User login
Reviews of Research on Health-Care Acquired Infections, Glucocorticoid Therapy in COPD, and Blood-Pressure Lowering in Intracerebral Hemorrhages
In This Edition
Literature At A Glance
A guide to this month’s studies
- Perioperative SSRI use associated with adverse surgical outcomes
- Copper-surfaced rooms reduce health-care-acquired infections
- Glucocorticoid therapy for five days not inferior to 14 days for COPD exacerbation
- Patient preference for participation in medical decision-making may be associated with increased resource utilization
- Early parenteral nutrition in critically ill adults does not significantly affect mortality or infection rates
- Aggressive fluid and sodium restriction in acute decompensated heart failure did not improve outcomes
- Lower rate of pacemaker, defibrillator device-pocket hematoma without anticoagulation interruption
- Prophylactic penicillin decreased risk of recurrent leg cellulitis
- Universal ICU decolonization reduced rates of mrsa clinical isolates and bloodstream infection
- Intensive blood-pressure lowering in intracerebral hemorrhage did not reduce death or severe disability
Perioperative SSRI Use Associated with Adverse Surgical Outcomes
Clinical question: Does selective serotonin reuptake inhibitor (SSRI) use during hospitalization for surgery increase the risk of adverse perioperative outcomes?
Background: SSRIs commonly are prescribed but are associated with a small but higher risk for hemorrhage, arrhythmia, and sudden death. Single-site studies have described an association between SSRIs and adverse perioperative outcomes, but larger studies utilizing a broad range of surgical cases are lacking.
Study design: Retrospective cohort study.
Setting: Three hundred hospitals concentrated in the Southern U.S.
Synopsis: Using the “Perspective” database, this study examined 530,416 patients age >18 years undergoing major elective surgery, 72,540 (13.7%) of whom received an SSRI. Regression analysis showed patients receiving an SSRI had higher odds of mortality (OR 1.2, 95% CI [1.07-1.36]), higher odds of 30-day readmission (OR 1.22 [1.18-1.26]), and higher odds for bleeding (1.09 [1.04-1.15]). When the analysis was restricted to only patients with a diagnosis of depression, a higher risk of bleeding and readmission persisted.
This study reaffirms an association but does not establish a causal relationship between SSRI use and adverse perioperative outcomes. SSRI use may be a surrogate for other factors, including more severe mood disorders, poorer functional status, or chronic pain. Additionally, no information has been provided as to optimal duration of withholding SSRIs preoperatively. As such, it may be premature for hospitalists involved in perioperative care to modify recommendations based on this study.
Bottom line: Perioperative SSRI use is associated with an increased risk of bleeding and 30-day readmission.
Citation: Auerbach AD, Vittinghoff E, Maselli J, et al. Perioperative use of selective serotonin reuptake inhibitors and risks for adverse outcomes of surgery. JAMA Intern Med. 2013;173(12):1075-1081.
Copper-Surfaced Rooms Reduce Health-Care-Acquired Infections
Clinical question: Can copper alloy surfaces in ICU rooms lower rates of health-care-acquired infections (HAIs)?
Background: Environmental contamination is a potential source of HAIs. Copper has intrinsic broad-spectrum antimicrobial properties. This study tests the efficacy copper-surfaced items in hospital rooms have in preventing HAIs.
Study design: Randomized controlled trial.
Setting: Medical ICUs at Medical University of South Carolina and Ralph H. Johnson Veterans Affairs Medical Center in Charleston, and the Memorial Sloan Kettering Cancer Center in New York City.
Synopsis: Six hundred fifty ICU patients were randomized to receive care either in rooms with copper surfacing on commonly handled patient care objects or in traditional rooms. Patients were screened for methicillin-resistant Staphylococcus aureus (MRSA) or vancomycin-resistant enterococci (VRE) on admission. The proportion of patients that developed either an HAI and/or MRSA or VRE colonization was significantly lower among patients in rooms with the copper-surfaced items (0.071 vs. 0.128; P=0.02). The rate of HAIs alone was also lower in the rooms with the copper (0.034 vs. 0.081; P=0.013).
A potential limitation to this study is that the rooms with copper items appeared different than traditional rooms, and therefore might have changed the behavior of health-care workers. Further, it is unclear how much copper surfacing would be necessary on general wards, where patients are more mobile. Still, HAIs are associated with longer lengths of stay and higher 30-day readmission rates, so these encouraging results warrant additional investigation into antimicrobial copper-alloy surfaces.
Bottom line: Copper-surfaced objects reduce HAI rates in ICU patients.
Citation: Salgado CD, Sepkowitz, KA, John JF, et al. Copper surfaces reduce the rate of healthcare-acquired infections in the intensive care unit. Infect Control Hosp Epidemiol. 2013;34(5):479-486.
Glucocorticoid Therapy for Five Days Not Inferior to 14 Days for COPD Exacerbation
Clinical question: Do short-course glucocorticoids work as well as conventional long courses for COPD exacerbation?
Background: International guidelines advocate a seven- to 14-day treatment course with glucocorticoids for COPD exacerbation, but the optimal duration of treatment is not known, and there are potential risks associated with glucocorticoid exposure.
Study design: Randomized, noninferiority, multicenter trial.
Setting: Five Swiss teaching hospitals.
Synopsis: Three hundred fourteen patients presenting to the ED with acute COPD exacerbation and without a history of asthma were randomized to receive treatment with 40 mg prednisone daily for either five or 14 days in a placebo-controlled, double-blinded fashion. There was no significant difference in the primary endpoint of re-exacerbation within six months. Patients in the five-day glucocorticoid group compared with the 14-day group were exposed to significantly less glucocorticoid.
Bottom line: Treatment for five days with glucocorticoids was not inferior to 14 days for acute COPD exacerbations with regard to re-exacerbations within six months and resulted in less glucocorticoid exposure overall.
Citation: Leuppi JF, Schuetz P, Bingisser R, et al. Short-term vs. conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE Randomized Clinical Trial. JAMA. 2013;390(21):2223-2231.
Patient Preference for Participation in Medical Decision-Making May Be Associated with Increased Resource Utilization
Clinical question: Do patient preferences for participation in medical decision-making affect health-care utilization?
Background: Patient participation in medical decision-making has been associated with improved patient satisfaction and health outcomes. There is little evidence to support theories that patient preferences might decrease or increase health-care utilization.
Study design: Survey study in academic research setting.
Setting: University of Chicago Medical Center.
Synopsis: More than 21,700 patients admitted to a general internal-medicine service completed a survey that included questions regarding preferences about receiving medical information and participation in medical decision-making. Survey data were linked with administrative data, including length of stay and total hospitalization costs.
Most patients (96.3%) expressed interest in receiving information about their illness and treatment options, but the majority of patients (71.1%) also expressed a preference to leave medical decision-making to their physician. Patients who preferred to participate in medical decision-making had significantly longer hospital LOS and higher total hospitalization cost.
Bottom line: Participation in medical decision-making significantly increased LOS and total costs.
Citation: Tak HJ, Ruhnke GW, Meltzer DO. Association of patient preferences for participation in decision making with length of stay and costs among hospitalized patients. JAMA Intern Med. 2013;173(13):1195-1205. doi: 10.1001/jamainternmed.2013.6048.
Early Parenteral Nutrition in Critically Ill Adults Does Not Significantly Affect Mortality or Infection Rates
Clinical question: Does providing early parenteral nutrition to critically ill adults with short-term relative contraindications to early enteral nutrition affect outcomes?
Background: The appropriate use of parenteral nutrition in critically ill adults is controversial. A systematic review found that critically ill patients randomized to receive early parenteral nutrition had significantly lower mortality but increased infection rates compared with standard care. A large-scale randomized trial was necessary to confirm the results.
Study design: Multicenter, randomized, single-blinded, controlled trial.
Setting: ICUs in 31 tertiary-care and community hospitals in Australia and New Zealand.
Synopsis: Researchers randomized 1,372 critically ill adults with relative contraindications to early enteral nutrition upon admission to the ICU to receive early parenteral nutrition or standard care. Early parenteral nutrition was started an average of 44 minutes after randomization. Clinicians defined standard care, with most patients remaining unfed for 2.8 days after randomization. Results were analyzed by intention-to-treat analysis, and loss to follow-up was 1%.
There was no significant difference in the primary outcome of 60-day mortality. Early parenteral nutrition patients received significantly fewer days of invasive ventilation, but did not have shorter ICU or hospital stays. Early parenteral nutrition patients experienced significantly less muscle-wasting and fat loss. There was no significant difference in new infection rates.
Bottom line: Early parenteral nutrition in critically ill adults resulted in significantly fewer days of invasive mechanical ventilation but did not cause a significant difference in length of stay, infection rates, or 60-day mortality.
Citation: Doig GS, Simpson F, Sweetman EA, et al. Early parenteral nutrition in critically ill patients with short-term relative contraindications to early enteral nutrition. JAMA. 2013;309(20): 2130-2138.
Aggressive Fluid and Sodium Restriction in Acute Decompensated Heart Failure Did Not Improve Outcomes
Clinical question: Does aggressive fluid and sodium restriction in acute decompensated heart failure (ADHF) result in increased weight loss, improved clinical stability, or decreased 30-day readmission rate?
Background: Fluid and sodium restriction are standard nonpharmacologic measures used in the management of ADHF in hospitalized patients, despite an absence of data to support their efficacy.
Study design: Randomized, controlled clinical trial with blinded outcome assessments.
Setting: A public teaching hospital in Brazil.
Synopsis: Seventy-five patients hospitalized with ADHF were randomized to receive aggressive fluid (800 mL/day) and sodium restriction (800 mg/day) or liberal intake (at least 2.5 L/day fluid, 3 to 5 g/day sodium). There were no significant between-group differences in diuretic administration. The primary outcomes of weight loss and clinical stability at three days were not significantly different between the groups. The heart-failure-specific readmission rate at 30 days was not significantly different between the groups. The aggressive restriction group had significantly worse thirst.
The study is limited by the small fraction of patients enrolled (9.2% of 813 screened) and homogenous population. Additional confirmatory trials likely are needed to change the standard of care, but this study demonstrated that aggressive fluid and sodium restriction does not benefit hospitalized patients with ADHF.
Bottom line: Aggressive fluid and sodium restriction in hospitalized patients with ADHF does not result in improved short-term weight loss, clinical stability, or decreased 30-day readmission rate, but it does cause significantly worse thirst.
Citation: Aliti GB, Rabelo ER, Clausell N, et al. Aggressive fluid and sodium restriction in acute decompensated heart failure. JAMA Intern Med. 2013;173(12):1058-1064.
Lower Rate of Pacemaker, Defibrillator Device-Pocket Hematoma without Anticoagulation Interruption
Clinical question: Is it safer to place a pacemaker or implantable cardioverter-defibrillator (ICD) while on therapeutic warfarin versus bridging with heparin/low-molecular-weight heparin (LMWH)?
Background: Current guidelines recommend bridging with heparin or LMWH for patients at high risk for thromboembolic events around the time of pacemaker or ICD placement, but it is associated with significant risk of device-pocket hematoma. Some centers place pacemakers and ICDs without interruption of warfarin. However, there are limited data to support the safety of this approach.
Study design: Multicenter, single-blinded, randomized, controlled trial.
Setting: Seventeen centers in Canada and one center in Brazil.
Synopsis: Patients with a predicted annual risk of 5% of thromboembolism were randomized to continue anticoagulation with warfarin (median INR 2.3) or to bridge therapy with heparin or LMWH; they then evaluated the incidence of clinically significant hematoma requiring prolonged hospitalization, interruption of anticoagulation therapy, or further surgical intervention. After reviewing the data on 668 patients, the Data and Safety Monitoring Board recommended termination of the study given a significantly lower rate of device-pocket hematoma in the warfarin group (3.5%) compared with the bridge group (16%) with RR 0.19 (95% CI 0.10-0.36, P<0.001). Otherwise, major surgical and thromboembolic complications were rare and not significantly different in both groups.
Bottom line: Continued warfarin therapy was associated with significantly reduced incidence of device-pocket hematoma compared with bridge with heparin or LMWH.
Citation: Birnie DH, Healey JS, Wells GA, et al. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med. 2013;368(22):2084-2093.
Prophylactic Penicillin Decreased Risk of Recurrent Leg Cellulitis
Clinical question: Does prophylactic, low-dose penicillin prevent recurrent cellulitis in patients with a history of two or more episodes of cellulitis?
Background: Some guidelines recommend prophylactic antibiotics for recurrent leg cellulitis, but there is no large randomized trial to support this practice, and clinical opinion is mixed.
Study design: Double-blinded, randomized, controlled trial.
Setting: Twenty-eight hospitals in the United Kingdom and Ireland.
Synopsis: Researchers randomized 274 patients with recurrent episodes of leg cellulitis (at least two episodes within the previous three years) to low-dose penicillin (250 mg) or placebo for 12 months and followed them for more than three years. During the prophylactic period, the penicillin group had a 45% reduction in the risk of a repeat cellulitis as compared to placebo (22% vs. 37%), equivalent to a number needed to treat to prevent a first recurrent cellulitis of five. The number of repeat episodes of cellulitis was lower overall in penicillin compared with the placebo group (119 vs. 164, P=0.02), although no significant difference was noted during the three-year follow-up period.
Factors associated with prophylaxis failure included three or more previous episodes of cellulitis, body mass index of 33 kg/m² or higher, and the presence of edema. No significant difference in adverse events was noted between the groups. Complete follow-up data was not available for participants during the follow-up period. Further study is needed to assess the long-term adverse effects and the duration of prophylaxis needed.
Bottom line: Prophylactic penicillin was effective in preventing recurrent leg cellulitis without increasing adverse effects, but its protective effect gradually declined once discontinued.
Citation: Thomas KS, Crook AM, Nunn AJ, et al. Penicillin to prevent recurrent leg cellulitis. N Engl J Med. 2013;368(18):1695-1703.
Universal ICU Decolonization Reduced Rates of MRSA Clinical Isolates and Bloodstream Infection
Clinical question: What is the most effective decolonization strategy for reducing methicillin-resistant Staphylococcus aureus (MRSA) and other pathogens in ICUs?
Background: Studies have shown that daily chlorhexidine bathing of all patients in ICUs reduced MRSA acquisition and bloodstream infection from all pathogens. However, this universal strategy has not been compared to MRSA screening and contact precautions alone, or to targeted decolonization of MRSA carriers.
Study design: Cluster-randomized comparative-effectiveness trial.
Setting: Adult ICUs in 43 Hospital Corporation of America (HCA) hospitals in 16 states.
Synopsis: All adult ICUs in a given hospital were randomized to one of three infection prevention strategies: Group 1 continued MRSA screening and isolation; Group 2 performed screening, isolation, and decolonization of MRSA carriers; and Group 3 implemented universal decolonization with intranasal mupirocin and daily bathing with chlorhexidine-impregnated cloths but no screening.
Forty-three hospitals, including 74 ICUs and 74,256 patients, underwent randomization. Significant reductions in the primary outcome of ICU-attributable MRSA clinical isolates (excluding MRSA screening tests) and the secondary outcome of bloodstream infection due to any pathogen were demonstrated across the three groups. One bloodstream infection was prevented for every 54 patients who underwent decolonization. Formal cost-effectiveness analysis was not performed.
Bottom line: In the ICU, universal decolonization was more effective than screening and isolation or targeted decolonization in the reduction of clinical MRSA isolates and bloodstream infection due to any pathogen, although monitoring for emerging resistance is necessary.
Citation: Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368(24):2255-2265.
Intensive Blood-Pressure Lowering in Intracerebral Hemorrhage Did Not Reduce Death or Severe Disability
Clinical question: What is the efficacy and safety of early intensive blood-pressure lowering in patients with acute intracerebral hemorrhage?
Background: After intracerebral hemorrhage, blood pressure often becomes elevated and is a predictor of outcome. It is not known whether rapid lowering of blood pressure would improve outcome.
Study design: International, multicenter, prospective, randomized, open-treatment, blinded end-point trial.
Setting: One hundred forty-four hospitals in 21 countries.
Synopsis: Researchers randomly assigned 2,839 patients with intracerebral hemorrhage in the previous six hours to intensive blood-pressure lowering with target systolic blood pressure of <140 mmHg within one hour, or guideline-recommended treatment with target systolic blood pressure of <180 mmHg. The mean systolic blood pressure achieved was 150 mmHg in the intensive-treatment group and 164 mmHg in the standard-treatment group.
There was no significant difference between the two groups in the primary outcome of death or major disability. A pre-specified ordinal analysis of modified Rankin score (score of 0 indicates no symptoms; a score of 5 indicates severe disability) did show significantly lower modified Rankin scores with intensive treatment. There was no difference between the two groups in the rate of serious adverse events.
Bottom line: Early intensive blood-pressure lowering in patients with acute intracerebral hemorrhage did not reduce death or major disability, although there may be improved functional outcomes with intensive blood-pressure lowering.
Citation: Anderson CS, Heeley E, Huang Y, et al. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med. 2013;368(25):2355-2365.
In This Edition
Literature At A Glance
A guide to this month’s studies
- Perioperative SSRI use associated with adverse surgical outcomes
- Copper-surfaced rooms reduce health-care-acquired infections
- Glucocorticoid therapy for five days not inferior to 14 days for COPD exacerbation
- Patient preference for participation in medical decision-making may be associated with increased resource utilization
- Early parenteral nutrition in critically ill adults does not significantly affect mortality or infection rates
- Aggressive fluid and sodium restriction in acute decompensated heart failure did not improve outcomes
- Lower rate of pacemaker, defibrillator device-pocket hematoma without anticoagulation interruption
- Prophylactic penicillin decreased risk of recurrent leg cellulitis
- Universal ICU decolonization reduced rates of mrsa clinical isolates and bloodstream infection
- Intensive blood-pressure lowering in intracerebral hemorrhage did not reduce death or severe disability
Perioperative SSRI Use Associated with Adverse Surgical Outcomes
Clinical question: Does selective serotonin reuptake inhibitor (SSRI) use during hospitalization for surgery increase the risk of adverse perioperative outcomes?
Background: SSRIs commonly are prescribed but are associated with a small but higher risk for hemorrhage, arrhythmia, and sudden death. Single-site studies have described an association between SSRIs and adverse perioperative outcomes, but larger studies utilizing a broad range of surgical cases are lacking.
Study design: Retrospective cohort study.
Setting: Three hundred hospitals concentrated in the Southern U.S.
Synopsis: Using the “Perspective” database, this study examined 530,416 patients age >18 years undergoing major elective surgery, 72,540 (13.7%) of whom received an SSRI. Regression analysis showed patients receiving an SSRI had higher odds of mortality (OR 1.2, 95% CI [1.07-1.36]), higher odds of 30-day readmission (OR 1.22 [1.18-1.26]), and higher odds for bleeding (1.09 [1.04-1.15]). When the analysis was restricted to only patients with a diagnosis of depression, a higher risk of bleeding and readmission persisted.
This study reaffirms an association but does not establish a causal relationship between SSRI use and adverse perioperative outcomes. SSRI use may be a surrogate for other factors, including more severe mood disorders, poorer functional status, or chronic pain. Additionally, no information has been provided as to optimal duration of withholding SSRIs preoperatively. As such, it may be premature for hospitalists involved in perioperative care to modify recommendations based on this study.
Bottom line: Perioperative SSRI use is associated with an increased risk of bleeding and 30-day readmission.
Citation: Auerbach AD, Vittinghoff E, Maselli J, et al. Perioperative use of selective serotonin reuptake inhibitors and risks for adverse outcomes of surgery. JAMA Intern Med. 2013;173(12):1075-1081.
Copper-Surfaced Rooms Reduce Health-Care-Acquired Infections
Clinical question: Can copper alloy surfaces in ICU rooms lower rates of health-care-acquired infections (HAIs)?
Background: Environmental contamination is a potential source of HAIs. Copper has intrinsic broad-spectrum antimicrobial properties. This study tests the efficacy copper-surfaced items in hospital rooms have in preventing HAIs.
Study design: Randomized controlled trial.
Setting: Medical ICUs at Medical University of South Carolina and Ralph H. Johnson Veterans Affairs Medical Center in Charleston, and the Memorial Sloan Kettering Cancer Center in New York City.
Synopsis: Six hundred fifty ICU patients were randomized to receive care either in rooms with copper surfacing on commonly handled patient care objects or in traditional rooms. Patients were screened for methicillin-resistant Staphylococcus aureus (MRSA) or vancomycin-resistant enterococci (VRE) on admission. The proportion of patients that developed either an HAI and/or MRSA or VRE colonization was significantly lower among patients in rooms with the copper-surfaced items (0.071 vs. 0.128; P=0.02). The rate of HAIs alone was also lower in the rooms with the copper (0.034 vs. 0.081; P=0.013).
