User login
Dermatologists, rheumatologists differ in management of pediatric discoid lupus erythematosus
CHICAGO – With no consensus guidelines, rheumatologists and dermatologists have significant practice-based differences in their treatment of children with discoid lupus erythematosus, according to a survey of the two specialties.
The survey’s results from 57 pediatric dermatologists and 47 pediatric rheumatologists showed a lack of consensus between the two specialties in how to screen for systemic lupus erythematosus (SLE). The two specialties also differed in identification of which features of discoid lupus erythematosus (DLE) might predispose children to developing SLE, and in some therapy choices for DLE.
Although rare in children, DLE may develop into systemic lupus erythematosus in about 25%-30% of pediatric patients, according to Lisa Arkin, MD, who shared the survey results during a poster presentation at the World Congress of Pediatric Dermatology.
Dr. Arkin and her colleagues conducted a Web-based survey to examine differences in DLE treatment practice patterns between pediatric dermatologists and pediatric rheumatologists. They sent the survey to 292 members of the Childhood Arthritis and Rheumatology Research Alliance (CARRA), and to 200 members of the Pediatric Dermatology Research Alliance (PeDRA), and received responses from 44% of the rheumatologists and 56% of the dermatologists. Of those, 57 dermatologists and 47 rheumatologists met inclusion criteria for the study.
More than half of the respondents in each specialty had seen fewer than 10 patients with skin-limited DLE, and fewer than 10 patients with SLE and DLE, over the course of their careers, said Dr. Arkin, director of pediatric dermatology at the University of Wisconsin, Madison.
Consensus was defined by Dr. Arkin and her colleagues as greater than 70% agreement from both specialties, and 2-sided P values less than .05 showed practice differences between rheumatologists and dermatologists.
Clinicians reached a consensus that the presence of either arthritis or nephritis in a pediatric patient with DLE put the patient at high risk for SLE. Arthritis was identified as a high-risk feature by 41 of 57 dermatologists (72%) and 36 of 47 rheumatologists (77%), while nephritis was seen as a high-risk feature by 39 dermatologists (68%) and 37 rheumatologists (79%). However, said Dr. Arkin, “no other features from a list of 30 risk factors including demographics, clinical, or laboratory features achieved consensus.”
In deciding which laboratory studies to order to screen for SLE upon DLE diagnosis, 26 dermatologists (46%) and 38 rheumatologists (81%) choose a full screening panel, according to the survey results. That was a significant between-specialty difference (P less than .001). The full panel consisted of obtaining a complete blood count with differential, testing for renal and hepatic function, obtaining the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level, and doing urine studies. The panel also included testing for complements, autoantibodies including anti–double-stranded DNA, single-stranded A and B, ribonucleoprotein, anti-Smith, and antiphospholipid antibodies.
However, when individual laboratory studies were examined, several did achieve consensus for baseline screening. Those included the CBC with differential; urinalysis (but not urine protein creatinine), ESR (but not CRP); complement, renal, and hepatic function testing; and most autoantibody testing (but not antiphospholipid antibody testing). Where there were differences in likelihood to order a test, rheumatologists were more likely to order the test than dermatologists.
In deciding on initial treatment, “rheumatologists were more likely to always initiate hydroxychloroquine than dermatologists,” said Dr. Arkin. Of the rheumatologists, 49% always initiated hydroxychloroquine, compared with 14% of the dermatologists (P less than .001).
In contrast, “dermatologists were more likely to always initiate topical therapy than the rheumatologists,” said Dr. Arkin. Topical therapy was always started by 81% of the dermatologists and 33% of the rheumatologists (P less than .001).
Although the specialties differed in whether they always initiated a certain treatment, “hydroxychloroquine achieved consensus as first-line therapy,” Dr. Arkin noted, with 81% of dermatologists and 87% of rheumatologists choosing hydroxychloroquine when the survey asked for a first-line systemic therapy.
There was no consensus about which agents were best for add-on therapy. Of the dermatologists, 32% would sometimes use methotrexate, which was used by 21% of rheumatologists for refractory skin disease. Quinacrine was used as add-on therapy by 21% of dermatologists and 15% of rheumatologists. Rheumatologists were significantly more likely to add dapsone than dermatologists (28% vs. 5%, P = .002).
The survey points to the need to develop consensus guidelines in the treatment of pediatric DLE, said Dr. Arkin. “Knowledge gaps include risk factors for SLE, optimal screening, and therapy,” she explained. “Collection of robust longitudinal data will aid in developing pediatric consensus guidelines for DLE.”
Dr. Arkin had no conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
CHICAGO – With no consensus guidelines, rheumatologists and dermatologists have significant practice-based differences in their treatment of children with discoid lupus erythematosus, according to a survey of the two specialties.
The survey’s results from 57 pediatric dermatologists and 47 pediatric rheumatologists showed a lack of consensus between the two specialties in how to screen for systemic lupus erythematosus (SLE). The two specialties also differed in identification of which features of discoid lupus erythematosus (DLE) might predispose children to developing SLE, and in some therapy choices for DLE.
Although rare in children, DLE may develop into systemic lupus erythematosus in about 25%-30% of pediatric patients, according to Lisa Arkin, MD, who shared the survey results during a poster presentation at the World Congress of Pediatric Dermatology.
Dr. Arkin and her colleagues conducted a Web-based survey to examine differences in DLE treatment practice patterns between pediatric dermatologists and pediatric rheumatologists. They sent the survey to 292 members of the Childhood Arthritis and Rheumatology Research Alliance (CARRA), and to 200 members of the Pediatric Dermatology Research Alliance (PeDRA), and received responses from 44% of the rheumatologists and 56% of the dermatologists. Of those, 57 dermatologists and 47 rheumatologists met inclusion criteria for the study.
More than half of the respondents in each specialty had seen fewer than 10 patients with skin-limited DLE, and fewer than 10 patients with SLE and DLE, over the course of their careers, said Dr. Arkin, director of pediatric dermatology at the University of Wisconsin, Madison.
Consensus was defined by Dr. Arkin and her colleagues as greater than 70% agreement from both specialties, and 2-sided P values less than .05 showed practice differences between rheumatologists and dermatologists.
Clinicians reached a consensus that the presence of either arthritis or nephritis in a pediatric patient with DLE put the patient at high risk for SLE. Arthritis was identified as a high-risk feature by 41 of 57 dermatologists (72%) and 36 of 47 rheumatologists (77%), while nephritis was seen as a high-risk feature by 39 dermatologists (68%) and 37 rheumatologists (79%). However, said Dr. Arkin, “no other features from a list of 30 risk factors including demographics, clinical, or laboratory features achieved consensus.”
In deciding which laboratory studies to order to screen for SLE upon DLE diagnosis, 26 dermatologists (46%) and 38 rheumatologists (81%) choose a full screening panel, according to the survey results. That was a significant between-specialty difference (P less than .001). The full panel consisted of obtaining a complete blood count with differential, testing for renal and hepatic function, obtaining the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level, and doing urine studies. The panel also included testing for complements, autoantibodies including anti–double-stranded DNA, single-stranded A and B, ribonucleoprotein, anti-Smith, and antiphospholipid antibodies.
However, when individual laboratory studies were examined, several did achieve consensus for baseline screening. Those included the CBC with differential; urinalysis (but not urine protein creatinine), ESR (but not CRP); complement, renal, and hepatic function testing; and most autoantibody testing (but not antiphospholipid antibody testing). Where there were differences in likelihood to order a test, rheumatologists were more likely to order the test than dermatologists.
In deciding on initial treatment, “rheumatologists were more likely to always initiate hydroxychloroquine than dermatologists,” said Dr. Arkin. Of the rheumatologists, 49% always initiated hydroxychloroquine, compared with 14% of the dermatologists (P less than .001).
In contrast, “dermatologists were more likely to always initiate topical therapy than the rheumatologists,” said Dr. Arkin. Topical therapy was always started by 81% of the dermatologists and 33% of the rheumatologists (P less than .001).
Although the specialties differed in whether they always initiated a certain treatment, “hydroxychloroquine achieved consensus as first-line therapy,” Dr. Arkin noted, with 81% of dermatologists and 87% of rheumatologists choosing hydroxychloroquine when the survey asked for a first-line systemic therapy.
There was no consensus about which agents were best for add-on therapy. Of the dermatologists, 32% would sometimes use methotrexate, which was used by 21% of rheumatologists for refractory skin disease. Quinacrine was used as add-on therapy by 21% of dermatologists and 15% of rheumatologists. Rheumatologists were significantly more likely to add dapsone than dermatologists (28% vs. 5%, P = .002).
The survey points to the need to develop consensus guidelines in the treatment of pediatric DLE, said Dr. Arkin. “Knowledge gaps include risk factors for SLE, optimal screening, and therapy,” she explained. “Collection of robust longitudinal data will aid in developing pediatric consensus guidelines for DLE.”
Dr. Arkin had no conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
CHICAGO – With no consensus guidelines, rheumatologists and dermatologists have significant practice-based differences in their treatment of children with discoid lupus erythematosus, according to a survey of the two specialties.
The survey’s results from 57 pediatric dermatologists and 47 pediatric rheumatologists showed a lack of consensus between the two specialties in how to screen for systemic lupus erythematosus (SLE). The two specialties also differed in identification of which features of discoid lupus erythematosus (DLE) might predispose children to developing SLE, and in some therapy choices for DLE.
Although rare in children, DLE may develop into systemic lupus erythematosus in about 25%-30% of pediatric patients, according to Lisa Arkin, MD, who shared the survey results during a poster presentation at the World Congress of Pediatric Dermatology.
Dr. Arkin and her colleagues conducted a Web-based survey to examine differences in DLE treatment practice patterns between pediatric dermatologists and pediatric rheumatologists. They sent the survey to 292 members of the Childhood Arthritis and Rheumatology Research Alliance (CARRA), and to 200 members of the Pediatric Dermatology Research Alliance (PeDRA), and received responses from 44% of the rheumatologists and 56% of the dermatologists. Of those, 57 dermatologists and 47 rheumatologists met inclusion criteria for the study.
More than half of the respondents in each specialty had seen fewer than 10 patients with skin-limited DLE, and fewer than 10 patients with SLE and DLE, over the course of their careers, said Dr. Arkin, director of pediatric dermatology at the University of Wisconsin, Madison.
Consensus was defined by Dr. Arkin and her colleagues as greater than 70% agreement from both specialties, and 2-sided P values less than .05 showed practice differences between rheumatologists and dermatologists.
Clinicians reached a consensus that the presence of either arthritis or nephritis in a pediatric patient with DLE put the patient at high risk for SLE. Arthritis was identified as a high-risk feature by 41 of 57 dermatologists (72%) and 36 of 47 rheumatologists (77%), while nephritis was seen as a high-risk feature by 39 dermatologists (68%) and 37 rheumatologists (79%). However, said Dr. Arkin, “no other features from a list of 30 risk factors including demographics, clinical, or laboratory features achieved consensus.”
In deciding which laboratory studies to order to screen for SLE upon DLE diagnosis, 26 dermatologists (46%) and 38 rheumatologists (81%) choose a full screening panel, according to the survey results. That was a significant between-specialty difference (P less than .001). The full panel consisted of obtaining a complete blood count with differential, testing for renal and hepatic function, obtaining the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level, and doing urine studies. The panel also included testing for complements, autoantibodies including anti–double-stranded DNA, single-stranded A and B, ribonucleoprotein, anti-Smith, and antiphospholipid antibodies.
However, when individual laboratory studies were examined, several did achieve consensus for baseline screening. Those included the CBC with differential; urinalysis (but not urine protein creatinine), ESR (but not CRP); complement, renal, and hepatic function testing; and most autoantibody testing (but not antiphospholipid antibody testing). Where there were differences in likelihood to order a test, rheumatologists were more likely to order the test than dermatologists.
In deciding on initial treatment, “rheumatologists were more likely to always initiate hydroxychloroquine than dermatologists,” said Dr. Arkin. Of the rheumatologists, 49% always initiated hydroxychloroquine, compared with 14% of the dermatologists (P less than .001).
In contrast, “dermatologists were more likely to always initiate topical therapy than the rheumatologists,” said Dr. Arkin. Topical therapy was always started by 81% of the dermatologists and 33% of the rheumatologists (P less than .001).
Although the specialties differed in whether they always initiated a certain treatment, “hydroxychloroquine achieved consensus as first-line therapy,” Dr. Arkin noted, with 81% of dermatologists and 87% of rheumatologists choosing hydroxychloroquine when the survey asked for a first-line systemic therapy.
There was no consensus about which agents were best for add-on therapy. Of the dermatologists, 32% would sometimes use methotrexate, which was used by 21% of rheumatologists for refractory skin disease. Quinacrine was used as add-on therapy by 21% of dermatologists and 15% of rheumatologists. Rheumatologists were significantly more likely to add dapsone than dermatologists (28% vs. 5%, P = .002).
The survey points to the need to develop consensus guidelines in the treatment of pediatric DLE, said Dr. Arkin. “Knowledge gaps include risk factors for SLE, optimal screening, and therapy,” she explained. “Collection of robust longitudinal data will aid in developing pediatric consensus guidelines for DLE.”
Dr. Arkin had no conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
AT WCPD 2017
Key clinical point:
Major finding: The two specialties reached a consensus in identifying 2 of 30 potential high-risk features for the development of systemic lupus erythematosus.
Data source: Survey results from 57 pediatric dermatologists and 47 pediatric rheumatologists who treat children with DLE.Disclosures: Dr. Arkin had no relevant financial conflicts.
Targeted therapy may be possible for pityriasis rubra pilaris
CHICAGO – If you’ve ever found that making a diagnosis of pityriasis rubra pilaris is difficult, you’re not alone.
In a recent case series of 100 patients with a median age of 61 years, only 50 patients had an undeniable diagnosis of pityriasis rubra pilaris (PRP). Of those patients, only 26% were diagnosed at initial presentation. The mean delay to diagnosis was 29 months, and 54% required two or more biopsies (JAMA Derm. 2016 Jun 1;152[6]:670-5).
“This is one of those conditions that sometimes you’re going to follow patients and not know what it is right away,” Patricia M. Witman, MD, said at the World Congress of Pediatric Dermatology. Although eczema and contact dermatitis are common diseases where the diagnosis is missed, it’s easy to mistake pityriasis rubra pilaris for psoriasis.
“On the flip side, follicular psoriasis is easy to mistake for PRP,” said Dr. Witman, chief of the division of dermatology at Nationwide Children’s Hospital, Columbus, Ohio. “It’s an uncommon variant that occurs in only about 2.1% of all pediatric psoriasis. These patients can have keratoderma, but they have classic psoriasis on biopsy. Pediatric patients with this subtype do not typically have classic psoriasis plaques.”
PRP is an anti-inflammatory papulosquamous disease with an incidence that ranges between 1:3,500 and 1:400,000. It occurs equally in men and women and has a bimodal onset, with 52%-60% of cases occurring in the 6th or 7th decade of life, and 6%-12% occurring in the 1st or 2nd decade of life, with a mean age of 7 years. Characteristic clinical features include cephalocaudal spread, keratotic follicular papules, well-demarcated orange/red plaques, fine scale, islands of sparing, erythroderma, and palmoplantar keratoderma.
“Even though it’s characteristic, there is a wide spread in the type of disease manifestations,” Dr. Witman said. “There are six different types based on how old you are and on the presentation.”
She limited her presentation to a discussion of three types:
- Classic juvenile type. This type usually presents between ages 5 and 10 and resembles the type I, or classic adult, type, with cephalocaudal spread, follicular keratotic erythematous papules coalescing into plaques, islands of sparing, and typically keratoderma. The scalp dermatitis it causes “often has a little finer scale than the thick micaceous scale we see in psoriasis,” she noted. “Patients can have a photoaggravated presentation with relative sparing of areas protected from the sun.”
- Circumscribed juvenile type. This is the most common pediatric variant. It usually presents between the ages of 3 and 10, with a mean age of 6. It is characterized by sharply demarcated plaques on extensor elbows and knees, as well as follicular hyperkeratosis. About 70% will also have keratoderma. “The Achilles tendon involvement is considered to be fairly pathognomonic for this condition and helps differentiate it from psoriasis, as well as an orange to yellow color of the keratoderma,” Dr. Witman said.
- The atypical juvenile type, or type V. This the familial variant of PRP. It’s also the rarest, occurring in just 6.5% of cases. “It’s autosomal dominant and has incomplete penetrance and variable expression,” she said. “It typically presents in infancy or in the first few years of life. Patients with this form also tend to have a more sclerodermoid palmoplantar keratoderma and ichthyosiform features. It is typically a lifelong condition, but there have been occasional case reports of self-resolution.”
The clinical features of PRP often overlap with psoriasis.
“Although there are some things that are more pathognomonic for PRP, like the Achilles tendon involvement and the keratoderma, for psoriasis we have nail pitting, which we don’t usually see in PRP,” Dr. Witman said. “Sometimes, it’s the skin biopsy that helps us distinguish those defining features, and it may take more than one biopsy to make the diagnosis.”
Dermoscopy can also be helpful. One analysis found that dermoscopy features of PRP include multiple keratotic papules with peripheral rings of erythema that coalesce into a yellow-orange plaque, linear vessels at the periphery of papules, and papules centered on hair (J Am Acad Dermatol. 2015 Jan;72[1]:S58-9).
“Even when you have that definite diagnosis of PRP, you have to remember that PRP can be seen within the context of other disease,” Dr. Witman cautioned. “Malignancy is usually limited to our adult patients with PRP, but infection can certainly trigger PRP in our pediatric patients, most commonly streptococcus. Medication reactions, especially to the biologics, have also been reported to cause PRP-like reactions, as well as autoimmune disease.”
