User login
Management of Metastatic Gastric Cancer
INTRODUCTION
According to the Surveillance, Epidemiology and End Results database, in 2017 there were 28,000 new cases of gastric cancer, accounting for 1.8% of all malignancies in the United States, and an estimated 10,960 gastric cancer–related deaths.1 Worldwide, gastric cancer is the fifth most common malignancy and the third most common cause of death from any cancer.2 The incidence of gastric cancer varies significantly by geographic region, with countries in Eastern Asia (China, Japan), Eastern Europe, and Central and South America accounting for 50% of all new cases.3 Although the incidence of gastric cancer has declined in recent years, this decrease has not been observed consistently across all nations.2 In particular, the incidence of gastric cancers arising from the cardia has been increasing, which is perhaps due to a higher prevalence of obesity in Western societies.4
In this article, we review key aspects of management of metastatic gastric cancer, including selection of first- and second-line therapy, and discuss targeted agents and upcoming clinical trials.
EPIDEMIOLOGY AND RISK FACTORS
Chronic infection with Helicobacter pylori, a gram-negative bacterium, is a strong etiological factor for the development of gastric cancer, contributing to up to 70% of cases.2 The pathogen can colonize the gastric mucosa, leading to chronic inflammation. Although most patients remain asymptomatic, 1% to 3% develop gastric cancer and another 0.1% develop mucosa-associated lymphoid tissue lymphoma.5 H. pylori infection is more commonly associated with cancer of the gastric body than with cancer of the gastroesophageal junction (GEJ). The increased burden of gastric cancer in countries in Eastern Asia, Latin America, and Eastern Europe has been correlated to the prevalence of chronic H. pylori infection in these areas.
Carcinogenesis secondary to H. pylori infection may occur via several mechanisms. First, H. pylori can release virulence factors, such as cytotoxin-associated gene A, vacuolating cytotoxin, and outer membrane proteins, into the cytosol of host cells, leading to changes in patterns of cell proliferation and apoptosis.6 These virulence factors can modulate the host immune system, attenuating it to promote dysplasia. In addition, continued recognition of these factors by the immune system leads to a persistent inflammatory response, with the release of cytokines such as interleukin (IL) -1β, IL-6, and IL-8. This leads to chronic mucosal damage, further promoting dysplasia with eventual transformation into adenocarcinoma.7 In Japan and Korea, where screening for H. pylori infection is routinely performed, there have been improvements in overall survival (OS) rates for gastric cancer, with 5-year OS rates of 70%.8 The International Agency for Research on Cancer recommends further research into population-based screening and treatment programs for patients with chronic H. pylori infection. However, despite this recommendation, optimal screening strategies are not clearly defined.9
Other risk factors for the development of gastric cancer include chronic gastroesophageal reflux disease; smoking; alcohol use; exposure to radiation; diets high in fats, salt, and smoked items and low in fruits and vegetables; obesity; and exposure to chemotherapeutic agents such as procarbazine.10 Another pathogen suspected, but not proven, to be associated with increased risk for gastric cancer is the Epstein-Barr virus, a human herpesvirus found in 80% of all gastric carcinomas with lymphoid features.11 In addition, whether the use of medications such as statins and nonsteroidal anti-inflammatory drugs confers a decreased risk of gastric cancers remains unclear.10
EVALUATION
CASE PRESENTATION
A 55-year-old Caucasian man with a history of type 2 diabetes mellitus presents to the gastrointestinal (GI) clinic with a 6-month history of dysphagia. The dysphagia is worsened with ingestion of solids, particularly towards the end of the day. He states that the food often gets “stuck in the middle of the chest.” The patient denies any nausea or emesis but notes that he has a poor appetite. He reports having worsening mid-epigastric abdominal pain that is non-radiating, dull in character, and 6/10 in intensity. He also reports a 10-lb weight loss over the past 2 months. He has no previous history of reflux, chest pain, dyspnea, or cough. Review of systems is otherwise benign. Physical exam is within normal limits.
• Which tests should be conducted when gastric cancer is suspected?
Persistent epigastric abdominal pain and weight loss are the most common early symptoms of gastric cancer. Nausea, early satiety, dysphagia, and occult GI bleeding can be other presenting signs. Patients presenting with alarm symptoms of nausea, emesis, early satiety, abdominal pain, or weight loss should be fully evaluated with upper GI endoscopy. Early diagnosis of gastric cancer is essential in obtaining a curative resection. However, at least 40% of patients present with de novo metastatic disease at the time of initial diagnosis.12 Gastric cancer spreads by direct extension through the gastric wall, with the liver, peritoneum, and regional lymph nodes being the most common sites of metastatic deposits.13 Classically, Virchow’s node, the left supraclavicular lymph node, is involved with metastatic gastric cancer. Involvement of the left axillary lymph node (Irish node) or a periumbilical nodule (Sister Mary Joseph node) may also be observed. Other, less commonly noted sites of metastatic disease include the ovaries, central nervous system, bone, lung, and soft tissues.13
Upper GI endoscopy is the best method for determining tumor location and extent and obtaining a specimen for a definitive tissue diagnosis.14 It is essential to accurately identify the location of the tumor in the stomach and relative to the GEJ. The American Joint Committee on Cancer classification defines tumors involving the GEJ with an epicenter no more than 2 cm into the proximal stomach as esophageal cancers.15 Tumors of the GEJ with their epicenter more than 2 cm into the proximal stomach are defined as gastric cancers. If metastatic disease is suspected, computed tomography (CT) scan of the chest, abdomen, and pelvis with oral and intravenous contrast can be obtained to determine the extent of disease spread. In the absence of any metastatic disease, endoscopic ultrasound (EUS) should be conducted to determine the depth of tumor invasion (T staging) and lymph node status. In the era of targeted therapy, patients with metastatic disease should undergo testing for human epidermal growth factor-2 (HER-2) expression, microsatellite instability (MSI), and programmed death ligand 1 (PD-L1) expression. Patients should be staged according to the TNM staging system.
FIRST-LINE TREATMENT OPTIONS
CASE CONTINUED
The patient undergoes esophagoduodenoscopy (EGD) and is found to have a gastric cardia mass extending into the distal esophagus. EUS also demonstrates multiple abdominal and mediastinal lymph nodes. No gastric outlet obstruction is found. Biopsy shows poorly differentiated invasive adenocarcinoma. Warthin–Starry stain is negative for H. pylori organism. The tumor cells are positive for cytokeratin (CK7), CK19, and mucin-1 gene (MUC1); focally positive for CK20; and negative for MUC2. HER2 testing results are reported as immunohistochemistry (IHC) 3+, consistent with strongly positive HER2 protein expression. Further IHC testing for mismatch repair (MMR) proteins shows intact nuclear expression of MLH1, MSH2, MSH6, and PMS2 protein, consistent with a low probability of MSI-high tumor. The tumor is found to be PD-L1 positive. Imaging reveals abnormal mass-like nodular thickening of the gastric wall, with an infiltrative opacity within the pancreatico-duodenal groove, suspicious for tumor infiltration. Multiple metastatic deposits are noted in the liver, peritoneum, and bilateral lungs. There is extensive gastrohepatic ligament and periportal lymphadenopathy and mild enlargement of the pulmonary hilar lymph nodes. These findings are consistent with stage 4 (T4bN3aM1) gastric cancer. Given these findings, staging laparoscopy is deferred.
• What are the first-line treatment options for patients with metastatic gastric cancer?

Patients with metastatic gastric cancer have a poor prognosis, and management is stratified based on performance status (Figure). In patients with good performance status, systemic chemotherapy is the mainstay of treatment. The goal of therapy is not curative, but rather treatment focuses on palliation of symptoms arising from tumor spread. Given this treatment goal, there has been considerable interest in clarifying the utility of chemotherapy as opposed to best supportive care. In a recent Cochrane review of 64 randomized control trials involving 11,698 patients, chemotherapy was found to improve OS by 6.7 months as compared to best supportive care (hazard ratio [HR] 0.3 [95% confidence interval {CI} 0.24 to 0.55]).16 Five classes of cytotoxic chemotherapeutic agents have demonstrated activity in gastric cancer. These include fluoropyrimidine (either infusional fluorouracil or capecitabine), platinum agents (cisplatin or oxaliplatin), taxanes (docetaxel or paclitaxel), anthracyclines (epirubicin), and irinotecan.13 Treatment options are further divided based on whether the patient has HER2-overexpressing or non-expressing malignancy.
HER2-NEGATIVE DISEASE
For patients with HER2-negative disease, National Comprehensive Cancer Network (NCCN) guidelines recommend using 2-drug combination regimens rather than 3 drugs, given concern for increased toxicity with 3-drug regimens.17 For patients with a performance status of 0 to 1, utilization of a 3-drug regimen is a reasonable alternative. The combination of a fluoropyrimidine with a platinum agent is considered the standard of care, with regimens such as fluorouracil, leucovorin, and oxaliplatin (FOLFOX) being commonly used.
Epirubicin-containing regimens have also been extensively studied in advanced gastric cancer. In a study of 274 previously untreated patients with GEJ cancers, the combination of epirubicin, cisplatin, and fluorouracil (ECF) was compared to fluorouracil, doxorubicin, and methotrexate (FAMTX). There was an OS benefit favoring ECF (8.9 months versus 5.7 months) at 1 year (95% CI 27% to 45%, P = 0.0009). The ECF regimen was associated with an increased risk of nausea, emesis, and alopecia, while more hematologic toxicity and infections were noted with the FAMTX regimen.18 In addition, in a phase 3 trial, Van Cutsem and colleagues examined the role of docetaxel in combination with cisplatin and fluorouracil (DCF) compared to cisplatin and fluorouracil alone. Addition of docetaxel led to improved OS and time to progression (9.2 months versus 8.6 months for cisplatin and fluorouracil alone, P = 0.02) but with an increased risk of grade 3 and 4 toxicities (69% versus 59%). These adverse events included neutropenia (82% versus 57% of cisplatin and fluorouracil patients), diarrhea (19% versus 8%), stomatitis (21% versus 27%), and fatigue (19% versus 14%).19
The landmark phase 3 REAL-2 study compared 4 chemotherapy regimens in patients with untreated advanced esophagogastric cancer. This study was conducted to determine if the efficacy of cisplatin and oxaliplatin, a third-generation platinum agent, is equivalent to that of fluorouracil and capecitabine, an oral fluoropyrimidine. In this trial, a 2 × 2 design was used to compare 4 regimens: ECF versus epirubicin, cisplatin, and capecitabine (ECX) versus epirubicin, oxaliplatin, and fluorouracil (EOF) versus epirubicin, oxaliplatin, and capecitabine (EOX). The study found EOX to be noninferior to ECF, with a trend towards improved OS compared to other combination regimens (11.2 months versus 9.9 months, HR 0.80 [95% CI 0.66 to 0.97], P = 0.02).20 Thus, the study demonstrated that an oxaliplatin and capecitabine-based regimen could replace cisplatin and fluorouracil. Given that fluorouracil administration requires long continuous infusions, the oral-based capecitabine regimen is an attractive option for patients.
Several trials have demonstrated the equivalency of oxaliplatin with cisplatin in combination regimens for the treatment of advanced gastric cancer. Oxaliplatin has the benefit of an improved toxicity profile as compared to cisplatin, with the major dose-limiting toxicity being peripheral neuropathy
Given previous evidence that DCF (docetaxel, cisplatin, fluorouracil) is superior to cisplatin and fluorouracil alone, there was interest in determining if the addition of docetaxel to a backbone of fluorouracil, oxaliplatin, and leucovorin (FLO) could elicit a higher response rate. This concept was investigated in a phase 2 trial that assigned 54 patients with metastatic gastric or GEJ adenocarcinoma to receive biweekly infusions of oxaliplatin, leucovorin, fluorouracil, and docetaxel.21 Median time to response was 1.54 months, and the overall response rate was 57.7%. Median progression-free survival (PFS) was 5.2 months, and OS was 11.1 months. The most common grade 3 or 4 toxicities included neutropenia (48%), leukopenia (27.8%), diarrhea (14.8%), and fatigue (11.1%).
Irinotecan-based regimens have also been extensively studied in the first-line treatment of metastatic gastric cancer, particularly as an alternative to platinum-based therapy, but superiority has not been established. The combination of fluorouracil, leucovorin, and irinotecan (FOLFIRI) was compared to ECX in a phase 3 trial.22 The study enrolled 416 patients with locally advanced or metastatic gastric or GEJ cancer. At a median follow up of 31 months, the time to progression was longer in the FOLFIRI arm as compared to the ECX arm (5.1 months versus 4.2 months, P = 0.008), but there was no difference in OS (9.5 months versus 9.7 months, P = 0.95), median PFS (5.3 months versus 5.8 months, P = 0.96), or response rate (39.2% versus 37.8%). However, the FOLFIRI regimen had an improved toxicity profile, with a lower overall rate of grade 3 or 4 toxicity (69% versus 84%, P < 0.001). Given these findings, the FOLFIRI regimen is an acceptable alternative to platinum-based therapy in suitable patients.22
HER2-POSITIVE DISEASE
The HER2 proto-oncogene, initially described in breast cancer, has been implicated in several malignancies, including gastric and esophageal cancer. Overexpression or amplification of HER2 can be found in up to 30% of gastric cancers.23 For these patients, adding trastuzumab to a standard regimen of platinum and fluoropyrimidine is the standard of care. The prospective phase 3 Trastuzumab for Gastric Cancer (ToGA) trial randomly assigned 594 patients with HER2-positive gastric cancer to receive either cisplatin and fluorouracil or capecitabine and cisplatin with trastuzumab (n = 294) or without (n = 290) trastuzumab every 3 weeks for a total of 6 cycles, followed by maintenance trastuzumab until disease progression was noted.24 HER2 positivity was defined as HER2 protein overexpression by IHC (cutoff of 3+) or gene amplification by fluorescence in situ hybridization (FISH); tumors with IHC 2+ patterns were followed with FISH studies to confirm positivity. The study found a higher incidence of HER2-positive tumors in patients with GEJ tumors compared to patients with distal gastric cancers (33% versus 20%).24 In this trial, the addition of trastuzumab was associated with an improvement in OS: 13.5 months in the trastuzumab cohort versus 11.1 months in those receiving chemotherapy alone (HR 0.74, P = 0.0048). There was not a significant difference in toxicities between the 2 cohorts, with nausea, emesis, and neutropenia being the most common adverse events. Rates of overall grade 3 or 4 events were similar as well (68% in each cohort). Further exploratory analysis was also conducted according to HER2 status by dividing patients into a “high-expressor” group (n = 446), defined as patients with IHC 3+ tumors or IHC 2+ and FISH positivity, and a “low-expressor” group (n = 131), which included patients with IHC 0 or 1+ tumors. Analysis of patients in the 2 subgroups demonstrated an improved OS with the addition of trastuzumab for the high-expressor cohort, with a median OS of 16 months (HR 0.65 [95% CI 0.51 to 0.83]) compared to 11.8 months in those receiving only chemotherapy.
Dual HER2 blockade has been investigated in metastatic gastric cancer. The phase 3 randomized JACOB trial assigned 780 patients to receive either trastuzumab with a cisplatin/fluoropyrimidine regimen with or without the addition of pertuzumab; the primary end point was OS.25 A non-statistically significant trend towards improvement in OS was found in the pertuzumab arm (17.5 months) as compared with the standard of care arm (14.2 months, HR 0.84, P = 0.0565). The pertuzumab/trastuzumab/chemotherapy cohort experienced a higher incidence of diarrhea (61.6% versus 35.1% in control arm). Cardiac toxicity was comparable in the 2 cohorts.

The Table provides a summary of relevant clinical trials in metastatic gastric cancer.
SECOND-LINE THERAPY
CASE CONTINUED
The patient receives capecitabine, oxaliplatin, and trastuzumab therapy for 6 cycles, followed by trastuzumab for another 3 cycles. While on therapy, he develops a painful right clavicular lesion. He undergoes magnetic resonance imaging of the right clavicle, which shows a lesion in the distal two-thirds of the right clavicle measuring 9.7 × 3.7 × 3.8 cm. The patient is started on palliative radiation to the clavicle. However, repeat CT imaging shows progressive liver metastases.
• What is the approach to second-line therapy for metastatic gastric cancer?
Improvements in our understanding of the molecular pathways that lead to tumorigenesis have contributed to the development of several targeted agents whose efficacy in gastric cancer is being investigated. The NCCN guidelines recommend that for all patients who progress on frontline therapy, second-line therapy consists of a combination of ramucirumab and paclitaxel. Other options include single-agent docetaxel, paclitaxel, irinotecan, or ramucirumab. Combination therapy using irinotecan with either docetaxel, fluorouracil, or cisplatin may also be used.
Ramucirumab, a human IgG1 monoclonal antibody that targets the vascular endothelial growth factor receptor 2 (VEGFR2), was initially approved in 2014 as monotherapy for patients who had previously progressed on first-line chemotherapy. Its approval was based on the results of the phase 3 randomized, double-blind placebo-controlled REGARD study.26 The trial randomly assigned 355 patients with advanced gastric or GEJ adenocarcinoma and disease progression after first-line platinum-containing or fluoropyrimidine-containing chemotherapy to receive best supportive care plus either ramucirumab (n = 238) or placebo (n = 117). Monotherapy with ramucirumab significantly improved median OS compared with placebo (5.2 months versus. 3.8 months; HR 0.776 [95% CI 0.6 to 0.99], P = 0.047). There was also an improvement in PFS of 2.1 months in the ramucirumab cohort, as compared to 1.3 months in the placebo cohort (P < 0.0001). Patients in the ramucirumab arm experienced a higher incidence of hypertension (16% versus 8%), but all other adverse events occurred at comparable rates. Five deaths in the ramucirumab group were thought to be secondary to the study drug, as compared to 2 deaths in the placebo group.
In the subsequent phase 3 RAINBOW trial, the addition of ramucirumab to paclitaxel was investigated, with 330 patients assigned to the combination group and 335 to the paclitaxel-only group.27 The trial again showed that combination therapy afforded patients a significant survival advantage compared to paclitaxel alone, with a median OS of 9.6 months versus 7.4 months for the monotherapy group (HR 0.807 [95% CI 0.678 to 0.962], P = 0.017). A PFS benefit of 4.4 months was observed in the combination therapy groups, as compared with 2.9 months in the monotherapy group (HR 0.635, P < 0.0001). The ramucirumab/paclitaxel group also had a higher overall response rate of 28% versus 16%. The combination cohort had an increased incidence of grade 3 or higher adverse hypertensive events (14% versus 2%) and neutropenia (41% versus 19%), while the incidence of grade 3 febrile neutropenic events was similar between the groups (3% versus 2%).
The addition of bevacizumab, another monoclonal antibody against VEGF, to standard chemotherapy regimens has been explored, but studies have failed to show a survival benefit with this agent in the first-line treatment of advanced gastric cancer. The phase 3 Avastin in Gastric Cancer (AVAGAST) trial was a multinational, randomized study where patients received either bevacizumab (n = 387) or placebo (n = 387) in addition to cisplatin and capecitabine.28 The substitution of fluorouracil for capecitabine was permitted for patients who were unable to tolerate oral medications. Cisplatin was administered for a maximum of 6 cycles, while capecitabine and bevacizumab were administered until disease progression. The study failed to show an improvement in OS, with a median OS of 12.1 months noted in the bevacizumab cohort, as compared to 10.1 months in the placebo arm (HR 0.87 [95% CI 0.73 to 1.03], P = 0.1002). However, there was a modest improvement in median PFS (6.7 months versus 5.3 months; HR 0.80 [95% CI 0.68 to 0.93], P = 0.0037) and overall response rate (46% versus 37.4%, P = 0.0315). The most commonly reported grade 3 to 5 adverse events included neutropenia (35%), anemia (10%), and loss of appetite (8%). Interestingly, in a follow-up report, higher serum levels of VEGF-A were thought to correlate with an enhanced response to bevacizumab.29 However, the routine use of biomarker analysis in selecting patients for treatment with bevacizumab in metastatic gastric cancer remains to be further clarified.
Use of other agents with anti-HER2 activity in the second-line treatment of patients who have experienced progression while on trastuzumab remains unclear. In the recent T-ACT trial, patients with disease refractory to frontline therapy with combination trastuzumab and fluoropyrimidine/platinum agents were randomly assigned to receive either weekly paclitaxel (n = 45) or weekly paclitaxel plus trastuzumab (n = 44).30 Patients in the combination cohort received an initial dose of trastuzumab 8 mg/kg followed by 6 mg/kg every 3 weeks until progression. The study did not find a difference in either PFS (3.19 months versus 3.68 months; HR 0.91 [95% CI 0.67 to 1.22], P = 0.33) or OS (9.95 months versus 10.2 months; HR 1.23 [95% CI 0.75 to 1.99], P = 0.20). The study thus failed to show a benefit to continuing trastuzumab after progression in the first-line setting.
Lapatinib in combination with paclitaxel has been compared to paclitaxel alone for the treatment of advanced HER2-positive gastric cancer in an Asian population in the phase 3 TyTAN trial.31 With a primary end point of OS, the study randomly assigned 129 patients to receive paclitaxel alone and 132 patients to receive paclitaxel with lapatinib. There was a nonsignificant trend towards improvement in OS in the combination group (11 months) as compared to the paclitaxel-only group(8.9 months, P = 0.1044), with no significant difference in median PFS (5.4 months versus 4.4 months). However, it is important to note that only 15 patients in this trial had previously been exposed to trastuzumab. Another trial, the phase 3 GATSBY study, examined the efficacy of trastuzumab emtansine in the second-line setting compared to taxanes alone and failed to show any improvement in PFS or OS.32 Given these results, no alternative anti-HER2 therapy has been proven to be efficacious for patients who are trastuzumab refractory. Therefore, including anti-HER2 therapy in the second-line treatment of HER2-positive gastric cancer is not recommended.
IMMUNOTHERAPY AND OTHER TARGETED THERAPIES
Several other targeted therapies have been studied in advanced gastric cancer, without any demonstrable survival benefit. The PI3K/AKT/mTOR pathway is known to be involved in regulation of cell growth and angiogenesis, and the mTOR inhibitor everolimus is widely used to treat other malignancies, including breast cancer. The use of everolimus in the second-line setting was studied in the phase 3 GRANITE-1 trial, where it was compared to best supportive care and failed to provide any survival benefit.33 Cetuximab, a recombinant human and mouse chimeric monoclonal antibody, and panitumumab, a recombinant human antibody against the epidermal growth factor receptor (EGFR), have also been examined in gastric and GEJ cancer patients. However, the large phase 3 EXPAND and REAL-3 trials did not show a survival benefit when these agents were added to standard chemotherapy.34,35
Overexpression of MET, a proto-oncogene and tyrosine kinase receptor, has also been implicated in gastric cancer progression. The ligand for MET is the hepatocyte growth factor (HGF), and aberrant signaling of this pathway has been shown to correlate with an aggressive gastric cancer phenotype and poorer OS by promoting tumor growth and angiogenesis. However, no MET inhibitors thus far have been found to be clinically effective. RILOMET-1 and RILOMET-2 were phase 3 trials examining the efficacy of rilotumumab, a humanized anti-HGF antibody, in combination with chemotherapy (ECX and cisplatin with capecitabine, respectively) for the frontline treatment of MET-positive GEJ and gastric cancers. Both studies were discontinued due to a higher treatment-related mortality in patients receiving rilotumumab, with a higher incidence of adverse events due to disease progression being noted.36 Similarly, onartuzumab, a monovalent monoclonal antibody against the MET receptor, was investigated in the phase 3 METGastric trial in combination with modified FOLFOX6 as first-line therapy for HER2-negative, MET-positive metastatic GEJ and gastric cancers. The study did not demonstrate any significant improvements in OS or PFS.37
There has been significant interest in incorporating immunotherapy in the treatment of early and metastatic gastric cancer. Pembrolizumab is the first programmed death receptor (PD-1) inhibitor to be approved for treatment of patients with PD-L1−positive advanced gastric cancer who had previously received 2 or more lines of chemotherapy. Although earlier studies of pembrolizumab in lung cancer utilized the tumor proportion score (TPS) to determine PD-L1 positivity, this was not found to be applicable to gastric cancer. Instead, the combined positive score (CPS) is used in gastric cancer. The CPS evaluates the number of tumor cells and immune cells (macrophages and lymphocytes) that stain positive for PD-L1 relative to all viable tumor cells. Comparatively, the TPS only examines the percentage of viable tumor cells that show complete or partial positive staining for PD-L1. A CPS score of 1 or greater identifies patients who would be suitable candidates for pembrolizumab.
The approval of pembrolizumab was based on the positive findings from the recent KEYNOTE-059 trial.38 The study included 259 patients who had previously received either fluoropyrimidine, cisplatin, or anti-HER2 therapy, with 148 patients (55%) of these patients having PD-L1−positive tumors. The PD-L1 status was determined using a pharmDx Kit, which is now approved by the US Food and Drug Administration to select patients who could benefit from pembrolizumab treatment. CPS was calculated as the number of PD-L1−staining cells divided by the total number of evaluated cells. The study included patients with microsatellite stable (MSI-S), undetermined, or deficient MMR status. The overall response rate to pembrolizumab across all patients was 11.6%, median PFS was 2 months, and the 12-month OS rate was 23.4%. In the subset of patients with MSI-H tumors, the overall response rate was 57.1%, with a complete response rate of 14.3%; in those with MSI-S tumors, the overall response rate was 9% and the complete response rate was 2.4%. Among patients with PD-L1–positive tumors, the overall response rate was 15.5% (95% CI 10.1% to 22.4). Common adverse events included fatigue, hypothyroidism, nausea, diarrhea, and arthralgia.38
CASE CONCLUSION
This patient with metastatic gastric cancer receives second-line chemotherapy with ramucirumab and paclitaxel. Follow-up imaging shows persistent liver metastases and new lung metastasis. Because the tumor is PD-L1–positive, the patient receives 4 cycles of pembrolizumab, with no significant change noted in disease burden. He notes a significant decline in functional status with increased weight loss, nausea, emesis, and fatigue. The patient opts to forego any further therapy and instead chooses to pursue supportive care only.
SUMMARY
Gastric cancer is the third most common cause of cancer death worldwide. Common risk factors for developing gastric cancer include H. pylori infection, smoking, alcohol abuse, radiation exposure, high-fat diet, and obesity. Patients presenting with alarm symptoms of nausea, emesis, early satiety, abdominal pain, or weight loss should be fully evaluated with upper GI endoscopy. If there is suspicion for metastatic disease, CT evaluation of the chest, abdomen, and pelvis with oral and intravenous contrast should be obtained. Treatment of patients with metastatic gastric cancer is guided by their performance status at presentation. For patients with good performance status, a combination of platinum and fluoropyrimidine therapy, such as FOLFOX, can be considered. Doublet chemotherapy regimens are preferred over triplet chemotherapy regimens given their better tolerability. For patients with HER2-positive disease, the addition of trastuzumab to the platinum and fluoropyrimidine backbone is the standard of care in the first line.
Several targeted agents have been studied in patients progressing on initial therapy, with ramucirumab and paclitaxel being considered the regimen of choice in the second line. No anti-HER2 therapy has been approved for patients who are refractory to trastuzumab. Pembrolizumab is approved for use in patients who are PD-L1–positive and have previously progressed on at least 2 lines of chemotherapy. Pembrolizumab is also approved for the treatment of patients with unresectable or metastatic, MSI-H or MMR-deficient gastric cancers that have progressed after prior treatment and who have no satisfactory alternative treatment options.
1. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41
2. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Allen C, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol 2017;3:524–8.
3. Sitarz R, Skierucha M, Mielko J, et al. Gastric cancer: epidemiology, prevention, classification, and treatment. Cancer Manag Res 2018;10:239–48.
4. Olefson S, Moss SF. Obesity and related risk factors in gastric cardia adenocarcinoma. Gastric Cancer 2015;18:23–32.
5. Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett 2014;345:196–202.
6. Espinoza JL, Matsumoto A, Tanaka H, Matsumura I. Gastric microbiota: An emerging player in Helicobacter pylori-induced gastric malignancies. Cancer Lett 2018;414:147–52.
7. Chmiela M, Gonciarz W. Molecular mimicry in Helicobacter pylori infections. World J Gastroenterol 2017;23:3964–77.
8. Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
9. Ford A, Gurusamy KS, Delaney B, et al. Eradication therapy for peptic ulcer disease in Helicobacter pylori-positive patients. Cochrane Database Syst Rev 2004(4):CD003840.
10. Karimi P, Islami F, Anandasabapathy S, et al. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev 2014;23:700–13.
11. Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
12. Chan BA , Sim HW, Natori A, et al. Survival outcomes for de novo versus relapsed stage IV gastric and gastroesophageal junction (GEJ) adenocarcinoma [abstract]. J Clin Oncol 2018;36(no. 4 suppl):148.
13. DeVita VT, Lawrence TS, Rosenberg SA. DeVita, Hellman, and Rosenberg’s cancer: principles & practice of oncology. 9th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2011.
14. Siewert JR, Hölscher AH, Becker K, Gössner W. [Cardia cancer: attempt at a therapeutically relevant classification]. [Article in German.] Chirurg 1987;58:25–32.
15. Amin MB, Edge SB, Greene FL, et al, eds. AJCC cancer staging manual. 8th ed. New York: Springer; 2017.
16. Wagner AD, Syn NL, Moehler M, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev 2017;8:CD004064.
17. Qiu H, Zhou Z. [Updates and interpretation on NCCN clinical practice guidelines for gastric cancer 2017 version 5]. [Article in Chinese.] Zhonghua Wei Chang Wai Ke Za Zhi 2018;21:160–4.
18. Webb A, Cunningham D, Scarffe JH, et al. Randomized trial comparing epirubicin, cisplatin, and fluorouracil versus fluorouracil, doxorubicin, and methotrexate in advanced esophagogastric cancer. J Clin Oncol 1997;15:261–7.
19. Van Cutsem E, Moiseyenko VM, Tjulandin S, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 2006;24:4991–7.
20. Cunningham D, Okines AF, Ashley S. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2010;362:858–9.
21. Al-Batran SE, Hartmann JT, Hofheinz R, et al. Biweekly fluorouracil, leucovorin, oxaliplatin, and docetaxel (FLOT) for patients with metastatic adenocarcinoma of the stomach or esophagogastric junction: a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie. Ann Oncol 2008;19:1882–7.
22. Guimbaud R, Louvet C, Ries P, et al. Prospective, randomized, multicenter, phase III study of fluorouracil, leucovorin, and irinotecan versus epirubicin, cisplatin, and capecitabine in advanced gastric adenocarcinoma: a French intergroup (Federation Francophone de Cancerologie Digestive, Federation Nationale des Centres de Lutte Contre le Cancer, and Groupe Cooperateur Multidisciplinaire en Oncologie) study. J Clin Oncol 2014;32:3520–6.
23. Boku N. HER2-positive gastric cancer. Gastric Cancer 2014;17:1–12.
24. Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
25. Tabernero J, Hoff PM, Shen L, et al. Pertuzumab + trastuzumab + chemotherapy for HER2-positive metastatic gastric or gastro-oesophageal junction cancer: Final analysis of a Phase III study (JACOB) [abstract]. Ann Oncol 2017;28(suppl 5):6160.
26. Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014;383:31–9.
27. Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol 2014;15:1224–35.
28. Ohtsu A, Shah MA, Van Cutsem E, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 2011;29:3968–76.
29. Van Cutsem E, de Haas S, Kang YK, et al, Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a biomarker evaluation from the AVAGAST randomized phase III trial. J Clin Oncol 2012;30:2119–27.
30. Makiyama A, Sagara K, Kawada J, et al. A randomized phase II study of weekly paclitaxel ± trastuzumab in patients with HER2-positive advanced gastric or gastro-esophageal junction cancer refractory to trastuzumab combined with fluoropyrimidine and platinum: WJOG7112G (T-ACT) [abstract]. J Clin Oncol 2018;36(no. 15 suppl):4011.
31. Satoh T, Xu RH, Chung HC, et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN--a randomized, phase III study. J Clin Oncol 2014;32:2039–49.
32. Thuss-Patience PC, Shah MA, Ohtsu A, et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): an international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol 2017;18:640–53.
33. Ohtsu A, Ajani JA, Bai YX, et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1 study. J Clin Oncol 2013;31:3935–43.
34. Lordick F, Kang YK, Chung HC, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:490–9.
35. Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:481–9.
36. Catenacci DVT, Tebbutt NC, Davidenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1467–82.
37. Shah MA, Bang YJ, Lordick F, et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma: the METGastric randomized clinical trial. JAMA Oncol 2017;3:620–7.
38. Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol 2018;4(5):e180013.
INTRODUCTION
According to the Surveillance, Epidemiology and End Results database, in 2017 there were 28,000 new cases of gastric cancer, accounting for 1.8% of all malignancies in the United States, and an estimated 10,960 gastric cancer–related deaths.1 Worldwide, gastric cancer is the fifth most common malignancy and the third most common cause of death from any cancer.2 The incidence of gastric cancer varies significantly by geographic region, with countries in Eastern Asia (China, Japan), Eastern Europe, and Central and South America accounting for 50% of all new cases.3 Although the incidence of gastric cancer has declined in recent years, this decrease has not been observed consistently across all nations.2 In particular, the incidence of gastric cancers arising from the cardia has been increasing, which is perhaps due to a higher prevalence of obesity in Western societies.4
In this article, we review key aspects of management of metastatic gastric cancer, including selection of first- and second-line therapy, and discuss targeted agents and upcoming clinical trials.
EPIDEMIOLOGY AND RISK FACTORS
Chronic infection with Helicobacter pylori, a gram-negative bacterium, is a strong etiological factor for the development of gastric cancer, contributing to up to 70% of cases.2 The pathogen can colonize the gastric mucosa, leading to chronic inflammation. Although most patients remain asymptomatic, 1% to 3% develop gastric cancer and another 0.1% develop mucosa-associated lymphoid tissue lymphoma.5 H. pylori infection is more commonly associated with cancer of the gastric body than with cancer of the gastroesophageal junction (GEJ). The increased burden of gastric cancer in countries in Eastern Asia, Latin America, and Eastern Europe has been correlated to the prevalence of chronic H. pylori infection in these areas.
Carcinogenesis secondary to H. pylori infection may occur via several mechanisms. First, H. pylori can release virulence factors, such as cytotoxin-associated gene A, vacuolating cytotoxin, and outer membrane proteins, into the cytosol of host cells, leading to changes in patterns of cell proliferation and apoptosis.6 These virulence factors can modulate the host immune system, attenuating it to promote dysplasia. In addition, continued recognition of these factors by the immune system leads to a persistent inflammatory response, with the release of cytokines such as interleukin (IL) -1β, IL-6, and IL-8. This leads to chronic mucosal damage, further promoting dysplasia with eventual transformation into adenocarcinoma.7 In Japan and Korea, where screening for H. pylori infection is routinely performed, there have been improvements in overall survival (OS) rates for gastric cancer, with 5-year OS rates of 70%.8 The International Agency for Research on Cancer recommends further research into population-based screening and treatment programs for patients with chronic H. pylori infection. However, despite this recommendation, optimal screening strategies are not clearly defined.9
Other risk factors for the development of gastric cancer include chronic gastroesophageal reflux disease; smoking; alcohol use; exposure to radiation; diets high in fats, salt, and smoked items and low in fruits and vegetables; obesity; and exposure to chemotherapeutic agents such as procarbazine.10 Another pathogen suspected, but not proven, to be associated with increased risk for gastric cancer is the Epstein-Barr virus, a human herpesvirus found in 80% of all gastric carcinomas with lymphoid features.11 In addition, whether the use of medications such as statins and nonsteroidal anti-inflammatory drugs confers a decreased risk of gastric cancers remains unclear.10
EVALUATION
CASE PRESENTATION
A 55-year-old Caucasian man with a history of type 2 diabetes mellitus presents to the gastrointestinal (GI) clinic with a 6-month history of dysphagia. The dysphagia is worsened with ingestion of solids, particularly towards the end of the day. He states that the food often gets “stuck in the middle of the chest.” The patient denies any nausea or emesis but notes that he has a poor appetite. He reports having worsening mid-epigastric abdominal pain that is non-radiating, dull in character, and 6/10 in intensity. He also reports a 10-lb weight loss over the past 2 months. He has no previous history of reflux, chest pain, dyspnea, or cough. Review of systems is otherwise benign. Physical exam is within normal limits.
• Which tests should be conducted when gastric cancer is suspected?
Persistent epigastric abdominal pain and weight loss are the most common early symptoms of gastric cancer. Nausea, early satiety, dysphagia, and occult GI bleeding can be other presenting signs. Patients presenting with alarm symptoms of nausea, emesis, early satiety, abdominal pain, or weight loss should be fully evaluated with upper GI endoscopy. Early diagnosis of gastric cancer is essential in obtaining a curative resection. However, at least 40% of patients present with de novo metastatic disease at the time of initial diagnosis.12 Gastric cancer spreads by direct extension through the gastric wall, with the liver, peritoneum, and regional lymph nodes being the most common sites of metastatic deposits.13 Classically, Virchow’s node, the left supraclavicular lymph node, is involved with metastatic gastric cancer. Involvement of the left axillary lymph node (Irish node) or a periumbilical nodule (Sister Mary Joseph node) may also be observed. Other, less commonly noted sites of metastatic disease include the ovaries, central nervous system, bone, lung, and soft tissues.13
Upper GI endoscopy is the best method for determining tumor location and extent and obtaining a specimen for a definitive tissue diagnosis.14 It is essential to accurately identify the location of the tumor in the stomach and relative to the GEJ. The American Joint Committee on Cancer classification defines tumors involving the GEJ with an epicenter no more than 2 cm into the proximal stomach as esophageal cancers.15 Tumors of the GEJ with their epicenter more than 2 cm into the proximal stomach are defined as gastric cancers. If metastatic disease is suspected, computed tomography (CT) scan of the chest, abdomen, and pelvis with oral and intravenous contrast can be obtained to determine the extent of disease spread. In the absence of any metastatic disease, endoscopic ultrasound (EUS) should be conducted to determine the depth of tumor invasion (T staging) and lymph node status. In the era of targeted therapy, patients with metastatic disease should undergo testing for human epidermal growth factor-2 (HER-2) expression, microsatellite instability (MSI), and programmed death ligand 1 (PD-L1) expression. Patients should be staged according to the TNM staging system.
FIRST-LINE TREATMENT OPTIONS
CASE CONTINUED
The patient undergoes esophagoduodenoscopy (EGD) and is found to have a gastric cardia mass extending into the distal esophagus. EUS also demonstrates multiple abdominal and mediastinal lymph nodes. No gastric outlet obstruction is found. Biopsy shows poorly differentiated invasive adenocarcinoma. Warthin–Starry stain is negative for H. pylori organism. The tumor cells are positive for cytokeratin (CK7), CK19, and mucin-1 gene (MUC1); focally positive for CK20; and negative for MUC2. HER2 testing results are reported as immunohistochemistry (IHC) 3+, consistent with strongly positive HER2 protein expression. Further IHC testing for mismatch repair (MMR) proteins shows intact nuclear expression of MLH1, MSH2, MSH6, and PMS2 protein, consistent with a low probability of MSI-high tumor. The tumor is found to be PD-L1 positive. Imaging reveals abnormal mass-like nodular thickening of the gastric wall, with an infiltrative opacity within the pancreatico-duodenal groove, suspicious for tumor infiltration. Multiple metastatic deposits are noted in the liver, peritoneum, and bilateral lungs. There is extensive gastrohepatic ligament and periportal lymphadenopathy and mild enlargement of the pulmonary hilar lymph nodes. These findings are consistent with stage 4 (T4bN3aM1) gastric cancer. Given these findings, staging laparoscopy is deferred.
• What are the first-line treatment options for patients with metastatic gastric cancer?
Patients with metastatic gastric cancer have a poor prognosis, and management is stratified based on performance status (Figure). In patients with good performance status, systemic chemotherapy is the mainstay of treatment. The goal of therapy is not curative, but rather treatment focuses on palliation of symptoms arising from tumor spread. Given this treatment goal, there has been considerable interest in clarifying the utility of chemotherapy as opposed to best supportive care. In a recent Cochrane review of 64 randomized control trials involving 11,698 patients, chemotherapy was found to improve OS by 6.7 months as compared to best supportive care (hazard ratio [HR] 0.3 [95% confidence interval {CI} 0.24 to 0.55]).16 Five classes of cytotoxic chemotherapeutic agents have demonstrated activity in gastric cancer. These include fluoropyrimidine (either infusional fluorouracil or capecitabine), platinum agents (cisplatin or oxaliplatin), taxanes (docetaxel or paclitaxel), anthracyclines (epirubicin), and irinotecan.13 Treatment options are further divided based on whether the patient has HER2-overexpressing or non-expressing malignancy.
HER2-NEGATIVE DISEASE
For patients with HER2-negative disease, National Comprehensive Cancer Network (NCCN) guidelines recommend using 2-drug combination regimens rather than 3 drugs, given concern for increased toxicity with 3-drug regimens.17 For patients with a performance status of 0 to 1, utilization of a 3-drug regimen is a reasonable alternative. The combination of a fluoropyrimidine with a platinum agent is considered the standard of care, with regimens such as fluorouracil, leucovorin, and oxaliplatin (FOLFOX) being commonly used.
Epirubicin-containing regimens have also been extensively studied in advanced gastric cancer. In a study of 274 previously untreated patients with GEJ cancers, the combination of epirubicin, cisplatin, and fluorouracil (ECF) was compared to fluorouracil, doxorubicin, and methotrexate (FAMTX). There was an OS benefit favoring ECF (8.9 months versus 5.7 months) at 1 year (95% CI 27% to 45%, P = 0.0009). The ECF regimen was associated with an increased risk of nausea, emesis, and alopecia, while more hematologic toxicity and infections were noted with the FAMTX regimen.18 In addition, in a phase 3 trial, Van Cutsem and colleagues examined the role of docetaxel in combination with cisplatin and fluorouracil (DCF) compared to cisplatin and fluorouracil alone. Addition of docetaxel led to improved OS and time to progression (9.2 months versus 8.6 months for cisplatin and fluorouracil alone, P = 0.02) but with an increased risk of grade 3 and 4 toxicities (69% versus 59%). These adverse events included neutropenia (82% versus 57% of cisplatin and fluorouracil patients), diarrhea (19% versus 8%), stomatitis (21% versus 27%), and fatigue (19% versus 14%).19
The landmark phase 3 REAL-2 study compared 4 chemotherapy regimens in patients with untreated advanced esophagogastric cancer. This study was conducted to determine if the efficacy of cisplatin and oxaliplatin, a third-generation platinum agent, is equivalent to that of fluorouracil and capecitabine, an oral fluoropyrimidine. In this trial, a 2 × 2 design was used to compare 4 regimens: ECF versus epirubicin, cisplatin, and capecitabine (ECX) versus epirubicin, oxaliplatin, and fluorouracil (EOF) versus epirubicin, oxaliplatin, and capecitabine (EOX). The study found EOX to be noninferior to ECF, with a trend towards improved OS compared to other combination regimens (11.2 months versus 9.9 months, HR 0.80 [95% CI 0.66 to 0.97], P = 0.02).20 Thus, the study demonstrated that an oxaliplatin and capecitabine-based regimen could replace cisplatin and fluorouracil. Given that fluorouracil administration requires long continuous infusions, the oral-based capecitabine regimen is an attractive option for patients.
Several trials have demonstrated the equivalency of oxaliplatin with cisplatin in combination regimens for the treatment of advanced gastric cancer. Oxaliplatin has the benefit of an improved toxicity profile as compared to cisplatin, with the major dose-limiting toxicity being peripheral neuropathy
Given previous evidence that DCF (docetaxel, cisplatin, fluorouracil) is superior to cisplatin and fluorouracil alone, there was interest in determining if the addition of docetaxel to a backbone of fluorouracil, oxaliplatin, and leucovorin (FLO) could elicit a higher response rate. This concept was investigated in a phase 2 trial that assigned 54 patients with metastatic gastric or GEJ adenocarcinoma to receive biweekly infusions of oxaliplatin, leucovorin, fluorouracil, and docetaxel.21 Median time to response was 1.54 months, and the overall response rate was 57.7%. Median progression-free survival (PFS) was 5.2 months, and OS was 11.1 months. The most common grade 3 or 4 toxicities included neutropenia (48%), leukopenia (27.8%), diarrhea (14.8%), and fatigue (11.1%).
Irinotecan-based regimens have also been extensively studied in the first-line treatment of metastatic gastric cancer, particularly as an alternative to platinum-based therapy, but superiority has not been established. The combination of fluorouracil, leucovorin, and irinotecan (FOLFIRI) was compared to ECX in a phase 3 trial.22 The study enrolled 416 patients with locally advanced or metastatic gastric or GEJ cancer. At a median follow up of 31 months, the time to progression was longer in the FOLFIRI arm as compared to the ECX arm (5.1 months versus 4.2 months, P = 0.008), but there was no difference in OS (9.5 months versus 9.7 months, P = 0.95), median PFS (5.3 months versus 5.8 months, P = 0.96), or response rate (39.2% versus 37.8%). However, the FOLFIRI regimen had an improved toxicity profile, with a lower overall rate of grade 3 or 4 toxicity (69% versus 84%, P < 0.001). Given these findings, the FOLFIRI regimen is an acceptable alternative to platinum-based therapy in suitable patients.22
HER2-POSITIVE DISEASE
The HER2 proto-oncogene, initially described in breast cancer, has been implicated in several malignancies, including gastric and esophageal cancer. Overexpression or amplification of HER2 can be found in up to 30% of gastric cancers.23 For these patients, adding trastuzumab to a standard regimen of platinum and fluoropyrimidine is the standard of care. The prospective phase 3 Trastuzumab for Gastric Cancer (ToGA) trial randomly assigned 594 patients with HER2-positive gastric cancer to receive either cisplatin and fluorouracil or capecitabine and cisplatin with trastuzumab (n = 294) or without (n = 290) trastuzumab every 3 weeks for a total of 6 cycles, followed by maintenance trastuzumab until disease progression was noted.24 HER2 positivity was defined as HER2 protein overexpression by IHC (cutoff of 3+) or gene amplification by fluorescence in situ hybridization (FISH); tumors with IHC 2+ patterns were followed with FISH studies to confirm positivity. The study found a higher incidence of HER2-positive tumors in patients with GEJ tumors compared to patients with distal gastric cancers (33% versus 20%).24 In this trial, the addition of trastuzumab was associated with an improvement in OS: 13.5 months in the trastuzumab cohort versus 11.1 months in those receiving chemotherapy alone (HR 0.74, P = 0.0048). There was not a significant difference in toxicities between the 2 cohorts, with nausea, emesis, and neutropenia being the most common adverse events. Rates of overall grade 3 or 4 events were similar as well (68% in each cohort). Further exploratory analysis was also conducted according to HER2 status by dividing patients into a “high-expressor” group (n = 446), defined as patients with IHC 3+ tumors or IHC 2+ and FISH positivity, and a “low-expressor” group (n = 131), which included patients with IHC 0 or 1+ tumors. Analysis of patients in the 2 subgroups demonstrated an improved OS with the addition of trastuzumab for the high-expressor cohort, with a median OS of 16 months (HR 0.65 [95% CI 0.51 to 0.83]) compared to 11.8 months in those receiving only chemotherapy.
Dual HER2 blockade has been investigated in metastatic gastric cancer. The phase 3 randomized JACOB trial assigned 780 patients to receive either trastuzumab with a cisplatin/fluoropyrimidine regimen with or without the addition of pertuzumab; the primary end point was OS.25 A non-statistically significant trend towards improvement in OS was found in the pertuzumab arm (17.5 months) as compared with the standard of care arm (14.2 months, HR 0.84, P = 0.0565). The pertuzumab/trastuzumab/chemotherapy cohort experienced a higher incidence of diarrhea (61.6% versus 35.1% in control arm). Cardiac toxicity was comparable in the 2 cohorts.
The Table provides a summary of relevant clinical trials in metastatic gastric cancer.
SECOND-LINE THERAPY
CASE CONTINUED
The patient receives capecitabine, oxaliplatin, and trastuzumab therapy for 6 cycles, followed by trastuzumab for another 3 cycles. While on therapy, he develops a painful right clavicular lesion. He undergoes magnetic resonance imaging of the right clavicle, which shows a lesion in the distal two-thirds of the right clavicle measuring 9.7 × 3.7 × 3.8 cm. The patient is started on palliative radiation to the clavicle. However, repeat CT imaging shows progressive liver metastases.
• What is the approach to second-line therapy for metastatic gastric cancer?
Improvements in our understanding of the molecular pathways that lead to tumorigenesis have contributed to the development of several targeted agents whose efficacy in gastric cancer is being investigated. The NCCN guidelines recommend that for all patients who progress on frontline therapy, second-line therapy consists of a combination of ramucirumab and paclitaxel. Other options include single-agent docetaxel, paclitaxel, irinotecan, or ramucirumab. Combination therapy using irinotecan with either docetaxel, fluorouracil, or cisplatin may also be used.
Ramucirumab, a human IgG1 monoclonal antibody that targets the vascular endothelial growth factor receptor 2 (VEGFR2), was initially approved in 2014 as monotherapy for patients who had previously progressed on first-line chemotherapy. Its approval was based on the results of the phase 3 randomized, double-blind placebo-controlled REGARD study.26 The trial randomly assigned 355 patients with advanced gastric or GEJ adenocarcinoma and disease progression after first-line platinum-containing or fluoropyrimidine-containing chemotherapy to receive best supportive care plus either ramucirumab (n = 238) or placebo (n = 117). Monotherapy with ramucirumab significantly improved median OS compared with placebo (5.2 months versus. 3.8 months; HR 0.776 [95% CI 0.6 to 0.99], P = 0.047). There was also an improvement in PFS of 2.1 months in the ramucirumab cohort, as compared to 1.3 months in the placebo cohort (P < 0.0001). Patients in the ramucirumab arm experienced a higher incidence of hypertension (16% versus 8%), but all other adverse events occurred at comparable rates. Five deaths in the ramucirumab group were thought to be secondary to the study drug, as compared to 2 deaths in the placebo group.
In the subsequent phase 3 RAINBOW trial, the addition of ramucirumab to paclitaxel was investigated, with 330 patients assigned to the combination group and 335 to the paclitaxel-only group.27 The trial again showed that combination therapy afforded patients a significant survival advantage compared to paclitaxel alone, with a median OS of 9.6 months versus 7.4 months for the monotherapy group (HR 0.807 [95% CI 0.678 to 0.962], P = 0.017). A PFS benefit of 4.4 months was observed in the combination therapy groups, as compared with 2.9 months in the monotherapy group (HR 0.635, P < 0.0001). The ramucirumab/paclitaxel group also had a higher overall response rate of 28% versus 16%. The combination cohort had an increased incidence of grade 3 or higher adverse hypertensive events (14% versus 2%) and neutropenia (41% versus 19%), while the incidence of grade 3 febrile neutropenic events was similar between the groups (3% versus 2%).
The addition of bevacizumab, another monoclonal antibody against VEGF, to standard chemotherapy regimens has been explored, but studies have failed to show a survival benefit with this agent in the first-line treatment of advanced gastric cancer. The phase 3 Avastin in Gastric Cancer (AVAGAST) trial was a multinational, randomized study where patients received either bevacizumab (n = 387) or placebo (n = 387) in addition to cisplatin and capecitabine.28 The substitution of fluorouracil for capecitabine was permitted for patients who were unable to tolerate oral medications. Cisplatin was administered for a maximum of 6 cycles, while capecitabine and bevacizumab were administered until disease progression. The study failed to show an improvement in OS, with a median OS of 12.1 months noted in the bevacizumab cohort, as compared to 10.1 months in the placebo arm (HR 0.87 [95% CI 0.73 to 1.03], P = 0.1002). However, there was a modest improvement in median PFS (6.7 months versus 5.3 months; HR 0.80 [95% CI 0.68 to 0.93], P = 0.0037) and overall response rate (46% versus 37.4%, P = 0.0315). The most commonly reported grade 3 to 5 adverse events included neutropenia (35%), anemia (10%), and loss of appetite (8%). Interestingly, in a follow-up report, higher serum levels of VEGF-A were thought to correlate with an enhanced response to bevacizumab.29 However, the routine use of biomarker analysis in selecting patients for treatment with bevacizumab in metastatic gastric cancer remains to be further clarified.
Use of other agents with anti-HER2 activity in the second-line treatment of patients who have experienced progression while on trastuzumab remains unclear. In the recent T-ACT trial, patients with disease refractory to frontline therapy with combination trastuzumab and fluoropyrimidine/platinum agents were randomly assigned to receive either weekly paclitaxel (n = 45) or weekly paclitaxel plus trastuzumab (n = 44).30 Patients in the combination cohort received an initial dose of trastuzumab 8 mg/kg followed by 6 mg/kg every 3 weeks until progression. The study did not find a difference in either PFS (3.19 months versus 3.68 months; HR 0.91 [95% CI 0.67 to 1.22], P = 0.33) or OS (9.95 months versus 10.2 months; HR 1.23 [95% CI 0.75 to 1.99], P = 0.20). The study thus failed to show a benefit to continuing trastuzumab after progression in the first-line setting.
Lapatinib in combination with paclitaxel has been compared to paclitaxel alone for the treatment of advanced HER2-positive gastric cancer in an Asian population in the phase 3 TyTAN trial.31 With a primary end point of OS, the study randomly assigned 129 patients to receive paclitaxel alone and 132 patients to receive paclitaxel with lapatinib. There was a nonsignificant trend towards improvement in OS in the combination group (11 months) as compared to the paclitaxel-only group(8.9 months, P = 0.1044), with no significant difference in median PFS (5.4 months versus 4.4 months). However, it is important to note that only 15 patients in this trial had previously been exposed to trastuzumab. Another trial, the phase 3 GATSBY study, examined the efficacy of trastuzumab emtansine in the second-line setting compared to taxanes alone and failed to show any improvement in PFS or OS.32 Given these results, no alternative anti-HER2 therapy has been proven to be efficacious for patients who are trastuzumab refractory. Therefore, including anti-HER2 therapy in the second-line treatment of HER2-positive gastric cancer is not recommended.
IMMUNOTHERAPY AND OTHER TARGETED THERAPIES
Several other targeted therapies have been studied in advanced gastric cancer, without any demonstrable survival benefit. The PI3K/AKT/mTOR pathway is known to be involved in regulation of cell growth and angiogenesis, and the mTOR inhibitor everolimus is widely used to treat other malignancies, including breast cancer. The use of everolimus in the second-line setting was studied in the phase 3 GRANITE-1 trial, where it was compared to best supportive care and failed to provide any survival benefit.33 Cetuximab, a recombinant human and mouse chimeric monoclonal antibody, and panitumumab, a recombinant human antibody against the epidermal growth factor receptor (EGFR), have also been examined in gastric and GEJ cancer patients. However, the large phase 3 EXPAND and REAL-3 trials did not show a survival benefit when these agents were added to standard chemotherapy.34,35
Overexpression of MET, a proto-oncogene and tyrosine kinase receptor, has also been implicated in gastric cancer progression. The ligand for MET is the hepatocyte growth factor (HGF), and aberrant signaling of this pathway has been shown to correlate with an aggressive gastric cancer phenotype and poorer OS by promoting tumor growth and angiogenesis. However, no MET inhibitors thus far have been found to be clinically effective. RILOMET-1 and RILOMET-2 were phase 3 trials examining the efficacy of rilotumumab, a humanized anti-HGF antibody, in combination with chemotherapy (ECX and cisplatin with capecitabine, respectively) for the frontline treatment of MET-positive GEJ and gastric cancers. Both studies were discontinued due to a higher treatment-related mortality in patients receiving rilotumumab, with a higher incidence of adverse events due to disease progression being noted.36 Similarly, onartuzumab, a monovalent monoclonal antibody against the MET receptor, was investigated in the phase 3 METGastric trial in combination with modified FOLFOX6 as first-line therapy for HER2-negative, MET-positive metastatic GEJ and gastric cancers. The study did not demonstrate any significant improvements in OS or PFS.37
There has been significant interest in incorporating immunotherapy in the treatment of early and metastatic gastric cancer. Pembrolizumab is the first programmed death receptor (PD-1) inhibitor to be approved for treatment of patients with PD-L1−positive advanced gastric cancer who had previously received 2 or more lines of chemotherapy. Although earlier studies of pembrolizumab in lung cancer utilized the tumor proportion score (TPS) to determine PD-L1 positivity, this was not found to be applicable to gastric cancer. Instead, the combined positive score (CPS) is used in gastric cancer. The CPS evaluates the number of tumor cells and immune cells (macrophages and lymphocytes) that stain positive for PD-L1 relative to all viable tumor cells. Comparatively, the TPS only examines the percentage of viable tumor cells that show complete or partial positive staining for PD-L1. A CPS score of 1 or greater identifies patients who would be suitable candidates for pembrolizumab.
The approval of pembrolizumab was based on the positive findings from the recent KEYNOTE-059 trial.38 The study included 259 patients who had previously received either fluoropyrimidine, cisplatin, or anti-HER2 therapy, with 148 patients (55%) of these patients having PD-L1−positive tumors. The PD-L1 status was determined using a pharmDx Kit, which is now approved by the US Food and Drug Administration to select patients who could benefit from pembrolizumab treatment. CPS was calculated as the number of PD-L1−staining cells divided by the total number of evaluated cells. The study included patients with microsatellite stable (MSI-S), undetermined, or deficient MMR status. The overall response rate to pembrolizumab across all patients was 11.6%, median PFS was 2 months, and the 12-month OS rate was 23.4%. In the subset of patients with MSI-H tumors, the overall response rate was 57.1%, with a complete response rate of 14.3%; in those with MSI-S tumors, the overall response rate was 9% and the complete response rate was 2.4%. Among patients with PD-L1–positive tumors, the overall response rate was 15.5% (95% CI 10.1% to 22.4). Common adverse events included fatigue, hypothyroidism, nausea, diarrhea, and arthralgia.38
CASE CONCLUSION
This patient with metastatic gastric cancer receives second-line chemotherapy with ramucirumab and paclitaxel. Follow-up imaging shows persistent liver metastases and new lung metastasis. Because the tumor is PD-L1–positive, the patient receives 4 cycles of pembrolizumab, with no significant change noted in disease burden. He notes a significant decline in functional status with increased weight loss, nausea, emesis, and fatigue. The patient opts to forego any further therapy and instead chooses to pursue supportive care only.
SUMMARY
Gastric cancer is the third most common cause of cancer death worldwide. Common risk factors for developing gastric cancer include H. pylori infection, smoking, alcohol abuse, radiation exposure, high-fat diet, and obesity. Patients presenting with alarm symptoms of nausea, emesis, early satiety, abdominal pain, or weight loss should be fully evaluated with upper GI endoscopy. If there is suspicion for metastatic disease, CT evaluation of the chest, abdomen, and pelvis with oral and intravenous contrast should be obtained. Treatment of patients with metastatic gastric cancer is guided by their performance status at presentation. For patients with good performance status, a combination of platinum and fluoropyrimidine therapy, such as FOLFOX, can be considered. Doublet chemotherapy regimens are preferred over triplet chemotherapy regimens given their better tolerability. For patients with HER2-positive disease, the addition of trastuzumab to the platinum and fluoropyrimidine backbone is the standard of care in the first line.
Several targeted agents have been studied in patients progressing on initial therapy, with ramucirumab and paclitaxel being considered the regimen of choice in the second line. No anti-HER2 therapy has been approved for patients who are refractory to trastuzumab. Pembrolizumab is approved for use in patients who are PD-L1–positive and have previously progressed on at least 2 lines of chemotherapy. Pembrolizumab is also approved for the treatment of patients with unresectable or metastatic, MSI-H or MMR-deficient gastric cancers that have progressed after prior treatment and who have no satisfactory alternative treatment options.
INTRODUCTION
According to the Surveillance, Epidemiology and End Results database, in 2017 there were 28,000 new cases of gastric cancer, accounting for 1.8% of all malignancies in the United States, and an estimated 10,960 gastric cancer–related deaths.1 Worldwide, gastric cancer is the fifth most common malignancy and the third most common cause of death from any cancer.2 The incidence of gastric cancer varies significantly by geographic region, with countries in Eastern Asia (China, Japan), Eastern Europe, and Central and South America accounting for 50% of all new cases.3 Although the incidence of gastric cancer has declined in recent years, this decrease has not been observed consistently across all nations.2 In particular, the incidence of gastric cancers arising from the cardia has been increasing, which is perhaps due to a higher prevalence of obesity in Western societies.4
In this article, we review key aspects of management of metastatic gastric cancer, including selection of first- and second-line therapy, and discuss targeted agents and upcoming clinical trials.
EPIDEMIOLOGY AND RISK FACTORS
Chronic infection with Helicobacter pylori, a gram-negative bacterium, is a strong etiological factor for the development of gastric cancer, contributing to up to 70% of cases.2 The pathogen can colonize the gastric mucosa, leading to chronic inflammation. Although most patients remain asymptomatic, 1% to 3% develop gastric cancer and another 0.1% develop mucosa-associated lymphoid tissue lymphoma.5 H. pylori infection is more commonly associated with cancer of the gastric body than with cancer of the gastroesophageal junction (GEJ). The increased burden of gastric cancer in countries in Eastern Asia, Latin America, and Eastern Europe has been correlated to the prevalence of chronic H. pylori infection in these areas.
Carcinogenesis secondary to H. pylori infection may occur via several mechanisms. First, H. pylori can release virulence factors, such as cytotoxin-associated gene A, vacuolating cytotoxin, and outer membrane proteins, into the cytosol of host cells, leading to changes in patterns of cell proliferation and apoptosis.6 These virulence factors can modulate the host immune system, attenuating it to promote dysplasia. In addition, continued recognition of these factors by the immune system leads to a persistent inflammatory response, with the release of cytokines such as interleukin (IL) -1β, IL-6, and IL-8. This leads to chronic mucosal damage, further promoting dysplasia with eventual transformation into adenocarcinoma.7 In Japan and Korea, where screening for H. pylori infection is routinely performed, there have been improvements in overall survival (OS) rates for gastric cancer, with 5-year OS rates of 70%.8 The International Agency for Research on Cancer recommends further research into population-based screening and treatment programs for patients with chronic H. pylori infection. However, despite this recommendation, optimal screening strategies are not clearly defined.9
Other risk factors for the development of gastric cancer include chronic gastroesophageal reflux disease; smoking; alcohol use; exposure to radiation; diets high in fats, salt, and smoked items and low in fruits and vegetables; obesity; and exposure to chemotherapeutic agents such as procarbazine.10 Another pathogen suspected, but not proven, to be associated with increased risk for gastric cancer is the Epstein-Barr virus, a human herpesvirus found in 80% of all gastric carcinomas with lymphoid features.11 In addition, whether the use of medications such as statins and nonsteroidal anti-inflammatory drugs confers a decreased risk of gastric cancers remains unclear.10
EVALUATION
CASE PRESENTATION
A 55-year-old Caucasian man with a history of type 2 diabetes mellitus presents to the gastrointestinal (GI) clinic with a 6-month history of dysphagia. The dysphagia is worsened with ingestion of solids, particularly towards the end of the day. He states that the food often gets “stuck in the middle of the chest.” The patient denies any nausea or emesis but notes that he has a poor appetite. He reports having worsening mid-epigastric abdominal pain that is non-radiating, dull in character, and 6/10 in intensity. He also reports a 10-lb weight loss over the past 2 months. He has no previous history of reflux, chest pain, dyspnea, or cough. Review of systems is otherwise benign. Physical exam is within normal limits.
• Which tests should be conducted when gastric cancer is suspected?
Persistent epigastric abdominal pain and weight loss are the most common early symptoms of gastric cancer. Nausea, early satiety, dysphagia, and occult GI bleeding can be other presenting signs. Patients presenting with alarm symptoms of nausea, emesis, early satiety, abdominal pain, or weight loss should be fully evaluated with upper GI endoscopy. Early diagnosis of gastric cancer is essential in obtaining a curative resection. However, at least 40% of patients present with de novo metastatic disease at the time of initial diagnosis.12 Gastric cancer spreads by direct extension through the gastric wall, with the liver, peritoneum, and regional lymph nodes being the most common sites of metastatic deposits.13 Classically, Virchow’s node, the left supraclavicular lymph node, is involved with metastatic gastric cancer. Involvement of the left axillary lymph node (Irish node) or a periumbilical nodule (Sister Mary Joseph node) may also be observed. Other, less commonly noted sites of metastatic disease include the ovaries, central nervous system, bone, lung, and soft tissues.13
Upper GI endoscopy is the best method for determining tumor location and extent and obtaining a specimen for a definitive tissue diagnosis.14 It is essential to accurately identify the location of the tumor in the stomach and relative to the GEJ. The American Joint Committee on Cancer classification defines tumors involving the GEJ with an epicenter no more than 2 cm into the proximal stomach as esophageal cancers.15 Tumors of the GEJ with their epicenter more than 2 cm into the proximal stomach are defined as gastric cancers. If metastatic disease is suspected, computed tomography (CT) scan of the chest, abdomen, and pelvis with oral and intravenous contrast can be obtained to determine the extent of disease spread. In the absence of any metastatic disease, endoscopic ultrasound (EUS) should be conducted to determine the depth of tumor invasion (T staging) and lymph node status. In the era of targeted therapy, patients with metastatic disease should undergo testing for human epidermal growth factor-2 (HER-2) expression, microsatellite instability (MSI), and programmed death ligand 1 (PD-L1) expression. Patients should be staged according to the TNM staging system.
FIRST-LINE TREATMENT OPTIONS
CASE CONTINUED
The patient undergoes esophagoduodenoscopy (EGD) and is found to have a gastric cardia mass extending into the distal esophagus. EUS also demonstrates multiple abdominal and mediastinal lymph nodes. No gastric outlet obstruction is found. Biopsy shows poorly differentiated invasive adenocarcinoma. Warthin–Starry stain is negative for H. pylori organism. The tumor cells are positive for cytokeratin (CK7), CK19, and mucin-1 gene (MUC1); focally positive for CK20; and negative for MUC2. HER2 testing results are reported as immunohistochemistry (IHC) 3+, consistent with strongly positive HER2 protein expression. Further IHC testing for mismatch repair (MMR) proteins shows intact nuclear expression of MLH1, MSH2, MSH6, and PMS2 protein, consistent with a low probability of MSI-high tumor. The tumor is found to be PD-L1 positive. Imaging reveals abnormal mass-like nodular thickening of the gastric wall, with an infiltrative opacity within the pancreatico-duodenal groove, suspicious for tumor infiltration. Multiple metastatic deposits are noted in the liver, peritoneum, and bilateral lungs. There is extensive gastrohepatic ligament and periportal lymphadenopathy and mild enlargement of the pulmonary hilar lymph nodes. These findings are consistent with stage 4 (T4bN3aM1) gastric cancer. Given these findings, staging laparoscopy is deferred.
• What are the first-line treatment options for patients with metastatic gastric cancer?
Patients with metastatic gastric cancer have a poor prognosis, and management is stratified based on performance status (Figure). In patients with good performance status, systemic chemotherapy is the mainstay of treatment. The goal of therapy is not curative, but rather treatment focuses on palliation of symptoms arising from tumor spread. Given this treatment goal, there has been considerable interest in clarifying the utility of chemotherapy as opposed to best supportive care. In a recent Cochrane review of 64 randomized control trials involving 11,698 patients, chemotherapy was found to improve OS by 6.7 months as compared to best supportive care (hazard ratio [HR] 0.3 [95% confidence interval {CI} 0.24 to 0.55]).16 Five classes of cytotoxic chemotherapeutic agents have demonstrated activity in gastric cancer. These include fluoropyrimidine (either infusional fluorouracil or capecitabine), platinum agents (cisplatin or oxaliplatin), taxanes (docetaxel or paclitaxel), anthracyclines (epirubicin), and irinotecan.13 Treatment options are further divided based on whether the patient has HER2-overexpressing or non-expressing malignancy.
HER2-NEGATIVE DISEASE
For patients with HER2-negative disease, National Comprehensive Cancer Network (NCCN) guidelines recommend using 2-drug combination regimens rather than 3 drugs, given concern for increased toxicity with 3-drug regimens.17 For patients with a performance status of 0 to 1, utilization of a 3-drug regimen is a reasonable alternative. The combination of a fluoropyrimidine with a platinum agent is considered the standard of care, with regimens such as fluorouracil, leucovorin, and oxaliplatin (FOLFOX) being commonly used.
Epirubicin-containing regimens have also been extensively studied in advanced gastric cancer. In a study of 274 previously untreated patients with GEJ cancers, the combination of epirubicin, cisplatin, and fluorouracil (ECF) was compared to fluorouracil, doxorubicin, and methotrexate (FAMTX). There was an OS benefit favoring ECF (8.9 months versus 5.7 months) at 1 year (95% CI 27% to 45%, P = 0.0009). The ECF regimen was associated with an increased risk of nausea, emesis, and alopecia, while more hematologic toxicity and infections were noted with the FAMTX regimen.18 In addition, in a phase 3 trial, Van Cutsem and colleagues examined the role of docetaxel in combination with cisplatin and fluorouracil (DCF) compared to cisplatin and fluorouracil alone. Addition of docetaxel led to improved OS and time to progression (9.2 months versus 8.6 months for cisplatin and fluorouracil alone, P = 0.02) but with an increased risk of grade 3 and 4 toxicities (69% versus 59%). These adverse events included neutropenia (82% versus 57% of cisplatin and fluorouracil patients), diarrhea (19% versus 8%), stomatitis (21% versus 27%), and fatigue (19% versus 14%).19
The landmark phase 3 REAL-2 study compared 4 chemotherapy regimens in patients with untreated advanced esophagogastric cancer. This study was conducted to determine if the efficacy of cisplatin and oxaliplatin, a third-generation platinum agent, is equivalent to that of fluorouracil and capecitabine, an oral fluoropyrimidine. In this trial, a 2 × 2 design was used to compare 4 regimens: ECF versus epirubicin, cisplatin, and capecitabine (ECX) versus epirubicin, oxaliplatin, and fluorouracil (EOF) versus epirubicin, oxaliplatin, and capecitabine (EOX). The study found EOX to be noninferior to ECF, with a trend towards improved OS compared to other combination regimens (11.2 months versus 9.9 months, HR 0.80 [95% CI 0.66 to 0.97], P = 0.02).20 Thus, the study demonstrated that an oxaliplatin and capecitabine-based regimen could replace cisplatin and fluorouracil. Given that fluorouracil administration requires long continuous infusions, the oral-based capecitabine regimen is an attractive option for patients.
Several trials have demonstrated the equivalency of oxaliplatin with cisplatin in combination regimens for the treatment of advanced gastric cancer. Oxaliplatin has the benefit of an improved toxicity profile as compared to cisplatin, with the major dose-limiting toxicity being peripheral neuropathy
Given previous evidence that DCF (docetaxel, cisplatin, fluorouracil) is superior to cisplatin and fluorouracil alone, there was interest in determining if the addition of docetaxel to a backbone of fluorouracil, oxaliplatin, and leucovorin (FLO) could elicit a higher response rate. This concept was investigated in a phase 2 trial that assigned 54 patients with metastatic gastric or GEJ adenocarcinoma to receive biweekly infusions of oxaliplatin, leucovorin, fluorouracil, and docetaxel.21 Median time to response was 1.54 months, and the overall response rate was 57.7%. Median progression-free survival (PFS) was 5.2 months, and OS was 11.1 months. The most common grade 3 or 4 toxicities included neutropenia (48%), leukopenia (27.8%), diarrhea (14.8%), and fatigue (11.1%).
Irinotecan-based regimens have also been extensively studied in the first-line treatment of metastatic gastric cancer, particularly as an alternative to platinum-based therapy, but superiority has not been established. The combination of fluorouracil, leucovorin, and irinotecan (FOLFIRI) was compared to ECX in a phase 3 trial.22 The study enrolled 416 patients with locally advanced or metastatic gastric or GEJ cancer. At a median follow up of 31 months, the time to progression was longer in the FOLFIRI arm as compared to the ECX arm (5.1 months versus 4.2 months, P = 0.008), but there was no difference in OS (9.5 months versus 9.7 months, P = 0.95), median PFS (5.3 months versus 5.8 months, P = 0.96), or response rate (39.2% versus 37.8%). However, the FOLFIRI regimen had an improved toxicity profile, with a lower overall rate of grade 3 or 4 toxicity (69% versus 84%, P < 0.001). Given these findings, the FOLFIRI regimen is an acceptable alternative to platinum-based therapy in suitable patients.22
HER2-POSITIVE DISEASE
The HER2 proto-oncogene, initially described in breast cancer, has been implicated in several malignancies, including gastric and esophageal cancer. Overexpression or amplification of HER2 can be found in up to 30% of gastric cancers.23 For these patients, adding trastuzumab to a standard regimen of platinum and fluoropyrimidine is the standard of care. The prospective phase 3 Trastuzumab for Gastric Cancer (ToGA) trial randomly assigned 594 patients with HER2-positive gastric cancer to receive either cisplatin and fluorouracil or capecitabine and cisplatin with trastuzumab (n = 294) or without (n = 290) trastuzumab every 3 weeks for a total of 6 cycles, followed by maintenance trastuzumab until disease progression was noted.24 HER2 positivity was defined as HER2 protein overexpression by IHC (cutoff of 3+) or gene amplification by fluorescence in situ hybridization (FISH); tumors with IHC 2+ patterns were followed with FISH studies to confirm positivity. The study found a higher incidence of HER2-positive tumors in patients with GEJ tumors compared to patients with distal gastric cancers (33% versus 20%).24 In this trial, the addition of trastuzumab was associated with an improvement in OS: 13.5 months in the trastuzumab cohort versus 11.1 months in those receiving chemotherapy alone (HR 0.74, P = 0.0048). There was not a significant difference in toxicities between the 2 cohorts, with nausea, emesis, and neutropenia being the most common adverse events. Rates of overall grade 3 or 4 events were similar as well (68% in each cohort). Further exploratory analysis was also conducted according to HER2 status by dividing patients into a “high-expressor” group (n = 446), defined as patients with IHC 3+ tumors or IHC 2+ and FISH positivity, and a “low-expressor” group (n = 131), which included patients with IHC 0 or 1+ tumors. Analysis of patients in the 2 subgroups demonstrated an improved OS with the addition of trastuzumab for the high-expressor cohort, with a median OS of 16 months (HR 0.65 [95% CI 0.51 to 0.83]) compared to 11.8 months in those receiving only chemotherapy.
Dual HER2 blockade has been investigated in metastatic gastric cancer. The phase 3 randomized JACOB trial assigned 780 patients to receive either trastuzumab with a cisplatin/fluoropyrimidine regimen with or without the addition of pertuzumab; the primary end point was OS.25 A non-statistically significant trend towards improvement in OS was found in the pertuzumab arm (17.5 months) as compared with the standard of care arm (14.2 months, HR 0.84, P = 0.0565). The pertuzumab/trastuzumab/chemotherapy cohort experienced a higher incidence of diarrhea (61.6% versus 35.1% in control arm). Cardiac toxicity was comparable in the 2 cohorts.
The Table provides a summary of relevant clinical trials in metastatic gastric cancer.
SECOND-LINE THERAPY
CASE CONTINUED
The patient receives capecitabine, oxaliplatin, and trastuzumab therapy for 6 cycles, followed by trastuzumab for another 3 cycles. While on therapy, he develops a painful right clavicular lesion. He undergoes magnetic resonance imaging of the right clavicle, which shows a lesion in the distal two-thirds of the right clavicle measuring 9.7 × 3.7 × 3.8 cm. The patient is started on palliative radiation to the clavicle. However, repeat CT imaging shows progressive liver metastases.
• What is the approach to second-line therapy for metastatic gastric cancer?
Improvements in our understanding of the molecular pathways that lead to tumorigenesis have contributed to the development of several targeted agents whose efficacy in gastric cancer is being investigated. The NCCN guidelines recommend that for all patients who progress on frontline therapy, second-line therapy consists of a combination of ramucirumab and paclitaxel. Other options include single-agent docetaxel, paclitaxel, irinotecan, or ramucirumab. Combination therapy using irinotecan with either docetaxel, fluorouracil, or cisplatin may also be used.
Ramucirumab, a human IgG1 monoclonal antibody that targets the vascular endothelial growth factor receptor 2 (VEGFR2), was initially approved in 2014 as monotherapy for patients who had previously progressed on first-line chemotherapy. Its approval was based on the results of the phase 3 randomized, double-blind placebo-controlled REGARD study.26 The trial randomly assigned 355 patients with advanced gastric or GEJ adenocarcinoma and disease progression after first-line platinum-containing or fluoropyrimidine-containing chemotherapy to receive best supportive care plus either ramucirumab (n = 238) or placebo (n = 117). Monotherapy with ramucirumab significantly improved median OS compared with placebo (5.2 months versus. 3.8 months; HR 0.776 [95% CI 0.6 to 0.99], P = 0.047). There was also an improvement in PFS of 2.1 months in the ramucirumab cohort, as compared to 1.3 months in the placebo cohort (P < 0.0001). Patients in the ramucirumab arm experienced a higher incidence of hypertension (16% versus 8%), but all other adverse events occurred at comparable rates. Five deaths in the ramucirumab group were thought to be secondary to the study drug, as compared to 2 deaths in the placebo group.
In the subsequent phase 3 RAINBOW trial, the addition of ramucirumab to paclitaxel was investigated, with 330 patients assigned to the combination group and 335 to the paclitaxel-only group.27 The trial again showed that combination therapy afforded patients a significant survival advantage compared to paclitaxel alone, with a median OS of 9.6 months versus 7.4 months for the monotherapy group (HR 0.807 [95% CI 0.678 to 0.962], P = 0.017). A PFS benefit of 4.4 months was observed in the combination therapy groups, as compared with 2.9 months in the monotherapy group (HR 0.635, P < 0.0001). The ramucirumab/paclitaxel group also had a higher overall response rate of 28% versus 16%. The combination cohort had an increased incidence of grade 3 or higher adverse hypertensive events (14% versus 2%) and neutropenia (41% versus 19%), while the incidence of grade 3 febrile neutropenic events was similar between the groups (3% versus 2%).
The addition of bevacizumab, another monoclonal antibody against VEGF, to standard chemotherapy regimens has been explored, but studies have failed to show a survival benefit with this agent in the first-line treatment of advanced gastric cancer. The phase 3 Avastin in Gastric Cancer (AVAGAST) trial was a multinational, randomized study where patients received either bevacizumab (n = 387) or placebo (n = 387) in addition to cisplatin and capecitabine.28 The substitution of fluorouracil for capecitabine was permitted for patients who were unable to tolerate oral medications. Cisplatin was administered for a maximum of 6 cycles, while capecitabine and bevacizumab were administered until disease progression. The study failed to show an improvement in OS, with a median OS of 12.1 months noted in the bevacizumab cohort, as compared to 10.1 months in the placebo arm (HR 0.87 [95% CI 0.73 to 1.03], P = 0.1002). However, there was a modest improvement in median PFS (6.7 months versus 5.3 months; HR 0.80 [95% CI 0.68 to 0.93], P = 0.0037) and overall response rate (46% versus 37.4%, P = 0.0315). The most commonly reported grade 3 to 5 adverse events included neutropenia (35%), anemia (10%), and loss of appetite (8%). Interestingly, in a follow-up report, higher serum levels of VEGF-A were thought to correlate with an enhanced response to bevacizumab.29 However, the routine use of biomarker analysis in selecting patients for treatment with bevacizumab in metastatic gastric cancer remains to be further clarified.
Use of other agents with anti-HER2 activity in the second-line treatment of patients who have experienced progression while on trastuzumab remains unclear. In the recent T-ACT trial, patients with disease refractory to frontline therapy with combination trastuzumab and fluoropyrimidine/platinum agents were randomly assigned to receive either weekly paclitaxel (n = 45) or weekly paclitaxel plus trastuzumab (n = 44).30 Patients in the combination cohort received an initial dose of trastuzumab 8 mg/kg followed by 6 mg/kg every 3 weeks until progression. The study did not find a difference in either PFS (3.19 months versus 3.68 months; HR 0.91 [95% CI 0.67 to 1.22], P = 0.33) or OS (9.95 months versus 10.2 months; HR 1.23 [95% CI 0.75 to 1.99], P = 0.20). The study thus failed to show a benefit to continuing trastuzumab after progression in the first-line setting.
Lapatinib in combination with paclitaxel has been compared to paclitaxel alone for the treatment of advanced HER2-positive gastric cancer in an Asian population in the phase 3 TyTAN trial.31 With a primary end point of OS, the study randomly assigned 129 patients to receive paclitaxel alone and 132 patients to receive paclitaxel with lapatinib. There was a nonsignificant trend towards improvement in OS in the combination group (11 months) as compared to the paclitaxel-only group(8.9 months, P = 0.1044), with no significant difference in median PFS (5.4 months versus 4.4 months). However, it is important to note that only 15 patients in this trial had previously been exposed to trastuzumab. Another trial, the phase 3 GATSBY study, examined the efficacy of trastuzumab emtansine in the second-line setting compared to taxanes alone and failed to show any improvement in PFS or OS.32 Given these results, no alternative anti-HER2 therapy has been proven to be efficacious for patients who are trastuzumab refractory. Therefore, including anti-HER2 therapy in the second-line treatment of HER2-positive gastric cancer is not recommended.
IMMUNOTHERAPY AND OTHER TARGETED THERAPIES
Several other targeted therapies have been studied in advanced gastric cancer, without any demonstrable survival benefit. The PI3K/AKT/mTOR pathway is known to be involved in regulation of cell growth and angiogenesis, and the mTOR inhibitor everolimus is widely used to treat other malignancies, including breast cancer. The use of everolimus in the second-line setting was studied in the phase 3 GRANITE-1 trial, where it was compared to best supportive care and failed to provide any survival benefit.33 Cetuximab, a recombinant human and mouse chimeric monoclonal antibody, and panitumumab, a recombinant human antibody against the epidermal growth factor receptor (EGFR), have also been examined in gastric and GEJ cancer patients. However, the large phase 3 EXPAND and REAL-3 trials did not show a survival benefit when these agents were added to standard chemotherapy.34,35
Overexpression of MET, a proto-oncogene and tyrosine kinase receptor, has also been implicated in gastric cancer progression. The ligand for MET is the hepatocyte growth factor (HGF), and aberrant signaling of this pathway has been shown to correlate with an aggressive gastric cancer phenotype and poorer OS by promoting tumor growth and angiogenesis. However, no MET inhibitors thus far have been found to be clinically effective. RILOMET-1 and RILOMET-2 were phase 3 trials examining the efficacy of rilotumumab, a humanized anti-HGF antibody, in combination with chemotherapy (ECX and cisplatin with capecitabine, respectively) for the frontline treatment of MET-positive GEJ and gastric cancers. Both studies were discontinued due to a higher treatment-related mortality in patients receiving rilotumumab, with a higher incidence of adverse events due to disease progression being noted.36 Similarly, onartuzumab, a monovalent monoclonal antibody against the MET receptor, was investigated in the phase 3 METGastric trial in combination with modified FOLFOX6 as first-line therapy for HER2-negative, MET-positive metastatic GEJ and gastric cancers. The study did not demonstrate any significant improvements in OS or PFS.37
There has been significant interest in incorporating immunotherapy in the treatment of early and metastatic gastric cancer. Pembrolizumab is the first programmed death receptor (PD-1) inhibitor to be approved for treatment of patients with PD-L1−positive advanced gastric cancer who had previously received 2 or more lines of chemotherapy. Although earlier studies of pembrolizumab in lung cancer utilized the tumor proportion score (TPS) to determine PD-L1 positivity, this was not found to be applicable to gastric cancer. Instead, the combined positive score (CPS) is used in gastric cancer. The CPS evaluates the number of tumor cells and immune cells (macrophages and lymphocytes) that stain positive for PD-L1 relative to all viable tumor cells. Comparatively, the TPS only examines the percentage of viable tumor cells that show complete or partial positive staining for PD-L1. A CPS score of 1 or greater identifies patients who would be suitable candidates for pembrolizumab.
The approval of pembrolizumab was based on the positive findings from the recent KEYNOTE-059 trial.38 The study included 259 patients who had previously received either fluoropyrimidine, cisplatin, or anti-HER2 therapy, with 148 patients (55%) of these patients having PD-L1−positive tumors. The PD-L1 status was determined using a pharmDx Kit, which is now approved by the US Food and Drug Administration to select patients who could benefit from pembrolizumab treatment. CPS was calculated as the number of PD-L1−staining cells divided by the total number of evaluated cells. The study included patients with microsatellite stable (MSI-S), undetermined, or deficient MMR status. The overall response rate to pembrolizumab across all patients was 11.6%, median PFS was 2 months, and the 12-month OS rate was 23.4%. In the subset of patients with MSI-H tumors, the overall response rate was 57.1%, with a complete response rate of 14.3%; in those with MSI-S tumors, the overall response rate was 9% and the complete response rate was 2.4%. Among patients with PD-L1–positive tumors, the overall response rate was 15.5% (95% CI 10.1% to 22.4). Common adverse events included fatigue, hypothyroidism, nausea, diarrhea, and arthralgia.38
CASE CONCLUSION
This patient with metastatic gastric cancer receives second-line chemotherapy with ramucirumab and paclitaxel. Follow-up imaging shows persistent liver metastases and new lung metastasis. Because the tumor is PD-L1–positive, the patient receives 4 cycles of pembrolizumab, with no significant change noted in disease burden. He notes a significant decline in functional status with increased weight loss, nausea, emesis, and fatigue. The patient opts to forego any further therapy and instead chooses to pursue supportive care only.
SUMMARY
Gastric cancer is the third most common cause of cancer death worldwide. Common risk factors for developing gastric cancer include H. pylori infection, smoking, alcohol abuse, radiation exposure, high-fat diet, and obesity. Patients presenting with alarm symptoms of nausea, emesis, early satiety, abdominal pain, or weight loss should be fully evaluated with upper GI endoscopy. If there is suspicion for metastatic disease, CT evaluation of the chest, abdomen, and pelvis with oral and intravenous contrast should be obtained. Treatment of patients with metastatic gastric cancer is guided by their performance status at presentation. For patients with good performance status, a combination of platinum and fluoropyrimidine therapy, such as FOLFOX, can be considered. Doublet chemotherapy regimens are preferred over triplet chemotherapy regimens given their better tolerability. For patients with HER2-positive disease, the addition of trastuzumab to the platinum and fluoropyrimidine backbone is the standard of care in the first line.
Several targeted agents have been studied in patients progressing on initial therapy, with ramucirumab and paclitaxel being considered the regimen of choice in the second line. No anti-HER2 therapy has been approved for patients who are refractory to trastuzumab. Pembrolizumab is approved for use in patients who are PD-L1–positive and have previously progressed on at least 2 lines of chemotherapy. Pembrolizumab is also approved for the treatment of patients with unresectable or metastatic, MSI-H or MMR-deficient gastric cancers that have progressed after prior treatment and who have no satisfactory alternative treatment options.
1. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41
2. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Allen C, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol 2017;3:524–8.
3. Sitarz R, Skierucha M, Mielko J, et al. Gastric cancer: epidemiology, prevention, classification, and treatment. Cancer Manag Res 2018;10:239–48.
4. Olefson S, Moss SF. Obesity and related risk factors in gastric cardia adenocarcinoma. Gastric Cancer 2015;18:23–32.
5. Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett 2014;345:196–202.
6. Espinoza JL, Matsumoto A, Tanaka H, Matsumura I. Gastric microbiota: An emerging player in Helicobacter pylori-induced gastric malignancies. Cancer Lett 2018;414:147–52.
7. Chmiela M, Gonciarz W. Molecular mimicry in Helicobacter pylori infections. World J Gastroenterol 2017;23:3964–77.
8. Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
9. Ford A, Gurusamy KS, Delaney B, et al. Eradication therapy for peptic ulcer disease in Helicobacter pylori-positive patients. Cochrane Database Syst Rev 2004(4):CD003840.
10. Karimi P, Islami F, Anandasabapathy S, et al. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev 2014;23:700–13.
11. Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
12. Chan BA , Sim HW, Natori A, et al. Survival outcomes for de novo versus relapsed stage IV gastric and gastroesophageal junction (GEJ) adenocarcinoma [abstract]. J Clin Oncol 2018;36(no. 4 suppl):148.
13. DeVita VT, Lawrence TS, Rosenberg SA. DeVita, Hellman, and Rosenberg’s cancer: principles & practice of oncology. 9th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2011.
14. Siewert JR, Hölscher AH, Becker K, Gössner W. [Cardia cancer: attempt at a therapeutically relevant classification]. [Article in German.] Chirurg 1987;58:25–32.
15. Amin MB, Edge SB, Greene FL, et al, eds. AJCC cancer staging manual. 8th ed. New York: Springer; 2017.
16. Wagner AD, Syn NL, Moehler M, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev 2017;8:CD004064.
17. Qiu H, Zhou Z. [Updates and interpretation on NCCN clinical practice guidelines for gastric cancer 2017 version 5]. [Article in Chinese.] Zhonghua Wei Chang Wai Ke Za Zhi 2018;21:160–4.
18. Webb A, Cunningham D, Scarffe JH, et al. Randomized trial comparing epirubicin, cisplatin, and fluorouracil versus fluorouracil, doxorubicin, and methotrexate in advanced esophagogastric cancer. J Clin Oncol 1997;15:261–7.