A potential limitation to this study is that the rooms with copper items appeared different than traditional rooms, and therefore might have changed the behavior of health-care workers. Further, it is unclear how much copper surfacing would be necessary on general wards, where patients are more mobile. Still, HAIs are associated with longer lengths of stay and higher 30-day readmission rates, so these encouraging results warrant additional investigation into antimicrobial copper-alloy surfaces.
Bottom line: Copper-surfaced objects reduce HAI rates in ICU patients.
Citation: Salgado CD, Sepkowitz, KA, John JF, et al. Copper surfaces reduce the rate of healthcare-acquired infections in the intensive care unit. Infect Control Hosp Epidemiol. 2013;34(5):479-486.
Glucocorticoid Therapy for Five Days Not Inferior to 14 Days for COPD Exacerbation
Clinical question: Do short-course glucocorticoids work as well as conventional long courses for COPD exacerbation?
Background: International guidelines advocate a seven- to 14-day treatment course with glucocorticoids for COPD exacerbation, but the optimal duration of treatment is not known, and there are potential risks associated with glucocorticoid exposure.
Study design: Randomized, noninferiority, multicenter trial.
Setting: Five Swiss teaching hospitals.
Synopsis: Three hundred fourteen patients presenting to the ED with acute COPD exacerbation and without a history of asthma were randomized to receive treatment with 40 mg prednisone daily for either five or 14 days in a placebo-controlled, double-blinded fashion. There was no significant difference in the primary endpoint of re-exacerbation within six months. Patients in the five-day glucocorticoid group compared with the 14-day group were exposed to significantly less glucocorticoid.
Bottom line: Treatment for five days with glucocorticoids was not inferior to 14 days for acute COPD exacerbations with regard to re-exacerbations within six months and resulted in less glucocorticoid exposure overall.
Citation: Leuppi JF, Schuetz P, Bingisser R, et al. Short-term vs. conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE Randomized Clinical Trial. JAMA. 2013;390(21):2223-2231.
Patient Preference for Participation in Medical Decision-Making May Be Associated with Increased Resource Utilization
Clinical question: Do patient preferences for participation in medical decision-making affect health-care utilization?
Background: Patient participation in medical decision-making has been associated with improved patient satisfaction and health outcomes. There is little evidence to support theories that patient preferences might decrease or increase health-care utilization.
Study design: Survey study in academic research setting.
Setting: University of Chicago Medical Center.
Synopsis: More than 21,700 patients admitted to a general internal-medicine service completed a survey that included questions regarding preferences about receiving medical information and participation in medical decision-making. Survey data were linked with administrative data, including length of stay and total hospitalization costs.
Most patients (96.3%) expressed interest in receiving information about their illness and treatment options, but the majority of patients (71.1%) also expressed a preference to leave medical decision-making to their physician. Patients who preferred to participate in medical decision-making had significantly longer hospital LOS and higher total hospitalization cost.
Bottom line: Participation in medical decision-making significantly increased LOS and total costs.
Citation: Tak HJ, Ruhnke GW, Meltzer DO. Association of patient preferences for participation in decision making with length of stay and costs among hospitalized patients. JAMA Intern Med. 2013;173(13):1195-1205. doi: 10.1001/jamainternmed.2013.6048.
Early Parenteral Nutrition in Critically Ill Adults Does Not Significantly Affect Mortality or Infection Rates
Clinical question: Does providing early parenteral nutrition to critically ill adults with short-term relative contraindications to early enteral nutrition affect outcomes?
Background: The appropriate use of parenteral nutrition in critically ill adults is controversial. A systematic review found that critically ill patients randomized to receive early parenteral nutrition had significantly lower mortality but increased infection rates compared with standard care. A large-scale randomized trial was necessary to confirm the results.
Study design: Multicenter, randomized, single-blinded, controlled trial.
Setting: ICUs in 31 tertiary-care and community hospitals in Australia and New Zealand.
Synopsis: Researchers randomized 1,372 critically ill adults with relative contraindications to early enteral nutrition upon admission to the ICU to receive early parenteral nutrition or standard care. Early parenteral nutrition was started an average of 44 minutes after randomization. Clinicians defined standard care, with most patients remaining unfed for 2.8 days after randomization. Results were analyzed by intention-to-treat analysis, and loss to follow-up was 1%.
There was no significant difference in the primary outcome of 60-day mortality. Early parenteral nutrition patients received significantly fewer days of invasive ventilation, but did not have shorter ICU or hospital stays. Early parenteral nutrition patients experienced significantly less muscle-wasting and fat loss. There was no significant difference in new infection rates.
Bottom line: Early parenteral nutrition in critically ill adults resulted in significantly fewer days of invasive mechanical ventilation but did not cause a significant difference in length of stay, infection rates, or 60-day mortality.
Citation: Doig GS, Simpson F, Sweetman EA, et al. Early parenteral nutrition in critically ill patients with short-term relative contraindications to early enteral nutrition. JAMA. 2013;309(20): 2130-2138.
Aggressive Fluid and Sodium Restriction in Acute Decompensated Heart Failure Did Not Improve Outcomes
Clinical question: Does aggressive fluid and sodium restriction in acute decompensated heart failure (ADHF) result in increased weight loss, improved clinical stability, or decreased 30-day readmission rate?
Background: Fluid and sodium restriction are standard nonpharmacologic measures used in the management of ADHF in hospitalized patients, despite an absence of data to support their efficacy.
Study design: Randomized, controlled clinical trial with blinded outcome assessments.
Setting: A public teaching hospital in Brazil.
Synopsis: Seventy-five patients hospitalized with ADHF were randomized to receive aggressive fluid (800 mL/day) and sodium restriction (800 mg/day) or liberal intake (at least 2.5 L/day fluid, 3 to 5 g/day sodium). There were no significant between-group differences in diuretic administration. The primary outcomes of weight loss and clinical stability at three days were not significantly different between the groups. The heart-failure-specific readmission rate at 30 days was not significantly different between the groups. The aggressive restriction group had significantly worse thirst.
The study is limited by the small fraction of patients enrolled (9.2% of 813 screened) and homogenous population. Additional confirmatory trials likely are needed to change the standard of care, but this study demonstrated that aggressive fluid and sodium restriction does not benefit hospitalized patients with ADHF.
Bottom line: Aggressive fluid and sodium restriction in hospitalized patients with ADHF does not result in improved short-term weight loss, clinical stability, or decreased 30-day readmission rate, but it does cause significantly worse thirst.
Citation: Aliti GB, Rabelo ER, Clausell N, et al. Aggressive fluid and sodium restriction in acute decompensated heart failure. JAMA Intern Med. 2013;173(12):1058-1064.
Lower Rate of Pacemaker, Defibrillator Device-Pocket Hematoma without Anticoagulation Interruption
Clinical question: Is it safer to place a pacemaker or implantable cardioverter-defibrillator (ICD) while on therapeutic warfarin versus bridging with heparin/low-molecular-weight heparin (LMWH)?
Background: Current guidelines recommend bridging with heparin or LMWH for patients at high risk for thromboembolic events around the time of pacemaker or ICD placement, but it is associated with significant risk of device-pocket hematoma. Some centers place pacemakers and ICDs without interruption of warfarin. However, there are limited data to support the safety of this approach.
Study design: Multicenter, single-blinded, randomized, controlled trial.
Setting: Seventeen centers in Canada and one center in Brazil.
Synopsis: Patients with a predicted annual risk of 5% of thromboembolism were randomized to continue anticoagulation with warfarin (median INR 2.3) or to bridge therapy with heparin or LMWH; they then evaluated the incidence of clinically significant hematoma requiring prolonged hospitalization, interruption of anticoagulation therapy, or further surgical intervention. After reviewing the data on 668 patients, the Data and Safety Monitoring Board recommended termination of the study given a significantly lower rate of device-pocket hematoma in the warfarin group (3.5%) compared with the bridge group (16%) with RR 0.19 (95% CI 0.10-0.36, P<0.001). Otherwise, major surgical and thromboembolic complications were rare and not significantly different in both groups.
Bottom line: Continued warfarin therapy was associated with significantly reduced incidence of device-pocket hematoma compared with bridge with heparin or LMWH.
Citation: Birnie DH, Healey JS, Wells GA, et al. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med. 2013;368(22):2084-2093.
Prophylactic Penicillin Decreased Risk of Recurrent Leg Cellulitis
Clinical question: Does prophylactic, low-dose penicillin prevent recurrent cellulitis in patients with a history of two or more episodes of cellulitis?
Background: Some guidelines recommend prophylactic antibiotics for recurrent leg cellulitis, but there is no large randomized trial to support this practice, and clinical opinion is mixed.
Study design: Double-blinded, randomized, controlled trial.
Setting: Twenty-eight hospitals in the United Kingdom and Ireland.
Synopsis: Researchers randomized 274 patients with recurrent episodes of leg cellulitis (at least two episodes within the previous three years) to low-dose penicillin (250 mg) or placebo for 12 months and followed them for more than three years. During the prophylactic period, the penicillin group had a 45% reduction in the risk of a repeat cellulitis as compared to placebo (22% vs. 37%), equivalent to a number needed to treat to prevent a first recurrent cellulitis of five. The number of repeat episodes of cellulitis was lower overall in penicillin compared with the placebo group (119 vs. 164, P=0.02), although no significant difference was noted during the three-year follow-up period.
Factors associated with prophylaxis failure included three or more previous episodes of cellulitis, body mass index of 33 kg/m² or higher, and the presence of edema. No significant difference in adverse events was noted between the groups. Complete follow-up data was not available for participants during the follow-up period. Further study is needed to assess the long-term adverse effects and the duration of prophylaxis needed.
Bottom line: Prophylactic penicillin was effective in preventing recurrent leg cellulitis without increasing adverse effects, but its protective effect gradually declined once discontinued.
Citation: Thomas KS, Crook AM, Nunn AJ, et al. Penicillin to prevent recurrent leg cellulitis. N Engl J Med. 2013;368(18):1695-1703.
Universal ICU Decolonization Reduced Rates of MRSA Clinical Isolates and Bloodstream Infection
Clinical question: What is the most effective decolonization strategy for reducing methicillin-resistant Staphylococcus aureus (MRSA) and other pathogens in ICUs?
Background: Studies have shown that daily chlorhexidine bathing of all patients in ICUs reduced MRSA acquisition and bloodstream infection from all pathogens. However, this universal strategy has not been compared to MRSA screening and contact precautions alone, or to targeted decolonization of MRSA carriers.
Study design: Cluster-randomized comparative-effectiveness trial.
Setting: Adult ICUs in 43 Hospital Corporation of America (HCA) hospitals in 16 states.
Synopsis: All adult ICUs in a given hospital were randomized to one of three infection prevention strategies: Group 1 continued MRSA screening and isolation; Group 2 performed screening, isolation, and decolonization of MRSA carriers; and Group 3 implemented universal decolonization with intranasal mupirocin and daily bathing with chlorhexidine-impregnated cloths but no screening.
Forty-three hospitals, including 74 ICUs and 74,256 patients, underwent randomization. Significant reductions in the primary outcome of ICU-attributable MRSA clinical isolates (excluding MRSA screening tests) and the secondary outcome of bloodstream infection due to any pathogen were demonstrated across the three groups. One bloodstream infection was prevented for every 54 patients who underwent decolonization. Formal cost-effectiveness analysis was not performed.
Bottom line: In the ICU, universal decolonization was more effective than screening and isolation or targeted decolonization in the reduction of clinical MRSA isolates and bloodstream infection due to any pathogen, although monitoring for emerging resistance is necessary.
Citation: Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368(24):2255-2265.
Intensive Blood-Pressure Lowering in Intracerebral Hemorrhage Did Not Reduce Death or Severe Disability
Clinical question: What is the efficacy and safety of early intensive blood-pressure lowering in patients with acute intracerebral hemorrhage?
Background: After intracerebral hemorrhage, blood pressure often becomes elevated and is a predictor of outcome. It is not known whether rapid lowering of blood pressure would improve outcome.
Study design: International, multicenter, prospective, randomized, open-treatment, blinded end-point trial.
Setting: One hundred forty-four hospitals in 21 countries.
Synopsis: Researchers randomly assigned 2,839 patients with intracerebral hemorrhage in the previous six hours to intensive blood-pressure lowering with target systolic blood pressure of <140 mmHg within one hour, or guideline-recommended treatment with target systolic blood pressure of <180 mmHg. The mean systolic blood pressure achieved was 150 mmHg in the intensive-treatment group and 164 mmHg in the standard-treatment group.
There was no significant difference between the two groups in the primary outcome of death or major disability. A pre-specified ordinal analysis of modified Rankin score (score of 0 indicates no symptoms; a score of 5 indicates severe disability) did show significantly lower modified Rankin scores with intensive treatment. There was no difference between the two groups in the rate of serious adverse events.
Bottom line: Early intensive blood-pressure lowering in patients with acute intracerebral hemorrhage did not reduce death or major disability, although there may be improved functional outcomes with intensive blood-pressure lowering.
Citation: Anderson CS, Heeley E, Huang Y, et al. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med. 2013;368(25):2355-2365.
In This Edition
Literature At A Glance
A guide to this month’s studies
- Perioperative SSRI use associated with adverse surgical outcomes
- Copper-surfaced rooms reduce health-care-acquired infections
- Glucocorticoid therapy for five days not inferior to 14 days for COPD exacerbation
- Patient preference for participation in medical decision-making may be associated with increased resource utilization
- Early parenteral nutrition in critically ill adults does not significantly affect mortality or infection rates
- Aggressive fluid and sodium restriction in acute decompensated heart failure did not improve outcomes
- Lower rate of pacemaker, defibrillator device-pocket hematoma without anticoagulation interruption
- Prophylactic penicillin decreased risk of recurrent leg cellulitis
- Universal ICU decolonization reduced rates of mrsa clinical isolates and bloodstream infection
- Intensive blood-pressure lowering in intracerebral hemorrhage did not reduce death or severe disability
Perioperative SSRI Use Associated with Adverse Surgical Outcomes
Clinical question: Does selective serotonin reuptake inhibitor (SSRI) use during hospitalization for surgery increase the risk of adverse perioperative outcomes?
Background: SSRIs commonly are prescribed but are associated with a small but higher risk for hemorrhage, arrhythmia, and sudden death. Single-site studies have described an association between SSRIs and adverse perioperative outcomes, but larger studies utilizing a broad range of surgical cases are lacking.
Study design: Retrospective cohort study.
Setting: Three hundred hospitals concentrated in the Southern U.S.
Synopsis: Using the “Perspective” database, this study examined 530,416 patients age >18 years undergoing major elective surgery, 72,540 (13.7%) of whom received an SSRI. Regression analysis showed patients receiving an SSRI had higher odds of mortality (OR 1.2, 95% CI [1.07-1.36]), higher odds of 30-day readmission (OR 1.22 [1.18-1.26]), and higher odds for bleeding (1.09 [1.04-1.15]). When the analysis was restricted to only patients with a diagnosis of depression, a higher risk of bleeding and readmission persisted.
This study reaffirms an association but does not establish a causal relationship between SSRI use and adverse perioperative outcomes. SSRI use may be a surrogate for other factors, including more severe mood disorders, poorer functional status, or chronic pain. Additionally, no information has been provided as to optimal duration of withholding SSRIs preoperatively. As such, it may be premature for hospitalists involved in perioperative care to modify recommendations based on this study.
Bottom line: Perioperative SSRI use is associated with an increased risk of bleeding and 30-day readmission.
Citation: Auerbach AD, Vittinghoff E, Maselli J, et al. Perioperative use of selective serotonin reuptake inhibitors and risks for adverse outcomes of surgery. JAMA Intern Med. 2013;173(12):1075-1081.
Copper-Surfaced Rooms Reduce Health-Care-Acquired Infections
Clinical question: Can copper alloy surfaces in ICU rooms lower rates of health-care-acquired infections (HAIs)?
Background: Environmental contamination is a potential source of HAIs. Copper has intrinsic broad-spectrum antimicrobial properties. This study tests the efficacy copper-surfaced items in hospital rooms have in preventing HAIs.
Study design: Randomized controlled trial.
Setting: Medical ICUs at Medical University of South Carolina and Ralph H. Johnson Veterans Affairs Medical Center in Charleston, and the Memorial Sloan Kettering Cancer Center in New York City.
Synopsis: Six hundred fifty ICU patients were randomized to receive care either in rooms with copper surfacing on commonly handled patient care objects or in traditional rooms. Patients were screened for methicillin-resistant Staphylococcus aureus (MRSA) or vancomycin-resistant enterococci (VRE) on admission. The proportion of patients that developed either an HAI and/or MRSA or VRE colonization was significantly lower among patients in rooms with the copper-surfaced items (0.071 vs. 0.128; P=0.02). The rate of HAIs alone was also lower in the rooms with the copper (0.034 vs. 0.081; P=0.013).
A potential limitation to this study is that the rooms with copper items appeared different than traditional rooms, and therefore might have changed the behavior of health-care workers. Further, it is unclear how much copper surfacing would be necessary on general wards, where patients are more mobile. Still, HAIs are associated with longer lengths of stay and higher 30-day readmission rates, so these encouraging results warrant additional investigation into antimicrobial copper-alloy surfaces.
Bottom line: Copper-surfaced objects reduce HAI rates in ICU patients.
Citation: Salgado CD, Sepkowitz, KA, John JF, et al. Copper surfaces reduce the rate of healthcare-acquired infections in the intensive care unit. Infect Control Hosp Epidemiol. 2013;34(5):479-486.
Glucocorticoid Therapy for Five Days Not Inferior to 14 Days for COPD Exacerbation
Clinical question: Do short-course glucocorticoids work as well as conventional long courses for COPD exacerbation?
Background: International guidelines advocate a seven- to 14-day treatment course with glucocorticoids for COPD exacerbation, but the optimal duration of treatment is not known, and there are potential risks associated with glucocorticoid exposure.
Study design: Randomized, noninferiority, multicenter trial.
Setting: Five Swiss teaching hospitals.
Synopsis: Three hundred fourteen patients presenting to the ED with acute COPD exacerbation and without a history of asthma were randomized to receive treatment with 40 mg prednisone daily for either five or 14 days in a placebo-controlled, double-blinded fashion. There was no significant difference in the primary endpoint of re-exacerbation within six months. Patients in the five-day glucocorticoid group compared with the 14-day group were exposed to significantly less glucocorticoid.
Bottom line: Treatment for five days with glucocorticoids was not inferior to 14 days for acute COPD exacerbations with regard to re-exacerbations within six months and resulted in less glucocorticoid exposure overall.
Citation: Leuppi JF, Schuetz P, Bingisser R, et al. Short-term vs. conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE Randomized Clinical Trial. JAMA. 2013;390(21):2223-2231.
Patient Preference for Participation in Medical Decision-Making May Be Associated with Increased Resource Utilization
Clinical question: Do patient preferences for participation in medical decision-making affect health-care utilization?
Background: Patient participation in medical decision-making has been associated with improved patient satisfaction and health outcomes. There is little evidence to support theories that patient preferences might decrease or increase health-care utilization.
Study design: Survey study in academic research setting.
Setting: University of Chicago Medical Center.
Synopsis: More than 21,700 patients admitted to a general internal-medicine service completed a survey that included questions regarding preferences about receiving medical information and participation in medical decision-making. Survey data were linked with administrative data, including length of stay and total hospitalization costs.
Most patients (96.3%) expressed interest in receiving information about their illness and treatment options, but the majority of patients (71.1%) also expressed a preference to leave medical decision-making to their physician. Patients who preferred to participate in medical decision-making had significantly longer hospital LOS and higher total hospitalization cost.
Bottom line: Participation in medical decision-making significantly increased LOS and total costs.
Citation: Tak HJ, Ruhnke GW, Meltzer DO. Association of patient preferences for participation in decision making with length of stay and costs among hospitalized patients. JAMA Intern Med. 2013;173(13):1195-1205. doi: 10.1001/jamainternmed.2013.6048.
Early Parenteral Nutrition in Critically Ill Adults Does Not Significantly Affect Mortality or Infection Rates
Clinical question: Does providing early parenteral nutrition to critically ill adults with short-term relative contraindications to early enteral nutrition affect outcomes?
Background: The appropriate use of parenteral nutrition in critically ill adults is controversial. A systematic review found that critically ill patients randomized to receive early parenteral nutrition had significantly lower mortality but increased infection rates compared with standard care. A large-scale randomized trial was necessary to confirm the results.
Study design: Multicenter, randomized, single-blinded, controlled trial.
