User login
Leveling the Playing Field: Accounting for Academic Productivity During the COVID-19 Pandemic
Professional upheavals caused by the coronavirus disease 2019 (COVID-19) pandemic have affected the academic productivity of many physicians. This is due in part to rapid changes in clinical care and medical education: physician-researchers have been redeployed to frontline clinical care; clinician-educators have been forced to rapidly transition in-person curricula to virtual platforms; and primary care physicians and subspecialists have been forced to transition to telehealth-based practices. In addition to these changes in clinical and educational responsibilities, the COVID-19 pandemic has substantially altered the personal lives of physicians. During the height of the pandemic, clinicians simultaneously wrestled with a lack of available childcare, unexpected home-schooling responsibilities, decreased income, and many other COVID-19-related stresses.1 Additionally, the ever-present “second pandemic” of structural racism, persistent health disparities, and racial inequity has further increased the personal and professional demands facing academic faculty.2
In particular, the pandemic has placed personal and professional pressure on female and minority faculty members. In spite of these pressures, however, the academic promotions process still requires rigid accounting of scholarly productivity. As the focus of academic practices has shifted to support clinical care during the pandemic, scholarly productivity has suffered for clinicians on the frontline. As a result, academic clinical faculty have expressed significant stress and concerns about failing to meet benchmarks for promotion (eg, publications, curricula development, national presentations). To counter these shifts (and the inherent inequity that they create for female clinicians and for men and women who are Black, Indigenous, and/or of color), academic institutions should not only recognize the effects the COVID-19 pandemic has had on faculty, but also adopt immediate solutions to more equitably account for such disruptions to academic portfolios. In this paper, we explore populations whose career trajectories are most at-risk and propose a framework to capture novel and nontraditional contributions while also acknowledging the rapid changes the COVID-19 pandemic has brought to academic medicine.
POPULATIONS AT RISK FOR CAREER DISRUPTION
Even before the COVID-19 pandemic, physician mothers, underrepresented racial/ethnic minority groups, and junior faculty were most at-risk for career disruptions. The closure of daycare facilities and schools and shift to online learning resulting from the pandemic, along with the common challenges of parenting, have taken a significant toll on the lives of working parents. Because women tend to carry a disproportionate share of childcare and household responsibilities, these changes have inequitably leveraged themselves as a “mommy tax” on working women.3,4
As underrepresented medicine faculty (particularly Black, Hispanic, Latino, and Native American clinicians) comprise only 8% of the academic medical workforce,they currently face a variety of personal and professional challenges.5 This is especially true for Black and Latinx physicians who have been experiencing an increased COVID-19 burden in their communities, while concurrently fighting entrenched structural racism and police violence. In academia, these challenges have worsened because of the “minority tax”—the toll of often uncompensated extra responsibilities (time or money) placed on minority faculty in the name of achieving diversity. The unintended consequences of these responsibilities result in having fewer mentors,6 caring for underserved populations,7 and performing more clinical care8 than non-underrepresented minority faculty. Because minority faculty are unlikely to be in leadership positions, it is reasonable to conclude they have been shouldering heavier clinical obligations and facing greater career disruption of scholarly work due to the COVID-19 pandemic.
Junior faculty (eg, instructors and assistant professors) also remain professionally vulnerable during the COVID-19 pandemic. Because junior faculty are often more clinically focused and less likely to hold leadership positions than senior faculty, they are more likely to have assumed frontline clinical positions, which come at the expense of academic work. Junior faculty are also at a critical building phase in their academic career—a time when they benefit from the opportunity to share their scholarly work and network at conferences. Unfortunately, many conferences have been canceled or moved to a virtual platform. Given that some institutions may be freezing academic funding for conferences due to budgetary shortfalls from the pandemic, junior faculty may be particularly at risk if they are not able to present their work. In addition, junior faculty often face disproportionate struggles at home, trying to balance demands of work and caring for young children. Considering the unique needs of each of these groups, it is especially important to consider intersectionality, or the compounded issues for individuals who exist in multiple disproportionately affected groups (eg, a Black female junior faculty member who is also a mother).
THE COVID-19-CURRICULUM VITAE MATRIX
The typical format of a professional curriculum vitae (CV) at most academic institutions does not allow one to document potential disruptions or novel contributions, including those that occurred during the COVID-19 pandemic. As a group of academic clinicians, educators, and researchers whose careers have been affected by the pandemic, we created a COVID-19 CV matrix, a potential framework to serve as a supplement for faculty. In this matrix, faculty members may document their contributions, disruptions that affected their work, and caregiving responsibilities during this time period, while also providing a rubric for promotions and tenure committees to equitably evaluate the pandemic period on an academic CV. Our COVID-19 CV matrix consists of six domains: (1) clinical care, (2) research, (3) education, (4) service, (5) advocacy/media, and (6) social media. These domains encompass traditional and nontraditional contributions made by healthcare professionals during the pandemic (Table). This matrix broadens the ability of both faculty and institutions to determine the actual impact of individuals during the pandemic.

ACCOUNT FOR YOUR (NEW) IMPACT
Throughout the COVID-19 pandemic, academic faculty have been innovative, contributing in novel ways not routinely captured by promotions committees—eg, the digital health researcher who now directs the telemedicine response for their institution and the health disparities researcher who now leads daily webinar sessions on structural racism to medical students. Other novel contributions include advancing COVID-19 innovations and engaging in media and community advocacy (eg, organizing large-scale donations of equipment and funds to support organizations in need). While such nontraditional contributions may not have been readily captured or thought “CV worthy” in the past, faculty should now account for them. More importantly, promotions committees need to recognize that these pivots or alterations in career paths are not signals of professional failure, but rather evidence of a shifting landscape and the respective response of the individual. Furthermore, because these pivots often help fulfill an institutional mission, they are impactful.
ACKNOWLEDGE THE DISRUPTION
It is important for promotions and tenure committees to recognize the impact and disruption COVID-19 has had on traditional academic work, acknowledging the time and energy required for a faculty member to make needed work adjustments. This enables a leader to better assess how a faculty member’s academic portfolio has been affected. For example, researchers have had to halt studies, medical educators have had to redevelop and transition curricula to virtual platforms, and physicians have had to discontinue clinician quality improvement initiatives due to competing hospital priorities. Faculty members who document such unintentional alterations in their academic career path can explain to their institution how they have continued to positively influence their field and the community during the pandemic. This approach is analogous to the current model of accounting for clinical time when judging faculty members’ contributions in scholarly achievement.
The COVID-19 CV matrix has the potential to be annotated to explain the burden of one’s personal situation, which is often “invisible” in the professional environment. For example, many physicians have had to assume additional childcare responsibilities, tend to sick family members, friends, and even themselves. It is also possible that a faculty member has a partner who is also an essential worker, one who had to self-isolate due to COVID-19 exposure or illness, or who has been working overtime due to high patient volumes.
INSTITUTIONAL RESPONSE
How can institutions respond to the altered academic landscape caused by the COVID-19 pandemic? Promotions committees typically have two main tools at their disposal: adjusting the tenure clock or the benchmarks. Extending the period of time available to qualify for tenure is commonplace in the “publish-or-perish” academic tracks of university research professors. Clock adjustments are typically granted to faculty following the birth of a child or for other specific family- or health-related hardships, in accordance with the Family and Medical Leave Act. Unfortunately, tenure-clock extensions for female faculty members can exacerbate gender inequity: Data on tenure-clock extensions show a higher rate of tenure granted to male faculty compared to female faculty.9 For this reason, it is also important to explore adjustments or modifications to benchmark criteria. This could be accomplished by broadening the criteria for promotion, recognizing that impact occurs in many forms, thereby enabling meeting a benchmark. It can also occur by examining the trajectory of an individual within a promotion pathway before it was disrupted to determine impact. To avoid exacerbating social and gender inequities within academia, institutions should use these professional levers and create new ones to provide parity and equality across the promotional playing field. While the CV matrix openly acknowledges the disruptions and tangents the COVID-19 pandemic has had on academic careers, it remains important for academic institutions to recognize these disruptions and innovate the manner in which they acknowledge scholarly contributions.
Conclusion
While academic rigidity and known social taxes (minority and mommy taxes) are particularly problematic in the current climate, these issues have always been at play in evaluating academic success. Improved documentation of novel contributions, disruptions, caregiving, and other challenges can enable more holistic and timely professional advancement for all faculty, regardless of their sex, race, ethnicity, or social background. Ultimately, we hope this framework initiates further conversations among academic institutions on how to define productivity in an age where journal impact factor or number of publications is not the fullest measure of one’s impact in their field.
1. Jones Y, Durand V, Morton K, et al; ADVANCE PHM Steering Committee. Collateral damage: how covid-19 is adversely impacting women physicians. J Hosp Med. 2020;15(8):507-509. https://doi.org/10.12788/jhm.3470
2. Manning KD. When grief and crises intersect: perspectives of a black physician in the time of two pandemics. J Hosp Med. 2020;15(9):566-567. https://doi.org/10.12788/jhm.3481
3. Cohen P, Hsu T. Pandemic could scar a generation of working mothers. New York Times. Published June 3, 2020. Updated June 30, 2020. Accessed November 11, 2020. https://www.nytimes.com/2020/06/03/business/economy/coronavirus-working-women.html
4. Cain Miller C. Nearly half of men say they do most of the home schooling. 3 percent of women agree. Published May 6, 2020. Updated May 8, 2020. Accessed November 11, 2020. New York Times. https://www.nytimes.com/2020/05/06/upshot/pandemic-chores-homeschooling-gender.html
5. Rodríguez JE, Campbell KM, Pololi LH. Addressing disparities in academic medicine: what of the minority tax? BMC Med Educ. 2015;15:6. https://doi.org/10.1186/s12909-015-0290-9
6. Lewellen-Williams C, Johnson VA, Deloney LA, Thomas BR, Goyol A, Henry-Tillman R. The POD: a new model for mentoring underrepresented minority faculty. Acad Med. 2006;81(3):275-279. https://doi.org/10.1097/00001888-200603000-00020
7. Pololi LH, Evans AT, Gibbs BK, Krupat E, Brennan RT, Civian JT. The experience of minority faculty who are underrepresented in medicine, at 26 representative U.S. medical schools. Acad Med. 2013;88(9):1308-1314. https://doi.org/10.1097/acm.0b013e31829eefff
8. Richert A, Campbell K, Rodríguez J, Borowsky IW, Parikh R, Colwell A. ACU workforce column: expanding and supporting the health care workforce. J Health Care Poor Underserved. 2013;24(4):1423-1431. https://doi.org/10.1353/hpu.2013.0162
9. Woitowich NC, Jain S, Arora VM, Joffe H. COVID-19 threatens progress toward gender equity within academic medicine. Acad Med. 2020;29:10.1097/ACM.0000000000003782. https://doi.org/10.1097/acm.0000000000003782
Professional upheavals caused by the coronavirus disease 2019 (COVID-19) pandemic have affected the academic productivity of many physicians. This is due in part to rapid changes in clinical care and medical education: physician-researchers have been redeployed to frontline clinical care; clinician-educators have been forced to rapidly transition in-person curricula to virtual platforms; and primary care physicians and subspecialists have been forced to transition to telehealth-based practices. In addition to these changes in clinical and educational responsibilities, the COVID-19 pandemic has substantially altered the personal lives of physicians. During the height of the pandemic, clinicians simultaneously wrestled with a lack of available childcare, unexpected home-schooling responsibilities, decreased income, and many other COVID-19-related stresses.1 Additionally, the ever-present “second pandemic” of structural racism, persistent health disparities, and racial inequity has further increased the personal and professional demands facing academic faculty.2
In particular, the pandemic has placed personal and professional pressure on female and minority faculty members. In spite of these pressures, however, the academic promotions process still requires rigid accounting of scholarly productivity. As the focus of academic practices has shifted to support clinical care during the pandemic, scholarly productivity has suffered for clinicians on the frontline. As a result, academic clinical faculty have expressed significant stress and concerns about failing to meet benchmarks for promotion (eg, publications, curricula development, national presentations). To counter these shifts (and the inherent inequity that they create for female clinicians and for men and women who are Black, Indigenous, and/or of color), academic institutions should not only recognize the effects the COVID-19 pandemic has had on faculty, but also adopt immediate solutions to more equitably account for such disruptions to academic portfolios. In this paper, we explore populations whose career trajectories are most at-risk and propose a framework to capture novel and nontraditional contributions while also acknowledging the rapid changes the COVID-19 pandemic has brought to academic medicine.