One such entity is referred to as “Wong-like” dermatomyositis, in which patients present with a rash that looks identical to PRP (Ped Dermatol. 2007;24[2]:155-6). “It can occur in both the juvenile and adult populations,” she said. “It has clinical and histopathologic features of PRP, yet it may precede or occur concurrently with a diagnosis of dermatomyositis.”
In 2012, a group of researchers discovered that a gain of function mutation in CARD14 leads to atypical juvenile-type PRP (Am J Hum Genet. 2012 Jul 13;91:163-70). CARD14 is a member of a protein family known as caspase recruitment domain, family member 14, which also is mutated in a variant of familial psoriasis.
“It’s a protein that’s predominantly expressed in the skin, and it’s a known activator of transcription factor nuclear factor kappa light chain enhancer in activated B cells [NFkB], which is responsible for inflammation in the epidermis,” Dr. Witman explained. “What we know is that if you have a gain of function mutation in CARD14, we think that this activates the NFkB pathway and leads to increased inflammation of the skin. The same process would be expected in cases of familial psoriasis.”
Current treatment of PRP is challenging, he said. A recent survey of patients found that 76% found emollients most effective, followed by topical steroids (50%) and salicylic acid (45%) (JAMA Derm. 2016 Jun 1;152[6]:670-5). When it came to systemic therapies, 59% found retinoids most effective, followed by methotrexate (42%) and tumor necrosis factor inhibitors (40%). Only 8% found phototherapy helpful.
Dr. Witman noted that the discovery of the CARD14 mutation as the cause of the familial variant “brings us closer to an understanding of juvenile PRP,” and to the potential for targeted therapy.
“This really raises the question: Do ustekinumab and similar drugs have a future role in the treatment of PRP?” she asked. “We do see anecdotal evidence of clearance in adults using ustekinumab, both those with and without type V PRP and a CARD14 mutation. It has been tried in adult patients, predominantly in those who have failed multiple therapies. But this is anecdotal evidence with isolated case reports. These patients did respond to dosing that is typical for psoriasis.
“At this point, it is not approved for PRP, nor is it approved in children – but it’s something to think about once we have more information,” Dr. Witman noted. “The newer biologics targeting IL[interleukin]-23 and IL-17 may hold promise for PRP, based on what we have learned in psoriasis. But at this point, the safest and most effective therapy for the different variants of PRP in our pediatric patients remains to be seen.”
Dr. Witman reported having no relevant financial disclosures.
CHICAGO – If you’ve ever found that making a diagnosis of pityriasis rubra pilaris is difficult, you’re not alone.
In a recent case series of 100 patients with a median age of 61 years, only 50 patients had an undeniable diagnosis of pityriasis rubra pilaris (PRP). Of those patients, only 26% were diagnosed at initial presentation. The mean delay to diagnosis was 29 months, and 54% required two or more biopsies (JAMA Derm. 2016 Jun 1;152[6]:670-5).
“This is one of those conditions that sometimes you’re going to follow patients and not know what it is right away,” Patricia M. Witman, MD, said at the World Congress of Pediatric Dermatology. Although eczema and contact dermatitis are common diseases where the diagnosis is missed, it’s easy to mistake pityriasis rubra pilaris for psoriasis.
“On the flip side, follicular psoriasis is easy to mistake for PRP,” said Dr. Witman, chief of the division of dermatology at Nationwide Children’s Hospital, Columbus, Ohio. “It’s an uncommon variant that occurs in only about 2.1% of all pediatric psoriasis. These patients can have keratoderma, but they have classic psoriasis on biopsy. Pediatric patients with this subtype do not typically have classic psoriasis plaques.”
PRP is an anti-inflammatory papulosquamous disease with an incidence that ranges between 1:3,500 and 1:400,000. It occurs equally in men and women and has a bimodal onset, with 52%-60% of cases occurring in the 6th or 7th decade of life, and 6%-12% occurring in the 1st or 2nd decade of life, with a mean age of 7 years. Characteristic clinical features include cephalocaudal spread, keratotic follicular papules, well-demarcated orange/red plaques, fine scale, islands of sparing, erythroderma, and palmoplantar keratoderma.
“Even though it’s characteristic, there is a wide spread in the type of disease manifestations,” Dr. Witman said. “There are six different types based on how old you are and on the presentation.”
She limited her presentation to a discussion of three types:
- Classic juvenile type. This type usually presents between ages 5 and 10 and resembles the type I, or classic adult, type, with cephalocaudal spread, follicular keratotic erythematous papules coalescing into plaques, islands of sparing, and typically keratoderma. The scalp dermatitis it causes “often has a little finer scale than the thick micaceous scale we see in psoriasis,” she noted. “Patients can have a photoaggravated presentation with relative sparing of areas protected from the sun.”
- Circumscribed juvenile type. This is the most common pediatric variant. It usually presents between the ages of 3 and 10, with a mean age of 6. It is characterized by sharply demarcated plaques on extensor elbows and knees, as well as follicular hyperkeratosis. About 70% will also have keratoderma. “The Achilles tendon involvement is considered to be fairly pathognomonic for this condition and helps differentiate it from psoriasis, as well as an orange to yellow color of the keratoderma,” Dr. Witman said.
- The atypical juvenile type, or type V. This the familial variant of PRP. It’s also the rarest, occurring in just 6.5% of cases. “It’s autosomal dominant and has incomplete penetrance and variable expression,” she said. “It typically presents in infancy or in the first few years of life. Patients with this form also tend to have a more sclerodermoid palmoplantar keratoderma and ichthyosiform features. It is typically a lifelong condition, but there have been occasional case reports of self-resolution.”
The clinical features of PRP often overlap with psoriasis.
“Although there are some things that are more pathognomonic for PRP, like the Achilles tendon involvement and the keratoderma, for psoriasis we have nail pitting, which we don’t usually see in PRP,” Dr. Witman said. “Sometimes, it’s the skin biopsy that helps us distinguish those defining features, and it may take more than one biopsy to make the diagnosis.”
Dermoscopy can also be helpful. One analysis found that dermoscopy features of PRP include multiple keratotic papules with peripheral rings of erythema that coalesce into a yellow-orange plaque, linear vessels at the periphery of papules, and papules centered on hair (J Am Acad Dermatol. 2015 Jan;72[1]:S58-9).
“Even when you have that definite diagnosis of PRP, you have to remember that PRP can be seen within the context of other disease,” Dr. Witman cautioned. “Malignancy is usually limited to our adult patients with PRP, but infection can certainly trigger PRP in our pediatric patients, most commonly streptococcus. Medication reactions, especially to the biologics, have also been reported to cause PRP-like reactions, as well as autoimmune disease.”
One such entity is referred to as “Wong-like” dermatomyositis, in which patients present with a rash that looks identical to PRP (Ped Dermatol. 2007;24[2]:155-6). “It can occur in both the juvenile and adult populations,” she said. “It has clinical and histopathologic features of PRP, yet it may precede or occur concurrently with a diagnosis of dermatomyositis.”
In 2012, a group of researchers discovered that a gain of function mutation in CARD14 leads to atypical juvenile-type PRP (Am J Hum Genet. 2012 Jul 13;91:163-70). CARD14 is a member of a protein family known as caspase recruitment domain, family member 14, which also is mutated in a variant of familial psoriasis.
“It’s a protein that’s predominantly expressed in the skin, and it’s a known activator of transcription factor nuclear factor kappa light chain enhancer in activated B cells [NFkB], which is responsible for inflammation in the epidermis,” Dr. Witman explained. “What we know is that if you have a gain of function mutation in CARD14, we think that this activates the NFkB pathway and leads to increased inflammation of the skin. The same process would be expected in cases of familial psoriasis.”
Current treatment of PRP is challenging, he said. A recent survey of patients found that 76% found emollients most effective, followed by topical steroids (50%) and salicylic acid (45%) (JAMA Derm. 2016 Jun 1;152[6]:670-5). When it came to systemic therapies, 59% found retinoids most effective, followed by methotrexate (42%) and tumor necrosis factor inhibitors (40%). Only 8% found phototherapy helpful.
Dr. Witman noted that the discovery of the CARD14 mutation as the cause of the familial variant “brings us closer to an understanding of juvenile PRP,” and to the potential for targeted therapy.
“This really raises the question: Do ustekinumab and similar drugs have a future role in the treatment of PRP?” she asked. “We do see anecdotal evidence of clearance in adults using ustekinumab, both those with and without type V PRP and a CARD14 mutation. It has been tried in adult patients, predominantly in those who have failed multiple therapies. But this is anecdotal evidence with isolated case reports. These patients did respond to dosing that is typical for psoriasis.
“At this point, it is not approved for PRP, nor is it approved in children – but it’s something to think about once we have more information,” Dr. Witman noted. “The newer biologics targeting IL[interleukin]-23 and IL-17 may hold promise for PRP, based on what we have learned in psoriasis. But at this point, the safest and most effective therapy for the different variants of PRP in our pediatric patients remains to be seen.”
Dr. Witman reported having no relevant financial disclosures.
CHICAGO – If you’ve ever found that making a diagnosis of pityriasis rubra pilaris is difficult, you’re not alone.
In a recent case series of 100 patients with a median age of 61 years, only 50 patients had an undeniable diagnosis of pityriasis rubra pilaris (PRP). Of those patients, only 26% were diagnosed at initial presentation. The mean delay to diagnosis was 29 months, and 54% required two or more biopsies (JAMA Derm. 2016 Jun 1;152[6]:670-5).
“This is one of those conditions that sometimes you’re going to follow patients and not know what it is right away,” Patricia M. Witman, MD, said at the World Congress of Pediatric Dermatology. Although eczema and contact dermatitis are common diseases where the diagnosis is missed, it’s easy to mistake pityriasis rubra pilaris for psoriasis.
“On the flip side, follicular psoriasis is easy to mistake for PRP,” said Dr. Witman, chief of the division of dermatology at Nationwide Children’s Hospital, Columbus, Ohio. “It’s an uncommon variant that occurs in only about 2.1% of all pediatric psoriasis. These patients can have keratoderma, but they have classic psoriasis on biopsy. Pediatric patients with this subtype do not typically have classic psoriasis plaques.”
PRP is an anti-inflammatory papulosquamous disease with an incidence that ranges between 1:3,500 and 1:400,000. It occurs equally in men and women and has a bimodal onset, with 52%-60% of cases occurring in the 6th or 7th decade of life, and 6%-12% occurring in the 1st or 2nd decade of life, with a mean age of 7 years. Characteristic clinical features include cephalocaudal spread, keratotic follicular papules, well-demarcated orange/red plaques, fine scale, islands of sparing, erythroderma, and palmoplantar keratoderma.
“Even though it’s characteristic, there is a wide spread in the type of disease manifestations,” Dr. Witman said. “There are six different types based on how old you are and on the presentation.”
She limited her presentation to a discussion of three types:
- Classic juvenile type. This type usually presents between ages 5 and 10 and resembles the type I, or classic adult, type, with cephalocaudal spread, follicular keratotic erythematous papules coalescing into plaques, islands of sparing, and typically keratoderma. The scalp dermatitis it causes “often has a little finer scale than the thick micaceous scale we see in psoriasis,” she noted. “Patients can have a photoaggravated presentation with relative sparing of areas protected from the sun.”
- Circumscribed juvenile type. This is the most common pediatric variant. It usually presents between the ages of 3 and 10, with a mean age of 6. It is characterized by sharply demarcated plaques on extensor elbows and knees, as well as follicular hyperkeratosis. About 70% will also have keratoderma. “The Achilles tendon involvement is considered to be fairly pathognomonic for this condition and helps differentiate it from psoriasis, as well as an orange to yellow color of the keratoderma,” Dr. Witman said.
- The atypical juvenile type, or type V. This the familial variant of PRP. It’s also the rarest, occurring in just 6.5% of cases. “It’s autosomal dominant and has incomplete penetrance and variable expression,” she said. “It typically presents in infancy or in the first few years of life. Patients with this form also tend to have a more sclerodermoid palmoplantar keratoderma and ichthyosiform features. It is typically a lifelong condition, but there have been occasional case reports of self-resolution.”
The clinical features of PRP often overlap with psoriasis.
“Although there are some things that are more pathognomonic for PRP, like the Achilles tendon involvement and the keratoderma, for psoriasis we have nail pitting, which we don’t usually see in PRP,” Dr. Witman said. “Sometimes, it’s the skin biopsy that helps us distinguish those defining features, and it may take more than one biopsy to make the diagnosis.”
Dermoscopy can also be helpful. One analysis found that dermoscopy features of PRP include multiple keratotic papules with peripheral rings of erythema that coalesce into a yellow-orange plaque, linear vessels at the periphery of papules, and papules centered on hair (J Am Acad Dermatol. 2015 Jan;72[1]:S58-9).
“Even when you have that definite diagnosis of PRP, you have to remember that PRP can be seen within the context of other disease,” Dr. Witman cautioned. “Malignancy is usually limited to our adult patients with PRP, but infection can certainly trigger PRP in our pediatric patients, most commonly streptococcus. Medication reactions, especially to the biologics, have also been reported to cause PRP-like reactions, as well as autoimmune disease.”
One such entity is referred to as “Wong-like” dermatomyositis, in which patients present with a rash that looks identical to PRP (Ped Dermatol. 2007;24[2]:155-6). “It can occur in both the juvenile and adult populations,” she said. “It has clinical and histopathologic features of PRP, yet it may precede or occur concurrently with a diagnosis of dermatomyositis.”
In 2012, a group of researchers discovered that a gain of function mutation in CARD14 leads to atypical juvenile-type PRP (Am J Hum Genet. 2012 Jul 13;91:163-70). CARD14 is a member of a protein family known as caspase recruitment domain, family member 14, which also is mutated in a variant of familial psoriasis.
“It’s a protein that’s predominantly expressed in the skin, and it’s a known activator of transcription factor nuclear factor kappa light chain enhancer in activated B cells [NFkB], which is responsible for inflammation in the epidermis,” Dr. Witman explained. “What we know is that if you have a gain of function mutation in CARD14, we think that this activates the NFkB pathway and leads to increased inflammation of the skin. The same process would be expected in cases of familial psoriasis.”
Current treatment of PRP is challenging, he said. A recent survey of patients found that 76% found emollients most effective, followed by topical steroids (50%) and salicylic acid (45%) (JAMA Derm. 2016 Jun 1;152[6]:670-5). When it came to systemic therapies, 59% found retinoids most effective, followed by methotrexate (42%) and tumor necrosis factor inhibitors (40%). Only 8% found phototherapy helpful.
Dr. Witman noted that the discovery of the CARD14 mutation as the cause of the familial variant “brings us closer to an understanding of juvenile PRP,” and to the potential for targeted therapy.
“This really raises the question: Do ustekinumab and similar drugs have a future role in the treatment of PRP?” she asked. “We do see anecdotal evidence of clearance in adults using ustekinumab, both those with and without type V PRP and a CARD14 mutation. It has been tried in adult patients, predominantly in those who have failed multiple therapies. But this is anecdotal evidence with isolated case reports. These patients did respond to dosing that is typical for psoriasis.
“At this point, it is not approved for PRP, nor is it approved in children – but it’s something to think about once we have more information,” Dr. Witman noted. “The newer biologics targeting IL[interleukin]-23 and IL-17 may hold promise for PRP, based on what we have learned in psoriasis. But at this point, the safest and most effective therapy for the different variants of PRP in our pediatric patients remains to be seen.”
Dr. Witman reported having no relevant financial disclosures.
AT WCPD 2017
Expert shares tips for spotting allergic contact dermatitis in children
CHICAGO – If severe eczema persists in a pediatric patient despite your best treatment efforts, think allergic contact dermatitis.
“Or, if your eczema patients tell you that they have a cream that’s making things worse, you should think about a contact allergen,” Catalina Matiz, MD, said at the World Congress of Pediatric Dermatology.
Allergic contact dermatitis (ACD) is a type IV delayed-type hypersensitivity reaction to haptens that come into contact with the skin. Poison ivy is a common plant-based culprit, while nickel is the most common metal allergen in adults and children. “The skin barrier also plays a role,” said Dr. Matiz of the department of dermatology at Rady Children’s Hospital–San Diego, and the University of California, San Diego. “Compared with adults, children have a thinner stratum corneum, and some haptens can penetrate the skin. Some studies suggest that patients with atopic dermatitis may have increased rates of allergic sensitization, and filaggrin mutations have been found in patients with atopic dermatitis and in patients with ACD to nickel. Filaggrin helps to aggregate the cytoskeletal proteins that form the cornified cell envelope. Without filaggrin, the skin barrier is defective.”
The top 10 pediatric allergens found in personal hygiene products across five studies in the medical literature include neomycin, balsam of Peru, fragrance mix, benzalkonium chloride, lanolin, cocamidopropyl betaine, formaldehyde, methylchloroisothiazolinone/methylisothiazolinone (MCI/MI), propylene glycol, and corticosteroids. Dr. Matiz makes it practice to patch test as a last resort. “I always try to get a history, try to improve their symptoms, and have them start avoidance first, following the preemptive avoidance list,” she said (Expert Rev Clin Immunol. 2016;12[5]:551-61).
The T.R.U.E. test includes 35 allergens. “The T.R.U.E test is a good tool, which can capture up to 70% of relevant reactions in children with the inconvenience that some of the allergens in the test are not that relevant in children, and it’s not yet [Food and Drug Administration] approved to use in children,” she noted. The comprehensive chamber test allows you to select from unlimited number of allergens, “but that’s difficult. You have to have specialized staff to help you make the cells.”
