User login
EULAR, ACR present preliminary recommendations for rare genetic autoinflammatory diseases
As researchers learn more about the genetic etiology of immunopathology, they have been able to more clearly understand rare but debilitating autoinflammatory conditions in ways that have improved identification and management of these diseases. At this year’s European Congress of Rheumatology, two researchers outlined new recommendations from the European Alliance of Associations for Rheumatology (EULAR) and the American College of Rheumatology (ACR) for the management of two groups of such autoinflammatory diseases: interleukin-1-mediated and Type-I interferonopathies, and suspected macrophage activation syndrome and hemophagocytic lymphohistiocytosis.

These are the first recommendations from EULAR for these diseases, according to Loreto Carmona, MD, PhD, chair of the EULAR scientific program committee and scientific director of the Institute for Musculoskeletal Health in Madrid.
“They are rare diseases and there is a great need to standardize diagnosis and care for the safety and outcome of the patients,” Dr. Carmona said in an interview. “These diseases need deep expertise and so the experts are trying, they are still preliminary, to add clarity to their management.” Dr. Carmona was not involved with the development of the guidelines and moderated the session during which they were presented.
“The rapidly emerging knowledge of the genetic causes of novel systemic autoinflammatory diseases, which present typically in early childhood with severe and chronic systemic and organ-specific inflammation, linked the disease pathogenesis to the pathologic production of major proinflammatory cytokines,” presenter Raphaela Goldbach-Mansky, MD, a senior investigator and chief of the translational autoinflammatory disease studies unit of the U.S. National Institute of Allergy and Infectious Diseases, told congress attendees. This greater understanding led to the “targeted and anticytokine treatments that have changed patients’ lives,” she said.
The guidelines relied on the products of three working groups for each disease type. After meeting to come up with clinical questions, the groups each conducted systematic literature reviews through EMBASE, PubMed, and the Cochrane Library for publications dated from 1970 to August 2020 that excluded non-English-language studies, case reports, and animal model or basic science studies. They then met again to develop final consensus statements.
The interferonopathy and interleukin (IL)-1-mediated systemic autoinflammatory diseases (SAIDs) working groups met throughout 2020, and the hemophagocytic lymphohistiocytosis (HLH)/ macrophage activation syndrome (MAS) working group met in March and April of 2021.
“One needs a lot of experience with these diseases to even think about them,” Dr. Carmona said. “We haven’t been presented yet with all the details of the recommendations, but we hope they are clear because they are much needed.”
She noted that these preliminary recommendations are based on the best available evidence to date along with expertise from multidisciplinary panels.
“We need to be acquainted with these recommendations, as the majority of us, either if we are pediatric or adult rheumatologists, will face some problem with these diseases at some point,” Dr. Carmona said.
IL-1-mediated SAIDs
Recommendations for IL-1-mediated SAIDs focused on mevalonate kinase deficiency (MKD), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), cryopyrinopathies (CAPS), and deficiency of the IL-1 receptor antagonist (DIRA). Presentation of these conditions involves chronic or intermittent flares of systemic and organ inflammation that can cause progressive organ damage, morbidity, and increased mortality if not treated. Diagnosis requires a multidisciplinary team whose evaluation should include disease-related complications and long-term care plans.

Diagnostic workup should include genetic testing using next-generation sequencing as this “facilitates initiation of targeted treatments, genetic counseling, and informs prognosis” for patients with CAPS, TRAPS, MKD, and DIRA, Erkan Demirkaya, MD, a scientist at the Children’s Health Research Institute and professor of pediatric rheumatology at the University of Western Ontario in London, Canada, told attendees. Evaluation should also include clinical workup that focuses on the extent of inflammatory organ involvement, and screening for disease- and treatment-related comorbidities.
“The goal of therapy is to control clinical signs and symptoms and normalize laboratory biomarkers of systemic inflammation,” Dr. Demirkaya said. Long-term monitoring goals should focus on the following:
- “Adequate treatment adjusted to the needs of the growing child and prevention of systemic and organ-specific inflammatory manifestations;
- Fostering of self-management skills and medical decision-making;
- Initiating a transition program to adult specialist care in adolescent patients.”
