patient.”
—Francis Peabody, physician
Patients may quickly forget what we say to them or even what we do for them, but they will never forget how we made them feel. Observe intently, listen carefully, talk less. Most people do not listen with the intent to understand. Rather, they listen with the intent to reply. We need to break this pattern by learning to listen with our heart. In fact, the quieter we become, the more we can hear. There is great symbolism in the fact that we have two ears and only one mouth.
“You got to know when to hold ‘em, know when to fold ‘em.”
—From “The Gambler” as sung by Kenny Rogers
Sometimes the best medicine is no medicine at all, but rather a soft shoulder, an open ear, a kind heart, and a compassionate soul.
“Do small things with great love.”
—Mother Teresa, Catholic missionary
The vast majority of us will not rise to lofty political or administrative positions or ever achieve celebrity status. We are unlikely to win the Nobel Prize and unlikely to find the cure for cancer or preeclampsia. However, we can work diligently to complete each small task with precision so that, like a great artist views his or her work, we, too, will want to sign our name to the patient care plan we have created and implemented.
“Earn this.”
—From Saving Private Ryan, a Steven Spielberg movie
At the end of this movie, the mortally wounded infantry captain (played by Tom Hanks) looks up at Private Ryan (played by Matt Damon) and says, “Earn this,” meaning make sure that you live your life in a way to justify the sacrifices so many made to save you. Like Private Ryan, we have to recognize that our MD degree does not constitute a lifetime entitlement to respect and honor. Rather, we have to practice each day so we continue to earn the respect of our patients, students, and colleagues and, so that, with confidence, we can then say to our patients, “How can I be of help to you?” ●

Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
In the first 2 years of medical school, the most common reasons for unsuccessful performance are a deficiency in cognitive knowledge, inefficient time management, and poor study skills. Thereafter, however, the principal reasons for poor performance in training or practice are personality issues and/or unprofessional behavior.
In this article, I review the attributes expected of a physician and the factors that undermine professionalism. I then offer suggestions for smoothing the pathway for personal and professional success. I crafted these suggestions with the “help” of some unlikely medical philosophers. (Note: Some variations of the cited quotations may exist.) I have tempered their guidance with my own personal experiences as a spouse, parent, and grandparent and my professional experiences over almost 50 years, during which I served as a career military officer, student clerkship director, residency program director, fellowship program director, and associate dean for student affairs. I readily acknowledge that, as major league baseball player Yogi Berra reputedly said, “I made too many wrong mistakes,” and that bad experiences are a tough way to ultimately learn good judgment. I hope these suggestions will help you avoid many of my “wrong mistakes.”
High expectations for the medical professional
“To whom much is given, much shall be required.”
—Luke 12:48
Medicine is a higher calling. It is not the usual type of business, and our patients certainly are not just customers or clients. In the unique moment of personal contact, we are asked to put the interest and well-being of our patient above all else. Our patients rightly have high expectations for what type of person their physician should be. The personal strengths expected of a physician include:
- humility
- honesty—personal and fiscal
- integrity
- strong moral compass
- fairness
- responsible
- diligent
- accountable
- insightful
- wise
- technically competent
- perseverant
- sympathetic
- empathetic
- inspiring.
To exhibit all these characteristics consistently is a herculean task and one that is impossible to fulfill. Many factors conspire to undermine our ability to steadfastly be all that we can be. Among these factors are:
- time constraints
- financial pressures
- physical illness
- emotional illness
- the explosion of information technology and scientific knowledge
- bureaucratic inefficiencies.
Therefore, we need to acknowledge with the philosopher Voltaire that “Perfect is the enemy of good.” We need to set our performance bar at excellence, not perfection. If we expect perfection of ourselves, we are destined to be consistently disappointed.
What follows is a series of well-intentioned and good-natured suggestions for keeping ourselves on an even keel, personally and professionally, and maintaining our compass setting on true north.
Continue to: Practical suggestions...
Practical suggestions
“It may not be that the race always goes to the swift nor the battle to the strong, but that is the way to bet.”
—Damon Runyon, journalist
The message is to study hard, work hard, practice our technical skills, and stay on top of our game. We must commit ourselves to a lifetime of learning.
“Chance favors the prepared mind.”
—Louis Pasteur, scientist
One of the best examples of this adage is Alexander Fleming’s “chance” discovery of the bactericidal effect of a mold growing on a culture plate in his laboratory. This observation led to the development of penicillin, an amazing antibiotic that, over the course of the past century, has saved the lives of literally hundreds of thousands of patients. We need to sustain our scientific curiosity throughout our careers and always remain open to new discoveries. Moreover, we need to maintain our capacity for awe and wonder as we consider the exquisite beauty of the scientific world.
“I have a dream.”
—Martin Luther King Jr, civil rights leader
Like Reverend King, we must aspire to a world where civility, peace, and social justice prevail, a world where we embrace diversity and inclusiveness and eschew prejudice, mean-spiritedness, and narrow-mindedness. We must acknowledge that some truths and moral principles are absolute, not relative.
“Once you learn to quit, it becomes a habit.”
—Vince Lombardi, professional football coach
Our lesson: Never quit. We must be fiercely determined to do the right thing, even in troubled and confusing times.
“A pessimist sees the difficulty in every opportunity; an optimist sees the opportunity in every difficulty.”
—Winston Churchill, British prime minister
Until proven wrong, always think the best of everyone. The bright side is far superior to the dark side. We must strive to consistently have a positive attitude and to be part of the solution to a problem, not the problem itself.
“It’s all such a delicate balance.”
—From “It’s a Delicate Balance” by Tom Dundee, folk singer and songwriter
Our top 3 priorities should always be our own emotional and physical well-being, the well-being and security of our loved ones, and the well-being of our patients. The order of these priorities may change, depending upon circumstances. When urgent patient care demands our presence and we miss a birthday celebration, anniversary dinner, soccer game, or dance recital, we need to make certain that, the next time a conflict arises, we arrange to have a colleague cover our clinical or administrative responsibilities.
We must learn to say no when our plate is too full. Failure to say no inevitably leads to life-work imbalance. It is always flattering to be asked to make a presentation, serve on a committee, or prepare a textbook chapter, and it is natural to be concerned that, if we decline, we will not be invited again. However, that concern is unwarranted. Rather, others will respect us for acknowledging when we are too busy and will be grateful that we did not accept an invitation and then miss important deadlines. Conversely, when we do say yes, we need to honor that commitment in a timely manner.
Continue to: The importance of time...
The importance of time
Perhaps the most common complaints that patients have with respect to their interactions with physicians are that they were forced to wait too long and then felt rushed through their appointment. Therefore:
- We must respect our patients’ time and recognize that their time is as valuable as ours.
- We must schedule our patient appointments appropriately and allow different amounts of time depending upon the complexity of a patient’s condition. We should not consistently overschedule. We need to offer a genuine apology when we keep a patient waiting for more than 15 minutes in the absence of an outright emergency that requires our attention elsewhere.
- When we interact with patients, we should sit down, establish eye-to-eye contact, and never appear hurried.
“You don’t make your character in a crisis; you exhibit it.”
—Oren Arnold, journalist and novelist
In the often-chaotic environment of the operating room or the labor and delivery suite, we must be the calm voice of reason at the center of the storm. We should not yell and make demands of others. We must strive to be unflappable. The other members of the team will be appreciative if they recognize that we have a steady hand on the tiller.
“To do good is noble. To teach others to do good is nobler—and less trouble.”
—Mark Twain, humorist
We need to teach our patients about their condition(s) so that they can assume more responsibility for their own care. We also need to teach our students and colleagues so that they can help us provide the best possible care for our patients. Being a good teacher is inherent in being a good physician. As the famous scientist Albert Einstein said, “If you cannot explain it simply, you do not understand it well enough.”
“It ain’t the things you don’t know that get you. It’s the things you think you know that ain’t so.”
—Artemus Ward, humorist
We must constantly strive to practice evidence-based medicine. We should not be the first to embrace the new or the last to give up the old. In medicine, as opposed to the highway, the best place to be is usually in the middle of the road. However, our commitment to evidence-based medicine cannot be absolute. In fact, no more than half of all our present treatment guidelines are based on level 1 evidence. At times, good old-fashioned common sense tempered by years of sobering experience should carry the day.
“We may be lost, but we’re making good time.”
—Yogi Berra, major league baseball player
In my experience, only the minority of mistakes in medicine result from lack of fundamental knowledge or a deficiency in technical skill. Rather, most result from imprudent haste and/or attempts to multitask. Therefore, our lesson is to slow down, concentrate on one task at a time, complete that task, and then refocus on the next challenge.
“The single greatest problem in communication is the illusion that it has taken place.”
—George Bernard Shaw, playwright
We must be sure that we always “close the loop” in our written and verbal communication so that we can avoid misunderstandings that threaten personal relationships and/or patient safety.
“You raise me up so I can stand on mountains.”
—From “You Raise Me Up” as sung by Josh Groban
All of us need a mentor to raise us up. We must choose our mentors carefully and recognize that we may need different mentors at different stages of our career. As we benefit from effective mentoring, we must pay it forward and be a good mentor to others.
“Worrying is a total waste of time. It accomplishes nothing, changes nothing, and robs you of joy. It is like paying a debt that you don’t owe.”
—Mark Twain, humorist
We have to assiduously cultivate the strength of resilience. We must accept that mistakes inevitably will occur and that perfection in practice is simply not possible, despite our best intentions. We then have to learn from these errors and ensure that they never occur again. We need to apologize for our mistakes and move on. If we carry our last strikeout into our next at bat, we are likely doomed to more misfortune.
“Feeling gratitude and not expressing it is like wrapping a present and not giving it.”
—William Arthur Ward, motivational writer
Our lesson is to be keenly aware of the importance of showing gratitude to those around us. The height of our success will depend directly on the depth of our gratitude. The higher we rise in the hierarchy of the medical profession, the more gracious and kind we need to be.
“Kindness is the language which the deaf can hear and the blind can see.”
—Mark Twain, humorist
“Kindness is the only service that will stand the storm of life and not wash out.”
—Abraham Lincoln, American president
There is never an excuse for rudeness or hubris. We should never teach or conduct business by intimidation. The words please, thank you, and I’m sorry should be front and center in our vocabulary. We must learn not to take ourselves too seriously, to remember that the best part of life is the laughter, and to always strive for grace and humility.
“The secret of the care of the patient is in caring for the patient.”
—Francis Peabody, physician
Patients may quickly forget what we say to them or even what we do for them, but they will never forget how we made them feel. Observe intently, listen carefully, talk less. Most people do not listen with the intent to understand. Rather, they listen with the intent to reply. We need to break this pattern by learning to listen with our heart. In fact, the quieter we become, the more we can hear. There is great symbolism in the fact that we have two ears and only one mouth.
“You got to know when to hold ‘em, know when to fold ‘em.”
—From “The Gambler” as sung by Kenny Rogers
Sometimes the best medicine is no medicine at all, but rather a soft shoulder, an open ear, a kind heart, and a compassionate soul.
“Do small things with great love.”
—Mother Teresa, Catholic missionary
The vast majority of us will not rise to lofty political or administrative positions or ever achieve celebrity status. We are unlikely to win the Nobel Prize and unlikely to find the cure for cancer or preeclampsia. However, we can work diligently to complete each small task with precision so that, like a great artist views his or her work, we, too, will want to sign our name to the patient care plan we have created and implemented.
“Earn this.”
—From Saving Private Ryan, a Steven Spielberg movie
At the end of this movie, the mortally wounded infantry captain (played by Tom Hanks) looks up at Private Ryan (played by Matt Damon) and says, “Earn this,” meaning make sure that you live your life in a way to justify the sacrifices so many made to save you. Like Private Ryan, we have to recognize that our MD degree does not constitute a lifetime entitlement to respect and honor. Rather, we have to practice each day so we continue to earn the respect of our patients, students, and colleagues and, so that, with confidence, we can then say to our patients, “How can I be of help to you?” ●
In the first 2 years of medical school, the most common reasons for unsuccessful performance are a deficiency in cognitive knowledge, inefficient time management, and poor study skills. Thereafter, however, the principal reasons for poor performance in training or practice are personality issues and/or unprofessional behavior.
In this article, I review the attributes expected of a physician and the factors that undermine professionalism. I then offer suggestions for smoothing the pathway for personal and professional success. I crafted these suggestions with the “help” of some unlikely medical philosophers. (Note: Some variations of the cited quotations may exist.) I have tempered their guidance with my own personal experiences as a spouse, parent, and grandparent and my professional experiences over almost 50 years, during which I served as a career military officer, student clerkship director, residency program director, fellowship program director, and associate dean for student affairs. I readily acknowledge that, as major league baseball player Yogi Berra reputedly said, “I made too many wrong mistakes,” and that bad experiences are a tough way to ultimately learn good judgment. I hope these suggestions will help you avoid many of my “wrong mistakes.”
High expectations for the medical professional
“To whom much is given, much shall be required.”
—Luke 12:48
Medicine is a higher calling. It is not the usual type of business, and our patients certainly are not just customers or clients. In the unique moment of personal contact, we are asked to put the interest and well-being of our patient above all else. Our patients rightly have high expectations for what type of person their physician should be. The personal strengths expected of a physician include:
- humility
- honesty—personal and fiscal
- integrity
- strong moral compass
- fairness
- responsible
- diligent
- accountable
- insightful
- wise
- technically competent
- perseverant
- sympathetic
- empathetic
- inspiring.
To exhibit all these characteristics consistently is a herculean task and one that is impossible to fulfill. Many factors conspire to undermine our ability to steadfastly be all that we can be. Among these factors are:
- time constraints
- financial pressures
- physical illness
- emotional illness
- the explosion of information technology and scientific knowledge
- bureaucratic inefficiencies.
Therefore, we need to acknowledge with the philosopher Voltaire that “Perfect is the enemy of good.” We need to set our performance bar at excellence, not perfection. If we expect perfection of ourselves, we are destined to be consistently disappointed.
What follows is a series of well-intentioned and good-natured suggestions for keeping ourselves on an even keel, personally and professionally, and maintaining our compass setting on true north.
Continue to: Practical suggestions...
Practical suggestions
“It may not be that the race always goes to the swift nor the battle to the strong, but that is the way to bet.”
—Damon Runyon, journalist
The message is to study hard, work hard, practice our technical skills, and stay on top of our game. We must commit ourselves to a lifetime of learning.
“Chance favors the prepared mind.”
—Louis Pasteur, scientist
One of the best examples of this adage is Alexander Fleming’s “chance” discovery of the bactericidal effect of a mold growing on a culture plate in his laboratory. This observation led to the development of penicillin, an amazing antibiotic that, over the course of the past century, has saved the lives of literally hundreds of thousands of patients. We need to sustain our scientific curiosity throughout our careers and always remain open to new discoveries. Moreover, we need to maintain our capacity for awe and wonder as we consider the exquisite beauty of the scientific world.
“I have a dream.”
—Martin Luther King Jr, civil rights leader
Like Reverend King, we must aspire to a world where civility, peace, and social justice prevail, a world where we embrace diversity and inclusiveness and eschew prejudice, mean-spiritedness, and narrow-mindedness. We must acknowledge that some truths and moral principles are absolute, not relative.
“Once you learn to quit, it becomes a habit.”
—Vince Lombardi, professional football coach
Our lesson: Never quit. We must be fiercely determined to do the right thing, even in troubled and confusing times.
“A pessimist sees the difficulty in every opportunity; an optimist sees the opportunity in every difficulty.”
—Winston Churchill, British prime minister
Until proven wrong, always think the best of everyone. The bright side is far superior to the dark side. We must strive to consistently have a positive attitude and to be part of the solution to a problem, not the problem itself.
“It’s all such a delicate balance.”
—From “It’s a Delicate Balance” by Tom Dundee, folk singer and songwriter
Our top 3 priorities should always be our own emotional and physical well-being, the well-being and security of our loved ones, and the well-being of our patients. The order of these priorities may change, depending upon circumstances. When urgent patient care demands our presence and we miss a birthday celebration, anniversary dinner, soccer game, or dance recital, we need to make certain that, the next time a conflict arises, we arrange to have a colleague cover our clinical or administrative responsibilities.
We must learn to say no when our plate is too full. Failure to say no inevitably leads to life-work imbalance. It is always flattering to be asked to make a presentation, serve on a committee, or prepare a textbook chapter, and it is natural to be concerned that, if we decline, we will not be invited again. However, that concern is unwarranted. Rather, others will respect us for acknowledging when we are too busy and will be grateful that we did not accept an invitation and then miss important deadlines. Conversely, when we do say yes, we need to honor that commitment in a timely manner.
Continue to: The importance of time...
The importance of time
Perhaps the most common complaints that patients have with respect to their interactions with physicians are that they were forced to wait too long and then felt rushed through their appointment. Therefore:
- We must respect our patients’ time and recognize that their time is as valuable as ours.
- We must schedule our patient appointments appropriately and allow different amounts of time depending upon the complexity of a patient’s condition. We should not consistently overschedule. We need to offer a genuine apology when we keep a patient waiting for more than 15 minutes in the absence of an outright emergency that requires our attention elsewhere.
- When we interact with patients, we should sit down, establish eye-to-eye contact, and never appear hurried.
“You don’t make your character in a crisis; you exhibit it.”
—Oren Arnold, journalist and novelist
In the often-chaotic environment of the operating room or the labor and delivery suite, we must be the calm voice of reason at the center of the storm. We should not yell and make demands of others. We must strive to be unflappable. The other members of the team will be appreciative if they recognize that we have a steady hand on the tiller.
“To do good is noble. To teach others to do good is nobler—and less trouble.”
—Mark Twain, humorist
We need to teach our patients about their condition(s) so that they can assume more responsibility for their own care. We also need to teach our students and colleagues so that they can help us provide the best possible care for our patients. Being a good teacher is inherent in being a good physician. As the famous scientist Albert Einstein said, “If you cannot explain it simply, you do not understand it well enough.”
“It ain’t the things you don’t know that get you. It’s the things you think you know that ain’t so.”
—Artemus Ward, humorist
We must constantly strive to practice evidence-based medicine. We should not be the first to embrace the new or the last to give up the old. In medicine, as opposed to the highway, the best place to be is usually in the middle of the road. However, our commitment to evidence-based medicine cannot be absolute. In fact, no more than half of all our present treatment guidelines are based on level 1 evidence. At times, good old-fashioned common sense tempered by years of sobering experience should carry the day.
“We may be lost, but we’re making good time.”
—Yogi Berra, major league baseball player
In my experience, only the minority of mistakes in medicine result from lack of fundamental knowledge or a deficiency in technical skill. Rather, most result from imprudent haste and/or attempts to multitask. Therefore, our lesson is to slow down, concentrate on one task at a time, complete that task, and then refocus on the next challenge.
“The single greatest problem in communication is the illusion that it has taken place.”
—George Bernard Shaw, playwright
We must be sure that we always “close the loop” in our written and verbal communication so that we can avoid misunderstandings that threaten personal relationships and/or patient safety.
“You raise me up so I can stand on mountains.”
—From “You Raise Me Up” as sung by Josh Groban
All of us need a mentor to raise us up. We must choose our mentors carefully and recognize that we may need different mentors at different stages of our career. As we benefit from effective mentoring, we must pay it forward and be a good mentor to others.
“Worrying is a total waste of time. It accomplishes nothing, changes nothing, and robs you of joy. It is like paying a debt that you don’t owe.”
—Mark Twain, humorist
We have to assiduously cultivate the strength of resilience. We must accept that mistakes inevitably will occur and that perfection in practice is simply not possible, despite our best intentions. We then have to learn from these errors and ensure that they never occur again. We need to apologize for our mistakes and move on. If we carry our last strikeout into our next at bat, we are likely doomed to more misfortune.
“Feeling gratitude and not expressing it is like wrapping a present and not giving it.”
—William Arthur Ward, motivational writer
Our lesson is to be keenly aware of the importance of showing gratitude to those around us. The height of our success will depend directly on the depth of our gratitude. The higher we rise in the hierarchy of the medical profession, the more gracious and kind we need to be.
“Kindness is the language which the deaf can hear and the blind can see.”
—Mark Twain, humorist
“Kindness is the only service that will stand the storm of life and not wash out.”
—Abraham Lincoln, American president
There is never an excuse for rudeness or hubris. We should never teach or conduct business by intimidation. The words please, thank you, and I’m sorry should be front and center in our vocabulary. We must learn not to take ourselves too seriously, to remember that the best part of life is the laughter, and to always strive for grace and humility.
“The secret of the care of the patient is in caring for the patient.”
—Francis Peabody, physician
Patients may quickly forget what we say to them or even what we do for them, but they will never forget how we made them feel. Observe intently, listen carefully, talk less. Most people do not listen with the intent to understand. Rather, they listen with the intent to reply. We need to break this pattern by learning to listen with our heart. In fact, the quieter we become, the more we can hear. There is great symbolism in the fact that we have two ears and only one mouth.
“You got to know when to hold ‘em, know when to fold ‘em.”
—From “The Gambler” as sung by Kenny Rogers
Sometimes the best medicine is no medicine at all, but rather a soft shoulder, an open ear, a kind heart, and a compassionate soul.
“Do small things with great love.”
—Mother Teresa, Catholic missionary
The vast majority of us will not rise to lofty political or administrative positions or ever achieve celebrity status. We are unlikely to win the Nobel Prize and unlikely to find the cure for cancer or preeclampsia. However, we can work diligently to complete each small task with precision so that, like a great artist views his or her work, we, too, will want to sign our name to the patient care plan we have created and implemented.
“Earn this.”
—From Saving Private Ryan, a Steven Spielberg movie
At the end of this movie, the mortally wounded infantry captain (played by Tom Hanks) looks up at Private Ryan (played by Matt Damon) and says, “Earn this,” meaning make sure that you live your life in a way to justify the sacrifices so many made to save you. Like Private Ryan, we have to recognize that our MD degree does not constitute a lifetime entitlement to respect and honor. Rather, we have to practice each day so we continue to earn the respect of our patients, students, and colleagues and, so that, with confidence, we can then say to our patients, “How can I be of help to you?” ●
RSV vaccination during pregnancy: Finally ready for prime time
A 28-year-old primigravid woman at 30 weeks’ gestation inquires about the new vaccine to protect her newborn baby against respiratory syncytial virus infection (RSV). Her neighbor’s daughter recently was hospitalized for the treatment of RSV, and she is understandably concerned about her own newborn. The patient is healthy, and she has never had any serious respiratory infection. She is taking no medications other than prenatal vitamins.
What advice should you give her?
If you decide to administer this vaccine, what is the appropriate timing of administration?
Are there any maternal or fetal safety concerns related to use of this vaccine in pregnancy?
Respiratory syncytial virus (RSV) is a member of the Paramyxoviridae family. It is an enveloped, single-stranded RNA virus that is 150-300 nm in size. The virus codes for 10 virus-specific proteins. The 2 most important are the G protein, which enables the virus to attach to host cells, and the F protein, which facilitates the entry of the virus into the host cell by fusing the host and viral membranes. Two distinct subtypes exist: A and B. There is genetic variation within each subtype and between subtypes. These subtle genetic variations create the potential for reinfections, and hence, research has focused on development of a vaccine that covers both subtypes.1
RSV is the most common cause of acute lower respiratory tract infection in infants younger than 6 months of age. In these children, RSV is one of the most prominent causes of death, with mortality particularly marked in low- and middle-resource countries as well as in children who were born premature and/or who are immunocompromised. RSV has its greatest impact during winter epidemics in temperate climates and during the rainy seasons in tropical climates. The virus rarely is encountered in the summer.1 Among young children, RSV primarily is transmitted via close contact with contaminated fingers or fomites and by self-inoculation of the conjunctiva or anterior nares. The incubation period of the infection is 4 to 6 days, and viral shedding may persist for 2 weeks or longer. Most patients gradually recover within 1 to 2 weeks.1 Adults who contract RSV usually have symptoms suggestive of a common cold; however, in older adults or those who have comorbidities, serious and potentially life-threatening lower respiratory tract infections may develop.
Recently, there have been 2 main approaches to the prevention and treatment of RSV in infants. One has been the development of monoclonal antibodies such as motavizumab, palivizumab, and nirsevimab. The other has been the development of a vaccine that could be administered to pregnant women and which could provide protection for the neonate in the early months of life.2,3
In late August 2023, the US Food and Drug Administration (FDA) announced the approval of a new bivalent RSV prefusion F vaccine (ABRYSVO, Pfizer) intended for administration to pregnant women.4 Of note, previous efforts to develop whole-virus vaccines either have been ineffective or have potentiated the disease in infants who became infected; development of an effective vaccine had eluded scientists and clinicians for nearly 50 years.2 Thus, the new vaccine that targets the F protein of the virus represents a major and welcomed breakthrough.
This article reviews the 3 most recent investigations that preceded the ultimate approval of this vaccine and discusses specific logistical issues related to vaccine administration.
Continue to: First step toward vaccine approval...
First step toward vaccine approval
Madhi and colleagues5 were among the first to conduct a large well-designed study to evaluate the effectiveness of maternal vaccination in preventing neonatal infection in the first few months of life. The authors enrolled more than 4,500 healthy pregnant women at 28 to 36 weeks of gestation and assigned them to receive either a single intramuscular dose of an RSV fusion (F) protein vaccine or placebo in a ratio of 2:1. The primary end point was a “medically significant lower respiratory tract infection” within the first 90 days of life. The percentage of infants who met the primary end point was low in both groups: 1.5% in the vaccine group and 2.4% in the placebo group (efficacy 39.4%). The efficacy of the vaccine in preventing lower respiratory tract infection with severe hypoxemia was 48.3% and 44.4% in preventing hospitalization. Although there were differences between the 2 groups, they did not meet the prespecified success criterion for efficacy. Vaccine recipients had more local injection site reactions (40.7% vs 9.9%); however, there was no difference in the frequency of other adverse effects.
Intermediate step: Continued assessment of vaccine safety and immunogenicity
The next important step in the development of the RSV vaccine was a study by Simoes et al,6 who conducted a phase 2b trial to determine the safety and immunogenicity of the RSVpreF vaccine. The authors randomly assigned pregnant women at 24 to 36 weeks of gestation to receive either 120 or 240 µg of RSVpreF vaccine or placebo. The key endpoints were the following: maternal and infant safety; the maternal-to-infant transplacental transfer ratio; and the presence of RSV A, B, and combined A/B neutralizing antibody in maternal serum and umbilical cord blood at delivery. The authors conducted a planned interim analysis that included 327 mothers who received the vaccine. The incidence of adverse effects was similar in mothers and infants in the vaccine compared with the placebo group. None of the adverse effects were judged to be serious. The transplacental neutralizing antibody transfer ratios ranged from 1.4 to 2.1 across a range of gestational ages. The vaccine elicited meaningful neutralizing titers of antibody in maternal serum even up to 7 weeks after immunization. The levels of neutralizing antibodies in umbilical cord blood did not vary substantially with respect to gestational age. A post hoc analysis showed that the transferred antibodies prevented medically-attended RSV-associated lower respiratory tract illnesses in the infants.
Final step: Convincing proof of efficacy
The most recent of the 3 studies, and the one that had the greatest impact in convincing the FDA to approve the vaccine, was the report by Kampmann and colleagues.7 The authors conducted a phase 3 prospective, randomized, double-blind trial in 18 different countries over 4 RSV seasons: 2 in the northern hemisphere and 2 in the southern hemisphere. They enrolled healthy pregnant women with singleton gestations at 24 to 36 weeks of gestation and assigned them in a 1:1 ratio to a single intramuscular injection of 120 µg of a bivalent RSV prefusion F protein-based (RSVpreF) vaccine or placebo. They excluded patients with any recognized risk factor for an adverse pregnancy outcome, including preterm labor. The 2 primary efficacy endpoints were a medically-attended severe RSV–lower respiratory tract infection and any medically attended RSV-associated lower respiratory tract illness in infants within 90, 120, 150, and 180 days after birth.
The efficacy of the vaccine in preventing severe lower respiratory tract illness within 90 days of delivery was 81.8% (99.5% confidence interval [CI], 40.6–96.3). The efficacy within 180 days of delivery was 69.4% (97.58% CI, 44.3–84.1). These differences reached the study’s pre-established statistical criteria for success. The overall rate of lower respiratory tract infections was not significantly different. The frequencies of adverse effects in mothers and infants were similar in the vaccine and placebo groups. In particular, the frequency of preterm delivery in the vaccine group was 0.8%, compared with 0.6% in the placebo group (P = NS).
In previous reports to the FDA,4 the frequency rate of preterm delivery in RSV vaccine recipients was slightly increased in vaccine recipients compared with patients who received placebo. The difference among the groups was too small to infer a causal relationship; however, as a condition of vaccine approval, the FDA has required Pfizer to conduct a postmarketing study to be certain that administration of the vaccine does not increase the risk for preterm delivery.
Practical details
The new vaccine is a bivalent recombinant vaccine that elicits a robust antibody response against the F (fusion) protein of the virus. In addition to the F antigen, the vaccine contains the following buffer ingredients: tromethamine, sucrose, mannitol, polysorbate, and sodium chloride.8 There are no preservatives in the vaccine.
The vaccine should be administered in a single, 0.5 mL, intramuscular injection at 32 to 36 weeks of gestation. Patients who are allergic to any of the components of the vaccine should not be vaccinated. Patients with a mild upper respiratory tract infection may receive the vaccine. Administration should be delayed in patients who are moderately to severely ill. The vaccine may be administered at the same time as other vaccines, such as influenza or Tdap.
The most common side effects of the vaccine are local injection site reactions, such as pain, redness, or swelling. Some patients may experience mild systemic manifestations, including fatigue, fever, headache, nausea, diarrhea, arthralgias, and myalgias. According to the Centers for Disease Control and Prevention, the approximate wholesale acquisition cost of the vaccine is $320 for 1 injection.
CASE Resolution
This patient is healthy and has no contraindication to the new RSV vaccine. According to the FDA, the optimal time for administration of the vaccine is 32 to 36 weeks of gestation. The patient should anticipate very few side effects following the vaccination, and the vaccine has approximately 80% efficacy in preventing severe lower respiratory tract infection in her neonate. ●
- RSV is the most common cause of acute lower respiratory tract infection in infants younger than 6 months of age.
- In low- and middle-resource countries, RSV is a leading cause of infant death.
- In late August 2023, the FDA approved the first RSV vaccine that can be administered to pregnant women to provide protection for the infant in the first few months of life.
- The vaccine specifically targets the F protein of the virus, a protein which is essential for facilitating fusion between the viral and host cell membranes, resulting in penetration of the virus into the host cell.
- The vaccine should be administered as a single intramuscular injection at 32 to 36 weeks’ gestation.
- The vaccine is approximately 82% effective in preventing severe lower respiratory tract infection in infants within the first 6 months of life.
- To exercise an abundance of caution, because of a possible association between administration of the vaccine and an increased risk for preterm delivery, vaccination should be delayed until 36 weeks in patients clearly identified as at-risk for preterm delivery.
- Dolin R. Common viral respiratory infections. In, Isselbacher KJ, Braunwald E, Wilson JD, et al, eds. Harrison’s Principles of Internal Medicine. 13th ed. McGraw-Hill; 1994:805-806.
- Mazur N, Terstappen J, Baral R, et al. Respiratory syncytial virus prevention within reach: the vaccine and monoclonal antibody landscape. Lancet Infect Dis. 2023;23:E2-E21.
- Hammitt LL, Dagan R, Yuan Y, et al. Nirsevimab for prevention of RSV in healthy late-preterm and term infants. N Engl J Med. 2022;386:837-846.
- US Food and Drug Administration News Release. August 21, 2023. Accessed October 26, 2023. https://www.fda.gov/news -events/press-announcements/fda-approves-first-vaccine -pregnant-individuals-prevent-rsv-infants
- Madhi SA, Polack FP, Piedra PA, et al. Respiratory syncytial virus vaccination during pregnancy and effects in infants. N Engl J Med. 2020;383:426-439.
- Simoes EAF, Center KJ, Tita ATN, et al. Prefusion F proteinbased respiratory syncytial virus immunization in pregnancy. N Eng J Med. 2022;386:1615-1626.
- Kampmann B, Madhi SA, Munjal I, et al. Bivalent prefusion F vaccine in pregnancy to prevent RSV illness in infants. N Engl J Med. 2023;388:1451-1464.
- Centers for Disease Control and Prevention. Vaccine Information Statement. Respiratory Syncytial Virus (RSV) Vaccine VIS. October 19, 2023. Accessed October 26, 2023. https://www. cdc.gov/vaccines/hcp/vis/vis-statements/rsv.html

Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
A 28-year-old primigravid woman at 30 weeks’ gestation inquires about the new vaccine to protect her newborn baby against respiratory syncytial virus infection (RSV). Her neighbor’s daughter recently was hospitalized for the treatment of RSV, and she is understandably concerned about her own newborn. The patient is healthy, and she has never had any serious respiratory infection. She is taking no medications other than prenatal vitamins.
What advice should you give her?
If you decide to administer this vaccine, what is the appropriate timing of administration?
Are there any maternal or fetal safety concerns related to use of this vaccine in pregnancy?
Respiratory syncytial virus (RSV) is a member of the Paramyxoviridae family. It is an enveloped, single-stranded RNA virus that is 150-300 nm in size. The virus codes for 10 virus-specific proteins. The 2 most important are the G protein, which enables the virus to attach to host cells, and the F protein, which facilitates the entry of the virus into the host cell by fusing the host and viral membranes. Two distinct subtypes exist: A and B. There is genetic variation within each subtype and between subtypes. These subtle genetic variations create the potential for reinfections, and hence, research has focused on development of a vaccine that covers both subtypes.1
RSV is the most common cause of acute lower respiratory tract infection in infants younger than 6 months of age. In these children, RSV is one of the most prominent causes of death, with mortality particularly marked in low- and middle-resource countries as well as in children who were born premature and/or who are immunocompromised. RSV has its greatest impact during winter epidemics in temperate climates and during the rainy seasons in tropical climates. The virus rarely is encountered in the summer.1 Among young children, RSV primarily is transmitted via close contact with contaminated fingers or fomites and by self-inoculation of the conjunctiva or anterior nares. The incubation period of the infection is 4 to 6 days, and viral shedding may persist for 2 weeks or longer. Most patients gradually recover within 1 to 2 weeks.1 Adults who contract RSV usually have symptoms suggestive of a common cold; however, in older adults or those who have comorbidities, serious and potentially life-threatening lower respiratory tract infections may develop.
Recently, there have been 2 main approaches to the prevention and treatment of RSV in infants. One has been the development of monoclonal antibodies such as motavizumab, palivizumab, and nirsevimab. The other has been the development of a vaccine that could be administered to pregnant women and which could provide protection for the neonate in the early months of life.2,3
In late August 2023, the US Food and Drug Administration (FDA) announced the approval of a new bivalent RSV prefusion F vaccine (ABRYSVO, Pfizer) intended for administration to pregnant women.4 Of note, previous efforts to develop whole-virus vaccines either have been ineffective or have potentiated the disease in infants who became infected; development of an effective vaccine had eluded scientists and clinicians for nearly 50 years.2 Thus, the new vaccine that targets the F protein of the virus represents a major and welcomed breakthrough.
This article reviews the 3 most recent investigations that preceded the ultimate approval of this vaccine and discusses specific logistical issues related to vaccine administration.
Continue to: First step toward vaccine approval...
First step toward vaccine approval
Madhi and colleagues5 were among the first to conduct a large well-designed study to evaluate the effectiveness of maternal vaccination in preventing neonatal infection in the first few months of life. The authors enrolled more than 4,500 healthy pregnant women at 28 to 36 weeks of gestation and assigned them to receive either a single intramuscular dose of an RSV fusion (F) protein vaccine or placebo in a ratio of 2:1. The primary end point was a “medically significant lower respiratory tract infection” within the first 90 days of life. The percentage of infants who met the primary end point was low in both groups: 1.5% in the vaccine group and 2.4% in the placebo group (efficacy 39.4%). The efficacy of the vaccine in preventing lower respiratory tract infection with severe hypoxemia was 48.3% and 44.4% in preventing hospitalization. Although there were differences between the 2 groups, they did not meet the prespecified success criterion for efficacy. Vaccine recipients had more local injection site reactions (40.7% vs 9.9%); however, there was no difference in the frequency of other adverse effects.
Intermediate step: Continued assessment of vaccine safety and immunogenicity
The next important step in the development of the RSV vaccine was a study by Simoes et al,6 who conducted a phase 2b trial to determine the safety and immunogenicity of the RSVpreF vaccine. The authors randomly assigned pregnant women at 24 to 36 weeks of gestation to receive either 120 or 240 µg of RSVpreF vaccine or placebo. The key endpoints were the following: maternal and infant safety; the maternal-to-infant transplacental transfer ratio; and the presence of RSV A, B, and combined A/B neutralizing antibody in maternal serum and umbilical cord blood at delivery. The authors conducted a planned interim analysis that included 327 mothers who received the vaccine. The incidence of adverse effects was similar in mothers and infants in the vaccine compared with the placebo group. None of the adverse effects were judged to be serious. The transplacental neutralizing antibody transfer ratios ranged from 1.4 to 2.1 across a range of gestational ages. The vaccine elicited meaningful neutralizing titers of antibody in maternal serum even up to 7 weeks after immunization. The levels of neutralizing antibodies in umbilical cord blood did not vary substantially with respect to gestational age. A post hoc analysis showed that the transferred antibodies prevented medically-attended RSV-associated lower respiratory tract illnesses in the infants.
Final step: Convincing proof of efficacy
The most recent of the 3 studies, and the one that had the greatest impact in convincing the FDA to approve the vaccine, was the report by Kampmann and colleagues.7 The authors conducted a phase 3 prospective, randomized, double-blind trial in 18 different countries over 4 RSV seasons: 2 in the northern hemisphere and 2 in the southern hemisphere. They enrolled healthy pregnant women with singleton gestations at 24 to 36 weeks of gestation and assigned them in a 1:1 ratio to a single intramuscular injection of 120 µg of a bivalent RSV prefusion F protein-based (RSVpreF) vaccine or placebo. They excluded patients with any recognized risk factor for an adverse pregnancy outcome, including preterm labor. The 2 primary efficacy endpoints were a medically-attended severe RSV–lower respiratory tract infection and any medically attended RSV-associated lower respiratory tract illness in infants within 90, 120, 150, and 180 days after birth.
The efficacy of the vaccine in preventing severe lower respiratory tract illness within 90 days of delivery was 81.8% (99.5% confidence interval [CI], 40.6–96.3). The efficacy within 180 days of delivery was 69.4% (97.58% CI, 44.3–84.1). These differences reached the study’s pre-established statistical criteria for success. The overall rate of lower respiratory tract infections was not significantly different. The frequencies of adverse effects in mothers and infants were similar in the vaccine and placebo groups. In particular, the frequency of preterm delivery in the vaccine group was 0.8%, compared with 0.6% in the placebo group (P = NS).
In previous reports to the FDA,4 the frequency rate of preterm delivery in RSV vaccine recipients was slightly increased in vaccine recipients compared with patients who received placebo. The difference among the groups was too small to infer a causal relationship; however, as a condition of vaccine approval, the FDA has required Pfizer to conduct a postmarketing study to be certain that administration of the vaccine does not increase the risk for preterm delivery.
Practical details
The new vaccine is a bivalent recombinant vaccine that elicits a robust antibody response against the F (fusion) protein of the virus. In addition to the F antigen, the vaccine contains the following buffer ingredients: tromethamine, sucrose, mannitol, polysorbate, and sodium chloride.8 There are no preservatives in the vaccine.
The vaccine should be administered in a single, 0.5 mL, intramuscular injection at 32 to 36 weeks of gestation. Patients who are allergic to any of the components of the vaccine should not be vaccinated. Patients with a mild upper respiratory tract infection may receive the vaccine. Administration should be delayed in patients who are moderately to severely ill. The vaccine may be administered at the same time as other vaccines, such as influenza or Tdap.
The most common side effects of the vaccine are local injection site reactions, such as pain, redness, or swelling. Some patients may experience mild systemic manifestations, including fatigue, fever, headache, nausea, diarrhea, arthralgias, and myalgias. According to the Centers for Disease Control and Prevention, the approximate wholesale acquisition cost of the vaccine is $320 for 1 injection.
CASE Resolution
This patient is healthy and has no contraindication to the new RSV vaccine. According to the FDA, the optimal time for administration of the vaccine is 32 to 36 weeks of gestation. The patient should anticipate very few side effects following the vaccination, and the vaccine has approximately 80% efficacy in preventing severe lower respiratory tract infection in her neonate. ●
- RSV is the most common cause of acute lower respiratory tract infection in infants younger than 6 months of age.
- In low- and middle-resource countries, RSV is a leading cause of infant death.
- In late August 2023, the FDA approved the first RSV vaccine that can be administered to pregnant women to provide protection for the infant in the first few months of life.
- The vaccine specifically targets the F protein of the virus, a protein which is essential for facilitating fusion between the viral and host cell membranes, resulting in penetration of the virus into the host cell.
- The vaccine should be administered as a single intramuscular injection at 32 to 36 weeks’ gestation.
- The vaccine is approximately 82% effective in preventing severe lower respiratory tract infection in infants within the first 6 months of life.
- To exercise an abundance of caution, because of a possible association between administration of the vaccine and an increased risk for preterm delivery, vaccination should be delayed until 36 weeks in patients clearly identified as at-risk for preterm delivery.
A 28-year-old primigravid woman at 30 weeks’ gestation inquires about the new vaccine to protect her newborn baby against respiratory syncytial virus infection (RSV). Her neighbor’s daughter recently was hospitalized for the treatment of RSV, and she is understandably concerned about her own newborn. The patient is healthy, and she has never had any serious respiratory infection. She is taking no medications other than prenatal vitamins.
What advice should you give her?
If you decide to administer this vaccine, what is the appropriate timing of administration?
Are there any maternal or fetal safety concerns related to use of this vaccine in pregnancy?
Respiratory syncytial virus (RSV) is a member of the Paramyxoviridae family. It is an enveloped, single-stranded RNA virus that is 150-300 nm in size. The virus codes for 10 virus-specific proteins. The 2 most important are the G protein, which enables the virus to attach to host cells, and the F protein, which facilitates the entry of the virus into the host cell by fusing the host and viral membranes. Two distinct subtypes exist: A and B. There is genetic variation within each subtype and between subtypes. These subtle genetic variations create the potential for reinfections, and hence, research has focused on development of a vaccine that covers both subtypes.1
RSV is the most common cause of acute lower respiratory tract infection in infants younger than 6 months of age. In these children, RSV is one of the most prominent causes of death, with mortality particularly marked in low- and middle-resource countries as well as in children who were born premature and/or who are immunocompromised. RSV has its greatest impact during winter epidemics in temperate climates and during the rainy seasons in tropical climates. The virus rarely is encountered in the summer.1 Among young children, RSV primarily is transmitted via close contact with contaminated fingers or fomites and by self-inoculation of the conjunctiva or anterior nares. The incubation period of the infection is 4 to 6 days, and viral shedding may persist for 2 weeks or longer. Most patients gradually recover within 1 to 2 weeks.1 Adults who contract RSV usually have symptoms suggestive of a common cold; however, in older adults or those who have comorbidities, serious and potentially life-threatening lower respiratory tract infections may develop.
Recently, there have been 2 main approaches to the prevention and treatment of RSV in infants. One has been the development of monoclonal antibodies such as motavizumab, palivizumab, and nirsevimab. The other has been the development of a vaccine that could be administered to pregnant women and which could provide protection for the neonate in the early months of life.2,3
In late August 2023, the US Food and Drug Administration (FDA) announced the approval of a new bivalent RSV prefusion F vaccine (ABRYSVO, Pfizer) intended for administration to pregnant women.4 Of note, previous efforts to develop whole-virus vaccines either have been ineffective or have potentiated the disease in infants who became infected; development of an effective vaccine had eluded scientists and clinicians for nearly 50 years.2 Thus, the new vaccine that targets the F protein of the virus represents a major and welcomed breakthrough.
This article reviews the 3 most recent investigations that preceded the ultimate approval of this vaccine and discusses specific logistical issues related to vaccine administration.
Continue to: First step toward vaccine approval...
First step toward vaccine approval
Madhi and colleagues5 were among the first to conduct a large well-designed study to evaluate the effectiveness of maternal vaccination in preventing neonatal infection in the first few months of life. The authors enrolled more than 4,500 healthy pregnant women at 28 to 36 weeks of gestation and assigned them to receive either a single intramuscular dose of an RSV fusion (F) protein vaccine or placebo in a ratio of 2:1. The primary end point was a “medically significant lower respiratory tract infection” within the first 90 days of life. The percentage of infants who met the primary end point was low in both groups: 1.5% in the vaccine group and 2.4% in the placebo group (efficacy 39.4%). The efficacy of the vaccine in preventing lower respiratory tract infection with severe hypoxemia was 48.3% and 44.4% in preventing hospitalization. Although there were differences between the 2 groups, they did not meet the prespecified success criterion for efficacy. Vaccine recipients had more local injection site reactions (40.7% vs 9.9%); however, there was no difference in the frequency of other adverse effects.
Intermediate step: Continued assessment of vaccine safety and immunogenicity
The next important step in the development of the RSV vaccine was a study by Simoes et al,6 who conducted a phase 2b trial to determine the safety and immunogenicity of the RSVpreF vaccine. The authors randomly assigned pregnant women at 24 to 36 weeks of gestation to receive either 120 or 240 µg of RSVpreF vaccine or placebo. The key endpoints were the following: maternal and infant safety; the maternal-to-infant transplacental transfer ratio; and the presence of RSV A, B, and combined A/B neutralizing antibody in maternal serum and umbilical cord blood at delivery. The authors conducted a planned interim analysis that included 327 mothers who received the vaccine. The incidence of adverse effects was similar in mothers and infants in the vaccine compared with the placebo group. None of the adverse effects were judged to be serious. The transplacental neutralizing antibody transfer ratios ranged from 1.4 to 2.1 across a range of gestational ages. The vaccine elicited meaningful neutralizing titers of antibody in maternal serum even up to 7 weeks after immunization. The levels of neutralizing antibodies in umbilical cord blood did not vary substantially with respect to gestational age. A post hoc analysis showed that the transferred antibodies prevented medically-attended RSV-associated lower respiratory tract illnesses in the infants.
Final step: Convincing proof of efficacy
The most recent of the 3 studies, and the one that had the greatest impact in convincing the FDA to approve the vaccine, was the report by Kampmann and colleagues.7 The authors conducted a phase 3 prospective, randomized, double-blind trial in 18 different countries over 4 RSV seasons: 2 in the northern hemisphere and 2 in the southern hemisphere. They enrolled healthy pregnant women with singleton gestations at 24 to 36 weeks of gestation and assigned them in a 1:1 ratio to a single intramuscular injection of 120 µg of a bivalent RSV prefusion F protein-based (RSVpreF) vaccine or placebo. They excluded patients with any recognized risk factor for an adverse pregnancy outcome, including preterm labor. The 2 primary efficacy endpoints were a medically-attended severe RSV–lower respiratory tract infection and any medically attended RSV-associated lower respiratory tract illness in infants within 90, 120, 150, and 180 days after birth.
The efficacy of the vaccine in preventing severe lower respiratory tract illness within 90 days of delivery was 81.8% (99.5% confidence interval [CI], 40.6–96.3). The efficacy within 180 days of delivery was 69.4% (97.58% CI, 44.3–84.1). These differences reached the study’s pre-established statistical criteria for success. The overall rate of lower respiratory tract infections was not significantly different. The frequencies of adverse effects in mothers and infants were similar in the vaccine and placebo groups. In particular, the frequency of preterm delivery in the vaccine group was 0.8%, compared with 0.6% in the placebo group (P = NS).
In previous reports to the FDA,4 the frequency rate of preterm delivery in RSV vaccine recipients was slightly increased in vaccine recipients compared with patients who received placebo. The difference among the groups was too small to infer a causal relationship; however, as a condition of vaccine approval, the FDA has required Pfizer to conduct a postmarketing study to be certain that administration of the vaccine does not increase the risk for preterm delivery.
Practical details
The new vaccine is a bivalent recombinant vaccine that elicits a robust antibody response against the F (fusion) protein of the virus. In addition to the F antigen, the vaccine contains the following buffer ingredients: tromethamine, sucrose, mannitol, polysorbate, and sodium chloride.8 There are no preservatives in the vaccine.
The vaccine should be administered in a single, 0.5 mL, intramuscular injection at 32 to 36 weeks of gestation. Patients who are allergic to any of the components of the vaccine should not be vaccinated. Patients with a mild upper respiratory tract infection may receive the vaccine. Administration should be delayed in patients who are moderately to severely ill. The vaccine may be administered at the same time as other vaccines, such as influenza or Tdap.
The most common side effects of the vaccine are local injection site reactions, such as pain, redness, or swelling. Some patients may experience mild systemic manifestations, including fatigue, fever, headache, nausea, diarrhea, arthralgias, and myalgias. According to the Centers for Disease Control and Prevention, the approximate wholesale acquisition cost of the vaccine is $320 for 1 injection.
CASE Resolution
This patient is healthy and has no contraindication to the new RSV vaccine. According to the FDA, the optimal time for administration of the vaccine is 32 to 36 weeks of gestation. The patient should anticipate very few side effects following the vaccination, and the vaccine has approximately 80% efficacy in preventing severe lower respiratory tract infection in her neonate. ●
- RSV is the most common cause of acute lower respiratory tract infection in infants younger than 6 months of age.
- In low- and middle-resource countries, RSV is a leading cause of infant death.
- In late August 2023, the FDA approved the first RSV vaccine that can be administered to pregnant women to provide protection for the infant in the first few months of life.
- The vaccine specifically targets the F protein of the virus, a protein which is essential for facilitating fusion between the viral and host cell membranes, resulting in penetration of the virus into the host cell.
- The vaccine should be administered as a single intramuscular injection at 32 to 36 weeks’ gestation.
- The vaccine is approximately 82% effective in preventing severe lower respiratory tract infection in infants within the first 6 months of life.
- To exercise an abundance of caution, because of a possible association between administration of the vaccine and an increased risk for preterm delivery, vaccination should be delayed until 36 weeks in patients clearly identified as at-risk for preterm delivery.
- Dolin R. Common viral respiratory infections. In, Isselbacher KJ, Braunwald E, Wilson JD, et al, eds. Harrison’s Principles of Internal Medicine. 13th ed. McGraw-Hill; 1994:805-806.
- Mazur N, Terstappen J, Baral R, et al. Respiratory syncytial virus prevention within reach: the vaccine and monoclonal antibody landscape. Lancet Infect Dis. 2023;23:E2-E21.
- Hammitt LL, Dagan R, Yuan Y, et al. Nirsevimab for prevention of RSV in healthy late-preterm and term infants. N Engl J Med. 2022;386:837-846.
- US Food and Drug Administration News Release. August 21, 2023. Accessed October 26, 2023. https://www.fda.gov/news -events/press-announcements/fda-approves-first-vaccine -pregnant-individuals-prevent-rsv-infants
- Madhi SA, Polack FP, Piedra PA, et al. Respiratory syncytial virus vaccination during pregnancy and effects in infants. N Engl J Med. 2020;383:426-439.
- Simoes EAF, Center KJ, Tita ATN, et al. Prefusion F proteinbased respiratory syncytial virus immunization in pregnancy. N Eng J Med. 2022;386:1615-1626.
- Kampmann B, Madhi SA, Munjal I, et al. Bivalent prefusion F vaccine in pregnancy to prevent RSV illness in infants. N Engl J Med. 2023;388:1451-1464.
- Centers for Disease Control and Prevention. Vaccine Information Statement. Respiratory Syncytial Virus (RSV) Vaccine VIS. October 19, 2023. Accessed October 26, 2023. https://www. cdc.gov/vaccines/hcp/vis/vis-statements/rsv.html
- Dolin R. Common viral respiratory infections. In, Isselbacher KJ, Braunwald E, Wilson JD, et al, eds. Harrison’s Principles of Internal Medicine. 13th ed. McGraw-Hill; 1994:805-806.
- Mazur N, Terstappen J, Baral R, et al. Respiratory syncytial virus prevention within reach: the vaccine and monoclonal antibody landscape. Lancet Infect Dis. 2023;23:E2-E21.
- Hammitt LL, Dagan R, Yuan Y, et al. Nirsevimab for prevention of RSV in healthy late-preterm and term infants. N Engl J Med. 2022;386:837-846.
- US Food and Drug Administration News Release. August 21, 2023. Accessed October 26, 2023. https://www.fda.gov/news -events/press-announcements/fda-approves-first-vaccine -pregnant-individuals-prevent-rsv-infants
- Madhi SA, Polack FP, Piedra PA, et al. Respiratory syncytial virus vaccination during pregnancy and effects in infants. N Engl J Med. 2020;383:426-439.
- Simoes EAF, Center KJ, Tita ATN, et al. Prefusion F proteinbased respiratory syncytial virus immunization in pregnancy. N Eng J Med. 2022;386:1615-1626.
- Kampmann B, Madhi SA, Munjal I, et al. Bivalent prefusion F vaccine in pregnancy to prevent RSV illness in infants. N Engl J Med. 2023;388:1451-1464.
- Centers for Disease Control and Prevention. Vaccine Information Statement. Respiratory Syncytial Virus (RSV) Vaccine VIS. October 19, 2023. Accessed October 26, 2023. https://www. cdc.gov/vaccines/hcp/vis/vis-statements/rsv.html
The challenges of managing CMV infection during pregnancy
CASE Anomalous findings on fetal anatomic survey
A 27-year-old previously healthy primigravid woman is at 18 weeks’ gestation. She is a first-grade schoolteacher. On her fetal anatomic survey, the estimated fetal weight was in the eighth percentile. Echogenic bowel and a small amount of ascitic fluid were noted in the fetal abdomen. The lateral and third ventricles were mildly dilated, the head circumference was 2 standard deviations below normal, and the placenta was slightly thickened and edematous.
What is the most likely diagnosis?
What diagnostic tests are indicated?
What management options are available for this patient?
Cytomegalovirus (CMV) is the most common of the perinatally transmitted infections, affecting 1% to 4% of all pregnancies. Although the virus typically causes either asymptomatic infection or only mild illness in immunocompetent individuals, it can cause life-threatening disease in immunocompromised persons and in the developing fetus. In this article, we review the virology and epidemiology of CMV infection and then focus on the key methods to diagnose infection in the mother and fetus. We conclude by considering measures that may be of at least modest value in treating CMV in pregnancy.
Virology of CMV infection
Cytomegalovirus is a double-stranded DNA virus in the Herpesviridae family. This ubiquitous virus is present in virtually all secretions and excretions of an infected host, including blood, urine, saliva, breast milk, genital secretions, and tissues and organs used for donation. Infection is transmitted through direct contact with any of the substances listed; contact with infected urine or saliva is the most common mode of transmission. Disease occurrence does not show seasonal variation.
After exposure, an incubation period of 28 to 60 days ensues, followed by development of viremia and clinical symptoms. In the majority of exposed individuals, CMV establishes a lifelong latent infection, and recurrent episodes of illness can occur as a result of reactivation of latent virus (also known as secondary infection) or, more rarely, infection with a new viral strain. In fact, most CMV illness episodes in pregnancy represent a reactivation of a previous infection rather than a new infection.
Following initial infection, both IgM (immunoglobulin M) and IgG (immunoglobulin G) antibodies develop rapidly and can be detected in blood within 1 to 2 weeks. IgM levels typically wane within 30 to 60 days, although persistence for several months is not unusual, and levels also can increase with viral reactivation (secondary infection). IgG antibodies typically persist for many years after a primary infection.
Intrauterine CMV infection occurs through hematogenous transplacental passage during maternal viremia. The risk of transmission and severity of fetal effects depend on whether or not the infection is primary or secondary in nature as well as the gestational age at fetal exposure.1,2
Additionally, postnatal vertical transmission can occur through exposure to viral particles in genital secretions as well as breast milk. CMV acquired in the postnatal period rarely produces severe sequelae in a healthy term neonate, but it has been associated with an increased rate of complications in very low birth weight and premature newborns.3
Continue to: Who is at risk...
Who is at risk
Congenital CMV, which occurs in 2.1 to 7.7 per 10,000 live births in the United States, is both the most common congenital infection and the leading cause of nonhereditary congenital hearing loss in children.4,5 The main reservoir of CMV in the United States is young children in day care settings, with approximately 50% of this population showing evidence of viral shedding in saliva.1 Adult populations in North America have a high prevalence of CMV IgG antibodies indicative of prior infection, with rates reaching 50% to 80%. Among seronegative individuals aged 12 to 49, the rate of seroconversion is approximately 1 in 60 annually.6 Significant racial disparities have been noted in rates of seroprevalence and seroconversion, with higher rates of infection in non-Hispanic Black and Mexican American individuals.6 Overall, the rate of new CMV infection among pregnant women in the United States is 0.7% to 4%.7
Clinical manifestations
Manifestations of infection differ depending on whether or not infection is primary or recurrent (secondary) and whether or not the host is immunocompetent or has a compromised immune system. Unique manifestations develop in the fetus.
CMV infection in children and adults. Among individuals with a normal immune response, the typical course of CMV is either no symptoms or a mononucleosis-like illness. In symptomatic patients, the most common symptoms include malaise, fever, and night sweats, and the most common associated laboratory abnormalities are elevation in liver function tests and a decreased white blood cell count, with a predominance of lymphocytes.8
Immunocompromised individuals are at risk for significant morbidity and mortality resulting from CMV. Illness may be the result of reactivation of latent infection due to decreased immune function or may be acquired as a result of treatment such as transplantation of CMV-positive organs or tissues, including bone marrow. Virtually any organ system can be affected, with potential for permanent organ damage and death. Severe systemic infection also can occur.
CMV infection in the fetus and neonate. As noted previously, fetal infection develops as a result of transplacental passage coincident with maternal infection. The risk of CMV transmission to the fetus and the severity of fetal injury vary based on gestational age at fetal infection and whether or not maternal infection is primary or secondary.
In most studies, primary maternal infections are associated with higher rates of fetal infection and more severe fetal and neonatal disease manifestations.2,7,9,10 Primary infections carry an overall 30% to 40% risk of transmission to the fetus.7,11 The risk of fetal transmission is much lower with a recurrent infection and is usually less than 2%.11 Due to their greater overall incidence, secondary infections account for the majority of cases of fetal and neonatal CMV disease.7 Importantly, although secondary infections generally have been regarded as having a lower risk and lower severity of fetal and neonatal disease, several recent studies have demonstrated rates of complications similar to, and even exceeding, those of primary infections.12-15 The TABLE provides a summary of the risks of fetal transmission and symptomatic fetal infection based on trimester of pregnancy.2,11,16-18
In the fetus, CMV may affect multiple organ systems. Among sonographic and magnetic resonance imaging (MRI) findings, central nervous system (CNS) anomalies are the most common.19,20 These can include microcephaly, ventriculomegaly, and periventricular calcifications. The gastrointestinal system also is frequently affected, and findings include echogenic bowel, hepatosplenomegaly, and liver calcifications. Lastly, isolated effusions, placentomegaly, fetal growth restriction, and even frank hydrops can develop. More favorable neurologic outcomes have been demonstrated in infants with no prenatal brain imaging abnormalities.20,21 However, the role of MRI in prenatal prognosis currently is not well defined.
FIGURE 1 illustrates selected sonographic findings associated with fetal CMV infection.
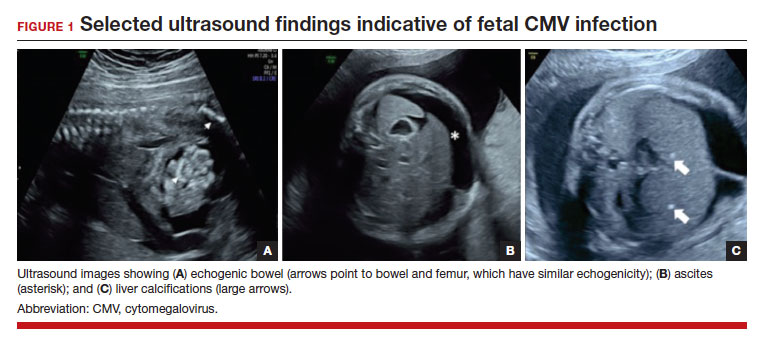
About 85% to 90% of infants with congenital CMV that results from primary maternal infection have no symptoms at birth. Among the 10% to 15% of infants that do have symptoms, petechial rash, jaundice, and hepatosplenomegaly are the most common manifestations (“blueberry muffin baby”). Approximately 10% to 20% of infants in this group have evidence of chorioretinitis on ophthalmologic examination, and 50% show either microcephaly or low birth weight.22Among survivors of symptomatic congenital CMV, more than 50% have long-term neurologic morbidities that may include sensorineural hearing loss, seizures, vision impairment, and developmental disabilities. Note that even when neonates appear asymptomatic at birth (regardless of whether infection is primary or secondary), 5% may develop microcephaly and motor deficits, 10% go on to develop sensorineural hearing loss, and the overall rate of neurologic morbidity reaches 13% to 15%.12,23 Some of the observed deficits manifest at several years of age, and, currently, no models exist for prediction of outcome.
Continue to: Diagnosing CMV infection...
Diagnosing CMV infection
Maternal infection
If maternal CMV infection is suspected based on a symptomatic illness or an abnormal fetal ultrasound exam, the first diagnostic test should be an assessment of IgM and IgG serology. If the former test results are positive and the latter negative, the diagnosis of acute CMV infection is confirmed. A positive serum CMV DNA polymerase chain reaction (PCR) test adds additional assurance that the diagnosis is correct. Primary infection, as noted above, poses the greatest risk of serious injury to the fetus.1
A frequent diagnostic dilemma arises when both the IgM and IgG antibody are positive. Remember that CMV IgM antibody can remain positive for 9 to 12 months after a primary infection and can reappear in the maternal serum in the face of a recurrent or reactivated infection. When confronted by both a positive IgM and positive IgG result, the clinician should then order IgG avidity testing. If the avidity is low to moderate, which reflects poor binding of antibody to the virus, the patient likely has an acute infection. If the avidity is high, which reflects enhanced binding of antibody to virus, the patient probably has a recurrent or reactivated infection; this scenario poses less danger to the developing fetus. The presence of CMV DNA in serum is also more consistent with acute infection, although viremia still can occur with recurrent infection. FIGURE 2 presents a suggested algorithm for the diagnosis of CMV in the pregnant patient.1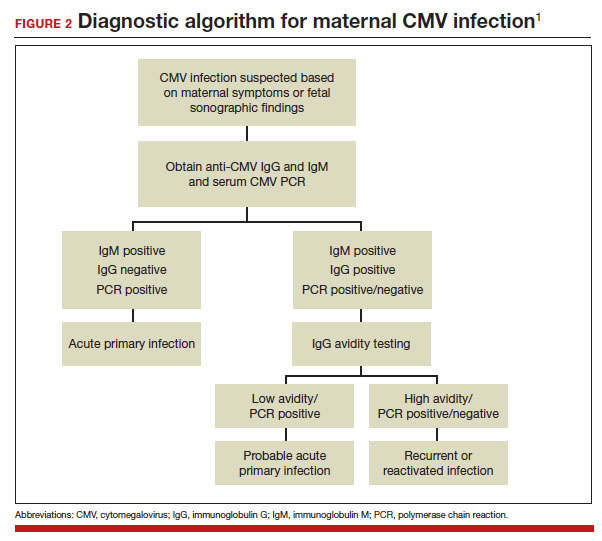
If a diagnosis of maternal CMV infection is confirmed, liver function tests should be obtained to determine if CMV hepatitis is present. If the liver function tests are abnormal, a coagulation profile also should be performed to identify the mother who might be at risk for peripartum hemorrhage.
Fetal infection
The single best test for confirmation of congenital CMV infection is detection of viral DNA and quantitation of viral load in the amniotic fluid by PCR. If the amniocentesis is performed prior to 20 weeks’ gestation and is negative, the test should be repeated in approximately 4 weeks.1,19,24
Detection of viral DNA indicates congenital infection. The ultimate task, however, is to determine if the infection has injured the fetus. Detailed ultrasound examination is the key to identifying fetal injury. As noted previously, the principal ultrasonographic findings that suggest congenital CMV infection include2,19,20,21,25:
- hydropic placenta
- fetal growth restriction
- microcephaly (head circumference more than 3 standard deviations below the mean)
- periventricular calcifications
- enlarged liver
- echogenic bowel
- ascites
- fetal hydrops.
Management: Evidence on CMV hyperimmune globulin, valacyclovir
If the immunocompetent mother has clinical manifestations of infection, she should receive symptomatic treatment. She should be encouraged to rest as much as possible, stay well hydrated, and use acetaminophen (1,000 mg every 6 to 8 hours) as needed for malaise and fever.
However, if the mother is immunocompromised and has signs of serious complications, such as chorioretinitis, hepatitis, or pneumonia, more aggressive therapy is indicated. Drugs used in this setting include foscarnet and ganciclovir and are best prescribed in consultation with a medical infectious disease specialist.
At this time, no consistently effective therapy for congenital infection is available. Therefore, if a patient has primary CMV infection in the first half of pregnancy, particularly in the first trimester, she should be counseled that the risk of fetal infection is approximately 40% and that approximately 5% to 15% of infants will be severely affected at birth. Given this information, some patients may opt for pregnancy termination.
In 2005, a report from Nigro and colleagues stimulated great hope that CMV-specific hyperimmune globulin (CytoGam) might be of value for both treatment and prophylaxis for congenital infection.26 These authors studied 157 women with confirmed primary CMV infection. One-hundred forty-eight women were asymptomatic and were identified by routine serologic screening, 8 had symptomatic infection, and 1 was identified because of abnormal fetal ultrasound findings. Forty-five women had CMV detected in amniotic fluid by PCR or culture more than 6 weeks before study enrollment. Thirty-one of these women were treated with intravenous hyperimmune globulin (200 U or 200 mg/kg maternal body weight); 14 declined treatment. Seven of the latter women had infants who were acutely symptomatic at the time of delivery; only 1 of the 31 treated women had an affected neonate (adjusted odds ratio [OR], 0.02; P<.001). In this same study, 84 women did not have a diagnostic amniocentesis because their infection occurred within 6 weeks of enrollment, their gestational age was less than 20 weeks, or they declined the procedure. Thirty-seven of these women received hyperimmune globulin (100 U or 100 mg/kg) every month until delivery, and 47 declined treatment. Six of the treated women delivered infected infants compared with 19 of the untreated women (adjusted OR, 0.32; P<.04).
Although these results were quite encouraging, several problems existed with the study’s design, as noted in an editorial that accompanied the study’s publication.27 First, the study was not randomized or placebo controlled. Second, patients were not stratified based on the severity of fetal ultrasound abnormalities. Third, the dosing of hyperimmune globulin varied; 9 of the 31 patients in the treatment group received additional infusions of drug into either the amniotic fluid or fetal umbilical vein. Moreover, patients in the prophylaxis group actually received a higher cumulative dose of hyperimmune globulin than patients in the treatment group.
Two subsequent investigations that were better designed were unable to verify the effectiveness of hyperimmune globulin. In 2014, Revello and colleagues reported the results of a prospective, randomized, placebo-controlled, double-blinded study of 124 women at 5 to 26 weeks’ gestation with confirmed primary CMV infection.28 The rate of congenital infection was 30% in the group treated with hyperimmune globulin and 44% in the placebo group (P=.13). There also was no significant difference in the concentration of serum CMV DNA in treated versus untreated mothers. Moreover, the number of adverse obstetric events (preterm delivery, fetal growth restriction, intrahepatic cholestasis of pregnancy, and postpartum preeclampsia) in the treatment group was higher than in the placebo group, 13% versus 2%.
In 2021, Hughes and colleagues published the results of a multicenter, double-blind trial in 399 women who had a diagnosis of primary CMV infection before 23 weeks’ gestation.29 The primary outcome was defined as a composite of congenital CMV infection or fetal/neonatal death. An adverse primary outcome occurred in 22.7% of the patients who received hyperimmune globulin and 19.4% of those who received placebo (relative risk, 1.17; 95% confidence interval [CI], 0.80–1.72; P=.42).
Continue to: Jacquemard and colleagues...
Jacquemard and colleagues then proposed a different approach.30 In a small pilot study of 20 patients, these authors used high doses of oral valacylovir (2 g 4 times daily) and documented therapeutic drug concentrations and a decline in CMV viral load in fetal serum. Patients were not stratified by severity of fetal injury at onset of treatment, so the authors were unable to define which fetuses were most likely to benefit from treatment.
In a follow-up investigation, Leruez-Ville and colleagues reported another small series in which high-dose oral valacyclovir (8 g daily) was used for treatment.31 They excluded fetuses with severe brain anomalies and fetuses with no sonographic evidence of injury. The median gestational age at diagnosis was 26 weeks. Thirty-four of 43 treated fetuses were free of injury at birth. In addition, the viral load in the neonate’s serum decreased significantly after treatment, and the platelet count increased. The authors then compared these outcomes to a historical cohort and confirmed that treatment increased the proportion of asymptomatic neonates from 43% without treatment to 82% with treatment (P<.05 with no overlapping confidence intervals).
We conclude from these investigations that hyperimmune globulin is unlikely to be of value in treating congenital CMV infection, especially if the fetus already has sonographic findings of severe injury. High-dose oral valacyclovir also is unlikely to be of value in severely affected fetuses, particularly those with evidence of CNS injury. However, antiviral therapy may be of modest value in situations when the fetus is less severely injured.
Preventive measures
Since no definitive treatment is available for congenital CMV infection, our efforts as clinicians should focus on measures that may prevent transmission of infection to the pregnant patient. These measures include:
- Encouraging patients to use careful handwashing techniques when handling infant diapers and toys.
- Encouraging patients to adopt safe sexual practices if not already engaged in a mutually faithful, monogamous relationship.
- Using CMV-negative blood when transfusing a pregnant woman or a fetus.
At the present time, unfortunately, a readily available and highly effective therapy for prevention of CMV infection is not available.
CASE Congenital infection diagnosed
The ultrasound findings are most consistent with congenital CMV infection, especially given the patient’s work as an elementary schoolteacher. The diagnosis of maternal infection is best established by conventional serology (positive IgM, negative IgM) and detection of viral DNA in maternal blood by PCR testing. The diagnosis of congenital infection is best confirmed by documentation of viral DNA in the amniotic fluid by PCR testing. Given that this fetus already has evidence of moderate to severe injury, no treatment is likely to be effective in reversing the abnormal ultrasound findings. Pregnancy termination may be an option, depending upon the patient’s desires and the legal restrictions prevalent in the patient’s geographic area. ●
- Cytomegalovirus infection is the most common of the perinatally transmitted infections.
- Maternal infection is often asymptomatic. When symptoms are present, they resemble those of an influenza-like illness. In immunocompromised persons, however, CMV may cause serious complications, including pneumonia, hepatitis, and chorioretinitis.
- The virus is transmitted by contact with contaminated body fluids, such as saliva, urine, blood, and genital secretions.
- The greatest risk of severe fetal injury results from primary maternal infection in the first trimester of pregnancy.
- Manifestations of severe congenital CMV infection include growth restriction, microcephaly, ventriculomegaly, hepatosplenomegaly, ascites, chorioretinitis, thrombocytopenia, purpura, and hydrops (“blueberry muffin baby”).
- Late manifestations of infection, which usually follow recurrent maternal infection, may appear as a child enters elementary school and include visual and auditory deficits, developmental delays, and learning disabilities.
- The diagnosis of maternal infection is confirmed by serology and detection of viral DNA in the serum by PCR testing.
- The diagnosis of fetal infection is best made by a combination of abnormal ultrasound findings and detection of CMV DNA in amniotic fluid. The characteristic ultrasound findings include placentomegaly, microcephaly, ventriculomegaly, growth restriction, echogenic bowel, and serous effusions/hydrops.
- Treatment of the mother with antiviral medications such as valacyclovir may be of modest value in reducing placental edema, decreasing viral load in the fetus, and hastening the resolution of some ultrasound findings, such as echogenic bowel.
- While initial studies seemed promising, the use of hyperimmune globulin has not proven to be consistently effective in treating congenital infection.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik’s Maternal Fetal Medicine: Principles and Practice. 8th ed. 2019:888-890.
- Chatzakis C, Ville Y, Makrydimas G, et al. Timing of primary maternal cytomegalovirus infection and rates of vertical transmission and fetal consequences. Am J Obstet Gynecol. 2020;223:870-883.e11. doi:10.1016/j.ajog.2020.05.038
- Kelly MS, Benjamin DK, Puopolo KM, et al. Postnatal cytomegalovirus infection and the risk for bronchopulmonary dysplasia. JAMA Pediatr. 2015;169:e153785. doi:10.1001 /jamapediatrics.2015.3785
- Messinger CJ, Lipsitch M, Bateman BT, et al. Association between congenital cytomegalovirus and the prevalence at birth of microcephaly in the United States. JAMA Pediatr. 2020;174:1159-1167. doi:10.1001/jamapediatrics.2020.3009
- De Cuyper E, Acke F, Keymeulen A, et al. Risk factors for hearing loss at birth in newborns with congenital cytomegalovirus infection. JAMA Otolaryngol Head Neck Surg. 2023;149:122-130. doi:10.1001/jamaoto.2022.4109
- Colugnati FA, Staras SA, Dollard SC, et al. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis. 2007;7:71. doi:10.1186/1471-2334-7-71
- Stagno S, Pass RF, Cloud G, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA. 1986;256:1904-1908.
- Wreghitt TG, Teare EL, Sule O, et al. Cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 2003;37:1603-1606. doi:10.1086/379711
- Fowler KB, Stagno S, Pass RF, et al. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326:663-667. doi:10.1056 /NEJM199203053261003
- Faure-Bardon V, Magny JF, Parodi M, et al. Sequelae of congenital cytomegalovirus following maternal primary infections are limited to those acquired in the first trimester of pregnancy. Clin Infect Dis. 2019;69:1526-1532. doi:10.1093/ cid/ciy1128
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253-276. doi:10.1002/ rmv.535
- Boppana SB, Pass RF, Britt WJ, et al. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J. 1992;11:93-99. doi:10.1097/00006454-199202000-00007
- Ross SA, Fowler KB, Ashrith G, et al. Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J Pediatr. 2006;148:332-336. doi:10.1016/j.jpeds.2005.09.003
- Zalel Y, Gilboa Y, Berkenshtat M, et al. Secondary cytomegalovirus infection can cause severe fetal sequelae despite maternal preconceptional immunity. Ultrasound Obstet Gynecol. 31:417-420. doi:10.1002/uog.5255
- Scaramuzzino F, Di Pastena M, Chiurchiu S, et al. Secondary cytomegalovirus infections: how much do we still not know? Comparison of children with symptomatic congenital cytomegalovirus born to mothers with primary and secondary infection. Front Pediatr. 2022;10:885926. doi:10.3389/fped.2022.885926
- Gindes L, Teperberg-Oikawa M, Sherman D, et al. Congenital cytomegalovirus infection following primary maternal infection in the third trimester. BJOG. 2008;115:830-835. doi:10.1111/j.1471-0528.2007.01651.x
- Hadar E, Dorfman E, Bardin R, et al. Symptomatic congenital cytomegalovirus disease following non-primary maternal infection: a retrospective cohort study. BMC Infect Dis. 2017;17:31. doi:10.1186/s12879-016-2161-3
- Elkan Miller T, Weisz B, Yinon Y, et al. Congenital cytomegalovirus infection following second and third trimester maternal infection is associated with mild childhood adverse outcome not predicted by prenatal imaging. J Pediatric Infect Dis Soc. 2021;10:562-568. doi:10.1093/jpids/ piaa154
- Lipitz S, Yinon Y, Malinger G, et al. Risk of cytomegalovirusassociated sequelae in relation to time of infection and findings on prenatal imaging. Ultrasound Obstet Gynecol. 2013;41:508-514. doi:10.1002/uog.12377
- Lipitz S, Elkan Miller T, Yinon Y, et al. Revisiting short- and long-term outcome after fetal first-trimester primary cytomegalovirus infection in relation to prenatal imaging findings. Ultrasound Obstet Gynecol. 2020;56:572-578. doi:10.1002/uog.21946
- Buca D, Di Mascio D, Rizzo G, et al. Outcome of fetuses with congenital cytomegalovirus infection and normal ultrasound at diagnosis: systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2021;57:551-559. doi:10.1002/uog.23143
- Boppana SB, Ross SA, Fowler KB. Congenital cytomegalovirus infection: clinical outcome. Clin Infect Dis. 2013;57 (suppl 4):S178-S181. doi:10.1093/cid/cit629
- Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17:355-363. doi:10.1002/rmv.544
- Hughes BL, Gyamfi-Bannerman C. Diagnosis and antenatal management of congenital cytomegalovirus infection. Am J Obstet Gynecol. 2016;214:B5-11. doi:10.1016 /j.ajog.2016.02.042
- Rouse DJ, Fette LM, Hughes BL, et al. Noninvasive prediction of congenital cytomegalovirus infection after maternal primary infection. Obstet Gynecol. 2022;139:400-406. doi:10.1097/AOG.0000000000004691
- Nigro G, Adler SP, La Torre R, et al; Congenital Cytomegalovirus Collaborating Group. Passive immunization during pregnancy for congenital cytomegalovirus infection. N Engl J Med. 2005;353:1350-1362. doi:10.1056/NEJMoa043337
- Duff P. Immunotherapy for congenital cytomegalovirus infection. N Engl J Med. 2005;355:1402-1404. doi:10.1056 /NEJMe058172
- Revello MG, Lazzarotto T, Guerra B, et al. A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N Engl J Med. 2014;370:1316-1326. doi:10.1056/NEJMoa1310214
- Hughes BL, Clifton RG, Rouse DJ, et al. A trial of hyperimmune globulin to prevent congenital cytomegalovirus infection. N Engl J Med. 2021;385:436-444. doi:10.1056/NEJMoa1913569
- Jacquemard F, Yamamoto M, Costa JM, et al. Maternal administration of valaciclovir in symptomatic intrauterine cytomegalovirus infection. BJOG. 2007;114:1113-1121. doi:10.1111/j.1471-0528.2007.01308.x
- Leruez-Ville M, Ghout I, Bussières L, et al. In utero treatment of congenital cytomegalovirus infection with valacyclovir in a multicenter, open-label, phase II study. Am J Obstet Gynecol. 2016;215:462.e1-462.e10. doi:10.1016/j.ajog.2016.04.003

Dr. Berwick is a first-year Maternal-Fetal Medicine Fellow, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.