19. Van Cutsem E, Moiseyenko VM, Tjulandin S, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 2006;24:4991–7.
20. Cunningham D, Okines AF, Ashley S. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2010;362:858–9.
21. Al-Batran SE, Hartmann JT, Hofheinz R, et al. Biweekly fluorouracil, leucovorin, oxaliplatin, and docetaxel (FLOT) for patients with metastatic adenocarcinoma of the stomach or esophagogastric junction: a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie. Ann Oncol 2008;19:1882–7.
22. Guimbaud R, Louvet C, Ries P, et al. Prospective, randomized, multicenter, phase III study of fluorouracil, leucovorin, and irinotecan versus epirubicin, cisplatin, and capecitabine in advanced gastric adenocarcinoma: a French intergroup (Federation Francophone de Cancerologie Digestive, Federation Nationale des Centres de Lutte Contre le Cancer, and Groupe Cooperateur Multidisciplinaire en Oncologie) study. J Clin Oncol 2014;32:3520–6.
23. Boku N. HER2-positive gastric cancer. Gastric Cancer 2014;17:1–12.
24. Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
25. Tabernero J, Hoff PM, Shen L, et al. Pertuzumab + trastuzumab + chemotherapy for HER2-positive metastatic gastric or gastro-oesophageal junction cancer: Final analysis of a Phase III study (JACOB) [abstract]. Ann Oncol 2017;28(suppl 5):6160.
26. Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014;383:31–9.
27. Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol 2014;15:1224–35.
28. Ohtsu A, Shah MA, Van Cutsem E, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 2011;29:3968–76.
29. Van Cutsem E, de Haas S, Kang YK, et al, Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a biomarker evaluation from the AVAGAST randomized phase III trial. J Clin Oncol 2012;30:2119–27.
30. Makiyama A, Sagara K, Kawada J, et al. A randomized phase II study of weekly paclitaxel ± trastuzumab in patients with HER2-positive advanced gastric or gastro-esophageal junction cancer refractory to trastuzumab combined with fluoropyrimidine and platinum: WJOG7112G (T-ACT) [abstract]. J Clin Oncol 2018;36(no. 15 suppl):4011.
31. Satoh T, Xu RH, Chung HC, et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN--a randomized, phase III study. J Clin Oncol 2014;32:2039–49.
32. Thuss-Patience PC, Shah MA, Ohtsu A, et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): an international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol 2017;18:640–53.
33. Ohtsu A, Ajani JA, Bai YX, et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1 study. J Clin Oncol 2013;31:3935–43.
34. Lordick F, Kang YK, Chung HC, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:490–9.
35. Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:481–9.
36. Catenacci DVT, Tebbutt NC, Davidenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1467–82.
37. Shah MA, Bang YJ, Lordick F, et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma: the METGastric randomized clinical trial. JAMA Oncol 2017;3:620–7.
38. Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol 2018;4(5):e180013.
1. Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41
2. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Allen C, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol 2017;3:524–8.
3. Sitarz R, Skierucha M, Mielko J, et al. Gastric cancer: epidemiology, prevention, classification, and treatment. Cancer Manag Res 2018;10:239–48.
4. Olefson S, Moss SF. Obesity and related risk factors in gastric cardia adenocarcinoma. Gastric Cancer 2015;18:23–32.
5. Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett 2014;345:196–202.
6. Espinoza JL, Matsumoto A, Tanaka H, Matsumura I. Gastric microbiota: An emerging player in Helicobacter pylori-induced gastric malignancies. Cancer Lett 2018;414:147–52.
7. Chmiela M, Gonciarz W. Molecular mimicry in Helicobacter pylori infections. World J Gastroenterol 2017;23:3964–77.
8. Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
9. Ford A, Gurusamy KS, Delaney B, et al. Eradication therapy for peptic ulcer disease in Helicobacter pylori-positive patients. Cochrane Database Syst Rev 2004(4):CD003840.
10. Karimi P, Islami F, Anandasabapathy S, et al. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev 2014;23:700–13.
11. Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
12. Chan BA , Sim HW, Natori A, et al. Survival outcomes for de novo versus relapsed stage IV gastric and gastroesophageal junction (GEJ) adenocarcinoma [abstract]. J Clin Oncol 2018;36(no. 4 suppl):148.
13. DeVita VT, Lawrence TS, Rosenberg SA. DeVita, Hellman, and Rosenberg’s cancer: principles & practice of oncology. 9th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2011.
14. Siewert JR, Hölscher AH, Becker K, Gössner W. [Cardia cancer: attempt at a therapeutically relevant classification]. [Article in German.] Chirurg 1987;58:25–32.
15. Amin MB, Edge SB, Greene FL, et al, eds. AJCC cancer staging manual. 8th ed. New York: Springer; 2017.
16. Wagner AD, Syn NL, Moehler M, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev 2017;8:CD004064.
17. Qiu H, Zhou Z. [Updates and interpretation on NCCN clinical practice guidelines for gastric cancer 2017 version 5]. [Article in Chinese.] Zhonghua Wei Chang Wai Ke Za Zhi 2018;21:160–4.
18. Webb A, Cunningham D, Scarffe JH, et al. Randomized trial comparing epirubicin, cisplatin, and fluorouracil versus fluorouracil, doxorubicin, and methotrexate in advanced esophagogastric cancer. J Clin Oncol 1997;15:261–7.
19. Van Cutsem E, Moiseyenko VM, Tjulandin S, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 2006;24:4991–7.
20. Cunningham D, Okines AF, Ashley S. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2010;362:858–9.
21. Al-Batran SE, Hartmann JT, Hofheinz R, et al. Biweekly fluorouracil, leucovorin, oxaliplatin, and docetaxel (FLOT) for patients with metastatic adenocarcinoma of the stomach or esophagogastric junction: a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie. Ann Oncol 2008;19:1882–7.
22. Guimbaud R, Louvet C, Ries P, et al. Prospective, randomized, multicenter, phase III study of fluorouracil, leucovorin, and irinotecan versus epirubicin, cisplatin, and capecitabine in advanced gastric adenocarcinoma: a French intergroup (Federation Francophone de Cancerologie Digestive, Federation Nationale des Centres de Lutte Contre le Cancer, and Groupe Cooperateur Multidisciplinaire en Oncologie) study. J Clin Oncol 2014;32:3520–6.
23. Boku N. HER2-positive gastric cancer. Gastric Cancer 2014;17:1–12.
24. Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
25. Tabernero J, Hoff PM, Shen L, et al. Pertuzumab + trastuzumab + chemotherapy for HER2-positive metastatic gastric or gastro-oesophageal junction cancer: Final analysis of a Phase III study (JACOB) [abstract]. Ann Oncol 2017;28(suppl 5):6160.
26. Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014;383:31–9.
27. Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol 2014;15:1224–35.
28. Ohtsu A, Shah MA, Van Cutsem E, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 2011;29:3968–76.
29. Van Cutsem E, de Haas S, Kang YK, et al, Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a biomarker evaluation from the AVAGAST randomized phase III trial. J Clin Oncol 2012;30:2119–27.
30. Makiyama A, Sagara K, Kawada J, et al. A randomized phase II study of weekly paclitaxel ± trastuzumab in patients with HER2-positive advanced gastric or gastro-esophageal junction cancer refractory to trastuzumab combined with fluoropyrimidine and platinum: WJOG7112G (T-ACT) [abstract]. J Clin Oncol 2018;36(no. 15 suppl):4011.
31. Satoh T, Xu RH, Chung HC, et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN--a randomized, phase III study. J Clin Oncol 2014;32:2039–49.
32. Thuss-Patience PC, Shah MA, Ohtsu A, et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): an international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol 2017;18:640–53.
33. Ohtsu A, Ajani JA, Bai YX, et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1 study. J Clin Oncol 2013;31:3935–43.
34. Lordick F, Kang YK, Chung HC, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:490–9.
35. Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:481–9.
36. Catenacci DVT, Tebbutt NC, Davidenko I, et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1467–82.
37. Shah MA, Bang YJ, Lordick F, et al. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-negative, MET-positive gastroesophageal adenocarcinoma: the METGastric randomized clinical trial. JAMA Oncol 2017;3:620–7.
38. Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol 2018;4(5):e180013.
Transfusion Medicine
INTRODUCTION
Transfusion therapy is an essential part of hematology practice, allowing for curative therapy of diseases such as leukemia, aplastic anemia, and aggressive lymphomas. Nonetheless, transfusions are associated with significant risks, including transmission of infections and transfusion-related reactions. Controversy remains about key issues in transfusion therapy, such as triggers for red cell transfusions. This article reviews the available blood products (Table 1) and indications for transfusion along with the associated risks, and also discusses specific clinical situations, such as massive transfusion.

BLOOD PRODUCTS
WHOLE BLOOD
Whole blood is the product of 1 unit of donated blood plus anticoagulant/preservative, and by definition contains 1 unit of plasma and red cells. Whole blood can be stored for 5 weeks. Although it was the standard product in the past, whole blood is rarely used since 1 unit of donated blood can now be fractionated into 1 unit of red blood cells (RBC), 1 unit of platelets, and 1 unit of fresh frozen plasma (FFP). Thus, the use of whole blood for just a single transfusion represents a waste of resources. There are 2 exceptions. One is autologous blood donations, which are whole blood units. Second, whole blood is increasingly being used in massive transfusions for trauma patients, with the rationale being that all essential blood components are being transfused at once.1
PACKED RED CELLS
The remaining red cell mass after most of the plasma is removed is called the “packed” red cell unit (hematocrit = 70%–80%), and so red cells are often called “packed” red cells, or PRBC. A preservative is added to improve the flow of blood and to provide “nutrients” for the red cells, and this reduces the hematocrit to approximately 60%. The volume of a red cell unit is approximately 340 mL. In the average adult, 1 unit of RBC raises the hematocrit by 3%. The indications for transfusion of red cells are to increase red cell mass, and thus oxygen delivery, in patients who are compromised by their anemia.
Several randomized trials have helped define the indications for red cell transfusions and justify lower hematocrit thresholds for initiating transfusion. The TRICC (Transfusion Requirements in Critical Care Investigators for the Canadian Critical Care Trials Group) trial showed that in critical care patients (30-day mortality, 18.7%–23.3%), a conservative transfusion strategy of waiting until the hematocrit was below 21% had the same outcomes as transfusing at a threshold of 24%.2 The TRACS (Transfusion Requirements After Cardiac Surgery) trial showed that a hematocrit target of 24% had the same benefit as a target of 30% in patients who had undergone cardiac bypass surgery.3 For patients with acute myocardial infarction, the outcomes were worse with aggressive transfusion at a hematocrit of 30% compared to 24%.4 In patients with upper gastrointestinal bleeding, a hemoglobin transfusion trigger of 7 g/dL was associated with a lower mortality than a trigger of 9 g/dL (5% versus 9%).5 Finally, the FOCUS (Transfusion Trigger Trial for Functional Outcomes in Cardiovascular Patients Undergoing Surgical Hip Fracture Repair) trial showed that in older patients (average age 80 years) who had undergone hip fracture surgery, transfusions based on symptoms and not a fixed trigger of 30% had the same outcomes but considerable savings in blood products.6 Based on these trials, decisions regarding when to transfuse patients should be based on symptoms and not “numbers.” Young patients, especially those with reversible anemias, can tolerate low blood counts and should not be transfused based on an arbitrary number.
PLATELETS
Several types of platelet products exist. One unit of platelet concentrate is derived from 1 unit of donor blood. Plateletpheresis from volunteer donors is also used to harvest platelets, with the resulting product referred to as plateletpheresis platelets. One unit of single-donor (pheresis) platelets is equivalent to 6 platelet concentrates. Finally, HLA-matched platelets are single-donor pheresis units that are obtained from an HLA-matched donor. This product should be ordered only if there is evidence of HLA antibodies (see Platelet Alloimmunization section).
The dose of platelets for the average patient is 6 units of platelet concentrate or 1 pheresis unit. In theory, 1 unit of platelet concentrate can raise the count by 5 to 7 × 103/µL, but often this response is blunted by concurrent illness or bleeding. In patients who appear to have a poor response, the platelet count can be checked 15 minutes after platelet infusion. No rise or a minimal rise (< 2 × 103/µL) in the platelet count is suggestive of platelet refractoriness, while a good 15-minute response but poor 24-hour count is more suggestive of consumption—fever, sepsis, drug, or splenomegaly—and not refractoriness.
The indication for platelet transfusion depends on the clinical situation. For patients with immune thrombocytopenia, platelets should not be transfused unless there is life-threatening bleeding. For stable patients with marrow aplasia from chemotherapy, a cut-off of a morning platelet count of less than 10 × 103/µL has been shown to be as safe as higher levels for prophylactic transfusions.7 For patients with active bleeding, the platelet count should be kept above 50 × 103/µL. Patients with acquired or inherited platelet dysfunction may benefit from transfusion no matter the platelet count.
Platelet Alloimmunization
Patients exposed to transfused white cells with different HLA antigens can develop antibodies to these antigens.8 Anti-HLA antibodies are common in patients who previously have received transfused blood that is not leukodepleted and in patients who have been pregnant. Since platelets carry class I HLA antigens, they will be rapidly destroyed by anti-HLA antibodies when transfused into these patients. In patients transfused for aplastic anemia or myelodysplasia, as many as 90% will become HLA-immunized. The incidence is lower in patients receiving chemotherapy but still can be as high as 60% to 90%.9,10 Patients who have developed anti-HLA antibodies can respond to transfused platelets matched for HLA antigens. Unfortunately, some patients will either be a rare HLA type or be so heavily immunized that they will not respond to any platelet transfusion.
The significance of alloimmunization centers on 2 concepts: recognition and avoidance. Patients with HLA antibodies will fail to have an increment of their platelet counts with transfusions. Accordingly, patients who do not experience an increase in their count 15 minutes after the transfusion may have HLA antibodies. One can test for the presence of anti-HLA antibodies, although some patients instead have specific antiplatelet antibodies that will not respond to HLA-matched platelets. In patients who have been pregnant or previously transfused and are scheduled to undergo transplant or aggressive chemotherapy, it is wise to test for anti-HLA antibodies in order to plan their transfusion needs. The evidence suggests that transfused white cells are responsible for initiating the anti-HLA response. Trials have shown that giving leukodepleted blood products may reduce the incidence of alloimmunization, so patients who are not HLA-alloimmunized should receive only leukodepleted products.11A difficult problem is bleeding in patients who are refractory to platelet transfusion.12 Patients who test positive for the presence of anti-HLA antibodies can receive transfusions of HLA-matched platelets.13 Unfortunately, matched platelet transfusions are not effective in 20% to 70% of these patients. Also, since some loci are difficult to match, effective products may be unavailable. Finally, as many as 25% of patients have antiplatelet antibodies in which HLA-matched products will be ineffective. Platelet cross-matching can be performed to find compatible units for these patients, but this may not always be successful. In the patient who is totally refractory to platelet transfusion, consider drugs as an etiology of antiplatelet antibodies (especially vancomycin).14 Use of antifibrinolytic agents such as epsilon-aminocaproic acid or tranexamic acid may decrease the incidence of minor bleeding, but these are ineffective for major bleeding. “Platelet drips”—infusing either a platelet concentrate per hour or 1 plateletpheresis unit every 6 hours—may be given as a continuous infusion, but there is no evidence that this is helpful.15
FRESH FROZEN PLASMA
FFP is made from 1 unit of donated whole blood, with an average volume of 225 mL per unit. One unit of FFP can increase coagulation factor levels by 5% and fibrinogen by 10 mg/dL in the average stable patient. FFP can take 20 to 30 minutes to thaw before use, so in situations where FFP is needed quickly, the blood bank must be informed to “keep ahead” some units. Units of FFP that have been thawed but not used can be stored refrigerated for 5 days to prevent wasting blood products.
The indications for FFP are limited to several situations. These include a documented coagulation defect that can be corrected by a reasonable amount of FFP, such as factor V deficiency and factor XI deficiency, disseminated intravascular coagulation (DIC), reversal of warfarin, and massive transfusions. FFP is also used for the therapy of thrombotic thrombocytopenic purpura.
There is little justification for FFP transfusion in many of the clinical settings in which it is commonly used. For example, FFP is given for minor elevations of the INR in patients with liver disease, despite literature showing not only that the INR rise is not reflective of coagulation defects, but also that patients with liver disease may even be thrombophilic.16,17 Reviews of FFP use found limited evidence-based indications for its use.18,19 Also, several studies have shown that transfusion of FFP is not effective at reversing minor elevations of the INR (1.3–1.8).20 In a meta-analysis, FFP was associated with increased risk for lung injury and a trend toward increased mortality.18
CRYOPRECIPITATE
Cryoprecipitate is produced from 1 unit of FFP that is thawed at 4°C. The precipitate is resuspended with 10 mL of saline or FFP and refrozen for storage. One unit contains at least 150 mg of fibrinogen and 80 units of factor VIII, along with von Willebrand factor. Thawing time for cryoprecipitate is approximately 20 minutes.
Cryoprecipitate is used to raise the fibrinogen level in patients with DIC or massive transfusion with hemodilution. It is third-line therapy in the treatment of type 1 von Willebrand disease and is second-line therapy in the treatment of patients with other types of von Willebrand disease. Currently, von Willebrand factor concentrates are the preferred replacement product for von Willebrand disease. Cryoprecipitate can be used as a source of factor VIII for hemophiliacs, but the preferred product for these patients is the super pure factor VIII concentrates or recombinant products. Cryoprecipitate can also be used to shorten the bleeding time of uremic patients, but its effectiveness for this is controversial.
GRANULOCYTES
Granulocytes are harvested by leukopheresis of normal donors, with a target yield of 1010 granulocytes from each donor. To reach this target, the donors are often “stimulated” with neutrophil growth factors. The harvesting procedure can take 3 hours and is associated with some risks to the donor (eg, citrate toxicity). The current indications for granulocytes are very limited since the advent of neutrophil growth factors and improved antimicrobials.21 They can be useful in the neutropenic patient with a documented bacterial infection in whom the white blood cell count is not expected to recover in the near future. Given the difficulty of keeping the count up, these transfusions have been mainly used in treating small children.
SPECIAL BLOOD PRODUCTS
IRRADIATED BLOOD PRODUCTS
Irradiation of blood is performed for only one reason: to prevent transfusion-related graft-versus-host disease (TGVHD) (Table 2).22 The irradiation can be performed at the blood center or in the transfusion service of larger hospitals. The units are not radioactive and can be transfused safely to other patients. There is increased leakage of potassium in irradiated units of blood, so the units need to be transfused within 14 days; in patients potentially sensitive to potassium (eg, neonates), the units must be transfused within 24 hours. Patients undergoing stem cell transplant, those receiving either interuterine transfusions or products from relatives, any patient with Hodgkin disease or receiving purine analogs or alemtuzumab, and patients with severe congenital immune deficiencies should receive irradiated blood. Most would also advocate that patients with hematologic malignances receiving chemotherapy receive irradiated products, but this is more controversial.

LEUKPDEPLETED BLOOD
Contamination of blood products by white blood cells is increasingly being recognized as a possible cause of adverse effects in transfused patients, including febrile transfusion reactions, inducing HLA alloimmunization, immunosuppression, disease transmission, and TGVHD. Reducing white cells can reduce the incidence of all of these complications except TGVHD. Currently, white cells are removed by infusion through filters that trap the cells. This can be done either at the bedside, in the blood bank, or at the donor center. The majority of red cells provided by blood centers in many areas of the country are already leukoreduced, eliminating the need for labor-intensive filtration at the transfusion center or bedside. Platelets collected by plateletpheresis methods can also be made leukocyte-poor. The current indications for leukodepleted productions are:
- Prevention of febrile transfusion reactions in patients with previous documented reactions
- Prevention of HLA alloimmunization (ineffective if patient has received 1 or more blood products not leukodepleted or is already HLA immunized)
- Prevention of cytomegalovirus (CMV) infection
CMV-NEGATIVE BLOOD
CMV can be transmitted through any cellular blood product—red cells and platelets. For patients who are CMV-negative and receiving transplants, especially stem cell transplants, a new CMV infection can be devastating.21 For years only blood from CMV-negative donors was used to transfuse CMV-negative patients. This policy is effective in preventing CMV infection, but because 50% of the population is positive for CMV antibodies, it may potentially lead to shortages of products that could be transfused to the patient. Currently, leukoreduced blood products are used since leukofiltration of the blood is just as effective as transfusion of CMV-negative blood in preventing infections and allows greater use of all blood products.23
COMPLICATIONS OF TRANSFUSIONS
HEMOLYTIC TRANSFUSION REACTION
There are 2 forms of hemolytic reactions—immediate and delayed.24 The immediate reaction is associated with fevers, hypotension, back pain, and oliguria. In severe cases, DIC and renal failure may occur. The immediate reaction is due to transfusion of blood that reacts with the recipient’s preformed high-titer blood antigen antibodies, most often to ABO. This is fatal 2% of the time and occurs almost always as a result of errors in correct identification of the patient. Reactions are due to recipient antibodies attacking donated RBCs, resulting in release of hemoglobin and red cell membrane–antigen complexes. These complexes are believed to lead to the hypotension, fevers, chills, and renal damage associated with the hemolytic reaction. Treatment consists of immediately stopping the transfusion, notifying the blood bank, vigorous intravenous hydration to keep the urine output over 100 mL/hr, and supportive therapy.
The delayed reaction can range in severity from an abrupt drop in the hematocrit to normal response to transfusion but the patient developing a positive Coombs’ test. The delayed response is due to an anamnestic response to blood-group antigens. When the patient is exposed to the same antigen, there is a rise in antibody titer leading to the reaction. Some alloantibodies can lead to a brisk reaction, most often anti-Kidd. The frequency with which delayed transfusion reactions occur is underestimated because mild reactions often do not get worked up or even discovered.
ALLERGIC REACTIONS
Allergic reactions are common (1%–3% of transfusions) and occur in patients having antibodies to proteins in donor blood, which can lead to hives and itching with transfusions. Most of the time these allergic reactions are mild and can be treated with antihistamines. Prophylaxis with antihistamines is not indicated for future transfusions unless the reactions are frequent. Rarely these reactions can be associated with shock and hypotension. Patients who are immunoglobulin (Ig) A–deficient can develop anaphylactic reactions to IgA-containing blood products. Patients with severe allergic reactions need to have their IgA measured and, if deficient, receive only washed units or plasma from IgA-deficient donors to prevent future severe reactions.
FEBRILE REACTIONS
The most common transfusion reaction is a febrile reaction that occurs after the transfusion starts and that sometimes can be complicated by chills. This reaction often occurs due to the presence of leukocyte debris and cytokines in the donated blood. Therapy is supportive and involves stopping the transfusion and administering acetaminophen, but since hemolytic transfusion reactions can present with fever all patients need to be thoroughly evaluated. The incidence of reactions can be decreased by using leukodepleted blood and plateletpheresis platelets. Most patients do not benefit from receiving prophylactic acetaminophen for future transfusion unless they have multiple reactions.
TRANSFUSION-RELATED ACUTE LUNG INJURY
Once thought a rare complication, transfusion-related acute lung injury (TRALI) is increasingly being recognized, with an incidence of approximately 1:5000 patients; it is now the most frequent cause of transfusion-related death.24,25 TRALI is noncardiac pulmonary edema and typically manifests clinically with hypoxemia, fever, bilateral infiltrates, and hypotension 2 to 6 hours after blood is given. Ventilatory support is often required. Recovery is usually rapid (24–48 hours) and complete. The etiology is complex. In many cases, transfused anti-HLA antibodies react with the recipient’s white cells leading to pulmonary damage. Another theory is that transfusion of preformed cytokines leads to pulmonary damage. Because plasma products from multiparous women are most often associated with anti-HLA antibodies, the restricted use of blood products from women has decreased the incidence of TRALI over the past few years.26
TRANSFUSION-ASSOCIATED CIRCULATORY OVERLOAD
Increasingly it being recognized that volume overload resulting from transfusions can lead to significant morbidity.27 Patients with heart or renal disease or patients who already have compromised fluid status are at risk for transfusion-associated circulatory overload (TACO). Another risk factor is transfusion of multiple blood products. Patients with TACO develop dyspnea within 6 hours of transfusion, but do not have fever or rash with the dyspnea. The diagnosis is made by demonstrating circulatory overload (eg, high venous pressure, B-type natriuretic peptide). Treatment is aggressive diuresis. Strategies to prevent TACO include judicious use of blood products, especially in patients at risk for TACO, and the use of prophylactic diuretics, especially with red cell or plasma transfusions.28
TRANSFUSION-RELATED GRAFT-VERSUS-HOST DISEASE
TGVHD is a rare reaction, but one that is most often fatal.29 TGVHD occurs when donor lymphocytes attack the blood recipient’s organs—skin, liver, intestines, and marrow. This is very rare in the normal blood recipient unless the donor and recipient share some HLA haplotypes.30 In immunosuppressed patients, TGVHD can occur with lesser degrees of HLA similarity, with cases reported in blood recipients who are mainly patients with Hodgkin disease or acute leukemia undergoing chemotherapy, and in patients receiving purine analogs. TGVHD had not been reported in AIDS patients despite profound immunosuppression, perhaps because the milieu of the patient does not allow lymphocyte expansion. Symptoms of TGVHD are an erythematous rash that may progress to epidermal toxic necrolysis, liver dysfunction, diarrhea, and pancytopenia. TGVHD is prevented by irradiating blood products given to at-risk patients with 2500 to 3500 rads. Directed blood donation from all blood relatives should also be irradiated. TGVHD cannot be prevented by leukopoor blood because the minute amount of lymphocytes that are not filtered still can lead to these complications.
POST-TRANSFUSION PURPURA
Patients with post-transfusion purpura (PTP) develop severe thrombocytopenia (< 10 × 103/µL) with often severe bleeding 1 to 2 weeks after receiving any type of blood product.31 Patients who develop PTP most often lack platelet antigen PLA1 or other platelet antigens. For unknown reasons, exposure to the antigens from the transfusion leads to rapid destruction of the patient’s own platelets. The diagnostic clue is thrombocytopenia in a patient, typically female, who has received a red cell or platelet blood product in the past 7 to 10 days. Treatment consists of intravenous immunoglobulin32 and plasmapheresis to remove the offending antibody. If patients with a history of PTP require further transfusions, only PLA1-negative platelets should be given.
IRON OVERLOAD
Every transfusion of red cells delivers approximately 250 mg of iron to the recipient. Since there is no natural way of ridding the body of iron, heavily transfused patients are at risk of iron overload. This is most often seen in children heavily transfused for thalassemia. Starting in the second decade of life, these individuals will develop endocrinopathies due to iron overload, liver problems, and often fatal cardiomyopathies. Studies have shown that chelation of iron with deferoxamine can be effective in preventing this fatal complication.33 Oral iron chelators such as deferasirox and deferiprone are also effective. The risk of iron overload in heavily transfused patients with myelodysplasia or other transfusion-dependent anemias is unclear, and uncertainty exists about the need for chelation.34
Young patients who face years of transfusions should be started on iron chelation to avoid iron overload. For older patients with transfusion-dependent anemia, iron chelation therapy should be considered if their life expectancy is long (years to decades) or special studies such as T2-weighted cardiac magnetic resonance imaging showing iron overloading.35
INFECTIOUS COMPLICATIONS
Concern over transmission of HIV infection via blood products in the late 1980s led to both a reduction in blood product use and a greater awareness of infectious complications of transfusion and their prevention. However, no blood product can ever be assumed to be safe for 2 reasons. One is that blood products can transmit infections during a “window period”—the time before a contaminated product can be detected by testing. The second is that blood is not screened for all potential infections (eg, babesiosis or new infections such as West Nile virus at the start of the outbreak). Risk of infection is reduced in 2 ways: deferral of potential infectious donors and blood product testing.
As part of the donation process, potential blood donors are asked a series of questions to see if they have risk factors for infections (eg, recent travel to malarious areas, recent tattoos), and if they answer positive are deferred from donating blood. Blood products are then tested for infectious agents by a combination of methods including detection of viral antigen, antibody response to infections, and more recently polymerase chain reaction (PCR).36 Current screening includes syphilis testing; testing for antibodies to HIV, HTLV (human T-lymphotropic virus), hepatitis C virus, hepatitis B core antigen (HBcAg), hepatitis B surface antigen, and PCR for HIV, hepatitis B virus, HCV, and West Nile virus. Some centers also test for Trypanosoma cruzi, the cause of Chagas disease.
In the past, the numerically most common transfusion-related disease was hepatitis, first B and then C.37,38 The first step in eliminating these infections was to stop paying donors for blood products. With the introduction of effective testing for hepatitis B and then C, the incidence of transfusion-related hepatitis has plummeted.36 For example, with the introduction of a diagnostic test for hepatitis C, the estimated risk has fallen from 5% to less than 1 per million. Currently, the risk of transmission of hepatitis B and C, HIV, and HTLV is less than 1 in a million.38
Despite this testing, blood transfusions can transmit a variety of infections, including malaria and babesiosis.39 Any new blood-borne infection introduced into the population can get into the blood supply as well. For example, at the start of the West Nile virus epidemic, there was a cluster of transfusion-transmitted cases that resulted in severe and sometimes fatal illness in immunosuppressed patients, but this issue has been addressed with the development of a PCR assay for screening blood.40 The rate of transfusion-related babesiosis has been increasing and screening for the causative parasite is being considered.
MASSIVE TRANSFUSIONS
Acutely bleeding patients can require large amounts of transfusion products. Early data showed high mortality rates with transfusion of more than 20 units of blood,41 but with modern blood banking techniques and improved laboratory testing, this rate has decreased dramatically, with survival rates of 43% to 70% in patients transfused with more than 50 units of blood.42
The basic approach to massive transfusions is to first transfuse the patient to maintain hemodynamic stability while specific blood tests are being obtained, and then to use the results of these early tests to guide the rest of the resuscitation. An important component is the ability to rapidly deliver standard packages of red cells, usually 6 to 10 units at a time, to the bleeding patient. To avoid delay while the patient’s blood is being typed, the first products delivered are blood group O Rh-positive units. Given the shortage of Rh-negative blood, this should be reserved for only empiric therapy of women of child-bearing age. Once the blood type is known, the patient can be switched over to type-specific blood.
In the past decade, there has been a shift toward increasing the amount of plasma given to patients receiving massive transfusions. This shift has occurred for 2 reasons. One is that modeling of coagulation changes in massive bleeding suggests the need for larger amounts of plasma to correct defects than have previously been recommended.43 The other reason is based on analysis of resuscitation protocols used in military and civilian trauma centers showing that giving red cells and plasma units in a 1:1 ratio appears to be associated with improved outcomes in massive transfusion. Several studies have extended this concept to platelets, again suggesting improved survival with 1 unit of random donor platelets given 1:1 with red cells and plasma units. The PROPPR (Prospective Observational Multicenter Major Trauma Transfusion) study compared a 1:1:1 to 1:1:2 ratio in patients with severe trauma and major bleeding and found less exsanguination and faster achievement of hemostasis in the first 24 hours.44 This has led to the widespread adoption of the 1:1 ratio by most trauma centers, and by default to other massive transfusion situations despite the lack of clinical trial data.45
One barrier to increased use is that plasma is kept frozen and requires 20 minutes to thaw. Many institutions are now keeping inventories of thawed plasma available for immediate use, ranging from 2 to 4 units of group AB plasma to keeping their entire inventory as liquid plasma.46 Plasma that is thawed but not used can be relabeled as “thawed plasma” and kept for up to 5 days. Also, many centers now use group A plasma for massive transfusions as this rarely leads to transfusion reactions and is much more available.47 Research is currently under way on lyophilized plasma, which can be stored at room temperature and can be rapidly reconstituted for emergency use.

The standard approach for laboratory testing is obtaining 5 tests: hematocrit, platelet count, INR/prothrombin time, activated partial thromboplastin time (aPTT), and fibrinogen.48 Product selection is guided by these tests, and they are repeated at regular intervals during the massive transfusion. A typical protocol is shown in Table 3. It is important as part of any protocol to have a flow chart that records laboratory results and products given that any member of the team can easily view.
The transfusion threshold for a low hematocrit depends on the stability of the patient. If the hematocrit is below 30% and the patient is bleeding or hemodynamically unstable, one should transfuse packed red cells. Stable patients can tolerate lower hematocrits, and an aggressive transfusion policy may even be detrimental.2,49 If the patient is bleeding, has florid DIC, or has received platelet aggregation inhibitors, then keeping the platelet count above 50 ×
While in the past fibrinogen targets of 50 to 100 mg/dL were recommended, recent data indicate that a target of 150 mg/dL or higher may be more appropriate.51–53 Severe fibrinolysis may occur in certain clinical situations such as brain injuries, hepatic trauma, or ischemic limb reperfusion, and the use of large amounts of cryoprecipitate can be anticipated. In patients with an INR greater than 2 and an abnormal aPTT, one can give 2 to 4 units of FFP. For an aPTT greater than 1.5 times normal, 2 to 4 units of plasma should be given. Elevation of the aPTT above 1.8 times normal control is associated with microvascular bleeding in trauma patients.54 Patients with marked abnormalities (eg, anaPTT more than 2 times normal) may require aggressive therapy with at least 15 to 30 mL/kg (4–8 units for an average adult) of plasma.55
Recently there has been increasing interest in the use of thromboelastography (TEG) in massive transfusion.56 This is a point-of-care assay performed on fresh whole blood that can assess multiple facets of hemostasis, including coagulation, platelet function, and fibrinolysis.57,58 TEG is performed by placing a 0.35-mL sample of whole blood into an oscillating container with a sensor pin that measures the force of thrombus formation. TEG measures 5 parameters:
- r time: time from starting TEG until clot formation
- K time: time needed for tracing to go from 2 mm to 20 mm
- alpha angle: slope of tracing between r and K time
- MA: greatest amplitude of TEG tracing
- Whole blood lysis index: amplitude of tracing 60 minutes after MA.
Several centers have incorporated TEG into resuscitation protocols that include standardized strategies for responding to abnormalities. Data suggest that use of TEG may decrease the use of blood products, especially in cardiac surgery, but this has not been prospectively studied in massive transfusions.56,59
COMPLICATIONS OF MASSIVE TRANSFUSIONS
Electrolyte abnormalities are unusual even in patients who receive massive transfusions.60 Platelet concentrates and plasma contain citrate that can chelate calcium. However, the citrate is rapidly metabolized, and it is rare to see clinically significant hypocalcemia. Although empiric calcium replacement is often recommended, one study suggests that this is associated with a worse outcome and should not be done.61 If hypocalcemia is a clinical concern, then levels should be drawn to guide therapy. Stored blood is acidic, with a pH of 6.5 to 6.9. However, acidosis attributed solely to transfused blood is rare and most often is a reflection of the patient’s stability. Empirical bicarbonate replacement has been associated with severe alkalosis and is not recom mended.62,63 Although potassium leaks out of stored red cells, even older units of blood contain only 8 mEq/L of potassium, so hyperkalemia is usually not a concern.
PATIENTS WITH AUTOIMMUNE HEMOLYTIC ANEMIA
Patients with autoimmune hemolytic anemia can be difficult to transfuse,64 because the autoantibody can interfere with several aspects of the transfusion services evaluation. In some patients the autoantibody can be so strong that the patient’s blood type cannot be determined. In most patients, the final step of the cross-match—mixing the donor blood with recipient plasma—will show noncompatibility due to the autoantibodies reacting with any red cells.
The first step when transfusing a patient with autoimmune hemolytic anemia is to draw several tubes of blood for the transfusion service before any potential transfusions. This allows the transfusion service to remove the autoantibodies so they can screen for underlying alloantibodies. Second, if the patient requires immediate transfusion, then type-specific or O-negative blood should be given. If the patient has not been recently (months) transfused, the incidence of a severe transfusion reaction is low. The first unit should be infused slowly with close observation of the patient. For patients who have been multiply transfused, the use of an “in-vivo” cross-match may be helpful. This is where the patient is slowly transfused 10 to 15 mL of blood over 15 minutes. The the plasma and urine are then assessed for signs of hemolysis and, if negative, the remaining product is given.
REFUSAL OF BLOOD PRODUCTS
The initial step in managing patients who refuse blood products is to find out why they are refusing them. Many patients have an exaggerated fear of HIV and other infectious agents, so discussing the very low risk for infection transmission can often resolve the situation. The most common reason for refusal of blood products is religious belief. Jehovah’s Witness patients will refuse blood products due to their interpretation of the Bible.65 All members will refuse red cells, plasma, and platelets, while decisions about “derived” blood products—products made by manipulation of the original donated units—are a matter of conscience. These include cryoprecipitate, intravenous gammaglobulin, and albumin.
In an elective situation, the first step is to discuss with the patient those products that are a matter of conscience and clearly document this. The patient’s blood count and iron stores should be assessed to identify any correctible causes of anemia or low iron stores before surgery. The use of erythropoietin to correct blood counts before surgery is controversial, as this may increase thrombosis risk and is contraindicated in patients with curable tumors.
For patients with acute blood loss, use of intravenous iron combined with high-dose erythropoietin is the most common approach to raise the blood count.65 A recommended erythropoietin dose is 300 units/kg 3 times a week, dropping to 100 units/kg 3 times weekly until the goal hematocrit is reached. Another often overlooked step is to consolidate and minimize laboratory testing. The most important step is to be respectful of the patient and their beliefs. Many larger cities have liaisons that can help with interactions between Jehovah’s Witness patients and the health care system.
NON-TRANSFUSION THERAPIES FOR ACUTE BLEEDING
DESMOPRESSIN
Desmopressin (DDAVP) is a synthetic analog of antidiuretic hormone that raises the levels of both factor VIII and von Willebrand protein severalfold.66 Desmopressin is effective in supporting hemostasis in patients with a wide variety of congenital and acquired bleeding disorders. However, desmopressin does not reduce blood loss before routine surgery in a healthy patient and should not be used for this purpose.67
TRANEXAMIC ACID
Tranexamic acid is an antifibrinolytic agent that blocks the binding of plasmin to fibrin.68 This agent was first shown to be useful in disorders that involve excessive fibrinolysis69–73 or as adjunctive therapy for oral or dental procedures in patients with a bleeding diathesis. In patients with severe thrombocytopenia, the use of antifibrinolytic agents may reduce bleeding. Increasing data shows that tranexamic acid can prevent blood loss in a variety of surgeries including heart bypass, liver transplantation, and orthopedic surgery.74 Patients across these settings have decreased blood loss and need for transfusion with no increased risk of thrombosis. The CRASH-2 study showed that the use of tranexamic acid significantly reduced mortality in trauma patients.75 The WOMEN trial demonstrated that 1 g of tranexamic acid given to women with blood loss of more than 500 mL after vaginal delivery or 1000 mL after cesarean section has a risk reduction of death of 0.81 with no increased risk of thrombosis.76 Given this abundant data, it is clear tranexamic acid needs to be part of any massive transfusion protocol.77
RECOMBINANT FACTOR VIIa
Recombinant factor VIIa (rVIIa) was originally developed as a “bypass” agent to support hemostasis in hemophiliacs.78 However, the use of rVIIa for a wide array of bleeding disorders, including patients with factor VII and XI deficiency and Glanzmann thrombasthenia, has been reported.79 Increasingly, rVIIa is being used as a “universal hemostatic agent” for patients with uncontrolled bleeding from any mechanism.80 Multiple case reports have described the use of rVIIa for bleeding in cardiac surgery patients, obstetrical bleeding, reversal of anticoagulation, and trauma.81 Unfortunately, little formal trial data exists to put these anecdotes into perspective, and formal review of clinical trial results has shown no benefit.82,83 However, when used in older patients, especially those with vascular risk factors, the risk of arterial thrombosis appears to increase.84 In the trials for intracranial hemorrhage, the thrombosis rate was 5% to 9%, and rates up to 10% for arterial events were seen in older patients in a review of all trials.85–87 Given the lack of data but the evidence of risk, rVIIa use should be restricted to patients with documented bleeding disorders that have been shown to benefit by its use.
1. Yazer MH, Cap AP, Spinella PC, et al. How do I implement a whole blood program for massively bleeding patients? Transfusion 2018;58:622–8.
2. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999;340:409–17.
3. Hajjar LA, Vincent JL, Galas FR, et al. Tranfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA 2010;304:1559–67.
4. Cooper HA, Rao SV, Greenberg MD, et al. Conservative versus liberal red cell transfusion in acute myocardial infarction (the CRIT Randomized Pilot Study). Am J Cardiol 2011;108:1108–11.
5. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
6. Carson JL, Terrin ML, Noveck H, et al; the FOCUS Investigators. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med 2011 Dec 4. [Epub ahead of print]
7. Schiffer CA, Anderson KC, Bennett CL, et al. Platelet transfusion for patients with cancer: clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol 2001;19:1519–38.
8. Schiffer CA. Prevention of alloimmunization against platelets. Blood 1991;77:1–4.
9. Hod E, Schwartz J. Platelet transfusion refractoriness. Br J Haematol 2008;142:348–60.
10. Novotny VMJ, Van Doorn R, Witvliet MD, et al. Occurrence of allogeneic HLA and non-HLA antibodies after transfusion of prestorage filtered platelets and red blood cells: A prospective study. Blood 1995;85:1736–41.
11. McFarland J, Menitove J, Kagen L et al. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med 1997;337:1861–9.
12. Juskewitch JE, Norgan AP, De Goey SR, et al. How do I … manage the platelet transfusion-refractory patient? Transfusion 2017;57:2828–35.
13. Schiffer CA. Diagnosis and management of refractoriness to platelet transfusion. Blood Rev 2001;15:175–80.
14. Christie DJ, van Buren N, Lennon SS, Putnam JL. Vancomycin-dependent antibodies associated with thrombocytopenia and refractoriness to platelet transfusion in patients with leukemia. Blood 1990;75:518–23.
15. Dzik S. How I do it: platelet support for refractory patients. Transfusion 2007;47:374–8.
16. Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med 2011;365:147–56.
17. Lisman T, Porte RJ. Pathogenesis, prevention, and management of bleeding and thrombosis in patients with liver diseases. Res Pract Thromb Haemost 2017;1:150–61.
18. Murad MH, Stubbs JR, Gandhi MJ, et al. The effect of plasma transfusion on morbidity and mortality: a systematic review and meta-analysis. Transfusion 2010;50:1370–83.
19. Green L, Bolton-Maggs P, Beattie C, et al. British Society of Haematology Guidelines on the spectrum of fresh frozen plasma and cryoprecipitate products: their handling and use in various patient groups in the absence of major bleeding. Br J Haematol 2018;181:54–67.
20. Abdel-Wahab OI, Healy B, Dzik WH. Effect of fresh-frozen plasma transfusion on prothrombin time and bleeding in patients with mild coagulation abnormalities. Transfusion 2006;46:1279–85.
21. Price TH. Granulocyte transfusion: current status. Semin Hematol 2007;44:15–23.
22. Treleaven J, Gennery A, Marsh J, et al. Guidelines on the use of irradiated blood components prepared by the British Committee for Standards in Haematology blood transfusion task force. Br J Haematol 2011;152:35–51.
23. Thiele T, Kruger W, Zimmermann K, et al. Transmission of cytomegalovirus (CMV) infection by leukoreduced blood products not tested for CMV antibodies: a single-center prospective study in high-risk patients undergoing allogeneic hematopoietic stem cell transplantation (CME). Transfusion 2011;51:2620–6.
24. Delaney M, Wendel S, Bercovitz RS, et al; Biomedical Excellence for Safer Transfusion (BEST) Collaborative. Transfusion reactions: prevention, diagnosis, and treatment. Lancet 2016;388:2825–36.
25. Toy P, Gajic O, Bacchetti P, et al. Transfusion related acute lung injury: incidence and risk factors. Blood 2011 Nov 23. [Epub ahead of print]
26. Wiersum-Osselton JC, Middelburg RA, Beckers EA, et al. Male-only fresh-frozen plasma for transfusion-related acute lung injury prevention: before-and-after comparative cohort study. Transfusion 2011;51:1278–83.
27. Friedman T, Javidroozi M, Lobel G, Shander A. Complications of allogeneic blood product administration, with emphasis on transfusion-related acute lung injury and transfusion-associated circulatory overload. Adv Anesth 2017;35:159–73.
28. Lin CR, Armali C, Callum J, et al. Transfusion-associated circulatory overload prevention: a retrospective observational study of diuretic use. Vox Sang 2018;113:386–92.
29. Sun X, Yu H, Xu Z, et al. Transfusion-associated graft-versus-host-disease: case report and review of literature. Transfus Apher Sci 2010;43:331–4.
30. Petz LD, Calhoun L, Yam P, et al. Transfusion-associated graft-versus-host disease in immunocompetent patients: report of a fatal case associated with transfusion of blood from a second-degree relative, and a survey of predisposing factors. Transfusion 1993;33:742–50.
31. Mueller-Eckhardt C. Post-transfusion purpura. Br J Hematol 1986;64:419–24.
32. Mueller-Eckhardt C, Kiefel V. High-dose IgG for posttransfusion purpura-revisited. Blut 1988;57:163–7.
33. Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet 2000;355(9220):2051–2.
34. Zeidan AM, Griffiths EA. To chelate or not to chelate in MDS: That is the question! Blood Rev 2018. pii: S0268-960X(17)30128-5.
35. Konen E, Ghoti H, Goitein O, et al. No evidence for myocardial iron overload in multitransfused patients with myelodysplastic syndrome using cardiac magnetic resonance T2 technique. Am J Hematol 2007;82:1013–16.
36. Squires JE. Risks of transfusion. South Med J 2011;104:762–9.
37. Sharma S, Sharma P, Tyler LN. Transfusion of blood and blood products: indications and complications. Am Fam Physician 2011;83:719–24.
38. Jacquot C, Delaney M. Efforts toward elimination of infectious agents in blood products. J Intensive Care Med 2018 Jan 1:885066618756589 [Epub ahead of print].
39. Herwaldt BL, Linden JV, Bosserman E, et al. Transfusion-associated babesiosis in the United States: a description of cases. Ann Intern Med 2011;155:509–19.
40. Biggerstaff BJ, Petersen LR. Estimated risk of West Nile virus transmission through blood transfusion during an epidemic in Queens, New York City. Transfusion 2002;42:1019–26.
41. Wilson RF, Mammen E, Walt AJ. Eight years of experience with massive blood transfusions. J Trauma 1971;11:275–85.
42. Wade CE, del Junco DJ, Holcomb JB, et al. Variations between level I trauma centers in 24-hour mortality in severely injured patients requiring a massive transfusion. J Trauma 2011;71(2 Suppl 3):S389–S393.
43. Hirshberg A, Dugas M, Banez EI, et al. Minimizing dilutional coagulopathy in exsanguinating hemorrhage: a computer simulation. J Trauma 2003;54:454–63.
44. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
45. Pasquier P, Gayat E, Rackelboom T, et al. An observational study of the fresh frozen plasma: red blood cell ratio in postpartum hemorrhage. Anesth Analg 2013;116:155–61.
46. Yuan S, Ziman A, Anthony MA, et al. How do we provide blood products to trauma patients? Transfusion 2009;49:1045–9.
47. Stevens WT, Morse BC, Bernard A, et al. Incompatible type A plasma transfusion in patients requiring massive transfusion protocol: Outcomes of an Eastern Association for the Surgery of Trauma multicenter study. J Trauma Acute Care Surg 2017;83:25–9.
48. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
49. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
50. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
51. Gerlach R, Tolle F, Raabe A, et al. Increased risk for postoperative hemorrhage after intracranial surgery in patients with decreased factor XIII activity: implications of a prospective study. Stroke 2002;33:1618–23.
52. Fenger-Eriksen C, Lindberg-Larsen M, Christensen AQ, et al. Fibrinogen concentrate substitution therapy in patients with massive haemorrhage and low plasma fibrinogen concentrations. Br J Anaesth 2008;101:769–73.
53. Charbit B, Mandelbrot L, Samain E, et al. The decrease of fibrinogen is an early predictor of the severity of postpartum hemorrhage. J Thromb Haemost 2007;5:266–73.
54. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
55. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
56. Curry NS, Davenport R, Pavord S, et al.The use of viscoelastic haemostatic assays in the management of major bleeding: A British Society for Haematology Guideline. Br J Haematol 2018. doi: 10.1111/bjh.15524. [Epub ahead of print].
57. Kashuk JL, Moore EE, Sawyer M, et al. Postinjury coagulopathy management: goal directed resuscitation via POC thrombelastography. Ann Surg 2010;251:604–14.
58. Whitten CW, Greilich PE. Thromboelastography: past, present, and future. Anesthesiology 2000;92:1223–5.
59. Girdauskas E, Kempfert J, Kuntze T, et al. Thromboelastometrically guided transfusion protocol during aortic surgery with circulatory arrest: a prospective, randomized trial. J Thorac Cardiovasc Surg 2010;140:1117–24.
60. Goskowicz R. The complications of massive tranfusion. Anesthesiology Clin North Am 1999;17:959–78.
61. Howland WS, Schwiezer O, Boyan CP. Massive blood replacement without calcuim administration. Surg Gynecol Obstet 1964;159:171–7.
62. Miller RD, Tong MJ, Robbins TO. Effects of massive transfusion of blood on acid-base balance. JAMA 1971;216:1762–5.
63. Collins JA. Problems associated with the massive transfusion of stored blood. Surgery 1974;75:274–95.
64. Petz LD. A physician’s guide to transfusion in autoimmune haemolytic anaemia. Br J Haematol 2004;124:712–6.
65. Scharman CD, Burger D, Shatzel JJ, et al. Treatment of individuals who cannot receive blood products for religious or other reasons. Am J Hematol 2017;92:1370–81
66. Leissinger C, Carcao M, Gill JC, et al. Desmopressin (DDAVP) in the management of patients with congenital bleeding disorders. Haemophilia 2014;20:158–67.
67. Desborough MJ, Oakland KA, Landoni G, et al. Desmopressin for treatment of platelet dysfunction and reversal of antiplatelet agents: a systematic review and meta-analysis of randomized controlled trials. J Thromb Haemost 2017;15:263–72.
68. Ng W, Jerath A, Wa˛sowicz M. Tranexamic acid: a clinical review. Anaesthesiol Intensive Ther 2015;47:339–50.
69. Amitrano L, Guardascione MA, Brancaccio V, Balzano A. Coagulation disorders in liver disease. Semin Liver Dis 2002;22:83–96.
70. Chang JC, Kane KK. Pathologic hyperfibrinolysis associated with amyloidosis: clinical response to epsilon amino caproic acid. Am J Clin Pathol 1984;81:382–7.
71. Anonymous. Tranexamic acid. Med Letter Drugs Therapeutics 1987;29:89–90.
72. Schwartz BS, Williams EC, Conlan MG, Mosher DF. Epsilon-aminocaproic acid in the treatment of patients with acute promyelocytic leukemia and acquired alpha-2-plasmin inhibitor defiency. Ann Intern Med 1986;105:873–7.
73. Takahashi H, Tatewaki W, Wada K, et al. Fibrinolysis and fibrinogenolysis in liver disease. Am J Hematol 1990;34:241-–5.
74. Ker K, Edwards P, Perel P, et al. Effect of tranexamic acid on surgical bleeding: systematic review and cumulative meta-analysis. BMJ 2012;344:e3054.
75. Shakur H, Roberts I, Bautista R, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376(9734):23–32.
76. WOMAN Trial Collaborators. Effect of early tranexamic acid administration on mortality, hysterectomy, and other morbidities in women with post-partum haemorrhage (WOMAN): an international, randomised, double-blind, placebo-controlled trial. Lancet 2017;389:2105–16.
77. Godbey EA, Schwartz J. ‘Massive transfusion protocols and the use of tranexamic acid’. Curr Opin Hematol 2018 Aug 16. doi: 10.1097/MOH.0000000000000457. [Epub ahead of print]
78. Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost 1997;78:1463–7.
79. DeLoughery TG. Management of bleeding emergencies: when to use recombinant activated factor VII. Expert Opin Pharmacother 2006;7:25–34.
80. Aledort L. Recombinant factor VIIa Is a pan-hemostatic agent? Thromb Haemost 2000;83:637–8.
81. Logan AC, Yank V, Stafford RS. Off-label use of recombinant factor VIIa in U.S. hospitals: analysis of hospital records. Ann Intern Med 2011;154:516–22.
82. Lin Y, Stanworth S, Birchall J, Doree C, Hyde C. Use of recombinant factor VIIa for the prevention and treatment of bleeding in patients without hemophilia: a systematic review and meta-analysis. CMAJ 2011;183:E9–19.
83. Yank V, Tuohy CV, Logan AC et al. Systematic review: benefits and harms of in-hospital use of recombinant factor VIIa for off-label indications. Ann Intern Med 2011;154:529–40.
84. Pavese P, Bonadona A, Beaubien J, et al. FVIIa corrects the coagulopathy of fulminant hepatic failure but may be associated with thrombosis: a report of four cases. Can J Anaesth 2005;52:26–29.
85. Mayer SA, Brun NC, Begtrup K, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med 2005;352:777–85.
86. Mayer SA, Brun NC, Begtrup K, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med 2008;358:2127–37.
87. Levi M, Levy JH, Andersen HF, Truloff D. Safety of recombinant activated factor VII in randomized clinical trials. N Engl J Med 2010;363:1791–1800.
INTRODUCTION
Transfusion therapy is an essential part of hematology practice, allowing for curative therapy of diseases such as leukemia, aplastic anemia, and aggressive lymphomas. Nonetheless, transfusions are associated with significant risks, including transmission of infections and transfusion-related reactions. Controversy remains about key issues in transfusion therapy, such as triggers for red cell transfusions. This article reviews the available blood products (Table 1) and indications for transfusion along with the associated risks, and also discusses specific clinical situations, such as massive transfusion.
BLOOD PRODUCTS
WHOLE BLOOD
Whole blood is the product of 1 unit of donated blood plus anticoagulant/preservative, and by definition contains 1 unit of plasma and red cells. Whole blood can be stored for 5 weeks. Although it was the standard product in the past, whole blood is rarely used since 1 unit of donated blood can now be fractionated into 1 unit of red blood cells (RBC), 1 unit of platelets, and 1 unit of fresh frozen plasma (FFP). Thus, the use of whole blood for just a single transfusion represents a waste of resources. There are 2 exceptions. One is autologous blood donations, which are whole blood units. Second, whole blood is increasingly being used in massive transfusions for trauma patients, with the rationale being that all essential blood components are being transfused at once.1
PACKED RED CELLS
The remaining red cell mass after most of the plasma is removed is called the “packed” red cell unit (hematocrit = 70%–80%), and so red cells are often called “packed” red cells, or PRBC. A preservative is added to improve the flow of blood and to provide “nutrients” for the red cells, and this reduces the hematocrit to approximately 60%. The volume of a red cell unit is approximately 340 mL. In the average adult, 1 unit of RBC raises the hematocrit by 3%. The indications for transfusion of red cells are to increase red cell mass, and thus oxygen delivery, in patients who are compromised by their anemia.
Several randomized trials have helped define the indications for red cell transfusions and justify lower hematocrit thresholds for initiating transfusion. The TRICC (Transfusion Requirements in Critical Care Investigators for the Canadian Critical Care Trials Group) trial showed that in critical care patients (30-day mortality, 18.7%–23.3%), a conservative transfusion strategy of waiting until the hematocrit was below 21% had the same outcomes as transfusing at a threshold of 24%.2 The TRACS (Transfusion Requirements After Cardiac Surgery) trial showed that a hematocrit target of 24% had the same benefit as a target of 30% in patients who had undergone cardiac bypass surgery.3 For patients with acute myocardial infarction, the outcomes were worse with aggressive transfusion at a hematocrit of 30% compared to 24%.4 In patients with upper gastrointestinal bleeding, a hemoglobin transfusion trigger of 7 g/dL was associated with a lower mortality than a trigger of 9 g/dL (5% versus 9%).5 Finally, the FOCUS (Transfusion Trigger Trial for Functional Outcomes in Cardiovascular Patients Undergoing Surgical Hip Fracture Repair) trial showed that in older patients (average age 80 years) who had undergone hip fracture surgery, transfusions based on symptoms and not a fixed trigger of 30% had the same outcomes but considerable savings in blood products.6 Based on these trials, decisions regarding when to transfuse patients should be based on symptoms and not “numbers.” Young patients, especially those with reversible anemias, can tolerate low blood counts and should not be transfused based on an arbitrary number.
PLATELETS
Several types of platelet products exist. One unit of platelet concentrate is derived from 1 unit of donor blood. Plateletpheresis from volunteer donors is also used to harvest platelets, with the resulting product referred to as plateletpheresis platelets. One unit of single-donor (pheresis) platelets is equivalent to 6 platelet concentrates. Finally, HLA-matched platelets are single-donor pheresis units that are obtained from an HLA-matched donor. This product should be ordered only if there is evidence of HLA antibodies (see Platelet Alloimmunization section).
The dose of platelets for the average patient is 6 units of platelet concentrate or 1 pheresis unit. In theory, 1 unit of platelet concentrate can raise the count by 5 to 7 × 103/µL, but often this response is blunted by concurrent illness or bleeding. In patients who appear to have a poor response, the platelet count can be checked 15 minutes after platelet infusion. No rise or a minimal rise (< 2 × 103/µL) in the platelet count is suggestive of platelet refractoriness, while a good 15-minute response but poor 24-hour count is more suggestive of consumption—fever, sepsis, drug, or splenomegaly—and not refractoriness.
The indication for platelet transfusion depends on the clinical situation. For patients with immune thrombocytopenia, platelets should not be transfused unless there is life-threatening bleeding. For stable patients with marrow aplasia from chemotherapy, a cut-off of a morning platelet count of less than 10 × 103/µL has been shown to be as safe as higher levels for prophylactic transfusions.7 For patients with active bleeding, the platelet count should be kept above 50 × 103/µL. Patients with acquired or inherited platelet dysfunction may benefit from transfusion no matter the platelet count.
Platelet Alloimmunization
Patients exposed to transfused white cells with different HLA antigens can develop antibodies to these antigens.8 Anti-HLA antibodies are common in patients who previously have received transfused blood that is not leukodepleted and in patients who have been pregnant. Since platelets carry class I HLA antigens, they will be rapidly destroyed by anti-HLA antibodies when transfused into these patients. In patients transfused for aplastic anemia or myelodysplasia, as many as 90% will become HLA-immunized. The incidence is lower in patients receiving chemotherapy but still can be as high as 60% to 90%.9,10 Patients who have developed anti-HLA antibodies can respond to transfused platelets matched for HLA antigens. Unfortunately, some patients will either be a rare HLA type or be so heavily immunized that they will not respond to any platelet transfusion.
The significance of alloimmunization centers on 2 concepts: recognition and avoidance. Patients with HLA antibodies will fail to have an increment of their platelet counts with transfusions. Accordingly, patients who do not experience an increase in their count 15 minutes after the transfusion may have HLA antibodies. One can test for the presence of anti-HLA antibodies, although some patients instead have specific antiplatelet antibodies that will not respond to HLA-matched platelets. In patients who have been pregnant or previously transfused and are scheduled to undergo transplant or aggressive chemotherapy, it is wise to test for anti-HLA antibodies in order to plan their transfusion needs. The evidence suggests that transfused white cells are responsible for initiating the anti-HLA response. Trials have shown that giving leukodepleted blood products may reduce the incidence of alloimmunization, so patients who are not HLA-alloimmunized should receive only leukodepleted products.11A difficult problem is bleeding in patients who are refractory to platelet transfusion.12 Patients who test positive for the presence of anti-HLA antibodies can receive transfusions of HLA-matched platelets.13 Unfortunately, matched platelet transfusions are not effective in 20% to 70% of these patients. Also, since some loci are difficult to match, effective products may be unavailable. Finally, as many as 25% of patients have antiplatelet antibodies in which HLA-matched products will be ineffective. Platelet cross-matching can be performed to find compatible units for these patients, but this may not always be successful. In the patient who is totally refractory to platelet transfusion, consider drugs as an etiology of antiplatelet antibodies (especially vancomycin).14 Use of antifibrinolytic agents such as epsilon-aminocaproic acid or tranexamic acid may decrease the incidence of minor bleeding, but these are ineffective for major bleeding. “Platelet drips”—infusing either a platelet concentrate per hour or 1 plateletpheresis unit every 6 hours—may be given as a continuous infusion, but there is no evidence that this is helpful.15
FRESH FROZEN PLASMA
FFP is made from 1 unit of donated whole blood, with an average volume of 225 mL per unit. One unit of FFP can increase coagulation factor levels by 5% and fibrinogen by 10 mg/dL in the average stable patient. FFP can take 20 to 30 minutes to thaw before use, so in situations where FFP is needed quickly, the blood bank must be informed to “keep ahead” some units. Units of FFP that have been thawed but not used can be stored refrigerated for 5 days to prevent wasting blood products.
The indications for FFP are limited to several situations. These include a documented coagulation defect that can be corrected by a reasonable amount of FFP, such as factor V deficiency and factor XI deficiency, disseminated intravascular coagulation (DIC), reversal of warfarin, and massive transfusions. FFP is also used for the therapy of thrombotic thrombocytopenic purpura.
There is little justification for FFP transfusion in many of the clinical settings in which it is commonly used. For example, FFP is given for minor elevations of the INR in patients with liver disease, despite literature showing not only that the INR rise is not reflective of coagulation defects, but also that patients with liver disease may even be thrombophilic.16,17 Reviews of FFP use found limited evidence-based indications for its use.18,19 Also, several studies have shown that transfusion of FFP is not effective at reversing minor elevations of the INR (1.3–1.8).20 In a meta-analysis, FFP was associated with increased risk for lung injury and a trend toward increased mortality.18
CRYOPRECIPITATE
Cryoprecipitate is produced from 1 unit of FFP that is thawed at 4°C. The precipitate is resuspended with 10 mL of saline or FFP and refrozen for storage. One unit contains at least 150 mg of fibrinogen and 80 units of factor VIII, along with von Willebrand factor. Thawing time for cryoprecipitate is approximately 20 minutes.
Cryoprecipitate is used to raise the fibrinogen level in patients with DIC or massive transfusion with hemodilution. It is third-line therapy in the treatment of type 1 von Willebrand disease and is second-line therapy in the treatment of patients with other types of von Willebrand disease. Currently, von Willebrand factor concentrates are the preferred replacement product for von Willebrand disease. Cryoprecipitate can be used as a source of factor VIII for hemophiliacs, but the preferred product for these patients is the super pure factor VIII concentrates or recombinant products. Cryoprecipitate can also be used to shorten the bleeding time of uremic patients, but its effectiveness for this is controversial.
GRANULOCYTES
Granulocytes are harvested by leukopheresis of normal donors, with a target yield of 1010 granulocytes from each donor. To reach this target, the donors are often “stimulated” with neutrophil growth factors. The harvesting procedure can take 3 hours and is associated with some risks to the donor (eg, citrate toxicity). The current indications for granulocytes are very limited since the advent of neutrophil growth factors and improved antimicrobials.21 They can be useful in the neutropenic patient with a documented bacterial infection in whom the white blood cell count is not expected to recover in the near future. Given the difficulty of keeping the count up, these transfusions have been mainly used in treating small children.
SPECIAL BLOOD PRODUCTS
IRRADIATED BLOOD PRODUCTS
Irradiation of blood is performed for only one reason: to prevent transfusion-related graft-versus-host disease (TGVHD) (Table 2).22 The irradiation can be performed at the blood center or in the transfusion service of larger hospitals. The units are not radioactive and can be transfused safely to other patients. There is increased leakage of potassium in irradiated units of blood, so the units need to be transfused within 14 days; in patients potentially sensitive to potassium (eg, neonates), the units must be transfused within 24 hours. Patients undergoing stem cell transplant, those receiving either interuterine transfusions or products from relatives, any patient with Hodgkin disease or receiving purine analogs or alemtuzumab, and patients with severe congenital immune deficiencies should receive irradiated blood. Most would also advocate that patients with hematologic malignances receiving chemotherapy receive irradiated products, but this is more controversial.
LEUKPDEPLETED BLOOD
Contamination of blood products by white blood cells is increasingly being recognized as a possible cause of adverse effects in transfused patients, including febrile transfusion reactions, inducing HLA alloimmunization, immunosuppression, disease transmission, and TGVHD. Reducing white cells can reduce the incidence of all of these complications except TGVHD. Currently, white cells are removed by infusion through filters that trap the cells. This can be done either at the bedside, in the blood bank, or at the donor center. The majority of red cells provided by blood centers in many areas of the country are already leukoreduced, eliminating the need for labor-intensive filtration at the transfusion center or bedside. Platelets collected by plateletpheresis methods can also be made leukocyte-poor. The current indications for leukodepleted productions are:
- Prevention of febrile transfusion reactions in patients with previous documented reactions
- Prevention of HLA alloimmunization (ineffective if patient has received 1 or more blood products not leukodepleted or is already HLA immunized)
- Prevention of cytomegalovirus (CMV) infection
CMV-NEGATIVE BLOOD
CMV can be transmitted through any cellular blood product—red cells and platelets. For patients who are CMV-negative and receiving transplants, especially stem cell transplants, a new CMV infection can be devastating.21 For years only blood from CMV-negative donors was used to transfuse CMV-negative patients. This policy is effective in preventing CMV infection, but because 50% of the population is positive for CMV antibodies, it may potentially lead to shortages of products that could be transfused to the patient. Currently, leukoreduced blood products are used since leukofiltration of the blood is just as effective as transfusion of CMV-negative blood in preventing infections and allows greater use of all blood products.23
COMPLICATIONS OF TRANSFUSIONS
HEMOLYTIC TRANSFUSION REACTION
There are 2 forms of hemolytic reactions—immediate and delayed.24 The immediate reaction is associated with fevers, hypotension, back pain, and oliguria. In severe cases, DIC and renal failure may occur. The immediate reaction is due to transfusion of blood that reacts with the recipient’s preformed high-titer blood antigen antibodies, most often to ABO. This is fatal 2% of the time and occurs almost always as a result of errors in correct identification of the patient. Reactions are due to recipient antibodies attacking donated RBCs, resulting in release of hemoglobin and red cell membrane–antigen complexes. These complexes are believed to lead to the hypotension, fevers, chills, and renal damage associated with the hemolytic reaction. Treatment consists of immediately stopping the transfusion, notifying the blood bank, vigorous intravenous hydration to keep the urine output over 100 mL/hr, and supportive therapy.
The delayed reaction can range in severity from an abrupt drop in the hematocrit to normal response to transfusion but the patient developing a positive Coombs’ test. The delayed response is due to an anamnestic response to blood-group antigens. When the patient is exposed to the same antigen, there is a rise in antibody titer leading to the reaction. Some alloantibodies can lead to a brisk reaction, most often anti-Kidd. The frequency with which delayed transfusion reactions occur is underestimated because mild reactions often do not get worked up or even discovered.
ALLERGIC REACTIONS
Allergic reactions are common (1%–3% of transfusions) and occur in patients having antibodies to proteins in donor blood, which can lead to hives and itching with transfusions. Most of the time these allergic reactions are mild and can be treated with antihistamines. Prophylaxis with antihistamines is not indicated for future transfusions unless the reactions are frequent. Rarely these reactions can be associated with shock and hypotension. Patients who are immunoglobulin (Ig) A–deficient can develop anaphylactic reactions to IgA-containing blood products. Patients with severe allergic reactions need to have their IgA measured and, if deficient, receive only washed units or plasma from IgA-deficient donors to prevent future severe reactions.
FEBRILE REACTIONS
The most common transfusion reaction is a febrile reaction that occurs after the transfusion starts and that sometimes can be complicated by chills. This reaction often occurs due to the presence of leukocyte debris and cytokines in the donated blood. Therapy is supportive and involves stopping the transfusion and administering acetaminophen, but since hemolytic transfusion reactions can present with fever all patients need to be thoroughly evaluated. The incidence of reactions can be decreased by using leukodepleted blood and plateletpheresis platelets. Most patients do not benefit from receiving prophylactic acetaminophen for future transfusion unless they have multiple reactions.
TRANSFUSION-RELATED ACUTE LUNG INJURY
Once thought a rare complication, transfusion-related acute lung injury (TRALI) is increasingly being recognized, with an incidence of approximately 1:5000 patients; it is now the most frequent cause of transfusion-related death.24,25 TRALI is noncardiac pulmonary edema and typically manifests clinically with hypoxemia, fever, bilateral infiltrates, and hypotension 2 to 6 hours after blood is given. Ventilatory support is often required. Recovery is usually rapid (24–48 hours) and complete. The etiology is complex. In many cases, transfused anti-HLA antibodies react with the recipient’s white cells leading to pulmonary damage. Another theory is that transfusion of preformed cytokines leads to pulmonary damage. Because plasma products from multiparous women are most often associated with anti-HLA antibodies, the restricted use of blood products from women has decreased the incidence of TRALI over the past few years.26
TRANSFUSION-ASSOCIATED CIRCULATORY OVERLOAD
Increasingly it being recognized that volume overload resulting from transfusions can lead to significant morbidity.27 Patients with heart or renal disease or patients who already have compromised fluid status are at risk for transfusion-associated circulatory overload (TACO). Another risk factor is transfusion of multiple blood products. Patients with TACO develop dyspnea within 6 hours of transfusion, but do not have fever or rash with the dyspnea. The diagnosis is made by demonstrating circulatory overload (eg, high venous pressure, B-type natriuretic peptide). Treatment is aggressive diuresis. Strategies to prevent TACO include judicious use of blood products, especially in patients at risk for TACO, and the use of prophylactic diuretics, especially with red cell or plasma transfusions.28
TRANSFUSION-RELATED GRAFT-VERSUS-HOST DISEASE
TGVHD is a rare reaction, but one that is most often fatal.29 TGVHD occurs when donor lymphocytes attack the blood recipient’s organs—skin, liver, intestines, and marrow. This is very rare in the normal blood recipient unless the donor and recipient share some HLA haplotypes.30 In immunosuppressed patients, TGVHD can occur with lesser degrees of HLA similarity, with cases reported in blood recipients who are mainly patients with Hodgkin disease or acute leukemia undergoing chemotherapy, and in patients receiving purine analogs. TGVHD had not been reported in AIDS patients despite profound immunosuppression, perhaps because the milieu of the patient does not allow lymphocyte expansion. Symptoms of TGVHD are an erythematous rash that may progress to epidermal toxic necrolysis, liver dysfunction, diarrhea, and pancytopenia. TGVHD is prevented by irradiating blood products given to at-risk patients with 2500 to 3500 rads. Directed blood donation from all blood relatives should also be irradiated. TGVHD cannot be prevented by leukopoor blood because the minute amount of lymphocytes that are not filtered still can lead to these complications.
POST-TRANSFUSION PURPURA
Patients with post-transfusion purpura (PTP) develop severe thrombocytopenia (< 10 × 103/µL) with often severe bleeding 1 to 2 weeks after receiving any type of blood product.31 Patients who develop PTP most often lack platelet antigen PLA1 or other platelet antigens. For unknown reasons, exposure to the antigens from the transfusion leads to rapid destruction of the patient’s own platelets. The diagnostic clue is thrombocytopenia in a patient, typically female, who has received a red cell or platelet blood product in the past 7 to 10 days. Treatment consists of intravenous immunoglobulin32 and plasmapheresis to remove the offending antibody. If patients with a history of PTP require further transfusions, only PLA1-negative platelets should be given.
IRON OVERLOAD
Every transfusion of red cells delivers approximately 250 mg of iron to the recipient. Since there is no natural way of ridding the body of iron, heavily transfused patients are at risk of iron overload. This is most often seen in children heavily transfused for thalassemia. Starting in the second decade of life, these individuals will develop endocrinopathies due to iron overload, liver problems, and often fatal cardiomyopathies. Studies have shown that chelation of iron with deferoxamine can be effective in preventing this fatal complication.33 Oral iron chelators such as deferasirox and deferiprone are also effective. The risk of iron overload in heavily transfused patients with myelodysplasia or other transfusion-dependent anemias is unclear, and uncertainty exists about the need for chelation.34
Young patients who face years of transfusions should be started on iron chelation to avoid iron overload. For older patients with transfusion-dependent anemia, iron chelation therapy should be considered if their life expectancy is long (years to decades) or special studies such as T2-weighted cardiac magnetic resonance imaging showing iron overloading.35
INFECTIOUS COMPLICATIONS
Concern over transmission of HIV infection via blood products in the late 1980s led to both a reduction in blood product use and a greater awareness of infectious complications of transfusion and their prevention. However, no blood product can ever be assumed to be safe for 2 reasons. One is that blood products can transmit infections during a “window period”—the time before a contaminated product can be detected by testing. The second is that blood is not screened for all potential infections (eg, babesiosis or new infections such as West Nile virus at the start of the outbreak). Risk of infection is reduced in 2 ways: deferral of potential infectious donors and blood product testing.
As part of the donation process, potential blood donors are asked a series of questions to see if they have risk factors for infections (eg, recent travel to malarious areas, recent tattoos), and if they answer positive are deferred from donating blood. Blood products are then tested for infectious agents by a combination of methods including detection of viral antigen, antibody response to infections, and more recently polymerase chain reaction (PCR).36 Current screening includes syphilis testing; testing for antibodies to HIV, HTLV (human T-lymphotropic virus), hepatitis C virus, hepatitis B core antigen (HBcAg), hepatitis B surface antigen, and PCR for HIV, hepatitis B virus, HCV, and West Nile virus. Some centers also test for Trypanosoma cruzi, the cause of Chagas disease.
In the past, the numerically most common transfusion-related disease was hepatitis, first B and then C.37,38 The first step in eliminating these infections was to stop paying donors for blood products. With the introduction of effective testing for hepatitis B and then C, the incidence of transfusion-related hepatitis has plummeted.36 For example, with the introduction of a diagnostic test for hepatitis C, the estimated risk has fallen from 5% to less than 1 per million. Currently, the risk of transmission of hepatitis B and C, HIV, and HTLV is less than 1 in a million.38
Despite this testing, blood transfusions can transmit a variety of infections, including malaria and babesiosis.39 Any new blood-borne infection introduced into the population can get into the blood supply as well. For example, at the start of the West Nile virus epidemic, there was a cluster of transfusion-transmitted cases that resulted in severe and sometimes fatal illness in immunosuppressed patients, but this issue has been addressed with the development of a PCR assay for screening blood.40 The rate of transfusion-related babesiosis has been increasing and screening for the causative parasite is being considered.
MASSIVE TRANSFUSIONS
Acutely bleeding patients can require large amounts of transfusion products. Early data showed high mortality rates with transfusion of more than 20 units of blood,41 but with modern blood banking techniques and improved laboratory testing, this rate has decreased dramatically, with survival rates of 43% to 70% in patients transfused with more than 50 units of blood.42
The basic approach to massive transfusions is to first transfuse the patient to maintain hemodynamic stability while specific blood tests are being obtained, and then to use the results of these early tests to guide the rest of the resuscitation. An important component is the ability to rapidly deliver standard packages of red cells, usually 6 to 10 units at a time, to the bleeding patient. To avoid delay while the patient’s blood is being typed, the first products delivered are blood group O Rh-positive units. Given the shortage of Rh-negative blood, this should be reserved for only empiric therapy of women of child-bearing age. Once the blood type is known, the patient can be switched over to type-specific blood.
In the past decade, there has been a shift toward increasing the amount of plasma given to patients receiving massive transfusions. This shift has occurred for 2 reasons. One is that modeling of coagulation changes in massive bleeding suggests the need for larger amounts of plasma to correct defects than have previously been recommended.43 The other reason is based on analysis of resuscitation protocols used in military and civilian trauma centers showing that giving red cells and plasma units in a 1:1 ratio appears to be associated with improved outcomes in massive transfusion. Several studies have extended this concept to platelets, again suggesting improved survival with 1 unit of random donor platelets given 1:1 with red cells and plasma units. The PROPPR (Prospective Observational Multicenter Major Trauma Transfusion) study compared a 1:1:1 to 1:1:2 ratio in patients with severe trauma and major bleeding and found less exsanguination and faster achievement of hemostasis in the first 24 hours.44 This has led to the widespread adoption of the 1:1 ratio by most trauma centers, and by default to other massive transfusion situations despite the lack of clinical trial data.45
One barrier to increased use is that plasma is kept frozen and requires 20 minutes to thaw. Many institutions are now keeping inventories of thawed plasma available for immediate use, ranging from 2 to 4 units of group AB plasma to keeping their entire inventory as liquid plasma.46 Plasma that is thawed but not used can be relabeled as “thawed plasma” and kept for up to 5 days. Also, many centers now use group A plasma for massive transfusions as this rarely leads to transfusion reactions and is much more available.47 Research is currently under way on lyophilized plasma, which can be stored at room temperature and can be rapidly reconstituted for emergency use.
The standard approach for laboratory testing is obtaining 5 tests: hematocrit, platelet count, INR/prothrombin time, activated partial thromboplastin time (aPTT), and fibrinogen.48 Product selection is guided by these tests, and they are repeated at regular intervals during the massive transfusion. A typical protocol is shown in Table 3. It is important as part of any protocol to have a flow chart that records laboratory results and products given that any member of the team can easily view.
The transfusion threshold for a low hematocrit depends on the stability of the patient. If the hematocrit is below 30% and the patient is bleeding or hemodynamically unstable, one should transfuse packed red cells. Stable patients can tolerate lower hematocrits, and an aggressive transfusion policy may even be detrimental.2,49 If the patient is bleeding, has florid DIC, or has received platelet aggregation inhibitors, then keeping the platelet count above 50 ×
While in the past fibrinogen targets of 50 to 100 mg/dL were recommended, recent data indicate that a target of 150 mg/dL or higher may be more appropriate.51–53 Severe fibrinolysis may occur in certain clinical situations such as brain injuries, hepatic trauma, or ischemic limb reperfusion, and the use of large amounts of cryoprecipitate can be anticipated. In patients with an INR greater than 2 and an abnormal aPTT, one can give 2 to 4 units of FFP. For an aPTT greater than 1.5 times normal, 2 to 4 units of plasma should be given. Elevation of the aPTT above 1.8 times normal control is associated with microvascular bleeding in trauma patients.54 Patients with marked abnormalities (eg, anaPTT more than 2 times normal) may require aggressive therapy with at least 15 to 30 mL/kg (4–8 units for an average adult) of plasma.55
Recently there has been increasing interest in the use of thromboelastography (TEG) in massive transfusion.56 This is a point-of-care assay performed on fresh whole blood that can assess multiple facets of hemostasis, including coagulation, platelet function, and fibrinolysis.57,58 TEG is performed by placing a 0.35-mL sample of whole blood into an oscillating container with a sensor pin that measures the force of thrombus formation. TEG measures 5 parameters:
- r time: time from starting TEG until clot formation
- K time: time needed for tracing to go from 2 mm to 20 mm
- alpha angle: slope of tracing between r and K time
- MA: greatest amplitude of TEG tracing
- Whole blood lysis index: amplitude of tracing 60 minutes after MA.
Several centers have incorporated TEG into resuscitation protocols that include standardized strategies for responding to abnormalities. Data suggest that use of TEG may decrease the use of blood products, especially in cardiac surgery, but this has not been prospectively studied in massive transfusions.56,59
COMPLICATIONS OF MASSIVE TRANSFUSIONS
Electrolyte abnormalities are unusual even in patients who receive massive transfusions.60 Platelet concentrates and plasma contain citrate that can chelate calcium. However, the citrate is rapidly metabolized, and it is rare to see clinically significant hypocalcemia. Although empiric calcium replacement is often recommended, one study suggests that this is associated with a worse outcome and should not be done.61 If hypocalcemia is a clinical concern, then levels should be drawn to guide therapy. Stored blood is acidic, with a pH of 6.5 to 6.9. However, acidosis attributed solely to transfused blood is rare and most often is a reflection of the patient’s stability. Empirical bicarbonate replacement has been associated with severe alkalosis and is not recom mended.62,63 Although potassium leaks out of stored red cells, even older units of blood contain only 8 mEq/L of potassium, so hyperkalemia is usually not a concern.
PATIENTS WITH AUTOIMMUNE HEMOLYTIC ANEMIA
Patients with autoimmune hemolytic anemia can be difficult to transfuse,64 because the autoantibody can interfere with several aspects of the transfusion services evaluation. In some patients the autoantibody can be so strong that the patient’s blood type cannot be determined. In most patients, the final step of the cross-match—mixing the donor blood with recipient plasma—will show noncompatibility due to the autoantibodies reacting with any red cells.
The first step when transfusing a patient with autoimmune hemolytic anemia is to draw several tubes of blood for the transfusion service before any potential transfusions. This allows the transfusion service to remove the autoantibodies so they can screen for underlying alloantibodies. Second, if the patient requires immediate transfusion, then type-specific or O-negative blood should be given. If the patient has not been recently (months) transfused, the incidence of a severe transfusion reaction is low. The first unit should be infused slowly with close observation of the patient. For patients who have been multiply transfused, the use of an “in-vivo” cross-match may be helpful. This is where the patient is slowly transfused 10 to 15 mL of blood over 15 minutes. The the plasma and urine are then assessed for signs of hemolysis and, if negative, the remaining product is given.
REFUSAL OF BLOOD PRODUCTS
The initial step in managing patients who refuse blood products is to find out why they are refusing them. Many patients have an exaggerated fear of HIV and other infectious agents, so discussing the very low risk for infection transmission can often resolve the situation. The most common reason for refusal of blood products is religious belief. Jehovah’s Witness patients will refuse blood products due to their interpretation of the Bible.65 All members will refuse red cells, plasma, and platelets, while decisions about “derived” blood products—products made by manipulation of the original donated units—are a matter of conscience. These include cryoprecipitate, intravenous gammaglobulin, and albumin.
In an elective situation, the first step is to discuss with the patient those products that are a matter of conscience and clearly document this. The patient’s blood count and iron stores should be assessed to identify any correctible causes of anemia or low iron stores before surgery. The use of erythropoietin to correct blood counts before surgery is controversial, as this may increase thrombosis risk and is contraindicated in patients with curable tumors.
For patients with acute blood loss, use of intravenous iron combined with high-dose erythropoietin is the most common approach to raise the blood count.65 A recommended erythropoietin dose is 300 units/kg 3 times a week, dropping to 100 units/kg 3 times weekly until the goal hematocrit is reached. Another often overlooked step is to consolidate and minimize laboratory testing. The most important step is to be respectful of the patient and their beliefs. Many larger cities have liaisons that can help with interactions between Jehovah’s Witness patients and the health care system.
NON-TRANSFUSION THERAPIES FOR ACUTE BLEEDING
DESMOPRESSIN
Desmopressin (DDAVP) is a synthetic analog of antidiuretic hormone that raises the levels of both factor VIII and von Willebrand protein severalfold.66 Desmopressin is effective in supporting hemostasis in patients with a wide variety of congenital and acquired bleeding disorders. However, desmopressin does not reduce blood loss before routine surgery in a healthy patient and should not be used for this purpose.67
TRANEXAMIC ACID
Tranexamic acid is an antifibrinolytic agent that blocks the binding of plasmin to fibrin.68 This agent was first shown to be useful in disorders that involve excessive fibrinolysis69–73 or as adjunctive therapy for oral or dental procedures in patients with a bleeding diathesis. In patients with severe thrombocytopenia, the use of antifibrinolytic agents may reduce bleeding. Increasing data shows that tranexamic acid can prevent blood loss in a variety of surgeries including heart bypass, liver transplantation, and orthopedic surgery.74 Patients across these settings have decreased blood loss and need for transfusion with no increased risk of thrombosis. The CRASH-2 study showed that the use of tranexamic acid significantly reduced mortality in trauma patients.75 The WOMEN trial demonstrated that 1 g of tranexamic acid given to women with blood loss of more than 500 mL after vaginal delivery or 1000 mL after cesarean section has a risk reduction of death of 0.81 with no increased risk of thrombosis.76 Given this abundant data, it is clear tranexamic acid needs to be part of any massive transfusion protocol.77
RECOMBINANT FACTOR VIIa
Recombinant factor VIIa (rVIIa) was originally developed as a “bypass” agent to support hemostasis in hemophiliacs.78 However, the use of rVIIa for a wide array of bleeding disorders, including patients with factor VII and XI deficiency and Glanzmann thrombasthenia, has been reported.79 Increasingly, rVIIa is being used as a “universal hemostatic agent” for patients with uncontrolled bleeding from any mechanism.80 Multiple case reports have described the use of rVIIa for bleeding in cardiac surgery patients, obstetrical bleeding, reversal of anticoagulation, and trauma.81 Unfortunately, little formal trial data exists to put these anecdotes into perspective, and formal review of clinical trial results has shown no benefit.82,83 However, when used in older patients, especially those with vascular risk factors, the risk of arterial thrombosis appears to increase.84 In the trials for intracranial hemorrhage, the thrombosis rate was 5% to 9%, and rates up to 10% for arterial events were seen in older patients in a review of all trials.85–87 Given the lack of data but the evidence of risk, rVIIa use should be restricted to patients with documented bleeding disorders that have been shown to benefit by its use.
INTRODUCTION
Transfusion therapy is an essential part of hematology practice, allowing for curative therapy of diseases such as leukemia, aplastic anemia, and aggressive lymphomas. Nonetheless, transfusions are associated with significant risks, including transmission of infections and transfusion-related reactions. Controversy remains about key issues in transfusion therapy, such as triggers for red cell transfusions. This article reviews the available blood products (Table 1) and indications for transfusion along with the associated risks, and also discusses specific clinical situations, such as massive transfusion.
BLOOD PRODUCTS
WHOLE BLOOD
Whole blood is the product of 1 unit of donated blood plus anticoagulant/preservative, and by definition contains 1 unit of plasma and red cells. Whole blood can be stored for 5 weeks. Although it was the standard product in the past, whole blood is rarely used since 1 unit of donated blood can now be fractionated into 1 unit of red blood cells (RBC), 1 unit of platelets, and 1 unit of fresh frozen plasma (FFP). Thus, the use of whole blood for just a single transfusion represents a waste of resources. There are 2 exceptions. One is autologous blood donations, which are whole blood units. Second, whole blood is increasingly being used in massive transfusions for trauma patients, with the rationale being that all essential blood components are being transfused at once.1
PACKED RED CELLS
The remaining red cell mass after most of the plasma is removed is called the “packed” red cell unit (hematocrit = 70%–80%), and so red cells are often called “packed” red cells, or PRBC. A preservative is added to improve the flow of blood and to provide “nutrients” for the red cells, and this reduces the hematocrit to approximately 60%. The volume of a red cell unit is approximately 340 mL. In the average adult, 1 unit of RBC raises the hematocrit by 3%. The indications for transfusion of red cells are to increase red cell mass, and thus oxygen delivery, in patients who are compromised by their anemia.
Several randomized trials have helped define the indications for red cell transfusions and justify lower hematocrit thresholds for initiating transfusion. The TRICC (Transfusion Requirements in Critical Care Investigators for the Canadian Critical Care Trials Group) trial showed that in critical care patients (30-day mortality, 18.7%–23.3%), a conservative transfusion strategy of waiting until the hematocrit was below 21% had the same outcomes as transfusing at a threshold of 24%.2 The TRACS (Transfusion Requirements After Cardiac Surgery) trial showed that a hematocrit target of 24% had the same benefit as a target of 30% in patients who had undergone cardiac bypass surgery.3 For patients with acute myocardial infarction, the outcomes were worse with aggressive transfusion at a hematocrit of 30% compared to 24%.4 In patients with upper gastrointestinal bleeding, a hemoglobin transfusion trigger of 7 g/dL was associated with a lower mortality than a trigger of 9 g/dL (5% versus 9%).5 Finally, the FOCUS (Transfusion Trigger Trial for Functional Outcomes in Cardiovascular Patients Undergoing Surgical Hip Fracture Repair) trial showed that in older patients (average age 80 years) who had undergone hip fracture surgery, transfusions based on symptoms and not a fixed trigger of 30% had the same outcomes but considerable savings in blood products.6 Based on these trials, decisions regarding when to transfuse patients should be based on symptoms and not “numbers.” Young patients, especially those with reversible anemias, can tolerate low blood counts and should not be transfused based on an arbitrary number.
PLATELETS
Several types of platelet products exist. One unit of platelet concentrate is derived from 1 unit of donor blood. Plateletpheresis from volunteer donors is also used to harvest platelets, with the resulting product referred to as plateletpheresis platelets. One unit of single-donor (pheresis) platelets is equivalent to 6 platelet concentrates. Finally, HLA-matched platelets are single-donor pheresis units that are obtained from an HLA-matched donor. This product should be ordered only if there is evidence of HLA antibodies (see Platelet Alloimmunization section).
The dose of platelets for the average patient is 6 units of platelet concentrate or 1 pheresis unit. In theory, 1 unit of platelet concentrate can raise the count by 5 to 7 × 103/µL, but often this response is blunted by concurrent illness or bleeding. In patients who appear to have a poor response, the platelet count can be checked 15 minutes after platelet infusion. No rise or a minimal rise (< 2 × 103/µL) in the platelet count is suggestive of platelet refractoriness, while a good 15-minute response but poor 24-hour count is more suggestive of consumption—fever, sepsis, drug, or splenomegaly—and not refractoriness.
The indication for platelet transfusion depends on the clinical situation. For patients with immune thrombocytopenia, platelets should not be transfused unless there is life-threatening bleeding. For stable patients with marrow aplasia from chemotherapy, a cut-off of a morning platelet count of less than 10 × 103/µL has been shown to be as safe as higher levels for prophylactic transfusions.7 For patients with active bleeding, the platelet count should be kept above 50 × 103/µL. Patients with acquired or inherited platelet dysfunction may benefit from transfusion no matter the platelet count.