Type-1 interferonopathies
The recommendations for this disease group focused on chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures (CANDLE)/proteasome-associated autoinflammatory syndromes (PRAAS), STING-associated vasculopathy with onset in infancy (SAVI), and Aicardi-Goutières syndrome (AGS).
These patients similarly present with chronic and organ-specific inflammation that leads to progressive organ damage, morbidity, and higher mortality risk when not managed. Each of these diseases requires a confirmed genetic diagnosis so that treatments can be targeted and the patient receives appropriate genetic counseling, screening for complications, and information on prognosis, Dr. Goldbach-Mansky said.
Treatment goals for type-1 interferonopathies are to “reduce systematic and organ inflammation to prevent or limit the development of progression of organ injury or damage and to improve quality of life,” Dr. Goldbach-Mansky told attendees.
Each patient requires a multidisciplinary care provider team that conducts long-term monitoring of disease activity, damage to specific organs, and any treatment-related complications.
Management of HLH/MAS
Early recognition and management of HLH and MAS can be challenging because systemic hyperinflammation exists along an immunopathologic continuum with typically nonspecific clinical and laboratory findings, Dr. Goldbach-Mansky said, but holistic, longitudinal consideration of these findings “are recognizable and warrant prompt diagnostic evaluation.” Even if the patient does not meet all specific diagnostic criteria for HLH/MAS, it may be necessary to begin therapies, she said.
One important point to consider is that “systemic hyperinflammation can be associated with hyperferritinemia and can progress to life-threatening HLH/MAS,” Dr. Goldbach-Mansky said. Further, although “systemic hyperinflammation and HLH/MAS can occur in nearly any inflammatory state,” certain common triggers and predisposing conditions can indicate the need to consider these conditions and begin appropriate treatment if needed. Part of effective management of systemic hyperinflammation and HLH/MAS is determining any modifiable factors contributing to the disease and mitigating or treating those.
HLH/MAS requires urgent intervention based on the patient’s degree of inflammation and extent of organ dysfunction, the recommendations state. Treatment goals include preventing or limiting immunopathology, preserving the integrity of the diagnostic workup, and minimizing therapy-related toxicity.
Dr. Carmona, Dr. Goldbach-Mansky, and Dr. Demirkaya have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
As researchers learn more about the genetic etiology of immunopathology, they have been able to more clearly understand rare but debilitating autoinflammatory conditions in ways that have improved identification and management of these diseases. At this year’s European Congress of Rheumatology, two researchers outlined new recommendations from the European Alliance of Associations for Rheumatology (EULAR) and the American College of Rheumatology (ACR) for the management of two groups of such autoinflammatory diseases: interleukin-1-mediated and Type-I interferonopathies, and suspected macrophage activation syndrome and hemophagocytic lymphohistiocytosis.
These are the first recommendations from EULAR for these diseases, according to Loreto Carmona, MD, PhD, chair of the EULAR scientific program committee and scientific director of the Institute for Musculoskeletal Health in Madrid.
“They are rare diseases and there is a great need to standardize diagnosis and care for the safety and outcome of the patients,” Dr. Carmona said in an interview. “These diseases need deep expertise and so the experts are trying, they are still preliminary, to add clarity to their management.” Dr. Carmona was not involved with the development of the guidelines and moderated the session during which they were presented.
“The rapidly emerging knowledge of the genetic causes of novel systemic autoinflammatory diseases, which present typically in early childhood with severe and chronic systemic and organ-specific inflammation, linked the disease pathogenesis to the pathologic production of major proinflammatory cytokines,” presenter Raphaela Goldbach-Mansky, MD, a senior investigator and chief of the translational autoinflammatory disease studies unit of the U.S. National Institute of Allergy and Infectious Diseases, told congress attendees. This greater understanding led to the “targeted and anticytokine treatments that have changed patients’ lives,” she said.
The guidelines relied on the products of three working groups for each disease type. After meeting to come up with clinical questions, the groups each conducted systematic literature reviews through EMBASE, PubMed, and the Cochrane Library for publications dated from 1970 to August 2020 that excluded non-English-language studies, case reports, and animal model or basic science studies. They then met again to develop final consensus statements.