POPULATIONS AT RISK FOR CAREER DISRUPTION
Even before the COVID-19 pandemic, physician mothers, underrepresented racial/ethnic minority groups, and junior faculty were most at-risk for career disruptions. The closure of daycare facilities and schools and shift to online learning resulting from the pandemic, along with the common challenges of parenting, have taken a significant toll on the lives of working parents. Because women tend to carry a disproportionate share of childcare and household responsibilities, these changes have inequitably leveraged themselves as a “mommy tax” on working women.3,4
As underrepresented medicine faculty (particularly Black, Hispanic, Latino, and Native American clinicians) comprise only 8% of the academic medical workforce,they currently face a variety of personal and professional challenges.5 This is especially true for Black and Latinx physicians who have been experiencing an increased COVID-19 burden in their communities, while concurrently fighting entrenched structural racism and police violence. In academia, these challenges have worsened because of the “minority tax”—the toll of often uncompensated extra responsibilities (time or money) placed on minority faculty in the name of achieving diversity. The unintended consequences of these responsibilities result in having fewer mentors,6 caring for underserved populations,7 and performing more clinical care8 than non-underrepresented minority faculty. Because minority faculty are unlikely to be in leadership positions, it is reasonable to conclude they have been shouldering heavier clinical obligations and facing greater career disruption of scholarly work due to the COVID-19 pandemic.
Junior faculty (eg, instructors and assistant professors) also remain professionally vulnerable during the COVID-19 pandemic. Because junior faculty are often more clinically focused and less likely to hold leadership positions than senior faculty, they are more likely to have assumed frontline clinical positions, which come at the expense of academic work. Junior faculty are also at a critical building phase in their academic career—a time when they benefit from the opportunity to share their scholarly work and network at conferences. Unfortunately, many conferences have been canceled or moved to a virtual platform. Given that some institutions may be freezing academic funding for conferences due to budgetary shortfalls from the pandemic, junior faculty may be particularly at risk if they are not able to present their work. In addition, junior faculty often face disproportionate struggles at home, trying to balance demands of work and caring for young children. Considering the unique needs of each of these groups, it is especially important to consider intersectionality, or the compounded issues for individuals who exist in multiple disproportionately affected groups (eg, a Black female junior faculty member who is also a mother).
THE COVID-19-CURRICULUM VITAE MATRIX
The typical format of a professional curriculum vitae (CV) at most academic institutions does not allow one to document potential disruptions or novel contributions, including those that occurred during the COVID-19 pandemic. As a group of academic clinicians, educators, and researchers whose careers have been affected by the pandemic, we created a COVID-19 CV matrix, a potential framework to serve as a supplement for faculty. In this matrix, faculty members may document their contributions, disruptions that affected their work, and caregiving responsibilities during this time period, while also providing a rubric for promotions and tenure committees to equitably evaluate the pandemic period on an academic CV. Our COVID-19 CV matrix consists of six domains: (1) clinical care, (2) research, (3) education, (4) service, (5) advocacy/media, and (6) social media. These domains encompass traditional and nontraditional contributions made by healthcare professionals during the pandemic (Table). This matrix broadens the ability of both faculty and institutions to determine the actual impact of individuals during the pandemic.
ACCOUNT FOR YOUR (NEW) IMPACT
Throughout the COVID-19 pandemic, academic faculty have been innovative, contributing in novel ways not routinely captured by promotions committees—eg, the digital health researcher who now directs the telemedicine response for their institution and the health disparities researcher who now leads daily webinar sessions on structural racism to medical students. Other novel contributions include advancing COVID-19 innovations and engaging in media and community advocacy (eg, organizing large-scale donations of equipment and funds to support organizations in need). While such nontraditional contributions may not have been readily captured or thought “CV worthy” in the past, faculty should now account for them. More importantly, promotions committees need to recognize that these pivots or alterations in career paths are not signals of professional failure, but rather evidence of a shifting landscape and the respective response of the individual. Furthermore, because these pivots often help fulfill an institutional mission, they are impactful.
ACKNOWLEDGE THE DISRUPTION
It is important for promotions and tenure committees to recognize the impact and disruption COVID-19 has had on traditional academic work, acknowledging the time and energy required for a faculty member to make needed work adjustments. This enables a leader to better assess how a faculty member’s academic portfolio has been affected. For example, researchers have had to halt studies, medical educators have had to redevelop and transition curricula to virtual platforms, and physicians have had to discontinue clinician quality improvement initiatives due to competing hospital priorities. Faculty members who document such unintentional alterations in their academic career path can explain to their institution how they have continued to positively influence their field and the community during the pandemic. This approach is analogous to the current model of accounting for clinical time when judging faculty members’ contributions in scholarly achievement.
The COVID-19 CV matrix has the potential to be annotated to explain the burden of one’s personal situation, which is often “invisible” in the professional environment. For example, many physicians have had to assume additional childcare responsibilities, tend to sick family members, friends, and even themselves. It is also possible that a faculty member has a partner who is also an essential worker, one who had to self-isolate due to COVID-19 exposure or illness, or who has been working overtime due to high patient volumes.
INSTITUTIONAL RESPONSE
How can institutions respond to the altered academic landscape caused by the COVID-19 pandemic? Promotions committees typically have two main tools at their disposal: adjusting the tenure clock or the benchmarks. Extending the period of time available to qualify for tenure is commonplace in the “publish-or-perish” academic tracks of university research professors. Clock adjustments are typically granted to faculty following the birth of a child or for other specific family- or health-related hardships, in accordance with the Family and Medical Leave Act. Unfortunately, tenure-clock extensions for female faculty members can exacerbate gender inequity: Data on tenure-clock extensions show a higher rate of tenure granted to male faculty compared to female faculty.9 For this reason, it is also important to explore adjustments or modifications to benchmark criteria. This could be accomplished by broadening the criteria for promotion, recognizing that impact occurs in many forms, thereby enabling meeting a benchmark. It can also occur by examining the trajectory of an individual within a promotion pathway before it was disrupted to determine impact. To avoid exacerbating social and gender inequities within academia, institutions should use these professional levers and create new ones to provide parity and equality across the promotional playing field. While the CV matrix openly acknowledges the disruptions and tangents the COVID-19 pandemic has had on academic careers, it remains important for academic institutions to recognize these disruptions and innovate the manner in which they acknowledge scholarly contributions.
Conclusion
While academic rigidity and known social taxes (minority and mommy taxes) are particularly problematic in the current climate, these issues have always been at play in evaluating academic success. Improved documentation of novel contributions, disruptions, caregiving, and other challenges can enable more holistic and timely professional advancement for all faculty, regardless of their sex, race, ethnicity, or social background. Ultimately, we hope this framework initiates further conversations among academic institutions on how to define productivity in an age where journal impact factor or number of publications is not the fullest measure of one’s impact in their field.
Professional upheavals caused by the coronavirus disease 2019 (COVID-19) pandemic have affected the academic productivity of many physicians. This is due in part to rapid changes in clinical care and medical education: physician-researchers have been redeployed to frontline clinical care; clinician-educators have been forced to rapidly transition in-person curricula to virtual platforms; and primary care physicians and subspecialists have been forced to transition to telehealth-based practices. In addition to these changes in clinical and educational responsibilities, the COVID-19 pandemic has substantially altered the personal lives of physicians. During the height of the pandemic, clinicians simultaneously wrestled with a lack of available childcare, unexpected home-schooling responsibilities, decreased income, and many other COVID-19-related stresses.1 Additionally, the ever-present “second pandemic” of structural racism, persistent health disparities, and racial inequity has further increased the personal and professional demands facing academic faculty.2
In particular, the pandemic has placed personal and professional pressure on female and minority faculty members. In spite of these pressures, however, the academic promotions process still requires rigid accounting of scholarly productivity. As the focus of academic practices has shifted to support clinical care during the pandemic, scholarly productivity has suffered for clinicians on the frontline. As a result, academic clinical faculty have expressed significant stress and concerns about failing to meet benchmarks for promotion (eg, publications, curricula development, national presentations). To counter these shifts (and the inherent inequity that they create for female clinicians and for men and women who are Black, Indigenous, and/or of color), academic institutions should not only recognize the effects the COVID-19 pandemic has had on faculty, but also adopt immediate solutions to more equitably account for such disruptions to academic portfolios. In this paper, we explore populations whose career trajectories are most at-risk and propose a framework to capture novel and nontraditional contributions while also acknowledging the rapid changes the COVID-19 pandemic has brought to academic medicine.
POPULATIONS AT RISK FOR CAREER DISRUPTION
Even before the COVID-19 pandemic, physician mothers, underrepresented racial/ethnic minority groups, and junior faculty were most at-risk for career disruptions. The closure of daycare facilities and schools and shift to online learning resulting from the pandemic, along with the common challenges of parenting, have taken a significant toll on the lives of working parents. Because women tend to carry a disproportionate share of childcare and household responsibilities, these changes have inequitably leveraged themselves as a “mommy tax” on working women.3,4
As underrepresented medicine faculty (particularly Black, Hispanic, Latino, and Native American clinicians) comprise only 8% of the academic medical workforce,they currently face a variety of personal and professional challenges.5 This is especially true for Black and Latinx physicians who have been experiencing an increased COVID-19 burden in their communities, while concurrently fighting entrenched structural racism and police violence. In academia, these challenges have worsened because of the “minority tax”—the toll of often uncompensated extra responsibilities (time or money) placed on minority faculty in the name of achieving diversity. The unintended consequences of these responsibilities result in having fewer mentors,6 caring for underserved populations,7 and performing more clinical care8 than non-underrepresented minority faculty. Because minority faculty are unlikely to be in leadership positions, it is reasonable to conclude they have been shouldering heavier clinical obligations and facing greater career disruption of scholarly work due to the COVID-19 pandemic.
Junior faculty (eg, instructors and assistant professors) also remain professionally vulnerable during the COVID-19 pandemic. Because junior faculty are often more clinically focused and less likely to hold leadership positions than senior faculty, they are more likely to have assumed frontline clinical positions, which come at the expense of academic work. Junior faculty are also at a critical building phase in their academic career—a time when they benefit from the opportunity to share their scholarly work and network at conferences. Unfortunately, many conferences have been canceled or moved to a virtual platform. Given that some institutions may be freezing academic funding for conferences due to budgetary shortfalls from the pandemic, junior faculty may be particularly at risk if they are not able to present their work. In addition, junior faculty often face disproportionate struggles at home, trying to balance demands of work and caring for young children. Considering the unique needs of each of these groups, it is especially important to consider intersectionality, or the compounded issues for individuals who exist in multiple disproportionately affected groups (eg, a Black female junior faculty member who is also a mother).
THE COVID-19-CURRICULUM VITAE MATRIX
The typical format of a professional curriculum vitae (CV) at most academic institutions does not allow one to document potential disruptions or novel contributions, including those that occurred during the COVID-19 pandemic. As a group of academic clinicians, educators, and researchers whose careers have been affected by the pandemic, we created a COVID-19 CV matrix, a potential framework to serve as a supplement for faculty. In this matrix, faculty members may document their contributions, disruptions that affected their work, and caregiving responsibilities during this time period, while also providing a rubric for promotions and tenure committees to equitably evaluate the pandemic period on an academic CV. Our COVID-19 CV matrix consists of six domains: (1) clinical care, (2) research, (3) education, (4) service, (5) advocacy/media, and (6) social media. These domains encompass traditional and nontraditional contributions made by healthcare professionals during the pandemic (Table). This matrix broadens the ability of both faculty and institutions to determine the actual impact of individuals during the pandemic.
ACCOUNT FOR YOUR (NEW) IMPACT
Throughout the COVID-19 pandemic, academic faculty have been innovative, contributing in novel ways not routinely captured by promotions committees—eg, the digital health researcher who now directs the telemedicine response for their institution and the health disparities researcher who now leads daily webinar sessions on structural racism to medical students. Other novel contributions include advancing COVID-19 innovations and engaging in media and community advocacy (eg, organizing large-scale donations of equipment and funds to support organizations in need). While such nontraditional contributions may not have been readily captured or thought “CV worthy” in the past, faculty should now account for them. More importantly, promotions committees need to recognize that these pivots or alterations in career paths are not signals of professional failure, but rather evidence of a shifting landscape and the respective response of the individual. Furthermore, because these pivots often help fulfill an institutional mission, they are impactful.
ACKNOWLEDGE THE DISRUPTION
It is important for promotions and tenure committees to recognize the impact and disruption COVID-19 has had on traditional academic work, acknowledging the time and energy required for a faculty member to make needed work adjustments. This enables a leader to better assess how a faculty member’s academic portfolio has been affected. For example, researchers have had to halt studies, medical educators have had to redevelop and transition curricula to virtual platforms, and physicians have had to discontinue clinician quality improvement initiatives due to competing hospital priorities. Faculty members who document such unintentional alterations in their academic career path can explain to their institution how they have continued to positively influence their field and the community during the pandemic. This approach is analogous to the current model of accounting for clinical time when judging faculty members’ contributions in scholarly achievement.