Dr. Duff is Professor, Division of MaternalFetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Dr. Berwick is a first-year Maternal-Fetal Medicine Fellow, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor, Division of MaternalFetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Dr. Berwick is a first-year Maternal-Fetal Medicine Fellow, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor, Division of MaternalFetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
CASE Anomalous findings on fetal anatomic survey
A 27-year-old previously healthy primigravid woman is at 18 weeks’ gestation. She is a first-grade schoolteacher. On her fetal anatomic survey, the estimated fetal weight was in the eighth percentile. Echogenic bowel and a small amount of ascitic fluid were noted in the fetal abdomen. The lateral and third ventricles were mildly dilated, the head circumference was 2 standard deviations below normal, and the placenta was slightly thickened and edematous.
What is the most likely diagnosis?
What diagnostic tests are indicated?
What management options are available for this patient?
Cytomegalovirus (CMV) is the most common of the perinatally transmitted infections, affecting 1% to 4% of all pregnancies. Although the virus typically causes either asymptomatic infection or only mild illness in immunocompetent individuals, it can cause life-threatening disease in immunocompromised persons and in the developing fetus. In this article, we review the virology and epidemiology of CMV infection and then focus on the key methods to diagnose infection in the mother and fetus. We conclude by considering measures that may be of at least modest value in treating CMV in pregnancy.
Virology of CMV infection
Cytomegalovirus is a double-stranded DNA virus in the Herpesviridae family. This ubiquitous virus is present in virtually all secretions and excretions of an infected host, including blood, urine, saliva, breast milk, genital secretions, and tissues and organs used for donation. Infection is transmitted through direct contact with any of the substances listed; contact with infected urine or saliva is the most common mode of transmission. Disease occurrence does not show seasonal variation.
After exposure, an incubation period of 28 to 60 days ensues, followed by development of viremia and clinical symptoms. In the majority of exposed individuals, CMV establishes a lifelong latent infection, and recurrent episodes of illness can occur as a result of reactivation of latent virus (also known as secondary infection) or, more rarely, infection with a new viral strain. In fact, most CMV illness episodes in pregnancy represent a reactivation of a previous infection rather than a new infection.
Following initial infection, both IgM (immunoglobulin M) and IgG (immunoglobulin G) antibodies develop rapidly and can be detected in blood within 1 to 2 weeks. IgM levels typically wane within 30 to 60 days, although persistence for several months is not unusual, and levels also can increase with viral reactivation (secondary infection). IgG antibodies typically persist for many years after a primary infection.
Intrauterine CMV infection occurs through hematogenous transplacental passage during maternal viremia. The risk of transmission and severity of fetal effects depend on whether or not the infection is primary or secondary in nature as well as the gestational age at fetal exposure.1,2
Additionally, postnatal vertical transmission can occur through exposure to viral particles in genital secretions as well as breast milk. CMV acquired in the postnatal period rarely produces severe sequelae in a healthy term neonate, but it has been associated with an increased rate of complications in very low birth weight and premature newborns.3
Continue to: Who is at risk...
Who is at risk
Congenital CMV, which occurs in 2.1 to 7.7 per 10,000 live births in the United States, is both the most common congenital infection and the leading cause of nonhereditary congenital hearing loss in children.4,5 The main reservoir of CMV in the United States is young children in day care settings, with approximately 50% of this population showing evidence of viral shedding in saliva.1 Adult populations in North America have a high prevalence of CMV IgG antibodies indicative of prior infection, with rates reaching 50% to 80%. Among seronegative individuals aged 12 to 49, the rate of seroconversion is approximately 1 in 60 annually.6 Significant racial disparities have been noted in rates of seroprevalence and seroconversion, with higher rates of infection in non-Hispanic Black and Mexican American individuals.6 Overall, the rate of new CMV infection among pregnant women in the United States is 0.7% to 4%.7
Clinical manifestations
Manifestations of infection differ depending on whether or not infection is primary or recurrent (secondary) and whether or not the host is immunocompetent or has a compromised immune system. Unique manifestations develop in the fetus.
CMV infection in children and adults. Among individuals with a normal immune response, the typical course of CMV is either no symptoms or a mononucleosis-like illness. In symptomatic patients, the most common symptoms include malaise, fever, and night sweats, and the most common associated laboratory abnormalities are elevation in liver function tests and a decreased white blood cell count, with a predominance of lymphocytes.8
Immunocompromised individuals are at risk for significant morbidity and mortality resulting from CMV. Illness may be the result of reactivation of latent infection due to decreased immune function or may be acquired as a result of treatment such as transplantation of CMV-positive organs or tissues, including bone marrow. Virtually any organ system can be affected, with potential for permanent organ damage and death. Severe systemic infection also can occur.
CMV infection in the fetus and neonate. As noted previously, fetal infection develops as a result of transplacental passage coincident with maternal infection. The risk of CMV transmission to the fetus and the severity of fetal injury vary based on gestational age at fetal infection and whether or not maternal infection is primary or secondary.
In most studies, primary maternal infections are associated with higher rates of fetal infection and more severe fetal and neonatal disease manifestations.2,7,9,10 Primary infections carry an overall 30% to 40% risk of transmission to the fetus.7,11 The risk of fetal transmission is much lower with a recurrent infection and is usually less than 2%.11 Due to their greater overall incidence, secondary infections account for the majority of cases of fetal and neonatal CMV disease.7 Importantly, although secondary infections generally have been regarded as having a lower risk and lower severity of fetal and neonatal disease, several recent studies have demonstrated rates of complications similar to, and even exceeding, those of primary infections.12-15 The TABLE provides a summary of the risks of fetal transmission and symptomatic fetal infection based on trimester of pregnancy.2,11,16-18
In the fetus, CMV may affect multiple organ systems. Among sonographic and magnetic resonance imaging (MRI) findings, central nervous system (CNS) anomalies are the most common.19,20 These can include microcephaly, ventriculomegaly, and periventricular calcifications. The gastrointestinal system also is frequently affected, and findings include echogenic bowel, hepatosplenomegaly, and liver calcifications. Lastly, isolated effusions, placentomegaly, fetal growth restriction, and even frank hydrops can develop. More favorable neurologic outcomes have been demonstrated in infants with no prenatal brain imaging abnormalities.20,21 However, the role of MRI in prenatal prognosis currently is not well defined.
FIGURE 1 illustrates selected sonographic findings associated with fetal CMV infection.
About 85% to 90% of infants with congenital CMV that results from primary maternal infection have no symptoms at birth. Among the 10% to 15% of infants that do have symptoms, petechial rash, jaundice, and hepatosplenomegaly are the most common manifestations (“blueberry muffin baby”). Approximately 10% to 20% of infants in this group have evidence of chorioretinitis on ophthalmologic examination, and 50% show either microcephaly or low birth weight.22Among survivors of symptomatic congenital CMV, more than 50% have long-term neurologic morbidities that may include sensorineural hearing loss, seizures, vision impairment, and developmental disabilities. Note that even when neonates appear asymptomatic at birth (regardless of whether infection is primary or secondary), 5% may develop microcephaly and motor deficits, 10% go on to develop sensorineural hearing loss, and the overall rate of neurologic morbidity reaches 13% to 15%.12,23 Some of the observed deficits manifest at several years of age, and, currently, no models exist for prediction of outcome.
Continue to: Diagnosing CMV infection...
Diagnosing CMV infection
Maternal infection
If maternal CMV infection is suspected based on a symptomatic illness or an abnormal fetal ultrasound exam, the first diagnostic test should be an assessment of IgM and IgG serology. If the former test results are positive and the latter negative, the diagnosis of acute CMV infection is confirmed. A positive serum CMV DNA polymerase chain reaction (PCR) test adds additional assurance that the diagnosis is correct. Primary infection, as noted above, poses the greatest risk of serious injury to the fetus.1
A frequent diagnostic dilemma arises when both the IgM and IgG antibody are positive. Remember that CMV IgM antibody can remain positive for 9 to 12 months after a primary infection and can reappear in the maternal serum in the face of a recurrent or reactivated infection. When confronted by both a positive IgM and positive IgG result, the clinician should then order IgG avidity testing. If the avidity is low to moderate, which reflects poor binding of antibody to the virus, the patient likely has an acute infection. If the avidity is high, which reflects enhanced binding of antibody to virus, the patient probably has a recurrent or reactivated infection; this scenario poses less danger to the developing fetus. The presence of CMV DNA in serum is also more consistent with acute infection, although viremia still can occur with recurrent infection. FIGURE 2 presents a suggested algorithm for the diagnosis of CMV in the pregnant patient.1
If a diagnosis of maternal CMV infection is confirmed, liver function tests should be obtained to determine if CMV hepatitis is present. If the liver function tests are abnormal, a coagulation profile also should be performed to identify the mother who might be at risk for peripartum hemorrhage.
Fetal infection
The single best test for confirmation of congenital CMV infection is detection of viral DNA and quantitation of viral load in the amniotic fluid by PCR. If the amniocentesis is performed prior to 20 weeks’ gestation and is negative, the test should be repeated in approximately 4 weeks.1,19,24
Detection of viral DNA indicates congenital infection. The ultimate task, however, is to determine if the infection has injured the fetus. Detailed ultrasound examination is the key to identifying fetal injury. As noted previously, the principal ultrasonographic findings that suggest congenital CMV infection include2,19,20,21,25:
- hydropic placenta
- fetal growth restriction
- microcephaly (head circumference more than 3 standard deviations below the mean)
- periventricular calcifications
- enlarged liver
- echogenic bowel
- ascites
- fetal hydrops.
Management: Evidence on CMV hyperimmune globulin, valacyclovir
If the immunocompetent mother has clinical manifestations of infection, she should receive symptomatic treatment. She should be encouraged to rest as much as possible, stay well hydrated, and use acetaminophen (1,000 mg every 6 to 8 hours) as needed for malaise and fever.
However, if the mother is immunocompromised and has signs of serious complications, such as chorioretinitis, hepatitis, or pneumonia, more aggressive therapy is indicated. Drugs used in this setting include foscarnet and ganciclovir and are best prescribed in consultation with a medical infectious disease specialist.
At this time, no consistently effective therapy for congenital infection is available. Therefore, if a patient has primary CMV infection in the first half of pregnancy, particularly in the first trimester, she should be counseled that the risk of fetal infection is approximately 40% and that approximately 5% to 15% of infants will be severely affected at birth. Given this information, some patients may opt for pregnancy termination.
In 2005, a report from Nigro and colleagues stimulated great hope that CMV-specific hyperimmune globulin (CytoGam) might be of value for both treatment and prophylaxis for congenital infection.26 These authors studied 157 women with confirmed primary CMV infection. One-hundred forty-eight women were asymptomatic and were identified by routine serologic screening, 8 had symptomatic infection, and 1 was identified because of abnormal fetal ultrasound findings. Forty-five women had CMV detected in amniotic fluid by PCR or culture more than 6 weeks before study enrollment. Thirty-one of these women were treated with intravenous hyperimmune globulin (200 U or 200 mg/kg maternal body weight); 14 declined treatment. Seven of the latter women had infants who were acutely symptomatic at the time of delivery; only 1 of the 31 treated women had an affected neonate (adjusted odds ratio [OR], 0.02; P<.001). In this same study, 84 women did not have a diagnostic amniocentesis because their infection occurred within 6 weeks of enrollment, their gestational age was less than 20 weeks, or they declined the procedure. Thirty-seven of these women received hyperimmune globulin (100 U or 100 mg/kg) every month until delivery, and 47 declined treatment. Six of the treated women delivered infected infants compared with 19 of the untreated women (adjusted OR, 0.32; P<.04).
Although these results were quite encouraging, several problems existed with the study’s design, as noted in an editorial that accompanied the study’s publication.27 First, the study was not randomized or placebo controlled. Second, patients were not stratified based on the severity of fetal ultrasound abnormalities. Third, the dosing of hyperimmune globulin varied; 9 of the 31 patients in the treatment group received additional infusions of drug into either the amniotic fluid or fetal umbilical vein. Moreover, patients in the prophylaxis group actually received a higher cumulative dose of hyperimmune globulin than patients in the treatment group.
Two subsequent investigations that were better designed were unable to verify the effectiveness of hyperimmune globulin. In 2014, Revello and colleagues reported the results of a prospective, randomized, placebo-controlled, double-blinded study of 124 women at 5 to 26 weeks’ gestation with confirmed primary CMV infection.28 The rate of congenital infection was 30% in the group treated with hyperimmune globulin and 44% in the placebo group (P=.13). There also was no significant difference in the concentration of serum CMV DNA in treated versus untreated mothers. Moreover, the number of adverse obstetric events (preterm delivery, fetal growth restriction, intrahepatic cholestasis of pregnancy, and postpartum preeclampsia) in the treatment group was higher than in the placebo group, 13% versus 2%.
In 2021, Hughes and colleagues published the results of a multicenter, double-blind trial in 399 women who had a diagnosis of primary CMV infection before 23 weeks’ gestation.29 The primary outcome was defined as a composite of congenital CMV infection or fetal/neonatal death. An adverse primary outcome occurred in 22.7% of the patients who received hyperimmune globulin and 19.4% of those who received placebo (relative risk, 1.17; 95% confidence interval [CI], 0.80–1.72; P=.42).
Continue to: Jacquemard and colleagues...
Jacquemard and colleagues then proposed a different approach.30 In a small pilot study of 20 patients, these authors used high doses of oral valacylovir (2 g 4 times daily) and documented therapeutic drug concentrations and a decline in CMV viral load in fetal serum. Patients were not stratified by severity of fetal injury at onset of treatment, so the authors were unable to define which fetuses were most likely to benefit from treatment.
In a follow-up investigation, Leruez-Ville and colleagues reported another small series in which high-dose oral valacyclovir (8 g daily) was used for treatment.31 They excluded fetuses with severe brain anomalies and fetuses with no sonographic evidence of injury. The median gestational age at diagnosis was 26 weeks. Thirty-four of 43 treated fetuses were free of injury at birth. In addition, the viral load in the neonate’s serum decreased significantly after treatment, and the platelet count increased. The authors then compared these outcomes to a historical cohort and confirmed that treatment increased the proportion of asymptomatic neonates from 43% without treatment to 82% with treatment (P<.05 with no overlapping confidence intervals).
We conclude from these investigations that hyperimmune globulin is unlikely to be of value in treating congenital CMV infection, especially if the fetus already has sonographic findings of severe injury. High-dose oral valacyclovir also is unlikely to be of value in severely affected fetuses, particularly those with evidence of CNS injury. However, antiviral therapy may be of modest value in situations when the fetus is less severely injured.
Preventive measures
Since no definitive treatment is available for congenital CMV infection, our efforts as clinicians should focus on measures that may prevent transmission of infection to the pregnant patient. These measures include:
- Encouraging patients to use careful handwashing techniques when handling infant diapers and toys.
- Encouraging patients to adopt safe sexual practices if not already engaged in a mutually faithful, monogamous relationship.
- Using CMV-negative blood when transfusing a pregnant woman or a fetus.
At the present time, unfortunately, a readily available and highly effective therapy for prevention of CMV infection is not available.
CASE Congenital infection diagnosed
The ultrasound findings are most consistent with congenital CMV infection, especially given the patient’s work as an elementary schoolteacher. The diagnosis of maternal infection is best established by conventional serology (positive IgM, negative IgM) and detection of viral DNA in maternal blood by PCR testing. The diagnosis of congenital infection is best confirmed by documentation of viral DNA in the amniotic fluid by PCR testing. Given that this fetus already has evidence of moderate to severe injury, no treatment is likely to be effective in reversing the abnormal ultrasound findings. Pregnancy termination may be an option, depending upon the patient’s desires and the legal restrictions prevalent in the patient’s geographic area. ●
- Cytomegalovirus infection is the most common of the perinatally transmitted infections.
- Maternal infection is often asymptomatic. When symptoms are present, they resemble those of an influenza-like illness. In immunocompromised persons, however, CMV may cause serious complications, including pneumonia, hepatitis, and chorioretinitis.
- The virus is transmitted by contact with contaminated body fluids, such as saliva, urine, blood, and genital secretions.
- The greatest risk of severe fetal injury results from primary maternal infection in the first trimester of pregnancy.
- Manifestations of severe congenital CMV infection include growth restriction, microcephaly, ventriculomegaly, hepatosplenomegaly, ascites, chorioretinitis, thrombocytopenia, purpura, and hydrops (“blueberry muffin baby”).
- Late manifestations of infection, which usually follow recurrent maternal infection, may appear as a child enters elementary school and include visual and auditory deficits, developmental delays, and learning disabilities.
- The diagnosis of maternal infection is confirmed by serology and detection of viral DNA in the serum by PCR testing.
- The diagnosis of fetal infection is best made by a combination of abnormal ultrasound findings and detection of CMV DNA in amniotic fluid. The characteristic ultrasound findings include placentomegaly, microcephaly, ventriculomegaly, growth restriction, echogenic bowel, and serous effusions/hydrops.
- Treatment of the mother with antiviral medications such as valacyclovir may be of modest value in reducing placental edema, decreasing viral load in the fetus, and hastening the resolution of some ultrasound findings, such as echogenic bowel.
- While initial studies seemed promising, the use of hyperimmune globulin has not proven to be consistently effective in treating congenital infection.
CASE Anomalous findings on fetal anatomic survey
A 27-year-old previously healthy primigravid woman is at 18 weeks’ gestation. She is a first-grade schoolteacher. On her fetal anatomic survey, the estimated fetal weight was in the eighth percentile. Echogenic bowel and a small amount of ascitic fluid were noted in the fetal abdomen. The lateral and third ventricles were mildly dilated, the head circumference was 2 standard deviations below normal, and the placenta was slightly thickened and edematous.
What is the most likely diagnosis?
What diagnostic tests are indicated?
What management options are available for this patient?
Cytomegalovirus (CMV) is the most common of the perinatally transmitted infections, affecting 1% to 4% of all pregnancies. Although the virus typically causes either asymptomatic infection or only mild illness in immunocompetent individuals, it can cause life-threatening disease in immunocompromised persons and in the developing fetus. In this article, we review the virology and epidemiology of CMV infection and then focus on the key methods to diagnose infection in the mother and fetus. We conclude by considering measures that may be of at least modest value in treating CMV in pregnancy.
Virology of CMV infection
Cytomegalovirus is a double-stranded DNA virus in the Herpesviridae family. This ubiquitous virus is present in virtually all secretions and excretions of an infected host, including blood, urine, saliva, breast milk, genital secretions, and tissues and organs used for donation. Infection is transmitted through direct contact with any of the substances listed; contact with infected urine or saliva is the most common mode of transmission. Disease occurrence does not show seasonal variation.
After exposure, an incubation period of 28 to 60 days ensues, followed by development of viremia and clinical symptoms. In the majority of exposed individuals, CMV establishes a lifelong latent infection, and recurrent episodes of illness can occur as a result of reactivation of latent virus (also known as secondary infection) or, more rarely, infection with a new viral strain. In fact, most CMV illness episodes in pregnancy represent a reactivation of a previous infection rather than a new infection.
Following initial infection, both IgM (immunoglobulin M) and IgG (immunoglobulin G) antibodies develop rapidly and can be detected in blood within 1 to 2 weeks. IgM levels typically wane within 30 to 60 days, although persistence for several months is not unusual, and levels also can increase with viral reactivation (secondary infection). IgG antibodies typically persist for many years after a primary infection.
Intrauterine CMV infection occurs through hematogenous transplacental passage during maternal viremia. The risk of transmission and severity of fetal effects depend on whether or not the infection is primary or secondary in nature as well as the gestational age at fetal exposure.1,2
Additionally, postnatal vertical transmission can occur through exposure to viral particles in genital secretions as well as breast milk. CMV acquired in the postnatal period rarely produces severe sequelae in a healthy term neonate, but it has been associated with an increased rate of complications in very low birth weight and premature newborns.3
Continue to: Who is at risk...
Who is at risk
Congenital CMV, which occurs in 2.1 to 7.7 per 10,000 live births in the United States, is both the most common congenital infection and the leading cause of nonhereditary congenital hearing loss in children.4,5 The main reservoir of CMV in the United States is young children in day care settings, with approximately 50% of this population showing evidence of viral shedding in saliva.1 Adult populations in North America have a high prevalence of CMV IgG antibodies indicative of prior infection, with rates reaching 50% to 80%. Among seronegative individuals aged 12 to 49, the rate of seroconversion is approximately 1 in 60 annually.6 Significant racial disparities have been noted in rates of seroprevalence and seroconversion, with higher rates of infection in non-Hispanic Black and Mexican American individuals.6 Overall, the rate of new CMV infection among pregnant women in the United States is 0.7% to 4%.7
Clinical manifestations
Manifestations of infection differ depending on whether or not infection is primary or recurrent (secondary) and whether or not the host is immunocompetent or has a compromised immune system. Unique manifestations develop in the fetus.
CMV infection in children and adults. Among individuals with a normal immune response, the typical course of CMV is either no symptoms or a mononucleosis-like illness. In symptomatic patients, the most common symptoms include malaise, fever, and night sweats, and the most common associated laboratory abnormalities are elevation in liver function tests and a decreased white blood cell count, with a predominance of lymphocytes.8
Immunocompromised individuals are at risk for significant morbidity and mortality resulting from CMV. Illness may be the result of reactivation of latent infection due to decreased immune function or may be acquired as a result of treatment such as transplantation of CMV-positive organs or tissues, including bone marrow. Virtually any organ system can be affected, with potential for permanent organ damage and death. Severe systemic infection also can occur.
CMV infection in the fetus and neonate. As noted previously, fetal infection develops as a result of transplacental passage coincident with maternal infection. The risk of CMV transmission to the fetus and the severity of fetal injury vary based on gestational age at fetal infection and whether or not maternal infection is primary or secondary.
In most studies, primary maternal infections are associated with higher rates of fetal infection and more severe fetal and neonatal disease manifestations.2,7,9,10 Primary infections carry an overall 30% to 40% risk of transmission to the fetus.7,11 The risk of fetal transmission is much lower with a recurrent infection and is usually less than 2%.11 Due to their greater overall incidence, secondary infections account for the majority of cases of fetal and neonatal CMV disease.7 Importantly, although secondary infections generally have been regarded as having a lower risk and lower severity of fetal and neonatal disease, several recent studies have demonstrated rates of complications similar to, and even exceeding, those of primary infections.12-15 The TABLE provides a summary of the risks of fetal transmission and symptomatic fetal infection based on trimester of pregnancy.2,11,16-18
In the fetus, CMV may affect multiple organ systems. Among sonographic and magnetic resonance imaging (MRI) findings, central nervous system (CNS) anomalies are the most common.19,20 These can include microcephaly, ventriculomegaly, and periventricular calcifications. The gastrointestinal system also is frequently affected, and findings include echogenic bowel, hepatosplenomegaly, and liver calcifications. Lastly, isolated effusions, placentomegaly, fetal growth restriction, and even frank hydrops can develop. More favorable neurologic outcomes have been demonstrated in infants with no prenatal brain imaging abnormalities.20,21 However, the role of MRI in prenatal prognosis currently is not well defined.
FIGURE 1 illustrates selected sonographic findings associated with fetal CMV infection.
About 85% to 90% of infants with congenital CMV that results from primary maternal infection have no symptoms at birth. Among the 10% to 15% of infants that do have symptoms, petechial rash, jaundice, and hepatosplenomegaly are the most common manifestations (“blueberry muffin baby”). Approximately 10% to 20% of infants in this group have evidence of chorioretinitis on ophthalmologic examination, and 50% show either microcephaly or low birth weight.22Among survivors of symptomatic congenital CMV, more than 50% have long-term neurologic morbidities that may include sensorineural hearing loss, seizures, vision impairment, and developmental disabilities. Note that even when neonates appear asymptomatic at birth (regardless of whether infection is primary or secondary), 5% may develop microcephaly and motor deficits, 10% go on to develop sensorineural hearing loss, and the overall rate of neurologic morbidity reaches 13% to 15%.12,23 Some of the observed deficits manifest at several years of age, and, currently, no models exist for prediction of outcome.
Continue to: Diagnosing CMV infection...
Diagnosing CMV infection
Maternal infection
If maternal CMV infection is suspected based on a symptomatic illness or an abnormal fetal ultrasound exam, the first diagnostic test should be an assessment of IgM and IgG serology. If the former test results are positive and the latter negative, the diagnosis of acute CMV infection is confirmed. A positive serum CMV DNA polymerase chain reaction (PCR) test adds additional assurance that the diagnosis is correct. Primary infection, as noted above, poses the greatest risk of serious injury to the fetus.1
A frequent diagnostic dilemma arises when both the IgM and IgG antibody are positive. Remember that CMV IgM antibody can remain positive for 9 to 12 months after a primary infection and can reappear in the maternal serum in the face of a recurrent or reactivated infection. When confronted by both a positive IgM and positive IgG result, the clinician should then order IgG avidity testing. If the avidity is low to moderate, which reflects poor binding of antibody to the virus, the patient likely has an acute infection. If the avidity is high, which reflects enhanced binding of antibody to virus, the patient probably has a recurrent or reactivated infection; this scenario poses less danger to the developing fetus. The presence of CMV DNA in serum is also more consistent with acute infection, although viremia still can occur with recurrent infection. FIGURE 2 presents a suggested algorithm for the diagnosis of CMV in the pregnant patient.1
If a diagnosis of maternal CMV infection is confirmed, liver function tests should be obtained to determine if CMV hepatitis is present. If the liver function tests are abnormal, a coagulation profile also should be performed to identify the mother who might be at risk for peripartum hemorrhage.
Fetal infection
The single best test for confirmation of congenital CMV infection is detection of viral DNA and quantitation of viral load in the amniotic fluid by PCR. If the amniocentesis is performed prior to 20 weeks’ gestation and is negative, the test should be repeated in approximately 4 weeks.1,19,24
Detection of viral DNA indicates congenital infection. The ultimate task, however, is to determine if the infection has injured the fetus. Detailed ultrasound examination is the key to identifying fetal injury. As noted previously, the principal ultrasonographic findings that suggest congenital CMV infection include2,19,20,21,25:
- hydropic placenta
- fetal growth restriction
- microcephaly (head circumference more than 3 standard deviations below the mean)
- periventricular calcifications
- enlarged liver
- echogenic bowel
- ascites
- fetal hydrops.
Management: Evidence on CMV hyperimmune globulin, valacyclovir
If the immunocompetent mother has clinical manifestations of infection, she should receive symptomatic treatment. She should be encouraged to rest as much as possible, stay well hydrated, and use acetaminophen (1,000 mg every 6 to 8 hours) as needed for malaise and fever.
However, if the mother is immunocompromised and has signs of serious complications, such as chorioretinitis, hepatitis, or pneumonia, more aggressive therapy is indicated. Drugs used in this setting include foscarnet and ganciclovir and are best prescribed in consultation with a medical infectious disease specialist.
At this time, no consistently effective therapy for congenital infection is available. Therefore, if a patient has primary CMV infection in the first half of pregnancy, particularly in the first trimester, she should be counseled that the risk of fetal infection is approximately 40% and that approximately 5% to 15% of infants will be severely affected at birth. Given this information, some patients may opt for pregnancy termination.
In 2005, a report from Nigro and colleagues stimulated great hope that CMV-specific hyperimmune globulin (CytoGam) might be of value for both treatment and prophylaxis for congenital infection.26 These authors studied 157 women with confirmed primary CMV infection. One-hundred forty-eight women were asymptomatic and were identified by routine serologic screening, 8 had symptomatic infection, and 1 was identified because of abnormal fetal ultrasound findings. Forty-five women had CMV detected in amniotic fluid by PCR or culture more than 6 weeks before study enrollment. Thirty-one of these women were treated with intravenous hyperimmune globulin (200 U or 200 mg/kg maternal body weight); 14 declined treatment. Seven of the latter women had infants who were acutely symptomatic at the time of delivery; only 1 of the 31 treated women had an affected neonate (adjusted odds ratio [OR], 0.02; P<.001). In this same study, 84 women did not have a diagnostic amniocentesis because their infection occurred within 6 weeks of enrollment, their gestational age was less than 20 weeks, or they declined the procedure. Thirty-seven of these women received hyperimmune globulin (100 U or 100 mg/kg) every month until delivery, and 47 declined treatment. Six of the treated women delivered infected infants compared with 19 of the untreated women (adjusted OR, 0.32; P<.04).
Although these results were quite encouraging, several problems existed with the study’s design, as noted in an editorial that accompanied the study’s publication.27 First, the study was not randomized or placebo controlled. Second, patients were not stratified based on the severity of fetal ultrasound abnormalities. Third, the dosing of hyperimmune globulin varied; 9 of the 31 patients in the treatment group received additional infusions of drug into either the amniotic fluid or fetal umbilical vein. Moreover, patients in the prophylaxis group actually received a higher cumulative dose of hyperimmune globulin than patients in the treatment group.
Two subsequent investigations that were better designed were unable to verify the effectiveness of hyperimmune globulin. In 2014, Revello and colleagues reported the results of a prospective, randomized, placebo-controlled, double-blinded study of 124 women at 5 to 26 weeks’ gestation with confirmed primary CMV infection.28 The rate of congenital infection was 30% in the group treated with hyperimmune globulin and 44% in the placebo group (P=.13). There also was no significant difference in the concentration of serum CMV DNA in treated versus untreated mothers. Moreover, the number of adverse obstetric events (preterm delivery, fetal growth restriction, intrahepatic cholestasis of pregnancy, and postpartum preeclampsia) in the treatment group was higher than in the placebo group, 13% versus 2%.
In 2021, Hughes and colleagues published the results of a multicenter, double-blind trial in 399 women who had a diagnosis of primary CMV infection before 23 weeks’ gestation.29 The primary outcome was defined as a composite of congenital CMV infection or fetal/neonatal death. An adverse primary outcome occurred in 22.7% of the patients who received hyperimmune globulin and 19.4% of those who received placebo (relative risk, 1.17; 95% confidence interval [CI], 0.80–1.72; P=.42).
Continue to: Jacquemard and colleagues...
Jacquemard and colleagues then proposed a different approach.30 In a small pilot study of 20 patients, these authors used high doses of oral valacylovir (2 g 4 times daily) and documented therapeutic drug concentrations and a decline in CMV viral load in fetal serum. Patients were not stratified by severity of fetal injury at onset of treatment, so the authors were unable to define which fetuses were most likely to benefit from treatment.
In a follow-up investigation, Leruez-Ville and colleagues reported another small series in which high-dose oral valacyclovir (8 g daily) was used for treatment.31 They excluded fetuses with severe brain anomalies and fetuses with no sonographic evidence of injury. The median gestational age at diagnosis was 26 weeks. Thirty-four of 43 treated fetuses were free of injury at birth. In addition, the viral load in the neonate’s serum decreased significantly after treatment, and the platelet count increased. The authors then compared these outcomes to a historical cohort and confirmed that treatment increased the proportion of asymptomatic neonates from 43% without treatment to 82% with treatment (P<.05 with no overlapping confidence intervals).
We conclude from these investigations that hyperimmune globulin is unlikely to be of value in treating congenital CMV infection, especially if the fetus already has sonographic findings of severe injury. High-dose oral valacyclovir also is unlikely to be of value in severely affected fetuses, particularly those with evidence of CNS injury. However, antiviral therapy may be of modest value in situations when the fetus is less severely injured.
Preventive measures
Since no definitive treatment is available for congenital CMV infection, our efforts as clinicians should focus on measures that may prevent transmission of infection to the pregnant patient. These measures include:
- Encouraging patients to use careful handwashing techniques when handling infant diapers and toys.
- Encouraging patients to adopt safe sexual practices if not already engaged in a mutually faithful, monogamous relationship.
- Using CMV-negative blood when transfusing a pregnant woman or a fetus.
At the present time, unfortunately, a readily available and highly effective therapy for prevention of CMV infection is not available.
CASE Congenital infection diagnosed
The ultrasound findings are most consistent with congenital CMV infection, especially given the patient’s work as an elementary schoolteacher. The diagnosis of maternal infection is best established by conventional serology (positive IgM, negative IgM) and detection of viral DNA in maternal blood by PCR testing. The diagnosis of congenital infection is best confirmed by documentation of viral DNA in the amniotic fluid by PCR testing. Given that this fetus already has evidence of moderate to severe injury, no treatment is likely to be effective in reversing the abnormal ultrasound findings. Pregnancy termination may be an option, depending upon the patient’s desires and the legal restrictions prevalent in the patient’s geographic area. ●
- Cytomegalovirus infection is the most common of the perinatally transmitted infections.
- Maternal infection is often asymptomatic. When symptoms are present, they resemble those of an influenza-like illness. In immunocompromised persons, however, CMV may cause serious complications, including pneumonia, hepatitis, and chorioretinitis.
- The virus is transmitted by contact with contaminated body fluids, such as saliva, urine, blood, and genital secretions.
- The greatest risk of severe fetal injury results from primary maternal infection in the first trimester of pregnancy.
- Manifestations of severe congenital CMV infection include growth restriction, microcephaly, ventriculomegaly, hepatosplenomegaly, ascites, chorioretinitis, thrombocytopenia, purpura, and hydrops (“blueberry muffin baby”).
- Late manifestations of infection, which usually follow recurrent maternal infection, may appear as a child enters elementary school and include visual and auditory deficits, developmental delays, and learning disabilities.
- The diagnosis of maternal infection is confirmed by serology and detection of viral DNA in the serum by PCR testing.
- The diagnosis of fetal infection is best made by a combination of abnormal ultrasound findings and detection of CMV DNA in amniotic fluid. The characteristic ultrasound findings include placentomegaly, microcephaly, ventriculomegaly, growth restriction, echogenic bowel, and serous effusions/hydrops.
- Treatment of the mother with antiviral medications such as valacyclovir may be of modest value in reducing placental edema, decreasing viral load in the fetus, and hastening the resolution of some ultrasound findings, such as echogenic bowel.
- While initial studies seemed promising, the use of hyperimmune globulin has not proven to be consistently effective in treating congenital infection.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik’s Maternal Fetal Medicine: Principles and Practice. 8th ed. 2019:888-890.
- Chatzakis C, Ville Y, Makrydimas G, et al. Timing of primary maternal cytomegalovirus infection and rates of vertical transmission and fetal consequences. Am J Obstet Gynecol. 2020;223:870-883.e11. doi:10.1016/j.ajog.2020.05.038
- Kelly MS, Benjamin DK, Puopolo KM, et al. Postnatal cytomegalovirus infection and the risk for bronchopulmonary dysplasia. JAMA Pediatr. 2015;169:e153785. doi:10.1001 /jamapediatrics.2015.3785
- Messinger CJ, Lipsitch M, Bateman BT, et al. Association between congenital cytomegalovirus and the prevalence at birth of microcephaly in the United States. JAMA Pediatr. 2020;174:1159-1167. doi:10.1001/jamapediatrics.2020.3009
- De Cuyper E, Acke F, Keymeulen A, et al. Risk factors for hearing loss at birth in newborns with congenital cytomegalovirus infection. JAMA Otolaryngol Head Neck Surg. 2023;149:122-130. doi:10.1001/jamaoto.2022.4109
- Colugnati FA, Staras SA, Dollard SC, et al. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis. 2007;7:71. doi:10.1186/1471-2334-7-71
- Stagno S, Pass RF, Cloud G, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA. 1986;256:1904-1908.
- Wreghitt TG, Teare EL, Sule O, et al. Cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 2003;37:1603-1606. doi:10.1086/379711
- Fowler KB, Stagno S, Pass RF, et al. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326:663-667. doi:10.1056 /NEJM199203053261003
- Faure-Bardon V, Magny JF, Parodi M, et al. Sequelae of congenital cytomegalovirus following maternal primary infections are limited to those acquired in the first trimester of pregnancy. Clin Infect Dis. 2019;69:1526-1532. doi:10.1093/ cid/ciy1128
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253-276. doi:10.1002/ rmv.535
- Boppana SB, Pass RF, Britt WJ, et al. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J. 1992;11:93-99. doi:10.1097/00006454-199202000-00007
- Ross SA, Fowler KB, Ashrith G, et al. Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J Pediatr. 2006;148:332-336. doi:10.1016/j.jpeds.2005.09.003
- Zalel Y, Gilboa Y, Berkenshtat M, et al. Secondary cytomegalovirus infection can cause severe fetal sequelae despite maternal preconceptional immunity. Ultrasound Obstet Gynecol. 31:417-420. doi:10.1002/uog.5255
- Scaramuzzino F, Di Pastena M, Chiurchiu S, et al. Secondary cytomegalovirus infections: how much do we still not know? Comparison of children with symptomatic congenital cytomegalovirus born to mothers with primary and secondary infection. Front Pediatr. 2022;10:885926. doi:10.3389/fped.2022.885926
- Gindes L, Teperberg-Oikawa M, Sherman D, et al. Congenital cytomegalovirus infection following primary maternal infection in the third trimester. BJOG. 2008;115:830-835. doi:10.1111/j.1471-0528.2007.01651.x
- Hadar E, Dorfman E, Bardin R, et al. Symptomatic congenital cytomegalovirus disease following non-primary maternal infection: a retrospective cohort study. BMC Infect Dis. 2017;17:31. doi:10.1186/s12879-016-2161-3
- Elkan Miller T, Weisz B, Yinon Y, et al. Congenital cytomegalovirus infection following second and third trimester maternal infection is associated with mild childhood adverse outcome not predicted by prenatal imaging. J Pediatric Infect Dis Soc. 2021;10:562-568. doi:10.1093/jpids/ piaa154
- Lipitz S, Yinon Y, Malinger G, et al. Risk of cytomegalovirusassociated sequelae in relation to time of infection and findings on prenatal imaging. Ultrasound Obstet Gynecol. 2013;41:508-514. doi:10.1002/uog.12377
- Lipitz S, Elkan Miller T, Yinon Y, et al. Revisiting short- and long-term outcome after fetal first-trimester primary cytomegalovirus infection in relation to prenatal imaging findings. Ultrasound Obstet Gynecol. 2020;56:572-578. doi:10.1002/uog.21946
- Buca D, Di Mascio D, Rizzo G, et al. Outcome of fetuses with congenital cytomegalovirus infection and normal ultrasound at diagnosis: systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2021;57:551-559. doi:10.1002/uog.23143
- Boppana SB, Ross SA, Fowler KB. Congenital cytomegalovirus infection: clinical outcome. Clin Infect Dis. 2013;57 (suppl 4):S178-S181. doi:10.1093/cid/cit629
- Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17:355-363. doi:10.1002/rmv.544
- Hughes BL, Gyamfi-Bannerman C. Diagnosis and antenatal management of congenital cytomegalovirus infection. Am J Obstet Gynecol. 2016;214:B5-11. doi:10.1016 /j.ajog.2016.02.042
- Rouse DJ, Fette LM, Hughes BL, et al. Noninvasive prediction of congenital cytomegalovirus infection after maternal primary infection. Obstet Gynecol. 2022;139:400-406. doi:10.1097/AOG.0000000000004691
- Nigro G, Adler SP, La Torre R, et al; Congenital Cytomegalovirus Collaborating Group. Passive immunization during pregnancy for congenital cytomegalovirus infection. N Engl J Med. 2005;353:1350-1362. doi:10.1056/NEJMoa043337
- Duff P. Immunotherapy for congenital cytomegalovirus infection. N Engl J Med. 2005;355:1402-1404. doi:10.1056 /NEJMe058172
- Revello MG, Lazzarotto T, Guerra B, et al. A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N Engl J Med. 2014;370:1316-1326. doi:10.1056/NEJMoa1310214
- Hughes BL, Clifton RG, Rouse DJ, et al. A trial of hyperimmune globulin to prevent congenital cytomegalovirus infection. N Engl J Med. 2021;385:436-444. doi:10.1056/NEJMoa1913569
- Jacquemard F, Yamamoto M, Costa JM, et al. Maternal administration of valaciclovir in symptomatic intrauterine cytomegalovirus infection. BJOG. 2007;114:1113-1121. doi:10.1111/j.1471-0528.2007.01308.x
- Leruez-Ville M, Ghout I, Bussières L, et al. In utero treatment of congenital cytomegalovirus infection with valacyclovir in a multicenter, open-label, phase II study. Am J Obstet Gynecol. 2016;215:462.e1-462.e10. doi:10.1016/j.ajog.2016.04.003
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik’s Maternal Fetal Medicine: Principles and Practice. 8th ed. 2019:888-890.
- Chatzakis C, Ville Y, Makrydimas G, et al. Timing of primary maternal cytomegalovirus infection and rates of vertical transmission and fetal consequences. Am J Obstet Gynecol. 2020;223:870-883.e11. doi:10.1016/j.ajog.2020.05.038
- Kelly MS, Benjamin DK, Puopolo KM, et al. Postnatal cytomegalovirus infection and the risk for bronchopulmonary dysplasia. JAMA Pediatr. 2015;169:e153785. doi:10.1001 /jamapediatrics.2015.3785
- Messinger CJ, Lipsitch M, Bateman BT, et al. Association between congenital cytomegalovirus and the prevalence at birth of microcephaly in the United States. JAMA Pediatr. 2020;174:1159-1167. doi:10.1001/jamapediatrics.2020.3009
- De Cuyper E, Acke F, Keymeulen A, et al. Risk factors for hearing loss at birth in newborns with congenital cytomegalovirus infection. JAMA Otolaryngol Head Neck Surg. 2023;149:122-130. doi:10.1001/jamaoto.2022.4109
- Colugnati FA, Staras SA, Dollard SC, et al. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis. 2007;7:71. doi:10.1186/1471-2334-7-71
- Stagno S, Pass RF, Cloud G, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA. 1986;256:1904-1908.
- Wreghitt TG, Teare EL, Sule O, et al. Cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 2003;37:1603-1606. doi:10.1086/379711
- Fowler KB, Stagno S, Pass RF, et al. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326:663-667. doi:10.1056 /NEJM199203053261003
- Faure-Bardon V, Magny JF, Parodi M, et al. Sequelae of congenital cytomegalovirus following maternal primary infections are limited to those acquired in the first trimester of pregnancy. Clin Infect Dis. 2019;69:1526-1532. doi:10.1093/ cid/ciy1128
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253-276. doi:10.1002/ rmv.535
- Boppana SB, Pass RF, Britt WJ, et al. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J. 1992;11:93-99. doi:10.1097/00006454-199202000-00007
- Ross SA, Fowler KB, Ashrith G, et al. Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J Pediatr. 2006;148:332-336. doi:10.1016/j.jpeds.2005.09.003
- Zalel Y, Gilboa Y, Berkenshtat M, et al. Secondary cytomegalovirus infection can cause severe fetal sequelae despite maternal preconceptional immunity. Ultrasound Obstet Gynecol. 31:417-420. doi:10.1002/uog.5255
- Scaramuzzino F, Di Pastena M, Chiurchiu S, et al. Secondary cytomegalovirus infections: how much do we still not know? Comparison of children with symptomatic congenital cytomegalovirus born to mothers with primary and secondary infection. Front Pediatr. 2022;10:885926. doi:10.3389/fped.2022.885926
- Gindes L, Teperberg-Oikawa M, Sherman D, et al. Congenital cytomegalovirus infection following primary maternal infection in the third trimester. BJOG. 2008;115:830-835. doi:10.1111/j.1471-0528.2007.01651.x
- Hadar E, Dorfman E, Bardin R, et al. Symptomatic congenital cytomegalovirus disease following non-primary maternal infection: a retrospective cohort study. BMC Infect Dis. 2017;17:31. doi:10.1186/s12879-016-2161-3
- Elkan Miller T, Weisz B, Yinon Y, et al. Congenital cytomegalovirus infection following second and third trimester maternal infection is associated with mild childhood adverse outcome not predicted by prenatal imaging. J Pediatric Infect Dis Soc. 2021;10:562-568. doi:10.1093/jpids/ piaa154
- Lipitz S, Yinon Y, Malinger G, et al. Risk of cytomegalovirusassociated sequelae in relation to time of infection and findings on prenatal imaging. Ultrasound Obstet Gynecol. 2013;41:508-514. doi:10.1002/uog.12377
- Lipitz S, Elkan Miller T, Yinon Y, et al. Revisiting short- and long-term outcome after fetal first-trimester primary cytomegalovirus infection in relation to prenatal imaging findings. Ultrasound Obstet Gynecol. 2020;56:572-578. doi:10.1002/uog.21946
- Buca D, Di Mascio D, Rizzo G, et al. Outcome of fetuses with congenital cytomegalovirus infection and normal ultrasound at diagnosis: systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2021;57:551-559. doi:10.1002/uog.23143
- Boppana SB, Ross SA, Fowler KB. Congenital cytomegalovirus infection: clinical outcome. Clin Infect Dis. 2013;57 (suppl 4):S178-S181. doi:10.1093/cid/cit629
- Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17:355-363. doi:10.1002/rmv.544
- Hughes BL, Gyamfi-Bannerman C. Diagnosis and antenatal management of congenital cytomegalovirus infection. Am J Obstet Gynecol. 2016;214:B5-11. doi:10.1016 /j.ajog.2016.02.042
- Rouse DJ, Fette LM, Hughes BL, et al. Noninvasive prediction of congenital cytomegalovirus infection after maternal primary infection. Obstet Gynecol. 2022;139:400-406. doi:10.1097/AOG.0000000000004691
- Nigro G, Adler SP, La Torre R, et al; Congenital Cytomegalovirus Collaborating Group. Passive immunization during pregnancy for congenital cytomegalovirus infection. N Engl J Med. 2005;353:1350-1362. doi:10.1056/NEJMoa043337
- Duff P. Immunotherapy for congenital cytomegalovirus infection. N Engl J Med. 2005;355:1402-1404. doi:10.1056 /NEJMe058172
- Revello MG, Lazzarotto T, Guerra B, et al. A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N Engl J Med. 2014;370:1316-1326. doi:10.1056/NEJMoa1310214
- Hughes BL, Clifton RG, Rouse DJ, et al. A trial of hyperimmune globulin to prevent congenital cytomegalovirus infection. N Engl J Med. 2021;385:436-444. doi:10.1056/NEJMoa1913569
- Jacquemard F, Yamamoto M, Costa JM, et al. Maternal administration of valaciclovir in symptomatic intrauterine cytomegalovirus infection. BJOG. 2007;114:1113-1121. doi:10.1111/j.1471-0528.2007.01308.x
- Leruez-Ville M, Ghout I, Bussières L, et al. In utero treatment of congenital cytomegalovirus infection with valacyclovir in a multicenter, open-label, phase II study. Am J Obstet Gynecol. 2016;215:462.e1-462.e10. doi:10.1016/j.ajog.2016.04.003
The HPV vaccine: Time for ObGyn physicians to up our game
CASE Sexually active woman asks about the HPV vaccine
A 26-year-old woman delivered her first child 4 weeks ago. She has had 3 lifetime sexual partners and is now in a mutually faithful monogamous relationship with her partner. She has no known history of sexually transmissible infections. She received only one Pap test 3 years ago, and the cytology showed no abnormal cells. This cervical specimen was not tested for human papillomavirus (HPV) DNA. At the time of her postpartum appointment, she inquires whether she is a candidate for the HPV vaccine.
What should be your response?
Genital HPV infection is the most common sexually transmissible infection in the United States. This virus is the cause of multiple genital malignancies, including cancers of the vagina, vulva, penis, anus, and cervix. The organism is also now the major cause of oropharyngeal cancer.
Of the more than 200 different HPV types that have been identified, 12 have been defined as oncogenic (high risk), and 8 to 12 types have been defined as possibly or probably oncogenic. The HPV strain with the highest risk of progression to cancer is HPV 16. The strains HPV 16 and 18 are responsible for approximately 70% of cases of cervical cancer. Each year in the United States, approximately 11,500 new cases of invasive cervical cancer occur. Unfortunately, this malignancy is responsible for about 4,000 deaths annually. Worldwide, HPV causes approximately 690,000 cancers each year.1
To a large extent, most cases of HPV infection would be preventable if patients were to take advantage of the remarkably effective HPV vaccine that is now available. However, acceptance of the vaccine has been disappointing. In 2020, only about half of adolescents, age 13 to 15, had received the appropriate number of vaccine doses.1
As ObGyn physicians, we can take several measures, in concert with our pediatrician colleagues, to improve HPV vaccination rates. In this article, I review the development of the HPV vaccine and describe the components, indications, dosing schedules, contraindications, adverse effects, and cost of the vaccine.
HPV vaccine development and expansion
The first HPV vaccine introduced in the United States was the recombinant quadrivalent vaccine (Gardasil; Merck); it was approved by the US Food and Drug Administration (FDA) in 2006. This vaccine is composed of viral-like particles unique to HPV 16 and 18 (the 2 most common causes of cervical, penile, anal, and oropharyngeal cancer) and HPV 6 and 11 (the 2 most common causes of genital warts). The formulation is prepared in baker’s yeast, and it elicits a robust production of neutralizing antibodies.2
In 2009, the FDA approved the bivalent vaccine (Cervarix; GlaxoSmithKline Biologicals). This vaccine contains viral-like particles unique to HPV 16 and 18, and it also induces a robust immune response. The vaccine is prepared in insect viral vectors.2
Both the quadrivalent and bivalent vaccines are no longer available in the United States. The only HPV vaccine currently marketed is the recombinant 9-valent vaccine (Gardasil 9; Merck), which was approved by the FDA in 2014. This newer vaccine targets the original 4 viral HPV strains in the quadrivalent vaccine (16, 18, 6, 11) plus 5 additional oncogenic strains: 31, 33, 45, 52, 58.2-4 The HPV strains targeted by this vaccine are responsible for approximately 90% of all cancers caused by HPV.
The 9-valent HPV vaccine, like the other 2, is highly effective in preventing cancers of the cervix, vagina, vulva, anus, penis; oropharyngeal cancers; and precancerous lesions such as genital warts.2-5 It will not, however, prevent the progression of preexisting infection or clear an infection that is already present at the time of vaccination.1
Although the original protocol for administration of the vaccine provided for 3 doses, recent studies indicate that 2 doses may be as effective as 3 in eliciting a favorable antibody response.6 There also is evidence that even a single dose of the vaccine can elicit a protective immune response.7 This encouraging finding is particularly important to public health officials responsible for developing HPV vaccination programs in low- and middle-resource countries.
Continue to: Target groups for the HPV vaccine...
Target groups for the HPV vaccine
The primary target group for the HPV vaccine is girls and boys who are aged 11 to 12 years. The key strategy is to immunize these individuals before they become sexually active. The vaccine also should be offered to children who are aged 9 to 10 years of age if they are judged to be at unusual risk, such as because of concern about sexual molestation. Children in these 2 age groups should receive 2 doses of the vaccine, with the second dose administered 6 to 12 months after the first dose.
The second target group for vaccination is individuals who are aged 13 to 26 years who have never been vaccinated. They should be offered catch-up vaccination. If older than age 15, they should receive 3 doses of the vaccine, with the second dose administered 1 to 2 months after the first dose and the third dose administered 6 months after the first dose.1
A third target group is individuals who are aged 27 to 45 years and who, in their own opinion or in the opinion of their physician, are at new or increased risk for HPV infection. These individuals should receive the 3-dose vaccine series as outlined above.1
Patients in any age range who are immunocompromised, for example, due to HIV infection, should receive the 3-dose series.1
The approximate retail cost of a single 0.5-mL intramuscular dose of the 9-valent vaccine is $240 (www.goodrx.com).
Vaccine adverse effects
The most common reactions to the HPV vaccine are inflammation at the site of injection, fatigue, headache, fever, gastrointestinal upset, vertigo, cough, and oropharyngeal discomfort. The most serious reaction—which fortunately is very rare—is anaphylaxis.1
Contraindications to the vaccine
The HPV vaccine should not be used in any patient who is hypersensitive to any component of the vaccine, including yeast. It should not be given to a patient who is moderately or severely ill at the time of the scheduled administration. Because of an abundance of caution, the manufacturer also recommends that the vaccine not be given to pregnant women even though the agent does not contain live virus.1
Of note, a study by Scheller and colleagues was very reassuring about the lack of adverse effects of HPV vaccine administration in pregnancy.8 The authors evaluated a large cohort of pregnant women in Demark and found that exposure to the vaccine was not associated with an increase in the frequency of major birth defects, spontaneous abortion, preterm delivery, low birthweight, fetal growth restriction, or stillbirth.8
Barriers to vaccination
One important barrier to HPV vaccination is patient apprehension that the vaccine may cause genital tract or oropharyngeal cancer. The patient should be reassured that the vaccine does not contain infectious viral particles and does not transmit infection. Rather, it builds robust immunity to infection.
Another important barrier is the misconception that the vaccine will promote sexual promiscuity in preteenagers and teenagers. Absolutely no evidence supports this belief. Multiple studies have demonstrated that teenagers do not engage in more high-risk sexual behavior following vaccination.
A specific barrier related to vaccination of young boys is the philosophical viewpoint that, “Why should my young male child be vaccinated to protect against a disease (specifically cervical cancer) that occurs only in girls and women?” The appropriate answer to this question is that the vaccine also protects against penile cancer, anal cancer, oropharyngeal cancer, and genital warts. While penile and anal cancers are rare, the other 2 conditions are not. In fact, oropharyngeal cancer is significantly more common in males than females.
A final important barrier to HPV vaccination is cost. The new evidence that demonstrated the effectiveness of a 2-dose vaccine series, and even single-dose vaccination, is of great importance in minimizing cost of the HPV vaccine series, in the absence of full reimbursement by public and private insurance agencies.
Continue to: Creating an effective vaccination program...
Creating an effective vaccination program
The following commonsense guidelines, which we have implemented at our medical center, should be helpful in organizing an effective HPV vaccination program for your office or department4,9,10:
- One clinician in the department or practice should be designated the “vaccination champion.” This individual should provide colleagues with periodic updates, emphasizing the importance of the HPV vaccine and other vaccines, such as Tdap (tetanus, diphtheria, pertussis), influenza, COVID, pneumococcal, hepatitis B, herpes zoster (shingles), and RSV (respiratory syncytial virus).
- One staff member in the practice or department should be designated as the go-to person for all logistical matters related to vaccines. This individual should be responsible for estimating usage, ordering vaccines, and storing them properly. He or she also should be knowledgeable about the cost of the vaccines and insurance reimbursement for the vaccines.
- Signs and educational materials should be posted in strategic locations in the office, advising patients of the importance of timely vaccination for themselves and their adolescent children.
- At every encounter, patients should be encouraged to receive the HPV vaccine series if they are in the appropriate age range and social situation for vaccination. They should not be required to have HPV testing before vaccine administration.
- Key leaders in the department or practice should lobby effectively with their pediatrician colleagues and with public and private insurance companies to encourage timely administration and proper coverage of this important immunization.
Other measures to reduce the risk of HPV-mediated malignancies
Practitioners should advise their patients to:
- Be circumspect in selection of sexual partners.
- Use male or female condoms when engaging in vaginal, anal, and/or oral sex with multiple partners, particularly those who may have genital or oral condylomas.
- Have regular Pap tests, every 3 to 5 years, depending upon age. More frequent testing may be indicated if there is a history of previous abnormal testing.
- Seek prompt medical or surgical treatment for genital or oral condylomas.
CASE Resolved with HPV vaccination
This patient is an excellent candidate for catch-up vaccination. She should receive the first dose of the 9-valent HPV vaccine at the time of her postpartum appointment. The second dose should be administered 1 to 2 months later. The third dose should be administered 6 months after the first dose. She also should have a Pap test, either cytology alone or cytology plus HPV screening. If the latter test is chosen and is reassuring, she will not need retesting for 5 years. If the former test is chosen, she should have a repeat test in 3 years. ●

- The overwhelming majority of precancerous lesions and overt malignancies of the genital tract and oropharynx are caused by oncogenic strains of HPV.
- Most of these cancers could be prevented if patients were vaccinated with the 9-valent HPV vaccine.
- The HPV vaccine should be offered to all children beginning at age 11 and to selected high-risk children at age 9. For children aged 14 years and younger, 2 doses of the vaccine are sufficient to induce a robust immune response. The second dose should be administered 6 to 12 months after the first dose.
- Individuals in the age range 13 to 26 years should be offered catch-up vaccination if they have not been previously vaccinated.
- Persons in the age range 27 to 45 years also should be offered vaccination if they have developed a new high-risk profile.
- Persons older than age 15, or those of any age with immunocompromising conditions, should receive 3 doses of the vaccine. The second dose should be administered 1 to 2 months after the first dose, and the third dose should be given 6 months after the first dose.
- The vaccine does not prevent the progression of preexisting infection or clear an infection that is already present at the time of vaccination.
- As a general rule, the vaccine should be deferred during pregnancy, although no adverse effects have been documented when the vaccine has been administered to pregnant women.
- Markowitz LE, Unger ER. Human papilloma virus vaccination. N Engl J Med. 2023;388:1790-1798.
- Schiller JT, Castellsague X, Garland SM. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine. 2012;30(suppl 5): F123-F138.
- Lei J, Ploner A, Elfstrom KM, et al. HPV vaccination and the risk of invasive cervical cancer. N Engl J Med. 2020;383: 1340-1348.
- ACOG Committee Opinion Summary No. 809. Human papillomavirus vaccination. Obstet Gynecol. 2020;136:435-436.
- Barbieri RL. 9vHPV vaccine: prevention of oropharyngeal cancer. OBG Manag. 2020;32:9, 14-15.
- Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316:2411-2421.
- Watson-Jones D, Changalucha J, Whitworth H, et al. Immunogenicity and safety of one-dose human papillomavirus vaccine compared with two or three doses in Tanzanian girls (DoRIS): an open-label, randomised noninferiority trial. Lancet Glob Health. 2022;10:e1473-e1484.
- Scheller NM, Pasternak B, Molgaard-Nielsen D, et al. Quadrivalent HPV vaccination and the risk of adverse pregnancy outcomes. N Engl J Med. 2017;376:1223-1233.
- ACOG Committee Opinion Summary No. 641. Human papillomavirus vaccination. Obstet Gynecol. 2015;126:693.
- Boitano TKL, Ketch PW, Scarinci IC, et al. An update on human papillomavirus vaccination in the United States. Obstet Gynecol. 2023;141:324-330.

Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
CASE Sexually active woman asks about the HPV vaccine
A 26-year-old woman delivered her first child 4 weeks ago. She has had 3 lifetime sexual partners and is now in a mutually faithful monogamous relationship with her partner. She has no known history of sexually transmissible infections. She received only one Pap test 3 years ago, and the cytology showed no abnormal cells. This cervical specimen was not tested for human papillomavirus (HPV) DNA. At the time of her postpartum appointment, she inquires whether she is a candidate for the HPV vaccine.
What should be your response?
Genital HPV infection is the most common sexually transmissible infection in the United States. This virus is the cause of multiple genital malignancies, including cancers of the vagina, vulva, penis, anus, and cervix. The organism is also now the major cause of oropharyngeal cancer.
Of the more than 200 different HPV types that have been identified, 12 have been defined as oncogenic (high risk), and 8 to 12 types have been defined as possibly or probably oncogenic. The HPV strain with the highest risk of progression to cancer is HPV 16. The strains HPV 16 and 18 are responsible for approximately 70% of cases of cervical cancer. Each year in the United States, approximately 11,500 new cases of invasive cervical cancer occur. Unfortunately, this malignancy is responsible for about 4,000 deaths annually. Worldwide, HPV causes approximately 690,000 cancers each year.1
To a large extent, most cases of HPV infection would be preventable if patients were to take advantage of the remarkably effective HPV vaccine that is now available. However, acceptance of the vaccine has been disappointing. In 2020, only about half of adolescents, age 13 to 15, had received the appropriate number of vaccine doses.1
As ObGyn physicians, we can take several measures, in concert with our pediatrician colleagues, to improve HPV vaccination rates. In this article, I review the development of the HPV vaccine and describe the components, indications, dosing schedules, contraindications, adverse effects, and cost of the vaccine.
HPV vaccine development and expansion
The first HPV vaccine introduced in the United States was the recombinant quadrivalent vaccine (Gardasil; Merck); it was approved by the US Food and Drug Administration (FDA) in 2006. This vaccine is composed of viral-like particles unique to HPV 16 and 18 (the 2 most common causes of cervical, penile, anal, and oropharyngeal cancer) and HPV 6 and 11 (the 2 most common causes of genital warts). The formulation is prepared in baker’s yeast, and it elicits a robust production of neutralizing antibodies.2
In 2009, the FDA approved the bivalent vaccine (Cervarix; GlaxoSmithKline Biologicals). This vaccine contains viral-like particles unique to HPV 16 and 18, and it also induces a robust immune response. The vaccine is prepared in insect viral vectors.2
Both the quadrivalent and bivalent vaccines are no longer available in the United States. The only HPV vaccine currently marketed is the recombinant 9-valent vaccine (Gardasil 9; Merck), which was approved by the FDA in 2014. This newer vaccine targets the original 4 viral HPV strains in the quadrivalent vaccine (16, 18, 6, 11) plus 5 additional oncogenic strains: 31, 33, 45, 52, 58.2-4 The HPV strains targeted by this vaccine are responsible for approximately 90% of all cancers caused by HPV.
The 9-valent HPV vaccine, like the other 2, is highly effective in preventing cancers of the cervix, vagina, vulva, anus, penis; oropharyngeal cancers; and precancerous lesions such as genital warts.2-5 It will not, however, prevent the progression of preexisting infection or clear an infection that is already present at the time of vaccination.1
Although the original protocol for administration of the vaccine provided for 3 doses, recent studies indicate that 2 doses may be as effective as 3 in eliciting a favorable antibody response.6 There also is evidence that even a single dose of the vaccine can elicit a protective immune response.7 This encouraging finding is particularly important to public health officials responsible for developing HPV vaccination programs in low- and middle-resource countries.
Continue to: Target groups for the HPV vaccine...
Target groups for the HPV vaccine
The primary target group for the HPV vaccine is girls and boys who are aged 11 to 12 years. The key strategy is to immunize these individuals before they become sexually active. The vaccine also should be offered to children who are aged 9 to 10 years of age if they are judged to be at unusual risk, such as because of concern about sexual molestation. Children in these 2 age groups should receive 2 doses of the vaccine, with the second dose administered 6 to 12 months after the first dose.
The second target group for vaccination is individuals who are aged 13 to 26 years who have never been vaccinated. They should be offered catch-up vaccination. If older than age 15, they should receive 3 doses of the vaccine, with the second dose administered 1 to 2 months after the first dose and the third dose administered 6 months after the first dose.1
A third target group is individuals who are aged 27 to 45 years and who, in their own opinion or in the opinion of their physician, are at new or increased risk for HPV infection. These individuals should receive the 3-dose vaccine series as outlined above.1
Patients in any age range who are immunocompromised, for example, due to HIV infection, should receive the 3-dose series.1
The approximate retail cost of a single 0.5-mL intramuscular dose of the 9-valent vaccine is $240 (www.goodrx.com).
Vaccine adverse effects
The most common reactions to the HPV vaccine are inflammation at the site of injection, fatigue, headache, fever, gastrointestinal upset, vertigo, cough, and oropharyngeal discomfort. The most serious reaction—which fortunately is very rare—is anaphylaxis.1
Contraindications to the vaccine
The HPV vaccine should not be used in any patient who is hypersensitive to any component of the vaccine, including yeast. It should not be given to a patient who is moderately or severely ill at the time of the scheduled administration. Because of an abundance of caution, the manufacturer also recommends that the vaccine not be given to pregnant women even though the agent does not contain live virus.1
Of note, a study by Scheller and colleagues was very reassuring about the lack of adverse effects of HPV vaccine administration in pregnancy.8 The authors evaluated a large cohort of pregnant women in Demark and found that exposure to the vaccine was not associated with an increase in the frequency of major birth defects, spontaneous abortion, preterm delivery, low birthweight, fetal growth restriction, or stillbirth.8
Barriers to vaccination
One important barrier to HPV vaccination is patient apprehension that the vaccine may cause genital tract or oropharyngeal cancer. The patient should be reassured that the vaccine does not contain infectious viral particles and does not transmit infection. Rather, it builds robust immunity to infection.
Another important barrier is the misconception that the vaccine will promote sexual promiscuity in preteenagers and teenagers. Absolutely no evidence supports this belief. Multiple studies have demonstrated that teenagers do not engage in more high-risk sexual behavior following vaccination.
A specific barrier related to vaccination of young boys is the philosophical viewpoint that, “Why should my young male child be vaccinated to protect against a disease (specifically cervical cancer) that occurs only in girls and women?” The appropriate answer to this question is that the vaccine also protects against penile cancer, anal cancer, oropharyngeal cancer, and genital warts. While penile and anal cancers are rare, the other 2 conditions are not. In fact, oropharyngeal cancer is significantly more common in males than females.
A final important barrier to HPV vaccination is cost. The new evidence that demonstrated the effectiveness of a 2-dose vaccine series, and even single-dose vaccination, is of great importance in minimizing cost of the HPV vaccine series, in the absence of full reimbursement by public and private insurance agencies.
Continue to: Creating an effective vaccination program...
Creating an effective vaccination program
The following commonsense guidelines, which we have implemented at our medical center, should be helpful in organizing an effective HPV vaccination program for your office or department4,9,10:
- One clinician in the department or practice should be designated the “vaccination champion.” This individual should provide colleagues with periodic updates, emphasizing the importance of the HPV vaccine and other vaccines, such as Tdap (tetanus, diphtheria, pertussis), influenza, COVID, pneumococcal, hepatitis B, herpes zoster (shingles), and RSV (respiratory syncytial virus).
- One staff member in the practice or department should be designated as the go-to person for all logistical matters related to vaccines. This individual should be responsible for estimating usage, ordering vaccines, and storing them properly. He or she also should be knowledgeable about the cost of the vaccines and insurance reimbursement for the vaccines.
- Signs and educational materials should be posted in strategic locations in the office, advising patients of the importance of timely vaccination for themselves and their adolescent children.
- At every encounter, patients should be encouraged to receive the HPV vaccine series if they are in the appropriate age range and social situation for vaccination. They should not be required to have HPV testing before vaccine administration.
- Key leaders in the department or practice should lobby effectively with their pediatrician colleagues and with public and private insurance companies to encourage timely administration and proper coverage of this important immunization.
Other measures to reduce the risk of HPV-mediated malignancies
Practitioners should advise their patients to:
- Be circumspect in selection of sexual partners.
- Use male or female condoms when engaging in vaginal, anal, and/or oral sex with multiple partners, particularly those who may have genital or oral condylomas.
- Have regular Pap tests, every 3 to 5 years, depending upon age. More frequent testing may be indicated if there is a history of previous abnormal testing.
- Seek prompt medical or surgical treatment for genital or oral condylomas.
CASE Resolved with HPV vaccination
This patient is an excellent candidate for catch-up vaccination. She should receive the first dose of the 9-valent HPV vaccine at the time of her postpartum appointment. The second dose should be administered 1 to 2 months later. The third dose should be administered 6 months after the first dose. She also should have a Pap test, either cytology alone or cytology plus HPV screening. If the latter test is chosen and is reassuring, she will not need retesting for 5 years. If the former test is chosen, she should have a repeat test in 3 years. ●
- The overwhelming majority of precancerous lesions and overt malignancies of the genital tract and oropharynx are caused by oncogenic strains of HPV.
- Most of these cancers could be prevented if patients were vaccinated with the 9-valent HPV vaccine.
- The HPV vaccine should be offered to all children beginning at age 11 and to selected high-risk children at age 9. For children aged 14 years and younger, 2 doses of the vaccine are sufficient to induce a robust immune response. The second dose should be administered 6 to 12 months after the first dose.
- Individuals in the age range 13 to 26 years should be offered catch-up vaccination if they have not been previously vaccinated.
- Persons in the age range 27 to 45 years also should be offered vaccination if they have developed a new high-risk profile.
- Persons older than age 15, or those of any age with immunocompromising conditions, should receive 3 doses of the vaccine. The second dose should be administered 1 to 2 months after the first dose, and the third dose should be given 6 months after the first dose.
- The vaccine does not prevent the progression of preexisting infection or clear an infection that is already present at the time of vaccination.
- As a general rule, the vaccine should be deferred during pregnancy, although no adverse effects have been documented when the vaccine has been administered to pregnant women.
CASE Sexually active woman asks about the HPV vaccine
A 26-year-old woman delivered her first child 4 weeks ago. She has had 3 lifetime sexual partners and is now in a mutually faithful monogamous relationship with her partner. She has no known history of sexually transmissible infections. She received only one Pap test 3 years ago, and the cytology showed no abnormal cells. This cervical specimen was not tested for human papillomavirus (HPV) DNA. At the time of her postpartum appointment, she inquires whether she is a candidate for the HPV vaccine.
What should be your response?
Genital HPV infection is the most common sexually transmissible infection in the United States. This virus is the cause of multiple genital malignancies, including cancers of the vagina, vulva, penis, anus, and cervix. The organism is also now the major cause of oropharyngeal cancer.
Of the more than 200 different HPV types that have been identified, 12 have been defined as oncogenic (high risk), and 8 to 12 types have been defined as possibly or probably oncogenic. The HPV strain with the highest risk of progression to cancer is HPV 16. The strains HPV 16 and 18 are responsible for approximately 70% of cases of cervical cancer. Each year in the United States, approximately 11,500 new cases of invasive cervical cancer occur. Unfortunately, this malignancy is responsible for about 4,000 deaths annually. Worldwide, HPV causes approximately 690,000 cancers each year.1
To a large extent, most cases of HPV infection would be preventable if patients were to take advantage of the remarkably effective HPV vaccine that is now available. However, acceptance of the vaccine has been disappointing. In 2020, only about half of adolescents, age 13 to 15, had received the appropriate number of vaccine doses.1
As ObGyn physicians, we can take several measures, in concert with our pediatrician colleagues, to improve HPV vaccination rates. In this article, I review the development of the HPV vaccine and describe the components, indications, dosing schedules, contraindications, adverse effects, and cost of the vaccine.
HPV vaccine development and expansion
The first HPV vaccine introduced in the United States was the recombinant quadrivalent vaccine (Gardasil; Merck); it was approved by the US Food and Drug Administration (FDA) in 2006. This vaccine is composed of viral-like particles unique to HPV 16 and 18 (the 2 most common causes of cervical, penile, anal, and oropharyngeal cancer) and HPV 6 and 11 (the 2 most common causes of genital warts). The formulation is prepared in baker’s yeast, and it elicits a robust production of neutralizing antibodies.2
In 2009, the FDA approved the bivalent vaccine (Cervarix; GlaxoSmithKline Biologicals). This vaccine contains viral-like particles unique to HPV 16 and 18, and it also induces a robust immune response. The vaccine is prepared in insect viral vectors.2
Both the quadrivalent and bivalent vaccines are no longer available in the United States. The only HPV vaccine currently marketed is the recombinant 9-valent vaccine (Gardasil 9; Merck), which was approved by the FDA in 2014. This newer vaccine targets the original 4 viral HPV strains in the quadrivalent vaccine (16, 18, 6, 11) plus 5 additional oncogenic strains: 31, 33, 45, 52, 58.2-4 The HPV strains targeted by this vaccine are responsible for approximately 90% of all cancers caused by HPV.
The 9-valent HPV vaccine, like the other 2, is highly effective in preventing cancers of the cervix, vagina, vulva, anus, penis; oropharyngeal cancers; and precancerous lesions such as genital warts.2-5 It will not, however, prevent the progression of preexisting infection or clear an infection that is already present at the time of vaccination.1
Although the original protocol for administration of the vaccine provided for 3 doses, recent studies indicate that 2 doses may be as effective as 3 in eliciting a favorable antibody response.6 There also is evidence that even a single dose of the vaccine can elicit a protective immune response.7 This encouraging finding is particularly important to public health officials responsible for developing HPV vaccination programs in low- and middle-resource countries.
Continue to: Target groups for the HPV vaccine...
Target groups for the HPV vaccine
The primary target group for the HPV vaccine is girls and boys who are aged 11 to 12 years. The key strategy is to immunize these individuals before they become sexually active. The vaccine also should be offered to children who are aged 9 to 10 years of age if they are judged to be at unusual risk, such as because of concern about sexual molestation. Children in these 2 age groups should receive 2 doses of the vaccine, with the second dose administered 6 to 12 months after the first dose.
The second target group for vaccination is individuals who are aged 13 to 26 years who have never been vaccinated. They should be offered catch-up vaccination. If older than age 15, they should receive 3 doses of the vaccine, with the second dose administered 1 to 2 months after the first dose and the third dose administered 6 months after the first dose.1
A third target group is individuals who are aged 27 to 45 years and who, in their own opinion or in the opinion of their physician, are at new or increased risk for HPV infection. These individuals should receive the 3-dose vaccine series as outlined above.1
Patients in any age range who are immunocompromised, for example, due to HIV infection, should receive the 3-dose series.1
The approximate retail cost of a single 0.5-mL intramuscular dose of the 9-valent vaccine is $240 (www.goodrx.com).
Vaccine adverse effects
The most common reactions to the HPV vaccine are inflammation at the site of injection, fatigue, headache, fever, gastrointestinal upset, vertigo, cough, and oropharyngeal discomfort. The most serious reaction—which fortunately is very rare—is anaphylaxis.1
Contraindications to the vaccine
The HPV vaccine should not be used in any patient who is hypersensitive to any component of the vaccine, including yeast. It should not be given to a patient who is moderately or severely ill at the time of the scheduled administration. Because of an abundance of caution, the manufacturer also recommends that the vaccine not be given to pregnant women even though the agent does not contain live virus.1
Of note, a study by Scheller and colleagues was very reassuring about the lack of adverse effects of HPV vaccine administration in pregnancy.8 The authors evaluated a large cohort of pregnant women in Demark and found that exposure to the vaccine was not associated with an increase in the frequency of major birth defects, spontaneous abortion, preterm delivery, low birthweight, fetal growth restriction, or stillbirth.8
Barriers to vaccination
One important barrier to HPV vaccination is patient apprehension that the vaccine may cause genital tract or oropharyngeal cancer. The patient should be reassured that the vaccine does not contain infectious viral particles and does not transmit infection. Rather, it builds robust immunity to infection.
Another important barrier is the misconception that the vaccine will promote sexual promiscuity in preteenagers and teenagers. Absolutely no evidence supports this belief. Multiple studies have demonstrated that teenagers do not engage in more high-risk sexual behavior following vaccination.
A specific barrier related to vaccination of young boys is the philosophical viewpoint that, “Why should my young male child be vaccinated to protect against a disease (specifically cervical cancer) that occurs only in girls and women?” The appropriate answer to this question is that the vaccine also protects against penile cancer, anal cancer, oropharyngeal cancer, and genital warts. While penile and anal cancers are rare, the other 2 conditions are not. In fact, oropharyngeal cancer is significantly more common in males than females.
A final important barrier to HPV vaccination is cost. The new evidence that demonstrated the effectiveness of a 2-dose vaccine series, and even single-dose vaccination, is of great importance in minimizing cost of the HPV vaccine series, in the absence of full reimbursement by public and private insurance agencies.
Continue to: Creating an effective vaccination program...
Creating an effective vaccination program
The following commonsense guidelines, which we have implemented at our medical center, should be helpful in organizing an effective HPV vaccination program for your office or department4,9,10:
- One clinician in the department or practice should be designated the “vaccination champion.” This individual should provide colleagues with periodic updates, emphasizing the importance of the HPV vaccine and other vaccines, such as Tdap (tetanus, diphtheria, pertussis), influenza, COVID, pneumococcal, hepatitis B, herpes zoster (shingles), and RSV (respiratory syncytial virus).
- One staff member in the practice or department should be designated as the go-to person for all logistical matters related to vaccines. This individual should be responsible for estimating usage, ordering vaccines, and storing them properly. He or she also should be knowledgeable about the cost of the vaccines and insurance reimbursement for the vaccines.
- Signs and educational materials should be posted in strategic locations in the office, advising patients of the importance of timely vaccination for themselves and their adolescent children.
- At every encounter, patients should be encouraged to receive the HPV vaccine series if they are in the appropriate age range and social situation for vaccination. They should not be required to have HPV testing before vaccine administration.
- Key leaders in the department or practice should lobby effectively with their pediatrician colleagues and with public and private insurance companies to encourage timely administration and proper coverage of this important immunization.
Other measures to reduce the risk of HPV-mediated malignancies
Practitioners should advise their patients to:
- Be circumspect in selection of sexual partners.
- Use male or female condoms when engaging in vaginal, anal, and/or oral sex with multiple partners, particularly those who may have genital or oral condylomas.
- Have regular Pap tests, every 3 to 5 years, depending upon age. More frequent testing may be indicated if there is a history of previous abnormal testing.
- Seek prompt medical or surgical treatment for genital or oral condylomas.
CASE Resolved with HPV vaccination
This patient is an excellent candidate for catch-up vaccination. She should receive the first dose of the 9-valent HPV vaccine at the time of her postpartum appointment. The second dose should be administered 1 to 2 months later. The third dose should be administered 6 months after the first dose. She also should have a Pap test, either cytology alone or cytology plus HPV screening. If the latter test is chosen and is reassuring, she will not need retesting for 5 years. If the former test is chosen, she should have a repeat test in 3 years. ●
- The overwhelming majority of precancerous lesions and overt malignancies of the genital tract and oropharynx are caused by oncogenic strains of HPV.
- Most of these cancers could be prevented if patients were vaccinated with the 9-valent HPV vaccine.
- The HPV vaccine should be offered to all children beginning at age 11 and to selected high-risk children at age 9. For children aged 14 years and younger, 2 doses of the vaccine are sufficient to induce a robust immune response. The second dose should be administered 6 to 12 months after the first dose.
- Individuals in the age range 13 to 26 years should be offered catch-up vaccination if they have not been previously vaccinated.
- Persons in the age range 27 to 45 years also should be offered vaccination if they have developed a new high-risk profile.
- Persons older than age 15, or those of any age with immunocompromising conditions, should receive 3 doses of the vaccine. The second dose should be administered 1 to 2 months after the first dose, and the third dose should be given 6 months after the first dose.
- The vaccine does not prevent the progression of preexisting infection or clear an infection that is already present at the time of vaccination.
- As a general rule, the vaccine should be deferred during pregnancy, although no adverse effects have been documented when the vaccine has been administered to pregnant women.
- Markowitz LE, Unger ER. Human papilloma virus vaccination. N Engl J Med. 2023;388:1790-1798.
- Schiller JT, Castellsague X, Garland SM. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine. 2012;30(suppl 5): F123-F138.
- Lei J, Ploner A, Elfstrom KM, et al. HPV vaccination and the risk of invasive cervical cancer. N Engl J Med. 2020;383: 1340-1348.
- ACOG Committee Opinion Summary No. 809. Human papillomavirus vaccination. Obstet Gynecol. 2020;136:435-436.
- Barbieri RL. 9vHPV vaccine: prevention of oropharyngeal cancer. OBG Manag. 2020;32:9, 14-15.
- Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316:2411-2421.
- Watson-Jones D, Changalucha J, Whitworth H, et al. Immunogenicity and safety of one-dose human papillomavirus vaccine compared with two or three doses in Tanzanian girls (DoRIS): an open-label, randomised noninferiority trial. Lancet Glob Health. 2022;10:e1473-e1484.
- Scheller NM, Pasternak B, Molgaard-Nielsen D, et al. Quadrivalent HPV vaccination and the risk of adverse pregnancy outcomes. N Engl J Med. 2017;376:1223-1233.
- ACOG Committee Opinion Summary No. 641. Human papillomavirus vaccination. Obstet Gynecol. 2015;126:693.
- Boitano TKL, Ketch PW, Scarinci IC, et al. An update on human papillomavirus vaccination in the United States. Obstet Gynecol. 2023;141:324-330.
- Markowitz LE, Unger ER. Human papilloma virus vaccination. N Engl J Med. 2023;388:1790-1798.
- Schiller JT, Castellsague X, Garland SM. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine. 2012;30(suppl 5): F123-F138.
- Lei J, Ploner A, Elfstrom KM, et al. HPV vaccination and the risk of invasive cervical cancer. N Engl J Med. 2020;383: 1340-1348.
- ACOG Committee Opinion Summary No. 809. Human papillomavirus vaccination. Obstet Gynecol. 2020;136:435-436.
- Barbieri RL. 9vHPV vaccine: prevention of oropharyngeal cancer. OBG Manag. 2020;32:9, 14-15.
- Iversen OE, Miranda MJ, Ulied A, et al. Immunogenicity of the 9-valent HPV vaccine using 2-dose regimens in girls and boys vs a 3-dose regimen in women. JAMA. 2016;316:2411-2421.
- Watson-Jones D, Changalucha J, Whitworth H, et al. Immunogenicity and safety of one-dose human papillomavirus vaccine compared with two or three doses in Tanzanian girls (DoRIS): an open-label, randomised noninferiority trial. Lancet Glob Health. 2022;10:e1473-e1484.
- Scheller NM, Pasternak B, Molgaard-Nielsen D, et al. Quadrivalent HPV vaccination and the risk of adverse pregnancy outcomes. N Engl J Med. 2017;376:1223-1233.
- ACOG Committee Opinion Summary No. 641. Human papillomavirus vaccination. Obstet Gynecol. 2015;126:693.
- Boitano TKL, Ketch PW, Scarinci IC, et al. An update on human papillomavirus vaccination in the United States. Obstet Gynecol. 2023;141:324-330.