Platelet Alloimmunization
Patients exposed to transfused white cells with different HLA antigens can develop antibodies to these antigens.8 Anti-HLA antibodies are common in patients who previously have received transfused blood that is not leukodepleted and in patients who have been pregnant. Since platelets carry class I HLA antigens, they will be rapidly destroyed by anti-HLA antibodies when transfused into these patients. In patients transfused for aplastic anemia or myelodysplasia, as many as 90% will become HLA-immunized. The incidence is lower in patients receiving chemotherapy but still can be as high as 60% to 90%.9,10 Patients who have developed anti-HLA antibodies can respond to transfused platelets matched for HLA antigens. Unfortunately, some patients will either be a rare HLA type or be so heavily immunized that they will not respond to any platelet transfusion.
The significance of alloimmunization centers on 2 concepts: recognition and avoidance. Patients with HLA antibodies will fail to have an increment of their platelet counts with transfusions. Accordingly, patients who do not experience an increase in their count 15 minutes after the transfusion may have HLA antibodies. One can test for the presence of anti-HLA antibodies, although some patients instead have specific antiplatelet antibodies that will not respond to HLA-matched platelets. In patients who have been pregnant or previously transfused and are scheduled to undergo transplant or aggressive chemotherapy, it is wise to test for anti-HLA antibodies in order to plan their transfusion needs. The evidence suggests that transfused white cells are responsible for initiating the anti-HLA response. Trials have shown that giving leukodepleted blood products may reduce the incidence of alloimmunization, so patients who are not HLA-alloimmunized should receive only leukodepleted products.11A difficult problem is bleeding in patients who are refractory to platelet transfusion.12 Patients who test positive for the presence of anti-HLA antibodies can receive transfusions of HLA-matched platelets.13 Unfortunately, matched platelet transfusions are not effective in 20% to 70% of these patients. Also, since some loci are difficult to match, effective products may be unavailable. Finally, as many as 25% of patients have antiplatelet antibodies in which HLA-matched products will be ineffective. Platelet cross-matching can be performed to find compatible units for these patients, but this may not always be successful. In the patient who is totally refractory to platelet transfusion, consider drugs as an etiology of antiplatelet antibodies (especially vancomycin).14 Use of antifibrinolytic agents such as epsilon-aminocaproic acid or tranexamic acid may decrease the incidence of minor bleeding, but these are ineffective for major bleeding. “Platelet drips”—infusing either a platelet concentrate per hour or 1 plateletpheresis unit every 6 hours—may be given as a continuous infusion, but there is no evidence that this is helpful.15
FRESH FROZEN PLASMA
FFP is made from 1 unit of donated whole blood, with an average volume of 225 mL per unit. One unit of FFP can increase coagulation factor levels by 5% and fibrinogen by 10 mg/dL in the average stable patient. FFP can take 20 to 30 minutes to thaw before use, so in situations where FFP is needed quickly, the blood bank must be informed to “keep ahead” some units. Units of FFP that have been thawed but not used can be stored refrigerated for 5 days to prevent wasting blood products.
The indications for FFP are limited to several situations. These include a documented coagulation defect that can be corrected by a reasonable amount of FFP, such as factor V deficiency and factor XI deficiency, disseminated intravascular coagulation (DIC), reversal of warfarin, and massive transfusions. FFP is also used for the therapy of thrombotic thrombocytopenic purpura.
There is little justification for FFP transfusion in many of the clinical settings in which it is commonly used. For example, FFP is given for minor elevations of the INR in patients with liver disease, despite literature showing not only that the INR rise is not reflective of coagulation defects, but also that patients with liver disease may even be thrombophilic.16,17 Reviews of FFP use found limited evidence-based indications for its use.18,19 Also, several studies have shown that transfusion of FFP is not effective at reversing minor elevations of the INR (1.3–1.8).20 In a meta-analysis, FFP was associated with increased risk for lung injury and a trend toward increased mortality.18
CRYOPRECIPITATE
Cryoprecipitate is produced from 1 unit of FFP that is thawed at 4°C. The precipitate is resuspended with 10 mL of saline or FFP and refrozen for storage. One unit contains at least 150 mg of fibrinogen and 80 units of factor VIII, along with von Willebrand factor. Thawing time for cryoprecipitate is approximately 20 minutes.
Cryoprecipitate is used to raise the fibrinogen level in patients with DIC or massive transfusion with hemodilution. It is third-line therapy in the treatment of type 1 von Willebrand disease and is second-line therapy in the treatment of patients with other types of von Willebrand disease. Currently, von Willebrand factor concentrates are the preferred replacement product for von Willebrand disease. Cryoprecipitate can be used as a source of factor VIII for hemophiliacs, but the preferred product for these patients is the super pure factor VIII concentrates or recombinant products. Cryoprecipitate can also be used to shorten the bleeding time of uremic patients, but its effectiveness for this is controversial.
GRANULOCYTES
Granulocytes are harvested by leukopheresis of normal donors, with a target yield of 1010 granulocytes from each donor. To reach this target, the donors are often “stimulated” with neutrophil growth factors. The harvesting procedure can take 3 hours and is associated with some risks to the donor (eg, citrate toxicity). The current indications for granulocytes are very limited since the advent of neutrophil growth factors and improved antimicrobials.21 They can be useful in the neutropenic patient with a documented bacterial infection in whom the white blood cell count is not expected to recover in the near future. Given the difficulty of keeping the count up, these transfusions have been mainly used in treating small children.
SPECIAL BLOOD PRODUCTS
IRRADIATED BLOOD PRODUCTS
Irradiation of blood is performed for only one reason: to prevent transfusion-related graft-versus-host disease (TGVHD) (Table 2).22 The irradiation can be performed at the blood center or in the transfusion service of larger hospitals. The units are not radioactive and can be transfused safely to other patients. There is increased leakage of potassium in irradiated units of blood, so the units need to be transfused within 14 days; in patients potentially sensitive to potassium (eg, neonates), the units must be transfused within 24 hours. Patients undergoing stem cell transplant, those receiving either interuterine transfusions or products from relatives, any patient with Hodgkin disease or receiving purine analogs or alemtuzumab, and patients with severe congenital immune deficiencies should receive irradiated blood. Most would also advocate that patients with hematologic malignances receiving chemotherapy receive irradiated products, but this is more controversial.
LEUKPDEPLETED BLOOD
Contamination of blood products by white blood cells is increasingly being recognized as a possible cause of adverse effects in transfused patients, including febrile transfusion reactions, inducing HLA alloimmunization, immunosuppression, disease transmission, and TGVHD. Reducing white cells can reduce the incidence of all of these complications except TGVHD. Currently, white cells are removed by infusion through filters that trap the cells. This can be done either at the bedside, in the blood bank, or at the donor center. The majority of red cells provided by blood centers in many areas of the country are already leukoreduced, eliminating the need for labor-intensive filtration at the transfusion center or bedside. Platelets collected by plateletpheresis methods can also be made leukocyte-poor. The current indications for leukodepleted productions are:
- Prevention of febrile transfusion reactions in patients with previous documented reactions
- Prevention of HLA alloimmunization (ineffective if patient has received 1 or more blood products not leukodepleted or is already HLA immunized)
- Prevention of cytomegalovirus (CMV) infection
CMV-NEGATIVE BLOOD
CMV can be transmitted through any cellular blood product—red cells and platelets. For patients who are CMV-negative and receiving transplants, especially stem cell transplants, a new CMV infection can be devastating.21 For years only blood from CMV-negative donors was used to transfuse CMV-negative patients. This policy is effective in preventing CMV infection, but because 50% of the population is positive for CMV antibodies, it may potentially lead to shortages of products that could be transfused to the patient. Currently, leukoreduced blood products are used since leukofiltration of the blood is just as effective as transfusion of CMV-negative blood in preventing infections and allows greater use of all blood products.23
COMPLICATIONS OF TRANSFUSIONS
HEMOLYTIC TRANSFUSION REACTION
There are 2 forms of hemolytic reactions—immediate and delayed.24 The immediate reaction is associated with fevers, hypotension, back pain, and oliguria. In severe cases, DIC and renal failure may occur. The immediate reaction is due to transfusion of blood that reacts with the recipient’s preformed high-titer blood antigen antibodies, most often to ABO. This is fatal 2% of the time and occurs almost always as a result of errors in correct identification of the patient. Reactions are due to recipient antibodies attacking donated RBCs, resulting in release of hemoglobin and red cell membrane–antigen complexes. These complexes are believed to lead to the hypotension, fevers, chills, and renal damage associated with the hemolytic reaction. Treatment consists of immediately stopping the transfusion, notifying the blood bank, vigorous intravenous hydration to keep the urine output over 100 mL/hr, and supportive therapy.
The delayed reaction can range in severity from an abrupt drop in the hematocrit to normal response to transfusion but the patient developing a positive Coombs’ test. The delayed response is due to an anamnestic response to blood-group antigens. When the patient is exposed to the same antigen, there is a rise in antibody titer leading to the reaction. Some alloantibodies can lead to a brisk reaction, most often anti-Kidd. The frequency with which delayed transfusion reactions occur is underestimated because mild reactions often do not get worked up or even discovered.
ALLERGIC REACTIONS
Allergic reactions are common (1%–3% of transfusions) and occur in patients having antibodies to proteins in donor blood, which can lead to hives and itching with transfusions. Most of the time these allergic reactions are mild and can be treated with antihistamines. Prophylaxis with antihistamines is not indicated for future transfusions unless the reactions are frequent. Rarely these reactions can be associated with shock and hypotension. Patients who are immunoglobulin (Ig) A–deficient can develop anaphylactic reactions to IgA-containing blood products. Patients with severe allergic reactions need to have their IgA measured and, if deficient, receive only washed units or plasma from IgA-deficient donors to prevent future severe reactions.
FEBRILE REACTIONS
The most common transfusion reaction is a febrile reaction that occurs after the transfusion starts and that sometimes can be complicated by chills. This reaction often occurs due to the presence of leukocyte debris and cytokines in the donated blood. Therapy is supportive and involves stopping the transfusion and administering acetaminophen, but since hemolytic transfusion reactions can present with fever all patients need to be thoroughly evaluated. The incidence of reactions can be decreased by using leukodepleted blood and plateletpheresis platelets. Most patients do not benefit from receiving prophylactic acetaminophen for future transfusion unless they have multiple reactions.
TRANSFUSION-RELATED ACUTE LUNG INJURY
Once thought a rare complication, transfusion-related acute lung injury (TRALI) is increasingly being recognized, with an incidence of approximately 1:5000 patients; it is now the most frequent cause of transfusion-related death.24,25 TRALI is noncardiac pulmonary edema and typically manifests clinically with hypoxemia, fever, bilateral infiltrates, and hypotension 2 to 6 hours after blood is given. Ventilatory support is often required. Recovery is usually rapid (24–48 hours) and complete. The etiology is complex. In many cases, transfused anti-HLA antibodies react with the recipient’s white cells leading to pulmonary damage. Another theory is that transfusion of preformed cytokines leads to pulmonary damage. Because plasma products from multiparous women are most often associated with anti-HLA antibodies, the restricted use of blood products from women has decreased the incidence of TRALI over the past few years.26
TRANSFUSION-ASSOCIATED CIRCULATORY OVERLOAD
Increasingly it being recognized that volume overload resulting from transfusions can lead to significant morbidity.27 Patients with heart or renal disease or patients who already have compromised fluid status are at risk for transfusion-associated circulatory overload (TACO). Another risk factor is transfusion of multiple blood products. Patients with TACO develop dyspnea within 6 hours of transfusion, but do not have fever or rash with the dyspnea. The diagnosis is made by demonstrating circulatory overload (eg, high venous pressure, B-type natriuretic peptide). Treatment is aggressive diuresis. Strategies to prevent TACO include judicious use of blood products, especially in patients at risk for TACO, and the use of prophylactic diuretics, especially with red cell or plasma transfusions.28
TRANSFUSION-RELATED GRAFT-VERSUS-HOST DISEASE
TGVHD is a rare reaction, but one that is most often fatal.29 TGVHD occurs when donor lymphocytes attack the blood recipient’s organs—skin, liver, intestines, and marrow. This is very rare in the normal blood recipient unless the donor and recipient share some HLA haplotypes.30 In immunosuppressed patients, TGVHD can occur with lesser degrees of HLA similarity, with cases reported in blood recipients who are mainly patients with Hodgkin disease or acute leukemia undergoing chemotherapy, and in patients receiving purine analogs. TGVHD had not been reported in AIDS patients despite profound immunosuppression, perhaps because the milieu of the patient does not allow lymphocyte expansion. Symptoms of TGVHD are an erythematous rash that may progress to epidermal toxic necrolysis, liver dysfunction, diarrhea, and pancytopenia. TGVHD is prevented by irradiating blood products given to at-risk patients with 2500 to 3500 rads. Directed blood donation from all blood relatives should also be irradiated. TGVHD cannot be prevented by leukopoor blood because the minute amount of lymphocytes that are not filtered still can lead to these complications.
POST-TRANSFUSION PURPURA
Patients with post-transfusion purpura (PTP) develop severe thrombocytopenia (< 10 × 103/µL) with often severe bleeding 1 to 2 weeks after receiving any type of blood product.31 Patients who develop PTP most often lack platelet antigen PLA1 or other platelet antigens. For unknown reasons, exposure to the antigens from the transfusion leads to rapid destruction of the patient’s own platelets. The diagnostic clue is thrombocytopenia in a patient, typically female, who has received a red cell or platelet blood product in the past 7 to 10 days. Treatment consists of intravenous immunoglobulin32 and plasmapheresis to remove the offending antibody. If patients with a history of PTP require further transfusions, only PLA1-negative platelets should be given.
IRON OVERLOAD
Every transfusion of red cells delivers approximately 250 mg of iron to the recipient. Since there is no natural way of ridding the body of iron, heavily transfused patients are at risk of iron overload. This is most often seen in children heavily transfused for thalassemia. Starting in the second decade of life, these individuals will develop endocrinopathies due to iron overload, liver problems, and often fatal cardiomyopathies. Studies have shown that chelation of iron with deferoxamine can be effective in preventing this fatal complication.33 Oral iron chelators such as deferasirox and deferiprone are also effective. The risk of iron overload in heavily transfused patients with myelodysplasia or other transfusion-dependent anemias is unclear, and uncertainty exists about the need for chelation.34
Young patients who face years of transfusions should be started on iron chelation to avoid iron overload. For older patients with transfusion-dependent anemia, iron chelation therapy should be considered if their life expectancy is long (years to decades) or special studies such as T2-weighted cardiac magnetic resonance imaging showing iron overloading.35
INFECTIOUS COMPLICATIONS
Concern over transmission of HIV infection via blood products in the late 1980s led to both a reduction in blood product use and a greater awareness of infectious complications of transfusion and their prevention. However, no blood product can ever be assumed to be safe for 2 reasons. One is that blood products can transmit infections during a “window period”—the time before a contaminated product can be detected by testing. The second is that blood is not screened for all potential infections (eg, babesiosis or new infections such as West Nile virus at the start of the outbreak). Risk of infection is reduced in 2 ways: deferral of potential infectious donors and blood product testing.
As part of the donation process, potential blood donors are asked a series of questions to see if they have risk factors for infections (eg, recent travel to malarious areas, recent tattoos), and if they answer positive are deferred from donating blood. Blood products are then tested for infectious agents by a combination of methods including detection of viral antigen, antibody response to infections, and more recently polymerase chain reaction (PCR).36 Current screening includes syphilis testing; testing for antibodies to HIV, HTLV (human T-lymphotropic virus), hepatitis C virus, hepatitis B core antigen (HBcAg), hepatitis B surface antigen, and PCR for HIV, hepatitis B virus, HCV, and West Nile virus. Some centers also test for Trypanosoma cruzi, the cause of Chagas disease.
In the past, the numerically most common transfusion-related disease was hepatitis, first B and then C.37,38 The first step in eliminating these infections was to stop paying donors for blood products. With the introduction of effective testing for hepatitis B and then C, the incidence of transfusion-related hepatitis has plummeted.36 For example, with the introduction of a diagnostic test for hepatitis C, the estimated risk has fallen from 5% to less than 1 per million. Currently, the risk of transmission of hepatitis B and C, HIV, and HTLV is less than 1 in a million.38
Despite this testing, blood transfusions can transmit a variety of infections, including malaria and babesiosis.39 Any new blood-borne infection introduced into the population can get into the blood supply as well. For example, at the start of the West Nile virus epidemic, there was a cluster of transfusion-transmitted cases that resulted in severe and sometimes fatal illness in immunosuppressed patients, but this issue has been addressed with the development of a PCR assay for screening blood.40 The rate of transfusion-related babesiosis has been increasing and screening for the causative parasite is being considered.
MASSIVE TRANSFUSIONS
Acutely bleeding patients can require large amounts of transfusion products. Early data showed high mortality rates with transfusion of more than 20 units of blood,41 but with modern blood banking techniques and improved laboratory testing, this rate has decreased dramatically, with survival rates of 43% to 70% in patients transfused with more than 50 units of blood.42
The basic approach to massive transfusions is to first transfuse the patient to maintain hemodynamic stability while specific blood tests are being obtained, and then to use the results of these early tests to guide the rest of the resuscitation. An important component is the ability to rapidly deliver standard packages of red cells, usually 6 to 10 units at a time, to the bleeding patient. To avoid delay while the patient’s blood is being typed, the first products delivered are blood group O Rh-positive units. Given the shortage of Rh-negative blood, this should be reserved for only empiric therapy of women of child-bearing age. Once the blood type is known, the patient can be switched over to type-specific blood.
In the past decade, there has been a shift toward increasing the amount of plasma given to patients receiving massive transfusions. This shift has occurred for 2 reasons. One is that modeling of coagulation changes in massive bleeding suggests the need for larger amounts of plasma to correct defects than have previously been recommended.43 The other reason is based on analysis of resuscitation protocols used in military and civilian trauma centers showing that giving red cells and plasma units in a 1:1 ratio appears to be associated with improved outcomes in massive transfusion. Several studies have extended this concept to platelets, again suggesting improved survival with 1 unit of random donor platelets given 1:1 with red cells and plasma units. The PROPPR (Prospective Observational Multicenter Major Trauma Transfusion) study compared a 1:1:1 to 1:1:2 ratio in patients with severe trauma and major bleeding and found less exsanguination and faster achievement of hemostasis in the first 24 hours.44 This has led to the widespread adoption of the 1:1 ratio by most trauma centers, and by default to other massive transfusion situations despite the lack of clinical trial data.45
One barrier to increased use is that plasma is kept frozen and requires 20 minutes to thaw. Many institutions are now keeping inventories of thawed plasma available for immediate use, ranging from 2 to 4 units of group AB plasma to keeping their entire inventory as liquid plasma.46 Plasma that is thawed but not used can be relabeled as “thawed plasma” and kept for up to 5 days. Also, many centers now use group A plasma for massive transfusions as this rarely leads to transfusion reactions and is much more available.47 Research is currently under way on lyophilized plasma, which can be stored at room temperature and can be rapidly reconstituted for emergency use.
The standard approach for laboratory testing is obtaining 5 tests: hematocrit, platelet count, INR/prothrombin time, activated partial thromboplastin time (aPTT), and fibrinogen.48 Product selection is guided by these tests, and they are repeated at regular intervals during the massive transfusion. A typical protocol is shown in Table 3. It is important as part of any protocol to have a flow chart that records laboratory results and products given that any member of the team can easily view.
The transfusion threshold for a low hematocrit depends on the stability of the patient. If the hematocrit is below 30% and the patient is bleeding or hemodynamically unstable, one should transfuse packed red cells. Stable patients can tolerate lower hematocrits, and an aggressive transfusion policy may even be detrimental.2,49 If the patient is bleeding, has florid DIC, or has received platelet aggregation inhibitors, then keeping the platelet count above 50 ×
While in the past fibrinogen targets of 50 to 100 mg/dL were recommended, recent data indicate that a target of 150 mg/dL or higher may be more appropriate.51–53 Severe fibrinolysis may occur in certain clinical situations such as brain injuries, hepatic trauma, or ischemic limb reperfusion, and the use of large amounts of cryoprecipitate can be anticipated. In patients with an INR greater than 2 and an abnormal aPTT, one can give 2 to 4 units of FFP. For an aPTT greater than 1.5 times normal, 2 to 4 units of plasma should be given. Elevation of the aPTT above 1.8 times normal control is associated with microvascular bleeding in trauma patients.54 Patients with marked abnormalities (eg, anaPTT more than 2 times normal) may require aggressive therapy with at least 15 to 30 mL/kg (4–8 units for an average adult) of plasma.55
Recently there has been increasing interest in the use of thromboelastography (TEG) in massive transfusion.56 This is a point-of-care assay performed on fresh whole blood that can assess multiple facets of hemostasis, including coagulation, platelet function, and fibrinolysis.57,58 TEG is performed by placing a 0.35-mL sample of whole blood into an oscillating container with a sensor pin that measures the force of thrombus formation. TEG measures 5 parameters:
- r time: time from starting TEG until clot formation
- K time: time needed for tracing to go from 2 mm to 20 mm
- alpha angle: slope of tracing between r and K time
- MA: greatest amplitude of TEG tracing
- Whole blood lysis index: amplitude of tracing 60 minutes after MA.
Several centers have incorporated TEG into resuscitation protocols that include standardized strategies for responding to abnormalities. Data suggest that use of TEG may decrease the use of blood products, especially in cardiac surgery, but this has not been prospectively studied in massive transfusions.56,59
COMPLICATIONS OF MASSIVE TRANSFUSIONS
Electrolyte abnormalities are unusual even in patients who receive massive transfusions.60 Platelet concentrates and plasma contain citrate that can chelate calcium. However, the citrate is rapidly metabolized, and it is rare to see clinically significant hypocalcemia. Although empiric calcium replacement is often recommended, one study suggests that this is associated with a worse outcome and should not be done.61 If hypocalcemia is a clinical concern, then levels should be drawn to guide therapy. Stored blood is acidic, with a pH of 6.5 to 6.9. However, acidosis attributed solely to transfused blood is rare and most often is a reflection of the patient’s stability. Empirical bicarbonate replacement has been associated with severe alkalosis and is not recom mended.62,63 Although potassium leaks out of stored red cells, even older units of blood contain only 8 mEq/L of potassium, so hyperkalemia is usually not a concern.
PATIENTS WITH AUTOIMMUNE HEMOLYTIC ANEMIA
Patients with autoimmune hemolytic anemia can be difficult to transfuse,64 because the autoantibody can interfere with several aspects of the transfusion services evaluation. In some patients the autoantibody can be so strong that the patient’s blood type cannot be determined. In most patients, the final step of the cross-match—mixing the donor blood with recipient plasma—will show noncompatibility due to the autoantibodies reacting with any red cells.
The first step when transfusing a patient with autoimmune hemolytic anemia is to draw several tubes of blood for the transfusion service before any potential transfusions. This allows the transfusion service to remove the autoantibodies so they can screen for underlying alloantibodies. Second, if the patient requires immediate transfusion, then type-specific or O-negative blood should be given. If the patient has not been recently (months) transfused, the incidence of a severe transfusion reaction is low. The first unit should be infused slowly with close observation of the patient. For patients who have been multiply transfused, the use of an “in-vivo” cross-match may be helpful. This is where the patient is slowly transfused 10 to 15 mL of blood over 15 minutes. The the plasma and urine are then assessed for signs of hemolysis and, if negative, the remaining product is given.
REFUSAL OF BLOOD PRODUCTS
The initial step in managing patients who refuse blood products is to find out why they are refusing them. Many patients have an exaggerated fear of HIV and other infectious agents, so discussing the very low risk for infection transmission can often resolve the situation. The most common reason for refusal of blood products is religious belief. Jehovah’s Witness patients will refuse blood products due to their interpretation of the Bible.65 All members will refuse red cells, plasma, and platelets, while decisions about “derived” blood products—products made by manipulation of the original donated units—are a matter of conscience. These include cryoprecipitate, intravenous gammaglobulin, and albumin.
In an elective situation, the first step is to discuss with the patient those products that are a matter of conscience and clearly document this. The patient’s blood count and iron stores should be assessed to identify any correctible causes of anemia or low iron stores before surgery. The use of erythropoietin to correct blood counts before surgery is controversial, as this may increase thrombosis risk and is contraindicated in patients with curable tumors.
For patients with acute blood loss, use of intravenous iron combined with high-dose erythropoietin is the most common approach to raise the blood count.65 A recommended erythropoietin dose is 300 units/kg 3 times a week, dropping to 100 units/kg 3 times weekly until the goal hematocrit is reached. Another often overlooked step is to consolidate and minimize laboratory testing. The most important step is to be respectful of the patient and their beliefs. Many larger cities have liaisons that can help with interactions between Jehovah’s Witness patients and the health care system.
NON-TRANSFUSION THERAPIES FOR ACUTE BLEEDING
DESMOPRESSIN
Desmopressin (DDAVP) is a synthetic analog of antidiuretic hormone that raises the levels of both factor VIII and von Willebrand protein severalfold.66 Desmopressin is effective in supporting hemostasis in patients with a wide variety of congenital and acquired bleeding disorders. However, desmopressin does not reduce blood loss before routine surgery in a healthy patient and should not be used for this purpose.67
TRANEXAMIC ACID
Tranexamic acid is an antifibrinolytic agent that blocks the binding of plasmin to fibrin.68 This agent was first shown to be useful in disorders that involve excessive fibrinolysis69–73 or as adjunctive therapy for oral or dental procedures in patients with a bleeding diathesis. In patients with severe thrombocytopenia, the use of antifibrinolytic agents may reduce bleeding. Increasing data shows that tranexamic acid can prevent blood loss in a variety of surgeries including heart bypass, liver transplantation, and orthopedic surgery.74 Patients across these settings have decreased blood loss and need for transfusion with no increased risk of thrombosis. The CRASH-2 study showed that the use of tranexamic acid significantly reduced mortality in trauma patients.75 The WOMEN trial demonstrated that 1 g of tranexamic acid given to women with blood loss of more than 500 mL after vaginal delivery or 1000 mL after cesarean section has a risk reduction of death of 0.81 with no increased risk of thrombosis.76 Given this abundant data, it is clear tranexamic acid needs to be part of any massive transfusion protocol.77
RECOMBINANT FACTOR VIIa
Recombinant factor VIIa (rVIIa) was originally developed as a “bypass” agent to support hemostasis in hemophiliacs.78 However, the use of rVIIa for a wide array of bleeding disorders, including patients with factor VII and XI deficiency and Glanzmann thrombasthenia, has been reported.79 Increasingly, rVIIa is being used as a “universal hemostatic agent” for patients with uncontrolled bleeding from any mechanism.80 Multiple case reports have described the use of rVIIa for bleeding in cardiac surgery patients, obstetrical bleeding, reversal of anticoagulation, and trauma.81 Unfortunately, little formal trial data exists to put these anecdotes into perspective, and formal review of clinical trial results has shown no benefit.82,83 However, when used in older patients, especially those with vascular risk factors, the risk of arterial thrombosis appears to increase.84 In the trials for intracranial hemorrhage, the thrombosis rate was 5% to 9%, and rates up to 10% for arterial events were seen in older patients in a review of all trials.85–87 Given the lack of data but the evidence of risk, rVIIa use should be restricted to patients with documented bleeding disorders that have been shown to benefit by its use.
1. Yazer MH, Cap AP, Spinella PC, et al. How do I implement a whole blood program for massively bleeding patients? Transfusion 2018;58:622–8.
2. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999;340:409–17.
3. Hajjar LA, Vincent JL, Galas FR, et al. Tranfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA 2010;304:1559–67.
4. Cooper HA, Rao SV, Greenberg MD, et al. Conservative versus liberal red cell transfusion in acute myocardial infarction (the CRIT Randomized Pilot Study). Am J Cardiol 2011;108:1108–11.
5. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
6. Carson JL, Terrin ML, Noveck H, et al; the FOCUS Investigators. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med 2011 Dec 4. [Epub ahead of print]
7. Schiffer CA, Anderson KC, Bennett CL, et al. Platelet transfusion for patients with cancer: clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol 2001;19:1519–38.
8. Schiffer CA. Prevention of alloimmunization against platelets. Blood 1991;77:1–4.
9. Hod E, Schwartz J. Platelet transfusion refractoriness. Br J Haematol 2008;142:348–60.
10. Novotny VMJ, Van Doorn R, Witvliet MD, et al. Occurrence of allogeneic HLA and non-HLA antibodies after transfusion of prestorage filtered platelets and red blood cells: A prospective study. Blood 1995;85:1736–41.
11. McFarland J, Menitove J, Kagen L et al. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med 1997;337:1861–9.
12. Juskewitch JE, Norgan AP, De Goey SR, et al. How do I … manage the platelet transfusion-refractory patient? Transfusion 2017;57:2828–35.
13. Schiffer CA. Diagnosis and management of refractoriness to platelet transfusion. Blood Rev 2001;15:175–80.
14. Christie DJ, van Buren N, Lennon SS, Putnam JL. Vancomycin-dependent antibodies associated with thrombocytopenia and refractoriness to platelet transfusion in patients with leukemia. Blood 1990;75:518–23.
15. Dzik S. How I do it: platelet support for refractory patients. Transfusion 2007;47:374–8.
16. Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med 2011;365:147–56.
17. Lisman T, Porte RJ. Pathogenesis, prevention, and management of bleeding and thrombosis in patients with liver diseases. Res Pract Thromb Haemost 2017;1:150–61.
18. Murad MH, Stubbs JR, Gandhi MJ, et al. The effect of plasma transfusion on morbidity and mortality: a systematic review and meta-analysis. Transfusion 2010;50:1370–83.
19. Green L, Bolton-Maggs P, Beattie C, et al. British Society of Haematology Guidelines on the spectrum of fresh frozen plasma and cryoprecipitate products: their handling and use in various patient groups in the absence of major bleeding. Br J Haematol 2018;181:54–67.
20. Abdel-Wahab OI, Healy B, Dzik WH. Effect of fresh-frozen plasma transfusion on prothrombin time and bleeding in patients with mild coagulation abnormalities. Transfusion 2006;46:1279–85.
21. Price TH. Granulocyte transfusion: current status. Semin Hematol 2007;44:15–23.
22. Treleaven J, Gennery A, Marsh J, et al. Guidelines on the use of irradiated blood components prepared by the British Committee for Standards in Haematology blood transfusion task force. Br J Haematol 2011;152:35–51.
23. Thiele T, Kruger W, Zimmermann K, et al. Transmission of cytomegalovirus (CMV) infection by leukoreduced blood products not tested for CMV antibodies: a single-center prospective study in high-risk patients undergoing allogeneic hematopoietic stem cell transplantation (CME). Transfusion 2011;51:2620–6.
24. Delaney M, Wendel S, Bercovitz RS, et al; Biomedical Excellence for Safer Transfusion (BEST) Collaborative. Transfusion reactions: prevention, diagnosis, and treatment. Lancet 2016;388:2825–36.
25. Toy P, Gajic O, Bacchetti P, et al. Transfusion related acute lung injury: incidence and risk factors. Blood 2011 Nov 23. [Epub ahead of print]
26. Wiersum-Osselton JC, Middelburg RA, Beckers EA, et al. Male-only fresh-frozen plasma for transfusion-related acute lung injury prevention: before-and-after comparative cohort study. Transfusion 2011;51:1278–83.
27. Friedman T, Javidroozi M, Lobel G, Shander A. Complications of allogeneic blood product administration, with emphasis on transfusion-related acute lung injury and transfusion-associated circulatory overload. Adv Anesth 2017;35:159–73.
28. Lin CR, Armali C, Callum J, et al. Transfusion-associated circulatory overload prevention: a retrospective observational study of diuretic use. Vox Sang 2018;113:386–92.
29. Sun X, Yu H, Xu Z, et al. Transfusion-associated graft-versus-host-disease: case report and review of literature. Transfus Apher Sci 2010;43:331–4.
30. Petz LD, Calhoun L, Yam P, et al. Transfusion-associated graft-versus-host disease in immunocompetent patients: report of a fatal case associated with transfusion of blood from a second-degree relative, and a survey of predisposing factors. Transfusion 1993;33:742–50.
31. Mueller-Eckhardt C. Post-transfusion purpura. Br J Hematol 1986;64:419–24.
32. Mueller-Eckhardt C, Kiefel V. High-dose IgG for posttransfusion purpura-revisited. Blut 1988;57:163–7.
33. Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet 2000;355(9220):2051–2.
34. Zeidan AM, Griffiths EA. To chelate or not to chelate in MDS: That is the question! Blood Rev 2018. pii: S0268-960X(17)30128-5.
35. Konen E, Ghoti H, Goitein O, et al. No evidence for myocardial iron overload in multitransfused patients with myelodysplastic syndrome using cardiac magnetic resonance T2 technique. Am J Hematol 2007;82:1013–16.
36. Squires JE. Risks of transfusion. South Med J 2011;104:762–9.
37. Sharma S, Sharma P, Tyler LN. Transfusion of blood and blood products: indications and complications. Am Fam Physician 2011;83:719–24.
38. Jacquot C, Delaney M. Efforts toward elimination of infectious agents in blood products. J Intensive Care Med 2018 Jan 1:885066618756589 [Epub ahead of print].
39. Herwaldt BL, Linden JV, Bosserman E, et al. Transfusion-associated babesiosis in the United States: a description of cases. Ann Intern Med 2011;155:509–19.
40. Biggerstaff BJ, Petersen LR. Estimated risk of West Nile virus transmission through blood transfusion during an epidemic in Queens, New York City. Transfusion 2002;42:1019–26.
41. Wilson RF, Mammen E, Walt AJ. Eight years of experience with massive blood transfusions. J Trauma 1971;11:275–85.
42. Wade CE, del Junco DJ, Holcomb JB, et al. Variations between level I trauma centers in 24-hour mortality in severely injured patients requiring a massive transfusion. J Trauma 2011;71(2 Suppl 3):S389–S393.
43. Hirshberg A, Dugas M, Banez EI, et al. Minimizing dilutional coagulopathy in exsanguinating hemorrhage: a computer simulation. J Trauma 2003;54:454–63.
44. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
45. Pasquier P, Gayat E, Rackelboom T, et al. An observational study of the fresh frozen plasma: red blood cell ratio in postpartum hemorrhage. Anesth Analg 2013;116:155–61.
46. Yuan S, Ziman A, Anthony MA, et al. How do we provide blood products to trauma patients? Transfusion 2009;49:1045–9.
47. Stevens WT, Morse BC, Bernard A, et al. Incompatible type A plasma transfusion in patients requiring massive transfusion protocol: Outcomes of an Eastern Association for the Surgery of Trauma multicenter study. J Trauma Acute Care Surg 2017;83:25–9.
48. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
49. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
50. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
51. Gerlach R, Tolle F, Raabe A, et al. Increased risk for postoperative hemorrhage after intracranial surgery in patients with decreased factor XIII activity: implications of a prospective study. Stroke 2002;33:1618–23.
52. Fenger-Eriksen C, Lindberg-Larsen M, Christensen AQ, et al. Fibrinogen concentrate substitution therapy in patients with massive haemorrhage and low plasma fibrinogen concentrations. Br J Anaesth 2008;101:769–73.
53. Charbit B, Mandelbrot L, Samain E, et al. The decrease of fibrinogen is an early predictor of the severity of postpartum hemorrhage. J Thromb Haemost 2007;5:266–73.
54. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
55. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
56. Curry NS, Davenport R, Pavord S, et al.The use of viscoelastic haemostatic assays in the management of major bleeding: A British Society for Haematology Guideline. Br J Haematol 2018. doi: 10.1111/bjh.15524. [Epub ahead of print].
57. Kashuk JL, Moore EE, Sawyer M, et al. Postinjury coagulopathy management: goal directed resuscitation via POC thrombelastography. Ann Surg 2010;251:604–14.
58. Whitten CW, Greilich PE. Thromboelastography: past, present, and future. Anesthesiology 2000;92:1223–5.
59. Girdauskas E, Kempfert J, Kuntze T, et al. Thromboelastometrically guided transfusion protocol during aortic surgery with circulatory arrest: a prospective, randomized trial. J Thorac Cardiovasc Surg 2010;140:1117–24.
60. Goskowicz R. The complications of massive tranfusion. Anesthesiology Clin North Am 1999;17:959–78.
61. Howland WS, Schwiezer O, Boyan CP. Massive blood replacement without calcuim administration. Surg Gynecol Obstet 1964;159:171–7.
62. Miller RD, Tong MJ, Robbins TO. Effects of massive transfusion of blood on acid-base balance. JAMA 1971;216:1762–5.
63. Collins JA. Problems associated with the massive transfusion of stored blood. Surgery 1974;75:274–95.
64. Petz LD. A physician’s guide to transfusion in autoimmune haemolytic anaemia. Br J Haematol 2004;124:712–6.
65. Scharman CD, Burger D, Shatzel JJ, et al. Treatment of individuals who cannot receive blood products for religious or other reasons. Am J Hematol 2017;92:1370–81
66. Leissinger C, Carcao M, Gill JC, et al. Desmopressin (DDAVP) in the management of patients with congenital bleeding disorders. Haemophilia 2014;20:158–67.
67. Desborough MJ, Oakland KA, Landoni G, et al. Desmopressin for treatment of platelet dysfunction and reversal of antiplatelet agents: a systematic review and meta-analysis of randomized controlled trials. J Thromb Haemost 2017;15:263–72.
68. Ng W, Jerath A, Wa˛sowicz M. Tranexamic acid: a clinical review. Anaesthesiol Intensive Ther 2015;47:339–50.
69. Amitrano L, Guardascione MA, Brancaccio V, Balzano A. Coagulation disorders in liver disease. Semin Liver Dis 2002;22:83–96.
70. Chang JC, Kane KK. Pathologic hyperfibrinolysis associated with amyloidosis: clinical response to epsilon amino caproic acid. Am J Clin Pathol 1984;81:382–7.
71. Anonymous. Tranexamic acid. Med Letter Drugs Therapeutics 1987;29:89–90.
72. Schwartz BS, Williams EC, Conlan MG, Mosher DF. Epsilon-aminocaproic acid in the treatment of patients with acute promyelocytic leukemia and acquired alpha-2-plasmin inhibitor defiency. Ann Intern Med 1986;105:873–7.
73. Takahashi H, Tatewaki W, Wada K, et al. Fibrinolysis and fibrinogenolysis in liver disease. Am J Hematol 1990;34:241-–5.
74. Ker K, Edwards P, Perel P, et al. Effect of tranexamic acid on surgical bleeding: systematic review and cumulative meta-analysis. BMJ 2012;344:e3054.
75. Shakur H, Roberts I, Bautista R, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376(9734):23–32.
76. WOMAN Trial Collaborators. Effect of early tranexamic acid administration on mortality, hysterectomy, and other morbidities in women with post-partum haemorrhage (WOMAN): an international, randomised, double-blind, placebo-controlled trial. Lancet 2017;389:2105–16.
77. Godbey EA, Schwartz J. ‘Massive transfusion protocols and the use of tranexamic acid’. Curr Opin Hematol 2018 Aug 16. doi: 10.1097/MOH.0000000000000457. [Epub ahead of print]
78. Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost 1997;78:1463–7.
79. DeLoughery TG. Management of bleeding emergencies: when to use recombinant activated factor VII. Expert Opin Pharmacother 2006;7:25–34.
80. Aledort L. Recombinant factor VIIa Is a pan-hemostatic agent? Thromb Haemost 2000;83:637–8.
81. Logan AC, Yank V, Stafford RS. Off-label use of recombinant factor VIIa in U.S. hospitals: analysis of hospital records. Ann Intern Med 2011;154:516–22.
82. Lin Y, Stanworth S, Birchall J, Doree C, Hyde C. Use of recombinant factor VIIa for the prevention and treatment of bleeding in patients without hemophilia: a systematic review and meta-analysis. CMAJ 2011;183:E9–19.
83. Yank V, Tuohy CV, Logan AC et al. Systematic review: benefits and harms of in-hospital use of recombinant factor VIIa for off-label indications. Ann Intern Med 2011;154:529–40.
84. Pavese P, Bonadona A, Beaubien J, et al. FVIIa corrects the coagulopathy of fulminant hepatic failure but may be associated with thrombosis: a report of four cases. Can J Anaesth 2005;52:26–29.
85. Mayer SA, Brun NC, Begtrup K, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med 2005;352:777–85.
86. Mayer SA, Brun NC, Begtrup K, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med 2008;358:2127–37.
87. Levi M, Levy JH, Andersen HF, Truloff D. Safety of recombinant activated factor VII in randomized clinical trials. N Engl J Med 2010;363:1791–1800.
1. Yazer MH, Cap AP, Spinella PC, et al. How do I implement a whole blood program for massively bleeding patients? Transfusion 2018;58:622–8.
2. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999;340:409–17.
3. Hajjar LA, Vincent JL, Galas FR, et al. Tranfusion requirements after cardiac surgery: the TRACS randomized controlled trial. JAMA 2010;304:1559–67.
4. Cooper HA, Rao SV, Greenberg MD, et al. Conservative versus liberal red cell transfusion in acute myocardial infarction (the CRIT Randomized Pilot Study). Am J Cardiol 2011;108:1108–11.
5. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
6. Carson JL, Terrin ML, Noveck H, et al; the FOCUS Investigators. Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med 2011 Dec 4. [Epub ahead of print]
7. Schiffer CA, Anderson KC, Bennett CL, et al. Platelet transfusion for patients with cancer: clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol 2001;19:1519–38.
8. Schiffer CA. Prevention of alloimmunization against platelets. Blood 1991;77:1–4.
9. Hod E, Schwartz J. Platelet transfusion refractoriness. Br J Haematol 2008;142:348–60.
10. Novotny VMJ, Van Doorn R, Witvliet MD, et al. Occurrence of allogeneic HLA and non-HLA antibodies after transfusion of prestorage filtered platelets and red blood cells: A prospective study. Blood 1995;85:1736–41.
11. McFarland J, Menitove J, Kagen L et al. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med 1997;337:1861–9.
12. Juskewitch JE, Norgan AP, De Goey SR, et al. How do I … manage the platelet transfusion-refractory patient? Transfusion 2017;57:2828–35.
13. Schiffer CA. Diagnosis and management of refractoriness to platelet transfusion. Blood Rev 2001;15:175–80.
14. Christie DJ, van Buren N, Lennon SS, Putnam JL. Vancomycin-dependent antibodies associated with thrombocytopenia and refractoriness to platelet transfusion in patients with leukemia. Blood 1990;75:518–23.
15. Dzik S. How I do it: platelet support for refractory patients. Transfusion 2007;47:374–8.
16. Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med 2011;365:147–56.
17. Lisman T, Porte RJ. Pathogenesis, prevention, and management of bleeding and thrombosis in patients with liver diseases. Res Pract Thromb Haemost 2017;1:150–61.
18. Murad MH, Stubbs JR, Gandhi MJ, et al. The effect of plasma transfusion on morbidity and mortality: a systematic review and meta-analysis. Transfusion 2010;50:1370–83.
19. Green L, Bolton-Maggs P, Beattie C, et al. British Society of Haematology Guidelines on the spectrum of fresh frozen plasma and cryoprecipitate products: their handling and use in various patient groups in the absence of major bleeding. Br J Haematol 2018;181:54–67.
20. Abdel-Wahab OI, Healy B, Dzik WH. Effect of fresh-frozen plasma transfusion on prothrombin time and bleeding in patients with mild coagulation abnormalities. Transfusion 2006;46:1279–85.
21. Price TH. Granulocyte transfusion: current status. Semin Hematol 2007;44:15–23.
22. Treleaven J, Gennery A, Marsh J, et al. Guidelines on the use of irradiated blood components prepared by the British Committee for Standards in Haematology blood transfusion task force. Br J Haematol 2011;152:35–51.
23. Thiele T, Kruger W, Zimmermann K, et al. Transmission of cytomegalovirus (CMV) infection by leukoreduced blood products not tested for CMV antibodies: a single-center prospective study in high-risk patients undergoing allogeneic hematopoietic stem cell transplantation (CME). Transfusion 2011;51:2620–6.
24. Delaney M, Wendel S, Bercovitz RS, et al; Biomedical Excellence for Safer Transfusion (BEST) Collaborative. Transfusion reactions: prevention, diagnosis, and treatment. Lancet 2016;388:2825–36.
25. Toy P, Gajic O, Bacchetti P, et al. Transfusion related acute lung injury: incidence and risk factors. Blood 2011 Nov 23. [Epub ahead of print]
26. Wiersum-Osselton JC, Middelburg RA, Beckers EA, et al. Male-only fresh-frozen plasma for transfusion-related acute lung injury prevention: before-and-after comparative cohort study. Transfusion 2011;51:1278–83.
27. Friedman T, Javidroozi M, Lobel G, Shander A. Complications of allogeneic blood product administration, with emphasis on transfusion-related acute lung injury and transfusion-associated circulatory overload. Adv Anesth 2017;35:159–73.
28. Lin CR, Armali C, Callum J, et al. Transfusion-associated circulatory overload prevention: a retrospective observational study of diuretic use. Vox Sang 2018;113:386–92.
29. Sun X, Yu H, Xu Z, et al. Transfusion-associated graft-versus-host-disease: case report and review of literature. Transfus Apher Sci 2010;43:331–4.
30. Petz LD, Calhoun L, Yam P, et al. Transfusion-associated graft-versus-host disease in immunocompetent patients: report of a fatal case associated with transfusion of blood from a second-degree relative, and a survey of predisposing factors. Transfusion 1993;33:742–50.
31. Mueller-Eckhardt C. Post-transfusion purpura. Br J Hematol 1986;64:419–24.
32. Mueller-Eckhardt C, Kiefel V. High-dose IgG for posttransfusion purpura-revisited. Blut 1988;57:163–7.
33. Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet 2000;355(9220):2051–2.
34. Zeidan AM, Griffiths EA. To chelate or not to chelate in MDS: That is the question! Blood Rev 2018. pii: S0268-960X(17)30128-5.
35. Konen E, Ghoti H, Goitein O, et al. No evidence for myocardial iron overload in multitransfused patients with myelodysplastic syndrome using cardiac magnetic resonance T2 technique. Am J Hematol 2007;82:1013–16.
36. Squires JE. Risks of transfusion. South Med J 2011;104:762–9.
37. Sharma S, Sharma P, Tyler LN. Transfusion of blood and blood products: indications and complications. Am Fam Physician 2011;83:719–24.
38. Jacquot C, Delaney M. Efforts toward elimination of infectious agents in blood products. J Intensive Care Med 2018 Jan 1:885066618756589 [Epub ahead of print].
39. Herwaldt BL, Linden JV, Bosserman E, et al. Transfusion-associated babesiosis in the United States: a description of cases. Ann Intern Med 2011;155:509–19.
40. Biggerstaff BJ, Petersen LR. Estimated risk of West Nile virus transmission through blood transfusion during an epidemic in Queens, New York City. Transfusion 2002;42:1019–26.
41. Wilson RF, Mammen E, Walt AJ. Eight years of experience with massive blood transfusions. J Trauma 1971;11:275–85.
42. Wade CE, del Junco DJ, Holcomb JB, et al. Variations between level I trauma centers in 24-hour mortality in severely injured patients requiring a massive transfusion. J Trauma 2011;71(2 Suppl 3):S389–S393.
43. Hirshberg A, Dugas M, Banez EI, et al. Minimizing dilutional coagulopathy in exsanguinating hemorrhage: a computer simulation. J Trauma 2003;54:454–63.
44. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
45. Pasquier P, Gayat E, Rackelboom T, et al. An observational study of the fresh frozen plasma: red blood cell ratio in postpartum hemorrhage. Anesth Analg 2013;116:155–61.
46. Yuan S, Ziman A, Anthony MA, et al. How do we provide blood products to trauma patients? Transfusion 2009;49:1045–9.
47. Stevens WT, Morse BC, Bernard A, et al. Incompatible type A plasma transfusion in patients requiring massive transfusion protocol: Outcomes of an Eastern Association for the Surgery of Trauma multicenter study. J Trauma Acute Care Surg 2017;83:25–9.
48. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
49. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
50. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
51. Gerlach R, Tolle F, Raabe A, et al. Increased risk for postoperative hemorrhage after intracranial surgery in patients with decreased factor XIII activity: implications of a prospective study. Stroke 2002;33:1618–23.
52. Fenger-Eriksen C, Lindberg-Larsen M, Christensen AQ, et al. Fibrinogen concentrate substitution therapy in patients with massive haemorrhage and low plasma fibrinogen concentrations. Br J Anaesth 2008;101:769–73.
53. Charbit B, Mandelbrot L, Samain E, et al. The decrease of fibrinogen is an early predictor of the severity of postpartum hemorrhage. J Thromb Haemost 2007;5:266–73.
54. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
55. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
56. Curry NS, Davenport R, Pavord S, et al.The use of viscoelastic haemostatic assays in the management of major bleeding: A British Society for Haematology Guideline. Br J Haematol 2018. doi: 10.1111/bjh.15524. [Epub ahead of print].
57. Kashuk JL, Moore EE, Sawyer M, et al. Postinjury coagulopathy management: goal directed resuscitation via POC thrombelastography. Ann Surg 2010;251:604–14.
58. Whitten CW, Greilich PE. Thromboelastography: past, present, and future. Anesthesiology 2000;92:1223–5.
59. Girdauskas E, Kempfert J, Kuntze T, et al. Thromboelastometrically guided transfusion protocol during aortic surgery with circulatory arrest: a prospective, randomized trial. J Thorac Cardiovasc Surg 2010;140:1117–24.
60. Goskowicz R. The complications of massive tranfusion. Anesthesiology Clin North Am 1999;17:959–78.
61. Howland WS, Schwiezer O, Boyan CP. Massive blood replacement without calcuim administration. Surg Gynecol Obstet 1964;159:171–7.
62. Miller RD, Tong MJ, Robbins TO. Effects of massive transfusion of blood on acid-base balance. JAMA 1971;216:1762–5.
63. Collins JA. Problems associated with the massive transfusion of stored blood. Surgery 1974;75:274–95.
64. Petz LD. A physician’s guide to transfusion in autoimmune haemolytic anaemia. Br J Haematol 2004;124:712–6.
65. Scharman CD, Burger D, Shatzel JJ, et al. Treatment of individuals who cannot receive blood products for religious or other reasons. Am J Hematol 2017;92:1370–81
66. Leissinger C, Carcao M, Gill JC, et al. Desmopressin (DDAVP) in the management of patients with congenital bleeding disorders. Haemophilia 2014;20:158–67.
67. Desborough MJ, Oakland KA, Landoni G, et al. Desmopressin for treatment of platelet dysfunction and reversal of antiplatelet agents: a systematic review and meta-analysis of randomized controlled trials. J Thromb Haemost 2017;15:263–72.
68. Ng W, Jerath A, Wa˛sowicz M. Tranexamic acid: a clinical review. Anaesthesiol Intensive Ther 2015;47:339–50.
69. Amitrano L, Guardascione MA, Brancaccio V, Balzano A. Coagulation disorders in liver disease. Semin Liver Dis 2002;22:83–96.
70. Chang JC, Kane KK. Pathologic hyperfibrinolysis associated with amyloidosis: clinical response to epsilon amino caproic acid. Am J Clin Pathol 1984;81:382–7.
71. Anonymous. Tranexamic acid. Med Letter Drugs Therapeutics 1987;29:89–90.
72. Schwartz BS, Williams EC, Conlan MG, Mosher DF. Epsilon-aminocaproic acid in the treatment of patients with acute promyelocytic leukemia and acquired alpha-2-plasmin inhibitor defiency. Ann Intern Med 1986;105:873–7.
73. Takahashi H, Tatewaki W, Wada K, et al. Fibrinolysis and fibrinogenolysis in liver disease. Am J Hematol 1990;34:241-–5.
74. Ker K, Edwards P, Perel P, et al. Effect of tranexamic acid on surgical bleeding: systematic review and cumulative meta-analysis. BMJ 2012;344:e3054.
75. Shakur H, Roberts I, Bautista R, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376(9734):23–32.
76. WOMAN Trial Collaborators. Effect of early tranexamic acid administration on mortality, hysterectomy, and other morbidities in women with post-partum haemorrhage (WOMAN): an international, randomised, double-blind, placebo-controlled trial. Lancet 2017;389:2105–16.
77. Godbey EA, Schwartz J. ‘Massive transfusion protocols and the use of tranexamic acid’. Curr Opin Hematol 2018 Aug 16. doi: 10.1097/MOH.0000000000000457. [Epub ahead of print]
78. Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost 1997;78:1463–7.
79. DeLoughery TG. Management of bleeding emergencies: when to use recombinant activated factor VII. Expert Opin Pharmacother 2006;7:25–34.
80. Aledort L. Recombinant factor VIIa Is a pan-hemostatic agent? Thromb Haemost 2000;83:637–8.
81. Logan AC, Yank V, Stafford RS. Off-label use of recombinant factor VIIa in U.S. hospitals: analysis of hospital records. Ann Intern Med 2011;154:516–22.
82. Lin Y, Stanworth S, Birchall J, Doree C, Hyde C. Use of recombinant factor VIIa for the prevention and treatment of bleeding in patients without hemophilia: a systematic review and meta-analysis. CMAJ 2011;183:E9–19.
83. Yank V, Tuohy CV, Logan AC et al. Systematic review: benefits and harms of in-hospital use of recombinant factor VIIa for off-label indications. Ann Intern Med 2011;154:529–40.
84. Pavese P, Bonadona A, Beaubien J, et al. FVIIa corrects the coagulopathy of fulminant hepatic failure but may be associated with thrombosis: a report of four cases. Can J Anaesth 2005;52:26–29.
85. Mayer SA, Brun NC, Begtrup K, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med 2005;352:777–85.
86. Mayer SA, Brun NC, Begtrup K, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med 2008;358:2127–37.
87. Levi M, Levy JH, Andersen HF, Truloff D. Safety of recombinant activated factor VII in randomized clinical trials. N Engl J Med 2010;363:1791–1800.
Acute Myeloid Leukemia
Introduction
Acute myeloid leukemia (AML) comprises a heterogeneous group of disorders characterized by proliferation of clonal, abnormally differentiated hematopoietic progenitor cells of myeloid lineage that infiltrate the bone marrow, blood, and other tissues.1 In most cases, AML is rapidly fatal if left untreated. Over the past 2 decades, our understanding of the underlying disease biology responsible for the development of AML has improved substantially. We have learned that biological differences drive the various clinical, cytogenetic, and molecular subentities of AML; distinguishing among these subentities helps to identify optimal therapies, while offering improved clinical outcomes for select groups. After years of stagnation in therapeutic advances, 4 new drugs for treating AML were approved by the US Food and Drug Administration (FDA) in 2017. In this article, we review key features of AML diagnosis and management in the context of 2 case presentations.
Epidemiology and Risk Factors
An estimated 21,380 new cases of AML were diagnosed in the United States in 2017, constituting roughly 1.3% of all new cases of cancer.2 Approximately 10,590 patients died of AML in 2017. The median age of patients at the time of diagnosis is 68 years, and the incidence is approximately 4.2 per 100,000 persons per year. The 5-year survival for AML has steadily risen from a meager 6.3% in 1975 to 17.3% in 1995 and 28.1% in 2009.2 The cure rates for AML vary drastically with age. Long-term survival is achieved in approximately 35% to 40% of adults who present at age 60 years or younger, but only 5% to 15% of those older than 60 years at presentation will achieve long-term survival.3
Most cases of AML occur in the absence of any known risk factors. High-dose radiation exposure, chronic benzene exposure, chronic tobacco smoking, and certain chemotherapeutics are known to increase the risk for AML.4 Inconsistent correlations have also been made between exposure to organic solvents, petroleum products, radon, pesticides, and herbicides and the development of AML.4 Obesity may also increase AML risk.4
Two distinct subcategories of therapy-related AML (t-AML) are known. Patients who have been exposed to alkylating chemotherapeutics (eg, melphalan, cyclophosphamide, and nitrogen mustard) can develop t-AML with chromosomal 5 and/or 7 abnormalities after a latency period of approximately 4 to 8 years.5 In contrast, patients exposed to topoisomerase II inhibitors (notably etoposide) develop AML with abnormalities of 11q23 (leading to MLL gene rearrangement) or 21q22 (RUNX1) after a latency period of about 1 to 3 years.6 AML can also arise out of other myeloid disorders such as myelodysplastic syndrome and myeloproliferative neoplasms, and other bone marrow failure syndromes such as aplastic anemia.4 Various inherited or congenital conditions such as Down syndrome, Bloom syndrome, Fanconi anemia, neurofibromatosis 1, and dyskeratosis congenita can also predispose to the development of AML. A more detailed listing of conditions associated with AML can be found elsewhere.4
Molecular Landscape
The first cancer genome sequence was reported in an AML patient in 2008.7 Since then, various elegantly conducted studies have expanded our understanding of the molecular abnormalities in AML. The Cancer Genome Atlas Research Network analyzed the genomes of 200 cases of de novo AML in adults.8 Only 13 mutations were found on average, much fewer than the number of mutations in most adult cancers. Twenty-three genes were commonly mutated, and another 237 were mutated in 2 or more cases. Essentially, all cases had at least 1 nonsynonymous mutation in 1 of 9 categories of genes: transcription-factor fusions (18%), the gene encoding nucleophosmin (NPM1) (27%), tumor-suppressor genes (16%), DNA-methylation–related genes (44%), signaling genes (59%), chromatin-modifying genes (30%), myeloid transcription-factor genes (22%), spliceosome-complex genes (14%), and cohesin-complex genes (13%).
In another study, samples from 1540 patients from 3 prospective trials of intensive chemotherapy were analyzed to understand how genetic diversity defines the pathophysiology of AML.9 The study authors identified 5234 driver mutations from 76 genes or genomic regions, with 2 or more drivers identified in 86% of the samples. Eleven classes of mutational events, each with distinct diagnostic features and clinical outcomes, were identified. Acting as an internal positive control in this analysis, previously recognized mutational and cytogenetic groups emerged as distinct entities, including the groups with biallelic CEBPA mutations, mutations in NPM1, MLL fusions, and the cytogenetic entities t(6;9), inv(3), t(8;21), t(15;17), and inv(16). Three additional categories emerged as distinct entities: AML with mutations in genes encoding chromatin, RNA splicing regulators, or both (18% of patients); AML with TP53 mutations, chromosomal aneuploidies, or both (13%); and, provisionally, AML with IDH2R172 mutations (1%). An additional level of complexity was also revealed within the subgroup of patients with NPM1 mutations, where gene–gene interactions identified co-mutational events associated with both favorable or adverse prognosis.
Further supporting this molecular classification of AML, a study that performed targeted mutational analysis of 194 patients with defined secondary AML (s-AML) or t-AML and 105 unselected AML patients found that the presence of mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 (all members of the chromatin or RNA splicing families) was highly specific for the diagnosis of s-AML.10 These findings are particularly clinically useful in those without a known history of antecedent hematologic disorder. These mutations defining the AML ontogeny were found to occur early in leukemogenesis, persist in clonal remissions, and predict worse clinical outcomes. Mutations in genes involved in regulation of DNA modification and of chromatin state (commonly DNMT3A, ASXL1, and TET2) have also been shown to be present in preleukemic stem or progenitor cells and to occur early in leukemogenesis.3 Unsurprisingly, some of these same mutations, including those in epigenetic regulators (DNMT3A, ASXL1, and TET2) and less frequently in splicing factor genes (SF3B1, SRSF2), have been associated with clonal hematopoietic expansion in elderly, seemingly healthy adults, a condition termed clonal hematopoiesis of indeterminate potential (CHIP).3,11,12 The presence of CHIP is associated with increased risk of hematologic neoplasms and all-cause mortality, the latter being possibly driven by a near doubling in the risk of coronary heart disease in humans and by accelerated atherosclerosis in a mouse model.11,13,14
Clinical Presentation and Work-up
Case Patient 1
A 57-year-old woman with a history of hypertension presents to the emergency department with complaints of productive cough and fevers for the previous 3 days. Examination reveals conjunctival pallor, gingival hyperplasia, and decreased breath sounds at the posterior right lung field. Investigations reveal a white blood cell (WBC) count of 51,000/µL with 15% blasts, a hemoglobin of 7.8 g/dL, and a platelet count of 56 × 103/µL. Peripheral blood smear is notable for large myeloblasts with occasional Auer rods. Chest radiograph shows a consolidation in the right lower lobe.
Case Patient 2
A 69-year-old man presents to his primary care physician for evaluation of worsening fatigue for the previous 4 months. Ten years prior to presentation, he had received 6 cycles of RCHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) as treatment for diffuse large B-cell lymphoma. Conjunctival pallor, patches of purpura over the extremities, and mucosal petechiae are noted on examination. Laboratory analyisis reveals a WBC count of 2400/µL with 12% blasts, hemoglobin of 9.0 g/dL, and platelet count of 10 × 103/µL. Peripheral smear shows dysplastic myeloid cells and blasts.
Clinical Features
Patients with AML typically present with features secondary to proliferation of blasts (ie, findings of bone marrow failure and end organ damage).4,5 Fatigue, pallor, dizziness, dyspnea, and headaches occur secondary to anemia. Easy and prolonged bruising, petechiae, epistaxis, gingival bleeding, and conjunctival hemorrhages result from thrombocytopenia. Bleeding from other sites such as the central nervous system and gastrointestinal tract occurs but is uncommon. Patients may also present with infections resulting from unrecognized neutropenia. Constitutional symptoms including anorexia, fevers, and weight loss are frequently reported, while organomegaly (hepatomegaly and/or splenomegaly) is seen in about a quarter of patients.4 Infiltration of blasts into almost every organ has been noted, a condition known as myeloid (or granulocytic) sarcoma.15 This condition is more commonly found in patients with blastic, monoblastic, or myelomonocytic variants of AML, and is known as isolated myeloid sarcoma if no concurrent marrow or blood involvement is identified. In the absence of induction chemotherapy, systemic involvement occurs in a matter of weeks to months following such presentation.16
Laboratory analysis will usually demonstrate derangements in peripheral blood cell lines. At least half of patients have a total WBC count less than 5000/µL, a platelet count less than 50 × 103/µL, or both at the time of diagnosis.4,17 Approximately 10% of patients present with hyperleukocytosis and a WBC count greater than 100,000/µL, which can be associated with leukostasis.5 Additionally, spontaneous electrolyte derangement consistent with tumor lysis syndrome and coagulation abnormalities found in disseminated intravascular coagulation may be noted, even before initiation of therapy.
Work-Up of Suspected AML
Bone marrow biopsy and aspirate, along with touch preparations of the core biopsy sample, are crucial in the workup of suspected AML. At least 200 WBCs on blood smears and 500 nucleated cells on spiculated marrow smears should be counted.3 Reactivity with specific histochemical stains (myeloperoxidase, Sudan black B, or naphthyl AS-D-chloroacetate), presence of Auer rods, and reactivity to monoclonal antibodies against epitopes present on myeloblasts (eg, CD13, CD33, CD117) help distinguish myeloblasts from lymphoblasts.4 Flow cytometric analysis helps in confirming myeloid lineage; blasts generally express CD34 and HLA-DR, markers of immature hematopoietic precursors, and dim CD45 (common leukocyte antigen). One or more lymphoid antigens may be aberrantly expressed as well. Of note, in about 2% to 3% of acute leukemia cases, immunohistochemistry and/or flow cytometry findings demonstrate immature cells with features of both myeloid and lymphoid lineages (biphenotypic) or different populations of myeloid and lymphoid leukemia cells (bilineal). These leukemias are termed mixed-phenotype acute leukemia and are typically treated with either AML or acute lymphoblastic leukemia regimens.18
Cytogenetics, as assessed through conventional karyotype and fluorescence in situ hybridization (FISH), constitutes an essential part of the work-up. Eight balanced translocations and inversions and their variants are included in the World Health Organization (WHO) category “AML with recurrent genetic abnormalities,” while 9 balanced rearrangements and multiple unbalanced abnormalities in the presence of a blast count ≥ 20% are sufficient to establish the diagnosis of “AML with myelodysplasia-related changes.”3,19 Various other gene rearrangements thought to represent disease-initiating events are recognized as well, but these rearrangements do not yet formally define WHO disease categories.3 FISH can help detect RUNX1-RUNX1T1, CBFB-MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, as well as chromosomal changes like 5q, 7q, or 17p, especially when fewer than 20 metaphases are assessable (due to failure of culture) by conventional cytogenetic methods.3
As certain molecular markers help with disease prognosis and the selection of personalized therapies, testing for these markers is recommended as part of a complete work-up of AML. The current standard of care is to test for nucleophosmin (NPM1), fms-like tyrosine kinase 3 (FLT3), and CEBPA mutations in all newly diagnosed patients.1RUNX1 mutation analysis should also be considered as its presence defines a provisional WHO subcategory.19 In the case of FLT3, the analysis should include both internal tandem duplications (FLT3-ITD, associated with worse prognosis especially at high allelic ratio) and tyrosine-kinase domain mutations (FLT3-TKD; D835 and I836), especially now that FLT3 inhibitors are regularly used.20 Most academic centers now routinely use next-generation sequencing–based panels to assess multiple mutations. 
Diagnosis and Classification
A marrow or blood blast (myeloblasts, monoblasts, megakaryoblasts, or promonocytes [considered blast equivalents]) count of ≥ 20% is required for AML diagnosis.3,19 The presence of t(15;17), t(8;21), inv(16), or t(16;16), however, is considered diagnostic of AML irrespective of blast count.3,19 The previously used French-American-British (FAB) classification scheme has been replaced by the WHO classification (Table 2), which takes into account the morphologic, cytogenetic, genetic, and clinical features of the leukemia. 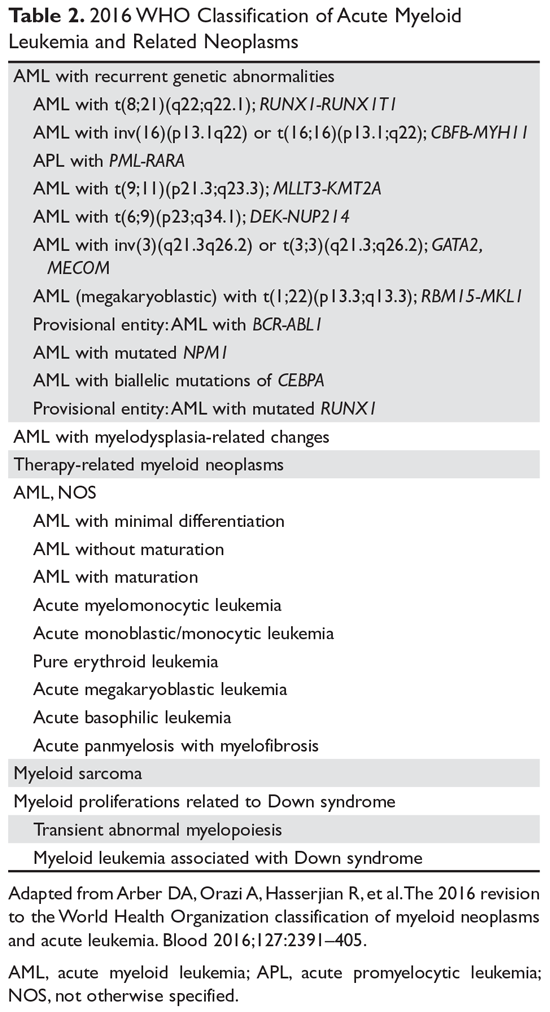
The category “AML with myelodysplasia-related changes” includes AML that has evolved out of an antecedent myelodysplastic syndrome, has ≥ 50% dysplasia in 2 or more lineages, or has myelodysplasia-related cytogenetic changes (eg, –5/del(5q), –7/del(7q), ≥ 3 cytogenetic abnormalities).19 “Therapy-related myeloid neoplasm,” or therapy-related AML, is diagnosed when the patient has previously received cytotoxic agents or ionizing radiation.19
Cases which do not meet the criteria for 1 of the previously mentioned categories are currently classified as “AML, not otherwise specified.” Further subclassification is pursued as per the older FAB scheme; however, no additional prognostic information is obtained in doing so.3,19 Myeloid sarcoma is strictly not a subcategory of AML. Rather, it is an extramedullary mass of myeloid blasts that effaces the normal tissue architecture.16 Rarely, myeloid sarcoma can be present without systemic disease involvement; it is important to note that management of such cases is identical to management of overt AML.16
Finally, myeloid proliferations related to Down syndrome include 2 entities seen in children with Down syndrome.19 Transient abnormal myelopoiesis, seen in 10% to 30% of newborns with Down syndrome, presents with circulating blasts that resolve in a couple of months. Myeloid leukemia associated with Down syndrome is AML that occurs usually in the first 3 years of life and persists if not treated.19
Case 1 Continued
The presence of 15% blasts in the peripheral blood is concerning for, but not diagnostic of, AML. On the other hand, the presence of Auer rods is virtually pathognomonic for AML. Gingival hyperplasia in this patient may be reflective of extramedullary disease. Cytogenetics from the peripheral blood and marrow aspirate show inv(16) in 20 of 20 cells. Molecular panel is notable for mutation in c-KIT. As such, the patient is diagnosed with core-binding factor AML, which per the ELN classification is considered a favorable-risk AML. The presence of c-KIT mutation, however, confers a relatively worse outcome.
Case 2 Continued
Presence of pancytopenia in a patient who previously received cytotoxic chemotherapy is highly concerning for therapy-related myeloid neoplasm. The presence of 12% blasts in the peripheral blood does not meet the criteria for diagnosis of AML. However, marrow specimens show 40% blasts, thus meeting the criteria for an AML diagnosis. Additionally, cytogenetics are notable for the presence of monosomy 7, while a next-generation sequencing panel shows a mutation in TP53. Put together, this patient meets the criteria for therapy-related AML which is an adverse-risk AML according to the ELN classification.
Management
The 2 most significant factors that must be considered when selecting AML therapies are the patient’s suitability for intensive chemotherapy and the biological characteristics of the AML. The former is a nuanced decision that incorporates age, performance status, and existing comorbidities. Treatment-related mortality calculators can guide physicians when making therapy decisions, especially in older patients (≥ 65 years). Retrospective evidence from various studies suggests that older, medically fit patients may derive clinically comparable benefits from intensive and less intensive induction therapies.25–27 The biological characteristics of the leukemia can be suggested by morphologic findings, cytogenetics, and molecular information, in addition to a history of antecedent myeloid neoplasms. Recently, an AML composite model incorporating an augmented Hematopoietic Cell Transplantation–specific Comorbidity Index (HCT-CI) score, age, and cytogenetic/molecular risks was shown to improve treatment decision-making about AML; this model potentially could be used to guide patient stratification in clinical trials as well.28 The overall treatment model of AML is largely unchanged otherwise. It is generally divided into induction, consolidation, and maintenance therapies.
Induction Therapy
In patients who can tolerate intensive therapies, the role of anthracycline- and cytarabine-based treatment is well established. However, the choice of specific anthracycline is not well established. One study concluded that idarubicin and mitoxantrone led to better outcomes as compared to daunorubicin, while another showed no difference between these agents.29,30 A pooled study of AML trials conducted in patients aged 50 years and older showed that while idarubicin led to a higher complete remission rate (69% versus 61%), the overall survival (OS) did not differ significantly.31 As for dosing, daunorubicin given at 45 mg/m2 daily for 3 days has been shown to have lower complete remission rates and higher relapse rates than a dose of 90 mg/m2 daily for 3 days in younger patients.32–34 However, it is not clear whether the 90 mg/m2 dose is superior to the frequently used dose of 60 mg/m2.35 A French study has shown comparable rates of complete remission, relapse, and OS between the 60 mg/m2 and 90 mg/m2 doses in patients with intermediate or unfavorable cytogenetics.36
If idarubicin is used, a dose of 12 mg/m2 for 3 days is considered the standard. In patients aged 50 to 70 years, there were no statistically significant differences in rates of relapse or OS between daunorubicin 80 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 4 days.37 As for cytarabine, the bulk of the evidence indicates that a dose of 1000 mg/m2 or higher should not be used.38 As such, the typical induction chemotherapy regimen of choice is 3 days of anthracycline (daunorubicin or idarubicin) and 7 days of cytarabine (100–200 mg/m2 continuous infusion), also known as the 7+3 regimen, which was first pioneered in the 1970s. In a recent phase 3 trial, 309 patients aged 60 to 75 years with high-risk AML (AML with myelodysplasia-related changes or t-AML) were randomly assigned to either the 7+3 regimen or CPX-351 (ie, nano-liposomal encapsulation of cytarabine and daunorubicin in a 5:1 molar ratio).39 A higher composite complete response rate (47.7% versus 33.3%; P = 0.016) and improved survival (9.56 months versus 5.95 months; hazard ratio [HR] 0.69, P = 0.005) were seen with CPX-351, leading to its approval by the FDA in patients with high-risk AML.
The 7+3 regimen has served as a backbone onto which other drugs have been added in clinical trials—the majority without any clinical benefits—for patients who can tolerate intensive therapy. In this context, the role of 2 therapies recently approved by the FDA must be discussed. In the RATIFY trial, 717 patients aged 18 to 59 years with AML and a FLT3 mutation were randomly assigned to receive standard chemotherapy (induction and consolidation therapy) plus either midostaurin or placebo; those who were in remission after consolidation therapy received either midostaurin or placebo in the maintenance phase.40 The primary endpoint was met as midostaurin improved OS (HR 0.78, P = 0.009). The benefit of midostaurin was consistent across all FLT3 subtypes and mutant allele burdens, regardless of whether patients proceeded to allogeneic stem cell transplant (allo-SCT). Based on the results of RATIFY, midostaurin was approved by the FDA for treatment of AML patients who are positive for the FLT3 mutation. Whether more potent and selective FLT3 inhibitors like gilteritinib, quizartinib, or crenolanib improve the outcomes is currently under investigation in various clinical trials.20
The development of gemtuzumab ozogamicin (GO) has been more complicated. GO, an antibody-drug conjugate comprised of a CD33-directed humanized monoclonal antibody linked covalently to the cytotoxic agent calicheamicin, binds CD33 present on the surface of myeloid leukemic blasts and immature normal cells of myelomonocytic lineage.41 The drug first received an accelerated approval in 2000 as monotherapy (2 doses of 9 mg/m2 14 days apart) for the treatment of patients 60 years of age and older with CD33-positive AML in first relapse based on the results of 3 open-label multicenter trials.41,42 However, a confirmatory S0106 trial in which GO 6 mg/m2 was added on day 4 in newly diagnosed AML patients was terminated early when an interim analysis showed an increased rate of death in induction (6% versus 1%) and lack of improvement in complete response, disease-free survival, or OS with the addition of GO.43 This study led to the withdrawal of GO from the US market in 2010. However, 2 randomized trials that studied GO using a different dose and schedule suggested that the addition of GO to intensive chemotherapy improved survival outcomes in patients with favorable and intermediate-risk cytogenetics.44,45 The results of the multicenter, open-label phase 3 ALFA-0701 trial, which randomly assigned 271 patients aged 50 to 70 years with newly diagnosed AML to daunorubicin and cytarabine alone or in combination with GO (3 mg/m2 on days 1, 4, and 7 during induction and day 1 of 2 consolidation courses), showed a statistically significant improvement in event-free survival (17.3 months versus 9.5 months; HR 0.56 [95% confidence interval 0.42 to 0.76]).45 Again, the survival benefits were more pronounced in patients with favorable or intermediate-risk cytogenetics than in those with unfavorable cytogenetics. The results of this trial led to the re-approval of GO in newly diagnosed AML patients.
For patients who cannot tolerate intensive therapies, the 2 main therapeutic options are low-dose cytarabine (LDAC) and the hypomethylating agents (HMA) azacitidine and decitabine. A phase 3 trial of decitabine versus mostly LDAC (or best supportive care, BSC) demonstrated favorable survival with decitabine (7.7 months versus 5.0 months).46 In the AZA-AML-001 trial, azacitidine improved median survival (10.4 months versus 6.5 months) in comparison to the control arm (LDAC, 7+3, BSC).47 Emerging data has also suggested that HMAs may be particularly active in patients with unfavorable-risk AML, a group for which LDAC has been shown to be especially useless.48 As such, HMA therapies are generally preferred over LDAC in practice. Finally, it is pertinent to note that GO can also be used as monotherapy based on the results of the open-label phase 3 AML-19 study in which GO demonstrated a survival advantage over BSC (4.9 months versus 3.6 months, P = 0.005).49
Postremission or Consolidation Therapy
There is no standard consolidation therapy for AML at present. In general, for patients who received HMA in the induction phase, the same HMA should be continued indefinitely until disease progression or allo-SCT.3 For those who received intensive chemotherapy in the induction phase, the consensus is to use cytarabine-based consolidation therapies. Cytarabine given as a single agent in high-doses has generally led to similar outcomes as multiagent chemotherapy.50 In this regard, cytarabine regimens, with or without anthracycline, at 3000 mg/m2 have similar efficacy as an intermediate dose of 1000 mg/m2.38 A total of 2 to 4 cycles of post-remission therapy is considered standard.3 Intensified post-remission chemotherapy has not been associated with consistent benefit in older AML patients or those with poor-risk disease. In recent years, measurable residual disease (MRD) assessment has emerged as a potentially useful tool in risk stratification and treatment planning, with various studies suggesting that MRD status in complete remission is one of the most important prognostic factors.51 Prospective studies confirming the significance of MRD as a marker for therapy selection are awaited. Finally, maintenance chemotherapy is not part of standard AML treatment.3
Role of Stem Cell Transplant
AML is the most common indication for allo-SCT. The availability of alternative donor strategies, which include mismatched, unrelated, haplo-identical, and cord blood donor sources, and the development of non-myeloablative and reduced-intensity conditioning (RIC) regimens (which take advantage of graft-versus-leukemia effect while decreasing cytotoxicity from myeloablative regimens) have expanded the possibility of allo-SCT to most patients under the age of 75 years.3 The decision to perform transplant is now largely based upon assessment of the risk (nonrelapse mortality) to benefit (reduction in risk of relapse) ratio, as determined by both disease-related features (cytogenetics, molecular profile) and clinical characteristics of the donor (type, availability, match) and the recipient (comorbidities, performance status).3 In a meta-analysis of 24 prospective trials involving more than 6000 AML patients in first complete remission, allo-SCT was associated with a significant survival benefit in patients with intermediate- and poor-risk AML but not in patients with good-risk AML.52 In line with this, good-risk AML patients are generally not recommended for transplant in first complete remission. For patients with normal karyotype who were said to have de novo AML (historically an intermediate-risk AML group), superior OS was demonstrated with transplant over intensive chemotherapy in those patients with either FLT3-ITD mutations or those with the molecular profile characterized by negativity for mutations in NPM1/CEBPA/FLT3.53 For patients with primary refractory disease and high-risk AML, transplant is probably the only curative option.
The choice of conditioning regimen is guided by several factors, including the subtype of AML, disease status, donor-recipient genetic disparity, graft source, comorbidities in the recipient (ie, tolerability for intensive conditioning regimen), as well as the reliance on graft-versus-leukemia effect as compared to cytotoxic effect of the regimen. The BMT CTN 0901 trial, which randomly assigned 218 patients aged 18 to 65 years to RIC (typically fludarabine/busulfan) or myeloablative regimens, showed an advantage for myeloablative regimens.54 The trial demonstrated a lower risk of relapse (13.5% versus 48.3%, P < 0.01) and higher rates of relapse-free survival (67.7% versus 47.3%, P < 0.01) and OS (67.7% versus. 77.4%, P = 0.07) at 18 months despite higher treatment-related mortality (15.8% versus 4.4%, P = 0.02) and a higher rate of grade 2 to 4 acute graft-versus-host disease (44.7% versus 31.6%, P = 0.024). At present, a RIC regimen is generally recommended for older patients or those with a higher comorbidity burden, while the myeloablative regimen is recommended for younger, fit patients.
Relapsed/Refractory Disease
The treatment of relapsed and refractory AML constitutes a major challenge, with OS estimated around 10% at 3 years.55 Currently, there is no standard salvage therapy in this setting, thus underscoring the need for clinical trials. For younger, fitter patients, the typical approach is to use intensive chemotherapy to achieve a second complete remission followed by a stem cell transplant. In younger patients, a second complete remission is achievable in about 55% of patients, although this rate is lower (~20%–30%) in more unselected patients.56,57 About two thirds of those who achieve complete remission may be able to proceed to transplant.57 For older patients where transplant is not possible, the goal is to use less intensive therapies that help with palliation. HMAs (azacitidine, decitabine) are used and have complete remission rates of 16% to 21% and median survival of 6 to 9 months in older patients.3 LDAC is another option in this setting. The recent approval of GO in this setting has further expanded the options. This approval was based on the outcomes of the phase 2 single-arm MyloFrance-1 study in which single-agent GO administered at 3 mg/m2 on days 1, 4, and 7 led to complete remission in 15 of 57 patients.58
With greater elucidation of the molecular characteristics of AML, the emergence of more effective targeted therapies is possible. Enasidenib, an inhibitor of mutant isocitrate dehydrogenase 2 (IDH2) protein that promotes differentiation of leukemic myeloblasts, recently received regulatory approval based on a single-arm trial. The overall response rate in this study was 38.5%, including a composite complete remission rate of 26.6% at a dose of 100 mg daily.59 IDH differentiation syndrome, akin to the differentiation syndrome seen in acute promyelocytic leukemia, occurred in approximately 12% of the patients, with the most frequent manifestations being dyspnea, fever, pulmonary infiltrates, and hypoxia.60
Survival of patients who relapse following transplant is particularly poor. A recent Center for International Blood and Marrow Transplant Research study found a 3-year OS ranging from a dismal 4% for those who present with early relapses (within 1 to 6 months) post-transplant to a more modest 38% for those who relapsed ≥ 3 years after their first transplant.61 The German Cooperative Transplant Study Group have suggested that azacitidine or chemotherapy followed by donor-lymphocyte infusions might improve responses over chemotherapy alone.62 Ipilimumab-based CTLA-4 blockade was reported to produce responses in a small cohort of patients, which was particularly notable in patients presenting with extramedullary manifestations of relapse.63 In patients who are otherwise fit but have a florid relapse, a second transplant can sometimes be sought, but the value of a different donor for second transplant is unclear.3
Case 1 Conclusion
Given his relatively young age, suitability for intensive therapy, and the presence of a core- binding factor abnormality, the patient is treated with an induction regimen containing daunorubicin, cytarabine, and GO (7+3 + GO). He achieves complete remission. This is followed by consolidation chemotherapy with high-dose cytarabine and GO. Allo-SCT is reserved for later should the AML relapse. Note that dasatinib, a c-KIT inhibitor, can be added to the treatment regimens as per the results of the CALGB 10801 protocol.64 Also, autologous SCT, instead of allo-SCT, can be considered in rare situations with relapsed core-binding factor AML (especially with inv(16) AML, younger patients, longer time in complete remission prior to relapse, and use of GO).
Case 2 Conclusion
The patient is deemed suitable for intensive chemotherapy. As such, CPX-351 is given in induction and consolidation and complete remission is achieved. Because he has adverse-risk AML, an allo-SCT is planned, but the patient relapses before it can be performed. Following 3 courses of decitabine therapy, the patient achieves complete remission once again but declines transplant. He maintains remission for an additional 4 months but then the leukemia progresses. Clinical trials are recommended to the patient, but he decides to pursue hospice care.
Conclusion
AML is the most common acute leukemia in adults. As defined currently, AML represents a group of related but distinct myeloid disorders that are characterized by various chromosomal, genetic, and epigenetic alterations. Early diagnosis and treatment can help prevent the emergence or manage the detrimental effects of its various complications such as leukostasis and tumor lysis syndrome. Improvements in supportive care, incremental treatment advances, and the wide adoption of allo-SCT for less than favorable cases have significantly improved survival of AML patients since the initial design of combinatorial (7+3) induction chemotherapy, particularly in patients presenting at a younger age. HMAs and the emergence of targeted therapies like FLT-3 and IDH2 inhibitors have added to our therapeutic armamentarium. Despite these advances, long-term survival rates in AML patients continue to be only approximately 40% to 50%. Older patients (particularly those over age 65 at the time of diagnosis), those with relapsed disease, and those with AML with certain unfavorable genetic abnormalities continue to have dismal outcomes. The design of newer targeted therapies, epigenetic agents, and immunotherapies will hopefully address this unmet need.
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–52.
2. National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts. Leukemia: Acute Myeloid Leukemia (AML). 2018;2018.
3. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47.
4. Liesveld JL, Lichtman MA. Acute myelogenous leukemia. In: Kaushansky K, Lichtman MA, Prchal JT, et al, eds. New York: Williams Hematology. 9th ed. New York: McGraw-Hill Education; 2015.
5. Randhawa JK, Khoury J, Ravandi-Kashani F. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 3rd ed. New York: McGraw-Hill Medical; 2016.
6. Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002;30:41–7.
7. Graubert TA, Mardis ER. Genomics of acute myeloid leukemia. Cancer J 2011;17:487–91.
8. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74.
9. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21.
10. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015;125:1367–76.
11. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98.
12. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16.
13. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–21.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87.
15. Pileri S, Ascani S, Cox M, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia 2007;21:340–50.
16. Vachhani P, Bose P. Isolated gastric myeloid sarcoma: a case report and review of the literature. Case Rep Hematol 2014;2014:541807.
17. Rowe JM. Clinical and laboratory features of the myeloid and lymphocytic leukemias. Am J Med Technol 1983;49:103–9.
18. Wolach O, Stone RM. Mixed-phenotype acute leukemia: current challenges in diagnosis and therapy. Curr Opin Hematol 2017;24:139–45.
19. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
20. Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol 2018;93:553–63.
21. Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89.
22. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33.
23. Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 2012;119:34–43.
24. Cairoli R, Beghini A, Grillo G, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 2006;107:3463–8.
25. Sorror ML, Storer BE, Elsawy M, et al. Intensive versus non-intensive induction therapy for patients (Pts) with newly diagnosed acute myeloid leukemia (AML) using two different novel prognostic models [abstract]. Blood 2016;128(22):216.
26. Quintás-Cardama A, Ravandi F, Liu-Dumlao T, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood 2012;120;4840-5.
27. Gupta N, Miller A, Gandhi Set al. Comparison of epigenetic versus standard induction chemotherapy for newly diagnosed acute myeloid leukemia patients ≥60 years old.Am J Hematol 2015;90:639-46.
28. Sorror ML, Storer BE, Fathi AT, et al. Development and validation of a novel acute myeloid leukemia-composite model to estimate risks of mortality. JAMA Oncol 2017;3:1675–82.
29. Rowe JM, Neuberg D, Friedenberg W, et al. A phase 3 study of three induction regimens and of priming with GM-CSF in older adults with acute myeloid leukemia: a trial by the Eastern Cooperative Oncology Group. Blood 2004;103:479–85.
30. Mandelli F, Vignetti M, Suciu S, et al. Daunorubicin versus mitoxantrone versus idarubicin as induction and consolidation chemotherapy for adults with acute myeloid leukemia: the EORTC and GIMEMA Groups Study AML-10. J Clin Oncol 2009;27:5397–403.
31. Gardin C, Chevret S, Pautas C, et al. Superior long-term outcome with idarubicin compared with high-dose daunorubicin in patients with acute myeloid leukemia age 50 years and older. J Clin Oncol 2013;31:321–7.
32. Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009;361:1249–59.
33. Lee JH, Joo YD, Kim H, et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood 2011;118:3832–41.
34. Lowenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009;361:1235–48.
35. Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood 2015;125:3878–85.
36. Devillier R, Bertoli S, Prebet T, et al. Comparison of 60 or 90 mg/m(2) of daunorubicin in induction therapy for acute myeloid leukemia with intermediate or unfavorable cytogenetics. Am J Hematol 2015;90:E29–30.
37. Pautas C, Merabet F, Thomas X, et al. Randomized study of intensified anthracycline doses for induction and recombinant interleukin-2 for maintenance in patients with acute myeloid leukemia age 50 to 70 years: results of the ALFA-9801 study. J Clin Oncol 2010;28:808–14.
38. Lowenberg B. Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 2013;121:26–8.
39. Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7 + 3 in older patients with newly diagnosed high risk (secondary) AML [abstract]. J Clin Oncol 2016;34(15_suppl):7000-7000.
40. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64.
41. Jen EY, Ko CW, Lee JE, et al. FDA approval: Gemtuzumab ozogamicin for the treatment of adults with newly-diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res 2018; doi: 10.1158/1078-0432. CCR-17-3179.
42. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol 2001;19:3244–54.
43. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013;121:4854–60.
44. Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol 2012;30:3924–31.
45. Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012;379:1508–16.
46. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291–9.
48. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 2016;375:2023–36.
49. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol 2016;34:972–9.
50. Miyawaki S, Ohtake S, Fujisawa S, et al. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood 2011;117:2366–72.
51. Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood 2018;131:1275–91.
52. Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301:2349–61.
53. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008;358:1909–18.
54. Pasquini MC, Logan B, Wu J, et al. Results of a phase III randomized, multi-center study of allogeneic stem cell transplantation after high versus reduced intensity conditioning in patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML): Blood and Marrow Transplant Clinical Trials Network (BMT CTN) 0901. Blood 2015;126:LBA–8.
55. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol 2017;18:17,017-0456-2.
56. Burnett AK, Goldstone A, Hills RK, et al. Curability of patients with acute myeloid leukemia who did not undergo transplantation in first remission. J Clin Oncol 2013;31:1293–301.
57. Ravandi F, Ritchie EK, Sayar H, et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): a randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol 2015;16:1025–36.
58. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 2007;21:66–71.
59. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–31.
60. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol 2018;doi: 10.1001/jamaoncol.2017.4695.
61. Bejanyan N, Weisdorf DJ, Logan BR, et al. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant 2015;21:454–9.
62. Schroeder T, Rachlis E, Bug G, et al. Treatment of acute myeloid leukemia or myelodysplastic syndrome relapse after allogeneic stem cell transplantation with azacitidine and donor lymphocyte infusions--a retrospective multicenter analysis from the German Cooperative Transplant Study Group. Biol Blood Marrow Transplant 2015;21:653–60.
63. Davids MS, Kim HT, Bachireddy P, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med 2016;375:143–53.
64. Marcucci G, Geyer S, Zhao W, et al. Adding KIT inhibitor dasatinib (DAS) to chemotherapy overcomes the negative impact of KIT mutation/over-expression in core binding factor (CBF) acute myeloid leukemia (AML): results from CALGB 10801 (Alliance) [abstract]. Blood 2014;124:8.
Introduction
Acute myeloid leukemia (AML) comprises a heterogeneous group of disorders characterized by proliferation of clonal, abnormally differentiated hematopoietic progenitor cells of myeloid lineage that infiltrate the bone marrow, blood, and other tissues.1 In most cases, AML is rapidly fatal if left untreated. Over the past 2 decades, our understanding of the underlying disease biology responsible for the development of AML has improved substantially. We have learned that biological differences drive the various clinical, cytogenetic, and molecular subentities of AML; distinguishing among these subentities helps to identify optimal therapies, while offering improved clinical outcomes for select groups. After years of stagnation in therapeutic advances, 4 new drugs for treating AML were approved by the US Food and Drug Administration (FDA) in 2017. In this article, we review key features of AML diagnosis and management in the context of 2 case presentations.
Epidemiology and Risk Factors
An estimated 21,380 new cases of AML were diagnosed in the United States in 2017, constituting roughly 1.3% of all new cases of cancer.2 Approximately 10,590 patients died of AML in 2017. The median age of patients at the time of diagnosis is 68 years, and the incidence is approximately 4.2 per 100,000 persons per year. The 5-year survival for AML has steadily risen from a meager 6.3% in 1975 to 17.3% in 1995 and 28.1% in 2009.2 The cure rates for AML vary drastically with age. Long-term survival is achieved in approximately 35% to 40% of adults who present at age 60 years or younger, but only 5% to 15% of those older than 60 years at presentation will achieve long-term survival.3
Most cases of AML occur in the absence of any known risk factors. High-dose radiation exposure, chronic benzene exposure, chronic tobacco smoking, and certain chemotherapeutics are known to increase the risk for AML.4 Inconsistent correlations have also been made between exposure to organic solvents, petroleum products, radon, pesticides, and herbicides and the development of AML.4 Obesity may also increase AML risk.4
Two distinct subcategories of therapy-related AML (t-AML) are known. Patients who have been exposed to alkylating chemotherapeutics (eg, melphalan, cyclophosphamide, and nitrogen mustard) can develop t-AML with chromosomal 5 and/or 7 abnormalities after a latency period of approximately 4 to 8 years.5 In contrast, patients exposed to topoisomerase II inhibitors (notably etoposide) develop AML with abnormalities of 11q23 (leading to MLL gene rearrangement) or 21q22 (RUNX1) after a latency period of about 1 to 3 years.6 AML can also arise out of other myeloid disorders such as myelodysplastic syndrome and myeloproliferative neoplasms, and other bone marrow failure syndromes such as aplastic anemia.4 Various inherited or congenital conditions such as Down syndrome, Bloom syndrome, Fanconi anemia, neurofibromatosis 1, and dyskeratosis congenita can also predispose to the development of AML. A more detailed listing of conditions associated with AML can be found elsewhere.4
Molecular Landscape
The first cancer genome sequence was reported in an AML patient in 2008.7 Since then, various elegantly conducted studies have expanded our understanding of the molecular abnormalities in AML. The Cancer Genome Atlas Research Network analyzed the genomes of 200 cases of de novo AML in adults.8 Only 13 mutations were found on average, much fewer than the number of mutations in most adult cancers. Twenty-three genes were commonly mutated, and another 237 were mutated in 2 or more cases. Essentially, all cases had at least 1 nonsynonymous mutation in 1 of 9 categories of genes: transcription-factor fusions (18%), the gene encoding nucleophosmin (NPM1) (27%), tumor-suppressor genes (16%), DNA-methylation–related genes (44%), signaling genes (59%), chromatin-modifying genes (30%), myeloid transcription-factor genes (22%), spliceosome-complex genes (14%), and cohesin-complex genes (13%).
In another study, samples from 1540 patients from 3 prospective trials of intensive chemotherapy were analyzed to understand how genetic diversity defines the pathophysiology of AML.9 The study authors identified 5234 driver mutations from 76 genes or genomic regions, with 2 or more drivers identified in 86% of the samples. Eleven classes of mutational events, each with distinct diagnostic features and clinical outcomes, were identified. Acting as an internal positive control in this analysis, previously recognized mutational and cytogenetic groups emerged as distinct entities, including the groups with biallelic CEBPA mutations, mutations in NPM1, MLL fusions, and the cytogenetic entities t(6;9), inv(3), t(8;21), t(15;17), and inv(16). Three additional categories emerged as distinct entities: AML with mutations in genes encoding chromatin, RNA splicing regulators, or both (18% of patients); AML with TP53 mutations, chromosomal aneuploidies, or both (13%); and, provisionally, AML with IDH2R172 mutations (1%). An additional level of complexity was also revealed within the subgroup of patients with NPM1 mutations, where gene–gene interactions identified co-mutational events associated with both favorable or adverse prognosis.
Further supporting this molecular classification of AML, a study that performed targeted mutational analysis of 194 patients with defined secondary AML (s-AML) or t-AML and 105 unselected AML patients found that the presence of mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 (all members of the chromatin or RNA splicing families) was highly specific for the diagnosis of s-AML.10 These findings are particularly clinically useful in those without a known history of antecedent hematologic disorder. These mutations defining the AML ontogeny were found to occur early in leukemogenesis, persist in clonal remissions, and predict worse clinical outcomes. Mutations in genes involved in regulation of DNA modification and of chromatin state (commonly DNMT3A, ASXL1, and TET2) have also been shown to be present in preleukemic stem or progenitor cells and to occur early in leukemogenesis.3 Unsurprisingly, some of these same mutations, including those in epigenetic regulators (DNMT3A, ASXL1, and TET2) and less frequently in splicing factor genes (SF3B1, SRSF2), have been associated with clonal hematopoietic expansion in elderly, seemingly healthy adults, a condition termed clonal hematopoiesis of indeterminate potential (CHIP).3,11,12 The presence of CHIP is associated with increased risk of hematologic neoplasms and all-cause mortality, the latter being possibly driven by a near doubling in the risk of coronary heart disease in humans and by accelerated atherosclerosis in a mouse model.11,13,14
Clinical Presentation and Work-up
Case Patient 1
A 57-year-old woman with a history of hypertension presents to the emergency department with complaints of productive cough and fevers for the previous 3 days. Examination reveals conjunctival pallor, gingival hyperplasia, and decreased breath sounds at the posterior right lung field. Investigations reveal a white blood cell (WBC) count of 51,000/µL with 15% blasts, a hemoglobin of 7.8 g/dL, and a platelet count of 56 × 103/µL. Peripheral blood smear is notable for large myeloblasts with occasional Auer rods. Chest radiograph shows a consolidation in the right lower lobe.
Case Patient 2
A 69-year-old man presents to his primary care physician for evaluation of worsening fatigue for the previous 4 months. Ten years prior to presentation, he had received 6 cycles of RCHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) as treatment for diffuse large B-cell lymphoma. Conjunctival pallor, patches of purpura over the extremities, and mucosal petechiae are noted on examination. Laboratory analyisis reveals a WBC count of 2400/µL with 12% blasts, hemoglobin of 9.0 g/dL, and platelet count of 10 × 103/µL. Peripheral smear shows dysplastic myeloid cells and blasts.
Clinical Features
Patients with AML typically present with features secondary to proliferation of blasts (ie, findings of bone marrow failure and end organ damage).4,5 Fatigue, pallor, dizziness, dyspnea, and headaches occur secondary to anemia. Easy and prolonged bruising, petechiae, epistaxis, gingival bleeding, and conjunctival hemorrhages result from thrombocytopenia. Bleeding from other sites such as the central nervous system and gastrointestinal tract occurs but is uncommon. Patients may also present with infections resulting from unrecognized neutropenia. Constitutional symptoms including anorexia, fevers, and weight loss are frequently reported, while organomegaly (hepatomegaly and/or splenomegaly) is seen in about a quarter of patients.4 Infiltration of blasts into almost every organ has been noted, a condition known as myeloid (or granulocytic) sarcoma.15 This condition is more commonly found in patients with blastic, monoblastic, or myelomonocytic variants of AML, and is known as isolated myeloid sarcoma if no concurrent marrow or blood involvement is identified. In the absence of induction chemotherapy, systemic involvement occurs in a matter of weeks to months following such presentation.16
Laboratory analysis will usually demonstrate derangements in peripheral blood cell lines. At least half of patients have a total WBC count less than 5000/µL, a platelet count less than 50 × 103/µL, or both at the time of diagnosis.4,17 Approximately 10% of patients present with hyperleukocytosis and a WBC count greater than 100,000/µL, which can be associated with leukostasis.5 Additionally, spontaneous electrolyte derangement consistent with tumor lysis syndrome and coagulation abnormalities found in disseminated intravascular coagulation may be noted, even before initiation of therapy.
Work-Up of Suspected AML
Bone marrow biopsy and aspirate, along with touch preparations of the core biopsy sample, are crucial in the workup of suspected AML. At least 200 WBCs on blood smears and 500 nucleated cells on spiculated marrow smears should be counted.3 Reactivity with specific histochemical stains (myeloperoxidase, Sudan black B, or naphthyl AS-D-chloroacetate), presence of Auer rods, and reactivity to monoclonal antibodies against epitopes present on myeloblasts (eg, CD13, CD33, CD117) help distinguish myeloblasts from lymphoblasts.4 Flow cytometric analysis helps in confirming myeloid lineage; blasts generally express CD34 and HLA-DR, markers of immature hematopoietic precursors, and dim CD45 (common leukocyte antigen). One or more lymphoid antigens may be aberrantly expressed as well. Of note, in about 2% to 3% of acute leukemia cases, immunohistochemistry and/or flow cytometry findings demonstrate immature cells with features of both myeloid and lymphoid lineages (biphenotypic) or different populations of myeloid and lymphoid leukemia cells (bilineal). These leukemias are termed mixed-phenotype acute leukemia and are typically treated with either AML or acute lymphoblastic leukemia regimens.18
Cytogenetics, as assessed through conventional karyotype and fluorescence in situ hybridization (FISH), constitutes an essential part of the work-up. Eight balanced translocations and inversions and their variants are included in the World Health Organization (WHO) category “AML with recurrent genetic abnormalities,” while 9 balanced rearrangements and multiple unbalanced abnormalities in the presence of a blast count ≥ 20% are sufficient to establish the diagnosis of “AML with myelodysplasia-related changes.”3,19 Various other gene rearrangements thought to represent disease-initiating events are recognized as well, but these rearrangements do not yet formally define WHO disease categories.3 FISH can help detect RUNX1-RUNX1T1, CBFB-MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, as well as chromosomal changes like 5q, 7q, or 17p, especially when fewer than 20 metaphases are assessable (due to failure of culture) by conventional cytogenetic methods.3
As certain molecular markers help with disease prognosis and the selection of personalized therapies, testing for these markers is recommended as part of a complete work-up of AML. The current standard of care is to test for nucleophosmin (NPM1), fms-like tyrosine kinase 3 (FLT3), and CEBPA mutations in all newly diagnosed patients.1RUNX1 mutation analysis should also be considered as its presence defines a provisional WHO subcategory.19 In the case of FLT3, the analysis should include both internal tandem duplications (FLT3-ITD, associated with worse prognosis especially at high allelic ratio) and tyrosine-kinase domain mutations (FLT3-TKD; D835 and I836), especially now that FLT3 inhibitors are regularly used.20 Most academic centers now routinely use next-generation sequencing–based panels to assess multiple mutations.
Diagnosis and Classification
A marrow or blood blast (myeloblasts, monoblasts, megakaryoblasts, or promonocytes [considered blast equivalents]) count of ≥ 20% is required for AML diagnosis.3,19 The presence of t(15;17), t(8;21), inv(16), or t(16;16), however, is considered diagnostic of AML irrespective of blast count.3,19 The previously used French-American-British (FAB) classification scheme has been replaced by the WHO classification (Table 2), which takes into account the morphologic, cytogenetic, genetic, and clinical features of the leukemia.
The category “AML with myelodysplasia-related changes” includes AML that has evolved out of an antecedent myelodysplastic syndrome, has ≥ 50% dysplasia in 2 or more lineages, or has myelodysplasia-related cytogenetic changes (eg, –5/del(5q), –7/del(7q), ≥ 3 cytogenetic abnormalities).19 “Therapy-related myeloid neoplasm,” or therapy-related AML, is diagnosed when the patient has previously received cytotoxic agents or ionizing radiation.19
Cases which do not meet the criteria for 1 of the previously mentioned categories are currently classified as “AML, not otherwise specified.” Further subclassification is pursued as per the older FAB scheme; however, no additional prognostic information is obtained in doing so.3,19 Myeloid sarcoma is strictly not a subcategory of AML. Rather, it is an extramedullary mass of myeloid blasts that effaces the normal tissue architecture.16 Rarely, myeloid sarcoma can be present without systemic disease involvement; it is important to note that management of such cases is identical to management of overt AML.16
Finally, myeloid proliferations related to Down syndrome include 2 entities seen in children with Down syndrome.19 Transient abnormal myelopoiesis, seen in 10% to 30% of newborns with Down syndrome, presents with circulating blasts that resolve in a couple of months. Myeloid leukemia associated with Down syndrome is AML that occurs usually in the first 3 years of life and persists if not treated.19
Case 1 Continued
The presence of 15% blasts in the peripheral blood is concerning for, but not diagnostic of, AML. On the other hand, the presence of Auer rods is virtually pathognomonic for AML. Gingival hyperplasia in this patient may be reflective of extramedullary disease. Cytogenetics from the peripheral blood and marrow aspirate show inv(16) in 20 of 20 cells. Molecular panel is notable for mutation in c-KIT. As such, the patient is diagnosed with core-binding factor AML, which per the ELN classification is considered a favorable-risk AML. The presence of c-KIT mutation, however, confers a relatively worse outcome.
Case 2 Continued
Presence of pancytopenia in a patient who previously received cytotoxic chemotherapy is highly concerning for therapy-related myeloid neoplasm. The presence of 12% blasts in the peripheral blood does not meet the criteria for diagnosis of AML. However, marrow specimens show 40% blasts, thus meeting the criteria for an AML diagnosis. Additionally, cytogenetics are notable for the presence of monosomy 7, while a next-generation sequencing panel shows a mutation in TP53. Put together, this patient meets the criteria for therapy-related AML which is an adverse-risk AML according to the ELN classification.
Management
The 2 most significant factors that must be considered when selecting AML therapies are the patient’s suitability for intensive chemotherapy and the biological characteristics of the AML. The former is a nuanced decision that incorporates age, performance status, and existing comorbidities. Treatment-related mortality calculators can guide physicians when making therapy decisions, especially in older patients (≥ 65 years). Retrospective evidence from various studies suggests that older, medically fit patients may derive clinically comparable benefits from intensive and less intensive induction therapies.25–27 The biological characteristics of the leukemia can be suggested by morphologic findings, cytogenetics, and molecular information, in addition to a history of antecedent myeloid neoplasms. Recently, an AML composite model incorporating an augmented Hematopoietic Cell Transplantation–specific Comorbidity Index (HCT-CI) score, age, and cytogenetic/molecular risks was shown to improve treatment decision-making about AML; this model potentially could be used to guide patient stratification in clinical trials as well.28 The overall treatment model of AML is largely unchanged otherwise. It is generally divided into induction, consolidation, and maintenance therapies.
Induction Therapy
In patients who can tolerate intensive therapies, the role of anthracycline- and cytarabine-based treatment is well established. However, the choice of specific anthracycline is not well established. One study concluded that idarubicin and mitoxantrone led to better outcomes as compared to daunorubicin, while another showed no difference between these agents.29,30 A pooled study of AML trials conducted in patients aged 50 years and older showed that while idarubicin led to a higher complete remission rate (69% versus 61%), the overall survival (OS) did not differ significantly.31 As for dosing, daunorubicin given at 45 mg/m2 daily for 3 days has been shown to have lower complete remission rates and higher relapse rates than a dose of 90 mg/m2 daily for 3 days in younger patients.32–34 However, it is not clear whether the 90 mg/m2 dose is superior to the frequently used dose of 60 mg/m2.35 A French study has shown comparable rates of complete remission, relapse, and OS between the 60 mg/m2 and 90 mg/m2 doses in patients with intermediate or unfavorable cytogenetics.36
If idarubicin is used, a dose of 12 mg/m2 for 3 days is considered the standard. In patients aged 50 to 70 years, there were no statistically significant differences in rates of relapse or OS between daunorubicin 80 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 4 days.37 As for cytarabine, the bulk of the evidence indicates that a dose of 1000 mg/m2 or higher should not be used.38 As such, the typical induction chemotherapy regimen of choice is 3 days of anthracycline (daunorubicin or idarubicin) and 7 days of cytarabine (100–200 mg/m2 continuous infusion), also known as the 7+3 regimen, which was first pioneered in the 1970s. In a recent phase 3 trial, 309 patients aged 60 to 75 years with high-risk AML (AML with myelodysplasia-related changes or t-AML) were randomly assigned to either the 7+3 regimen or CPX-351 (ie, nano-liposomal encapsulation of cytarabine and daunorubicin in a 5:1 molar ratio).39 A higher composite complete response rate (47.7% versus 33.3%; P = 0.016) and improved survival (9.56 months versus 5.95 months; hazard ratio [HR] 0.69, P = 0.005) were seen with CPX-351, leading to its approval by the FDA in patients with high-risk AML.
The 7+3 regimen has served as a backbone onto which other drugs have been added in clinical trials—the majority without any clinical benefits—for patients who can tolerate intensive therapy. In this context, the role of 2 therapies recently approved by the FDA must be discussed. In the RATIFY trial, 717 patients aged 18 to 59 years with AML and a FLT3 mutation were randomly assigned to receive standard chemotherapy (induction and consolidation therapy) plus either midostaurin or placebo; those who were in remission after consolidation therapy received either midostaurin or placebo in the maintenance phase.40 The primary endpoint was met as midostaurin improved OS (HR 0.78, P = 0.009). The benefit of midostaurin was consistent across all FLT3 subtypes and mutant allele burdens, regardless of whether patients proceeded to allogeneic stem cell transplant (allo-SCT). Based on the results of RATIFY, midostaurin was approved by the FDA for treatment of AML patients who are positive for the FLT3 mutation. Whether more potent and selective FLT3 inhibitors like gilteritinib, quizartinib, or crenolanib improve the outcomes is currently under investigation in various clinical trials.20
The development of gemtuzumab ozogamicin (GO) has been more complicated. GO, an antibody-drug conjugate comprised of a CD33-directed humanized monoclonal antibody linked covalently to the cytotoxic agent calicheamicin, binds CD33 present on the surface of myeloid leukemic blasts and immature normal cells of myelomonocytic lineage.41 The drug first received an accelerated approval in 2000 as monotherapy (2 doses of 9 mg/m2 14 days apart) for the treatment of patients 60 years of age and older with CD33-positive AML in first relapse based on the results of 3 open-label multicenter trials.41,42 However, a confirmatory S0106 trial in which GO 6 mg/m2 was added on day 4 in newly diagnosed AML patients was terminated early when an interim analysis showed an increased rate of death in induction (6% versus 1%) and lack of improvement in complete response, disease-free survival, or OS with the addition of GO.43 This study led to the withdrawal of GO from the US market in 2010. However, 2 randomized trials that studied GO using a different dose and schedule suggested that the addition of GO to intensive chemotherapy improved survival outcomes in patients with favorable and intermediate-risk cytogenetics.44,45 The results of the multicenter, open-label phase 3 ALFA-0701 trial, which randomly assigned 271 patients aged 50 to 70 years with newly diagnosed AML to daunorubicin and cytarabine alone or in combination with GO (3 mg/m2 on days 1, 4, and 7 during induction and day 1 of 2 consolidation courses), showed a statistically significant improvement in event-free survival (17.3 months versus 9.5 months; HR 0.56 [95% confidence interval 0.42 to 0.76]).45 Again, the survival benefits were more pronounced in patients with favorable or intermediate-risk cytogenetics than in those with unfavorable cytogenetics. The results of this trial led to the re-approval of GO in newly diagnosed AML patients.
For patients who cannot tolerate intensive therapies, the 2 main therapeutic options are low-dose cytarabine (LDAC) and the hypomethylating agents (HMA) azacitidine and decitabine. A phase 3 trial of decitabine versus mostly LDAC (or best supportive care, BSC) demonstrated favorable survival with decitabine (7.7 months versus 5.0 months).46 In the AZA-AML-001 trial, azacitidine improved median survival (10.4 months versus 6.5 months) in comparison to the control arm (LDAC, 7+3, BSC).47 Emerging data has also suggested that HMAs may be particularly active in patients with unfavorable-risk AML, a group for which LDAC has been shown to be especially useless.48 As such, HMA therapies are generally preferred over LDAC in practice. Finally, it is pertinent to note that GO can also be used as monotherapy based on the results of the open-label phase 3 AML-19 study in which GO demonstrated a survival advantage over BSC (4.9 months versus 3.6 months, P = 0.005).49
Postremission or Consolidation Therapy
There is no standard consolidation therapy for AML at present. In general, for patients who received HMA in the induction phase, the same HMA should be continued indefinitely until disease progression or allo-SCT.3 For those who received intensive chemotherapy in the induction phase, the consensus is to use cytarabine-based consolidation therapies. Cytarabine given as a single agent in high-doses has generally led to similar outcomes as multiagent chemotherapy.50 In this regard, cytarabine regimens, with or without anthracycline, at 3000 mg/m2 have similar efficacy as an intermediate dose of 1000 mg/m2.38 A total of 2 to 4 cycles of post-remission therapy is considered standard.3 Intensified post-remission chemotherapy has not been associated with consistent benefit in older AML patients or those with poor-risk disease. In recent years, measurable residual disease (MRD) assessment has emerged as a potentially useful tool in risk stratification and treatment planning, with various studies suggesting that MRD status in complete remission is one of the most important prognostic factors.51 Prospective studies confirming the significance of MRD as a marker for therapy selection are awaited. Finally, maintenance chemotherapy is not part of standard AML treatment.3
Role of Stem Cell Transplant
AML is the most common indication for allo-SCT. The availability of alternative donor strategies, which include mismatched, unrelated, haplo-identical, and cord blood donor sources, and the development of non-myeloablative and reduced-intensity conditioning (RIC) regimens (which take advantage of graft-versus-leukemia effect while decreasing cytotoxicity from myeloablative regimens) have expanded the possibility of allo-SCT to most patients under the age of 75 years.3 The decision to perform transplant is now largely based upon assessment of the risk (nonrelapse mortality) to benefit (reduction in risk of relapse) ratio, as determined by both disease-related features (cytogenetics, molecular profile) and clinical characteristics of the donor (type, availability, match) and the recipient (comorbidities, performance status).3 In a meta-analysis of 24 prospective trials involving more than 6000 AML patients in first complete remission, allo-SCT was associated with a significant survival benefit in patients with intermediate- and poor-risk AML but not in patients with good-risk AML.52 In line with this, good-risk AML patients are generally not recommended for transplant in first complete remission. For patients with normal karyotype who were said to have de novo AML (historically an intermediate-risk AML group), superior OS was demonstrated with transplant over intensive chemotherapy in those patients with either FLT3-ITD mutations or those with the molecular profile characterized by negativity for mutations in NPM1/CEBPA/FLT3.53 For patients with primary refractory disease and high-risk AML, transplant is probably the only curative option.
The choice of conditioning regimen is guided by several factors, including the subtype of AML, disease status, donor-recipient genetic disparity, graft source, comorbidities in the recipient (ie, tolerability for intensive conditioning regimen), as well as the reliance on graft-versus-leukemia effect as compared to cytotoxic effect of the regimen. The BMT CTN 0901 trial, which randomly assigned 218 patients aged 18 to 65 years to RIC (typically fludarabine/busulfan) or myeloablative regimens, showed an advantage for myeloablative regimens.54 The trial demonstrated a lower risk of relapse (13.5% versus 48.3%, P < 0.01) and higher rates of relapse-free survival (67.7% versus 47.3%, P < 0.01) and OS (67.7% versus. 77.4%, P = 0.07) at 18 months despite higher treatment-related mortality (15.8% versus 4.4%, P = 0.02) and a higher rate of grade 2 to 4 acute graft-versus-host disease (44.7% versus 31.6%, P = 0.024). At present, a RIC regimen is generally recommended for older patients or those with a higher comorbidity burden, while the myeloablative regimen is recommended for younger, fit patients.
Relapsed/Refractory Disease
The treatment of relapsed and refractory AML constitutes a major challenge, with OS estimated around 10% at 3 years.55 Currently, there is no standard salvage therapy in this setting, thus underscoring the need for clinical trials. For younger, fitter patients, the typical approach is to use intensive chemotherapy to achieve a second complete remission followed by a stem cell transplant. In younger patients, a second complete remission is achievable in about 55% of patients, although this rate is lower (~20%–30%) in more unselected patients.56,57 About two thirds of those who achieve complete remission may be able to proceed to transplant.57 For older patients where transplant is not possible, the goal is to use less intensive therapies that help with palliation. HMAs (azacitidine, decitabine) are used and have complete remission rates of 16% to 21% and median survival of 6 to 9 months in older patients.3 LDAC is another option in this setting. The recent approval of GO in this setting has further expanded the options. This approval was based on the outcomes of the phase 2 single-arm MyloFrance-1 study in which single-agent GO administered at 3 mg/m2 on days 1, 4, and 7 led to complete remission in 15 of 57 patients.58
With greater elucidation of the molecular characteristics of AML, the emergence of more effective targeted therapies is possible. Enasidenib, an inhibitor of mutant isocitrate dehydrogenase 2 (IDH2) protein that promotes differentiation of leukemic myeloblasts, recently received regulatory approval based on a single-arm trial. The overall response rate in this study was 38.5%, including a composite complete remission rate of 26.6% at a dose of 100 mg daily.59 IDH differentiation syndrome, akin to the differentiation syndrome seen in acute promyelocytic leukemia, occurred in approximately 12% of the patients, with the most frequent manifestations being dyspnea, fever, pulmonary infiltrates, and hypoxia.60
Survival of patients who relapse following transplant is particularly poor. A recent Center for International Blood and Marrow Transplant Research study found a 3-year OS ranging from a dismal 4% for those who present with early relapses (within 1 to 6 months) post-transplant to a more modest 38% for those who relapsed ≥ 3 years after their first transplant.61 The German Cooperative Transplant Study Group have suggested that azacitidine or chemotherapy followed by donor-lymphocyte infusions might improve responses over chemotherapy alone.62 Ipilimumab-based CTLA-4 blockade was reported to produce responses in a small cohort of patients, which was particularly notable in patients presenting with extramedullary manifestations of relapse.63 In patients who are otherwise fit but have a florid relapse, a second transplant can sometimes be sought, but the value of a different donor for second transplant is unclear.3
Case 1 Conclusion
Given his relatively young age, suitability for intensive therapy, and the presence of a core- binding factor abnormality, the patient is treated with an induction regimen containing daunorubicin, cytarabine, and GO (7+3 + GO). He achieves complete remission. This is followed by consolidation chemotherapy with high-dose cytarabine and GO. Allo-SCT is reserved for later should the AML relapse. Note that dasatinib, a c-KIT inhibitor, can be added to the treatment regimens as per the results of the CALGB 10801 protocol.64 Also, autologous SCT, instead of allo-SCT, can be considered in rare situations with relapsed core-binding factor AML (especially with inv(16) AML, younger patients, longer time in complete remission prior to relapse, and use of GO).
Case 2 Conclusion
The patient is deemed suitable for intensive chemotherapy. As such, CPX-351 is given in induction and consolidation and complete remission is achieved. Because he has adverse-risk AML, an allo-SCT is planned, but the patient relapses before it can be performed. Following 3 courses of decitabine therapy, the patient achieves complete remission once again but declines transplant. He maintains remission for an additional 4 months but then the leukemia progresses. Clinical trials are recommended to the patient, but he decides to pursue hospice care.
Conclusion
AML is the most common acute leukemia in adults. As defined currently, AML represents a group of related but distinct myeloid disorders that are characterized by various chromosomal, genetic, and epigenetic alterations. Early diagnosis and treatment can help prevent the emergence or manage the detrimental effects of its various complications such as leukostasis and tumor lysis syndrome. Improvements in supportive care, incremental treatment advances, and the wide adoption of allo-SCT for less than favorable cases have significantly improved survival of AML patients since the initial design of combinatorial (7+3) induction chemotherapy, particularly in patients presenting at a younger age. HMAs and the emergence of targeted therapies like FLT-3 and IDH2 inhibitors have added to our therapeutic armamentarium. Despite these advances, long-term survival rates in AML patients continue to be only approximately 40% to 50%. Older patients (particularly those over age 65 at the time of diagnosis), those with relapsed disease, and those with AML with certain unfavorable genetic abnormalities continue to have dismal outcomes. The design of newer targeted therapies, epigenetic agents, and immunotherapies will hopefully address this unmet need.
Introduction
Acute myeloid leukemia (AML) comprises a heterogeneous group of disorders characterized by proliferation of clonal, abnormally differentiated hematopoietic progenitor cells of myeloid lineage that infiltrate the bone marrow, blood, and other tissues.1 In most cases, AML is rapidly fatal if left untreated. Over the past 2 decades, our understanding of the underlying disease biology responsible for the development of AML has improved substantially. We have learned that biological differences drive the various clinical, cytogenetic, and molecular subentities of AML; distinguishing among these subentities helps to identify optimal therapies, while offering improved clinical outcomes for select groups. After years of stagnation in therapeutic advances, 4 new drugs for treating AML were approved by the US Food and Drug Administration (FDA) in 2017. In this article, we review key features of AML diagnosis and management in the context of 2 case presentations.
Epidemiology and Risk Factors
An estimated 21,380 new cases of AML were diagnosed in the United States in 2017, constituting roughly 1.3% of all new cases of cancer.2 Approximately 10,590 patients died of AML in 2017. The median age of patients at the time of diagnosis is 68 years, and the incidence is approximately 4.2 per 100,000 persons per year. The 5-year survival for AML has steadily risen from a meager 6.3% in 1975 to 17.3% in 1995 and 28.1% in 2009.2 The cure rates for AML vary drastically with age. Long-term survival is achieved in approximately 35% to 40% of adults who present at age 60 years or younger, but only 5% to 15% of those older than 60 years at presentation will achieve long-term survival.3
Most cases of AML occur in the absence of any known risk factors. High-dose radiation exposure, chronic benzene exposure, chronic tobacco smoking, and certain chemotherapeutics are known to increase the risk for AML.4 Inconsistent correlations have also been made between exposure to organic solvents, petroleum products, radon, pesticides, and herbicides and the development of AML.4 Obesity may also increase AML risk.4
Two distinct subcategories of therapy-related AML (t-AML) are known. Patients who have been exposed to alkylating chemotherapeutics (eg, melphalan, cyclophosphamide, and nitrogen mustard) can develop t-AML with chromosomal 5 and/or 7 abnormalities after a latency period of approximately 4 to 8 years.5 In contrast, patients exposed to topoisomerase II inhibitors (notably etoposide) develop AML with abnormalities of 11q23 (leading to MLL gene rearrangement) or 21q22 (RUNX1) after a latency period of about 1 to 3 years.6 AML can also arise out of other myeloid disorders such as myelodysplastic syndrome and myeloproliferative neoplasms, and other bone marrow failure syndromes such as aplastic anemia.4 Various inherited or congenital conditions such as Down syndrome, Bloom syndrome, Fanconi anemia, neurofibromatosis 1, and dyskeratosis congenita can also predispose to the development of AML. A more detailed listing of conditions associated with AML can be found elsewhere.4
Molecular Landscape
The first cancer genome sequence was reported in an AML patient in 2008.7 Since then, various elegantly conducted studies have expanded our understanding of the molecular abnormalities in AML. The Cancer Genome Atlas Research Network analyzed the genomes of 200 cases of de novo AML in adults.8 Only 13 mutations were found on average, much fewer than the number of mutations in most adult cancers. Twenty-three genes were commonly mutated, and another 237 were mutated in 2 or more cases. Essentially, all cases had at least 1 nonsynonymous mutation in 1 of 9 categories of genes: transcription-factor fusions (18%), the gene encoding nucleophosmin (NPM1) (27%), tumor-suppressor genes (16%), DNA-methylation–related genes (44%), signaling genes (59%), chromatin-modifying genes (30%), myeloid transcription-factor genes (22%), spliceosome-complex genes (14%), and cohesin-complex genes (13%).
In another study, samples from 1540 patients from 3 prospective trials of intensive chemotherapy were analyzed to understand how genetic diversity defines the pathophysiology of AML.9 The study authors identified 5234 driver mutations from 76 genes or genomic regions, with 2 or more drivers identified in 86% of the samples. Eleven classes of mutational events, each with distinct diagnostic features and clinical outcomes, were identified. Acting as an internal positive control in this analysis, previously recognized mutational and cytogenetic groups emerged as distinct entities, including the groups with biallelic CEBPA mutations, mutations in NPM1, MLL fusions, and the cytogenetic entities t(6;9), inv(3), t(8;21), t(15;17), and inv(16). Three additional categories emerged as distinct entities: AML with mutations in genes encoding chromatin, RNA splicing regulators, or both (18% of patients); AML with TP53 mutations, chromosomal aneuploidies, or both (13%); and, provisionally, AML with IDH2R172 mutations (1%). An additional level of complexity was also revealed within the subgroup of patients with NPM1 mutations, where gene–gene interactions identified co-mutational events associated with both favorable or adverse prognosis.
Further supporting this molecular classification of AML, a study that performed targeted mutational analysis of 194 patients with defined secondary AML (s-AML) or t-AML and 105 unselected AML patients found that the presence of mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 (all members of the chromatin or RNA splicing families) was highly specific for the diagnosis of s-AML.10 These findings are particularly clinically useful in those without a known history of antecedent hematologic disorder. These mutations defining the AML ontogeny were found to occur early in leukemogenesis, persist in clonal remissions, and predict worse clinical outcomes. Mutations in genes involved in regulation of DNA modification and of chromatin state (commonly DNMT3A, ASXL1, and TET2) have also been shown to be present in preleukemic stem or progenitor cells and to occur early in leukemogenesis.3 Unsurprisingly, some of these same mutations, including those in epigenetic regulators (DNMT3A, ASXL1, and TET2) and less frequently in splicing factor genes (SF3B1, SRSF2), have been associated with clonal hematopoietic expansion in elderly, seemingly healthy adults, a condition termed clonal hematopoiesis of indeterminate potential (CHIP).3,11,12 The presence of CHIP is associated with increased risk of hematologic neoplasms and all-cause mortality, the latter being possibly driven by a near doubling in the risk of coronary heart disease in humans and by accelerated atherosclerosis in a mouse model.11,13,14
Clinical Presentation and Work-up
Case Patient 1
A 57-year-old woman with a history of hypertension presents to the emergency department with complaints of productive cough and fevers for the previous 3 days. Examination reveals conjunctival pallor, gingival hyperplasia, and decreased breath sounds at the posterior right lung field. Investigations reveal a white blood cell (WBC) count of 51,000/µL with 15% blasts, a hemoglobin of 7.8 g/dL, and a platelet count of 56 × 103/µL. Peripheral blood smear is notable for large myeloblasts with occasional Auer rods. Chest radiograph shows a consolidation in the right lower lobe.
Case Patient 2
A 69-year-old man presents to his primary care physician for evaluation of worsening fatigue for the previous 4 months. Ten years prior to presentation, he had received 6 cycles of RCHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) as treatment for diffuse large B-cell lymphoma. Conjunctival pallor, patches of purpura over the extremities, and mucosal petechiae are noted on examination. Laboratory analyisis reveals a WBC count of 2400/µL with 12% blasts, hemoglobin of 9.0 g/dL, and platelet count of 10 × 103/µL. Peripheral smear shows dysplastic myeloid cells and blasts.
Clinical Features
Patients with AML typically present with features secondary to proliferation of blasts (ie, findings of bone marrow failure and end organ damage).4,5 Fatigue, pallor, dizziness, dyspnea, and headaches occur secondary to anemia. Easy and prolonged bruising, petechiae, epistaxis, gingival bleeding, and conjunctival hemorrhages result from thrombocytopenia. Bleeding from other sites such as the central nervous system and gastrointestinal tract occurs but is uncommon. Patients may also present with infections resulting from unrecognized neutropenia. Constitutional symptoms including anorexia, fevers, and weight loss are frequently reported, while organomegaly (hepatomegaly and/or splenomegaly) is seen in about a quarter of patients.4 Infiltration of blasts into almost every organ has been noted, a condition known as myeloid (or granulocytic) sarcoma.15 This condition is more commonly found in patients with blastic, monoblastic, or myelomonocytic variants of AML, and is known as isolated myeloid sarcoma if no concurrent marrow or blood involvement is identified. In the absence of induction chemotherapy, systemic involvement occurs in a matter of weeks to months following such presentation.16
Laboratory analysis will usually demonstrate derangements in peripheral blood cell lines. At least half of patients have a total WBC count less than 5000/µL, a platelet count less than 50 × 103/µL, or both at the time of diagnosis.4,17 Approximately 10% of patients present with hyperleukocytosis and a WBC count greater than 100,000/µL, which can be associated with leukostasis.5 Additionally, spontaneous electrolyte derangement consistent with tumor lysis syndrome and coagulation abnormalities found in disseminated intravascular coagulation may be noted, even before initiation of therapy.
Work-Up of Suspected AML
Bone marrow biopsy and aspirate, along with touch preparations of the core biopsy sample, are crucial in the workup of suspected AML. At least 200 WBCs on blood smears and 500 nucleated cells on spiculated marrow smears should be counted.3 Reactivity with specific histochemical stains (myeloperoxidase, Sudan black B, or naphthyl AS-D-chloroacetate), presence of Auer rods, and reactivity to monoclonal antibodies against epitopes present on myeloblasts (eg, CD13, CD33, CD117) help distinguish myeloblasts from lymphoblasts.4 Flow cytometric analysis helps in confirming myeloid lineage; blasts generally express CD34 and HLA-DR, markers of immature hematopoietic precursors, and dim CD45 (common leukocyte antigen). One or more lymphoid antigens may be aberrantly expressed as well. Of note, in about 2% to 3% of acute leukemia cases, immunohistochemistry and/or flow cytometry findings demonstrate immature cells with features of both myeloid and lymphoid lineages (biphenotypic) or different populations of myeloid and lymphoid leukemia cells (bilineal). These leukemias are termed mixed-phenotype acute leukemia and are typically treated with either AML or acute lymphoblastic leukemia regimens.18
Cytogenetics, as assessed through conventional karyotype and fluorescence in situ hybridization (FISH), constitutes an essential part of the work-up. Eight balanced translocations and inversions and their variants are included in the World Health Organization (WHO) category “AML with recurrent genetic abnormalities,” while 9 balanced rearrangements and multiple unbalanced abnormalities in the presence of a blast count ≥ 20% are sufficient to establish the diagnosis of “AML with myelodysplasia-related changes.”3,19 Various other gene rearrangements thought to represent disease-initiating events are recognized as well, but these rearrangements do not yet formally define WHO disease categories.3 FISH can help detect RUNX1-RUNX1T1, CBFB-MYH11, KMT2A (MLL), and MECOM (EVI1) gene fusions, as well as chromosomal changes like 5q, 7q, or 17p, especially when fewer than 20 metaphases are assessable (due to failure of culture) by conventional cytogenetic methods.3
As certain molecular markers help with disease prognosis and the selection of personalized therapies, testing for these markers is recommended as part of a complete work-up of AML. The current standard of care is to test for nucleophosmin (NPM1), fms-like tyrosine kinase 3 (FLT3), and CEBPA mutations in all newly diagnosed patients.1RUNX1 mutation analysis should also be considered as its presence defines a provisional WHO subcategory.19 In the case of FLT3, the analysis should include both internal tandem duplications (FLT3-ITD, associated with worse prognosis especially at high allelic ratio) and tyrosine-kinase domain mutations (FLT3-TKD; D835 and I836), especially now that FLT3 inhibitors are regularly used.20 Most academic centers now routinely use next-generation sequencing–based panels to assess multiple mutations.
Diagnosis and Classification
A marrow or blood blast (myeloblasts, monoblasts, megakaryoblasts, or promonocytes [considered blast equivalents]) count of ≥ 20% is required for AML diagnosis.3,19 The presence of t(15;17), t(8;21), inv(16), or t(16;16), however, is considered diagnostic of AML irrespective of blast count.3,19 The previously used French-American-British (FAB) classification scheme has been replaced by the WHO classification (Table 2), which takes into account the morphologic, cytogenetic, genetic, and clinical features of the leukemia.
The category “AML with myelodysplasia-related changes” includes AML that has evolved out of an antecedent myelodysplastic syndrome, has ≥ 50% dysplasia in 2 or more lineages, or has myelodysplasia-related cytogenetic changes (eg, –5/del(5q), –7/del(7q), ≥ 3 cytogenetic abnormalities).19 “Therapy-related myeloid neoplasm,” or therapy-related AML, is diagnosed when the patient has previously received cytotoxic agents or ionizing radiation.19
Cases which do not meet the criteria for 1 of the previously mentioned categories are currently classified as “AML, not otherwise specified.” Further subclassification is pursued as per the older FAB scheme; however, no additional prognostic information is obtained in doing so.3,19 Myeloid sarcoma is strictly not a subcategory of AML. Rather, it is an extramedullary mass of myeloid blasts that effaces the normal tissue architecture.16 Rarely, myeloid sarcoma can be present without systemic disease involvement; it is important to note that management of such cases is identical to management of overt AML.16
Finally, myeloid proliferations related to Down syndrome include 2 entities seen in children with Down syndrome.19 Transient abnormal myelopoiesis, seen in 10% to 30% of newborns with Down syndrome, presents with circulating blasts that resolve in a couple of months. Myeloid leukemia associated with Down syndrome is AML that occurs usually in the first 3 years of life and persists if not treated.19
Case 1 Continued
The presence of 15% blasts in the peripheral blood is concerning for, but not diagnostic of, AML. On the other hand, the presence of Auer rods is virtually pathognomonic for AML. Gingival hyperplasia in this patient may be reflective of extramedullary disease. Cytogenetics from the peripheral blood and marrow aspirate show inv(16) in 20 of 20 cells. Molecular panel is notable for mutation in c-KIT. As such, the patient is diagnosed with core-binding factor AML, which per the ELN classification is considered a favorable-risk AML. The presence of c-KIT mutation, however, confers a relatively worse outcome.
Case 2 Continued
Presence of pancytopenia in a patient who previously received cytotoxic chemotherapy is highly concerning for therapy-related myeloid neoplasm. The presence of 12% blasts in the peripheral blood does not meet the criteria for diagnosis of AML. However, marrow specimens show 40% blasts, thus meeting the criteria for an AML diagnosis. Additionally, cytogenetics are notable for the presence of monosomy 7, while a next-generation sequencing panel shows a mutation in TP53. Put together, this patient meets the criteria for therapy-related AML which is an adverse-risk AML according to the ELN classification.
Management
The 2 most significant factors that must be considered when selecting AML therapies are the patient’s suitability for intensive chemotherapy and the biological characteristics of the AML. The former is a nuanced decision that incorporates age, performance status, and existing comorbidities. Treatment-related mortality calculators can guide physicians when making therapy decisions, especially in older patients (≥ 65 years). Retrospective evidence from various studies suggests that older, medically fit patients may derive clinically comparable benefits from intensive and less intensive induction therapies.25–27 The biological characteristics of the leukemia can be suggested by morphologic findings, cytogenetics, and molecular information, in addition to a history of antecedent myeloid neoplasms. Recently, an AML composite model incorporating an augmented Hematopoietic Cell Transplantation–specific Comorbidity Index (HCT-CI) score, age, and cytogenetic/molecular risks was shown to improve treatment decision-making about AML; this model potentially could be used to guide patient stratification in clinical trials as well.28 The overall treatment model of AML is largely unchanged otherwise. It is generally divided into induction, consolidation, and maintenance therapies.
Induction Therapy
In patients who can tolerate intensive therapies, the role of anthracycline- and cytarabine-based treatment is well established. However, the choice of specific anthracycline is not well established. One study concluded that idarubicin and mitoxantrone led to better outcomes as compared to daunorubicin, while another showed no difference between these agents.29,30 A pooled study of AML trials conducted in patients aged 50 years and older showed that while idarubicin led to a higher complete remission rate (69% versus 61%), the overall survival (OS) did not differ significantly.31 As for dosing, daunorubicin given at 45 mg/m2 daily for 3 days has been shown to have lower complete remission rates and higher relapse rates than a dose of 90 mg/m2 daily for 3 days in younger patients.32–34 However, it is not clear whether the 90 mg/m2 dose is superior to the frequently used dose of 60 mg/m2.35 A French study has shown comparable rates of complete remission, relapse, and OS between the 60 mg/m2 and 90 mg/m2 doses in patients with intermediate or unfavorable cytogenetics.36
If idarubicin is used, a dose of 12 mg/m2 for 3 days is considered the standard. In patients aged 50 to 70 years, there were no statistically significant differences in rates of relapse or OS between daunorubicin 80 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 3 days versus idarubicin 12 mg/m2 for 4 days.37 As for cytarabine, the bulk of the evidence indicates that a dose of 1000 mg/m2 or higher should not be used.38 As such, the typical induction chemotherapy regimen of choice is 3 days of anthracycline (daunorubicin or idarubicin) and 7 days of cytarabine (100–200 mg/m2 continuous infusion), also known as the 7+3 regimen, which was first pioneered in the 1970s. In a recent phase 3 trial, 309 patients aged 60 to 75 years with high-risk AML (AML with myelodysplasia-related changes or t-AML) were randomly assigned to either the 7+3 regimen or CPX-351 (ie, nano-liposomal encapsulation of cytarabine and daunorubicin in a 5:1 molar ratio).39 A higher composite complete response rate (47.7% versus 33.3%; P = 0.016) and improved survival (9.56 months versus 5.95 months; hazard ratio [HR] 0.69, P = 0.005) were seen with CPX-351, leading to its approval by the FDA in patients with high-risk AML.
The 7+3 regimen has served as a backbone onto which other drugs have been added in clinical trials—the majority without any clinical benefits—for patients who can tolerate intensive therapy. In this context, the role of 2 therapies recently approved by the FDA must be discussed. In the RATIFY trial, 717 patients aged 18 to 59 years with AML and a FLT3 mutation were randomly assigned to receive standard chemotherapy (induction and consolidation therapy) plus either midostaurin or placebo; those who were in remission after consolidation therapy received either midostaurin or placebo in the maintenance phase.40 The primary endpoint was met as midostaurin improved OS (HR 0.78, P = 0.009). The benefit of midostaurin was consistent across all FLT3 subtypes and mutant allele burdens, regardless of whether patients proceeded to allogeneic stem cell transplant (allo-SCT). Based on the results of RATIFY, midostaurin was approved by the FDA for treatment of AML patients who are positive for the FLT3 mutation. Whether more potent and selective FLT3 inhibitors like gilteritinib, quizartinib, or crenolanib improve the outcomes is currently under investigation in various clinical trials.20
The development of gemtuzumab ozogamicin (GO) has been more complicated. GO, an antibody-drug conjugate comprised of a CD33-directed humanized monoclonal antibody linked covalently to the cytotoxic agent calicheamicin, binds CD33 present on the surface of myeloid leukemic blasts and immature normal cells of myelomonocytic lineage.41 The drug first received an accelerated approval in 2000 as monotherapy (2 doses of 9 mg/m2 14 days apart) for the treatment of patients 60 years of age and older with CD33-positive AML in first relapse based on the results of 3 open-label multicenter trials.41,42 However, a confirmatory S0106 trial in which GO 6 mg/m2 was added on day 4 in newly diagnosed AML patients was terminated early when an interim analysis showed an increased rate of death in induction (6% versus 1%) and lack of improvement in complete response, disease-free survival, or OS with the addition of GO.43 This study led to the withdrawal of GO from the US market in 2010. However, 2 randomized trials that studied GO using a different dose and schedule suggested that the addition of GO to intensive chemotherapy improved survival outcomes in patients with favorable and intermediate-risk cytogenetics.44,45 The results of the multicenter, open-label phase 3 ALFA-0701 trial, which randomly assigned 271 patients aged 50 to 70 years with newly diagnosed AML to daunorubicin and cytarabine alone or in combination with GO (3 mg/m2 on days 1, 4, and 7 during induction and day 1 of 2 consolidation courses), showed a statistically significant improvement in event-free survival (17.3 months versus 9.5 months; HR 0.56 [95% confidence interval 0.42 to 0.76]).45 Again, the survival benefits were more pronounced in patients with favorable or intermediate-risk cytogenetics than in those with unfavorable cytogenetics. The results of this trial led to the re-approval of GO in newly diagnosed AML patients.
For patients who cannot tolerate intensive therapies, the 2 main therapeutic options are low-dose cytarabine (LDAC) and the hypomethylating agents (HMA) azacitidine and decitabine. A phase 3 trial of decitabine versus mostly LDAC (or best supportive care, BSC) demonstrated favorable survival with decitabine (7.7 months versus 5.0 months).46 In the AZA-AML-001 trial, azacitidine improved median survival (10.4 months versus 6.5 months) in comparison to the control arm (LDAC, 7+3, BSC).47 Emerging data has also suggested that HMAs may be particularly active in patients with unfavorable-risk AML, a group for which LDAC has been shown to be especially useless.48 As such, HMA therapies are generally preferred over LDAC in practice. Finally, it is pertinent to note that GO can also be used as monotherapy based on the results of the open-label phase 3 AML-19 study in which GO demonstrated a survival advantage over BSC (4.9 months versus 3.6 months, P = 0.005).49
Postremission or Consolidation Therapy
There is no standard consolidation therapy for AML at present. In general, for patients who received HMA in the induction phase, the same HMA should be continued indefinitely until disease progression or allo-SCT.3 For those who received intensive chemotherapy in the induction phase, the consensus is to use cytarabine-based consolidation therapies. Cytarabine given as a single agent in high-doses has generally led to similar outcomes as multiagent chemotherapy.50 In this regard, cytarabine regimens, with or without anthracycline, at 3000 mg/m2 have similar efficacy as an intermediate dose of 1000 mg/m2.38 A total of 2 to 4 cycles of post-remission therapy is considered standard.3 Intensified post-remission chemotherapy has not been associated with consistent benefit in older AML patients or those with poor-risk disease. In recent years, measurable residual disease (MRD) assessment has emerged as a potentially useful tool in risk stratification and treatment planning, with various studies suggesting that MRD status in complete remission is one of the most important prognostic factors.51 Prospective studies confirming the significance of MRD as a marker for therapy selection are awaited. Finally, maintenance chemotherapy is not part of standard AML treatment.3
Role of Stem Cell Transplant
AML is the most common indication for allo-SCT. The availability of alternative donor strategies, which include mismatched, unrelated, haplo-identical, and cord blood donor sources, and the development of non-myeloablative and reduced-intensity conditioning (RIC) regimens (which take advantage of graft-versus-leukemia effect while decreasing cytotoxicity from myeloablative regimens) have expanded the possibility of allo-SCT to most patients under the age of 75 years.3 The decision to perform transplant is now largely based upon assessment of the risk (nonrelapse mortality) to benefit (reduction in risk of relapse) ratio, as determined by both disease-related features (cytogenetics, molecular profile) and clinical characteristics of the donor (type, availability, match) and the recipient (comorbidities, performance status).3 In a meta-analysis of 24 prospective trials involving more than 6000 AML patients in first complete remission, allo-SCT was associated with a significant survival benefit in patients with intermediate- and poor-risk AML but not in patients with good-risk AML.52 In line with this, good-risk AML patients are generally not recommended for transplant in first complete remission. For patients with normal karyotype who were said to have de novo AML (historically an intermediate-risk AML group), superior OS was demonstrated with transplant over intensive chemotherapy in those patients with either FLT3-ITD mutations or those with the molecular profile characterized by negativity for mutations in NPM1/CEBPA/FLT3.53 For patients with primary refractory disease and high-risk AML, transplant is probably the only curative option.
The choice of conditioning regimen is guided by several factors, including the subtype of AML, disease status, donor-recipient genetic disparity, graft source, comorbidities in the recipient (ie, tolerability for intensive conditioning regimen), as well as the reliance on graft-versus-leukemia effect as compared to cytotoxic effect of the regimen. The BMT CTN 0901 trial, which randomly assigned 218 patients aged 18 to 65 years to RIC (typically fludarabine/busulfan) or myeloablative regimens, showed an advantage for myeloablative regimens.54 The trial demonstrated a lower risk of relapse (13.5% versus 48.3%, P < 0.01) and higher rates of relapse-free survival (67.7% versus 47.3%, P < 0.01) and OS (67.7% versus. 77.4%, P = 0.07) at 18 months despite higher treatment-related mortality (15.8% versus 4.4%, P = 0.02) and a higher rate of grade 2 to 4 acute graft-versus-host disease (44.7% versus 31.6%, P = 0.024). At present, a RIC regimen is generally recommended for older patients or those with a higher comorbidity burden, while the myeloablative regimen is recommended for younger, fit patients.
Relapsed/Refractory Disease
The treatment of relapsed and refractory AML constitutes a major challenge, with OS estimated around 10% at 3 years.55 Currently, there is no standard salvage therapy in this setting, thus underscoring the need for clinical trials. For younger, fitter patients, the typical approach is to use intensive chemotherapy to achieve a second complete remission followed by a stem cell transplant. In younger patients, a second complete remission is achievable in about 55% of patients, although this rate is lower (~20%–30%) in more unselected patients.56,57 About two thirds of those who achieve complete remission may be able to proceed to transplant.57 For older patients where transplant is not possible, the goal is to use less intensive therapies that help with palliation. HMAs (azacitidine, decitabine) are used and have complete remission rates of 16% to 21% and median survival of 6 to 9 months in older patients.3 LDAC is another option in this setting. The recent approval of GO in this setting has further expanded the options. This approval was based on the outcomes of the phase 2 single-arm MyloFrance-1 study in which single-agent GO administered at 3 mg/m2 on days 1, 4, and 7 led to complete remission in 15 of 57 patients.58
With greater elucidation of the molecular characteristics of AML, the emergence of more effective targeted therapies is possible. Enasidenib, an inhibitor of mutant isocitrate dehydrogenase 2 (IDH2) protein that promotes differentiation of leukemic myeloblasts, recently received regulatory approval based on a single-arm trial. The overall response rate in this study was 38.5%, including a composite complete remission rate of 26.6% at a dose of 100 mg daily.59 IDH differentiation syndrome, akin to the differentiation syndrome seen in acute promyelocytic leukemia, occurred in approximately 12% of the patients, with the most frequent manifestations being dyspnea, fever, pulmonary infiltrates, and hypoxia.60
Survival of patients who relapse following transplant is particularly poor. A recent Center for International Blood and Marrow Transplant Research study found a 3-year OS ranging from a dismal 4% for those who present with early relapses (within 1 to 6 months) post-transplant to a more modest 38% for those who relapsed ≥ 3 years after their first transplant.61 The German Cooperative Transplant Study Group have suggested that azacitidine or chemotherapy followed by donor-lymphocyte infusions might improve responses over chemotherapy alone.62 Ipilimumab-based CTLA-4 blockade was reported to produce responses in a small cohort of patients, which was particularly notable in patients presenting with extramedullary manifestations of relapse.63 In patients who are otherwise fit but have a florid relapse, a second transplant can sometimes be sought, but the value of a different donor for second transplant is unclear.3
Case 1 Conclusion
Given his relatively young age, suitability for intensive therapy, and the presence of a core- binding factor abnormality, the patient is treated with an induction regimen containing daunorubicin, cytarabine, and GO (7+3 + GO). He achieves complete remission. This is followed by consolidation chemotherapy with high-dose cytarabine and GO. Allo-SCT is reserved for later should the AML relapse. Note that dasatinib, a c-KIT inhibitor, can be added to the treatment regimens as per the results of the CALGB 10801 protocol.64 Also, autologous SCT, instead of allo-SCT, can be considered in rare situations with relapsed core-binding factor AML (especially with inv(16) AML, younger patients, longer time in complete remission prior to relapse, and use of GO).
Case 2 Conclusion
The patient is deemed suitable for intensive chemotherapy. As such, CPX-351 is given in induction and consolidation and complete remission is achieved. Because he has adverse-risk AML, an allo-SCT is planned, but the patient relapses before it can be performed. Following 3 courses of decitabine therapy, the patient achieves complete remission once again but declines transplant. He maintains remission for an additional 4 months but then the leukemia progresses. Clinical trials are recommended to the patient, but he decides to pursue hospice care.
Conclusion
AML is the most common acute leukemia in adults. As defined currently, AML represents a group of related but distinct myeloid disorders that are characterized by various chromosomal, genetic, and epigenetic alterations. Early diagnosis and treatment can help prevent the emergence or manage the detrimental effects of its various complications such as leukostasis and tumor lysis syndrome. Improvements in supportive care, incremental treatment advances, and the wide adoption of allo-SCT for less than favorable cases have significantly improved survival of AML patients since the initial design of combinatorial (7+3) induction chemotherapy, particularly in patients presenting at a younger age. HMAs and the emergence of targeted therapies like FLT-3 and IDH2 inhibitors have added to our therapeutic armamentarium. Despite these advances, long-term survival rates in AML patients continue to be only approximately 40% to 50%. Older patients (particularly those over age 65 at the time of diagnosis), those with relapsed disease, and those with AML with certain unfavorable genetic abnormalities continue to have dismal outcomes. The design of newer targeted therapies, epigenetic agents, and immunotherapies will hopefully address this unmet need.
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–52.
2. National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts. Leukemia: Acute Myeloid Leukemia (AML). 2018;2018.
3. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47.
4. Liesveld JL, Lichtman MA. Acute myelogenous leukemia. In: Kaushansky K, Lichtman MA, Prchal JT, et al, eds. New York: Williams Hematology. 9th ed. New York: McGraw-Hill Education; 2015.
5. Randhawa JK, Khoury J, Ravandi-Kashani F. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 3rd ed. New York: McGraw-Hill Medical; 2016.
6. Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002;30:41–7.
7. Graubert TA, Mardis ER. Genomics of acute myeloid leukemia. Cancer J 2011;17:487–91.
8. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74.
9. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21.
10. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015;125:1367–76.
11. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98.
12. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16.
13. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–21.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87.
15. Pileri S, Ascani S, Cox M, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia 2007;21:340–50.
16. Vachhani P, Bose P. Isolated gastric myeloid sarcoma: a case report and review of the literature. Case Rep Hematol 2014;2014:541807.
17. Rowe JM. Clinical and laboratory features of the myeloid and lymphocytic leukemias. Am J Med Technol 1983;49:103–9.
18. Wolach O, Stone RM. Mixed-phenotype acute leukemia: current challenges in diagnosis and therapy. Curr Opin Hematol 2017;24:139–45.
19. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
20. Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol 2018;93:553–63.
21. Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89.
22. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33.
23. Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 2012;119:34–43.
24. Cairoli R, Beghini A, Grillo G, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 2006;107:3463–8.
25. Sorror ML, Storer BE, Elsawy M, et al. Intensive versus non-intensive induction therapy for patients (Pts) with newly diagnosed acute myeloid leukemia (AML) using two different novel prognostic models [abstract]. Blood 2016;128(22):216.
26. Quintás-Cardama A, Ravandi F, Liu-Dumlao T, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood 2012;120;4840-5.
27. Gupta N, Miller A, Gandhi Set al. Comparison of epigenetic versus standard induction chemotherapy for newly diagnosed acute myeloid leukemia patients ≥60 years old.Am J Hematol 2015;90:639-46.
28. Sorror ML, Storer BE, Fathi AT, et al. Development and validation of a novel acute myeloid leukemia-composite model to estimate risks of mortality. JAMA Oncol 2017;3:1675–82.
29. Rowe JM, Neuberg D, Friedenberg W, et al. A phase 3 study of three induction regimens and of priming with GM-CSF in older adults with acute myeloid leukemia: a trial by the Eastern Cooperative Oncology Group. Blood 2004;103:479–85.
30. Mandelli F, Vignetti M, Suciu S, et al. Daunorubicin versus mitoxantrone versus idarubicin as induction and consolidation chemotherapy for adults with acute myeloid leukemia: the EORTC and GIMEMA Groups Study AML-10. J Clin Oncol 2009;27:5397–403.
31. Gardin C, Chevret S, Pautas C, et al. Superior long-term outcome with idarubicin compared with high-dose daunorubicin in patients with acute myeloid leukemia age 50 years and older. J Clin Oncol 2013;31:321–7.
32. Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009;361:1249–59.
33. Lee JH, Joo YD, Kim H, et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood 2011;118:3832–41.
34. Lowenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009;361:1235–48.
35. Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood 2015;125:3878–85.
36. Devillier R, Bertoli S, Prebet T, et al. Comparison of 60 or 90 mg/m(2) of daunorubicin in induction therapy for acute myeloid leukemia with intermediate or unfavorable cytogenetics. Am J Hematol 2015;90:E29–30.
37. Pautas C, Merabet F, Thomas X, et al. Randomized study of intensified anthracycline doses for induction and recombinant interleukin-2 for maintenance in patients with acute myeloid leukemia age 50 to 70 years: results of the ALFA-9801 study. J Clin Oncol 2010;28:808–14.
38. Lowenberg B. Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 2013;121:26–8.
39. Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7 + 3 in older patients with newly diagnosed high risk (secondary) AML [abstract]. J Clin Oncol 2016;34(15_suppl):7000-7000.
40. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64.
41. Jen EY, Ko CW, Lee JE, et al. FDA approval: Gemtuzumab ozogamicin for the treatment of adults with newly-diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res 2018; doi: 10.1158/1078-0432. CCR-17-3179.
42. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol 2001;19:3244–54.
43. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013;121:4854–60.
44. Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol 2012;30:3924–31.
45. Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012;379:1508–16.
46. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291–9.
48. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 2016;375:2023–36.
49. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol 2016;34:972–9.
50. Miyawaki S, Ohtake S, Fujisawa S, et al. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood 2011;117:2366–72.
51. Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood 2018;131:1275–91.
52. Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301:2349–61.
53. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008;358:1909–18.
54. Pasquini MC, Logan B, Wu J, et al. Results of a phase III randomized, multi-center study of allogeneic stem cell transplantation after high versus reduced intensity conditioning in patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML): Blood and Marrow Transplant Clinical Trials Network (BMT CTN) 0901. Blood 2015;126:LBA–8.
55. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol 2017;18:17,017-0456-2.
56. Burnett AK, Goldstone A, Hills RK, et al. Curability of patients with acute myeloid leukemia who did not undergo transplantation in first remission. J Clin Oncol 2013;31:1293–301.
57. Ravandi F, Ritchie EK, Sayar H, et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): a randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol 2015;16:1025–36.
58. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 2007;21:66–71.
59. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–31.
60. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol 2018;doi: 10.1001/jamaoncol.2017.4695.
61. Bejanyan N, Weisdorf DJ, Logan BR, et al. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant 2015;21:454–9.
62. Schroeder T, Rachlis E, Bug G, et al. Treatment of acute myeloid leukemia or myelodysplastic syndrome relapse after allogeneic stem cell transplantation with azacitidine and donor lymphocyte infusions--a retrospective multicenter analysis from the German Cooperative Transplant Study Group. Biol Blood Marrow Transplant 2015;21:653–60.
63. Davids MS, Kim HT, Bachireddy P, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med 2016;375:143–53.
64. Marcucci G, Geyer S, Zhao W, et al. Adding KIT inhibitor dasatinib (DAS) to chemotherapy overcomes the negative impact of KIT mutation/over-expression in core binding factor (CBF) acute myeloid leukemia (AML): results from CALGB 10801 (Alliance) [abstract]. Blood 2014;124:8.
1. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015;373:1136–52.
2. National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts. Leukemia: Acute Myeloid Leukemia (AML). 2018;2018.
3. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47.
4. Liesveld JL, Lichtman MA. Acute myelogenous leukemia. In: Kaushansky K, Lichtman MA, Prchal JT, et al, eds. New York: Williams Hematology. 9th ed. New York: McGraw-Hill Education; 2015.
5. Randhawa JK, Khoury J, Ravandi-Kashani F. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 3rd ed. New York: McGraw-Hill Medical; 2016.
6. Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002;30:41–7.
7. Graubert TA, Mardis ER. Genomics of acute myeloid leukemia. Cancer J 2011;17:487–91.
8. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74.
9. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21.
10. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015;125:1367–76.
11. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98.
12. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16.
13. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–21.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87.
15. Pileri S, Ascani S, Cox M, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia 2007;21:340–50.
16. Vachhani P, Bose P. Isolated gastric myeloid sarcoma: a case report and review of the literature. Case Rep Hematol 2014;2014:541807.
17. Rowe JM. Clinical and laboratory features of the myeloid and lymphocytic leukemias. Am J Med Technol 1983;49:103–9.
18. Wolach O, Stone RM. Mixed-phenotype acute leukemia: current challenges in diagnosis and therapy. Curr Opin Hematol 2017;24:139–45.
19. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405.
20. Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol 2018;93:553–63.
21. Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89.
22. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010;363:2424–33.
23. Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 2012;119:34–43.
24. Cairoli R, Beghini A, Grillo G, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 2006;107:3463–8.
25. Sorror ML, Storer BE, Elsawy M, et al. Intensive versus non-intensive induction therapy for patients (Pts) with newly diagnosed acute myeloid leukemia (AML) using two different novel prognostic models [abstract]. Blood 2016;128(22):216.
26. Quintás-Cardama A, Ravandi F, Liu-Dumlao T, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood 2012;120;4840-5.
27. Gupta N, Miller A, Gandhi Set al. Comparison of epigenetic versus standard induction chemotherapy for newly diagnosed acute myeloid leukemia patients ≥60 years old.Am J Hematol 2015;90:639-46.
28. Sorror ML, Storer BE, Fathi AT, et al. Development and validation of a novel acute myeloid leukemia-composite model to estimate risks of mortality. JAMA Oncol 2017;3:1675–82.
29. Rowe JM, Neuberg D, Friedenberg W, et al. A phase 3 study of three induction regimens and of priming with GM-CSF in older adults with acute myeloid leukemia: a trial by the Eastern Cooperative Oncology Group. Blood 2004;103:479–85.
30. Mandelli F, Vignetti M, Suciu S, et al. Daunorubicin versus mitoxantrone versus idarubicin as induction and consolidation chemotherapy for adults with acute myeloid leukemia: the EORTC and GIMEMA Groups Study AML-10. J Clin Oncol 2009;27:5397–403.
31. Gardin C, Chevret S, Pautas C, et al. Superior long-term outcome with idarubicin compared with high-dose daunorubicin in patients with acute myeloid leukemia age 50 years and older. J Clin Oncol 2013;31:321–7.
32. Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009;361:1249–59.
33. Lee JH, Joo YD, Kim H, et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood 2011;118:3832–41.
34. Lowenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009;361:1235–48.
35. Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood 2015;125:3878–85.
36. Devillier R, Bertoli S, Prebet T, et al. Comparison of 60 or 90 mg/m(2) of daunorubicin in induction therapy for acute myeloid leukemia with intermediate or unfavorable cytogenetics. Am J Hematol 2015;90:E29–30.
37. Pautas C, Merabet F, Thomas X, et al. Randomized study of intensified anthracycline doses for induction and recombinant interleukin-2 for maintenance in patients with acute myeloid leukemia age 50 to 70 years: results of the ALFA-9801 study. J Clin Oncol 2010;28:808–14.
38. Lowenberg B. Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 2013;121:26–8.
39. Lancet JE, Uy GL, Cortes JE, et al. Final results of a phase III randomized trial of CPX-351 versus 7 + 3 in older patients with newly diagnosed high risk (secondary) AML [abstract]. J Clin Oncol 2016;34(15_suppl):7000-7000.
40. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64.
41. Jen EY, Ko CW, Lee JE, et al. FDA approval: Gemtuzumab ozogamicin for the treatment of adults with newly-diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res 2018; doi: 10.1158/1078-0432. CCR-17-3179.
42. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol 2001;19:3244–54.
43. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013;121:4854–60.
44. Burnett AK, Russell NH, Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol 2012;30:3924–31.
45. Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012;379:1508–16.
46. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291–9.
48. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 2016;375:2023–36.
49. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol 2016;34:972–9.
50. Miyawaki S, Ohtake S, Fujisawa S, et al. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood 2011;117:2366–72.
51. Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood 2018;131:1275–91.
52. Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 2009;301:2349–61.
53. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008;358:1909–18.
54. Pasquini MC, Logan B, Wu J, et al. Results of a phase III randomized, multi-center study of allogeneic stem cell transplantation after high versus reduced intensity conditioning in patients with myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML): Blood and Marrow Transplant Clinical Trials Network (BMT CTN) 0901. Blood 2015;126:LBA–8.
55. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol 2017;18:17,017-0456-2.
56. Burnett AK, Goldstone A, Hills RK, et al. Curability of patients with acute myeloid leukemia who did not undergo transplantation in first remission. J Clin Oncol 2013;31:1293–301.
57. Ravandi F, Ritchie EK, Sayar H, et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): a randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol 2015;16:1025–36.
58. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 2007;21:66–71.
59. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–31.
60. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol 2018;doi: 10.1001/jamaoncol.2017.4695.
61. Bejanyan N, Weisdorf DJ, Logan BR, et al. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant 2015;21:454–9.
62. Schroeder T, Rachlis E, Bug G, et al. Treatment of acute myeloid leukemia or myelodysplastic syndrome relapse after allogeneic stem cell transplantation with azacitidine and donor lymphocyte infusions--a retrospective multicenter analysis from the German Cooperative Transplant Study Group. Biol Blood Marrow Transplant 2015;21:653–60.
63. Davids MS, Kim HT, Bachireddy P, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med 2016;375:143–53.
64. Marcucci G, Geyer S, Zhao W, et al. Adding KIT inhibitor dasatinib (DAS) to chemotherapy overcomes the negative impact of KIT mutation/over-expression in core binding factor (CBF) acute myeloid leukemia (AML): results from CALGB 10801 (Alliance) [abstract]. Blood 2014;124:8.
Management of Colorectal Cancer in Older Adults
Introduction
Colorectal cancer (CRC) is the fourth most common cancer in the United States and has a high prevalence among the older population.1 In 2017, there were an estimated 135,430 new cases of CRC and 50,260 deaths due to CRC. It is the second leading cause of cancer death in the United States, and the death rate for patients with CRC increases with age (Figure).2 
Although elderly persons are more frequently diagnosed with CRC, they are underrepresented in clinical trials. This may be due in part to stringent eligibility criteria in prospective randomized controlled trials that exclude older patients with certain comorbidities and decreased functional status. Hutchins and colleagues compared the proportion of persons aged 65 years and older enrolled in Southwest Oncology Group (SWOG) clinical trials and the proportion of persons in this age group in the US population with the same cancer diagnoses.5 They found that while 72% of the US population with CRC were aged ≥ 65 years, persons in this age group comprised only 40% of patients enrolled in SWOG trials. An update on this study performed after Medicare policy changed in 2000 to include coverage of costs incurred due to clinical trials showed an upward trend in the accrual of older patients in SWOG trials, from 25% during the period 1993–1996 to 38% during the period 2001–2003; however, the percentage of older patients with CRC on clinical trials overall remained stable from 1993 to 2003.6
The underrepresentation of older adults with CRC in clinical trials presents oncologists with a challenging task when practicing evidence-based medicine in this patient population. Analysis of a large claims database demonstrated that the use of multi-agent chemotherapy for the treatment of metastatic CRC in older adults increased over time, while the use of single-agent 5-fluorouracil (5-FU) decreased.7 However, the adoption of combination therapy with irinotecan or oxaliplatin in older adults lagged behind the initial adoption of these agents in younger patients. This data demonstrates that as the field of medical oncology evolves, providers are becoming more comfortable treating older patients with multiple medical problems using standard approved regimens.
Geriatric Assessment
Before treating older patients with cancer, it is necessary to define the patient’s physiological age, ideally through a multidisciplinary team evaluation.
The Eastern Cooperative Oncology Group performance status (ECOG PS) and Karnofsky Performance Status (KPS) are crude measures of functional status.12 Generally, elderly patients with good ECOG PS or KPS scores are considered fit enough to receive standard therapy similar to their younger counterparts. Evaluation of functional status using these performance scores is often suboptimal, resulting in patients with a normal or adequate performance status score who may still experience poor outcomes, including decreased survival and inability to tolerate treatment. A study that explored parameters among older patients that predict for increased risk of chemotherapy-related toxicities found that physician-rated KPS score did not accurately predict the risk for adverse events.13 Therefore, a CGA represents a better way to evaluate functional status and other domains.
Functional status can also be evaluated by self-reported tools such as activities of daily living, which refer to basic self-care, and instrumental activities of daily living (IADLs), which are essential for independent living in the community.14,15 Mobility, gait, and balance can also be measured using the “Timed Get Up and Go” test and gait speed. Klepin et al found that faster gait speed was associated with overall survival (OS) in patients with metastatic cancer.16
Cognitive function is an important component of the geriatric assessment in older patients with cancer, as dementia is a prognostic factor for survival in the overall geriatric population. In a retrospective review, patients with dementia were less likely to have a biopsy-proven diagnosis and were twice as likely to have their CRC diagnosed postmortem.17 In addition, establishing that the patient has intact cognitive function prior to initiating treatment is essential to ensure that the patient can comply with treatment and understands when to report adverse effects. Nutritional status is an important portion of the geriatric assessment because malnutrition is associated with increased mortality and decreased tolerance for chemotherapy.18–20 Evaluating the patient’s psychosocial support is crucial as well because older patients are at greater risk of social isolation and depression.21 While the incidence of depression is lower in older adults with cancer than in younger adults with cancer, clinically significant depression is still noted in 3% to 25% of elderly cancer patients.22 Other critical components of the CGA are review of the patient’s comorbidities and medications to avoid complications of polypharmacy.
Both the Cancer and Aging Research Group (CARG) and Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) toxicity tools are valuable tools, as they predict chemotherapy tolerance in elderly patients.13,23 These tools can help guide discussions between oncologists and patients as well as the formulation of an appropriate treatment plan.24 Although toxicity tools can help to determine which patients are at risk for severe toxicity secondary to treatment, these tools do not replace the CGA. A prospective cohort study that evaluated the impact of CGA on tolerance to chemotherapy in older patients with cancer compared patients aged ≥ 70 years at the start of their treatment with chemotherapy (± radiation therapy) using geriatrician-delivered CGA versus standard care given by oncology.8 Patients who received geriatrician-guided CGA interventions tolerated chemotherapy better and completed treatments as planned (odds ratio 4.14 [95% confidence interval {CI} 1.50 to 11.42], P = 0.006) with fewer treatment modifications.
Unfortunately, the CGA is time-consuming to administer and difficult to incorporate into a busy oncology practice. Therefore, other screening models are used to identify patients who may benefit from a full CGA. The International Society of Geriatric Oncology performed a systematic review of screening tools used to identify older cancer patients in need of geriatric assessment and found that the 3 most studied screening tools are the G8, the Vulnerable Elders Survey-13 (VES-13), and the Flemish version of the Triage Risk Screening Tool.25 Another study found that the G8 was more sensitive than the VES-13 (76.5% versus 68.7%, P = 0.0046), whereas the VES-13 was more specific than the G8 (74.3% versus 64.4%, P < 0.0001).26 In addition to providing guidance to initiate a full geriatric assessment, these screening tools may assist in decision making for older cancer patients, especially those with advanced disease.
Surgery
Early-Stage Disease
When possible, surgical resection of colorectal tumors is the primary treatment in both the curative setting and to avoid complications, such as obstruction or perforation.27 Multiple studies have shown that fit elderly patients benefit from curative surgery similarly to their younger counterparts.27–29 With the growing population of persons aged 65 years or older, surgeons are becoming more comfortable with operating on the elderly.4 However, a large systematic review of 28 independent studies with a total of 34,194 patients showed that older patients were less likely to undergo curative surgery.30 Eligibility for surgery should not be determined by age alone, but rather should be based on a full assessment of the patient’s health, including comorbidities, functional status, nutrition, cognition, social support, and psychological status. The impact of age on short-term outcomes after colorectal surgery in terms of 30-day postoperative morbidity and mortality rates was explored in a study that divided patients into 2 groups: those aged ≥ 80 years (mean age 85) and those aged < 80 years (mean age 55.3).31 There were no statistical differences in 30-day postoperative morbidity and mortality rates between the 2 groups, and preexisting comorbidities and urgent nature of surgery were important predictors of colorectal surgery outcomes in the older adults, results that have been seen in several other studies.28,30 When possible, laparoscopic surgery is preferred as it is associated with less intraoperative blood loss, less postoperative pain, reduced postoperative ileus, a shorter hospital stay, and fewer cardiovascular and pulmonary complications.32 The Preoperative Assessment of Cancer in the Elderly (PACE), which combines surgical risk assessment tools with CGA tools, can assist surgeons in determining candidacy for surgery and help decrease unequal access to surgery in the geriatric population.33
Metastasectomy
A large international multicenter cohort study explored the outcomes of patients aged ≥ 70 years who underwent liver resection of colorectal metastases. The study investigatorsfound that neoadjuvant chemotherapy was used less frequently and less extensive surgery was performed in elderly patients than in younger patients.34 Sixty-day postoperative mortality was slightly higher (3.8% versus 1.6%, P < 0.001) and 3-year OS was slightly lower (57.1% versus 60.2%, P < 0.001) in the elderly group as compared to their younger counterparts, but overall the outcomes after liver surgery were similar. Therefore, the management of liver metastases in oligometastatic disease in elderly patients fit for surgery should be the same as that offered to younger patients. Since outcomes are comparable, older patients should be offered neoadjuvant chemotherapy, as several studies have shown similar response rates and OS in younger and older patients.35,36
Rectal Cancer
The standard of care for locally advanced rectal cancer is combined modality treatment with radiation and chemotherapy followed by total mesorectal excision. However, given conflicting data regarding the ability of elderly patients to tolerate neoadjuvant 5-FU-based chemotherapy and radiation, elderly patients are treated with trimodality therapy less often than their younger counterparts.37,38 A systematic review of 22 randomized trials involving 8507 patients with rectal cancer showed that adjuvant radiation therapy could reduce the risk of local recurrence and death from rectal cancer in patients of all ages.39 However, the risk of noncancer-related death was increased in the older population. The Stockholm II trial showed similar benefits of preoperative radiation overall, but this benefit did not extend to patients older than 68 years because of an increased risk of morbidity and mortality.40 In older patients, mortality from noncancer causes within the first 6 months after surgery was higher in the group that received perioperative radiation than in the group that did not receive radiation. Elderly patients (age > 68 years) accounted for most of the mortality, which was predominantly due to cardiovascular disease.
A retrospective study of 36 patients aged ≥ 70 years with rectal cancer evaluated the toxicity and feasibility of neoadjuvant 5-FU combined with pelvic radiation for treating locally advanced rectal cancer. Patients were classified as healthy and “fit” or “vulnerable” based on the presence of comorbidities.41 This study demonstrated that tolerability and response to neoadjuvant chemotherapy and radiation as well as ability to undergo surgery were similar in “vulnerable” patients and “fit” patients. Conversely, Margalit and colleagues studied the rate of treatment deviations in elderly patients with rectal cancer treated with combined modality therapy and found that most patients required early termination of treatment, treatment interruptions, or dose reductions.42 While trimodality treatment is the standard of care in rectal cancer, there is conflicting data from retrospective studies regarding the tolerability and feasibility of this approach. It is important to proceed with caution but to still consider fit older patients with locally advanced rectal cancer for neoadjuvant chemotherapy and radiation followed by surgery.
In patients who have a complete response (CR) to neoadjuvant chemoradiation, watchful waiting rather than proceeding to surgery may be a reasonable strategy, especially in older patients. A systematic review of 867 patients with locally advanced rectal cancer showed no statistically significant difference in OS between patients who were observed with watchful waiting and those who underwent surgery.43 The International Watch and Wait Database includes 679 patients who were managed with a watch-and-wait regimen because they had a clinical CR after chemoradiation. An outcomes analysis of these patients showed that 25% had local regrowth, with 3-year OS of 91% overall and 87% in patients with local regrowth.44 In most patients (84%), regrowth of the tumor occurred within the first 2 years of follow up.
In frail older adults, for whom longer courses of treatment are not feasible or chemotherapy is contraindicated, short-course radiation therapy can be considered either in the neoadjuvant setting or alone for palliation.45 A randomized trial of short-course radiation versus long-course chemoradiation in patients with T3 rectal cancer found that the difference in 3-year local recurrence rates was not statistically significant.46
Chemotherapy
An expected natural decline in function occurs with age, but given the great variability that exists between patients, it is important to focus on physiologic age rather than chronologic age to determine ability to receive and tolerate anticancer treatment. Decreases in renal and hepatic function, cognitive impairment, changes in gastrointestinal motility, decrements in cardiac and bone marrow reserves, as well as comorbidities and polypharmacy affect a patient’s ability to tolerate chemotherapy.47,48 Toxicity tools such as CARG and CRASH can help to predict severity of toxicity with chemotherapy.13,23 The information provided by these tools can help guide conversations between the oncologist and patient regarding treatment plans.
Adjuvant Chemotherapy for Early-Stage Disease
Stage II Disease
Defining treatment guidelines for older patients with stage II colon cancer is difficult due to lack of data that shows benefit in this population. The QUASAR (Quick and Simple and Reliable) group’s prospective study of adjuvant single-agent 5-FU in stage II colon cancer patients showed an absolute improvement in survival of 3.6% when 5-FU was given after surgery (95% CI 1.0 to 6.0).49 The subgroup analysis of patients aged ≥ 70 years showed a limited benefit of adjuvant 5-FU (hazard ratio [HR] 1.13 [95% CI 0.74 to 1.75]). Given the limited benefit, adjuvant 5-FU for elderly patients with stage II colon cancer should be used judiciously as patients may have competing causes of morbidity or mortality.
The use of oxaliplatin-based therapy in the adjuvant setting for stage II disease was evaluated in a subgroup analysis of the MOSAIC study (Multicenter International Study of Oxaliplatin/5-FU/Leucovorin in the Adjuvant Treatment of Colon Cancer).50 Adjuvant oxaliplatin-based treatment may be offered to patients with stage II colon cancer that carries high-risk features (poorly differentiated histology, lymphovascular invasion, bowel obstruction and/or perforation, < 12 lymph nodes sampled, perineural invasion, or indeterminate or positive margins) due to a trend toward improved disease-free survival (DFS) at 5 years. Patients in this group who received adjuvant FOLFOX (leucovorin, oxaliplatin, 5-FU) versus 5-FU/leucovorin had a DFS of 82.3% versus 74.6%, respectively (HR 0.72 [95% CI 0.50 to 1.02]), a difference that was not statistically significant. A subgroup analysis of 315 patients aged 70 to 75 years with stage II colon cancer enrolled in the MOSAIC study found no statistically significant DFS or OS benefit with the addition of oxaliplatin to 5-FU/leucovorin.51 Therefore, use of this platinum/fluoropyrimidine combination for adjuvant therapy for high-risk stage II disease in older patients remains controversial given its associated risks and the lack of definitive data demonstrating a benefit in this patient group. Decisions regarding this therapy should be made through a shared discussion with patients about its risks and benefits.
Microsatellite status is an important biomarker in the evaluation of stage II CRC. Microsatellite stability is a marker of a functioning DNA mismatch repair system. In patients with colon cancer, tumor microsatellite stability is classified based on the percentage of abnormal microsatellite regions.52 Several studies have shown that patients with tumors that display high microsatellite instability (MSI-H) have an improved prognosis over patients with microsatellite stable tumors.53,54 While patients with stage II MSI-H colon cancer have better outcomes, MSI is associated with a reduced response to treatment with fluoropyrimidines, as demonstrated in a systematic review that found that patients with tumors with MSI obtained no benefit from adjuvant 5-FU (HR 1.24 [95% CI 0.72 to 2.14]).55 Aparicio and colleagues reported an increased prevalence of MSI-H tumors with increasing age.56 Therefore, mismatch repair phenotype should be considered when making adjuvant chemotherapy decisions in the older adult with colon cancer, as it may affect the decision to recommend single-agent 5-FU treatment.
Stage III Disease
The use of single-agent 5-FU for stage III resected CRC has been evaluated in multiple studies. Sargent et al performed a pooled analysis of 3351 patients from 7 randomized phase 3 trials comparing surgery and adjuvant 5-FU-based chemotherapy versus surgery alone in stage II or III colon cancer patients.57 Adjuvant chemotherapy was associated with improvement in both OS and time to tumor recurrence (HR 0.76 and 0.68, respectively). The 5-year OS was 71% for those who received adjuvant treatment and 64% for those who were treated with surgery alone. The benefit of adjuvant treatment was independent of age, and there was no difference in toxicity across age groups, except for 1 study which showed increased rates of leukopenia in the elderly. The oral fluoropyrimidine capecitabine was shown to be an effective alternative to 5-FU plus leucovorin as adjuvant treatment for those with resected stage III colon cancer.58 However, in the subgroup analysis of DFS in the intention-to-treat group, the improvement in DFS was not statistically significant in those aged ≥ 70 years. This study justified the phase 3 Xeloda in Adjuvant Colon Cancer Therapy (X-ACT) trial, which compared capecitabine and 5-FU/leucovorin as adjuvant therapy in patients with resected stage III colon cancer.59 The X-ACT trial showed no significant effect of age on DFS or OS.
The addition of oxaliplatin to 5-FU in the adjuvant setting for stage III tumors has been studied and debated in the elderly population in multiple trials. The MOSAIC trial investigated FOLFOX versus 5-FU/leucovorin in the adjuvant setting.50 The addition of oxaliplatin was associated with a DFS and OS benefit, with a 20% reduction in risk of colon cancer recurrence and 16% reduction in risk of death in all patients. The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-07 trial then studied 2409 patients with stage II or III colon cancer treated with weekly bolus 5-FU/leucovorin with or without oxaliplatin.60 In this study, OS was significantly improved with the addition of oxaliplatin in patients younger than 70 years, but OS at 5 years was 4.7% worse for patients aged ≥ 70 years treated with weekly 5-FU/leucovorin and oxaliplatin compared with those treated with weekly 5-FU/leucovorin (71.6% versus 76.3%, respectively). In contrast, the XELOXA trial (NO16968), which randomly assigned stage III colon cancer patients to capecitabine and oxaliplatin (XELOX) or bolus 5-FU/leucovorin (standard of care at study start), showed an efficacy benefit, albeit not statistically significant, in patients aged ≥ 70 years (HR 0.87 [95% CI 0.63 to 1.18]).61–63
The Adjuvant Colon Cancer Endpoints (ACCENT) database included 7 randomized trials totaling 14,528 patients with stage II or III colon cancer treated with adjuvant 5-FU with or without oxaliplatin or irinotecan.64 Subgroup analysis of patients aged ≥ 70 years (n = 2575) showed no benefit with an oxaliplatin-based regimen in DFS (HR 0.94 [95% CI 0.78 to 1.13]) or OS (HR 1.04 [95% CI, 0.85 to 1.27]). Based on these studies and the increased toxicity with oxaliplatin, oxaliplatin-based adjuvant chemotherapy is utilized less often than single-agent 5-FU in geriatric patients with early-stage colon cancer.65 Conversely, a recent pooled analysis of individual patient data from 4 randomized trials (NSABP C-08, XELOXA, X-ACT, and AVANT) showed improved DFS and OS with adjuvant XELOX or FOLFOX over single-agent 5-FU in patients aged ≥ 70 years (DFS HR 0.77 [95% CI 0.62 to 0.95], P = 0.014; OS HR 0.78 [95% CI 0.61 to 0.99], P = 0.045).66 This analysis also showed that grade 3 and 4 adverse events related to oxaliplatin were similar across age groups.
These data come from post-hoc analyses, and there is no prospective data to steer decision making in elderly patients with early-stage CRC (Table). 
It is well established that patients with stage III colon cancer benefit from oxaliplatin-based adjuvant chemotherapy after curative surgical resection.68 However, older patients are less likely to be referred to oncology as compared with their younger counterparts, due to the conflicting data regarding the benefit of this approach in older adults. Studies have shown that when the referral is placed, the geriatric population is less likely to receive chemotherapy.69 Sanoff et al analyzed 4 data sets (SEER-Medicare, National Comprehensive Cancer Network, New York State Cancer Registry, and Cancer Care Outcomes Research and Surveillance Consortium) to assess the benefit of adjuvant chemotherapy for resected stage III CRC among patients aged ≥ 75 years. Their analysis showed that only 40% of patients evaluated received adjuvant chemotherapy for stage III CRC after surgical resection.70
Summary
Prospective data to guide the treatment of older patients with early-stage CRC in the adjuvant setting is lacking. For fit older patients with stage II disease, limited benefit will be derived from single-agent 5-FU. For those with stage III CRC, the benefit and toxicities of fluoropyrimidines as adjuvant therapy appear to be similar regardless of age. The addition of oxaliplatin to fluoropyrimidines in patients aged ≥ 70 years has not been proven to improve DFS or OS and could result in an incremental toxicity profile. Therefore, treatment plans must be individualized, and decisions should be made through an informed discussion evaluating the overall risk/benefit ratio of each approach.