The COVID-19 CV matrix has the potential to be annotated to explain the burden of one’s personal situation, which is often “invisible” in the professional environment. For example, many physicians have had to assume additional childcare responsibilities, tend to sick family members, friends, and even themselves. It is also possible that a faculty member has a partner who is also an essential worker, one who had to self-isolate due to COVID-19 exposure or illness, or who has been working overtime due to high patient volumes.
INSTITUTIONAL RESPONSE
How can institutions respond to the altered academic landscape caused by the COVID-19 pandemic? Promotions committees typically have two main tools at their disposal: adjusting the tenure clock or the benchmarks. Extending the period of time available to qualify for tenure is commonplace in the “publish-or-perish” academic tracks of university research professors. Clock adjustments are typically granted to faculty following the birth of a child or for other specific family- or health-related hardships, in accordance with the Family and Medical Leave Act. Unfortunately, tenure-clock extensions for female faculty members can exacerbate gender inequity: Data on tenure-clock extensions show a higher rate of tenure granted to male faculty compared to female faculty.9 For this reason, it is also important to explore adjustments or modifications to benchmark criteria. This could be accomplished by broadening the criteria for promotion, recognizing that impact occurs in many forms, thereby enabling meeting a benchmark. It can also occur by examining the trajectory of an individual within a promotion pathway before it was disrupted to determine impact. To avoid exacerbating social and gender inequities within academia, institutions should use these professional levers and create new ones to provide parity and equality across the promotional playing field. While the CV matrix openly acknowledges the disruptions and tangents the COVID-19 pandemic has had on academic careers, it remains important for academic institutions to recognize these disruptions and innovate the manner in which they acknowledge scholarly contributions.
Conclusion
While academic rigidity and known social taxes (minority and mommy taxes) are particularly problematic in the current climate, these issues have always been at play in evaluating academic success. Improved documentation of novel contributions, disruptions, caregiving, and other challenges can enable more holistic and timely professional advancement for all faculty, regardless of their sex, race, ethnicity, or social background. Ultimately, we hope this framework initiates further conversations among academic institutions on how to define productivity in an age where journal impact factor or number of publications is not the fullest measure of one’s impact in their field.
1. Jones Y, Durand V, Morton K, et al; ADVANCE PHM Steering Committee. Collateral damage: how covid-19 is adversely impacting women physicians. J Hosp Med. 2020;15(8):507-509. https://doi.org/10.12788/jhm.3470
2. Manning KD. When grief and crises intersect: perspectives of a black physician in the time of two pandemics. J Hosp Med. 2020;15(9):566-567. https://doi.org/10.12788/jhm.3481
3. Cohen P, Hsu T. Pandemic could scar a generation of working mothers. New York Times. Published June 3, 2020. Updated June 30, 2020. Accessed November 11, 2020. https://www.nytimes.com/2020/06/03/business/economy/coronavirus-working-women.html
4. Cain Miller C. Nearly half of men say they do most of the home schooling. 3 percent of women agree. Published May 6, 2020. Updated May 8, 2020. Accessed November 11, 2020. New York Times. https://www.nytimes.com/2020/05/06/upshot/pandemic-chores-homeschooling-gender.html
5. Rodríguez JE, Campbell KM, Pololi LH. Addressing disparities in academic medicine: what of the minority tax? BMC Med Educ. 2015;15:6. https://doi.org/10.1186/s12909-015-0290-9
6. Lewellen-Williams C, Johnson VA, Deloney LA, Thomas BR, Goyol A, Henry-Tillman R. The POD: a new model for mentoring underrepresented minority faculty. Acad Med. 2006;81(3):275-279. https://doi.org/10.1097/00001888-200603000-00020
7. Pololi LH, Evans AT, Gibbs BK, Krupat E, Brennan RT, Civian JT. The experience of minority faculty who are underrepresented in medicine, at 26 representative U.S. medical schools. Acad Med. 2013;88(9):1308-1314. https://doi.org/10.1097/acm.0b013e31829eefff
8. Richert A, Campbell K, Rodríguez J, Borowsky IW, Parikh R, Colwell A. ACU workforce column: expanding and supporting the health care workforce. J Health Care Poor Underserved. 2013;24(4):1423-1431. https://doi.org/10.1353/hpu.2013.0162
9. Woitowich NC, Jain S, Arora VM, Joffe H. COVID-19 threatens progress toward gender equity within academic medicine. Acad Med. 2020;29:10.1097/ACM.0000000000003782. https://doi.org/10.1097/acm.0000000000003782
1. Jones Y, Durand V, Morton K, et al; ADVANCE PHM Steering Committee. Collateral damage: how covid-19 is adversely impacting women physicians. J Hosp Med. 2020;15(8):507-509. https://doi.org/10.12788/jhm.3470
2. Manning KD. When grief and crises intersect: perspectives of a black physician in the time of two pandemics. J Hosp Med. 2020;15(9):566-567. https://doi.org/10.12788/jhm.3481
3. Cohen P, Hsu T. Pandemic could scar a generation of working mothers. New York Times. Published June 3, 2020. Updated June 30, 2020. Accessed November 11, 2020. https://www.nytimes.com/2020/06/03/business/economy/coronavirus-working-women.html
4. Cain Miller C. Nearly half of men say they do most of the home schooling. 3 percent of women agree. Published May 6, 2020. Updated May 8, 2020. Accessed November 11, 2020. New York Times. https://www.nytimes.com/2020/05/06/upshot/pandemic-chores-homeschooling-gender.html
5. Rodríguez JE, Campbell KM, Pololi LH. Addressing disparities in academic medicine: what of the minority tax? BMC Med Educ. 2015;15:6. https://doi.org/10.1186/s12909-015-0290-9
6. Lewellen-Williams C, Johnson VA, Deloney LA, Thomas BR, Goyol A, Henry-Tillman R. The POD: a new model for mentoring underrepresented minority faculty. Acad Med. 2006;81(3):275-279. https://doi.org/10.1097/00001888-200603000-00020
7. Pololi LH, Evans AT, Gibbs BK, Krupat E, Brennan RT, Civian JT. The experience of minority faculty who are underrepresented in medicine, at 26 representative U.S. medical schools. Acad Med. 2013;88(9):1308-1314. https://doi.org/10.1097/acm.0b013e31829eefff
8. Richert A, Campbell K, Rodríguez J, Borowsky IW, Parikh R, Colwell A. ACU workforce column: expanding and supporting the health care workforce. J Health Care Poor Underserved. 2013;24(4):1423-1431. https://doi.org/10.1353/hpu.2013.0162
9. Woitowich NC, Jain S, Arora VM, Joffe H. COVID-19 threatens progress toward gender equity within academic medicine. Acad Med. 2020;29:10.1097/ACM.0000000000003782. https://doi.org/10.1097/acm.0000000000003782
© 2021 Society of Hospital Medicine
Use of Fluorodeoxyglucose-Positron Emission Tomography in the Diagnosis of Intravascular Diffuse Large B-Cell Lymphoma
Patient 1
A white man aged 67 years, with diastolic heart failure and chronic obstructive pulmonary disease, presented to the emergency department (ED) with shortness of breath. The initial laboratory results were significant for a newly elevated creatinine level of 2.06 mg/dL and a brain natriuretic peptide level of 648 pg/mL.
Imaging studies included a chest radiograph, a ventilation/perfusion scan, and an echocardiogram, as well as a right heart catheterization. All were nondiagnostic. 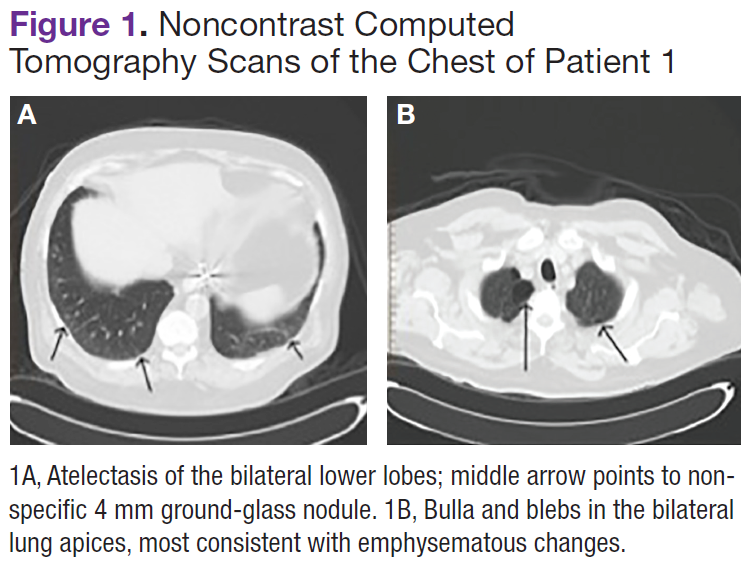
The patient's shortness of breath persisted despite treatment with diuretics, antibiotics, and steroids. Further laboratory workup revealed an elevated lactate dehydrogenase (LDH) level of 1,338 IU/L. A bone marrow biopsy performed because of concern about malignancy was unremarkable. Flow cytometry of the bone marrow aspirate did not reveal clonal B- or T-cell populations. Immunohistochemical staining was not performed. During this hospitalization for shortness of breath, the patient's mental staus began to decline, and his oxygen requirements increased. The patient was intubated but expired 48 hours after mechanical ventilation was initiated.
Patient 2
A white woman aged 67 years presented to the ED with generalized weakness, fatigue, and nausea. The patient’s medical history was significant for a diagnosis of stage IIIa ovarian cancer. She was treated with surgical resection and completed 6 cycles of adjuvant carboplatin and paclitaxel 3 months prior to this presentation. She had good response to treatment with normalization of CA-125. 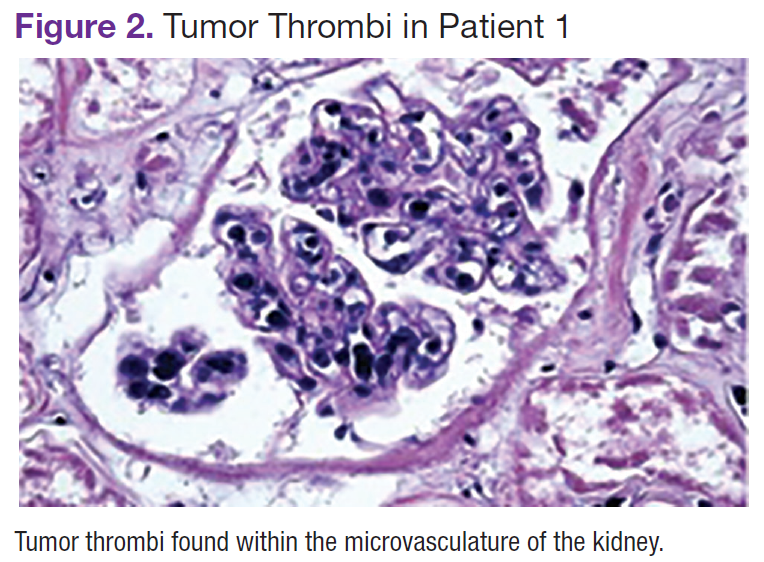
After completion of chemotherapy, the patient was found to have persistent anemia and thrombocytopenia. Admission laboratory results were significant for a hemoglobin level of 8.4 g/dL, a platelet count of 20,000/μL, and an LDH level of 1,220 IU/L.