The interferonopathy and interleukin (IL)-1-mediated systemic autoinflammatory diseases (SAIDs) working groups met throughout 2020, and the hemophagocytic lymphohistiocytosis (HLH)/ macrophage activation syndrome (MAS) working group met in March and April of 2021.
“One needs a lot of experience with these diseases to even think about them,” Dr. Carmona said. “We haven’t been presented yet with all the details of the recommendations, but we hope they are clear because they are much needed.”
She noted that these preliminary recommendations are based on the best available evidence to date along with expertise from multidisciplinary panels.
“We need to be acquainted with these recommendations, as the majority of us, either if we are pediatric or adult rheumatologists, will face some problem with these diseases at some point,” Dr. Carmona said.
IL-1-mediated SAIDs
Recommendations for IL-1-mediated SAIDs focused on mevalonate kinase deficiency (MKD), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), cryopyrinopathies (CAPS), and deficiency of the IL-1 receptor antagonist (DIRA). Presentation of these conditions involves chronic or intermittent flares of systemic and organ inflammation that can cause progressive organ damage, morbidity, and increased mortality if not treated. Diagnosis requires a multidisciplinary team whose evaluation should include disease-related complications and long-term care plans.
Diagnostic workup should include genetic testing using next-generation sequencing as this “facilitates initiation of targeted treatments, genetic counseling, and informs prognosis” for patients with CAPS, TRAPS, MKD, and DIRA, Erkan Demirkaya, MD, a scientist at the Children’s Health Research Institute and professor of pediatric rheumatology at the University of Western Ontario in London, Canada, told attendees. Evaluation should also include clinical workup that focuses on the extent of inflammatory organ involvement, and screening for disease- and treatment-related comorbidities.
“The goal of therapy is to control clinical signs and symptoms and normalize laboratory biomarkers of systemic inflammation,” Dr. Demirkaya said. Long-term monitoring goals should focus on the following:
- “Adequate treatment adjusted to the needs of the growing child and prevention of systemic and organ-specific inflammatory manifestations;
- Fostering of self-management skills and medical decision-making;
- Initiating a transition program to adult specialist care in adolescent patients.”
Type-1 interferonopathies
The recommendations for this disease group focused on chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures (CANDLE)/proteasome-associated autoinflammatory syndromes (PRAAS), STING-associated vasculopathy with onset in infancy (SAVI), and Aicardi-Goutières syndrome (AGS).
These patients similarly present with chronic and organ-specific inflammation that leads to progressive organ damage, morbidity, and higher mortality risk when not managed. Each of these diseases requires a confirmed genetic diagnosis so that treatments can be targeted and the patient receives appropriate genetic counseling, screening for complications, and information on prognosis, Dr. Goldbach-Mansky said.
Treatment goals for type-1 interferonopathies are to “reduce systematic and organ inflammation to prevent or limit the development of progression of organ injury or damage and to improve quality of life,” Dr. Goldbach-Mansky told attendees.
Each patient requires a multidisciplinary care provider team that conducts long-term monitoring of disease activity, damage to specific organs, and any treatment-related complications.
Management of HLH/MAS
Early recognition and management of HLH and MAS can be challenging because systemic hyperinflammation exists along an immunopathologic continuum with typically nonspecific clinical and laboratory findings, Dr. Goldbach-Mansky said, but holistic, longitudinal consideration of these findings “are recognizable and warrant prompt diagnostic evaluation.” Even if the patient does not meet all specific diagnostic criteria for HLH/MAS, it may be necessary to begin therapies, she said.
One important point to consider is that “systemic hyperinflammation can be associated with hyperferritinemia and can progress to life-threatening HLH/MAS,” Dr. Goldbach-Mansky said. Further, although “systemic hyperinflammation and HLH/MAS can occur in nearly any inflammatory state,” certain common triggers and predisposing conditions can indicate the need to consider these conditions and begin appropriate treatment if needed. Part of effective management of systemic hyperinflammation and HLH/MAS is determining any modifiable factors contributing to the disease and mitigating or treating those.