Metastatic Disease
Palliative Chemotherapy
Approximately 20% of patients with CRC are diagnosed with metastatic disease at presentation, and 35% to 40% develop metastatic disease following surgery and adjuvant therapy.2 The mainstay of treatment in this population is systemic therapy in the form of chemotherapy with or without biologic agents. In this setting, several prospective studies specific to older adults have been completed, providing more evidence-based guidance to oncologists who see these patients. Folprecht et al retrospectively reviewed data from 22 clinical trials evaluating 5-FU-based palliative chemotherapy in 3825 patients with metastatic CRC, including 629 patients aged ≥ 70 years.71 OS in elderly patients (10.8 months [95% CI 9.7 to 11.8]) was equivalent to that in younger patients (11.3 months [95% CI 10.9 to 11.7], P = 0.31). Similarly, relative risk and progression-free survival (PFS) were comparable irrespective of age.
Standard of care for most patients with metastatic colon cancer consists of 5-FU/leucovorin in combination with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) with a monoclonal antibody.72 A retrospective pooled analysis of patients with metastatic CRC compared the safety and efficacy of FOLFOX4 in patients aged < 70 years versus those aged ≥ 70 years.73 While age ≥ 70 years was associated with an increased rate of grade ≥ 3 hematologic toxicity, it was not associated with increased rates of severe neurologic events, diarrhea, nausea, vomiting, infection, 60-day mortality, or overall incidence of grade ≥ 3 toxicity. The benefit of treatment was consistent across both age groups; therefore, age alone should not exclude an otherwise healthy individual from receiving FOLFOX.
These post-hoc analyses show that fit older patients who were candidates for trial participation tolerated these treatments well; however, these treatments may be more challenging for less fit older adults. The UK Medical Research Council FOCUS2 (Fluorouracil, Oxaliplatin, CPT11 [irinotecan]: Use and Sequencing) study was a prospective phase 3 trial that included 459 patients with metastatic CRC who were deemed too frail or not fit enough for standard-dose chemotherapy by their oncologists.74 In this group, 43% of patients were older than 75 years and 13% were older than 80 years. Patients were randomly assigned to receive infusional 5-FU with levofolinate; oxaliplatin and 5-FU; capecitabine; or oxaliplatin and capecitabine; all regimens were initiated with an empiric 20% dose reduction. The addition of oxaliplatin suggested some improvement in PFS, but this was not significant (5.8 months versus 4.5 months, HR 0.84 [95% CI 0.69 to 1.01], P = 0.07). Oxaliplatin was not associated with increased grade 3 or 4 toxicities. Capecitabine is often viewed as less toxic because it is taken by mouth, but this study found that replacement of 5-FU with capecitabine did not improve quality of life. Grade 3 or 4 toxicities were seen more frequently in those receiving capecitabine than in those receiving 5-FU (40% versus 30%, P = 0.03) in this older and frailer group of patients. As the patients on this study were frail and treatment dose was reduced, this data may not apply to fit older adults who are candidates for standard therapy.
When managing an older patient with metastatic CRC, it is important to tailor therapy based on goals of care, toxicity of proposed treatment, other comorbidities, and the patient’s functional status. One approach to minimizing toxicity in the older population is the stop-and-go strategy. The OPTIMOX1 study showed that stopping oxaliplatin after 6 cycles of FOLFOX7 and continuing maintenance therapy with infusional 5-FU/leucovorin alone for 12 cycles prior to reintroducing FOLFOX7 achieved efficacy similar to continuous FOLFOX4 with decreased toxicity.75 Figer et al studied an exploratory cohort of 37 patients aged 76 to 80 years who were included in the OPTIMOX1 study.76 The overall relative risk, median PFS, and median OS did not differ between the older patients in this cohort and younger patients studied in the original study. Older patients did experience more neutropenia, neurotoxicity, and overall grade 3 to 4 toxicity, but there were no toxic deaths in patients older than 75 years. The approach of giving treatment breaks, as in OPTIMOX2, may also provide patients with better quality of life, but perhaps at the expense of cancer-related survival.77
The combination of irinotecan and 5-FU has also been studied as treatment for patients with metastatic CRC. A pooled analysis of 2691 patients aged ≥ 70 years with metastatic CRC across 4 phase 3 randomized trials investigating irinotecan and 5-FU demonstrated that irinotecan-containing chemotherapy provided similar benefits to both older and younger patients with similar risk of toxicity.78 A phase 2 trial studying FOLFIRI as first-line treatment in older metastatic CRC patients showed this to be a safe and active regimen with manageable toxicity.79 Another randomized phase 3 trial for older patients compared 5-FU/leucovorin with or without irinotecan for first-line treatment of metastatic CRC (FFCD 2001-02).80 The study accrued 282 patients aged ≥ 75 years (median age 80 years), and found that the addition of irinotecan to infusional 5-FU–based chemotherapy did not significantly increase either PFS or OS. Aparicio et al performed a substudy of baseline geriatric evaluation prior to treatment in the FFCD 2001-02 study and assessed the value of geriatric parameters for predicting outcomes (objective response rate [ORR], PFS, and OS).81 Multivariate analysis showed that none of the geriatric parameters were predictive of ORR or PFS but that normal IADL was associated with better OS. This combination may still be appropriate for some older patients with metastatic disease, while single- agent 5-FU may be more appropriate in frail patients.
Biologic Agents
VEGF Inhibitors
Targeted biologic agents have been studied in the treatment of metastatic CRC. Bevacizumab is a recombinant, humanized monoclonal antibody against vascular endothelial growth factor (VEGF) that is approved in the first-line setting for treatment of metastatic CRC. A pooled analysis examined 439 patients 65 years of age and older with metastatic CRC who received bevacizumab plus chemotherapy versus placebo plus chemotherapy.82 In this analysis, the addition of bevacizumab was associated with an improvement in OS (19.3 months versus 14.3 months, HR 0.7 [95% CI 0.55 to 0.90], P = 0.006) and in PFS (9.2 months versus 6.2 months, HR 0.52 [95% CI 0.40 to 0.67], P < 0.0001). Known adverse events associated with bevacizumab were seen in the bevacizumab plus chemotherapy group but not at increased rates in the older population compared to their younger counterparts. Conversely, another pooled analysis found that while there was a PFS and OS benefit in older patients receiving bevacizumab, there was an increased incidence of thrombotic events in patients older than 65 years.83 The BEAT (Bevacizumab Expanded Access Trial) and BRiTE (Bevacizumab Regimens Investigation of Treatment Effects) studies showed similar clinical outcomes across all age groups.84,85 While older patients experienced more arterial thromboembolic events with the addition of bevacizumab, other factors such as ECOG PS, prior anticoagulation, and history of arterial disease were more predictive of these adverse events than age.
The randomized phase 3 AVEX study explored the efficacy and tolerability of capecitabine plus bevacizumab versus capecitabine alone in 280 frail patients aged ≥ 70 years.86 PFS in the capecitabine/bevacizumab arm was 9.1 months versus 5.1 months in the capecitabine alone arm. While the OS difference was not statistically significant, patients in the capecitabine/bevacizumab arm had an OS of 20.7 months versus 16.8 months in the capecitabine alone group. As reported in prior studies, patients in the capecitabine/bevacizumab arm had increased rates of toxic events (40%) compared with those who received capecitabine alone (22%), with reports of hypertension, hand-foot syndrome, bleeding, and thrombotic events. More recently, the phase 2 PRODIGE 20 trial studied the addition of bevacizumab to chemotherapy (5-FU, FOLFOX, or FOLFIRI) based on physician choice in untreated metastatic CRC patients aged ≥ 75 years (median age 80 years).87 They found that the addition of bevacizumab to standard of care chemotherapy was both safe and effective. The adverse events seen with bevacizumab, such as hypertension and thrombotic events, were consistent with prior studies.
A newer antiangiogenic agent, ziv-aflibercept, has been approved for the second-line treatment of metastatic CRC. The VELOUR trial demonstrated that the addition of ziv-aflibercept to FOLFIRI benefited patients across all age groups compared with FOLFIRI plus placebo in patients who had failed prior oxaliplatin-based chemotherapy.88,89 Ramucirumab is a human IgG-1 monoclonal antibody approved in second-line treatment in combination with FOLFIRI. A subgroup analysis of the RAISE study showed that the survival benefit was similar in patients aged ≥ 65 years versus those < 65 years.90 Based on the above data, the use of a VEGF inhibitor in combination with chemotherapy should be considered in older patients with metastatic CRC. Furthermore, based on the conflicting data regarding the benefit of FOLFOX/FOLFIRI over single-agent 5-FU discussed above, the combination of capecitabine plus bevacizumab may be considered a front-line treatment option in older patients based on the AVEX study.
EGFR Inhibitors
Cetuximab and panitumumab are anti-epidermal growth factor receptor (EGFR) antibodies approved for the treatment of RAS wild-type metastatic CRC. Data regarding the use of EGFR inhibitors in the geriatric population is scarce and the data that does exist is conflicting.91,92 The PRIME study demonstrated that panitumumab plus FOLFOX had a PFS benefit compared to FOLFOX alone in KRAS wild-type metastatic CRC patients.92 While the study met its primary endpoint, the benefit did not translate to patients aged ≥ 65 years in subgroup analysis. Conversely, a retrospective study of the efficacy and safety of cetuximab in elderly patients with heavily pretreated metastatic CRC found similar efficacy in older and younger patients as well as no increased adverse events in the older population.91 A phase 2 trial investigating cetuximab as single-agent first-line treatment of metastatic CRC in fit older patients found cetuximab to be safe with moderate activity in this population, but did not support the use of cetuximab as first-line single-agent treatment in fit geriatric patients who may be candidates for combination therapy.93 Our group studied the patterns of use and tolerance of anti-EGFR antibodies in 117 older adults with metastatic CRC with a median age of 73 years.94 The study showed that older age at the time of treatment was associated with administration of anti-EGFR antibody as monotherapy rather than in combination with chemotherapy (P = 0.0009). We found no association between age and presence of grade 3 or higher toxicity. In addition, the toxicity profile seen in older patients was similar to what has been demonstrated in prior studies involving a younger patient population. Given the discordance seen between studies, additional prospective trials are needed to elucidate the efficacy and safety of EGFR inhibitors in the geriatric population.
Other Agents
Two newer agents approved in the treatment of metastatic CRC are regorafenib, a multikinase inhibitor, and trifluridine/tipiracil (TFD/TPI), a nucleoside analog combined with an inhibitor of thymidine phosphorylase. The phase 3 CORRECT trial studied regorafenib as monotherapy in previously treated metastatic CRC and found an OS benefit of 1.4 months and minimal PFS benefit.95 Van Cutsem et al performed a subgroup analysis by age and found similar OS benefit in patients < 65 years of age and ≥ 65 years.96 The most frequent adverse events grade 3 or higher were hand-foot syndrome, fatigue, diarrhea, hypertension, and desquamation/rash, which were seen at similar rates in both age groups. More recently, the phase 2 Regorafenib Dose Optimization Study (ReDOS) found that weekly dose escalation of regorafenib from 80 mg to 160 mg daily over 3 weeks was superior to the standard 160 mg daily dosing in patients with metastatic CRC.97 The dose escalation group had a longer median OS, although this difference was not statistically significant, as well as a more favorable toxicity profile. Therefore, this new dosing strategy may be a reasonable option for older patients with pretreated metastatic CRC. A study of TFD/TPI versus placebo in refractory metastatic CRC found an OS benefit of 7.1 months versus 5.3 months.98 In subgroup analyses, the OS benefit extended to both patients < 65 years and ≥ 65 years. Given the sparse data on these newer agents in the geriatric population and the modest benefit they provide to those with refractory metastatic CRC, more data is needed to determine their utility in elderly patients. The decision to use these agents in the older patients warrants a thorough discussion with the patient regarding risks, benefit, and treatment goals.
Immunotherapy
Between 3.5% and 6.5% of stage IV colorectal cancers are MSI-H and have deficient mismatch repair (dMMR).99–101 A recent phase 2 trial studied the use of pembrolizumab, an IgG4 monoclonal antibody against PD-1 (programmed cell death-1), in heavily pretreated patients with dMMR metastatic CRC, MMR-proficient (pMMR) metastatic CRC, and noncolorectal dMMR metastatic cancer.102 Patients with dMMR metastatic CRC had a 50% ORR and 89% disease control rate (DCR), as compared with an ORR of 0% and DCR of 16% in patients with pMMR metastatic CRC. There was also an OS and PFS benefit seen in the dMMR CRC group as compared with the pMMR CRC group. Another phase 2 study, CheckMate 142, studied the anti-PD-1 monoclonal antibody nivolumab with or without ipilimumab (a monoclonal antibody against cytotoxic T-lymphocyte antigen 4) in patients with dMMR and pMMR metastatic CRC.103 In the interim analysis, nivolumab was found to provide both disease control and durable response in patients with dMMR metastatic CRC.
While these studies led to the FDA approval of pembrolizumab and nivolumab for management of previously treated MSI-H or dMMR metastatic CRC, data on the use of immunotherapy in older adults is scarce. Immunosenescence, or the gradual deterioration of the immune system that comes with aging, may impact the efficacy of immune checkpoint inhibitors (ICI) in older patients with advanced cancer.104 There is conflicting data on the efficacy of PD-1 and programmed death ligand-1) PD-L1 inhibitors in older patients across different cancers. A meta-analysis of immunotherapy in older adults with a variety of malignancies showed overall efficacy comparable to that seen in adults younger than 65 years.105 However, another review found ICIs to be less effective in older patients with head and neck, non-small cell lung cancer, and renal cell carcinoma compared with their younger counterparts.104 Regarding the toxicity profile of ICIs in the elderly, similar rates of grade 3 or higher adverse events in patients younger than 65 years and older than 65 years have been reported.106 However, patients aged ≥ 70 years had increased rates of grade 3 to 5 adverse events as compared to patients younger than 65 years (71.7% versus 58.4%, respectively). Given the scant data on ICIs in older patients with MSI-H or dMMR metastatic CRC, more clinical trials inclusive of this population are needed in order to determine the efficacy and safety of immunotherapy.
Palliative Care
The incorporation of palliative care early following the diagnosis of cancer has been shown to improve quality of life, decrease depression, and help with symptom management.107 The triggers for geriatric patients to initiate palliative care may be different from those of younger patients, as older patients may have different goals of care.108 Older patients will often choose quality over quantity of life when making treatment decisions.109 The ideal medical treatment for the frail patient with colorectal cancer would focus on treating disease while providing palliative measures to help support the patient and improve quality of life. It is paramount that patients maintain functional independence as loss of independence is recognized as a major threat to an older patient’s quality of life.110 The optimal way to achieve these goals is through the efforts of a multidisciplinary care team including not only physicians and nurses, but also social workers, nutritionists, physical therapists, and family who can provide support for the patient’s psychosocial, cognitive, and medical needs.111 Although cancer and noncancer–related death occur more frequently in the geriatric population, data to guide a specific palliative care approach to the elderly population is lacking.108
Conclusion
Colorectal cancer is a disease of older adults with a median age at diagnosis of 67 years.1 With the aging population, oncologists will be faced with treating increasing numbers of older patients, and must adjust their practice to accommodate this population of patients. Treating geriatric patients is challenging given the lack of available data to guide the treatment approach. Although several prospective elderly-specific studies have been conducted evaluating treatments for metastatic CRC, most treatment decisions are made based on the available retrospective studies and pooled analyses. Oncologists must carefully consider and evaluate each patient based on physiologic age rather than chronologic age.112 Overall, older patients should be given the opportunity to receive standard of care treatments in the appropriate setting. The decision to modify treatment plans should be made after a thorough evaluation by a multidisciplinary team and a discussion with the patient regarding their goals and the risks and benefits of the treatment. Geriatric assessment tools can help the care team identify patients with various geriatric syndromes that may not be detected on routine oncology evaluation. This type of evaluation is time consuming and is rarely done in a busy oncology practice. Ongoing studies are aiming to develop a method to incorporate geriatric assessments into the care of older adults.Additional prospective trials targeting older, more frail patients are essential to improve upon our knowledge so we can provide best care for this growing elderly population.
1. National Cancer Institute. SEER cancer stat facts: colorectal cancer. http://seer.cancer.gov/statfacts/html/colorect.html. Accessed March 1, 2018.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
3. Kochanek KD, Murphy S, Xu J, Arias E. Mortality in the United States, 2016. NCHS Data Brief 2017:1-8.
4. Ortman JM, Velkoff VA, Hogan H. An aging nation: the older population in the United States, current population reports, P25-1140. Washington, DC: U.S. Census Bureau; 2014.
5. Hutchins LF, Unger JM, Crowley JJ, et al. Underrepresentation of patients 65 years of age or older in cancer-treatment trials. N Engl J Med 1999;341:2061–7.
6. Unger JM, Coltman CA Jr, Crowley JJ, et al. Impact of the year 2000 Medicare policy change on older patient enrollment to cancer clinical trials. J Clin Oncol 2006;24:141–4.
7. Vijayvergia N, Li T, Wong YN, et al. Chemotherapy use and adoption of new agents is affected by age and comorbidities in patients with metastatic colorectal cancer. Cancer 2016;122:3191–8.
8. Kalsi T, Babic-Illman G, Ross PJ, et al. The impact of comprehensive geriatric assessment interventions on tolerance to chemotherapy in older people. Br J Cancer 2015;112:1435–44.
9. National Comprehensive Cancer Network. Older adult oncology (Version 2.2017). Accessed March 1, 2018,
10. Balducci L. Frailty: a common pathway in aging and cancer. Interdiscip Top Gerontol 2013;38:61–72.
11. Baijal P, Periyakoil V. Understanding frailty in cancer patients. Cancer J 2014;20:358–66.
12. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982;5:649–55.
13. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
14. Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist 1969;9:179–86.
15. Katz S, Downs TD, Cash HR, Grotz RC. Progress in development of the index of ADL. Gerontologist 1970;10:20–30.
16. Klepin HD, Geiger AM, Tooze JA, et al. Physical performance and subsequent disability and survival in older adults with malignancy: results from the health, aging and body composition study. J Am Geriatr Soc 2010;58:76–82.
17. Gupta SK, Lamont EB. Patterns of presentation, diagnosis, and treatment in older patients with colon cancer and comorbid dementia. J Am Geriatr Soc 2004;52:1681–7.
18. Dewys WD, Begg C, Lavin PT, et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med 1980;69:491–7.
19. Aaldriks AA, van der Geest LG, Giltay EJ, et al. Frailty and malnutrition predictive of mortality risk in older patients with advanced colorectal cancer receiving chemotherapy. J Geriatr Oncol 2013;4:218–26.
20. Martucci RB, Barbosa MV, D’Almeida CA, et al. Undernutrition as independent predictor of early mortality in elderly cancer patients. Nutrition 2017;34:65–70.
21. Naeim A, Aapro M, Subbarao R, Balducci L. Supportive care considerations for older adults with cancer. J Clin Oncol 2014;32:2627–34.
22. Kua J. The prevalence of psychological and psychiatric sequelae of cancer in the elderly - how much do we know? Ann Acad Med Singapore 2005;34:250–6.
23. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118:3377–86.
24. Kim J, Hurria A. Determining chemotherapy tolerance in older patients with cancer. J Natl Compr Canc Netw 2013;11:1494-502.
25. Decoster L, Van Puyvelde K, Mohile S, et al. Screening tools for multidimensional health problems warranting a geriatric assessment in older cancer patients: an update on SIOG recommendations. Ann Oncol 2015;26:288–300.
26. Soubeyran P, Bellera C, Goyard J, et al. Screening for vulnerability in older cancer patients: the ONCODAGE Prospective Multicenter Cohort Study. PLoS One 2014; 9:e115060.
27. Gurevitch AJ, Davidovitch B, Kashtan H. Outcome of right colectomy for cancer in octogenarians. J Gastrointest Surg 2009;13:100–4.
28. Schiffmann L, Ozcan S, Schwarz F, et al. Colorectal cancer in the elderly: surgical treatment and long-term survival. Int J Colorectal Dis 2008;23:601–10.
29. Ong ES, Alassas M, Dunn KB, Rajput A. Colorectal cancer surgery in the elderly: acceptable morbidity? Am J Surg 2008;195:344–8.
30. Surgery for colorectal cancer in elderly patients: a systematic review. Colorectal Cancer Collaborative Group. Lancet 2000;356:968–74.
31. Shalaby M, Di Lorenzo N, Franceschilli L, et al. Outcome of colorectal surgery in elderly populations. Ann Coloproctol 2016;32:139–43.
32. Frasson M, Braga M, Vignali A, et al. Benefits of laparoscopic colorectal resection are more pronounced in elderly patients. Dis Colon Rectum 2008;51:296–300.
33. PACE participants, Audisio RA, Pope D, et al. Shall we operate? Preoperative assessment in elderly cancer patients (PACE) can help. A SIOG surgical task force prospective study. Crit Rev Oncol Hematol 2008;65:156–63.
34. Adam R, Frilling A, Elias D, et al. Liver resection of colorectal metastases in elderly patients. Br J Surg 2010;97:366–76.
35. de Liguori Carino N, van Leeuwen BL, Ghaneh P, et al. Liver resection for colorectal liver metastases in older patients. Crit Rev Oncol Hematol 2008;67:273–8.
36. Tamandl D, Gruenberger B, Herberger B, et al. Surgery after neoadjuvant chemotherapy for colorectal liver metastases is safe and feasible in elderly patients. J Surg Oncol 2009;100:364–71.
37. Shahir MA, Lemmens VE, van de Poll-Franse LV, et al. Elderly patients with rectal cancer have a higher risk of treatment-related complications and a poorer prognosis than younger patients: a population-based study. Eur J Cancer 2006;42:3015–21.
38. Chang GJ, Skibber JM, Feig BW, Rodriguez-Bigas M. Are we undertreating rectal cancer in the elderly? An epidemiologic study. Ann Surg 2007;246:215–21.
39. Colorectal Cancer Collaborative Group. Adjuvant radiotherapy for rectal cancer: a systematic overview of 8,507 patients from 22 randomised trials. Lancet 2001;358:1291–304.
40. Martling A, Holm T, Johansson H, et al, Stockholm Colorectal Cancer Study Group. The Stockholm II trial on preoperative radiotherapy in rectal carcinoma: long-term follow-up of a population-based study. Cancer 2001;92:896–902.
41. Pasetto LM, Friso ML, Pucciarelli S, et al. Rectal cancer neoadjuvant treatment in elderly patients. Anticancer Res 2006;26:3913–23.
42. Margalit DN, Mamon HJ, Ancukiewicz M, et al. Tolerability of combined modality therapy for rectal cancer in elderly patients aged 75 years and older. Int J Radiat Oncol Biol Phys 2011;81:e735–41.
43. Dossa F, Chesney TR, Acuna SA, Baxter NN. A watch-and-wait approach for locally advanced rectal cancer after a clinical complete response following neoadjuvant chemoradiation: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2017;2:501–13.
44. van der Valk M. The International Watch & Wait database (IWWD) for rectal cancer: An update. J Clin Oncol 2017;35 suppl:521.
45. Donato V, Valeriani M, Zurlo A. Short course radiation therapy for elderly cancer patients. Evidences from the literature review. Crit Rev Oncol Hematol 2003;45:305–11.
46. Ngan SY, Burmeister B, Fisher RJ, et al. Randomized trial of short-course radiotherapy versus long-course chemoradiation comparing rates of local recurrence in patients with T3 rectal cancer: Trans-Tasman Radiation Oncology Group trial 01.04. J Clin Oncol 2012;30:3827–33.
47. McCleary NJ, Dotan E, Browner I. Refining the chemotherapy approach for older patients with colon cancer. J Clin Oncol 2014;32:2570–80.
48. Millan M, Merino S, Caro A, et al. Treatment of colorectal cancer in the elderly. World J Gastrointest Oncol 2015;7:204–20.
49. Quasar Collaborative Group, Gray R, Barnwell J, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370:2020–9.
50. Andre T, Boni C, Navarro M, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 2009;27:3109–16.
51. Tournigand C, Andre T, Bonnetain F, et al. Adjuvant therapy with fluorouracil and oxaliplatin in stage II and elderly patients (between ages 70 and 75 years) with colon cancer: subgroup analyses of the Multicenter International Study of Oxaliplatin, Fluorouracil, and Leucovorin in the Adjuvant Treatment of Colon Cancer trial. J Clin Oncol 2012;30:3353–60.
52. Winder T, Lenz HJ. Molecular predictive and prognostic markers in colon cancer. Cancer Treat Rev 2010;36:550–6.
53. Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
54. Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
55. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
56. Aparicio T, Schischmanoff O, Poupardin C, et al. Deficient mismatch repair phenotype is a prognostic factor for colorectal cancer in elderly patients. Dig Liver Dis 2013;45:245–50.
57. Sargent DJ, Goldberg RM, Jacobson SD, et al. A pooled analysis of adjuvant chemotherapy for resected colon cancer in elderly patients. N Engl J Med 2001;345:1091–7.
58. Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
59. Twelves C, Scheithauer W, McKendrick J, et al. Capecitabine versus 5-fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results from the X-ACT trial with analysis by age and preliminary evidence of a pharmacodynamic marker of efficacy. Ann Oncol 2012;23:1190–7.
60. Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses. J Clin Oncol 2011;29:3768–74.
61. Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
62. Haller DG, Cassidy J, Tabernero J, et al. Efficacy findings from a randomized phase III trial of capecitabine plus oxaliplatin versus bolus 5-FU/LV for stage III colon cancer (NO16968): impact of age on disease-free survival (DFS) [abstract]. J Clin Oncol 2010;28:3521.
63. Schmoll HJ, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
64. McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
65. Kahn KL, Adams JL, Weeks JC, et al. Adjuvant chemotherapy use and adverse events among older patients with stage III colon cancer. JAMA 2010;303:1037–45.
66. Haller DG, O’Connell MJ, Cartwright TH, et al. Impact of age and medical comorbidity on adjuvant treatment outcomes for stage III colon cancer: a pooled analysis of individual patient data from four randomized, controlled trials. Ann Oncol 2015;26:715-24.
67. Aparicio T, Francois E, Cristol-Dalstein L, et al. PRODIGE 34-FFCD 1402-ADAGE: Adjuvant chemotherapy in elderly patients with resected stage III colon cancer: A randomized phase 3 trial. Dig Liver Dis 2016;48:206–7.
68. Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
69. Mahoney T, Kuo YH, Topilow A, Davis JM. Stage III colon cancers: why adjuvant chemotherapy is not offered to elderly patients. Arch Surg 2000;135:182–5.
70. Sanoff HK, Carpenter WR, Sturmer T, et al. Effect of adjuvant chemotherapy on survival of patients with stage III colon cancer diagnosed after age 75 years. J Clin Oncol 2012;30:2624–34.
71. Folprecht G, Cunningham D, Ross P, et al. Efficacy of 5-fluorouracil-based chemotherapy in elderly patients with metastatic colorectal cancer: a pooled analysis of clinical trials. Ann Oncol 2004;15:1330–8.
72. Van Cutsem E, Cervantes A, Nordlinger B, Arnold D, ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 Suppl 3:iii1–9.
73. Goldberg RM, Tabah-Fisch I, Bleiberg H, et al. Pooled analysis of safety and efficacy of oxaliplatin plus fluorouracil/leucovorin administered bimonthly in elderly patients with colorectal cancer. J Clin Oncol 2006;24:4085–91.
74. Seymour MT, Thompson LC, Wasan HS, et al. Chemotherapy options in elderly and frail patients with metastatic colorectal cancer (MRC FOCUS2): an open-label, randomised factorial trial. Lancet 2011;377:1749–59.
75. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer--a GERCOR study. J Clin Oncol 2006;24:394–400.
76. Figer A, Perez-Staub N, Carola E, et al. FOLFOX in patients aged between 76 and 80 years with metastatic colorectal cancer: an exploratory cohort of the OPTIMOX1 study. Cancer 2007;110:2666–71.
77. Chibaudel B, Maindrault-Goebel F, Lledo G, et al. Can chemotherapy be discontinued in unresectable metastatic colorectal cancer? The GERCOR OPTIMOX2 Study. J Clin Oncol 2009;27:5727–33.
78. Folprecht G, Seymour MT, Saltz L, et al. Irinotecan/fluorouracil combination in first-line therapy of older and younger patients with metastatic colorectal cancer: combined analysis of 2,691 patients in randomized controlled trials. J Clin Oncol 2008;26:1443–51.
79. Souglakos J, Pallis A, Kakolyris S, et al. Combination of irinotecan (CPT-11) plus 5-fluorouracil and leucovorin (FOLFIRI regimen) as first line treatment for elderly patients with metastatic colorectal cancer: a phase II trial. Oncology 2005;69:384–90.
80. Aparicio T, Lavau-Denes S, Phelip JM, et al. Randomized phase III trial in elderly patients comparing LV5FU2 with or without irinotecan for first-line treatment of metastatic colorectal cancer (FFCD 2001-02). Ann Oncol 2016;27:121–7.
81. Aparicio T, Gargot D, Teillet L, et al. Geriatric factors analyses from FFCD 2001-02 phase III study of first-line chemotherapy for elderly metastatic colorectal cancer patients. Eur J Cancer 2017;74:98–108.
82. Kabbinavar FF, Hurwitz HI, Yi J, et al. Addition of bevacizumab to fluorouracil-based first-line treatment of metastatic colorectal cancer: pooled analysis of cohorts of older patients from two randomized clinical trials. J Clin Oncol 2009;27:199–205.
83. Cassidy J, Saltz LB, Giantonio BJ, et al. Effect of bevacizumab in older patients with metastatic colorectal cancer: pooled analysis of four randomized studies. J Cancer Res Clin Oncol 2010;136:737–43.
84. Van Cutsem E, Rivera F, Berry S, et al. Safety and efficacy of first-line bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in metastatic colorectal cancer: the BEAT study. Ann Oncol 2009;20:1842–7.
85. Kozloff MF, Berlin J, Flynn PJ, et al. Clinical outcomes in elderly patients with metastatic colorectal cancer receiving bevacizumab and chemotherapy: results from the BRiTE observational cohort study. Oncology 2010;78:329–39.
86. Cunningham D, Lang I, Marcuello E, et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): an open-label, randomised phase 3 trial. Lancet Oncol 2013;14:1077–85.
87. Aparicio T, Bouche O, Taieb J, et al. Bevacizumab+chemotherapy versus chemotherapy alone in elderly patients with untreated metastatic colorectal cancer: a randomized phase II trial-PRODIGE 20 study results. Ann Oncol 2018;29:133–8.
88. Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 2012;30:3499–506.
89. Ruff P, Van Cutsem E, Lakomy R, et al. Observed benefit and safety of aflibercept in elderly patients with metastatic colorectal cancer: An age-based analysis from the randomized placebo-controlled phase III VELOUR trial. J Geriatr Oncol 2018;9:32–9.
90. Obermannova R, Van Cutsem E, Yoshino T, et al. Subgroup analysis in RAISE: a randomized, double-blind phase III study of irinotecan, folinic acid, and 5-fluorouracil (FOLFIRI) plus ramucirumab or placebo in patients with metastatic colorectal carcinoma progression. Ann Oncol 2016;27:2082–90.
91. Bouchahda M, Macarulla T, Spano JP, et al. Cetuximab efficacy and safety in a retrospective cohort of elderly patients with heavily pretreated metastatic colorectal cancer. Crit Rev Oncol Hematol 2008;67:255-62.
92. Douillard JY, Siena S, Cassidy J, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol 2014;25:1346–55.
93. Sastre J, Gravalos C, Rivera F, et al. First-line cetuximab plus capecitabine in elderly patients with advanced colorectal cancer: clinical outcome and subgroup analysis according to KRAS status from a Spanish TTD Group Study. Oncologist 2012;17:339–45.
94. Dotan E, Devarajan K, D’Silva AJ, et al. Patterns of use and tolerance of anti-epidermal growth factor receptor antibodies in older adults with metastatic colorectal cancer. Clin Colorectal Cancer 2014;13:192–8.
95. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:303–12.
96. Van Cutsem E, Sobrero A, Siena S, et al. Regorafenib (REG) in progressive metastatic colorectal cancer (mCRC): Analysis of age subgroups in the phase III CORRECT trial [abstract]. J Clin Oncol 2013;31(15 suppl):3636-3636.
97. Bekaii-Saab TS, Ou FS, Anderson DM, et al. Regorafenib dose optimization study (ReDOS): Randomized phase II trial to evaluate dosing strategies for regorafenib in refractory metastatic colorectal cancer (mCRC): an ACCRU Network study [abstract]. J Clin Oncol 2018;36(4 suppl):611-611.
98. Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909–19.
99. Koopman M, Kortman GA, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer 2009;100:266–73.
100. Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res 2014;20:5322–30.
101. Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
102. Le DT, Uram JN, Wang H, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20.
103. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–91.
104. Daste A, Domblides C, Gross-Goupil M, et al. Immune checkpoint inhibitors and elderly people: A review. Eur J Cancer 2017;82:155–66.
105. Elias R, Giobbie-Hurder A, McCleary NJ, et al. Efficacy of PD-1 & PD-L1 inhibitors in older adults: a meta-analysis. J Immunother Cancer 2018;6:26.
106. Singh H, Kim G, Maher VE, et al. FDA subset analysis of the safety of nivolumab in elderly patients with advanced cancers [abstract]. J Clin Oncol 2016;34(15 suppl):10010-10010.
107. Temel JS, Greer JA, El-Jawahri A, et al. Effects of early integrated palliative care in patients with lung and GI cancer: a randomized clinical trial. J Clin Oncol 2017;35:834–41.
108. Brighi N, Balducci L, Biasco G. Cancer in the elderly: is it time for palliative care in geriatric oncology? J Geriatr Oncol 2014;5:197–203.
109. Meropol NJ, Egleston BL, Buzaglo JS, et al. Cancer patient preferences for quality and length of life. Cancer 2008;113:3459–66.
110. Bagshaw SM, Stelfox HT, Johnson JA, et al. Long-term association between frailty and health-related quality of life among survivors of critical illness: a prospective multicenter cohort study. Crit Care Med 2015;43:973–82.
111. Lynch MP, Marcone D, Kagan SH. Developing a multidisciplinary geriatric oncology program in a community cancer center. Clin J Oncol Nurs 2007;11:929–33.
112. Sheridan J, Walsh P, Kevans D, et al. Determinants of short- and long-term survival from colorectal cancer in very elderly patients. J Geriatr Oncol 2014;5:376–83.
Introduction
Colorectal cancer (CRC) is the fourth most common cancer in the United States and has a high prevalence among the older population.1 In 2017, there were an estimated 135,430 new cases of CRC and 50,260 deaths due to CRC. It is the second leading cause of cancer death in the United States, and the death rate for patients with CRC increases with age (Figure).2
Although elderly persons are more frequently diagnosed with CRC, they are underrepresented in clinical trials. This may be due in part to stringent eligibility criteria in prospective randomized controlled trials that exclude older patients with certain comorbidities and decreased functional status. Hutchins and colleagues compared the proportion of persons aged 65 years and older enrolled in Southwest Oncology Group (SWOG) clinical trials and the proportion of persons in this age group in the US population with the same cancer diagnoses.5 They found that while 72% of the US population with CRC were aged ≥ 65 years, persons in this age group comprised only 40% of patients enrolled in SWOG trials. An update on this study performed after Medicare policy changed in 2000 to include coverage of costs incurred due to clinical trials showed an upward trend in the accrual of older patients in SWOG trials, from 25% during the period 1993–1996 to 38% during the period 2001–2003; however, the percentage of older patients with CRC on clinical trials overall remained stable from 1993 to 2003.6
The underrepresentation of older adults with CRC in clinical trials presents oncologists with a challenging task when practicing evidence-based medicine in this patient population. Analysis of a large claims database demonstrated that the use of multi-agent chemotherapy for the treatment of metastatic CRC in older adults increased over time, while the use of single-agent 5-fluorouracil (5-FU) decreased.7 However, the adoption of combination therapy with irinotecan or oxaliplatin in older adults lagged behind the initial adoption of these agents in younger patients. This data demonstrates that as the field of medical oncology evolves, providers are becoming more comfortable treating older patients with multiple medical problems using standard approved regimens.
Geriatric Assessment
Before treating older patients with cancer, it is necessary to define the patient’s physiological age, ideally through a multidisciplinary team evaluation.
The Eastern Cooperative Oncology Group performance status (ECOG PS) and Karnofsky Performance Status (KPS) are crude measures of functional status.12 Generally, elderly patients with good ECOG PS or KPS scores are considered fit enough to receive standard therapy similar to their younger counterparts. Evaluation of functional status using these performance scores is often suboptimal, resulting in patients with a normal or adequate performance status score who may still experience poor outcomes, including decreased survival and inability to tolerate treatment. A study that explored parameters among older patients that predict for increased risk of chemotherapy-related toxicities found that physician-rated KPS score did not accurately predict the risk for adverse events.13 Therefore, a CGA represents a better way to evaluate functional status and other domains.
Functional status can also be evaluated by self-reported tools such as activities of daily living, which refer to basic self-care, and instrumental activities of daily living (IADLs), which are essential for independent living in the community.14,15 Mobility, gait, and balance can also be measured using the “Timed Get Up and Go” test and gait speed. Klepin et al found that faster gait speed was associated with overall survival (OS) in patients with metastatic cancer.16
Cognitive function is an important component of the geriatric assessment in older patients with cancer, as dementia is a prognostic factor for survival in the overall geriatric population. In a retrospective review, patients with dementia were less likely to have a biopsy-proven diagnosis and were twice as likely to have their CRC diagnosed postmortem.17 In addition, establishing that the patient has intact cognitive function prior to initiating treatment is essential to ensure that the patient can comply with treatment and understands when to report adverse effects. Nutritional status is an important portion of the geriatric assessment because malnutrition is associated with increased mortality and decreased tolerance for chemotherapy.18–20 Evaluating the patient’s psychosocial support is crucial as well because older patients are at greater risk of social isolation and depression.21 While the incidence of depression is lower in older adults with cancer than in younger adults with cancer, clinically significant depression is still noted in 3% to 25% of elderly cancer patients.22 Other critical components of the CGA are review of the patient’s comorbidities and medications to avoid complications of polypharmacy.
Both the Cancer and Aging Research Group (CARG) and Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) toxicity tools are valuable tools, as they predict chemotherapy tolerance in elderly patients.13,23 These tools can help guide discussions between oncologists and patients as well as the formulation of an appropriate treatment plan.24 Although toxicity tools can help to determine which patients are at risk for severe toxicity secondary to treatment, these tools do not replace the CGA. A prospective cohort study that evaluated the impact of CGA on tolerance to chemotherapy in older patients with cancer compared patients aged ≥ 70 years at the start of their treatment with chemotherapy (± radiation therapy) using geriatrician-delivered CGA versus standard care given by oncology.8 Patients who received geriatrician-guided CGA interventions tolerated chemotherapy better and completed treatments as planned (odds ratio 4.14 [95% confidence interval {CI} 1.50 to 11.42], P = 0.006) with fewer treatment modifications.
Unfortunately, the CGA is time-consuming to administer and difficult to incorporate into a busy oncology practice. Therefore, other screening models are used to identify patients who may benefit from a full CGA. The International Society of Geriatric Oncology performed a systematic review of screening tools used to identify older cancer patients in need of geriatric assessment and found that the 3 most studied screening tools are the G8, the Vulnerable Elders Survey-13 (VES-13), and the Flemish version of the Triage Risk Screening Tool.25 Another study found that the G8 was more sensitive than the VES-13 (76.5% versus 68.7%, P = 0.0046), whereas the VES-13 was more specific than the G8 (74.3% versus 64.4%, P < 0.0001).26 In addition to providing guidance to initiate a full geriatric assessment, these screening tools may assist in decision making for older cancer patients, especially those with advanced disease.
Surgery
Early-Stage Disease
When possible, surgical resection of colorectal tumors is the primary treatment in both the curative setting and to avoid complications, such as obstruction or perforation.27 Multiple studies have shown that fit elderly patients benefit from curative surgery similarly to their younger counterparts.27–29 With the growing population of persons aged 65 years or older, surgeons are becoming more comfortable with operating on the elderly.4 However, a large systematic review of 28 independent studies with a total of 34,194 patients showed that older patients were less likely to undergo curative surgery.30 Eligibility for surgery should not be determined by age alone, but rather should be based on a full assessment of the patient’s health, including comorbidities, functional status, nutrition, cognition, social support, and psychological status. The impact of age on short-term outcomes after colorectal surgery in terms of 30-day postoperative morbidity and mortality rates was explored in a study that divided patients into 2 groups: those aged ≥ 80 years (mean age 85) and those aged < 80 years (mean age 55.3).31 There were no statistical differences in 30-day postoperative morbidity and mortality rates between the 2 groups, and preexisting comorbidities and urgent nature of surgery were important predictors of colorectal surgery outcomes in the older adults, results that have been seen in several other studies.28,30 When possible, laparoscopic surgery is preferred as it is associated with less intraoperative blood loss, less postoperative pain, reduced postoperative ileus, a shorter hospital stay, and fewer cardiovascular and pulmonary complications.32 The Preoperative Assessment of Cancer in the Elderly (PACE), which combines surgical risk assessment tools with CGA tools, can assist surgeons in determining candidacy for surgery and help decrease unequal access to surgery in the geriatric population.33
Metastasectomy
A large international multicenter cohort study explored the outcomes of patients aged ≥ 70 years who underwent liver resection of colorectal metastases. The study investigatorsfound that neoadjuvant chemotherapy was used less frequently and less extensive surgery was performed in elderly patients than in younger patients.34 Sixty-day postoperative mortality was slightly higher (3.8% versus 1.6%, P < 0.001) and 3-year OS was slightly lower (57.1% versus 60.2%, P < 0.001) in the elderly group as compared to their younger counterparts, but overall the outcomes after liver surgery were similar. Therefore, the management of liver metastases in oligometastatic disease in elderly patients fit for surgery should be the same as that offered to younger patients. Since outcomes are comparable, older patients should be offered neoadjuvant chemotherapy, as several studies have shown similar response rates and OS in younger and older patients.35,36
Rectal Cancer
The standard of care for locally advanced rectal cancer is combined modality treatment with radiation and chemotherapy followed by total mesorectal excision. However, given conflicting data regarding the ability of elderly patients to tolerate neoadjuvant 5-FU-based chemotherapy and radiation, elderly patients are treated with trimodality therapy less often than their younger counterparts.37,38 A systematic review of 22 randomized trials involving 8507 patients with rectal cancer showed that adjuvant radiation therapy could reduce the risk of local recurrence and death from rectal cancer in patients of all ages.39 However, the risk of noncancer-related death was increased in the older population. The Stockholm II trial showed similar benefits of preoperative radiation overall, but this benefit did not extend to patients older than 68 years because of an increased risk of morbidity and mortality.40 In older patients, mortality from noncancer causes within the first 6 months after surgery was higher in the group that received perioperative radiation than in the group that did not receive radiation. Elderly patients (age > 68 years) accounted for most of the mortality, which was predominantly due to cardiovascular disease.
A retrospective study of 36 patients aged ≥ 70 years with rectal cancer evaluated the toxicity and feasibility of neoadjuvant 5-FU combined with pelvic radiation for treating locally advanced rectal cancer. Patients were classified as healthy and “fit” or “vulnerable” based on the presence of comorbidities.41 This study demonstrated that tolerability and response to neoadjuvant chemotherapy and radiation as well as ability to undergo surgery were similar in “vulnerable” patients and “fit” patients. Conversely, Margalit and colleagues studied the rate of treatment deviations in elderly patients with rectal cancer treated with combined modality therapy and found that most patients required early termination of treatment, treatment interruptions, or dose reductions.42 While trimodality treatment is the standard of care in rectal cancer, there is conflicting data from retrospective studies regarding the tolerability and feasibility of this approach. It is important to proceed with caution but to still consider fit older patients with locally advanced rectal cancer for neoadjuvant chemotherapy and radiation followed by surgery.
In patients who have a complete response (CR) to neoadjuvant chemoradiation, watchful waiting rather than proceeding to surgery may be a reasonable strategy, especially in older patients. A systematic review of 867 patients with locally advanced rectal cancer showed no statistically significant difference in OS between patients who were observed with watchful waiting and those who underwent surgery.43 The International Watch and Wait Database includes 679 patients who were managed with a watch-and-wait regimen because they had a clinical CR after chemoradiation. An outcomes analysis of these patients showed that 25% had local regrowth, with 3-year OS of 91% overall and 87% in patients with local regrowth.44 In most patients (84%), regrowth of the tumor occurred within the first 2 years of follow up.
In frail older adults, for whom longer courses of treatment are not feasible or chemotherapy is contraindicated, short-course radiation therapy can be considered either in the neoadjuvant setting or alone for palliation.45 A randomized trial of short-course radiation versus long-course chemoradiation in patients with T3 rectal cancer found that the difference in 3-year local recurrence rates was not statistically significant.46
Chemotherapy
An expected natural decline in function occurs with age, but given the great variability that exists between patients, it is important to focus on physiologic age rather than chronologic age to determine ability to receive and tolerate anticancer treatment. Decreases in renal and hepatic function, cognitive impairment, changes in gastrointestinal motility, decrements in cardiac and bone marrow reserves, as well as comorbidities and polypharmacy affect a patient’s ability to tolerate chemotherapy.47,48 Toxicity tools such as CARG and CRASH can help to predict severity of toxicity with chemotherapy.13,23 The information provided by these tools can help guide conversations between the oncologist and patient regarding treatment plans.
Adjuvant Chemotherapy for Early-Stage Disease
Stage II Disease
Defining treatment guidelines for older patients with stage II colon cancer is difficult due to lack of data that shows benefit in this population. The QUASAR (Quick and Simple and Reliable) group’s prospective study of adjuvant single-agent 5-FU in stage II colon cancer patients showed an absolute improvement in survival of 3.6% when 5-FU was given after surgery (95% CI 1.0 to 6.0).49 The subgroup analysis of patients aged ≥ 70 years showed a limited benefit of adjuvant 5-FU (hazard ratio [HR] 1.13 [95% CI 0.74 to 1.75]). Given the limited benefit, adjuvant 5-FU for elderly patients with stage II colon cancer should be used judiciously as patients may have competing causes of morbidity or mortality.
The use of oxaliplatin-based therapy in the adjuvant setting for stage II disease was evaluated in a subgroup analysis of the MOSAIC study (Multicenter International Study of Oxaliplatin/5-FU/Leucovorin in the Adjuvant Treatment of Colon Cancer).50 Adjuvant oxaliplatin-based treatment may be offered to patients with stage II colon cancer that carries high-risk features (poorly differentiated histology, lymphovascular invasion, bowel obstruction and/or perforation, < 12 lymph nodes sampled, perineural invasion, or indeterminate or positive margins) due to a trend toward improved disease-free survival (DFS) at 5 years. Patients in this group who received adjuvant FOLFOX (leucovorin, oxaliplatin, 5-FU) versus 5-FU/leucovorin had a DFS of 82.3% versus 74.6%, respectively (HR 0.72 [95% CI 0.50 to 1.02]), a difference that was not statistically significant. A subgroup analysis of 315 patients aged 70 to 75 years with stage II colon cancer enrolled in the MOSAIC study found no statistically significant DFS or OS benefit with the addition of oxaliplatin to 5-FU/leucovorin.51 Therefore, use of this platinum/fluoropyrimidine combination for adjuvant therapy for high-risk stage II disease in older patients remains controversial given its associated risks and the lack of definitive data demonstrating a benefit in this patient group. Decisions regarding this therapy should be made through a shared discussion with patients about its risks and benefits.
Microsatellite status is an important biomarker in the evaluation of stage II CRC. Microsatellite stability is a marker of a functioning DNA mismatch repair system. In patients with colon cancer, tumor microsatellite stability is classified based on the percentage of abnormal microsatellite regions.52 Several studies have shown that patients with tumors that display high microsatellite instability (MSI-H) have an improved prognosis over patients with microsatellite stable tumors.53,54 While patients with stage II MSI-H colon cancer have better outcomes, MSI is associated with a reduced response to treatment with fluoropyrimidines, as demonstrated in a systematic review that found that patients with tumors with MSI obtained no benefit from adjuvant 5-FU (HR 1.24 [95% CI 0.72 to 2.14]).55 Aparicio and colleagues reported an increased prevalence of MSI-H tumors with increasing age.56 Therefore, mismatch repair phenotype should be considered when making adjuvant chemotherapy decisions in the older adult with colon cancer, as it may affect the decision to recommend single-agent 5-FU treatment.
Stage III Disease
The use of single-agent 5-FU for stage III resected CRC has been evaluated in multiple studies. Sargent et al performed a pooled analysis of 3351 patients from 7 randomized phase 3 trials comparing surgery and adjuvant 5-FU-based chemotherapy versus surgery alone in stage II or III colon cancer patients.57 Adjuvant chemotherapy was associated with improvement in both OS and time to tumor recurrence (HR 0.76 and 0.68, respectively). The 5-year OS was 71% for those who received adjuvant treatment and 64% for those who were treated with surgery alone. The benefit of adjuvant treatment was independent of age, and there was no difference in toxicity across age groups, except for 1 study which showed increased rates of leukopenia in the elderly. The oral fluoropyrimidine capecitabine was shown to be an effective alternative to 5-FU plus leucovorin as adjuvant treatment for those with resected stage III colon cancer.58 However, in the subgroup analysis of DFS in the intention-to-treat group, the improvement in DFS was not statistically significant in those aged ≥ 70 years. This study justified the phase 3 Xeloda in Adjuvant Colon Cancer Therapy (X-ACT) trial, which compared capecitabine and 5-FU/leucovorin as adjuvant therapy in patients with resected stage III colon cancer.59 The X-ACT trial showed no significant effect of age on DFS or OS.
The addition of oxaliplatin to 5-FU in the adjuvant setting for stage III tumors has been studied and debated in the elderly population in multiple trials. The MOSAIC trial investigated FOLFOX versus 5-FU/leucovorin in the adjuvant setting.50 The addition of oxaliplatin was associated with a DFS and OS benefit, with a 20% reduction in risk of colon cancer recurrence and 16% reduction in risk of death in all patients. The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-07 trial then studied 2409 patients with stage II or III colon cancer treated with weekly bolus 5-FU/leucovorin with or without oxaliplatin.60 In this study, OS was significantly improved with the addition of oxaliplatin in patients younger than 70 years, but OS at 5 years was 4.7% worse for patients aged ≥ 70 years treated with weekly 5-FU/leucovorin and oxaliplatin compared with those treated with weekly 5-FU/leucovorin (71.6% versus 76.3%, respectively). In contrast, the XELOXA trial (NO16968), which randomly assigned stage III colon cancer patients to capecitabine and oxaliplatin (XELOX) or bolus 5-FU/leucovorin (standard of care at study start), showed an efficacy benefit, albeit not statistically significant, in patients aged ≥ 70 years (HR 0.87 [95% CI 0.63 to 1.18]).61–63
The Adjuvant Colon Cancer Endpoints (ACCENT) database included 7 randomized trials totaling 14,528 patients with stage II or III colon cancer treated with adjuvant 5-FU with or without oxaliplatin or irinotecan.64 Subgroup analysis of patients aged ≥ 70 years (n = 2575) showed no benefit with an oxaliplatin-based regimen in DFS (HR 0.94 [95% CI 0.78 to 1.13]) or OS (HR 1.04 [95% CI, 0.85 to 1.27]). Based on these studies and the increased toxicity with oxaliplatin, oxaliplatin-based adjuvant chemotherapy is utilized less often than single-agent 5-FU in geriatric patients with early-stage colon cancer.65 Conversely, a recent pooled analysis of individual patient data from 4 randomized trials (NSABP C-08, XELOXA, X-ACT, and AVANT) showed improved DFS and OS with adjuvant XELOX or FOLFOX over single-agent 5-FU in patients aged ≥ 70 years (DFS HR 0.77 [95% CI 0.62 to 0.95], P = 0.014; OS HR 0.78 [95% CI 0.61 to 0.99], P = 0.045).66 This analysis also showed that grade 3 and 4 adverse events related to oxaliplatin were similar across age groups.
These data come from post-hoc analyses, and there is no prospective data to steer decision making in elderly patients with early-stage CRC (Table).
It is well established that patients with stage III colon cancer benefit from oxaliplatin-based adjuvant chemotherapy after curative surgical resection.68 However, older patients are less likely to be referred to oncology as compared with their younger counterparts, due to the conflicting data regarding the benefit of this approach in older adults. Studies have shown that when the referral is placed, the geriatric population is less likely to receive chemotherapy.69 Sanoff et al analyzed 4 data sets (SEER-Medicare, National Comprehensive Cancer Network, New York State Cancer Registry, and Cancer Care Outcomes Research and Surveillance Consortium) to assess the benefit of adjuvant chemotherapy for resected stage III CRC among patients aged ≥ 75 years. Their analysis showed that only 40% of patients evaluated received adjuvant chemotherapy for stage III CRC after surgical resection.70
Summary
Prospective data to guide the treatment of older patients with early-stage CRC in the adjuvant setting is lacking. For fit older patients with stage II disease, limited benefit will be derived from single-agent 5-FU. For those with stage III CRC, the benefit and toxicities of fluoropyrimidines as adjuvant therapy appear to be similar regardless of age. The addition of oxaliplatin to fluoropyrimidines in patients aged ≥ 70 years has not been proven to improve DFS or OS and could result in an incremental toxicity profile. Therefore, treatment plans must be individualized, and decisions should be made through an informed discussion evaluating the overall risk/benefit ratio of each approach.
Metastatic Disease
Palliative Chemotherapy
Approximately 20% of patients with CRC are diagnosed with metastatic disease at presentation, and 35% to 40% develop metastatic disease following surgery and adjuvant therapy.2 The mainstay of treatment in this population is systemic therapy in the form of chemotherapy with or without biologic agents. In this setting, several prospective studies specific to older adults have been completed, providing more evidence-based guidance to oncologists who see these patients. Folprecht et al retrospectively reviewed data from 22 clinical trials evaluating 5-FU-based palliative chemotherapy in 3825 patients with metastatic CRC, including 629 patients aged ≥ 70 years.71 OS in elderly patients (10.8 months [95% CI 9.7 to 11.8]) was equivalent to that in younger patients (11.3 months [95% CI 10.9 to 11.7], P = 0.31). Similarly, relative risk and progression-free survival (PFS) were comparable irrespective of age.
Standard of care for most patients with metastatic colon cancer consists of 5-FU/leucovorin in combination with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) with a monoclonal antibody.72 A retrospective pooled analysis of patients with metastatic CRC compared the safety and efficacy of FOLFOX4 in patients aged < 70 years versus those aged ≥ 70 years.73 While age ≥ 70 years was associated with an increased rate of grade ≥ 3 hematologic toxicity, it was not associated with increased rates of severe neurologic events, diarrhea, nausea, vomiting, infection, 60-day mortality, or overall incidence of grade ≥ 3 toxicity. The benefit of treatment was consistent across both age groups; therefore, age alone should not exclude an otherwise healthy individual from receiving FOLFOX.
These post-hoc analyses show that fit older patients who were candidates for trial participation tolerated these treatments well; however, these treatments may be more challenging for less fit older adults. The UK Medical Research Council FOCUS2 (Fluorouracil, Oxaliplatin, CPT11 [irinotecan]: Use and Sequencing) study was a prospective phase 3 trial that included 459 patients with metastatic CRC who were deemed too frail or not fit enough for standard-dose chemotherapy by their oncologists.74 In this group, 43% of patients were older than 75 years and 13% were older than 80 years. Patients were randomly assigned to receive infusional 5-FU with levofolinate; oxaliplatin and 5-FU; capecitabine; or oxaliplatin and capecitabine; all regimens were initiated with an empiric 20% dose reduction. The addition of oxaliplatin suggested some improvement in PFS, but this was not significant (5.8 months versus 4.5 months, HR 0.84 [95% CI 0.69 to 1.01], P = 0.07). Oxaliplatin was not associated with increased grade 3 or 4 toxicities. Capecitabine is often viewed as less toxic because it is taken by mouth, but this study found that replacement of 5-FU with capecitabine did not improve quality of life. Grade 3 or 4 toxicities were seen more frequently in those receiving capecitabine than in those receiving 5-FU (40% versus 30%, P = 0.03) in this older and frailer group of patients. As the patients on this study were frail and treatment dose was reduced, this data may not apply to fit older adults who are candidates for standard therapy.
When managing an older patient with metastatic CRC, it is important to tailor therapy based on goals of care, toxicity of proposed treatment, other comorbidities, and the patient’s functional status. One approach to minimizing toxicity in the older population is the stop-and-go strategy. The OPTIMOX1 study showed that stopping oxaliplatin after 6 cycles of FOLFOX7 and continuing maintenance therapy with infusional 5-FU/leucovorin alone for 12 cycles prior to reintroducing FOLFOX7 achieved efficacy similar to continuous FOLFOX4 with decreased toxicity.75 Figer et al studied an exploratory cohort of 37 patients aged 76 to 80 years who were included in the OPTIMOX1 study.76 The overall relative risk, median PFS, and median OS did not differ between the older patients in this cohort and younger patients studied in the original study. Older patients did experience more neutropenia, neurotoxicity, and overall grade 3 to 4 toxicity, but there were no toxic deaths in patients older than 75 years. The approach of giving treatment breaks, as in OPTIMOX2, may also provide patients with better quality of life, but perhaps at the expense of cancer-related survival.77
The combination of irinotecan and 5-FU has also been studied as treatment for patients with metastatic CRC. A pooled analysis of 2691 patients aged ≥ 70 years with metastatic CRC across 4 phase 3 randomized trials investigating irinotecan and 5-FU demonstrated that irinotecan-containing chemotherapy provided similar benefits to both older and younger patients with similar risk of toxicity.78 A phase 2 trial studying FOLFIRI as first-line treatment in older metastatic CRC patients showed this to be a safe and active regimen with manageable toxicity.79 Another randomized phase 3 trial for older patients compared 5-FU/leucovorin with or without irinotecan for first-line treatment of metastatic CRC (FFCD 2001-02).80 The study accrued 282 patients aged ≥ 75 years (median age 80 years), and found that the addition of irinotecan to infusional 5-FU–based chemotherapy did not significantly increase either PFS or OS. Aparicio et al performed a substudy of baseline geriatric evaluation prior to treatment in the FFCD 2001-02 study and assessed the value of geriatric parameters for predicting outcomes (objective response rate [ORR], PFS, and OS).81 Multivariate analysis showed that none of the geriatric parameters were predictive of ORR or PFS but that normal IADL was associated with better OS. This combination may still be appropriate for some older patients with metastatic disease, while single- agent 5-FU may be more appropriate in frail patients.
Biologic Agents
VEGF Inhibitors
Targeted biologic agents have been studied in the treatment of metastatic CRC. Bevacizumab is a recombinant, humanized monoclonal antibody against vascular endothelial growth factor (VEGF) that is approved in the first-line setting for treatment of metastatic CRC. A pooled analysis examined 439 patients 65 years of age and older with metastatic CRC who received bevacizumab plus chemotherapy versus placebo plus chemotherapy.82 In this analysis, the addition of bevacizumab was associated with an improvement in OS (19.3 months versus 14.3 months, HR 0.7 [95% CI 0.55 to 0.90], P = 0.006) and in PFS (9.2 months versus 6.2 months, HR 0.52 [95% CI 0.40 to 0.67], P < 0.0001). Known adverse events associated with bevacizumab were seen in the bevacizumab plus chemotherapy group but not at increased rates in the older population compared to their younger counterparts. Conversely, another pooled analysis found that while there was a PFS and OS benefit in older patients receiving bevacizumab, there was an increased incidence of thrombotic events in patients older than 65 years.83 The BEAT (Bevacizumab Expanded Access Trial) and BRiTE (Bevacizumab Regimens Investigation of Treatment Effects) studies showed similar clinical outcomes across all age groups.84,85 While older patients experienced more arterial thromboembolic events with the addition of bevacizumab, other factors such as ECOG PS, prior anticoagulation, and history of arterial disease were more predictive of these adverse events than age.
The randomized phase 3 AVEX study explored the efficacy and tolerability of capecitabine plus bevacizumab versus capecitabine alone in 280 frail patients aged ≥ 70 years.86 PFS in the capecitabine/bevacizumab arm was 9.1 months versus 5.1 months in the capecitabine alone arm. While the OS difference was not statistically significant, patients in the capecitabine/bevacizumab arm had an OS of 20.7 months versus 16.8 months in the capecitabine alone group. As reported in prior studies, patients in the capecitabine/bevacizumab arm had increased rates of toxic events (40%) compared with those who received capecitabine alone (22%), with reports of hypertension, hand-foot syndrome, bleeding, and thrombotic events. More recently, the phase 2 PRODIGE 20 trial studied the addition of bevacizumab to chemotherapy (5-FU, FOLFOX, or FOLFIRI) based on physician choice in untreated metastatic CRC patients aged ≥ 75 years (median age 80 years).87 They found that the addition of bevacizumab to standard of care chemotherapy was both safe and effective. The adverse events seen with bevacizumab, such as hypertension and thrombotic events, were consistent with prior studies.
A newer antiangiogenic agent, ziv-aflibercept, has been approved for the second-line treatment of metastatic CRC. The VELOUR trial demonstrated that the addition of ziv-aflibercept to FOLFIRI benefited patients across all age groups compared with FOLFIRI plus placebo in patients who had failed prior oxaliplatin-based chemotherapy.88,89 Ramucirumab is a human IgG-1 monoclonal antibody approved in second-line treatment in combination with FOLFIRI. A subgroup analysis of the RAISE study showed that the survival benefit was similar in patients aged ≥ 65 years versus those < 65 years.90 Based on the above data, the use of a VEGF inhibitor in combination with chemotherapy should be considered in older patients with metastatic CRC. Furthermore, based on the conflicting data regarding the benefit of FOLFOX/FOLFIRI over single-agent 5-FU discussed above, the combination of capecitabine plus bevacizumab may be considered a front-line treatment option in older patients based on the AVEX study.
EGFR Inhibitors
Cetuximab and panitumumab are anti-epidermal growth factor receptor (EGFR) antibodies approved for the treatment of RAS wild-type metastatic CRC. Data regarding the use of EGFR inhibitors in the geriatric population is scarce and the data that does exist is conflicting.91,92 The PRIME study demonstrated that panitumumab plus FOLFOX had a PFS benefit compared to FOLFOX alone in KRAS wild-type metastatic CRC patients.92 While the study met its primary endpoint, the benefit did not translate to patients aged ≥ 65 years in subgroup analysis. Conversely, a retrospective study of the efficacy and safety of cetuximab in elderly patients with heavily pretreated metastatic CRC found similar efficacy in older and younger patients as well as no increased adverse events in the older population.91 A phase 2 trial investigating cetuximab as single-agent first-line treatment of metastatic CRC in fit older patients found cetuximab to be safe with moderate activity in this population, but did not support the use of cetuximab as first-line single-agent treatment in fit geriatric patients who may be candidates for combination therapy.93 Our group studied the patterns of use and tolerance of anti-EGFR antibodies in 117 older adults with metastatic CRC with a median age of 73 years.94 The study showed that older age at the time of treatment was associated with administration of anti-EGFR antibody as monotherapy rather than in combination with chemotherapy (P = 0.0009). We found no association between age and presence of grade 3 or higher toxicity. In addition, the toxicity profile seen in older patients was similar to what has been demonstrated in prior studies involving a younger patient population. Given the discordance seen between studies, additional prospective trials are needed to elucidate the efficacy and safety of EGFR inhibitors in the geriatric population.
Other Agents
Two newer agents approved in the treatment of metastatic CRC are regorafenib, a multikinase inhibitor, and trifluridine/tipiracil (TFD/TPI), a nucleoside analog combined with an inhibitor of thymidine phosphorylase. The phase 3 CORRECT trial studied regorafenib as monotherapy in previously treated metastatic CRC and found an OS benefit of 1.4 months and minimal PFS benefit.95 Van Cutsem et al performed a subgroup analysis by age and found similar OS benefit in patients < 65 years of age and ≥ 65 years.96 The most frequent adverse events grade 3 or higher were hand-foot syndrome, fatigue, diarrhea, hypertension, and desquamation/rash, which were seen at similar rates in both age groups. More recently, the phase 2 Regorafenib Dose Optimization Study (ReDOS) found that weekly dose escalation of regorafenib from 80 mg to 160 mg daily over 3 weeks was superior to the standard 160 mg daily dosing in patients with metastatic CRC.97 The dose escalation group had a longer median OS, although this difference was not statistically significant, as well as a more favorable toxicity profile. Therefore, this new dosing strategy may be a reasonable option for older patients with pretreated metastatic CRC. A study of TFD/TPI versus placebo in refractory metastatic CRC found an OS benefit of 7.1 months versus 5.3 months.98 In subgroup analyses, the OS benefit extended to both patients < 65 years and ≥ 65 years. Given the sparse data on these newer agents in the geriatric population and the modest benefit they provide to those with refractory metastatic CRC, more data is needed to determine their utility in elderly patients. The decision to use these agents in the older patients warrants a thorough discussion with the patient regarding risks, benefit, and treatment goals.
Immunotherapy
Between 3.5% and 6.5% of stage IV colorectal cancers are MSI-H and have deficient mismatch repair (dMMR).99–101 A recent phase 2 trial studied the use of pembrolizumab, an IgG4 monoclonal antibody against PD-1 (programmed cell death-1), in heavily pretreated patients with dMMR metastatic CRC, MMR-proficient (pMMR) metastatic CRC, and noncolorectal dMMR metastatic cancer.102 Patients with dMMR metastatic CRC had a 50% ORR and 89% disease control rate (DCR), as compared with an ORR of 0% and DCR of 16% in patients with pMMR metastatic CRC. There was also an OS and PFS benefit seen in the dMMR CRC group as compared with the pMMR CRC group. Another phase 2 study, CheckMate 142, studied the anti-PD-1 monoclonal antibody nivolumab with or without ipilimumab (a monoclonal antibody against cytotoxic T-lymphocyte antigen 4) in patients with dMMR and pMMR metastatic CRC.103 In the interim analysis, nivolumab was found to provide both disease control and durable response in patients with dMMR metastatic CRC.
While these studies led to the FDA approval of pembrolizumab and nivolumab for management of previously treated MSI-H or dMMR metastatic CRC, data on the use of immunotherapy in older adults is scarce. Immunosenescence, or the gradual deterioration of the immune system that comes with aging, may impact the efficacy of immune checkpoint inhibitors (ICI) in older patients with advanced cancer.104 There is conflicting data on the efficacy of PD-1 and programmed death ligand-1) PD-L1 inhibitors in older patients across different cancers. A meta-analysis of immunotherapy in older adults with a variety of malignancies showed overall efficacy comparable to that seen in adults younger than 65 years.105 However, another review found ICIs to be less effective in older patients with head and neck, non-small cell lung cancer, and renal cell carcinoma compared with their younger counterparts.104 Regarding the toxicity profile of ICIs in the elderly, similar rates of grade 3 or higher adverse events in patients younger than 65 years and older than 65 years have been reported.106 However, patients aged ≥ 70 years had increased rates of grade 3 to 5 adverse events as compared to patients younger than 65 years (71.7% versus 58.4%, respectively). Given the scant data on ICIs in older patients with MSI-H or dMMR metastatic CRC, more clinical trials inclusive of this population are needed in order to determine the efficacy and safety of immunotherapy.
Palliative Care
The incorporation of palliative care early following the diagnosis of cancer has been shown to improve quality of life, decrease depression, and help with symptom management.107 The triggers for geriatric patients to initiate palliative care may be different from those of younger patients, as older patients may have different goals of care.108 Older patients will often choose quality over quantity of life when making treatment decisions.109 The ideal medical treatment for the frail patient with colorectal cancer would focus on treating disease while providing palliative measures to help support the patient and improve quality of life. It is paramount that patients maintain functional independence as loss of independence is recognized as a major threat to an older patient’s quality of life.110 The optimal way to achieve these goals is through the efforts of a multidisciplinary care team including not only physicians and nurses, but also social workers, nutritionists, physical therapists, and family who can provide support for the patient’s psychosocial, cognitive, and medical needs.111 Although cancer and noncancer–related death occur more frequently in the geriatric population, data to guide a specific palliative care approach to the elderly population is lacking.108
Conclusion
Colorectal cancer is a disease of older adults with a median age at diagnosis of 67 years.1 With the aging population, oncologists will be faced with treating increasing numbers of older patients, and must adjust their practice to accommodate this population of patients. Treating geriatric patients is challenging given the lack of available data to guide the treatment approach. Although several prospective elderly-specific studies have been conducted evaluating treatments for metastatic CRC, most treatment decisions are made based on the available retrospective studies and pooled analyses. Oncologists must carefully consider and evaluate each patient based on physiologic age rather than chronologic age.112 Overall, older patients should be given the opportunity to receive standard of care treatments in the appropriate setting. The decision to modify treatment plans should be made after a thorough evaluation by a multidisciplinary team and a discussion with the patient regarding their goals and the risks and benefits of the treatment. Geriatric assessment tools can help the care team identify patients with various geriatric syndromes that may not be detected on routine oncology evaluation. This type of evaluation is time consuming and is rarely done in a busy oncology practice. Ongoing studies are aiming to develop a method to incorporate geriatric assessments into the care of older adults.Additional prospective trials targeting older, more frail patients are essential to improve upon our knowledge so we can provide best care for this growing elderly population.
Introduction
Colorectal cancer (CRC) is the fourth most common cancer in the United States and has a high prevalence among the older population.1 In 2017, there were an estimated 135,430 new cases of CRC and 50,260 deaths due to CRC. It is the second leading cause of cancer death in the United States, and the death rate for patients with CRC increases with age (Figure).2
Although elderly persons are more frequently diagnosed with CRC, they are underrepresented in clinical trials. This may be due in part to stringent eligibility criteria in prospective randomized controlled trials that exclude older patients with certain comorbidities and decreased functional status. Hutchins and colleagues compared the proportion of persons aged 65 years and older enrolled in Southwest Oncology Group (SWOG) clinical trials and the proportion of persons in this age group in the US population with the same cancer diagnoses.5 They found that while 72% of the US population with CRC were aged ≥ 65 years, persons in this age group comprised only 40% of patients enrolled in SWOG trials. An update on this study performed after Medicare policy changed in 2000 to include coverage of costs incurred due to clinical trials showed an upward trend in the accrual of older patients in SWOG trials, from 25% during the period 1993–1996 to 38% during the period 2001–2003; however, the percentage of older patients with CRC on clinical trials overall remained stable from 1993 to 2003.6
The underrepresentation of older adults with CRC in clinical trials presents oncologists with a challenging task when practicing evidence-based medicine in this patient population. Analysis of a large claims database demonstrated that the use of multi-agent chemotherapy for the treatment of metastatic CRC in older adults increased over time, while the use of single-agent 5-fluorouracil (5-FU) decreased.7 However, the adoption of combination therapy with irinotecan or oxaliplatin in older adults lagged behind the initial adoption of these agents in younger patients. This data demonstrates that as the field of medical oncology evolves, providers are becoming more comfortable treating older patients with multiple medical problems using standard approved regimens.
Geriatric Assessment
Before treating older patients with cancer, it is necessary to define the patient’s physiological age, ideally through a multidisciplinary team evaluation.
The Eastern Cooperative Oncology Group performance status (ECOG PS) and Karnofsky Performance Status (KPS) are crude measures of functional status.12 Generally, elderly patients with good ECOG PS or KPS scores are considered fit enough to receive standard therapy similar to their younger counterparts. Evaluation of functional status using these performance scores is often suboptimal, resulting in patients with a normal or adequate performance status score who may still experience poor outcomes, including decreased survival and inability to tolerate treatment. A study that explored parameters among older patients that predict for increased risk of chemotherapy-related toxicities found that physician-rated KPS score did not accurately predict the risk for adverse events.13 Therefore, a CGA represents a better way to evaluate functional status and other domains.
Functional status can also be evaluated by self-reported tools such as activities of daily living, which refer to basic self-care, and instrumental activities of daily living (IADLs), which are essential for independent living in the community.14,15 Mobility, gait, and balance can also be measured using the “Timed Get Up and Go” test and gait speed. Klepin et al found that faster gait speed was associated with overall survival (OS) in patients with metastatic cancer.16
Cognitive function is an important component of the geriatric assessment in older patients with cancer, as dementia is a prognostic factor for survival in the overall geriatric population. In a retrospective review, patients with dementia were less likely to have a biopsy-proven diagnosis and were twice as likely to have their CRC diagnosed postmortem.17 In addition, establishing that the patient has intact cognitive function prior to initiating treatment is essential to ensure that the patient can comply with treatment and understands when to report adverse effects. Nutritional status is an important portion of the geriatric assessment because malnutrition is associated with increased mortality and decreased tolerance for chemotherapy.18–20 Evaluating the patient’s psychosocial support is crucial as well because older patients are at greater risk of social isolation and depression.21 While the incidence of depression is lower in older adults with cancer than in younger adults with cancer, clinically significant depression is still noted in 3% to 25% of elderly cancer patients.22 Other critical components of the CGA are review of the patient’s comorbidities and medications to avoid complications of polypharmacy.
Both the Cancer and Aging Research Group (CARG) and Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) toxicity tools are valuable tools, as they predict chemotherapy tolerance in elderly patients.13,23 These tools can help guide discussions between oncologists and patients as well as the formulation of an appropriate treatment plan.24 Although toxicity tools can help to determine which patients are at risk for severe toxicity secondary to treatment, these tools do not replace the CGA. A prospective cohort study that evaluated the impact of CGA on tolerance to chemotherapy in older patients with cancer compared patients aged ≥ 70 years at the start of their treatment with chemotherapy (± radiation therapy) using geriatrician-delivered CGA versus standard care given by oncology.8 Patients who received geriatrician-guided CGA interventions tolerated chemotherapy better and completed treatments as planned (odds ratio 4.14 [95% confidence interval {CI} 1.50 to 11.42], P = 0.006) with fewer treatment modifications.
Unfortunately, the CGA is time-consuming to administer and difficult to incorporate into a busy oncology practice. Therefore, other screening models are used to identify patients who may benefit from a full CGA. The International Society of Geriatric Oncology performed a systematic review of screening tools used to identify older cancer patients in need of geriatric assessment and found that the 3 most studied screening tools are the G8, the Vulnerable Elders Survey-13 (VES-13), and the Flemish version of the Triage Risk Screening Tool.25 Another study found that the G8 was more sensitive than the VES-13 (76.5% versus 68.7%, P = 0.0046), whereas the VES-13 was more specific than the G8 (74.3% versus 64.4%, P < 0.0001).26 In addition to providing guidance to initiate a full geriatric assessment, these screening tools may assist in decision making for older cancer patients, especially those with advanced disease.
Surgery
Early-Stage Disease
When possible, surgical resection of colorectal tumors is the primary treatment in both the curative setting and to avoid complications, such as obstruction or perforation.27 Multiple studies have shown that fit elderly patients benefit from curative surgery similarly to their younger counterparts.27–29 With the growing population of persons aged 65 years or older, surgeons are becoming more comfortable with operating on the elderly.4 However, a large systematic review of 28 independent studies with a total of 34,194 patients showed that older patients were less likely to undergo curative surgery.30 Eligibility for surgery should not be determined by age alone, but rather should be based on a full assessment of the patient’s health, including comorbidities, functional status, nutrition, cognition, social support, and psychological status. The impact of age on short-term outcomes after colorectal surgery in terms of 30-day postoperative morbidity and mortality rates was explored in a study that divided patients into 2 groups: those aged ≥ 80 years (mean age 85) and those aged < 80 years (mean age 55.3).31 There were no statistical differences in 30-day postoperative morbidity and mortality rates between the 2 groups, and preexisting comorbidities and urgent nature of surgery were important predictors of colorectal surgery outcomes in the older adults, results that have been seen in several other studies.28,30 When possible, laparoscopic surgery is preferred as it is associated with less intraoperative blood loss, less postoperative pain, reduced postoperative ileus, a shorter hospital stay, and fewer cardiovascular and pulmonary complications.32 The Preoperative Assessment of Cancer in the Elderly (PACE), which combines surgical risk assessment tools with CGA tools, can assist surgeons in determining candidacy for surgery and help decrease unequal access to surgery in the geriatric population.33
Metastasectomy
A large international multicenter cohort study explored the outcomes of patients aged ≥ 70 years who underwent liver resection of colorectal metastases. The study investigatorsfound that neoadjuvant chemotherapy was used less frequently and less extensive surgery was performed in elderly patients than in younger patients.34 Sixty-day postoperative mortality was slightly higher (3.8% versus 1.6%, P < 0.001) and 3-year OS was slightly lower (57.1% versus 60.2%, P < 0.001) in the elderly group as compared to their younger counterparts, but overall the outcomes after liver surgery were similar. Therefore, the management of liver metastases in oligometastatic disease in elderly patients fit for surgery should be the same as that offered to younger patients. Since outcomes are comparable, older patients should be offered neoadjuvant chemotherapy, as several studies have shown similar response rates and OS in younger and older patients.35,36
Rectal Cancer
The standard of care for locally advanced rectal cancer is combined modality treatment with radiation and chemotherapy followed by total mesorectal excision. However, given conflicting data regarding the ability of elderly patients to tolerate neoadjuvant 5-FU-based chemotherapy and radiation, elderly patients are treated with trimodality therapy less often than their younger counterparts.37,38 A systematic review of 22 randomized trials involving 8507 patients with rectal cancer showed that adjuvant radiation therapy could reduce the risk of local recurrence and death from rectal cancer in patients of all ages.39 However, the risk of noncancer-related death was increased in the older population. The Stockholm II trial showed similar benefits of preoperative radiation overall, but this benefit did not extend to patients older than 68 years because of an increased risk of morbidity and mortality.40 In older patients, mortality from noncancer causes within the first 6 months after surgery was higher in the group that received perioperative radiation than in the group that did not receive radiation. Elderly patients (age > 68 years) accounted for most of the mortality, which was predominantly due to cardiovascular disease.
A retrospective study of 36 patients aged ≥ 70 years with rectal cancer evaluated the toxicity and feasibility of neoadjuvant 5-FU combined with pelvic radiation for treating locally advanced rectal cancer. Patients were classified as healthy and “fit” or “vulnerable” based on the presence of comorbidities.41 This study demonstrated that tolerability and response to neoadjuvant chemotherapy and radiation as well as ability to undergo surgery were similar in “vulnerable” patients and “fit” patients. Conversely, Margalit and colleagues studied the rate of treatment deviations in elderly patients with rectal cancer treated with combined modality therapy and found that most patients required early termination of treatment, treatment interruptions, or dose reductions.42 While trimodality treatment is the standard of care in rectal cancer, there is conflicting data from retrospective studies regarding the tolerability and feasibility of this approach. It is important to proceed with caution but to still consider fit older patients with locally advanced rectal cancer for neoadjuvant chemotherapy and radiation followed by surgery.
In patients who have a complete response (CR) to neoadjuvant chemoradiation, watchful waiting rather than proceeding to surgery may be a reasonable strategy, especially in older patients. A systematic review of 867 patients with locally advanced rectal cancer showed no statistically significant difference in OS between patients who were observed with watchful waiting and those who underwent surgery.43 The International Watch and Wait Database includes 679 patients who were managed with a watch-and-wait regimen because they had a clinical CR after chemoradiation. An outcomes analysis of these patients showed that 25% had local regrowth, with 3-year OS of 91% overall and 87% in patients with local regrowth.44 In most patients (84%), regrowth of the tumor occurred within the first 2 years of follow up.
In frail older adults, for whom longer courses of treatment are not feasible or chemotherapy is contraindicated, short-course radiation therapy can be considered either in the neoadjuvant setting or alone for palliation.45 A randomized trial of short-course radiation versus long-course chemoradiation in patients with T3 rectal cancer found that the difference in 3-year local recurrence rates was not statistically significant.46
Chemotherapy
An expected natural decline in function occurs with age, but given the great variability that exists between patients, it is important to focus on physiologic age rather than chronologic age to determine ability to receive and tolerate anticancer treatment. Decreases in renal and hepatic function, cognitive impairment, changes in gastrointestinal motility, decrements in cardiac and bone marrow reserves, as well as comorbidities and polypharmacy affect a patient’s ability to tolerate chemotherapy.47,48 Toxicity tools such as CARG and CRASH can help to predict severity of toxicity with chemotherapy.13,23 The information provided by these tools can help guide conversations between the oncologist and patient regarding treatment plans.
Adjuvant Chemotherapy for Early-Stage Disease
Stage II Disease
Defining treatment guidelines for older patients with stage II colon cancer is difficult due to lack of data that shows benefit in this population. The QUASAR (Quick and Simple and Reliable) group’s prospective study of adjuvant single-agent 5-FU in stage II colon cancer patients showed an absolute improvement in survival of 3.6% when 5-FU was given after surgery (95% CI 1.0 to 6.0).49 The subgroup analysis of patients aged ≥ 70 years showed a limited benefit of adjuvant 5-FU (hazard ratio [HR] 1.13 [95% CI 0.74 to 1.75]). Given the limited benefit, adjuvant 5-FU for elderly patients with stage II colon cancer should be used judiciously as patients may have competing causes of morbidity or mortality.
The use of oxaliplatin-based therapy in the adjuvant setting for stage II disease was evaluated in a subgroup analysis of the MOSAIC study (Multicenter International Study of Oxaliplatin/5-FU/Leucovorin in the Adjuvant Treatment of Colon Cancer).50 Adjuvant oxaliplatin-based treatment may be offered to patients with stage II colon cancer that carries high-risk features (poorly differentiated histology, lymphovascular invasion, bowel obstruction and/or perforation, < 12 lymph nodes sampled, perineural invasion, or indeterminate or positive margins) due to a trend toward improved disease-free survival (DFS) at 5 years. Patients in this group who received adjuvant FOLFOX (leucovorin, oxaliplatin, 5-FU) versus 5-FU/leucovorin had a DFS of 82.3% versus 74.6%, respectively (HR 0.72 [95% CI 0.50 to 1.02]), a difference that was not statistically significant. A subgroup analysis of 315 patients aged 70 to 75 years with stage II colon cancer enrolled in the MOSAIC study found no statistically significant DFS or OS benefit with the addition of oxaliplatin to 5-FU/leucovorin.51 Therefore, use of this platinum/fluoropyrimidine combination for adjuvant therapy for high-risk stage II disease in older patients remains controversial given its associated risks and the lack of definitive data demonstrating a benefit in this patient group. Decisions regarding this therapy should be made through a shared discussion with patients about its risks and benefits.
Microsatellite status is an important biomarker in the evaluation of stage II CRC. Microsatellite stability is a marker of a functioning DNA mismatch repair system. In patients with colon cancer, tumor microsatellite stability is classified based on the percentage of abnormal microsatellite regions.52 Several studies have shown that patients with tumors that display high microsatellite instability (MSI-H) have an improved prognosis over patients with microsatellite stable tumors.53,54 While patients with stage II MSI-H colon cancer have better outcomes, MSI is associated with a reduced response to treatment with fluoropyrimidines, as demonstrated in a systematic review that found that patients with tumors with MSI obtained no benefit from adjuvant 5-FU (HR 1.24 [95% CI 0.72 to 2.14]).55 Aparicio and colleagues reported an increased prevalence of MSI-H tumors with increasing age.56 Therefore, mismatch repair phenotype should be considered when making adjuvant chemotherapy decisions in the older adult with colon cancer, as it may affect the decision to recommend single-agent 5-FU treatment.
Stage III Disease
The use of single-agent 5-FU for stage III resected CRC has been evaluated in multiple studies. Sargent et al performed a pooled analysis of 3351 patients from 7 randomized phase 3 trials comparing surgery and adjuvant 5-FU-based chemotherapy versus surgery alone in stage II or III colon cancer patients.57 Adjuvant chemotherapy was associated with improvement in both OS and time to tumor recurrence (HR 0.76 and 0.68, respectively). The 5-year OS was 71% for those who received adjuvant treatment and 64% for those who were treated with surgery alone. The benefit of adjuvant treatment was independent of age, and there was no difference in toxicity across age groups, except for 1 study which showed increased rates of leukopenia in the elderly. The oral fluoropyrimidine capecitabine was shown to be an effective alternative to 5-FU plus leucovorin as adjuvant treatment for those with resected stage III colon cancer.58 However, in the subgroup analysis of DFS in the intention-to-treat group, the improvement in DFS was not statistically significant in those aged ≥ 70 years. This study justified the phase 3 Xeloda in Adjuvant Colon Cancer Therapy (X-ACT) trial, which compared capecitabine and 5-FU/leucovorin as adjuvant therapy in patients with resected stage III colon cancer.59 The X-ACT trial showed no significant effect of age on DFS or OS.
The addition of oxaliplatin to 5-FU in the adjuvant setting for stage III tumors has been studied and debated in the elderly population in multiple trials. The MOSAIC trial investigated FOLFOX versus 5-FU/leucovorin in the adjuvant setting.50 The addition of oxaliplatin was associated with a DFS and OS benefit, with a 20% reduction in risk of colon cancer recurrence and 16% reduction in risk of death in all patients. The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-07 trial then studied 2409 patients with stage II or III colon cancer treated with weekly bolus 5-FU/leucovorin with or without oxaliplatin.60 In this study, OS was significantly improved with the addition of oxaliplatin in patients younger than 70 years, but OS at 5 years was 4.7% worse for patients aged ≥ 70 years treated with weekly 5-FU/leucovorin and oxaliplatin compared with those treated with weekly 5-FU/leucovorin (71.6% versus 76.3%, respectively). In contrast, the XELOXA trial (NO16968), which randomly assigned stage III colon cancer patients to capecitabine and oxaliplatin (XELOX) or bolus 5-FU/leucovorin (standard of care at study start), showed an efficacy benefit, albeit not statistically significant, in patients aged ≥ 70 years (HR 0.87 [95% CI 0.63 to 1.18]).61–63
The Adjuvant Colon Cancer Endpoints (ACCENT) database included 7 randomized trials totaling 14,528 patients with stage II or III colon cancer treated with adjuvant 5-FU with or without oxaliplatin or irinotecan.64 Subgroup analysis of patients aged ≥ 70 years (n = 2575) showed no benefit with an oxaliplatin-based regimen in DFS (HR 0.94 [95% CI 0.78 to 1.13]) or OS (HR 1.04 [95% CI, 0.85 to 1.27]). Based on these studies and the increased toxicity with oxaliplatin, oxaliplatin-based adjuvant chemotherapy is utilized less often than single-agent 5-FU in geriatric patients with early-stage colon cancer.65 Conversely, a recent pooled analysis of individual patient data from 4 randomized trials (NSABP C-08, XELOXA, X-ACT, and AVANT) showed improved DFS and OS with adjuvant XELOX or FOLFOX over single-agent 5-FU in patients aged ≥ 70 years (DFS HR 0.77 [95% CI 0.62 to 0.95], P = 0.014; OS HR 0.78 [95% CI 0.61 to 0.99], P = 0.045).66 This analysis also showed that grade 3 and 4 adverse events related to oxaliplatin were similar across age groups.
These data come from post-hoc analyses, and there is no prospective data to steer decision making in elderly patients with early-stage CRC (Table).
It is well established that patients with stage III colon cancer benefit from oxaliplatin-based adjuvant chemotherapy after curative surgical resection.68 However, older patients are less likely to be referred to oncology as compared with their younger counterparts, due to the conflicting data regarding the benefit of this approach in older adults. Studies have shown that when the referral is placed, the geriatric population is less likely to receive chemotherapy.69 Sanoff et al analyzed 4 data sets (SEER-Medicare, National Comprehensive Cancer Network, New York State Cancer Registry, and Cancer Care Outcomes Research and Surveillance Consortium) to assess the benefit of adjuvant chemotherapy for resected stage III CRC among patients aged ≥ 75 years. Their analysis showed that only 40% of patients evaluated received adjuvant chemotherapy for stage III CRC after surgical resection.70
Summary
Prospective data to guide the treatment of older patients with early-stage CRC in the adjuvant setting is lacking. For fit older patients with stage II disease, limited benefit will be derived from single-agent 5-FU. For those with stage III CRC, the benefit and toxicities of fluoropyrimidines as adjuvant therapy appear to be similar regardless of age. The addition of oxaliplatin to fluoropyrimidines in patients aged ≥ 70 years has not been proven to improve DFS or OS and could result in an incremental toxicity profile. Therefore, treatment plans must be individualized, and decisions should be made through an informed discussion evaluating the overall risk/benefit ratio of each approach.
Metastatic Disease
Palliative Chemotherapy
Approximately 20% of patients with CRC are diagnosed with metastatic disease at presentation, and 35% to 40% develop metastatic disease following surgery and adjuvant therapy.2 The mainstay of treatment in this population is systemic therapy in the form of chemotherapy with or without biologic agents. In this setting, several prospective studies specific to older adults have been completed, providing more evidence-based guidance to oncologists who see these patients. Folprecht et al retrospectively reviewed data from 22 clinical trials evaluating 5-FU-based palliative chemotherapy in 3825 patients with metastatic CRC, including 629 patients aged ≥ 70 years.71 OS in elderly patients (10.8 months [95% CI 9.7 to 11.8]) was equivalent to that in younger patients (11.3 months [95% CI 10.9 to 11.7], P = 0.31). Similarly, relative risk and progression-free survival (PFS) were comparable irrespective of age.