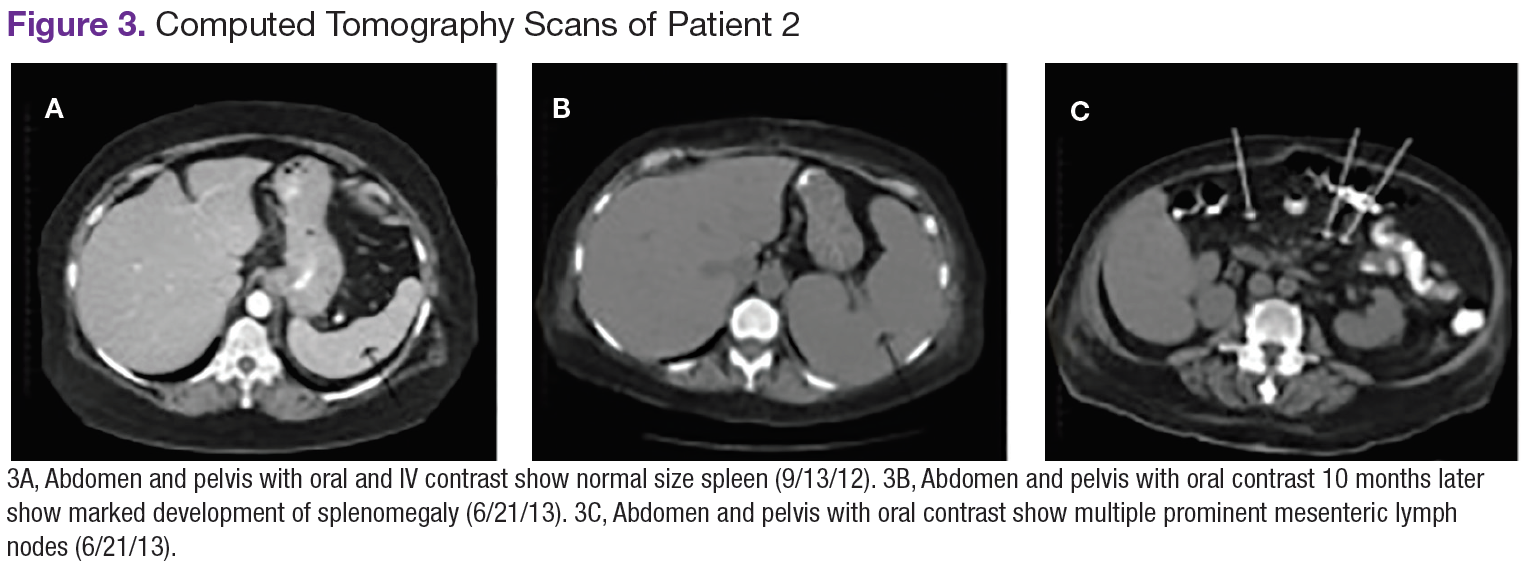
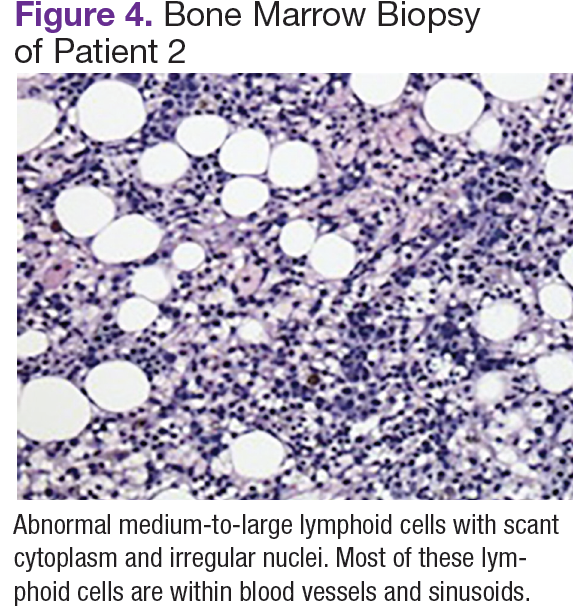
Chest, abdomen, and pelvis CT scans showed mesenteric adenopathy and splenomegaly (Figures 3A, 3B, and 3C) compared with prior imaging. Bone marrow biopsy revealed large lymphoid cells with scant cytoplasm and irregular nuclei, primarily within blood vessels and sinusoids consistent with IVLBCL (Figure 4). Flow cytometry of the bone marrow specimen showed an abnormal B-cell population with expression CD20, CD19, FMC-7, and dim κ light chain restriction. The cells were negative for CD5 and CD10. Immunohistochemical staining was positive for CD20, CD79a, PAX5, BCL-2 , and MUM1.
The patient was treated with 4 cycles of cyclophosphamide, doxorubicin, vincristine, prednisone, and rituximab, plus intrathecal methotrexate. The chemotherapy dose was reduced in the final cycle because of neuropathy in the hands and feet. The patient had undergone autologous stem-cell transplantation to allow high-dose chemotherapy. She was doing well more than 5 months after her transplant without evidence of recurrent disease.
Patient 3
A white man aged 76 years presented to the ED with cutaneous nodules, weight loss, fatigue, fevers, and epigastric pain. The patient’s medical history was significant for asymptomatic lymphoplasmacytic lymphoma diagnosed 2 months earlier, which had not required treatment. Laboratory results on admission revealed transaminitis, mild anemia with a hemoglobin level of 11 g/dL, and LDH level of 497 IU/L.
Chest, abdomen, and pelvis CT scans showed a 1.7cm hepatic lesion and mesenteric adenopathy. A bone marrow biopsy was unchanged from prior studies and showed minimal involvement (5%) of marrow space by low grade B-cell lymphoma.
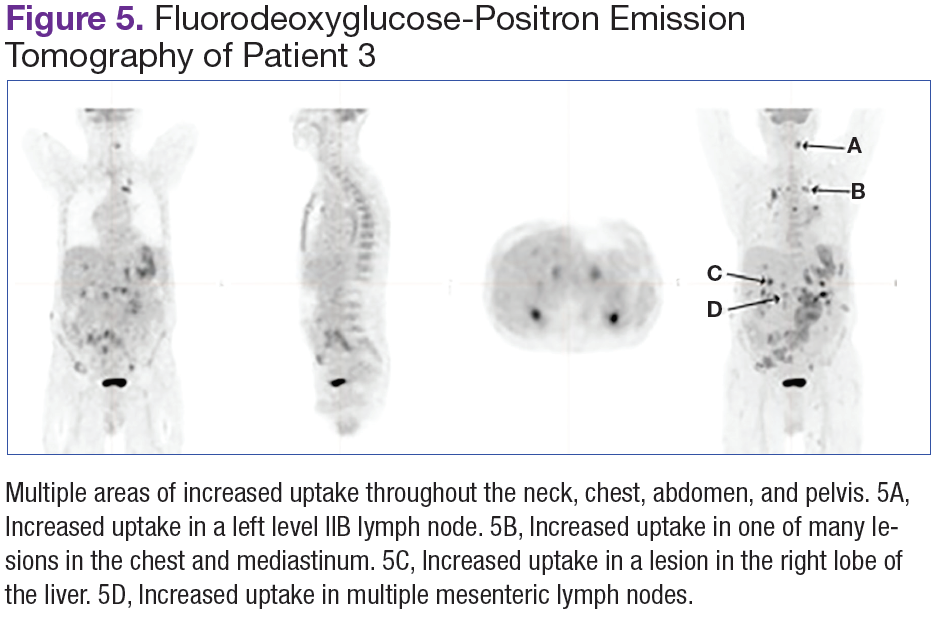
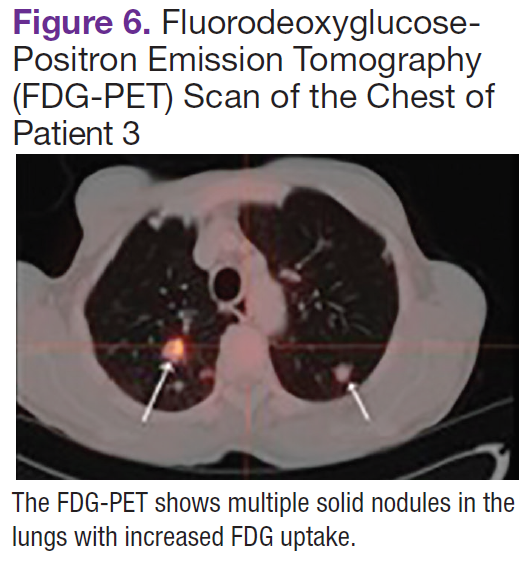
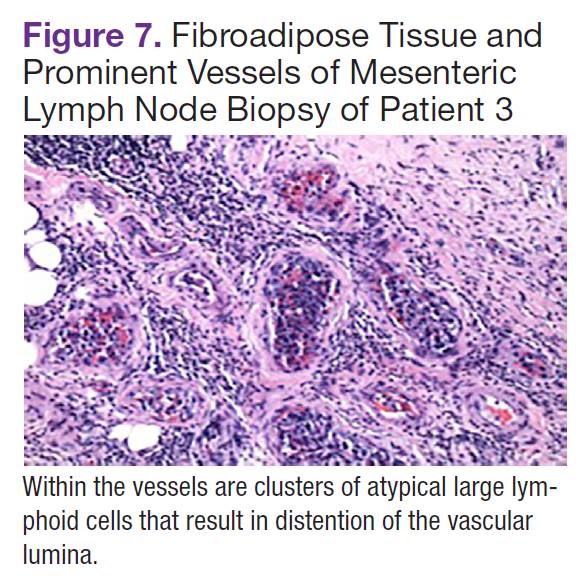
Fluorodeoxyglucose-positron emission tomography (FDG-PET) scans showed multiple areas of uptake in the neck, chest, abdomen, and pelvis (Figures 5 and 6). No increased uptake in the subcutaneous nodules was noted on examination. Laparoscopic biopsy of FDG-avid mesenteric nodes showed clusters of atypical large lymphoid cells resulting in distention of the vascular lumina, resulting in the diagnosis of IVLBCL (Figure 7).
Immunohistochemical stains showed that the intravascular lymphocytes were strongly positive for CD20 and BCL-2 and negative for CD5 and CD10. Flow cytometry on the sample was limited by a low cell count and could not be assessed for clonality. The patient completed 6 cycles of rituximab as well as intrathecal methotrexate. Restaging studies showed a complete remission.
Two months later, the patient developed a skin nodule on the right shoulder. A repeat FDG-PET scan showed increased uptake, and fine-needle biopsy confirmed recurrent disease. The patient is undergoing treatment with ifosfamide, carboplatin, etoposide, and rituximab, as well as workup for autologous stem-cell transplant.
Discussion
Intravascular large B-cell lymphoma, a subtype of diffuse large B-cell lymphoma, is unique because it is primarily extranodal and typically without significant tumor burden.1-4 Standard imaging modalities, therefore, are often nonspecific and do not aid clinicians in establishing a diagnosis. Fluorodeoxyglucose-positron emission tomography has a known role in the assessment of diffuse large B-cell lymphoma, both at time of diagnosis and in monitoring response to treatment.5 However, the use of FDGPET in the diagnosis and management of IVLBCL has not been clearly established.
In a review of the literature, 26 English-language case reports and small case series reporting individual centers’ experience with the use of this imaging modality in the diagnosis of IVLBCL were identified. Two cases were eliminated from review because they did not discuss the use of FDG-PET in relationship to diagnosis. Of the remaining 24 cases, 21 underwent initial imaging with 1 or more of the following imaging modalities: CT, magnetic resonance, ultrasound, bone scan, and gallium scintigraphy, all of which were nonspecific and did not lead to a definitive diagnosis.3,6-25 Each of the 21 cases was followed up by FDG-PET; in 19, the FDG-PET scan was positive and resulted in a diagnosis of IVLBCL. In 2 cases, the FDG-PET scan was nonrevealing and was not considered helpful in diagnosis.11,18 In 3 of the 21 cases, the FDG-PET scan was the primary imaging modality.6,14,25
In this review, all 3 patients had initial imaging with CT scans of anatomic locations that were largely unrevealing, although later histologic examination showed them to be locations of active disease either by biopsy or on autopsy. One patient who underwent early FDG-PET was found to have increased uptake in the mesenteric lymph nodes, which were later biopsied, as well as uptake in the bilateral adrenal glands, lungs, and bone.
Several characteristic FDG-PET findings that have been described in the literature have been identified in patients with IVLBCL, including diffuse accumulation in bilateral lung fields, accumulation in the renal cortex or adrenal glands, diffuse bony involvement, and hypometabolism in the brain.7,10,12,13,17,23,25 These findings show that organs with the richest blood supply, specifically the lungs and kidneys, often are affected. The brain, an obligate glucose metabolizer, would be expected to have high uptake; however, with tumor thrombi occluding small intracranial vessels, micro infarcts ensue and are evidenced by areas of low uptake on FDG-PET scans in patients with IVLBCL.7 These characteristic patterns seen on FDG-PET scans can help to support a diagnosis of IVLBCL when clinical suspicion is high. Further, clinicians may be able to use imaging results to guide an appropriate site for biopsy to confirm diagnosis.
Conclusion
Intravascular large B-cell lymphoma remains a diagnostic challenge for clinicians. Prognosis is generally poor and likely related to frequent delays in diagnosis.1 Clinicians continue to work toward improving their ability to diagnose this disease in its early stages. New diagnostic algorithms and the use of random skin biopsies have shown some promise in improving diagnostic efficiency.26-28 Based on the authors’ experience and review of the literature, FDG-PET may be another promising tool to aid early diagnosis. Characteristic FDG-PET findings have been well described and may help to support the diagnosis of IVLBCL and guide an appropriate biopsy site when clinical suspicion for IVLBCL exists.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
Patient 1
A white man aged 67 years, with diastolic heart failure and chronic obstructive pulmonary disease, presented to the emergency department (ED) with shortness of breath. The initial laboratory results were significant for a newly elevated creatinine level of 2.06 mg/dL and a brain natriuretic peptide level of 648 pg/mL.
Imaging studies included a chest radiograph, a ventilation/perfusion scan, and an echocardiogram, as well as a right heart catheterization. All were nondiagnostic.