HLH/MAS requires urgent intervention based on the patient’s degree of inflammation and extent of organ dysfunction, the recommendations state. Treatment goals include preventing or limiting immunopathology, preserving the integrity of the diagnostic workup, and minimizing therapy-related toxicity.
Dr. Carmona, Dr. Goldbach-Mansky, and Dr. Demirkaya have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
As researchers learn more about the genetic etiology of immunopathology, they have been able to more clearly understand rare but debilitating autoinflammatory conditions in ways that have improved identification and management of these diseases. At this year’s European Congress of Rheumatology, two researchers outlined new recommendations from the European Alliance of Associations for Rheumatology (EULAR) and the American College of Rheumatology (ACR) for the management of two groups of such autoinflammatory diseases: interleukin-1-mediated and Type-I interferonopathies, and suspected macrophage activation syndrome and hemophagocytic lymphohistiocytosis.
These are the first recommendations from EULAR for these diseases, according to Loreto Carmona, MD, PhD, chair of the EULAR scientific program committee and scientific director of the Institute for Musculoskeletal Health in Madrid.
“They are rare diseases and there is a great need to standardize diagnosis and care for the safety and outcome of the patients,” Dr. Carmona said in an interview. “These diseases need deep expertise and so the experts are trying, they are still preliminary, to add clarity to their management.” Dr. Carmona was not involved with the development of the guidelines and moderated the session during which they were presented.
“The rapidly emerging knowledge of the genetic causes of novel systemic autoinflammatory diseases, which present typically in early childhood with severe and chronic systemic and organ-specific inflammation, linked the disease pathogenesis to the pathologic production of major proinflammatory cytokines,” presenter Raphaela Goldbach-Mansky, MD, a senior investigator and chief of the translational autoinflammatory disease studies unit of the U.S. National Institute of Allergy and Infectious Diseases, told congress attendees. This greater understanding led to the “targeted and anticytokine treatments that have changed patients’ lives,” she said.
The guidelines relied on the products of three working groups for each disease type. After meeting to come up with clinical questions, the groups each conducted systematic literature reviews through EMBASE, PubMed, and the Cochrane Library for publications dated from 1970 to August 2020 that excluded non-English-language studies, case reports, and animal model or basic science studies. They then met again to develop final consensus statements.
The interferonopathy and interleukin (IL)-1-mediated systemic autoinflammatory diseases (SAIDs) working groups met throughout 2020, and the hemophagocytic lymphohistiocytosis (HLH)/ macrophage activation syndrome (MAS) working group met in March and April of 2021.
“One needs a lot of experience with these diseases to even think about them,” Dr. Carmona said. “We haven’t been presented yet with all the details of the recommendations, but we hope they are clear because they are much needed.”
She noted that these preliminary recommendations are based on the best available evidence to date along with expertise from multidisciplinary panels.
“We need to be acquainted with these recommendations, as the majority of us, either if we are pediatric or adult rheumatologists, will face some problem with these diseases at some point,” Dr. Carmona said.
IL-1-mediated SAIDs
Recommendations for IL-1-mediated SAIDs focused on mevalonate kinase deficiency (MKD), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), cryopyrinopathies (CAPS), and deficiency of the IL-1 receptor antagonist (DIRA). Presentation of these conditions involves chronic or intermittent flares of systemic and organ inflammation that can cause progressive organ damage, morbidity, and increased mortality if not treated. Diagnosis requires a multidisciplinary team whose evaluation should include disease-related complications and long-term care plans.
Diagnostic workup should include genetic testing using next-generation sequencing as this “facilitates initiation of targeted treatments, genetic counseling, and informs prognosis” for patients with CAPS, TRAPS, MKD, and DIRA, Erkan Demirkaya, MD, a scientist at the Children’s Health Research Institute and professor of pediatric rheumatology at the University of Western Ontario in London, Canada, told attendees. Evaluation should also include clinical workup that focuses on the extent of inflammatory organ involvement, and screening for disease- and treatment-related comorbidities.