Standard of care for most patients with metastatic colon cancer consists of 5-FU/leucovorin in combination with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) with a monoclonal antibody.72 A retrospective pooled analysis of patients with metastatic CRC compared the safety and efficacy of FOLFOX4 in patients aged < 70 years versus those aged ≥ 70 years.73 While age ≥ 70 years was associated with an increased rate of grade ≥ 3 hematologic toxicity, it was not associated with increased rates of severe neurologic events, diarrhea, nausea, vomiting, infection, 60-day mortality, or overall incidence of grade ≥ 3 toxicity. The benefit of treatment was consistent across both age groups; therefore, age alone should not exclude an otherwise healthy individual from receiving FOLFOX.
These post-hoc analyses show that fit older patients who were candidates for trial participation tolerated these treatments well; however, these treatments may be more challenging for less fit older adults. The UK Medical Research Council FOCUS2 (Fluorouracil, Oxaliplatin, CPT11 [irinotecan]: Use and Sequencing) study was a prospective phase 3 trial that included 459 patients with metastatic CRC who were deemed too frail or not fit enough for standard-dose chemotherapy by their oncologists.74 In this group, 43% of patients were older than 75 years and 13% were older than 80 years. Patients were randomly assigned to receive infusional 5-FU with levofolinate; oxaliplatin and 5-FU; capecitabine; or oxaliplatin and capecitabine; all regimens were initiated with an empiric 20% dose reduction. The addition of oxaliplatin suggested some improvement in PFS, but this was not significant (5.8 months versus 4.5 months, HR 0.84 [95% CI 0.69 to 1.01], P = 0.07). Oxaliplatin was not associated with increased grade 3 or 4 toxicities. Capecitabine is often viewed as less toxic because it is taken by mouth, but this study found that replacement of 5-FU with capecitabine did not improve quality of life. Grade 3 or 4 toxicities were seen more frequently in those receiving capecitabine than in those receiving 5-FU (40% versus 30%, P = 0.03) in this older and frailer group of patients. As the patients on this study were frail and treatment dose was reduced, this data may not apply to fit older adults who are candidates for standard therapy.
When managing an older patient with metastatic CRC, it is important to tailor therapy based on goals of care, toxicity of proposed treatment, other comorbidities, and the patient’s functional status. One approach to minimizing toxicity in the older population is the stop-and-go strategy. The OPTIMOX1 study showed that stopping oxaliplatin after 6 cycles of FOLFOX7 and continuing maintenance therapy with infusional 5-FU/leucovorin alone for 12 cycles prior to reintroducing FOLFOX7 achieved efficacy similar to continuous FOLFOX4 with decreased toxicity.75 Figer et al studied an exploratory cohort of 37 patients aged 76 to 80 years who were included in the OPTIMOX1 study.76 The overall relative risk, median PFS, and median OS did not differ between the older patients in this cohort and younger patients studied in the original study. Older patients did experience more neutropenia, neurotoxicity, and overall grade 3 to 4 toxicity, but there were no toxic deaths in patients older than 75 years. The approach of giving treatment breaks, as in OPTIMOX2, may also provide patients with better quality of life, but perhaps at the expense of cancer-related survival.77
The combination of irinotecan and 5-FU has also been studied as treatment for patients with metastatic CRC. A pooled analysis of 2691 patients aged ≥ 70 years with metastatic CRC across 4 phase 3 randomized trials investigating irinotecan and 5-FU demonstrated that irinotecan-containing chemotherapy provided similar benefits to both older and younger patients with similar risk of toxicity.78 A phase 2 trial studying FOLFIRI as first-line treatment in older metastatic CRC patients showed this to be a safe and active regimen with manageable toxicity.79 Another randomized phase 3 trial for older patients compared 5-FU/leucovorin with or without irinotecan for first-line treatment of metastatic CRC (FFCD 2001-02).80 The study accrued 282 patients aged ≥ 75 years (median age 80 years), and found that the addition of irinotecan to infusional 5-FU–based chemotherapy did not significantly increase either PFS or OS. Aparicio et al performed a substudy of baseline geriatric evaluation prior to treatment in the FFCD 2001-02 study and assessed the value of geriatric parameters for predicting outcomes (objective response rate [ORR], PFS, and OS).81 Multivariate analysis showed that none of the geriatric parameters were predictive of ORR or PFS but that normal IADL was associated with better OS. This combination may still be appropriate for some older patients with metastatic disease, while single- agent 5-FU may be more appropriate in frail patients.
Biologic Agents
VEGF Inhibitors
Targeted biologic agents have been studied in the treatment of metastatic CRC. Bevacizumab is a recombinant, humanized monoclonal antibody against vascular endothelial growth factor (VEGF) that is approved in the first-line setting for treatment of metastatic CRC. A pooled analysis examined 439 patients 65 years of age and older with metastatic CRC who received bevacizumab plus chemotherapy versus placebo plus chemotherapy.82 In this analysis, the addition of bevacizumab was associated with an improvement in OS (19.3 months versus 14.3 months, HR 0.7 [95% CI 0.55 to 0.90], P = 0.006) and in PFS (9.2 months versus 6.2 months, HR 0.52 [95% CI 0.40 to 0.67], P < 0.0001). Known adverse events associated with bevacizumab were seen in the bevacizumab plus chemotherapy group but not at increased rates in the older population compared to their younger counterparts. Conversely, another pooled analysis found that while there was a PFS and OS benefit in older patients receiving bevacizumab, there was an increased incidence of thrombotic events in patients older than 65 years.83 The BEAT (Bevacizumab Expanded Access Trial) and BRiTE (Bevacizumab Regimens Investigation of Treatment Effects) studies showed similar clinical outcomes across all age groups.84,85 While older patients experienced more arterial thromboembolic events with the addition of bevacizumab, other factors such as ECOG PS, prior anticoagulation, and history of arterial disease were more predictive of these adverse events than age.
The randomized phase 3 AVEX study explored the efficacy and tolerability of capecitabine plus bevacizumab versus capecitabine alone in 280 frail patients aged ≥ 70 years.86 PFS in the capecitabine/bevacizumab arm was 9.1 months versus 5.1 months in the capecitabine alone arm. While the OS difference was not statistically significant, patients in the capecitabine/bevacizumab arm had an OS of 20.7 months versus 16.8 months in the capecitabine alone group. As reported in prior studies, patients in the capecitabine/bevacizumab arm had increased rates of toxic events (40%) compared with those who received capecitabine alone (22%), with reports of hypertension, hand-foot syndrome, bleeding, and thrombotic events. More recently, the phase 2 PRODIGE 20 trial studied the addition of bevacizumab to chemotherapy (5-FU, FOLFOX, or FOLFIRI) based on physician choice in untreated metastatic CRC patients aged ≥ 75 years (median age 80 years).87 They found that the addition of bevacizumab to standard of care chemotherapy was both safe and effective. The adverse events seen with bevacizumab, such as hypertension and thrombotic events, were consistent with prior studies.
A newer antiangiogenic agent, ziv-aflibercept, has been approved for the second-line treatment of metastatic CRC. The VELOUR trial demonstrated that the addition of ziv-aflibercept to FOLFIRI benefited patients across all age groups compared with FOLFIRI plus placebo in patients who had failed prior oxaliplatin-based chemotherapy.88,89 Ramucirumab is a human IgG-1 monoclonal antibody approved in second-line treatment in combination with FOLFIRI. A subgroup analysis of the RAISE study showed that the survival benefit was similar in patients aged ≥ 65 years versus those < 65 years.90 Based on the above data, the use of a VEGF inhibitor in combination with chemotherapy should be considered in older patients with metastatic CRC. Furthermore, based on the conflicting data regarding the benefit of FOLFOX/FOLFIRI over single-agent 5-FU discussed above, the combination of capecitabine plus bevacizumab may be considered a front-line treatment option in older patients based on the AVEX study.
EGFR Inhibitors
Cetuximab and panitumumab are anti-epidermal growth factor receptor (EGFR) antibodies approved for the treatment of RAS wild-type metastatic CRC. Data regarding the use of EGFR inhibitors in the geriatric population is scarce and the data that does exist is conflicting.91,92 The PRIME study demonstrated that panitumumab plus FOLFOX had a PFS benefit compared to FOLFOX alone in KRAS wild-type metastatic CRC patients.92 While the study met its primary endpoint, the benefit did not translate to patients aged ≥ 65 years in subgroup analysis. Conversely, a retrospective study of the efficacy and safety of cetuximab in elderly patients with heavily pretreated metastatic CRC found similar efficacy in older and younger patients as well as no increased adverse events in the older population.91 A phase 2 trial investigating cetuximab as single-agent first-line treatment of metastatic CRC in fit older patients found cetuximab to be safe with moderate activity in this population, but did not support the use of cetuximab as first-line single-agent treatment in fit geriatric patients who may be candidates for combination therapy.93 Our group studied the patterns of use and tolerance of anti-EGFR antibodies in 117 older adults with metastatic CRC with a median age of 73 years.94 The study showed that older age at the time of treatment was associated with administration of anti-EGFR antibody as monotherapy rather than in combination with chemotherapy (P = 0.0009). We found no association between age and presence of grade 3 or higher toxicity. In addition, the toxicity profile seen in older patients was similar to what has been demonstrated in prior studies involving a younger patient population. Given the discordance seen between studies, additional prospective trials are needed to elucidate the efficacy and safety of EGFR inhibitors in the geriatric population.
Other Agents
Two newer agents approved in the treatment of metastatic CRC are regorafenib, a multikinase inhibitor, and trifluridine/tipiracil (TFD/TPI), a nucleoside analog combined with an inhibitor of thymidine phosphorylase. The phase 3 CORRECT trial studied regorafenib as monotherapy in previously treated metastatic CRC and found an OS benefit of 1.4 months and minimal PFS benefit.95 Van Cutsem et al performed a subgroup analysis by age and found similar OS benefit in patients < 65 years of age and ≥ 65 years.96 The most frequent adverse events grade 3 or higher were hand-foot syndrome, fatigue, diarrhea, hypertension, and desquamation/rash, which were seen at similar rates in both age groups. More recently, the phase 2 Regorafenib Dose Optimization Study (ReDOS) found that weekly dose escalation of regorafenib from 80 mg to 160 mg daily over 3 weeks was superior to the standard 160 mg daily dosing in patients with metastatic CRC.97 The dose escalation group had a longer median OS, although this difference was not statistically significant, as well as a more favorable toxicity profile. Therefore, this new dosing strategy may be a reasonable option for older patients with pretreated metastatic CRC. A study of TFD/TPI versus placebo in refractory metastatic CRC found an OS benefit of 7.1 months versus 5.3 months.98 In subgroup analyses, the OS benefit extended to both patients < 65 years and ≥ 65 years. Given the sparse data on these newer agents in the geriatric population and the modest benefit they provide to those with refractory metastatic CRC, more data is needed to determine their utility in elderly patients. The decision to use these agents in the older patients warrants a thorough discussion with the patient regarding risks, benefit, and treatment goals.
Immunotherapy
Between 3.5% and 6.5% of stage IV colorectal cancers are MSI-H and have deficient mismatch repair (dMMR).99–101 A recent phase 2 trial studied the use of pembrolizumab, an IgG4 monoclonal antibody against PD-1 (programmed cell death-1), in heavily pretreated patients with dMMR metastatic CRC, MMR-proficient (pMMR) metastatic CRC, and noncolorectal dMMR metastatic cancer.102 Patients with dMMR metastatic CRC had a 50% ORR and 89% disease control rate (DCR), as compared with an ORR of 0% and DCR of 16% in patients with pMMR metastatic CRC. There was also an OS and PFS benefit seen in the dMMR CRC group as compared with the pMMR CRC group. Another phase 2 study, CheckMate 142, studied the anti-PD-1 monoclonal antibody nivolumab with or without ipilimumab (a monoclonal antibody against cytotoxic T-lymphocyte antigen 4) in patients with dMMR and pMMR metastatic CRC.103 In the interim analysis, nivolumab was found to provide both disease control and durable response in patients with dMMR metastatic CRC.
While these studies led to the FDA approval of pembrolizumab and nivolumab for management of previously treated MSI-H or dMMR metastatic CRC, data on the use of immunotherapy in older adults is scarce. Immunosenescence, or the gradual deterioration of the immune system that comes with aging, may impact the efficacy of immune checkpoint inhibitors (ICI) in older patients with advanced cancer.104 There is conflicting data on the efficacy of PD-1 and programmed death ligand-1) PD-L1 inhibitors in older patients across different cancers. A meta-analysis of immunotherapy in older adults with a variety of malignancies showed overall efficacy comparable to that seen in adults younger than 65 years.105 However, another review found ICIs to be less effective in older patients with head and neck, non-small cell lung cancer, and renal cell carcinoma compared with their younger counterparts.104 Regarding the toxicity profile of ICIs in the elderly, similar rates of grade 3 or higher adverse events in patients younger than 65 years and older than 65 years have been reported.106 However, patients aged ≥ 70 years had increased rates of grade 3 to 5 adverse events as compared to patients younger than 65 years (71.7% versus 58.4%, respectively). Given the scant data on ICIs in older patients with MSI-H or dMMR metastatic CRC, more clinical trials inclusive of this population are needed in order to determine the efficacy and safety of immunotherapy.
Palliative Care
The incorporation of palliative care early following the diagnosis of cancer has been shown to improve quality of life, decrease depression, and help with symptom management.107 The triggers for geriatric patients to initiate palliative care may be different from those of younger patients, as older patients may have different goals of care.108 Older patients will often choose quality over quantity of life when making treatment decisions.109 The ideal medical treatment for the frail patient with colorectal cancer would focus on treating disease while providing palliative measures to help support the patient and improve quality of life. It is paramount that patients maintain functional independence as loss of independence is recognized as a major threat to an older patient’s quality of life.110 The optimal way to achieve these goals is through the efforts of a multidisciplinary care team including not only physicians and nurses, but also social workers, nutritionists, physical therapists, and family who can provide support for the patient’s psychosocial, cognitive, and medical needs.111 Although cancer and noncancer–related death occur more frequently in the geriatric population, data to guide a specific palliative care approach to the elderly population is lacking.108
Conclusion
Colorectal cancer is a disease of older adults with a median age at diagnosis of 67 years.1 With the aging population, oncologists will be faced with treating increasing numbers of older patients, and must adjust their practice to accommodate this population of patients. Treating geriatric patients is challenging given the lack of available data to guide the treatment approach. Although several prospective elderly-specific studies have been conducted evaluating treatments for metastatic CRC, most treatment decisions are made based on the available retrospective studies and pooled analyses. Oncologists must carefully consider and evaluate each patient based on physiologic age rather than chronologic age.112 Overall, older patients should be given the opportunity to receive standard of care treatments in the appropriate setting. The decision to modify treatment plans should be made after a thorough evaluation by a multidisciplinary team and a discussion with the patient regarding their goals and the risks and benefits of the treatment. Geriatric assessment tools can help the care team identify patients with various geriatric syndromes that may not be detected on routine oncology evaluation. This type of evaluation is time consuming and is rarely done in a busy oncology practice. Ongoing studies are aiming to develop a method to incorporate geriatric assessments into the care of older adults.Additional prospective trials targeting older, more frail patients are essential to improve upon our knowledge so we can provide best care for this growing elderly population.
1. National Cancer Institute. SEER cancer stat facts: colorectal cancer. http://seer.cancer.gov/statfacts/html/colorect.html. Accessed March 1, 2018.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
3. Kochanek KD, Murphy S, Xu J, Arias E. Mortality in the United States, 2016. NCHS Data Brief 2017:1-8.
4. Ortman JM, Velkoff VA, Hogan H. An aging nation: the older population in the United States, current population reports, P25-1140. Washington, DC: U.S. Census Bureau; 2014.
5. Hutchins LF, Unger JM, Crowley JJ, et al. Underrepresentation of patients 65 years of age or older in cancer-treatment trials. N Engl J Med 1999;341:2061–7.
6. Unger JM, Coltman CA Jr, Crowley JJ, et al. Impact of the year 2000 Medicare policy change on older patient enrollment to cancer clinical trials. J Clin Oncol 2006;24:141–4.
7. Vijayvergia N, Li T, Wong YN, et al. Chemotherapy use and adoption of new agents is affected by age and comorbidities in patients with metastatic colorectal cancer. Cancer 2016;122:3191–8.
8. Kalsi T, Babic-Illman G, Ross PJ, et al. The impact of comprehensive geriatric assessment interventions on tolerance to chemotherapy in older people. Br J Cancer 2015;112:1435–44.
9. National Comprehensive Cancer Network. Older adult oncology (Version 2.2017). Accessed March 1, 2018,
10. Balducci L. Frailty: a common pathway in aging and cancer. Interdiscip Top Gerontol 2013;38:61–72.
11. Baijal P, Periyakoil V. Understanding frailty in cancer patients. Cancer J 2014;20:358–66.
12. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982;5:649–55.
13. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
14. Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist 1969;9:179–86.
15. Katz S, Downs TD, Cash HR, Grotz RC. Progress in development of the index of ADL. Gerontologist 1970;10:20–30.
16. Klepin HD, Geiger AM, Tooze JA, et al. Physical performance and subsequent disability and survival in older adults with malignancy: results from the health, aging and body composition study. J Am Geriatr Soc 2010;58:76–82.
17. Gupta SK, Lamont EB. Patterns of presentation, diagnosis, and treatment in older patients with colon cancer and comorbid dementia. J Am Geriatr Soc 2004;52:1681–7.
18. Dewys WD, Begg C, Lavin PT, et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med 1980;69:491–7.
19. Aaldriks AA, van der Geest LG, Giltay EJ, et al. Frailty and malnutrition predictive of mortality risk in older patients with advanced colorectal cancer receiving chemotherapy. J Geriatr Oncol 2013;4:218–26.
20. Martucci RB, Barbosa MV, D’Almeida CA, et al. Undernutrition as independent predictor of early mortality in elderly cancer patients. Nutrition 2017;34:65–70.
21. Naeim A, Aapro M, Subbarao R, Balducci L. Supportive care considerations for older adults with cancer. J Clin Oncol 2014;32:2627–34.
22. Kua J. The prevalence of psychological and psychiatric sequelae of cancer in the elderly - how much do we know? Ann Acad Med Singapore 2005;34:250–6.
23. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118:3377–86.
24. Kim J, Hurria A. Determining chemotherapy tolerance in older patients with cancer. J Natl Compr Canc Netw 2013;11:1494-502.
25. Decoster L, Van Puyvelde K, Mohile S, et al. Screening tools for multidimensional health problems warranting a geriatric assessment in older cancer patients: an update on SIOG recommendations. Ann Oncol 2015;26:288–300.
26. Soubeyran P, Bellera C, Goyard J, et al. Screening for vulnerability in older cancer patients: the ONCODAGE Prospective Multicenter Cohort Study. PLoS One 2014; 9:e115060.
27. Gurevitch AJ, Davidovitch B, Kashtan H. Outcome of right colectomy for cancer in octogenarians. J Gastrointest Surg 2009;13:100–4.
28. Schiffmann L, Ozcan S, Schwarz F, et al. Colorectal cancer in the elderly: surgical treatment and long-term survival. Int J Colorectal Dis 2008;23:601–10.
29. Ong ES, Alassas M, Dunn KB, Rajput A. Colorectal cancer surgery in the elderly: acceptable morbidity? Am J Surg 2008;195:344–8.
30. Surgery for colorectal cancer in elderly patients: a systematic review. Colorectal Cancer Collaborative Group. Lancet 2000;356:968–74.
31. Shalaby M, Di Lorenzo N, Franceschilli L, et al. Outcome of colorectal surgery in elderly populations. Ann Coloproctol 2016;32:139–43.
32. Frasson M, Braga M, Vignali A, et al. Benefits of laparoscopic colorectal resection are more pronounced in elderly patients. Dis Colon Rectum 2008;51:296–300.
33. PACE participants, Audisio RA, Pope D, et al. Shall we operate? Preoperative assessment in elderly cancer patients (PACE) can help. A SIOG surgical task force prospective study. Crit Rev Oncol Hematol 2008;65:156–63.
34. Adam R, Frilling A, Elias D, et al. Liver resection of colorectal metastases in elderly patients. Br J Surg 2010;97:366–76.
35. de Liguori Carino N, van Leeuwen BL, Ghaneh P, et al. Liver resection for colorectal liver metastases in older patients. Crit Rev Oncol Hematol 2008;67:273–8.
36. Tamandl D, Gruenberger B, Herberger B, et al. Surgery after neoadjuvant chemotherapy for colorectal liver metastases is safe and feasible in elderly patients. J Surg Oncol 2009;100:364–71.
37. Shahir MA, Lemmens VE, van de Poll-Franse LV, et al. Elderly patients with rectal cancer have a higher risk of treatment-related complications and a poorer prognosis than younger patients: a population-based study. Eur J Cancer 2006;42:3015–21.
38. Chang GJ, Skibber JM, Feig BW, Rodriguez-Bigas M. Are we undertreating rectal cancer in the elderly? An epidemiologic study. Ann Surg 2007;246:215–21.
39. Colorectal Cancer Collaborative Group. Adjuvant radiotherapy for rectal cancer: a systematic overview of 8,507 patients from 22 randomised trials. Lancet 2001;358:1291–304.
40. Martling A, Holm T, Johansson H, et al, Stockholm Colorectal Cancer Study Group. The Stockholm II trial on preoperative radiotherapy in rectal carcinoma: long-term follow-up of a population-based study. Cancer 2001;92:896–902.
41. Pasetto LM, Friso ML, Pucciarelli S, et al. Rectal cancer neoadjuvant treatment in elderly patients. Anticancer Res 2006;26:3913–23.
42. Margalit DN, Mamon HJ, Ancukiewicz M, et al. Tolerability of combined modality therapy for rectal cancer in elderly patients aged 75 years and older. Int J Radiat Oncol Biol Phys 2011;81:e735–41.
43. Dossa F, Chesney TR, Acuna SA, Baxter NN. A watch-and-wait approach for locally advanced rectal cancer after a clinical complete response following neoadjuvant chemoradiation: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2017;2:501–13.
44. van der Valk M. The International Watch & Wait database (IWWD) for rectal cancer: An update. J Clin Oncol 2017;35 suppl:521.
45. Donato V, Valeriani M, Zurlo A. Short course radiation therapy for elderly cancer patients. Evidences from the literature review. Crit Rev Oncol Hematol 2003;45:305–11.
46. Ngan SY, Burmeister B, Fisher RJ, et al. Randomized trial of short-course radiotherapy versus long-course chemoradiation comparing rates of local recurrence in patients with T3 rectal cancer: Trans-Tasman Radiation Oncology Group trial 01.04. J Clin Oncol 2012;30:3827–33.
47. McCleary NJ, Dotan E, Browner I. Refining the chemotherapy approach for older patients with colon cancer. J Clin Oncol 2014;32:2570–80.
48. Millan M, Merino S, Caro A, et al. Treatment of colorectal cancer in the elderly. World J Gastrointest Oncol 2015;7:204–20.
49. Quasar Collaborative Group, Gray R, Barnwell J, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370:2020–9.
50. Andre T, Boni C, Navarro M, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 2009;27:3109–16.
51. Tournigand C, Andre T, Bonnetain F, et al. Adjuvant therapy with fluorouracil and oxaliplatin in stage II and elderly patients (between ages 70 and 75 years) with colon cancer: subgroup analyses of the Multicenter International Study of Oxaliplatin, Fluorouracil, and Leucovorin in the Adjuvant Treatment of Colon Cancer trial. J Clin Oncol 2012;30:3353–60.
52. Winder T, Lenz HJ. Molecular predictive and prognostic markers in colon cancer. Cancer Treat Rev 2010;36:550–6.
53. Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
54. Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
55. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
56. Aparicio T, Schischmanoff O, Poupardin C, et al. Deficient mismatch repair phenotype is a prognostic factor for colorectal cancer in elderly patients. Dig Liver Dis 2013;45:245–50.
57. Sargent DJ, Goldberg RM, Jacobson SD, et al. A pooled analysis of adjuvant chemotherapy for resected colon cancer in elderly patients. N Engl J Med 2001;345:1091–7.
58. Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
59. Twelves C, Scheithauer W, McKendrick J, et al. Capecitabine versus 5-fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results from the X-ACT trial with analysis by age and preliminary evidence of a pharmacodynamic marker of efficacy. Ann Oncol 2012;23:1190–7.
60. Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses. J Clin Oncol 2011;29:3768–74.
61. Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
62. Haller DG, Cassidy J, Tabernero J, et al. Efficacy findings from a randomized phase III trial of capecitabine plus oxaliplatin versus bolus 5-FU/LV for stage III colon cancer (NO16968): impact of age on disease-free survival (DFS) [abstract]. J Clin Oncol 2010;28:3521.
63. Schmoll HJ, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
64. McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
65. Kahn KL, Adams JL, Weeks JC, et al. Adjuvant chemotherapy use and adverse events among older patients with stage III colon cancer. JAMA 2010;303:1037–45.
66. Haller DG, O’Connell MJ, Cartwright TH, et al. Impact of age and medical comorbidity on adjuvant treatment outcomes for stage III colon cancer: a pooled analysis of individual patient data from four randomized, controlled trials. Ann Oncol 2015;26:715-24.
67. Aparicio T, Francois E, Cristol-Dalstein L, et al. PRODIGE 34-FFCD 1402-ADAGE: Adjuvant chemotherapy in elderly patients with resected stage III colon cancer: A randomized phase 3 trial. Dig Liver Dis 2016;48:206–7.
68. Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
69. Mahoney T, Kuo YH, Topilow A, Davis JM. Stage III colon cancers: why adjuvant chemotherapy is not offered to elderly patients. Arch Surg 2000;135:182–5.
70. Sanoff HK, Carpenter WR, Sturmer T, et al. Effect of adjuvant chemotherapy on survival of patients with stage III colon cancer diagnosed after age 75 years. J Clin Oncol 2012;30:2624–34.
71. Folprecht G, Cunningham D, Ross P, et al. Efficacy of 5-fluorouracil-based chemotherapy in elderly patients with metastatic colorectal cancer: a pooled analysis of clinical trials. Ann Oncol 2004;15:1330–8.
72. Van Cutsem E, Cervantes A, Nordlinger B, Arnold D, ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 Suppl 3:iii1–9.
73. Goldberg RM, Tabah-Fisch I, Bleiberg H, et al. Pooled analysis of safety and efficacy of oxaliplatin plus fluorouracil/leucovorin administered bimonthly in elderly patients with colorectal cancer. J Clin Oncol 2006;24:4085–91.
74. Seymour MT, Thompson LC, Wasan HS, et al. Chemotherapy options in elderly and frail patients with metastatic colorectal cancer (MRC FOCUS2): an open-label, randomised factorial trial. Lancet 2011;377:1749–59.
75. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer--a GERCOR study. J Clin Oncol 2006;24:394–400.
76. Figer A, Perez-Staub N, Carola E, et al. FOLFOX in patients aged between 76 and 80 years with metastatic colorectal cancer: an exploratory cohort of the OPTIMOX1 study. Cancer 2007;110:2666–71.
77. Chibaudel B, Maindrault-Goebel F, Lledo G, et al. Can chemotherapy be discontinued in unresectable metastatic colorectal cancer? The GERCOR OPTIMOX2 Study. J Clin Oncol 2009;27:5727–33.
78. Folprecht G, Seymour MT, Saltz L, et al. Irinotecan/fluorouracil combination in first-line therapy of older and younger patients with metastatic colorectal cancer: combined analysis of 2,691 patients in randomized controlled trials. J Clin Oncol 2008;26:1443–51.
79. Souglakos J, Pallis A, Kakolyris S, et al. Combination of irinotecan (CPT-11) plus 5-fluorouracil and leucovorin (FOLFIRI regimen) as first line treatment for elderly patients with metastatic colorectal cancer: a phase II trial. Oncology 2005;69:384–90.
80. Aparicio T, Lavau-Denes S, Phelip JM, et al. Randomized phase III trial in elderly patients comparing LV5FU2 with or without irinotecan for first-line treatment of metastatic colorectal cancer (FFCD 2001-02). Ann Oncol 2016;27:121–7.
81. Aparicio T, Gargot D, Teillet L, et al. Geriatric factors analyses from FFCD 2001-02 phase III study of first-line chemotherapy for elderly metastatic colorectal cancer patients. Eur J Cancer 2017;74:98–108.
82. Kabbinavar FF, Hurwitz HI, Yi J, et al. Addition of bevacizumab to fluorouracil-based first-line treatment of metastatic colorectal cancer: pooled analysis of cohorts of older patients from two randomized clinical trials. J Clin Oncol 2009;27:199–205.
83. Cassidy J, Saltz LB, Giantonio BJ, et al. Effect of bevacizumab in older patients with metastatic colorectal cancer: pooled analysis of four randomized studies. J Cancer Res Clin Oncol 2010;136:737–43.
84. Van Cutsem E, Rivera F, Berry S, et al. Safety and efficacy of first-line bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in metastatic colorectal cancer: the BEAT study. Ann Oncol 2009;20:1842–7.
85. Kozloff MF, Berlin J, Flynn PJ, et al. Clinical outcomes in elderly patients with metastatic colorectal cancer receiving bevacizumab and chemotherapy: results from the BRiTE observational cohort study. Oncology 2010;78:329–39.
86. Cunningham D, Lang I, Marcuello E, et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): an open-label, randomised phase 3 trial. Lancet Oncol 2013;14:1077–85.
87. Aparicio T, Bouche O, Taieb J, et al. Bevacizumab+chemotherapy versus chemotherapy alone in elderly patients with untreated metastatic colorectal cancer: a randomized phase II trial-PRODIGE 20 study results. Ann Oncol 2018;29:133–8.
88. Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 2012;30:3499–506.
89. Ruff P, Van Cutsem E, Lakomy R, et al. Observed benefit and safety of aflibercept in elderly patients with metastatic colorectal cancer: An age-based analysis from the randomized placebo-controlled phase III VELOUR trial. J Geriatr Oncol 2018;9:32–9.
90. Obermannova R, Van Cutsem E, Yoshino T, et al. Subgroup analysis in RAISE: a randomized, double-blind phase III study of irinotecan, folinic acid, and 5-fluorouracil (FOLFIRI) plus ramucirumab or placebo in patients with metastatic colorectal carcinoma progression. Ann Oncol 2016;27:2082–90.
91. Bouchahda M, Macarulla T, Spano JP, et al. Cetuximab efficacy and safety in a retrospective cohort of elderly patients with heavily pretreated metastatic colorectal cancer. Crit Rev Oncol Hematol 2008;67:255-62.
92. Douillard JY, Siena S, Cassidy J, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol 2014;25:1346–55.
93. Sastre J, Gravalos C, Rivera F, et al. First-line cetuximab plus capecitabine in elderly patients with advanced colorectal cancer: clinical outcome and subgroup analysis according to KRAS status from a Spanish TTD Group Study. Oncologist 2012;17:339–45.
94. Dotan E, Devarajan K, D’Silva AJ, et al. Patterns of use and tolerance of anti-epidermal growth factor receptor antibodies in older adults with metastatic colorectal cancer. Clin Colorectal Cancer 2014;13:192–8.
95. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:303–12.
96. Van Cutsem E, Sobrero A, Siena S, et al. Regorafenib (REG) in progressive metastatic colorectal cancer (mCRC): Analysis of age subgroups in the phase III CORRECT trial [abstract]. J Clin Oncol 2013;31(15 suppl):3636-3636.
97. Bekaii-Saab TS, Ou FS, Anderson DM, et al. Regorafenib dose optimization study (ReDOS): Randomized phase II trial to evaluate dosing strategies for regorafenib in refractory metastatic colorectal cancer (mCRC): an ACCRU Network study [abstract]. J Clin Oncol 2018;36(4 suppl):611-611.
98. Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909–19.
99. Koopman M, Kortman GA, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer 2009;100:266–73.
100. Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res 2014;20:5322–30.
101. Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
102. Le DT, Uram JN, Wang H, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20.
103. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–91.
104. Daste A, Domblides C, Gross-Goupil M, et al. Immune checkpoint inhibitors and elderly people: A review. Eur J Cancer 2017;82:155–66.
105. Elias R, Giobbie-Hurder A, McCleary NJ, et al. Efficacy of PD-1 & PD-L1 inhibitors in older adults: a meta-analysis. J Immunother Cancer 2018;6:26.
106. Singh H, Kim G, Maher VE, et al. FDA subset analysis of the safety of nivolumab in elderly patients with advanced cancers [abstract]. J Clin Oncol 2016;34(15 suppl):10010-10010.
107. Temel JS, Greer JA, El-Jawahri A, et al. Effects of early integrated palliative care in patients with lung and GI cancer: a randomized clinical trial. J Clin Oncol 2017;35:834–41.
108. Brighi N, Balducci L, Biasco G. Cancer in the elderly: is it time for palliative care in geriatric oncology? J Geriatr Oncol 2014;5:197–203.
109. Meropol NJ, Egleston BL, Buzaglo JS, et al. Cancer patient preferences for quality and length of life. Cancer 2008;113:3459–66.
110. Bagshaw SM, Stelfox HT, Johnson JA, et al. Long-term association between frailty and health-related quality of life among survivors of critical illness: a prospective multicenter cohort study. Crit Care Med 2015;43:973–82.
111. Lynch MP, Marcone D, Kagan SH. Developing a multidisciplinary geriatric oncology program in a community cancer center. Clin J Oncol Nurs 2007;11:929–33.
112. Sheridan J, Walsh P, Kevans D, et al. Determinants of short- and long-term survival from colorectal cancer in very elderly patients. J Geriatr Oncol 2014;5:376–83.
1. National Cancer Institute. SEER cancer stat facts: colorectal cancer. http://seer.cancer.gov/statfacts/html/colorect.html. Accessed March 1, 2018.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
3. Kochanek KD, Murphy S, Xu J, Arias E. Mortality in the United States, 2016. NCHS Data Brief 2017:1-8.
4. Ortman JM, Velkoff VA, Hogan H. An aging nation: the older population in the United States, current population reports, P25-1140. Washington, DC: U.S. Census Bureau; 2014.
5. Hutchins LF, Unger JM, Crowley JJ, et al. Underrepresentation of patients 65 years of age or older in cancer-treatment trials. N Engl J Med 1999;341:2061–7.
6. Unger JM, Coltman CA Jr, Crowley JJ, et al. Impact of the year 2000 Medicare policy change on older patient enrollment to cancer clinical trials. J Clin Oncol 2006;24:141–4.
7. Vijayvergia N, Li T, Wong YN, et al. Chemotherapy use and adoption of new agents is affected by age and comorbidities in patients with metastatic colorectal cancer. Cancer 2016;122:3191–8.
8. Kalsi T, Babic-Illman G, Ross PJ, et al. The impact of comprehensive geriatric assessment interventions on tolerance to chemotherapy in older people. Br J Cancer 2015;112:1435–44.
9. National Comprehensive Cancer Network. Older adult oncology (Version 2.2017). Accessed March 1, 2018,
10. Balducci L. Frailty: a common pathway in aging and cancer. Interdiscip Top Gerontol 2013;38:61–72.
11. Baijal P, Periyakoil V. Understanding frailty in cancer patients. Cancer J 2014;20:358–66.
12. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982;5:649–55.
13. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol 2011;29:3457–65.
14. Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist 1969;9:179–86.
15. Katz S, Downs TD, Cash HR, Grotz RC. Progress in development of the index of ADL. Gerontologist 1970;10:20–30.
16. Klepin HD, Geiger AM, Tooze JA, et al. Physical performance and subsequent disability and survival in older adults with malignancy: results from the health, aging and body composition study. J Am Geriatr Soc 2010;58:76–82.
17. Gupta SK, Lamont EB. Patterns of presentation, diagnosis, and treatment in older patients with colon cancer and comorbid dementia. J Am Geriatr Soc 2004;52:1681–7.
18. Dewys WD, Begg C, Lavin PT, et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med 1980;69:491–7.
19. Aaldriks AA, van der Geest LG, Giltay EJ, et al. Frailty and malnutrition predictive of mortality risk in older patients with advanced colorectal cancer receiving chemotherapy. J Geriatr Oncol 2013;4:218–26.
20. Martucci RB, Barbosa MV, D’Almeida CA, et al. Undernutrition as independent predictor of early mortality in elderly cancer patients. Nutrition 2017;34:65–70.
21. Naeim A, Aapro M, Subbarao R, Balducci L. Supportive care considerations for older adults with cancer. J Clin Oncol 2014;32:2627–34.
22. Kua J. The prevalence of psychological and psychiatric sequelae of cancer in the elderly - how much do we know? Ann Acad Med Singapore 2005;34:250–6.
23. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH) score. Cancer 2012;118:3377–86.
24. Kim J, Hurria A. Determining chemotherapy tolerance in older patients with cancer. J Natl Compr Canc Netw 2013;11:1494-502.
25. Decoster L, Van Puyvelde K, Mohile S, et al. Screening tools for multidimensional health problems warranting a geriatric assessment in older cancer patients: an update on SIOG recommendations. Ann Oncol 2015;26:288–300.
26. Soubeyran P, Bellera C, Goyard J, et al. Screening for vulnerability in older cancer patients: the ONCODAGE Prospective Multicenter Cohort Study. PLoS One 2014; 9:e115060.
27. Gurevitch AJ, Davidovitch B, Kashtan H. Outcome of right colectomy for cancer in octogenarians. J Gastrointest Surg 2009;13:100–4.
28. Schiffmann L, Ozcan S, Schwarz F, et al. Colorectal cancer in the elderly: surgical treatment and long-term survival. Int J Colorectal Dis 2008;23:601–10.
29. Ong ES, Alassas M, Dunn KB, Rajput A. Colorectal cancer surgery in the elderly: acceptable morbidity? Am J Surg 2008;195:344–8.
30. Surgery for colorectal cancer in elderly patients: a systematic review. Colorectal Cancer Collaborative Group. Lancet 2000;356:968–74.
31. Shalaby M, Di Lorenzo N, Franceschilli L, et al. Outcome of colorectal surgery in elderly populations. Ann Coloproctol 2016;32:139–43.
32. Frasson M, Braga M, Vignali A, et al. Benefits of laparoscopic colorectal resection are more pronounced in elderly patients. Dis Colon Rectum 2008;51:296–300.
33. PACE participants, Audisio RA, Pope D, et al. Shall we operate? Preoperative assessment in elderly cancer patients (PACE) can help. A SIOG surgical task force prospective study. Crit Rev Oncol Hematol 2008;65:156–63.
34. Adam R, Frilling A, Elias D, et al. Liver resection of colorectal metastases in elderly patients. Br J Surg 2010;97:366–76.
35. de Liguori Carino N, van Leeuwen BL, Ghaneh P, et al. Liver resection for colorectal liver metastases in older patients. Crit Rev Oncol Hematol 2008;67:273–8.
36. Tamandl D, Gruenberger B, Herberger B, et al. Surgery after neoadjuvant chemotherapy for colorectal liver metastases is safe and feasible in elderly patients. J Surg Oncol 2009;100:364–71.
37. Shahir MA, Lemmens VE, van de Poll-Franse LV, et al. Elderly patients with rectal cancer have a higher risk of treatment-related complications and a poorer prognosis than younger patients: a population-based study. Eur J Cancer 2006;42:3015–21.
38. Chang GJ, Skibber JM, Feig BW, Rodriguez-Bigas M. Are we undertreating rectal cancer in the elderly? An epidemiologic study. Ann Surg 2007;246:215–21.
39. Colorectal Cancer Collaborative Group. Adjuvant radiotherapy for rectal cancer: a systematic overview of 8,507 patients from 22 randomised trials. Lancet 2001;358:1291–304.
40. Martling A, Holm T, Johansson H, et al, Stockholm Colorectal Cancer Study Group. The Stockholm II trial on preoperative radiotherapy in rectal carcinoma: long-term follow-up of a population-based study. Cancer 2001;92:896–902.
41. Pasetto LM, Friso ML, Pucciarelli S, et al. Rectal cancer neoadjuvant treatment in elderly patients. Anticancer Res 2006;26:3913–23.
42. Margalit DN, Mamon HJ, Ancukiewicz M, et al. Tolerability of combined modality therapy for rectal cancer in elderly patients aged 75 years and older. Int J Radiat Oncol Biol Phys 2011;81:e735–41.
43. Dossa F, Chesney TR, Acuna SA, Baxter NN. A watch-and-wait approach for locally advanced rectal cancer after a clinical complete response following neoadjuvant chemoradiation: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2017;2:501–13.
44. van der Valk M. The International Watch & Wait database (IWWD) for rectal cancer: An update. J Clin Oncol 2017;35 suppl:521.
45. Donato V, Valeriani M, Zurlo A. Short course radiation therapy for elderly cancer patients. Evidences from the literature review. Crit Rev Oncol Hematol 2003;45:305–11.
46. Ngan SY, Burmeister B, Fisher RJ, et al. Randomized trial of short-course radiotherapy versus long-course chemoradiation comparing rates of local recurrence in patients with T3 rectal cancer: Trans-Tasman Radiation Oncology Group trial 01.04. J Clin Oncol 2012;30:3827–33.
47. McCleary NJ, Dotan E, Browner I. Refining the chemotherapy approach for older patients with colon cancer. J Clin Oncol 2014;32:2570–80.
48. Millan M, Merino S, Caro A, et al. Treatment of colorectal cancer in the elderly. World J Gastrointest Oncol 2015;7:204–20.
49. Quasar Collaborative Group, Gray R, Barnwell J, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370:2020–9.
50. Andre T, Boni C, Navarro M, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol 2009;27:3109–16.
51. Tournigand C, Andre T, Bonnetain F, et al. Adjuvant therapy with fluorouracil and oxaliplatin in stage II and elderly patients (between ages 70 and 75 years) with colon cancer: subgroup analyses of the Multicenter International Study of Oxaliplatin, Fluorouracil, and Leucovorin in the Adjuvant Treatment of Colon Cancer trial. J Clin Oncol 2012;30:3353–60.
52. Winder T, Lenz HJ. Molecular predictive and prognostic markers in colon cancer. Cancer Treat Rev 2010;36:550–6.
53. Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
54. Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
55. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
56. Aparicio T, Schischmanoff O, Poupardin C, et al. Deficient mismatch repair phenotype is a prognostic factor for colorectal cancer in elderly patients. Dig Liver Dis 2013;45:245–50.
57. Sargent DJ, Goldberg RM, Jacobson SD, et al. A pooled analysis of adjuvant chemotherapy for resected colon cancer in elderly patients. N Engl J Med 2001;345:1091–7.
58. Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
59. Twelves C, Scheithauer W, McKendrick J, et al. Capecitabine versus 5-fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results from the X-ACT trial with analysis by age and preliminary evidence of a pharmacodynamic marker of efficacy. Ann Oncol 2012;23:1190–7.
60. Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses. J Clin Oncol 2011;29:3768–74.
61. Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
62. Haller DG, Cassidy J, Tabernero J, et al. Efficacy findings from a randomized phase III trial of capecitabine plus oxaliplatin versus bolus 5-FU/LV for stage III colon cancer (NO16968): impact of age on disease-free survival (DFS) [abstract]. J Clin Oncol 2010;28:3521.
63. Schmoll HJ, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
64. McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
65. Kahn KL, Adams JL, Weeks JC, et al. Adjuvant chemotherapy use and adverse events among older patients with stage III colon cancer. JAMA 2010;303:1037–45.
66. Haller DG, O’Connell MJ, Cartwright TH, et al. Impact of age and medical comorbidity on adjuvant treatment outcomes for stage III colon cancer: a pooled analysis of individual patient data from four randomized, controlled trials. Ann Oncol 2015;26:715-24.
67. Aparicio T, Francois E, Cristol-Dalstein L, et al. PRODIGE 34-FFCD 1402-ADAGE: Adjuvant chemotherapy in elderly patients with resected stage III colon cancer: A randomized phase 3 trial. Dig Liver Dis 2016;48:206–7.
68. Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
69. Mahoney T, Kuo YH, Topilow A, Davis JM. Stage III colon cancers: why adjuvant chemotherapy is not offered to elderly patients. Arch Surg 2000;135:182–5.
70. Sanoff HK, Carpenter WR, Sturmer T, et al. Effect of adjuvant chemotherapy on survival of patients with stage III colon cancer diagnosed after age 75 years. J Clin Oncol 2012;30:2624–34.
71. Folprecht G, Cunningham D, Ross P, et al. Efficacy of 5-fluorouracil-based chemotherapy in elderly patients with metastatic colorectal cancer: a pooled analysis of clinical trials. Ann Oncol 2004;15:1330–8.
72. Van Cutsem E, Cervantes A, Nordlinger B, Arnold D, ESMO Guidelines Working Group. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 Suppl 3:iii1–9.
73. Goldberg RM, Tabah-Fisch I, Bleiberg H, et al. Pooled analysis of safety and efficacy of oxaliplatin plus fluorouracil/leucovorin administered bimonthly in elderly patients with colorectal cancer. J Clin Oncol 2006;24:4085–91.
74. Seymour MT, Thompson LC, Wasan HS, et al. Chemotherapy options in elderly and frail patients with metastatic colorectal cancer (MRC FOCUS2): an open-label, randomised factorial trial. Lancet 2011;377:1749–59.
75. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer--a GERCOR study. J Clin Oncol 2006;24:394–400.
76. Figer A, Perez-Staub N, Carola E, et al. FOLFOX in patients aged between 76 and 80 years with metastatic colorectal cancer: an exploratory cohort of the OPTIMOX1 study. Cancer 2007;110:2666–71.
77. Chibaudel B, Maindrault-Goebel F, Lledo G, et al. Can chemotherapy be discontinued in unresectable metastatic colorectal cancer? The GERCOR OPTIMOX2 Study. J Clin Oncol 2009;27:5727–33.
78. Folprecht G, Seymour MT, Saltz L, et al. Irinotecan/fluorouracil combination in first-line therapy of older and younger patients with metastatic colorectal cancer: combined analysis of 2,691 patients in randomized controlled trials. J Clin Oncol 2008;26:1443–51.
79. Souglakos J, Pallis A, Kakolyris S, et al. Combination of irinotecan (CPT-11) plus 5-fluorouracil and leucovorin (FOLFIRI regimen) as first line treatment for elderly patients with metastatic colorectal cancer: a phase II trial. Oncology 2005;69:384–90.
80. Aparicio T, Lavau-Denes S, Phelip JM, et al. Randomized phase III trial in elderly patients comparing LV5FU2 with or without irinotecan for first-line treatment of metastatic colorectal cancer (FFCD 2001-02). Ann Oncol 2016;27:121–7.
81. Aparicio T, Gargot D, Teillet L, et al. Geriatric factors analyses from FFCD 2001-02 phase III study of first-line chemotherapy for elderly metastatic colorectal cancer patients. Eur J Cancer 2017;74:98–108.
82. Kabbinavar FF, Hurwitz HI, Yi J, et al. Addition of bevacizumab to fluorouracil-based first-line treatment of metastatic colorectal cancer: pooled analysis of cohorts of older patients from two randomized clinical trials. J Clin Oncol 2009;27:199–205.
83. Cassidy J, Saltz LB, Giantonio BJ, et al. Effect of bevacizumab in older patients with metastatic colorectal cancer: pooled analysis of four randomized studies. J Cancer Res Clin Oncol 2010;136:737–43.
84. Van Cutsem E, Rivera F, Berry S, et al. Safety and efficacy of first-line bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in metastatic colorectal cancer: the BEAT study. Ann Oncol 2009;20:1842–7.
85. Kozloff MF, Berlin J, Flynn PJ, et al. Clinical outcomes in elderly patients with metastatic colorectal cancer receiving bevacizumab and chemotherapy: results from the BRiTE observational cohort study. Oncology 2010;78:329–39.
86. Cunningham D, Lang I, Marcuello E, et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): an open-label, randomised phase 3 trial. Lancet Oncol 2013;14:1077–85.
87. Aparicio T, Bouche O, Taieb J, et al. Bevacizumab+chemotherapy versus chemotherapy alone in elderly patients with untreated metastatic colorectal cancer: a randomized phase II trial-PRODIGE 20 study results. Ann Oncol 2018;29:133–8.
88. Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 2012;30:3499–506.
89. Ruff P, Van Cutsem E, Lakomy R, et al. Observed benefit and safety of aflibercept in elderly patients with metastatic colorectal cancer: An age-based analysis from the randomized placebo-controlled phase III VELOUR trial. J Geriatr Oncol 2018;9:32–9.
90. Obermannova R, Van Cutsem E, Yoshino T, et al. Subgroup analysis in RAISE: a randomized, double-blind phase III study of irinotecan, folinic acid, and 5-fluorouracil (FOLFIRI) plus ramucirumab or placebo in patients with metastatic colorectal carcinoma progression. Ann Oncol 2016;27:2082–90.
91. Bouchahda M, Macarulla T, Spano JP, et al. Cetuximab efficacy and safety in a retrospective cohort of elderly patients with heavily pretreated metastatic colorectal cancer. Crit Rev Oncol Hematol 2008;67:255-62.
92. Douillard JY, Siena S, Cassidy J, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol 2014;25:1346–55.
93. Sastre J, Gravalos C, Rivera F, et al. First-line cetuximab plus capecitabine in elderly patients with advanced colorectal cancer: clinical outcome and subgroup analysis according to KRAS status from a Spanish TTD Group Study. Oncologist 2012;17:339–45.
94. Dotan E, Devarajan K, D’Silva AJ, et al. Patterns of use and tolerance of anti-epidermal growth factor receptor antibodies in older adults with metastatic colorectal cancer. Clin Colorectal Cancer 2014;13:192–8.
95. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:303–12.
96. Van Cutsem E, Sobrero A, Siena S, et al. Regorafenib (REG) in progressive metastatic colorectal cancer (mCRC): Analysis of age subgroups in the phase III CORRECT trial [abstract]. J Clin Oncol 2013;31(15 suppl):3636-3636.
97. Bekaii-Saab TS, Ou FS, Anderson DM, et al. Regorafenib dose optimization study (ReDOS): Randomized phase II trial to evaluate dosing strategies for regorafenib in refractory metastatic colorectal cancer (mCRC): an ACCRU Network study [abstract]. J Clin Oncol 2018;36(4 suppl):611-611.
98. Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909–19.
99. Koopman M, Kortman GA, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer 2009;100:266–73.
100. Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res 2014;20:5322–30.
101. Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
102. Le DT, Uram JN, Wang H, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20.
103. Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182–91.
104. Daste A, Domblides C, Gross-Goupil M, et al. Immune checkpoint inhibitors and elderly people: A review. Eur J Cancer 2017;82:155–66.
105. Elias R, Giobbie-Hurder A, McCleary NJ, et al. Efficacy of PD-1 & PD-L1 inhibitors in older adults: a meta-analysis. J Immunother Cancer 2018;6:26.
106. Singh H, Kim G, Maher VE, et al. FDA subset analysis of the safety of nivolumab in elderly patients with advanced cancers [abstract]. J Clin Oncol 2016;34(15 suppl):10010-10010.
107. Temel JS, Greer JA, El-Jawahri A, et al. Effects of early integrated palliative care in patients with lung and GI cancer: a randomized clinical trial. J Clin Oncol 2017;35:834–41.
108. Brighi N, Balducci L, Biasco G. Cancer in the elderly: is it time for palliative care in geriatric oncology? J Geriatr Oncol 2014;5:197–203.
109. Meropol NJ, Egleston BL, Buzaglo JS, et al. Cancer patient preferences for quality and length of life. Cancer 2008;113:3459–66.
110. Bagshaw SM, Stelfox HT, Johnson JA, et al. Long-term association between frailty and health-related quality of life among survivors of critical illness: a prospective multicenter cohort study. Crit Care Med 2015;43:973–82.
111. Lynch MP, Marcone D, Kagan SH. Developing a multidisciplinary geriatric oncology program in a community cancer center. Clin J Oncol Nurs 2007;11:929–33.
112. Sheridan J, Walsh P, Kevans D, et al. Determinants of short- and long-term survival from colorectal cancer in very elderly patients. J Geriatr Oncol 2014;5:376–83.
Urothelial Carcinoma: Muscle-Invasive and Metastatic Disease
Introduction
Bladder cancer is by far the most common cancer of the urinary system. Worldwide, approximately 450,000 new cases are diagnosed and 165,000 deaths are caused by bladder cancer each year.1 In the United States and in Europe, the most common type of bladder cancer is urothelial carcinoma (also referred to as transitional cell carcinoma), which accounts for more than 90% of all bladder cancers in these regions of the world. The remainder of bladder cancers are divided among squamous cell carcinomas, adenocarcinomas, small cell carcinomas, and, even more rarely, between various other nonepithelial tumors (eg, sarcoma).
Bladder cancer is classically thought of as a disease of the elderly, with a median age at diagnosis of 69 years in men and 71 years in women.2 The incidence of bladder cancer increases with age: in persons aged 65 to 69 years, incidence is 142 per 100,000 men and 33 per 100,000 women, and in those older than 85 years the rate doubles to 296 per 100,000 men and 74 per 100,000 women.3 The incidence is 3 times greater in men than in women.4
Urothelial carcinoma is traditionally categorized by its degree of invasion into the bladder wall: superficial (non-muscle-invasive), muscle-invasive, or metastatic disease. At the time of diagnosis, most patients have non-muscle-invasive disease (~60%); about 4% of all patients present initially with metastatic disease.5 This article focuses on metastatic bladder cancer, but muscle-invasive disease is discussed as well.
The most important factor contributing to the development of urothelial carcinoma is tobacco smoking. The risk of developing bladder cancer is 4 to 5 times higher in smokers as compared to nonsmokers, with some variation according to sex.6 Quantity of smoking exposure also plays a role, with heavy smokers demonstrating a higher likelihood for high-grade tumors with muscle invasion (or beyond) when compared to light smokers.7 Another important risk factor is occupational exposure to industrial materials, such as carpets, paints, plastics, and industrial chemicals. This type of exposure may be responsible for, or at least contribute to, the development of approximately 20% of urothelial carcinomas. Other risk factors for urothelial carcinoma include but are not limited to prior radiation to the pelvis, prior upper tract urothelial malignancy, human papillomavirus infection, and prior bladder augmentation.
Diagnosis and Staging
Case Presentation
A 63-year-old man with a past medical history of diabetes, deep vein thrombosis, occasional alcohol use, and regular pipe tobacco use presents to his primary care physician with complaints of hematuria. He reports that his urine was a dark red color that morning, which had never happened before. The patient is hemodynamically stable upon evaluation in the office, and a point-of-care urinalysis dipstick is strongly positive for blood. He is referred to a urologist for further evaluation.
In the urology office, urine microscopy is notable for more than 50 red blood cells (RBCs) per high-power field with normal RBC morphology. Flexible cystoscopy performed in the office reveals a single 2-cm, sessile, verrucous, nodular lesion located on the anterior bladder wall. A urine sample and a bladder wash specimen are sent for cytology evaluation. The patient is scheduled to undergo a complete transurethral resection of bladder tumor (TURBT) later that week with samples sent to pathology for evaluation.
- What are the clinical features of bladder cancer?
Hematuria is the most common presentation of bladder cancer, although its specificity is far lower than traditionally thought. In fact, only about 2% to 20% of cases that present with hematuria are found to be caused by malignancy. However, the incidence of genitourinary tract malignancy is much higher in patients presenting with gross hematuria (10%–20%)8–10 than in patients with microscopic hematuria alone.8,10–14 Typically, hematuria associated with malignancy is painless. Multiple studies have shown, however, that hematuria can be a normal variant, with one study demonstrating that up to 61% of patients with hematuria had no identifiable abnormality.8,10,11,13
Abdominal pain, flank pain, dysuria, urinary frequency/urgency, or other irritative voiding symptoms in the absence of hematuria can be presenting symptoms of bladder cancer as well. In these settings, discomfort typically suggests more advanced malignancy with at least local involvement or obstruction. Suprapubic pain may herald invasion into perivesical tissues and nerves, while involvement of the obturator fossa, perirectal fat, urogenital diaphragm, or presacral nerves can often present with perineal or rectal pain. Similarly, lower abdominal pain may represent involvement of lymph nodes, and right upper quadrant pain may signal liver metastasis. Cough or shortness of breath may signify metastatic disease in the lung. Finally, back, rib, or other boney pain may suggest distant metastasis.
- What next steps are required to complete this patient’s staging?
White light cystoscopy remains the gold standard for diagnosis and initial staging of bladder cancer. Additional tools include urine cytology and upper tract studies, including renal computed tomography (CT) urograms. Full urologic evaluation with all 3 modalities (cystourethroscopy, urinary cytology, and upper tract evaluation) is warranted for patients with a high suspicion for malignant etiology of hematuria. CT urograms are particularly useful for upper tract evaluation because they can be used to visualize kidney parenchyma, both renal pelvises and ureters, and pertinent abdominal and pelvic lymph nodes. Initial staging is completed through TURBT, which should ideally contain a segment of muscularis propria to distinguish between Ta (noninvasive), T1, and T2 tumors (Figure 1). 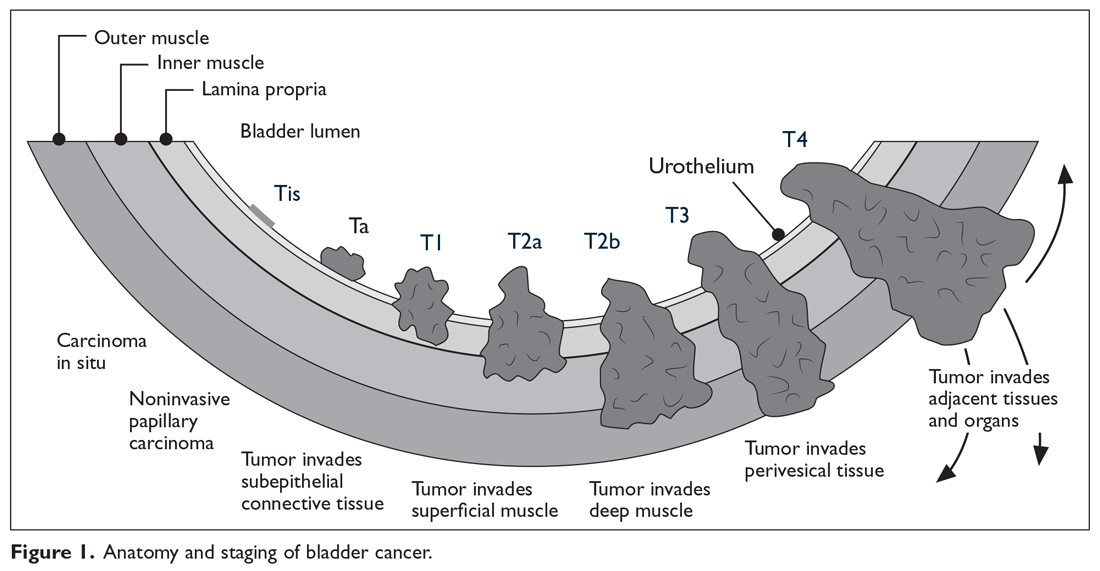
Regarding staging, T1 tumors are distinguished from Ta malignancies by their involvement in the urothelial basement membrane. Tumor invasion into the muscularis propria indicates T2 tumors, while T3 tumors extend through the muscle into the serosa and involve the complete thickness of the bladder wall. Involvement of nearby structures defines T4 bladder cancers, with T4a malignancies involving adjacent organs (prostate, vagina, uterus, or bowel) and T4b tumors involving the abdominal wall, pelvic wall, or other more distant organs. According to the American Joint Committee on Cancer’s most recent TNM staging system (Table 1),16 lymph node involvement in the true pelvis (that is, N1–N3) with T1 to T4a disease is now classified as stage III disease. 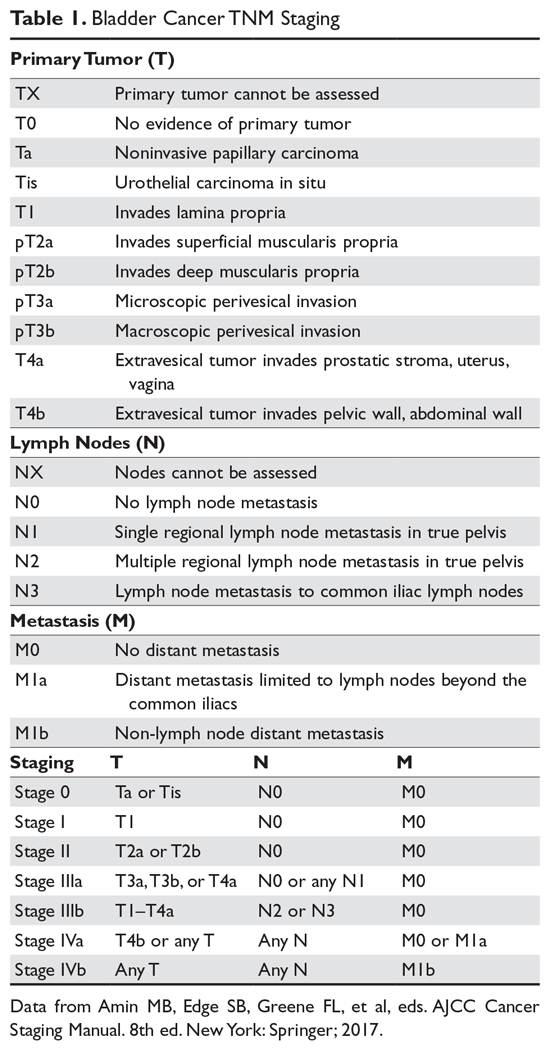
Bladder cancer is often broadly categorized as either non-muscle-invasive or muscle-invasive (which can include metastatic disease). This classification has important implications for treatment. As such, all diagnostic biopsies should be performed with the goal of reaching at least the depth of the muscularis propria in order to accurately detect potential muscularis invasion. If no muscle is detected in the initial specimen, re-resection is recommended if safe and feasible. In cases where muscle cannot be obtained, imaging evidence of T3 disease from CT or magnetic resonance imaging may be used as a surrogate indicator. Once muscle-invasive disease is confirmed, CT evaluation of the chest is also recommended, as bladder cancer can metastasize to the lungs; furthermore, patients are often at risk for secondary concomitant lung cancers given that smoking is the most prevalent risk factor for both. However, patients with small, indeterminate lung nodules not amenable to biopsy should not be denied curative intent treatment given the high likelihood that they represent benign findings.17
Pathogenesis
Because non-muscle-invasive and muscle-invasive tumors behave so differently, they are thought to arise from 2 distinct mechanisms. Although there is overlap and non-muscle-invasive cancer can certainly progress to a high-grade, invasive type of malignancy over time, current theory proposes that non-muscle-invasive bladder cancer predominantly develops just from urothelial hyperplasia, which then recruits branching vasculature to grow slowly. More aggressive urothelial carcinomas, including muscle-invasive and metastatic disease, are instead thought to arise directly from flat dysplasia that progresses to carcinoma in situ, and is much more prone to invasive growth and distant spread.18
Regardless of grade and stage, the most commonly identified genomic alterations in urothelial carcinoma are mutations in the promoter region of the telomerase reverse transcriptase (TERT) gene, which have been identified in approximately 70% of cases.19 Mutations in TERT can be readily detected in urine sediments and may ultimately have implications for diagnosis and early detection.20,21 In current practice, however, the clinical relevance of these observations remains under development. Other genomic alterations that may contribute to the development of urothelial carcinoma, and also provide new potential therapeutic targets, include alterations in the TP53 gene, the RB (retinoblastoma) gene, and the FGFR3 (fibroblast growth factor receptor) gene. FGFR3 has particular significance as it appears to be relatively common in non-muscle invasive disease (up to 60%–70%) and is likely an actionable driver mutation that may define a particular molecular subset of urothelial carcinoma; thus, it may have important implications for treatment decisions.22
Treatment
Case Continued
Pathologic evaluation of the specimen reveals a high-grade urothelial carcinoma with tumor invasion into the muscularis propria. A CT urogram is performed and does not reveal any notably enlarged pelvic nodes or suspicious lesions in the upper urinary tract. CT chest does not reveal any evidence of distant metastatic disease. Given the presence of muscle-invasive disease, the patient agrees to proceed with neoadjuvant chemotherapy and radical cystoprostatectomy with pelvic node dissection. He undergoes treatment with dose-dense (accelerated) MVAC (methotrexate, vinblastine, doxorubicin, and cisplatin) for 3 cycles, followed by surgery with cystoprostatectomy. Overall, he tolerates the procedure well and recovers quickly. Pathology reveals the presence of disease in 2 regional nodes, consistent with T4a (stage III) disease, and a small degree of residual disease in the bladder. He is followed closely in the oncology clinic, returning for urine cytology, liver and renal function tests, and imaging with CT of chest, abdomen, and pelvis every 3 months.
- What is the first-line approach to management in patients with muscle-invasive disease?
- How would the treatment strategy differ if the patient had presented with metastatic disease (stage IV)?
First-Line Management for Curative Intent: Muscle-Invasive Disease
Muscle-invasive urothelial carcinoma (including T2, T3, or T4 disease) is typically treated in a multidisciplinary fashion with neoadjuvant cisplatin-based chemotherapy followed by radical cystectomy. This approach is recommended over radical cystectomy alone because of high relapse rates following cystectomy alone, even in the setting of bilateral pelvic lymphadenectomy.23 However, because of the associated short- and long-term toxicity of cisplatin-based regimens, this optimal treatment paradigm is reserved for patients deemed cisplatin-eligible.
Medical fitness to receive cisplatin-based chemotherapy is assessed by a number of factors and varies by institution, but most frequently consider functional status (Eastern Cooperative Oncology Group [ECOG] performance status or Karnofsky Performance Status), creatinine clearance, hearing preservation, peripheral neuropathy, and cardiac function.24 Many programs will elect to defer cisplatin-based chemotherapy in patients with low performance status (ie, < 60–70 on Karnofsky scale or > 2 on ECOG scale), creatinine clearance below 60 mL/min, or significant heart failure (NHYA class III or worse). Cisplatin-based chemotherapy may worsen hearing loss in those with hearing loss of 25 dB from baseline at 2 continuous frequencies and also may worsen neuropathy in those with baseline grade 1 peripheral neuropathy. However, these adverse outcomes must be balanced against the curative intent of the multimodality systemic approach.
In patients with renal insufficiency, caution must be taken with regard to cisplatin. Percutaneous nephrostomy placement or ureteral stenting should be attempted to relieve any ureteral outlet obstruction and restore kidney function if a patient’s renal insufficiency has resulted from this obstruction. If medical renal disease or long-term renal insufficiency is present, however, patients should instead be referred for immediate cystectomy or for a bladder-preserving approach. Generally, a creatinine clearance of 60 mL/min is required to safely receive cisplatin-based chemotherapy, although some advocate for treatment with a creatinine clearance as low as 50 mL/min. When this extended criterion is used, the dose of cisplatin may be split over 2 days to minimize renal toxicity and maximize hydration. Analysis of renal function utilizing a 24-hour urine collection should be incorporated whenever possible, as estimates of creatinine clearance have been demonstrated to be inaccurate in some instances.25
For cisplatin-eligible patients, neoadjuvant chemotherapy with a cisplatin base has consistently demonstrated a survival benefit when given prior to surgery.26,27 Historically, several different platinum-based regimens have been studied, with none showing superior effectiveness in a randomized trial over the others in the neoadjuvant setting. These regimens have included classic MVAC, dose-dense MVAC (MVAC with pegfilgrastim), GC (gemcitabine and cisplatin), and CMV (methotrexate, vinblastine, cisplatin, and leucovorin).
While classic MVAC was preferred in the 1990s and early 2000s,28,29 the availability of growth factor, such as pegfilgrastim, has made dose-dense MVAC (otherwise referred to as accelerated MVAC or ddMVAC) widely preferred and universally recommended over classic MVAC. The ddMVAC regimen with the addition of a synthetic granulocyte colony-stimulating factor (G-CSF) is substantially better tolerated than classic MVAC, as the G-CSF support minimizes the severe toxicities of classic MVAC, such as myelosuppression and mucositis, and allows for the administration of drugs in a dose-dense fashion.30,31
Both ddMVAC and GC are considered reasonable options for neoadjuvant chemotherapy and are the predominant choices for cisplatin-eligible patients (Table 2). 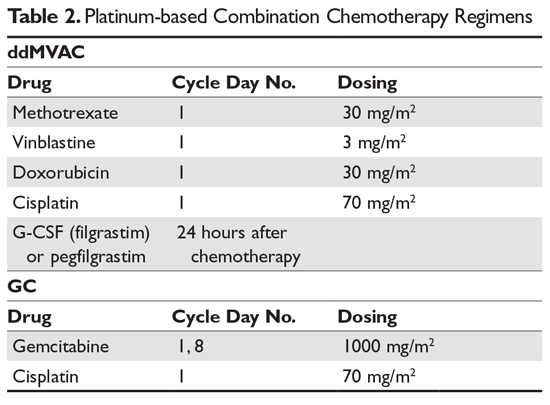
Prospective data defining the role of adjuvant chemotherapy for patients after cystectomy has been fraught by a variety of factors, including the known benefit of neoadjuvant chemotherapy, the high complication rate of cystectomy making chemotherapy infeasible, and clinician bias that has hampered accrual in prior trials. Thus, no level 1 evidence exists defining the benefit of adjuvant chemotherapy in patients who did not receive neoadjuvant therapy. In a report of the largest study performed in this setting, there was a statistically significant benefit in PFS but not in OS.36 Criticisms of this trial include its lack of statistical power due to a failure to accrue the targeted goal and the preponderance of node-positive patients. Regardless, for patients who have pT2–4, N1 disease after radical cystectomy and remain cisplatin-eligible after not receiving neoadjuvant chemotherapy, this remains an option.
Despite the established clinical dogma surrounding neoadjuvant chemotherapy followed by surgery, some patients are either not eligible for or decline to receive radical cystectomy, while others are not candidates for neoadjuvant cisplatin-based chemotherapy for the reasons outlined above. For patients who are surgical candidates but unable to receive neoadjuvant chemotherapy due to renal or cardiac function, they may proceed directly to surgery. For patients unable or unwilling to proceed to radical cystectomy regardless, bladder preservation strategies exist. Maximal TURBT may be an option for some patients, but, as outlined above, used alone this would be likely to lead to a high degree of local and distant failure. Combined modality chemoradiotherapy as consolidation after maximal TURBT is an established option for patients unable to undergo surgery or seeking bladder preservation. Several trials have demonstrated encouraging outcomes with this approach and were highlighted in a large meta-analysis.37 Various chemosensitizing chemotherapeutic regimens have been evaluated, including cisplatin alone or as a doublet, gemcitabine alone, and 5-fluouracil plus mitomycin C, but no randomized studies have compared these regimens to each other, nor have they been compared to surgical approaches. However, this strategy remains an option as an alternative to surgery.
First-Line Management: Metastatic Disease
The approach to therapy in patients who present with metastatic urothelial carcinoma is very similar to that used in neoadjuvant perioperative chemotherapy. The consensus first-line treatment in medically appropriate patients is cisplatin-based chemotherapy with either GC or ddMVAC (both category 1 National Comprehensive Cancer Network [NCCN] recommendations; Figure 2).30,31,38–40 
Head-to-head studies specifically comparing ddMVAC and GC have been limited. GC has been compared to classic MVAC, with results showing equivalent efficacy but improved tolerability, as expected.38,40 ddMVAC was compared with a modified version of GC (termed “dose-dense GC”) in a phase 3 study from Greece, which demonstrated similar outcomes.41
Surgical intervention with radical cystectomy and regional lymph node dissection is typically deferred for patients who present with distant metastatic disease, unlike those who present with locally advanced disease. Radical cystectomy has traditionally been thought of as overly aggressive without sufficient benefit, although evidence to guide this approach remains sparse.42 As such, most expert recommendations and consensus statements simply recommend against surgical intervention and leave the decision between ddMVAC and GC up to the individual clinician.
In patients who are not eligible for cisplatin therapy, it is reasonable to consider chemotherapy with a combination of gemcitabine and carboplatin. This combination has been shown to be equivalent to MCAVI (methotrexate, carboplatin, vinblastine) in terms of overall survival (OS; 9 months versus 8 months) and progression-free survival (PFS; 6 months versus 4 months) with significantly fewer serious toxicities (9% versus 21%).43
The advent of immunotherapy in recent years has provided several new alternatives for cisplatin-ineligible patients. While immunotherapies such as pembrolizumab or atezolizumab are not yet recommended as first-line therapy for cisplatin-eligible patients, these 2 drugs are approved as options for first-line therapy in cisplatin-ineligible patients with metastatic disease. In a recent phase 2 trial (IMvigor210) involving 119 patients who were given atezolizumab as first-line therapy, median PFS was 2.7 months and median OS was 15.9 months.44 Another trial using data from patients in the KEYNOTE-052 study who received pembrolizumab as first-line therapy demonstrated antitumor activity with pembrolizumab and acceptable tolerability in cisplatin-ineligible patients with advanced urothelial carcinoma.45 The primary endpoint was objective response (either complete or partial response), which was achieved in 24% of the intention-to-treat population. Median PFS was 2 months, and 6-month OS was observed in 67% of patients. Both atezolizumab and pembrolizumab were given accelerated approval based on these single-arm studies in this setting. However, due to inferior outcomes in subsequent trials that included single-agent immunotherapy arms for patients in the first-line setting, the US Food and Drug Administration (FDA) has clarified the approval. In the subsequent trials, patients with a low PD-L1 biomarker based on the individual assay used for each drug did worse on immunotherapy alone (compared to chemotherapy or both combined), and the single-therapy arms were stopped early. Thus, the FDA now recommends that pembrolizumab or atezolizumab be used in the first line only for cisplatin-ineligible patients who have PD-L1 expression on tumor cells above the threshold studied on each individual assay, or are unfit for any platinum-based chemotherapy. Further study regarding the optimal role of biomarkers and chemotherapy-immunotherapy combinations is ongoing.
Case Continued
Ten months after his procedure, the patient is found to have prominent retroperitoneal lymphadenopathy and a 1.0-cm liver nodule suspicious for malignancy is noted on surveillance imaging. CT-guided biopsy of the liver reveals high-grade urothelial carcinoma, consistent with both recurrence and distant metastasis. The patient is informed that he needs to resume systemic therapy for recurrent metastatic disease. The options discussed include salvage single-agent chemotherapy with gemcitabine or immunotherapy with pembrolizumab. He elects to move forward with immunotherapy and is scheduled to begin pembrolizumab.
- What other immunotherapies might this patient consider for second-line therapy?
- Is chemotherapy a second-line option for this patient?
Second-Line Therapies and Management of Progressive Disease
Disease progression is unfortunately seen in the majority of cases of advanced urothelial carcinoma.46 New second-line therapies have recently been approved by the FDA in the form of monoclonal antibodies targeting programmed death 1 (PD-1) and a PD-1 ligand (PD-L1) (Figure 3). 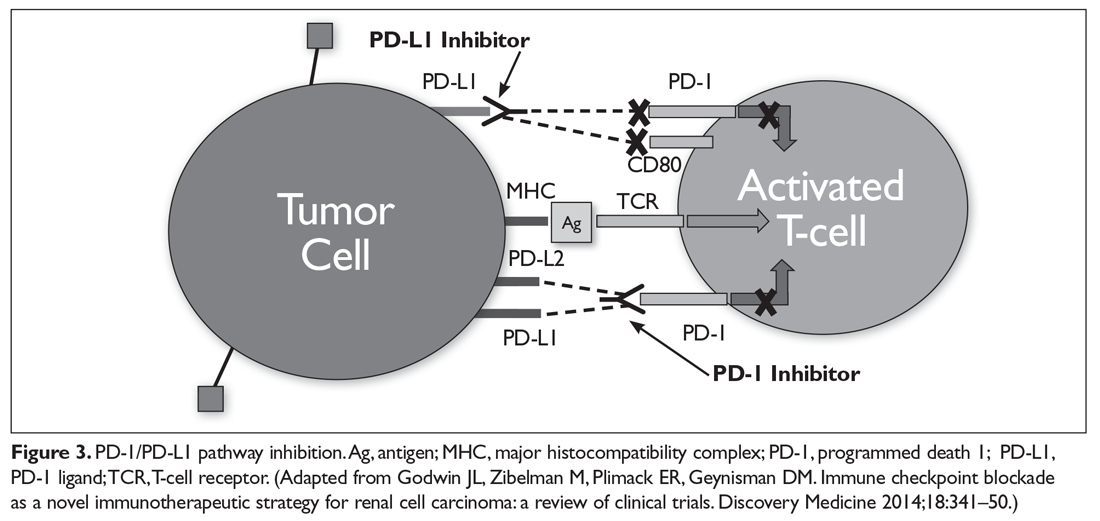
Approval of pembrolizumab, a PD-1 inhibitor, was largely supported by the Keynote-045 trial,47,48 which looked at 542 patients who had progressed or recurred after platinum-based chemotherapy. These patients were randomly assigned to either pembrolizumab or investigator’s choice of chemotherapy (paclitaxel, docetaxel, or vinflunine). Patients treated with pembrolizumab had a significantly improved OS (median of 10.3 months versus 7.4 months), but no statistically significant difference in PFS (2.1 months versus 3.3 months). Interestingly, the rate of responses of 12 months or longer was higher with pembrolizumab than with more traditional second-line chemotherapy (68% versus 35%). The strength of this data has led to a category 1 recommendation in the most recent NCCN guidelines.39
The approval of atezolizumab, a PD-L1 inhibitor, as a second-line therapy for advanced urothelial carcinoma is largely supported by data from IMvigor211, a phase 3 trial that studied 931 patients randomly assigned to atezolizumab or investigator’s choice chemotherapy. OS did not differ significantly between patients in the atezolizumab group who had ≥ 5% expression of PD-L1 on tumor-infiltrating immune cells and patients in the chemotherapy group (11.1 months versus 10.6 months), but mean duration of response was longer (15.9 months versus 8.3 months).49 Therapy with atezolizumab had significantly fewer toxicities than chemotherapy (grade 3 or 4 toxicities of 20% versus 43%).
Phase 3 studies of nivolumab (PD-1 inhibitor), avelumab (PD-L1 inhibitor), and durvalumab (PD-L1 inhibitor) have not yet been published. These agents have received accelerated approval, however, as second-line treatment of advanced urothelial carcinoma based on promising data from phase 1 and phase 2 studies.50–52
Second-line chemotherapy is also an option for patients who do not qualify for immunotherapy or who progress during or after immunotherapy. Although there has been a great deal of excitement about new developments with immunotherapy and the survival benefit seen compared to investigator’s choice chemotherapy, the fact remains that most patients do not respond to immunotherapy. Still, some patients do derive benefit from single-agent chemotherapy in the platinum-refractory setting. Options based on primarily single-arm studies include gemcitabine, paclitaxel, docetaxel, pemetrexed, ifosfamide, oxaliplatin, and eribulin (Figure 2). In a randomized phase 3 trial, vinflunine demonstrated an OS benefit in platinum-refractory patients compared to best supportive care; it subsequently received approval by the European Medicines Agency.53 More recently in the phase 3 RANGE trial, docetaxel plus ramucirumab (a monoclonal antibody targeting vascular endothelial growth factor receptor 2) was compared to docetaxel plus placebo and met its primary endpoint of an improvement in PFS (median 4.07 months versus 2.76 months, P = 0.0118).54 OS has not been reported and this regimen has not yet received regulatory approval, however. Unfortunately, trials comparing these regimens are lacking, and response rates and survival remain modest. Clearly, better therapies and biomarkers to help personalize treatment options are needed.
Further investigations are underway with alternative regimens, including but not limited to targeted therapy in the setting of specific genetic and epigenetic alterations. These include mutations affecting tyrosine kinase receptors (eg, RAS/RAF, PI3K, AKT, and mTOR), cell cycle regulators (eg, TP53 or RB1), FGFR3 mutations, PTEN deletions, gene amplifications (eg, FGFR1, CCND1, and MDM2), or changes in genes responsible for chromatin remodeling (eg, UTX, CHD6, or ARID1A). As noted, there is particular excitement regarding FGFR3 inhibitors, which have shown compelling efficacy in phase 1 and 2 single-arm trials. Several agents are being evaluated in randomized trials and represent a potential path to the first targeted therapeutic class with a role in urothelial malignancies.
Surgical resection of metastases may be considered in very select cases.55 Surgery may have a role in limiting metastatic complications and improving cancer control, but this should be discussed at length with the patient using a multidisciplinary approach with careful restaging prior to surgery.
Case Continued
The patient remains on pembrolizumab every 3 weeks as per protocol with regular surveillance imaging. His disease stabilizes as the nodule in his liver and the retroperitoneal lymph nodes, all representing metastatic disease, became slightly smaller in size without evidence of any new disease. He continues to follow up closely with his genitourinary oncologist, undergoing regular surveillance and imaging every 3 months without evidence of disease progression.
Approximately 12 months into therapy, the patient notices a nonproductive cough with progressive and rapidly worsening shortness of breath. He is noted to be hypoxic with oxygen saturation levels to 79% in clinic and is sent immediately to the emergency department by his oncologist. Diffuse bilateral reticular opacities are noted on chest radiograph. Non-contrast CT scan demonstrates diffuse ground-glass opacities consistent with acute respiratory distress syndrome–pattern pneumonitis. He is admitted to the intensive care unit.
The patient is aggressively treated with high-flow nasal oxygen supplementation, intravenous steroids, and empiric antibiotics. He slowly improves on high-dose steroids (methylprednisolone 1 mg/kg/day) without requiring intubation or infliximab therapy and is discharged home in stable condition after 10 days. Oral steroid therapy is continued with a long taper over 6 weeks. In the setting of his grade 3 pneumonitis, pembrolizumab is discontinued and the patient is scheduled for a follow-up appointment with his oncologist to discuss next steps.
- In addition to pneumonitis, what other toxicities should you monitor for in patients treated with an immune checkpoint inhibitor?
- Is this patient a candidate to receive immunotherapy again in the future?
Treatment Toxicities
As use of immune checkpoint inhibitors has become more prevalent, the medical community has become increasingly aware of various immune-related adverse effects (irAE) associated with these drugs. These toxicities can be seen in virtually any organ system, and even vague complaints that arise years after therapy initiation should be treated with a high level of suspicion. The most commonly affected organ systems include the skin, gastrointestinal (GI) tract, lungs, liver, and endocrine system, although all other organ systems can be involved (Table 3) and toxicities appear to be similar across individual drugs. 
The American Society of Clinical Oncology recently published a complete set of recommendations to guide clinicians on appropriate treatment strategies for each manifestation of immunotherapy-related toxicity.56 The details of these recommendations largely fall outside the purview of this article, but the mainstays of management are worth noting. These include high-dose systemic glucocorticoids, along with supportive care and cessation of immunotherapy in grade 3 or 4 toxicities. Infliximab is frequently recommended as an adjunct in severe or refractory cases.
Chemotherapy-related toxicities, on the other hand, are well-described and tend to be more familiar to patients and clinicians (Table 3). Classic MVAC, which has now been largely replaced by ddMVAC, was notoriously difficult to tolerate. It was known for a high rate of serious (grade 3 or 4) myelosuppressive complications as well as frequent GI toxicities. These complications include neutropenia (57%), stomatitis (10%), and nausea and vomiting (6%).23 ddMVAC with growth factor support is much better tolerated than classic MVAC. Prominent complaints with ddMVAC still can include nausea, GI distress, mucositis, and fatigue, but the incidence of myelosuppressive complications in particular has markedly decreased. GC is largely well tolerated, with minimal nausea and manageable myelotoxicity, but it is associated with an increased risk of venous thromboembolism.38
Prognosis
Case Conclusion
After returning home, the patient discusses his complicated medical course with his oncologist. Given his continued high quality of life with good functional status, he requests to continue with therapy for his metastatic bladder cancer and is interested in joining a clinical trial. He is referred to a nearby academic center with openings in a clinical trial for which he would be eligible. In the meantime, his oncologist guides him through filling out an advance directive and recommends that he make an appointment with palliative care services to ensure adequate home support for any future needs he may have.
- What is the estimated 5-year survival rate for patients with metastatic bladder cancer?
Overall, prognosis in patients with metastatic bladder cancer remains poor. Median survival in patients being treated with multi-agent chemotherapy is approximately 15 months,38,40 with an expected 5-year survival of just 15%. This is much improved, however, as prior to the advent of modern chemotherapy estimated survival was just 6 months with metastatic bladder cancer. Importantly, these figures do not take into account the recent advancements with immunotherapy, and thus it is reasonable to assume survival rates may continue to improve. In light of these recent advances, it is strongly recommended that whenever possible patients and clinicians consider participation in clinical trials to continue uncovering new and better therapies moving forward.
A number of tools have been developed to help risk stratify patients based on comorbidity, performance status, and other characteristics, but none have been universally adopted.57–60 As with many other malignancies, performance status is an important predictor of clinical outcomes in these patients.61–63 Sites of metastasis also may serve to suggest the course of disease. Patients with visceral metastases typically exhibit significantly worse disease with a shortened survival. The role of molecular factors as prognostic markers in bladder cancer is still under investigation. Many biomarkers are being considered (including mutations and polymorphisms in p53, ERCC1, and ERCC2), and evidence suggests some may have a role in prognosis; thus far, none have been validated as prognostic or predictive tools in urothelial carcinoma.
Conclusion
Bladder cancer includes an aggressive group of genitourinary tract malignancies, of which urothelial carcinoma is by far the most common in the Western world. Cisplatin-based therapy remains a mainstay of treatment for eligible patients with both localized and metastatic disease, but immunotherapies have provided a new and promising tool to use in the setting of progressing malignancy. The individual impact of these agents on OS is still being examined. Further studies and ongoing participation in clinical trials whenever possible continue to be essential to the discovery of future treatment options for this highly aggressive disease.
1. Stewart BW, Kleihues P. World cancer report. IARCPress. 2003.
2. Scosyrev E, Noyes K, Feng C, Messing E. Sex and racial differences in bladder cancer presentation and mortality in the US. Cancer 2009;115:68–74.
3. Hinotsu S, Akaza H, Miki T, et al. Bladder cancer develops 6 years earlier in current smokers: Analysis of bladder cancer registry data collected by the cancer registration committee of the Japanese Urological Association. Int J Urol 2009;16:64–9.
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
5. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: bladder cancer. 2018. https://seer.cancer.gov/statfacts/html/urinb.html. Accessed May 5, 2018.
6. Freedman ND, Silverman DT, Hollenbeck AR, et al. Association between smoking and risk of bladder cancer among men and women. JAMA 2011;306:737–45.
7. Pietzak EJ, Mucksavage P, Guzzo TJ, Malkowicz SB. Heavy cigarette smoking and aggressive bladder cancer at initial presentation. Urology 2015;86:968–73.
8. Khadra MH, Pickard RS, Charlton M, et al. A prospective analysis of 1,930 patients with hematuria to evaluate current diagnostic practice. J Urol 2000;163:524–7.
9. Grossman HB, Messing E, Soloway M, et al. Detection of bladder cancer using a point-of-care proteomic assay. JAMA 2005;293:810–16.
10. Mariani AJ, Mariani MC, Macchioni C, et al. The significance of adult hematuria: 1,000 hematuria evaluations including a risk-benefit and cost-effectiveness analysis. J Urol 1989;141:350–5.
11. Grossfeld GD, Litwin MS, Wolf JS, et al. Evaluation of asymptomatic microscopic hematuria in adults: the American Urological Association best practice policy--part II: patient evaluation, cytology, voided markers, imaging, cystoscopy, nephrology evaluation, and follow-up. Urology 2001;57:604–10.
12. Grossfeld GD, Litwin MS, Wolf JS, et al. Evaluation of asymptomatic microscopic hematuria in adults: the American Urological Association best practice policy--part I: definition, detection, prevalence, and etiology. Urology 2001;57:599–603.
13. Mohr DN, Offord KP, Owen RA, Melton LJ. Asymptomatic microhematuria and urologic disease. A population-based study. JAMA 1986;256:224–9.
14. Messing EM, Young TB, Hunt VB, et al. Home screening for hematuria: results of a multiclinic study. J Urol 1992;148:289–92.
15. Gray PJ, Lin CC, Jemal A, et al. Clinical–pathologic stage discrepancy in bladder cancer patients treated with radical cystectomy: results from the National Cancer Data Base. Int J Radiat Oncol 2014;88:1048–56.
16. Bochner B, Hansel D, Efstathiou J, et al. Urinary bladder. In: Amin M, ed. AJCC cancer staging manual. 8th. New York: Springer; 2017:757.
17. Cahn DB, McGreen B, Lee A, et al. Clinical destiny of indeterminate pulmonary nodules in patients undergoing radical cystectomy for urothelial carcinoma of the bladder [abstract]. J Clin Oncol 2017;35(6 suppl):297-297.
18. Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer 2015;15:25–41.
19. Kurtis B, Zhuge J, Ojaimi C, et al. Recurrent TERT promoter mutations in urothelial carcinoma and potential clinical applications. Ann Diagn Pathol 2016;21:7–11.
20. Ito H, Kyo S, Kanaya T, et al. Detection of human telomerase reverse transcriptase messenger RNA in voided urine samples as a useful diagnostic tool for bladder cancer. Clin Cancer Res 1998;4:2807–10.