A list of the minimum 20 allergens you should test for in children and the recommended supplemental allergens depending on history and locations of their dermatitis can be found in the following article: Curr Allergy Asthma Rep 2014;14[6]:444. “I always tell patients when they come for consultations to bring in everything they’re using: their shampoos, creams, and medications, because we want to see what they’re exposed to, so we can select the right allergens and also test their own products,” Dr. Matiz said. She recommends avoiding testing for strong sensitizers such as paraphenylenediamine, in children younger than 12 years of age who don’t have a history of exposure.
Testing tips for children younger than age 5 include decreasing concentrations to half for nickel, formaldehyde, and rubber accelerators. “Don’t test for paraphenylenediamine unless there is high suspicion,” she said. “Consider removing patches by 24 hours in the very young.”
The best antidote to contact dermatitis is avoidance of the known trigger. “You want to spend a lot of time with patients and parents on this,” she advised. “Give a list of safe products to use from the American Contact Dermatitis Society’s Contact Allergen Management Program [www.contactderm.org], and provide handouts about the location and history of positive allergens [www.truetest.com].” And, she added, “make a plan of treatment and follow-up in 6 weeks.”
Dr. Matiz disclosed that she is a subinvestigator in the Clinical Evaluation of T.R.U.E Test Panel 3.3 in Children and Adolescents study.
CHICAGO – If severe eczema persists in a pediatric patient despite your best treatment efforts, think allergic contact dermatitis.
“Or, if your eczema patients tell you that they have a cream that’s making things worse, you should think about a contact allergen,” Catalina Matiz, MD, said at the World Congress of Pediatric Dermatology.
Allergic contact dermatitis (ACD) is a type IV delayed-type hypersensitivity reaction to haptens that come into contact with the skin. Poison ivy is a common plant-based culprit, while nickel is the most common metal allergen in adults and children. “The skin barrier also plays a role,” said Dr. Matiz of the department of dermatology at Rady Children’s Hospital–San Diego, and the University of California, San Diego. “Compared with adults, children have a thinner stratum corneum, and some haptens can penetrate the skin. Some studies suggest that patients with atopic dermatitis may have increased rates of allergic sensitization, and filaggrin mutations have been found in patients with atopic dermatitis and in patients with ACD to nickel. Filaggrin helps to aggregate the cytoskeletal proteins that form the cornified cell envelope. Without filaggrin, the skin barrier is defective.”
The top 10 pediatric allergens found in personal hygiene products across five studies in the medical literature include neomycin, balsam of Peru, fragrance mix, benzalkonium chloride, lanolin, cocamidopropyl betaine, formaldehyde, methylchloroisothiazolinone/methylisothiazolinone (MCI/MI), propylene glycol, and corticosteroids. Dr. Matiz makes it practice to patch test as a last resort. “I always try to get a history, try to improve their symptoms, and have them start avoidance first, following the preemptive avoidance list,” she said (Expert Rev Clin Immunol. 2016;12[5]:551-61).
The T.R.U.E. test includes 35 allergens. “The T.R.U.E test is a good tool, which can capture up to 70% of relevant reactions in children with the inconvenience that some of the allergens in the test are not that relevant in children, and it’s not yet [Food and Drug Administration] approved to use in children,” she noted. The comprehensive chamber test allows you to select from unlimited number of allergens, “but that’s difficult. You have to have specialized staff to help you make the cells.”
A list of the minimum 20 allergens you should test for in children and the recommended supplemental allergens depending on history and locations of their dermatitis can be found in the following article: Curr Allergy Asthma Rep 2014;14[6]:444. “I always tell patients when they come for consultations to bring in everything they’re using: their shampoos, creams, and medications, because we want to see what they’re exposed to, so we can select the right allergens and also test their own products,” Dr. Matiz said. She recommends avoiding testing for strong sensitizers such as paraphenylenediamine, in children younger than 12 years of age who don’t have a history of exposure.
Testing tips for children younger than age 5 include decreasing concentrations to half for nickel, formaldehyde, and rubber accelerators. “Don’t test for paraphenylenediamine unless there is high suspicion,” she said. “Consider removing patches by 24 hours in the very young.”
The best antidote to contact dermatitis is avoidance of the known trigger. “You want to spend a lot of time with patients and parents on this,” she advised. “Give a list of safe products to use from the American Contact Dermatitis Society’s Contact Allergen Management Program [www.contactderm.org], and provide handouts about the location and history of positive allergens [www.truetest.com].” And, she added, “make a plan of treatment and follow-up in 6 weeks.”
Dr. Matiz disclosed that she is a subinvestigator in the Clinical Evaluation of T.R.U.E Test Panel 3.3 in Children and Adolescents study.
CHICAGO – If severe eczema persists in a pediatric patient despite your best treatment efforts, think allergic contact dermatitis.
“Or, if your eczema patients tell you that they have a cream that’s making things worse, you should think about a contact allergen,” Catalina Matiz, MD, said at the World Congress of Pediatric Dermatology.
Allergic contact dermatitis (ACD) is a type IV delayed-type hypersensitivity reaction to haptens that come into contact with the skin. Poison ivy is a common plant-based culprit, while nickel is the most common metal allergen in adults and children. “The skin barrier also plays a role,” said Dr. Matiz of the department of dermatology at Rady Children’s Hospital–San Diego, and the University of California, San Diego. “Compared with adults, children have a thinner stratum corneum, and some haptens can penetrate the skin. Some studies suggest that patients with atopic dermatitis may have increased rates of allergic sensitization, and filaggrin mutations have been found in patients with atopic dermatitis and in patients with ACD to nickel. Filaggrin helps to aggregate the cytoskeletal proteins that form the cornified cell envelope. Without filaggrin, the skin barrier is defective.”
The top 10 pediatric allergens found in personal hygiene products across five studies in the medical literature include neomycin, balsam of Peru, fragrance mix, benzalkonium chloride, lanolin, cocamidopropyl betaine, formaldehyde, methylchloroisothiazolinone/methylisothiazolinone (MCI/MI), propylene glycol, and corticosteroids. Dr. Matiz makes it practice to patch test as a last resort. “I always try to get a history, try to improve their symptoms, and have them start avoidance first, following the preemptive avoidance list,” she said (Expert Rev Clin Immunol. 2016;12[5]:551-61).
The T.R.U.E. test includes 35 allergens. “The T.R.U.E test is a good tool, which can capture up to 70% of relevant reactions in children with the inconvenience that some of the allergens in the test are not that relevant in children, and it’s not yet [Food and Drug Administration] approved to use in children,” she noted. The comprehensive chamber test allows you to select from unlimited number of allergens, “but that’s difficult. You have to have specialized staff to help you make the cells.”
A list of the minimum 20 allergens you should test for in children and the recommended supplemental allergens depending on history and locations of their dermatitis can be found in the following article: Curr Allergy Asthma Rep 2014;14[6]:444. “I always tell patients when they come for consultations to bring in everything they’re using: their shampoos, creams, and medications, because we want to see what they’re exposed to, so we can select the right allergens and also test their own products,” Dr. Matiz said. She recommends avoiding testing for strong sensitizers such as paraphenylenediamine, in children younger than 12 years of age who don’t have a history of exposure.
Testing tips for children younger than age 5 include decreasing concentrations to half for nickel, formaldehyde, and rubber accelerators. “Don’t test for paraphenylenediamine unless there is high suspicion,” she said. “Consider removing patches by 24 hours in the very young.”
The best antidote to contact dermatitis is avoidance of the known trigger. “You want to spend a lot of time with patients and parents on this,” she advised. “Give a list of safe products to use from the American Contact Dermatitis Society’s Contact Allergen Management Program [www.contactderm.org], and provide handouts about the location and history of positive allergens [www.truetest.com].” And, she added, “make a plan of treatment and follow-up in 6 weeks.”
Dr. Matiz disclosed that she is a subinvestigator in the Clinical Evaluation of T.R.U.E Test Panel 3.3 in Children and Adolescents study.
AT WCPD 2017
Multimodal approach is state of the art for ulcerated infantile hemangiomas
CHICAGO –
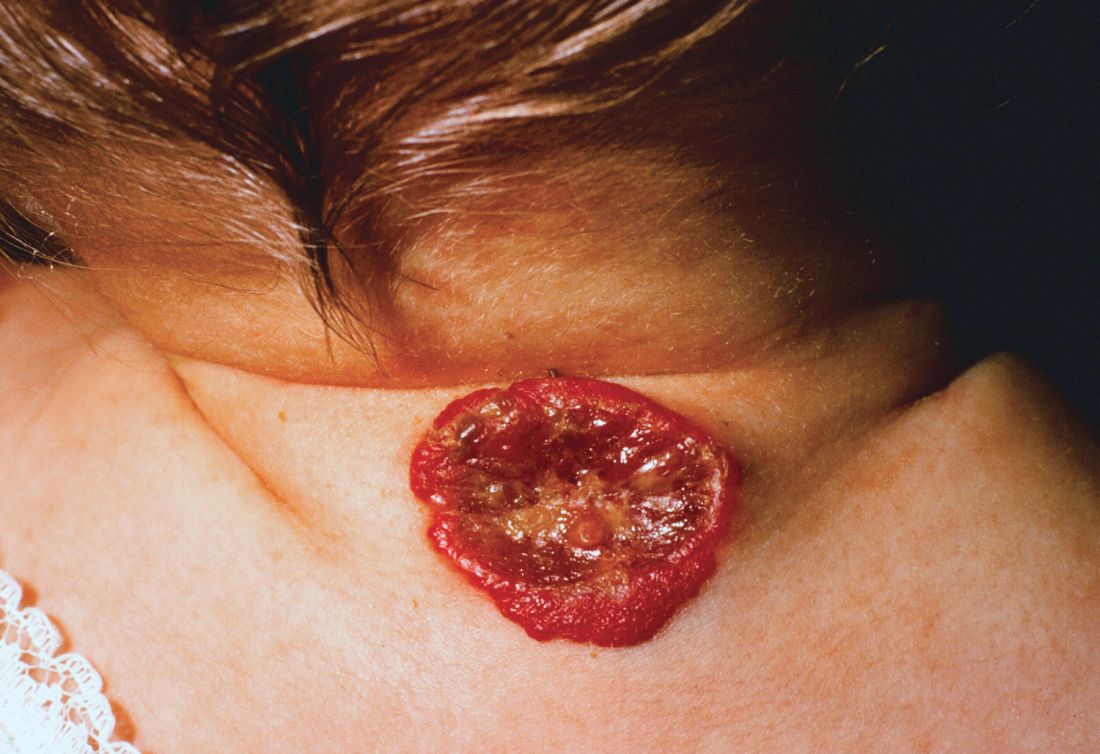
About 16% of infantile hemangiomas become ulcerated at some point during their proliferative phase, said Kate Puttgen, MD, during a talk at the World Congress of Pediatric Dermatology.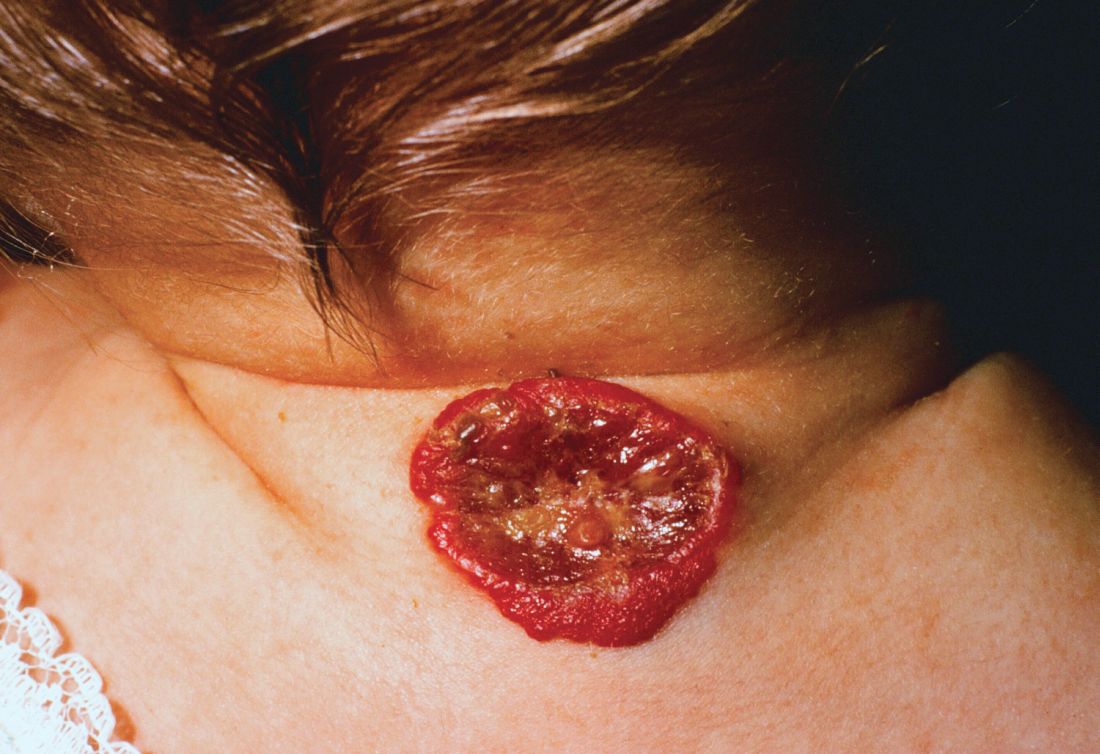
One clinical clue to picking up an infantile hemangioma (IH) that’s destined to ulcerate is an early grayish to white discoloration of the lesion, said Dr. Puttgen, chief of the division of pediatric dermatology at Johns Hopkins Medicine, Baltimore.
“Multimodal therapy is an absolute necessity” in treating an ulcerated IH, said Dr. Puttgen. Using an “all hands on deck” approach – a combination of topical and systemic modalities – can help bring the lesion under control.
Beta-blockers are first-line therapy to manage complicated IHs, with propranolol yielding a 98% response rate for all complicated IHs in the literature, said Dr. Puttgen.
Propranolol can decrease the volume and color of IHs and speed involution, in part by its ability to continue working after the proliferative growth phase of an IH. It’s also been shown to reduce the need for surgery in nasal IH, and it’s well tolerated, she added.
Evidence-based therapies for ulcerated hemangiomas include systemic propranolol at 1-3 mg/kg per day. That protocol will result in a healed ulcer within 2-6 weeks in most of the published case series, Dr. Puttgen noted.
Topical timolol also has evidence supporting its use for an ulcerated IH, and it has been found generally safe. In one study of 30 patients with IH, she said, three had mild adverse events consisting of sleep disturbance, diarrhea, and acrocyanosis. Another study reported success when brimonidine 0.2% and timolol 0.5% were used together. It’s possible, said Dr. Puttgen, that there’s a synergistic effect when combining the selective alpha-2 adrenergic agonist effect of brimonidine with timolol, which provides nonselective beta adrenergic blockade. However, she said, there has been an isolated report of brimonidine toxicity.
The ulcerated IHs need wound care, Dr. Puttgen added, with barrier creams and dressings. Pain management should be considered, because an ulcerated IH may have a large, friable, bleeding area. Pulsed-dye laser can also be a useful treatment modality for an ulcerating IH.
Going beyond the treatments for which the evidence is strongest and moving into more “state-of-the-art” treatments, “there may be a niche role for oral corticosteroids” as combination systemic therapy with propranolol, Dr. Puttgen said.
She shared images from a recently published report, in which she’s the senior author, showing the progression of an ulcerated IH. The hemangioma had received wound care and pulsed-dye laser treatment, and the infant was started on systemic propranolol. After 2 weeks, the IH had decreased significantly in volume, but the ulcerated area had actually increased. With the addition of oral corticosteroids, there was a reduction in ulceration after 2 weeks; and after 5 weeks of prednisolone, “the ulceration resolved without rebound,” said Dr. Puttgen. The corticosteroid was then tapered and propranolol was continued for an additional 2 months, then tapered. By 10 months, the IH had almost completely resolved (Br J Dermatol. 2017 Apr;176[4]:1064-7).
If a corticosteroid is added to propranolol, there may be benefit to a slower propranolol dose, Dr. Puttgen said. She suggests an altered dosing schedule, beginning with 1 mg/kg per day in two or three divided doses. Then, over a period of 2-7 days, the total daily dose can be increased to 1.5 mg/kg per day. Bumping the dose up to 2 mg/kg per day or higher should not happen until after 2 weeks at the reduced dosing schedule, she explained.
Dr. Puttgen disclosed that she is on the advisory board and has received honoraria from Pierre Fabre Dermatologie.
koakes@frontlinemedcom.com
On Twitter @karioakes
CHICAGO –
About 16% of infantile hemangiomas become ulcerated at some point during their proliferative phase, said Kate Puttgen, MD, during a talk at the World Congress of Pediatric Dermatology.
One clinical clue to picking up an infantile hemangioma (IH) that’s destined to ulcerate is an early grayish to white discoloration of the lesion, said Dr. Puttgen, chief of the division of pediatric dermatology at Johns Hopkins Medicine, Baltimore.
“Multimodal therapy is an absolute necessity” in treating an ulcerated IH, said Dr. Puttgen. Using an “all hands on deck” approach – a combination of topical and systemic modalities – can help bring the lesion under control.