“The goal of therapy is to control clinical signs and symptoms and normalize laboratory biomarkers of systemic inflammation,” Dr. Demirkaya said. Long-term monitoring goals should focus on the following:
- “Adequate treatment adjusted to the needs of the growing child and prevention of systemic and organ-specific inflammatory manifestations;
- Fostering of self-management skills and medical decision-making;
- Initiating a transition program to adult specialist care in adolescent patients.”
Type-1 interferonopathies
The recommendations for this disease group focused on chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures (CANDLE)/proteasome-associated autoinflammatory syndromes (PRAAS), STING-associated vasculopathy with onset in infancy (SAVI), and Aicardi-Goutières syndrome (AGS).
These patients similarly present with chronic and organ-specific inflammation that leads to progressive organ damage, morbidity, and higher mortality risk when not managed. Each of these diseases requires a confirmed genetic diagnosis so that treatments can be targeted and the patient receives appropriate genetic counseling, screening for complications, and information on prognosis, Dr. Goldbach-Mansky said.
Treatment goals for type-1 interferonopathies are to “reduce systematic and organ inflammation to prevent or limit the development of progression of organ injury or damage and to improve quality of life,” Dr. Goldbach-Mansky told attendees.
Each patient requires a multidisciplinary care provider team that conducts long-term monitoring of disease activity, damage to specific organs, and any treatment-related complications.
Management of HLH/MAS
Early recognition and management of HLH and MAS can be challenging because systemic hyperinflammation exists along an immunopathologic continuum with typically nonspecific clinical and laboratory findings, Dr. Goldbach-Mansky said, but holistic, longitudinal consideration of these findings “are recognizable and warrant prompt diagnostic evaluation.” Even if the patient does not meet all specific diagnostic criteria for HLH/MAS, it may be necessary to begin therapies, she said.
One important point to consider is that “systemic hyperinflammation can be associated with hyperferritinemia and can progress to life-threatening HLH/MAS,” Dr. Goldbach-Mansky said. Further, although “systemic hyperinflammation and HLH/MAS can occur in nearly any inflammatory state,” certain common triggers and predisposing conditions can indicate the need to consider these conditions and begin appropriate treatment if needed. Part of effective management of systemic hyperinflammation and HLH/MAS is determining any modifiable factors contributing to the disease and mitigating or treating those.
HLH/MAS requires urgent intervention based on the patient’s degree of inflammation and extent of organ dysfunction, the recommendations state. Treatment goals include preventing or limiting immunopathology, preserving the integrity of the diagnostic workup, and minimizing therapy-related toxicity.
Dr. Carmona, Dr. Goldbach-Mansky, and Dr. Demirkaya have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE EULAR 2021 CONGRESS
Gene variant confirmed as strong predictor of lung disease in RA
Carriers have more than twofold greater risk
Patients with rheumatoid arthritis who carry a specific allele of the gene MUC5B have about double the risk of developing interstitial lung disease when compared with noncarriers, according to a large Finnish biobank study presented at the annual European Congress of Rheumatology.
“The risk difference [or carriers relative to noncarriers] started at about age 65, with a bigger difference [for] men than women,” reported Antti Palomäki, MD, PhD, of the center for rheumatology and clinical immunology at Turku (Finland) University.
The gain-of-function MUC5B variant, which encodes mucin 5B, was first linked to RA-associated interstitial lung disease (ILD) more than 3 years ago. At that time, it was already a known genetic risk factor for idiopathic pulmonary fibrosis in the general population. The new data confirm the association in a longitudinal analysis of a large biobank and suggest the association might have clinical utility.
“This is not ready for clinical practice at the moment. We do not yet know whether we can change therapy to reduce risk,” Dr. Palomäki said, adding “in the future we can look.”
One question that might be asked in clinical studies using MUC5B as a tool to assess and modify risk of ILD in patients with RA is whether one therapy is better than another in avoiding or delaying development of lung fibrosis. Dr. Palomäki noted that biologics, for example, might be a more favorable choice in patients with RA who are at high risk of developing ILD.