Setting: ICUs in 31 tertiary-care and community hospitals in Australia and New Zealand.
Synopsis: Researchers randomized 1,372 critically ill adults with relative contraindications to early enteral nutrition upon admission to the ICU to receive early parenteral nutrition or standard care. Early parenteral nutrition was started an average of 44 minutes after randomization. Clinicians defined standard care, with most patients remaining unfed for 2.8 days after randomization. Results were analyzed by intention-to-treat analysis, and loss to follow-up was 1%.
There was no significant difference in the primary outcome of 60-day mortality. Early parenteral nutrition patients received significantly fewer days of invasive ventilation, but did not have shorter ICU or hospital stays. Early parenteral nutrition patients experienced significantly less muscle-wasting and fat loss. There was no significant difference in new infection rates.
Bottom line: Early parenteral nutrition in critically ill adults resulted in significantly fewer days of invasive mechanical ventilation but did not cause a significant difference in length of stay, infection rates, or 60-day mortality.
Citation: Doig GS, Simpson F, Sweetman EA, et al. Early parenteral nutrition in critically ill patients with short-term relative contraindications to early enteral nutrition. JAMA. 2013;309(20): 2130-2138.
Aggressive Fluid and Sodium Restriction in Acute Decompensated Heart Failure Did Not Improve Outcomes
Clinical question: Does aggressive fluid and sodium restriction in acute decompensated heart failure (ADHF) result in increased weight loss, improved clinical stability, or decreased 30-day readmission rate?
Background: Fluid and sodium restriction are standard nonpharmacologic measures used in the management of ADHF in hospitalized patients, despite an absence of data to support their efficacy.
Study design: Randomized, controlled clinical trial with blinded outcome assessments.
Setting: A public teaching hospital in Brazil.
Synopsis: Seventy-five patients hospitalized with ADHF were randomized to receive aggressive fluid (800 mL/day) and sodium restriction (800 mg/day) or liberal intake (at least 2.5 L/day fluid, 3 to 5 g/day sodium). There were no significant between-group differences in diuretic administration. The primary outcomes of weight loss and clinical stability at three days were not significantly different between the groups. The heart-failure-specific readmission rate at 30 days was not significantly different between the groups. The aggressive restriction group had significantly worse thirst.
The study is limited by the small fraction of patients enrolled (9.2% of 813 screened) and homogenous population. Additional confirmatory trials likely are needed to change the standard of care, but this study demonstrated that aggressive fluid and sodium restriction does not benefit hospitalized patients with ADHF.
Bottom line: Aggressive fluid and sodium restriction in hospitalized patients with ADHF does not result in improved short-term weight loss, clinical stability, or decreased 30-day readmission rate, but it does cause significantly worse thirst.
Citation: Aliti GB, Rabelo ER, Clausell N, et al. Aggressive fluid and sodium restriction in acute decompensated heart failure. JAMA Intern Med. 2013;173(12):1058-1064.
Lower Rate of Pacemaker, Defibrillator Device-Pocket Hematoma without Anticoagulation Interruption
Clinical question: Is it safer to place a pacemaker or implantable cardioverter-defibrillator (ICD) while on therapeutic warfarin versus bridging with heparin/low-molecular-weight heparin (LMWH)?
Background: Current guidelines recommend bridging with heparin or LMWH for patients at high risk for thromboembolic events around the time of pacemaker or ICD placement, but it is associated with significant risk of device-pocket hematoma. Some centers place pacemakers and ICDs without interruption of warfarin. However, there are limited data to support the safety of this approach.
Study design: Multicenter, single-blinded, randomized, controlled trial.
Setting: Seventeen centers in Canada and one center in Brazil.
Synopsis: Patients with a predicted annual risk of 5% of thromboembolism were randomized to continue anticoagulation with warfarin (median INR 2.3) or to bridge therapy with heparin or LMWH; they then evaluated the incidence of clinically significant hematoma requiring prolonged hospitalization, interruption of anticoagulation therapy, or further surgical intervention. After reviewing the data on 668 patients, the Data and Safety Monitoring Board recommended termination of the study given a significantly lower rate of device-pocket hematoma in the warfarin group (3.5%) compared with the bridge group (16%) with RR 0.19 (95% CI 0.10-0.36, P<0.001). Otherwise, major surgical and thromboembolic complications were rare and not significantly different in both groups.
Bottom line: Continued warfarin therapy was associated with significantly reduced incidence of device-pocket hematoma compared with bridge with heparin or LMWH.
Citation: Birnie DH, Healey JS, Wells GA, et al. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med. 2013;368(22):2084-2093.
Prophylactic Penicillin Decreased Risk of Recurrent Leg Cellulitis
Clinical question: Does prophylactic, low-dose penicillin prevent recurrent cellulitis in patients with a history of two or more episodes of cellulitis?
Background: Some guidelines recommend prophylactic antibiotics for recurrent leg cellulitis, but there is no large randomized trial to support this practice, and clinical opinion is mixed.
Study design: Double-blinded, randomized, controlled trial.
Setting: Twenty-eight hospitals in the United Kingdom and Ireland.
Synopsis: Researchers randomized 274 patients with recurrent episodes of leg cellulitis (at least two episodes within the previous three years) to low-dose penicillin (250 mg) or placebo for 12 months and followed them for more than three years. During the prophylactic period, the penicillin group had a 45% reduction in the risk of a repeat cellulitis as compared to placebo (22% vs. 37%), equivalent to a number needed to treat to prevent a first recurrent cellulitis of five. The number of repeat episodes of cellulitis was lower overall in penicillin compared with the placebo group (119 vs. 164, P=0.02), although no significant difference was noted during the three-year follow-up period.
Factors associated with prophylaxis failure included three or more previous episodes of cellulitis, body mass index of 33 kg/m² or higher, and the presence of edema. No significant difference in adverse events was noted between the groups. Complete follow-up data was not available for participants during the follow-up period. Further study is needed to assess the long-term adverse effects and the duration of prophylaxis needed.
Bottom line: Prophylactic penicillin was effective in preventing recurrent leg cellulitis without increasing adverse effects, but its protective effect gradually declined once discontinued.
Citation: Thomas KS, Crook AM, Nunn AJ, et al. Penicillin to prevent recurrent leg cellulitis. N Engl J Med. 2013;368(18):1695-1703.
Universal ICU Decolonization Reduced Rates of MRSA Clinical Isolates and Bloodstream Infection
Clinical question: What is the most effective decolonization strategy for reducing methicillin-resistant Staphylococcus aureus (MRSA) and other pathogens in ICUs?
Background: Studies have shown that daily chlorhexidine bathing of all patients in ICUs reduced MRSA acquisition and bloodstream infection from all pathogens. However, this universal strategy has not been compared to MRSA screening and contact precautions alone, or to targeted decolonization of MRSA carriers.
Study design: Cluster-randomized comparative-effectiveness trial.
Setting: Adult ICUs in 43 Hospital Corporation of America (HCA) hospitals in 16 states.
Synopsis: All adult ICUs in a given hospital were randomized to one of three infection prevention strategies: Group 1 continued MRSA screening and isolation; Group 2 performed screening, isolation, and decolonization of MRSA carriers; and Group 3 implemented universal decolonization with intranasal mupirocin and daily bathing with chlorhexidine-impregnated cloths but no screening.
Forty-three hospitals, including 74 ICUs and 74,256 patients, underwent randomization. Significant reductions in the primary outcome of ICU-attributable MRSA clinical isolates (excluding MRSA screening tests) and the secondary outcome of bloodstream infection due to any pathogen were demonstrated across the three groups. One bloodstream infection was prevented for every 54 patients who underwent decolonization. Formal cost-effectiveness analysis was not performed.
Bottom line: In the ICU, universal decolonization was more effective than screening and isolation or targeted decolonization in the reduction of clinical MRSA isolates and bloodstream infection due to any pathogen, although monitoring for emerging resistance is necessary.
Citation: Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. N Engl J Med. 2013;368(24):2255-2265.
Intensive Blood-Pressure Lowering in Intracerebral Hemorrhage Did Not Reduce Death or Severe Disability
Clinical question: What is the efficacy and safety of early intensive blood-pressure lowering in patients with acute intracerebral hemorrhage?
Background: After intracerebral hemorrhage, blood pressure often becomes elevated and is a predictor of outcome. It is not known whether rapid lowering of blood pressure would improve outcome.
Study design: International, multicenter, prospective, randomized, open-treatment, blinded end-point trial.
Setting: One hundred forty-four hospitals in 21 countries.
Synopsis: Researchers randomly assigned 2,839 patients with intracerebral hemorrhage in the previous six hours to intensive blood-pressure lowering with target systolic blood pressure of <140 mmHg within one hour, or guideline-recommended treatment with target systolic blood pressure of <180 mmHg. The mean systolic blood pressure achieved was 150 mmHg in the intensive-treatment group and 164 mmHg in the standard-treatment group.
There was no significant difference between the two groups in the primary outcome of death or major disability. A pre-specified ordinal analysis of modified Rankin score (score of 0 indicates no symptoms; a score of 5 indicates severe disability) did show significantly lower modified Rankin scores with intensive treatment. There was no difference between the two groups in the rate of serious adverse events.
Bottom line: Early intensive blood-pressure lowering in patients with acute intracerebral hemorrhage did not reduce death or major disability, although there may be improved functional outcomes with intensive blood-pressure lowering.
Citation: Anderson CS, Heeley E, Huang Y, et al. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med. 2013;368(25):2355-2365.
How Should a Patient with Cocaine-Associated Chest Pain be Treated?

Case
A 38-year-old man with a history of tobacco use presents to the emergency department complaining of constant substernal chest pain for three hours. His temperature is 37.7°C, his heart rate is 110 beats per minute, and his blood pressure is 155/95 mmHg. He appears anxious and diaphoretic but examination is otherwise unremarkable. He admits to cocaine use one hour before the onset of symptoms. What are the appropriate treatments for his condition?
Overview
Cocaine is the second-most-commonly used illicit drug in the U.S. and represents 31% of all ED visits related to substance abuse.1,2 According to recent survey results, 2.1 million people report recent cocaine use, and 1.6 million engage in cocaine abuse or dependence.2 Acute cardiopulmonary complaints are common in individuals who present to the ED after cocaine use, with chest pain being the most frequently reported symptom in 40%.3
Numerous etiologies for cocaine-associated chest pain (CACP) have been discovered, including musculoskeletal pain, pulmonary hypertension, cardiomyopathy, arrhythmias, and endocarditis.4 Only 0.5% of patients with aortic dissection over a four-year period had a recent history of cocaine use, making cocaine a rare cause of a rare condition.5 Cardiac chest pain remains the most frequent underlying etiology, resulting in the most common complication of myocardial infarction (MI) in up to 6% of patients.6,7
The ways in which cocaine use can cause myocardial ischemia and MI are multifactorial. A vigorous central sympathomimetic effect, coronary artery vasoconstriction, stimulation of platelets, and enhanced atherosclerosis all lead to a myocardial oxygen supply-demand imbalance.8 Other key interactions in the cardiovascular system are displayed in Figure 1. Understanding the role of these mechanisms in CACP is crucial to patient care.
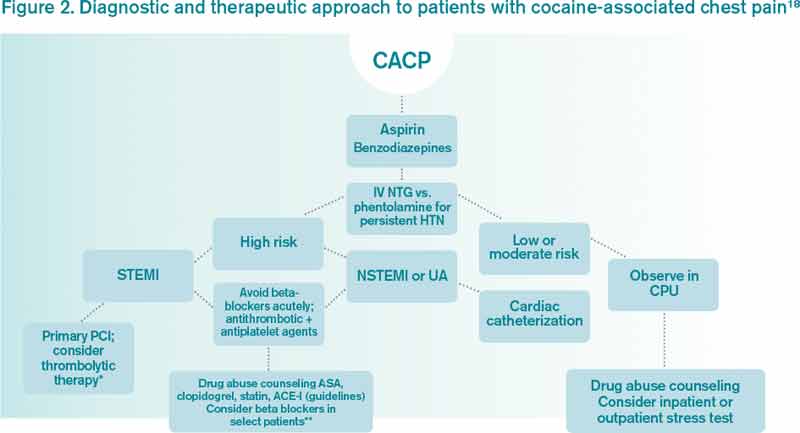
Clinician goals in the management of CACP are to rapidly and accurately exclude life-threatening etiologies; assess the need for urgent acute coronary syndrome (ACS) evaluation; risk-stratify patients and ensure appropriate disposition; normalize the toxic effects of cocaine; treat resultant organ damage; and prevent long-term complications. An algorithm detailing this approach is provided in Figure 2.

Review of the Data
Diagnostic evaluation. Given potential differences in treatment regimens, it is imperative to differentiate patients who present with CACP from those whose chest pain is not associated with cocaine either by direct questioning or by screening of urine for cocaine metabolites. Once the presence of cocaine has been confirmed, guideline-based evaluation for potential ACS with serial electrocardiograms (ECG), cardiac biomarkers, and close monitoring of cardiac rhythms and hemodynamics is largely similar to standard management of all patients presenting with chest pain, with a few caveats.
Interpretation of the ECG can be challenging in the setting of cocaine. Studies have shown “abnormal” ECGs in 56% to 84% of patients, with many representing early repolarization or left ventricular hypertrophy.9,10 Likewise, patients with MI are as likely to present with normal or nonspecific ECG findings as with ischemic findings.7,11 ECG interpretation to diagnose ischemia or infarction in patients with CACP yields a sensitivity of 36% and specificity of 90%.7
Creatine kinase (CK), CK-MB fraction, and myoglobin have low specificity for the diagnosis of ischemia, as cocaine can induce skeletal muscle injury and rhabdomyolysis.9,12 Cardiac troponins demonstrate a superior specificity compared to CK and CK-MB and are thus the preferred cardiac biomarkers in diagnosing cocaine-associated MI.12
Initial management and disposition. Patients at high risk for cardiovascular events are generally admitted to a monitored bed.13 Immediate reperfusion therapy with primary percutaneous coronary intervention is recommended in patients with ST-elevation MI (STEMI). Treatment with thrombolytic agents is associated with an increased risk of intracerebral hemorrhage and lacks documented efficacy in patients with CACP. Thrombolysis should therefore only be utilized if the diagnosis of STEMI is unequivocal and an experienced cardiac catheterization laboratory is unavailable.14,15
Patients with unstable angina (UA) or non-ST-elevation MI (NSTEMI) are at higher risk for further cardiac events in a similar manner to those with ACS unrelated to cocaine. These cases might benefit from early cardiac catheterization and revascularization.16 Because of the increased risk of stent thrombosis in cocaine-users, thought to be due to recidivism, a detailed risk-benefit analysis should be undertaken prior to the implantation of cardiac stents.
Other diagnostic tests, such as stress testing and myocardial imaging, have not shown significant accuracy in diagnosing MI in this setting; moreover, these patients are at low overall risk for cardiac events and mortality. Consequently, an extensive diagnostic evaluation might not be cost-effective.7,10,13,17 Patients who have CACP without MI have a very low frequency of delayed complications.3,17 As such, cost-effective evaluation strategies, such as nine- or 12-hour observation periods in a chest pain unit, are appropriate for many of these low- to moderate-risk patients.13 For all CACP patients, the most critical post-discharge interventions are cardiac risk modification and cocaine cessation.13
Normalizing the toxic effects of cocaine with medications.
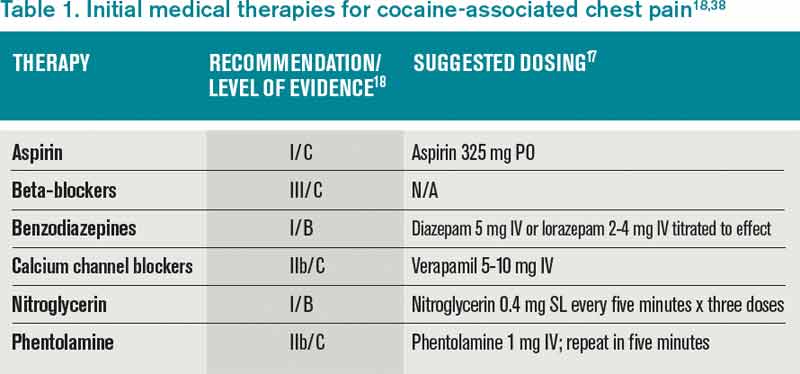
Aspirin: While no specific study has been performed in patients with CACP and aspirin, CACP guidelines, based on data supporting ACS guidelines for all patients, recommend administration of full-dose aspirin given its associated reduction in morbidity and mortality.18,19 Furthermore, given the platelet-stimulating effects of cocaine, using aspirin in this setting seems very reasonable.
Benzodiazepines: CACP guidelines support the use of benzodiazepines early in management to indirectly combat the agitation, hypertension, and tachycardia resulting from the stimulatory effects of cocaine.18,20 These recommendations are based on several animal and human studies that demonstrate significant reduction in heart rate and systemic arterial pressure with the use of these agents.21,22
Nitroglycerin: Cardiac catheterization studies have shown reversal of vasoconstriction with administration of nitroglycerin. One study demonstrated a benefit of the drug in 49% of participants.23 Additional investigation into the benefit of benzodiazepine and nitroglycerin combination therapy revealed mixed results. In one study, lorazepam plus nitroglycerin was found to be more efficacious than nitroglycerin alone.24 In another, however, use of diazepam in combination with nitroglycerin did not show benefit when evaluating pain relief, cardiac dynamics, and left ventricular function.25
Phentolamine: Phentolamine administration has been studied much less in the literature. This nonselective alpha-adrenergic antagonist exerts a dose-dependent reversal of cocaine’s vasoconstrictive properties in monkeys and humans.26,27 International guidelines for Emergency Cardiovascular Care recommend its use in treatment of cocaine-associated ACS;27 however, the AHA recommends it less strongly.18
Calcium channel blockers: Calcium channel blockers (CCBs) have not shown promise as first-line agents. While catheterization studies demonstrate the vasodilatory properties of verapamil, larger studies looking at all-cause mortality conclude that CCBs might worsen mortality rates,28 and animal studies indicate an increased risk of seizures.29 At this time, CCBs are recommended only if cardiac symptoms continue after both benzodiazepines and nitroglycerin are administered.18
The beta-blocker controversy: The use of beta-blockers in patients with CACP remains controversial given the theoretical risk of unopposed alpha-adrenergic activation. Coronary vasospasm, decreased myocardial oxygen delivery, and increased systemic vascular resistance can result from their use.30
Propranolol, a nonselective beta-blocker, was shown in catheterization studies to potentiate the coronary vasoconstriction of cocaine.31 Labetalol, a combined alpha/beta-blocker, reduced mean arterial pressure after cocaine administration during cardiac catheterization but did not reverse coronary vasoconstriction.32 This was attributed to the predominating beta greater than alpha blockade at doses administered. The selective beta-1 antagonists esmolol and metoprolol have shown no benefit in CACP.33 Carvedilol, a combined alpha/beta-blocker with both peripheral and central nervous system activity, has potential to attenuate both physiologic and behavioral response to cocaine, but it has not been well studied in this patient subset.34
The 2005 ACC/AHA STEMI guidelines recommended against beta-blockers in the setting of STEMI precipitated by cocaine use due to the potential of exacerbating coronary vasoconstriction.35 The 2007 ACC/AHA UA/NSTEMI guidelines stated that the use of a combined alpha/beta-blocker in patients with cocaine-induced ACS may be reasonable for patients with hypertension or tachycardia if pre-treated with a vasodilator.19 The 2008 ACC/AHA guidelines on the management of cocaine-related chest pain and MI recommended against the use of beta-blockers in the acute setting given the low incidence of cocaine-related MI and death.18
In a more recent study, Dattilo et al showed that beta-blockers administered to patients admitted with positive urine toxicology for cocaine significantly reduced MI and in-hospital mortality. Reduction of MI was of borderline significance in those admitted with a chief complaint of chest pain.36 Limitations of this study include unknown time of cocaine ingestion, lack of follow-up on discharge mortality, and a small sample size of 348 patients lacking statistical power.
Another retrospective cohort study examined patients admitted with chest pain and urine toxicology positive for cocaine and found that beta-blocker administration during hospitalization was not associated with increased incident mortality. Further, after a mean follow-up of 2.5 years, there was a statistically significant decrease in cardiovascular death.37 Drawbacks of this study included an older patient population, greater proportion of coronary artery disease, and higher follow-up of cardiovascular mortality rates than in previous studies, suggesting this subset might have received greater benefit from beta-blockers as a result of these characteristics.