Beta-blockers are first-line therapy to manage complicated IHs, with propranolol yielding a 98% response rate for all complicated IHs in the literature, said Dr. Puttgen.
Propranolol can decrease the volume and color of IHs and speed involution, in part by its ability to continue working after the proliferative growth phase of an IH. It’s also been shown to reduce the need for surgery in nasal IH, and it’s well tolerated, she added.
Evidence-based therapies for ulcerated hemangiomas include systemic propranolol at 1-3 mg/kg per day. That protocol will result in a healed ulcer within 2-6 weeks in most of the published case series, Dr. Puttgen noted.
Topical timolol also has evidence supporting its use for an ulcerated IH, and it has been found generally safe. In one study of 30 patients with IH, she said, three had mild adverse events consisting of sleep disturbance, diarrhea, and acrocyanosis. Another study reported success when brimonidine 0.2% and timolol 0.5% were used together. It’s possible, said Dr. Puttgen, that there’s a synergistic effect when combining the selective alpha-2 adrenergic agonist effect of brimonidine with timolol, which provides nonselective beta adrenergic blockade. However, she said, there has been an isolated report of brimonidine toxicity.
The ulcerated IHs need wound care, Dr. Puttgen added, with barrier creams and dressings. Pain management should be considered, because an ulcerated IH may have a large, friable, bleeding area. Pulsed-dye laser can also be a useful treatment modality for an ulcerating IH.
Going beyond the treatments for which the evidence is strongest and moving into more “state-of-the-art” treatments, “there may be a niche role for oral corticosteroids” as combination systemic therapy with propranolol, Dr. Puttgen said.
She shared images from a recently published report, in which she’s the senior author, showing the progression of an ulcerated IH. The hemangioma had received wound care and pulsed-dye laser treatment, and the infant was started on systemic propranolol. After 2 weeks, the IH had decreased significantly in volume, but the ulcerated area had actually increased. With the addition of oral corticosteroids, there was a reduction in ulceration after 2 weeks; and after 5 weeks of prednisolone, “the ulceration resolved without rebound,” said Dr. Puttgen. The corticosteroid was then tapered and propranolol was continued for an additional 2 months, then tapered. By 10 months, the IH had almost completely resolved (Br J Dermatol. 2017 Apr;176[4]:1064-7).
If a corticosteroid is added to propranolol, there may be benefit to a slower propranolol dose, Dr. Puttgen said. She suggests an altered dosing schedule, beginning with 1 mg/kg per day in two or three divided doses. Then, over a period of 2-7 days, the total daily dose can be increased to 1.5 mg/kg per day. Bumping the dose up to 2 mg/kg per day or higher should not happen until after 2 weeks at the reduced dosing schedule, she explained.
Dr. Puttgen disclosed that she is on the advisory board and has received honoraria from Pierre Fabre Dermatologie.
koakes@frontlinemedcom.com
On Twitter @karioakes
CHICAGO –
About 16% of infantile hemangiomas become ulcerated at some point during their proliferative phase, said Kate Puttgen, MD, during a talk at the World Congress of Pediatric Dermatology.
One clinical clue to picking up an infantile hemangioma (IH) that’s destined to ulcerate is an early grayish to white discoloration of the lesion, said Dr. Puttgen, chief of the division of pediatric dermatology at Johns Hopkins Medicine, Baltimore.
“Multimodal therapy is an absolute necessity” in treating an ulcerated IH, said Dr. Puttgen. Using an “all hands on deck” approach – a combination of topical and systemic modalities – can help bring the lesion under control.
Beta-blockers are first-line therapy to manage complicated IHs, with propranolol yielding a 98% response rate for all complicated IHs in the literature, said Dr. Puttgen.
Propranolol can decrease the volume and color of IHs and speed involution, in part by its ability to continue working after the proliferative growth phase of an IH. It’s also been shown to reduce the need for surgery in nasal IH, and it’s well tolerated, she added.
Evidence-based therapies for ulcerated hemangiomas include systemic propranolol at 1-3 mg/kg per day. That protocol will result in a healed ulcer within 2-6 weeks in most of the published case series, Dr. Puttgen noted.
Topical timolol also has evidence supporting its use for an ulcerated IH, and it has been found generally safe. In one study of 30 patients with IH, she said, three had mild adverse events consisting of sleep disturbance, diarrhea, and acrocyanosis. Another study reported success when brimonidine 0.2% and timolol 0.5% were used together. It’s possible, said Dr. Puttgen, that there’s a synergistic effect when combining the selective alpha-2 adrenergic agonist effect of brimonidine with timolol, which provides nonselective beta adrenergic blockade. However, she said, there has been an isolated report of brimonidine toxicity.
The ulcerated IHs need wound care, Dr. Puttgen added, with barrier creams and dressings. Pain management should be considered, because an ulcerated IH may have a large, friable, bleeding area. Pulsed-dye laser can also be a useful treatment modality for an ulcerating IH.
Going beyond the treatments for which the evidence is strongest and moving into more “state-of-the-art” treatments, “there may be a niche role for oral corticosteroids” as combination systemic therapy with propranolol, Dr. Puttgen said.
She shared images from a recently published report, in which she’s the senior author, showing the progression of an ulcerated IH. The hemangioma had received wound care and pulsed-dye laser treatment, and the infant was started on systemic propranolol. After 2 weeks, the IH had decreased significantly in volume, but the ulcerated area had actually increased. With the addition of oral corticosteroids, there was a reduction in ulceration after 2 weeks; and after 5 weeks of prednisolone, “the ulceration resolved without rebound,” said Dr. Puttgen. The corticosteroid was then tapered and propranolol was continued for an additional 2 months, then tapered. By 10 months, the IH had almost completely resolved (Br J Dermatol. 2017 Apr;176[4]:1064-7).
If a corticosteroid is added to propranolol, there may be benefit to a slower propranolol dose, Dr. Puttgen said. She suggests an altered dosing schedule, beginning with 1 mg/kg per day in two or three divided doses. Then, over a period of 2-7 days, the total daily dose can be increased to 1.5 mg/kg per day. Bumping the dose up to 2 mg/kg per day or higher should not happen until after 2 weeks at the reduced dosing schedule, she explained.
Dr. Puttgen disclosed that she is on the advisory board and has received honoraria from Pierre Fabre Dermatologie.
koakes@frontlinemedcom.com
On Twitter @karioakes
EXPERT ANALYSIS FROM WCPD 2017
New genetic causes of ichthyoses likely to emerge
CHICAGO– The way Keith Choate, MD, PhD, sees it, he and other clinicians are only beginning to scratch the surface on their understanding of the genetic causes of ichthyosis and ichthyosis syndromes.
“Despite the fact that we now understand that there are about 21,000 genes in the genome, we have very superficial understanding and functions known for only about 4,000 genes,” Dr. Choate of the departments of dermatology and genetics at Yale University, New Haven, Conn., said at the World Congress of Pediatric Dermatology. “Clinical insight is what’s driving all of the discovery. We continue to find new disorders, and these next-generation technologies really permit us to find the genetic basis for those disorders. I like to say that it’s the disorders that we don’t read about in the textbook that end up being the ones that are most interesting.”
Ichthyoses are cardinal disorders that occur when the normal pattern of epidermal differentiation is disrupted and leads to compensatory hyperproliferation. Clinically, ichthyoses present in a variety of ways, and more than 50 genes can cause them.
Dr. Choate is the principal investigator of the National Registry for Ichthyosis and Related Skin Types, which has been recruiting kindreds of ichthyosis patients within the United States and internationally. To date, they have provided genetic diagnoses for 674 of the 880 cases enrolled. The process involves phenotyping with a clinical history and photography, obtaining DNA from blood or saliva, and prescreening the DNA samples for mutations in 51 genes currently implicated in ichthyosis. Subjects without known mutations undergo whole exome or genome sequencing.
“We have created a unique resource in doing this,” he said. “Genotyped/phenotyped patients provide a resource for clinical and translational studies in disorders of keratinization.”
When the researchers examined patients from the registry who have epidermolytic presentations, 100% had mutations in the known genes, “so the biopsy is diagnostic,” Dr. Choate said. “About 80% have mutations in keratin 10, about 13% have mutations in keratin 1, and another 6% have mutations in keratin 2.”
About 85% of patients with recessive and syndromic disorders have mutations in this same 51-gene panel; 15% of cases don’t have mutations in those genes. “This is a similar fraction to what my colleagues have found at a variety of institutions around the world,” Dr. Choate said. “What’s fascinating is that this 15% has been the source of remarkable discovery.”
He then discussed three cases of a novel erythrokeratoderma phenotype that were referred to the registry. In one case, a boy had pervasive intellectual disability, congenital alopecia, and absence of the eyebrows. “Within the first days of life, he developed a significant erythroderma with copious scaling of the skin that persisted throughout life and that was unresponsive to a variety of different therapeutic interventions, including immunosuppressant medications,” Dr. Choate said. “He had nail dystrophy and progressive enamel decay with severe caries, leading to loss of all of his teeth by the age of 6.”
Another case was a child who died of cardiomyopathy at about 3 years of age. He had congenital absence of the eyebrows and eyelashes, nail dystrophy, and scaling. “About 2 weeks before his death, he had a skin biopsy that we would ultimately repurpose to identify a new genetic cause of what we would call the erythrokeratodermia-cardiomyopathy syndrome,” Dr. Choate said. “It included features of congenital erythroderma, defective dental enamel, abnormal nails, and progressive and lethal cardiomyopathy. When we did exome sequencing, we found that all three of these patients showed tightly clustered de novo mutations in a gene called desmoplakin (DSP). Other DSP mutations do not cause erythrokeratoderma.”
Electron microscopy showed aggregates of desmosomes, normal corneodesmosomes, widening of intercellular spaces, and abnormal lipid secretion. The finding led the researchers to conclude that clustered DSP mutations cause a novel cutaneous phenotype with erythrokeratoderma and progressive cardiomyopathy. “The next time an insurer refuses to do genetic testing for you in a patient who has erythrokeratoderma, this is the disorder that you want to cite as the reason why you need to do genetic testing,” he said.
In a recent study, Dr. Choate and his associates identified the genetic cause for a rare subtype of progressive symmetric erythrokeratoderma (PSEK), a disorder that features thick facial plaques and thickened palms and soles (Am J Hum Genet. 2017 Jun 1;100[6]:978-84). Histology reveals a thickened epidermis, loss of granular layer, and retention of nuclei in the stratum corneum. They discovered that PSEK was caused by mutations in 3-ketodihydrosphingosine reductase (KDSR), an enzyme that is central to de novo ceramides in skin.
“Ceramides are secreted by keratinocytes with cholesterol and free fatty acids to form the cutaneous lipid barrier,” he explained. “They also regulate cutaneous proliferation and differentiation. One of the things this story in particular told us was, when you find just one mutation and a compelling candidate gene and can’t find the other, it’s often because of how you’re approaching detection. In two of our cases, genome sequencing was necessary to find a large inversion, which disrupted the encoded protein. Finally, the challenge of studying ceramides is that it’s hard to get cells in culture to produce them. Therefore, we had to work with collaborators in yeast biology to prove pathogenesis.”
Dr. Choate cited other recent developments, including the discovery that familial pityriasis rubra pilaris is caused by mutations in CARD14, which is a known activator of nuclear factor kappa B signaling (Am J Hum Genet. 2012 Jul 13;91[1]:163-70). This led to the subsequent use of ustekinumab for patients with familial pityriasis rubra pilaris. Another group of researchers found that SULT2B1 encodes sulfotransferase family 2B member 1 and is central to epidermal cholesterol metabolism (Am J Hum Genet. 2017 Jun 1;100[6]:926-39).
“There are still new genetic causes of ichthyosis to be found, particularly cases that don’t meet the textbook criteria for the disorder,” he concluded. “Severe, dominant disorders primarily due to de novo mutations are fertile ground for discovery. Genetic investigation is critical to our understanding of disease biology and biology of the skin. It’s also potentially relevant to outcomes of therapy. [Erythrokeratodermia-cardiomyopathy syndrome] highlights the potential for comorbidities, and the efficacy of ustekinumab in familial [pityriasis rubra pilaris] highlights the therapeutic importance of understanding the pathway underlying the disease.”
Dr. Choate reported having no financial disclosures.
CHICAGO– The way Keith Choate, MD, PhD, sees it, he and other clinicians are only beginning to scratch the surface on their understanding of the genetic causes of ichthyosis and ichthyosis syndromes.
“Despite the fact that we now understand that there are about 21,000 genes in the genome, we have very superficial understanding and functions known for only about 4,000 genes,” Dr. Choate of the departments of dermatology and genetics at Yale University, New Haven, Conn., said at the World Congress of Pediatric Dermatology. “Clinical insight is what’s driving all of the discovery. We continue to find new disorders, and these next-generation technologies really permit us to find the genetic basis for those disorders. I like to say that it’s the disorders that we don’t read about in the textbook that end up being the ones that are most interesting.”
Ichthyoses are cardinal disorders that occur when the normal pattern of epidermal differentiation is disrupted and leads to compensatory hyperproliferation. Clinically, ichthyoses present in a variety of ways, and more than 50 genes can cause them.
Dr. Choate is the principal investigator of the National Registry for Ichthyosis and Related Skin Types, which has been recruiting kindreds of ichthyosis patients within the United States and internationally. To date, they have provided genetic diagnoses for 674 of the 880 cases enrolled. The process involves phenotyping with a clinical history and photography, obtaining DNA from blood or saliva, and prescreening the DNA samples for mutations in 51 genes currently implicated in ichthyosis. Subjects without known mutations undergo whole exome or genome sequencing.
“We have created a unique resource in doing this,” he said. “Genotyped/phenotyped patients provide a resource for clinical and translational studies in disorders of keratinization.”
When the researchers examined patients from the registry who have epidermolytic presentations, 100% had mutations in the known genes, “so the biopsy is diagnostic,” Dr. Choate said. “About 80% have mutations in keratin 10, about 13% have mutations in keratin 1, and another 6% have mutations in keratin 2.”
About 85% of patients with recessive and syndromic disorders have mutations in this same 51-gene panel; 15% of cases don’t have mutations in those genes. “This is a similar fraction to what my colleagues have found at a variety of institutions around the world,” Dr. Choate said. “What’s fascinating is that this 15% has been the source of remarkable discovery.”
He then discussed three cases of a novel erythrokeratoderma phenotype that were referred to the registry. In one case, a boy had pervasive intellectual disability, congenital alopecia, and absence of the eyebrows. “Within the first days of life, he developed a significant erythroderma with copious scaling of the skin that persisted throughout life and that was unresponsive to a variety of different therapeutic interventions, including immunosuppressant medications,” Dr. Choate said. “He had nail dystrophy and progressive enamel decay with severe caries, leading to loss of all of his teeth by the age of 6.”
Another case was a child who died of cardiomyopathy at about 3 years of age. He had congenital absence of the eyebrows and eyelashes, nail dystrophy, and scaling. “About 2 weeks before his death, he had a skin biopsy that we would ultimately repurpose to identify a new genetic cause of what we would call the erythrokeratodermia-cardiomyopathy syndrome,” Dr. Choate said. “It included features of congenital erythroderma, defective dental enamel, abnormal nails, and progressive and lethal cardiomyopathy. When we did exome sequencing, we found that all three of these patients showed tightly clustered de novo mutations in a gene called desmoplakin (DSP). Other DSP mutations do not cause erythrokeratoderma.”
Electron microscopy showed aggregates of desmosomes, normal corneodesmosomes, widening of intercellular spaces, and abnormal lipid secretion. The finding led the researchers to conclude that clustered DSP mutations cause a novel cutaneous phenotype with erythrokeratoderma and progressive cardiomyopathy. “The next time an insurer refuses to do genetic testing for you in a patient who has erythrokeratoderma, this is the disorder that you want to cite as the reason why you need to do genetic testing,” he said.
In a recent study, Dr. Choate and his associates identified the genetic cause for a rare subtype of progressive symmetric erythrokeratoderma (PSEK), a disorder that features thick facial plaques and thickened palms and soles (Am J Hum Genet. 2017 Jun 1;100[6]:978-84). Histology reveals a thickened epidermis, loss of granular layer, and retention of nuclei in the stratum corneum. They discovered that PSEK was caused by mutations in 3-ketodihydrosphingosine reductase (KDSR), an enzyme that is central to de novo ceramides in skin.
“Ceramides are secreted by keratinocytes with cholesterol and free fatty acids to form the cutaneous lipid barrier,” he explained. “They also regulate cutaneous proliferation and differentiation. One of the things this story in particular told us was, when you find just one mutation and a compelling candidate gene and can’t find the other, it’s often because of how you’re approaching detection. In two of our cases, genome sequencing was necessary to find a large inversion, which disrupted the encoded protein. Finally, the challenge of studying ceramides is that it’s hard to get cells in culture to produce them. Therefore, we had to work with collaborators in yeast biology to prove pathogenesis.”
Dr. Choate cited other recent developments, including the discovery that familial pityriasis rubra pilaris is caused by mutations in CARD14, which is a known activator of nuclear factor kappa B signaling (Am J Hum Genet. 2012 Jul 13;91[1]:163-70). This led to the subsequent use of ustekinumab for patients with familial pityriasis rubra pilaris. Another group of researchers found that SULT2B1 encodes sulfotransferase family 2B member 1 and is central to epidermal cholesterol metabolism (Am J Hum Genet. 2017 Jun 1;100[6]:926-39).