The association of the MUC5B variant with increased ILD incidence in patients with RA was drawn from a data set known as FinnGen, a biobank collection of epidemiologic cohorts and hospital samples with genotypes of about 10% of the Finnish population. Follow-up extends to 46 years in some of these individuals.
When 248,4000 individuals in this data set were evaluated, 5,534 had a diagnosis of RA. Of these, 178 (3.2%) developed ILD. About 20% of both those with and without RA were MUC5B variant carriers, meaning the remainder were not.
Sex and age factor into lifetime risk
In patients with RA, the lifetime rate of ILD among MUC5B variant carriers was 16.8% versus only 6.1% among noncarriers. This finding translated into a hazard ratio for ILD of 2.27 (95% confidence interval, 1.75–2.96) for variant carriers versus noncarriers.
The lifetime rate of ILD in patients with RA was greater in men versus women regardless of carrier status (18.5% vs. 8.5%). For women, the lifetime rate was lower for carriers, although the difference relative to female noncarriers was greater (14.5% vs. 4.7%).
ILD, whether in the general population or in patients with RA, is a disease of advancing age. When Dr. Palomäki showed a graph, the rise in ILD incidence did not start in any population, whether those with or without RA and regardless of carrier status, until about age 55. In those without RA and in noncarriers of the variant, ILD incidence remained low and began a discernible climb at around age 70.
In those who did not have RA but were positive for the variant, the rates rose more than twice as fast, particularly after age 70. In people who had RA but not the variant, the rate of ILD was greater than in patients who carried the variant without RA, starting the climb earlier and rising more steeply with age. In those with RA and the variant, the climb in ILD incidence rose rapidly after age 65 years even though the incidence remained fairly similar between all of these groups at age 60.
Putting the findings into context
The need to develop ways to prevent ILD in RA is urgent. ILD is one of the most common extraarticular manifestations of RA, developing in up to 60% of patients with RA in older age groups when evaluated with imaging, according to Dr. Palomäki. Although it develops into a clinically significant complication in only about 10% of these patients, ILD still is a significant cause of illness and death in elderly patients with RA.
In the 2018 study that first linked the MUC5B variant to RA-ILD, the investigators also found that the variant was associated with an increased likelihood of developing the usual interstitial pneumonia type of ILD on imaging. David Schwartz, MD, professor of medicine, pulmonary sciences, and critical care and chair of the department of medicine at the University of Colorado at Denver, Aurora, was a senior author of that study. He said these findings build on the 2018 study.
“While the gain-of-function MUC5B promoter variant is important in predicting who will develop RA-ILD, these findings also suggest that MUC5B may be involved in the etiology of RA-ILD, at least for those with the MUC5B variant,” he said.
“The study also raises the possibility that there are several subtypes of RA-ILD, and the subtype that is driven by MUC5B may respond differently to RA biologics or therapeutic agents to treat ILD,” he added.
In the discussion following the presentation by Dr. Palomäki, others agreed, with that statement including Dr. Palomäki. He expressed interest in clinical studies comparing different classes of RA therapies for their relative impact on the risk of developing ILD.Dr. Palomäki reported financial relationships with AbbVie, Merck, Pfizer, and Sanofi. Dr. Schwartz is the founder of Eleven P15, which is developing methods for early diagnosis and treatment of pulmonary fibrosis.
Carriers have more than twofold greater risk
Carriers have more than twofold greater risk
Patients with rheumatoid arthritis who carry a specific allele of the gene MUC5B have about double the risk of developing interstitial lung disease when compared with noncarriers, according to a large Finnish biobank study presented at the annual European Congress of Rheumatology.
“The risk difference [or carriers relative to noncarriers] started at about age 65, with a bigger difference [for] men than women,” reported Antti Palomäki, MD, PhD, of the center for rheumatology and clinical immunology at Turku (Finland) University.
The gain-of-function MUC5B variant, which encodes mucin 5B, was first linked to RA-associated interstitial lung disease (ILD) more than 3 years ago. At that time, it was already a known genetic risk factor for idiopathic pulmonary fibrosis in the general population. The new data confirm the association in a longitudinal analysis of a large biobank and suggest the association might have clinical utility.