The 2008 ACC/AHA guidelines instruct individualized consideration of the risk/benefit ratio for beta-blocker use in patients with CACP given the high rate of recidivism in cocaine abusers. The strongest indication is given to those with documented MI, left ventricular systolic dysfunction, or ventricular arrhythmias.18
It is important to note that these recommendations are based on cardiac catheterization laboratory studies, case reports, retrospective analyses, and animal experiments. No prospective controlled trials evaluating the role of beta-blockers in CACP and MI exist, and no trials regarding therapies to improve outcomes of patients sustaining a cocaine-associated MI have been reported.18
Back to the Case
This patient was experiencing cocaine-associated chest pain, which was confirmed with positive urine toxicology. Initial diagnostic workup with basic laboratory studies and cardiac biomarkers showed mild elevation in CK with troponin levels within normal limits. His ECG showed changes consistent with left ventricular hypertrophy. Chest radiograph was unremarkable.
He received aspirin, benzodiazepines, and nitroglycerin with normalization of vital signs, as well as subjective improvement in chest pain and anxiety. He was deemed to be at low risk for potential cardiac complications; thus, further cardiac testing was not pursued. Rather, he was admitted to an overnight observation unit with telemetry monitoring, where his chest pain did not recur.
He was seen in consultation with social work staff who arranged for drug abuse counseling after discharge. Given the uncertainty of relapse to cocaine use, as well as lack of known cardiac risk factors, he was not discharged on any new medications.
Bottom Line
The treatment of CACP includes normalizing the toxic systemic effects of the drug and minimizing the direct ischemic damage to the myocardium. Management varies slightly from traditional chest pain algorithms and includes benzodiazepines as well as antiplatelet agents and vasodilators to achieve this goal. Initial therapy with beta-blockers remains undefined and is largely discouraged in the acute setting. The role of beta-blockade upon discharge, however, can be beneficial in specific populations, especially those found to have underlying coronary disease.
Dr. Houchens and Dr. Czarnik are clinical instructors and Dr. Mack is a clinical lecturer at the University of Michigan Health System in Ann Arbor.
References
- Hughes A, Sathe N, Spagnola K. State Estimates of Substance Use from the 2005-2006 National Surveys on Drug Use and Health. DHHS Publication No. SMA 08-4311, NSDUH Series H-33. Rockville, MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2008.
- Volkow ND. Cocaine: Abuse and Addiction. National Institute on Drug Abuse. Washington, DC: U.S. Department of Health and Human Services; 2009.
- Brody SL, Slovis CM, Wrenn KD. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325-331.
- Levis JT, Garmel GM. Cocaine-associated chest pain. Emerg Med Clin North Am. 2005;23:1083-1103.
- Eagle KA, Isselbacher EM, DeSanctis RW. Cocaine-related aortic dissection in perspective. Circulation. 2002;105:1529-1530.
- Feldman JA, Fish SS, Beshansky JR, Griffith JL, Woolard RH, Selker HP. Acute cardiac ischemia in patients with cocaine-associated complaints: results of a multicenter trial. Ann Emerg Med. 2000;36:469-476.
- Hollander JE, Hoffman RS, Gennis P, et al. Prospective multicenter evaluation of cocaine associated chest pain. Cocaine Associated Chest Pain (COCHPA) Study Group. Acad Emerg Med. 1994;1:330-339.
- Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558-2569.
- Gitter MJ, Goldsmith SR, Dunbar DN, et al. Cocaine and chest pain: clinical features and outcomes of patients hospitalized to rule out myocardial infarction. Ann Intern Med. 1991;115:277-282.
- Amin M, Gabelman G, Karpel J, et al. Acute myocardial infarction and chest pain syndromes after cocaine use. Am J Cardiol. 1990;66:1434-1437.
- Tokarski GF, Paganussi P, Urbanski R, et al. An evaluation of cocaine-induced chest pain. Ann Emerg Med. 1990;19:1088-1092.
- Hollander JE, Levitt MA, Young GP, Briglia E, Wetli CV, Gawad Y. Effect of recent cocaine use on the specificity of cardiac markers for diagnosis of acute myocardial infarction. Am Heart J. 1998;135(2 Pt 1):245-252.
- Weber JE, Shofer FS, Larkin GL, Kalaria AS, Hollander JE. Validation of a brief observation period for patients with cocaine-associated chest pain. N Engl J Med. 2003;348:510-517.
- Hahn IH, Hoffman RS. Diagnosis and treatment of acute myocardial infarction: cocaine use and acute myocardial infarction. Emerg Med Clin North Am. 2001;19(2):1-18.
- Hoffman RS, Hollander JE. Evaluation of patients with chest pain after cocaine use. Crit Care Clin. 1997;13:809-828. Cannon CP, Weintraub WS, Demopoulos LA, et al.
- Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879-1887.
- Hollander JE, Hoffman RS. Cocaine-induced myocardial infarction: an analysis and review of the literature. J Emerg Med. 1992;10:169-177.
- McCord J, Jneid H, Hollander JE, et al. Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897-1907.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol. 2007;50:E1-E157.
- Hollander JE. Management of cocaine-associated myocardial ischemia. N Engl J Med. 1995;333:1267-1272.
- Brubacher JR, Hoffman RS. Cocaine toxicity. Top Emerg Med. 1997;19(4):1-16.
- Catavas JD, Waters IW. Acute cocaine intoxication in the conscious dog: studies on the mechanism of lethality. J Pharmacol Exp Ther. 1981;217:350-356.
- Hollander JE, Hoffman RS, Gennis P, et al. Nitroglycerin in the treatment of cocaine associated chest pain—clinical safety and efficacy. J Toxicol Clin Toxicol. 1994;32(3): 243-256.
- Honderick T, Williams D, Seaberg D, Wears R. A prospective, randomized, controlled trial of benzodiazepines and nitroglycerin or nitroglycerin alone in the treatment of cocaine-associated acute coronary syndromes. Am J Emerg Med. 2003;21(1):39-42.
- Baumann BM, Perrone J, Hornig SE, Shofer FS, Hollander JE. Randomized, double-blind, placebo-controlled trial of diazepam, nitroglycerin, or both for treatment of patients with potential cocaine-associated acute coronary syndromes. Acad Emerg Med. 2000;7:878-885.
- Schindler CW, Tella SR, Goldberg SR. Adrenoceptor mechanisms in the cardiovascular effects of cocaine in conscious squirrel monkeys. Life Sci. 1992;51(9):653-660.
- Lange RA, Cigarroa RG, Yancy CW Jr., et al. Cocaine-induced coronary-artery vasoconstriction. N Engl J Med. 1989;321(23):1557-1562.
- Furberg CD, Psaty BM, Meyer JV. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326-1331.
- Derlet RW, Albertson TE. Potentiation of cocaine toxicity with calcium channel blockers. Am J Emerg Med. 1989;7:464-468.
- Lange RA, Hillis LD. Cardiovascular complications of cocaine use. N Engl J Med. 2001;345:351-358.
- Lange RA, Cigarroa RG, Flores ED, et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112:897-903.
- Boehrer JD, Moliterno DJ, Willard JE, Hillis LD, Lange RA. Influence of labetalol on cocaine-induced coronary vasoconstriction in humans. Am J Med. 1993;94:608-610.
- Sand IC, Brody SL, Wrenn KD, Slovis CM. Experience with esmolol for the treatment of cocaine-associated cardiovascular complications. Am J Emerg Med. 1991;9:161-163.
- Sofuoglo M, Brown S, Babb DA, Pentel PR, Hatsukami DK. Carvedilol affects the physiological and behavioral response to smoked cocaine in humans. Drug Alcohol Depend. 2000;60:69-76.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force of Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:E1-E211.
- Dattilo PB, Hailpern SM, Fearon K, Sohal D, Nordin C. Beta-blockers are associated with reduced risk of myocardial infarction after cocaine use. Ann Emerg Med. 2008;51:117-125.
- Rangel C, Shu RG, Lazar LD, Vittinghoff E, Hsue PY, Marcus GM. Beta-blockers for chest pain associated with recent cocaine use. Arch Intern Med. 2010;170:874-879.
Case
A 38-year-old man with a history of tobacco use presents to the emergency department complaining of constant substernal chest pain for three hours. His temperature is 37.7°C, his heart rate is 110 beats per minute, and his blood pressure is 155/95 mmHg. He appears anxious and diaphoretic but examination is otherwise unremarkable. He admits to cocaine use one hour before the onset of symptoms. What are the appropriate treatments for his condition?
Overview
Cocaine is the second-most-commonly used illicit drug in the U.S. and represents 31% of all ED visits related to substance abuse.1,2 According to recent survey results, 2.1 million people report recent cocaine use, and 1.6 million engage in cocaine abuse or dependence.2 Acute cardiopulmonary complaints are common in individuals who present to the ED after cocaine use, with chest pain being the most frequently reported symptom in 40%.3
Numerous etiologies for cocaine-associated chest pain (CACP) have been discovered, including musculoskeletal pain, pulmonary hypertension, cardiomyopathy, arrhythmias, and endocarditis.4 Only 0.5% of patients with aortic dissection over a four-year period had a recent history of cocaine use, making cocaine a rare cause of a rare condition.5 Cardiac chest pain remains the most frequent underlying etiology, resulting in the most common complication of myocardial infarction (MI) in up to 6% of patients.6,7
The ways in which cocaine use can cause myocardial ischemia and MI are multifactorial. A vigorous central sympathomimetic effect, coronary artery vasoconstriction, stimulation of platelets, and enhanced atherosclerosis all lead to a myocardial oxygen supply-demand imbalance.8 Other key interactions in the cardiovascular system are displayed in Figure 1. Understanding the role of these mechanisms in CACP is crucial to patient care.
Clinician goals in the management of CACP are to rapidly and accurately exclude life-threatening etiologies; assess the need for urgent acute coronary syndrome (ACS) evaluation; risk-stratify patients and ensure appropriate disposition; normalize the toxic effects of cocaine; treat resultant organ damage; and prevent long-term complications. An algorithm detailing this approach is provided in Figure 2.
Review of the Data
Diagnostic evaluation. Given potential differences in treatment regimens, it is imperative to differentiate patients who present with CACP from those whose chest pain is not associated with cocaine either by direct questioning or by screening of urine for cocaine metabolites. Once the presence of cocaine has been confirmed, guideline-based evaluation for potential ACS with serial electrocardiograms (ECG), cardiac biomarkers, and close monitoring of cardiac rhythms and hemodynamics is largely similar to standard management of all patients presenting with chest pain, with a few caveats.
Interpretation of the ECG can be challenging in the setting of cocaine. Studies have shown “abnormal” ECGs in 56% to 84% of patients, with many representing early repolarization or left ventricular hypertrophy.9,10 Likewise, patients with MI are as likely to present with normal or nonspecific ECG findings as with ischemic findings.7,11 ECG interpretation to diagnose ischemia or infarction in patients with CACP yields a sensitivity of 36% and specificity of 90%.7
Creatine kinase (CK), CK-MB fraction, and myoglobin have low specificity for the diagnosis of ischemia, as cocaine can induce skeletal muscle injury and rhabdomyolysis.9,12 Cardiac troponins demonstrate a superior specificity compared to CK and CK-MB and are thus the preferred cardiac biomarkers in diagnosing cocaine-associated MI.12
Initial management and disposition. Patients at high risk for cardiovascular events are generally admitted to a monitored bed.13 Immediate reperfusion therapy with primary percutaneous coronary intervention is recommended in patients with ST-elevation MI (STEMI). Treatment with thrombolytic agents is associated with an increased risk of intracerebral hemorrhage and lacks documented efficacy in patients with CACP. Thrombolysis should therefore only be utilized if the diagnosis of STEMI is unequivocal and an experienced cardiac catheterization laboratory is unavailable.14,15
Patients with unstable angina (UA) or non-ST-elevation MI (NSTEMI) are at higher risk for further cardiac events in a similar manner to those with ACS unrelated to cocaine. These cases might benefit from early cardiac catheterization and revascularization.16 Because of the increased risk of stent thrombosis in cocaine-users, thought to be due to recidivism, a detailed risk-benefit analysis should be undertaken prior to the implantation of cardiac stents.
Other diagnostic tests, such as stress testing and myocardial imaging, have not shown significant accuracy in diagnosing MI in this setting; moreover, these patients are at low overall risk for cardiac events and mortality. Consequently, an extensive diagnostic evaluation might not be cost-effective.7,10,13,17 Patients who have CACP without MI have a very low frequency of delayed complications.3,17 As such, cost-effective evaluation strategies, such as nine- or 12-hour observation periods in a chest pain unit, are appropriate for many of these low- to moderate-risk patients.13 For all CACP patients, the most critical post-discharge interventions are cardiac risk modification and cocaine cessation.13
Normalizing the toxic effects of cocaine with medications.
Aspirin: While no specific study has been performed in patients with CACP and aspirin, CACP guidelines, based on data supporting ACS guidelines for all patients, recommend administration of full-dose aspirin given its associated reduction in morbidity and mortality.18,19 Furthermore, given the platelet-stimulating effects of cocaine, using aspirin in this setting seems very reasonable.
Benzodiazepines: CACP guidelines support the use of benzodiazepines early in management to indirectly combat the agitation, hypertension, and tachycardia resulting from the stimulatory effects of cocaine.18,20 These recommendations are based on several animal and human studies that demonstrate significant reduction in heart rate and systemic arterial pressure with the use of these agents.21,22
Nitroglycerin: Cardiac catheterization studies have shown reversal of vasoconstriction with administration of nitroglycerin. One study demonstrated a benefit of the drug in 49% of participants.23 Additional investigation into the benefit of benzodiazepine and nitroglycerin combination therapy revealed mixed results. In one study, lorazepam plus nitroglycerin was found to be more efficacious than nitroglycerin alone.24 In another, however, use of diazepam in combination with nitroglycerin did not show benefit when evaluating pain relief, cardiac dynamics, and left ventricular function.25
Phentolamine: Phentolamine administration has been studied much less in the literature. This nonselective alpha-adrenergic antagonist exerts a dose-dependent reversal of cocaine’s vasoconstrictive properties in monkeys and humans.26,27 International guidelines for Emergency Cardiovascular Care recommend its use in treatment of cocaine-associated ACS;27 however, the AHA recommends it less strongly.18
Calcium channel blockers: Calcium channel blockers (CCBs) have not shown promise as first-line agents. While catheterization studies demonstrate the vasodilatory properties of verapamil, larger studies looking at all-cause mortality conclude that CCBs might worsen mortality rates,28 and animal studies indicate an increased risk of seizures.29 At this time, CCBs are recommended only if cardiac symptoms continue after both benzodiazepines and nitroglycerin are administered.18
The beta-blocker controversy: The use of beta-blockers in patients with CACP remains controversial given the theoretical risk of unopposed alpha-adrenergic activation. Coronary vasospasm, decreased myocardial oxygen delivery, and increased systemic vascular resistance can result from their use.30
Propranolol, a nonselective beta-blocker, was shown in catheterization studies to potentiate the coronary vasoconstriction of cocaine.31 Labetalol, a combined alpha/beta-blocker, reduced mean arterial pressure after cocaine administration during cardiac catheterization but did not reverse coronary vasoconstriction.32 This was attributed to the predominating beta greater than alpha blockade at doses administered. The selective beta-1 antagonists esmolol and metoprolol have shown no benefit in CACP.33 Carvedilol, a combined alpha/beta-blocker with both peripheral and central nervous system activity, has potential to attenuate both physiologic and behavioral response to cocaine, but it has not been well studied in this patient subset.34
The 2005 ACC/AHA STEMI guidelines recommended against beta-blockers in the setting of STEMI precipitated by cocaine use due to the potential of exacerbating coronary vasoconstriction.35 The 2007 ACC/AHA UA/NSTEMI guidelines stated that the use of a combined alpha/beta-blocker in patients with cocaine-induced ACS may be reasonable for patients with hypertension or tachycardia if pre-treated with a vasodilator.19 The 2008 ACC/AHA guidelines on the management of cocaine-related chest pain and MI recommended against the use of beta-blockers in the acute setting given the low incidence of cocaine-related MI and death.18
In a more recent study, Dattilo et al showed that beta-blockers administered to patients admitted with positive urine toxicology for cocaine significantly reduced MI and in-hospital mortality. Reduction of MI was of borderline significance in those admitted with a chief complaint of chest pain.36 Limitations of this study include unknown time of cocaine ingestion, lack of follow-up on discharge mortality, and a small sample size of 348 patients lacking statistical power.
Another retrospective cohort study examined patients admitted with chest pain and urine toxicology positive for cocaine and found that beta-blocker administration during hospitalization was not associated with increased incident mortality. Further, after a mean follow-up of 2.5 years, there was a statistically significant decrease in cardiovascular death.37 Drawbacks of this study included an older patient population, greater proportion of coronary artery disease, and higher follow-up of cardiovascular mortality rates than in previous studies, suggesting this subset might have received greater benefit from beta-blockers as a result of these characteristics.
The 2008 ACC/AHA guidelines instruct individualized consideration of the risk/benefit ratio for beta-blocker use in patients with CACP given the high rate of recidivism in cocaine abusers. The strongest indication is given to those with documented MI, left ventricular systolic dysfunction, or ventricular arrhythmias.18
It is important to note that these recommendations are based on cardiac catheterization laboratory studies, case reports, retrospective analyses, and animal experiments. No prospective controlled trials evaluating the role of beta-blockers in CACP and MI exist, and no trials regarding therapies to improve outcomes of patients sustaining a cocaine-associated MI have been reported.18
Back to the Case
This patient was experiencing cocaine-associated chest pain, which was confirmed with positive urine toxicology. Initial diagnostic workup with basic laboratory studies and cardiac biomarkers showed mild elevation in CK with troponin levels within normal limits. His ECG showed changes consistent with left ventricular hypertrophy. Chest radiograph was unremarkable.
He received aspirin, benzodiazepines, and nitroglycerin with normalization of vital signs, as well as subjective improvement in chest pain and anxiety. He was deemed to be at low risk for potential cardiac complications; thus, further cardiac testing was not pursued. Rather, he was admitted to an overnight observation unit with telemetry monitoring, where his chest pain did not recur.
He was seen in consultation with social work staff who arranged for drug abuse counseling after discharge. Given the uncertainty of relapse to cocaine use, as well as lack of known cardiac risk factors, he was not discharged on any new medications.
Bottom Line
The treatment of CACP includes normalizing the toxic systemic effects of the drug and minimizing the direct ischemic damage to the myocardium. Management varies slightly from traditional chest pain algorithms and includes benzodiazepines as well as antiplatelet agents and vasodilators to achieve this goal. Initial therapy with beta-blockers remains undefined and is largely discouraged in the acute setting. The role of beta-blockade upon discharge, however, can be beneficial in specific populations, especially those found to have underlying coronary disease.
Dr. Houchens and Dr. Czarnik are clinical instructors and Dr. Mack is a clinical lecturer at the University of Michigan Health System in Ann Arbor.
References
- Hughes A, Sathe N, Spagnola K. State Estimates of Substance Use from the 2005-2006 National Surveys on Drug Use and Health. DHHS Publication No. SMA 08-4311, NSDUH Series H-33. Rockville, MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2008.
- Volkow ND. Cocaine: Abuse and Addiction. National Institute on Drug Abuse. Washington, DC: U.S. Department of Health and Human Services; 2009.
- Brody SL, Slovis CM, Wrenn KD. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325-331.
- Levis JT, Garmel GM. Cocaine-associated chest pain. Emerg Med Clin North Am. 2005;23:1083-1103.
- Eagle KA, Isselbacher EM, DeSanctis RW. Cocaine-related aortic dissection in perspective. Circulation. 2002;105:1529-1530.
- Feldman JA, Fish SS, Beshansky JR, Griffith JL, Woolard RH, Selker HP. Acute cardiac ischemia in patients with cocaine-associated complaints: results of a multicenter trial. Ann Emerg Med. 2000;36:469-476.
- Hollander JE, Hoffman RS, Gennis P, et al. Prospective multicenter evaluation of cocaine associated chest pain. Cocaine Associated Chest Pain (COCHPA) Study Group. Acad Emerg Med. 1994;1:330-339.
- Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558-2569.
- Gitter MJ, Goldsmith SR, Dunbar DN, et al. Cocaine and chest pain: clinical features and outcomes of patients hospitalized to rule out myocardial infarction. Ann Intern Med. 1991;115:277-282.
- Amin M, Gabelman G, Karpel J, et al. Acute myocardial infarction and chest pain syndromes after cocaine use. Am J Cardiol. 1990;66:1434-1437.
- Tokarski GF, Paganussi P, Urbanski R, et al. An evaluation of cocaine-induced chest pain. Ann Emerg Med. 1990;19:1088-1092.