Hepatitis B infection in pregnancy: Essentials of antiviral therapy and immunoprophylaxis
Hepatitis B is one of the more common infections encountered in the daily practice of obstetrics. It is responsible for 40% to 45% of all cases of viral hepatitis.1,2 Hepatitis B may cause serious complications in both the infected mother and neonate.
In this article, I review the virology, epidemiology, and clinical presentation of hepatitis B and then discuss the key diagnostic tests and, subsequently, the clinical management for both the mother and neonate. I focus particular attention on relatively new information about the value of specific antiviral medication to enhance the protective effect of conventional neonatal immunoprophylaxis.
To set the framework for the discussion, consider the following 2 case studies.
CASE 1 Undetectable level of hepatitis B surface antibody in a pregnant woman
A 25-year-old healthy primigravid woman at 10 weeks’ gestation had a series of laboratory studies that included a test for hepatitis B surface antigen (HBsAg) and hepatitis B surface antibody (HBsAb). The test for the surface antigen was negative. The test for the surface antibody was below the level of detection. Upon questioning, the patient indicates that she received the 3-dose hepatitis B vaccine when she was age 13 years.
- What treatment, if any, is indicated for this patient?
- What treatment is indicated for her neonate?
CASE 2 Pregnant woman tests positive for hepatitis B surface antigen
A 31-year-old woman (G3P2002) at 12 weeks’ gestation tested positive for HBsAg. She indicates that she never has had symptomatic hepatitis and that she considers herself to be in excellent health.
- What additional laboratory tests are indicated at this time?
- What additional laboratory test should be performed at the end of the second trimester?
- What treatment is indicated for the mother and neonate?
Virology and epidemiology of hepatitis B
Hepatitis B is caused by a double-stranded, enveloped DNA virus. The virus has 10 genotypes and 24 subtypes.3 The organism contains 3 major antigens. Detection of these antigens and their corresponding antibodies is an essential step in the diagnostic workup of patients who may be infected.
The surface antigen (HBsAg) confers infectivity and is the most valuable serologic marker of infection. The e antigen (HBeAg) is not present in every infected patient. It is secreted from infected cells, but it is not incorporated into the viral particle. When present, it denotes a high level of viral replication and exceptionally high infectivity. The core antigen (HBcAg) is a valuable serologic marker for distinguishing between acute and chronic infection.1-3
Hepatitis B is highly infectious, much more so than HIV or hepatitis C. The virus has an incubation period of 4 weeks to 6 months, and the duration of incubation is inversely related to the size of the viral inoculum. The virus is transmitted in 3 principal ways: sexual contact with contaminated genital tract secretions, contact with infected blood from sharing contaminated drug-injecting paraphernalia or from receiving a blood transfusion (extremely rare today), and transmission from an infected mother to her neonate. Perinatal transmission occurs primarily during the delivery process as opposed to transplacental infection. Transmission also can occur by more casual household contact, such as sharing eating utensils, kissing, and handling an infant.1,2,4,5
Worldwide, more than 400 million people have chronic hepatitis B infection. In the United States, approximately 1.25 to 1.5 million individuals are infected. Several groups are at particularly high risk for being infected, including1-3:
- Asians
- Alaska Natives
- sub-Saharan Africans
- sex workers
- intravenous drug users
- individuals with hemophilia
- international travelers
- staff and residents of long-term care facilities
- tattoo recipients.
Continue to: Clinical presentation...
Clinical presentation
Approximately 90% of adult patients who contract hepatitis B, either symptomatically or asymptomatically, will develop protective levels of antibody and clear the virus from their system. They will then have lifelong immunity to reinfection. Approximately 10% of patients will fail to develop protective levels of antibody and will become chronically infected, posing a risk to their household members, sexual contacts, and their fetus if they become pregnant. Persistence of the surface antigen in the patient’s serum for more than 6 months denotes chronic infection. A very small number of individuals—less than 1%—will develop acute liver failure and experience a fatal outcome.1-3,5
In the United States, the prevalence of acute hepatitis B in pregnancy is 1 to 2 per 1,000. Clinical manifestations typically include anorexia, nausea, low-grade fever, right upper quadrant pain and tenderness, passage of clay-colored stools, and jaundice.
The prevalence of chronic infection in pregnancy is significantly higher, approximately 5 to 15 per 1,000. Over the long term, patients with chronic infection are at risk for progressive liver injury, including cirrhosis and even hepatocellular carcinoma. These serious sequelae are particularly likely to occur when the patient is co-infected with hepatitis C, D, or both. The overall risk of progression to chronic cirrhosis is approximately 15% to 30%. In patients who progress to cirrhosis, the annual incidence of hepatocellular carcinoma is 10%.1-3
Diagnosis of hepatitis B infection
Patients with acute hepatitis B will test positive for HBsAg and immunoglobulin M (IgM) antibody to the core antigen. Some patients will also test positive for HBeAg. Assessment of the patient’s serum by polymerase chain reaction (PCR) allows quantitation of the viral load, which often is expressed as viral copies per milliliter. Alternatively, the quantitative hepatitis B DNA concentration may be expressed as international units per milliliter (IU/mL). The World Health Organization recommends this latter quantitative method. Multiplying the DNA in IU/mL by 5.6 provides the conversion to viral copies per milliliter.
Patients with chronic hepatitis B infection will test positive for the HBsAg and for immunoglobulin G (IgG) antibody to the core antigen. They may also have a positive test for the HBeAg, and PCR may be used to quantify the viral load.1-3
Managing hepatitis B infection in pregnancy
General supportive measures. All pregnant patients should be tested for the HBsAg and HBsAb at the time of the first prenatal appointment. The tests should be repeated at the beginning of the third trimester in high-risk patients. Seropositive patients should have a hepatitis B genotype, a test for the e antigen, and tests for other sexually transmissible infections (gonorrhea, chlamydia, syphilis, HIV) and for hepatitis C and D. Liver function tests should be performed to assess for elevations in the alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. Patients with elevated transaminase enzymes should have a coagulation profile to be certain they are not at risk for a coagulopathy.
At the end of the second trimester, patients should have a PCR assessment to determine the viral load. This assessment will be important for deciding if specific antiviral therapy is indicated during the third trimester to enhance the effects of neonatal immunoprophylaxis (see below). Of note, patients who are positive for the e antigen may have a very high viral load and yet have normal or near-normal transaminase levels. This seemingly paradoxical finding reflects the non-cytopathic nature of hepatitis B.
The patient should optimize her nutrition and sleep. She should avoid, or at least minimize, medications such as acetaminophen that could cause further liver injury. Without question, she should refrain from consuming even small amounts of alcohol. She should be tested for immunity to hepatitis A; if found to be susceptible, she should be vaccinated with the hepatitis A vaccine. This agent is an inactivated vaccine and is safe for administration at any time in pregnancy.1,2,5
Household contacts. In addition to the measures outlined above, the patient’s household contacts, particularly her sexual partner(s), should be tested for immunity to hepatitis B. If they do not have immunity by virtue of natural infection or previous vaccination, they should receive the hepatitis B vaccine series. It is also prudent to provide the sexual partner(s) with an initial dose of hepatitis B immune globulin (HBIG) to provide a temporary level of passive immunity.
Postdelivery care. After delivery, the patient should be referred to an infectious disease specialist or hepatologist for consideration of long-term treatment with antiviral agents, such as interferon alfa, pegylated interferon alfa, lamivudine, adefovir, entecavir, telbivudine, or tenofovir.6 The principal candidates for treatment are those who have cirrhosis and detectable levels of hepatitis B DNA. The ultimate goal of treatment is to reduce the serum hepatitis B DNA concentration to an undetectable level. Once the surface antigenemia is cleared, treatment can be stopped. A cure is defined when the absence of hepa-titis B DNA in the serum is sustained.
- Hepatitis B is a DNA virus that is transmitted via sexual contact, exposure to infected blood, and from an infected mother to her fetus.
- Most patients in our practice will most likely have chronic, asymptomatic infection, and the diagnosis will be established by detection of HBsAg in the patient’s serum.
- All obstetric patients should be tested for both HBsAg and HBsAb.
- Patients who are positive for the surface antigen should be tested for HIV infection and hepatitis C and D. They also should have a determination of the hepatitis B genotype and viral load and assessment of liver function (ALT, AST).
- Patients who are chronically infected with hepatitis B should be vaccinated against hepatitis A to prevent further liver injury. They also should avoid medications that might cause hepatic injury.
- Patients who have a viral DNA concentration greater than 200,000 IU/mL or a viral load greater than 1,120,000 million copies/mL should be treated with tenofovir, 300 mg daily, from week 28 until 4 to 8 weeks after delivery.
- Infants delivered to infected mothers should receive HBIG within 12 hours of birth and then begin the 3-dose hepatitis B vaccine series. The first dose should be administered prior to hospital discharge.
- Infants delivered to mothers who are negative for the surface antigen should begin the hepatitis B vaccine series prior to discharge from the hospital.
- Mothers who test negative for HBsAb should be questioned about prior vaccination. If they have never been vaccinated, they should receive the 3-dose vaccine series. If they have been vaccinated, they should receive a single hepatitis B vaccine booster. The vaccine is safe for administration at any time during pregnancy.
- Infected mothers may breastfeed as long as they do not have cracked or bleeding nipples or exudative skin lesions near the nipple(s).
Neonatal immunoprophylaxis
The Centers for Disease Control and Prevention recommends universal hepatitis B vaccination for all newborns. The first dose of the vaccine should be administered prior to hospital discharge. The second and third doses should be administered 1 and 6 months later.1,2,5 There are few, if any, medical contraindications to neonatal vaccination. For the vast majority of infants, the immunity induced by vaccination is lifelong. For a small number, immunity may wane over time. Thus, reassessment of the HBsAb concentration is indicated in selected situations, for example, acute high-risk exposure to an infected person, development of an immunosuppressive disorder, or pregnancy.
Infants delivered to mothers who are infected with hepatitis B also should receive HBIG in addition to the vaccine. HBIG provides passive immunization to counteract the high viral inoculum encountered by the neonate during delivery. This preparation should be administered within 12 hours of birth.1,2,5
In the absence of immunoprophylaxis, a neonate delivered to a mother who is seropositive for HBsAg has a 20% to 30% probability of becoming chronically infected. If the mother is positive for both the surface antigen and the e antigen, the risk of chronic infection increases to almost 90%. Approximately 90% of infants who are infected in the perinatal period subsequently develop chronic infection. However, with appropriate immunoprophylaxis in the neonatal period, the risk of perinatal transmission is reduced by 85% to 95%.1,2,5
Cesarean delivery offers no additional protection beyond that provided by immunoprophylaxis. Moreover, because immunoprophylaxis is so effective, infected mothers may breastfeed without fear of transmitting infection to their infant. Shi and colleagues published a systematic review and meta-analysis of the risk associated with breastfeeding in hepatitis B–infected mothers.7 Infants who breastfed did not have a higher rate of mother-to-child transmission, regardless of whether they received combined immunoprophylaxis or only hepatitis B vaccine and regardless of whether the HBsAg was detected in the mother’s breast milk. The only precaution is the need to avoid breastfeeding if the nipples are cracked or bleeding or if exudative lesions are present on the skin near the nipple.
Continue to: Maternal antiviral therapy...
Maternal antiviral therapy
As noted above, neonatal immunoprophylaxis is 85% to 95% effective in preventing perinatal transmission of hepatitis B infection. Failures of prophylaxis are primarily due to antenatal transmission in patients who have exceptionally high viral loads. Several cutoffs have been used to define “high viral load,” including greater than 1 to 2 million copies/mL and a hepatitis B DNA concentration greater than 200,000 IU/mL. There is not a perfect consensus on the appropriate cutoff.
In essence, 2 different approaches have been tried to further reduce the risk of perinatal transmission in these high-risk patients.8 The first major initiative was administration of HBIG (100–200 IU) intramuscularly to the patient at 28, 32, and 36 weeks. The outcomes with this approach have been inconsistent, due, at least in part, to varying doses of the agent and various cutoffs for defining “high risk,” and this intervention is no longer recommended.1,2
The second major approach is administration of specific antiviral drugs to the mother during the third trimester. The first agent widely used in clinical practice was lamivudine. In a systematic review and meta-analysis, Shi and colleagues reported that, in infants whose mothers received lamivudine plus conventional neonatal immunuprophylaxis, the risk of perinatal infection was significantly reduced compared with infants who received only immunoprophylaxis.9
Although lamivudine is effective, there is considerable concern about the rapid development of viral resistance to the medication. Accordingly, most attention today is focused on the use of tenofovir to prevent perinatal transmission.
In an important early investigation, Pan and colleagues reported the results of a randomized controlled trial conducted in China in women with a hepatitis B DNA concentration greater than 200,000 IU/mL (viral load > 1,120,000 copies/mL).10 Patients also were positive for the e antigen. Ninety-two patients were assigned to tenofovir disoproxil fumarate (TDF), 300 mg daily, from 30 to 32 weeks until postpartum week 4 plus conventional neonatal immunoprophylaxis, and 100 patients were assigned to immunoprophylaxis alone. In the intention-to-treat analysis, 18 neonates in the control group were infected compared with 5 in the treatment group (P = .007). In the per-protocol analysis, 7 neonates in the control group were infected compared with 0 in the treatment group (P = .01). No clinically significant adverse maternal or neonatal effects occurred in the treatment group.
Subsequently, Jourdain and colleagues reported a multicenter, double-blind trial conducted in 17 public health hospitals in Thailand.11 TDF (300 mg daily) or placebo was administered from 28 weeks’ gestation until 8 weeks postpartum. Patients in both arms of the study were positive for the e antigen; 87% to 90% of the patients had a serum hepatitis B DNA concentration greater than 200,000 IU/mL.Following birth, infants in both groups received an injection of HBIG and then 4 doses of hepatitis B vaccine (0, 1, 2, 4, and 6 months). Both the HBIG and hepatitis B vaccine were administered very promptly after birth (median time, 1.2–1.3 hours).
At 6 months after delivery, 2% of infants in the placebo group (3 of 147) were HBsAg-positive compared with none of the infants in the treatment arm.11 No serious adverse effects occurred in infants in the TDF group. This difference in outcome was not statistically significant, but the overall rate of infection was so low in both groups that the sample size was definitely too small to exclude a type 2 statistical error. Moreover, the fourth dose of neonatal hepatitis B vaccine may have contributed to the surprisingly low rate of perinatal transmission. Of note, the serum hepatitis B DNA concentration in the TDF group declined from a mean of 7.6 log10 IU/mL to a mean of 4.0 log10 IU/mL at delivery.
In the most recent report, Wang and colleagues reported the results of a prospective cohort study in patients with a hepatitis B virus DNA concentration greater than 200,000 IU/mL.12 Beginning at either 24 or 32 weeks, patients were assigned to treatment with either oral TDF (300 mg daily) or oral telbivudine (LdT, 600 mg daily). The medications were continued for 4 weeks postpartum. In the intention-to-treat analysis, the rates of perinatal transmission were comparable, 1.5% versus 1.8%. In the per-protocol analysis, no infants in either group were infected. However, the predelivery decline in hepatitis Bvirus DNA concentration was greater in the TDF group. The ALT elevation rate was also lower in the TDF group. Patients in the LdT group had fewer problems with anorexia but more instances of arthralgia compared with those in the TDF group.
Based primarily on these 3 investigations, I recommend that all infected patients with a hepatitis B DNA concentration greater than 200,000 IU/mL or a viral load greater than 1,120,000 million copies/mL receive oral TDF, 300 mg daily, from 28 weeks until at least 4 to 8 weeks postpartum. The decision about duration of postpartum treatment should be made in consultation with an infectious disease specialist or hepatologist.
Case studies resolved
CASE 1 No protective level of surface antibody
This patient should promptly receive a single booster dose of the hepatitis B vaccine. The vaccine is an inactivated agent and is safe for administration at any time in pregnancy. Following delivery and prior to discharge from the hospital, the neonate should receive the first dose of the hepatitis B vaccine. A second dose should be administered 1 month later, and a third dose should be administered 6 months after the first dose.
CASE 2 Mother is seropositive for HBsAg
This patient should be tested immediately for HIV infection and hepatitis C and D. The hepatitis B viral genotype should be determined. She also should have a panel of liver function tests. If any of these tests are abnormal, a coagulation profile should be obtained to be certain that the patient is not at risk for a coagulopathy. Near the end of the second trimester, a hepatitis B viral load should be obtained. If the viral DNA concentration is greater than 200,000 IU/mLor a viral load greater than 1,120,000 million copies/mL, the patient should be treated with tenofovir, 300 mg daily, from week 28 until at least 4 weeks after delivery. The neonate should receive an injection of HBIG within 12 hours of birth and the first dose of the hepatitis B vaccine prior to discharge from the hospital. Two additional doses of the vaccine should be administered 1 and 6 months later. ●
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:862.
- Bernstein HB, Lee MJ. Maternal and perinatal infection in pregnancy: viral. In: Landon MB, Galan HL, Jauniaux ERM, et al, eds. Gabbe’s Obstetrics. Normal and Problem Pregnancies. 8th ed. Elsevier; 2021;1092.
- Dusheiko G, Agarwal K, Maini MK. New approaches to chronic hepatitis B. N Engl J Med. 2023;388:55-69.
- Ma L, Alla NR, Li X, et al. Mother to child transmission of HBV: review of current clinical management and prevention strategies. Rev Med Virol. 2014; 24: 396-406.
- Society for Maternal-Fetal Medicine; Dionne-Odom J, Tita ATN, Silverman NS. SMFM consult: preventing vertical transmission of hepatitis B. Contemporary OB/GYN. September 22, 2015. Accessed August 21, 2023. https://www .contemporaryobgyn.net/view/smfm-consult-preventing -vertical-transmission-hepatitis-b
- Lok ASF. The maze of treatments for hepatitis B. N Engl J Med. 2005;352:2743-2746.
- Shi Z, Yang Y, Wang H, et al. Breastfeeding of newborns by mothers carrying hepatitis B virus: a meta-analysis and systematic review. Arch Pediatr Adolesc Med. 2011;165:837-846.
- Dusheiko G. A shift in thinking to reduce mother-to-infant transmission of hepatitis B. N Engl J Med. 2018;378:952-953.
- Shi Z, Yang Y, Ma L, et al. Lamivudine in late pregnancy to interrupt in utero transmission of hepatitis B virus: a systematic review and meta-analysis. Obstet Gynecol. 2010;116:147-159.
- Pan C, Duan Z, Dai E, et al; China Study Group for the Motherto-Child Transmission of Hepatitis B. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med. 2016;374:2324-2334.
- Jourdain G, Ngo-Giang-Huong N, Harrison L, et al. Tenofovir versus placebo to prevent perinatal transmission of hepatitis B. N Engl J Med. 2018;378:911-923.
- Wang M, Ran R, Zhu Y, et al. Comparison of tenofovir disoproxil fumarate and telbivudine in preventing hepatitis B transmission in mothers with high viral load. Int J Gynaecol Obstet. 2023:160:646-652.

Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The author reports no financial relationships relevant to this article.
Hepatitis B is one of the more common infections encountered in the daily practice of obstetrics. It is responsible for 40% to 45% of all cases of viral hepatitis.1,2 Hepatitis B may cause serious complications in both the infected mother and neonate.
In this article, I review the virology, epidemiology, and clinical presentation of hepatitis B and then discuss the key diagnostic tests and, subsequently, the clinical management for both the mother and neonate. I focus particular attention on relatively new information about the value of specific antiviral medication to enhance the protective effect of conventional neonatal immunoprophylaxis.
To set the framework for the discussion, consider the following 2 case studies.
CASE 1 Undetectable level of hepatitis B surface antibody in a pregnant woman
A 25-year-old healthy primigravid woman at 10 weeks’ gestation had a series of laboratory studies that included a test for hepatitis B surface antigen (HBsAg) and hepatitis B surface antibody (HBsAb). The test for the surface antigen was negative. The test for the surface antibody was below the level of detection. Upon questioning, the patient indicates that she received the 3-dose hepatitis B vaccine when she was age 13 years.
- What treatment, if any, is indicated for this patient?
- What treatment is indicated for her neonate?
CASE 2 Pregnant woman tests positive for hepatitis B surface antigen
A 31-year-old woman (G3P2002) at 12 weeks’ gestation tested positive for HBsAg. She indicates that she never has had symptomatic hepatitis and that she considers herself to be in excellent health.
- What additional laboratory tests are indicated at this time?
- What additional laboratory test should be performed at the end of the second trimester?
- What treatment is indicated for the mother and neonate?
Virology and epidemiology of hepatitis B
Hepatitis B is caused by a double-stranded, enveloped DNA virus. The virus has 10 genotypes and 24 subtypes.3 The organism contains 3 major antigens. Detection of these antigens and their corresponding antibodies is an essential step in the diagnostic workup of patients who may be infected.
The surface antigen (HBsAg) confers infectivity and is the most valuable serologic marker of infection. The e antigen (HBeAg) is not present in every infected patient. It is secreted from infected cells, but it is not incorporated into the viral particle. When present, it denotes a high level of viral replication and exceptionally high infectivity. The core antigen (HBcAg) is a valuable serologic marker for distinguishing between acute and chronic infection.1-3
Hepatitis B is highly infectious, much more so than HIV or hepatitis C. The virus has an incubation period of 4 weeks to 6 months, and the duration of incubation is inversely related to the size of the viral inoculum. The virus is transmitted in 3 principal ways: sexual contact with contaminated genital tract secretions, contact with infected blood from sharing contaminated drug-injecting paraphernalia or from receiving a blood transfusion (extremely rare today), and transmission from an infected mother to her neonate. Perinatal transmission occurs primarily during the delivery process as opposed to transplacental infection. Transmission also can occur by more casual household contact, such as sharing eating utensils, kissing, and handling an infant.1,2,4,5
Worldwide, more than 400 million people have chronic hepatitis B infection. In the United States, approximately 1.25 to 1.5 million individuals are infected. Several groups are at particularly high risk for being infected, including1-3:
- Asians
- Alaska Natives
- sub-Saharan Africans
- sex workers
- intravenous drug users
- individuals with hemophilia
- international travelers
- staff and residents of long-term care facilities
- tattoo recipients.
Continue to: Clinical presentation...
Clinical presentation
Approximately 90% of adult patients who contract hepatitis B, either symptomatically or asymptomatically, will develop protective levels of antibody and clear the virus from their system. They will then have lifelong immunity to reinfection. Approximately 10% of patients will fail to develop protective levels of antibody and will become chronically infected, posing a risk to their household members, sexual contacts, and their fetus if they become pregnant. Persistence of the surface antigen in the patient’s serum for more than 6 months denotes chronic infection. A very small number of individuals—less than 1%—will develop acute liver failure and experience a fatal outcome.1-3,5
In the United States, the prevalence of acute hepatitis B in pregnancy is 1 to 2 per 1,000. Clinical manifestations typically include anorexia, nausea, low-grade fever, right upper quadrant pain and tenderness, passage of clay-colored stools, and jaundice.
The prevalence of chronic infection in pregnancy is significantly higher, approximately 5 to 15 per 1,000. Over the long term, patients with chronic infection are at risk for progressive liver injury, including cirrhosis and even hepatocellular carcinoma. These serious sequelae are particularly likely to occur when the patient is co-infected with hepatitis C, D, or both. The overall risk of progression to chronic cirrhosis is approximately 15% to 30%. In patients who progress to cirrhosis, the annual incidence of hepatocellular carcinoma is 10%.1-3
Diagnosis of hepatitis B infection
Patients with acute hepatitis B will test positive for HBsAg and immunoglobulin M (IgM) antibody to the core antigen. Some patients will also test positive for HBeAg. Assessment of the patient’s serum by polymerase chain reaction (PCR) allows quantitation of the viral load, which often is expressed as viral copies per milliliter. Alternatively, the quantitative hepatitis B DNA concentration may be expressed as international units per milliliter (IU/mL). The World Health Organization recommends this latter quantitative method. Multiplying the DNA in IU/mL by 5.6 provides the conversion to viral copies per milliliter.
Patients with chronic hepatitis B infection will test positive for the HBsAg and for immunoglobulin G (IgG) antibody to the core antigen. They may also have a positive test for the HBeAg, and PCR may be used to quantify the viral load.1-3
Managing hepatitis B infection in pregnancy
General supportive measures. All pregnant patients should be tested for the HBsAg and HBsAb at the time of the first prenatal appointment. The tests should be repeated at the beginning of the third trimester in high-risk patients. Seropositive patients should have a hepatitis B genotype, a test for the e antigen, and tests for other sexually transmissible infections (gonorrhea, chlamydia, syphilis, HIV) and for hepatitis C and D. Liver function tests should be performed to assess for elevations in the alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. Patients with elevated transaminase enzymes should have a coagulation profile to be certain they are not at risk for a coagulopathy.
At the end of the second trimester, patients should have a PCR assessment to determine the viral load. This assessment will be important for deciding if specific antiviral therapy is indicated during the third trimester to enhance the effects of neonatal immunoprophylaxis (see below). Of note, patients who are positive for the e antigen may have a very high viral load and yet have normal or near-normal transaminase levels. This seemingly paradoxical finding reflects the non-cytopathic nature of hepatitis B.
The patient should optimize her nutrition and sleep. She should avoid, or at least minimize, medications such as acetaminophen that could cause further liver injury. Without question, she should refrain from consuming even small amounts of alcohol. She should be tested for immunity to hepatitis A; if found to be susceptible, she should be vaccinated with the hepatitis A vaccine. This agent is an inactivated vaccine and is safe for administration at any time in pregnancy.1,2,5
Household contacts. In addition to the measures outlined above, the patient’s household contacts, particularly her sexual partner(s), should be tested for immunity to hepatitis B. If they do not have immunity by virtue of natural infection or previous vaccination, they should receive the hepatitis B vaccine series. It is also prudent to provide the sexual partner(s) with an initial dose of hepatitis B immune globulin (HBIG) to provide a temporary level of passive immunity.
Postdelivery care. After delivery, the patient should be referred to an infectious disease specialist or hepatologist for consideration of long-term treatment with antiviral agents, such as interferon alfa, pegylated interferon alfa, lamivudine, adefovir, entecavir, telbivudine, or tenofovir.6 The principal candidates for treatment are those who have cirrhosis and detectable levels of hepatitis B DNA. The ultimate goal of treatment is to reduce the serum hepatitis B DNA concentration to an undetectable level. Once the surface antigenemia is cleared, treatment can be stopped. A cure is defined when the absence of hepa-titis B DNA in the serum is sustained.
- Hepatitis B is a DNA virus that is transmitted via sexual contact, exposure to infected blood, and from an infected mother to her fetus.
- Most patients in our practice will most likely have chronic, asymptomatic infection, and the diagnosis will be established by detection of HBsAg in the patient’s serum.
- All obstetric patients should be tested for both HBsAg and HBsAb.
- Patients who are positive for the surface antigen should be tested for HIV infection and hepatitis C and D. They also should have a determination of the hepatitis B genotype and viral load and assessment of liver function (ALT, AST).
- Patients who are chronically infected with hepatitis B should be vaccinated against hepatitis A to prevent further liver injury. They also should avoid medications that might cause hepatic injury.
- Patients who have a viral DNA concentration greater than 200,000 IU/mL or a viral load greater than 1,120,000 million copies/mL should be treated with tenofovir, 300 mg daily, from week 28 until 4 to 8 weeks after delivery.
- Infants delivered to infected mothers should receive HBIG within 12 hours of birth and then begin the 3-dose hepatitis B vaccine series. The first dose should be administered prior to hospital discharge.
- Infants delivered to mothers who are negative for the surface antigen should begin the hepatitis B vaccine series prior to discharge from the hospital.
- Mothers who test negative for HBsAb should be questioned about prior vaccination. If they have never been vaccinated, they should receive the 3-dose vaccine series. If they have been vaccinated, they should receive a single hepatitis B vaccine booster. The vaccine is safe for administration at any time during pregnancy.
- Infected mothers may breastfeed as long as they do not have cracked or bleeding nipples or exudative skin lesions near the nipple(s).
Neonatal immunoprophylaxis
The Centers for Disease Control and Prevention recommends universal hepatitis B vaccination for all newborns. The first dose of the vaccine should be administered prior to hospital discharge. The second and third doses should be administered 1 and 6 months later.1,2,5 There are few, if any, medical contraindications to neonatal vaccination. For the vast majority of infants, the immunity induced by vaccination is lifelong. For a small number, immunity may wane over time. Thus, reassessment of the HBsAb concentration is indicated in selected situations, for example, acute high-risk exposure to an infected person, development of an immunosuppressive disorder, or pregnancy.
Infants delivered to mothers who are infected with hepatitis B also should receive HBIG in addition to the vaccine. HBIG provides passive immunization to counteract the high viral inoculum encountered by the neonate during delivery. This preparation should be administered within 12 hours of birth.1,2,5
In the absence of immunoprophylaxis, a neonate delivered to a mother who is seropositive for HBsAg has a 20% to 30% probability of becoming chronically infected. If the mother is positive for both the surface antigen and the e antigen, the risk of chronic infection increases to almost 90%. Approximately 90% of infants who are infected in the perinatal period subsequently develop chronic infection. However, with appropriate immunoprophylaxis in the neonatal period, the risk of perinatal transmission is reduced by 85% to 95%.1,2,5
Cesarean delivery offers no additional protection beyond that provided by immunoprophylaxis. Moreover, because immunoprophylaxis is so effective, infected mothers may breastfeed without fear of transmitting infection to their infant. Shi and colleagues published a systematic review and meta-analysis of the risk associated with breastfeeding in hepatitis B–infected mothers.7 Infants who breastfed did not have a higher rate of mother-to-child transmission, regardless of whether they received combined immunoprophylaxis or only hepatitis B vaccine and regardless of whether the HBsAg was detected in the mother’s breast milk. The only precaution is the need to avoid breastfeeding if the nipples are cracked or bleeding or if exudative lesions are present on the skin near the nipple.
Continue to: Maternal antiviral therapy...
Maternal antiviral therapy
As noted above, neonatal immunoprophylaxis is 85% to 95% effective in preventing perinatal transmission of hepatitis B infection. Failures of prophylaxis are primarily due to antenatal transmission in patients who have exceptionally high viral loads. Several cutoffs have been used to define “high viral load,” including greater than 1 to 2 million copies/mL and a hepatitis B DNA concentration greater than 200,000 IU/mL. There is not a perfect consensus on the appropriate cutoff.
In essence, 2 different approaches have been tried to further reduce the risk of perinatal transmission in these high-risk patients.8 The first major initiative was administration of HBIG (100–200 IU) intramuscularly to the patient at 28, 32, and 36 weeks. The outcomes with this approach have been inconsistent, due, at least in part, to varying doses of the agent and various cutoffs for defining “high risk,” and this intervention is no longer recommended.1,2
The second major approach is administration of specific antiviral drugs to the mother during the third trimester. The first agent widely used in clinical practice was lamivudine. In a systematic review and meta-analysis, Shi and colleagues reported that, in infants whose mothers received lamivudine plus conventional neonatal immunuprophylaxis, the risk of perinatal infection was significantly reduced compared with infants who received only immunoprophylaxis.9
Although lamivudine is effective, there is considerable concern about the rapid development of viral resistance to the medication. Accordingly, most attention today is focused on the use of tenofovir to prevent perinatal transmission.
In an important early investigation, Pan and colleagues reported the results of a randomized controlled trial conducted in China in women with a hepatitis B DNA concentration greater than 200,000 IU/mL (viral load > 1,120,000 copies/mL).10 Patients also were positive for the e antigen. Ninety-two patients were assigned to tenofovir disoproxil fumarate (TDF), 300 mg daily, from 30 to 32 weeks until postpartum week 4 plus conventional neonatal immunoprophylaxis, and 100 patients were assigned to immunoprophylaxis alone. In the intention-to-treat analysis, 18 neonates in the control group were infected compared with 5 in the treatment group (P = .007). In the per-protocol analysis, 7 neonates in the control group were infected compared with 0 in the treatment group (P = .01). No clinically significant adverse maternal or neonatal effects occurred in the treatment group.
Subsequently, Jourdain and colleagues reported a multicenter, double-blind trial conducted in 17 public health hospitals in Thailand.11 TDF (300 mg daily) or placebo was administered from 28 weeks’ gestation until 8 weeks postpartum. Patients in both arms of the study were positive for the e antigen; 87% to 90% of the patients had a serum hepatitis B DNA concentration greater than 200,000 IU/mL.Following birth, infants in both groups received an injection of HBIG and then 4 doses of hepatitis B vaccine (0, 1, 2, 4, and 6 months). Both the HBIG and hepatitis B vaccine were administered very promptly after birth (median time, 1.2–1.3 hours).
At 6 months after delivery, 2% of infants in the placebo group (3 of 147) were HBsAg-positive compared with none of the infants in the treatment arm.11 No serious adverse effects occurred in infants in the TDF group. This difference in outcome was not statistically significant, but the overall rate of infection was so low in both groups that the sample size was definitely too small to exclude a type 2 statistical error. Moreover, the fourth dose of neonatal hepatitis B vaccine may have contributed to the surprisingly low rate of perinatal transmission. Of note, the serum hepatitis B DNA concentration in the TDF group declined from a mean of 7.6 log10 IU/mL to a mean of 4.0 log10 IU/mL at delivery.
In the most recent report, Wang and colleagues reported the results of a prospective cohort study in patients with a hepatitis B virus DNA concentration greater than 200,000 IU/mL.12 Beginning at either 24 or 32 weeks, patients were assigned to treatment with either oral TDF (300 mg daily) or oral telbivudine (LdT, 600 mg daily). The medications were continued for 4 weeks postpartum. In the intention-to-treat analysis, the rates of perinatal transmission were comparable, 1.5% versus 1.8%. In the per-protocol analysis, no infants in either group were infected. However, the predelivery decline in hepatitis Bvirus DNA concentration was greater in the TDF group. The ALT elevation rate was also lower in the TDF group. Patients in the LdT group had fewer problems with anorexia but more instances of arthralgia compared with those in the TDF group.
Based primarily on these 3 investigations, I recommend that all infected patients with a hepatitis B DNA concentration greater than 200,000 IU/mL or a viral load greater than 1,120,000 million copies/mL receive oral TDF, 300 mg daily, from 28 weeks until at least 4 to 8 weeks postpartum. The decision about duration of postpartum treatment should be made in consultation with an infectious disease specialist or hepatologist.
Case studies resolved
CASE 1 No protective level of surface antibody
This patient should promptly receive a single booster dose of the hepatitis B vaccine. The vaccine is an inactivated agent and is safe for administration at any time in pregnancy. Following delivery and prior to discharge from the hospital, the neonate should receive the first dose of the hepatitis B vaccine. A second dose should be administered 1 month later, and a third dose should be administered 6 months after the first dose.
CASE 2 Mother is seropositive for HBsAg
This patient should be tested immediately for HIV infection and hepatitis C and D. The hepatitis B viral genotype should be determined. She also should have a panel of liver function tests. If any of these tests are abnormal, a coagulation profile should be obtained to be certain that the patient is not at risk for a coagulopathy. Near the end of the second trimester, a hepatitis B viral load should be obtained. If the viral DNA concentration is greater than 200,000 IU/mLor a viral load greater than 1,120,000 million copies/mL, the patient should be treated with tenofovir, 300 mg daily, from week 28 until at least 4 weeks after delivery. The neonate should receive an injection of HBIG within 12 hours of birth and the first dose of the hepatitis B vaccine prior to discharge from the hospital. Two additional doses of the vaccine should be administered 1 and 6 months later. ●
Hepatitis B is one of the more common infections encountered in the daily practice of obstetrics. It is responsible for 40% to 45% of all cases of viral hepatitis.1,2 Hepatitis B may cause serious complications in both the infected mother and neonate.
In this article, I review the virology, epidemiology, and clinical presentation of hepatitis B and then discuss the key diagnostic tests and, subsequently, the clinical management for both the mother and neonate. I focus particular attention on relatively new information about the value of specific antiviral medication to enhance the protective effect of conventional neonatal immunoprophylaxis.
To set the framework for the discussion, consider the following 2 case studies.
CASE 1 Undetectable level of hepatitis B surface antibody in a pregnant woman
A 25-year-old healthy primigravid woman at 10 weeks’ gestation had a series of laboratory studies that included a test for hepatitis B surface antigen (HBsAg) and hepatitis B surface antibody (HBsAb). The test for the surface antigen was negative. The test for the surface antibody was below the level of detection. Upon questioning, the patient indicates that she received the 3-dose hepatitis B vaccine when she was age 13 years.
- What treatment, if any, is indicated for this patient?
- What treatment is indicated for her neonate?
CASE 2 Pregnant woman tests positive for hepatitis B surface antigen
A 31-year-old woman (G3P2002) at 12 weeks’ gestation tested positive for HBsAg. She indicates that she never has had symptomatic hepatitis and that she considers herself to be in excellent health.
- What additional laboratory tests are indicated at this time?
- What additional laboratory test should be performed at the end of the second trimester?
- What treatment is indicated for the mother and neonate?
Virology and epidemiology of hepatitis B
Hepatitis B is caused by a double-stranded, enveloped DNA virus. The virus has 10 genotypes and 24 subtypes.3 The organism contains 3 major antigens. Detection of these antigens and their corresponding antibodies is an essential step in the diagnostic workup of patients who may be infected.
The surface antigen (HBsAg) confers infectivity and is the most valuable serologic marker of infection. The e antigen (HBeAg) is not present in every infected patient. It is secreted from infected cells, but it is not incorporated into the viral particle. When present, it denotes a high level of viral replication and exceptionally high infectivity. The core antigen (HBcAg) is a valuable serologic marker for distinguishing between acute and chronic infection.1-3
Hepatitis B is highly infectious, much more so than HIV or hepatitis C. The virus has an incubation period of 4 weeks to 6 months, and the duration of incubation is inversely related to the size of the viral inoculum. The virus is transmitted in 3 principal ways: sexual contact with contaminated genital tract secretions, contact with infected blood from sharing contaminated drug-injecting paraphernalia or from receiving a blood transfusion (extremely rare today), and transmission from an infected mother to her neonate. Perinatal transmission occurs primarily during the delivery process as opposed to transplacental infection. Transmission also can occur by more casual household contact, such as sharing eating utensils, kissing, and handling an infant.1,2,4,5
Worldwide, more than 400 million people have chronic hepatitis B infection. In the United States, approximately 1.25 to 1.5 million individuals are infected. Several groups are at particularly high risk for being infected, including1-3:
- Asians
- Alaska Natives
- sub-Saharan Africans
- sex workers
- intravenous drug users
- individuals with hemophilia
- international travelers
- staff and residents of long-term care facilities
- tattoo recipients.
Continue to: Clinical presentation...
Clinical presentation
Approximately 90% of adult patients who contract hepatitis B, either symptomatically or asymptomatically, will develop protective levels of antibody and clear the virus from their system. They will then have lifelong immunity to reinfection. Approximately 10% of patients will fail to develop protective levels of antibody and will become chronically infected, posing a risk to their household members, sexual contacts, and their fetus if they become pregnant. Persistence of the surface antigen in the patient’s serum for more than 6 months denotes chronic infection. A very small number of individuals—less than 1%—will develop acute liver failure and experience a fatal outcome.1-3,5
In the United States, the prevalence of acute hepatitis B in pregnancy is 1 to 2 per 1,000. Clinical manifestations typically include anorexia, nausea, low-grade fever, right upper quadrant pain and tenderness, passage of clay-colored stools, and jaundice.
The prevalence of chronic infection in pregnancy is significantly higher, approximately 5 to 15 per 1,000. Over the long term, patients with chronic infection are at risk for progressive liver injury, including cirrhosis and even hepatocellular carcinoma. These serious sequelae are particularly likely to occur when the patient is co-infected with hepatitis C, D, or both. The overall risk of progression to chronic cirrhosis is approximately 15% to 30%. In patients who progress to cirrhosis, the annual incidence of hepatocellular carcinoma is 10%.1-3
Diagnosis of hepatitis B infection
Patients with acute hepatitis B will test positive for HBsAg and immunoglobulin M (IgM) antibody to the core antigen. Some patients will also test positive for HBeAg. Assessment of the patient’s serum by polymerase chain reaction (PCR) allows quantitation of the viral load, which often is expressed as viral copies per milliliter. Alternatively, the quantitative hepatitis B DNA concentration may be expressed as international units per milliliter (IU/mL). The World Health Organization recommends this latter quantitative method. Multiplying the DNA in IU/mL by 5.6 provides the conversion to viral copies per milliliter.
Patients with chronic hepatitis B infection will test positive for the HBsAg and for immunoglobulin G (IgG) antibody to the core antigen. They may also have a positive test for the HBeAg, and PCR may be used to quantify the viral load.1-3
Managing hepatitis B infection in pregnancy
General supportive measures. All pregnant patients should be tested for the HBsAg and HBsAb at the time of the first prenatal appointment. The tests should be repeated at the beginning of the third trimester in high-risk patients. Seropositive patients should have a hepatitis B genotype, a test for the e antigen, and tests for other sexually transmissible infections (gonorrhea, chlamydia, syphilis, HIV) and for hepatitis C and D. Liver function tests should be performed to assess for elevations in the alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels. Patients with elevated transaminase enzymes should have a coagulation profile to be certain they are not at risk for a coagulopathy.
At the end of the second trimester, patients should have a PCR assessment to determine the viral load. This assessment will be important for deciding if specific antiviral therapy is indicated during the third trimester to enhance the effects of neonatal immunoprophylaxis (see below). Of note, patients who are positive for the e antigen may have a very high viral load and yet have normal or near-normal transaminase levels. This seemingly paradoxical finding reflects the non-cytopathic nature of hepatitis B.
The patient should optimize her nutrition and sleep. She should avoid, or at least minimize, medications such as acetaminophen that could cause further liver injury. Without question, she should refrain from consuming even small amounts of alcohol. She should be tested for immunity to hepatitis A; if found to be susceptible, she should be vaccinated with the hepatitis A vaccine. This agent is an inactivated vaccine and is safe for administration at any time in pregnancy.1,2,5
Household contacts. In addition to the measures outlined above, the patient’s household contacts, particularly her sexual partner(s), should be tested for immunity to hepatitis B. If they do not have immunity by virtue of natural infection or previous vaccination, they should receive the hepatitis B vaccine series. It is also prudent to provide the sexual partner(s) with an initial dose of hepatitis B immune globulin (HBIG) to provide a temporary level of passive immunity.
Postdelivery care. After delivery, the patient should be referred to an infectious disease specialist or hepatologist for consideration of long-term treatment with antiviral agents, such as interferon alfa, pegylated interferon alfa, lamivudine, adefovir, entecavir, telbivudine, or tenofovir.6 The principal candidates for treatment are those who have cirrhosis and detectable levels of hepatitis B DNA. The ultimate goal of treatment is to reduce the serum hepatitis B DNA concentration to an undetectable level. Once the surface antigenemia is cleared, treatment can be stopped. A cure is defined when the absence of hepa-titis B DNA in the serum is sustained.
- Hepatitis B is a DNA virus that is transmitted via sexual contact, exposure to infected blood, and from an infected mother to her fetus.
- Most patients in our practice will most likely have chronic, asymptomatic infection, and the diagnosis will be established by detection of HBsAg in the patient’s serum.
- All obstetric patients should be tested for both HBsAg and HBsAb.
- Patients who are positive for the surface antigen should be tested for HIV infection and hepatitis C and D. They also should have a determination of the hepatitis B genotype and viral load and assessment of liver function (ALT, AST).
- Patients who are chronically infected with hepatitis B should be vaccinated against hepatitis A to prevent further liver injury. They also should avoid medications that might cause hepatic injury.
- Patients who have a viral DNA concentration greater than 200,000 IU/mL or a viral load greater than 1,120,000 million copies/mL should be treated with tenofovir, 300 mg daily, from week 28 until 4 to 8 weeks after delivery.
- Infants delivered to infected mothers should receive HBIG within 12 hours of birth and then begin the 3-dose hepatitis B vaccine series. The first dose should be administered prior to hospital discharge.
- Infants delivered to mothers who are negative for the surface antigen should begin the hepatitis B vaccine series prior to discharge from the hospital.
- Mothers who test negative for HBsAb should be questioned about prior vaccination. If they have never been vaccinated, they should receive the 3-dose vaccine series. If they have been vaccinated, they should receive a single hepatitis B vaccine booster. The vaccine is safe for administration at any time during pregnancy.
- Infected mothers may breastfeed as long as they do not have cracked or bleeding nipples or exudative skin lesions near the nipple(s).
Neonatal immunoprophylaxis
The Centers for Disease Control and Prevention recommends universal hepatitis B vaccination for all newborns. The first dose of the vaccine should be administered prior to hospital discharge. The second and third doses should be administered 1 and 6 months later.1,2,5 There are few, if any, medical contraindications to neonatal vaccination. For the vast majority of infants, the immunity induced by vaccination is lifelong. For a small number, immunity may wane over time. Thus, reassessment of the HBsAb concentration is indicated in selected situations, for example, acute high-risk exposure to an infected person, development of an immunosuppressive disorder, or pregnancy.
Infants delivered to mothers who are infected with hepatitis B also should receive HBIG in addition to the vaccine. HBIG provides passive immunization to counteract the high viral inoculum encountered by the neonate during delivery. This preparation should be administered within 12 hours of birth.1,2,5
In the absence of immunoprophylaxis, a neonate delivered to a mother who is seropositive for HBsAg has a 20% to 30% probability of becoming chronically infected. If the mother is positive for both the surface antigen and the e antigen, the risk of chronic infection increases to almost 90%. Approximately 90% of infants who are infected in the perinatal period subsequently develop chronic infection. However, with appropriate immunoprophylaxis in the neonatal period, the risk of perinatal transmission is reduced by 85% to 95%.1,2,5
Cesarean delivery offers no additional protection beyond that provided by immunoprophylaxis. Moreover, because immunoprophylaxis is so effective, infected mothers may breastfeed without fear of transmitting infection to their infant. Shi and colleagues published a systematic review and meta-analysis of the risk associated with breastfeeding in hepatitis B–infected mothers.7 Infants who breastfed did not have a higher rate of mother-to-child transmission, regardless of whether they received combined immunoprophylaxis or only hepatitis B vaccine and regardless of whether the HBsAg was detected in the mother’s breast milk. The only precaution is the need to avoid breastfeeding if the nipples are cracked or bleeding or if exudative lesions are present on the skin near the nipple.
Continue to: Maternal antiviral therapy...
Maternal antiviral therapy
As noted above, neonatal immunoprophylaxis is 85% to 95% effective in preventing perinatal transmission of hepatitis B infection. Failures of prophylaxis are primarily due to antenatal transmission in patients who have exceptionally high viral loads. Several cutoffs have been used to define “high viral load,” including greater than 1 to 2 million copies/mL and a hepatitis B DNA concentration greater than 200,000 IU/mL. There is not a perfect consensus on the appropriate cutoff.
In essence, 2 different approaches have been tried to further reduce the risk of perinatal transmission in these high-risk patients.8 The first major initiative was administration of HBIG (100–200 IU) intramuscularly to the patient at 28, 32, and 36 weeks. The outcomes with this approach have been inconsistent, due, at least in part, to varying doses of the agent and various cutoffs for defining “high risk,” and this intervention is no longer recommended.1,2
The second major approach is administration of specific antiviral drugs to the mother during the third trimester. The first agent widely used in clinical practice was lamivudine. In a systematic review and meta-analysis, Shi and colleagues reported that, in infants whose mothers received lamivudine plus conventional neonatal immunuprophylaxis, the risk of perinatal infection was significantly reduced compared with infants who received only immunoprophylaxis.9
Although lamivudine is effective, there is considerable concern about the rapid development of viral resistance to the medication. Accordingly, most attention today is focused on the use of tenofovir to prevent perinatal transmission.
In an important early investigation, Pan and colleagues reported the results of a randomized controlled trial conducted in China in women with a hepatitis B DNA concentration greater than 200,000 IU/mL (viral load > 1,120,000 copies/mL).10 Patients also were positive for the e antigen. Ninety-two patients were assigned to tenofovir disoproxil fumarate (TDF), 300 mg daily, from 30 to 32 weeks until postpartum week 4 plus conventional neonatal immunoprophylaxis, and 100 patients were assigned to immunoprophylaxis alone. In the intention-to-treat analysis, 18 neonates in the control group were infected compared with 5 in the treatment group (P = .007). In the per-protocol analysis, 7 neonates in the control group were infected compared with 0 in the treatment group (P = .01). No clinically significant adverse maternal or neonatal effects occurred in the treatment group.
Subsequently, Jourdain and colleagues reported a multicenter, double-blind trial conducted in 17 public health hospitals in Thailand.11 TDF (300 mg daily) or placebo was administered from 28 weeks’ gestation until 8 weeks postpartum. Patients in both arms of the study were positive for the e antigen; 87% to 90% of the patients had a serum hepatitis B DNA concentration greater than 200,000 IU/mL.Following birth, infants in both groups received an injection of HBIG and then 4 doses of hepatitis B vaccine (0, 1, 2, 4, and 6 months). Both the HBIG and hepatitis B vaccine were administered very promptly after birth (median time, 1.2–1.3 hours).
At 6 months after delivery, 2% of infants in the placebo group (3 of 147) were HBsAg-positive compared with none of the infants in the treatment arm.11 No serious adverse effects occurred in infants in the TDF group. This difference in outcome was not statistically significant, but the overall rate of infection was so low in both groups that the sample size was definitely too small to exclude a type 2 statistical error. Moreover, the fourth dose of neonatal hepatitis B vaccine may have contributed to the surprisingly low rate of perinatal transmission. Of note, the serum hepatitis B DNA concentration in the TDF group declined from a mean of 7.6 log10 IU/mL to a mean of 4.0 log10 IU/mL at delivery.
In the most recent report, Wang and colleagues reported the results of a prospective cohort study in patients with a hepatitis B virus DNA concentration greater than 200,000 IU/mL.12 Beginning at either 24 or 32 weeks, patients were assigned to treatment with either oral TDF (300 mg daily) or oral telbivudine (LdT, 600 mg daily). The medications were continued for 4 weeks postpartum. In the intention-to-treat analysis, the rates of perinatal transmission were comparable, 1.5% versus 1.8%. In the per-protocol analysis, no infants in either group were infected. However, the predelivery decline in hepatitis Bvirus DNA concentration was greater in the TDF group. The ALT elevation rate was also lower in the TDF group. Patients in the LdT group had fewer problems with anorexia but more instances of arthralgia compared with those in the TDF group.
Based primarily on these 3 investigations, I recommend that all infected patients with a hepatitis B DNA concentration greater than 200,000 IU/mL or a viral load greater than 1,120,000 million copies/mL receive oral TDF, 300 mg daily, from 28 weeks until at least 4 to 8 weeks postpartum. The decision about duration of postpartum treatment should be made in consultation with an infectious disease specialist or hepatologist.
Case studies resolved
CASE 1 No protective level of surface antibody
This patient should promptly receive a single booster dose of the hepatitis B vaccine. The vaccine is an inactivated agent and is safe for administration at any time in pregnancy. Following delivery and prior to discharge from the hospital, the neonate should receive the first dose of the hepatitis B vaccine. A second dose should be administered 1 month later, and a third dose should be administered 6 months after the first dose.
CASE 2 Mother is seropositive for HBsAg
This patient should be tested immediately for HIV infection and hepatitis C and D. The hepatitis B viral genotype should be determined. She also should have a panel of liver function tests. If any of these tests are abnormal, a coagulation profile should be obtained to be certain that the patient is not at risk for a coagulopathy. Near the end of the second trimester, a hepatitis B viral load should be obtained. If the viral DNA concentration is greater than 200,000 IU/mLor a viral load greater than 1,120,000 million copies/mL, the patient should be treated with tenofovir, 300 mg daily, from week 28 until at least 4 weeks after delivery. The neonate should receive an injection of HBIG within 12 hours of birth and the first dose of the hepatitis B vaccine prior to discharge from the hospital. Two additional doses of the vaccine should be administered 1 and 6 months later. ●
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:862.
- Bernstein HB, Lee MJ. Maternal and perinatal infection in pregnancy: viral. In: Landon MB, Galan HL, Jauniaux ERM, et al, eds. Gabbe’s Obstetrics. Normal and Problem Pregnancies. 8th ed. Elsevier; 2021;1092.
- Dusheiko G, Agarwal K, Maini MK. New approaches to chronic hepatitis B. N Engl J Med. 2023;388:55-69.
- Ma L, Alla NR, Li X, et al. Mother to child transmission of HBV: review of current clinical management and prevention strategies. Rev Med Virol. 2014; 24: 396-406.
- Society for Maternal-Fetal Medicine; Dionne-Odom J, Tita ATN, Silverman NS. SMFM consult: preventing vertical transmission of hepatitis B. Contemporary OB/GYN. September 22, 2015. Accessed August 21, 2023. https://www .contemporaryobgyn.net/view/smfm-consult-preventing -vertical-transmission-hepatitis-b
- Lok ASF. The maze of treatments for hepatitis B. N Engl J Med. 2005;352:2743-2746.
- Shi Z, Yang Y, Wang H, et al. Breastfeeding of newborns by mothers carrying hepatitis B virus: a meta-analysis and systematic review. Arch Pediatr Adolesc Med. 2011;165:837-846.
- Dusheiko G. A shift in thinking to reduce mother-to-infant transmission of hepatitis B. N Engl J Med. 2018;378:952-953.
- Shi Z, Yang Y, Ma L, et al. Lamivudine in late pregnancy to interrupt in utero transmission of hepatitis B virus: a systematic review and meta-analysis. Obstet Gynecol. 2010;116:147-159.
- Pan C, Duan Z, Dai E, et al; China Study Group for the Motherto-Child Transmission of Hepatitis B. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med. 2016;374:2324-2334.
- Jourdain G, Ngo-Giang-Huong N, Harrison L, et al. Tenofovir versus placebo to prevent perinatal transmission of hepatitis B. N Engl J Med. 2018;378:911-923.
- Wang M, Ran R, Zhu Y, et al. Comparison of tenofovir disoproxil fumarate and telbivudine in preventing hepatitis B transmission in mothers with high viral load. Int J Gynaecol Obstet. 2023:160:646-652.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:862.
- Bernstein HB, Lee MJ. Maternal and perinatal infection in pregnancy: viral. In: Landon MB, Galan HL, Jauniaux ERM, et al, eds. Gabbe’s Obstetrics. Normal and Problem Pregnancies. 8th ed. Elsevier; 2021;1092.
- Dusheiko G, Agarwal K, Maini MK. New approaches to chronic hepatitis B. N Engl J Med. 2023;388:55-69.
- Ma L, Alla NR, Li X, et al. Mother to child transmission of HBV: review of current clinical management and prevention strategies. Rev Med Virol. 2014; 24: 396-406.
- Society for Maternal-Fetal Medicine; Dionne-Odom J, Tita ATN, Silverman NS. SMFM consult: preventing vertical transmission of hepatitis B. Contemporary OB/GYN. September 22, 2015. Accessed August 21, 2023. https://www .contemporaryobgyn.net/view/smfm-consult-preventing -vertical-transmission-hepatitis-b
- Lok ASF. The maze of treatments for hepatitis B. N Engl J Med. 2005;352:2743-2746.
- Shi Z, Yang Y, Wang H, et al. Breastfeeding of newborns by mothers carrying hepatitis B virus: a meta-analysis and systematic review. Arch Pediatr Adolesc Med. 2011;165:837-846.
- Dusheiko G. A shift in thinking to reduce mother-to-infant transmission of hepatitis B. N Engl J Med. 2018;378:952-953.
- Shi Z, Yang Y, Ma L, et al. Lamivudine in late pregnancy to interrupt in utero transmission of hepatitis B virus: a systematic review and meta-analysis. Obstet Gynecol. 2010;116:147-159.
- Pan C, Duan Z, Dai E, et al; China Study Group for the Motherto-Child Transmission of Hepatitis B. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med. 2016;374:2324-2334.
- Jourdain G, Ngo-Giang-Huong N, Harrison L, et al. Tenofovir versus placebo to prevent perinatal transmission of hepatitis B. N Engl J Med. 2018;378:911-923.
- Wang M, Ran R, Zhu Y, et al. Comparison of tenofovir disoproxil fumarate and telbivudine in preventing hepatitis B transmission in mothers with high viral load. Int J Gynaecol Obstet. 2023:160:646-652.
Is azithromycin prophylaxis appropriate for vaginal delivery in low- and middle-resource populations?
Tita ATN, Carlo WA, McClure EM, et al; for the A-PLUS Trial Group. Azithromycin to prevent sepsis or death in women planning a vaginal birth. N Engl J Med. 2023;388:1161-1170. doi:10:1056/NEJMoa2212111.
EXPERT COMMENTARY
Maternal peripartum infection is 1 of the top 5 causes of maternal death, accounting for about 10% of cases of maternal mortality. Cesarean delivery (CD), of course, is the most important risk factor for puerperal infection. However, even vaginal delivery, particularly in low- to middle-resource countries, where deliveries often occur under less-than-optimal conditions, may be associated with a surprisingly high frequency of both maternal and neonatal infections. The beneficial effect of prophylactic antibiotics for CD is well established. An important remaining question is whether similar benefit can be achieved with prophylaxis for women planning to have a vaginal birth.
In 2017, Oluwalana and colleagues conducted a prospective, randomized, double-blind, placebo-controlled trial of a single 2-g oral dose of azithromycin in Gambian women undergoing labor.1 During the 8 weeks after delivery, maternal infections were lower in the azithromycin group, 3.6% versus 9.2% (relative risk [RR], 0.40; 95% confidence interval [CI], 0.22–0.71; P=.002). Infections also were lower in the newborns, 18.1% versus 23.8% (RR, 0.76; 95% CI, 0.58–0.99; P=.052), delivered to mothers who received azithromycin. The greatest impact on neonatal infections was the reduced frequency of skin infections.
In 2021, Subramaniam and colleagues evaluated the effect of a single dose of oral azithromycin with, or without, amoxicillin on the prevalence of peripartum infection in laboring women in Cameroon.2 Patients and their newborns were followed for 6 weeks after delivery. Unlike the previous investigation, the authors were unable to show a protective effect of prophylaxis on either maternal or neonatal infection.
Against this backdrop, Tita and colleagues conducted a remarkably large, well-designed, randomized, placebo-controlled study of azithromycin prophylaxis in women at 8 different sites in 7 low- or middle-income countries (the A-PLUS investigation).3
Details of the study
The investigators randomly assigned 29,278 patients at or beyond 28 weeks’ gestation to receive either a 2-g oral dose of azithromycin or placebo during labor. This particular drug was chosen because it is readily available, inexpensive, well tolerated, and has a broad range of activity against many important pelvic pathogens, including genital mycoplasmas. Some patients also received other antibiotics, for example, for group B streptococcal (GBS) prophylaxis or for CD prophylaxis if abdominal delivery was indicated.
The 2 primary outcomes were a composite of maternal sepsis or death and a composite of stillbirth or neonatal death or sepsis within 4 weeks of delivery. Secondary outcomes included individual components of the primary outcomes.
Results. The results of the investigation were compelling, and the data safety monitoring committee recommended stopping the trial early because of clear maternal benefit. The groups were well balanced with respect to important characteristics, such as incidence of CD, receipt of other prophylactic antibiotics, and median time between randomization and delivery.
The incidence of maternal sepsis or death was lower in the azithromycin group (1.6% vs 2.4%; RR, 0.67; 95% CI, 0.56–0.79; P<.001). The key effect was on the frequency of maternal sepsis because the incidence of maternal death was very low in both groups, 0.1%. With respect to secondary outcomes, prophylaxis was effective in reducing the frequency of endometritis (RR, 0.66; 95% CI, 0.55–0.79) and perineal and incisional infection (RR, 0.71; 95% CI, 0.56–0.85).
There was no difference in the frequency of neonatal sepsis or death. There also was no difference in the frequency of adverse drug effects in either group. Of note, more cases of neonatal pyloric stenosis were observed in the azithromycin group, but the overall incidence was lower than the expected background rate. This possible “signal” is important because this effect has been noted with increased frequency in neonates who received this antibiotic. ●
I believe that Tita and colleagues are quite correct in concluding that the simple, inexpensive intervention of azithromycin prophylaxis should be used routinely in patient populations similar to those included in this investigation and that the intervention can be invaluable in advancing the World Health Organization’s campaign to reduce the rate of maternal mortality in low- and middleresource nations.
What is not clear, however, is whether this same intervention would be effective in high-resource countries in which the level of skill of the obstetric providers is higher and more uniform; deliveries occur under more optimal sanitary conditions; treatment and prophylaxis for infections such as gonorrhea, chlamydia, chorioamnionitis, and GBS is more consistent; and early neonatal care is more robust. A similar large trial in wellresourced nations would indeed be welcome, particularly if the trial also addressed the possibility of an adverse effect on the neonatal microbiome if a policy of nearly universal antibiotic prophylaxis was adopted.
In the interim, we should focus our attention on the key interventions that are of proven value in decreasing the risk of peripartum maternal and neonatal infection:
- consistently screening for GBS colonization and administering intrapartum antibiotic prophylaxis to patients who test positive
- consistently screening for gonococcal and chlamydia infection in the antepartum period and treating infected patients with appropriate antibiotics
- minimizing the number of internal vaginal examinations during labor, particularly following rupture of membranes
- promptly identifying patients with chorioamnionitis and treating with antibiotics that specifically target GBS and Escherichia coli, the 2 most likely causes of neonatal sepsis, pneumonia, and meningitis
- administering preoperative prophylactic antibiotics (cefazolin plus azithromycin) to women who require CD.
PATRICK DUFF, MD
- Oluwalana C, Camara B, Bottomley C, et al. Azithromycin in labor lowers clinical infections in mothers and newborns: a double-blind trial. Pediatrics. 2017;139:e20162281. doi:10.1542/peds.2016-2281.
- Subramaniam A, Ye Y, Mbah R, et al. Single dose of oral azithromycin with or without amoxicillin to prevent peripartum infection in laboring, high-risk women in Cameroon: a randomized controlled trial. Obstet Gynecol. 2021;138:703-713. doi:10.1097/AOG.0000000000004565.
- Tita ATN, Carlo WA, McClure EM, et al; for the A-PLUS Trial Group. Azithromycin to prevent sepsis or death in women planning a vaginal birth. N Engl J Med. 2023;388:1161-1170. doi:10:1056/NEJMoa2212111.
Tita ATN, Carlo WA, McClure EM, et al; for the A-PLUS Trial Group. Azithromycin to prevent sepsis or death in women planning a vaginal birth. N Engl J Med. 2023;388:1161-1170. doi:10:1056/NEJMoa2212111.
EXPERT COMMENTARY
Maternal peripartum infection is 1 of the top 5 causes of maternal death, accounting for about 10% of cases of maternal mortality. Cesarean delivery (CD), of course, is the most important risk factor for puerperal infection. However, even vaginal delivery, particularly in low- to middle-resource countries, where deliveries often occur under less-than-optimal conditions, may be associated with a surprisingly high frequency of both maternal and neonatal infections. The beneficial effect of prophylactic antibiotics for CD is well established. An important remaining question is whether similar benefit can be achieved with prophylaxis for women planning to have a vaginal birth.
In 2017, Oluwalana and colleagues conducted a prospective, randomized, double-blind, placebo-controlled trial of a single 2-g oral dose of azithromycin in Gambian women undergoing labor.1 During the 8 weeks after delivery, maternal infections were lower in the azithromycin group, 3.6% versus 9.2% (relative risk [RR], 0.40; 95% confidence interval [CI], 0.22–0.71; P=.002). Infections also were lower in the newborns, 18.1% versus 23.8% (RR, 0.76; 95% CI, 0.58–0.99; P=.052), delivered to mothers who received azithromycin. The greatest impact on neonatal infections was the reduced frequency of skin infections.
In 2021, Subramaniam and colleagues evaluated the effect of a single dose of oral azithromycin with, or without, amoxicillin on the prevalence of peripartum infection in laboring women in Cameroon.2 Patients and their newborns were followed for 6 weeks after delivery. Unlike the previous investigation, the authors were unable to show a protective effect of prophylaxis on either maternal or neonatal infection.
Against this backdrop, Tita and colleagues conducted a remarkably large, well-designed, randomized, placebo-controlled study of azithromycin prophylaxis in women at 8 different sites in 7 low- or middle-income countries (the A-PLUS investigation).3
Details of the study
The investigators randomly assigned 29,278 patients at or beyond 28 weeks’ gestation to receive either a 2-g oral dose of azithromycin or placebo during labor. This particular drug was chosen because it is readily available, inexpensive, well tolerated, and has a broad range of activity against many important pelvic pathogens, including genital mycoplasmas. Some patients also received other antibiotics, for example, for group B streptococcal (GBS) prophylaxis or for CD prophylaxis if abdominal delivery was indicated.
The 2 primary outcomes were a composite of maternal sepsis or death and a composite of stillbirth or neonatal death or sepsis within 4 weeks of delivery. Secondary outcomes included individual components of the primary outcomes.
Results. The results of the investigation were compelling, and the data safety monitoring committee recommended stopping the trial early because of clear maternal benefit. The groups were well balanced with respect to important characteristics, such as incidence of CD, receipt of other prophylactic antibiotics, and median time between randomization and delivery.
The incidence of maternal sepsis or death was lower in the azithromycin group (1.6% vs 2.4%; RR, 0.67; 95% CI, 0.56–0.79; P<.001). The key effect was on the frequency of maternal sepsis because the incidence of maternal death was very low in both groups, 0.1%. With respect to secondary outcomes, prophylaxis was effective in reducing the frequency of endometritis (RR, 0.66; 95% CI, 0.55–0.79) and perineal and incisional infection (RR, 0.71; 95% CI, 0.56–0.85).
There was no difference in the frequency of neonatal sepsis or death. There also was no difference in the frequency of adverse drug effects in either group. Of note, more cases of neonatal pyloric stenosis were observed in the azithromycin group, but the overall incidence was lower than the expected background rate. This possible “signal” is important because this effect has been noted with increased frequency in neonates who received this antibiotic. ●
I believe that Tita and colleagues are quite correct in concluding that the simple, inexpensive intervention of azithromycin prophylaxis should be used routinely in patient populations similar to those included in this investigation and that the intervention can be invaluable in advancing the World Health Organization’s campaign to reduce the rate of maternal mortality in low- and middleresource nations.
What is not clear, however, is whether this same intervention would be effective in high-resource countries in which the level of skill of the obstetric providers is higher and more uniform; deliveries occur under more optimal sanitary conditions; treatment and prophylaxis for infections such as gonorrhea, chlamydia, chorioamnionitis, and GBS is more consistent; and early neonatal care is more robust. A similar large trial in wellresourced nations would indeed be welcome, particularly if the trial also addressed the possibility of an adverse effect on the neonatal microbiome if a policy of nearly universal antibiotic prophylaxis was adopted.
In the interim, we should focus our attention on the key interventions that are of proven value in decreasing the risk of peripartum maternal and neonatal infection:
- consistently screening for GBS colonization and administering intrapartum antibiotic prophylaxis to patients who test positive
- consistently screening for gonococcal and chlamydia infection in the antepartum period and treating infected patients with appropriate antibiotics
- minimizing the number of internal vaginal examinations during labor, particularly following rupture of membranes
- promptly identifying patients with chorioamnionitis and treating with antibiotics that specifically target GBS and Escherichia coli, the 2 most likely causes of neonatal sepsis, pneumonia, and meningitis
- administering preoperative prophylactic antibiotics (cefazolin plus azithromycin) to women who require CD.
PATRICK DUFF, MD
Tita ATN, Carlo WA, McClure EM, et al; for the A-PLUS Trial Group. Azithromycin to prevent sepsis or death in women planning a vaginal birth. N Engl J Med. 2023;388:1161-1170. doi:10:1056/NEJMoa2212111.
EXPERT COMMENTARY
Maternal peripartum infection is 1 of the top 5 causes of maternal death, accounting for about 10% of cases of maternal mortality. Cesarean delivery (CD), of course, is the most important risk factor for puerperal infection. However, even vaginal delivery, particularly in low- to middle-resource countries, where deliveries often occur under less-than-optimal conditions, may be associated with a surprisingly high frequency of both maternal and neonatal infections. The beneficial effect of prophylactic antibiotics for CD is well established. An important remaining question is whether similar benefit can be achieved with prophylaxis for women planning to have a vaginal birth.
In 2017, Oluwalana and colleagues conducted a prospective, randomized, double-blind, placebo-controlled trial of a single 2-g oral dose of azithromycin in Gambian women undergoing labor.1 During the 8 weeks after delivery, maternal infections were lower in the azithromycin group, 3.6% versus 9.2% (relative risk [RR], 0.40; 95% confidence interval [CI], 0.22–0.71; P=.002). Infections also were lower in the newborns, 18.1% versus 23.8% (RR, 0.76; 95% CI, 0.58–0.99; P=.052), delivered to mothers who received azithromycin. The greatest impact on neonatal infections was the reduced frequency of skin infections.
In 2021, Subramaniam and colleagues evaluated the effect of a single dose of oral azithromycin with, or without, amoxicillin on the prevalence of peripartum infection in laboring women in Cameroon.2 Patients and their newborns were followed for 6 weeks after delivery. Unlike the previous investigation, the authors were unable to show a protective effect of prophylaxis on either maternal or neonatal infection.
Against this backdrop, Tita and colleagues conducted a remarkably large, well-designed, randomized, placebo-controlled study of azithromycin prophylaxis in women at 8 different sites in 7 low- or middle-income countries (the A-PLUS investigation).3
Details of the study
The investigators randomly assigned 29,278 patients at or beyond 28 weeks’ gestation to receive either a 2-g oral dose of azithromycin or placebo during labor. This particular drug was chosen because it is readily available, inexpensive, well tolerated, and has a broad range of activity against many important pelvic pathogens, including genital mycoplasmas. Some patients also received other antibiotics, for example, for group B streptococcal (GBS) prophylaxis or for CD prophylaxis if abdominal delivery was indicated.
The 2 primary outcomes were a composite of maternal sepsis or death and a composite of stillbirth or neonatal death or sepsis within 4 weeks of delivery. Secondary outcomes included individual components of the primary outcomes.
Results. The results of the investigation were compelling, and the data safety monitoring committee recommended stopping the trial early because of clear maternal benefit. The groups were well balanced with respect to important characteristics, such as incidence of CD, receipt of other prophylactic antibiotics, and median time between randomization and delivery.
The incidence of maternal sepsis or death was lower in the azithromycin group (1.6% vs 2.4%; RR, 0.67; 95% CI, 0.56–0.79; P<.001). The key effect was on the frequency of maternal sepsis because the incidence of maternal death was very low in both groups, 0.1%. With respect to secondary outcomes, prophylaxis was effective in reducing the frequency of endometritis (RR, 0.66; 95% CI, 0.55–0.79) and perineal and incisional infection (RR, 0.71; 95% CI, 0.56–0.85).
There was no difference in the frequency of neonatal sepsis or death. There also was no difference in the frequency of adverse drug effects in either group. Of note, more cases of neonatal pyloric stenosis were observed in the azithromycin group, but the overall incidence was lower than the expected background rate. This possible “signal” is important because this effect has been noted with increased frequency in neonates who received this antibiotic. ●
I believe that Tita and colleagues are quite correct in concluding that the simple, inexpensive intervention of azithromycin prophylaxis should be used routinely in patient populations similar to those included in this investigation and that the intervention can be invaluable in advancing the World Health Organization’s campaign to reduce the rate of maternal mortality in low- and middleresource nations.
What is not clear, however, is whether this same intervention would be effective in high-resource countries in which the level of skill of the obstetric providers is higher and more uniform; deliveries occur under more optimal sanitary conditions; treatment and prophylaxis for infections such as gonorrhea, chlamydia, chorioamnionitis, and GBS is more consistent; and early neonatal care is more robust. A similar large trial in wellresourced nations would indeed be welcome, particularly if the trial also addressed the possibility of an adverse effect on the neonatal microbiome if a policy of nearly universal antibiotic prophylaxis was adopted.
In the interim, we should focus our attention on the key interventions that are of proven value in decreasing the risk of peripartum maternal and neonatal infection:
- consistently screening for GBS colonization and administering intrapartum antibiotic prophylaxis to patients who test positive
- consistently screening for gonococcal and chlamydia infection in the antepartum period and treating infected patients with appropriate antibiotics
- minimizing the number of internal vaginal examinations during labor, particularly following rupture of membranes
- promptly identifying patients with chorioamnionitis and treating with antibiotics that specifically target GBS and Escherichia coli, the 2 most likely causes of neonatal sepsis, pneumonia, and meningitis
- administering preoperative prophylactic antibiotics (cefazolin plus azithromycin) to women who require CD.
PATRICK DUFF, MD
- Oluwalana C, Camara B, Bottomley C, et al. Azithromycin in labor lowers clinical infections in mothers and newborns: a double-blind trial. Pediatrics. 2017;139:e20162281. doi:10.1542/peds.2016-2281.
- Subramaniam A, Ye Y, Mbah R, et al. Single dose of oral azithromycin with or without amoxicillin to prevent peripartum infection in laboring, high-risk women in Cameroon: a randomized controlled trial. Obstet Gynecol. 2021;138:703-713. doi:10.1097/AOG.0000000000004565.
- Tita ATN, Carlo WA, McClure EM, et al; for the A-PLUS Trial Group. Azithromycin to prevent sepsis or death in women planning a vaginal birth. N Engl J Med. 2023;388:1161-1170. doi:10:1056/NEJMoa2212111.
- Oluwalana C, Camara B, Bottomley C, et al. Azithromycin in labor lowers clinical infections in mothers and newborns: a double-blind trial. Pediatrics. 2017;139:e20162281. doi:10.1542/peds.2016-2281.
- Subramaniam A, Ye Y, Mbah R, et al. Single dose of oral azithromycin with or without amoxicillin to prevent peripartum infection in laboring, high-risk women in Cameroon: a randomized controlled trial. Obstet Gynecol. 2021;138:703-713. doi:10.1097/AOG.0000000000004565.
- Tita ATN, Carlo WA, McClure EM, et al; for the A-PLUS Trial Group. Azithromycin to prevent sepsis or death in women planning a vaginal birth. N Engl J Med. 2023;388:1161-1170. doi:10:1056/NEJMoa2212111.
Listeria infection in pregnancy: A potentially serious foodborne illness
CASE Pregnant patient with concerning symptoms of infection
A 28-year-old primigravid woman at 26 weeks’ gestation requests evaluation because of a 3-day history of low-grade fever (38.3 °C), chills, malaise, myalgias, pain in her upper back, nausea, diarrhea, and intermittent uterine contractions. Her symptoms began 2 days after she and her husband dined at a local Mexican restaurant. She specifically recalls eating unpasteurized cheese (queso fresco). Her husband also is experiencing similar symptoms.
- What is the most likely diagnosis?
- What tests should be performed to confirm the diagnosis?
- Does this infection pose a risk to the fetus?
- How should this patient be treated?
Listeriosis, a potentially serious foodborne illness, is an unusual infection in pregnancy. It can cause a number of adverse effects in both the pregnant woman and her fetus, including fetal death in utero. In this article, we review the microbiology and epidemiology of Listeria infection, consider the important steps in diagnosis, and discuss treatment options and prevention measures.
The causative organism in listeriosis
Listeriosis is caused by Listeria monocytogenes, a gram-positive, non–spore-forming bacillus. The organism is catalase positive and oxidase negative, and it exhibits tumbling motility when grown in culture. It can grow at temperatures less than 4 °C, which facilitates foodborne transmission of the bacterium despite adequate refrigeration. Of the 13 serotypes of L monocytogenes, the 1/2a, 1/2b, and 4b are most likely to be associated with human infection. The major virulence factors of L monocytogenes are the internalin surface proteins and the pore-forming listeriolysin O (LLO) cytotoxin. These factors enable the organism to effectively invade host cells.1
The pathogen uses several mechanisms to evade gastrointestinal defenses prior to entry into the bloodstream. It avoids destruction in the stomach by using proton pump inhibitors to elevate the pH of gastric acid. In the duodenum, it survives the antibacterial properties of bile by secreting bile salt hydrolases, which catabolize bile salts. In addition, the cytotoxin listeriolysin S (LLS) disrupts the protective barrier created by the normal gut flora. Once the organism penetrates the gastrointestinal barriers, it disseminates through the blood and lymphatics and then infects other tissues, such as the brain and placenta.1,2
Pathogenesis of infection
The primary reservoir of Listeria is soil and decaying vegetable matter. The organism also has been isolated from animal feed, water, sewage, and many animal species. With rare exceptions, most infections in adults result from inadvertent ingestion of the organism in contaminated food. In certain high-risk occupations, such as veterinary medicine, farming, and laboratory work, infection of the skin or eye can result from direct contact with an infected animal.3
Of note, foodborne illness caused by Listeria has the third highest mortality rate of any foodborne infection, 16% compared with 35% for Vibrio vulnificus and 17% for Clostridium botulinum.2,3 The principal foods that have been linked to listeriosis include:
- soft cheeses, particularly those made from unpasteurized milk
- melon
- hot dogs
- lunch meat, such as bologna
- deli meat, especially chicken
- canned foods, such as smoked seafood, and pâté or meat spreads that are labeled “keep refrigerated”
- unpasteurized milk
- sprouts
- hummus.
In healthy adults, listeriosis is usually a short-lived illness. However, in older adults, immunocompromised patients, and pregnant women, the infection can be devastating. Infection in the pregnant woman also poses major danger to the developing fetus because the organism has a special predilection for placental and fetal tissue.1,3,4
Immunity to Listeria infection depends primarily on T-cell lymphokine activation of macrophages. These latter cells are responsible for clearing the bacterium from the blood. As noted above, the principal virulence factor of L monocytogenes is listeriolysin O, a cholesterol-dependent cytolysin. This substance induces T-cell receptor unresponsiveness, thus interfering with the host immune response to the invading pathogen.1,3-5
Continue to: Clinical manifestations of listeriosis...
Clinical manifestations of listeriosis
Listeria infections may present with various manifestations, depending on the degree of exposure and the underlying immunocompetence of the host (FIGURE). In its most common and simplest form, listeriosis presents as a mild to moderate gastroenteritis following exposure to contaminated food. Symptoms typically develop within 24 hours of exposure and include fever, myalgias, abdominal or back pain, nausea, vomiting, and diarrhea.5
Conversely, in immunocompromised patients, including pregnant women, listeriosis can present as life-threatening sepsis and/or central nervous system (CNS) infection (invasive infection). In this clinical setting, the mean incubation period is 11 days. The manifestations of CNS infection include meningoencephalitis, cerebritis, rhombencephalitis (infection and inflammation of the brain stem), brain abscess, and spinal cord abscess.5
In addition to these 2 clinical presentations, listeriosis can cause unusual focal infections as illustrated in the FIGURE. Some of these infections have unique clinical associations. For example, skin or eye infections may occur as a result of direct inoculation in veterinarians, farmers, and laboratory workers. Listeria peritonitis may occur in patients who are receiving peritoneal dialysis and in those who have cirrhosis. Prosthetic joint and graft infections, of course, may occur in patients who have had invasive procedures for implantation of grafts or prosthetic devices.5
Listeriosis is especially dangerous in pregnancy because it not only can cause serious injury to the mother and even death but it also may pose a major risk to fetal well-being. Possible perinatal complications include fetal death; preterm labor and delivery; and neonatal sepsis, meningitis, and death.5-8
Making the diagnosis
Diagnosis begins with a thorough and focused history to assess for characteristic symptoms and possible Listeria exposure. Exposure should be presumed for patients who report consuming high-risk foods, especially foods recently recalled by the US Food and Drug Administration.
In the asymptomatic pregnant patient, diagnostic testing can be deferred, and the patient should be instructed to return for evaluation if symptoms develop within 2 months of exposure. However, symptomatic, febrile patients require testing. The most valuable testing modality is Gram stain and culture of blood. Gram stain typically will show gram-positive pleomorphic rods with rounded ends. Amniocentesis may be indicated if blood cultures are not definitive. Meconium staining of the amniotic fluid and a positive Gram stain are highly indicative of fetal infection. Cultures of the cerebrospinal fluid are indicated in any individual with focal neurologic findings. Stool cultures are rarely indicated.
When obtaining any of the cultures noted above, the clinician should alert the microbiologist of the concern for listeriosis because L monocytogenes can be confused with common contaminants, such as diphtheroids.5-9
Treatment and follow-up
The treatment of listeriosis in pregnancy depends on the severity of the infection and the immune status of the mother. The TABLE offers several different clinical scenarios and the appropriate treatment for each. As noted, several scenarios may require cultures of the blood, cerebrospinal fluid, and amniotic fluid.7,9,10
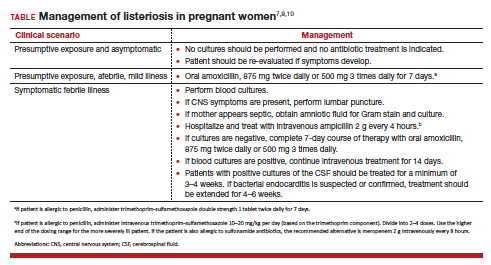
Following treatment of the mother, serial ultrasound examinations should be performed to monitor fetal growth, CNS anatomy, placental morphology, amniotic fluid volume, and umbilical artery Doppler velocimetry. In the presence of fetal growth restriction, oligohydramnios, or abnormal Doppler velocimetry, biophysical profile testing should be performed. After delivery, the placenta should be examined carefully for histologic evidence of Listeria infection, such as miliary abscesses, and cultured for the bacterium.7-9
Prevention measures
Conservative measures for prevention of Listeria infection in pregnant women include the following7,10-12:
- Refrigerate milk and milk products at 40 °F (4.4 °C).
- Thoroughly cook raw food from animal sources.
- Wash raw vegetables carefully before eating.
- Keep uncooked meats separate from cooked meats and vegetables.
- Do not consume any beverages or foods made from unpasteurized milk.
- After handling uncooked foods, carefully wash all utensils and hands.
- Avoid all soft cheeses, such as Mexican-style feta, Brie, Camembert, and blue cheese, even if they are supposedly made from pasteurized milk.
- Reheat until steaming hot all leftover foods or ready-to-eat foods, such as hot dogs.
- Do not let juice from hot dogs or lunch meat packages drip onto other foods, utensils, or food preparation surfaces.
- Do not store opened hot dog packages in the refrigerator for more than 1 week. Do not store unopened packages for longer than 2 weeks.
- Do not store unopened lunch and deli meat packages in the refrigerator for longer than 2 weeks. Do not store opened packages for longer than 3 to 5 days.
- If other immunosuppressive conditions are present in combination with pregnancy, thoroughly heat cold cuts before eating.
- Do not eat raw or even lightly cooked sprouts of any kind. Cook sprouts thoroughly. Rinsing sprouts will not remove Listeria organisms.
- Do not eat refrigerated pâté or meat spreads from a deli counter or the refrigerated section of a grocery store.
- Canned or shelf-stable pâté and meat spreads are safe to eat, but be sure to refrigerate them after opening the packages.
- Do not eat refrigerated smoked seafood. Canned or shelf-stable seafood, particularly when incorporated into a casserole, is safe to eat.
- Eat cut melon immediately. Refrigerate uneaten melon quickly if not eaten. Discard cut melon that is left at room temperature for more than 4 hours.
CASE Diagnosis made and prompt treatment initiated
The most likely diagnosis in this patient is listeriosis. Because the patient is moderately ill and experiencing uterine contractions, she should be hospitalized and monitored for progressive cervical dilation. Blood cultures should be obtained to identify L monocytogenes. In addition, an amniocentesis should be performed, and the amniotic fluid should be cultured for this microorganism. Stool culture and culture of the cerebrospinal fluid are not indicated. The patient should be treated with intravenous ampicillin, 2 g every 4 hours for 14 days. If she is allergic to penicillin, the alternative drug is trimethoprim-sulfamethoxazole, 8 to 10 mg/kg per day in 2 divided doses, for 14 days. Prompt and effective treatment of the mother should prevent infection in the fetus and newborn. ●
- Listeriosis is primarily a foodborne illness caused by Listeria monocytogenes, a gram-positive bacillus.
- Pregnant women, particularly those who are immunocompromised, are especially susceptible to Listeria infection.
- Foods that pose particular risk of transmitting infection include fresh unpasteurized cheeses, processed meats such as hot dogs, refrigerated pâté and meat spreads, refrigerated smoked seafood, unpasteurized milk, and unwashed raw produce.
- The infection may range from a mild gastroenteritis to life-threatening sepsis and meningitis.
- Listeriosis may cause early and late-onset neonatal infection that presents as either meningitis or sepsis.
- Blood and amniotic fluid cultures are essential to diagnose maternal infection. Stool cultures usually are not indicated.
- Mildly symptomatic but afebrile patients do not require treatment.
- Febrile symptomatic patients should be treated with either intravenous ampicillin or trimethoprim-sulfamethoxazole.
- Radoshevich L, Cossart P. Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat Rev Microbiol. 2018;16:32-46. doi:10.1038/nnrmicro.2017.126.
- Johnson JE, Mylonakis E. Listeria monocytogenes. In: Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 9th ed. Elsevier; 2020:2543-2549.
- Gelfand MS, Swamy GK, Thompson JL. Epidemiology and pathogenesis of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 9, 2022. https://www.uptodate.com/contents/epidemiology-and-pathogenesis-of-listeria-monocytogenes-infection?sectionName=CLINICAL%20EPIDEMIOLOGY&topicRef=1277&anchor=H4&source=see_link#H4
- Cherubin CE, Appleman MD, Heseltine PN, et al. Epidemiological spectrum and current treatment of listeriosis. Rev Infect Dis. 1991;13:1108-1114.
- Gelfand MS, Swamy GK, Thompson JL. Clinical manifestations and diagnosis of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 7, 2022. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-listeriamonocytogenes-infection
- Boucher M, Yonekura ML. Perinatal listeriosis (early-onset): correlation of antenatal manifestations and neonatal outcome. Obstet Gynecol. 1986;68:593-597.
- American College of Obstetricians and Gynecologists. ACOG committee opinion no. 614: management of pregnant women with presumptive exposure to Listeria monocytogenes. Obstet Gynecol. 2014;124:1241-1244.
- Rouse DJ, Keimig TW, Riley LE, et al. Case 16-2016. A 31-year-old pregnant woman with fever. N Engl J Med. 2016;374:2076-2083.
- Craig AM, Dotters-Katz S, Kuller JA, et al. Listeriosis in pregnancy: a review. Obstet Gynecol Surv. 2019;74: 362-368.
- Gelfand MS, Thompson JL, Swamy GK. Treatment and prevention of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 9, 2022. https://www.uptodate.com/contents/treatment-and-prevention-of-listeria-monocytogenes-infection?topicRef=1280&source=see_link
- Voetsch AC, Angulo FJ, Jones TF, et al; Centers for Disease Control and Prevention Emerging Infections Program Foodborne Diseases Active Surveillance Networking Group. Reduction in the incidence of invasive listeriosis in Foodborne Diseases Active Surveillance Network sites, 1996-2003. Clin Infect Dis. 2007;44:513-520.
- MacDonald PDM, Whitwan RE, Boggs JD, et al. Outbreak of listeriosis among Mexican immigrants as a result of consumption of illicitly produced Mexican-style cheese. Clin Infect Dis. 2005;40:677-682.

Ms. Stennett is a third-year medical student, University of Florida College of Medicine, Gainesville.

Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Ms. Stennett is a third-year medical student, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Ms. Stennett is a third-year medical student, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
CASE Pregnant patient with concerning symptoms of infection
A 28-year-old primigravid woman at 26 weeks’ gestation requests evaluation because of a 3-day history of low-grade fever (38.3 °C), chills, malaise, myalgias, pain in her upper back, nausea, diarrhea, and intermittent uterine contractions. Her symptoms began 2 days after she and her husband dined at a local Mexican restaurant. She specifically recalls eating unpasteurized cheese (queso fresco). Her husband also is experiencing similar symptoms.
- What is the most likely diagnosis?
- What tests should be performed to confirm the diagnosis?
- Does this infection pose a risk to the fetus?
- How should this patient be treated?
Listeriosis, a potentially serious foodborne illness, is an unusual infection in pregnancy. It can cause a number of adverse effects in both the pregnant woman and her fetus, including fetal death in utero. In this article, we review the microbiology and epidemiology of Listeria infection, consider the important steps in diagnosis, and discuss treatment options and prevention measures.
The causative organism in listeriosis
Listeriosis is caused by Listeria monocytogenes, a gram-positive, non–spore-forming bacillus. The organism is catalase positive and oxidase negative, and it exhibits tumbling motility when grown in culture. It can grow at temperatures less than 4 °C, which facilitates foodborne transmission of the bacterium despite adequate refrigeration. Of the 13 serotypes of L monocytogenes, the 1/2a, 1/2b, and 4b are most likely to be associated with human infection. The major virulence factors of L monocytogenes are the internalin surface proteins and the pore-forming listeriolysin O (LLO) cytotoxin. These factors enable the organism to effectively invade host cells.1
The pathogen uses several mechanisms to evade gastrointestinal defenses prior to entry into the bloodstream. It avoids destruction in the stomach by using proton pump inhibitors to elevate the pH of gastric acid. In the duodenum, it survives the antibacterial properties of bile by secreting bile salt hydrolases, which catabolize bile salts. In addition, the cytotoxin listeriolysin S (LLS) disrupts the protective barrier created by the normal gut flora. Once the organism penetrates the gastrointestinal barriers, it disseminates through the blood and lymphatics and then infects other tissues, such as the brain and placenta.1,2
Pathogenesis of infection
The primary reservoir of Listeria is soil and decaying vegetable matter. The organism also has been isolated from animal feed, water, sewage, and many animal species. With rare exceptions, most infections in adults result from inadvertent ingestion of the organism in contaminated food. In certain high-risk occupations, such as veterinary medicine, farming, and laboratory work, infection of the skin or eye can result from direct contact with an infected animal.3
Of note, foodborne illness caused by Listeria has the third highest mortality rate of any foodborne infection, 16% compared with 35% for Vibrio vulnificus and 17% for Clostridium botulinum.2,3 The principal foods that have been linked to listeriosis include:
- soft cheeses, particularly those made from unpasteurized milk
- melon
- hot dogs
- lunch meat, such as bologna
- deli meat, especially chicken
- canned foods, such as smoked seafood, and pâté or meat spreads that are labeled “keep refrigerated”
- unpasteurized milk
- sprouts
- hummus.
In healthy adults, listeriosis is usually a short-lived illness. However, in older adults, immunocompromised patients, and pregnant women, the infection can be devastating. Infection in the pregnant woman also poses major danger to the developing fetus because the organism has a special predilection for placental and fetal tissue.1,3,4
Immunity to Listeria infection depends primarily on T-cell lymphokine activation of macrophages. These latter cells are responsible for clearing the bacterium from the blood. As noted above, the principal virulence factor of L monocytogenes is listeriolysin O, a cholesterol-dependent cytolysin. This substance induces T-cell receptor unresponsiveness, thus interfering with the host immune response to the invading pathogen.1,3-5
Continue to: Clinical manifestations of listeriosis...
Clinical manifestations of listeriosis
Listeria infections may present with various manifestations, depending on the degree of exposure and the underlying immunocompetence of the host (FIGURE). In its most common and simplest form, listeriosis presents as a mild to moderate gastroenteritis following exposure to contaminated food. Symptoms typically develop within 24 hours of exposure and include fever, myalgias, abdominal or back pain, nausea, vomiting, and diarrhea.5
Conversely, in immunocompromised patients, including pregnant women, listeriosis can present as life-threatening sepsis and/or central nervous system (CNS) infection (invasive infection). In this clinical setting, the mean incubation period is 11 days. The manifestations of CNS infection include meningoencephalitis, cerebritis, rhombencephalitis (infection and inflammation of the brain stem), brain abscess, and spinal cord abscess.5
In addition to these 2 clinical presentations, listeriosis can cause unusual focal infections as illustrated in the FIGURE. Some of these infections have unique clinical associations. For example, skin or eye infections may occur as a result of direct inoculation in veterinarians, farmers, and laboratory workers. Listeria peritonitis may occur in patients who are receiving peritoneal dialysis and in those who have cirrhosis. Prosthetic joint and graft infections, of course, may occur in patients who have had invasive procedures for implantation of grafts or prosthetic devices.5
Listeriosis is especially dangerous in pregnancy because it not only can cause serious injury to the mother and even death but it also may pose a major risk to fetal well-being. Possible perinatal complications include fetal death; preterm labor and delivery; and neonatal sepsis, meningitis, and death.5-8
Making the diagnosis
Diagnosis begins with a thorough and focused history to assess for characteristic symptoms and possible Listeria exposure. Exposure should be presumed for patients who report consuming high-risk foods, especially foods recently recalled by the US Food and Drug Administration.
In the asymptomatic pregnant patient, diagnostic testing can be deferred, and the patient should be instructed to return for evaluation if symptoms develop within 2 months of exposure. However, symptomatic, febrile patients require testing. The most valuable testing modality is Gram stain and culture of blood. Gram stain typically will show gram-positive pleomorphic rods with rounded ends. Amniocentesis may be indicated if blood cultures are not definitive. Meconium staining of the amniotic fluid and a positive Gram stain are highly indicative of fetal infection. Cultures of the cerebrospinal fluid are indicated in any individual with focal neurologic findings. Stool cultures are rarely indicated.
When obtaining any of the cultures noted above, the clinician should alert the microbiologist of the concern for listeriosis because L monocytogenes can be confused with common contaminants, such as diphtheroids.5-9
Treatment and follow-up
The treatment of listeriosis in pregnancy depends on the severity of the infection and the immune status of the mother. The TABLE offers several different clinical scenarios and the appropriate treatment for each. As noted, several scenarios may require cultures of the blood, cerebrospinal fluid, and amniotic fluid.7,9,10
Following treatment of the mother, serial ultrasound examinations should be performed to monitor fetal growth, CNS anatomy, placental morphology, amniotic fluid volume, and umbilical artery Doppler velocimetry. In the presence of fetal growth restriction, oligohydramnios, or abnormal Doppler velocimetry, biophysical profile testing should be performed. After delivery, the placenta should be examined carefully for histologic evidence of Listeria infection, such as miliary abscesses, and cultured for the bacterium.7-9
Prevention measures
Conservative measures for prevention of Listeria infection in pregnant women include the following7,10-12:
- Refrigerate milk and milk products at 40 °F (4.4 °C).
- Thoroughly cook raw food from animal sources.
- Wash raw vegetables carefully before eating.
- Keep uncooked meats separate from cooked meats and vegetables.
- Do not consume any beverages or foods made from unpasteurized milk.
- After handling uncooked foods, carefully wash all utensils and hands.
- Avoid all soft cheeses, such as Mexican-style feta, Brie, Camembert, and blue cheese, even if they are supposedly made from pasteurized milk.
- Reheat until steaming hot all leftover foods or ready-to-eat foods, such as hot dogs.
- Do not let juice from hot dogs or lunch meat packages drip onto other foods, utensils, or food preparation surfaces.
- Do not store opened hot dog packages in the refrigerator for more than 1 week. Do not store unopened packages for longer than 2 weeks.
- Do not store unopened lunch and deli meat packages in the refrigerator for longer than 2 weeks. Do not store opened packages for longer than 3 to 5 days.
- If other immunosuppressive conditions are present in combination with pregnancy, thoroughly heat cold cuts before eating.
- Do not eat raw or even lightly cooked sprouts of any kind. Cook sprouts thoroughly. Rinsing sprouts will not remove Listeria organisms.
- Do not eat refrigerated pâté or meat spreads from a deli counter or the refrigerated section of a grocery store.
- Canned or shelf-stable pâté and meat spreads are safe to eat, but be sure to refrigerate them after opening the packages.
- Do not eat refrigerated smoked seafood. Canned or shelf-stable seafood, particularly when incorporated into a casserole, is safe to eat.
- Eat cut melon immediately. Refrigerate uneaten melon quickly if not eaten. Discard cut melon that is left at room temperature for more than 4 hours.
CASE Diagnosis made and prompt treatment initiated
The most likely diagnosis in this patient is listeriosis. Because the patient is moderately ill and experiencing uterine contractions, she should be hospitalized and monitored for progressive cervical dilation. Blood cultures should be obtained to identify L monocytogenes. In addition, an amniocentesis should be performed, and the amniotic fluid should be cultured for this microorganism. Stool culture and culture of the cerebrospinal fluid are not indicated. The patient should be treated with intravenous ampicillin, 2 g every 4 hours for 14 days. If she is allergic to penicillin, the alternative drug is trimethoprim-sulfamethoxazole, 8 to 10 mg/kg per day in 2 divided doses, for 14 days. Prompt and effective treatment of the mother should prevent infection in the fetus and newborn. ●
- Listeriosis is primarily a foodborne illness caused by Listeria monocytogenes, a gram-positive bacillus.
- Pregnant women, particularly those who are immunocompromised, are especially susceptible to Listeria infection.
- Foods that pose particular risk of transmitting infection include fresh unpasteurized cheeses, processed meats such as hot dogs, refrigerated pâté and meat spreads, refrigerated smoked seafood, unpasteurized milk, and unwashed raw produce.
- The infection may range from a mild gastroenteritis to life-threatening sepsis and meningitis.
- Listeriosis may cause early and late-onset neonatal infection that presents as either meningitis or sepsis.
- Blood and amniotic fluid cultures are essential to diagnose maternal infection. Stool cultures usually are not indicated.
- Mildly symptomatic but afebrile patients do not require treatment.
- Febrile symptomatic patients should be treated with either intravenous ampicillin or trimethoprim-sulfamethoxazole.
CASE Pregnant patient with concerning symptoms of infection
A 28-year-old primigravid woman at 26 weeks’ gestation requests evaluation because of a 3-day history of low-grade fever (38.3 °C), chills, malaise, myalgias, pain in her upper back, nausea, diarrhea, and intermittent uterine contractions. Her symptoms began 2 days after she and her husband dined at a local Mexican restaurant. She specifically recalls eating unpasteurized cheese (queso fresco). Her husband also is experiencing similar symptoms.
- What is the most likely diagnosis?
- What tests should be performed to confirm the diagnosis?
- Does this infection pose a risk to the fetus?
- How should this patient be treated?
Listeriosis, a potentially serious foodborne illness, is an unusual infection in pregnancy. It can cause a number of adverse effects in both the pregnant woman and her fetus, including fetal death in utero. In this article, we review the microbiology and epidemiology of Listeria infection, consider the important steps in diagnosis, and discuss treatment options and prevention measures.
The causative organism in listeriosis
Listeriosis is caused by Listeria monocytogenes, a gram-positive, non–spore-forming bacillus. The organism is catalase positive and oxidase negative, and it exhibits tumbling motility when grown in culture. It can grow at temperatures less than 4 °C, which facilitates foodborne transmission of the bacterium despite adequate refrigeration. Of the 13 serotypes of L monocytogenes, the 1/2a, 1/2b, and 4b are most likely to be associated with human infection. The major virulence factors of L monocytogenes are the internalin surface proteins and the pore-forming listeriolysin O (LLO) cytotoxin. These factors enable the organism to effectively invade host cells.1
The pathogen uses several mechanisms to evade gastrointestinal defenses prior to entry into the bloodstream. It avoids destruction in the stomach by using proton pump inhibitors to elevate the pH of gastric acid. In the duodenum, it survives the antibacterial properties of bile by secreting bile salt hydrolases, which catabolize bile salts. In addition, the cytotoxin listeriolysin S (LLS) disrupts the protective barrier created by the normal gut flora. Once the organism penetrates the gastrointestinal barriers, it disseminates through the blood and lymphatics and then infects other tissues, such as the brain and placenta.1,2
Pathogenesis of infection
The primary reservoir of Listeria is soil and decaying vegetable matter. The organism also has been isolated from animal feed, water, sewage, and many animal species. With rare exceptions, most infections in adults result from inadvertent ingestion of the organism in contaminated food. In certain high-risk occupations, such as veterinary medicine, farming, and laboratory work, infection of the skin or eye can result from direct contact with an infected animal.3
Of note, foodborne illness caused by Listeria has the third highest mortality rate of any foodborne infection, 16% compared with 35% for Vibrio vulnificus and 17% for Clostridium botulinum.2,3 The principal foods that have been linked to listeriosis include:
- soft cheeses, particularly those made from unpasteurized milk
- melon
- hot dogs
- lunch meat, such as bologna
- deli meat, especially chicken
- canned foods, such as smoked seafood, and pâté or meat spreads that are labeled “keep refrigerated”
- unpasteurized milk
- sprouts
- hummus.
In healthy adults, listeriosis is usually a short-lived illness. However, in older adults, immunocompromised patients, and pregnant women, the infection can be devastating. Infection in the pregnant woman also poses major danger to the developing fetus because the organism has a special predilection for placental and fetal tissue.1,3,4
Immunity to Listeria infection depends primarily on T-cell lymphokine activation of macrophages. These latter cells are responsible for clearing the bacterium from the blood. As noted above, the principal virulence factor of L monocytogenes is listeriolysin O, a cholesterol-dependent cytolysin. This substance induces T-cell receptor unresponsiveness, thus interfering with the host immune response to the invading pathogen.1,3-5
Continue to: Clinical manifestations of listeriosis...
Clinical manifestations of listeriosis
Listeria infections may present with various manifestations, depending on the degree of exposure and the underlying immunocompetence of the host (FIGURE). In its most common and simplest form, listeriosis presents as a mild to moderate gastroenteritis following exposure to contaminated food. Symptoms typically develop within 24 hours of exposure and include fever, myalgias, abdominal or back pain, nausea, vomiting, and diarrhea.5
Conversely, in immunocompromised patients, including pregnant women, listeriosis can present as life-threatening sepsis and/or central nervous system (CNS) infection (invasive infection). In this clinical setting, the mean incubation period is 11 days. The manifestations of CNS infection include meningoencephalitis, cerebritis, rhombencephalitis (infection and inflammation of the brain stem), brain abscess, and spinal cord abscess.5
In addition to these 2 clinical presentations, listeriosis can cause unusual focal infections as illustrated in the FIGURE. Some of these infections have unique clinical associations. For example, skin or eye infections may occur as a result of direct inoculation in veterinarians, farmers, and laboratory workers. Listeria peritonitis may occur in patients who are receiving peritoneal dialysis and in those who have cirrhosis. Prosthetic joint and graft infections, of course, may occur in patients who have had invasive procedures for implantation of grafts or prosthetic devices.5
Listeriosis is especially dangerous in pregnancy because it not only can cause serious injury to the mother and even death but it also may pose a major risk to fetal well-being. Possible perinatal complications include fetal death; preterm labor and delivery; and neonatal sepsis, meningitis, and death.5-8
Making the diagnosis
Diagnosis begins with a thorough and focused history to assess for characteristic symptoms and possible Listeria exposure. Exposure should be presumed for patients who report consuming high-risk foods, especially foods recently recalled by the US Food and Drug Administration.
In the asymptomatic pregnant patient, diagnostic testing can be deferred, and the patient should be instructed to return for evaluation if symptoms develop within 2 months of exposure. However, symptomatic, febrile patients require testing. The most valuable testing modality is Gram stain and culture of blood. Gram stain typically will show gram-positive pleomorphic rods with rounded ends. Amniocentesis may be indicated if blood cultures are not definitive. Meconium staining of the amniotic fluid and a positive Gram stain are highly indicative of fetal infection. Cultures of the cerebrospinal fluid are indicated in any individual with focal neurologic findings. Stool cultures are rarely indicated.
When obtaining any of the cultures noted above, the clinician should alert the microbiologist of the concern for listeriosis because L monocytogenes can be confused with common contaminants, such as diphtheroids.5-9
Treatment and follow-up
The treatment of listeriosis in pregnancy depends on the severity of the infection and the immune status of the mother. The TABLE offers several different clinical scenarios and the appropriate treatment for each. As noted, several scenarios may require cultures of the blood, cerebrospinal fluid, and amniotic fluid.7,9,10
Following treatment of the mother, serial ultrasound examinations should be performed to monitor fetal growth, CNS anatomy, placental morphology, amniotic fluid volume, and umbilical artery Doppler velocimetry. In the presence of fetal growth restriction, oligohydramnios, or abnormal Doppler velocimetry, biophysical profile testing should be performed. After delivery, the placenta should be examined carefully for histologic evidence of Listeria infection, such as miliary abscesses, and cultured for the bacterium.7-9
Prevention measures
Conservative measures for prevention of Listeria infection in pregnant women include the following7,10-12:
- Refrigerate milk and milk products at 40 °F (4.4 °C).
- Thoroughly cook raw food from animal sources.
- Wash raw vegetables carefully before eating.
- Keep uncooked meats separate from cooked meats and vegetables.
- Do not consume any beverages or foods made from unpasteurized milk.
- After handling uncooked foods, carefully wash all utensils and hands.
- Avoid all soft cheeses, such as Mexican-style feta, Brie, Camembert, and blue cheese, even if they are supposedly made from pasteurized milk.
- Reheat until steaming hot all leftover foods or ready-to-eat foods, such as hot dogs.
- Do not let juice from hot dogs or lunch meat packages drip onto other foods, utensils, or food preparation surfaces.
- Do not store opened hot dog packages in the refrigerator for more than 1 week. Do not store unopened packages for longer than 2 weeks.
- Do not store unopened lunch and deli meat packages in the refrigerator for longer than 2 weeks. Do not store opened packages for longer than 3 to 5 days.
- If other immunosuppressive conditions are present in combination with pregnancy, thoroughly heat cold cuts before eating.
- Do not eat raw or even lightly cooked sprouts of any kind. Cook sprouts thoroughly. Rinsing sprouts will not remove Listeria organisms.
- Do not eat refrigerated pâté or meat spreads from a deli counter or the refrigerated section of a grocery store.
- Canned or shelf-stable pâté and meat spreads are safe to eat, but be sure to refrigerate them after opening the packages.
- Do not eat refrigerated smoked seafood. Canned or shelf-stable seafood, particularly when incorporated into a casserole, is safe to eat.
- Eat cut melon immediately. Refrigerate uneaten melon quickly if not eaten. Discard cut melon that is left at room temperature for more than 4 hours.
CASE Diagnosis made and prompt treatment initiated
The most likely diagnosis in this patient is listeriosis. Because the patient is moderately ill and experiencing uterine contractions, she should be hospitalized and monitored for progressive cervical dilation. Blood cultures should be obtained to identify L monocytogenes. In addition, an amniocentesis should be performed, and the amniotic fluid should be cultured for this microorganism. Stool culture and culture of the cerebrospinal fluid are not indicated. The patient should be treated with intravenous ampicillin, 2 g every 4 hours for 14 days. If she is allergic to penicillin, the alternative drug is trimethoprim-sulfamethoxazole, 8 to 10 mg/kg per day in 2 divided doses, for 14 days. Prompt and effective treatment of the mother should prevent infection in the fetus and newborn. ●
- Listeriosis is primarily a foodborne illness caused by Listeria monocytogenes, a gram-positive bacillus.
- Pregnant women, particularly those who are immunocompromised, are especially susceptible to Listeria infection.
- Foods that pose particular risk of transmitting infection include fresh unpasteurized cheeses, processed meats such as hot dogs, refrigerated pâté and meat spreads, refrigerated smoked seafood, unpasteurized milk, and unwashed raw produce.
- The infection may range from a mild gastroenteritis to life-threatening sepsis and meningitis.
- Listeriosis may cause early and late-onset neonatal infection that presents as either meningitis or sepsis.
- Blood and amniotic fluid cultures are essential to diagnose maternal infection. Stool cultures usually are not indicated.
- Mildly symptomatic but afebrile patients do not require treatment.
- Febrile symptomatic patients should be treated with either intravenous ampicillin or trimethoprim-sulfamethoxazole.
- Radoshevich L, Cossart P. Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat Rev Microbiol. 2018;16:32-46. doi:10.1038/nnrmicro.2017.126.
- Johnson JE, Mylonakis E. Listeria monocytogenes. In: Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 9th ed. Elsevier; 2020:2543-2549.
- Gelfand MS, Swamy GK, Thompson JL. Epidemiology and pathogenesis of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 9, 2022. https://www.uptodate.com/contents/epidemiology-and-pathogenesis-of-listeria-monocytogenes-infection?sectionName=CLINICAL%20EPIDEMIOLOGY&topicRef=1277&anchor=H4&source=see_link#H4
- Cherubin CE, Appleman MD, Heseltine PN, et al. Epidemiological spectrum and current treatment of listeriosis. Rev Infect Dis. 1991;13:1108-1114.
- Gelfand MS, Swamy GK, Thompson JL. Clinical manifestations and diagnosis of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 7, 2022. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-listeriamonocytogenes-infection
- Boucher M, Yonekura ML. Perinatal listeriosis (early-onset): correlation of antenatal manifestations and neonatal outcome. Obstet Gynecol. 1986;68:593-597.
- American College of Obstetricians and Gynecologists. ACOG committee opinion no. 614: management of pregnant women with presumptive exposure to Listeria monocytogenes. Obstet Gynecol. 2014;124:1241-1244.
- Rouse DJ, Keimig TW, Riley LE, et al. Case 16-2016. A 31-year-old pregnant woman with fever. N Engl J Med. 2016;374:2076-2083.
- Craig AM, Dotters-Katz S, Kuller JA, et al. Listeriosis in pregnancy: a review. Obstet Gynecol Surv. 2019;74: 362-368.
- Gelfand MS, Thompson JL, Swamy GK. Treatment and prevention of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 9, 2022. https://www.uptodate.com/contents/treatment-and-prevention-of-listeria-monocytogenes-infection?topicRef=1280&source=see_link
- Voetsch AC, Angulo FJ, Jones TF, et al; Centers for Disease Control and Prevention Emerging Infections Program Foodborne Diseases Active Surveillance Networking Group. Reduction in the incidence of invasive listeriosis in Foodborne Diseases Active Surveillance Network sites, 1996-2003. Clin Infect Dis. 2007;44:513-520.
- MacDonald PDM, Whitwan RE, Boggs JD, et al. Outbreak of listeriosis among Mexican immigrants as a result of consumption of illicitly produced Mexican-style cheese. Clin Infect Dis. 2005;40:677-682.
- Radoshevich L, Cossart P. Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat Rev Microbiol. 2018;16:32-46. doi:10.1038/nnrmicro.2017.126.
- Johnson JE, Mylonakis E. Listeria monocytogenes. In: Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 9th ed. Elsevier; 2020:2543-2549.
- Gelfand MS, Swamy GK, Thompson JL. Epidemiology and pathogenesis of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 9, 2022. https://www.uptodate.com/contents/epidemiology-and-pathogenesis-of-listeria-monocytogenes-infection?sectionName=CLINICAL%20EPIDEMIOLOGY&topicRef=1277&anchor=H4&source=see_link#H4
- Cherubin CE, Appleman MD, Heseltine PN, et al. Epidemiological spectrum and current treatment of listeriosis. Rev Infect Dis. 1991;13:1108-1114.
- Gelfand MS, Swamy GK, Thompson JL. Clinical manifestations and diagnosis of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 7, 2022. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-listeriamonocytogenes-infection
- Boucher M, Yonekura ML. Perinatal listeriosis (early-onset): correlation of antenatal manifestations and neonatal outcome. Obstet Gynecol. 1986;68:593-597.
- American College of Obstetricians and Gynecologists. ACOG committee opinion no. 614: management of pregnant women with presumptive exposure to Listeria monocytogenes. Obstet Gynecol. 2014;124:1241-1244.
- Rouse DJ, Keimig TW, Riley LE, et al. Case 16-2016. A 31-year-old pregnant woman with fever. N Engl J Med. 2016;374:2076-2083.
- Craig AM, Dotters-Katz S, Kuller JA, et al. Listeriosis in pregnancy: a review. Obstet Gynecol Surv. 2019;74: 362-368.
- Gelfand MS, Thompson JL, Swamy GK. Treatment and prevention of Listeria monocytogenes infection. UpToDate. Updated August 23, 2022. Accessed November 9, 2022. https://www.uptodate.com/contents/treatment-and-prevention-of-listeria-monocytogenes-infection?topicRef=1280&source=see_link
- Voetsch AC, Angulo FJ, Jones TF, et al; Centers for Disease Control and Prevention Emerging Infections Program Foodborne Diseases Active Surveillance Networking Group. Reduction in the incidence of invasive listeriosis in Foodborne Diseases Active Surveillance Network sites, 1996-2003. Clin Infect Dis. 2007;44:513-520.
- MacDonald PDM, Whitwan RE, Boggs JD, et al. Outbreak of listeriosis among Mexican immigrants as a result of consumption of illicitly produced Mexican-style cheese. Clin Infect Dis. 2005;40:677-682.
Chagas disease: An unusual and dangerous infection for both mother and baby
CASE Pregnant woman with a suspected parasitic infection
A 20-year-old, previously healthy, primigravid woman at 24 weeks’ gestation immigrated from Bolivia to the United States 3 days ago. On the morning of her international flight, she awoke to discover a small insect bite just below her left eye. She sought medical evaluation because her eyelid is now significantly swollen, and she has a headache, anorexia, fatigue, and a fever of 38.4° C. The examining physician ordered a polymerase chain reaction (PCR) test for Trypanosoma cruzi, and the test is positive.
- How should this patient be treated during, and after, her delivery?
- Does this infection pose a risk to the newborn baby?
- What type of surveillance and treatment is indicated for the baby?
Chagas disease is common in South America, Central America, and Mexico and is well known to physicians in those countries. Clinicians who practice in the United States are much less familiar with the condition, but it is becoming increasingly common as a result of international travel within the Americas.
In this article, we review the interesting microbiology and epidemiology of Chagas disease, focus on its clinical manifestations, and discuss the most useful diagnostic tests for the illness. We conclude with a summary of preventive and treatment measures, with particular emphasis on managing the disease in pregnancy.
How Chagas disease is transmitted and who is at risk
Chagas disease was named in honor of a Brazilian physician, Carlos Chagas, who first described the condition in 1909. The disease is endemic in South America, Central America, and Mexico, and, recently, its prevalence has increased in the southern United States. Approximately 300,000 people in the United States are infected.1,2
The illness is caused by the parasite Trypanosoma cruzi, and it is also known as American trypanosomiasis. The parasite is spread primarily by the bite of triatomine insects (“kissing bugs”). Approximately 60% of these insects are infected with the parasite. The insects live and thrive in the interspaces of mud walls (adobe homes) and thatched roofs. At night, the insects leave their darkened spaces and feed on the exposed skin of sleeping persons. They are particularly likely to bite the moist skin surfaces near the eye and mouth, and, as they do, they defecate and excrete the parasite into the blood vessels beneath the skin. Within the blood, the trypomastigotes invade various host cells. Inside the host cells, the organism transforms into an amastigote, which is the replicative form of the parasite. After several rounds of replication, the amastigote transforms back into a trypomastigote, bursts from the cell, and goes on to infect other host cells.1
In addition to transmission by the insect vector, the parasite also can be transmitted by blood transfusion and organ donation. When contaminated blood is transfused, the risk of transmission is approximately 10% to 25% for each unit. Following implementation of effective screening programs by blood banks in Central America, South America, Mexico, and the United States, the risk of transmission from undetected infection is now approximately 1:200,000 per unit.
When a transplant procedure with an infected heart is performed, the risk of transmission is 75% to 100%. For liver transplants, the frequency of transmission is 0% to 29%; for kidney transplants, the risk of transmission is 0% to 19%.
Consumption of contaminated food or drink, particularly nonpasteurized items sold by street vendors, is also an important mechanism of transmission. In addition, transmission can occur as a result of laboratory exposure and by exposure to wild animals (racoons, opossums, marmosets, bats, armadillos) in forested areas. Finally, perinatal transmission now accounts for about 22% of infections. As effective vector control programs have been introduced in endemic areas, the proportion of cases caused by the insect vector has steadily decreased1-3 (FIGURE 1).
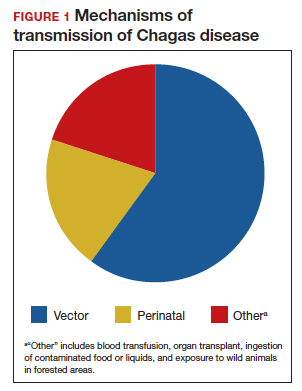
Continue to: Clinical manifestations of Chagas disease...
Clinical manifestations of Chagas disease
Chagas disease occurs in 2 stages, acute and chronic.1,2,4 In patients who are infected via an insect vector, the acute stage typically begins 1 to 2 weeks after the insect bite. This phase of the illness usually lasts 4 to 8 weeks and almost always resolves without treatment.
Some infected patients will be completely free of symptoms. Others will have manifestations such as:
- fever
- malaise
- headache
- hepatosplenomegaly
- lymphadenopathy
- swollen nodule at the site of infection
—Romaña’s sign, when the lesion is on the eyelid
—Chagoma, when the lesion is elsewhere on the skin.
Fortunately, less than 5% of patients will have severe illness, manifested by myocarditis, pericarditis, encephalitis, or meningitis.
People infected by ingestion of the parasite in food or drink often become more severely ill within 3 weeks. Their clinical manifestations include fever, vomiting, dyspnea, cough, chest pain, abdominal pain, and myalgias. Individuals infected through organ transplant or blood transfusion present more like those infected by the insect vector, but their illness may not develop until several weeks to 5 months after exposure.
In the absence of effective treatment, approximately 40% of patients with acute infection will develop chronic infection, often several decades later. The most common, and most ominous, feature of chronic illness is cardiac disease, experienced by about 30% of patients. Cardiac disease may be manifested as a serious arrhythmia, chest pain, congestive heart failure, or thromboembolism.
The other organ system that is likely to be adversely affected in patients with chronic disease is the gastrointestinal (GI) system, and approximately 10% of chronically infected patients experience this complication. Patients may develop a dilated esophagus, which leads to odynophagia and dysphagia. Diminished motility in other areas of the GI tract also may result in chronic constipation and even bowel obstruction. Chronically infected patients who are immunosuppressed due to HIV infection may become gravely ill as a result of encephalitis and brain abscesses. Cardiac and GI dysfunction is due to the parasite’s massive destruction of nerve endings.
Continue to: Making the diagnosis...
Making the diagnosis
The diagnosis of Chagas disease begins with screening patients who have epidemiologic risk factors that place them at high risk for contracting the infection and at significantly increased risk for morbidity and mortality as a result of either the acute infection or the later chronic stage of infection. A thorough history is vital in the evaluation because the acute illness can have such vague clinical manifestations, and many patients remain asymptomatic until signs of chronic infection appear.
Risk factors that warrant screening include being born in a country endemic for Chagas disease, living in an endemic country for more than 6 months, living with someone who has a confirmed diagnosis, residing in a house made of natural materials (mud walls, thatched roof) in an endemic area, and a history of discovering the triatomine bug in the household.
Screening options include serology, microscopy, and PCR testing. Screening with a single, highly sensitive immunoglobulin G (IgG) serologic test is recommended for nonendemic clinical or community settings. In patients who were born in or who lived in an endemic area for more than 6 months, special consideration should be given to screening women of reproductive age, patients of all ages who were born to a mother with a confirmed diagnosis, individuals who were exposed to a triatomine insect, and people who are immunocompromised.5
A positive serologic test should be confirmed with a second assay based on a different antigen. Currently, 4 IgG tests have US Food and Drug Administration (FDA) approval for diagnosis. If a patient has 2 positive serologic tests, the diagnosis is confirmed, regardless of clinical presentation. Discordant results warrant a third test to differentiate between positive and negative results (FIGURE 2).5 All patients with a confirmed diagnosis should have an electrocardiogram, echocardiogram, and abdominal computed tomography (CT) scan to assess for cardiac or GI abnormalities.
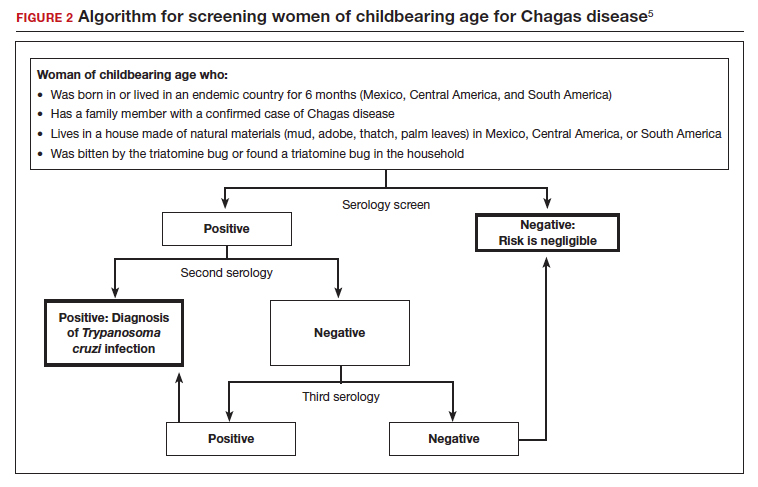
Neonates and infants of mothers with suspected or confirmed infection merit special attention. These children may demonstrate hepatomegaly, splenomegaly, anemia, thrombocytopenia, pneumonitis, heart failure, cardiac arrhythmias, or meningoencephalitis. Newborns delivered to infected mothers will invariably have positive tests for IgG antibody because of transplacental transfer of maternal antibody. Therefore, they should be evaluated by PCR or by direct microscopic examination of the blood for trypomastigotes. In neonates with a negative initial result, repeat testing should be performed by PCR at 4 to 6 weeks of age. Even if the second screening test is negative, the infant should be retested at 9 to 12 months. At this point, maternal IgG no longer should be circulating in the infant’s blood. Three negative tests should effectively rule out T cruzi infection (FIGURE 3).5-7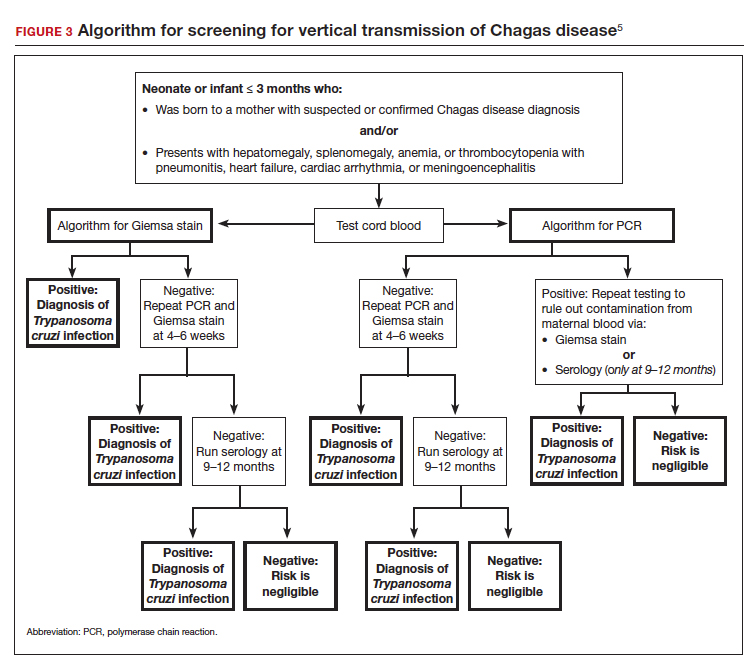
Organ recipients merit special consideration because, in these individuals, the late stages of Chagas disease may be fatal. In these patients, the preferred diagnostic test is PCR. For transplant patients, monitoring should occur every week for 2 months, bimonthly for the third month, and monthly for 6 months after transplantation. Routine monitoring is not recommended in patients with HIV infection who show no clinical signs of Chagas disease and who are not from endemic areas.
Treatment options
No vaccine or hyperimmune globulin can be used to treat Chagas disease. At this time, 2 antiparasitic drugs are available to treat the condition. One is benznidazole, which inhibits DNA, RNA, and protein synthesis within the microorganism. The medication is given in a dose of 5 to 8 mg/kg per day, divided into 2 doses, for 60 days. Benznidazole is FDA approved for the treatment of individuals older than age 2. It has been used off-label in children younger than 2 years of age. The drug is commercially available at http://www.benznidazoletablets.com.
Benznidazole causes multiple minor side effects and several very serious adverse effects. The serious adverse effects include acute generalized exanthematous pustulosis, toxic epidermal necrolysis, peripheral neuropathy, marrow suppression, and hepatotoxicity. Benznidazole has been teratogenic and carcinogenic in animal studies and should not be used in pregnancy.1,3,6
The second drug is nifurtimox. This drug is FDA approved for the treatment of Chagas disease in adults and for newborns and young children. It is commercially available for pharmacies to purchase from several drug wholesalers. Nifurtimox produces reactive oxygen species and toxic intermediates that induce DNA damage and cause cell death of the microorganism. The appropriate oral dose is 8 to 10 mg/kg per day, divided into 3 to 4 equal doses. The duration of treatment is 60 to 90 days, depending on the patient’s response. Like benznidazole, nifurtimox also is highly toxic. Severe adverse effects include a hypersensitivity reaction, anaphylaxis, angioedema, syncope, seizures, and psychosis. Nifurtimox also is teratogenic and is contraindicated in pregnancy.1,3,6
Clinicians who have questions about the use of either of these medications should contact the Centers for Disease Control and Prevention, Division of Parasitic Diseases public inquiries telephone line at (404) 718-4745.
Potential for cure. When either benznidazole or nifurtimox is administered early in the course of a patient’s acute infection, the chance for complete cure is excellent. The same is true for early treatment of the infected neonate. When treatment is delayed, or if it cannot be completed because of intolerable adverse effects, the prognosis for complete cure is diminished.
In adults who have chronic disease, antiparasitic treatment is unlikely to be effective. In such a situation, secondary treatment is directed toward correction of heart failure, control of cardiac rhythm disturbances, and control of GI motility disorders. For both cardiac and GI conditions, medication and surgery may be indicated. Antiparasitic treatment is more effective in children with chronic disease but it is still not uniformly effective.1,3,5,6
Preventing infection
Vector control is the key to preventing infection in areas where Chagas disease is endemic. One important, but often financially unaffordable, measure is construction of homes with building materials that do not support the growth of the triatomine insects that transmit the disease. A second critical preventive measure is the spraying of mud and thatched homes and surrounding areas with long-lasting insecticides. Pyrethroids are the preferred agents today. Alternative agents include fenitrothion and bendiocarb.1
Other important preventive measures include:
- screening the blood supply for T cruzi and eliminating units contaminated with the parasite
- screening for the parasite in organs targeted for transplant
- screening infected women of reproductive age in endemic areas and treating those who are positive before they become pregnant; this measure may be almost 95% effective in preventing congenital infection
- using mosquito netting when housing is insecure and air conditioning is not available
- in endemic areas, avoiding unpasteurized fruit drinks and unwashed fruits and vegetables.
Unique considerations in pregnancy
Chagas disease does not cause specific anatomic birth defects. However, infected women are more likely to experience spontaneous abortion, preterm premature rupture of membranes, preterm labor, and fetal growth restriction. Overall, the risk of perinatal transmission is approximately 5%, but it may be higher in women who have a very high parasite load. Infected neonates who remain untreated are at risk for developing the serious sequelae of chronic infection. At least half of neonates who are infected will initially be asymptomatic. Therefore, screening of at-risk neonates is essential in order to implement effective treatment.3,6
As noted earlier, the usual drugs used for treating Chagas disease should not be used in pregnancy. Nevertheless, it is still important to screen certain individuals for infection and, subsequently, target them and their neonates for treatment immediately following delivery. The following pregnant patients should be screened5,6:
- women with clinical manifestations that suggest acute or chronic infection
- women from areas of the world in which Chagas disease is endemic, namely, from the southern United States to northern Chile and Argentina. Although the disease is endemic in 21 countries, the countries with the highest prevalence are Bolivia, Argentina, and Paraguay.
- newborns delivered to mothers who have been identified as infected.
As mentioned, several tests are available for screening: PCR, antibody assays, and examination of peripheral blood smears. At least 2 test results should be positive to confirm the diagnosis of infection. Neonates should be followed for 9 to 12 months after delivery to determine if perinatal transmission has occurred. Treatment with antiparasitic drugs is indicated for all infected children.5
CASE Continue surveillance during pregnancy, treat after delivery
This patient should not be treated during pregnancy because the 2 major antiparasitic drugs are teratogenic. Antenatally, she should be followed for evidence of preterm labor and fetal growth restriction. She also should have an electrocardiogram and echocardiogram to evaluate for cardiac disease. Immediately after delivery, the patient should be treated with benznidazole for 60 days. Breastfeeding is acceptable. Her neonate should be screened for infection for up to 9 months, following the algorithm outlined earlier (FIGURE 3), and treated if the surveillance tests are positive. ●
- Chagas disease is caused by the parasite Trypanosoma cruzi, which is spread by the bite of the triatomine insect (the “kissing bug”).
- The condition is widespread among impoverished populations in South America, Central America, and Mexico, but it is rare in the United States except in individuals who immigrated here from endemic areas.
- Chagas disease evolves through 2 phases: acute and chronic. Manifestations of acute infection include fever, malaise, headache, hepatosplenomegaly, lymphadenopathy, and swelling at the site of the insect bite. The chronic phase is manifested by serious cardiac and gastrointestinal dysfunction.
- The diagnosis can be established by identifying the organism in a blood smear and by detecting antibody or antigen in the blood.
- The 2 drugs of choice for treatment of Chagas disease are benznidazole and nifurtimox. These drugs are teratogenic and are contraindicated in pregnancy.
- Women at risk for infection should be screened prior to, or during, pregnancy. Infants of infected mothers should be screened for infection for up to 9 to 12 months after delivery and treated if they test positive. Treatment of the infant is almost 100% effective in preventing chronic illness.
- Bern C. Chagas disease: epidemiology, screening, and prevention. UpToDate. Updated April 8, 2022. Accessed October 6, 2022. https://www.uptodate.com/contents /chagas-disease-epidemiology-screening-and-prevention
- Chagas disease. Cleveland Clinic. Reviewed October 8, 2021. Accessed October 6, 2022. https://my.clevelandclinic.org /health/diseases/21876-chagas-disease
- Howard EJ, Xiong X, Carlier Y, et al. Frequency of the congenital transmission of Trypanosoma cruzi: a systematic review and meta-analysis. BJOG. 2014;121:22-33.
- Chagas disease. Mayo Clinic. November 12, 2020. Accessed October 6, 2022. https://www.mayoclinic.org/diseases -conditions/chagas-disease/symptoms-causes/syc-20356212
- Forsyth CJ, Manne-Goehler J, Bern C, et al. Recommendations for screening and diagnosis of Chagas disease in the United States. J Infect Dis. 2022;225:1601-1610.
- Torrico F, Alonso-Vega C, Suarez E. et al. Maternal Trypanosoma cruzi infection, pregnancy outcome, morbidity, and mortality of congenitally infected and non-infected newborns in Bolivia. Am J Trop Med Hyg. 2004;70:201-209.
- Messenger LA, Bern C. Congenital Chagas disease: current diagnostics, limitations and future perspectives. Curr Opin Infect Dis. 2018;31:415-421.

Ms. Drew is a third-year medical student at the University of Florida, Gainesville.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Ms. Drew is a third-year medical student at the University of Florida, Gainesville.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Ms. Drew is a third-year medical student at the University of Florida, Gainesville.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
CASE Pregnant woman with a suspected parasitic infection
A 20-year-old, previously healthy, primigravid woman at 24 weeks’ gestation immigrated from Bolivia to the United States 3 days ago. On the morning of her international flight, she awoke to discover a small insect bite just below her left eye. She sought medical evaluation because her eyelid is now significantly swollen, and she has a headache, anorexia, fatigue, and a fever of 38.4° C. The examining physician ordered a polymerase chain reaction (PCR) test for Trypanosoma cruzi, and the test is positive.
- How should this patient be treated during, and after, her delivery?
- Does this infection pose a risk to the newborn baby?
- What type of surveillance and treatment is indicated for the baby?
Chagas disease is common in South America, Central America, and Mexico and is well known to physicians in those countries. Clinicians who practice in the United States are much less familiar with the condition, but it is becoming increasingly common as a result of international travel within the Americas.
In this article, we review the interesting microbiology and epidemiology of Chagas disease, focus on its clinical manifestations, and discuss the most useful diagnostic tests for the illness. We conclude with a summary of preventive and treatment measures, with particular emphasis on managing the disease in pregnancy.
How Chagas disease is transmitted and who is at risk
Chagas disease was named in honor of a Brazilian physician, Carlos Chagas, who first described the condition in 1909. The disease is endemic in South America, Central America, and Mexico, and, recently, its prevalence has increased in the southern United States. Approximately 300,000 people in the United States are infected.1,2
The illness is caused by the parasite Trypanosoma cruzi, and it is also known as American trypanosomiasis. The parasite is spread primarily by the bite of triatomine insects (“kissing bugs”). Approximately 60% of these insects are infected with the parasite. The insects live and thrive in the interspaces of mud walls (adobe homes) and thatched roofs. At night, the insects leave their darkened spaces and feed on the exposed skin of sleeping persons. They are particularly likely to bite the moist skin surfaces near the eye and mouth, and, as they do, they defecate and excrete the parasite into the blood vessels beneath the skin. Within the blood, the trypomastigotes invade various host cells. Inside the host cells, the organism transforms into an amastigote, which is the replicative form of the parasite. After several rounds of replication, the amastigote transforms back into a trypomastigote, bursts from the cell, and goes on to infect other host cells.1
In addition to transmission by the insect vector, the parasite also can be transmitted by blood transfusion and organ donation. When contaminated blood is transfused, the risk of transmission is approximately 10% to 25% for each unit. Following implementation of effective screening programs by blood banks in Central America, South America, Mexico, and the United States, the risk of transmission from undetected infection is now approximately 1:200,000 per unit.
When a transplant procedure with an infected heart is performed, the risk of transmission is 75% to 100%. For liver transplants, the frequency of transmission is 0% to 29%; for kidney transplants, the risk of transmission is 0% to 19%.
Consumption of contaminated food or drink, particularly nonpasteurized items sold by street vendors, is also an important mechanism of transmission. In addition, transmission can occur as a result of laboratory exposure and by exposure to wild animals (racoons, opossums, marmosets, bats, armadillos) in forested areas. Finally, perinatal transmission now accounts for about 22% of infections. As effective vector control programs have been introduced in endemic areas, the proportion of cases caused by the insect vector has steadily decreased1-3 (FIGURE 1).
Continue to: Clinical manifestations of Chagas disease...
Clinical manifestations of Chagas disease
Chagas disease occurs in 2 stages, acute and chronic.1,2,4 In patients who are infected via an insect vector, the acute stage typically begins 1 to 2 weeks after the insect bite. This phase of the illness usually lasts 4 to 8 weeks and almost always resolves without treatment.
Some infected patients will be completely free of symptoms. Others will have manifestations such as:
- fever
- malaise
- headache
- hepatosplenomegaly
- lymphadenopathy
- swollen nodule at the site of infection
—Romaña’s sign, when the lesion is on the eyelid
—Chagoma, when the lesion is elsewhere on the skin.
Fortunately, less than 5% of patients will have severe illness, manifested by myocarditis, pericarditis, encephalitis, or meningitis.
People infected by ingestion of the parasite in food or drink often become more severely ill within 3 weeks. Their clinical manifestations include fever, vomiting, dyspnea, cough, chest pain, abdominal pain, and myalgias. Individuals infected through organ transplant or blood transfusion present more like those infected by the insect vector, but their illness may not develop until several weeks to 5 months after exposure.
In the absence of effective treatment, approximately 40% of patients with acute infection will develop chronic infection, often several decades later. The most common, and most ominous, feature of chronic illness is cardiac disease, experienced by about 30% of patients. Cardiac disease may be manifested as a serious arrhythmia, chest pain, congestive heart failure, or thromboembolism.
The other organ system that is likely to be adversely affected in patients with chronic disease is the gastrointestinal (GI) system, and approximately 10% of chronically infected patients experience this complication. Patients may develop a dilated esophagus, which leads to odynophagia and dysphagia. Diminished motility in other areas of the GI tract also may result in chronic constipation and even bowel obstruction. Chronically infected patients who are immunosuppressed due to HIV infection may become gravely ill as a result of encephalitis and brain abscesses. Cardiac and GI dysfunction is due to the parasite’s massive destruction of nerve endings.
Continue to: Making the diagnosis...
Making the diagnosis
The diagnosis of Chagas disease begins with screening patients who have epidemiologic risk factors that place them at high risk for contracting the infection and at significantly increased risk for morbidity and mortality as a result of either the acute infection or the later chronic stage of infection. A thorough history is vital in the evaluation because the acute illness can have such vague clinical manifestations, and many patients remain asymptomatic until signs of chronic infection appear.
Risk factors that warrant screening include being born in a country endemic for Chagas disease, living in an endemic country for more than 6 months, living with someone who has a confirmed diagnosis, residing in a house made of natural materials (mud walls, thatched roof) in an endemic area, and a history of discovering the triatomine bug in the household.
Screening options include serology, microscopy, and PCR testing. Screening with a single, highly sensitive immunoglobulin G (IgG) serologic test is recommended for nonendemic clinical or community settings. In patients who were born in or who lived in an endemic area for more than 6 months, special consideration should be given to screening women of reproductive age, patients of all ages who were born to a mother with a confirmed diagnosis, individuals who were exposed to a triatomine insect, and people who are immunocompromised.5
A positive serologic test should be confirmed with a second assay based on a different antigen. Currently, 4 IgG tests have US Food and Drug Administration (FDA) approval for diagnosis. If a patient has 2 positive serologic tests, the diagnosis is confirmed, regardless of clinical presentation. Discordant results warrant a third test to differentiate between positive and negative results (FIGURE 2).5 All patients with a confirmed diagnosis should have an electrocardiogram, echocardiogram, and abdominal computed tomography (CT) scan to assess for cardiac or GI abnormalities.
Neonates and infants of mothers with suspected or confirmed infection merit special attention. These children may demonstrate hepatomegaly, splenomegaly, anemia, thrombocytopenia, pneumonitis, heart failure, cardiac arrhythmias, or meningoencephalitis. Newborns delivered to infected mothers will invariably have positive tests for IgG antibody because of transplacental transfer of maternal antibody. Therefore, they should be evaluated by PCR or by direct microscopic examination of the blood for trypomastigotes. In neonates with a negative initial result, repeat testing should be performed by PCR at 4 to 6 weeks of age. Even if the second screening test is negative, the infant should be retested at 9 to 12 months. At this point, maternal IgG no longer should be circulating in the infant’s blood. Three negative tests should effectively rule out T cruzi infection (FIGURE 3).5-7
Organ recipients merit special consideration because, in these individuals, the late stages of Chagas disease may be fatal. In these patients, the preferred diagnostic test is PCR. For transplant patients, monitoring should occur every week for 2 months, bimonthly for the third month, and monthly for 6 months after transplantation. Routine monitoring is not recommended in patients with HIV infection who show no clinical signs of Chagas disease and who are not from endemic areas.
Treatment options
No vaccine or hyperimmune globulin can be used to treat Chagas disease. At this time, 2 antiparasitic drugs are available to treat the condition. One is benznidazole, which inhibits DNA, RNA, and protein synthesis within the microorganism. The medication is given in a dose of 5 to 8 mg/kg per day, divided into 2 doses, for 60 days. Benznidazole is FDA approved for the treatment of individuals older than age 2. It has been used off-label in children younger than 2 years of age. The drug is commercially available at http://www.benznidazoletablets.com.
Benznidazole causes multiple minor side effects and several very serious adverse effects. The serious adverse effects include acute generalized exanthematous pustulosis, toxic epidermal necrolysis, peripheral neuropathy, marrow suppression, and hepatotoxicity. Benznidazole has been teratogenic and carcinogenic in animal studies and should not be used in pregnancy.1,3,6
The second drug is nifurtimox. This drug is FDA approved for the treatment of Chagas disease in adults and for newborns and young children. It is commercially available for pharmacies to purchase from several drug wholesalers. Nifurtimox produces reactive oxygen species and toxic intermediates that induce DNA damage and cause cell death of the microorganism. The appropriate oral dose is 8 to 10 mg/kg per day, divided into 3 to 4 equal doses. The duration of treatment is 60 to 90 days, depending on the patient’s response. Like benznidazole, nifurtimox also is highly toxic. Severe adverse effects include a hypersensitivity reaction, anaphylaxis, angioedema, syncope, seizures, and psychosis. Nifurtimox also is teratogenic and is contraindicated in pregnancy.1,3,6
Clinicians who have questions about the use of either of these medications should contact the Centers for Disease Control and Prevention, Division of Parasitic Diseases public inquiries telephone line at (404) 718-4745.
Potential for cure. When either benznidazole or nifurtimox is administered early in the course of a patient’s acute infection, the chance for complete cure is excellent. The same is true for early treatment of the infected neonate. When treatment is delayed, or if it cannot be completed because of intolerable adverse effects, the prognosis for complete cure is diminished.
In adults who have chronic disease, antiparasitic treatment is unlikely to be effective. In such a situation, secondary treatment is directed toward correction of heart failure, control of cardiac rhythm disturbances, and control of GI motility disorders. For both cardiac and GI conditions, medication and surgery may be indicated. Antiparasitic treatment is more effective in children with chronic disease but it is still not uniformly effective.1,3,5,6
Preventing infection
Vector control is the key to preventing infection in areas where Chagas disease is endemic. One important, but often financially unaffordable, measure is construction of homes with building materials that do not support the growth of the triatomine insects that transmit the disease. A second critical preventive measure is the spraying of mud and thatched homes and surrounding areas with long-lasting insecticides. Pyrethroids are the preferred agents today. Alternative agents include fenitrothion and bendiocarb.1
Other important preventive measures include:
- screening the blood supply for T cruzi and eliminating units contaminated with the parasite
- screening for the parasite in organs targeted for transplant
- screening infected women of reproductive age in endemic areas and treating those who are positive before they become pregnant; this measure may be almost 95% effective in preventing congenital infection
- using mosquito netting when housing is insecure and air conditioning is not available
- in endemic areas, avoiding unpasteurized fruit drinks and unwashed fruits and vegetables.
Unique considerations in pregnancy
Chagas disease does not cause specific anatomic birth defects. However, infected women are more likely to experience spontaneous abortion, preterm premature rupture of membranes, preterm labor, and fetal growth restriction. Overall, the risk of perinatal transmission is approximately 5%, but it may be higher in women who have a very high parasite load. Infected neonates who remain untreated are at risk for developing the serious sequelae of chronic infection. At least half of neonates who are infected will initially be asymptomatic. Therefore, screening of at-risk neonates is essential in order to implement effective treatment.3,6
As noted earlier, the usual drugs used for treating Chagas disease should not be used in pregnancy. Nevertheless, it is still important to screen certain individuals for infection and, subsequently, target them and their neonates for treatment immediately following delivery. The following pregnant patients should be screened5,6:
- women with clinical manifestations that suggest acute or chronic infection
- women from areas of the world in which Chagas disease is endemic, namely, from the southern United States to northern Chile and Argentina. Although the disease is endemic in 21 countries, the countries with the highest prevalence are Bolivia, Argentina, and Paraguay.
- newborns delivered to mothers who have been identified as infected.
As mentioned, several tests are available for screening: PCR, antibody assays, and examination of peripheral blood smears. At least 2 test results should be positive to confirm the diagnosis of infection. Neonates should be followed for 9 to 12 months after delivery to determine if perinatal transmission has occurred. Treatment with antiparasitic drugs is indicated for all infected children.5
CASE Continue surveillance during pregnancy, treat after delivery
This patient should not be treated during pregnancy because the 2 major antiparasitic drugs are teratogenic. Antenatally, she should be followed for evidence of preterm labor and fetal growth restriction. She also should have an electrocardiogram and echocardiogram to evaluate for cardiac disease. Immediately after delivery, the patient should be treated with benznidazole for 60 days. Breastfeeding is acceptable. Her neonate should be screened for infection for up to 9 months, following the algorithm outlined earlier (FIGURE 3), and treated if the surveillance tests are positive. ●
- Chagas disease is caused by the parasite Trypanosoma cruzi, which is spread by the bite of the triatomine insect (the “kissing bug”).
- The condition is widespread among impoverished populations in South America, Central America, and Mexico, but it is rare in the United States except in individuals who immigrated here from endemic areas.
- Chagas disease evolves through 2 phases: acute and chronic. Manifestations of acute infection include fever, malaise, headache, hepatosplenomegaly, lymphadenopathy, and swelling at the site of the insect bite. The chronic phase is manifested by serious cardiac and gastrointestinal dysfunction.
- The diagnosis can be established by identifying the organism in a blood smear and by detecting antibody or antigen in the blood.
- The 2 drugs of choice for treatment of Chagas disease are benznidazole and nifurtimox. These drugs are teratogenic and are contraindicated in pregnancy.
- Women at risk for infection should be screened prior to, or during, pregnancy. Infants of infected mothers should be screened for infection for up to 9 to 12 months after delivery and treated if they test positive. Treatment of the infant is almost 100% effective in preventing chronic illness.
CASE Pregnant woman with a suspected parasitic infection
A 20-year-old, previously healthy, primigravid woman at 24 weeks’ gestation immigrated from Bolivia to the United States 3 days ago. On the morning of her international flight, she awoke to discover a small insect bite just below her left eye. She sought medical evaluation because her eyelid is now significantly swollen, and she has a headache, anorexia, fatigue, and a fever of 38.4° C. The examining physician ordered a polymerase chain reaction (PCR) test for Trypanosoma cruzi, and the test is positive.
- How should this patient be treated during, and after, her delivery?
- Does this infection pose a risk to the newborn baby?
- What type of surveillance and treatment is indicated for the baby?
Chagas disease is common in South America, Central America, and Mexico and is well known to physicians in those countries. Clinicians who practice in the United States are much less familiar with the condition, but it is becoming increasingly common as a result of international travel within the Americas.
In this article, we review the interesting microbiology and epidemiology of Chagas disease, focus on its clinical manifestations, and discuss the most useful diagnostic tests for the illness. We conclude with a summary of preventive and treatment measures, with particular emphasis on managing the disease in pregnancy.
How Chagas disease is transmitted and who is at risk
Chagas disease was named in honor of a Brazilian physician, Carlos Chagas, who first described the condition in 1909. The disease is endemic in South America, Central America, and Mexico, and, recently, its prevalence has increased in the southern United States. Approximately 300,000 people in the United States are infected.1,2
The illness is caused by the parasite Trypanosoma cruzi, and it is also known as American trypanosomiasis. The parasite is spread primarily by the bite of triatomine insects (“kissing bugs”). Approximately 60% of these insects are infected with the parasite. The insects live and thrive in the interspaces of mud walls (adobe homes) and thatched roofs. At night, the insects leave their darkened spaces and feed on the exposed skin of sleeping persons. They are particularly likely to bite the moist skin surfaces near the eye and mouth, and, as they do, they defecate and excrete the parasite into the blood vessels beneath the skin. Within the blood, the trypomastigotes invade various host cells. Inside the host cells, the organism transforms into an amastigote, which is the replicative form of the parasite. After several rounds of replication, the amastigote transforms back into a trypomastigote, bursts from the cell, and goes on to infect other host cells.1
In addition to transmission by the insect vector, the parasite also can be transmitted by blood transfusion and organ donation. When contaminated blood is transfused, the risk of transmission is approximately 10% to 25% for each unit. Following implementation of effective screening programs by blood banks in Central America, South America, Mexico, and the United States, the risk of transmission from undetected infection is now approximately 1:200,000 per unit.
When a transplant procedure with an infected heart is performed, the risk of transmission is 75% to 100%. For liver transplants, the frequency of transmission is 0% to 29%; for kidney transplants, the risk of transmission is 0% to 19%.
Consumption of contaminated food or drink, particularly nonpasteurized items sold by street vendors, is also an important mechanism of transmission. In addition, transmission can occur as a result of laboratory exposure and by exposure to wild animals (racoons, opossums, marmosets, bats, armadillos) in forested areas. Finally, perinatal transmission now accounts for about 22% of infections. As effective vector control programs have been introduced in endemic areas, the proportion of cases caused by the insect vector has steadily decreased1-3 (FIGURE 1).
Continue to: Clinical manifestations of Chagas disease...
Clinical manifestations of Chagas disease
Chagas disease occurs in 2 stages, acute and chronic.1,2,4 In patients who are infected via an insect vector, the acute stage typically begins 1 to 2 weeks after the insect bite. This phase of the illness usually lasts 4 to 8 weeks and almost always resolves without treatment.
Some infected patients will be completely free of symptoms. Others will have manifestations such as:
- fever
- malaise
- headache
- hepatosplenomegaly
- lymphadenopathy
- swollen nodule at the site of infection
—Romaña’s sign, when the lesion is on the eyelid
—Chagoma, when the lesion is elsewhere on the skin.
Fortunately, less than 5% of patients will have severe illness, manifested by myocarditis, pericarditis, encephalitis, or meningitis.
People infected by ingestion of the parasite in food or drink often become more severely ill within 3 weeks. Their clinical manifestations include fever, vomiting, dyspnea, cough, chest pain, abdominal pain, and myalgias. Individuals infected through organ transplant or blood transfusion present more like those infected by the insect vector, but their illness may not develop until several weeks to 5 months after exposure.
In the absence of effective treatment, approximately 40% of patients with acute infection will develop chronic infection, often several decades later. The most common, and most ominous, feature of chronic illness is cardiac disease, experienced by about 30% of patients. Cardiac disease may be manifested as a serious arrhythmia, chest pain, congestive heart failure, or thromboembolism.
The other organ system that is likely to be adversely affected in patients with chronic disease is the gastrointestinal (GI) system, and approximately 10% of chronically infected patients experience this complication. Patients may develop a dilated esophagus, which leads to odynophagia and dysphagia. Diminished motility in other areas of the GI tract also may result in chronic constipation and even bowel obstruction. Chronically infected patients who are immunosuppressed due to HIV infection may become gravely ill as a result of encephalitis and brain abscesses. Cardiac and GI dysfunction is due to the parasite’s massive destruction of nerve endings.
Continue to: Making the diagnosis...
Making the diagnosis
The diagnosis of Chagas disease begins with screening patients who have epidemiologic risk factors that place them at high risk for contracting the infection and at significantly increased risk for morbidity and mortality as a result of either the acute infection or the later chronic stage of infection. A thorough history is vital in the evaluation because the acute illness can have such vague clinical manifestations, and many patients remain asymptomatic until signs of chronic infection appear.
Risk factors that warrant screening include being born in a country endemic for Chagas disease, living in an endemic country for more than 6 months, living with someone who has a confirmed diagnosis, residing in a house made of natural materials (mud walls, thatched roof) in an endemic area, and a history of discovering the triatomine bug in the household.
Screening options include serology, microscopy, and PCR testing. Screening with a single, highly sensitive immunoglobulin G (IgG) serologic test is recommended for nonendemic clinical or community settings. In patients who were born in or who lived in an endemic area for more than 6 months, special consideration should be given to screening women of reproductive age, patients of all ages who were born to a mother with a confirmed diagnosis, individuals who were exposed to a triatomine insect, and people who are immunocompromised.5
A positive serologic test should be confirmed with a second assay based on a different antigen. Currently, 4 IgG tests have US Food and Drug Administration (FDA) approval for diagnosis. If a patient has 2 positive serologic tests, the diagnosis is confirmed, regardless of clinical presentation. Discordant results warrant a third test to differentiate between positive and negative results (FIGURE 2).5 All patients with a confirmed diagnosis should have an electrocardiogram, echocardiogram, and abdominal computed tomography (CT) scan to assess for cardiac or GI abnormalities.
Neonates and infants of mothers with suspected or confirmed infection merit special attention. These children may demonstrate hepatomegaly, splenomegaly, anemia, thrombocytopenia, pneumonitis, heart failure, cardiac arrhythmias, or meningoencephalitis. Newborns delivered to infected mothers will invariably have positive tests for IgG antibody because of transplacental transfer of maternal antibody. Therefore, they should be evaluated by PCR or by direct microscopic examination of the blood for trypomastigotes. In neonates with a negative initial result, repeat testing should be performed by PCR at 4 to 6 weeks of age. Even if the second screening test is negative, the infant should be retested at 9 to 12 months. At this point, maternal IgG no longer should be circulating in the infant’s blood. Three negative tests should effectively rule out T cruzi infection (FIGURE 3).5-7
Organ recipients merit special consideration because, in these individuals, the late stages of Chagas disease may be fatal. In these patients, the preferred diagnostic test is PCR. For transplant patients, monitoring should occur every week for 2 months, bimonthly for the third month, and monthly for 6 months after transplantation. Routine monitoring is not recommended in patients with HIV infection who show no clinical signs of Chagas disease and who are not from endemic areas.
Treatment options
No vaccine or hyperimmune globulin can be used to treat Chagas disease. At this time, 2 antiparasitic drugs are available to treat the condition. One is benznidazole, which inhibits DNA, RNA, and protein synthesis within the microorganism. The medication is given in a dose of 5 to 8 mg/kg per day, divided into 2 doses, for 60 days. Benznidazole is FDA approved for the treatment of individuals older than age 2. It has been used off-label in children younger than 2 years of age. The drug is commercially available at http://www.benznidazoletablets.com.
Benznidazole causes multiple minor side effects and several very serious adverse effects. The serious adverse effects include acute generalized exanthematous pustulosis, toxic epidermal necrolysis, peripheral neuropathy, marrow suppression, and hepatotoxicity. Benznidazole has been teratogenic and carcinogenic in animal studies and should not be used in pregnancy.1,3,6
The second drug is nifurtimox. This drug is FDA approved for the treatment of Chagas disease in adults and for newborns and young children. It is commercially available for pharmacies to purchase from several drug wholesalers. Nifurtimox produces reactive oxygen species and toxic intermediates that induce DNA damage and cause cell death of the microorganism. The appropriate oral dose is 8 to 10 mg/kg per day, divided into 3 to 4 equal doses. The duration of treatment is 60 to 90 days, depending on the patient’s response. Like benznidazole, nifurtimox also is highly toxic. Severe adverse effects include a hypersensitivity reaction, anaphylaxis, angioedema, syncope, seizures, and psychosis. Nifurtimox also is teratogenic and is contraindicated in pregnancy.1,3,6
Clinicians who have questions about the use of either of these medications should contact the Centers for Disease Control and Prevention, Division of Parasitic Diseases public inquiries telephone line at (404) 718-4745.
Potential for cure. When either benznidazole or nifurtimox is administered early in the course of a patient’s acute infection, the chance for complete cure is excellent. The same is true for early treatment of the infected neonate. When treatment is delayed, or if it cannot be completed because of intolerable adverse effects, the prognosis for complete cure is diminished.
In adults who have chronic disease, antiparasitic treatment is unlikely to be effective. In such a situation, secondary treatment is directed toward correction of heart failure, control of cardiac rhythm disturbances, and control of GI motility disorders. For both cardiac and GI conditions, medication and surgery may be indicated. Antiparasitic treatment is more effective in children with chronic disease but it is still not uniformly effective.1,3,5,6
Preventing infection
Vector control is the key to preventing infection in areas where Chagas disease is endemic. One important, but often financially unaffordable, measure is construction of homes with building materials that do not support the growth of the triatomine insects that transmit the disease. A second critical preventive measure is the spraying of mud and thatched homes and surrounding areas with long-lasting insecticides. Pyrethroids are the preferred agents today. Alternative agents include fenitrothion and bendiocarb.1
Other important preventive measures include:
- screening the blood supply for T cruzi and eliminating units contaminated with the parasite
- screening for the parasite in organs targeted for transplant
- screening infected women of reproductive age in endemic areas and treating those who are positive before they become pregnant; this measure may be almost 95% effective in preventing congenital infection
- using mosquito netting when housing is insecure and air conditioning is not available
- in endemic areas, avoiding unpasteurized fruit drinks and unwashed fruits and vegetables.
Unique considerations in pregnancy
Chagas disease does not cause specific anatomic birth defects. However, infected women are more likely to experience spontaneous abortion, preterm premature rupture of membranes, preterm labor, and fetal growth restriction. Overall, the risk of perinatal transmission is approximately 5%, but it may be higher in women who have a very high parasite load. Infected neonates who remain untreated are at risk for developing the serious sequelae of chronic infection. At least half of neonates who are infected will initially be asymptomatic. Therefore, screening of at-risk neonates is essential in order to implement effective treatment.3,6
As noted earlier, the usual drugs used for treating Chagas disease should not be used in pregnancy. Nevertheless, it is still important to screen certain individuals for infection and, subsequently, target them and their neonates for treatment immediately following delivery. The following pregnant patients should be screened5,6:
- women with clinical manifestations that suggest acute or chronic infection
- women from areas of the world in which Chagas disease is endemic, namely, from the southern United States to northern Chile and Argentina. Although the disease is endemic in 21 countries, the countries with the highest prevalence are Bolivia, Argentina, and Paraguay.
- newborns delivered to mothers who have been identified as infected.
As mentioned, several tests are available for screening: PCR, antibody assays, and examination of peripheral blood smears. At least 2 test results should be positive to confirm the diagnosis of infection. Neonates should be followed for 9 to 12 months after delivery to determine if perinatal transmission has occurred. Treatment with antiparasitic drugs is indicated for all infected children.5
CASE Continue surveillance during pregnancy, treat after delivery
This patient should not be treated during pregnancy because the 2 major antiparasitic drugs are teratogenic. Antenatally, she should be followed for evidence of preterm labor and fetal growth restriction. She also should have an electrocardiogram and echocardiogram to evaluate for cardiac disease. Immediately after delivery, the patient should be treated with benznidazole for 60 days. Breastfeeding is acceptable. Her neonate should be screened for infection for up to 9 months, following the algorithm outlined earlier (FIGURE 3), and treated if the surveillance tests are positive. ●
- Chagas disease is caused by the parasite Trypanosoma cruzi, which is spread by the bite of the triatomine insect (the “kissing bug”).
- The condition is widespread among impoverished populations in South America, Central America, and Mexico, but it is rare in the United States except in individuals who immigrated here from endemic areas.
- Chagas disease evolves through 2 phases: acute and chronic. Manifestations of acute infection include fever, malaise, headache, hepatosplenomegaly, lymphadenopathy, and swelling at the site of the insect bite. The chronic phase is manifested by serious cardiac and gastrointestinal dysfunction.
- The diagnosis can be established by identifying the organism in a blood smear and by detecting antibody or antigen in the blood.
- The 2 drugs of choice for treatment of Chagas disease are benznidazole and nifurtimox. These drugs are teratogenic and are contraindicated in pregnancy.
- Women at risk for infection should be screened prior to, or during, pregnancy. Infants of infected mothers should be screened for infection for up to 9 to 12 months after delivery and treated if they test positive. Treatment of the infant is almost 100% effective in preventing chronic illness.
- Bern C. Chagas disease: epidemiology, screening, and prevention. UpToDate. Updated April 8, 2022. Accessed October 6, 2022. https://www.uptodate.com/contents /chagas-disease-epidemiology-screening-and-prevention
- Chagas disease. Cleveland Clinic. Reviewed October 8, 2021. Accessed October 6, 2022. https://my.clevelandclinic.org /health/diseases/21876-chagas-disease
- Howard EJ, Xiong X, Carlier Y, et al. Frequency of the congenital transmission of Trypanosoma cruzi: a systematic review and meta-analysis. BJOG. 2014;121:22-33.
- Chagas disease. Mayo Clinic. November 12, 2020. Accessed October 6, 2022. https://www.mayoclinic.org/diseases -conditions/chagas-disease/symptoms-causes/syc-20356212
- Forsyth CJ, Manne-Goehler J, Bern C, et al. Recommendations for screening and diagnosis of Chagas disease in the United States. J Infect Dis. 2022;225:1601-1610.
- Torrico F, Alonso-Vega C, Suarez E. et al. Maternal Trypanosoma cruzi infection, pregnancy outcome, morbidity, and mortality of congenitally infected and non-infected newborns in Bolivia. Am J Trop Med Hyg. 2004;70:201-209.
- Messenger LA, Bern C. Congenital Chagas disease: current diagnostics, limitations and future perspectives. Curr Opin Infect Dis. 2018;31:415-421.
- Bern C. Chagas disease: epidemiology, screening, and prevention. UpToDate. Updated April 8, 2022. Accessed October 6, 2022. https://www.uptodate.com/contents /chagas-disease-epidemiology-screening-and-prevention
- Chagas disease. Cleveland Clinic. Reviewed October 8, 2021. Accessed October 6, 2022. https://my.clevelandclinic.org /health/diseases/21876-chagas-disease
- Howard EJ, Xiong X, Carlier Y, et al. Frequency of the congenital transmission of Trypanosoma cruzi: a systematic review and meta-analysis. BJOG. 2014;121:22-33.
- Chagas disease. Mayo Clinic. November 12, 2020. Accessed October 6, 2022. https://www.mayoclinic.org/diseases -conditions/chagas-disease/symptoms-causes/syc-20356212
- Forsyth CJ, Manne-Goehler J, Bern C, et al. Recommendations for screening and diagnosis of Chagas disease in the United States. J Infect Dis. 2022;225:1601-1610.
- Torrico F, Alonso-Vega C, Suarez E. et al. Maternal Trypanosoma cruzi infection, pregnancy outcome, morbidity, and mortality of congenitally infected and non-infected newborns in Bolivia. Am J Trop Med Hyg. 2004;70:201-209.
- Messenger LA, Bern C. Congenital Chagas disease: current diagnostics, limitations and future perspectives. Curr Opin Infect Dis. 2018;31:415-421.