“There are still new genetic causes of ichthyosis to be found, particularly cases that don’t meet the textbook criteria for the disorder,” he concluded. “Severe, dominant disorders primarily due to de novo mutations are fertile ground for discovery. Genetic investigation is critical to our understanding of disease biology and biology of the skin. It’s also potentially relevant to outcomes of therapy. [Erythrokeratodermia-cardiomyopathy syndrome] highlights the potential for comorbidities, and the efficacy of ustekinumab in familial [pityriasis rubra pilaris] highlights the therapeutic importance of understanding the pathway underlying the disease.”
Dr. Choate reported having no financial disclosures.
CHICAGO– The way Keith Choate, MD, PhD, sees it, he and other clinicians are only beginning to scratch the surface on their understanding of the genetic causes of ichthyosis and ichthyosis syndromes.
“Despite the fact that we now understand that there are about 21,000 genes in the genome, we have very superficial understanding and functions known for only about 4,000 genes,” Dr. Choate of the departments of dermatology and genetics at Yale University, New Haven, Conn., said at the World Congress of Pediatric Dermatology. “Clinical insight is what’s driving all of the discovery. We continue to find new disorders, and these next-generation technologies really permit us to find the genetic basis for those disorders. I like to say that it’s the disorders that we don’t read about in the textbook that end up being the ones that are most interesting.”
Ichthyoses are cardinal disorders that occur when the normal pattern of epidermal differentiation is disrupted and leads to compensatory hyperproliferation. Clinically, ichthyoses present in a variety of ways, and more than 50 genes can cause them.
Dr. Choate is the principal investigator of the National Registry for Ichthyosis and Related Skin Types, which has been recruiting kindreds of ichthyosis patients within the United States and internationally. To date, they have provided genetic diagnoses for 674 of the 880 cases enrolled. The process involves phenotyping with a clinical history and photography, obtaining DNA from blood or saliva, and prescreening the DNA samples for mutations in 51 genes currently implicated in ichthyosis. Subjects without known mutations undergo whole exome or genome sequencing.
“We have created a unique resource in doing this,” he said. “Genotyped/phenotyped patients provide a resource for clinical and translational studies in disorders of keratinization.”
When the researchers examined patients from the registry who have epidermolytic presentations, 100% had mutations in the known genes, “so the biopsy is diagnostic,” Dr. Choate said. “About 80% have mutations in keratin 10, about 13% have mutations in keratin 1, and another 6% have mutations in keratin 2.”
About 85% of patients with recessive and syndromic disorders have mutations in this same 51-gene panel; 15% of cases don’t have mutations in those genes. “This is a similar fraction to what my colleagues have found at a variety of institutions around the world,” Dr. Choate said. “What’s fascinating is that this 15% has been the source of remarkable discovery.”
He then discussed three cases of a novel erythrokeratoderma phenotype that were referred to the registry. In one case, a boy had pervasive intellectual disability, congenital alopecia, and absence of the eyebrows. “Within the first days of life, he developed a significant erythroderma with copious scaling of the skin that persisted throughout life and that was unresponsive to a variety of different therapeutic interventions, including immunosuppressant medications,” Dr. Choate said. “He had nail dystrophy and progressive enamel decay with severe caries, leading to loss of all of his teeth by the age of 6.”
Another case was a child who died of cardiomyopathy at about 3 years of age. He had congenital absence of the eyebrows and eyelashes, nail dystrophy, and scaling. “About 2 weeks before his death, he had a skin biopsy that we would ultimately repurpose to identify a new genetic cause of what we would call the erythrokeratodermia-cardiomyopathy syndrome,” Dr. Choate said. “It included features of congenital erythroderma, defective dental enamel, abnormal nails, and progressive and lethal cardiomyopathy. When we did exome sequencing, we found that all three of these patients showed tightly clustered de novo mutations in a gene called desmoplakin (DSP). Other DSP mutations do not cause erythrokeratoderma.”
Electron microscopy showed aggregates of desmosomes, normal corneodesmosomes, widening of intercellular spaces, and abnormal lipid secretion. The finding led the researchers to conclude that clustered DSP mutations cause a novel cutaneous phenotype with erythrokeratoderma and progressive cardiomyopathy. “The next time an insurer refuses to do genetic testing for you in a patient who has erythrokeratoderma, this is the disorder that you want to cite as the reason why you need to do genetic testing,” he said.
In a recent study, Dr. Choate and his associates identified the genetic cause for a rare subtype of progressive symmetric erythrokeratoderma (PSEK), a disorder that features thick facial plaques and thickened palms and soles (Am J Hum Genet. 2017 Jun 1;100[6]:978-84). Histology reveals a thickened epidermis, loss of granular layer, and retention of nuclei in the stratum corneum. They discovered that PSEK was caused by mutations in 3-ketodihydrosphingosine reductase (KDSR), an enzyme that is central to de novo ceramides in skin.
“Ceramides are secreted by keratinocytes with cholesterol and free fatty acids to form the cutaneous lipid barrier,” he explained. “They also regulate cutaneous proliferation and differentiation. One of the things this story in particular told us was, when you find just one mutation and a compelling candidate gene and can’t find the other, it’s often because of how you’re approaching detection. In two of our cases, genome sequencing was necessary to find a large inversion, which disrupted the encoded protein. Finally, the challenge of studying ceramides is that it’s hard to get cells in culture to produce them. Therefore, we had to work with collaborators in yeast biology to prove pathogenesis.”
Dr. Choate cited other recent developments, including the discovery that familial pityriasis rubra pilaris is caused by mutations in CARD14, which is a known activator of nuclear factor kappa B signaling (Am J Hum Genet. 2012 Jul 13;91[1]:163-70). This led to the subsequent use of ustekinumab for patients with familial pityriasis rubra pilaris. Another group of researchers found that SULT2B1 encodes sulfotransferase family 2B member 1 and is central to epidermal cholesterol metabolism (Am J Hum Genet. 2017 Jun 1;100[6]:926-39).
“There are still new genetic causes of ichthyosis to be found, particularly cases that don’t meet the textbook criteria for the disorder,” he concluded. “Severe, dominant disorders primarily due to de novo mutations are fertile ground for discovery. Genetic investigation is critical to our understanding of disease biology and biology of the skin. It’s also potentially relevant to outcomes of therapy. [Erythrokeratodermia-cardiomyopathy syndrome] highlights the potential for comorbidities, and the efficacy of ustekinumab in familial [pityriasis rubra pilaris] highlights the therapeutic importance of understanding the pathway underlying the disease.”
Dr. Choate reported having no financial disclosures.
AT WCPD 2017
Screening MRI misses Sturge-Weber in babies with port-wine stain
CHICAGO – Screening infants with a port-wine stain for Sturge-Weber syndrome (SWS) with a magnetic resonance imaging brain scan had a 23% false-negative rate and actually delayed seizure detection, according to a recent study.
When infants with port-wine stains receive a dermatology consult, they may also be screened for SWS by means of MRI and by electroencephalography, particularly if their lesion phenotype puts them at higher risk for SWS. But the accuracy and benefit of the screenings has not been well established, said Michaela Zallmann, MD, the study’s first author. Hemifacial lesions that involve both the forehead and cheek and median lesions that are centered around the facial midline are both considered high-risk lesions.
Dr. Zallmann, a dermatologist at Monash University and the Royal Children’s Hospital, Melbourne, and her coinvestigators examined data on 126 patients with facial port-wine stains who came to a laser clinic over a 12-month period. Of these, 32 (25.4%) had a high-risk port-wine stain, and 9 of those 32 (28.1%) had a capillary-venous malformation characteristic of SWS. Of the high-risk patients, 14 received a screening MRI or EEG before having had a first seizure. Of those 14 scans, 1 resulted in a diagnosis of SWS; of the 13 patients with a negative MRI screen, 3 (23.1%) were later found to have SWS when their parents or caregivers detected seizures. Thus, a total of four of the high-risk infants who were screened eventually were diagnosed with SWS.
Of the 18 high-risk patients who did not receive a screening MRI, 3 (16.7%) developed seizures, while 2 (11.1%) were seizure free but developed glaucoma severe enough to require treatment. One patient who was also seizure free developed an autism spectrum disorder.
Two patients who were in the high-risk group received screening EEGs that detected abnormalities that were not yet clinically evident. These included sub-clinical seizures and posterior-quadrant focal slowing. Both of these patients had initial negative screening MRIs.
Scanning early in life, using inappropriate imaging protocols, and having an inexperienced radiologist were all factors associated with a higher probability of false negative screening MRI, according to the researchers’ analysis, which was presented in a poster session at the World Congress of Pediatric Dermatology.
All of the false negative MRIs in the study cohort were conducted in infants younger than 9 weeks old. But whether it is useful to reserve imaging for later in infancy is debatable. “While later imaging may have improved sensitivity, 75% of infants with SWS will have already had their first seizure by 12 months of age,” wrote Dr. Zallmann and her colleagues.
Of the infants involved in the study, two of the three patients with false negative scans did not receive a referral to a neurologist, nor did their parents receive seizure education. “False reassurance may delay seizure detection,” Dr. Zallmann said.
For infants with positive MRIs who went on to develop seizures, the mean age of when they experienced their first documented seizure was 10 weeks. For those who did not receive an MRI, the mean age was 14 months, compared with 28 months for patients who had received a false negative MRI.
In discussing the findings, Dr. Zallmann and her colleagues made the point that early referral to a pediatric neurologist is important, especially for infants with the higher-risk port-wine stain patterns of hemifacial and median lesions. Seizure education can help parents detect the often subtle signs of seizures in infants with SWS, which can include staring spells, subtle limb twitching, and lip smacking.
The fact that seizures were detected an average of 14 months later in patients with negative screening MRIs may mean that such subtle signs were missed. “False reassurance may delay the recognition and treatment of seizures and impact neurodevelopmental outcomes,” Dr. Zallmann and her colleagues wrote in the abstract that accompanied the poster.
The study, while small, helps fill in some knowledge gaps, the researchers pointed out; they noted that there is no consensus on what level or type of facial involvement warrants screening, which protocols are best for MRI and EEG, or even whether the screening will improve seizure detection or outcomes.
“Currently there is no evidence that screening improves neurodevelopmental outcomes,” they said. “Conversely, there is a role for early neurological referral, symptom education, and potentially of EEGs in the prevention of complications related to SWS.”
Dr. Zallmann reported no conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
CHICAGO – Screening infants with a port-wine stain for Sturge-Weber syndrome (SWS) with a magnetic resonance imaging brain scan had a 23% false-negative rate and actually delayed seizure detection, according to a recent study.
When infants with port-wine stains receive a dermatology consult, they may also be screened for SWS by means of MRI and by electroencephalography, particularly if their lesion phenotype puts them at higher risk for SWS. But the accuracy and benefit of the screenings has not been well established, said Michaela Zallmann, MD, the study’s first author. Hemifacial lesions that involve both the forehead and cheek and median lesions that are centered around the facial midline are both considered high-risk lesions.
Dr. Zallmann, a dermatologist at Monash University and the Royal Children’s Hospital, Melbourne, and her coinvestigators examined data on 126 patients with facial port-wine stains who came to a laser clinic over a 12-month period. Of these, 32 (25.4%) had a high-risk port-wine stain, and 9 of those 32 (28.1%) had a capillary-venous malformation characteristic of SWS. Of the high-risk patients, 14 received a screening MRI or EEG before having had a first seizure. Of those 14 scans, 1 resulted in a diagnosis of SWS; of the 13 patients with a negative MRI screen, 3 (23.1%) were later found to have SWS when their parents or caregivers detected seizures. Thus, a total of four of the high-risk infants who were screened eventually were diagnosed with SWS.
Of the 18 high-risk patients who did not receive a screening MRI, 3 (16.7%) developed seizures, while 2 (11.1%) were seizure free but developed glaucoma severe enough to require treatment. One patient who was also seizure free developed an autism spectrum disorder.
Two patients who were in the high-risk group received screening EEGs that detected abnormalities that were not yet clinically evident. These included sub-clinical seizures and posterior-quadrant focal slowing. Both of these patients had initial negative screening MRIs.
Scanning early in life, using inappropriate imaging protocols, and having an inexperienced radiologist were all factors associated with a higher probability of false negative screening MRI, according to the researchers’ analysis, which was presented in a poster session at the World Congress of Pediatric Dermatology.
All of the false negative MRIs in the study cohort were conducted in infants younger than 9 weeks old. But whether it is useful to reserve imaging for later in infancy is debatable. “While later imaging may have improved sensitivity, 75% of infants with SWS will have already had their first seizure by 12 months of age,” wrote Dr. Zallmann and her colleagues.
Of the infants involved in the study, two of the three patients with false negative scans did not receive a referral to a neurologist, nor did their parents receive seizure education. “False reassurance may delay seizure detection,” Dr. Zallmann said.
For infants with positive MRIs who went on to develop seizures, the mean age of when they experienced their first documented seizure was 10 weeks. For those who did not receive an MRI, the mean age was 14 months, compared with 28 months for patients who had received a false negative MRI.
In discussing the findings, Dr. Zallmann and her colleagues made the point that early referral to a pediatric neurologist is important, especially for infants with the higher-risk port-wine stain patterns of hemifacial and median lesions. Seizure education can help parents detect the often subtle signs of seizures in infants with SWS, which can include staring spells, subtle limb twitching, and lip smacking.
The fact that seizures were detected an average of 14 months later in patients with negative screening MRIs may mean that such subtle signs were missed. “False reassurance may delay the recognition and treatment of seizures and impact neurodevelopmental outcomes,” Dr. Zallmann and her colleagues wrote in the abstract that accompanied the poster.
The study, while small, helps fill in some knowledge gaps, the researchers pointed out; they noted that there is no consensus on what level or type of facial involvement warrants screening, which protocols are best for MRI and EEG, or even whether the screening will improve seizure detection or outcomes.
“Currently there is no evidence that screening improves neurodevelopmental outcomes,” they said. “Conversely, there is a role for early neurological referral, symptom education, and potentially of EEGs in the prevention of complications related to SWS.”
Dr. Zallmann reported no conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
CHICAGO – Screening infants with a port-wine stain for Sturge-Weber syndrome (SWS) with a magnetic resonance imaging brain scan had a 23% false-negative rate and actually delayed seizure detection, according to a recent study.
When infants with port-wine stains receive a dermatology consult, they may also be screened for SWS by means of MRI and by electroencephalography, particularly if their lesion phenotype puts them at higher risk for SWS. But the accuracy and benefit of the screenings has not been well established, said Michaela Zallmann, MD, the study’s first author. Hemifacial lesions that involve both the forehead and cheek and median lesions that are centered around the facial midline are both considered high-risk lesions.
Dr. Zallmann, a dermatologist at Monash University and the Royal Children’s Hospital, Melbourne, and her coinvestigators examined data on 126 patients with facial port-wine stains who came to a laser clinic over a 12-month period. Of these, 32 (25.4%) had a high-risk port-wine stain, and 9 of those 32 (28.1%) had a capillary-venous malformation characteristic of SWS. Of the high-risk patients, 14 received a screening MRI or EEG before having had a first seizure. Of those 14 scans, 1 resulted in a diagnosis of SWS; of the 13 patients with a negative MRI screen, 3 (23.1%) were later found to have SWS when their parents or caregivers detected seizures. Thus, a total of four of the high-risk infants who were screened eventually were diagnosed with SWS.
Of the 18 high-risk patients who did not receive a screening MRI, 3 (16.7%) developed seizures, while 2 (11.1%) were seizure free but developed glaucoma severe enough to require treatment. One patient who was also seizure free developed an autism spectrum disorder.
Two patients who were in the high-risk group received screening EEGs that detected abnormalities that were not yet clinically evident. These included sub-clinical seizures and posterior-quadrant focal slowing. Both of these patients had initial negative screening MRIs.
Scanning early in life, using inappropriate imaging protocols, and having an inexperienced radiologist were all factors associated with a higher probability of false negative screening MRI, according to the researchers’ analysis, which was presented in a poster session at the World Congress of Pediatric Dermatology.
All of the false negative MRIs in the study cohort were conducted in infants younger than 9 weeks old. But whether it is useful to reserve imaging for later in infancy is debatable. “While later imaging may have improved sensitivity, 75% of infants with SWS will have already had their first seizure by 12 months of age,” wrote Dr. Zallmann and her colleagues.
Of the infants involved in the study, two of the three patients with false negative scans did not receive a referral to a neurologist, nor did their parents receive seizure education. “False reassurance may delay seizure detection,” Dr. Zallmann said.
For infants with positive MRIs who went on to develop seizures, the mean age of when they experienced their first documented seizure was 10 weeks. For those who did not receive an MRI, the mean age was 14 months, compared with 28 months for patients who had received a false negative MRI.
In discussing the findings, Dr. Zallmann and her colleagues made the point that early referral to a pediatric neurologist is important, especially for infants with the higher-risk port-wine stain patterns of hemifacial and median lesions. Seizure education can help parents detect the often subtle signs of seizures in infants with SWS, which can include staring spells, subtle limb twitching, and lip smacking.
The fact that seizures were detected an average of 14 months later in patients with negative screening MRIs may mean that such subtle signs were missed. “False reassurance may delay the recognition and treatment of seizures and impact neurodevelopmental outcomes,” Dr. Zallmann and her colleagues wrote in the abstract that accompanied the poster.
The study, while small, helps fill in some knowledge gaps, the researchers pointed out; they noted that there is no consensus on what level or type of facial involvement warrants screening, which protocols are best for MRI and EEG, or even whether the screening will improve seizure detection or outcomes.
“Currently there is no evidence that screening improves neurodevelopmental outcomes,” they said. “Conversely, there is a role for early neurological referral, symptom education, and potentially of EEGs in the prevention of complications related to SWS.”