- Hollander JE, Levitt MA, Young GP, Briglia E, Wetli CV, Gawad Y. Effect of recent cocaine use on the specificity of cardiac markers for diagnosis of acute myocardial infarction. Am Heart J. 1998;135(2 Pt 1):245-252.
- Weber JE, Shofer FS, Larkin GL, Kalaria AS, Hollander JE. Validation of a brief observation period for patients with cocaine-associated chest pain. N Engl J Med. 2003;348:510-517.
- Hahn IH, Hoffman RS. Diagnosis and treatment of acute myocardial infarction: cocaine use and acute myocardial infarction. Emerg Med Clin North Am. 2001;19(2):1-18.
- Hoffman RS, Hollander JE. Evaluation of patients with chest pain after cocaine use. Crit Care Clin. 1997;13:809-828. Cannon CP, Weintraub WS, Demopoulos LA, et al.
- Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879-1887.
- Hollander JE, Hoffman RS. Cocaine-induced myocardial infarction: an analysis and review of the literature. J Emerg Med. 1992;10:169-177.
- McCord J, Jneid H, Hollander JE, et al. Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897-1907.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol. 2007;50:E1-E157.
- Hollander JE. Management of cocaine-associated myocardial ischemia. N Engl J Med. 1995;333:1267-1272.
- Brubacher JR, Hoffman RS. Cocaine toxicity. Top Emerg Med. 1997;19(4):1-16.
- Catavas JD, Waters IW. Acute cocaine intoxication in the conscious dog: studies on the mechanism of lethality. J Pharmacol Exp Ther. 1981;217:350-356.
- Hollander JE, Hoffman RS, Gennis P, et al. Nitroglycerin in the treatment of cocaine associated chest pain—clinical safety and efficacy. J Toxicol Clin Toxicol. 1994;32(3): 243-256.
- Honderick T, Williams D, Seaberg D, Wears R. A prospective, randomized, controlled trial of benzodiazepines and nitroglycerin or nitroglycerin alone in the treatment of cocaine-associated acute coronary syndromes. Am J Emerg Med. 2003;21(1):39-42.
- Baumann BM, Perrone J, Hornig SE, Shofer FS, Hollander JE. Randomized, double-blind, placebo-controlled trial of diazepam, nitroglycerin, or both for treatment of patients with potential cocaine-associated acute coronary syndromes. Acad Emerg Med. 2000;7:878-885.
- Schindler CW, Tella SR, Goldberg SR. Adrenoceptor mechanisms in the cardiovascular effects of cocaine in conscious squirrel monkeys. Life Sci. 1992;51(9):653-660.
- Lange RA, Cigarroa RG, Yancy CW Jr., et al. Cocaine-induced coronary-artery vasoconstriction. N Engl J Med. 1989;321(23):1557-1562.
- Furberg CD, Psaty BM, Meyer JV. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326-1331.
- Derlet RW, Albertson TE. Potentiation of cocaine toxicity with calcium channel blockers. Am J Emerg Med. 1989;7:464-468.
- Lange RA, Hillis LD. Cardiovascular complications of cocaine use. N Engl J Med. 2001;345:351-358.
- Lange RA, Cigarroa RG, Flores ED, et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112:897-903.
- Boehrer JD, Moliterno DJ, Willard JE, Hillis LD, Lange RA. Influence of labetalol on cocaine-induced coronary vasoconstriction in humans. Am J Med. 1993;94:608-610.
- Sand IC, Brody SL, Wrenn KD, Slovis CM. Experience with esmolol for the treatment of cocaine-associated cardiovascular complications. Am J Emerg Med. 1991;9:161-163.
- Sofuoglo M, Brown S, Babb DA, Pentel PR, Hatsukami DK. Carvedilol affects the physiological and behavioral response to smoked cocaine in humans. Drug Alcohol Depend. 2000;60:69-76.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force of Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:E1-E211.
- Dattilo PB, Hailpern SM, Fearon K, Sohal D, Nordin C. Beta-blockers are associated with reduced risk of myocardial infarction after cocaine use. Ann Emerg Med. 2008;51:117-125.
- Rangel C, Shu RG, Lazar LD, Vittinghoff E, Hsue PY, Marcus GM. Beta-blockers for chest pain associated with recent cocaine use. Arch Intern Med. 2010;170:874-879.
Case
A 38-year-old man with a history of tobacco use presents to the emergency department complaining of constant substernal chest pain for three hours. His temperature is 37.7°C, his heart rate is 110 beats per minute, and his blood pressure is 155/95 mmHg. He appears anxious and diaphoretic but examination is otherwise unremarkable. He admits to cocaine use one hour before the onset of symptoms. What are the appropriate treatments for his condition?
Overview
Cocaine is the second-most-commonly used illicit drug in the U.S. and represents 31% of all ED visits related to substance abuse.1,2 According to recent survey results, 2.1 million people report recent cocaine use, and 1.6 million engage in cocaine abuse or dependence.2 Acute cardiopulmonary complaints are common in individuals who present to the ED after cocaine use, with chest pain being the most frequently reported symptom in 40%.3
Numerous etiologies for cocaine-associated chest pain (CACP) have been discovered, including musculoskeletal pain, pulmonary hypertension, cardiomyopathy, arrhythmias, and endocarditis.4 Only 0.5% of patients with aortic dissection over a four-year period had a recent history of cocaine use, making cocaine a rare cause of a rare condition.5 Cardiac chest pain remains the most frequent underlying etiology, resulting in the most common complication of myocardial infarction (MI) in up to 6% of patients.6,7
The ways in which cocaine use can cause myocardial ischemia and MI are multifactorial. A vigorous central sympathomimetic effect, coronary artery vasoconstriction, stimulation of platelets, and enhanced atherosclerosis all lead to a myocardial oxygen supply-demand imbalance.8 Other key interactions in the cardiovascular system are displayed in Figure 1. Understanding the role of these mechanisms in CACP is crucial to patient care.
Clinician goals in the management of CACP are to rapidly and accurately exclude life-threatening etiologies; assess the need for urgent acute coronary syndrome (ACS) evaluation; risk-stratify patients and ensure appropriate disposition; normalize the toxic effects of cocaine; treat resultant organ damage; and prevent long-term complications. An algorithm detailing this approach is provided in Figure 2.
Review of the Data
Diagnostic evaluation. Given potential differences in treatment regimens, it is imperative to differentiate patients who present with CACP from those whose chest pain is not associated with cocaine either by direct questioning or by screening of urine for cocaine metabolites. Once the presence of cocaine has been confirmed, guideline-based evaluation for potential ACS with serial electrocardiograms (ECG), cardiac biomarkers, and close monitoring of cardiac rhythms and hemodynamics is largely similar to standard management of all patients presenting with chest pain, with a few caveats.
Interpretation of the ECG can be challenging in the setting of cocaine. Studies have shown “abnormal” ECGs in 56% to 84% of patients, with many representing early repolarization or left ventricular hypertrophy.9,10 Likewise, patients with MI are as likely to present with normal or nonspecific ECG findings as with ischemic findings.7,11 ECG interpretation to diagnose ischemia or infarction in patients with CACP yields a sensitivity of 36% and specificity of 90%.7
Creatine kinase (CK), CK-MB fraction, and myoglobin have low specificity for the diagnosis of ischemia, as cocaine can induce skeletal muscle injury and rhabdomyolysis.9,12 Cardiac troponins demonstrate a superior specificity compared to CK and CK-MB and are thus the preferred cardiac biomarkers in diagnosing cocaine-associated MI.12
Initial management and disposition. Patients at high risk for cardiovascular events are generally admitted to a monitored bed.13 Immediate reperfusion therapy with primary percutaneous coronary intervention is recommended in patients with ST-elevation MI (STEMI). Treatment with thrombolytic agents is associated with an increased risk of intracerebral hemorrhage and lacks documented efficacy in patients with CACP. Thrombolysis should therefore only be utilized if the diagnosis of STEMI is unequivocal and an experienced cardiac catheterization laboratory is unavailable.14,15
Patients with unstable angina (UA) or non-ST-elevation MI (NSTEMI) are at higher risk for further cardiac events in a similar manner to those with ACS unrelated to cocaine. These cases might benefit from early cardiac catheterization and revascularization.16 Because of the increased risk of stent thrombosis in cocaine-users, thought to be due to recidivism, a detailed risk-benefit analysis should be undertaken prior to the implantation of cardiac stents.
Other diagnostic tests, such as stress testing and myocardial imaging, have not shown significant accuracy in diagnosing MI in this setting; moreover, these patients are at low overall risk for cardiac events and mortality. Consequently, an extensive diagnostic evaluation might not be cost-effective.7,10,13,17 Patients who have CACP without MI have a very low frequency of delayed complications.3,17 As such, cost-effective evaluation strategies, such as nine- or 12-hour observation periods in a chest pain unit, are appropriate for many of these low- to moderate-risk patients.13 For all CACP patients, the most critical post-discharge interventions are cardiac risk modification and cocaine cessation.13
Normalizing the toxic effects of cocaine with medications.
Aspirin: While no specific study has been performed in patients with CACP and aspirin, CACP guidelines, based on data supporting ACS guidelines for all patients, recommend administration of full-dose aspirin given its associated reduction in morbidity and mortality.18,19 Furthermore, given the platelet-stimulating effects of cocaine, using aspirin in this setting seems very reasonable.
Benzodiazepines: CACP guidelines support the use of benzodiazepines early in management to indirectly combat the agitation, hypertension, and tachycardia resulting from the stimulatory effects of cocaine.18,20 These recommendations are based on several animal and human studies that demonstrate significant reduction in heart rate and systemic arterial pressure with the use of these agents.21,22
Nitroglycerin: Cardiac catheterization studies have shown reversal of vasoconstriction with administration of nitroglycerin. One study demonstrated a benefit of the drug in 49% of participants.23 Additional investigation into the benefit of benzodiazepine and nitroglycerin combination therapy revealed mixed results. In one study, lorazepam plus nitroglycerin was found to be more efficacious than nitroglycerin alone.24 In another, however, use of diazepam in combination with nitroglycerin did not show benefit when evaluating pain relief, cardiac dynamics, and left ventricular function.25
Phentolamine: Phentolamine administration has been studied much less in the literature. This nonselective alpha-adrenergic antagonist exerts a dose-dependent reversal of cocaine’s vasoconstrictive properties in monkeys and humans.26,27 International guidelines for Emergency Cardiovascular Care recommend its use in treatment of cocaine-associated ACS;27 however, the AHA recommends it less strongly.18
Calcium channel blockers: Calcium channel blockers (CCBs) have not shown promise as first-line agents. While catheterization studies demonstrate the vasodilatory properties of verapamil, larger studies looking at all-cause mortality conclude that CCBs might worsen mortality rates,28 and animal studies indicate an increased risk of seizures.29 At this time, CCBs are recommended only if cardiac symptoms continue after both benzodiazepines and nitroglycerin are administered.18
The beta-blocker controversy: The use of beta-blockers in patients with CACP remains controversial given the theoretical risk of unopposed alpha-adrenergic activation. Coronary vasospasm, decreased myocardial oxygen delivery, and increased systemic vascular resistance can result from their use.30
Propranolol, a nonselective beta-blocker, was shown in catheterization studies to potentiate the coronary vasoconstriction of cocaine.31 Labetalol, a combined alpha/beta-blocker, reduced mean arterial pressure after cocaine administration during cardiac catheterization but did not reverse coronary vasoconstriction.32 This was attributed to the predominating beta greater than alpha blockade at doses administered. The selective beta-1 antagonists esmolol and metoprolol have shown no benefit in CACP.33 Carvedilol, a combined alpha/beta-blocker with both peripheral and central nervous system activity, has potential to attenuate both physiologic and behavioral response to cocaine, but it has not been well studied in this patient subset.34
The 2005 ACC/AHA STEMI guidelines recommended against beta-blockers in the setting of STEMI precipitated by cocaine use due to the potential of exacerbating coronary vasoconstriction.35 The 2007 ACC/AHA UA/NSTEMI guidelines stated that the use of a combined alpha/beta-blocker in patients with cocaine-induced ACS may be reasonable for patients with hypertension or tachycardia if pre-treated with a vasodilator.19 The 2008 ACC/AHA guidelines on the management of cocaine-related chest pain and MI recommended against the use of beta-blockers in the acute setting given the low incidence of cocaine-related MI and death.18
In a more recent study, Dattilo et al showed that beta-blockers administered to patients admitted with positive urine toxicology for cocaine significantly reduced MI and in-hospital mortality. Reduction of MI was of borderline significance in those admitted with a chief complaint of chest pain.36 Limitations of this study include unknown time of cocaine ingestion, lack of follow-up on discharge mortality, and a small sample size of 348 patients lacking statistical power.
Another retrospective cohort study examined patients admitted with chest pain and urine toxicology positive for cocaine and found that beta-blocker administration during hospitalization was not associated with increased incident mortality. Further, after a mean follow-up of 2.5 years, there was a statistically significant decrease in cardiovascular death.37 Drawbacks of this study included an older patient population, greater proportion of coronary artery disease, and higher follow-up of cardiovascular mortality rates than in previous studies, suggesting this subset might have received greater benefit from beta-blockers as a result of these characteristics.
The 2008 ACC/AHA guidelines instruct individualized consideration of the risk/benefit ratio for beta-blocker use in patients with CACP given the high rate of recidivism in cocaine abusers. The strongest indication is given to those with documented MI, left ventricular systolic dysfunction, or ventricular arrhythmias.18
It is important to note that these recommendations are based on cardiac catheterization laboratory studies, case reports, retrospective analyses, and animal experiments. No prospective controlled trials evaluating the role of beta-blockers in CACP and MI exist, and no trials regarding therapies to improve outcomes of patients sustaining a cocaine-associated MI have been reported.18
Back to the Case
This patient was experiencing cocaine-associated chest pain, which was confirmed with positive urine toxicology. Initial diagnostic workup with basic laboratory studies and cardiac biomarkers showed mild elevation in CK with troponin levels within normal limits. His ECG showed changes consistent with left ventricular hypertrophy. Chest radiograph was unremarkable.
He received aspirin, benzodiazepines, and nitroglycerin with normalization of vital signs, as well as subjective improvement in chest pain and anxiety. He was deemed to be at low risk for potential cardiac complications; thus, further cardiac testing was not pursued. Rather, he was admitted to an overnight observation unit with telemetry monitoring, where his chest pain did not recur.
He was seen in consultation with social work staff who arranged for drug abuse counseling after discharge. Given the uncertainty of relapse to cocaine use, as well as lack of known cardiac risk factors, he was not discharged on any new medications.
Bottom Line
The treatment of CACP includes normalizing the toxic systemic effects of the drug and minimizing the direct ischemic damage to the myocardium. Management varies slightly from traditional chest pain algorithms and includes benzodiazepines as well as antiplatelet agents and vasodilators to achieve this goal. Initial therapy with beta-blockers remains undefined and is largely discouraged in the acute setting. The role of beta-blockade upon discharge, however, can be beneficial in specific populations, especially those found to have underlying coronary disease.
Dr. Houchens and Dr. Czarnik are clinical instructors and Dr. Mack is a clinical lecturer at the University of Michigan Health System in Ann Arbor.
References
- Hughes A, Sathe N, Spagnola K. State Estimates of Substance Use from the 2005-2006 National Surveys on Drug Use and Health. DHHS Publication No. SMA 08-4311, NSDUH Series H-33. Rockville, MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2008.
- Volkow ND. Cocaine: Abuse and Addiction. National Institute on Drug Abuse. Washington, DC: U.S. Department of Health and Human Services; 2009.
- Brody SL, Slovis CM, Wrenn KD. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325-331.
- Levis JT, Garmel GM. Cocaine-associated chest pain. Emerg Med Clin North Am. 2005;23:1083-1103.
- Eagle KA, Isselbacher EM, DeSanctis RW. Cocaine-related aortic dissection in perspective. Circulation. 2002;105:1529-1530.
- Feldman JA, Fish SS, Beshansky JR, Griffith JL, Woolard RH, Selker HP. Acute cardiac ischemia in patients with cocaine-associated complaints: results of a multicenter trial. Ann Emerg Med. 2000;36:469-476.
- Hollander JE, Hoffman RS, Gennis P, et al. Prospective multicenter evaluation of cocaine associated chest pain. Cocaine Associated Chest Pain (COCHPA) Study Group. Acad Emerg Med. 1994;1:330-339.
- Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558-2569.
- Gitter MJ, Goldsmith SR, Dunbar DN, et al. Cocaine and chest pain: clinical features and outcomes of patients hospitalized to rule out myocardial infarction. Ann Intern Med. 1991;115:277-282.
- Amin M, Gabelman G, Karpel J, et al. Acute myocardial infarction and chest pain syndromes after cocaine use. Am J Cardiol. 1990;66:1434-1437.
- Tokarski GF, Paganussi P, Urbanski R, et al. An evaluation of cocaine-induced chest pain. Ann Emerg Med. 1990;19:1088-1092.
- Hollander JE, Levitt MA, Young GP, Briglia E, Wetli CV, Gawad Y. Effect of recent cocaine use on the specificity of cardiac markers for diagnosis of acute myocardial infarction. Am Heart J. 1998;135(2 Pt 1):245-252.
- Weber JE, Shofer FS, Larkin GL, Kalaria AS, Hollander JE. Validation of a brief observation period for patients with cocaine-associated chest pain. N Engl J Med. 2003;348:510-517.
- Hahn IH, Hoffman RS. Diagnosis and treatment of acute myocardial infarction: cocaine use and acute myocardial infarction. Emerg Med Clin North Am. 2001;19(2):1-18.
- Hoffman RS, Hollander JE. Evaluation of patients with chest pain after cocaine use. Crit Care Clin. 1997;13:809-828. Cannon CP, Weintraub WS, Demopoulos LA, et al.
- Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879-1887.
- Hollander JE, Hoffman RS. Cocaine-induced myocardial infarction: an analysis and review of the literature. J Emerg Med. 1992;10:169-177.
- McCord J, Jneid H, Hollander JE, et al. Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897-1907.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol. 2007;50:E1-E157.
- Hollander JE. Management of cocaine-associated myocardial ischemia. N Engl J Med. 1995;333:1267-1272.
- Brubacher JR, Hoffman RS. Cocaine toxicity. Top Emerg Med. 1997;19(4):1-16.
- Catavas JD, Waters IW. Acute cocaine intoxication in the conscious dog: studies on the mechanism of lethality. J Pharmacol Exp Ther. 1981;217:350-356.
- Hollander JE, Hoffman RS, Gennis P, et al. Nitroglycerin in the treatment of cocaine associated chest pain—clinical safety and efficacy. J Toxicol Clin Toxicol. 1994;32(3): 243-256.
- Honderick T, Williams D, Seaberg D, Wears R. A prospective, randomized, controlled trial of benzodiazepines and nitroglycerin or nitroglycerin alone in the treatment of cocaine-associated acute coronary syndromes. Am J Emerg Med. 2003;21(1):39-42.
- Baumann BM, Perrone J, Hornig SE, Shofer FS, Hollander JE. Randomized, double-blind, placebo-controlled trial of diazepam, nitroglycerin, or both for treatment of patients with potential cocaine-associated acute coronary syndromes. Acad Emerg Med. 2000;7:878-885.
- Schindler CW, Tella SR, Goldberg SR. Adrenoceptor mechanisms in the cardiovascular effects of cocaine in conscious squirrel monkeys. Life Sci. 1992;51(9):653-660.
- Lange RA, Cigarroa RG, Yancy CW Jr., et al. Cocaine-induced coronary-artery vasoconstriction. N Engl J Med. 1989;321(23):1557-1562.
- Furberg CD, Psaty BM, Meyer JV. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326-1331.
- Derlet RW, Albertson TE. Potentiation of cocaine toxicity with calcium channel blockers. Am J Emerg Med. 1989;7:464-468.
- Lange RA, Hillis LD. Cardiovascular complications of cocaine use. N Engl J Med. 2001;345:351-358.
- Lange RA, Cigarroa RG, Flores ED, et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112:897-903.
- Boehrer JD, Moliterno DJ, Willard JE, Hillis LD, Lange RA. Influence of labetalol on cocaine-induced coronary vasoconstriction in humans. Am J Med. 1993;94:608-610.
- Sand IC, Brody SL, Wrenn KD, Slovis CM. Experience with esmolol for the treatment of cocaine-associated cardiovascular complications. Am J Emerg Med. 1991;9:161-163.
- Sofuoglo M, Brown S, Babb DA, Pentel PR, Hatsukami DK. Carvedilol affects the physiological and behavioral response to smoked cocaine in humans. Drug Alcohol Depend. 2000;60:69-76.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force of Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:E1-E211.