21. Utting M, Werner W, Dahse R, et al. Microsatellite analysis of free tumor DNA in urine, serum, and plasma of patients: a minimally invasive method for the detection of bladder cancer. Clin Cancer Res 2002;8:35–40.
22. Sethakorn N, O’Donnell PH. Spectrum of genomic alterations in FGFR3: current appraisal of the potential role of FGFR3 in advanced urothelial carcinoma. BJU Int 2016;118:681–91.
23. Grossman HB, Natale RB, Tangen CM, et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med 2003;349:859–66.
24. Galsky MD, Hahn NM, Rosenberg J, et al. Treatment of patients with metastatic urothelial cancer ‘unfit’ for cisplatin-based chemotherapy. J Clin Oncol 2011;29:2432–8.
25. Raj GV, Iasonos A, Herr H, Donat SM. Formulas calculating creatinine clearance are inadequate for determining eligibility for cisplatin-based chemotherapy in bladder cancer. J Clin Oncol 2006;24:3095–100.26. Advanced Bladder Cancer Meta-analysis Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer: a systematic review and meta-analysis. Lancet 2003;361:1927–34.
27. Advanced Bladder Cancer Meta-analysis Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data. Eur Urol 2005;48:202–6.
28. Sternberg CN. A critical review of the management of bladder cancer. Crit Rev Oncol Hematol 1999;31:193–207.
29. Sternberg CN, Yagoda A, Scher HI, et al. Preliminary results of M-VAC (methotrexate, vinblastine, doxorubicin and cisplatin) for transitional cell carcinoma of the urothelium. J Urol 1985;133:403–7.
30. Sternberg CN, de Mulder P, Schornagel JH, et al. Seven year update of an EORTC phase III trial of high-dose intensity M-VAC chemotherapy and G-CSF versus classic M-VAC in advanced urothelial tract tumours. Eur J Cancer 2006;42:50–4.
31. Sternberg CN, de Mulder PHM, Schornagel JH, et al. Randomized phase III trial of high–dose-intensity methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC) chemotherapy and recombinant human granulocyte colony-stimulating factor versus classic MVAC in advanced urothelial tract tumors: European Organization for Research and Treatment of Cancer Protocol No. 30924. J Clin Oncol 2001;19:2638–46.
32. Soloway MS, Einstein A, Corder MP, et al. A comparison of cisplatin and the combination of cisplatin and cyclophosphamide in advanced urothelial cancer. A National Bladder Cancer Collaborative Group A Study. Cancer 1983;52:767–72.
33. Plimack ER, Hoffman-Censits JH, Viterbo R, et al. Accelerated methotrexate, vinblastine, doxorubicin, and cisplatin is safe, effective, and efficient neoadjuvant treatment for muscle-invasive bladder cancer: results of a multicenter phase II study with molecular correlates of response and toxicity. J Clin Oncol 2014;32:1895–901.
34. Van Allen EM, Mouw KW, Kim P, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov 2014;4:1140–53.
35. Plimack ER, Dunbrack RL, Brennan TA, et al. Defects in DNA repair genes predict response to neoadjuvant cisplatin-based chemotherapy in muscle-invasive bladder cancer. Eur Urol 2015;68:959–67.
36. Sternberg CN, Skoneczna I, Kerst JM, et al. Immediate versus deferred chemotherapy after radical cystectomy in patients with pT3-pT4 or N+ M0 urothelial carcinoma of the bladder (EORTC 30994): an intergroup, open-label, randomised phase 3 trial. Lancet Oncol 2015;16:76–86.
37. Mak RH, Hunt D, Shipley WU, et al. Long-term outcomes in patients with muscle-invasive bladder cancer after selective bladder-preserving combined-modality therapy: a pooled analysis of Radiation Therapy Oncology Group protocols 8802, 8903, 9506, 9706, 9906, and 0233. J Clin Oncol 2014;32:3801–9.
38. von der Maase H, Hansen SW, Roberts JT, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol 2000;18:3068–77.
39. Flaig T, Spiess P, Agarwal N, et al. National Comprehensive Cancer Network. Bladder cancer (version 3.2018). 2018. www.nccn.org/professionals/physician_gls/pdf/bladder.pdf. Accessed May 5, 2018.
40. von der Maase H, Sengelov L, Roberts JT, et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol 2005;23:4602–8.
41. Bamias A, Dafni U, Karadimou A, et al. Prospective, open-label, randomized, phase III study of two dose-dense regimens MVAC versus gemcitabine/cisplatin in patients with inoperable, metastatic or relapsed urothelial cancer: a Hellenic Cooperative Oncology Group study (HE 16/03). Ann Oncol 2013;24:1011–7.
42. Li R, Metcalfe M, Kukreja J, Navai N. Role of radical cystectomy in non-organ confined bladder cancer: a systematic review. Bladder Cancer 2018;4:31–40.
43. De Santis M, Bellmunt J, Mead G, et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin-based chemotherapy: EORTC study 30986. J Clin Oncol 2012;30:191–9.
44. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 2017;389:67–76.
45. Balar A V, Castellano D, O’Donnell PH, et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): a multicentre, single-arm, phase 2 study. Lancet Oncol 2017;18:1483–92.
46. Manoharan M, Ayyathurai R, Soloway MS. Radical cystectomy for urothelial carcinoma of the bladder: an analysis of perioperative and survival outcome. BJU Int 2009;104:1227–32.
47. Bellmunt J, de Wit R, Vaughn D, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 2017;376:1015–26.
48. Bajorin D, de Wit R, Vaughn D, et al. Planned survival analysis from KEYNOTE-045: Phase 3, open-label study of pembrolizumab (pembro) versus paclitaxel, docetaxel, or vinflunine in recurrent, advanced urothelial cancer (UC). (Abstract 4501). J Clin Oncol 2017;35(15_suppl):4501-4501.
49. Powles T, Durán I, van der Heijden MS, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018;391:748–57.
50. Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 2017;18:312–22.
51. Patel MR, Ellerton J, Infante JR, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol 2018;19:51–64.
52. Powles T, O’Donnell PH, Massard C, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma. JAMA Oncol 2017;3:e172411.
53. Bellmunt J, Theodore C, Demkov T, et al. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum-containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract. J Clin Oncol 2009;27:4454–61.
54. Petrylak DP, de Wit R, Chi KN, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel in patients with locally advanced or metastatic urothelial carcinoma after platinum-based therapy (RANGE): a randomised, double-blind, phase 3 trial. Lancet 2017;390:2266–77.
55. Abufaraj M, Dalbagni G, Daneshmand S, et al. The role of surgery in metastatic bladder cancer: a systematic review. Eur Urol 2018;73:543–57.
56. Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2018;36:1714–68.
57. Bajorin DF, Dodd PM, Mazumdar M, et al. Long-term survival in metastatic transitional-cell carcinoma and prognostic factors predicting outcome of therapy. J Clin Oncol 1999;17:3173–81.
58. Mayr R, May M, Martini T, et al. Comorbidity and performance indices as predictors of cancer-independent mortality but not of cancer-specific mortality after radical cystectomy for urothelial carcinoma of the bladder. Eur Urol 2012;62:662–70.
59. Nakagawa T, Hara T, Kawahara T, et al. Prognostic risk stratification of patients with urothelial carcinoma of the bladder with recurrence after radical cystectomy. J Urol 2013;189:1275–81.
60. Ploeg M, Kums AC, Aben KK, et al. Prognostic factors for survival in patients with recurrence of muscle invasive bladder cancer after treatment with curative intent. Clin Genitourin Cancer 2011;9:14–21.
61. Saxman SB, Propert KJ, Einhorn LH, et al. Long-term follow-up of a phase III intergroup study of cisplatin alone or in combination with methotrexate, vinblastine, and doxorubicin in patients with metastatic urothelial carcinoma: a cooperative group study. J Clin Oncol 1997;15:2564–9.
62. Lin CC, Hsu CH, Huang CY, et al. Prognostic factors for metastatic urothelial carcinoma treated with cisplatin and 5-fluorouracil-based regimens. Urology 2007;69:479–84.
63. Schag CC, Heinrich RL, Ganz PA. Karnofsky performance status revisited: reliability, validity, and guidelines. J Clin Oncol 1984;2:187–93.
Introduction
Bladder cancer is by far the most common cancer of the urinary system. Worldwide, approximately 450,000 new cases are diagnosed and 165,000 deaths are caused by bladder cancer each year.1 In the United States and in Europe, the most common type of bladder cancer is urothelial carcinoma (also referred to as transitional cell carcinoma), which accounts for more than 90% of all bladder cancers in these regions of the world. The remainder of bladder cancers are divided among squamous cell carcinomas, adenocarcinomas, small cell carcinomas, and, even more rarely, between various other nonepithelial tumors (eg, sarcoma).
Bladder cancer is classically thought of as a disease of the elderly, with a median age at diagnosis of 69 years in men and 71 years in women.2 The incidence of bladder cancer increases with age: in persons aged 65 to 69 years, incidence is 142 per 100,000 men and 33 per 100,000 women, and in those older than 85 years the rate doubles to 296 per 100,000 men and 74 per 100,000 women.3 The incidence is 3 times greater in men than in women.4
Urothelial carcinoma is traditionally categorized by its degree of invasion into the bladder wall: superficial (non-muscle-invasive), muscle-invasive, or metastatic disease. At the time of diagnosis, most patients have non-muscle-invasive disease (~60%); about 4% of all patients present initially with metastatic disease.5 This article focuses on metastatic bladder cancer, but muscle-invasive disease is discussed as well.
The most important factor contributing to the development of urothelial carcinoma is tobacco smoking. The risk of developing bladder cancer is 4 to 5 times higher in smokers as compared to nonsmokers, with some variation according to sex.6 Quantity of smoking exposure also plays a role, with heavy smokers demonstrating a higher likelihood for high-grade tumors with muscle invasion (or beyond) when compared to light smokers.7 Another important risk factor is occupational exposure to industrial materials, such as carpets, paints, plastics, and industrial chemicals. This type of exposure may be responsible for, or at least contribute to, the development of approximately 20% of urothelial carcinomas. Other risk factors for urothelial carcinoma include but are not limited to prior radiation to the pelvis, prior upper tract urothelial malignancy, human papillomavirus infection, and prior bladder augmentation.
Diagnosis and Staging
Case Presentation
A 63-year-old man with a past medical history of diabetes, deep vein thrombosis, occasional alcohol use, and regular pipe tobacco use presents to his primary care physician with complaints of hematuria. He reports that his urine was a dark red color that morning, which had never happened before. The patient is hemodynamically stable upon evaluation in the office, and a point-of-care urinalysis dipstick is strongly positive for blood. He is referred to a urologist for further evaluation.
In the urology office, urine microscopy is notable for more than 50 red blood cells (RBCs) per high-power field with normal RBC morphology. Flexible cystoscopy performed in the office reveals a single 2-cm, sessile, verrucous, nodular lesion located on the anterior bladder wall. A urine sample and a bladder wash specimen are sent for cytology evaluation. The patient is scheduled to undergo a complete transurethral resection of bladder tumor (TURBT) later that week with samples sent to pathology for evaluation.
- What are the clinical features of bladder cancer?
Hematuria is the most common presentation of bladder cancer, although its specificity is far lower than traditionally thought. In fact, only about 2% to 20% of cases that present with hematuria are found to be caused by malignancy. However, the incidence of genitourinary tract malignancy is much higher in patients presenting with gross hematuria (10%–20%)8–10 than in patients with microscopic hematuria alone.8,10–14 Typically, hematuria associated with malignancy is painless. Multiple studies have shown, however, that hematuria can be a normal variant, with one study demonstrating that up to 61% of patients with hematuria had no identifiable abnormality.8,10,11,13
Abdominal pain, flank pain, dysuria, urinary frequency/urgency, or other irritative voiding symptoms in the absence of hematuria can be presenting symptoms of bladder cancer as well. In these settings, discomfort typically suggests more advanced malignancy with at least local involvement or obstruction. Suprapubic pain may herald invasion into perivesical tissues and nerves, while involvement of the obturator fossa, perirectal fat, urogenital diaphragm, or presacral nerves can often present with perineal or rectal pain. Similarly, lower abdominal pain may represent involvement of lymph nodes, and right upper quadrant pain may signal liver metastasis. Cough or shortness of breath may signify metastatic disease in the lung. Finally, back, rib, or other boney pain may suggest distant metastasis.
- What next steps are required to complete this patient’s staging?
White light cystoscopy remains the gold standard for diagnosis and initial staging of bladder cancer. Additional tools include urine cytology and upper tract studies, including renal computed tomography (CT) urograms. Full urologic evaluation with all 3 modalities (cystourethroscopy, urinary cytology, and upper tract evaluation) is warranted for patients with a high suspicion for malignant etiology of hematuria. CT urograms are particularly useful for upper tract evaluation because they can be used to visualize kidney parenchyma, both renal pelvises and ureters, and pertinent abdominal and pelvic lymph nodes. Initial staging is completed through TURBT, which should ideally contain a segment of muscularis propria to distinguish between Ta (noninvasive), T1, and T2 tumors (Figure 1).
Regarding staging, T1 tumors are distinguished from Ta malignancies by their involvement in the urothelial basement membrane. Tumor invasion into the muscularis propria indicates T2 tumors, while T3 tumors extend through the muscle into the serosa and involve the complete thickness of the bladder wall. Involvement of nearby structures defines T4 bladder cancers, with T4a malignancies involving adjacent organs (prostate, vagina, uterus, or bowel) and T4b tumors involving the abdominal wall, pelvic wall, or other more distant organs. According to the American Joint Committee on Cancer’s most recent TNM staging system (Table 1),16 lymph node involvement in the true pelvis (that is, N1–N3) with T1 to T4a disease is now classified as stage III disease.
Bladder cancer is often broadly categorized as either non-muscle-invasive or muscle-invasive (which can include metastatic disease). This classification has important implications for treatment. As such, all diagnostic biopsies should be performed with the goal of reaching at least the depth of the muscularis propria in order to accurately detect potential muscularis invasion. If no muscle is detected in the initial specimen, re-resection is recommended if safe and feasible. In cases where muscle cannot be obtained, imaging evidence of T3 disease from CT or magnetic resonance imaging may be used as a surrogate indicator. Once muscle-invasive disease is confirmed, CT evaluation of the chest is also recommended, as bladder cancer can metastasize to the lungs; furthermore, patients are often at risk for secondary concomitant lung cancers given that smoking is the most prevalent risk factor for both. However, patients with small, indeterminate lung nodules not amenable to biopsy should not be denied curative intent treatment given the high likelihood that they represent benign findings.17
Pathogenesis
Because non-muscle-invasive and muscle-invasive tumors behave so differently, they are thought to arise from 2 distinct mechanisms. Although there is overlap and non-muscle-invasive cancer can certainly progress to a high-grade, invasive type of malignancy over time, current theory proposes that non-muscle-invasive bladder cancer predominantly develops just from urothelial hyperplasia, which then recruits branching vasculature to grow slowly. More aggressive urothelial carcinomas, including muscle-invasive and metastatic disease, are instead thought to arise directly from flat dysplasia that progresses to carcinoma in situ, and is much more prone to invasive growth and distant spread.18
Regardless of grade and stage, the most commonly identified genomic alterations in urothelial carcinoma are mutations in the promoter region of the telomerase reverse transcriptase (TERT) gene, which have been identified in approximately 70% of cases.19 Mutations in TERT can be readily detected in urine sediments and may ultimately have implications for diagnosis and early detection.20,21 In current practice, however, the clinical relevance of these observations remains under development. Other genomic alterations that may contribute to the development of urothelial carcinoma, and also provide new potential therapeutic targets, include alterations in the TP53 gene, the RB (retinoblastoma) gene, and the FGFR3 (fibroblast growth factor receptor) gene. FGFR3 has particular significance as it appears to be relatively common in non-muscle invasive disease (up to 60%–70%) and is likely an actionable driver mutation that may define a particular molecular subset of urothelial carcinoma; thus, it may have important implications for treatment decisions.22
Treatment
Case Continued
Pathologic evaluation of the specimen reveals a high-grade urothelial carcinoma with tumor invasion into the muscularis propria. A CT urogram is performed and does not reveal any notably enlarged pelvic nodes or suspicious lesions in the upper urinary tract. CT chest does not reveal any evidence of distant metastatic disease. Given the presence of muscle-invasive disease, the patient agrees to proceed with neoadjuvant chemotherapy and radical cystoprostatectomy with pelvic node dissection. He undergoes treatment with dose-dense (accelerated) MVAC (methotrexate, vinblastine, doxorubicin, and cisplatin) for 3 cycles, followed by surgery with cystoprostatectomy. Overall, he tolerates the procedure well and recovers quickly. Pathology reveals the presence of disease in 2 regional nodes, consistent with T4a (stage III) disease, and a small degree of residual disease in the bladder. He is followed closely in the oncology clinic, returning for urine cytology, liver and renal function tests, and imaging with CT of chest, abdomen, and pelvis every 3 months.
- What is the first-line approach to management in patients with muscle-invasive disease?
- How would the treatment strategy differ if the patient had presented with metastatic disease (stage IV)?
First-Line Management for Curative Intent: Muscle-Invasive Disease
Muscle-invasive urothelial carcinoma (including T2, T3, or T4 disease) is typically treated in a multidisciplinary fashion with neoadjuvant cisplatin-based chemotherapy followed by radical cystectomy. This approach is recommended over radical cystectomy alone because of high relapse rates following cystectomy alone, even in the setting of bilateral pelvic lymphadenectomy.23 However, because of the associated short- and long-term toxicity of cisplatin-based regimens, this optimal treatment paradigm is reserved for patients deemed cisplatin-eligible.
Medical fitness to receive cisplatin-based chemotherapy is assessed by a number of factors and varies by institution, but most frequently consider functional status (Eastern Cooperative Oncology Group [ECOG] performance status or Karnofsky Performance Status), creatinine clearance, hearing preservation, peripheral neuropathy, and cardiac function.24 Many programs will elect to defer cisplatin-based chemotherapy in patients with low performance status (ie, < 60–70 on Karnofsky scale or > 2 on ECOG scale), creatinine clearance below 60 mL/min, or significant heart failure (NHYA class III or worse). Cisplatin-based chemotherapy may worsen hearing loss in those with hearing loss of 25 dB from baseline at 2 continuous frequencies and also may worsen neuropathy in those with baseline grade 1 peripheral neuropathy. However, these adverse outcomes must be balanced against the curative intent of the multimodality systemic approach.
In patients with renal insufficiency, caution must be taken with regard to cisplatin. Percutaneous nephrostomy placement or ureteral stenting should be attempted to relieve any ureteral outlet obstruction and restore kidney function if a patient’s renal insufficiency has resulted from this obstruction. If medical renal disease or long-term renal insufficiency is present, however, patients should instead be referred for immediate cystectomy or for a bladder-preserving approach. Generally, a creatinine clearance of 60 mL/min is required to safely receive cisplatin-based chemotherapy, although some advocate for treatment with a creatinine clearance as low as 50 mL/min. When this extended criterion is used, the dose of cisplatin may be split over 2 days to minimize renal toxicity and maximize hydration. Analysis of renal function utilizing a 24-hour urine collection should be incorporated whenever possible, as estimates of creatinine clearance have been demonstrated to be inaccurate in some instances.25
For cisplatin-eligible patients, neoadjuvant chemotherapy with a cisplatin base has consistently demonstrated a survival benefit when given prior to surgery.26,27 Historically, several different platinum-based regimens have been studied, with none showing superior effectiveness in a randomized trial over the others in the neoadjuvant setting. These regimens have included classic MVAC, dose-dense MVAC (MVAC with pegfilgrastim), GC (gemcitabine and cisplatin), and CMV (methotrexate, vinblastine, cisplatin, and leucovorin).
While classic MVAC was preferred in the 1990s and early 2000s,28,29 the availability of growth factor, such as pegfilgrastim, has made dose-dense MVAC (otherwise referred to as accelerated MVAC or ddMVAC) widely preferred and universally recommended over classic MVAC. The ddMVAC regimen with the addition of a synthetic granulocyte colony-stimulating factor (G-CSF) is substantially better tolerated than classic MVAC, as the G-CSF support minimizes the severe toxicities of classic MVAC, such as myelosuppression and mucositis, and allows for the administration of drugs in a dose-dense fashion.30,31
Both ddMVAC and GC are considered reasonable options for neoadjuvant chemotherapy and are the predominant choices for cisplatin-eligible patients (Table 2).
Prospective data defining the role of adjuvant chemotherapy for patients after cystectomy has been fraught by a variety of factors, including the known benefit of neoadjuvant chemotherapy, the high complication rate of cystectomy making chemotherapy infeasible, and clinician bias that has hampered accrual in prior trials. Thus, no level 1 evidence exists defining the benefit of adjuvant chemotherapy in patients who did not receive neoadjuvant therapy. In a report of the largest study performed in this setting, there was a statistically significant benefit in PFS but not in OS.36 Criticisms of this trial include its lack of statistical power due to a failure to accrue the targeted goal and the preponderance of node-positive patients. Regardless, for patients who have pT2–4, N1 disease after radical cystectomy and remain cisplatin-eligible after not receiving neoadjuvant chemotherapy, this remains an option.
Despite the established clinical dogma surrounding neoadjuvant chemotherapy followed by surgery, some patients are either not eligible for or decline to receive radical cystectomy, while others are not candidates for neoadjuvant cisplatin-based chemotherapy for the reasons outlined above. For patients who are surgical candidates but unable to receive neoadjuvant chemotherapy due to renal or cardiac function, they may proceed directly to surgery. For patients unable or unwilling to proceed to radical cystectomy regardless, bladder preservation strategies exist. Maximal TURBT may be an option for some patients, but, as outlined above, used alone this would be likely to lead to a high degree of local and distant failure. Combined modality chemoradiotherapy as consolidation after maximal TURBT is an established option for patients unable to undergo surgery or seeking bladder preservation. Several trials have demonstrated encouraging outcomes with this approach and were highlighted in a large meta-analysis.37 Various chemosensitizing chemotherapeutic regimens have been evaluated, including cisplatin alone or as a doublet, gemcitabine alone, and 5-fluouracil plus mitomycin C, but no randomized studies have compared these regimens to each other, nor have they been compared to surgical approaches. However, this strategy remains an option as an alternative to surgery.
First-Line Management: Metastatic Disease
The approach to therapy in patients who present with metastatic urothelial carcinoma is very similar to that used in neoadjuvant perioperative chemotherapy. The consensus first-line treatment in medically appropriate patients is cisplatin-based chemotherapy with either GC or ddMVAC (both category 1 National Comprehensive Cancer Network [NCCN] recommendations; Figure 2).30,31,38–40
Head-to-head studies specifically comparing ddMVAC and GC have been limited. GC has been compared to classic MVAC, with results showing equivalent efficacy but improved tolerability, as expected.38,40 ddMVAC was compared with a modified version of GC (termed “dose-dense GC”) in a phase 3 study from Greece, which demonstrated similar outcomes.41
Surgical intervention with radical cystectomy and regional lymph node dissection is typically deferred for patients who present with distant metastatic disease, unlike those who present with locally advanced disease. Radical cystectomy has traditionally been thought of as overly aggressive without sufficient benefit, although evidence to guide this approach remains sparse.42 As such, most expert recommendations and consensus statements simply recommend against surgical intervention and leave the decision between ddMVAC and GC up to the individual clinician.
In patients who are not eligible for cisplatin therapy, it is reasonable to consider chemotherapy with a combination of gemcitabine and carboplatin. This combination has been shown to be equivalent to MCAVI (methotrexate, carboplatin, vinblastine) in terms of overall survival (OS; 9 months versus 8 months) and progression-free survival (PFS; 6 months versus 4 months) with significantly fewer serious toxicities (9% versus 21%).43
The advent of immunotherapy in recent years has provided several new alternatives for cisplatin-ineligible patients. While immunotherapies such as pembrolizumab or atezolizumab are not yet recommended as first-line therapy for cisplatin-eligible patients, these 2 drugs are approved as options for first-line therapy in cisplatin-ineligible patients with metastatic disease. In a recent phase 2 trial (IMvigor210) involving 119 patients who were given atezolizumab as first-line therapy, median PFS was 2.7 months and median OS was 15.9 months.44 Another trial using data from patients in the KEYNOTE-052 study who received pembrolizumab as first-line therapy demonstrated antitumor activity with pembrolizumab and acceptable tolerability in cisplatin-ineligible patients with advanced urothelial carcinoma.45 The primary endpoint was objective response (either complete or partial response), which was achieved in 24% of the intention-to-treat population. Median PFS was 2 months, and 6-month OS was observed in 67% of patients. Both atezolizumab and pembrolizumab were given accelerated approval based on these single-arm studies in this setting. However, due to inferior outcomes in subsequent trials that included single-agent immunotherapy arms for patients in the first-line setting, the US Food and Drug Administration (FDA) has clarified the approval. In the subsequent trials, patients with a low PD-L1 biomarker based on the individual assay used for each drug did worse on immunotherapy alone (compared to chemotherapy or both combined), and the single-therapy arms were stopped early. Thus, the FDA now recommends that pembrolizumab or atezolizumab be used in the first line only for cisplatin-ineligible patients who have PD-L1 expression on tumor cells above the threshold studied on each individual assay, or are unfit for any platinum-based chemotherapy. Further study regarding the optimal role of biomarkers and chemotherapy-immunotherapy combinations is ongoing.
Case Continued
Ten months after his procedure, the patient is found to have prominent retroperitoneal lymphadenopathy and a 1.0-cm liver nodule suspicious for malignancy is noted on surveillance imaging. CT-guided biopsy of the liver reveals high-grade urothelial carcinoma, consistent with both recurrence and distant metastasis. The patient is informed that he needs to resume systemic therapy for recurrent metastatic disease. The options discussed include salvage single-agent chemotherapy with gemcitabine or immunotherapy with pembrolizumab. He elects to move forward with immunotherapy and is scheduled to begin pembrolizumab.
- What other immunotherapies might this patient consider for second-line therapy?
- Is chemotherapy a second-line option for this patient?
Second-Line Therapies and Management of Progressive Disease
Disease progression is unfortunately seen in the majority of cases of advanced urothelial carcinoma.46 New second-line therapies have recently been approved by the FDA in the form of monoclonal antibodies targeting programmed death 1 (PD-1) and a PD-1 ligand (PD-L1) (Figure 3).
Approval of pembrolizumab, a PD-1 inhibitor, was largely supported by the Keynote-045 trial,47,48 which looked at 542 patients who had progressed or recurred after platinum-based chemotherapy. These patients were randomly assigned to either pembrolizumab or investigator’s choice of chemotherapy (paclitaxel, docetaxel, or vinflunine). Patients treated with pembrolizumab had a significantly improved OS (median of 10.3 months versus 7.4 months), but no statistically significant difference in PFS (2.1 months versus 3.3 months). Interestingly, the rate of responses of 12 months or longer was higher with pembrolizumab than with more traditional second-line chemotherapy (68% versus 35%). The strength of this data has led to a category 1 recommendation in the most recent NCCN guidelines.39
The approval of atezolizumab, a PD-L1 inhibitor, as a second-line therapy for advanced urothelial carcinoma is largely supported by data from IMvigor211, a phase 3 trial that studied 931 patients randomly assigned to atezolizumab or investigator’s choice chemotherapy. OS did not differ significantly between patients in the atezolizumab group who had ≥ 5% expression of PD-L1 on tumor-infiltrating immune cells and patients in the chemotherapy group (11.1 months versus 10.6 months), but mean duration of response was longer (15.9 months versus 8.3 months).49 Therapy with atezolizumab had significantly fewer toxicities than chemotherapy (grade 3 or 4 toxicities of 20% versus 43%).
Phase 3 studies of nivolumab (PD-1 inhibitor), avelumab (PD-L1 inhibitor), and durvalumab (PD-L1 inhibitor) have not yet been published. These agents have received accelerated approval, however, as second-line treatment of advanced urothelial carcinoma based on promising data from phase 1 and phase 2 studies.50–52
Second-line chemotherapy is also an option for patients who do not qualify for immunotherapy or who progress during or after immunotherapy. Although there has been a great deal of excitement about new developments with immunotherapy and the survival benefit seen compared to investigator’s choice chemotherapy, the fact remains that most patients do not respond to immunotherapy. Still, some patients do derive benefit from single-agent chemotherapy in the platinum-refractory setting. Options based on primarily single-arm studies include gemcitabine, paclitaxel, docetaxel, pemetrexed, ifosfamide, oxaliplatin, and eribulin (Figure 2). In a randomized phase 3 trial, vinflunine demonstrated an OS benefit in platinum-refractory patients compared to best supportive care; it subsequently received approval by the European Medicines Agency.53 More recently in the phase 3 RANGE trial, docetaxel plus ramucirumab (a monoclonal antibody targeting vascular endothelial growth factor receptor 2) was compared to docetaxel plus placebo and met its primary endpoint of an improvement in PFS (median 4.07 months versus 2.76 months, P = 0.0118).54 OS has not been reported and this regimen has not yet received regulatory approval, however. Unfortunately, trials comparing these regimens are lacking, and response rates and survival remain modest. Clearly, better therapies and biomarkers to help personalize treatment options are needed.
Further investigations are underway with alternative regimens, including but not limited to targeted therapy in the setting of specific genetic and epigenetic alterations. These include mutations affecting tyrosine kinase receptors (eg, RAS/RAF, PI3K, AKT, and mTOR), cell cycle regulators (eg, TP53 or RB1), FGFR3 mutations, PTEN deletions, gene amplifications (eg, FGFR1, CCND1, and MDM2), or changes in genes responsible for chromatin remodeling (eg, UTX, CHD6, or ARID1A). As noted, there is particular excitement regarding FGFR3 inhibitors, which have shown compelling efficacy in phase 1 and 2 single-arm trials. Several agents are being evaluated in randomized trials and represent a potential path to the first targeted therapeutic class with a role in urothelial malignancies.
Surgical resection of metastases may be considered in very select cases.55 Surgery may have a role in limiting metastatic complications and improving cancer control, but this should be discussed at length with the patient using a multidisciplinary approach with careful restaging prior to surgery.
Case Continued
The patient remains on pembrolizumab every 3 weeks as per protocol with regular surveillance imaging. His disease stabilizes as the nodule in his liver and the retroperitoneal lymph nodes, all representing metastatic disease, became slightly smaller in size without evidence of any new disease. He continues to follow up closely with his genitourinary oncologist, undergoing regular surveillance and imaging every 3 months without evidence of disease progression.
Approximately 12 months into therapy, the patient notices a nonproductive cough with progressive and rapidly worsening shortness of breath. He is noted to be hypoxic with oxygen saturation levels to 79% in clinic and is sent immediately to the emergency department by his oncologist. Diffuse bilateral reticular opacities are noted on chest radiograph. Non-contrast CT scan demonstrates diffuse ground-glass opacities consistent with acute respiratory distress syndrome–pattern pneumonitis. He is admitted to the intensive care unit.
The patient is aggressively treated with high-flow nasal oxygen supplementation, intravenous steroids, and empiric antibiotics. He slowly improves on high-dose steroids (methylprednisolone 1 mg/kg/day) without requiring intubation or infliximab therapy and is discharged home in stable condition after 10 days. Oral steroid therapy is continued with a long taper over 6 weeks. In the setting of his grade 3 pneumonitis, pembrolizumab is discontinued and the patient is scheduled for a follow-up appointment with his oncologist to discuss next steps.
- In addition to pneumonitis, what other toxicities should you monitor for in patients treated with an immune checkpoint inhibitor?
- Is this patient a candidate to receive immunotherapy again in the future?
Treatment Toxicities
As use of immune checkpoint inhibitors has become more prevalent, the medical community has become increasingly aware of various immune-related adverse effects (irAE) associated with these drugs. These toxicities can be seen in virtually any organ system, and even vague complaints that arise years after therapy initiation should be treated with a high level of suspicion. The most commonly affected organ systems include the skin, gastrointestinal (GI) tract, lungs, liver, and endocrine system, although all other organ systems can be involved (Table 3) and toxicities appear to be similar across individual drugs.
The American Society of Clinical Oncology recently published a complete set of recommendations to guide clinicians on appropriate treatment strategies for each manifestation of immunotherapy-related toxicity.56 The details of these recommendations largely fall outside the purview of this article, but the mainstays of management are worth noting. These include high-dose systemic glucocorticoids, along with supportive care and cessation of immunotherapy in grade 3 or 4 toxicities. Infliximab is frequently recommended as an adjunct in severe or refractory cases.
Chemotherapy-related toxicities, on the other hand, are well-described and tend to be more familiar to patients and clinicians (Table 3). Classic MVAC, which has now been largely replaced by ddMVAC, was notoriously difficult to tolerate. It was known for a high rate of serious (grade 3 or 4) myelosuppressive complications as well as frequent GI toxicities. These complications include neutropenia (57%), stomatitis (10%), and nausea and vomiting (6%).23 ddMVAC with growth factor support is much better tolerated than classic MVAC. Prominent complaints with ddMVAC still can include nausea, GI distress, mucositis, and fatigue, but the incidence of myelosuppressive complications in particular has markedly decreased. GC is largely well tolerated, with minimal nausea and manageable myelotoxicity, but it is associated with an increased risk of venous thromboembolism.38
Prognosis
Case Conclusion
After returning home, the patient discusses his complicated medical course with his oncologist. Given his continued high quality of life with good functional status, he requests to continue with therapy for his metastatic bladder cancer and is interested in joining a clinical trial. He is referred to a nearby academic center with openings in a clinical trial for which he would be eligible. In the meantime, his oncologist guides him through filling out an advance directive and recommends that he make an appointment with palliative care services to ensure adequate home support for any future needs he may have.
- What is the estimated 5-year survival rate for patients with metastatic bladder cancer?
Overall, prognosis in patients with metastatic bladder cancer remains poor. Median survival in patients being treated with multi-agent chemotherapy is approximately 15 months,38,40 with an expected 5-year survival of just 15%. This is much improved, however, as prior to the advent of modern chemotherapy estimated survival was just 6 months with metastatic bladder cancer. Importantly, these figures do not take into account the recent advancements with immunotherapy, and thus it is reasonable to assume survival rates may continue to improve. In light of these recent advances, it is strongly recommended that whenever possible patients and clinicians consider participation in clinical trials to continue uncovering new and better therapies moving forward.
A number of tools have been developed to help risk stratify patients based on comorbidity, performance status, and other characteristics, but none have been universally adopted.57–60 As with many other malignancies, performance status is an important predictor of clinical outcomes in these patients.61–63 Sites of metastasis also may serve to suggest the course of disease. Patients with visceral metastases typically exhibit significantly worse disease with a shortened survival. The role of molecular factors as prognostic markers in bladder cancer is still under investigation. Many biomarkers are being considered (including mutations and polymorphisms in p53, ERCC1, and ERCC2), and evidence suggests some may have a role in prognosis; thus far, none have been validated as prognostic or predictive tools in urothelial carcinoma.
Conclusion
Bladder cancer includes an aggressive group of genitourinary tract malignancies, of which urothelial carcinoma is by far the most common in the Western world. Cisplatin-based therapy remains a mainstay of treatment for eligible patients with both localized and metastatic disease, but immunotherapies have provided a new and promising tool to use in the setting of progressing malignancy. The individual impact of these agents on OS is still being examined. Further studies and ongoing participation in clinical trials whenever possible continue to be essential to the discovery of future treatment options for this highly aggressive disease.
Introduction
Bladder cancer is by far the most common cancer of the urinary system. Worldwide, approximately 450,000 new cases are diagnosed and 165,000 deaths are caused by bladder cancer each year.1 In the United States and in Europe, the most common type of bladder cancer is urothelial carcinoma (also referred to as transitional cell carcinoma), which accounts for more than 90% of all bladder cancers in these regions of the world. The remainder of bladder cancers are divided among squamous cell carcinomas, adenocarcinomas, small cell carcinomas, and, even more rarely, between various other nonepithelial tumors (eg, sarcoma).
Bladder cancer is classically thought of as a disease of the elderly, with a median age at diagnosis of 69 years in men and 71 years in women.2 The incidence of bladder cancer increases with age: in persons aged 65 to 69 years, incidence is 142 per 100,000 men and 33 per 100,000 women, and in those older than 85 years the rate doubles to 296 per 100,000 men and 74 per 100,000 women.3 The incidence is 3 times greater in men than in women.4
Urothelial carcinoma is traditionally categorized by its degree of invasion into the bladder wall: superficial (non-muscle-invasive), muscle-invasive, or metastatic disease. At the time of diagnosis, most patients have non-muscle-invasive disease (~60%); about 4% of all patients present initially with metastatic disease.5 This article focuses on metastatic bladder cancer, but muscle-invasive disease is discussed as well.
The most important factor contributing to the development of urothelial carcinoma is tobacco smoking. The risk of developing bladder cancer is 4 to 5 times higher in smokers as compared to nonsmokers, with some variation according to sex.6 Quantity of smoking exposure also plays a role, with heavy smokers demonstrating a higher likelihood for high-grade tumors with muscle invasion (or beyond) when compared to light smokers.7 Another important risk factor is occupational exposure to industrial materials, such as carpets, paints, plastics, and industrial chemicals. This type of exposure may be responsible for, or at least contribute to, the development of approximately 20% of urothelial carcinomas. Other risk factors for urothelial carcinoma include but are not limited to prior radiation to the pelvis, prior upper tract urothelial malignancy, human papillomavirus infection, and prior bladder augmentation.
Diagnosis and Staging
Case Presentation
A 63-year-old man with a past medical history of diabetes, deep vein thrombosis, occasional alcohol use, and regular pipe tobacco use presents to his primary care physician with complaints of hematuria. He reports that his urine was a dark red color that morning, which had never happened before. The patient is hemodynamically stable upon evaluation in the office, and a point-of-care urinalysis dipstick is strongly positive for blood. He is referred to a urologist for further evaluation.
In the urology office, urine microscopy is notable for more than 50 red blood cells (RBCs) per high-power field with normal RBC morphology. Flexible cystoscopy performed in the office reveals a single 2-cm, sessile, verrucous, nodular lesion located on the anterior bladder wall. A urine sample and a bladder wash specimen are sent for cytology evaluation. The patient is scheduled to undergo a complete transurethral resection of bladder tumor (TURBT) later that week with samples sent to pathology for evaluation.
- What are the clinical features of bladder cancer?
Hematuria is the most common presentation of bladder cancer, although its specificity is far lower than traditionally thought. In fact, only about 2% to 20% of cases that present with hematuria are found to be caused by malignancy. However, the incidence of genitourinary tract malignancy is much higher in patients presenting with gross hematuria (10%–20%)8–10 than in patients with microscopic hematuria alone.8,10–14 Typically, hematuria associated with malignancy is painless. Multiple studies have shown, however, that hematuria can be a normal variant, with one study demonstrating that up to 61% of patients with hematuria had no identifiable abnormality.8,10,11,13
Abdominal pain, flank pain, dysuria, urinary frequency/urgency, or other irritative voiding symptoms in the absence of hematuria can be presenting symptoms of bladder cancer as well. In these settings, discomfort typically suggests more advanced malignancy with at least local involvement or obstruction. Suprapubic pain may herald invasion into perivesical tissues and nerves, while involvement of the obturator fossa, perirectal fat, urogenital diaphragm, or presacral nerves can often present with perineal or rectal pain. Similarly, lower abdominal pain may represent involvement of lymph nodes, and right upper quadrant pain may signal liver metastasis. Cough or shortness of breath may signify metastatic disease in the lung. Finally, back, rib, or other boney pain may suggest distant metastasis.
- What next steps are required to complete this patient’s staging?
White light cystoscopy remains the gold standard for diagnosis and initial staging of bladder cancer. Additional tools include urine cytology and upper tract studies, including renal computed tomography (CT) urograms. Full urologic evaluation with all 3 modalities (cystourethroscopy, urinary cytology, and upper tract evaluation) is warranted for patients with a high suspicion for malignant etiology of hematuria. CT urograms are particularly useful for upper tract evaluation because they can be used to visualize kidney parenchyma, both renal pelvises and ureters, and pertinent abdominal and pelvic lymph nodes. Initial staging is completed through TURBT, which should ideally contain a segment of muscularis propria to distinguish between Ta (noninvasive), T1, and T2 tumors (Figure 1).
Regarding staging, T1 tumors are distinguished from Ta malignancies by their involvement in the urothelial basement membrane. Tumor invasion into the muscularis propria indicates T2 tumors, while T3 tumors extend through the muscle into the serosa and involve the complete thickness of the bladder wall. Involvement of nearby structures defines T4 bladder cancers, with T4a malignancies involving adjacent organs (prostate, vagina, uterus, or bowel) and T4b tumors involving the abdominal wall, pelvic wall, or other more distant organs. According to the American Joint Committee on Cancer’s most recent TNM staging system (Table 1),16 lymph node involvement in the true pelvis (that is, N1–N3) with T1 to T4a disease is now classified as stage III disease.
Bladder cancer is often broadly categorized as either non-muscle-invasive or muscle-invasive (which can include metastatic disease). This classification has important implications for treatment. As such, all diagnostic biopsies should be performed with the goal of reaching at least the depth of the muscularis propria in order to accurately detect potential muscularis invasion. If no muscle is detected in the initial specimen, re-resection is recommended if safe and feasible. In cases where muscle cannot be obtained, imaging evidence of T3 disease from CT or magnetic resonance imaging may be used as a surrogate indicator. Once muscle-invasive disease is confirmed, CT evaluation of the chest is also recommended, as bladder cancer can metastasize to the lungs; furthermore, patients are often at risk for secondary concomitant lung cancers given that smoking is the most prevalent risk factor for both. However, patients with small, indeterminate lung nodules not amenable to biopsy should not be denied curative intent treatment given the high likelihood that they represent benign findings.17
Pathogenesis
Because non-muscle-invasive and muscle-invasive tumors behave so differently, they are thought to arise from 2 distinct mechanisms. Although there is overlap and non-muscle-invasive cancer can certainly progress to a high-grade, invasive type of malignancy over time, current theory proposes that non-muscle-invasive bladder cancer predominantly develops just from urothelial hyperplasia, which then recruits branching vasculature to grow slowly. More aggressive urothelial carcinomas, including muscle-invasive and metastatic disease, are instead thought to arise directly from flat dysplasia that progresses to carcinoma in situ, and is much more prone to invasive growth and distant spread.18
Regardless of grade and stage, the most commonly identified genomic alterations in urothelial carcinoma are mutations in the promoter region of the telomerase reverse transcriptase (TERT) gene, which have been identified in approximately 70% of cases.19 Mutations in TERT can be readily detected in urine sediments and may ultimately have implications for diagnosis and early detection.20,21 In current practice, however, the clinical relevance of these observations remains under development. Other genomic alterations that may contribute to the development of urothelial carcinoma, and also provide new potential therapeutic targets, include alterations in the TP53 gene, the RB (retinoblastoma) gene, and the FGFR3 (fibroblast growth factor receptor) gene. FGFR3 has particular significance as it appears to be relatively common in non-muscle invasive disease (up to 60%–70%) and is likely an actionable driver mutation that may define a particular molecular subset of urothelial carcinoma; thus, it may have important implications for treatment decisions.22
Treatment
Case Continued
Pathologic evaluation of the specimen reveals a high-grade urothelial carcinoma with tumor invasion into the muscularis propria. A CT urogram is performed and does not reveal any notably enlarged pelvic nodes or suspicious lesions in the upper urinary tract. CT chest does not reveal any evidence of distant metastatic disease. Given the presence of muscle-invasive disease, the patient agrees to proceed with neoadjuvant chemotherapy and radical cystoprostatectomy with pelvic node dissection. He undergoes treatment with dose-dense (accelerated) MVAC (methotrexate, vinblastine, doxorubicin, and cisplatin) for 3 cycles, followed by surgery with cystoprostatectomy. Overall, he tolerates the procedure well and recovers quickly. Pathology reveals the presence of disease in 2 regional nodes, consistent with T4a (stage III) disease, and a small degree of residual disease in the bladder. He is followed closely in the oncology clinic, returning for urine cytology, liver and renal function tests, and imaging with CT of chest, abdomen, and pelvis every 3 months.
- What is the first-line approach to management in patients with muscle-invasive disease?
- How would the treatment strategy differ if the patient had presented with metastatic disease (stage IV)?
First-Line Management for Curative Intent: Muscle-Invasive Disease
Muscle-invasive urothelial carcinoma (including T2, T3, or T4 disease) is typically treated in a multidisciplinary fashion with neoadjuvant cisplatin-based chemotherapy followed by radical cystectomy. This approach is recommended over radical cystectomy alone because of high relapse rates following cystectomy alone, even in the setting of bilateral pelvic lymphadenectomy.23 However, because of the associated short- and long-term toxicity of cisplatin-based regimens, this optimal treatment paradigm is reserved for patients deemed cisplatin-eligible.
Medical fitness to receive cisplatin-based chemotherapy is assessed by a number of factors and varies by institution, but most frequently consider functional status (Eastern Cooperative Oncology Group [ECOG] performance status or Karnofsky Performance Status), creatinine clearance, hearing preservation, peripheral neuropathy, and cardiac function.24 Many programs will elect to defer cisplatin-based chemotherapy in patients with low performance status (ie, < 60–70 on Karnofsky scale or > 2 on ECOG scale), creatinine clearance below 60 mL/min, or significant heart failure (NHYA class III or worse). Cisplatin-based chemotherapy may worsen hearing loss in those with hearing loss of 25 dB from baseline at 2 continuous frequencies and also may worsen neuropathy in those with baseline grade 1 peripheral neuropathy. However, these adverse outcomes must be balanced against the curative intent of the multimodality systemic approach.
In patients with renal insufficiency, caution must be taken with regard to cisplatin. Percutaneous nephrostomy placement or ureteral stenting should be attempted to relieve any ureteral outlet obstruction and restore kidney function if a patient’s renal insufficiency has resulted from this obstruction. If medical renal disease or long-term renal insufficiency is present, however, patients should instead be referred for immediate cystectomy or for a bladder-preserving approach. Generally, a creatinine clearance of 60 mL/min is required to safely receive cisplatin-based chemotherapy, although some advocate for treatment with a creatinine clearance as low as 50 mL/min. When this extended criterion is used, the dose of cisplatin may be split over 2 days to minimize renal toxicity and maximize hydration. Analysis of renal function utilizing a 24-hour urine collection should be incorporated whenever possible, as estimates of creatinine clearance have been demonstrated to be inaccurate in some instances.25
For cisplatin-eligible patients, neoadjuvant chemotherapy with a cisplatin base has consistently demonstrated a survival benefit when given prior to surgery.26,27 Historically, several different platinum-based regimens have been studied, with none showing superior effectiveness in a randomized trial over the others in the neoadjuvant setting. These regimens have included classic MVAC, dose-dense MVAC (MVAC with pegfilgrastim), GC (gemcitabine and cisplatin), and CMV (methotrexate, vinblastine, cisplatin, and leucovorin).
While classic MVAC was preferred in the 1990s and early 2000s,28,29 the availability of growth factor, such as pegfilgrastim, has made dose-dense MVAC (otherwise referred to as accelerated MVAC or ddMVAC) widely preferred and universally recommended over classic MVAC. The ddMVAC regimen with the addition of a synthetic granulocyte colony-stimulating factor (G-CSF) is substantially better tolerated than classic MVAC, as the G-CSF support minimizes the severe toxicities of classic MVAC, such as myelosuppression and mucositis, and allows for the administration of drugs in a dose-dense fashion.30,31
Both ddMVAC and GC are considered reasonable options for neoadjuvant chemotherapy and are the predominant choices for cisplatin-eligible patients (Table 2).
Prospective data defining the role of adjuvant chemotherapy for patients after cystectomy has been fraught by a variety of factors, including the known benefit of neoadjuvant chemotherapy, the high complication rate of cystectomy making chemotherapy infeasible, and clinician bias that has hampered accrual in prior trials. Thus, no level 1 evidence exists defining the benefit of adjuvant chemotherapy in patients who did not receive neoadjuvant therapy. In a report of the largest study performed in this setting, there was a statistically significant benefit in PFS but not in OS.36 Criticisms of this trial include its lack of statistical power due to a failure to accrue the targeted goal and the preponderance of node-positive patients. Regardless, for patients who have pT2–4, N1 disease after radical cystectomy and remain cisplatin-eligible after not receiving neoadjuvant chemotherapy, this remains an option.
Despite the established clinical dogma surrounding neoadjuvant chemotherapy followed by surgery, some patients are either not eligible for or decline to receive radical cystectomy, while others are not candidates for neoadjuvant cisplatin-based chemotherapy for the reasons outlined above. For patients who are surgical candidates but unable to receive neoadjuvant chemotherapy due to renal or cardiac function, they may proceed directly to surgery. For patients unable or unwilling to proceed to radical cystectomy regardless, bladder preservation strategies exist. Maximal TURBT may be an option for some patients, but, as outlined above, used alone this would be likely to lead to a high degree of local and distant failure. Combined modality chemoradiotherapy as consolidation after maximal TURBT is an established option for patients unable to undergo surgery or seeking bladder preservation. Several trials have demonstrated encouraging outcomes with this approach and were highlighted in a large meta-analysis.37 Various chemosensitizing chemotherapeutic regimens have been evaluated, including cisplatin alone or as a doublet, gemcitabine alone, and 5-fluouracil plus mitomycin C, but no randomized studies have compared these regimens to each other, nor have they been compared to surgical approaches. However, this strategy remains an option as an alternative to surgery.
First-Line Management: Metastatic Disease
The approach to therapy in patients who present with metastatic urothelial carcinoma is very similar to that used in neoadjuvant perioperative chemotherapy. The consensus first-line treatment in medically appropriate patients is cisplatin-based chemotherapy with either GC or ddMVAC (both category 1 National Comprehensive Cancer Network [NCCN] recommendations; Figure 2).30,31,38–40
Head-to-head studies specifically comparing ddMVAC and GC have been limited. GC has been compared to classic MVAC, with results showing equivalent efficacy but improved tolerability, as expected.38,40 ddMVAC was compared with a modified version of GC (termed “dose-dense GC”) in a phase 3 study from Greece, which demonstrated similar outcomes.41
Surgical intervention with radical cystectomy and regional lymph node dissection is typically deferred for patients who present with distant metastatic disease, unlike those who present with locally advanced disease. Radical cystectomy has traditionally been thought of as overly aggressive without sufficient benefit, although evidence to guide this approach remains sparse.42 As such, most expert recommendations and consensus statements simply recommend against surgical intervention and leave the decision between ddMVAC and GC up to the individual clinician.
In patients who are not eligible for cisplatin therapy, it is reasonable to consider chemotherapy with a combination of gemcitabine and carboplatin. This combination has been shown to be equivalent to MCAVI (methotrexate, carboplatin, vinblastine) in terms of overall survival (OS; 9 months versus 8 months) and progression-free survival (PFS; 6 months versus 4 months) with significantly fewer serious toxicities (9% versus 21%).43
The advent of immunotherapy in recent years has provided several new alternatives for cisplatin-ineligible patients. While immunotherapies such as pembrolizumab or atezolizumab are not yet recommended as first-line therapy for cisplatin-eligible patients, these 2 drugs are approved as options for first-line therapy in cisplatin-ineligible patients with metastatic disease. In a recent phase 2 trial (IMvigor210) involving 119 patients who were given atezolizumab as first-line therapy, median PFS was 2.7 months and median OS was 15.9 months.44 Another trial using data from patients in the KEYNOTE-052 study who received pembrolizumab as first-line therapy demonstrated antitumor activity with pembrolizumab and acceptable tolerability in cisplatin-ineligible patients with advanced urothelial carcinoma.45 The primary endpoint was objective response (either complete or partial response), which was achieved in 24% of the intention-to-treat population. Median PFS was 2 months, and 6-month OS was observed in 67% of patients. Both atezolizumab and pembrolizumab were given accelerated approval based on these single-arm studies in this setting. However, due to inferior outcomes in subsequent trials that included single-agent immunotherapy arms for patients in the first-line setting, the US Food and Drug Administration (FDA) has clarified the approval. In the subsequent trials, patients with a low PD-L1 biomarker based on the individual assay used for each drug did worse on immunotherapy alone (compared to chemotherapy or both combined), and the single-therapy arms were stopped early. Thus, the FDA now recommends that pembrolizumab or atezolizumab be used in the first line only for cisplatin-ineligible patients who have PD-L1 expression on tumor cells above the threshold studied on each individual assay, or are unfit for any platinum-based chemotherapy. Further study regarding the optimal role of biomarkers and chemotherapy-immunotherapy combinations is ongoing.
Case Continued
Ten months after his procedure, the patient is found to have prominent retroperitoneal lymphadenopathy and a 1.0-cm liver nodule suspicious for malignancy is noted on surveillance imaging. CT-guided biopsy of the liver reveals high-grade urothelial carcinoma, consistent with both recurrence and distant metastasis. The patient is informed that he needs to resume systemic therapy for recurrent metastatic disease. The options discussed include salvage single-agent chemotherapy with gemcitabine or immunotherapy with pembrolizumab. He elects to move forward with immunotherapy and is scheduled to begin pembrolizumab.
- What other immunotherapies might this patient consider for second-line therapy?
- Is chemotherapy a second-line option for this patient?
Second-Line Therapies and Management of Progressive Disease
Disease progression is unfortunately seen in the majority of cases of advanced urothelial carcinoma.46 New second-line therapies have recently been approved by the FDA in the form of monoclonal antibodies targeting programmed death 1 (PD-1) and a PD-1 ligand (PD-L1) (Figure 3).
Approval of pembrolizumab, a PD-1 inhibitor, was largely supported by the Keynote-045 trial,47,48 which looked at 542 patients who had progressed or recurred after platinum-based chemotherapy. These patients were randomly assigned to either pembrolizumab or investigator’s choice of chemotherapy (paclitaxel, docetaxel, or vinflunine). Patients treated with pembrolizumab had a significantly improved OS (median of 10.3 months versus 7.4 months), but no statistically significant difference in PFS (2.1 months versus 3.3 months). Interestingly, the rate of responses of 12 months or longer was higher with pembrolizumab than with more traditional second-line chemotherapy (68% versus 35%). The strength of this data has led to a category 1 recommendation in the most recent NCCN guidelines.39
The approval of atezolizumab, a PD-L1 inhibitor, as a second-line therapy for advanced urothelial carcinoma is largely supported by data from IMvigor211, a phase 3 trial that studied 931 patients randomly assigned to atezolizumab or investigator’s choice chemotherapy. OS did not differ significantly between patients in the atezolizumab group who had ≥ 5% expression of PD-L1 on tumor-infiltrating immune cells and patients in the chemotherapy group (11.1 months versus 10.6 months), but mean duration of response was longer (15.9 months versus 8.3 months).49 Therapy with atezolizumab had significantly fewer toxicities than chemotherapy (grade 3 or 4 toxicities of 20% versus 43%).
Phase 3 studies of nivolumab (PD-1 inhibitor), avelumab (PD-L1 inhibitor), and durvalumab (PD-L1 inhibitor) have not yet been published. These agents have received accelerated approval, however, as second-line treatment of advanced urothelial carcinoma based on promising data from phase 1 and phase 2 studies.50–52
Second-line chemotherapy is also an option for patients who do not qualify for immunotherapy or who progress during or after immunotherapy. Although there has been a great deal of excitement about new developments with immunotherapy and the survival benefit seen compared to investigator’s choice chemotherapy, the fact remains that most patients do not respond to immunotherapy. Still, some patients do derive benefit from single-agent chemotherapy in the platinum-refractory setting. Options based on primarily single-arm studies include gemcitabine, paclitaxel, docetaxel, pemetrexed, ifosfamide, oxaliplatin, and eribulin (Figure 2). In a randomized phase 3 trial, vinflunine demonstrated an OS benefit in platinum-refractory patients compared to best supportive care; it subsequently received approval by the European Medicines Agency.53 More recently in the phase 3 RANGE trial, docetaxel plus ramucirumab (a monoclonal antibody targeting vascular endothelial growth factor receptor 2) was compared to docetaxel plus placebo and met its primary endpoint of an improvement in PFS (median 4.07 months versus 2.76 months, P = 0.0118).54 OS has not been reported and this regimen has not yet received regulatory approval, however. Unfortunately, trials comparing these regimens are lacking, and response rates and survival remain modest. Clearly, better therapies and biomarkers to help personalize treatment options are needed.
Further investigations are underway with alternative regimens, including but not limited to targeted therapy in the setting of specific genetic and epigenetic alterations. These include mutations affecting tyrosine kinase receptors (eg, RAS/RAF, PI3K, AKT, and mTOR), cell cycle regulators (eg, TP53 or RB1), FGFR3 mutations, PTEN deletions, gene amplifications (eg, FGFR1, CCND1, and MDM2), or changes in genes responsible for chromatin remodeling (eg, UTX, CHD6, or ARID1A). As noted, there is particular excitement regarding FGFR3 inhibitors, which have shown compelling efficacy in phase 1 and 2 single-arm trials. Several agents are being evaluated in randomized trials and represent a potential path to the first targeted therapeutic class with a role in urothelial malignancies.
Surgical resection of metastases may be considered in very select cases.55 Surgery may have a role in limiting metastatic complications and improving cancer control, but this should be discussed at length with the patient using a multidisciplinary approach with careful restaging prior to surgery.
Case Continued
The patient remains on pembrolizumab every 3 weeks as per protocol with regular surveillance imaging. His disease stabilizes as the nodule in his liver and the retroperitoneal lymph nodes, all representing metastatic disease, became slightly smaller in size without evidence of any new disease. He continues to follow up closely with his genitourinary oncologist, undergoing regular surveillance and imaging every 3 months without evidence of disease progression.
Approximately 12 months into therapy, the patient notices a nonproductive cough with progressive and rapidly worsening shortness of breath. He is noted to be hypoxic with oxygen saturation levels to 79% in clinic and is sent immediately to the emergency department by his oncologist. Diffuse bilateral reticular opacities are noted on chest radiograph. Non-contrast CT scan demonstrates diffuse ground-glass opacities consistent with acute respiratory distress syndrome–pattern pneumonitis. He is admitted to the intensive care unit.
The patient is aggressively treated with high-flow nasal oxygen supplementation, intravenous steroids, and empiric antibiotics. He slowly improves on high-dose steroids (methylprednisolone 1 mg/kg/day) without requiring intubation or infliximab therapy and is discharged home in stable condition after 10 days. Oral steroid therapy is continued with a long taper over 6 weeks. In the setting of his grade 3 pneumonitis, pembrolizumab is discontinued and the patient is scheduled for a follow-up appointment with his oncologist to discuss next steps.
- In addition to pneumonitis, what other toxicities should you monitor for in patients treated with an immune checkpoint inhibitor?
- Is this patient a candidate to receive immunotherapy again in the future?
Treatment Toxicities
As use of immune checkpoint inhibitors has become more prevalent, the medical community has become increasingly aware of various immune-related adverse effects (irAE) associated with these drugs. These toxicities can be seen in virtually any organ system, and even vague complaints that arise years after therapy initiation should be treated with a high level of suspicion. The most commonly affected organ systems include the skin, gastrointestinal (GI) tract, lungs, liver, and endocrine system, although all other organ systems can be involved (Table 3) and toxicities appear to be similar across individual drugs.
The American Society of Clinical Oncology recently published a complete set of recommendations to guide clinicians on appropriate treatment strategies for each manifestation of immunotherapy-related toxicity.56 The details of these recommendations largely fall outside the purview of this article, but the mainstays of management are worth noting. These include high-dose systemic glucocorticoids, along with supportive care and cessation of immunotherapy in grade 3 or 4 toxicities. Infliximab is frequently recommended as an adjunct in severe or refractory cases.
Chemotherapy-related toxicities, on the other hand, are well-described and tend to be more familiar to patients and clinicians (Table 3). Classic MVAC, which has now been largely replaced by ddMVAC, was notoriously difficult to tolerate. It was known for a high rate of serious (grade 3 or 4) myelosuppressive complications as well as frequent GI toxicities. These complications include neutropenia (57%), stomatitis (10%), and nausea and vomiting (6%).23 ddMVAC with growth factor support is much better tolerated than classic MVAC. Prominent complaints with ddMVAC still can include nausea, GI distress, mucositis, and fatigue, but the incidence of myelosuppressive complications in particular has markedly decreased. GC is largely well tolerated, with minimal nausea and manageable myelotoxicity, but it is associated with an increased risk of venous thromboembolism.38
Prognosis
Case Conclusion
After returning home, the patient discusses his complicated medical course with his oncologist. Given his continued high quality of life with good functional status, he requests to continue with therapy for his metastatic bladder cancer and is interested in joining a clinical trial. He is referred to a nearby academic center with openings in a clinical trial for which he would be eligible. In the meantime, his oncologist guides him through filling out an advance directive and recommends that he make an appointment with palliative care services to ensure adequate home support for any future needs he may have.
- What is the estimated 5-year survival rate for patients with metastatic bladder cancer?
Overall, prognosis in patients with metastatic bladder cancer remains poor. Median survival in patients being treated with multi-agent chemotherapy is approximately 15 months,38,40 with an expected 5-year survival of just 15%. This is much improved, however, as prior to the advent of modern chemotherapy estimated survival was just 6 months with metastatic bladder cancer. Importantly, these figures do not take into account the recent advancements with immunotherapy, and thus it is reasonable to assume survival rates may continue to improve. In light of these recent advances, it is strongly recommended that whenever possible patients and clinicians consider participation in clinical trials to continue uncovering new and better therapies moving forward.
A number of tools have been developed to help risk stratify patients based on comorbidity, performance status, and other characteristics, but none have been universally adopted.57–60 As with many other malignancies, performance status is an important predictor of clinical outcomes in these patients.61–63 Sites of metastasis also may serve to suggest the course of disease. Patients with visceral metastases typically exhibit significantly worse disease with a shortened survival. The role of molecular factors as prognostic markers in bladder cancer is still under investigation. Many biomarkers are being considered (including mutations and polymorphisms in p53, ERCC1, and ERCC2), and evidence suggests some may have a role in prognosis; thus far, none have been validated as prognostic or predictive tools in urothelial carcinoma.
Conclusion
Bladder cancer includes an aggressive group of genitourinary tract malignancies, of which urothelial carcinoma is by far the most common in the Western world. Cisplatin-based therapy remains a mainstay of treatment for eligible patients with both localized and metastatic disease, but immunotherapies have provided a new and promising tool to use in the setting of progressing malignancy. The individual impact of these agents on OS is still being examined. Further studies and ongoing participation in clinical trials whenever possible continue to be essential to the discovery of future treatment options for this highly aggressive disease.
1. Stewart BW, Kleihues P. World cancer report. IARCPress. 2003.
2. Scosyrev E, Noyes K, Feng C, Messing E. Sex and racial differences in bladder cancer presentation and mortality in the US. Cancer 2009;115:68–74.
3. Hinotsu S, Akaza H, Miki T, et al. Bladder cancer develops 6 years earlier in current smokers: Analysis of bladder cancer registry data collected by the cancer registration committee of the Japanese Urological Association. Int J Urol 2009;16:64–9.
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
5. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: bladder cancer. 2018. https://seer.cancer.gov/statfacts/html/urinb.html. Accessed May 5, 2018.
6. Freedman ND, Silverman DT, Hollenbeck AR, et al. Association between smoking and risk of bladder cancer among men and women. JAMA 2011;306:737–45.
7. Pietzak EJ, Mucksavage P, Guzzo TJ, Malkowicz SB. Heavy cigarette smoking and aggressive bladder cancer at initial presentation. Urology 2015;86:968–73.
8. Khadra MH, Pickard RS, Charlton M, et al. A prospective analysis of 1,930 patients with hematuria to evaluate current diagnostic practice. J Urol 2000;163:524–7.
9. Grossman HB, Messing E, Soloway M, et al. Detection of bladder cancer using a point-of-care proteomic assay. JAMA 2005;293:810–16.
10. Mariani AJ, Mariani MC, Macchioni C, et al. The significance of adult hematuria: 1,000 hematuria evaluations including a risk-benefit and cost-effectiveness analysis. J Urol 1989;141:350–5.
11. Grossfeld GD, Litwin MS, Wolf JS, et al. Evaluation of asymptomatic microscopic hematuria in adults: the American Urological Association best practice policy--part II: patient evaluation, cytology, voided markers, imaging, cystoscopy, nephrology evaluation, and follow-up. Urology 2001;57:604–10.
12. Grossfeld GD, Litwin MS, Wolf JS, et al. Evaluation of asymptomatic microscopic hematuria in adults: the American Urological Association best practice policy--part I: definition, detection, prevalence, and etiology. Urology 2001;57:599–603.
13. Mohr DN, Offord KP, Owen RA, Melton LJ. Asymptomatic microhematuria and urologic disease. A population-based study. JAMA 1986;256:224–9.
14. Messing EM, Young TB, Hunt VB, et al. Home screening for hematuria: results of a multiclinic study. J Urol 1992;148:289–92.
15. Gray PJ, Lin CC, Jemal A, et al. Clinical–pathologic stage discrepancy in bladder cancer patients treated with radical cystectomy: results from the National Cancer Data Base. Int J Radiat Oncol 2014;88:1048–56.
16. Bochner B, Hansel D, Efstathiou J, et al. Urinary bladder. In: Amin M, ed. AJCC cancer staging manual. 8th. New York: Springer; 2017:757.
17. Cahn DB, McGreen B, Lee A, et al. Clinical destiny of indeterminate pulmonary nodules in patients undergoing radical cystectomy for urothelial carcinoma of the bladder [abstract]. J Clin Oncol 2017;35(6 suppl):297-297.
18. Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer 2015;15:25–41.
19. Kurtis B, Zhuge J, Ojaimi C, et al. Recurrent TERT promoter mutations in urothelial carcinoma and potential clinical applications. Ann Diagn Pathol 2016;21:7–11.
20. Ito H, Kyo S, Kanaya T, et al. Detection of human telomerase reverse transcriptase messenger RNA in voided urine samples as a useful diagnostic tool for bladder cancer. Clin Cancer Res 1998;4:2807–10.
21. Utting M, Werner W, Dahse R, et al. Microsatellite analysis of free tumor DNA in urine, serum, and plasma of patients: a minimally invasive method for the detection of bladder cancer. Clin Cancer Res 2002;8:35–40.
22. Sethakorn N, O’Donnell PH. Spectrum of genomic alterations in FGFR3: current appraisal of the potential role of FGFR3 in advanced urothelial carcinoma. BJU Int 2016;118:681–91.
23. Grossman HB, Natale RB, Tangen CM, et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med 2003;349:859–66.
24. Galsky MD, Hahn NM, Rosenberg J, et al. Treatment of patients with metastatic urothelial cancer ‘unfit’ for cisplatin-based chemotherapy. J Clin Oncol 2011;29:2432–8.
25. Raj GV, Iasonos A, Herr H, Donat SM. Formulas calculating creatinine clearance are inadequate for determining eligibility for cisplatin-based chemotherapy in bladder cancer. J Clin Oncol 2006;24:3095–100.26. Advanced Bladder Cancer Meta-analysis Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer: a systematic review and meta-analysis. Lancet 2003;361:1927–34.
27. Advanced Bladder Cancer Meta-analysis Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data. Eur Urol 2005;48:202–6.
28. Sternberg CN. A critical review of the management of bladder cancer. Crit Rev Oncol Hematol 1999;31:193–207.
29. Sternberg CN, Yagoda A, Scher HI, et al. Preliminary results of M-VAC (methotrexate, vinblastine, doxorubicin and cisplatin) for transitional cell carcinoma of the urothelium. J Urol 1985;133:403–7.
30. Sternberg CN, de Mulder P, Schornagel JH, et al. Seven year update of an EORTC phase III trial of high-dose intensity M-VAC chemotherapy and G-CSF versus classic M-VAC in advanced urothelial tract tumours. Eur J Cancer 2006;42:50–4.
31. Sternberg CN, de Mulder PHM, Schornagel JH, et al. Randomized phase III trial of high–dose-intensity methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC) chemotherapy and recombinant human granulocyte colony-stimulating factor versus classic MVAC in advanced urothelial tract tumors: European Organization for Research and Treatment of Cancer Protocol No. 30924. J Clin Oncol 2001;19:2638–46.
32. Soloway MS, Einstein A, Corder MP, et al. A comparison of cisplatin and the combination of cisplatin and cyclophosphamide in advanced urothelial cancer. A National Bladder Cancer Collaborative Group A Study. Cancer 1983;52:767–72.
33. Plimack ER, Hoffman-Censits JH, Viterbo R, et al. Accelerated methotrexate, vinblastine, doxorubicin, and cisplatin is safe, effective, and efficient neoadjuvant treatment for muscle-invasive bladder cancer: results of a multicenter phase II study with molecular correlates of response and toxicity. J Clin Oncol 2014;32:1895–901.
34. Van Allen EM, Mouw KW, Kim P, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov 2014;4:1140–53.
35. Plimack ER, Dunbrack RL, Brennan TA, et al. Defects in DNA repair genes predict response to neoadjuvant cisplatin-based chemotherapy in muscle-invasive bladder cancer. Eur Urol 2015;68:959–67.
36. Sternberg CN, Skoneczna I, Kerst JM, et al. Immediate versus deferred chemotherapy after radical cystectomy in patients with pT3-pT4 or N+ M0 urothelial carcinoma of the bladder (EORTC 30994): an intergroup, open-label, randomised phase 3 trial. Lancet Oncol 2015;16:76–86.
37. Mak RH, Hunt D, Shipley WU, et al. Long-term outcomes in patients with muscle-invasive bladder cancer after selective bladder-preserving combined-modality therapy: a pooled analysis of Radiation Therapy Oncology Group protocols 8802, 8903, 9506, 9706, 9906, and 0233. J Clin Oncol 2014;32:3801–9.
38. von der Maase H, Hansen SW, Roberts JT, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol 2000;18:3068–77.
39. Flaig T, Spiess P, Agarwal N, et al. National Comprehensive Cancer Network. Bladder cancer (version 3.2018). 2018. www.nccn.org/professionals/physician_gls/pdf/bladder.pdf. Accessed May 5, 2018.
40. von der Maase H, Sengelov L, Roberts JT, et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol 2005;23:4602–8.
41. Bamias A, Dafni U, Karadimou A, et al. Prospective, open-label, randomized, phase III study of two dose-dense regimens MVAC versus gemcitabine/cisplatin in patients with inoperable, metastatic or relapsed urothelial cancer: a Hellenic Cooperative Oncology Group study (HE 16/03). Ann Oncol 2013;24:1011–7.
42. Li R, Metcalfe M, Kukreja J, Navai N. Role of radical cystectomy in non-organ confined bladder cancer: a systematic review. Bladder Cancer 2018;4:31–40.
43. De Santis M, Bellmunt J, Mead G, et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin-based chemotherapy: EORTC study 30986. J Clin Oncol 2012;30:191–9.
44. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 2017;389:67–76.
45. Balar A V, Castellano D, O’Donnell PH, et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): a multicentre, single-arm, phase 2 study. Lancet Oncol 2017;18:1483–92.
46. Manoharan M, Ayyathurai R, Soloway MS. Radical cystectomy for urothelial carcinoma of the bladder: an analysis of perioperative and survival outcome. BJU Int 2009;104:1227–32.
47. Bellmunt J, de Wit R, Vaughn D, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 2017;376:1015–26.
48. Bajorin D, de Wit R, Vaughn D, et al. Planned survival analysis from KEYNOTE-045: Phase 3, open-label study of pembrolizumab (pembro) versus paclitaxel, docetaxel, or vinflunine in recurrent, advanced urothelial cancer (UC). (Abstract 4501). J Clin Oncol 2017;35(15_suppl):4501-4501.
49. Powles T, Durán I, van der Heijden MS, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018;391:748–57.
50. Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 2017;18:312–22.
51. Patel MR, Ellerton J, Infante JR, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol 2018;19:51–64.
52. Powles T, O’Donnell PH, Massard C, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma. JAMA Oncol 2017;3:e172411.
53. Bellmunt J, Theodore C, Demkov T, et al. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum-containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract. J Clin Oncol 2009;27:4454–61.
54. Petrylak DP, de Wit R, Chi KN, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel in patients with locally advanced or metastatic urothelial carcinoma after platinum-based therapy (RANGE): a randomised, double-blind, phase 3 trial. Lancet 2017;390:2266–77.
55. Abufaraj M, Dalbagni G, Daneshmand S, et al. The role of surgery in metastatic bladder cancer: a systematic review. Eur Urol 2018;73:543–57.
56. Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2018;36:1714–68.
57. Bajorin DF, Dodd PM, Mazumdar M, et al. Long-term survival in metastatic transitional-cell carcinoma and prognostic factors predicting outcome of therapy. J Clin Oncol 1999;17:3173–81.
58. Mayr R, May M, Martini T, et al. Comorbidity and performance indices as predictors of cancer-independent mortality but not of cancer-specific mortality after radical cystectomy for urothelial carcinoma of the bladder. Eur Urol 2012;62:662–70.
59. Nakagawa T, Hara T, Kawahara T, et al. Prognostic risk stratification of patients with urothelial carcinoma of the bladder with recurrence after radical cystectomy. J Urol 2013;189:1275–81.
60. Ploeg M, Kums AC, Aben KK, et al. Prognostic factors for survival in patients with recurrence of muscle invasive bladder cancer after treatment with curative intent. Clin Genitourin Cancer 2011;9:14–21.
61. Saxman SB, Propert KJ, Einhorn LH, et al. Long-term follow-up of a phase III intergroup study of cisplatin alone or in combination with methotrexate, vinblastine, and doxorubicin in patients with metastatic urothelial carcinoma: a cooperative group study. J Clin Oncol 1997;15:2564–9.
62. Lin CC, Hsu CH, Huang CY, et al. Prognostic factors for metastatic urothelial carcinoma treated with cisplatin and 5-fluorouracil-based regimens. Urology 2007;69:479–84.
63. Schag CC, Heinrich RL, Ganz PA. Karnofsky performance status revisited: reliability, validity, and guidelines. J Clin Oncol 1984;2:187–93.
1. Stewart BW, Kleihues P. World cancer report. IARCPress. 2003.
2. Scosyrev E, Noyes K, Feng C, Messing E. Sex and racial differences in bladder cancer presentation and mortality in the US. Cancer 2009;115:68–74.
3. Hinotsu S, Akaza H, Miki T, et al. Bladder cancer develops 6 years earlier in current smokers: Analysis of bladder cancer registry data collected by the cancer registration committee of the Japanese Urological Association. Int J Urol 2009;16:64–9.
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
5. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer stat facts: bladder cancer. 2018. https://seer.cancer.gov/statfacts/html/urinb.html. Accessed May 5, 2018.
6. Freedman ND, Silverman DT, Hollenbeck AR, et al. Association between smoking and risk of bladder cancer among men and women. JAMA 2011;306:737–45.
7. Pietzak EJ, Mucksavage P, Guzzo TJ, Malkowicz SB. Heavy cigarette smoking and aggressive bladder cancer at initial presentation. Urology 2015;86:968–73.
8. Khadra MH, Pickard RS, Charlton M, et al. A prospective analysis of 1,930 patients with hematuria to evaluate current diagnostic practice. J Urol 2000;163:524–7.
9. Grossman HB, Messing E, Soloway M, et al. Detection of bladder cancer using a point-of-care proteomic assay. JAMA 2005;293:810–16.
10. Mariani AJ, Mariani MC, Macchioni C, et al. The significance of adult hematuria: 1,000 hematuria evaluations including a risk-benefit and cost-effectiveness analysis. J Urol 1989;141:350–5.
11. Grossfeld GD, Litwin MS, Wolf JS, et al. Evaluation of asymptomatic microscopic hematuria in adults: the American Urological Association best practice policy--part II: patient evaluation, cytology, voided markers, imaging, cystoscopy, nephrology evaluation, and follow-up. Urology 2001;57:604–10.
12. Grossfeld GD, Litwin MS, Wolf JS, et al. Evaluation of asymptomatic microscopic hematuria in adults: the American Urological Association best practice policy--part I: definition, detection, prevalence, and etiology. Urology 2001;57:599–603.
13. Mohr DN, Offord KP, Owen RA, Melton LJ. Asymptomatic microhematuria and urologic disease. A population-based study. JAMA 1986;256:224–9.
14. Messing EM, Young TB, Hunt VB, et al. Home screening for hematuria: results of a multiclinic study. J Urol 1992;148:289–92.
15. Gray PJ, Lin CC, Jemal A, et al. Clinical–pathologic stage discrepancy in bladder cancer patients treated with radical cystectomy: results from the National Cancer Data Base. Int J Radiat Oncol 2014;88:1048–56.
16. Bochner B, Hansel D, Efstathiou J, et al. Urinary bladder. In: Amin M, ed. AJCC cancer staging manual. 8th. New York: Springer; 2017:757.
17. Cahn DB, McGreen B, Lee A, et al. Clinical destiny of indeterminate pulmonary nodules in patients undergoing radical cystectomy for urothelial carcinoma of the bladder [abstract]. J Clin Oncol 2017;35(6 suppl):297-297.
18. Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer 2015;15:25–41.
19. Kurtis B, Zhuge J, Ojaimi C, et al. Recurrent TERT promoter mutations in urothelial carcinoma and potential clinical applications. Ann Diagn Pathol 2016;21:7–11.
20. Ito H, Kyo S, Kanaya T, et al. Detection of human telomerase reverse transcriptase messenger RNA in voided urine samples as a useful diagnostic tool for bladder cancer. Clin Cancer Res 1998;4:2807–10.
21. Utting M, Werner W, Dahse R, et al. Microsatellite analysis of free tumor DNA in urine, serum, and plasma of patients: a minimally invasive method for the detection of bladder cancer. Clin Cancer Res 2002;8:35–40.
22. Sethakorn N, O’Donnell PH. Spectrum of genomic alterations in FGFR3: current appraisal of the potential role of FGFR3 in advanced urothelial carcinoma. BJU Int 2016;118:681–91.
23. Grossman HB, Natale RB, Tangen CM, et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med 2003;349:859–66.
24. Galsky MD, Hahn NM, Rosenberg J, et al. Treatment of patients with metastatic urothelial cancer ‘unfit’ for cisplatin-based chemotherapy. J Clin Oncol 2011;29:2432–8.
25. Raj GV, Iasonos A, Herr H, Donat SM. Formulas calculating creatinine clearance are inadequate for determining eligibility for cisplatin-based chemotherapy in bladder cancer. J Clin Oncol 2006;24:3095–100.26. Advanced Bladder Cancer Meta-analysis Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer: a systematic review and meta-analysis. Lancet 2003;361:1927–34.
27. Advanced Bladder Cancer Meta-analysis Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data. Eur Urol 2005;48:202–6.
28. Sternberg CN. A critical review of the management of bladder cancer. Crit Rev Oncol Hematol 1999;31:193–207.
29. Sternberg CN, Yagoda A, Scher HI, et al. Preliminary results of M-VAC (methotrexate, vinblastine, doxorubicin and cisplatin) for transitional cell carcinoma of the urothelium. J Urol 1985;133:403–7.
30. Sternberg CN, de Mulder P, Schornagel JH, et al. Seven year update of an EORTC phase III trial of high-dose intensity M-VAC chemotherapy and G-CSF versus classic M-VAC in advanced urothelial tract tumours. Eur J Cancer 2006;42:50–4.
31. Sternberg CN, de Mulder PHM, Schornagel JH, et al. Randomized phase III trial of high–dose-intensity methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC) chemotherapy and recombinant human granulocyte colony-stimulating factor versus classic MVAC in advanced urothelial tract tumors: European Organization for Research and Treatment of Cancer Protocol No. 30924. J Clin Oncol 2001;19:2638–46.
32. Soloway MS, Einstein A, Corder MP, et al. A comparison of cisplatin and the combination of cisplatin and cyclophosphamide in advanced urothelial cancer. A National Bladder Cancer Collaborative Group A Study. Cancer 1983;52:767–72.
33. Plimack ER, Hoffman-Censits JH, Viterbo R, et al. Accelerated methotrexate, vinblastine, doxorubicin, and cisplatin is safe, effective, and efficient neoadjuvant treatment for muscle-invasive bladder cancer: results of a multicenter phase II study with molecular correlates of response and toxicity. J Clin Oncol 2014;32:1895–901.
34. Van Allen EM, Mouw KW, Kim P, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov 2014;4:1140–53.
35. Plimack ER, Dunbrack RL, Brennan TA, et al. Defects in DNA repair genes predict response to neoadjuvant cisplatin-based chemotherapy in muscle-invasive bladder cancer. Eur Urol 2015;68:959–67.
36. Sternberg CN, Skoneczna I, Kerst JM, et al. Immediate versus deferred chemotherapy after radical cystectomy in patients with pT3-pT4 or N+ M0 urothelial carcinoma of the bladder (EORTC 30994): an intergroup, open-label, randomised phase 3 trial. Lancet Oncol 2015;16:76–86.
37. Mak RH, Hunt D, Shipley WU, et al. Long-term outcomes in patients with muscle-invasive bladder cancer after selective bladder-preserving combined-modality therapy: a pooled analysis of Radiation Therapy Oncology Group protocols 8802, 8903, 9506, 9706, 9906, and 0233. J Clin Oncol 2014;32:3801–9.
38. von der Maase H, Hansen SW, Roberts JT, et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: results of a large, randomized, multinational, multicenter, phase III study. J Clin Oncol 2000;18:3068–77.
39. Flaig T, Spiess P, Agarwal N, et al. National Comprehensive Cancer Network. Bladder cancer (version 3.2018). 2018. www.nccn.org/professionals/physician_gls/pdf/bladder.pdf. Accessed May 5, 2018.
40. von der Maase H, Sengelov L, Roberts JT, et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol 2005;23:4602–8.
41. Bamias A, Dafni U, Karadimou A, et al. Prospective, open-label, randomized, phase III study of two dose-dense regimens MVAC versus gemcitabine/cisplatin in patients with inoperable, metastatic or relapsed urothelial cancer: a Hellenic Cooperative Oncology Group study (HE 16/03). Ann Oncol 2013;24:1011–7.
42. Li R, Metcalfe M, Kukreja J, Navai N. Role of radical cystectomy in non-organ confined bladder cancer: a systematic review. Bladder Cancer 2018;4:31–40.
43. De Santis M, Bellmunt J, Mead G, et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin-based chemotherapy: EORTC study 30986. J Clin Oncol 2012;30:191–9.
44. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 2017;389:67–76.
45. Balar A V, Castellano D, O’Donnell PH, et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): a multicentre, single-arm, phase 2 study. Lancet Oncol 2017;18:1483–92.
46. Manoharan M, Ayyathurai R, Soloway MS. Radical cystectomy for urothelial carcinoma of the bladder: an analysis of perioperative and survival outcome. BJU Int 2009;104:1227–32.
47. Bellmunt J, de Wit R, Vaughn D, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 2017;376:1015–26.
48. Bajorin D, de Wit R, Vaughn D, et al. Planned survival analysis from KEYNOTE-045: Phase 3, open-label study of pembrolizumab (pembro) versus paclitaxel, docetaxel, or vinflunine in recurrent, advanced urothelial cancer (UC). (Abstract 4501). J Clin Oncol 2017;35(15_suppl):4501-4501.
49. Powles T, Durán I, van der Heijden MS, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018;391:748–57.
50. Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 2017;18:312–22.
51. Patel MR, Ellerton J, Infante JR, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol 2018;19:51–64.
52. Powles T, O’Donnell PH, Massard C, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma. JAMA Oncol 2017;3:e172411.
53. Bellmunt J, Theodore C, Demkov T, et al. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum-containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract. J Clin Oncol 2009;27:4454–61.
54. Petrylak DP, de Wit R, Chi KN, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel in patients with locally advanced or metastatic urothelial carcinoma after platinum-based therapy (RANGE): a randomised, double-blind, phase 3 trial. Lancet 2017;390:2266–77.
55. Abufaraj M, Dalbagni G, Daneshmand S, et al. The role of surgery in metastatic bladder cancer: a systematic review. Eur Urol 2018;73:543–57.
56. Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2018;36:1714–68.
57. Bajorin DF, Dodd PM, Mazumdar M, et al. Long-term survival in metastatic transitional-cell carcinoma and prognostic factors predicting outcome of therapy. J Clin Oncol 1999;17:3173–81.
58. Mayr R, May M, Martini T, et al. Comorbidity and performance indices as predictors of cancer-independent mortality but not of cancer-specific mortality after radical cystectomy for urothelial carcinoma of the bladder. Eur Urol 2012;62:662–70.
59. Nakagawa T, Hara T, Kawahara T, et al. Prognostic risk stratification of patients with urothelial carcinoma of the bladder with recurrence after radical cystectomy. J Urol 2013;189:1275–81.
60. Ploeg M, Kums AC, Aben KK, et al. Prognostic factors for survival in patients with recurrence of muscle invasive bladder cancer after treatment with curative intent. Clin Genitourin Cancer 2011;9:14–21.
61. Saxman SB, Propert KJ, Einhorn LH, et al. Long-term follow-up of a phase III intergroup study of cisplatin alone or in combination with methotrexate, vinblastine, and doxorubicin in patients with metastatic urothelial carcinoma: a cooperative group study. J Clin Oncol 1997;15:2564–9.
62. Lin CC, Hsu CH, Huang CY, et al. Prognostic factors for metastatic urothelial carcinoma treated with cisplatin and 5-fluorouracil-based regimens. Urology 2007;69:479–84.
63. Schag CC, Heinrich RL, Ganz PA. Karnofsky performance status revisited: reliability, validity, and guidelines. J Clin Oncol 1984;2:187–93.
Pancreatic Adenocarcinoma: Management of Advanced Unresectable and Metastatic Disease
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
Introduction
Pancreatic ductal adenocarcinoma is a challenging disease with a poor prognosis, with 5-year survival rates in the single digits (~8%).1 Survival rates in pancreatic cancer are low in part because most patients have advanced disease at the time of diagnosis and early development of systemic metastatic disease is common, with approximately 52% of patients with newly diagnosed pancreatic cancer having metastatic disease at diagnosis.1 Surgical resection with negative margins is the cornerstone of potentially curative therapy for localized disease, but only 15% to 20% of patients are eligible for resection at the time of initial diagnosis. Patients with unresectable and metastatic disease are offered palliative chemotherapy. Unfortunately, early recurrence is common in patients with resectable tumors who achieve a complete resection and are treated with adjuvant therapy (5-year recurrence rate ~80%).2,3 This article reviews the management of patients with unresectable and/or metastatic pancreatic cancer. A previous article reviewed the diagnosis and staging of pancreatic cancer and the approach to neoadjuvant and adjuvant therapy in patients with resectable and borderline-resectable disease.4
First-Line Systemic Treatment
Case Presentation
A 72-year-old man who underwent treatment for pancreatic adenocarcinoma 18 months ago presents to the emergency department after developing poor appetite, weight loss, and abdominal discomfort and fullness without diarrhea, which has been constant for the past 2 weeks even though he has been taking analgesics and pancreatic enzymes.
The patient was diagnosed with pancreatic cancer 18 months ago after presenting with yellowish skin and sclera color; abdominal and pelvis computed tomography (CT) with intravenous contrast showed a pancreatic head mass measuring 2.6 × 2.3 cm minimally abutting the anterior surface of the superior mesenteric vein. Endoscopic ultrasound confirmed an irregular mass at the head of the pancreas and sonographic evidence suggested invasion into the portal vein. Examination of a tissue sample obtained during the procedure showed that the mass was consistent with pancreatic adenocarcinoma. Magnetic resonance imaging (MRI) performed to define venous vasculature involvement revealed a pancreatic head mass measuring 3.0 × 2.7 cm without arterial or venous vasculature invasion. The mass was abutting the portal vein and superior mesenteric veins, and a nonspecific 8-mm aortocaval lymph node was noted. The tumor was deemed to be borderline resectable, and the patient received neoadjuvant therapy with gemcitabine and nab-paclitaxel. After 4 cycles, his carbohydrate antigen (CA) 19-9 level decreased, and MRI revealed a smaller head mass (1.3 × 1.4 cm) with stable effacement of the superior mesenteric vein and no portal vein involvement; the aortocaval lymph node remained stable. He was treated with gemcitabine chemoradiotherapy prior to undergoing an uncomplicated partial pancreaticoduodenectomy. Analysis of a surgical pathology specimen revealed T3N0 disease with a closest margin of 0.1 cm. Postsurgery, the patient completed 4 cycles of adjuvant chemotherapy with gemcitabine plus capecitabine.
At his current presentation, MRI of the abdomen and pelvis reveals a new liver mass and peritoneal thickness. Serology testing reveals a CA 19-9 level of 240 U/mL, and other liver function tests are within normal limits. Biopsy of the mass confirms recurrence.
- What systemic chemotherapy would you recommend for this patient with metastatic pancreatic adenocarcinoma?
Most cases of pancreatic cancer are unresectable and/or metastatic at the time of diagnosis. Identifying treatment endpoints and the patient’s goals of care is a critical step in management. Systemic chemotherapy can provide significant survival benefit in first-line and second-line treatment compared to best supportive care. Palliative interventions also include systemic therapy, which often improves pain control and other cancer related–symptoms and hence quality of life. Participation in clinical trials should be offered to all patients. Therapy selection depends on the patient’s performance status, comorbidities, and liver profile and the results of biomarker testing and mutation analysis.