Dr. Zallmann reported no conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
AT WCPD 2017
Key clinical point:
Major finding: Magnetic resonance imaging screening for Sturge-Weber syndrome resulted in a 23.2% false negative rate in babies with port-wine stain.
Data source: A review of screening and outcomes for 126 infants with port-wine stain seen in a laser clinic over a 12-month period. Disclosures: The lead author reported no disclosures.
Study aims to validate AAD criteria for diagnosing AD and create usable form
CHICAGO – A streamlined set of diagnostic criteria from the American Academy of Dermatology’s most recent consensus criteria for diagnosing atopic dermatitis (AD) produced a specificity of more than 95%, and was also highly sensitive, a prospective analysis found.
“Atopic dermatitis typically presents in childhood and is associated with a worsened quality of life, with severe itch and lack of sleep, and substantial health care costs due to therapeutic management and increased hospitalizations,” study author Jeremy Udkoff said at the World Congress of Pediatric Dermatology. “We also know that in order to treat the disease and to learn more about it, we have to have a good tool for diagnosing it. When it comes to clinical studies and research, we require a systematic and refined set of criteria.”
The next set of commonly used criteria to appear were created by the U.K. working party, for which researchers used logistic regression to systematically create a minimum set of effective criteria for AD (Br J Dermatol. 1994;131[3]:383-96). For these guidelines, meeting a diagnosis of AD requires an itchy skin condition, followed by three or more of the following: a history of flexural involvement; a personal history of asthma or hay fever; a history of general dry skin in the last year; visible flexural eczema, and onset under the age of 2 years. “A subsequent validation trial found [the U.K. working party criteria] to have a low sensitivity, which as you can imagine, could be a very large problem,” he said (Arch Dermatol. 1999;135[5]:514-6).
In 2001, the AAD consensus conference created revised hierarchical criteria known as the AAD consensus criteria (J Am Acad Dermatol. 2003;49[6]:1088-95). “These were initially created for more of a gestalt-type picture of AD in the clinic, but because it flows so well, it’s currently being used in about one-third of clinical trials,” Mr. Udkoff said. “However, [the AAD criteria] have not been validated, so we didn’t know its sensitivity or specificity. In addition, we didn’t have a ‘checkbox’ form that tells us how many of each of the criteria are required to make the diagnosis. We didn’t know how many ‘essential,’ ‘important,’ or ‘associated’ features we need to make this diagnosis.”
For the current study, he and his associates set out to determine how many “essential,” “important,” and “associated” criteria are necessary to make the AAD consensus criteria work. They also set out to create a usable checkbox form, validate the criteria, and compare it to the Hanifin-Rajka (HR) and U.K. criteria. To accomplish this, they created a questionnaire comprised of HR, U.K., and AAD criteria, examined the criteria on 60 subjects with and without AD, and compared the diagnostic features of each of those criteria against a gold standard dermatology diagnosis from one of seven pediatric dermatologists. Next, they ranked all 56 possible AAD criterion combinations based on their overall sensitivity and specificity, and chose the most predictive combination. “Once we had the optimal set of criteria, we validated it on a new cohort to determine its sensitivity and specificity, and compared it with the classic HR and U.K. criteria,” Mr. Udkoff explained.
Overall, the researchers evaluated findings from 100 subjects: 58 with AD, and 42 controls. Those with AD were about 3 years younger, compared with controls (a mean age of 5 years vs. about 8 years, respectively). About 40% of patients were Hispanic and about 30% were white. Mr. Udkoff and his associates confirmed the hierarchical structure of the AAD criteria and found that individual “essential” AAD criteria of pruritus, typical AD pattern, and chronic/relapsing course each had a sensitivity that exceeded 96%. This was followed by the “important” criteria of early age of onset, atopy, and xerosis, which had a sensitivity that ranged between 88% and 95%, while the associated criteria had a sensitivity that ranged between 50% and 85%.
Next, the researchers systematically tested all combinations of the AAD criteria and found that three “essential” AAD criteria, two or more of the “important” criteria, and one or more of the “associated” criteria were optimal in diagnosing AD. Mr. Udkoff noted that the findings can be translated into a simple “3-2-1 rule” that “is both practical and pragmatic,” he said. Using this rule, sensitivity was 91.4% and specificity was 95.2%.
Currently, the researchers are working to validate this criteria in different subgroups of patients. To date, they have found that children younger than 1.5 years get one bonus “essential” criteria for being an infant, so for that population a 2-2-1 rule would apply.
Mr. Udkoff reported that the research was supported by a training grant from the National Institutes of Health. He reported having no financial disclosures.
CHICAGO – A streamlined set of diagnostic criteria from the American Academy of Dermatology’s most recent consensus criteria for diagnosing atopic dermatitis (AD) produced a specificity of more than 95%, and was also highly sensitive, a prospective analysis found.
“Atopic dermatitis typically presents in childhood and is associated with a worsened quality of life, with severe itch and lack of sleep, and substantial health care costs due to therapeutic management and increased hospitalizations,” study author Jeremy Udkoff said at the World Congress of Pediatric Dermatology. “We also know that in order to treat the disease and to learn more about it, we have to have a good tool for diagnosing it. When it comes to clinical studies and research, we require a systematic and refined set of criteria.”
The next set of commonly used criteria to appear were created by the U.K. working party, for which researchers used logistic regression to systematically create a minimum set of effective criteria for AD (Br J Dermatol. 1994;131[3]:383-96). For these guidelines, meeting a diagnosis of AD requires an itchy skin condition, followed by three or more of the following: a history of flexural involvement; a personal history of asthma or hay fever; a history of general dry skin in the last year; visible flexural eczema, and onset under the age of 2 years. “A subsequent validation trial found [the U.K. working party criteria] to have a low sensitivity, which as you can imagine, could be a very large problem,” he said (Arch Dermatol. 1999;135[5]:514-6).
In 2001, the AAD consensus conference created revised hierarchical criteria known as the AAD consensus criteria (J Am Acad Dermatol. 2003;49[6]:1088-95). “These were initially created for more of a gestalt-type picture of AD in the clinic, but because it flows so well, it’s currently being used in about one-third of clinical trials,” Mr. Udkoff said. “However, [the AAD criteria] have not been validated, so we didn’t know its sensitivity or specificity. In addition, we didn’t have a ‘checkbox’ form that tells us how many of each of the criteria are required to make the diagnosis. We didn’t know how many ‘essential,’ ‘important,’ or ‘associated’ features we need to make this diagnosis.”
For the current study, he and his associates set out to determine how many “essential,” “important,” and “associated” criteria are necessary to make the AAD consensus criteria work. They also set out to create a usable checkbox form, validate the criteria, and compare it to the Hanifin-Rajka (HR) and U.K. criteria. To accomplish this, they created a questionnaire comprised of HR, U.K., and AAD criteria, examined the criteria on 60 subjects with and without AD, and compared the diagnostic features of each of those criteria against a gold standard dermatology diagnosis from one of seven pediatric dermatologists. Next, they ranked all 56 possible AAD criterion combinations based on their overall sensitivity and specificity, and chose the most predictive combination. “Once we had the optimal set of criteria, we validated it on a new cohort to determine its sensitivity and specificity, and compared it with the classic HR and U.K. criteria,” Mr. Udkoff explained.
Overall, the researchers evaluated findings from 100 subjects: 58 with AD, and 42 controls. Those with AD were about 3 years younger, compared with controls (a mean age of 5 years vs. about 8 years, respectively). About 40% of patients were Hispanic and about 30% were white. Mr. Udkoff and his associates confirmed the hierarchical structure of the AAD criteria and found that individual “essential” AAD criteria of pruritus, typical AD pattern, and chronic/relapsing course each had a sensitivity that exceeded 96%. This was followed by the “important” criteria of early age of onset, atopy, and xerosis, which had a sensitivity that ranged between 88% and 95%, while the associated criteria had a sensitivity that ranged between 50% and 85%.
Next, the researchers systematically tested all combinations of the AAD criteria and found that three “essential” AAD criteria, two or more of the “important” criteria, and one or more of the “associated” criteria were optimal in diagnosing AD. Mr. Udkoff noted that the findings can be translated into a simple “3-2-1 rule” that “is both practical and pragmatic,” he said. Using this rule, sensitivity was 91.4% and specificity was 95.2%.
Currently, the researchers are working to validate this criteria in different subgroups of patients. To date, they have found that children younger than 1.5 years get one bonus “essential” criteria for being an infant, so for that population a 2-2-1 rule would apply.
Mr. Udkoff reported that the research was supported by a training grant from the National Institutes of Health. He reported having no financial disclosures.
CHICAGO – A streamlined set of diagnostic criteria from the American Academy of Dermatology’s most recent consensus criteria for diagnosing atopic dermatitis (AD) produced a specificity of more than 95%, and was also highly sensitive, a prospective analysis found.
“Atopic dermatitis typically presents in childhood and is associated with a worsened quality of life, with severe itch and lack of sleep, and substantial health care costs due to therapeutic management and increased hospitalizations,” study author Jeremy Udkoff said at the World Congress of Pediatric Dermatology. “We also know that in order to treat the disease and to learn more about it, we have to have a good tool for diagnosing it. When it comes to clinical studies and research, we require a systematic and refined set of criteria.”
The next set of commonly used criteria to appear were created by the U.K. working party, for which researchers used logistic regression to systematically create a minimum set of effective criteria for AD (Br J Dermatol. 1994;131[3]:383-96). For these guidelines, meeting a diagnosis of AD requires an itchy skin condition, followed by three or more of the following: a history of flexural involvement; a personal history of asthma or hay fever; a history of general dry skin in the last year; visible flexural eczema, and onset under the age of 2 years. “A subsequent validation trial found [the U.K. working party criteria] to have a low sensitivity, which as you can imagine, could be a very large problem,” he said (Arch Dermatol. 1999;135[5]:514-6).
In 2001, the AAD consensus conference created revised hierarchical criteria known as the AAD consensus criteria (J Am Acad Dermatol. 2003;49[6]:1088-95). “These were initially created for more of a gestalt-type picture of AD in the clinic, but because it flows so well, it’s currently being used in about one-third of clinical trials,” Mr. Udkoff said. “However, [the AAD criteria] have not been validated, so we didn’t know its sensitivity or specificity. In addition, we didn’t have a ‘checkbox’ form that tells us how many of each of the criteria are required to make the diagnosis. We didn’t know how many ‘essential,’ ‘important,’ or ‘associated’ features we need to make this diagnosis.”
For the current study, he and his associates set out to determine how many “essential,” “important,” and “associated” criteria are necessary to make the AAD consensus criteria work. They also set out to create a usable checkbox form, validate the criteria, and compare it to the Hanifin-Rajka (HR) and U.K. criteria. To accomplish this, they created a questionnaire comprised of HR, U.K., and AAD criteria, examined the criteria on 60 subjects with and without AD, and compared the diagnostic features of each of those criteria against a gold standard dermatology diagnosis from one of seven pediatric dermatologists. Next, they ranked all 56 possible AAD criterion combinations based on their overall sensitivity and specificity, and chose the most predictive combination. “Once we had the optimal set of criteria, we validated it on a new cohort to determine its sensitivity and specificity, and compared it with the classic HR and U.K. criteria,” Mr. Udkoff explained.
Overall, the researchers evaluated findings from 100 subjects: 58 with AD, and 42 controls. Those with AD were about 3 years younger, compared with controls (a mean age of 5 years vs. about 8 years, respectively). About 40% of patients were Hispanic and about 30% were white. Mr. Udkoff and his associates confirmed the hierarchical structure of the AAD criteria and found that individual “essential” AAD criteria of pruritus, typical AD pattern, and chronic/relapsing course each had a sensitivity that exceeded 96%. This was followed by the “important” criteria of early age of onset, atopy, and xerosis, which had a sensitivity that ranged between 88% and 95%, while the associated criteria had a sensitivity that ranged between 50% and 85%.
Next, the researchers systematically tested all combinations of the AAD criteria and found that three “essential” AAD criteria, two or more of the “important” criteria, and one or more of the “associated” criteria were optimal in diagnosing AD. Mr. Udkoff noted that the findings can be translated into a simple “3-2-1 rule” that “is both practical and pragmatic,” he said. Using this rule, sensitivity was 91.4% and specificity was 95.2%.
Currently, the researchers are working to validate this criteria in different subgroups of patients. To date, they have found that children younger than 1.5 years get one bonus “essential” criteria for being an infant, so for that population a 2-2-1 rule would apply.
Mr. Udkoff reported that the research was supported by a training grant from the National Institutes of Health. He reported having no financial disclosures.
AT WCPD 2017
Key clinical point:
Major finding: The “important” AD criteria of early age of onset, atopy, and xerosis had a sensitivity that ranged between 88% and 95%.
Data source: An analysis of optimal AAD criteria for AD that included 58 patients with AD and 42 controls.
Disclosures: Mr. Udkoff reported that the research was supported by a training grant from the National Institutes of Health. He reported having no financial disclosures.
Tips for navigating pityriasis lichenoides in children
CHICAGO – Pityriasis lichenoides chronica (PLC) is a benign, chronic disorder that can last from 1 to 5 years, yet no formal treatment standards exist.
“Topical corticosteroids will not alter the course of the disease, but they provide symptomatic relief when there is pruritus,” Sibel Ersoy-Evans, MD, said at the World Congress of Pediatric Dermatology. “Oral antibiotics, especially erythromycin, and phototherapy are the most common modalities used in children. Systemic immunosuppressants are rarely needed.”
Dr. Ersoy-Evans, a dermatologist at Hacettepe University in Ankara, Turkey, described pityriasis lichenoides (PL) as a spectrum with two polar ends: PLC and pityriasis lichenoides et varioliformis acuta (PLEVA). Its incidence and prevalence are unknown, but age distribution peaks at 2, 5, and 10 years of age. The average age of onset is 6.5 years, it’s slightly more common in boys, and there is no racial or ethnic predilection.
PLEVA presents with symmetrical, reddish-brown papules that later evolve into vesicular, purpuric, necrotic papules. The scalp, face, mucosa, palms, and soles are spared. Resolution occurs with varioliform scarring and dyspigmentation in 2-18 months. On the other hand, chronic PL is characterized by asymptomatic scaly papules and plaques. The lesions start de novo or evolve from PLEVA lesions and they resolve with hypo-hyperpigmentation in about 8-20 months. In what is believed to be the largest study of its kind, Dr. Ersoy-Evans and her associates respectively reviewed 124 patients with PL. They observed that 57% of patients had PLEVA, primarily those who were white, and 37% had PLC (J Am Acad Dermatol. 2007;56[2]:205-10). The median age of onset was 6 years in PLC patients, versus 5 years in those with PLEVA. Age peaks were observed at 2-3 years and 5-7 years.
They also observed that 30% of cases had a history of recent infection prior to the eruption. “Most patients had generalized distribution, and there wasn’t a significant difference in duration according to the distribution of the lesions,” she said. “Postinflammatory pigmentary changes were observed in 90% of the cases, most commonly hypopigmentation. Pruritus was the most common symptom observed, and the disease was relapsing/recurring in 77% of patients. The duration was longer in PL patients.”
A separate study of 25 children and 32 adults with PL found that hypopigmentation was more common in children (72% vs. 19%, respectively), fewer children experienced remission (20% vs. 78%), and phototherapy was more effective than oral antibiotics in children (Br J Dermatol. 2007;157[5]:941-5).
PL runs a chronic, remitting course in 25%-77% of patients, but there are infrequent reports about progression into cutaneous T-cell lymphoma. “We have not observed any lymphoma development in our cohort with a 10-year follow-up, but long-term follow-up is necessary, especially if clonality is present,” Dr. Ersoy-Evans said. The diagnosis of PL is mostly clinical, but histologic examination may be necessary in some cases. “CD8-positive cells predominate in PLEVA, whereas CD4-positive cells predominate in PLC,” she said.
In the review of 124 patients that she and her associates published in 2007, erythromycin estolate or ethylsuccinate was administered to 80% of children, and 67% of these children showed at least a partial response, with a median response time of 2 months.
In a separate study of 24 children with PL, the response rate was highest (83%) after 3.8 months of erythromycin therapy (Ped Dermatol. 2012; 29[6]:719-24). Dr. Ersoy-Evans stated that oral erythromycin should be continued at least 2 months for a response.
A separate study evaluated the use of phototherapy in 18 children with PLC for a mean of 3.7 months (Ped Dermatol 2008;25[6]:599-605). Among 12 patients who received broadband ultraviolet B radiation for a mean of 3.7 months, the response rate was 83%, which was achieved after a mean of 18 sessions. All five patients who received narrow-band UVB cleared, and the mean number of sessions for a response was 22. One patient who received psoralen and ultraviolet A therapy had no response. A larger review of the published literature found that the delivery of narrow-band UVB has the lowest recurrence rate in PL patients treated with phototherapy (Am J Clin Dermatol. 2016;17:583-91). “Given the efficacy and favorable side effects, I think that phototherapy can be a promising option for PL in children,” Dr. Ersoy-Evans said.
She reported having no financial disclosures
CHICAGO – Pityriasis lichenoides chronica (PLC) is a benign, chronic disorder that can last from 1 to 5 years, yet no formal treatment standards exist.
“Topical corticosteroids will not alter the course of the disease, but they provide symptomatic relief when there is pruritus,” Sibel Ersoy-Evans, MD, said at the World Congress of Pediatric Dermatology. “Oral antibiotics, especially erythromycin, and phototherapy are the most common modalities used in children. Systemic immunosuppressants are rarely needed.”