- Dattilo PB, Hailpern SM, Fearon K, Sohal D, Nordin C. Beta-blockers are associated with reduced risk of myocardial infarction after cocaine use. Ann Emerg Med. 2008;51:117-125.
- Rangel C, Shu RG, Lazar LD, Vittinghoff E, Hsue PY, Marcus GM. Beta-blockers for chest pain associated with recent cocaine use. Arch Intern Med. 2010;170:874-879.
How Is SIADH Diagnosed and Managed?
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4

Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.

Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4
Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.
Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
Case
A 70-year-old woman with hypertension presents after a fall. Her medications include hydrochlorothiazide. Her blood pressure is 130/70 mm/Hg, with heart rate of 86. She has normal orthostatic vital signs. Her mucus membranes are moist and she has no jugular venous distension, edema, or ascites. Her plasma sodium (PNa) is 125 mmol/L, potassium 3.6 mmol/L, blood urea nitrogen (BUN) 30 mg/dL, and creatinine 0.8 mg/dL. Additional labs include serum thyroid stimulating hormone 1.12 mIU/L, cortisol 15 mcg/dL, serum osmolality 270 mOsm/kg, uric acid 4 mg/dL, urine osmolality 300 mOsm/kg, urine sodium (UNa) 40 mmol/L, fractional excretion of sodium 1.0%, and fractional excretion of urate (FEUrate) 13%. She receives 2 L isotonic saline intravenously over 24 hours, with resulting PNa of 127.
What is the cause of her hyponatremia, and how should her hyponatremia be managed?
Overview
Hyponatremia is one of the most common electrolyte abnormalities; it has a prevalence as high as 30% upon admission to the hospital.1 Hyponatremia is important clinically because of its high risk of mortality in the acute and symptomatic setting, and the risk of central pontine myelinolysis (CPM), or death with too rapid correction.2 Even so-called “asymptomatic” mild hyponatremia is associated with increased falls and impairments in gait and attention in the elderly.3
Hyponatremia is a state of excess water compared with the amount of solute in the extracellular fluid. To aid in diagnosing the etiology of hypotonic hyponatremia, the differential is traditionally divided into categories based on extracellular fluid volume (ECV) status, as shown in Table 1 (below), with syndrome of inappropriate antidiuretic hormone secretion (SIADH) being the most common cause of euvolemic hyponatremia.2 However, data show that clinical determination of volume status is often flawed,4 and an algorithmic approach to diagnosis and treatment yields improved results.5
Review of the Data
Diagnosis of SIADH. The original diagnostic criteria for SIADH, with minor modifications, are presented in Table 2, page 18).6,7,8 However, applying these criteria in clinical settings presents several difficulties, most notably a determination of ECV. The gold standard for assessing ECV status is by radioisotope, which is not practically feasible.9 Therefore, clinicians must rely on surrogate clinical markers of ECV (orthostatic hypotension, skin turgor, mucus membrane dryness, central venous pressure, BUN, BUN-creatinine ratio, and serum uric acid levels), which lack both sensitivity and specificity.4 Astoundingly, clinical assessment of ECV has been demonstrated to be accurate only 50% of the time when differentiating euvolemic patients from those with hypovolemia.4
Another challenge lies in the interpretation of UNa, which frequently is used as a surrogate for extra-arterial blood volume (EABV) status.10 Unfortunately, in the setting of diuretic use, UNa becomes inaccurate. The FEUrate, however, is unaffected by diuretic use and can be helpful in distinguishing between etiologies of hyponatremia with UNa greater than 30 mmol/L.11 The FEUrate is about 10% in normal euvolemic subjects and is reduced (usually <8%) in patients with low effective arterial blood volume.11,12 A trial of 86 patients demonstrated that a FEUrate of 12% had a specificity and positive predictive value of 100% in accurately identifying SIADH from diuretic-induced hyponatremia in patients on diuretics.11,12 Therefore, the UNa is a valid marker of EABV status when patients are not on diuretics; however, the FEUrate should be used in the setting of diuretic use.
Yet another pitfall is differentiating patients with salt depletion from those with SIADH. In these situations, measurement of the change in PNa concentration after a test infusion of isotonic saline is helpful. In salt depletion, PNa usually increases ≥5 mmol/L after 2 L saline infusion, which is not the case with SIADH.13 Incorrectly diagnosing renal salt wasting (RSW) as SIADH results in fluid restriction and, consequently, ECV depletion and increased morbidity.14 The persistence of hypouricemia and elevated FEUrate after correction of the hyponatremia in RSW differentiates it from SIADH.13, 14
Given these challenges, recommendations to use an algorithmic approach for the evaluation and diagnosis of hyponatremia have surfaced. In a study of 121 patients admitted with hyponatremia, an algorithm-based approach to the diagnosis of hyponatremia yielded an overall diagnostic accuracy of 71%, compared with an accuracy of 32% by experienced clinicians.5 This study also highlighted SIADH as the most frequent false-positive diagnosis that was expected whenever the combination of euvolemia and a UNa >30 mmol/L was present.5 Cases of diuretic-induced hyponatremia often were misclassified due to errors in the accurate assessment of ECV status, as most of these patients appeared clinically euvolemic or hypervolemic.5 Therefore, it is important to use an algorithm in identifying SIADH and to use one that does not rely solely on clinical estimation of ECV status (see Figure 1, below).
Management of acute and symptomatic hyponatremia. When hyponatremia develops acutely, urgent treatment is required (see Figure 2, below).15 Hyponatremia is considered acute when the onset is within 48 hours.15 Acute hyponatremia is most easily identified in the hospital and is commonly iatrogenic. Small case reviews in the 1980s began to associate postoperative deaths with the administration of hypotonic fluids.16 Asymptomatic patients with hyponatremia presenting from home should be considered chronic hyponatremias as the duration often is unclear.
Acute hyponatremia or neurologically symptomatic hyponatremia regardless of duration requires the use of hypertonic saline.15 Traditional sodium correction algorithms are based on early case series, which were focused on limiting neurologic complications from sodium overcorrection.17 This resulted in protocols recommending a conservative rate of correction spread over a 24- to 48-hour period.17 Infusing 3% saline at a rate of 1 ml/kg/hr to 2 ml/kg/hr results in a 1 mmol/L/hr to 2 mmol/L/hr increase in PNa.15 This simplified formula results in similar correction rates as more complex calculations.15 Correction should not exceed 8 mmol/L to 10 mmol/L within the first 24 hours, and 18 mmol/L to 25 mmol/L by 48 hours to avoid CPM.15 PNa should be checked every two hours to ensure that the correction rate is not exceeding the predicted rate, as the formulas do not take into account oral intake and ongoing losses.15
Recent observations focused on the initial four hours from onset of hyponatremia suggest a higher rate of correction can be tolerated without complications.18 Rapid sodium correction of 4 mmol/L to 6 mmol/L often is enough to stop neurologic complications.18 This can be accomplished with a bolus infusion of 100 mL of 3% saline.19 This may be repeated twice at 10-minute intervals until there is neurologic improvement.19 This might sound aggressive, but this would correspond to a rise in PNa of 5 mmol/L to 6 mmol/L in a 50 kg woman. Subsequent treatment with hypertonic fluid might not be needed if symptoms resolve.
Management of chronic hyponatremia. Hyponatremia secondary to SIADH improves with the treatment of the underlying cause, thus an active search for a causative medication or condition should be sought (see Table 1, p. 17).20
Water restriction. Restriction of fluid intake is the first-line treatment for SIADH in patients without hypovolemia. The severity of fluid restriction is guided by the concentration of the urinary solutes.15 Restriction of water intake to 500 ml/day to 1,000 ml/day is generally advised for many patients, as losses from the skin, lungs, and urine exceed this amount, leading to a gradual reduction in total body water.21 The main drawback of fluid restriction is poor compliance due to an intact thirst mechanism.
Saline infusion. The infusion of normal saline theoretically worsens hyponatremia due to SIADH because the water is retained while the salt is excreted. However, a trial of normal saline sometimes is attempted in patients in whom the differentiation between hypovolemia and euvolemia is difficult. From a study of a series of 17 patients with chronic SIADH, Musch and Decaux concluded that the infusion of intravenous normal (0.9%) saline raises PNa when the urine osmolality is less than 530 mosm/L.22
Oral solutes (urea and salt). The oral intake of salt augments water excretion23, and salt tablets are used as a second-line agent in patients with persistent hyponatremia despite fluid restriction.23 The oral administration of urea also results in increased free-water excretion via osmotic diuresis,24 but its poor palatability, lack of availability in the U.S., and limited user experience has restricted its usage.24
Demeclocycline. Demeclo-cycline is a tetracycline derivative that causes a partial nephrogenic diabetes insipidus.25 Its limitations include a slow onset of action (two to five days) and an unpredictable treatment effect with the possibility of causing profound polyuria and hypernatremia. It is also associated with reversible azotemia and sometimes nephrotoxicity, especially in patients with cirrhosis.
Lithium. Lithium also causes nephrogenic diabetes insipidus by downregulating vasopressin-stimulated aquaporin-2 expression and thus improves hyponatremia in SIADH.26 However, its use is significantly limited by its unpredictable response and the risks of interstitial nephritis and end-stage renal disease with chronic use. Therefore, it is no longer recommended for the treatment of SIADH.
Vasopressin receptor antagonists. Due to the role of excessive levels of vasopressin in the pathophysiology of most types of SIADH, antagonists of the vasopressin receptor were developed with the goal of preventing the excess water absorption that causes hyponatremia. Two vasopressin receptor antagonists, or vaptans, have been approved by the FDA for the treatment of nonemergent euvolemic and hypervolemic hyponatremia. Conivaptan is a nonselective vasopressin receptor antagonist that is for IV use only. Tolvaptan is a selective V2 receptor antagonist that is taken orally. Both conivaptan and tolvaptan successfully increase PNa levels while the drugs are being taken.27,28,29,30 Tolvaptan increases PNa levels in hyponatremia due to SIADH and CHF, and modestly so in cirrhosis.30
The most common side effects of the vaptans include dry mouth, increased thirst, and increased urination, although serious side effects (hypernatremia or too-rapid rate of increase in PNa) are possible.29 It is unclear if treating stable, asymptomatic hyponatremia with vaptans has any reduction in morbidity or mortality. One study found that tolvaptan increased the patients’ self-evaluations of mental functioning, but a study of tolvaptan used in combination with diuretics in the setting of CHF did not result in decreased mortality.29,31 Due to their expense, necessity of being started in the hospital, and unclear long-term benefit, the vaptans are only recommended when traditional measures such as fluid restriction and salt tablets have been unsuccessful.
Back to the Case
Our patient has hypotonic hyponatremia based on her low serum osmolality. The duration of her hyponatremia is unclear, but the patient is not experiencing seizures or coma. Therefore, her hyponatremia should be corrected slowly, and hypertonic saline is not indicated.
As is common in clinical practice, her true volume status is difficult to clinically ascertain. By physical exam, she appears euvolemic, but because she is on hydrochlorothiazide, she might be subtly hypovolemic. The UNa of 40 mmol/L is not consistent with hypovolemia, but its accuracy is limited in the setting of diuretics. The failure to improve her sodium by at least 5 mmol/L after a 2 L normal saline infusion argues against low effective arterial blood volume and indicates that the hydrochlorothiazide is unlikely to be the cause of her hyponatremia.
Therefore, the most likely cause of the hyponatremia is SIADH, a diagnosis further corroborated by the elevated FEUrate of 13%. Her chronic hyponatremia should be managed initially with fluid restriction while an investigation for an underlying cause of SIADH is initiated.
Bottom Line
The diagnosis of SIADH relies on the careful evaluation of laboratory values, use of an algorithm, and recognizing the limitations of clinically assessing volume status. The underlying cause of SIADH must also be sought and treated. TH
Dr. Grant is a clinical lecturer in internal medicine, Dr. Cho is a clinical instructor in internal medicine, and Dr. Nichani is an assistant professor of internal medicine at the University of Michigan Hospital and Health Systems in Ann Arbor.
References
- Upadhyay A, Jaber BL, Madias NE. Incidence and prevalence of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S30-35.
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-21.
- Renneboog B, Musch W, Vandemergel X, Manto MU, Decaux G. Mild chronic hyponatremia is associated with falls, unsteadiness, and attention deficits. Am J Med. 2006;119(1):71.e71-78.
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83(5):905-908.
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Störk S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123(7):652-657.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790-806.
- Smith DM, McKenna K, Thompson CJ. Hyponatraemia. Clin Endocrinol (Oxf). 2000;52(6):667-678.
- Verbalis JG. Hyponatraemia. Baillieres Clin Endocrinol Metab. Aug 1989;3(2):499-530.
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting? Kidney Int. 2009;76(9):934-938.
- Verbalis JG. Disorders of body water homeostasis. Best Pract Res Clin Endocrinol Metab. 2003;17(4):471-503.
- Fenske W, Störk S, Koschker AC, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991-2997.
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis. 1998;32(6):917-933.
- Milionis HJ, Liamis GL, Elisaf MS. The hyponatremic patient: a systematic approach to laboratory diagnosis. CMAJ. 2002;166(8):1056-1062.
- Bitew S, Imbriano L, Miyawaki N, Fishbane S, Maesaka JK. More on renal salt wasting without cerebral disease: response to saline infusion. Clin J Am Soc Nephrol. 2009;4(2):309-315.
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356(20):2064-2072.
- Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529-1535.
- Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190-1195.
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;29(3):282-299.
- Hew-Butler T, Ayus JC, Kipps C, et al. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;18(2):111-121.
- List AF, Hainsworth JD, Davis BW, Hande KR, Greco FA, Johnson DH. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4(8):1191-1198.
- Verbalis JG. Managing hyponatremia in patients with syndrome of inappropriate antidiuretic hormone secretion. J Hosp Med. 2010;5 Suppl 3:S18-S26.
- Musch W, Decaux G. Treating the syndrome of inappropriate ADH secretion with isotonic saline. QJM. 1998;91(11):749-753.
- Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19(6):1076-1078.
- Decaux G, Brimioulle S, Genette F, Mockel J. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone by urea. Am J Med. 1980;69(1):99-106.
- Forrest JN Jr., Cox M, Hong C, Morrison G, Bia M, Singer I. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298(4):173-177.
- Nielsen J, Hoffert JD, Knepper MA, Agre P, Nielsen S, Fenton RA. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc Natl Acad Sci U S A. 2008;105(9):3634-3639.
- Zeltser D, Rosansky S, van Rensburg H, Verbalis JG, Smith N. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27(5):447-457.
- Verbalis JG, Zeltser D, Smith N, Barve A, Andoh M. Assessment of the efficacy and safety of intravenous conivaptan in patients with euvolaemic hyponatraemia: subgroup analysis of a randomized, controlled study. Clin Endocrinol (Oxf). 2008;69(1):159-168.
- Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099-2112.
- Berl T, Quittnat-Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705-712.
- Konstam MA, Gheorghiade M, Burnett JC Jr., et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319-1331.
What Is the Best Treatment of an Adult Patient with Hypercalcemia of Malignancy?
Case
A 63-year-old man with hypertension, diabetes, and recently diagnosed squamous-cell lung cancer presents with diffuse abdominal pain and confusion of two-day duration. He weighs 105 Kg, his blood pressure is 105/65 mm/Hg, heart rate is 105 beats per minute, and temperature is 99.0 degrees Fahrenheit. His respirations are 18 breaths per minute, oxygen saturation is 95% on room air, and his orthostatics are positive. Dry mucus membranes with decreased skin turgor are noted on physical examination. Laboratory evaluation reveals a calcium level of 15.5 mg/dL, creatinine level of 1.2 mg/dL, albumin level of 4.3 g/dL, and a phosphorous level of 2.9 mg/dL.
What is the best treatment of this condition?
Overview
Calcium homeostasis involves complex interactions between the kidney, gastrointestinal (GI) tract, and the skeletal system via hormonal influences. Although 99% of the body’s calcium is stored in the bones, 50% of serum calcium is in the active ionized form, 40% is bound to albumin, and 10% is complexed with anions.1 It’s important to remember these percentages when evaluating a patient’s serum calcium; elevated serum calcium can be validated by using either a correction formula (corrected calcium=measured total calcium + [0.8 x (4.5-albumin)]) or by direct measurement of the ionized calcium, which is the physiologically active form.
Hypercalcemia of malignancy is the most common cause of hypercalcemia in the hospitalized patient. Twenty to 30% of patients with cancer will develop hypercalcemia at some point in their disease course.2 Overall, this portends a poor prognosis with a median survival of three to four months.3
Four general mechanisms are involved in the pathogenesis of malignant hypercalcemia; these mechanisms form the basis for available treatment strategies available:
- Osteolytic tumors, such as multiple myeloma, can directly act on bone, leading to osteoclast activation and release of calcium;
- Humoral mediators elaborated by malignant cells, such as parathyroid hormone-related peptide (PTH-RP), can effect activation of osteoclasts and decrease renal elimination of calcium, causing humoral hypercalcemia of malignancy;
- Some malignancies (most commonly lymphomas) can directly synthesize 1,25 (OH)2 vitamin D, leading to increased luminal absorption of both calcium and phosphorus from the GI tract; and
- Direct production of parathyroid hormone (PTH) by the malignant cells is rare, but has been reported.2
Other factors, including impaired mobility, might lead to further bone resorption and a worsening of the hypercalcemic state.
A patient with hypercalcemia must have a systematic workup, with knowledge of other causes of hypercalcemia that could be present, irrespective of malignancy. Examples include primary hyperparathyroidism, medications effect, and genetic etiologies. Although further discussion is beyond the scope of this article, a broad diagnostic approach is represented in Figure 1 (at right).
Effective management of hypercalcemia demands consideration of both the patient’s immediate, as well as longer-term, clinical situation in light of the patient’s prognosis. The primary aim in the acute management of hypercalcemia is to normalize serum values and decrease symptoms. However, this must be done with appreciation that the metabolic derangement was generated by an underlying malignancy. The main focus of clinical therapeutics should be aimed at this.
Review of the Data
Intravenous (IV) fluids. IV hydration with isotonic saline represents the most immediate and critical intervention in the acute management of malignant hypercalcemia. This condition has multiple, potentially deleterious effects on the kidney, including vasoconstriction, inhibition of salt absorption distally, and antagonism of anti-diuretic hormone (ADH), leading to both salt and water loss. The decrease in intravascular volume then potentiates increased sodium re-absorption proximally in the kidney.
Isotonic saline restores the volume depletion that invariably occurs in the setting of hypercalcemia-provoked urinary salt wasting. The restoration of intravascular volume results in an increase in the glomerular filtration rate and, thus, an increase in calcium filtration. Furthermore, proximal tubular sodium and calcium re-absorption decrease as the glomerular filtration rate increases. Additionally, an increase in sodium and water presentation to the distal renal tubular sites provokes a further calciuresis.
It is estimated that with saline hydration, the calcium concentration should decline, at least by the degree to which dehydration raised it, typically in the range of 1.6 mg to 2.4 mg per deciliter.4 Hydration alone, however, rarely leads to normalization of the serum calcium concentration in patients with severe hypercalcemia.
The rate of infusion is based on the severity of hypercalcemia, and the patient’s age and comorbidities, with particular attention to cardiac or renal disease. A standard approach for most patients without edema and without heart or renal failure is to begin a saline infusion at an initial rate between 200 mL/h to 300 mL/h. The goal is to maintain urine output at 100 mL/h to 150 mL/h.
Furosemide. Following the administration of intravenous fluids to re-establish a euvolemic state, furosemide historically has been used because it has a calcinuric effect with forced diuresis. It also is useful for managing and preventing the fluid overload that occurs with saline hydration. However, data does not support its routine use to lower calcium levels in hypercalcemic patients.
The majority of articles studying the use of furosemide were published in the 1970s and ’80s, and they involve a variety of doses and administration schedules ranging from 40 mg orally daily to 100 mg IV hourly with variable improvement in serum calcium levels and effects that were short-lived. Although some studies have shown that these high doses (2,400 mg/24 hours) of furosemide can decrease calcium levels, resultant severe metabolic derangements in other electrolytes were encountered. This approach required frequent and invasive monitoring to prevent such derangements.5 The clinical application of these studies have led to published recommendations that are as variable as the doses used in the initial studies more than 30 years ago.
This includes the consideration that, in light of the availability and efficacy of bisphosphonates, furosemide might no longer be clinically helpful in this endeavor.6 The current role of furosemide in the management in hypercalcemic patients remains on an as-needed basis for management of fluid overload states brought about after aggressive IV fluid resuscitation.
Bisphosphonates. Bisphosph-onates first became available for the management of hypercalcemia in the early 1990s and have dramatically changed the acute intervention and improved the long-term clinical course of patients with malignant hypercalcemia. Though first developed in the 19th century with industrial applications, it wasn’t until the 1960s that their role in bone metabolism was appreciated.