The patient's shortness of breath persisted despite treatment with diuretics, antibiotics, and steroids. Further laboratory workup revealed an elevated lactate dehydrogenase (LDH) level of 1,338 IU/L. A bone marrow biopsy performed because of concern about malignancy was unremarkable. Flow cytometry of the bone marrow aspirate did not reveal clonal B- or T-cell populations. Immunohistochemical staining was not performed. During this hospitalization for shortness of breath, the patient's mental staus began to decline, and his oxygen requirements increased. The patient was intubated but expired 48 hours after mechanical ventilation was initiated.
Patient 2
A white woman aged 67 years presented to the ED with generalized weakness, fatigue, and nausea. The patient’s medical history was significant for a diagnosis of stage IIIa ovarian cancer. She was treated with surgical resection and completed 6 cycles of adjuvant carboplatin and paclitaxel 3 months prior to this presentation. She had good response to treatment with normalization of CA-125.
After completion of chemotherapy, the patient was found to have persistent anemia and thrombocytopenia. Admission laboratory results were significant for a hemoglobin level of 8.4 g/dL, a platelet count of 20,000/μL, and an LDH level of 1,220 IU/L.
Chest, abdomen, and pelvis CT scans showed mesenteric adenopathy and splenomegaly (Figures 3A, 3B, and 3C) compared with prior imaging. Bone marrow biopsy revealed large lymphoid cells with scant cytoplasm and irregular nuclei, primarily within blood vessels and sinusoids consistent with IVLBCL (Figure 4). Flow cytometry of the bone marrow specimen showed an abnormal B-cell population with expression CD20, CD19, FMC-7, and dim κ light chain restriction. The cells were negative for CD5 and CD10. Immunohistochemical staining was positive for CD20, CD79a, PAX5, BCL-2 , and MUM1.
The patient was treated with 4 cycles of cyclophosphamide, doxorubicin, vincristine, prednisone, and rituximab, plus intrathecal methotrexate. The chemotherapy dose was reduced in the final cycle because of neuropathy in the hands and feet. The patient had undergone autologous stem-cell transplantation to allow high-dose chemotherapy. She was doing well more than 5 months after her transplant without evidence of recurrent disease.
Patient 3
A white man aged 76 years presented to the ED with cutaneous nodules, weight loss, fatigue, fevers, and epigastric pain. The patient’s medical history was significant for asymptomatic lymphoplasmacytic lymphoma diagnosed 2 months earlier, which had not required treatment. Laboratory results on admission revealed transaminitis, mild anemia with a hemoglobin level of 11 g/dL, and LDH level of 497 IU/L.
Chest, abdomen, and pelvis CT scans showed a 1.7cm hepatic lesion and mesenteric adenopathy. A bone marrow biopsy was unchanged from prior studies and showed minimal involvement (5%) of marrow space by low grade B-cell lymphoma.
Fluorodeoxyglucose-positron emission tomography (FDG-PET) scans showed multiple areas of uptake in the neck, chest, abdomen, and pelvis (Figures 5 and 6). No increased uptake in the subcutaneous nodules was noted on examination. Laparoscopic biopsy of FDG-avid mesenteric nodes showed clusters of atypical large lymphoid cells resulting in distention of the vascular lumina, resulting in the diagnosis of IVLBCL (Figure 7).
Immunohistochemical stains showed that the intravascular lymphocytes were strongly positive for CD20 and BCL-2 and negative for CD5 and CD10. Flow cytometry on the sample was limited by a low cell count and could not be assessed for clonality. The patient completed 6 cycles of rituximab as well as intrathecal methotrexate. Restaging studies showed a complete remission.
Two months later, the patient developed a skin nodule on the right shoulder. A repeat FDG-PET scan showed increased uptake, and fine-needle biopsy confirmed recurrent disease. The patient is undergoing treatment with ifosfamide, carboplatin, etoposide, and rituximab, as well as workup for autologous stem-cell transplant.
Discussion
Intravascular large B-cell lymphoma, a subtype of diffuse large B-cell lymphoma, is unique because it is primarily extranodal and typically without significant tumor burden.1-4 Standard imaging modalities, therefore, are often nonspecific and do not aid clinicians in establishing a diagnosis. Fluorodeoxyglucose-positron emission tomography has a known role in the assessment of diffuse large B-cell lymphoma, both at time of diagnosis and in monitoring response to treatment.5 However, the use of FDGPET in the diagnosis and management of IVLBCL has not been clearly established.
In a review of the literature, 26 English-language case reports and small case series reporting individual centers’ experience with the use of this imaging modality in the diagnosis of IVLBCL were identified. Two cases were eliminated from review because they did not discuss the use of FDG-PET in relationship to diagnosis. Of the remaining 24 cases, 21 underwent initial imaging with 1 or more of the following imaging modalities: CT, magnetic resonance, ultrasound, bone scan, and gallium scintigraphy, all of which were nonspecific and did not lead to a definitive diagnosis.3,6-25 Each of the 21 cases was followed up by FDG-PET; in 19, the FDG-PET scan was positive and resulted in a diagnosis of IVLBCL. In 2 cases, the FDG-PET scan was nonrevealing and was not considered helpful in diagnosis.11,18 In 3 of the 21 cases, the FDG-PET scan was the primary imaging modality.6,14,25
In this review, all 3 patients had initial imaging with CT scans of anatomic locations that were largely unrevealing, although later histologic examination showed them to be locations of active disease either by biopsy or on autopsy. One patient who underwent early FDG-PET was found to have increased uptake in the mesenteric lymph nodes, which were later biopsied, as well as uptake in the bilateral adrenal glands, lungs, and bone.
Several characteristic FDG-PET findings that have been described in the literature have been identified in patients with IVLBCL, including diffuse accumulation in bilateral lung fields, accumulation in the renal cortex or adrenal glands, diffuse bony involvement, and hypometabolism in the brain.7,10,12,13,17,23,25 These findings show that organs with the richest blood supply, specifically the lungs and kidneys, often are affected. The brain, an obligate glucose metabolizer, would be expected to have high uptake; however, with tumor thrombi occluding small intracranial vessels, micro infarcts ensue and are evidenced by areas of low uptake on FDG-PET scans in patients with IVLBCL.7 These characteristic patterns seen on FDG-PET scans can help to support a diagnosis of IVLBCL when clinical suspicion is high. Further, clinicians may be able to use imaging results to guide an appropriate site for biopsy to confirm diagnosis.
Conclusion
Intravascular large B-cell lymphoma remains a diagnostic challenge for clinicians. Prognosis is generally poor and likely related to frequent delays in diagnosis.1 Clinicians continue to work toward improving their ability to diagnose this disease in its early stages. New diagnostic algorithms and the use of random skin biopsies have shown some promise in improving diagnostic efficiency.26-28 Based on the authors’ experience and review of the literature, FDG-PET may be another promising tool to aid early diagnosis. Characteristic FDG-PET findings have been well described and may help to support the diagnosis of IVLBCL and guide an appropriate biopsy site when clinical suspicion for IVLBCL exists.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
Patient 1
A white man aged 67 years, with diastolic heart failure and chronic obstructive pulmonary disease, presented to the emergency department (ED) with shortness of breath. The initial laboratory results were significant for a newly elevated creatinine level of 2.06 mg/dL and a brain natriuretic peptide level of 648 pg/mL.
Imaging studies included a chest radiograph, a ventilation/perfusion scan, and an echocardiogram, as well as a right heart catheterization. All were nondiagnostic.
The patient's shortness of breath persisted despite treatment with diuretics, antibiotics, and steroids. Further laboratory workup revealed an elevated lactate dehydrogenase (LDH) level of 1,338 IU/L. A bone marrow biopsy performed because of concern about malignancy was unremarkable. Flow cytometry of the bone marrow aspirate did not reveal clonal B- or T-cell populations. Immunohistochemical staining was not performed. During this hospitalization for shortness of breath, the patient's mental staus began to decline, and his oxygen requirements increased. The patient was intubated but expired 48 hours after mechanical ventilation was initiated.
Patient 2
A white woman aged 67 years presented to the ED with generalized weakness, fatigue, and nausea. The patient’s medical history was significant for a diagnosis of stage IIIa ovarian cancer. She was treated with surgical resection and completed 6 cycles of adjuvant carboplatin and paclitaxel 3 months prior to this presentation. She had good response to treatment with normalization of CA-125.
After completion of chemotherapy, the patient was found to have persistent anemia and thrombocytopenia. Admission laboratory results were significant for a hemoglobin level of 8.4 g/dL, a platelet count of 20,000/μL, and an LDH level of 1,220 IU/L.
Chest, abdomen, and pelvis CT scans showed mesenteric adenopathy and splenomegaly (Figures 3A, 3B, and 3C) compared with prior imaging. Bone marrow biopsy revealed large lymphoid cells with scant cytoplasm and irregular nuclei, primarily within blood vessels and sinusoids consistent with IVLBCL (Figure 4). Flow cytometry of the bone marrow specimen showed an abnormal B-cell population with expression CD20, CD19, FMC-7, and dim κ light chain restriction. The cells were negative for CD5 and CD10. Immunohistochemical staining was positive for CD20, CD79a, PAX5, BCL-2 , and MUM1.
The patient was treated with 4 cycles of cyclophosphamide, doxorubicin, vincristine, prednisone, and rituximab, plus intrathecal methotrexate. The chemotherapy dose was reduced in the final cycle because of neuropathy in the hands and feet. The patient had undergone autologous stem-cell transplantation to allow high-dose chemotherapy. She was doing well more than 5 months after her transplant without evidence of recurrent disease.
Patient 3
A white man aged 76 years presented to the ED with cutaneous nodules, weight loss, fatigue, fevers, and epigastric pain. The patient’s medical history was significant for asymptomatic lymphoplasmacytic lymphoma diagnosed 2 months earlier, which had not required treatment. Laboratory results on admission revealed transaminitis, mild anemia with a hemoglobin level of 11 g/dL, and LDH level of 497 IU/L.
Chest, abdomen, and pelvis CT scans showed a 1.7cm hepatic lesion and mesenteric adenopathy. A bone marrow biopsy was unchanged from prior studies and showed minimal involvement (5%) of marrow space by low grade B-cell lymphoma.
Fluorodeoxyglucose-positron emission tomography (FDG-PET) scans showed multiple areas of uptake in the neck, chest, abdomen, and pelvis (Figures 5 and 6). No increased uptake in the subcutaneous nodules was noted on examination. Laparoscopic biopsy of FDG-avid mesenteric nodes showed clusters of atypical large lymphoid cells resulting in distention of the vascular lumina, resulting in the diagnosis of IVLBCL (Figure 7).
Immunohistochemical stains showed that the intravascular lymphocytes were strongly positive for CD20 and BCL-2 and negative for CD5 and CD10. Flow cytometry on the sample was limited by a low cell count and could not be assessed for clonality. The patient completed 6 cycles of rituximab as well as intrathecal methotrexate. Restaging studies showed a complete remission.
Two months later, the patient developed a skin nodule on the right shoulder. A repeat FDG-PET scan showed increased uptake, and fine-needle biopsy confirmed recurrent disease. The patient is undergoing treatment with ifosfamide, carboplatin, etoposide, and rituximab, as well as workup for autologous stem-cell transplant.
Discussion
Intravascular large B-cell lymphoma, a subtype of diffuse large B-cell lymphoma, is unique because it is primarily extranodal and typically without significant tumor burden.1-4 Standard imaging modalities, therefore, are often nonspecific and do not aid clinicians in establishing a diagnosis. Fluorodeoxyglucose-positron emission tomography has a known role in the assessment of diffuse large B-cell lymphoma, both at time of diagnosis and in monitoring response to treatment.5 However, the use of FDGPET in the diagnosis and management of IVLBCL has not been clearly established.
In a review of the literature, 26 English-language case reports and small case series reporting individual centers’ experience with the use of this imaging modality in the diagnosis of IVLBCL were identified. Two cases were eliminated from review because they did not discuss the use of FDG-PET in relationship to diagnosis. Of the remaining 24 cases, 21 underwent initial imaging with 1 or more of the following imaging modalities: CT, magnetic resonance, ultrasound, bone scan, and gallium scintigraphy, all of which were nonspecific and did not lead to a definitive diagnosis.3,6-25 Each of the 21 cases was followed up by FDG-PET; in 19, the FDG-PET scan was positive and resulted in a diagnosis of IVLBCL. In 2 cases, the FDG-PET scan was nonrevealing and was not considered helpful in diagnosis.11,18 In 3 of the 21 cases, the FDG-PET scan was the primary imaging modality.6,14,25
In this review, all 3 patients had initial imaging with CT scans of anatomic locations that were largely unrevealing, although later histologic examination showed them to be locations of active disease either by biopsy or on autopsy. One patient who underwent early FDG-PET was found to have increased uptake in the mesenteric lymph nodes, which were later biopsied, as well as uptake in the bilateral adrenal glands, lungs, and bone.