Several single-agents, including fluoropyrimidines, gemcitabine, irinotecan, platinum compounds, and taxanes, have minor objective response rates (< 10%) and a minimal survival benefit (~2 weeks) in metastatic pancreatic adenocarcinoma. Conversely, multi-agent therapies provide higher response rates and can extend overall survival (OS). Two combinations, nab-paclitaxel plus gemcitabine and FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and flourouracil), have significantly prolonged survival compared to best single-agent gemcitabine, as demonstrated in the MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) and PRODIGE 4/ACCORD 11 trials.5,6 Because both multi-agent regimens are also associated with a more toxic adverse effect profile, gemcitabine monotherapy continues to be a front-line therapy for patients with multiple comorbidities, elderly frail patients (> 80 years of age), or patients who cannot tolerate other combinations.7
Gemcitabine-Based Therapy
Gemcitabine became a standard of care treatment for pancreatic cancer in the mid-1990s, and was tested as a second-line therapy in a multicenter phase 2 clinical trial that accrued 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil therapy. In this trial, 27% of patients treated with gemcitabine achieved a clinical benefit response and the median OS was 3.85 months.8 The agent was generally well-tolerated with a low incidence of grade 3 or 4 toxicities. Subsequently, a randomized clinical trial compared gemcitabine to fluorouracil in the front-line setting in 126 patients with newly diagnosed advanced pancreatic cancer.9 Patients were randomly assigned to receive single-agent intravenous fluorouracil administered without leucovorin as a short-term infusion (600 mg/m2 once weekly) or gemcitabine (1000 mg/m2 weekly for up to 7 weeks followed by 1 week of rest, and then weekly for 3 out of every 4 weeks thereafter). A higher proportion of patients treated with gemcitabine had a clinical benefit response (23.8% versus 4.8%), with an improvement in a composite measure of pain (pain intensity and analgesic consumption) and performance status. Clinical responses assessed by a secondary measure, weight gain, were below 10% in both arms, but the median OS was significantly longer for the gemcitabine arm (5.65 months versus 4.4 months, P = 0.0025) and the 1-year OS rate also favored the gemcitabine arm (18% versus 2%). Grade 3/4 neutropenia was reported more frequently in the gemcitabine arm (23% versus 5%). There is no evidence that increasing the dose intensity of the fixed-dose rate of gemcitabine (1000 mg/m2 per week administered as a 30-minute infusion) leads to improved antitumor activity.
Following publication of the trial conducted by Burris and colleagues,9 a plethora of clinical trials have tried to outperform gemcitabine monotherapy, with all trials studying gemcitabine monotherapy compared with gemcitabine plus another agent (fluorouracil, cisplatin, oxaliplatin, irinotecan, pemetrexed, novel biologics including cetuximab, bevacizumab, axitinib, sorafenib, aflibercept). These combinations have failed to significantly extend OS compared to single-agent gemcitabine, although some showed a marginal clinical benefit:
- Capecitabine10 (hazard ratio [HR] 0.86 [95% confidence interval {CI} 0.75 to 0.98])
- Erlotinib11 (HR 0.81 [95% CI 0.69 to 0.99])
- Cisplatin, epirubicin, fluorouracil, gemcitabine12 (HR 0.65 [95% CI 0.43 to 0.99])
The best outcomes were obtained with gemcitabine plus nab-paclitaxel compared to gemcitabine monotherapy. The gemcitabine/nab-paclitaxel combination has not been compared to FOLFIRINOX in the front-line setting, as the ACCORD 11 and MPACT trials were ongoing simultaneously. However, a large retrospective trial that compared use of the regimens in the US Oncology Network in the United States demonstrated similar efficacy, although more patients treated with FOLFIRINOX needed white blood cell growth factor administration.13
Gemcitabine/nab-paclitaxel was studied in a phase 1/2 clinical trial with 67 untreated metastatic pancreatic cancer patients.14 Patients received nab-paclitaxel at doses of 100, 125, or 150 mg/m2 followed by gemcitabine 1000 mg/m2 on days 1, 8, and 15 every 28 days. The maximum tolerated dose (MTD) was 1000 mg/m2 of gemcitabine plus 125 mg/m2 of nab-paclitaxel once a week for 3 weeks every 28 days. Dose-limiting toxicities were sepsis and neutropenia. Patients who received the MTD had a response rate of 48%, median OS of 12.2 months, and a 1-year survival rate of 48%.
The landmark phase 3 MPACT trial confirmed that adding nab-paclitaxel to gemcitabine prolongs survival compared with gemcitabine monotherapy.5 This multinational randomized study included 861 treatment-naive patients with a Karnofsky performance score of 70 or higher. The median OS in the nab-paclitaxel/gemcitabine group was 8.5 months, as compared to 6.7 months in the gemcitabine monotherapy group (HR for death 0.72 [95% CI 0.62 to 0.83], P < 0.001). The survival rate was 35% in the nab-paclitaxel/gemcitabine group versus 22% in the gemcitabine group at 1 year, and 9% versus 4% at 2 years. Median progression-free survival (PFS) was 5.5 months in the nab-paclitaxel/gemcitabine group, compared to 3.7 months in the gemcitabine group (HR for disease progression or death 0.69 [95% CI 0.58 to 0.82], P < 0.001). The overall response rate according to independent review was 23% compared with 7% in the 2 groups, respectively (P < 0.001). The most common adverse events of grade 3 or higher were neutropenia (38% in the nab-paclitaxel/gemcitabine group versus 27% in the gemcitabine group), fatigue (17% versus 7%), and neuropathy (17% versus 1%). Febrile neutropenia occurred in 3% of the combination group versus 1% of the montherapy group. In the nab-paclitaxel/gemcitabine group, neuropathy of grade 3 or higher improved to grade 1 or lower a median of 29 days after discontinuation of nab-paclitaxel. In 2013, nab-paclitaxel in combination with gemcitabine received U.S. Food and Drug Administration (FDA) approval as first-line therapy for metastatic pancreatic cancer.
A pilot phase 1b/2 trial that added cisplatin to nab-paclitaxel and gemcitabine in treating 24 treatment-naive metastatic pancreatic adenocarcinoma patients showed impressive tumor response (complete response 8.3%, partial response 62.5%, stable disease 16.7%, progressive disease 12.5%) and extended median OS to 16.5 months.15 A phase 1b trial conducted in Europe added capecitabine to the cisplatin, nab-paclitaxel, and gemcitabine regimen, albeit with a different schedule and doses, in 24 patients with locally advanced and metastatic disease.16 This trial demonstrated an impressive overall response rate of 67%, with 43% of patients achieving a complete metabolic response on positron emission tomography scan and the CA 19-9 level decreasing by ≥ 49% in all 19 patients who had an elevated basal value. Moreover, PFS at 6 months was 96%. After chemotherapy 17 patients remained unresectable and 7 patients were taken to surgery; of the latter group, only 1 was determined to be unresectable at the time of surgery. This regimen is being explored in a larger study in patients with stage III and IV disease.
FOLFIRINOX
A randomized phase 2 clinical trial comparing FOLFIRINOX to gemcitabine monotherapy in 88 patients with treatment-naive metastatic pancreatic cancer revealed a high response rate for FOLFIRINOX (39% versus 11%, respectively) with a tolerable toxicity profile.17 FOLFIRINOX became the front-line standard of care therapy in pancreatic adenocarcinoma after the results of the subsequent phase 3 ACCORD 11 study preplanned interim analysis showed an unprecedented significantly improved OS benefit.6 The ACCORD 11 trial randomly assigned 342 patients with an Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 and a serum bilirubin level less than 1.5 times the upper limit of normal to receive FOLFIRINOX (oxaliplatin 85 mg/m2, irinotecan 180 mg/m2, leucovorin 400 mg/m2, and fluorouracil 400 mg/m2 given as a bolus followed by 2400 mg/m2 given as a 46-hour continuous infusion, every 2 weeks) or gemcitabine at a dose of 1000 mg/m2 weekly for 7 of 8 weeks and then weekly for 3 of 4 weeks. The median OS in the FOLFIRINOX group was 11.1 months as compared with 6.8 months in the gemcitabine group (HR 0.57 [95% CI 0.45 to 0.73], P < 0.001). The FOLFIRINOX group also had a longer median PFS (6.4 months versus 3.3 months, HR 0.47 [95% CI 0.37 to 0.59], P < 0.001) and higher objective response rate (31.6% versus 9.4%, P < 0.001). More adverse events were noted in the FOLFIRINOX group, including grade 3 or 4 neutropenia (46% versus 21%), febrile neutropenia (5.4% versus 1.2%), thrombocytopenia (9.1% versus 3.6%), sensory neuropathy (9% versus 0%), vomiting (15% versus 8%), fatigue (23% versus 18%), and diarrhea (13% versus 2%). Despite the greater toxicity, only 31% of the FOLFIRINOX group had a definitive degradation of quality of life, as compared to 66% in the gemcitabine group (HR 0.47 [95% CI 0.30 to 0.70], P < 0.001), thus indicating an improvement in quality of life.
Of note, combinations containing irinotecan require adequate biliary function for excretion of its active glucuronide metabolite, SN-38. Approximately 10% of patients in the United States are homozygous for the UGT1A1*28 allele polymorphism, which causes increased SN-38 bioavailability and hence a potential for severe toxicities (eg, life threatening-refractory diarrhea).18 Therefore, it is recommended that physicians start with a lower dose of irinotecan or choose a different regimen altogether in such patients.
Current Approach and Future Directions
Based on results of the ACCORD 11 and MPACT trials, both front-line regimens (nab-paclitaxel/gemcitabine and FOLFIRINOX) can be considered appropriate treatment options for treatment-naive patients with good performance status who have locally advanced unresectable or metastatic pancreatic adenocarcinoma. FOLFIRINOX has a higher objective response rate than nab-paclitaxel-gemcitabine (32% versus 23%, respectively), but the adverse effect profile favors the nab-paclitaxel/gemcitabine combination, acknowledging this conclusion is limited due to lack of a comparative trial. Modifications to both regimens have been presented at American Society of Clinical Oncology symposiums, with preliminary data showing an extended median OS and a more tolerable toxicity profile.19,20 In a recent retrospective observational cohort comparative analysis of nab-paclitaxel/gemcitabine versus FOLFIRINOX, results showed no statistical difference in median OS. The real-world data showed that gemcitabine-based therapy is being offered commonly to elderly patients and patients with poor performance status.13 There is no current research proposal for conducting a direct head-to-head comparison between these 2 regimens. Based on extrapolated data from the prior mentioned trials and retrospective analysis reviews, current guidelines recommend offering younger (< 65 years old), healthier (no comorbidity contraindication) patients with excellent performance status (ECOG 0) first-line FOLFIRINOX or gemcitabine/nab-paclitaxel. Elderly patients with stable comorbidities and good performance status (ECOG 1 or 2, Karnofsky performance status ≥ 70) could be preferably considered for treatment with nab-paclitaxel/gemcitabine as first-line or modified FOLFIRINOX if performance status is excellent. Patients with poor performance status (ECOG ≥ 2), advanced age, and significant comorbidities could still be considered candidates for gemcitabine monotherapy. However, there are promising indications that the combination of gemcitabine, nab-paclitaxel, and cisplatin could be a frontline therapy in advanced pancreaticobilliary malignancies in the future.
Second-Line Systemic Treatment
Case Continued
The patient and oncologist opt to begin treatment with modified FOLFIRINOX therapy, and after the patient completes 10 cycles CT scan shows progression of disease. His oncologist decides to refer the patient to a comprehensive cancer center for evaluation for participation in clinical trials, as his performance status remains very good (ECOG 1) and he would like to seek a novel therapy. His liver mass biopsy and blood liquid biopsy are sent for tumor mutational profile evaluation; results show a high tumor mutational burden and microsatellite instability.
- What are second-line treatment options for metastatic pancreatic cancer?
Second-line regimen recommendations for metastatic pancreatic cancer depend on which agents were used in first-line therapy and the patient’s performance status and comorbidities. Patients who progressed on first-line FOLFIRINOX and continue to have a good performance status (ECOG 0 or 1) may be considered for gemcitabine/nab-paclitaxel therapy; otherwise, they may be candidates for gemcitabine plus capecitabine or gemcitabine monotherapy based on performance status and goals of care. Patients who progressed on front-line gemcitabine/nab-paclitaxel may opt for FOLFIRINOX (or an oxaliplatin-based regimen [FOLFOX] or irinotecan-based regimen [FOLFIRI] if FOLFIRINOX is not tolerable), nanoliposomal irinotecan/fluorouracil/leucovorin, or a short-term infusional fluorouracil and leucovorin regimen. The preferences for second-line treatment are not well established, and patients should be encouraged to participate in clinical trials. Chemotherapy should be offered only to those patients who maintain good performance status after progression on first-line therapy. For patients with poor performance status (ECOG 3 or 4) or multiple comorbidities, a discussion about goals of care and palliative therapy is warranted.
Gemcitabine-Based Therapy
An AGEO prospective multicenter cohort assigned 57 patients with metastatic pancreatic adenocarcinoma who had disease progression on FOLFIRINOX therapy to receive gemcitabine/nab-paclitaxel (dose as per MPACT trial).21 The median OS was 8.8 months and median PFS was 5.1 months after FOLFIRINOX. There were reported manageable grade 3/4 toxicities in 40% of patients, which included neutropenia (12.5%), neurotoxicity (12.5%), asthenia (9%), and thrombocytopenia (6.5%). A phase 2 clinical trial that evaluated gemcitabine monotherapy in 74 patients with metastatic pancreatic cancer who had progressed on fluorouracil showed a 3.85-month survival benefit.22
Irinotecan-Based Regimens
The NAPOLI-1 (NAnoliPOsomaL Irinotecan) trial evaluated nanoliposomal irinotecan (MM-398, nal-IRI) and fluorouracil/leucovorin in patients with metastatic pancreatic cancer refractory to gemcitabine-based therapy.23 This global, open-label phase 3 trial initially randomly assigned and stratified 417 patients in a 1:1 fashion to receive either nanoliposomal irinotecan monotherapy (120 mg/m2 every 3 weeks, equivalent to 100 mg/m2 of irinotecan base) or fluorouracil/leucovorin combination. A third treatment arm consisting of nanoliposomal irinotecan (80 mg/m2, equivalent to 70 mg/m2 of irinotecan base) with fluorouracil and leucovorin every 2 weeks was added later in a 1:1:1 fashion. Patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin had a significantly improved OS of 6.1 months compared to 4.2 months with fluorouracil/leucovorin (HR 0.67 [95% CI 0.49 to 0.92], P = 0.012). The results of an intention-to-treat analysis favored the nanoliposomal irinotecan regimen, with a median OS of 8.9 months compared with 5.1 months (HR 0.57, P = 0.011). In addition, median PFS was improved in the nanoliposomal irinotecan arm (3.1 months versus 1.5 months; HR 0.56, P < 0.001), and median OS did not differ between patients treated with nanoliposomal irinotecan monotherapy and those treated with fluorouracil/leucovorin (4.9 months versus 4.2 months; HR 0.99 [95% CI 0.77 to 1.28], P = 0.94). The grade 3/4 adverse events that occurred most frequently in the 117 patients assigned to nanoliposomal irinotecan plus fluorouracil/leucovorin were neutropenia (27%), diarrhea (13%), vomiting (11%), and fatigue (14%). Nanoliposomal irinotecan combination provides another second-line treatment option for patients with metastatic pancreatic adenocarcinoma who have progressed on gemcitabine-based therapy but are not candidates for FOLFIRINOX.
Oxaliplatin-Based Regimens
Regimens that combine oxaliplatin with fluorouracil and leucovorin or capecitabine have shown superiority to fluorouracil/leucovorin or best supportive care (BSC). The CONKO study group compared oxaliplatin plus fluorouracil/leucovorin to BSC as second-line therapy in patients with advanced pancreatic cancer who progressed while on gemcitabine therapy (CONKO-003).24 In this phase 3 trial, patients were randomly assigned (1:1) and stratified based on duration of first-line therapy, performance status, and tumor stage to receive BSC alone or the OFF regimen, which consisted of oxaliplatin (85 mg/m2 on days 8 and 22) plus short-term infusional fluorouracil (2000 mg/m2 over 24 hours) and leucovorin (200 mg/m2 over 30 minutes), both given on days 1, 8, 15, and 22 of a 6-week cycle. This trial was terminated early according to predefined protocol regulations because of insufficient accrual (lack of acceptance of BSC by patients and physicians). Median second-line survival was 4.82 months for patients who received OFF treatment and 2.30 months for those who received BSC (HR 0.45 [95% CI 0.24 to 0.83], P = 0.008). Neurotoxicity (grade 1/2) and nausea, emesis, and diarrhea (grade 2/3) were worse in the chemotherapy arm; otherwise, the regimen was well tolerated.
A later modification of the CONKO-003 trial changed the comparison arm from BSC to fluorouracil/leucovorin.25 The median OS in the OFF group was 5.9 months versus 3.3 months in the fluorouracil/leucovorin group (HR 0.66 [95% CI 0.48 to 0.91], log-rank P = 0.010). Time to progression was significantly extended with OFF (2.9 months) as compared with fluorouracil/leucovorin (2.0 months; HR 0.68 [95% CI 0.50 to 0.94], log-rank P = 0.019). Rates of adverse events were similar between the treatment arms, with the exception of grades 1/2 neurotoxicity, which were reported in 38.2% and 7.1% of patients in the OFF and fluorouracil/leucovorin groups, respectively (P < 0.001).
The phase 3 PANCREOX trial failed to show superiority of modified FOLFOX6 (mFOLFOX6; infusional fluorouracil, leucovorin, and oxaliplatin) over fluorouracil/leucovorin.26 A phase 2 trial of oxaliplatin plus capecitabine for second-line therapy in gemcitabine-treated advanced pancreatic cancer patients with dose adjustments for performance status (ECOG 2) and age (> 65 years) showed a median OS of 5.7 months without a comparison.27 A modified oxaliplatin regimen may be a reasonable second-line therapy option for gemcitabine-treated patients who are not candidates for an irinotecan-based regimen (eg, elevated bilirubin) and continue to have an acceptable performance status.
Targeted Therapies
A variety of targeted therapies have failed to demonstrate major activity in metastatic pancreatic cancer, including bevacizumab targeting vascular endothelial growth factor, cetuximab targeting epidermal growth factor receptor, ruxolitinib targeting JAK pathway signaling, saridegib targeting the hedgehog pathway, and MK-0646 targeting insulin-like growth factor 1 receptor (IGFR). Other novel agents against targetable pathways that had promising early-phase results are currently being studied in ongoing clinical trials; these include JAK-2, PI3K, MEK, and BRAF inhibitors and immunotherapy.
Recent research efforts have focused on targeted testing of advanced pancreatic cancers for mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H) and for the germline and somatic BRCA1/2 or PALB2 mutations to determine potential eligibility for immunotherapy. Patients with these tumor characteristics and/or mutations might also be more sensitive to platinum-based chemotherapy agents or poly (ADP-ribose) polymerase (PARP) inhibitors. Germline mutations in BRCA 1/2 are present in 5% to 8% of patients with pancreatic cancer (up to 10%–15% in Ashkenazi Jewish population).28 A superior median OS was retrospectively observed for patients with advanced stage BRCA 1/2-associated pancreatic adenocarcinoma who were treated with platinum-based chemotherapy agents versus those treated with non-platinum-based agents (22 versus 9 months; P = 0.039).22 PARP inhibitors have shown activity in germline BRCA1/2-associated breast (off label) and ovarian cancers (approved by the FDA). The efficacy and safety of PARP inhibitors were evaluated in a phase 2 study of a spectrum of BRCA1/2-associated cancers, including pancreatic cancer. The results revealed a tumor response rate of 21.7% (5 of 23 patients with pancreatic cancer [95% CI 7.5 to 43.7]), and 35% of patients had stable disease for a duration of 8 weeks or more (95% CI 16.4 to 57.3) with good tolerability.29 Three novel PARP inhibitors are currently under clinical trial investigation in patients with germline BRCA 1/2- and PALB2-mutated metastatic pancreatic cancer: maintenance olaparib (NCT02184195) and rucaparib (NCT03140670) are both being studied as monotherapy in patients whose disease has not progressed on first-line platinum-based chemotherapy, and veliparib is being evaluated in a 3-arm study that includes gemcitabine and cisplatin with or without veliparib and single-agent maintenance veliparib (NCT01585805).
In 2017, the FDA granted accelerated approval to pembrolizumab for treatment of patients with unresectable or metastatic MSI-H or dMMR solid tumors whose disease progressed on prior treatments, making it the first oncology drug to be approved based on the genetic features of the tumor rather than its location in the body. This first tissue/site-agnostic approval was based on results from 5 single-arm trials involving 149 patients, including 5 patients with pancreatic cancer.30 The objective response rate with pembrolizumab was 39.6% (95% CI 31.7 to 47.9), including a 7.4% complete response rate and a 32.2% partial response rate. The median duration of response was not reached at the time of publication (range, 1.6+ months to 22.7+ months).
Palliative and Supportive Care
Case Continued
The patient opts to participate in a novel immunotherapy clinical trial and is currently on his second cycle. He continues to have right upper quadrant pain despite opioid analgesia, has not gained any weight, and noticed new right lower extremity swelling after a recent holiday vacation to Florida.
- What supportive measures should be in place for patients with metastatic adenocarcinoma?
Most patients with advanced pancreatic adenocarcinoma will require a palliative intervention. All new unresectable pancreatic cancer patients should have an early psychosocial evaluation; identification of symptoms and implementation of preventive interventions that would improve quality of life and reduce suffering are paramount. A multidisciplinary team including physician/nursing staff, nutritionist/dietitian, palliative service, a social worker, and a case manager should be involved in patient care. More than two-thirds of patients can develop symptomatic biliary obstruction.31 Bile duct obstruction due to locally advanced pancreatic adenocarcinoma causes hyperbilirubinemia, which requires endoscopic placement of a metallic or plastic stent; plastic stents have a higher rate of re-occlusion.32 Appropriate bile flow allows treatment with irinotecan-based regimens. Percutaneous biliary drainage may be necessary if endoscopic intervention is not feasible.
Approximately one quarter of patients may present with gastric outlet obstruction due to duodenal obstruction.31 Endoscopic placement of an enteral expandable metal stent is preferred. Alternatively, percutaneous endoscopic gastrostomy tube placement may give symptomatic relief. Palliative surgical interventions are reserved for patients with greater life expectancy and in whom all other interventions have failed or are not feasible.
Almost all patients with pancreatic adenocarcinoma will experience cancer-associated pain. Intractable pain should be treated with a celiac plexus block. Radiation therapy may be considered as an adjunct therapy for pain, bleeding, and/or local obstruction. The National Comprehensive Cancer Network guidelines recommend that patients who undergo a laparotomy for potentially resectable disease but are found to have unresectable disease at the time of surgery should undergo stenting, open biliary-enteric bypass with or without gastrojejunostomy, and/or celiac plexus neurolysis.33
Pancreatic exocrine enzyme insufficiency due to tumor extension, duct blockage, or surgical removal may cause malabsoprtive steatorrhea, contributing to cancer cachexia syndrome. Nutritional evaluation and daily oral pancreatic enzyme supplementation are recommended.34
Patients diagnosed with pancreatic adenocarcinoma have a venous thromboembolism (VTE) incidence of 20 per 100 person-years (5%–60% of patients) and are considered at very high risk for VTE based on the Khorana score.35 The preferred VTE treatment is low-molecular-weight heparin rather than warfarin based on the results of the CLOT study.36 There is no current evidence for routine prophylactic therapy or the use of direct oral anticoagulants.
Finally, a cancer diagnosis, particularly pancreatic cancer, causes a significant amount of psychosocial stress and requires active support and counseling from a professional.
Conclusion
Pancreatic adenocarcinoma is the most lethal of all the gastrointestinal malignancies. FOLFIRINOX and gemcitabine/nab-paclitaxel are superior to gemcitabine monotherapy for patients with advanced unresectable and/or metastatic pancreatic cancer who are candidates for more aggressive therapy and are considered first-line therapies. Early data on the gemcitabine, nab-paclitaxel, and cisplatin combination appears to show superior efficacy. Second-line therapies are selected based on the patient’s performance status, first-line regimen, and residual toxicities from the prior regimen; options include gemcitabine/nab-paclitaxel, FOLFIRINOX (± oxaliplatin or irinotecan), single-agent gemcitabine (elderly frail patients), fluorouracil and liposomal-irinotecan, or referral for a clinical trial. The main challenge with pancreatic cancer is the development of stroma around the tumor, which abrogates drug delivery, allows for tumor growth in a hypoxic microenvironment, alters the metabolomics, and causes an immunosuppressive microenvironment. Drugs that target the microenvironments, such as hedgehog pathway inhibitors, have failed to show any clinical benefit, and we hope to see more efficacious microenvironment-targeted novel drugs in the future. In addition, immunotherapy has not shown any significant efficacy in clinical trials and many trials are still ongoing.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
1. National Institutes of Health/National Cancer Institute. Surveillance, Epidemiology and End Results Program (SEER). Cancer stat facts: pancreatic cancer. seer.cancer. gov/statfacts/html/pancreas.html. Accessed April 20, 2018.
2. Allen PJ, Kuk D, Castillo CF, et al. Multi-institutional validation study of the American Joint Commission on Cancer (8th Edition) changes for T and N staging in patients with pancreatic adenocarcinoma. Ann Surg 2017;265:185–91.
3. Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
4. Recio-Boiles A, Babiker HM. Pancreatic adenocarcinoma: update on neoadjuvant and adjuvant treatment. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(2):25–38.
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
6. Conroy T, Desseigne F, Ychou M, et al, Groupe Tumeurs Digestives of Unicancer, PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
7. Vander Walde N, Jagsi R, Dotan E, et al. NCCN Guidelines insights: older adult oncology, version 2.2016. J Natl Compr Canc Netw 2016;14:1357–70.
8. Rothenberg ML, Moore MJ, Cripps MC, et al. A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
9. Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13.
10. Cunningham D, Chau I, Stocken DD, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009;27:5513–8.
11. Moore MJ, Goldstein D, Hamm J, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–6.
12. Reni M, Cordio S, Milandri C, et al. Gemcitabine versus cisplatin, epirubicin, fluorouracil, and gemcitabine in advanced pancreatic cancer: a randomised controlled multicentre phase III trial. Lancet Oncol 2005;6:369–76.
13. Cartwright TH, Parisi M, Espirito JL, et al. Treatment outcomes with first-line (1L) nab-paclitaxel + gemcitabine (AG) and FOLFIRINOX (FFX) in metastatic pancreatic adenocarcinoma (mPAC) [abstract]. J Clin Oncol 2017 35:15 suppl:e18147.
14. Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54.
15. Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer [abstract]. J Clin Oncol 2017 35:4_suppl:341.
16. Reni M, Balzano G, Zanon S, et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br J Cancer 2016;115:290–6.
17. Ychou M, Desseigne F, Guimbaud R, et al. Randomized phase II trial comparing folfirinox (5FU/leucovorin [LV], irinotecan [I]and oxaliplatin [O]) vs gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA). First results of the ACCORD 11 trial [abstract 4516]. J Clin Oncol 2007;25:210s.
18. Iyer L, Das S, Janisch L, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002;2:43–7.
19. Krishna K, Blazer MA, Wei L, et al. Modified gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer (MPC): A single-institution experience [abstract]. J Clin Oncol 201533; (suppl 3). Abstract 366.
20. Ueno M, Ozaka M, Ishii H, et al. Phase II study of modified FOLFIRINOX for chemotherapy-naive patients with metastatic pancreatic cancer [abstract]. J Clin Oncol 2016;34(suppl). Abstract 4111.
21. Portal A, Pernot S, Tougeron D, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic adenocarcinoma after Folfirinox failure: an AGEO prospective multicentre cohort. Br J Cancer 2015;113:989–95.
22. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8.
23. Wang-Gillam A, Li CP, Bodoky G, et al, NAPOLI-1 Study Group. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57.
24. Pelzer U, Schwaner I, Stieler J, et al. Best supportive care (BSC) versus oxaliplatin, folinic acid and 5-fluorouracil (OFF) plus BSC in patients for second-line advanced pancreatic cancer: a phase III-study from the German CONKO-study group. Eur J Cancer 011;47:1676–81.
25. Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J Clin Oncol 2014;32:2423–9.
26. Gill S, Ko YJ, Cripps C, et al. PANCREOX: a randomized phase III study of 5-fluorouracil/leucovorin with or without oxaliplatin for second-line advanced pancreatic cancer in patients who have received gemcitabine-based chemotherapy. J Clin Oncol 2016;34:3914–20.
27. Xiong HQ, Varadhachary GR, Blais JC, et al. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer 2008;113:2046–52.
28. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
29. Kaufman B, Shapira-Frommer R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–50.
30. Goldberg KB, Blumenthal GM, McKee AE, Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp Biol Med 2018;243:308–12.
31. House MG, Choti MA. Palliative therapy for pancreatic/biliary cancer. Surg Clin North Am 2005;85:359–71.
32. Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc 2006;63:986–95.
33. Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1028–61.
34. Landers A, Muircroft W, Brown H. Pancreatic enzyme replacement therapy (PERT) for malabsorption in patients with metastatic pancreatic cancer. BMJ Support Palliat Care 2016;6:75–9.
35. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008;111:4902–7.
36. Lee AY, Levine MN, Baker RI, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. N Engl J Med 2003;349:146–53.
Aggressive B-Cell Non-Hodgkin Lymphoma
Introduction
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features.1 While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women.2 NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides,3 immunosuppression associated with HIV infection,4 autoimmune disorders,5 iatrogenically induced immune suppression in the post-transplant and other settings,6 family history of NHL,7 and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL.8 In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma.9 Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas.10 Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes,11–13 including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Work-Up
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1. Both hepatitis B and C have been associated with increased risk of NHL. In addition, there is a risk of hepatitis B reactivation following certain NHL therapies. A contrast-enhanced computed tomography (CT) scan in addition to positron emission tomography (PET) is useful to define the extent of disease in situations needing greater definition (eg, lymphadenopathy close to the bowel, cervical and supraclavicular nodal involvement, and lymphadenopathy causing thrombosis or compression of nearby structures).14 In cases where it is apparent that the patient has advanced stage disease (Ann Arbor stage III/IV) based on imaging, bone marrow biopsy is unlikely to alter the treatment plan. For such patients, if the complete blood count is unremarkable, deferral of bone marrow biopsy may be reasonable. For new cases of DLBCL, assessment for MYC translocation by fluorescence in situ hybridization (FISH) is recommended. If a MYC translocation is identified, then testing for BCL2 and BCL6 translocations by FISH should be performed.
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL.14 This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases.2 It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes.15
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races.16 There is a slight male predominance (55%), median age at diagnosis is 65 years,16,17 and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue.18 Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis.19
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease.20 The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement,21 this feature does not carry the same poor prognosis associated with transformed disease22 or DLBCL involvement of the bone marrow.23
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein,24 and about 70% express the BCL-6 protein.25 C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL.26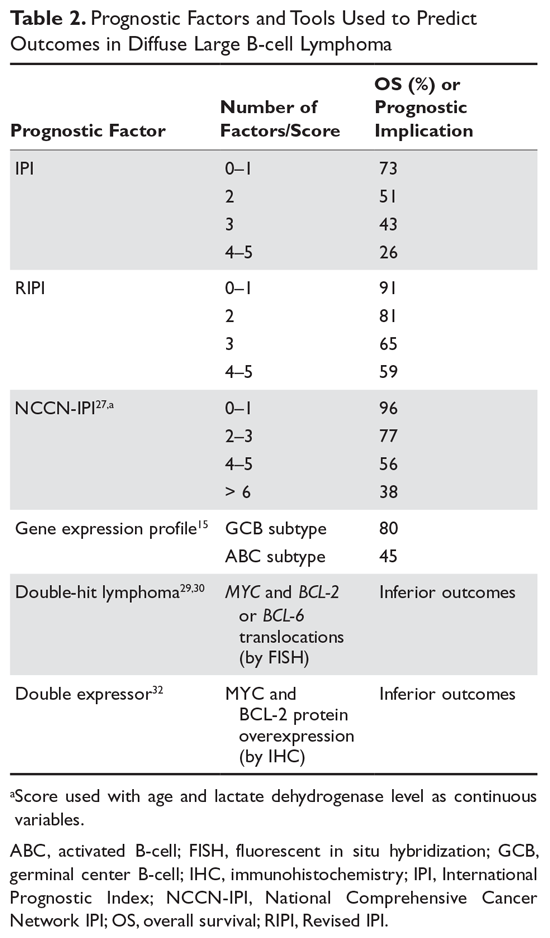
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2). Subsequently, modifications were made to adjust for age and stage, with the latest iteration being the NCCN (National Comprehensive Cancer Network) IPI.27 This tool uses age, performance status, LDH ratio (relative to the upper limit of normal), a more precise definition for presence of extranodal sites of disease (defined as lymphomatous involvement in the bone marrow, CNS, liver/GI tract, or lung), and Ann Arbor stage to stratify patients into 4 risk groups with significantly different 5-year OS, ranging from 38% to 96% based on the subgroup. Importantly, the NCCN-IPI was derived in a cohort of patients treated with rituximab-based therapy.
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP.28 The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis.29,30 Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma.1 In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL.31,32 Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy
DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years.33 The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS.34,35
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues36 studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms,37 with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT.36
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed.38 Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]);39 dose-dense therapy (R-CHOP-14);replacement of rituximab with obinutuzuimab;40 addition of novel agents such as bortezomib,41 lenalidomide,42 or ibrutinib43,44 to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide,45 enzastaurin,46 and everolimus.47 Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit.48 More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however.49 As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%).50,51 Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes,52 with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission.53
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively.54 A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF).55 In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months.56 For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series.57
Relapsed/Refractory Disease
Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype.58 However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone.59 The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort.60
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT.61 Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained.62 However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL.64 PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL.20 It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious.64 Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease,65 with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL.66 PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis.67 This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT.68–70 The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78%71 when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases.72,73 For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80%.74–76 In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease.77 This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe.78 MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease.79 Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases.80 In rare cases, CD5 or cyclin D1 may be negative.80 Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression.81 Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome.82 In cyclin D1–negative cases, SOX11 expression may help with diagnosis.83 A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range.84 In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range.85–87 Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004,88 and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010.89 The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients.90 The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement.91
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course,92 but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy
For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission.87,93,94 Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial.93 At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy.95 However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach.96 Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP.97,98
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years.99 In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib,100,101 the BTK inhibitors ibrutinib102,103 and acalabrutinib,104 and the immunomodulatory agent lenalidomide.105
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission.95 Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease.95,106 A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma.107 The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90%.107 Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients.108 In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT.109
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70%.110,111 High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse.112 However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease.113
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients.114
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.
Introduction
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features.1 While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women.2 NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides,3 immunosuppression associated with HIV infection,4 autoimmune disorders,5 iatrogenically induced immune suppression in the post-transplant and other settings,6 family history of NHL,7 and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL.8 In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma.9 Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas.10 Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes,11–13 including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Work-Up
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1. Both hepatitis B and C have been associated with increased risk of NHL. In addition, there is a risk of hepatitis B reactivation following certain NHL therapies. A contrast-enhanced computed tomography (CT) scan in addition to positron emission tomography (PET) is useful to define the extent of disease in situations needing greater definition (eg, lymphadenopathy close to the bowel, cervical and supraclavicular nodal involvement, and lymphadenopathy causing thrombosis or compression of nearby structures).14 In cases where it is apparent that the patient has advanced stage disease (Ann Arbor stage III/IV) based on imaging, bone marrow biopsy is unlikely to alter the treatment plan. For such patients, if the complete blood count is unremarkable, deferral of bone marrow biopsy may be reasonable. For new cases of DLBCL, assessment for MYC translocation by fluorescence in situ hybridization (FISH) is recommended. If a MYC translocation is identified, then testing for BCL2 and BCL6 translocations by FISH should be performed.
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL.14 This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases.2 It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes.15
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races.16 There is a slight male predominance (55%), median age at diagnosis is 65 years,16,17 and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue.18 Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis.19
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease.20 The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement,21 this feature does not carry the same poor prognosis associated with transformed disease22 or DLBCL involvement of the bone marrow.23
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein,24 and about 70% express the BCL-6 protein.25 C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL.26
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2). Subsequently, modifications were made to adjust for age and stage, with the latest iteration being the NCCN (National Comprehensive Cancer Network) IPI.27 This tool uses age, performance status, LDH ratio (relative to the upper limit of normal), a more precise definition for presence of extranodal sites of disease (defined as lymphomatous involvement in the bone marrow, CNS, liver/GI tract, or lung), and Ann Arbor stage to stratify patients into 4 risk groups with significantly different 5-year OS, ranging from 38% to 96% based on the subgroup. Importantly, the NCCN-IPI was derived in a cohort of patients treated with rituximab-based therapy.
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP.28 The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis.29,30 Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma.1 In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL.31,32 Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy
DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years.33 The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS.34,35
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues36 studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms,37 with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT.36
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed.38 Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]);39 dose-dense therapy (R-CHOP-14);replacement of rituximab with obinutuzuimab;40 addition of novel agents such as bortezomib,41 lenalidomide,42 or ibrutinib43,44 to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide,45 enzastaurin,46 and everolimus.47 Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit.48 More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however.49 As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%).50,51 Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes,52 with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission.53
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively.54 A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF).55 In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months.56 For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series.57
Relapsed/Refractory Disease
Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype.58 However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone.59 The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort.60
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT.61 Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained.62 However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL.64 PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL.20 It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious.64 Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease,65 with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL.66 PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis.67 This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT.68–70 The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78%71 when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases.72,73 For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80%.74–76 In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease.77 This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe.78 MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease.79 Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases.80 In rare cases, CD5 or cyclin D1 may be negative.80 Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression.81 Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome.82 In cyclin D1–negative cases, SOX11 expression may help with diagnosis.83 A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range.84 In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range.85–87 Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004,88 and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010.89 The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients.90 The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement.91
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course,92 but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy
For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission.87,93,94 Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial.93 At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy.95 However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach.96 Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP.97,98
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years.99 In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib,100,101 the BTK inhibitors ibrutinib102,103 and acalabrutinib,104 and the immunomodulatory agent lenalidomide.105
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission.95 Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease.95,106 A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma.107 The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90%.107 Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients.108 In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT.109
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70%.110,111 High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse.112 However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease.113
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients.114
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
Introduction
Non-Hodgkin lymphoma (NHL) comprises a wide variety of malignant hematologic disorders with varying clinical and biological features. The more than 60 separate NHL subtypes can be classified according to cell of origin (B cell versus T cell), anatomical location (eg, orbital, testicular, bone, central nervous system), clinical behavior (indolent versus aggressive), histological features, or cytogenetic abnormalities. Although various NHL classification schemes have been used over the years, the World Health Organization (WHO) classification is now widely accepted as the definitive pathologic classification system for lymphoproliferative disorders, incorporating morphologic, immunohistochemical, flow cytometric, cytogenetic, and molecular features.1 While the pathologic and molecular subclassification of NHL has become increasingly refined in recent years, from a management standpoint, classification based on clinical behavior remains very useful. This approach separates NHL subtypes into indolent versus aggressive categories. Whereas indolent NHLs may remain clinically insignificant for months to years, aggressive B-cell NHLs generally become life-threatening within weeks to months without treatment.
Epidemiology
Data from cancer registries show a steady, unexplainable increase in the incidence of NHL during the second half of the 20th century; the incidence has subsequently plateaued. There was a significant increase in NHL incidence between 1970 and 1995, which has been attributed in part to the HIV epidemic. More than 72,000 new cases of NHL were diagnosed in the United States in 2017, compared to just over 8000 cases of Hodgkin lymphoma, making NHL the sixth most common cancer in adult men and the fifth most common in adult women.2 NHL appears to occur more frequently in Western countries than in Asian populations.
Various factors associated with increased risk for B-cell NHL have been identified over the years, including occupational and environmental exposure to certain pesticides and herbicides,3 immunosuppression associated with HIV infection,4 autoimmune disorders,5 iatrogenically induced immune suppression in the post-transplant and other settings,6 family history of NHL,7 and a personal history of a prior cancer, including Hodgkin lymphoma and prior NHL.8 In terms of infectious agents associated with aggressive B-cell NHLs, Epstein-Barr virus (EBV) has a clear pathogenic role in Burkitt lymphoma, in many cases of post-transplant lymphoproliferative disorders, and in some cases of HIV-related aggressive B-cell lymphoma.9 Human herpesvirus-8 viral genomes have been found in virtually all cases of primary effusion lymphomas.10 Epidemiological studies also have linked hepatitis B and C to increased incidences of certain NHL subtypes,11–13 including primary hepatic diffuse large B-cell lymphoma (DLBCL). Similarly, Helicobacter pylori has been associated with gastric DLBCL.
Staging and Work-Up
A tissue biopsy is essential in the diagnosis and management of NHL. The most significant disadvantage of fine-needle aspiration cytology is the lack of histologic architecture. The optimal specimen is an excisional biopsy; when this cannot be performed, a core needle biopsy, ideally using a 16-gauge or larger caliber needle, is the next best choice.
The baseline tests appropriate for most cases of newly diagnosed aggressive B-cell NHL are listed in Table 1. Both hepatitis B and C have been associated with increased risk of NHL. In addition, there is a risk of hepatitis B reactivation following certain NHL therapies. A contrast-enhanced computed tomography (CT) scan in addition to positron emission tomography (PET) is useful to define the extent of disease in situations needing greater definition (eg, lymphadenopathy close to the bowel, cervical and supraclavicular nodal involvement, and lymphadenopathy causing thrombosis or compression of nearby structures).14 In cases where it is apparent that the patient has advanced stage disease (Ann Arbor stage III/IV) based on imaging, bone marrow biopsy is unlikely to alter the treatment plan. For such patients, if the complete blood count is unremarkable, deferral of bone marrow biopsy may be reasonable. For new cases of DLBCL, assessment for MYC translocation by fluorescence in situ hybridization (FISH) is recommended. If a MYC translocation is identified, then testing for BCL2 and BCL6 translocations by FISH should be performed.
Prior to the initiation of treatment, patients should always undergo a thorough cardiac and pulmonary evaluation, especially if the patient will be treated with an anthracycline or mediastinal irradiation. Central nervous system (CNS) evaluation with magnetic resonance imaging (MRI) and lumbar puncture is essential if there are neurological signs or symptoms. In addition, certain anatomical sites including the testicles, paranasal sinuses, kidney, adrenal glands, and epidural space have been associated with increased involvement of the CNS and may warrant MRI evaluation and lumbar puncture. Certain NHL subtypes like Burkitt lymphoma, high-grade NHL with translocations of MYC and BCL-2 or BCL-6 (double-hit lymphoma), blastoid mantle cell lymphoma, and lymphoblastic lymphoma have a high risk of CNS involvement, and patients with these subtypes need CNS evaluation.
The Lugano classification is used to stage patients with NHL.14 This classification is based on the Ann Arbor staging system and uses the distribution and number of tumor sites to stage disease. In general, this staging system in isolation is of limited value in predicting survival after treatment. However, the Ann Arbor stage does have prognostic impact when incorporated into risk scoring systems such as the International Prognostic Index (IPI). In clinical practice, the Ann Arbor stage is useful primarily to determine eligibility for localized therapy approaches. The absence or presence of systemic symptoms such as fevers, drenching night sweats, or weight loss (> 10% of baseline over 6 months or less) is designated by A or B, respectively.
Diffuse Large B-Cell Lymphoma
DLBCL is the most common lymphoid neoplasm in adults, accounting for about 25% of all NHL cases.2 It is increasingly clear that the diagnostic category of DLBCL is quite heterogeneous in terms of morphology, genetics, and biologic behavior. A number of clinicopathologic subtypes of DLBCL exist, such as T cell/histiocyte–rich large B-cell lymphoma, primary mediastinal large B-cell lymphoma, intravascular large B-cell lymphoma, DLBCL associated with chronic inflammation, lymphomatoid granulomatosis, and EBV-positive large B-cell lymphoma, among others. Gene expression profiling (GEP) can distinguish 2 cell of origin DLBCL subtypes: the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes.15
DLBCL may be primary (de novo) or may arise through the transformation of many different types of low-grade B-cell lymphomas. This latter scenario is referred to as histologic transformation or transformed lymphoma. In some cases, patients may have a previously diagnosed low-grade B-cell NHL; in other cases, both low-grade and aggressive B-cell NHL may be diagnosed concurrently. The presence of elements of both low-grade and aggressive B-cell NHL in the same biopsy specimen is sometimes referred to as a composite lymphoma.
In the United States, incidence varies by ethnicity, with DLBCL being more common in Caucasians than other races.16 There is a slight male predominance (55%), median age at diagnosis is 65 years,16,17 and the incidence increases with age.
Presentation, Pathology, and Prognostic Factors
The most common presentation of patients with DLBCL is rapidly enlarging lymphadenopathy, usually in the neck or abdomen. Extranodal/extramedullary presentation is seen in approximately 40% of cases, with the gastrointestinal (GI) tract being the most common site. However, extranodal DLBCL can arise in virtually any tissue.18 Nodal DLBCL presents with symptoms related to the sites of involvement (eg, shortness of breath or chest pain with mediastinal lymphadenopathy), while extranodal DLBCL typically presents with symptoms secondary to dysfunction at the site of origin. Up to one third of patients present with constitutional symptoms (B symptoms) and more than 50% have elevated serum lactate dehydrogenase (LDH) at diagnosis.19
Approximately 40% of patients present with stage I/II disease. Of these, only a subset present with stage I, or truly localized disease (defined as that which can be contained within 1 irradiation field). About 60% of patients present with advanced (stage III–IV) disease.20 The bone marrow is involved in about 15% to 30% of cases. DLBCL involvement of the bone marrow is associated with a less favorable prognosis. Patients with DLBCL elsewhere may have low-grade NHL involvement of the bone marrow. Referred to as discordant bone marrow involvement,21 this feature does not carry the same poor prognosis associated with transformed disease22 or DLBCL involvement of the bone marrow.23
DLBCL is defined as a neoplasm of large B-lymphoid cells with a diffuse growth pattern. The proliferative fraction of cells, as determined by Ki-67 staining, is usually greater than 40%, and may even exceed 90%. Lymph nodes usually demonstrate complete effacement of the normal architecture by sheets of atypical lymphoid cells. Tumor cells in DLBCL generally express pan B-cell antigens (CD19, CD20, CD22, CD79a, Pax-5) as well as CD45 and surface immunoglobulin. Between 20% and 37% of DLBCL cases express the BCL-2 protein,24 and about 70% express the BCL-6 protein.25 C-MYC protein expression is seen in a higher percentage (~ 30%–50%) of cases of DLBCL.26
Many factors are associated with outcome in DLBCL. The IPI score was developed in the pre-rituximab era and is a robust prognostic tool. This simple tool uses 5 easily obtained clinical factors (age > 60 years, impaired performance status, elevated LDH, > 1 extranodal site of disease, and stage III/IV disease). By summing these factors, 4 groups with distinct 5-year overall survival (OS) rates ranging from 26% to 73% were identified (Table 2). Subsequently, modifications were made to adjust for age and stage, with the latest iteration being the NCCN (National Comprehensive Cancer Network) IPI.27 This tool uses age, performance status, LDH ratio (relative to the upper limit of normal), a more precise definition for presence of extranodal sites of disease (defined as lymphomatous involvement in the bone marrow, CNS, liver/GI tract, or lung), and Ann Arbor stage to stratify patients into 4 risk groups with significantly different 5-year OS, ranging from 38% to 96% based on the subgroup. Importantly, the NCCN-IPI was derived in a cohort of patients treated with rituximab-based therapy.
Cytogenetic and molecular factors also predict outcome in DLBCL. The ABC subtype distinguished by GEP has consistently been shown to have inferior outcomes with first-line therapy. As GEP is not routinely available in clinical practice, immunohistochemical (IHC) approaches (eg, the Hans algorithm) have been developed that can approximate the GEP subtypes. These IHC approaches have approximately 80% concordance with GEP.28 The 3 most common chromosomal translocations in DLBCL involve BCL-2, BCL-6 and MYC. MYC-rearranged DLBCLs have a less favorable prognosis.29,30 Cases in which a MYC translocation occurs in combination with a BCL-2 or BCL-6 translocation are commonly referred to as double-hit lymphoma (DHL); cases with all 3 translocations are referred to as triple-hit lymphoma (THL). Both DHL and THL have a worse prognosis with standard DLBCL therapy compared to non-DHL/THL cases. In the 2016 revised WHO classification, DHL and THL are an entity technically distinct from DLBCL, referred to as high-grade B-cell lymphoma.1 In some cases, MYC and BCL-2 protein overexpression occurs in the absence of chromosomal translocations. Cases in which MYC and BCL-2 are overexpressed (by IHC) are referred to as double expressor lymphoma (DEL), and also have inferior outcome compared with non-DEL DLBCL.31,32 Interestingly, MYC protein expression alone does not confer inferior outcomes, unlike isolated MYC translocation, which is associated with inferior outcomes.
Treatment
First-Line Therapy
DLBCL is an aggressive disease and, in most cases, survival without treatment can be measured in weeks to months. The advent of combination chemotherapy (CHOP [cyclophosphamide, doxorubicin, vincristine, and prednisone] or CHOP-like regimens) led to disease-free survival (DFS) rates of 35% to 40% at 3 to 5 years.33 The addition of rituximab to CHOP (R-CHOP) has improved both progression-free surivial (PFS) and OS.34,35
Treatment options vary for patients with localized (stage I/II) and advanced (stage III/IV) disease. Options for limited-stage DLBCL include an abbreviated course of R-CHOP (3 or 4 cycles) with involved-field radiation therapy (IFRT) versus a full course (6–8 cycles) of R-CHOP without radiation therapy (RT). Most studies comparing combined modality therapy (chemotherapy plus RT) versus chemotherapy alone were conducted in the pre-rituximab era. With the introduction of rituximab, Persky and colleagues36 studied the use of 3 cycles of R-CHOP followed by RT, demonstrating a slightly improved OS of 92% at 4 years as compared to 88% in a historical cohort. The French LYSA/GOELAMS group performed the only direct comparison in the rituximab era (4 cycles of R-CHOP followed by RT versus 4 cycles of R-CHOP followed by 2 additional cycles of R-CHOP) and reported similar outcomes between both arms,37 with OS of 92% in the R-CHOP alone arm and 96% in the R-CHOP + RT arm (nonsignificant difference statistically). IFRT alone is not recommended other than for palliation in patients who cannot tolerate chemotherapy or combined modality therapy. Stage I and II patients with bulky disease (> 10 cm) have a prognosis similar to patients with advanced DLBCL and should be treated aggressively with 6 to 8 cycles of R-CHOP with or without RT.36
For patients with advanced stage disease, a full course of R-CHOP-21 (6–8 cycles given on a 21-day cycle) is the standard of care. This approach results in OS rates of 70% and 60% at 2 and 5 years, respectively. For older adults unable to tolerate full-dose R-CHOP, attenuated versions of R-CHOP with decreased dose density or decreased dose intensity have been developed.38 Numerous randomized trials have attempted to improve upon the results of R-CHOP-21 using strategies such as infusional chemotherapy (DA-EPOCH-R [etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab]);39 dose-dense therapy (R-CHOP-14);replacement of rituximab with obinutuzuimab;40 addition of novel agents such as bortezomib,41 lenalidomide,42 or ibrutinib43,44 to R-CHOP; and various maintenance strategies such as rituximab, lenalidomide,45 enzastaurin,46 and everolimus.47 Unfortunately, none of these strategies has been shown to improve OS in DLBCL. In part this appears to be due to the fact that inclusion/exclusion criteria for DLBCL trials have been too strict, such that the most severely ill DLBCL patients are typically not included. As a result, the results in the control arms have ended up better than what was expected based on historical data. Efforts are underway to include all patients in future first-line DLBCL studies.
Currently, autologous hematopoietic cell transplantation (auto-HCT) is not routinely used in the initial treatment of DLBCL. In the pre-rituximab era, numerous trials were conducted in DLBCL patients with high and/or high-intermediate risk disease based on the IPI score to determine if outcomes could be improved with high-dose therapy and auto-HCT as consolidation after patients achieved complete remission with first-line therapy. The results of these trials were conflicting. A 2003 meta-analysis of 11 such trials concluded that the results were very heterogeneous and showed no OS benefit.48 More recently, the Southwestern Oncology Group published the results of a prospective trial testing the impact of auto-HCT for consolidation of aggressive NHL patients with an IPI score of 3 to 5 who achieved complete remission with first-line therapy with CHOP or R-CHOP. In this study, 75% of the patients had DLBCL and, of the B-cell NHL patients, 47% received R-CHOP. A survival benefit was seen only in the subgroup that had an IPI score of 4 or 5; a subgroup analysis restricted to those receiving R-CHOP as induction was not performed, however.49 As a result, this area remains controversial, with most institutions not routinely performing auto-HCT for any DLBCL patients in first complete remission and some institutions considering auto-HCT in first complete remission for patients with an IPI score of 4 or 5. These studies all used the IPI score to identify high-risk patients. It is possible that the use of newer biomarkers or minimal-residual disease analysis will lead to a more robust algorithm for identifying high-risk patients and selecting patients who might benefit from consolidation of first complete remission with auto-HCT.
For patients with DHL or THL, long-term PFS with standard R-CHOP therapy is poor (20% to 40%).50,51 Treatment with more intensive first-line regimens such as DA-EPOCH-R, R-hyperCVAD (rituximab plus hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone), or CODOX-M/IVAC±R (cyclophosphamide, vincristine, doxorubicin, high‐dose methotrexate/ifosfamide, etoposide, high‐dose cytarabine ± rituximab), along with CNS prophylaxis, however, has been shown to produce superior outcomes,52 with 3-year relapse-free survival rates of 88% compared to 56% for R-CHOP. For patients who achieve a complete response by PET/CT scan after intensive induction, consolidation with auto-HCT has not been shown to improve outcomes based on retrospective analysis. However for DHL/THL patients who achieve complete response after R-CHOP, PFS was improved if auto-HCT was given as consolidation of first remission.53
Patients with DLBCL have an approximately 5% risk of subsequently developing CNS involvement. Historically (in the pre-rituximab era), patients who presented with multiple sites of extranodal disease and/or extensive bone marrow involvement and/or an elevated LDH had an increased risk (up to 20%–30%) of developing CNS involvement. In addition, patients with involvement of certain anatomical sites (testicular, paranasal sinuses, epidural space) had an increased risk of CNS disease. Several algorithms have been proposed to identify patients who should receive prophylactic CNS therapy. One of the most robust tools for this purpose is the CNS-IPI, which is a 6-point score consisting of the 5 IPI elements, plus 1 additional point if the adrenal glands or kidneys are involved. Importantly, the CNS-IPI was developed and validated in patients treated with R-CHOP-like therapy. Subsequent risk of CNS relapse was 0.6%, 3.4%, and 10.2% for those with low-, intermediate- and high-risk CNS-IPI scores, respectively.54 A reasonable strategy, therefore, is to perform CNS prophylaxis in those with a CNS-IPI score of 4 to 6. When CNS prophylaxis is used, intrathecal methotrexate or high-dose systemic methotrexate is most frequently given, with high-dose systemic methotrexate favored over intrathecal chemotherapy given that high-dose methotrexate penetrates the brain and spinal cord parenchyma, in addition to treating the cerebrospinal fluid (CSF).55 In contrast, intrathecal therapy only treats the CSF and requires repeated lumbar punctures or placement of an Ommaya reservoir. For DLBCL patients who present with active CSF involvement (known as lymphomatous meningitis), intrathecal chemotherapy treatments are typically given 2 or 3 times weekly until the CSF clears, followed by weekly intrathecal treatment for 4 weeks, and then monthly intrathecal treatment for 4 months.56 For those with concurrent systemic and brain parenchymal DLBCL, a strategy of alternating R-CHOP with mid-cycle high-dose methotrexate can be successful. In addition, consolidation with high-dose therapy and auto-HCT improved survival in such patients in 1 retrospective series.57
Relapsed/Refractory Disease
Between 30% and 40% of patients with advanced stage DLBCL will either fail to attain a remission with primary therapy (referred to as primary induction failure) or will relapse. In general, for those with progressive or relapsed disease, an updated tissue biopsy is recommended. This is especially true for patients who have had prior complete remission and have new lymph node enlargement, or those who have emergence of new sites of disease at the completion of first-line therapy.
Patients with relapsed disease are treated with systemic second-line platinum-based chemoimmunotherapy, with the usual goal of ultimately proceeding to auto-HCT. A number of platinum-based regimens have been used in this setting such as R-ICE, R-DHAP, R-GDP, R-Gem-Ox, and R-ESHAP. None of these regimens has been shown to be superior in terms of efficacy, and the choice of regimen is typically made based on the anticipated tolerance of the patient in light of comorbidities, laboratory studies, and physician preference. In the CORAL study, R-DHAP (rituximab, dexamethasone, high-dose cytarabine, cisplatin) seemed to show superior PFS in patients with the GCB subtype.58 However, this was an unplanned subgroup analysis and R-DHAP was associated with higher renal toxicity.
Several studies have demonstrated that long-term PFS can be observed for relapsed/refractory DLBCL patients who respond to second-line therapy and then undergo high-dose therapy with auto-HCT. The Parma trial remains the only published prospective randomized trial performed in relapsed DLBCL comparing a transplant strategy to a non-transplant strategy. This study, performed in the pre-rituximab era, clearly showed a benefit in terms of DFS and OS in favor of auto-HCT versus salvage therapy alone.59 The benefit of auto-HCT in patients treated in the rituximab era, even in patients who experience early failure (within 1 year of diagnosis), was confirmed in a retrospective analysis by the Center for International Blood and Marrow Transplant Research. In this study, a 44% 3-year PFS was seen in the early failure cohort versus 52% in the late failure cohort.60
Some DLBCL patients are very unlikely to benefit from auto-HCT. The REFINE study focused on patients with primary induction failure or early relapse within 6 months of completing first-line therapy. Among such patients, primary progressive disease (defined as progression while still receiving first-line therapy), a high NCCN-IPI score at relapse, and MYC rearrangement were risk factors for poor PFS following auto-HCT.61 Patients with 2 or 3 high-risk features had a 2-year OS of 10.7% compared to 74.3% for those without any high-risk features.
Allogeneic HCT (allo-HCT) is a treatment option for relapsed/refractory DLBCL. This option is more commonly considered for patients in whom an autotransplant has failed to achieve durable remission. For properly selected patients in this setting, a long-term PFS in the 30% to 40% range can be attained.62 However, in practice, only about 20% of patients who fail auto-HCT end up undergoing allo-HCT due to rapid progression of disease, age, poor performance status, or lack of suitable donor. It has been proposed that in the coming years, allo-HCT will be utilized less commonly in this setting due to the advent of chimeric antigen receptor T-cell (CAR T) therapy.
CAR T-cell therapy genetically modifies the patient’s own T lymphocytes with a gene that encodes an antigen receptor to direct the T cells against lymphoma cells. Typically, the T cells are genetically modified and expanded in a production facility and then infused back into the patient. Axicabtagene ciloleucel is directed against the CD-19 receptor and has been approved by the US Food and Drug Administration (FDA) for treatment of patients with DLBCL who have failed 2 or more lines of systemic therapy. Use of CAR-T therapy in such patients was examined in a multicenter trial (ZUMA-1), which reported a 54% complete response rate and 52% OS rate at 18 months.63 CAR-T therapy is associated with serious side effects such as cytokine release syndrome, neurological toxicities, and prolonged cytopenias. While there are now some patients with ongoing remission 2 or more years after undergoing CAR-T therapy, it remains uncertain what proportion of patients have been truly cured with this modality. Nevertheless, this new treatment option remains a source of optimism for relapsed and refractory DLBCL patients.
Primary Mediastinal Large B-Cell Lymphoma
Primary mediastinal large B-cell lymphoma (PMBCL) is a form of DLBCL arising in the mediastinum from the thymic B cell. It is an uncommon entity and has clinical and pathologic features distinct from systemic DLBCL.64 PMBCL accounts for 2% of all NHLs and about 7% of all DLBCL.20 It typically affects women in the third to fourth decade of life.
Presentation and Prognostic Features
PMBCL usually presents as a locally invasive anterior mediastinal mass, often with a superior vena cava syndrome which may or may not be clinically obvious.64 Other presentations include pericardial tamponade, thrombosis of neck veins, and acute airway obstruction. About 80% of patients present with bulky (> 10 cm) stage I or II disease,65 with distant spread uncommon on presentation. Morphologically and on GEP, PMBL has a profile more similar to classical Hodgkin lymphoma (cHL) than non-mediastinal DLBCL.66 PMBL is distinguished from cHL by immunophenotyping: unlike cHL, PMBCL has pan B cell markers, rarely expresses CD15, and has weak CD30.
Poor prognostic features in PMBCL are Eastern Cooperative Oncology Group (ECOG) performance status greater than 2, pericardial effusion, bulky disease, and elevated serum LDH. The diagnosis of PMBCL can be difficult because the tumor is often encased with extensive fibrosis and necrosis. As a result, a needle biopsy may not yield sufficient tissue, thus making a surgical biopsy often the only viable way to obtain sufficient tissue.
Treatment
Early series suggested that PMBCL is unusually aggressive, with a poor prognosis.67 This led to studies using more aggressive chemotherapy regimens (often in combination with mediastinal radiation) as well as upfront auto-HCT.68–70 The addition of rituximab to treatment regimens significantly improved outcomes in PMBCL. For example, a subgroup analysis of the PMBCL patients in the MinT trial revealed a 3-year event-free survival (EFS) of 78%71 when rituximab was combined with CHOP. Because of previous reports demonstrating radiosensitivity of PMBL, radiation was traditionally sequenced into treatment regimens for PMBL. However, this is associated with higher long-term toxicities, often a concern in PMBCL patients given that the disease frequently affects younger females, and given that breast tissue will be in the radiation field. For patients with a strong personal or family history of breast cancer or cardiovascular disease, these concerns are even more significant. More recently, the DA-EPOCH-R regimen has been shown to produce very high rates (80%–90%) of long-term DFS, without the need for mediastinal radiation in most cases.72,73 For patients receiving R-CHOP, consolidation with mediastinal radiation is still commonly given. This approach also leads to high rates of long-term remission and, although utilizing mediastinal radiation, allows for less intensive chemotherapy. Determining which approach is most appropriate for an individual patient requires an assessment of the risks of each treatment option for that patient. A randomized trial by the International Extranodal Lymphoma Study Group (IELSG37) is evaluating whether RT may be safely omitted in PMBCL patients who achieve a complete metabolic response after R-CHOP.
Most relapses of PMBCL occur within the first 1 to 2 years and often present with extranodal disease in various organs. For those with relapsed or refractory disease, high-dose chemotherapy followed by auto-HCT provides 5-year survival rates of 50% to 80%.74–76 In a phase 1b trial evaluating the role of pembrolizumab in relapsed/refractory patients (KEYNOTE-13), 7 of 17 PMBCL patients achieved responses, with an additional 6 demonstrating stable disease.77 This provides an additional option for patients who might be too weak to undergo auto-HCT or for those who relapse following auto-HCT.
Mantle Cell Lymphoma
The name mantle cell lymphoma (MCL) is based on the presumed normal cell counterpart to MCL, which is believed to be found in the mantle zone surrounding germinal center follicles. It represents approximately 6% of all NHL cases in the United States and Europe.78 MCL occurs at a median age of 63 to 68 years and has a male predominance.
Presentation and Prognostic Features
Patients can present with a broad spectrum of clinical features, and most patients (70%) present with advanced disease.79 Up to one third of patients have B symptoms, with most demonstrating lymphadenopathy and bone marrow involvement. Approximately 25% present with extranodal disease as the primary presentation (eg, GI tract, pleura, breast, or orbits). MCL can involve any part of the GI tract and often presents as polypoid lesions.
Histologically, the pattern of MCL may be diffuse, nodular, mantle zone, or a combination of the these; morphologically, MCL can range from small, more irregular lymphocytes to lymphoblast-like cells. Blastoid and pleomorphic variants of MCL have a higher proliferation index and a more aggressive clinical course than other variants. MCL is characterized by the expression of pan B cell antigens (CD19+, CD20+) with coexpression of the T-cell antigen CD5, lack of CD23 expression, and nuclear expression of cyclin D1. Nuclear staining for cyclin D1 is present in more than 98% of cases.80 In rare cases, CD5 or cyclin D1 may be negative.80 Most MCL cases have a unique translocation that fuses the immunoglobulin heavy chain gene promoter (14q32) to the promoter of the BCL-1 gene (11q13), which encodes the cyclin D1 protein. This translocation is not unique to MCL and can be present in multiple myeloma as well. Interestingly, cyclin D1 is overproduced in cases lacking t(11:14), likely from other point mutations resulting in its overexpression.81 Cyclin D1–negative tumors overexpress cyclin D2 or D3, with no apparent difference in clinical behavior or outcome.82 In cyclin D1–negative cases, SOX11 expression may help with diagnosis.83 A proliferation rate greater than 30% (as measured by Ki-67 staining), low SOX11 expression, and presence of p53 mutations have all been associated with adverse outcome.
In a minority of cases, MCL follows an indolent clinical course. For the remainder, however, MCL is an aggressive disease that generally requires treatment soon after diagnosis. When initially described in the 1980s and 1990s, treatment of MCL was characterized by low complete response rates, short durations of remission, repeated recurrences, and a median survival in the 2- to 5-year range.84 In recent years, intensive regimens incorporating rituximab and high-dose cytarabine with or without auto-HCT have been developed and are associated with high complete response rates and median duration of first remission in the 6- to 9-year range.85–87 Several prognostic indices have been applied to patients with MCL, including the IPI, the Follicular Lymphoma International Prognostic Index , and the Mantle Cell Lymphoma International Prognostic Index (MIPI). The MIPI was originally described based on a cohort from the period 1996 to 2004,88 and subsequently confirmed in a separate cohort of 958 patients with MCL treated on prospective trials between 2004 and 2010.89 The MIPI score can identify 3 risk groups with significant survival differences (83%, 63%, and 34% survival at 5 years). A refined version of the MIPI score, the combined MIPI or MIPI-c, incorporates proliferation rate and is better able to stratify patients.90 The blastoid variant of MCL follows a more aggressive clinical course and is associated with a high proliferation rate, shorter remissions, and a higher rate of CNS involvement.91
In most patients, MCL is an aggressive disease with a short OS without treatment. A subset of patients may have a more indolent course,92 but unfortunately reliable factors that identify this group at the time of diagnosis are not available. Pretreatment evaluation is as with other lymphomas, with lumbar puncture and MRI of the brain also recommended for patients with the blastoid variant. For those presenting with GI symptoms, endoscopy is recommended as part of the initial evaluation as well.
Treatment
First-line Therapy
For patients under age 65 to 70 years with a good performance status and few comorbidities, an intensive induction regimen (such as R-CHOP/R-DHAP, Maxi-R-CHOP/R-araC, or R-DHAP) followed by consolidation with auto-HCT is commonly given, with a goal of achieving a durable (6–9 year) first remission.87,93,94 Auto-HCT is now routinely followed by 3 years of maintenance rituximab based on the survival benefit seen in the recent LYSA trial.93 At many centers, auto-HCT in first remission is a standard of care, with the greatest benefit seen in patients who have achieved a complete remission with no more than 2 lines of chemotherapy.95 However, there remains some controversy about whether all patients truly benefit from auto-HCT in first remission, and current research efforts are focused on identifying patients most likely to benefit from auto-HCT and incorporation of new agents into first-line regimens. For patients who are not candidates for auto-HCT, bendamustine plus rituximab (BR) or R-CHOP alone or followed by maintenance rituximab is a reasonable approach.96 Based on the StiL and BRIGHT trials, BR seems to have less toxicity and higher rates of response with no difference in OS when compared to R-CHOP.97,98
In summary, dose-intense induction chemotherapy with consolidative auto-HCT results in high rates of long-term remission and can be considered in MCL patients who lack significant comorbidities and who understand the risks and benefits of this approach. For other patients, the less aggressive frontline approaches are more appropriate.
Relapsed/Refractory Disease
Despite initial high response rates, most patients with MCL will eventually relapse. For example, most patients given CHOP or R-CHOP alone as first-line therapy will relapse within 2 years.99 In recent years, a number of therapies have emerged for relapsed/refractory MCL; however, the optimal sequencing of these is unclear. FDA-approved options for relapsed/refractory MCL include the proteasome inhibitor bortezomib,100,101 the BTK inhibitors ibrutinib102,103 and acalabrutinib,104 and the immunomodulatory agent lenalidomide.105
Auto-HCT can be considered for patients who did not undergo auto-HCT as part of first-line therapy and who had a reasonably long first remission.95 Allo-HCT has curative potential in MCL with good evidence of a graft-versus-lymphoma effect. With a matched related or matched unrelated donor, the chance for treatment-related mortality is 15% to 25% at 1 to 2 years, with a 50% to 60% chance for long-term PFS. However, given the risk of treatment-related mortality and graft-versus-host disease, this option is typically reserved for patients with early relapse after auto-HCT, multiple relapses, or relatively chemotherapy-unresponsive disease.95,106 A number of clinical trials for relapsed/refractory MCL are ongoing, and participation in these is encouraged whenever possible.
Burkitt Lymphoma
Burkitt lymphoma is a rare, aggressive and highly curable subtype of NHL. It can occur at any age, although peak incidence is in the first decade of life. There are 3 distinct clinical forms of Burkitt lymphoma.107 The endemic form is common in African children and commonly involves the jaw and kidneys. The sporadic (nonendemic) form accounts for 1% to 2% of all lymphomas in the United States and Western Europe and usually has an abdominal presentation. The immunodeficiency-associated form is commonly seen in HIV patients with a relatively preserved CD4 cell count.
Patients typically present with rapidly growing masses and tumor lysis syndrome. CNS and bone marrow involvement are common. Burkitt lymphoma cells are high-grade, rapidly proliferating medium-sized cells with a monomorphic appearance. Biopsies show a classic histological appearance known as a “starry sky pattern” due to benign macrophages engulfing debris resulting from apoptosis. It is derived from a germinal center B cell and has distinct oncogenic pathways. Translocations such as t(8;14), t(2;8) or t(8;22) juxtapose the MYC locus with immunoglobulin heavy or light chain loci and result in MYC overexpression. Burkitt lymphoma is typically CD10-positive and BCL-2-negative, with a MYC translocation and a proliferation rate greater than 95%.
With conventional NHL regimens, Burkitt lymphoma had a poor prognosis, with complete remission in the 30% to 70% range and low rates of long-term remission. With the introduction of short-term, dose-intensive, multiagent chemotherapy regimens (adapted from pediatric acute lymphoblastic leukemia [ALL] regimens), the complete remission rate improved to 60% to 90%.107 Early stage disease (localized or completely resected intra-abdominal disease) can have complete remission rates of 100%, with 2- to 5-year freedom-from-progression rates of 95%. CNS prophylaxis, including high-dose methotrexate, high-dose cytarabine, and intrathecal chemotherapy, is a standard component of Burkitt lymphoma regimens (CNS relapse rates can reach 50% without prophylactic therapy). Crucially, relapse after 1 to 2 years is very rare following complete response to induction therapy. Classically, several intensive regimens have been used for Burkitt lymphoma. In recent years, the most commonly used regimens have been the modified Magrath regimen of R-CODOX-M/IVAC and R-hyperCVAD. DA-EPOCH-R has also been used, typically for older, more frail, or HIV-positive patients. However, at the American Society of Hematology 2017 annual meeting, results from the NCI 9177 trial were presented which validated, in a prospective multi-center fashion, the use of DA-EPOCH-R in all Burkitt lymphoma patients.108 In NCI 9177, low-risk patients (defined as normal LDH, ECOG performance score 0 or 1, ≤ stage II, and no tumor lesion > 7 cm) received 2 cycles of DA-EPOCH-R without intrathecal therapy followed by PET. If interim PET was negative, low-risk patients then received 1 more cycle of DA-EPOCH-R. High-risk patients with negative brain MRI and CSF cytology/flow cytometry received 2 cycles of DA-EPOCH-R with intrathecal therapy (2 doses per cycle) followed by PET. Unless interim PET showed progression, high-risk patients received 4 additional cycles of DA-EPOCH-R including methotrexate 12 mg intrathecally on days 1 and 5 (8 total doses). With a median follow-up of 36 months, this regimen resulted in an EFS of 85.7%. As expected, patients with CNS, marrow, or peripheral blood involvement fared worse. For those without CNS, marrow, or peripheral blood involvement, the results were excellent, with an EFS of 94.6% compared to 62.8% for those with CNS, bone marrow, or blood involvement at diagnosis.
Although no standard of care has been defined, patients with relapsed/refractory Burkitt lymphoma are often given standard second-line aggressive NHL regimens (eg, R-ICE); for those with chemosensitive disease, auto- or allo-HCT is often pursued, with long-term remissions possible following HCT.109
Lymphoblastic Lymphoma
Lymphoblastic lymphoma (LBL) is a rare disease postulated to arise from precursor B or T lymphoblasts at varying stages of differentiation. Accounting for approximately 2% of all NHLs, 85% to 90% of all cases have a T-cell phenotype, while B-cell LBL comprises approximately 10% to 15% of cases. LBL and ALL are thought to represent the same disease entity, but LBL has been arbitrarily defined as cases with lymph node or mediastinal disease. Those with significant (> 25%) bone marrow or peripheral blood involvement are classified as ALL.
Precursor T-cell LBL patients are usually adolescent and young males who commonly present with a mediastinal mass and peripheral lymphadenopathy. Precursor B-cell LBL patients are usually older (median age 39 years) with peripheral lymphadenopathy and extranodal involvement. Mediastinal involvement with B-cell LBL is uncommon, and there is no male predominance. LBL has a propensity for dissemination to the bone marrow and CNS.
Morphologically, the tumor cells are medium sized, with a scant cytoplasm and finely dispersed chromatin. Mitotic features and apoptotic bodies are present since it is a high-grade malignancy. The lymphoblasts are typically positive for CD7 and either surface or cytoplasmic CD3. Terminal deoxynucleotidyl transferase expression is a defining feature. Other markers such as CD19, CD22, CD20, CD79a, CD45, and CD10 are variably expressed. Poor prognostic factors in T-cell LBL are female gender, age greater than 35 years, complex cytogenetics, and lack of a matched sibling donor.
Regimens for LBL are based on dose-dense, multi-agent protocols used in ALL. Most of these regimens are characterized by intensive remission-induction chemotherapy, CNS prophylaxis, a phase of consolidation therapy, and a prolonged maintenance phase, often lasting for 12 to 18 months with long-term DFS rates of 40% to 70%.110,111 High-dose therapy with auto-HCT or allo-HCT in first complete response has been evaluated in an attempt to reduce the incidence of relapse.112 However, the intensity of primary chemotherapy appears to be a stronger determinant of long-term survival than the use of HCT as consolidation. As a result, HCT is not routinely applied to patients in first complete remission following modern induction regimens. After relapse, prognosis is poor, with median survival rates of 6 to 9 months with conventional chemotherapy, although long-term survival rates of 30% and 20%, respectively, are reported after HCT in relapsed and primary refractory disease.113
Treatment options in relapsed disease are limited. Nelarabine can produce responses in up to 40% of relapsed/refractory LBL/ALL patients.114
Summary
Aggressive NHLs are characterized by rapid clinical progression without therapy. However, a significant proportion of patients are cured with appropriate combination chemotherapy or combined modality (chemotherapy + RT) regimens. In contrast, the indolent lymphomas have a relatively good prognosis (median survival of 10 years or longer) but usually are not curable in advanced clinical stages. Overall 5-year survival for aggressive NHLs with current treatment is approximately 50% to 60%, with relapses typically occurring within the first 5 years. Treatment strategies for relapsed patients offer some potential for cure; however, clinical trial participation should be encouraged whenever possible to investigate new approaches for improving outcomes in this patient population.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.
1. Swerdlow, SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon, France: World Health Organization; 2017.
2. Surveillance, Epidemiology, and End Results (SEER) Program. www.seer.cancer.gov. Research Data 2017.
3. Boffetta P, de Vocht F. Occupation and the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev 2007;16:369–72.
4. Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol 2001;112:863–73.
5. Ekstrom Smedby K, Vajdic CM, Falster M, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 2008;111:4029–38.
6. Clarke CA, Morton LM, Lynch C, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer 2013;109:280–8.
7. Wang SS, Slager SL, Brennan P, et al. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10 211 cases and 11 905 controls from the International Lymphoma Epidemiology Consortium (InterLymph). Blood 2007;109:3479–88.
8. Dong C, Hemminki K. Second primary neoplasms among 53 159 haematolymphoproliferative malignancy patients in Sweden, 1958–1996: a search for common mechanisms. Br J Cancer 2001;85:997–1005.
9. Hummel M, Anagnostopoulos I, Korbjuhn P, Stein H. Epstein-Barr virus in B-cell non-Hodgkin’s lymphomas: unexpected infection patterns and different infection incidence in low- and high-grade types. J Pathol 1995;175:263–71.
10. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 1995;332:1186–91.
11. Viswanatha DS, Dogan A. Hepatitis C virus and lymphoma. J Clin Pathol 2007;60:1378–83.
12. Engels EA, Cho ER, Jee SH. Hepatitis B virus infection and risk of non-Hodgkin lymphoma in South Korea: a cohort study. Lancet Oncol 2010;11:827–34.
13. Marcucci F, Mele A. Hepatitis viruses and non-Hodgkin lymphoma: epidemiology, mechanisms of tumorigenesis, and therapeutic opportunities. Blood 2011;117:1792–8.
14. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
15. Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47.
16. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin 2016;66:443–59.
17. Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood 2006;107:265–76.
18. Møller MB, Pedersen NT, Christensen BE. Diffuse large B-cell lymphoma: clinical implications of extranodal versus nodal presentation--a population-based study of 1575 cases. Br J Haematol 2004;124:151–9.
19. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
20. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood 1997;89:3909–18.
21. Sehn LH, Scott DW, Chhanabhai M, et al. Impact of concordant and discordant bone marrow involvement on outcome in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2011;29:1452–7.
22. Fisher DE, Jacobson JO, Ault KA, Harris NL. Diffuse large cell lymphoma with discordant bone marrow histology. Clinical features and biological implications. Cancer 1989;64:1879–87.
23. Yao Z, Deng L, Xu-Monette ZY, et al. Concordant bone marrow involvement of diffuse large B-cell lymphoma represents a distinct clinical and biological entity in the era of immunotherapy. Leukemia 2018;32:353–63.
24. Gascoyne RD, Adomat SA, Krajewski S, et al. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin’s lymphoma. Blood 1997;90:244–51.
25. Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999;30:803–8.
26. Chisholm KM, Bangs CD, Bacchi CE, et al. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. Am J Surg Pathol 2015;39:294–303.
27. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 2014;123:837–42.
28. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–82.
29. Horn H, Ziepert M, Becher C, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–63.
30. Barrans S, Crouch S, Smith A, et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–5.
31. Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–31.
32. Green TM, Young KH, Visco C, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3460–7.
33. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med 1993;328:1002–6.
34. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol 2011;12:1013–22.
35. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–42.
36. Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol 2008;26:2258–63.
37. Lamy T, Damaj G, Soubeyran P, et al. R-CHOP 14 with or without radiotherapy in nonbulky limited-stage diffuse large B-cell lymphoma. Blood 2018;131:174–81.
38. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol 2011;12:460–8.
39. Wilson WH, sin-Ho J, Pitcher BN, et al. Phase III randomized study of R-CHOP versus DA-EPOCH-R and molecular analysis of untreated diffuse large B-cell lymphoma: CALGB/Alliance 50303. Blood 2016;128:469 LP-469. 38.
40. Vitolo U, Trne˘ný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3529–37.
41. Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol 2017;35:3538–46.
42. Nowakowski GS, LaPlant B, Macon WR, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 2015;33:251–7.
43. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014;15:1019–26.
44. Younes A, Zinzani PL, Sehn LH, et al. A randomized, double-blind, placebo-controlled phase 3 study of ibrutinib in combination with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in subjects with newly diagnosed nongerminal center B-cell subtype of diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2014;32(15_suppl):TPS8615.
45. Delarue R, Tilly H, Mounier N, et al. Dose-dense rituximab-CHOP compared with standard rituximab-CHOP in elderly patients with diffuse large B-cell lymphoma (the LNH03-6B study): a randomised phase 3 trial. Lancet Oncol 2013;14:525–33.
46. Leppä S, Fayad LE, Lee J-J, et al. A phase III study of enzastaurin in patients with high-risk diffuse large B cell lymphoma following response to primary treatment: the Prelude trial. Blood 2013;122:371 LP-371.
47. Witzig TE, Tobinai K, Rigacci L, et al. Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 2018;29:707–14.
48. Strehl J, Mey U, Glasmacher A, et al. High-dose chemotherapy followed by autologous stem cell transplantation as first-line therapy in aggressive non-Hodgkin’s lymphoma: a meta-analysis. Haematologica 2003;88:1304–15.
49. Stiff PJ, Unger JM, Cook JR, et al. Autologous transplantation as consolidation for aggressive non-Hodgkin’s lymphoma. N Engl J Med 2013;369:1681–90.
50. Oki Y, Noorani M, Lin P, et al. Double hit lymphoma: the MD Anderson Cancer Center clinical experience. Br J Haematol 2014;166:891–901.
51. Petrich AM, Gandhi M, Jovanovic B, et al. Impact of induction regimen and stem cell transplantation on outcomes in double-hit lymphoma: a multicenter retrospective analysis. Blood 2014;124:2354–61.
52. Howlett C, Snedecor SJ, Landsburg DJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol 2015;170:504–14.
53. Landsburg DJ, Falkiewicz MK, Maly J, et al. Outcomes of patients with double-hit lymphoma who achieve first complete remission. J Clin Oncol 2017;35:2260–7.
54. Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: a risk model for CNS relapse in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2016;34:3150–6.
55. Abramson JS, Hellmann M, Barnes JA, et al. Intravenous methotrexate as central nervous system (CNS) prophylaxis is associated with a low risk of CNS recurrence in high-risk patients with diffuse large B-cell lymphoma. Cancer 2010;116:4283–90.
56. Dunleavy K, Roschewski M, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: updated results of a multicenter prospective phase II study of DA-EPOCH-R. Hematol Oncol 2017;35(S2):133–4.
57. Damaj G, Ivanoff S, Coso D, et al. Concomitant systemic and central nervous system non-Hodgkin lymphoma: the role of consolidation in terms of high dose therapy and autologous stem cell transplantation. A 60-case retrospective study from LYSA and the LOC network. Haematologica 2015;100:1199–206.
58. Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol 2011;29:4079–87.
59. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with dalvage vhemotherapy in relapses of chemotherapy-densitive non-Hodgkin’s lymphoma. N Engl J Med 1995;333:1540–5.
60. Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2014;20:1729–36.
61. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: Ultra-high risk features and benchmarking for experimental therapies. Am J Hematol 2017;92:e24615.
62. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol 2016;174:235–48.
63. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44.
64. van Besien K, Kelta M, Bahaguna P. Primary mediastinal B-cell lymphoma: a review of pathology and management. J Clin Oncol 2001;19:1855–64.
65. Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol Off J Eur Soc Med Oncol 2006;17:123–30.
66. Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851–62.
67. Lavabre-Bertrand T, Donadio D, Fegueux N, et al. A study of 15 cases of primary mediastinal lymphoma of B-cell type. Cancer 1992;69:2561–6.
68. Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol 1997;15:1646–53.
69. Zinzani PL, Martelli M, Magagnoli M, et al. Treatment and clinical management of primary mediastinal large B-cell lymphoma with sclerosis: MACOP-B regimen and mediastinal radiotherapy monitored by (67)Gallium scan in 50 patients. Blood 1999;94:3289–93.
70. Todeschini G, Secchi S, Morra E, et al. Primary mediastinal large B-cell lymphoma (PMLBCL): long-term results from a retrospective multicentre Italian experience in 138 patients treated with CHOP or MACOP-B/VACOP-B. Br J Cancer 2004;90:372–6.
71. Rieger M, Osterborg A, Pettengell R, et al. Primary mediastinal B-cell lymphoma treated with CHOP-like chemotherapy with or without rituximab: results of the Mabthera International Trial Group study. Ann Oncol Off J Eur Soc Med Oncol 2011;22:664–70.
72. Shah NN, Szabo A, Huntington SF, et al. R-CHOP versus dose-adjusted R-EPOCH in frontline management of primary mediastinal B-cell lymphoma: a multi-centre analysis. Br J Haematol 2018;180:534–44.
73. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013;368:1408–16.
74. Aoki T, Shimada K, Suzuki R, et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J 2015;5:e372–e372.
75. Sehn LH, Antin JH, Shulman LN, et al. Primary diffuse large B-cell lymphoma of the mediastinum: outcome following high-dose chemotherapy and autologous hematopoietic cell transplantation. Blood 1998;91:717–23.
76. Kuruvilla J, Pintilie M, Tsang R, et al. Salvage chemotherapy and autologous stem cell transplantation are inferior for relapsed or refractory primary mediastinal large B-cell lymphoma compared with diffuse large B-cell lymphoma. Leuk Lymphoma 2008;49:1329–36.
77. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017;130:267–70.
78. Smith A, Howell D, Patmore R, et al. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer 2011;105:1684–92.
79. Argatoff LH, Connors JM, Klasa RJ, et al. Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997;89:2067–78.
80. Zukerberg LR, Yang WI, Arnold A, Harris NL. Cyclin D1 expression in non-Hodgkin’s lymphomas. Detection by immunohistochemistry. Am J Clin Pathol 1995;103:756–60.
81. Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007;109:4599–606.
82. Fu K, Weisenburger DD, Greiner TC, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood 2005;106:4315–21.
83. Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009;94:1555–62.
84. Norton AJ, Matthews J, Pappa V, et al. Mantle cell lymphoma: Natural history defined in a serially biopsied population over a 20-year period. Ann Oncol 1995;6:249–56.
85. Chihara D, Cheah CY, Westin JR, et al. Rituximab plus hyper-CVAD alternating with MTX/Ara-C in patients with newly diagnosed mantle cell lymphoma: 15-year follow-up of a phase II study from the MD Anderson Cancer Center. Br J Haematol 2016;172:80–8.
86. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
87. Eskelund CW, Kolstad A, Jerkeman M, et al. 15-year follow-up of the Second Nordic Mantle Cell Lymphoma trial (MCL2): prolonged remissions without survival plateau. Br J Haematol 2016;175:410–8.
88. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
89. Hoster E, Klapper W, Hermine O, et al. Confirmation of the mantle-cell lymphoma International Prognostic Index in randomized trials of the European Mantle-Cell Lymphoma Network. J Clin Oncol 2014;32:1338–46.
90. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: Results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol 2016;34:1386–94.
91. Bernard M, Gressin R, Lefrère F, et al. Blastic variant of mantle cell lymphoma: a rare but highly aggressive subtype. Leukemia 2001;15:1785–91.
92. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
93. Le Gouill S, Thieblemont C, Oberic L, et al. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017 Sep 28;377(13):1250–60.
94. Hermine O, Hoster E, Walewski J, et al. Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet 2016;388:565–75.
95. Fenske TS, Zhang M-J, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
96. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
97. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
98. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
99. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
100. Belch A, Kouroukis CT, Crump M, et al. A phase II study of bortezomib in mantle cell lymphoma: the National Cancer Institute of Canada Clinical Trials Group trial IND.150. Ann Oncol Off J Eur Soc Med Oncol 2007;18:116–21.
101. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
102. Dreyling M, Jurczak W, Jerkeman M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016;387:770–8.
103. Wang ML, Rule S, Martin P, Goy A, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
104. Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 2018;391:659–67.
105. Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
106. Khouri IF, Lee M-S, Saliba RM, et al. Nonablative allogeneic stem-cell transplantation for advanced/recurrent mantle-cell lymphoma. J Clin Oncol 2003;21:4407–12.
107. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood 2004;104:3009–20.
108. Roschewski M, Dunleavy K, Abramson JS, et al. Risk-adapted therapy in adults with Burkitt lymphoma: results of NCI 9177, a multicenter prospective phase II study of DA-EPOCH-R. Blood American Society of Hematology;2017;130(Suppl 1):188.
109. Maramattom L V, Hari PN, Burns LJ, et al. Autologous and allogeneic transplantation for burkitt lymphoma outcomes and changes in utilization: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2013;19:173–9.
110. Zinzani PL, Bendandi M, Visani G, et al. Adult lymphoblastic lymphoma: clinical features and prognostic factors in 53 patients. Leuk Lymphoma 1996;23:577–82.
111. Thomas DA, O’Brien S, Cortes J, et al. Outcome with the hyper-CVAD regimens in lymphoblastic lymphoma. Blood 2004;104:1624–30.
112. Aljurf M, Zaidi SZA. Chemotherapy and hematopoietic stem cell transplantation for adult T-cell lymphoblastic lymphoma: current status and controversies. Biol Blood Marrow Transplant 2005;11:739–54.
113. Sweetenham JW, Santini G, Qian W, et al. High-dose therapy and autologous stem-cell transplantation versus conventional-dose consolidation/maintenance therapy as postremission therapy for adult patients with lymphoblastic lymphoma: results of a randomized trial of the European Group for Blood and Marrow Transplantation and the United Kingdom Lymphoma Group. J Clin Oncol 2001;19:2927–36.
114. Zwaan CM, Kowalczyk J, Schmitt C, et al. Safety and efficacy of nelarabine in children and young adults with relapsed or refractory T-lineage acute lymphoblastic leukaemia or T-lineage lymphoblastic lymphoma: results of a phase 4 study. Br J Haematol 2017;179:284–93.