Viral threats to the fetus and mother: Parvovirus and varicella
We review 2 important viral infections in this article. One, parvovirus, poses a major threat to the fetus. The second, varicella, poses less risk to the fetus but significantly greater risk to the mother. We focus on the epidemiology, clinical presentation, diagnosis, and management of each infection.
Parvovirus infection and its risks to the fetus
CASE #1 Pregnant teacher exposed to fifth disease
A 28-year-old primigravid woman at 16 weeks’ gestation works as an elementary school teacher. Over the past 3 weeks, she has been exposed to 4 children who had fifth disease. She now requests evaluation because she has malaise, arthralgias, myalgias, fever of 38.2°C, and a fine lacelike erythematous rash on her trunk, arms, and cheeks.
- What is the most likely diagnosis?
- What diagnostic tests are indicated?
- Is her fetus at risk?
Epidemiology of parvovirus
Parvovirus B19 is a small, single-stranded DNA virus. It is highly contagious and is transmitted primarily by respiratory droplets. Transmission also can occur via infected blood, for example, through a blood transfusion. The incubation period is 10 to 20 days. Among adults, the individuals at greatest risk for infection are those who have close contact with young children, such as parents, day-care workers, and elementary school teachers. With sustained exposure in the household or classroom, the risk of seroconversion approaches 50%.1 Approximately 50% to 60% of reproductive-aged women have evidence of prior infection, and immunity is usually lifelong.
Clinical manifestations
The classic presentation of parvovirus infection is erythema infectiosum, also called fifth disease. This condition is characterized by a “slapped cheek” facial rash, malaise, myalgias, arthralgias, and low-grade fever. A fine lacelike rash often develops over the torso. In adults, the characteristic rash may be absent, and the most common presentation is a flu-like illness with joint pains.1,2 In children and in adults with an underlying hemoglobinopathy, parvovirus can cause transient aplastic crisis, and patients present with signs of a severe anemia, such as dyspnea, pallor, and fatigue.
Although parvovirus infection usually poses no serious risk in otherwise healthy children and adults, it can cause major fetal injury when the pregnant woman is infected early in pregnancy. The principal manifestation of fetal infection is hydrops. Hydrops primarily results when the virus crosses the placenta and attaches to the P antigen on the surface of red cell progenitors in the fetal marrow, causing an aplastic anemia with resultant high-output congestive heart failure. The virus also may directly injure the fetal myocardium, thus exacerbating heart failure. Other manifestations of congenital parvovirus include thrombocytopenia and hepatitis.3
The severity of fetal injury is inversely proportional to the gestational age at the time of maternal infection. When primary maternal infection occurs in the first trimester, the frequency of fetal hydrops is 5% to 10%. If infection develops in weeks 13 to 20, the risk of hydrops decreases to 5% or less. If infection develops beyond week 20, the incidence of fetal hydrops is 1% or lower.2
Continue to: Diagnostic steps...
Diagnostic steps
Appropriate diagnostic evaluation for a pregnant woman with exposure to parvovirus or clinical manifestations suggestive of parvovirus infection is outlined in FIGURE 1.
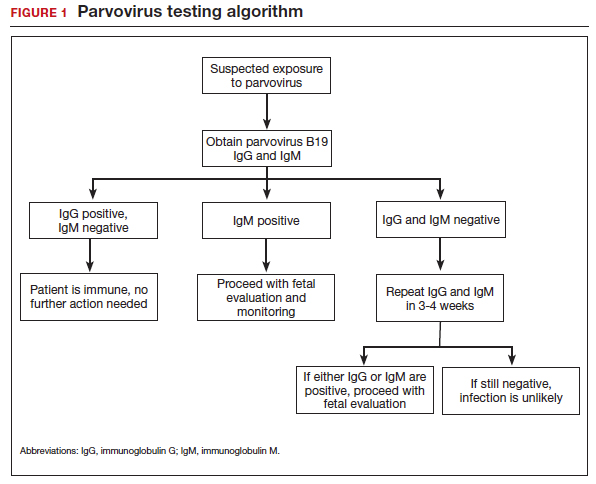
If infection is confirmed, serial ultrasound monitoring should be performed on a weekly to biweekly basis for 8 to 12 weeks, as delineated in FIGURE 2. Extended surveillance is necessary because the incubation period in the fetus is longer than that in the mother.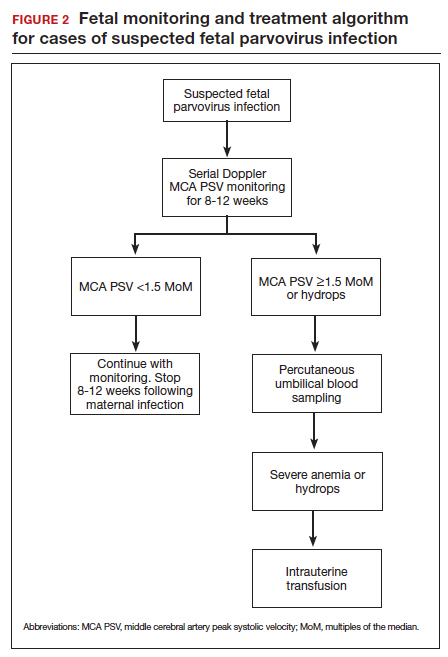
As the fetus develops anemia, peripheral tissues become hypoxic, leading to reflex peripheral vasoconstriction and increased cardiac output. At the same time, reduction in the number of fetal red blood cells decreases blood viscosity. The combination of these changes results in an increase in blood flow to the fetal brain, which can be detected by measuring the peak systolic velocity of flow in the middle cerebral artery (MCA PSV) with Doppler ultrasound imaging (FIGURE 3). The increase in MCA PSV parallels the decrease in fetal hematocrit and precedes the development of hydrops. In fact, signs of fetal hydrops do not usually develop until the fetal hematocrit falls to 15 to 20 vol%.
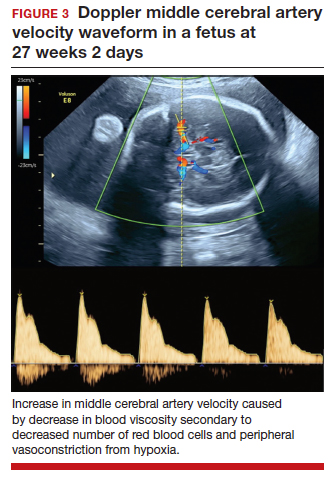
Management may necessitate intrauterine transfusion
Although some cases of fetal hydrops may resolve spontaneously, most authors agree that intrauterine transfusion is essential. In most instances, only a single intrauterine transfusion is necessary. In some fetuses, however, the infection is so prolonged and the anemia so severe that 2 to 3 transfusions may be required.
Infants who survive the intrauterine transfusion usually have an excellent long-term prognosis. However, isolated case reports have documented neurologic morbidity and prolonged transfusion-dependent anemia.4 In light of these reports, we recommend that a third trimester ultrasound exam be performed to assess fetal growth and evaluate the anatomy of the fetal brain. For the fetus with abnormal intracranial findings on ultrasonography, fetal magnetic resonance imaging is indicated.5
CASE #1 Diagnosis is probable parvovirus
The most likely diagnosis in this case is erythema infectiosum. This diagnosis can be confirmed by identifying positive immunogloblulin M (IgM) antibody and by detecting parvovirus in the maternal serum by polymerase chain reaction. Given the gestational age of 16 weeks, the risk of serious fetal injury should be less than 5%. Nevertheless, serial ultrasound examinations should be performed to assess for signs of fetal anemia.
Varicella exposure in pregnancy
CASE #2 Pregnant woman exposed to chickenpox has symptoms
Two weeks ago, a 32-year-old woman (G3P2002) at 24 weeks’ gestation was exposed to a neighbor’s child who had chickenpox. The patient has no history of natural infection or vaccination. She now has a fever of 38.6°C, malaise, headache, and a diffuse pruritic vesicular rash on her trunk and extremities. She also is experiencing a dry cough and mild dyspnea.
- What diagnostic tests are indicated?
- What treatment is indicated?
- What risk does this condition pose to the fetus?
Epidemiology of varicella
Varicella (chickenpox) is caused by the DNA varicella-zoster virus, an organism that is a member of the herpesvirus family. The disease occurs predominantly in children, and the infection is transmitted by respiratory droplets and by direct contact. Its incubation period is short (10–14 days), and it is highly contagious. More than 90% of susceptible close contacts will become infected after exposure to the index case. Like other herpesviruses, the varicella virus can establish a latent infection and then become manifest years later as herpes zoster (shingles).5,6
Continue to: Clinical manifestations...
Clinical manifestations
Patients with varicella usually have prodromal symptoms and signs that include malaise, fatigue, arthralgias, myalgias, and a low-grade fever. Varicella’s pathognomonic manifestation is a pruritic, macular rash that starts on the face and trunk and then spreads centripetally to the extremities. The lesions typically appear in “crops” and evolve through several distinct phases: macule, papule, vesicle, pustule, ulcer, and crust.5
In children, varicella is manifest almost entirely by mucocutaneous lesions. In adults, however, 2 serious and potentially life-threatening complications can occur. Approximately 1% of infected adults develop encephalitis and about 20% develop viral pneumonia, often accompanied by a severe superimposed bacterial pneumonia.5
When maternal infection develops in the first half of pregnancy, approximately 2% of fetuses will have evidence of congenital infection, usually manifested by circular, constricting scars on the extremities. These lesions typically occur in a dermatomal distribution. Spontaneous abortion and fetal death in utero also have been reported, but fortunately they are quite rare. When maternal infection occurs beyond 20 weeks of gestation, fetal injury is very uncommon.7
Interestingly, when maternal infection occurs at the time of delivery or shortly thereafter (from 5 days before until 2 days after delivery), neonatal varicella may develop. This infection may take 3 forms: disseminated mucocutaneous lesions, a deep-seated visceral infection, or severe pneumonia. In the era before the ready availability of antiviral agents, the case fatality rate from neonatal varicella was approximately 30%.5
Diagnosis is clinical
The diagnosis of varicella usually is established on the basis of clinical examination. It can be confirmed by identification of anti–varicella-zoster IgM.
Management includes assessing immunity
If a patient is seen for a preconception appointment, ask her whether she has ever had varicella or been vaccinated for this disease. If she is uncertain, a varicella-zoster immunoglobulin G (IgG) titer should be ordered. If the IgG titer is negative, denoting susceptibility to infection, the patient should be vaccinated before she tries to conceive (see below).8
If a patient has not had a preconception appointment and now presents for her first prenatal appointment, she should be asked about immunity to varicella. If she is uncertain, a varicella-zoster IgG assay should be obtained. Approximately 75% of patients who are uncertain about immunity will, in fact, be immune. Those who are not immune should be counseled to avoid exposure to individuals who may have varicella, and they should be targeted for vaccination immediately postpartum.5,9
If a susceptible pregnant patient has been exposed to an individual with varicella, she should receive 1 of 2 regimens within 72 to 96 hours to minimize the risk of maternal infection.5,9,10 One option is intramuscular varicella-zoster immune globulin (VariZIG), 125 U/10 kg body weight, with a maximum dose of 625 U (5 vials). The distributor of this agent is FFF Enterprises in Temucula, California (telephone: 800-843-7477). A company representative will assess the patient’s eligibility and deliver the drug within 24 hours if the patient is considered eligible. An alternative prophylactic regimen is oral acyclovir, 800 mg 5 times daily for 7 days, or oral valacyclovir, 1,000 mg 3 times daily for 7 days.
If, despite prophylaxis, the pregnant woman becomes infected, she should immediately be treated with 1 of the oral antiviral regimens described above. If she has evidence of encephalitis, pneumonia, or severe disseminated mucocutaneous infection, or if she is immunosuppressed, she should be hospitalized and treated with intravenous acyclovir, 10 mg/kg infused over 1 hour every 8 hours for 10 days.
Ultrasonography is the most valuable test to identify fetal infection. Key findings that suggest congenital varicella are fetal growth restriction, microcephaly, ventriculomegaly, echogenic foci in the liver, and limb abnormalities. There is no proven therapy for congenital varicella.
When a patient has varicella at the time of delivery, she should be isolated from her infant until all lesions have crusted over. In addition, the neonate should be treated with either VariZIG or an antiviral agent.5,9
Prevention with varicella vaccine
The varicella vaccine (Varivax) is a live-virus vaccine that is highly immunogenic. The vaccine is now part of the routine childhood immunization sequence. Children ages 1 to 12 years require only a single dose of the vaccine. Individuals older than 12 years of age require 2 doses, administered 4 to 6 weeks apart. The vaccine should not be administered during pregnancy. It also should not be administered to individuals who are severely immunocompromised, are receiving high-dose systemic steroids, have untreated tuberculosis, or have an allergy to neomycin, which is a component of the vaccine. The vaccine does not pose a risk to the breastfeeding infant.11
CASE #2 Hospitalization is recommended for this patient
The patient in this case developed acute varicella pneumonia as a result of her exposure to the neighbor’s child. The diagnosis can be confirmed by demonstrating a positive varicella-zoster IgM and by obtaining a chest x-ray that identifies the diffuse patchy infiltrates characteristic of viral pneumonia. Because this is such a potentially serious illness, the patient should be hospitalized and treated with intravenous acyclovir or valacyclovir. Antibiotics such as ceftriaxone and azithromycin may be indicated to treat superimposed bacterial pneumonia. Given the later gestational age, the fetus is at low risk for serious injury. ●
- Valeur-Jensen AK, Pedersen CB, Westergaard T, et al. Risk factors for parvovirus B19 infection in pregnancy. JAMA. 1999;281:1099-1105.
- Harger JH, Adler SP, Koch WC, et al. Prospective evaluation of 618 pregnant women exposed to parvovirus B19: risks and symptoms. Obstet Gynecol. 1998;91:413-420.
- Melamed N, Whittle W, Kelly EN, et al. Fetal thrombocytopenia in pregnancies with fetal human parvovirus-B19 infection. Am J Obstet Gynecol. 2015;212:793.e1-8.
- Nagel HTC, de Haan TR, Vandenbussche FPH, et al. Long-term outcome after fetal transfusion for hydrops associated with parvovirus B19 infection. Obstet Gynecol. 2007;109:42-47.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al (eds). Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:911-912.
- Cohen JI. Herpes zoster. N Engl J Med. 2013;369:255-263.
- Enders G, Miller E, Cradock-Watson J, et al. Consequences of varicella and herpes zoster in pregnancy: prospective study of 1739 cases. Lancet. 1994;343:1548-1551.
- Duff P. Varicella in pregnancy: five priorities for clinicians. Infect Dis Obstet Gynecol. 1994;1:163-165.
- Marin M, Guris D, Chaves SS, et al; Advisory Committee on Immunization Practices, Centers for Disease Control and Prevention. Prevention of varicella. MMWR Recommend Rep. 2007;56(RR-4):1-40.
- Swamy GK, Dotters-Katz SK. Safety and varicella outcomes after varicella zoster immune globulin administration in pregnancy. Am J Obstet Gynecol. 2019;221:655-656.
- Duff P. Varicella vaccine. Infect Dis Obstet Gynecol. 1996;4:63-65.

Dr. Berwick is Chief Resident, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.

Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Dr. Berwick is Chief Resident, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Dr. Berwick is Chief Resident, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
We review 2 important viral infections in this article. One, parvovirus, poses a major threat to the fetus. The second, varicella, poses less risk to the fetus but significantly greater risk to the mother. We focus on the epidemiology, clinical presentation, diagnosis, and management of each infection.
Parvovirus infection and its risks to the fetus
CASE #1 Pregnant teacher exposed to fifth disease
A 28-year-old primigravid woman at 16 weeks’ gestation works as an elementary school teacher. Over the past 3 weeks, she has been exposed to 4 children who had fifth disease. She now requests evaluation because she has malaise, arthralgias, myalgias, fever of 38.2°C, and a fine lacelike erythematous rash on her trunk, arms, and cheeks.
- What is the most likely diagnosis?
- What diagnostic tests are indicated?
- Is her fetus at risk?
Epidemiology of parvovirus
Parvovirus B19 is a small, single-stranded DNA virus. It is highly contagious and is transmitted primarily by respiratory droplets. Transmission also can occur via infected blood, for example, through a blood transfusion. The incubation period is 10 to 20 days. Among adults, the individuals at greatest risk for infection are those who have close contact with young children, such as parents, day-care workers, and elementary school teachers. With sustained exposure in the household or classroom, the risk of seroconversion approaches 50%.1 Approximately 50% to 60% of reproductive-aged women have evidence of prior infection, and immunity is usually lifelong.
Clinical manifestations
The classic presentation of parvovirus infection is erythema infectiosum, also called fifth disease. This condition is characterized by a “slapped cheek” facial rash, malaise, myalgias, arthralgias, and low-grade fever. A fine lacelike rash often develops over the torso. In adults, the characteristic rash may be absent, and the most common presentation is a flu-like illness with joint pains.1,2 In children and in adults with an underlying hemoglobinopathy, parvovirus can cause transient aplastic crisis, and patients present with signs of a severe anemia, such as dyspnea, pallor, and fatigue.
Although parvovirus infection usually poses no serious risk in otherwise healthy children and adults, it can cause major fetal injury when the pregnant woman is infected early in pregnancy. The principal manifestation of fetal infection is hydrops. Hydrops primarily results when the virus crosses the placenta and attaches to the P antigen on the surface of red cell progenitors in the fetal marrow, causing an aplastic anemia with resultant high-output congestive heart failure. The virus also may directly injure the fetal myocardium, thus exacerbating heart failure. Other manifestations of congenital parvovirus include thrombocytopenia and hepatitis.3
The severity of fetal injury is inversely proportional to the gestational age at the time of maternal infection. When primary maternal infection occurs in the first trimester, the frequency of fetal hydrops is 5% to 10%. If infection develops in weeks 13 to 20, the risk of hydrops decreases to 5% or less. If infection develops beyond week 20, the incidence of fetal hydrops is 1% or lower.2
Continue to: Diagnostic steps...
Diagnostic steps
Appropriate diagnostic evaluation for a pregnant woman with exposure to parvovirus or clinical manifestations suggestive of parvovirus infection is outlined in FIGURE 1.
If infection is confirmed, serial ultrasound monitoring should be performed on a weekly to biweekly basis for 8 to 12 weeks, as delineated in FIGURE 2. Extended surveillance is necessary because the incubation period in the fetus is longer than that in the mother.
As the fetus develops anemia, peripheral tissues become hypoxic, leading to reflex peripheral vasoconstriction and increased cardiac output. At the same time, reduction in the number of fetal red blood cells decreases blood viscosity. The combination of these changes results in an increase in blood flow to the fetal brain, which can be detected by measuring the peak systolic velocity of flow in the middle cerebral artery (MCA PSV) with Doppler ultrasound imaging (FIGURE 3). The increase in MCA PSV parallels the decrease in fetal hematocrit and precedes the development of hydrops. In fact, signs of fetal hydrops do not usually develop until the fetal hematocrit falls to 15 to 20 vol%.
Management may necessitate intrauterine transfusion
Although some cases of fetal hydrops may resolve spontaneously, most authors agree that intrauterine transfusion is essential. In most instances, only a single intrauterine transfusion is necessary. In some fetuses, however, the infection is so prolonged and the anemia so severe that 2 to 3 transfusions may be required.
Infants who survive the intrauterine transfusion usually have an excellent long-term prognosis. However, isolated case reports have documented neurologic morbidity and prolonged transfusion-dependent anemia.4 In light of these reports, we recommend that a third trimester ultrasound exam be performed to assess fetal growth and evaluate the anatomy of the fetal brain. For the fetus with abnormal intracranial findings on ultrasonography, fetal magnetic resonance imaging is indicated.5
CASE #1 Diagnosis is probable parvovirus
The most likely diagnosis in this case is erythema infectiosum. This diagnosis can be confirmed by identifying positive immunogloblulin M (IgM) antibody and by detecting parvovirus in the maternal serum by polymerase chain reaction. Given the gestational age of 16 weeks, the risk of serious fetal injury should be less than 5%. Nevertheless, serial ultrasound examinations should be performed to assess for signs of fetal anemia.
Varicella exposure in pregnancy
CASE #2 Pregnant woman exposed to chickenpox has symptoms
Two weeks ago, a 32-year-old woman (G3P2002) at 24 weeks’ gestation was exposed to a neighbor’s child who had chickenpox. The patient has no history of natural infection or vaccination. She now has a fever of 38.6°C, malaise, headache, and a diffuse pruritic vesicular rash on her trunk and extremities. She also is experiencing a dry cough and mild dyspnea.
- What diagnostic tests are indicated?
- What treatment is indicated?
- What risk does this condition pose to the fetus?
Epidemiology of varicella
Varicella (chickenpox) is caused by the DNA varicella-zoster virus, an organism that is a member of the herpesvirus family. The disease occurs predominantly in children, and the infection is transmitted by respiratory droplets and by direct contact. Its incubation period is short (10–14 days), and it is highly contagious. More than 90% of susceptible close contacts will become infected after exposure to the index case. Like other herpesviruses, the varicella virus can establish a latent infection and then become manifest years later as herpes zoster (shingles).5,6
Continue to: Clinical manifestations...
Clinical manifestations
Patients with varicella usually have prodromal symptoms and signs that include malaise, fatigue, arthralgias, myalgias, and a low-grade fever. Varicella’s pathognomonic manifestation is a pruritic, macular rash that starts on the face and trunk and then spreads centripetally to the extremities. The lesions typically appear in “crops” and evolve through several distinct phases: macule, papule, vesicle, pustule, ulcer, and crust.5
In children, varicella is manifest almost entirely by mucocutaneous lesions. In adults, however, 2 serious and potentially life-threatening complications can occur. Approximately 1% of infected adults develop encephalitis and about 20% develop viral pneumonia, often accompanied by a severe superimposed bacterial pneumonia.5
When maternal infection develops in the first half of pregnancy, approximately 2% of fetuses will have evidence of congenital infection, usually manifested by circular, constricting scars on the extremities. These lesions typically occur in a dermatomal distribution. Spontaneous abortion and fetal death in utero also have been reported, but fortunately they are quite rare. When maternal infection occurs beyond 20 weeks of gestation, fetal injury is very uncommon.7
Interestingly, when maternal infection occurs at the time of delivery or shortly thereafter (from 5 days before until 2 days after delivery), neonatal varicella may develop. This infection may take 3 forms: disseminated mucocutaneous lesions, a deep-seated visceral infection, or severe pneumonia. In the era before the ready availability of antiviral agents, the case fatality rate from neonatal varicella was approximately 30%.5
Diagnosis is clinical
The diagnosis of varicella usually is established on the basis of clinical examination. It can be confirmed by identification of anti–varicella-zoster IgM.
Management includes assessing immunity
If a patient is seen for a preconception appointment, ask her whether she has ever had varicella or been vaccinated for this disease. If she is uncertain, a varicella-zoster immunoglobulin G (IgG) titer should be ordered. If the IgG titer is negative, denoting susceptibility to infection, the patient should be vaccinated before she tries to conceive (see below).8
If a patient has not had a preconception appointment and now presents for her first prenatal appointment, she should be asked about immunity to varicella. If she is uncertain, a varicella-zoster IgG assay should be obtained. Approximately 75% of patients who are uncertain about immunity will, in fact, be immune. Those who are not immune should be counseled to avoid exposure to individuals who may have varicella, and they should be targeted for vaccination immediately postpartum.5,9
If a susceptible pregnant patient has been exposed to an individual with varicella, she should receive 1 of 2 regimens within 72 to 96 hours to minimize the risk of maternal infection.5,9,10 One option is intramuscular varicella-zoster immune globulin (VariZIG), 125 U/10 kg body weight, with a maximum dose of 625 U (5 vials). The distributor of this agent is FFF Enterprises in Temucula, California (telephone: 800-843-7477). A company representative will assess the patient’s eligibility and deliver the drug within 24 hours if the patient is considered eligible. An alternative prophylactic regimen is oral acyclovir, 800 mg 5 times daily for 7 days, or oral valacyclovir, 1,000 mg 3 times daily for 7 days.
If, despite prophylaxis, the pregnant woman becomes infected, she should immediately be treated with 1 of the oral antiviral regimens described above. If she has evidence of encephalitis, pneumonia, or severe disseminated mucocutaneous infection, or if she is immunosuppressed, she should be hospitalized and treated with intravenous acyclovir, 10 mg/kg infused over 1 hour every 8 hours for 10 days.
Ultrasonography is the most valuable test to identify fetal infection. Key findings that suggest congenital varicella are fetal growth restriction, microcephaly, ventriculomegaly, echogenic foci in the liver, and limb abnormalities. There is no proven therapy for congenital varicella.
When a patient has varicella at the time of delivery, she should be isolated from her infant until all lesions have crusted over. In addition, the neonate should be treated with either VariZIG or an antiviral agent.5,9
Prevention with varicella vaccine
The varicella vaccine (Varivax) is a live-virus vaccine that is highly immunogenic. The vaccine is now part of the routine childhood immunization sequence. Children ages 1 to 12 years require only a single dose of the vaccine. Individuals older than 12 years of age require 2 doses, administered 4 to 6 weeks apart. The vaccine should not be administered during pregnancy. It also should not be administered to individuals who are severely immunocompromised, are receiving high-dose systemic steroids, have untreated tuberculosis, or have an allergy to neomycin, which is a component of the vaccine. The vaccine does not pose a risk to the breastfeeding infant.11
CASE #2 Hospitalization is recommended for this patient
The patient in this case developed acute varicella pneumonia as a result of her exposure to the neighbor’s child. The diagnosis can be confirmed by demonstrating a positive varicella-zoster IgM and by obtaining a chest x-ray that identifies the diffuse patchy infiltrates characteristic of viral pneumonia. Because this is such a potentially serious illness, the patient should be hospitalized and treated with intravenous acyclovir or valacyclovir. Antibiotics such as ceftriaxone and azithromycin may be indicated to treat superimposed bacterial pneumonia. Given the later gestational age, the fetus is at low risk for serious injury. ●
We review 2 important viral infections in this article. One, parvovirus, poses a major threat to the fetus. The second, varicella, poses less risk to the fetus but significantly greater risk to the mother. We focus on the epidemiology, clinical presentation, diagnosis, and management of each infection.
Parvovirus infection and its risks to the fetus
CASE #1 Pregnant teacher exposed to fifth disease
A 28-year-old primigravid woman at 16 weeks’ gestation works as an elementary school teacher. Over the past 3 weeks, she has been exposed to 4 children who had fifth disease. She now requests evaluation because she has malaise, arthralgias, myalgias, fever of 38.2°C, and a fine lacelike erythematous rash on her trunk, arms, and cheeks.
- What is the most likely diagnosis?
- What diagnostic tests are indicated?
- Is her fetus at risk?
Epidemiology of parvovirus
Parvovirus B19 is a small, single-stranded DNA virus. It is highly contagious and is transmitted primarily by respiratory droplets. Transmission also can occur via infected blood, for example, through a blood transfusion. The incubation period is 10 to 20 days. Among adults, the individuals at greatest risk for infection are those who have close contact with young children, such as parents, day-care workers, and elementary school teachers. With sustained exposure in the household or classroom, the risk of seroconversion approaches 50%.1 Approximately 50% to 60% of reproductive-aged women have evidence of prior infection, and immunity is usually lifelong.
Clinical manifestations
The classic presentation of parvovirus infection is erythema infectiosum, also called fifth disease. This condition is characterized by a “slapped cheek” facial rash, malaise, myalgias, arthralgias, and low-grade fever. A fine lacelike rash often develops over the torso. In adults, the characteristic rash may be absent, and the most common presentation is a flu-like illness with joint pains.1,2 In children and in adults with an underlying hemoglobinopathy, parvovirus can cause transient aplastic crisis, and patients present with signs of a severe anemia, such as dyspnea, pallor, and fatigue.
Although parvovirus infection usually poses no serious risk in otherwise healthy children and adults, it can cause major fetal injury when the pregnant woman is infected early in pregnancy. The principal manifestation of fetal infection is hydrops. Hydrops primarily results when the virus crosses the placenta and attaches to the P antigen on the surface of red cell progenitors in the fetal marrow, causing an aplastic anemia with resultant high-output congestive heart failure. The virus also may directly injure the fetal myocardium, thus exacerbating heart failure. Other manifestations of congenital parvovirus include thrombocytopenia and hepatitis.3
The severity of fetal injury is inversely proportional to the gestational age at the time of maternal infection. When primary maternal infection occurs in the first trimester, the frequency of fetal hydrops is 5% to 10%. If infection develops in weeks 13 to 20, the risk of hydrops decreases to 5% or less. If infection develops beyond week 20, the incidence of fetal hydrops is 1% or lower.2
Continue to: Diagnostic steps...
Diagnostic steps
Appropriate diagnostic evaluation for a pregnant woman with exposure to parvovirus or clinical manifestations suggestive of parvovirus infection is outlined in FIGURE 1.
If infection is confirmed, serial ultrasound monitoring should be performed on a weekly to biweekly basis for 8 to 12 weeks, as delineated in FIGURE 2. Extended surveillance is necessary because the incubation period in the fetus is longer than that in the mother.
As the fetus develops anemia, peripheral tissues become hypoxic, leading to reflex peripheral vasoconstriction and increased cardiac output. At the same time, reduction in the number of fetal red blood cells decreases blood viscosity. The combination of these changes results in an increase in blood flow to the fetal brain, which can be detected by measuring the peak systolic velocity of flow in the middle cerebral artery (MCA PSV) with Doppler ultrasound imaging (FIGURE 3). The increase in MCA PSV parallels the decrease in fetal hematocrit and precedes the development of hydrops. In fact, signs of fetal hydrops do not usually develop until the fetal hematocrit falls to 15 to 20 vol%.
Management may necessitate intrauterine transfusion
Although some cases of fetal hydrops may resolve spontaneously, most authors agree that intrauterine transfusion is essential. In most instances, only a single intrauterine transfusion is necessary. In some fetuses, however, the infection is so prolonged and the anemia so severe that 2 to 3 transfusions may be required.
Infants who survive the intrauterine transfusion usually have an excellent long-term prognosis. However, isolated case reports have documented neurologic morbidity and prolonged transfusion-dependent anemia.4 In light of these reports, we recommend that a third trimester ultrasound exam be performed to assess fetal growth and evaluate the anatomy of the fetal brain. For the fetus with abnormal intracranial findings on ultrasonography, fetal magnetic resonance imaging is indicated.5
CASE #1 Diagnosis is probable parvovirus
The most likely diagnosis in this case is erythema infectiosum. This diagnosis can be confirmed by identifying positive immunogloblulin M (IgM) antibody and by detecting parvovirus in the maternal serum by polymerase chain reaction. Given the gestational age of 16 weeks, the risk of serious fetal injury should be less than 5%. Nevertheless, serial ultrasound examinations should be performed to assess for signs of fetal anemia.
Varicella exposure in pregnancy
CASE #2 Pregnant woman exposed to chickenpox has symptoms
Two weeks ago, a 32-year-old woman (G3P2002) at 24 weeks’ gestation was exposed to a neighbor’s child who had chickenpox. The patient has no history of natural infection or vaccination. She now has a fever of 38.6°C, malaise, headache, and a diffuse pruritic vesicular rash on her trunk and extremities. She also is experiencing a dry cough and mild dyspnea.
- What diagnostic tests are indicated?
- What treatment is indicated?
- What risk does this condition pose to the fetus?
Epidemiology of varicella
Varicella (chickenpox) is caused by the DNA varicella-zoster virus, an organism that is a member of the herpesvirus family. The disease occurs predominantly in children, and the infection is transmitted by respiratory droplets and by direct contact. Its incubation period is short (10–14 days), and it is highly contagious. More than 90% of susceptible close contacts will become infected after exposure to the index case. Like other herpesviruses, the varicella virus can establish a latent infection and then become manifest years later as herpes zoster (shingles).5,6
Continue to: Clinical manifestations...
Clinical manifestations
Patients with varicella usually have prodromal symptoms and signs that include malaise, fatigue, arthralgias, myalgias, and a low-grade fever. Varicella’s pathognomonic manifestation is a pruritic, macular rash that starts on the face and trunk and then spreads centripetally to the extremities. The lesions typically appear in “crops” and evolve through several distinct phases: macule, papule, vesicle, pustule, ulcer, and crust.5
In children, varicella is manifest almost entirely by mucocutaneous lesions. In adults, however, 2 serious and potentially life-threatening complications can occur. Approximately 1% of infected adults develop encephalitis and about 20% develop viral pneumonia, often accompanied by a severe superimposed bacterial pneumonia.5
When maternal infection develops in the first half of pregnancy, approximately 2% of fetuses will have evidence of congenital infection, usually manifested by circular, constricting scars on the extremities. These lesions typically occur in a dermatomal distribution. Spontaneous abortion and fetal death in utero also have been reported, but fortunately they are quite rare. When maternal infection occurs beyond 20 weeks of gestation, fetal injury is very uncommon.7
Interestingly, when maternal infection occurs at the time of delivery or shortly thereafter (from 5 days before until 2 days after delivery), neonatal varicella may develop. This infection may take 3 forms: disseminated mucocutaneous lesions, a deep-seated visceral infection, or severe pneumonia. In the era before the ready availability of antiviral agents, the case fatality rate from neonatal varicella was approximately 30%.5
Diagnosis is clinical
The diagnosis of varicella usually is established on the basis of clinical examination. It can be confirmed by identification of anti–varicella-zoster IgM.
Management includes assessing immunity
If a patient is seen for a preconception appointment, ask her whether she has ever had varicella or been vaccinated for this disease. If she is uncertain, a varicella-zoster immunoglobulin G (IgG) titer should be ordered. If the IgG titer is negative, denoting susceptibility to infection, the patient should be vaccinated before she tries to conceive (see below).8
If a patient has not had a preconception appointment and now presents for her first prenatal appointment, she should be asked about immunity to varicella. If she is uncertain, a varicella-zoster IgG assay should be obtained. Approximately 75% of patients who are uncertain about immunity will, in fact, be immune. Those who are not immune should be counseled to avoid exposure to individuals who may have varicella, and they should be targeted for vaccination immediately postpartum.5,9
If a susceptible pregnant patient has been exposed to an individual with varicella, she should receive 1 of 2 regimens within 72 to 96 hours to minimize the risk of maternal infection.5,9,10 One option is intramuscular varicella-zoster immune globulin (VariZIG), 125 U/10 kg body weight, with a maximum dose of 625 U (5 vials). The distributor of this agent is FFF Enterprises in Temucula, California (telephone: 800-843-7477). A company representative will assess the patient’s eligibility and deliver the drug within 24 hours if the patient is considered eligible. An alternative prophylactic regimen is oral acyclovir, 800 mg 5 times daily for 7 days, or oral valacyclovir, 1,000 mg 3 times daily for 7 days.
If, despite prophylaxis, the pregnant woman becomes infected, she should immediately be treated with 1 of the oral antiviral regimens described above. If she has evidence of encephalitis, pneumonia, or severe disseminated mucocutaneous infection, or if she is immunosuppressed, she should be hospitalized and treated with intravenous acyclovir, 10 mg/kg infused over 1 hour every 8 hours for 10 days.
Ultrasonography is the most valuable test to identify fetal infection. Key findings that suggest congenital varicella are fetal growth restriction, microcephaly, ventriculomegaly, echogenic foci in the liver, and limb abnormalities. There is no proven therapy for congenital varicella.
When a patient has varicella at the time of delivery, she should be isolated from her infant until all lesions have crusted over. In addition, the neonate should be treated with either VariZIG or an antiviral agent.5,9
Prevention with varicella vaccine
The varicella vaccine (Varivax) is a live-virus vaccine that is highly immunogenic. The vaccine is now part of the routine childhood immunization sequence. Children ages 1 to 12 years require only a single dose of the vaccine. Individuals older than 12 years of age require 2 doses, administered 4 to 6 weeks apart. The vaccine should not be administered during pregnancy. It also should not be administered to individuals who are severely immunocompromised, are receiving high-dose systemic steroids, have untreated tuberculosis, or have an allergy to neomycin, which is a component of the vaccine. The vaccine does not pose a risk to the breastfeeding infant.11
CASE #2 Hospitalization is recommended for this patient
The patient in this case developed acute varicella pneumonia as a result of her exposure to the neighbor’s child. The diagnosis can be confirmed by demonstrating a positive varicella-zoster IgM and by obtaining a chest x-ray that identifies the diffuse patchy infiltrates characteristic of viral pneumonia. Because this is such a potentially serious illness, the patient should be hospitalized and treated with intravenous acyclovir or valacyclovir. Antibiotics such as ceftriaxone and azithromycin may be indicated to treat superimposed bacterial pneumonia. Given the later gestational age, the fetus is at low risk for serious injury. ●
- Valeur-Jensen AK, Pedersen CB, Westergaard T, et al. Risk factors for parvovirus B19 infection in pregnancy. JAMA. 1999;281:1099-1105.
- Harger JH, Adler SP, Koch WC, et al. Prospective evaluation of 618 pregnant women exposed to parvovirus B19: risks and symptoms. Obstet Gynecol. 1998;91:413-420.
- Melamed N, Whittle W, Kelly EN, et al. Fetal thrombocytopenia in pregnancies with fetal human parvovirus-B19 infection. Am J Obstet Gynecol. 2015;212:793.e1-8.
- Nagel HTC, de Haan TR, Vandenbussche FPH, et al. Long-term outcome after fetal transfusion for hydrops associated with parvovirus B19 infection. Obstet Gynecol. 2007;109:42-47.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al (eds). Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:911-912.
- Cohen JI. Herpes zoster. N Engl J Med. 2013;369:255-263.
- Enders G, Miller E, Cradock-Watson J, et al. Consequences of varicella and herpes zoster in pregnancy: prospective study of 1739 cases. Lancet. 1994;343:1548-1551.
- Duff P. Varicella in pregnancy: five priorities for clinicians. Infect Dis Obstet Gynecol. 1994;1:163-165.
- Marin M, Guris D, Chaves SS, et al; Advisory Committee on Immunization Practices, Centers for Disease Control and Prevention. Prevention of varicella. MMWR Recommend Rep. 2007;56(RR-4):1-40.
- Swamy GK, Dotters-Katz SK. Safety and varicella outcomes after varicella zoster immune globulin administration in pregnancy. Am J Obstet Gynecol. 2019;221:655-656.
- Duff P. Varicella vaccine. Infect Dis Obstet Gynecol. 1996;4:63-65.
- Valeur-Jensen AK, Pedersen CB, Westergaard T, et al. Risk factors for parvovirus B19 infection in pregnancy. JAMA. 1999;281:1099-1105.
- Harger JH, Adler SP, Koch WC, et al. Prospective evaluation of 618 pregnant women exposed to parvovirus B19: risks and symptoms. Obstet Gynecol. 1998;91:413-420.
- Melamed N, Whittle W, Kelly EN, et al. Fetal thrombocytopenia in pregnancies with fetal human parvovirus-B19 infection. Am J Obstet Gynecol. 2015;212:793.e1-8.
- Nagel HTC, de Haan TR, Vandenbussche FPH, et al. Long-term outcome after fetal transfusion for hydrops associated with parvovirus B19 infection. Obstet Gynecol. 2007;109:42-47.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al (eds). Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:911-912.
- Cohen JI. Herpes zoster. N Engl J Med. 2013;369:255-263.
- Enders G, Miller E, Cradock-Watson J, et al. Consequences of varicella and herpes zoster in pregnancy: prospective study of 1739 cases. Lancet. 1994;343:1548-1551.
- Duff P. Varicella in pregnancy: five priorities for clinicians. Infect Dis Obstet Gynecol. 1994;1:163-165.
- Marin M, Guris D, Chaves SS, et al; Advisory Committee on Immunization Practices, Centers for Disease Control and Prevention. Prevention of varicella. MMWR Recommend Rep. 2007;56(RR-4):1-40.
- Swamy GK, Dotters-Katz SK. Safety and varicella outcomes after varicella zoster immune globulin administration in pregnancy. Am J Obstet Gynecol. 2019;221:655-656.
- Duff P. Varicella vaccine. Infect Dis Obstet Gynecol. 1996;4:63-65.
Monkeypox: Another emerging threat?
CASE Pregnant woman’s husband is ill after traveling
A 29-year-old primigravid woman at 18 weeks’ gestation just returned from a 10-day trip to Nigeria with her husband. While in Nigeria, the couple went on safari. On several occasions during the safari, they consumed bushmeat prepared by their guides. Her husband now has severe malaise, fever, chills, myalgias, cough, and prominent submandibular, cervical, and inguinal adenopathy. In addition, he has developed a diffuse papular-vesicular rash on his trunk and extremities.
- What is the most likely diagnosis?
- Does this condition pose a danger to his wife?
- What treatment is indicated for his wife?
What we know
In recent weeks, the specter of another poorly understood biological threat has emerged in the medical literature and lay press: monkeypox. This article will first review the epidemiology, clinical manifestations, and diagnosis of this infection, followed by a discussion of how to prevent and treat the condition, with special emphasis on the risks that this infection poses in pregnant women.
Virology
The monkeypox virus is a member of the orthopoxvirus genus. The variola (smallpox) virus and vaccinia virus are included in this genus. It is one of the largest of all viruses, measuring 200-250 nm. It is enveloped and contains double-stranded DNA. Its natural reservoir is probably African rodents. Two distinct strains of monkeypox exist in different geographical regions of Africa: the Central African clade and the West African clade. The Central African clade is significantly more virulent than the latter, with a mortality rate approaching 10%, versus 1% in the West African clade. The incubation period of the virus ranges from 4-20 days and averages 12 days.1,2
Epidemiology
Monkeypox was first discovered in 1958 by Preben von Magnus in a colony of research monkeys in Copenhagen, Denmark. The first case of monkeypox in humans occurred in the Democratic Republic of Congo in 1970 in a 9-year-old boy. Subsequently, cases were reported in the Ivory Coast, Liberia, Nigeria, and Sierra Leone. The infection was limited to the rain forests of central and western Africa until 2003. At that time, the first cases in the United States were reported. The US cases occurred in the Midwest and were traced to exposure to pet prairie dogs. These animals all came from a single distributor, and they apparently were infected when they were housed in the same space with Gambian rats, which are well recognized reservoirs of monkeypox in their native habitat in Africa.1-3
A limited outbreak of monkeypox occurred in the United Kingdom in 2018. Seventy-one cases, with no fatalities, were reported. In 2021 another US case of monkeypox was reported in Dallas, Texas, in an individual who had recently traveled to the United States from Nigeria. A second US case was reported in November 2021 from a patient in Maryland who had returned from a visit to Nigeria. Those were the only 2 reported cases of monkeypox in the United States in 2021.1-3
Then in early May 2022, the United Kingdom reported 9 cases of monkeypox. The first infected patient had recently traveled to Nigeria and, subsequently, infected 2 members of his family.4 On May 18, the Massachusetts Department of Public Health confirmed a case of monkeypox in an adult man who had recently traveled to Canada. As of July 7, 6,027 cases have been reported from at least 39 countries.
The current outbreak is unusual in that, previously, almost all cases occurred in western and central Africa in remote tropical rain forests. Infection usually resulted from close exposure to rats, rabbits, squirrels, monkeys, porcupines, and gazelles. Exposure occurred when persons captured, slaughtered, prepared, and then ate these animals for food without properly cooking the flesh.
The leading theory is that the present outbreak originated among men who had sex with men at 2 raves held in Spain and Belgium. The virus appears to have been spread by skin-to-skin contact, by respiratory droplets, by contact with contaminated bedding, and probably by sperm.2,4,6
Continue to: Clinical manifestations...
Clinical manifestations
Monkeypox evolves through 2 stages: a pre-eruptive stage and an eruptive stage. Prodromal symptoms include malaise, severe headache, myalgias, fever, drenching sweats, backache, fatigue, sore throat, dyspnea, and cough. Within 2-3 days, the characteristic skin eruption develops. The lesions usually begin on the face and then spread in a centrifugal manner to the trunk and extremities, including the palms of the hands and soles of the feet. The lesions typically progress from macules to papules to vesicles to pustules. They then crust and scab over. An interesting additional finding is the presence of prominent lymphadenopathy behind the ear, beneath the mandible, in the neck, and in the groin.1
Several different illnesses must be considered in the differential diagnosis of monkeypox infection. They include measles, scabies, secondary syphilis, and medication-associated allergic reactions. However, the 2 conditions most likely to be confused with monkeypox are chickenpox (varicella) and smallpox. Lymphadenopathy is much more prominent in monkeypox compared with chickenpox. Moreover, with monkeypox, all lesions tend to be at the same stage of evolution as opposed to appearing in crops as they do in chickenpox. Smallpox would be extremely unlikely in the absence of a recognized laboratory accident or a bioterrorism incident.7
Diagnosis
The presumptive diagnosis of monkeypox infection is made primarily based on clinical examination. However, laboratory testing is indicated to definitively differentiate monkeypox from other orthopoxvirus infections such as varicella and smallpox.
In specialized laboratories that employ highly trained personnel and maintain strict safety precautions, the virus can be isolated in mammalian cell cultures. Electron microscopy is a valuable tool for identifying the characteristic brick-shaped poxvirus virions. Routine histologic examination of a lesion will show ballooning degeneration of keratinocytes, prominent spongiosis, dermal edema, and acute inflammation, although these findings are not unique to monkeypox.1
The Centers for Disease Control and Prevention (CDC) has developed serologic tests that detect immunoglobulin (Ig) M- and IgG-specific antibody. However, the most useful and practical diagnostic test is assessment of a skin scraping by polymerase chain reaction (PCR). This test is more sensitive than assessment of serum PCR.1
When the diagnosis of monkeypox is being considered, the clinician should coordinate testing through the local and state public health departments and through the CDC. Effective communication with all agencies will ensure that laboratory specimens are processed in a timely and efficient manner. The CDC website presents information on specimen collection.8
How do we manage monkeypox?
Prevention
The first step in prevention of infection is to isolate infected individuals until all lesions have dried and crusted over. Susceptible people should avoid close contact with skin lesions, respiratory and genital secretions, and bedding of patients who are infected.
The ultimate preventive measure, however, is vaccination of susceptible people either immediately before exposure (eg, military personnel, first responders, infection control investigators, health care workers) or immediately after exposure (general population). Older individuals who received the original smallpox vaccine likely have immunity to monkeypox infection. Unfortunately, very few women who currently are of reproductive age received this vaccine because its use was discontinued in the United States in the early 1970s. Therefore, the vast majority of our patients are uniquely susceptible to this infection and should be vaccinated if there is an outbreak of monkeypox in their locality.7,9
The current preferred vaccine for prevention of both smallpox and monkeypox is the Jynneos (Bavarian Nordic A/S) vaccine.10 This agent incorporates a replication-deficient live virus and does not pose the same risk for adverse events as the original versions of the smallpox vaccine. Jynneos is administered subcutaneously rather than by scarification. Two 0.5-mL doses, delivered 28 days apart, are required for optimal effect. The vaccine must be obtained from local and state health departments, in consultation with the CDC.7,9
There is very little published information on the safety of the Jynneos vaccine in pregnant or lactating women, although animal data are reassuring. Moreover, the dangers of monkeypox infection are significant, and in the event of an outbreak, vaccination of susceptible individuals, including pregnant women, is indicated.
- Monkeypox is a member of the orthopoxvirus genus and is closely related to the smallpox virus. It is a large, double-stranded, enveloped DNA virus.
- The virus is transmitted primarily by close contact with infected animals or other humans or by consumption of contaminated bushmeat.
- The infection evolves in 2 phases. The pre-eruptive phase is characterized by severe flu-like symptoms and signs. The eruptive phase is distinguished by a diffuse papular-vesicular rash.
- The most valuable test for confirming the diagnosis is a polymerase chain reaction test of a fresh skin lesion.
- In women who are pregnant, monkeypox has been associated with spontaneous abortion and fetal death.
- Three antiviral agents may be of value in treating infected patients: cidofovir, brincidofovir, and tecovirimat. Only the latter has an acceptable safety profile for women who are pregnant or lactating.
- The new nonreplicating smallpox vaccine Jynneos (Bavarian Nordic A/S) is of great value for pre- and post-exposure prophylaxis.
Continue to: Treatment...
Treatment
Infected pregnant women should receive acetaminophen 1,000 mg orally every 8 hours, to control fever and provide analgesia. An antihistamine such as diphenhydramine 25 mg orally every 6-8 hours, may be used to control pruritus and provide mild sedation. Adequate fluid intake and optimal nutrition should be encouraged. Skin lesions should be inspected regularly to detect signs of superimposed bacterial infections. Small, localized bacterial skin infections can be treated with topical application of mupirocin ointment 2%, 3 times daily for 7-14 days. For diffuse and more severe bacterial skin infections, a systemic antibiotic may be necessary. Reasonable choices include amoxicillin-clavulanate 875 mg/125 mg orally every 12 hours, or trimethoprim-sulfamethoxazole double strength 800 mg/160 mg orally every 12 hours.11 The latter agent should be avoided in the first trimester of pregnancy because of potential teratogenic effects.
Several specific agents are available through the CDC for treatment of orthopoxvirus infections such as smallpox and monkeypox. Information about these agents is summarized in the TABLE.12-16
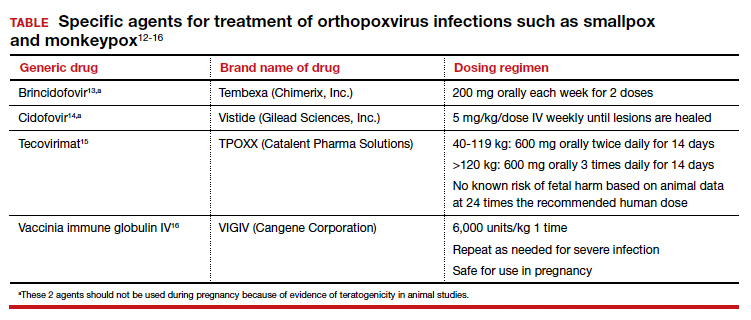
Unique considerations in pregnancy
Because monkeypox is so rare, there is very little information about the effects of this infection in pregnant women. The report most commonly cited in the literature is that by Mbala et al, which was published in 2017.17 These authors described 4 pregnant patients in the Democratic Republic of Congo who contracted monkeypox infection over a 4-year period. All 4 women were hospitalized and treated with systemic antibiotics, antiparasitic medications, and analgesics. One patient delivered a healthy infant. Two women had spontaneous abortions in the first trimester. The fourth patient experienced a stillbirth at 22 weeks’ gestation. At postmortem examination, the fetus had diffuse cutaneous lesions, prominent hepatomegaly, and hydrops. No structural malformations were noted. The placenta demonstrated numerous punctate hemorrhages, and high concentrations of virus were recovered from the placenta and from fetal tissue.
Although the information on pregnancy outcome is quite limited, it seems clear that the virus can cross the placenta and cause adverse effects such as spontaneous abortion and fetal death. Accordingly, I think the following guidelines are a reasonable approach to a pregnant patient who has been exposed to monkeypox or who has developed manifestations of infection.3,7,9
- In the event of a community outbreak, bioterrorism event, or exposure to a person with suspected or confirmed monkeypox infection, the pregnant patient should receive the Jynneos vaccine.
- The pregnant patient should be isolated from any individual with suspected or confirmed monkeypox.
- If infection develops despite these measures, the patient should be treated with either tecovirimat or vaccinia immune globulin IV. Hospitalization may be necessary for seriously ill individuals.
- Within 2 weeks of infection, a comprehensive ultrasound examination should be performed to assess for structural abnormalities in the fetus.
- Subsequently, serial ultrasound examinations should be performed at intervals of 4-6 weeks to assess fetal growth and re-evaluate fetal anatomy.
- Following delivery, a detailed neonatal examination should be performed to assess for evidence of viral injury. Neonatal skin lesions and neonatal serum can be assessed by PCR for monkeypox virus. The newborn should be isolated from the mother until all the mother’s lesions have dried and crusted over.
CASE Resolved
Given the husband’s recent travel to Nigeria and consumption of bushmeat, he most likely has monkeypox. The infection can be spread from person to person by close contact; thus, his wife is at risk. The couple should isolate until all of his lesions have dried and crusted over. The woman also should receive the Jynneos vaccine. If she becomes symptomatic, she should be treated with tecovirimat or vaccinia immune globulin IV. ●
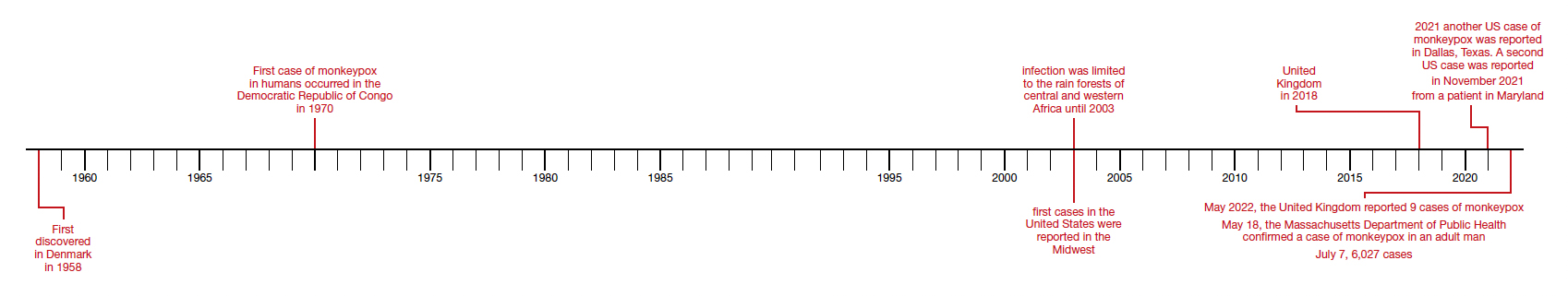
- Isaacs SN, Shenoy ES. Monkeypox. UpToDate. Updated June 28,2022. Accessed July 1, 2022. https://www.uptodate.com /contents/monkeypox?topicRef=8349&source=see_link
- Graham MB. Monkeypox. Medscape. Updated June 29, 2022. Accessed July 1, 2022. https://emedicine.medscape.com /article/1134714-overview.
- Khalil A, Samara A, O’Brien P, et al. Monkeypox and pregnancy: what do obstetricians need to know? Ultrasound Obstet Gynecol. 2022;60:22-27. doi:10.1002/uog.24968.
- World Health Organization. Monkeypox-United Kingdom of Great Britain and Northern Ireland. May 18, 2022. Accessed July 1, 2022. https://www.who.int/emergencies/diseaseoutbreak-news/item/2022-DON383.
- WHO reports two new monkeypox deaths, cases in new areas. Reuters. July 7, 2022. https://www.reuters.com/world /who-reports-two-new-monkeypox-deaths-2022-07-07/. Accessed July 19, 2022.
- World Health Organization. Multi-country monkeypox outbreak in non-endemic countries: update. May 29, 2022. Accessed July 1, 2022. https://www.who.int /emergencies/disease-outbreak-news/item/2022 -DON388#:~:text=Multi%2Dcountry%20monkeypox%20 outbreak%20in%20non%2Dendemic%20countries%3A%20 Update,-29%20May%202022&text=Since%2013%20 May%202022%2C%20monkeypox,Epidemiological%20 investigations%20are%20ongoing.
- Cono J, Cragan JD, Jamieson DJ, Rasmussen SA. Prophylaxis and treatment of pregnant women for emerging infections andbioterrorism emergencies. Emerg Infect Dis. 2006;12:16311637. doi:10.3201/eid1211.060618.
- Centers for Disease Control and Prevention. Preparation and collection of specimens. Reviewed June 29, 2022. Accessed July 6, 2022. https://www.cdc.gov/poxvirus /monkeypox/clinicians/prep-collection-specimens.html.
- Rao AK, Petersen BW, Whitehill F, et al. Monkeypox vaccination. MMWR Morb Mortal Wkly Rep. 2022;71:734-742. doi:10.15585/mmwr.mm7122e1.
- Smallpox and monkeypox vaccine, live, nonreplicating. Package insert. Bavarian Nordic A/S; 2021. Accessed July 1, 2022. https://www.fda.gov/media/131078/download.
- Duff P. Commonly used antibiotics in ObGyn practice. OBG Manag. 2022;34:29, 36-40. doi:10.12788/obgm.0191.
- Centers for Disease Control and Prevention. Treatment information for healthcare professionals: interim clinical guidance for the treatment of monkeypox. Reviewed June 17, 2022. Accessed July 1, 2022. https://www.cdc.gov/poxvirus /monkeypox/clinicians/treatment.html.
- Brincidofovir. Prescribing information. Chimerix, Inc.; 2021. Accessed July 1, 2022. https://www.accessdata.fda.gov /drugsatfda_docs/label/2021/214460s000,214461s000lbl.pdf.
- Cidofovir. Package insert. Gilead Sciences, Inc.; 2010. Accessed July 1, 2022. https://www.gilead.com/~/media /Files/pdfs/medicines/other/vistide/vistide.pdf.
- Tecovirimat. Prescribing information. Catalent Pharma Solutions; 2022. Accessed July 1, 2022. https://www.accessdata.fda.gov/drugsatfda_docs /label/2022/214518s000lbl.pdf.
- Vaccinia immune globulin IV. Prescribing information. Cangene Corporation; 2010. Accessed July 1, 2022. https: //www.fda.gov/media/77004/download.
- Mbala PK, Huggins JW, Riu-Rovira T, et al. Maternal and fetal outcomes among pregnant women with human monkeypox infection in the Democratic Republic of Congo. J Infect Dis. 2017;216:824-828. doi:10.1093/infdis/jix260.

Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville, Florida.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville, Florida.
The author reports no financial relationships relevant to this article.
Dr. Duff is Professor, Maternal-Fetal Medicine, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville, Florida.
The author reports no financial relationships relevant to this article.
CASE Pregnant woman’s husband is ill after traveling
A 29-year-old primigravid woman at 18 weeks’ gestation just returned from a 10-day trip to Nigeria with her husband. While in Nigeria, the couple went on safari. On several occasions during the safari, they consumed bushmeat prepared by their guides. Her husband now has severe malaise, fever, chills, myalgias, cough, and prominent submandibular, cervical, and inguinal adenopathy. In addition, he has developed a diffuse papular-vesicular rash on his trunk and extremities.
- What is the most likely diagnosis?
- Does this condition pose a danger to his wife?
- What treatment is indicated for his wife?
What we know
In recent weeks, the specter of another poorly understood biological threat has emerged in the medical literature and lay press: monkeypox. This article will first review the epidemiology, clinical manifestations, and diagnosis of this infection, followed by a discussion of how to prevent and treat the condition, with special emphasis on the risks that this infection poses in pregnant women.
Virology
The monkeypox virus is a member of the orthopoxvirus genus. The variola (smallpox) virus and vaccinia virus are included in this genus. It is one of the largest of all viruses, measuring 200-250 nm. It is enveloped and contains double-stranded DNA. Its natural reservoir is probably African rodents. Two distinct strains of monkeypox exist in different geographical regions of Africa: the Central African clade and the West African clade. The Central African clade is significantly more virulent than the latter, with a mortality rate approaching 10%, versus 1% in the West African clade. The incubation period of the virus ranges from 4-20 days and averages 12 days.1,2
Epidemiology
Monkeypox was first discovered in 1958 by Preben von Magnus in a colony of research monkeys in Copenhagen, Denmark. The first case of monkeypox in humans occurred in the Democratic Republic of Congo in 1970 in a 9-year-old boy. Subsequently, cases were reported in the Ivory Coast, Liberia, Nigeria, and Sierra Leone. The infection was limited to the rain forests of central and western Africa until 2003. At that time, the first cases in the United States were reported. The US cases occurred in the Midwest and were traced to exposure to pet prairie dogs. These animals all came from a single distributor, and they apparently were infected when they were housed in the same space with Gambian rats, which are well recognized reservoirs of monkeypox in their native habitat in Africa.1-3
A limited outbreak of monkeypox occurred in the United Kingdom in 2018. Seventy-one cases, with no fatalities, were reported. In 2021 another US case of monkeypox was reported in Dallas, Texas, in an individual who had recently traveled to the United States from Nigeria. A second US case was reported in November 2021 from a patient in Maryland who had returned from a visit to Nigeria. Those were the only 2 reported cases of monkeypox in the United States in 2021.1-3
Then in early May 2022, the United Kingdom reported 9 cases of monkeypox. The first infected patient had recently traveled to Nigeria and, subsequently, infected 2 members of his family.4 On May 18, the Massachusetts Department of Public Health confirmed a case of monkeypox in an adult man who had recently traveled to Canada. As of July 7, 6,027 cases have been reported from at least 39 countries.
The current outbreak is unusual in that, previously, almost all cases occurred in western and central Africa in remote tropical rain forests. Infection usually resulted from close exposure to rats, rabbits, squirrels, monkeys, porcupines, and gazelles. Exposure occurred when persons captured, slaughtered, prepared, and then ate these animals for food without properly cooking the flesh.
The leading theory is that the present outbreak originated among men who had sex with men at 2 raves held in Spain and Belgium. The virus appears to have been spread by skin-to-skin contact, by respiratory droplets, by contact with contaminated bedding, and probably by sperm.2,4,6
Continue to: Clinical manifestations...
Clinical manifestations
Monkeypox evolves through 2 stages: a pre-eruptive stage and an eruptive stage. Prodromal symptoms include malaise, severe headache, myalgias, fever, drenching sweats, backache, fatigue, sore throat, dyspnea, and cough. Within 2-3 days, the characteristic skin eruption develops. The lesions usually begin on the face and then spread in a centrifugal manner to the trunk and extremities, including the palms of the hands and soles of the feet. The lesions typically progress from macules to papules to vesicles to pustules. They then crust and scab over. An interesting additional finding is the presence of prominent lymphadenopathy behind the ear, beneath the mandible, in the neck, and in the groin.1
Several different illnesses must be considered in the differential diagnosis of monkeypox infection. They include measles, scabies, secondary syphilis, and medication-associated allergic reactions. However, the 2 conditions most likely to be confused with monkeypox are chickenpox (varicella) and smallpox. Lymphadenopathy is much more prominent in monkeypox compared with chickenpox. Moreover, with monkeypox, all lesions tend to be at the same stage of evolution as opposed to appearing in crops as they do in chickenpox. Smallpox would be extremely unlikely in the absence of a recognized laboratory accident or a bioterrorism incident.7
Diagnosis
The presumptive diagnosis of monkeypox infection is made primarily based on clinical examination. However, laboratory testing is indicated to definitively differentiate monkeypox from other orthopoxvirus infections such as varicella and smallpox.
In specialized laboratories that employ highly trained personnel and maintain strict safety precautions, the virus can be isolated in mammalian cell cultures. Electron microscopy is a valuable tool for identifying the characteristic brick-shaped poxvirus virions. Routine histologic examination of a lesion will show ballooning degeneration of keratinocytes, prominent spongiosis, dermal edema, and acute inflammation, although these findings are not unique to monkeypox.1
The Centers for Disease Control and Prevention (CDC) has developed serologic tests that detect immunoglobulin (Ig) M- and IgG-specific antibody. However, the most useful and practical diagnostic test is assessment of a skin scraping by polymerase chain reaction (PCR). This test is more sensitive than assessment of serum PCR.1
When the diagnosis of monkeypox is being considered, the clinician should coordinate testing through the local and state public health departments and through the CDC. Effective communication with all agencies will ensure that laboratory specimens are processed in a timely and efficient manner. The CDC website presents information on specimen collection.8
How do we manage monkeypox?
Prevention
The first step in prevention of infection is to isolate infected individuals until all lesions have dried and crusted over. Susceptible people should avoid close contact with skin lesions, respiratory and genital secretions, and bedding of patients who are infected.
The ultimate preventive measure, however, is vaccination of susceptible people either immediately before exposure (eg, military personnel, first responders, infection control investigators, health care workers) or immediately after exposure (general population). Older individuals who received the original smallpox vaccine likely have immunity to monkeypox infection. Unfortunately, very few women who currently are of reproductive age received this vaccine because its use was discontinued in the United States in the early 1970s. Therefore, the vast majority of our patients are uniquely susceptible to this infection and should be vaccinated if there is an outbreak of monkeypox in their locality.7,9
The current preferred vaccine for prevention of both smallpox and monkeypox is the Jynneos (Bavarian Nordic A/S) vaccine.10 This agent incorporates a replication-deficient live virus and does not pose the same risk for adverse events as the original versions of the smallpox vaccine. Jynneos is administered subcutaneously rather than by scarification. Two 0.5-mL doses, delivered 28 days apart, are required for optimal effect. The vaccine must be obtained from local and state health departments, in consultation with the CDC.7,9
There is very little published information on the safety of the Jynneos vaccine in pregnant or lactating women, although animal data are reassuring. Moreover, the dangers of monkeypox infection are significant, and in the event of an outbreak, vaccination of susceptible individuals, including pregnant women, is indicated.
- Monkeypox is a member of the orthopoxvirus genus and is closely related to the smallpox virus. It is a large, double-stranded, enveloped DNA virus.
- The virus is transmitted primarily by close contact with infected animals or other humans or by consumption of contaminated bushmeat.
- The infection evolves in 2 phases. The pre-eruptive phase is characterized by severe flu-like symptoms and signs. The eruptive phase is distinguished by a diffuse papular-vesicular rash.
- The most valuable test for confirming the diagnosis is a polymerase chain reaction test of a fresh skin lesion.
- In women who are pregnant, monkeypox has been associated with spontaneous abortion and fetal death.
- Three antiviral agents may be of value in treating infected patients: cidofovir, brincidofovir, and tecovirimat. Only the latter has an acceptable safety profile for women who are pregnant or lactating.
- The new nonreplicating smallpox vaccine Jynneos (Bavarian Nordic A/S) is of great value for pre- and post-exposure prophylaxis.
Continue to: Treatment...
Treatment
Infected pregnant women should receive acetaminophen 1,000 mg orally every 8 hours, to control fever and provide analgesia. An antihistamine such as diphenhydramine 25 mg orally every 6-8 hours, may be used to control pruritus and provide mild sedation. Adequate fluid intake and optimal nutrition should be encouraged. Skin lesions should be inspected regularly to detect signs of superimposed bacterial infections. Small, localized bacterial skin infections can be treated with topical application of mupirocin ointment 2%, 3 times daily for 7-14 days. For diffuse and more severe bacterial skin infections, a systemic antibiotic may be necessary. Reasonable choices include amoxicillin-clavulanate 875 mg/125 mg orally every 12 hours, or trimethoprim-sulfamethoxazole double strength 800 mg/160 mg orally every 12 hours.11 The latter agent should be avoided in the first trimester of pregnancy because of potential teratogenic effects.
Several specific agents are available through the CDC for treatment of orthopoxvirus infections such as smallpox and monkeypox. Information about these agents is summarized in the TABLE.12-16
Unique considerations in pregnancy
Because monkeypox is so rare, there is very little information about the effects of this infection in pregnant women. The report most commonly cited in the literature is that by Mbala et al, which was published in 2017.17 These authors described 4 pregnant patients in the Democratic Republic of Congo who contracted monkeypox infection over a 4-year period. All 4 women were hospitalized and treated with systemic antibiotics, antiparasitic medications, and analgesics. One patient delivered a healthy infant. Two women had spontaneous abortions in the first trimester. The fourth patient experienced a stillbirth at 22 weeks’ gestation. At postmortem examination, the fetus had diffuse cutaneous lesions, prominent hepatomegaly, and hydrops. No structural malformations were noted. The placenta demonstrated numerous punctate hemorrhages, and high concentrations of virus were recovered from the placenta and from fetal tissue.
Although the information on pregnancy outcome is quite limited, it seems clear that the virus can cross the placenta and cause adverse effects such as spontaneous abortion and fetal death. Accordingly, I think the following guidelines are a reasonable approach to a pregnant patient who has been exposed to monkeypox or who has developed manifestations of infection.3,7,9
- In the event of a community outbreak, bioterrorism event, or exposure to a person with suspected or confirmed monkeypox infection, the pregnant patient should receive the Jynneos vaccine.
- The pregnant patient should be isolated from any individual with suspected or confirmed monkeypox.
- If infection develops despite these measures, the patient should be treated with either tecovirimat or vaccinia immune globulin IV. Hospitalization may be necessary for seriously ill individuals.
- Within 2 weeks of infection, a comprehensive ultrasound examination should be performed to assess for structural abnormalities in the fetus.
- Subsequently, serial ultrasound examinations should be performed at intervals of 4-6 weeks to assess fetal growth and re-evaluate fetal anatomy.
- Following delivery, a detailed neonatal examination should be performed to assess for evidence of viral injury. Neonatal skin lesions and neonatal serum can be assessed by PCR for monkeypox virus. The newborn should be isolated from the mother until all the mother’s lesions have dried and crusted over.
CASE Resolved
Given the husband’s recent travel to Nigeria and consumption of bushmeat, he most likely has monkeypox. The infection can be spread from person to person by close contact; thus, his wife is at risk. The couple should isolate until all of his lesions have dried and crusted over. The woman also should receive the Jynneos vaccine. If she becomes symptomatic, she should be treated with tecovirimat or vaccinia immune globulin IV. ●
CASE Pregnant woman’s husband is ill after traveling
A 29-year-old primigravid woman at 18 weeks’ gestation just returned from a 10-day trip to Nigeria with her husband. While in Nigeria, the couple went on safari. On several occasions during the safari, they consumed bushmeat prepared by their guides. Her husband now has severe malaise, fever, chills, myalgias, cough, and prominent submandibular, cervical, and inguinal adenopathy. In addition, he has developed a diffuse papular-vesicular rash on his trunk and extremities.
- What is the most likely diagnosis?
- Does this condition pose a danger to his wife?
- What treatment is indicated for his wife?
What we know
In recent weeks, the specter of another poorly understood biological threat has emerged in the medical literature and lay press: monkeypox. This article will first review the epidemiology, clinical manifestations, and diagnosis of this infection, followed by a discussion of how to prevent and treat the condition, with special emphasis on the risks that this infection poses in pregnant women.
Virology
The monkeypox virus is a member of the orthopoxvirus genus. The variola (smallpox) virus and vaccinia virus are included in this genus. It is one of the largest of all viruses, measuring 200-250 nm. It is enveloped and contains double-stranded DNA. Its natural reservoir is probably African rodents. Two distinct strains of monkeypox exist in different geographical regions of Africa: the Central African clade and the West African clade. The Central African clade is significantly more virulent than the latter, with a mortality rate approaching 10%, versus 1% in the West African clade. The incubation period of the virus ranges from 4-20 days and averages 12 days.1,2
Epidemiology
Monkeypox was first discovered in 1958 by Preben von Magnus in a colony of research monkeys in Copenhagen, Denmark. The first case of monkeypox in humans occurred in the Democratic Republic of Congo in 1970 in a 9-year-old boy. Subsequently, cases were reported in the Ivory Coast, Liberia, Nigeria, and Sierra Leone. The infection was limited to the rain forests of central and western Africa until 2003. At that time, the first cases in the United States were reported. The US cases occurred in the Midwest and were traced to exposure to pet prairie dogs. These animals all came from a single distributor, and they apparently were infected when they were housed in the same space with Gambian rats, which are well recognized reservoirs of monkeypox in their native habitat in Africa.1-3
A limited outbreak of monkeypox occurred in the United Kingdom in 2018. Seventy-one cases, with no fatalities, were reported. In 2021 another US case of monkeypox was reported in Dallas, Texas, in an individual who had recently traveled to the United States from Nigeria. A second US case was reported in November 2021 from a patient in Maryland who had returned from a visit to Nigeria. Those were the only 2 reported cases of monkeypox in the United States in 2021.1-3
Then in early May 2022, the United Kingdom reported 9 cases of monkeypox. The first infected patient had recently traveled to Nigeria and, subsequently, infected 2 members of his family.4 On May 18, the Massachusetts Department of Public Health confirmed a case of monkeypox in an adult man who had recently traveled to Canada. As of July 7, 6,027 cases have been reported from at least 39 countries.
The current outbreak is unusual in that, previously, almost all cases occurred in western and central Africa in remote tropical rain forests. Infection usually resulted from close exposure to rats, rabbits, squirrels, monkeys, porcupines, and gazelles. Exposure occurred when persons captured, slaughtered, prepared, and then ate these animals for food without properly cooking the flesh.
The leading theory is that the present outbreak originated among men who had sex with men at 2 raves held in Spain and Belgium. The virus appears to have been spread by skin-to-skin contact, by respiratory droplets, by contact with contaminated bedding, and probably by sperm.2,4,6
Continue to: Clinical manifestations...
Clinical manifestations
Monkeypox evolves through 2 stages: a pre-eruptive stage and an eruptive stage. Prodromal symptoms include malaise, severe headache, myalgias, fever, drenching sweats, backache, fatigue, sore throat, dyspnea, and cough. Within 2-3 days, the characteristic skin eruption develops. The lesions usually begin on the face and then spread in a centrifugal manner to the trunk and extremities, including the palms of the hands and soles of the feet. The lesions typically progress from macules to papules to vesicles to pustules. They then crust and scab over. An interesting additional finding is the presence of prominent lymphadenopathy behind the ear, beneath the mandible, in the neck, and in the groin.1
Several different illnesses must be considered in the differential diagnosis of monkeypox infection. They include measles, scabies, secondary syphilis, and medication-associated allergic reactions. However, the 2 conditions most likely to be confused with monkeypox are chickenpox (varicella) and smallpox. Lymphadenopathy is much more prominent in monkeypox compared with chickenpox. Moreover, with monkeypox, all lesions tend to be at the same stage of evolution as opposed to appearing in crops as they do in chickenpox. Smallpox would be extremely unlikely in the absence of a recognized laboratory accident or a bioterrorism incident.7
Diagnosis
The presumptive diagnosis of monkeypox infection is made primarily based on clinical examination. However, laboratory testing is indicated to definitively differentiate monkeypox from other orthopoxvirus infections such as varicella and smallpox.
In specialized laboratories that employ highly trained personnel and maintain strict safety precautions, the virus can be isolated in mammalian cell cultures. Electron microscopy is a valuable tool for identifying the characteristic brick-shaped poxvirus virions. Routine histologic examination of a lesion will show ballooning degeneration of keratinocytes, prominent spongiosis, dermal edema, and acute inflammation, although these findings are not unique to monkeypox.1
The Centers for Disease Control and Prevention (CDC) has developed serologic tests that detect immunoglobulin (Ig) M- and IgG-specific antibody. However, the most useful and practical diagnostic test is assessment of a skin scraping by polymerase chain reaction (PCR). This test is more sensitive than assessment of serum PCR.1
When the diagnosis of monkeypox is being considered, the clinician should coordinate testing through the local and state public health departments and through the CDC. Effective communication with all agencies will ensure that laboratory specimens are processed in a timely and efficient manner. The CDC website presents information on specimen collection.8
How do we manage monkeypox?
Prevention
The first step in prevention of infection is to isolate infected individuals until all lesions have dried and crusted over. Susceptible people should avoid close contact with skin lesions, respiratory and genital secretions, and bedding of patients who are infected.
The ultimate preventive measure, however, is vaccination of susceptible people either immediately before exposure (eg, military personnel, first responders, infection control investigators, health care workers) or immediately after exposure (general population). Older individuals who received the original smallpox vaccine likely have immunity to monkeypox infection. Unfortunately, very few women who currently are of reproductive age received this vaccine because its use was discontinued in the United States in the early 1970s. Therefore, the vast majority of our patients are uniquely susceptible to this infection and should be vaccinated if there is an outbreak of monkeypox in their locality.7,9
The current preferred vaccine for prevention of both smallpox and monkeypox is the Jynneos (Bavarian Nordic A/S) vaccine.10 This agent incorporates a replication-deficient live virus and does not pose the same risk for adverse events as the original versions of the smallpox vaccine. Jynneos is administered subcutaneously rather than by scarification. Two 0.5-mL doses, delivered 28 days apart, are required for optimal effect. The vaccine must be obtained from local and state health departments, in consultation with the CDC.7,9
There is very little published information on the safety of the Jynneos vaccine in pregnant or lactating women, although animal data are reassuring. Moreover, the dangers of monkeypox infection are significant, and in the event of an outbreak, vaccination of susceptible individuals, including pregnant women, is indicated.
- Monkeypox is a member of the orthopoxvirus genus and is closely related to the smallpox virus. It is a large, double-stranded, enveloped DNA virus.
- The virus is transmitted primarily by close contact with infected animals or other humans or by consumption of contaminated bushmeat.
- The infection evolves in 2 phases. The pre-eruptive phase is characterized by severe flu-like symptoms and signs. The eruptive phase is distinguished by a diffuse papular-vesicular rash.
- The most valuable test for confirming the diagnosis is a polymerase chain reaction test of a fresh skin lesion.
- In women who are pregnant, monkeypox has been associated with spontaneous abortion and fetal death.
- Three antiviral agents may be of value in treating infected patients: cidofovir, brincidofovir, and tecovirimat. Only the latter has an acceptable safety profile for women who are pregnant or lactating.
- The new nonreplicating smallpox vaccine Jynneos (Bavarian Nordic A/S) is of great value for pre- and post-exposure prophylaxis.
Continue to: Treatment...
Treatment
Infected pregnant women should receive acetaminophen 1,000 mg orally every 8 hours, to control fever and provide analgesia. An antihistamine such as diphenhydramine 25 mg orally every 6-8 hours, may be used to control pruritus and provide mild sedation. Adequate fluid intake and optimal nutrition should be encouraged. Skin lesions should be inspected regularly to detect signs of superimposed bacterial infections. Small, localized bacterial skin infections can be treated with topical application of mupirocin ointment 2%, 3 times daily for 7-14 days. For diffuse and more severe bacterial skin infections, a systemic antibiotic may be necessary. Reasonable choices include amoxicillin-clavulanate 875 mg/125 mg orally every 12 hours, or trimethoprim-sulfamethoxazole double strength 800 mg/160 mg orally every 12 hours.11 The latter agent should be avoided in the first trimester of pregnancy because of potential teratogenic effects.
Several specific agents are available through the CDC for treatment of orthopoxvirus infections such as smallpox and monkeypox. Information about these agents is summarized in the TABLE.12-16
Unique considerations in pregnancy
Because monkeypox is so rare, there is very little information about the effects of this infection in pregnant women. The report most commonly cited in the literature is that by Mbala et al, which was published in 2017.17 These authors described 4 pregnant patients in the Democratic Republic of Congo who contracted monkeypox infection over a 4-year period. All 4 women were hospitalized and treated with systemic antibiotics, antiparasitic medications, and analgesics. One patient delivered a healthy infant. Two women had spontaneous abortions in the first trimester. The fourth patient experienced a stillbirth at 22 weeks’ gestation. At postmortem examination, the fetus had diffuse cutaneous lesions, prominent hepatomegaly, and hydrops. No structural malformations were noted. The placenta demonstrated numerous punctate hemorrhages, and high concentrations of virus were recovered from the placenta and from fetal tissue.
Although the information on pregnancy outcome is quite limited, it seems clear that the virus can cross the placenta and cause adverse effects such as spontaneous abortion and fetal death. Accordingly, I think the following guidelines are a reasonable approach to a pregnant patient who has been exposed to monkeypox or who has developed manifestations of infection.3,7,9
- In the event of a community outbreak, bioterrorism event, or exposure to a person with suspected or confirmed monkeypox infection, the pregnant patient should receive the Jynneos vaccine.
- The pregnant patient should be isolated from any individual with suspected or confirmed monkeypox.
- If infection develops despite these measures, the patient should be treated with either tecovirimat or vaccinia immune globulin IV. Hospitalization may be necessary for seriously ill individuals.
- Within 2 weeks of infection, a comprehensive ultrasound examination should be performed to assess for structural abnormalities in the fetus.
- Subsequently, serial ultrasound examinations should be performed at intervals of 4-6 weeks to assess fetal growth and re-evaluate fetal anatomy.
- Following delivery, a detailed neonatal examination should be performed to assess for evidence of viral injury. Neonatal skin lesions and neonatal serum can be assessed by PCR for monkeypox virus. The newborn should be isolated from the mother until all the mother’s lesions have dried and crusted over.
CASE Resolved
Given the husband’s recent travel to Nigeria and consumption of bushmeat, he most likely has monkeypox. The infection can be spread from person to person by close contact; thus, his wife is at risk. The couple should isolate until all of his lesions have dried and crusted over. The woman also should receive the Jynneos vaccine. If she becomes symptomatic, she should be treated with tecovirimat or vaccinia immune globulin IV. ●
- Isaacs SN, Shenoy ES. Monkeypox. UpToDate. Updated June 28,2022. Accessed July 1, 2022. https://www.uptodate.com /contents/monkeypox?topicRef=8349&source=see_link
- Graham MB. Monkeypox. Medscape. Updated June 29, 2022. Accessed July 1, 2022. https://emedicine.medscape.com /article/1134714-overview.
- Khalil A, Samara A, O’Brien P, et al. Monkeypox and pregnancy: what do obstetricians need to know? Ultrasound Obstet Gynecol. 2022;60:22-27. doi:10.1002/uog.24968.
- World Health Organization. Monkeypox-United Kingdom of Great Britain and Northern Ireland. May 18, 2022. Accessed July 1, 2022. https://www.who.int/emergencies/diseaseoutbreak-news/item/2022-DON383.
- WHO reports two new monkeypox deaths, cases in new areas. Reuters. July 7, 2022. https://www.reuters.com/world /who-reports-two-new-monkeypox-deaths-2022-07-07/. Accessed July 19, 2022.
- World Health Organization. Multi-country monkeypox outbreak in non-endemic countries: update. May 29, 2022. Accessed July 1, 2022. https://www.who.int /emergencies/disease-outbreak-news/item/2022 -DON388#:~:text=Multi%2Dcountry%20monkeypox%20 outbreak%20in%20non%2Dendemic%20countries%3A%20 Update,-29%20May%202022&text=Since%2013%20 May%202022%2C%20monkeypox,Epidemiological%20 investigations%20are%20ongoing.
- Cono J, Cragan JD, Jamieson DJ, Rasmussen SA. Prophylaxis and treatment of pregnant women for emerging infections andbioterrorism emergencies. Emerg Infect Dis. 2006;12:16311637. doi:10.3201/eid1211.060618.
- Centers for Disease Control and Prevention. Preparation and collection of specimens. Reviewed June 29, 2022. Accessed July 6, 2022. https://www.cdc.gov/poxvirus /monkeypox/clinicians/prep-collection-specimens.html.
- Rao AK, Petersen BW, Whitehill F, et al. Monkeypox vaccination. MMWR Morb Mortal Wkly Rep. 2022;71:734-742. doi:10.15585/mmwr.mm7122e1.
- Smallpox and monkeypox vaccine, live, nonreplicating. Package insert. Bavarian Nordic A/S; 2021. Accessed July 1, 2022. https://www.fda.gov/media/131078/download.
- Duff P. Commonly used antibiotics in ObGyn practice. OBG Manag. 2022;34:29, 36-40. doi:10.12788/obgm.0191.
- Centers for Disease Control and Prevention. Treatment information for healthcare professionals: interim clinical guidance for the treatment of monkeypox. Reviewed June 17, 2022. Accessed July 1, 2022. https://www.cdc.gov/poxvirus /monkeypox/clinicians/treatment.html.
- Brincidofovir. Prescribing information. Chimerix, Inc.; 2021. Accessed July 1, 2022. https://www.accessdata.fda.gov /drugsatfda_docs/label/2021/214460s000,214461s000lbl.pdf.
- Cidofovir. Package insert. Gilead Sciences, Inc.; 2010. Accessed July 1, 2022. https://www.gilead.com/~/media /Files/pdfs/medicines/other/vistide/vistide.pdf.
- Tecovirimat. Prescribing information. Catalent Pharma Solutions; 2022. Accessed July 1, 2022. https://www.accessdata.fda.gov/drugsatfda_docs /label/2022/214518s000lbl.pdf.
- Vaccinia immune globulin IV. Prescribing information. Cangene Corporation; 2010. Accessed July 1, 2022. https: //www.fda.gov/media/77004/download.
- Mbala PK, Huggins JW, Riu-Rovira T, et al. Maternal and fetal outcomes among pregnant women with human monkeypox infection in the Democratic Republic of Congo. J Infect Dis. 2017;216:824-828. doi:10.1093/infdis/jix260.
- Isaacs SN, Shenoy ES. Monkeypox. UpToDate. Updated June 28,2022. Accessed July 1, 2022. https://www.uptodate.com /contents/monkeypox?topicRef=8349&source=see_link
- Graham MB. Monkeypox. Medscape. Updated June 29, 2022. Accessed July 1, 2022. https://emedicine.medscape.com /article/1134714-overview.
- Khalil A, Samara A, O’Brien P, et al. Monkeypox and pregnancy: what do obstetricians need to know? Ultrasound Obstet Gynecol. 2022;60:22-27. doi:10.1002/uog.24968.
- World Health Organization. Monkeypox-United Kingdom of Great Britain and Northern Ireland. May 18, 2022. Accessed July 1, 2022. https://www.who.int/emergencies/diseaseoutbreak-news/item/2022-DON383.
- WHO reports two new monkeypox deaths, cases in new areas. Reuters. July 7, 2022. https://www.reuters.com/world /who-reports-two-new-monkeypox-deaths-2022-07-07/. Accessed July 19, 2022.
- World Health Organization. Multi-country monkeypox outbreak in non-endemic countries: update. May 29, 2022. Accessed July 1, 2022. https://www.who.int /emergencies/disease-outbreak-news/item/2022 -DON388#:~:text=Multi%2Dcountry%20monkeypox%20 outbreak%20in%20non%2Dendemic%20countries%3A%20 Update,-29%20May%202022&text=Since%2013%20 May%202022%2C%20monkeypox,Epidemiological%20 investigations%20are%20ongoing.
- Cono J, Cragan JD, Jamieson DJ, Rasmussen SA. Prophylaxis and treatment of pregnant women for emerging infections andbioterrorism emergencies. Emerg Infect Dis. 2006;12:16311637. doi:10.3201/eid1211.060618.
- Centers for Disease Control and Prevention. Preparation and collection of specimens. Reviewed June 29, 2022. Accessed July 6, 2022. https://www.cdc.gov/poxvirus /monkeypox/clinicians/prep-collection-specimens.html.
- Rao AK, Petersen BW, Whitehill F, et al. Monkeypox vaccination. MMWR Morb Mortal Wkly Rep. 2022;71:734-742. doi:10.15585/mmwr.mm7122e1.
- Smallpox and monkeypox vaccine, live, nonreplicating. Package insert. Bavarian Nordic A/S; 2021. Accessed July 1, 2022. https://www.fda.gov/media/131078/download.
- Duff P. Commonly used antibiotics in ObGyn practice. OBG Manag. 2022;34:29, 36-40. doi:10.12788/obgm.0191.
- Centers for Disease Control and Prevention. Treatment information for healthcare professionals: interim clinical guidance for the treatment of monkeypox. Reviewed June 17, 2022. Accessed July 1, 2022. https://www.cdc.gov/poxvirus /monkeypox/clinicians/treatment.html.
- Brincidofovir. Prescribing information. Chimerix, Inc.; 2021. Accessed July 1, 2022. https://www.accessdata.fda.gov /drugsatfda_docs/label/2021/214460s000,214461s000lbl.pdf.
- Cidofovir. Package insert. Gilead Sciences, Inc.; 2010. Accessed July 1, 2022. https://www.gilead.com/~/media /Files/pdfs/medicines/other/vistide/vistide.pdf.
- Tecovirimat. Prescribing information. Catalent Pharma Solutions; 2022. Accessed July 1, 2022. https://www.accessdata.fda.gov/drugsatfda_docs /label/2022/214518s000lbl.pdf.
- Vaccinia immune globulin IV. Prescribing information. Cangene Corporation; 2010. Accessed July 1, 2022. https: //www.fda.gov/media/77004/download.
- Mbala PK, Huggins JW, Riu-Rovira T, et al. Maternal and fetal outcomes among pregnant women with human monkeypox infection in the Democratic Republic of Congo. J Infect Dis. 2017;216:824-828. doi:10.1093/infdis/jix260.