Several characteristic FDG-PET findings that have been described in the literature have been identified in patients with IVLBCL, including diffuse accumulation in bilateral lung fields, accumulation in the renal cortex or adrenal glands, diffuse bony involvement, and hypometabolism in the brain.7,10,12,13,17,23,25 These findings show that organs with the richest blood supply, specifically the lungs and kidneys, often are affected. The brain, an obligate glucose metabolizer, would be expected to have high uptake; however, with tumor thrombi occluding small intracranial vessels, micro infarcts ensue and are evidenced by areas of low uptake on FDG-PET scans in patients with IVLBCL.7 These characteristic patterns seen on FDG-PET scans can help to support a diagnosis of IVLBCL when clinical suspicion is high. Further, clinicians may be able to use imaging results to guide an appropriate site for biopsy to confirm diagnosis.
Conclusion
Intravascular large B-cell lymphoma remains a diagnostic challenge for clinicians. Prognosis is generally poor and likely related to frequent delays in diagnosis.1 Clinicians continue to work toward improving their ability to diagnose this disease in its early stages. New diagnostic algorithms and the use of random skin biopsies have shown some promise in improving diagnostic efficiency.26-28 Based on the authors’ experience and review of the literature, FDG-PET may be another promising tool to aid early diagnosis. Characteristic FDG-PET findings have been well described and may help to support the diagnosis of IVLBCL and guide an appropriate biopsy site when clinical suspicion for IVLBCL exists.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
γ-δ T-Cell Lymphoma With Disseminated Intravascular Coagulation and Autoimmune Hemolytic Anemia
Gamma-delta (γ-δ) T-cell lymphomas (GDTCL) are rare and aggressive cancers with specific morphologic, phenotypic, and functional properties. When discovered in 1984, the T-cell receptor (TCR) was characterized as an alpha-beta (α-β) heterodimer. The γ-δ heterodimer was discovered later, when a third rearranging gene was recognized.1
Gaulard and colleagues described the first case of peripheral neoplasm with the γ-δ TCR.2 Now the present authors report the case of a patient with an autoimmune hemolytic anemia (AIHA) with both cold and warm antibodies—an atypical presentation of this rare form of TCL. Such a case has not been previously reported.
Clinical History
A 77-year-old woman with a past medical history of osteoarthritis, gout, mitral stenosis, bioprosthetic aortic valve replacement, and obesity presented to the emergency department (ED) reporting progressive weakness, confusion, and jaundice. She had been recently discharged from
another hospital after an 18-day stay for gangrenous cholecystitis and shingles. Her home medications were metronidazole and acyclovir. In the ED, she was febrile at 100.5°. Laboratory test results revealed anemia with a hemoglobin level of 50 g/L (83 g/L in clinic 2 weeks earlier) and neutropenia with an absolute neutrophilic count of 500 cells/μL (normal range 1,520-6,370 cells/μL). She also was thrombocytopenic with a platelet count of 71x109/L (normal range 150-450×109/L).
On admission, the hematology service was consulted for pancytopenia. The pertinent workup included a lactate dehydrogenase level of 31.16 μkat/L (normal range 1.7-3.4 μkat/L), a haptoglobin level of < 1,500 mg/L (normal range 260-1,850 mg/L), and a direct bilirubin level of 13.68 μmol/L (normal range 1.7-5.1 μmol/L). A peripheral blood smear was negative for schistocytes. Fibrin split products were 40 mg/L (normal < 10 mg/L), fibrinogen level was 6.94 μmol/L (normal range 5.8-11.8 μmol/L), prothrombin time was 14.6 seconds (normal range 10-14 sec), and international normalized ratio was 1.3 (normal < 1). The concomitant decrease in fibrinogen level and increase in fibrin split product titers were consistent with the diagnosis of acute disseminated intravascular coagulation. Iron studies were consistent with anemia of chronic disease (low reticulocyte count of 0.4%) and vitamin B12 deficiency (level 195). Coombs test results were positive for both cold and warm antibodies, with cold being more prominent. Abdominal ultrasonography revealed hepatosplenomegaly (HSM).
The patient was diagnosed with AIHA with no initial obvious underlying etiology. The differential diagnosis included autoimmune disorder, lymphoproliferative disease, and drug-induced process. She also was diagnosed with sepsis, which was thought to be contributing to the pancytopenia.
Broad-spectrum antibiotics (cefepime, metronidazole) and vitamin B12 supplements were started. After a blood transfusion, the patient developed fever and hypoxia, which required transfer to the medical intensive care unit. The differentials at this time included a transfusion reaction and/or transfusion-associated circulatory overload. Intravenous immunoglobulin was started at 1 g/kg to help with cold agglutinins. Prednisone 1 mg/kg was started as well. Peripheral blood flow cytometry results were positive for an abnormal T-cell population likely consistent with T-cell lineage lymphoma. Bone marrow biopsy results were consistent with GDTCL. Computed tomography (CT) of chest/abdomen/pelvis showed bilateral lung nodules < 1 cm, HSM with multiple spleen infarcts, and a 4.7-cm right adnexal soft-tissue lesion. Liver biopsy results were consistent with GDTCL. Results of a workup for cytomegalovirus and Epstein-Barr virus were negative, as was a mycoplasma screen. The patient was diagnosed with GDTCL with hepatic involvement, and CHOP (cyclophosphamide, hydroxydaunorubicin [doxorubicin], Oncovin [vincristine], prednisone) therapy was started.
Discussion
Peripheral TCL (PTCL) are a rare, typically extranodal group of malignancies. They are aggressive and generally have a poor outcome, with most patients dying of lymphoma within 2 years.3 T-cell lymphomas most commonly express the γ-δ TCR. About 2% to 4% of TCLs express the γ-δ TCR.4 In 2008, the World Health Organization recognized 2 distinct GDTCL subgroups: hepatosplenic GDTCL (HSGDTCL) and primary cutaneous GDTCL.5 As the patient presented with hepatic involvement, this discussion focused on HSGDTCL.
Hepatosplenic GDTCL are rare types of PTCL. First described as a separate TCL subgroup in the 1990 REAL (Revised European-American Lymphoma) classification,6 they are estimated to represent about 1.4% of all TCL, with about 100 cases reported in the literature.4
The GDTCL cells tend to live in mucosa, lymphoid tissue, epithelial-rich tissues (skin, gastrointestinal tract), and red pulp of spleen.7 They develop from thymic precursors in bone marrow and are CD4-/CD8- and thus known as double negative cells.8 They mimic natural killer cells, behave as cytotoxic cells, and are capable of TCR rearrangement as well as phagocytosis.9
Hepatosplenic GDTCL are usually phenotypically CD2+, CD3+, CD4-, CD5-, CD7+, CD8-, and TCR γ-δ+.10 They are rarely associated with Epstein-Barr virus infection; reported cases seem more common in Asia.11 Peak incidence is in young men (median age 20-25 years; male:female ratio 10:1). At-risk populations include the chronically immunosuppressed, including solid organ transplanted patients and patients under prolonged antigenic stimulation.12
The most common clinical features of HSGDTCL include B symptoms (fever of unknown origin, night sweats, loss of > 10% of body weight), marked HSM, and lack of lymphadenopathy. Patients often present with fever, weakness, and abdominal pain. Laboratory test results
typically show abnormal liver function and abnormal lactate dehydrogenase levels. Bone marrow is almost always involved, with possible trilineage cytopenia. Anemia and thrombocytopenia are reported in 75% and 85% of cases, respectively.13
Warm (70%) and cold auto-antibodies are the 2 classifications of AIHA.14 The AIHA can be primary, idiopathic, or a manifestation of underlying disease conditions, including non-Hodgkin lymphomas, systemic autoimmune diseases, chronic infections, postorgan transplantation, and solid tumors. It has also been reported as a complication of treatment with nucleoside analogues.15
Lacking specific symptoms, HSGDTCL is usually diagnosed late. The diagnosis should be suspected in young men who present with the aforementioned symptoms. However, not everyone with HSGDTCL falls in that group—the present patient was a 77-year-old woman.
Hepatosplenic GDTCL staging is similar to staging of other non-Hodgkin lymphomas. Total-body CT with contrast, bone marrow aspiration/biopsy, and direct lesion biopsy are required. Although positron emission tomography is generally thought to be as useful in TCL as in B-cell lymphomas, there is not enough evidence to support its use specifically in HSGDTCL.16 The staging classification follows the Ann Arbor system, with the majority of cases classified as stage IV.
Hepatosplenic GDTCL are aggressive tumors with a strong tendency to rapidly progress, and they are highly resistant to primary chemotherapy agents. Remission is rarely complete with use of conventional chemotherapy agents. Most patients die of the disease within 2 years of
diagnosis.12 Although the rarity of HSGDTCL has made it difficult to identify any clear prognostic factors, a correlation between thrombocytopenia severity and disease progression has been found in many studies.17 There is no standard treatment regimen. Proposed therapies
include splenectomy (for diagnosis or thrombocytopenia management), corticosteroids, alkylating agents, purine analogue, anthracycline-containing regimens, and cytarabine/cisplatin combinations. The anthracycline-based regimen most commonly used as first-line therapy is CHOP, or CHOP derivatives, with complete remission rates between 30% and 45%. However, long-term results remain disappointing (median relapse time 4 months).10 In 3 reviews, median survival was 16 months, 11 months, and 9.5 months.10,17,18 In the International T-Cell Lymphoma Project study, the 5-year failure-free survival rate was 0%, and the overall survival rate was 7%.4 In these studies, the majority of patients received some variation of CHOP-based therapy, and although positive responses were appreciated in many of the cases, they were generally short-lived.
These results have been disappointing, and other modalities have been tried—including high-dose cytarabine regimens, 2'-deoxycoformycin (pentostatin), and anti-CD52 monoclonal antibodies (alemtuzumab).19 In an HSGDTCL study, 2 of 21 patients treated with platinum/cytarabine-based induction regimens were still in remission at 42 and 52 months.17 Another study examined a variety of induction regimens used to treat HSGDTCL in 15 patients.18 Responses tended to be more durable in patients who received a dose-intense Hyper-CVIDDoxil regimen (fractionated cyclophosphamide, liposomal doxorubicin, vincristine, dexamethasone) alternated with methotrexate and cytarabine. Complete response was 50%, and median duration of complete response was 8 months. Over the past 10 years, a few case reports have described successful treatment with autologous or allogeneic stem cell transplantation.20
Conclusion
The present case represents a unique HSGDTCL presentation. To the authors’ knowledge, this is the first report of HSGDTCL presenting with acute disseminated intravascular coagulation and AIHA with both cold and warm antibodies.
Hepatosplenic GDTCL is a rare, novel disease. To understand more about this pathology, investigators need to better characterize the disease process and the manifestations. The hope is that more information will contribute to the development of more effective therapies. The unique presentation reported here may help in further characterizing and understanding this uncommon disease.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies.
Click here to read the digital edition.
1. Saito H, Kranz DM, Takagaki Y, Hayday AC, Eisen HN, Tonegawa S. A third rearranged and expressed gene in a clone of cytotoxic T lymphocytes. Nature. 1984;312(5989):36-40.
2. Gaulard P, Zafrani ES, Mavier P, et al. Peripheral T-cell lymphoma presenting as predominant liver disease: a report of three cases. Hepatology. 1986;6(5):864-868.
3. Gaulard P, de Leval L. Pathology of peripheral T-cell lymphomas: where do we stand? Semin Hematol. 2014;51(1):5-16.
4. Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124-4130.
5. The International Agency for Research on Cancer. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Vol 2. 4th ed. Lyon, France: IARC Press; 2008.
6. Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasm: a proposal from the International Lymphoma Study Group. Blood. 1994;84(5):1361-1392.
7. Farcet JP, Gaulard P, Marolleau JP, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75(11):2213-2219.
8. Bluestone JA, Khattri R, Sciammas R, Sperling AI. TCR gamma delta cells: a specialized T-cell subset in the immune system. Annu Rev Cell Dev Biol. 1995;11:307-353.
9. Holtmeier W, Kabelitz D. Gamma delta T cells link innate and adaptive immune responses. Chem Immunol Allergy. 2005;86:151-183.
10. Weidmann E. Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia. 2000;14(6):991-997.
11. Yu WW, Hsieh PP, Chuang SS. Cutaneous EBV-positive γδ T-cell lymphoma vs. extranodal NK/T-cell lymphoma: a case report and literature review. J Cutan Pathol. 2013;40(3):310-316.
12. Tripodo C, Iannitto E, Florena AM, et al. Gamma-delta T-cell lymphomas. Nat Rev Clin Oncol. 2009;6(12):707-717.
13. Foppoli M, Ferreri AJM. Gamma-delta T-cell lymphomas. Eur J Haematol. 2015;94(3):206-218.
14. Hoffbrand AV, Catovsky D, Tuddenham EGD, Green AR, eds. Postgraduate Haematology. 6th ed. Oxford, England: Wiley-Blackwell; 2011.