While their complex mechanism of action remains an issue of ongoing investigations, it is known that bisphosphonates are directed to the bones, where they inhibit an enzyme in the HMG-CoA reductase pathway and promote apoptotic cell death of osteoclasts.7 By blocking osteoclast-mediated bone resorption, the bisphosphonates are effective in treating the hypercalcemia that occurs with a variety of bone-resorbing disease processes, malignant hypercalcemia included. As relatively nontoxic compounds capable of conferring a profound and sustained diminution in serum calcium, these agents have become preferred in the management of acute and chronic hypercalcemia of malignancy.
There are five parenteral bisphosphonates available for the treatment of malignant hypercalcemia: pamidronate, zoledronic acid, ibandronate, etidronate, and clodronate. Etidronate and clodronate are first-generation agents, which are less potent and have more side effects than other agents and are not as commonly used. Ibandronate is a useful agent with a long half-life shown to be as effective as pamidronate, though it has not been as extensively studied as the other agents.
Pamidronate has been studied thoroughly in multiple observational and randomized trials, and has been shown to be highly efficacious and minimally toxic in the treatment of hypercalcemia due to multiple causes, including malignant hypercalcemia.8,9 A maximum calcium-lowering effect occurs at a dose of 90 mg, and the dose is often titrated based on the measured serum calcium. It is infused over two to four hours, effects a lowering of serum calcium within one to two days, and has a sustained effect lasting for up to two weeks or more.
As the most potent and most easily administered bisphosphonate, zoledronic acid is considered by many the agent of choice in the treatment of malignant hypercalcemia. It can be administered as a 4 mg-8 mg dose intravenously over 15 minutes (compared with two hours for pamidronate). Two Phase III trials comprising 275 patients have demonstrated zoledronic acid’s superior efficacy compared with pamidronate, with 88% of patients accomplishing a normalized serum calcium (compared with 70% of patients receiving a 90-mg dose of pamidronate).10
Even though these agents are relatively nontoxic, each can provoke a mild, transient flulike illness in recipients. Renal dysfunction has been noted rarely. These agents should be renally dosed and used with caution in patients with advanced renal insufficiency (serum creatinine >2.5). Osteonecrosis of the jaw has been observed in less than 2% of patients receiving IV bisphosphonates. Accordingly, it is recommended that patients undergo dental evaluation prior to receiving the agent (if feasible) and avoid invasive dental procedures around the time that they receive the agent.11

Other therapeutic interventions. The bisphosphonates represent the best studied and most efficacious pharmaceutical agents available to treat hypercalcemia. Straying from these agents should be considered only when they are contraindicated, in severe circumstances, or after the patient has failed to respond.
Calcitonin has long had FDA approval for treatment of hypercalcemia in adults. It has been shown in small, nonrandomized studies from the 1970s and ’80s to rapidly (within two hours) decrease calcium levels in hypercalcemic patients.12,13,14 However, these reductions are small (<10%) and transient (usually persisting up to 72 to 96 hours) due to the tachyphylaxsis noted with this medication. Nonetheless, calcitonin can be used as an adjuvant bridge to lower calcium levels in severely hypercalcemic patients for the first few days before other agents start taking effect.
Glucocorticoids have been used to treat hypercalcemia since the 1950s. Prednisone, dexamethasone, and methylprednisolone all carry FDA indications for hypercalcemia, but data are lacking and contradictory. A small (n=28) randomized controlled trial (RCT) conducted in 1984 showed no additional efficacy of glucocorticoids with IV fluids when compared with IV fluids alone.15 Another small (n=30) RCT done in 1992 on women with metastatic breast cancer showed a significant improvement in patients treated with prednisolone, IV fluids, and furosemide when compared with IV fluids and furosemide.16 Other nonrandomized trials have shown response to be unpredictable at best.17 Despite this, glucocorticoids likely retain a limited role for treatment in specific cases, including hypercalcemia induced by lymphomas elevating levels of 1,25(OH)2 vitamin D (as this interacts with a steroid-regulated receptor), or multiple myelomas where they potentially impact disease progression.
Gallium nitrate, an anhydrous salt of a heavy metal, has been shown in several randomized trials to be an effective therapeutic agent in lowering calcium levels in hypercalcemic patients.18,19 Furthermore, a double-blinded trial of 64 patients with hypercalcemia of malignancy showed gallium nitrate to be at least as effective as pamidronate for acute control of cancer-related hypercalcemia.20 However, the need for continuous infusion over a five-day period has limited the application of this agent.
Hemodialysis with a calcium-lacking dialysate has been shown in small, nonrandomized studies to be a temporarily effective method of reducing serum calcium levels.21,22 However, this treatment modality would best be reserved for patients with severe hypercalcemia, in whom aggressive intravascular volume repletion and bisphosphonates are not advisable (e.g. those with significant heart or kidney failure) and have an underlying etiology that is likely to be responsive to other treatment. Furthermore, consideration as to the appropriateness of such invasive temporizing procedures in patients with metastatic cancer should be undertaken.
Back to the Case
This patient had an ionized calcium level of 1.9 mmol/L (normal 1.1-1.4 mmol/L). He was started on aggressive IV hydration with normal saline and zoledronic acid. His home medications were reviewed, and it was confirmed that he was not taking such contraindicated medications as thiazides or calcium/vitamin D supplementation.
Further workup for the etiology of his hypercalcemia revealed an appropriately suppressed, intact PTH and normal 25 (OH) Vitamin D and 1,25 (OH)2 Vitamin D levels. His intact PTH-RP was elevated at 10pmol/L, and consistent with hypercalcemia of malignancy.
Oncology and palliative-care consults were requested to assist with coordination of the treatment of the patient’s underlying lung cancer; plans were made for systemic chemotherapy. His symptoms slowly improved, and 72 hours after admission, his serum calcium had normalized. He was discharged with a plan to initiate chemotherapy and continued follow-up with oncology.
Bottom Line
Acute management of hypercalcemia of malignancy focuses on lowering the serum calcium through a variety of pharmacologic agents. However, such long-term issues as treatment of the underlying malignancy and discussions about goals of care in this high-mortality patient population is paramount. TH
Dr. Hartley and Dr. Repaskey are clinical instructors in internal medicine at the University of Michigan Health System. Dr. Rohde is a clinical assistant professor of internal medicine at UMHS.
References
- Assadi F. Hypercalcemia: an evidence-based approach to clinical cases. Iran J Kidney Dis. 2009;3:(2):71-79.
- Stewart A. Hypercalcemia associated with cancer. N Engl J Med. 2005;542(4):373-379.
- Seccareccia D. Cancer-related hypercalcemia. Can Fam Physician. 2010;56:(3):244-246.
- Bilezikian JP. Management of acute hypercalcemia. N Engl J Med. 1992; 326(18):1196-1203.
- Suki WN, Yium JJ, VonMinden M, et al. Acute treatment of hypercalcemia with furosemide. N Engl J Med. 1970;283:836-840.
- LeGrand SB, Leskuski D, Zama I. Narrative review: furosemide for hypercalcemia: an unproven yet common practice. Ann Intern Med. 2008;149:259-263.
- Drake MT, Bart LC, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clinic Proc. 2008;83(9):1032-1045.
- Nussbaum SR, Younger J, Vandepol CJ, et al. Single-dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: comparison of 30-, 60-, and 90-mg dosages. Am J Med. 1993; 95(3):297-304.
- Gucalp R, Ritch P, Riernik PH, et al. Comparative study of pamidronate disodium and etidronate disodium in the treatment of cancer-related hypercalcemia. J Clin Oncol. 1992;10(1):134-142.
- Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol. 2001;19(2): 558-567.
- Tanvetyanon T. Management of the adverse effects associated with intravenous bisphosphonates. Ann Oncol. 2006;17(6):897-907.
- Wisneski LA, Croom WP, Silva OL, et al. Salmon calcitonin in hypercalcemia. Clin Pharmacol Ther. 1978; 24:219-222.
- Binstock ML, Mundy GR. Effect of calcitonin and glucocorticoids in combination on the hypercalcemia of malignancy. Ann Intern Med. 1980;93(2):269-272.
- Nilsson O, Almqvist S, Karlberg BE. Salmon calcitonin in the acute treatment of moderate and severe hypercalcemia in man. Acta Med Scand. 1978;204(4): 249-252.
- Percival RC, Yates AJ, Gray RE, et al. Role of glucocorticoids in management of malignant hypercalcemia. Br Med J. 1984;289(6440):287.
- Kristensen B, Ejlertsen B, Holmegaard SN, et al. Prednisolone in the treatment of severe malignant hypercalcemia in metastatic breast cancer: a randomized study. J Intern Med. 1992;232(3):237-245.
- Thalassinos NC, Joplin GF. Failure of corticosteroid therapy to correct the hypercalcemia of malignant disease. Lancet. 1970;2(7672):537-538.
- Warrell RP Jr, Murphy WK, Schulman P, et al. A randomized double-blind study of gallium nitrate compared with etidronate for acute control of cancer-related hypercalcemia. J Clin Oncol. 1991;9(8):1467-1475.
- Warrell RP Jr, Israel R, Frisone M, et al. Gallium nitrate for acute treatment of cancer-related hypercalcemia: a randomized, double-blinded comparison to calcitonin. Ann Intern Med. 1988;108:669-674.
- Cvitkovic F, Armand JP, Tubiana-Hulin M, et al. Randomized, double-blind, phase II trial of gallium nitrate compared with pamidronate for acute control of cancer-related hypercalcemia. Cancer J. 2006;12 (1):47-53.
- Cardella CJ, Birkin BL, Rapoport A. Role of dialysis in the treatment of severe hypercalcemia: report of two cases successfully treated with hemodialysis and review of the literature. Clin Nephrol. 1979; 12(6):285-290.
- Koo WS, Jeon DS, Ahn SJ, et al. Calcium-free hemodialysis for the management of hypercalcemia. Nephron. 1996;72(3):424-428.
Case
A 63-year-old man with hypertension, diabetes, and recently diagnosed squamous-cell lung cancer presents with diffuse abdominal pain and confusion of two-day duration. He weighs 105 Kg, his blood pressure is 105/65 mm/Hg, heart rate is 105 beats per minute, and temperature is 99.0 degrees Fahrenheit. His respirations are 18 breaths per minute, oxygen saturation is 95% on room air, and his orthostatics are positive. Dry mucus membranes with decreased skin turgor are noted on physical examination. Laboratory evaluation reveals a calcium level of 15.5 mg/dL, creatinine level of 1.2 mg/dL, albumin level of 4.3 g/dL, and a phosphorous level of 2.9 mg/dL.
What is the best treatment of this condition?
Overview
Calcium homeostasis involves complex interactions between the kidney, gastrointestinal (GI) tract, and the skeletal system via hormonal influences. Although 99% of the body’s calcium is stored in the bones, 50% of serum calcium is in the active ionized form, 40% is bound to albumin, and 10% is complexed with anions.1 It’s important to remember these percentages when evaluating a patient’s serum calcium; elevated serum calcium can be validated by using either a correction formula (corrected calcium=measured total calcium + [0.8 x (4.5-albumin)]) or by direct measurement of the ionized calcium, which is the physiologically active form.
Hypercalcemia of malignancy is the most common cause of hypercalcemia in the hospitalized patient. Twenty to 30% of patients with cancer will develop hypercalcemia at some point in their disease course.2 Overall, this portends a poor prognosis with a median survival of three to four months.3
Four general mechanisms are involved in the pathogenesis of malignant hypercalcemia; these mechanisms form the basis for available treatment strategies available:
- Osteolytic tumors, such as multiple myeloma, can directly act on bone, leading to osteoclast activation and release of calcium;
- Humoral mediators elaborated by malignant cells, such as parathyroid hormone-related peptide (PTH-RP), can effect activation of osteoclasts and decrease renal elimination of calcium, causing humoral hypercalcemia of malignancy;
- Some malignancies (most commonly lymphomas) can directly synthesize 1,25 (OH)2 vitamin D, leading to increased luminal absorption of both calcium and phosphorus from the GI tract; and
- Direct production of parathyroid hormone (PTH) by the malignant cells is rare, but has been reported.2
Other factors, including impaired mobility, might lead to further bone resorption and a worsening of the hypercalcemic state.
A patient with hypercalcemia must have a systematic workup, with knowledge of other causes of hypercalcemia that could be present, irrespective of malignancy. Examples include primary hyperparathyroidism, medications effect, and genetic etiologies. Although further discussion is beyond the scope of this article, a broad diagnostic approach is represented in Figure 1 (at right).
Effective management of hypercalcemia demands consideration of both the patient’s immediate, as well as longer-term, clinical situation in light of the patient’s prognosis. The primary aim in the acute management of hypercalcemia is to normalize serum values and decrease symptoms. However, this must be done with appreciation that the metabolic derangement was generated by an underlying malignancy. The main focus of clinical therapeutics should be aimed at this.
Review of the Data
Intravenous (IV) fluids. IV hydration with isotonic saline represents the most immediate and critical intervention in the acute management of malignant hypercalcemia. This condition has multiple, potentially deleterious effects on the kidney, including vasoconstriction, inhibition of salt absorption distally, and antagonism of anti-diuretic hormone (ADH), leading to both salt and water loss. The decrease in intravascular volume then potentiates increased sodium re-absorption proximally in the kidney.
Isotonic saline restores the volume depletion that invariably occurs in the setting of hypercalcemia-provoked urinary salt wasting. The restoration of intravascular volume results in an increase in the glomerular filtration rate and, thus, an increase in calcium filtration. Furthermore, proximal tubular sodium and calcium re-absorption decrease as the glomerular filtration rate increases. Additionally, an increase in sodium and water presentation to the distal renal tubular sites provokes a further calciuresis.
It is estimated that with saline hydration, the calcium concentration should decline, at least by the degree to which dehydration raised it, typically in the range of 1.6 mg to 2.4 mg per deciliter.4 Hydration alone, however, rarely leads to normalization of the serum calcium concentration in patients with severe hypercalcemia.
The rate of infusion is based on the severity of hypercalcemia, and the patient’s age and comorbidities, with particular attention to cardiac or renal disease. A standard approach for most patients without edema and without heart or renal failure is to begin a saline infusion at an initial rate between 200 mL/h to 300 mL/h. The goal is to maintain urine output at 100 mL/h to 150 mL/h.
Furosemide. Following the administration of intravenous fluids to re-establish a euvolemic state, furosemide historically has been used because it has a calcinuric effect with forced diuresis. It also is useful for managing and preventing the fluid overload that occurs with saline hydration. However, data does not support its routine use to lower calcium levels in hypercalcemic patients.
The majority of articles studying the use of furosemide were published in the 1970s and ’80s, and they involve a variety of doses and administration schedules ranging from 40 mg orally daily to 100 mg IV hourly with variable improvement in serum calcium levels and effects that were short-lived. Although some studies have shown that these high doses (2,400 mg/24 hours) of furosemide can decrease calcium levels, resultant severe metabolic derangements in other electrolytes were encountered. This approach required frequent and invasive monitoring to prevent such derangements.5 The clinical application of these studies have led to published recommendations that are as variable as the doses used in the initial studies more than 30 years ago.
This includes the consideration that, in light of the availability and efficacy of bisphosphonates, furosemide might no longer be clinically helpful in this endeavor.6 The current role of furosemide in the management in hypercalcemic patients remains on an as-needed basis for management of fluid overload states brought about after aggressive IV fluid resuscitation.
Bisphosphonates. Bisphosph-onates first became available for the management of hypercalcemia in the early 1990s and have dramatically changed the acute intervention and improved the long-term clinical course of patients with malignant hypercalcemia. Though first developed in the 19th century with industrial applications, it wasn’t until the 1960s that their role in bone metabolism was appreciated.
While their complex mechanism of action remains an issue of ongoing investigations, it is known that bisphosphonates are directed to the bones, where they inhibit an enzyme in the HMG-CoA reductase pathway and promote apoptotic cell death of osteoclasts.7 By blocking osteoclast-mediated bone resorption, the bisphosphonates are effective in treating the hypercalcemia that occurs with a variety of bone-resorbing disease processes, malignant hypercalcemia included. As relatively nontoxic compounds capable of conferring a profound and sustained diminution in serum calcium, these agents have become preferred in the management of acute and chronic hypercalcemia of malignancy.
There are five parenteral bisphosphonates available for the treatment of malignant hypercalcemia: pamidronate, zoledronic acid, ibandronate, etidronate, and clodronate. Etidronate and clodronate are first-generation agents, which are less potent and have more side effects than other agents and are not as commonly used. Ibandronate is a useful agent with a long half-life shown to be as effective as pamidronate, though it has not been as extensively studied as the other agents.
Pamidronate has been studied thoroughly in multiple observational and randomized trials, and has been shown to be highly efficacious and minimally toxic in the treatment of hypercalcemia due to multiple causes, including malignant hypercalcemia.8,9 A maximum calcium-lowering effect occurs at a dose of 90 mg, and the dose is often titrated based on the measured serum calcium. It is infused over two to four hours, effects a lowering of serum calcium within one to two days, and has a sustained effect lasting for up to two weeks or more.
As the most potent and most easily administered bisphosphonate, zoledronic acid is considered by many the agent of choice in the treatment of malignant hypercalcemia. It can be administered as a 4 mg-8 mg dose intravenously over 15 minutes (compared with two hours for pamidronate). Two Phase III trials comprising 275 patients have demonstrated zoledronic acid’s superior efficacy compared with pamidronate, with 88% of patients accomplishing a normalized serum calcium (compared with 70% of patients receiving a 90-mg dose of pamidronate).10
Even though these agents are relatively nontoxic, each can provoke a mild, transient flulike illness in recipients. Renal dysfunction has been noted rarely. These agents should be renally dosed and used with caution in patients with advanced renal insufficiency (serum creatinine >2.5). Osteonecrosis of the jaw has been observed in less than 2% of patients receiving IV bisphosphonates. Accordingly, it is recommended that patients undergo dental evaluation prior to receiving the agent (if feasible) and avoid invasive dental procedures around the time that they receive the agent.11
Other therapeutic interventions. The bisphosphonates represent the best studied and most efficacious pharmaceutical agents available to treat hypercalcemia. Straying from these agents should be considered only when they are contraindicated, in severe circumstances, or after the patient has failed to respond.
Calcitonin has long had FDA approval for treatment of hypercalcemia in adults. It has been shown in small, nonrandomized studies from the 1970s and ’80s to rapidly (within two hours) decrease calcium levels in hypercalcemic patients.12,13,14 However, these reductions are small (<10%) and transient (usually persisting up to 72 to 96 hours) due to the tachyphylaxsis noted with this medication. Nonetheless, calcitonin can be used as an adjuvant bridge to lower calcium levels in severely hypercalcemic patients for the first few days before other agents start taking effect.
Glucocorticoids have been used to treat hypercalcemia since the 1950s. Prednisone, dexamethasone, and methylprednisolone all carry FDA indications for hypercalcemia, but data are lacking and contradictory. A small (n=28) randomized controlled trial (RCT) conducted in 1984 showed no additional efficacy of glucocorticoids with IV fluids when compared with IV fluids alone.15 Another small (n=30) RCT done in 1992 on women with metastatic breast cancer showed a significant improvement in patients treated with prednisolone, IV fluids, and furosemide when compared with IV fluids and furosemide.16 Other nonrandomized trials have shown response to be unpredictable at best.17 Despite this, glucocorticoids likely retain a limited role for treatment in specific cases, including hypercalcemia induced by lymphomas elevating levels of 1,25(OH)2 vitamin D (as this interacts with a steroid-regulated receptor), or multiple myelomas where they potentially impact disease progression.
Gallium nitrate, an anhydrous salt of a heavy metal, has been shown in several randomized trials to be an effective therapeutic agent in lowering calcium levels in hypercalcemic patients.18,19 Furthermore, a double-blinded trial of 64 patients with hypercalcemia of malignancy showed gallium nitrate to be at least as effective as pamidronate for acute control of cancer-related hypercalcemia.20 However, the need for continuous infusion over a five-day period has limited the application of this agent.
Hemodialysis with a calcium-lacking dialysate has been shown in small, nonrandomized studies to be a temporarily effective method of reducing serum calcium levels.21,22 However, this treatment modality would best be reserved for patients with severe hypercalcemia, in whom aggressive intravascular volume repletion and bisphosphonates are not advisable (e.g. those with significant heart or kidney failure) and have an underlying etiology that is likely to be responsive to other treatment. Furthermore, consideration as to the appropriateness of such invasive temporizing procedures in patients with metastatic cancer should be undertaken.