Dr. Ersoy-Evans, a dermatologist at Hacettepe University in Ankara, Turkey, described pityriasis lichenoides (PL) as a spectrum with two polar ends: PLC and pityriasis lichenoides et varioliformis acuta (PLEVA). Its incidence and prevalence are unknown, but age distribution peaks at 2, 5, and 10 years of age. The average age of onset is 6.5 years, it’s slightly more common in boys, and there is no racial or ethnic predilection.
PLEVA presents with symmetrical, reddish-brown papules that later evolve into vesicular, purpuric, necrotic papules. The scalp, face, mucosa, palms, and soles are spared. Resolution occurs with varioliform scarring and dyspigmentation in 2-18 months. On the other hand, chronic PL is characterized by asymptomatic scaly papules and plaques. The lesions start de novo or evolve from PLEVA lesions and they resolve with hypo-hyperpigmentation in about 8-20 months. In what is believed to be the largest study of its kind, Dr. Ersoy-Evans and her associates respectively reviewed 124 patients with PL. They observed that 57% of patients had PLEVA, primarily those who were white, and 37% had PLC (J Am Acad Dermatol. 2007;56[2]:205-10). The median age of onset was 6 years in PLC patients, versus 5 years in those with PLEVA. Age peaks were observed at 2-3 years and 5-7 years.
They also observed that 30% of cases had a history of recent infection prior to the eruption. “Most patients had generalized distribution, and there wasn’t a significant difference in duration according to the distribution of the lesions,” she said. “Postinflammatory pigmentary changes were observed in 90% of the cases, most commonly hypopigmentation. Pruritus was the most common symptom observed, and the disease was relapsing/recurring in 77% of patients. The duration was longer in PL patients.”
A separate study of 25 children and 32 adults with PL found that hypopigmentation was more common in children (72% vs. 19%, respectively), fewer children experienced remission (20% vs. 78%), and phototherapy was more effective than oral antibiotics in children (Br J Dermatol. 2007;157[5]:941-5).
PL runs a chronic, remitting course in 25%-77% of patients, but there are infrequent reports about progression into cutaneous T-cell lymphoma. “We have not observed any lymphoma development in our cohort with a 10-year follow-up, but long-term follow-up is necessary, especially if clonality is present,” Dr. Ersoy-Evans said. The diagnosis of PL is mostly clinical, but histologic examination may be necessary in some cases. “CD8-positive cells predominate in PLEVA, whereas CD4-positive cells predominate in PLC,” she said.
In the review of 124 patients that she and her associates published in 2007, erythromycin estolate or ethylsuccinate was administered to 80% of children, and 67% of these children showed at least a partial response, with a median response time of 2 months.
In a separate study of 24 children with PL, the response rate was highest (83%) after 3.8 months of erythromycin therapy (Ped Dermatol. 2012; 29[6]:719-24). Dr. Ersoy-Evans stated that oral erythromycin should be continued at least 2 months for a response.
A separate study evaluated the use of phototherapy in 18 children with PLC for a mean of 3.7 months (Ped Dermatol 2008;25[6]:599-605). Among 12 patients who received broadband ultraviolet B radiation for a mean of 3.7 months, the response rate was 83%, which was achieved after a mean of 18 sessions. All five patients who received narrow-band UVB cleared, and the mean number of sessions for a response was 22. One patient who received psoralen and ultraviolet A therapy had no response. A larger review of the published literature found that the delivery of narrow-band UVB has the lowest recurrence rate in PL patients treated with phototherapy (Am J Clin Dermatol. 2016;17:583-91). “Given the efficacy and favorable side effects, I think that phototherapy can be a promising option for PL in children,” Dr. Ersoy-Evans said.
She reported having no financial disclosures
CHICAGO – Pityriasis lichenoides chronica (PLC) is a benign, chronic disorder that can last from 1 to 5 years, yet no formal treatment standards exist.
“Topical corticosteroids will not alter the course of the disease, but they provide symptomatic relief when there is pruritus,” Sibel Ersoy-Evans, MD, said at the World Congress of Pediatric Dermatology. “Oral antibiotics, especially erythromycin, and phototherapy are the most common modalities used in children. Systemic immunosuppressants are rarely needed.”
Dr. Ersoy-Evans, a dermatologist at Hacettepe University in Ankara, Turkey, described pityriasis lichenoides (PL) as a spectrum with two polar ends: PLC and pityriasis lichenoides et varioliformis acuta (PLEVA). Its incidence and prevalence are unknown, but age distribution peaks at 2, 5, and 10 years of age. The average age of onset is 6.5 years, it’s slightly more common in boys, and there is no racial or ethnic predilection.
PLEVA presents with symmetrical, reddish-brown papules that later evolve into vesicular, purpuric, necrotic papules. The scalp, face, mucosa, palms, and soles are spared. Resolution occurs with varioliform scarring and dyspigmentation in 2-18 months. On the other hand, chronic PL is characterized by asymptomatic scaly papules and plaques. The lesions start de novo or evolve from PLEVA lesions and they resolve with hypo-hyperpigmentation in about 8-20 months. In what is believed to be the largest study of its kind, Dr. Ersoy-Evans and her associates respectively reviewed 124 patients with PL. They observed that 57% of patients had PLEVA, primarily those who were white, and 37% had PLC (J Am Acad Dermatol. 2007;56[2]:205-10). The median age of onset was 6 years in PLC patients, versus 5 years in those with PLEVA. Age peaks were observed at 2-3 years and 5-7 years.
They also observed that 30% of cases had a history of recent infection prior to the eruption. “Most patients had generalized distribution, and there wasn’t a significant difference in duration according to the distribution of the lesions,” she said. “Postinflammatory pigmentary changes were observed in 90% of the cases, most commonly hypopigmentation. Pruritus was the most common symptom observed, and the disease was relapsing/recurring in 77% of patients. The duration was longer in PL patients.”
A separate study of 25 children and 32 adults with PL found that hypopigmentation was more common in children (72% vs. 19%, respectively), fewer children experienced remission (20% vs. 78%), and phototherapy was more effective than oral antibiotics in children (Br J Dermatol. 2007;157[5]:941-5).
PL runs a chronic, remitting course in 25%-77% of patients, but there are infrequent reports about progression into cutaneous T-cell lymphoma. “We have not observed any lymphoma development in our cohort with a 10-year follow-up, but long-term follow-up is necessary, especially if clonality is present,” Dr. Ersoy-Evans said. The diagnosis of PL is mostly clinical, but histologic examination may be necessary in some cases. “CD8-positive cells predominate in PLEVA, whereas CD4-positive cells predominate in PLC,” she said.
In the review of 124 patients that she and her associates published in 2007, erythromycin estolate or ethylsuccinate was administered to 80% of children, and 67% of these children showed at least a partial response, with a median response time of 2 months.
In a separate study of 24 children with PL, the response rate was highest (83%) after 3.8 months of erythromycin therapy (Ped Dermatol. 2012; 29[6]:719-24). Dr. Ersoy-Evans stated that oral erythromycin should be continued at least 2 months for a response.
A separate study evaluated the use of phototherapy in 18 children with PLC for a mean of 3.7 months (Ped Dermatol 2008;25[6]:599-605). Among 12 patients who received broadband ultraviolet B radiation for a mean of 3.7 months, the response rate was 83%, which was achieved after a mean of 18 sessions. All five patients who received narrow-band UVB cleared, and the mean number of sessions for a response was 22. One patient who received psoralen and ultraviolet A therapy had no response. A larger review of the published literature found that the delivery of narrow-band UVB has the lowest recurrence rate in PL patients treated with phototherapy (Am J Clin Dermatol. 2016;17:583-91). “Given the efficacy and favorable side effects, I think that phototherapy can be a promising option for PL in children,” Dr. Ersoy-Evans said.
She reported having no financial disclosures
AT WCPD 2017
Care of infants with ichthyosis requires ‘all hands on deck’
CHICAGO – The neonatal period and early infancy are especially critical for patients with ichthyosis, because compromised barrier function increases risk for morbidity and mortality.
“There are minimal data presently to guide management of patients with ichthyosis, making it a time of uncertainty,” Brittany Craiglow, MD, said at the World Congress of Pediatric Dermatology. “You’re going to want to get all hands on deck for the care of these patients. And don’t forget about the family – involve them in the care as much as possible. Reassure them; normalize their feelings, acknowledge them.”
There are six general phenotypes of ichthyosis that differ from the eventual “mature” phenotype and are associated with numerous genes: collodion baby, armor-like scale, exuberant vernix, erythroderma and scale, bullae and erosions, and generalized scale.
The collodion baby phenotype is characterized by a shiny parchment-like membrane that covers the baby’s body, ectropion, and fissures, and is commonly associated with autosomal recessive congenital ichthyosis (ARCI). “About 10% of babies with ARCI are self healing, so they’ll go on to have largely normal skin,” Dr. Craiglow said. Guidelines for managing this phenotype can be found in the Journal of the American Academy of Dermatology (2012 Dec;67[6]:1362-74).
Armor-like scale is pathognomonic for harlequin ichthyosis. “This condition is associated with the highest mortality in the neonatal period,” she said. “In addition to the potential complications associated with other phenotypes, babies with harlequin ichthyosis can also have issues related to constriction of movement and flexibility and digital ischemia.” Tips for practical management of this phenotype were published online in the journal Pediatrics (2017 Jan;139[1]).
The exuberant vernix/cephalic hyperkeratosis phenotype generally appears in children with keratitis-ichthyosis-deafness syndrome (KID) and ichthyosis prematurity syndrome (IPS). Special considerations in KID include a hearing test and ophthalmology exam, while special considerations in IPS include respiratory compromise and atopic diathesis. Electron microscopy is diagnostic, characterized by curvilinear bodies in the granular layer.
The erythroderma and scale phenotype occurs most commonly in ARCI and Netherton syndrome. “Special considerations in Netherton syndrome include failure to thrive/growth failure,” Dr. Craiglow said. “Hair shaft abnormalities are usually present later, and nutritional support is really important.”
Bullae and erosions are hallmark signs of epidermolytic/superficial ichthyosis. On biopsy, epidermolytic hyperkeratosis is diagnostic for this phenotype. At the same time, cases with normal skin or xerosis are suggestive of X-linked ichthyosis, ichthyosis vulgaris, erythrokeratodermas, and Sjögren-Larsson syndrome.
Genetic testing for ichthyosis is generally readily available, Dr. Craiglow said. She advised clinicians to obtain a sample soon after birth to confirm the clinical diagnosis, assist with assessing prognosis, and enable genetic counseling. “It’s important to help identify those at risk for systemic complications,” she said. “Obtaining insurance coverage may be easier when sent during hospital admission.”
Babies with moderate to severe congenital ichthyosis are typically cared for in the neonatal ICU of a tertiary care center by a multidisciplinary team consisting of neonatology, dermatology, nursing, nutrition, and genetics, as well as ophthalmology, otolaryngology, orthopedics, plastic surgery, and spiritual/religious services in many cases. “These babies often have impaired thermoregulation,” Dr. Craiglow said. “They need to be in an isolette, generally with humidity somewhere between 50% and 70% – you don’t want it too high, because they can overheat. It’s also important to get them out of the isolette and into an open crib when they’re ready. That can help with bonding and has been shown to decrease hospital stay.”
Infection is a common culprit for morbidity and mortality. “In general, there are not a lot of data to guide our management; but generally, we don’t recommend prophylactic antibiotics,” Dr. Craiglow said. “Some people do surveillance cultures just to know what microbes are there in case there are signs of infection. Look for level of alertness, because they’re not always going to have a fever. Look for hemodynamic instability, irritability, or poor feeding, and have a low threshold to do your cultures and treat if necessary.”
Pain control is an imperative aspect of pain management.
“Typical newborn pain parameters of facial expression and extremity tone may be hard to interpret,” she said. “Look at heart rate, blood pressure, crying, level of arousal, and have a low threshold to treat for pain, especially prior to changing dressings. Acetaminophen and NSAIDs and even opioids in some cases might be indicated. Families want to know that pain is being adequately controlled.”
Retinoids are generally used in patients with harlequin ichthyosis. “In the United States, we generally use acitretin, but there is no liquid formulation, so you have to enlist help from a compounding pharmacy to mix a formulation of 0.5-0.1 mg/kg per day,” Dr. Craiglow said. “You want to start as soon as you can. Topical retinoids such as tazarotene are also an option.”
Resources that she recommends for parents include the Foundation for Ichthyosis and Related Skin Types, the Ichthyosis Support Group, and the European Network for Ichthyosis.
Dr. Craiglow reported having no relevant financial disclosures.
CHICAGO – The neonatal period and early infancy are especially critical for patients with ichthyosis, because compromised barrier function increases risk for morbidity and mortality.
“There are minimal data presently to guide management of patients with ichthyosis, making it a time of uncertainty,” Brittany Craiglow, MD, said at the World Congress of Pediatric Dermatology. “You’re going to want to get all hands on deck for the care of these patients. And don’t forget about the family – involve them in the care as much as possible. Reassure them; normalize their feelings, acknowledge them.”
There are six general phenotypes of ichthyosis that differ from the eventual “mature” phenotype and are associated with numerous genes: collodion baby, armor-like scale, exuberant vernix, erythroderma and scale, bullae and erosions, and generalized scale.
The collodion baby phenotype is characterized by a shiny parchment-like membrane that covers the baby’s body, ectropion, and fissures, and is commonly associated with autosomal recessive congenital ichthyosis (ARCI). “About 10% of babies with ARCI are self healing, so they’ll go on to have largely normal skin,” Dr. Craiglow said. Guidelines for managing this phenotype can be found in the Journal of the American Academy of Dermatology (2012 Dec;67[6]:1362-74).
Armor-like scale is pathognomonic for harlequin ichthyosis. “This condition is associated with the highest mortality in the neonatal period,” she said. “In addition to the potential complications associated with other phenotypes, babies with harlequin ichthyosis can also have issues related to constriction of movement and flexibility and digital ischemia.” Tips for practical management of this phenotype were published online in the journal Pediatrics (2017 Jan;139[1]).
The exuberant vernix/cephalic hyperkeratosis phenotype generally appears in children with keratitis-ichthyosis-deafness syndrome (KID) and ichthyosis prematurity syndrome (IPS). Special considerations in KID include a hearing test and ophthalmology exam, while special considerations in IPS include respiratory compromise and atopic diathesis. Electron microscopy is diagnostic, characterized by curvilinear bodies in the granular layer.
The erythroderma and scale phenotype occurs most commonly in ARCI and Netherton syndrome. “Special considerations in Netherton syndrome include failure to thrive/growth failure,” Dr. Craiglow said. “Hair shaft abnormalities are usually present later, and nutritional support is really important.”
Bullae and erosions are hallmark signs of epidermolytic/superficial ichthyosis. On biopsy, epidermolytic hyperkeratosis is diagnostic for this phenotype. At the same time, cases with normal skin or xerosis are suggestive of X-linked ichthyosis, ichthyosis vulgaris, erythrokeratodermas, and Sjögren-Larsson syndrome.
Genetic testing for ichthyosis is generally readily available, Dr. Craiglow said. She advised clinicians to obtain a sample soon after birth to confirm the clinical diagnosis, assist with assessing prognosis, and enable genetic counseling. “It’s important to help identify those at risk for systemic complications,” she said. “Obtaining insurance coverage may be easier when sent during hospital admission.”
Babies with moderate to severe congenital ichthyosis are typically cared for in the neonatal ICU of a tertiary care center by a multidisciplinary team consisting of neonatology, dermatology, nursing, nutrition, and genetics, as well as ophthalmology, otolaryngology, orthopedics, plastic surgery, and spiritual/religious services in many cases. “These babies often have impaired thermoregulation,” Dr. Craiglow said. “They need to be in an isolette, generally with humidity somewhere between 50% and 70% – you don’t want it too high, because they can overheat. It’s also important to get them out of the isolette and into an open crib when they’re ready. That can help with bonding and has been shown to decrease hospital stay.”
Infection is a common culprit for morbidity and mortality. “In general, there are not a lot of data to guide our management; but generally, we don’t recommend prophylactic antibiotics,” Dr. Craiglow said. “Some people do surveillance cultures just to know what microbes are there in case there are signs of infection. Look for level of alertness, because they’re not always going to have a fever. Look for hemodynamic instability, irritability, or poor feeding, and have a low threshold to do your cultures and treat if necessary.”
Pain control is an imperative aspect of pain management.
“Typical newborn pain parameters of facial expression and extremity tone may be hard to interpret,” she said. “Look at heart rate, blood pressure, crying, level of arousal, and have a low threshold to treat for pain, especially prior to changing dressings. Acetaminophen and NSAIDs and even opioids in some cases might be indicated. Families want to know that pain is being adequately controlled.”
Retinoids are generally used in patients with harlequin ichthyosis. “In the United States, we generally use acitretin, but there is no liquid formulation, so you have to enlist help from a compounding pharmacy to mix a formulation of 0.5-0.1 mg/kg per day,” Dr. Craiglow said. “You want to start as soon as you can. Topical retinoids such as tazarotene are also an option.”
Resources that she recommends for parents include the Foundation for Ichthyosis and Related Skin Types, the Ichthyosis Support Group, and the European Network for Ichthyosis.
Dr. Craiglow reported having no relevant financial disclosures.
CHICAGO – The neonatal period and early infancy are especially critical for patients with ichthyosis, because compromised barrier function increases risk for morbidity and mortality.