15. Valent P, Lechner K. Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr. 2008;120(5-6):136-151.
16. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621.
17. Belhadj K, Reyes F, Farcet JP, et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. 2003;102(13):4261-4269.
18. Falchook GS, Vega F, Dang NH, et al. Hepatosplenic gamma-delta T-cell lymphoma: clinicopathological features and treatment. Ann Oncol. 2009;20(6):1080-1085.
19. Konuma T, Ooi J, Takahashi S, et al. Allogeneic stem cell transplantation for hepatosplenic
gammadelta T-cell lymphoma. Leuk Lymphoma. 2007;48(3):630-632.
20. Ferreri AJ, Govi S, Pileri SA. Hepatosplenic gamma-delta T-cell lymphoma. Crit
Rev Oncol Hematol. 2012;83(2):283-292.
Gamma-delta (γ-δ) T-cell lymphomas (GDTCL) are rare and aggressive cancers with specific morphologic, phenotypic, and functional properties. When discovered in 1984, the T-cell receptor (TCR) was characterized as an alpha-beta (α-β) heterodimer. The γ-δ heterodimer was discovered later, when a third rearranging gene was recognized.1
Gaulard and colleagues described the first case of peripheral neoplasm with the γ-δ TCR.2 Now the present authors report the case of a patient with an autoimmune hemolytic anemia (AIHA) with both cold and warm antibodies—an atypical presentation of this rare form of TCL. Such a case has not been previously reported.
Clinical History
A 77-year-old woman with a past medical history of osteoarthritis, gout, mitral stenosis, bioprosthetic aortic valve replacement, and obesity presented to the emergency department (ED) reporting progressive weakness, confusion, and jaundice. She had been recently discharged from
another hospital after an 18-day stay for gangrenous cholecystitis and shingles. Her home medications were metronidazole and acyclovir. In the ED, she was febrile at 100.5°. Laboratory test results revealed anemia with a hemoglobin level of 50 g/L (83 g/L in clinic 2 weeks earlier) and neutropenia with an absolute neutrophilic count of 500 cells/μL (normal range 1,520-6,370 cells/μL). She also was thrombocytopenic with a platelet count of 71x109/L (normal range 150-450×109/L).
On admission, the hematology service was consulted for pancytopenia. The pertinent workup included a lactate dehydrogenase level of 31.16 μkat/L (normal range 1.7-3.4 μkat/L), a haptoglobin level of < 1,500 mg/L (normal range 260-1,850 mg/L), and a direct bilirubin level of 13.68 μmol/L (normal range 1.7-5.1 μmol/L). A peripheral blood smear was negative for schistocytes. Fibrin split products were 40 mg/L (normal < 10 mg/L), fibrinogen level was 6.94 μmol/L (normal range 5.8-11.8 μmol/L), prothrombin time was 14.6 seconds (normal range 10-14 sec), and international normalized ratio was 1.3 (normal < 1). The concomitant decrease in fibrinogen level and increase in fibrin split product titers were consistent with the diagnosis of acute disseminated intravascular coagulation. Iron studies were consistent with anemia of chronic disease (low reticulocyte count of 0.4%) and vitamin B12 deficiency (level 195). Coombs test results were positive for both cold and warm antibodies, with cold being more prominent. Abdominal ultrasonography revealed hepatosplenomegaly (HSM).
The patient was diagnosed with AIHA with no initial obvious underlying etiology. The differential diagnosis included autoimmune disorder, lymphoproliferative disease, and drug-induced process. She also was diagnosed with sepsis, which was thought to be contributing to the pancytopenia.
Broad-spectrum antibiotics (cefepime, metronidazole) and vitamin B12 supplements were started. After a blood transfusion, the patient developed fever and hypoxia, which required transfer to the medical intensive care unit. The differentials at this time included a transfusion reaction and/or transfusion-associated circulatory overload. Intravenous immunoglobulin was started at 1 g/kg to help with cold agglutinins. Prednisone 1 mg/kg was started as well. Peripheral blood flow cytometry results were positive for an abnormal T-cell population likely consistent with T-cell lineage lymphoma. Bone marrow biopsy results were consistent with GDTCL. Computed tomography (CT) of chest/abdomen/pelvis showed bilateral lung nodules < 1 cm, HSM with multiple spleen infarcts, and a 4.7-cm right adnexal soft-tissue lesion. Liver biopsy results were consistent with GDTCL. Results of a workup for cytomegalovirus and Epstein-Barr virus were negative, as was a mycoplasma screen. The patient was diagnosed with GDTCL with hepatic involvement, and CHOP (cyclophosphamide, hydroxydaunorubicin [doxorubicin], Oncovin [vincristine], prednisone) therapy was started.
Discussion
Peripheral TCL (PTCL) are a rare, typically extranodal group of malignancies. They are aggressive and generally have a poor outcome, with most patients dying of lymphoma within 2 years.3 T-cell lymphomas most commonly express the γ-δ TCR. About 2% to 4% of TCLs express the γ-δ TCR.4 In 2008, the World Health Organization recognized 2 distinct GDTCL subgroups: hepatosplenic GDTCL (HSGDTCL) and primary cutaneous GDTCL.5 As the patient presented with hepatic involvement, this discussion focused on HSGDTCL.
Hepatosplenic GDTCL are rare types of PTCL. First described as a separate TCL subgroup in the 1990 REAL (Revised European-American Lymphoma) classification,6 they are estimated to represent about 1.4% of all TCL, with about 100 cases reported in the literature.4
The GDTCL cells tend to live in mucosa, lymphoid tissue, epithelial-rich tissues (skin, gastrointestinal tract), and red pulp of spleen.7 They develop from thymic precursors in bone marrow and are CD4-/CD8- and thus known as double negative cells.8 They mimic natural killer cells, behave as cytotoxic cells, and are capable of TCR rearrangement as well as phagocytosis.9
Hepatosplenic GDTCL are usually phenotypically CD2+, CD3+, CD4-, CD5-, CD7+, CD8-, and TCR γ-δ+.10 They are rarely associated with Epstein-Barr virus infection; reported cases seem more common in Asia.11 Peak incidence is in young men (median age 20-25 years; male:female ratio 10:1). At-risk populations include the chronically immunosuppressed, including solid organ transplanted patients and patients under prolonged antigenic stimulation.12
The most common clinical features of HSGDTCL include B symptoms (fever of unknown origin, night sweats, loss of > 10% of body weight), marked HSM, and lack of lymphadenopathy. Patients often present with fever, weakness, and abdominal pain. Laboratory test results
typically show abnormal liver function and abnormal lactate dehydrogenase levels. Bone marrow is almost always involved, with possible trilineage cytopenia. Anemia and thrombocytopenia are reported in 75% and 85% of cases, respectively.13
Warm (70%) and cold auto-antibodies are the 2 classifications of AIHA.14 The AIHA can be primary, idiopathic, or a manifestation of underlying disease conditions, including non-Hodgkin lymphomas, systemic autoimmune diseases, chronic infections, postorgan transplantation, and solid tumors. It has also been reported as a complication of treatment with nucleoside analogues.15
Lacking specific symptoms, HSGDTCL is usually diagnosed late. The diagnosis should be suspected in young men who present with the aforementioned symptoms. However, not everyone with HSGDTCL falls in that group—the present patient was a 77-year-old woman.
Hepatosplenic GDTCL staging is similar to staging of other non-Hodgkin lymphomas. Total-body CT with contrast, bone marrow aspiration/biopsy, and direct lesion biopsy are required. Although positron emission tomography is generally thought to be as useful in TCL as in B-cell lymphomas, there is not enough evidence to support its use specifically in HSGDTCL.16 The staging classification follows the Ann Arbor system, with the majority of cases classified as stage IV.
Hepatosplenic GDTCL are aggressive tumors with a strong tendency to rapidly progress, and they are highly resistant to primary chemotherapy agents. Remission is rarely complete with use of conventional chemotherapy agents. Most patients die of the disease within 2 years of
diagnosis.12 Although the rarity of HSGDTCL has made it difficult to identify any clear prognostic factors, a correlation between thrombocytopenia severity and disease progression has been found in many studies.17 There is no standard treatment regimen. Proposed therapies
include splenectomy (for diagnosis or thrombocytopenia management), corticosteroids, alkylating agents, purine analogue, anthracycline-containing regimens, and cytarabine/cisplatin combinations. The anthracycline-based regimen most commonly used as first-line therapy is CHOP, or CHOP derivatives, with complete remission rates between 30% and 45%. However, long-term results remain disappointing (median relapse time 4 months).10 In 3 reviews, median survival was 16 months, 11 months, and 9.5 months.10,17,18 In the International T-Cell Lymphoma Project study, the 5-year failure-free survival rate was 0%, and the overall survival rate was 7%.4 In these studies, the majority of patients received some variation of CHOP-based therapy, and although positive responses were appreciated in many of the cases, they were generally short-lived.
These results have been disappointing, and other modalities have been tried—including high-dose cytarabine regimens, 2'-deoxycoformycin (pentostatin), and anti-CD52 monoclonal antibodies (alemtuzumab).19 In an HSGDTCL study, 2 of 21 patients treated with platinum/cytarabine-based induction regimens were still in remission at 42 and 52 months.17 Another study examined a variety of induction regimens used to treat HSGDTCL in 15 patients.18 Responses tended to be more durable in patients who received a dose-intense Hyper-CVIDDoxil regimen (fractionated cyclophosphamide, liposomal doxorubicin, vincristine, dexamethasone) alternated with methotrexate and cytarabine. Complete response was 50%, and median duration of complete response was 8 months. Over the past 10 years, a few case reports have described successful treatment with autologous or allogeneic stem cell transplantation.20
Conclusion
The present case represents a unique HSGDTCL presentation. To the authors’ knowledge, this is the first report of HSGDTCL presenting with acute disseminated intravascular coagulation and AIHA with both cold and warm antibodies.
Hepatosplenic GDTCL is a rare, novel disease. To understand more about this pathology, investigators need to better characterize the disease process and the manifestations. The hope is that more information will contribute to the development of more effective therapies. The unique presentation reported here may help in further characterizing and understanding this uncommon disease.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies.
Click here to read the digital edition.
Gamma-delta (γ-δ) T-cell lymphomas (GDTCL) are rare and aggressive cancers with specific morphologic, phenotypic, and functional properties. When discovered in 1984, the T-cell receptor (TCR) was characterized as an alpha-beta (α-β) heterodimer. The γ-δ heterodimer was discovered later, when a third rearranging gene was recognized.1
Gaulard and colleagues described the first case of peripheral neoplasm with the γ-δ TCR.2 Now the present authors report the case of a patient with an autoimmune hemolytic anemia (AIHA) with both cold and warm antibodies—an atypical presentation of this rare form of TCL. Such a case has not been previously reported.
Clinical History
A 77-year-old woman with a past medical history of osteoarthritis, gout, mitral stenosis, bioprosthetic aortic valve replacement, and obesity presented to the emergency department (ED) reporting progressive weakness, confusion, and jaundice. She had been recently discharged from
another hospital after an 18-day stay for gangrenous cholecystitis and shingles. Her home medications were metronidazole and acyclovir. In the ED, she was febrile at 100.5°. Laboratory test results revealed anemia with a hemoglobin level of 50 g/L (83 g/L in clinic 2 weeks earlier) and neutropenia with an absolute neutrophilic count of 500 cells/μL (normal range 1,520-6,370 cells/μL). She also was thrombocytopenic with a platelet count of 71x109/L (normal range 150-450×109/L).
On admission, the hematology service was consulted for pancytopenia. The pertinent workup included a lactate dehydrogenase level of 31.16 μkat/L (normal range 1.7-3.4 μkat/L), a haptoglobin level of < 1,500 mg/L (normal range 260-1,850 mg/L), and a direct bilirubin level of 13.68 μmol/L (normal range 1.7-5.1 μmol/L). A peripheral blood smear was negative for schistocytes. Fibrin split products were 40 mg/L (normal < 10 mg/L), fibrinogen level was 6.94 μmol/L (normal range 5.8-11.8 μmol/L), prothrombin time was 14.6 seconds (normal range 10-14 sec), and international normalized ratio was 1.3 (normal < 1). The concomitant decrease in fibrinogen level and increase in fibrin split product titers were consistent with the diagnosis of acute disseminated intravascular coagulation. Iron studies were consistent with anemia of chronic disease (low reticulocyte count of 0.4%) and vitamin B12 deficiency (level 195). Coombs test results were positive for both cold and warm antibodies, with cold being more prominent. Abdominal ultrasonography revealed hepatosplenomegaly (HSM).
The patient was diagnosed with AIHA with no initial obvious underlying etiology. The differential diagnosis included autoimmune disorder, lymphoproliferative disease, and drug-induced process. She also was diagnosed with sepsis, which was thought to be contributing to the pancytopenia.