Back to the Case
This patient had an ionized calcium level of 1.9 mmol/L (normal 1.1-1.4 mmol/L). He was started on aggressive IV hydration with normal saline and zoledronic acid. His home medications were reviewed, and it was confirmed that he was not taking such contraindicated medications as thiazides or calcium/vitamin D supplementation.
Further workup for the etiology of his hypercalcemia revealed an appropriately suppressed, intact PTH and normal 25 (OH) Vitamin D and 1,25 (OH)2 Vitamin D levels. His intact PTH-RP was elevated at 10pmol/L, and consistent with hypercalcemia of malignancy.
Oncology and palliative-care consults were requested to assist with coordination of the treatment of the patient’s underlying lung cancer; plans were made for systemic chemotherapy. His symptoms slowly improved, and 72 hours after admission, his serum calcium had normalized. He was discharged with a plan to initiate chemotherapy and continued follow-up with oncology.
Bottom Line
Acute management of hypercalcemia of malignancy focuses on lowering the serum calcium through a variety of pharmacologic agents. However, such long-term issues as treatment of the underlying malignancy and discussions about goals of care in this high-mortality patient population is paramount. TH
Dr. Hartley and Dr. Repaskey are clinical instructors in internal medicine at the University of Michigan Health System. Dr. Rohde is a clinical assistant professor of internal medicine at UMHS.
References
- Assadi F. Hypercalcemia: an evidence-based approach to clinical cases. Iran J Kidney Dis. 2009;3:(2):71-79.
- Stewart A. Hypercalcemia associated with cancer. N Engl J Med. 2005;542(4):373-379.
- Seccareccia D. Cancer-related hypercalcemia. Can Fam Physician. 2010;56:(3):244-246.
- Bilezikian JP. Management of acute hypercalcemia. N Engl J Med. 1992; 326(18):1196-1203.
- Suki WN, Yium JJ, VonMinden M, et al. Acute treatment of hypercalcemia with furosemide. N Engl J Med. 1970;283:836-840.
- LeGrand SB, Leskuski D, Zama I. Narrative review: furosemide for hypercalcemia: an unproven yet common practice. Ann Intern Med. 2008;149:259-263.
- Drake MT, Bart LC, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clinic Proc. 2008;83(9):1032-1045.
- Nussbaum SR, Younger J, Vandepol CJ, et al. Single-dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: comparison of 30-, 60-, and 90-mg dosages. Am J Med. 1993; 95(3):297-304.
- Gucalp R, Ritch P, Riernik PH, et al. Comparative study of pamidronate disodium and etidronate disodium in the treatment of cancer-related hypercalcemia. J Clin Oncol. 1992;10(1):134-142.
- Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol. 2001;19(2): 558-567.
- Tanvetyanon T. Management of the adverse effects associated with intravenous bisphosphonates. Ann Oncol. 2006;17(6):897-907.
- Wisneski LA, Croom WP, Silva OL, et al. Salmon calcitonin in hypercalcemia. Clin Pharmacol Ther. 1978; 24:219-222.
- Binstock ML, Mundy GR. Effect of calcitonin and glucocorticoids in combination on the hypercalcemia of malignancy. Ann Intern Med. 1980;93(2):269-272.
- Nilsson O, Almqvist S, Karlberg BE. Salmon calcitonin in the acute treatment of moderate and severe hypercalcemia in man. Acta Med Scand. 1978;204(4): 249-252.
- Percival RC, Yates AJ, Gray RE, et al. Role of glucocorticoids in management of malignant hypercalcemia. Br Med J. 1984;289(6440):287.
- Kristensen B, Ejlertsen B, Holmegaard SN, et al. Prednisolone in the treatment of severe malignant hypercalcemia in metastatic breast cancer: a randomized study. J Intern Med. 1992;232(3):237-245.
- Thalassinos NC, Joplin GF. Failure of corticosteroid therapy to correct the hypercalcemia of malignant disease. Lancet. 1970;2(7672):537-538.
- Warrell RP Jr, Murphy WK, Schulman P, et al. A randomized double-blind study of gallium nitrate compared with etidronate for acute control of cancer-related hypercalcemia. J Clin Oncol. 1991;9(8):1467-1475.
- Warrell RP Jr, Israel R, Frisone M, et al. Gallium nitrate for acute treatment of cancer-related hypercalcemia: a randomized, double-blinded comparison to calcitonin. Ann Intern Med. 1988;108:669-674.
- Cvitkovic F, Armand JP, Tubiana-Hulin M, et al. Randomized, double-blind, phase II trial of gallium nitrate compared with pamidronate for acute control of cancer-related hypercalcemia. Cancer J. 2006;12 (1):47-53.
- Cardella CJ, Birkin BL, Rapoport A. Role of dialysis in the treatment of severe hypercalcemia: report of two cases successfully treated with hemodialysis and review of the literature. Clin Nephrol. 1979; 12(6):285-290.
- Koo WS, Jeon DS, Ahn SJ, et al. Calcium-free hemodialysis for the management of hypercalcemia. Nephron. 1996;72(3):424-428.
Case
A 63-year-old man with hypertension, diabetes, and recently diagnosed squamous-cell lung cancer presents with diffuse abdominal pain and confusion of two-day duration. He weighs 105 Kg, his blood pressure is 105/65 mm/Hg, heart rate is 105 beats per minute, and temperature is 99.0 degrees Fahrenheit. His respirations are 18 breaths per minute, oxygen saturation is 95% on room air, and his orthostatics are positive. Dry mucus membranes with decreased skin turgor are noted on physical examination. Laboratory evaluation reveals a calcium level of 15.5 mg/dL, creatinine level of 1.2 mg/dL, albumin level of 4.3 g/dL, and a phosphorous level of 2.9 mg/dL.
What is the best treatment of this condition?
Overview
Calcium homeostasis involves complex interactions between the kidney, gastrointestinal (GI) tract, and the skeletal system via hormonal influences. Although 99% of the body’s calcium is stored in the bones, 50% of serum calcium is in the active ionized form, 40% is bound to albumin, and 10% is complexed with anions.1 It’s important to remember these percentages when evaluating a patient’s serum calcium; elevated serum calcium can be validated by using either a correction formula (corrected calcium=measured total calcium + [0.8 x (4.5-albumin)]) or by direct measurement of the ionized calcium, which is the physiologically active form.
Hypercalcemia of malignancy is the most common cause of hypercalcemia in the hospitalized patient. Twenty to 30% of patients with cancer will develop hypercalcemia at some point in their disease course.2 Overall, this portends a poor prognosis with a median survival of three to four months.3
Four general mechanisms are involved in the pathogenesis of malignant hypercalcemia; these mechanisms form the basis for available treatment strategies available:
- Osteolytic tumors, such as multiple myeloma, can directly act on bone, leading to osteoclast activation and release of calcium;
- Humoral mediators elaborated by malignant cells, such as parathyroid hormone-related peptide (PTH-RP), can effect activation of osteoclasts and decrease renal elimination of calcium, causing humoral hypercalcemia of malignancy;
- Some malignancies (most commonly lymphomas) can directly synthesize 1,25 (OH)2 vitamin D, leading to increased luminal absorption of both calcium and phosphorus from the GI tract; and
- Direct production of parathyroid hormone (PTH) by the malignant cells is rare, but has been reported.2
Other factors, including impaired mobility, might lead to further bone resorption and a worsening of the hypercalcemic state.
A patient with hypercalcemia must have a systematic workup, with knowledge of other causes of hypercalcemia that could be present, irrespective of malignancy. Examples include primary hyperparathyroidism, medications effect, and genetic etiologies. Although further discussion is beyond the scope of this article, a broad diagnostic approach is represented in Figure 1 (at right).
Effective management of hypercalcemia demands consideration of both the patient’s immediate, as well as longer-term, clinical situation in light of the patient’s prognosis. The primary aim in the acute management of hypercalcemia is to normalize serum values and decrease symptoms. However, this must be done with appreciation that the metabolic derangement was generated by an underlying malignancy. The main focus of clinical therapeutics should be aimed at this.
Review of the Data
Intravenous (IV) fluids. IV hydration with isotonic saline represents the most immediate and critical intervention in the acute management of malignant hypercalcemia. This condition has multiple, potentially deleterious effects on the kidney, including vasoconstriction, inhibition of salt absorption distally, and antagonism of anti-diuretic hormone (ADH), leading to both salt and water loss. The decrease in intravascular volume then potentiates increased sodium re-absorption proximally in the kidney.
Isotonic saline restores the volume depletion that invariably occurs in the setting of hypercalcemia-provoked urinary salt wasting. The restoration of intravascular volume results in an increase in the glomerular filtration rate and, thus, an increase in calcium filtration. Furthermore, proximal tubular sodium and calcium re-absorption decrease as the glomerular filtration rate increases. Additionally, an increase in sodium and water presentation to the distal renal tubular sites provokes a further calciuresis.
It is estimated that with saline hydration, the calcium concentration should decline, at least by the degree to which dehydration raised it, typically in the range of 1.6 mg to 2.4 mg per deciliter.4 Hydration alone, however, rarely leads to normalization of the serum calcium concentration in patients with severe hypercalcemia.
The rate of infusion is based on the severity of hypercalcemia, and the patient’s age and comorbidities, with particular attention to cardiac or renal disease. A standard approach for most patients without edema and without heart or renal failure is to begin a saline infusion at an initial rate between 200 mL/h to 300 mL/h. The goal is to maintain urine output at 100 mL/h to 150 mL/h.
Furosemide. Following the administration of intravenous fluids to re-establish a euvolemic state, furosemide historically has been used because it has a calcinuric effect with forced diuresis. It also is useful for managing and preventing the fluid overload that occurs with saline hydration. However, data does not support its routine use to lower calcium levels in hypercalcemic patients.
The majority of articles studying the use of furosemide were published in the 1970s and ’80s, and they involve a variety of doses and administration schedules ranging from 40 mg orally daily to 100 mg IV hourly with variable improvement in serum calcium levels and effects that were short-lived. Although some studies have shown that these high doses (2,400 mg/24 hours) of furosemide can decrease calcium levels, resultant severe metabolic derangements in other electrolytes were encountered. This approach required frequent and invasive monitoring to prevent such derangements.5 The clinical application of these studies have led to published recommendations that are as variable as the doses used in the initial studies more than 30 years ago.
This includes the consideration that, in light of the availability and efficacy of bisphosphonates, furosemide might no longer be clinically helpful in this endeavor.6 The current role of furosemide in the management in hypercalcemic patients remains on an as-needed basis for management of fluid overload states brought about after aggressive IV fluid resuscitation.
Bisphosphonates. Bisphosph-onates first became available for the management of hypercalcemia in the early 1990s and have dramatically changed the acute intervention and improved the long-term clinical course of patients with malignant hypercalcemia. Though first developed in the 19th century with industrial applications, it wasn’t until the 1960s that their role in bone metabolism was appreciated.
While their complex mechanism of action remains an issue of ongoing investigations, it is known that bisphosphonates are directed to the bones, where they inhibit an enzyme in the HMG-CoA reductase pathway and promote apoptotic cell death of osteoclasts.7 By blocking osteoclast-mediated bone resorption, the bisphosphonates are effective in treating the hypercalcemia that occurs with a variety of bone-resorbing disease processes, malignant hypercalcemia included. As relatively nontoxic compounds capable of conferring a profound and sustained diminution in serum calcium, these agents have become preferred in the management of acute and chronic hypercalcemia of malignancy.
There are five parenteral bisphosphonates available for the treatment of malignant hypercalcemia: pamidronate, zoledronic acid, ibandronate, etidronate, and clodronate. Etidronate and clodronate are first-generation agents, which are less potent and have more side effects than other agents and are not as commonly used. Ibandronate is a useful agent with a long half-life shown to be as effective as pamidronate, though it has not been as extensively studied as the other agents.
Pamidronate has been studied thoroughly in multiple observational and randomized trials, and has been shown to be highly efficacious and minimally toxic in the treatment of hypercalcemia due to multiple causes, including malignant hypercalcemia.8,9 A maximum calcium-lowering effect occurs at a dose of 90 mg, and the dose is often titrated based on the measured serum calcium. It is infused over two to four hours, effects a lowering of serum calcium within one to two days, and has a sustained effect lasting for up to two weeks or more.
As the most potent and most easily administered bisphosphonate, zoledronic acid is considered by many the agent of choice in the treatment of malignant hypercalcemia. It can be administered as a 4 mg-8 mg dose intravenously over 15 minutes (compared with two hours for pamidronate). Two Phase III trials comprising 275 patients have demonstrated zoledronic acid’s superior efficacy compared with pamidronate, with 88% of patients accomplishing a normalized serum calcium (compared with 70% of patients receiving a 90-mg dose of pamidronate).10
Even though these agents are relatively nontoxic, each can provoke a mild, transient flulike illness in recipients. Renal dysfunction has been noted rarely. These agents should be renally dosed and used with caution in patients with advanced renal insufficiency (serum creatinine >2.5). Osteonecrosis of the jaw has been observed in less than 2% of patients receiving IV bisphosphonates. Accordingly, it is recommended that patients undergo dental evaluation prior to receiving the agent (if feasible) and avoid invasive dental procedures around the time that they receive the agent.11
Other therapeutic interventions. The bisphosphonates represent the best studied and most efficacious pharmaceutical agents available to treat hypercalcemia. Straying from these agents should be considered only when they are contraindicated, in severe circumstances, or after the patient has failed to respond.
Calcitonin has long had FDA approval for treatment of hypercalcemia in adults. It has been shown in small, nonrandomized studies from the 1970s and ’80s to rapidly (within two hours) decrease calcium levels in hypercalcemic patients.12,13,14 However, these reductions are small (<10%) and transient (usually persisting up to 72 to 96 hours) due to the tachyphylaxsis noted with this medication. Nonetheless, calcitonin can be used as an adjuvant bridge to lower calcium levels in severely hypercalcemic patients for the first few days before other agents start taking effect.
Glucocorticoids have been used to treat hypercalcemia since the 1950s. Prednisone, dexamethasone, and methylprednisolone all carry FDA indications for hypercalcemia, but data are lacking and contradictory. A small (n=28) randomized controlled trial (RCT) conducted in 1984 showed no additional efficacy of glucocorticoids with IV fluids when compared with IV fluids alone.15 Another small (n=30) RCT done in 1992 on women with metastatic breast cancer showed a significant improvement in patients treated with prednisolone, IV fluids, and furosemide when compared with IV fluids and furosemide.16 Other nonrandomized trials have shown response to be unpredictable at best.17 Despite this, glucocorticoids likely retain a limited role for treatment in specific cases, including hypercalcemia induced by lymphomas elevating levels of 1,25(OH)2 vitamin D (as this interacts with a steroid-regulated receptor), or multiple myelomas where they potentially impact disease progression.
Gallium nitrate, an anhydrous salt of a heavy metal, has been shown in several randomized trials to be an effective therapeutic agent in lowering calcium levels in hypercalcemic patients.18,19 Furthermore, a double-blinded trial of 64 patients with hypercalcemia of malignancy showed gallium nitrate to be at least as effective as pamidronate for acute control of cancer-related hypercalcemia.20 However, the need for continuous infusion over a five-day period has limited the application of this agent.
Hemodialysis with a calcium-lacking dialysate has been shown in small, nonrandomized studies to be a temporarily effective method of reducing serum calcium levels.21,22 However, this treatment modality would best be reserved for patients with severe hypercalcemia, in whom aggressive intravascular volume repletion and bisphosphonates are not advisable (e.g. those with significant heart or kidney failure) and have an underlying etiology that is likely to be responsive to other treatment. Furthermore, consideration as to the appropriateness of such invasive temporizing procedures in patients with metastatic cancer should be undertaken.
Back to the Case
This patient had an ionized calcium level of 1.9 mmol/L (normal 1.1-1.4 mmol/L). He was started on aggressive IV hydration with normal saline and zoledronic acid. His home medications were reviewed, and it was confirmed that he was not taking such contraindicated medications as thiazides or calcium/vitamin D supplementation.
Further workup for the etiology of his hypercalcemia revealed an appropriately suppressed, intact PTH and normal 25 (OH) Vitamin D and 1,25 (OH)2 Vitamin D levels. His intact PTH-RP was elevated at 10pmol/L, and consistent with hypercalcemia of malignancy.
Oncology and palliative-care consults were requested to assist with coordination of the treatment of the patient’s underlying lung cancer; plans were made for systemic chemotherapy. His symptoms slowly improved, and 72 hours after admission, his serum calcium had normalized. He was discharged with a plan to initiate chemotherapy and continued follow-up with oncology.
Bottom Line
Acute management of hypercalcemia of malignancy focuses on lowering the serum calcium through a variety of pharmacologic agents. However, such long-term issues as treatment of the underlying malignancy and discussions about goals of care in this high-mortality patient population is paramount. TH
Dr. Hartley and Dr. Repaskey are clinical instructors in internal medicine at the University of Michigan Health System. Dr. Rohde is a clinical assistant professor of internal medicine at UMHS.
References
- Assadi F. Hypercalcemia: an evidence-based approach to clinical cases. Iran J Kidney Dis. 2009;3:(2):71-79.
- Stewart A. Hypercalcemia associated with cancer. N Engl J Med. 2005;542(4):373-379.
- Seccareccia D. Cancer-related hypercalcemia. Can Fam Physician. 2010;56:(3):244-246.
- Bilezikian JP. Management of acute hypercalcemia. N Engl J Med. 1992; 326(18):1196-1203.
- Suki WN, Yium JJ, VonMinden M, et al. Acute treatment of hypercalcemia with furosemide. N Engl J Med. 1970;283:836-840.
- LeGrand SB, Leskuski D, Zama I. Narrative review: furosemide for hypercalcemia: an unproven yet common practice. Ann Intern Med. 2008;149:259-263.
- Drake MT, Bart LC, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clinic Proc. 2008;83(9):1032-1045.
- Nussbaum SR, Younger J, Vandepol CJ, et al. Single-dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: comparison of 30-, 60-, and 90-mg dosages. Am J Med. 1993; 95(3):297-304.
- Gucalp R, Ritch P, Riernik PH, et al. Comparative study of pamidronate disodium and etidronate disodium in the treatment of cancer-related hypercalcemia. J Clin Oncol. 1992;10(1):134-142.
- Major P, Lortholary A, Hon J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol. 2001;19(2): 558-567.
- Tanvetyanon T. Management of the adverse effects associated with intravenous bisphosphonates. Ann Oncol. 2006;17(6):897-907.
- Wisneski LA, Croom WP, Silva OL, et al. Salmon calcitonin in hypercalcemia. Clin Pharmacol Ther. 1978; 24:219-222.
- Binstock ML, Mundy GR. Effect of calcitonin and glucocorticoids in combination on the hypercalcemia of malignancy. Ann Intern Med. 1980;93(2):269-272.
- Nilsson O, Almqvist S, Karlberg BE. Salmon calcitonin in the acute treatment of moderate and severe hypercalcemia in man. Acta Med Scand. 1978;204(4): 249-252.
- Percival RC, Yates AJ, Gray RE, et al. Role of glucocorticoids in management of malignant hypercalcemia. Br Med J. 1984;289(6440):287.
- Kristensen B, Ejlertsen B, Holmegaard SN, et al. Prednisolone in the treatment of severe malignant hypercalcemia in metastatic breast cancer: a randomized study. J Intern Med. 1992;232(3):237-245.
- Thalassinos NC, Joplin GF. Failure of corticosteroid therapy to correct the hypercalcemia of malignant disease. Lancet. 1970;2(7672):537-538.
- Warrell RP Jr, Murphy WK, Schulman P, et al. A randomized double-blind study of gallium nitrate compared with etidronate for acute control of cancer-related hypercalcemia. J Clin Oncol. 1991;9(8):1467-1475.
- Warrell RP Jr, Israel R, Frisone M, et al. Gallium nitrate for acute treatment of cancer-related hypercalcemia: a randomized, double-blinded comparison to calcitonin. Ann Intern Med. 1988;108:669-674.
- Cvitkovic F, Armand JP, Tubiana-Hulin M, et al. Randomized, double-blind, phase II trial of gallium nitrate compared with pamidronate for acute control of cancer-related hypercalcemia. Cancer J. 2006;12 (1):47-53.
- Cardella CJ, Birkin BL, Rapoport A. Role of dialysis in the treatment of severe hypercalcemia: report of two cases successfully treated with hemodialysis and review of the literature. Clin Nephrol. 1979; 12(6):285-290.
- Koo WS, Jeon DS, Ahn SJ, et al. Calcium-free hemodialysis for the management of hypercalcemia. Nephron. 1996;72(3):424-428.