“There are minimal data presently to guide management of patients with ichthyosis, making it a time of uncertainty,” Brittany Craiglow, MD, said at the World Congress of Pediatric Dermatology. “You’re going to want to get all hands on deck for the care of these patients. And don’t forget about the family – involve them in the care as much as possible. Reassure them; normalize their feelings, acknowledge them.”
There are six general phenotypes of ichthyosis that differ from the eventual “mature” phenotype and are associated with numerous genes: collodion baby, armor-like scale, exuberant vernix, erythroderma and scale, bullae and erosions, and generalized scale.
The collodion baby phenotype is characterized by a shiny parchment-like membrane that covers the baby’s body, ectropion, and fissures, and is commonly associated with autosomal recessive congenital ichthyosis (ARCI). “About 10% of babies with ARCI are self healing, so they’ll go on to have largely normal skin,” Dr. Craiglow said. Guidelines for managing this phenotype can be found in the Journal of the American Academy of Dermatology (2012 Dec;67[6]:1362-74).
Armor-like scale is pathognomonic for harlequin ichthyosis. “This condition is associated with the highest mortality in the neonatal period,” she said. “In addition to the potential complications associated with other phenotypes, babies with harlequin ichthyosis can also have issues related to constriction of movement and flexibility and digital ischemia.” Tips for practical management of this phenotype were published online in the journal Pediatrics (2017 Jan;139[1]).
The exuberant vernix/cephalic hyperkeratosis phenotype generally appears in children with keratitis-ichthyosis-deafness syndrome (KID) and ichthyosis prematurity syndrome (IPS). Special considerations in KID include a hearing test and ophthalmology exam, while special considerations in IPS include respiratory compromise and atopic diathesis. Electron microscopy is diagnostic, characterized by curvilinear bodies in the granular layer.
The erythroderma and scale phenotype occurs most commonly in ARCI and Netherton syndrome. “Special considerations in Netherton syndrome include failure to thrive/growth failure,” Dr. Craiglow said. “Hair shaft abnormalities are usually present later, and nutritional support is really important.”
Bullae and erosions are hallmark signs of epidermolytic/superficial ichthyosis. On biopsy, epidermolytic hyperkeratosis is diagnostic for this phenotype. At the same time, cases with normal skin or xerosis are suggestive of X-linked ichthyosis, ichthyosis vulgaris, erythrokeratodermas, and Sjögren-Larsson syndrome.
Genetic testing for ichthyosis is generally readily available, Dr. Craiglow said. She advised clinicians to obtain a sample soon after birth to confirm the clinical diagnosis, assist with assessing prognosis, and enable genetic counseling. “It’s important to help identify those at risk for systemic complications,” she said. “Obtaining insurance coverage may be easier when sent during hospital admission.”
Babies with moderate to severe congenital ichthyosis are typically cared for in the neonatal ICU of a tertiary care center by a multidisciplinary team consisting of neonatology, dermatology, nursing, nutrition, and genetics, as well as ophthalmology, otolaryngology, orthopedics, plastic surgery, and spiritual/religious services in many cases. “These babies often have impaired thermoregulation,” Dr. Craiglow said. “They need to be in an isolette, generally with humidity somewhere between 50% and 70% – you don’t want it too high, because they can overheat. It’s also important to get them out of the isolette and into an open crib when they’re ready. That can help with bonding and has been shown to decrease hospital stay.”
Infection is a common culprit for morbidity and mortality. “In general, there are not a lot of data to guide our management; but generally, we don’t recommend prophylactic antibiotics,” Dr. Craiglow said. “Some people do surveillance cultures just to know what microbes are there in case there are signs of infection. Look for level of alertness, because they’re not always going to have a fever. Look for hemodynamic instability, irritability, or poor feeding, and have a low threshold to do your cultures and treat if necessary.”
Pain control is an imperative aspect of pain management.
“Typical newborn pain parameters of facial expression and extremity tone may be hard to interpret,” she said. “Look at heart rate, blood pressure, crying, level of arousal, and have a low threshold to treat for pain, especially prior to changing dressings. Acetaminophen and NSAIDs and even opioids in some cases might be indicated. Families want to know that pain is being adequately controlled.”
Retinoids are generally used in patients with harlequin ichthyosis. “In the United States, we generally use acitretin, but there is no liquid formulation, so you have to enlist help from a compounding pharmacy to mix a formulation of 0.5-0.1 mg/kg per day,” Dr. Craiglow said. “You want to start as soon as you can. Topical retinoids such as tazarotene are also an option.”
Resources that she recommends for parents include the Foundation for Ichthyosis and Related Skin Types, the Ichthyosis Support Group, and the European Network for Ichthyosis.
Dr. Craiglow reported having no relevant financial disclosures.
AT WCPD 2017
Precise cause of pityriasis rosea remains elusive
CHICAGO – Pityriasis rosea was recognized as early as 1798, yet its precise cause remains elusive.
“We still don’t have a lot of information on it, because it’s self limited and it resolves,” John C. Browning, MD, said at the World Congress of Pediatric Dermatology. “There hasn’t been quite as much of a burning push for research into pityriasis rosea as there has been for pityriasis rubra pilaris, for instance.”
Classic pityriasis rosea (PR) is characterized by oval scaly, erythematous lesions on the trunk and extremities, sparing the face, scalp, palms, and soles, said Dr. Browning, a pediatric dermatologist who is chief of dermatology at the Children’s Hospital of San Antonio, Texas. The hallmark sign is a so-called herald patch, an oval, slightly scaly patch with a pale center, which usually appears on the trunk and remains isolated for about 2 weeks before the generalized papulosquamous eruption begins. This typically lasts for about 45 days but can range from 2 weeks to 5 months.
Symptoms of PR may include malaise, nausea, loss of appetite, headache, difficulty concentrating, irritability, gastrointestinal upset, upper respiratory symptoms, joint pain, lymph node swelling, sore throat, and low-grade fever. Pruritus is variable, both in frequency and in intensity, and can be exacerbated by topical medications. Some studies have found a higher female-to-male ratio, while other studies have shown no such association.
“Only 6% of PR cases have been reported in children under 10 years of age,” Dr. Browning said. “PR in dark-skinned children tends to have more facial involvement and a scaly appearance, compared with lighter-skinned children.”
Human herpesvirus (HHV) 6 and HHV 7, members of the Roseolovirus genus of HHVs, have been implicated in triggering PR. These viruses cause a primary infection and can establish latent infection with reactivation if altered immunity develops.
“That’s probably why we don’t see PR in younger children, because that’s when the primary infection is happening,” Dr. Browning said. “These viruses are commonly acquired during childhood, with adult seroprevalence in the range of 80%-90%. Latency occurs in monocytes, bone marrow progenitor cells, in salivary glands, the brain, and in the kidneys, so it’s pretty widespread.”
He added that controversy exists as to whether HHV 6 and 7 cause PR, because older diagnostic methods only detected the presence of HHV DNA, rather than viral load. “HHV reactivation, rather than primary infection, is more likely the cause of PR as supported by sporadic occurrence of PR, reduced contagiousness, age of PR onset, possible relapse during a limited span of time, and frequent occurrence after stress or immunosuppressive states such as pregnancy,” Dr. Browning said.
Classical presentations of PR are not clinically worrisome, but atypical cases could indicate other triggers, such as drug-induced PR. “This is typically more itchy, can have mucous membrane involvement, and you’re not going to see the herald patch,” he said. “It’s more likely to be confluent with no prodromal symptoms.” Agents that have been implicated in drug-induced PR include isotretinoin, terbinafine, and adalimumab.
Other conditions that can trigger PR include secondary syphilis (characterized by involvement of the palms and soles, lymphadenopathy, and greater lesional infiltration); seborrheic dermatitis (characterized by greater involvement of the scalp and other hairy parts of the body); nummular eczema (more pruritic); and pityriasis lichenoides chronica, which involves more chronic and relapsing lesions. Histology of PR reveals focal parakeratosis in the epidermis in mounds with exocytosis of lymphocytes, variable spongiosis, mild acanthosis, and a thinned granular layer.
Dr. Browning noted that PR is more common in pregnancy. One study of 38 pregnant women with PR found that 13% miscarried before 16 weeks, compared with a normal miscarriage rate of 10% (J Am Acad Dermatol. 2008 May; 58[5 Suppl 1]:S78-83). “Neonatal hypotonia, weal motility, and hyporeactivity have been reported,” he said. “And with immunocompromised patients, you might see a longer, protracted course of PR.”
Although no treatment is recommended for classical cases of PR, Dr. Browning said that topical steroids “are widely employed because we all want to do something, especially if there’s some pruritus.” According to a position statement on the management of patients with PR, erythromycin has been reported to shorten the duration of rash and pruritus, but it can cause gastrointestinal disturbance (J Eur Acad Dermatol Venereol. 2016 Oct;30[10]:1670-81). Acyclovir has been reported to hasten clearance of PR in one placebo-controlled study, but PR has also been reported in another patient taking low doses of acyclovir. Phototherapy has also been found to be beneficial.
Dr. Browning reported having no financial disclosures.
CHICAGO – Pityriasis rosea was recognized as early as 1798, yet its precise cause remains elusive.
“We still don’t have a lot of information on it, because it’s self limited and it resolves,” John C. Browning, MD, said at the World Congress of Pediatric Dermatology. “There hasn’t been quite as much of a burning push for research into pityriasis rosea as there has been for pityriasis rubra pilaris, for instance.”
Classic pityriasis rosea (PR) is characterized by oval scaly, erythematous lesions on the trunk and extremities, sparing the face, scalp, palms, and soles, said Dr. Browning, a pediatric dermatologist who is chief of dermatology at the Children’s Hospital of San Antonio, Texas. The hallmark sign is a so-called herald patch, an oval, slightly scaly patch with a pale center, which usually appears on the trunk and remains isolated for about 2 weeks before the generalized papulosquamous eruption begins. This typically lasts for about 45 days but can range from 2 weeks to 5 months.
Symptoms of PR may include malaise, nausea, loss of appetite, headache, difficulty concentrating, irritability, gastrointestinal upset, upper respiratory symptoms, joint pain, lymph node swelling, sore throat, and low-grade fever. Pruritus is variable, both in frequency and in intensity, and can be exacerbated by topical medications. Some studies have found a higher female-to-male ratio, while other studies have shown no such association.
“Only 6% of PR cases have been reported in children under 10 years of age,” Dr. Browning said. “PR in dark-skinned children tends to have more facial involvement and a scaly appearance, compared with lighter-skinned children.”
Human herpesvirus (HHV) 6 and HHV 7, members of the Roseolovirus genus of HHVs, have been implicated in triggering PR. These viruses cause a primary infection and can establish latent infection with reactivation if altered immunity develops.
“That’s probably why we don’t see PR in younger children, because that’s when the primary infection is happening,” Dr. Browning said. “These viruses are commonly acquired during childhood, with adult seroprevalence in the range of 80%-90%. Latency occurs in monocytes, bone marrow progenitor cells, in salivary glands, the brain, and in the kidneys, so it’s pretty widespread.”
He added that controversy exists as to whether HHV 6 and 7 cause PR, because older diagnostic methods only detected the presence of HHV DNA, rather than viral load. “HHV reactivation, rather than primary infection, is more likely the cause of PR as supported by sporadic occurrence of PR, reduced contagiousness, age of PR onset, possible relapse during a limited span of time, and frequent occurrence after stress or immunosuppressive states such as pregnancy,” Dr. Browning said.
Classical presentations of PR are not clinically worrisome, but atypical cases could indicate other triggers, such as drug-induced PR. “This is typically more itchy, can have mucous membrane involvement, and you’re not going to see the herald patch,” he said. “It’s more likely to be confluent with no prodromal symptoms.” Agents that have been implicated in drug-induced PR include isotretinoin, terbinafine, and adalimumab.
Other conditions that can trigger PR include secondary syphilis (characterized by involvement of the palms and soles, lymphadenopathy, and greater lesional infiltration); seborrheic dermatitis (characterized by greater involvement of the scalp and other hairy parts of the body); nummular eczema (more pruritic); and pityriasis lichenoides chronica, which involves more chronic and relapsing lesions. Histology of PR reveals focal parakeratosis in the epidermis in mounds with exocytosis of lymphocytes, variable spongiosis, mild acanthosis, and a thinned granular layer.
Dr. Browning noted that PR is more common in pregnancy. One study of 38 pregnant women with PR found that 13% miscarried before 16 weeks, compared with a normal miscarriage rate of 10% (J Am Acad Dermatol. 2008 May; 58[5 Suppl 1]:S78-83). “Neonatal hypotonia, weal motility, and hyporeactivity have been reported,” he said. “And with immunocompromised patients, you might see a longer, protracted course of PR.”
Although no treatment is recommended for classical cases of PR, Dr. Browning said that topical steroids “are widely employed because we all want to do something, especially if there’s some pruritus.” According to a position statement on the management of patients with PR, erythromycin has been reported to shorten the duration of rash and pruritus, but it can cause gastrointestinal disturbance (J Eur Acad Dermatol Venereol. 2016 Oct;30[10]:1670-81). Acyclovir has been reported to hasten clearance of PR in one placebo-controlled study, but PR has also been reported in another patient taking low doses of acyclovir. Phototherapy has also been found to be beneficial.
Dr. Browning reported having no financial disclosures.
CHICAGO – Pityriasis rosea was recognized as early as 1798, yet its precise cause remains elusive.
“We still don’t have a lot of information on it, because it’s self limited and it resolves,” John C. Browning, MD, said at the World Congress of Pediatric Dermatology. “There hasn’t been quite as much of a burning push for research into pityriasis rosea as there has been for pityriasis rubra pilaris, for instance.”
Classic pityriasis rosea (PR) is characterized by oval scaly, erythematous lesions on the trunk and extremities, sparing the face, scalp, palms, and soles, said Dr. Browning, a pediatric dermatologist who is chief of dermatology at the Children’s Hospital of San Antonio, Texas. The hallmark sign is a so-called herald patch, an oval, slightly scaly patch with a pale center, which usually appears on the trunk and remains isolated for about 2 weeks before the generalized papulosquamous eruption begins. This typically lasts for about 45 days but can range from 2 weeks to 5 months.
Symptoms of PR may include malaise, nausea, loss of appetite, headache, difficulty concentrating, irritability, gastrointestinal upset, upper respiratory symptoms, joint pain, lymph node swelling, sore throat, and low-grade fever. Pruritus is variable, both in frequency and in intensity, and can be exacerbated by topical medications. Some studies have found a higher female-to-male ratio, while other studies have shown no such association.
“Only 6% of PR cases have been reported in children under 10 years of age,” Dr. Browning said. “PR in dark-skinned children tends to have more facial involvement and a scaly appearance, compared with lighter-skinned children.”
Human herpesvirus (HHV) 6 and HHV 7, members of the Roseolovirus genus of HHVs, have been implicated in triggering PR. These viruses cause a primary infection and can establish latent infection with reactivation if altered immunity develops.
“That’s probably why we don’t see PR in younger children, because that’s when the primary infection is happening,” Dr. Browning said. “These viruses are commonly acquired during childhood, with adult seroprevalence in the range of 80%-90%. Latency occurs in monocytes, bone marrow progenitor cells, in salivary glands, the brain, and in the kidneys, so it’s pretty widespread.”
He added that controversy exists as to whether HHV 6 and 7 cause PR, because older diagnostic methods only detected the presence of HHV DNA, rather than viral load. “HHV reactivation, rather than primary infection, is more likely the cause of PR as supported by sporadic occurrence of PR, reduced contagiousness, age of PR onset, possible relapse during a limited span of time, and frequent occurrence after stress or immunosuppressive states such as pregnancy,” Dr. Browning said.
Classical presentations of PR are not clinically worrisome, but atypical cases could indicate other triggers, such as drug-induced PR. “This is typically more itchy, can have mucous membrane involvement, and you’re not going to see the herald patch,” he said. “It’s more likely to be confluent with no prodromal symptoms.” Agents that have been implicated in drug-induced PR include isotretinoin, terbinafine, and adalimumab.
Other conditions that can trigger PR include secondary syphilis (characterized by involvement of the palms and soles, lymphadenopathy, and greater lesional infiltration); seborrheic dermatitis (characterized by greater involvement of the scalp and other hairy parts of the body); nummular eczema (more pruritic); and pityriasis lichenoides chronica, which involves more chronic and relapsing lesions. Histology of PR reveals focal parakeratosis in the epidermis in mounds with exocytosis of lymphocytes, variable spongiosis, mild acanthosis, and a thinned granular layer.
Dr. Browning noted that PR is more common in pregnancy. One study of 38 pregnant women with PR found that 13% miscarried before 16 weeks, compared with a normal miscarriage rate of 10% (J Am Acad Dermatol. 2008 May; 58[5 Suppl 1]:S78-83). “Neonatal hypotonia, weal motility, and hyporeactivity have been reported,” he said. “And with immunocompromised patients, you might see a longer, protracted course of PR.”
Although no treatment is recommended for classical cases of PR, Dr. Browning said that topical steroids “are widely employed because we all want to do something, especially if there’s some pruritus.” According to a position statement on the management of patients with PR, erythromycin has been reported to shorten the duration of rash and pruritus, but it can cause gastrointestinal disturbance (J Eur Acad Dermatol Venereol. 2016 Oct;30[10]:1670-81). Acyclovir has been reported to hasten clearance of PR in one placebo-controlled study, but PR has also been reported in another patient taking low doses of acyclovir. Phototherapy has also been found to be beneficial.
Dr. Browning reported having no financial disclosures.
EXPERT ANALYSIS FROM WCPD 2017