Broad-spectrum antibiotics (cefepime, metronidazole) and vitamin B12 supplements were started. After a blood transfusion, the patient developed fever and hypoxia, which required transfer to the medical intensive care unit. The differentials at this time included a transfusion reaction and/or transfusion-associated circulatory overload. Intravenous immunoglobulin was started at 1 g/kg to help with cold agglutinins. Prednisone 1 mg/kg was started as well. Peripheral blood flow cytometry results were positive for an abnormal T-cell population likely consistent with T-cell lineage lymphoma. Bone marrow biopsy results were consistent with GDTCL. Computed tomography (CT) of chest/abdomen/pelvis showed bilateral lung nodules < 1 cm, HSM with multiple spleen infarcts, and a 4.7-cm right adnexal soft-tissue lesion. Liver biopsy results were consistent with GDTCL. Results of a workup for cytomegalovirus and Epstein-Barr virus were negative, as was a mycoplasma screen. The patient was diagnosed with GDTCL with hepatic involvement, and CHOP (cyclophosphamide, hydroxydaunorubicin [doxorubicin], Oncovin [vincristine], prednisone) therapy was started.
Discussion
Peripheral TCL (PTCL) are a rare, typically extranodal group of malignancies. They are aggressive and generally have a poor outcome, with most patients dying of lymphoma within 2 years.3 T-cell lymphomas most commonly express the γ-δ TCR. About 2% to 4% of TCLs express the γ-δ TCR.4 In 2008, the World Health Organization recognized 2 distinct GDTCL subgroups: hepatosplenic GDTCL (HSGDTCL) and primary cutaneous GDTCL.5 As the patient presented with hepatic involvement, this discussion focused on HSGDTCL.
Hepatosplenic GDTCL are rare types of PTCL. First described as a separate TCL subgroup in the 1990 REAL (Revised European-American Lymphoma) classification,6 they are estimated to represent about 1.4% of all TCL, with about 100 cases reported in the literature.4
The GDTCL cells tend to live in mucosa, lymphoid tissue, epithelial-rich tissues (skin, gastrointestinal tract), and red pulp of spleen.7 They develop from thymic precursors in bone marrow and are CD4-/CD8- and thus known as double negative cells.8 They mimic natural killer cells, behave as cytotoxic cells, and are capable of TCR rearrangement as well as phagocytosis.9
Hepatosplenic GDTCL are usually phenotypically CD2+, CD3+, CD4-, CD5-, CD7+, CD8-, and TCR γ-δ+.10 They are rarely associated with Epstein-Barr virus infection; reported cases seem more common in Asia.11 Peak incidence is in young men (median age 20-25 years; male:female ratio 10:1). At-risk populations include the chronically immunosuppressed, including solid organ transplanted patients and patients under prolonged antigenic stimulation.12
The most common clinical features of HSGDTCL include B symptoms (fever of unknown origin, night sweats, loss of > 10% of body weight), marked HSM, and lack of lymphadenopathy. Patients often present with fever, weakness, and abdominal pain. Laboratory test results
typically show abnormal liver function and abnormal lactate dehydrogenase levels. Bone marrow is almost always involved, with possible trilineage cytopenia. Anemia and thrombocytopenia are reported in 75% and 85% of cases, respectively.13
Warm (70%) and cold auto-antibodies are the 2 classifications of AIHA.14 The AIHA can be primary, idiopathic, or a manifestation of underlying disease conditions, including non-Hodgkin lymphomas, systemic autoimmune diseases, chronic infections, postorgan transplantation, and solid tumors. It has also been reported as a complication of treatment with nucleoside analogues.15
Lacking specific symptoms, HSGDTCL is usually diagnosed late. The diagnosis should be suspected in young men who present with the aforementioned symptoms. However, not everyone with HSGDTCL falls in that group—the present patient was a 77-year-old woman.
Hepatosplenic GDTCL staging is similar to staging of other non-Hodgkin lymphomas. Total-body CT with contrast, bone marrow aspiration/biopsy, and direct lesion biopsy are required. Although positron emission tomography is generally thought to be as useful in TCL as in B-cell lymphomas, there is not enough evidence to support its use specifically in HSGDTCL.16 The staging classification follows the Ann Arbor system, with the majority of cases classified as stage IV.
Hepatosplenic GDTCL are aggressive tumors with a strong tendency to rapidly progress, and they are highly resistant to primary chemotherapy agents. Remission is rarely complete with use of conventional chemotherapy agents. Most patients die of the disease within 2 years of
diagnosis.12 Although the rarity of HSGDTCL has made it difficult to identify any clear prognostic factors, a correlation between thrombocytopenia severity and disease progression has been found in many studies.17 There is no standard treatment regimen. Proposed therapies
include splenectomy (for diagnosis or thrombocytopenia management), corticosteroids, alkylating agents, purine analogue, anthracycline-containing regimens, and cytarabine/cisplatin combinations. The anthracycline-based regimen most commonly used as first-line therapy is CHOP, or CHOP derivatives, with complete remission rates between 30% and 45%. However, long-term results remain disappointing (median relapse time 4 months).10 In 3 reviews, median survival was 16 months, 11 months, and 9.5 months.10,17,18 In the International T-Cell Lymphoma Project study, the 5-year failure-free survival rate was 0%, and the overall survival rate was 7%.4 In these studies, the majority of patients received some variation of CHOP-based therapy, and although positive responses were appreciated in many of the cases, they were generally short-lived.
These results have been disappointing, and other modalities have been tried—including high-dose cytarabine regimens, 2'-deoxycoformycin (pentostatin), and anti-CD52 monoclonal antibodies (alemtuzumab).19 In an HSGDTCL study, 2 of 21 patients treated with platinum/cytarabine-based induction regimens were still in remission at 42 and 52 months.17 Another study examined a variety of induction regimens used to treat HSGDTCL in 15 patients.18 Responses tended to be more durable in patients who received a dose-intense Hyper-CVIDDoxil regimen (fractionated cyclophosphamide, liposomal doxorubicin, vincristine, dexamethasone) alternated with methotrexate and cytarabine. Complete response was 50%, and median duration of complete response was 8 months. Over the past 10 years, a few case reports have described successful treatment with autologous or allogeneic stem cell transplantation.20
Conclusion
The present case represents a unique HSGDTCL presentation. To the authors’ knowledge, this is the first report of HSGDTCL presenting with acute disseminated intravascular coagulation and AIHA with both cold and warm antibodies.
Hepatosplenic GDTCL is a rare, novel disease. To understand more about this pathology, investigators need to better characterize the disease process and the manifestations. The hope is that more information will contribute to the development of more effective therapies. The unique presentation reported here may help in further characterizing and understanding this uncommon disease.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies.
Click here to read the digital edition.
1. Saito H, Kranz DM, Takagaki Y, Hayday AC, Eisen HN, Tonegawa S. A third rearranged and expressed gene in a clone of cytotoxic T lymphocytes. Nature. 1984;312(5989):36-40.
2. Gaulard P, Zafrani ES, Mavier P, et al. Peripheral T-cell lymphoma presenting as predominant liver disease: a report of three cases. Hepatology. 1986;6(5):864-868.
3. Gaulard P, de Leval L. Pathology of peripheral T-cell lymphomas: where do we stand? Semin Hematol. 2014;51(1):5-16.
4. Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124-4130.
5. The International Agency for Research on Cancer. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Vol 2. 4th ed. Lyon, France: IARC Press; 2008.
6. Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasm: a proposal from the International Lymphoma Study Group. Blood. 1994;84(5):1361-1392.
7. Farcet JP, Gaulard P, Marolleau JP, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75(11):2213-2219.
8. Bluestone JA, Khattri R, Sciammas R, Sperling AI. TCR gamma delta cells: a specialized T-cell subset in the immune system. Annu Rev Cell Dev Biol. 1995;11:307-353.
9. Holtmeier W, Kabelitz D. Gamma delta T cells link innate and adaptive immune responses. Chem Immunol Allergy. 2005;86:151-183.
10. Weidmann E. Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia. 2000;14(6):991-997.
11. Yu WW, Hsieh PP, Chuang SS. Cutaneous EBV-positive γδ T-cell lymphoma vs. extranodal NK/T-cell lymphoma: a case report and literature review. J Cutan Pathol. 2013;40(3):310-316.
12. Tripodo C, Iannitto E, Florena AM, et al. Gamma-delta T-cell lymphomas. Nat Rev Clin Oncol. 2009;6(12):707-717.
13. Foppoli M, Ferreri AJM. Gamma-delta T-cell lymphomas. Eur J Haematol. 2015;94(3):206-218.
14. Hoffbrand AV, Catovsky D, Tuddenham EGD, Green AR, eds. Postgraduate Haematology. 6th ed. Oxford, England: Wiley-Blackwell; 2011.
15. Valent P, Lechner K. Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr. 2008;120(5-6):136-151.
16. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621.
17. Belhadj K, Reyes F, Farcet JP, et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. 2003;102(13):4261-4269.
18. Falchook GS, Vega F, Dang NH, et al. Hepatosplenic gamma-delta T-cell lymphoma: clinicopathological features and treatment. Ann Oncol. 2009;20(6):1080-1085.
19. Konuma T, Ooi J, Takahashi S, et al. Allogeneic stem cell transplantation for hepatosplenic
gammadelta T-cell lymphoma. Leuk Lymphoma. 2007;48(3):630-632.
20. Ferreri AJ, Govi S, Pileri SA. Hepatosplenic gamma-delta T-cell lymphoma. Crit
Rev Oncol Hematol. 2012;83(2):283-292.
1. Saito H, Kranz DM, Takagaki Y, Hayday AC, Eisen HN, Tonegawa S. A third rearranged and expressed gene in a clone of cytotoxic T lymphocytes. Nature. 1984;312(5989):36-40.
2. Gaulard P, Zafrani ES, Mavier P, et al. Peripheral T-cell lymphoma presenting as predominant liver disease: a report of three cases. Hepatology. 1986;6(5):864-868.
3. Gaulard P, de Leval L. Pathology of peripheral T-cell lymphomas: where do we stand? Semin Hematol. 2014;51(1):5-16.
4. Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124-4130.
5. The International Agency for Research on Cancer. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Vol 2. 4th ed. Lyon, France: IARC Press; 2008.
6. Harris NL, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasm: a proposal from the International Lymphoma Study Group. Blood. 1994;84(5):1361-1392.
7. Farcet JP, Gaulard P, Marolleau JP, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75(11):2213-2219.
8. Bluestone JA, Khattri R, Sciammas R, Sperling AI. TCR gamma delta cells: a specialized T-cell subset in the immune system. Annu Rev Cell Dev Biol. 1995;11:307-353.
9. Holtmeier W, Kabelitz D. Gamma delta T cells link innate and adaptive immune responses. Chem Immunol Allergy. 2005;86:151-183.
10. Weidmann E. Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia. 2000;14(6):991-997.
11. Yu WW, Hsieh PP, Chuang SS. Cutaneous EBV-positive γδ T-cell lymphoma vs. extranodal NK/T-cell lymphoma: a case report and literature review. J Cutan Pathol. 2013;40(3):310-316.
12. Tripodo C, Iannitto E, Florena AM, et al. Gamma-delta T-cell lymphomas. Nat Rev Clin Oncol. 2009;6(12):707-717.
13. Foppoli M, Ferreri AJM. Gamma-delta T-cell lymphomas. Eur J Haematol. 2015;94(3):206-218.
14. Hoffbrand AV, Catovsky D, Tuddenham EGD, Green AR, eds. Postgraduate Haematology. 6th ed. Oxford, England: Wiley-Blackwell; 2011.
15. Valent P, Lechner K. Diagnosis and treatment of autoimmune haemolytic anaemias in adults: a clinical review. Wien Klin Wochenschr. 2008;120(5-6):136-151.
16. Khong PL, Pang CB, Liang R, Kwong YL, Au WY. Fluorine-18 fluorodeoxyglucose positron emission tomography in mature T-cell and natural killer cell malignancies. Ann Hematol. 2008;87(8):613-621.
17. Belhadj K, Reyes F, Farcet JP, et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. 2003;102(13):4261-4269.
18. Falchook GS, Vega F, Dang NH, et al. Hepatosplenic gamma-delta T-cell lymphoma: clinicopathological features and treatment. Ann Oncol. 2009;20(6):1080-1085.
19. Konuma T, Ooi J, Takahashi S, et al. Allogeneic stem cell transplantation for hepatosplenic
gammadelta T-cell lymphoma. Leuk Lymphoma. 2007;48(3):630-632.
20. Ferreri AJ, Govi S, Pileri SA. Hepatosplenic gamma-delta T-cell lymphoma. Crit
Rev Oncol Hematol. 2012;83(2):283-292.