“This is not ready for clinical practice at the moment. We do not yet know whether we can change therapy to reduce risk,” Dr. Palomäki said, adding “in the future we can look.”
One question that might be asked in clinical studies using MUC5B as a tool to assess and modify risk of ILD in patients with RA is whether one therapy is better than another in avoiding or delaying development of lung fibrosis. Dr. Palomäki noted that biologics, for example, might be a more favorable choice in patients with RA who are at high risk of developing ILD.
The association of the MUC5B variant with increased ILD incidence in patients with RA was drawn from a data set known as FinnGen, a biobank collection of epidemiologic cohorts and hospital samples with genotypes of about 10% of the Finnish population. Follow-up extends to 46 years in some of these individuals.
When 248,4000 individuals in this data set were evaluated, 5,534 had a diagnosis of RA. Of these, 178 (3.2%) developed ILD. About 20% of both those with and without RA were MUC5B variant carriers, meaning the remainder were not.
Sex and age factor into lifetime risk
In patients with RA, the lifetime rate of ILD among MUC5B variant carriers was 16.8% versus only 6.1% among noncarriers. This finding translated into a hazard ratio for ILD of 2.27 (95% confidence interval, 1.75–2.96) for variant carriers versus noncarriers.
The lifetime rate of ILD in patients with RA was greater in men versus women regardless of carrier status (18.5% vs. 8.5%). For women, the lifetime rate was lower for carriers, although the difference relative to female noncarriers was greater (14.5% vs. 4.7%).
ILD, whether in the general population or in patients with RA, is a disease of advancing age. When Dr. Palomäki showed a graph, the rise in ILD incidence did not start in any population, whether those with or without RA and regardless of carrier status, until about age 55. In those without RA and in noncarriers of the variant, ILD incidence remained low and began a discernible climb at around age 70.
In those who did not have RA but were positive for the variant, the rates rose more than twice as fast, particularly after age 70. In people who had RA but not the variant, the rate of ILD was greater than in patients who carried the variant without RA, starting the climb earlier and rising more steeply with age. In those with RA and the variant, the climb in ILD incidence rose rapidly after age 65 years even though the incidence remained fairly similar between all of these groups at age 60.
Putting the findings into context
The need to develop ways to prevent ILD in RA is urgent. ILD is one of the most common extraarticular manifestations of RA, developing in up to 60% of patients with RA in older age groups when evaluated with imaging, according to Dr. Palomäki. Although it develops into a clinically significant complication in only about 10% of these patients, ILD still is a significant cause of illness and death in elderly patients with RA.
In the 2018 study that first linked the MUC5B variant to RA-ILD, the investigators also found that the variant was associated with an increased likelihood of developing the usual interstitial pneumonia type of ILD on imaging. David Schwartz, MD, professor of medicine, pulmonary sciences, and critical care and chair of the department of medicine at the University of Colorado at Denver, Aurora, was a senior author of that study. He said these findings build on the 2018 study.
“While the gain-of-function MUC5B promoter variant is important in predicting who will develop RA-ILD, these findings also suggest that MUC5B may be involved in the etiology of RA-ILD, at least for those with the MUC5B variant,” he said.
“The study also raises the possibility that there are several subtypes of RA-ILD, and the subtype that is driven by MUC5B may respond differently to RA biologics or therapeutic agents to treat ILD,” he added.
In the discussion following the presentation by Dr. Palomäki, others agreed, with that statement including Dr. Palomäki. He expressed interest in clinical studies comparing different classes of RA therapies for their relative impact on the risk of developing ILD.Dr. Palomäki reported financial relationships with AbbVie, Merck, Pfizer, and Sanofi. Dr. Schwartz is the founder of Eleven P15, which is developing methods for early diagnosis and treatment of pulmonary fibrosis.
Patients with rheumatoid arthritis who carry a specific allele of the gene MUC5B have about double the risk of developing interstitial lung disease when compared with noncarriers, according to a large Finnish biobank study presented at the annual European Congress of Rheumatology.
“The risk difference [or carriers relative to noncarriers] started at about age 65, with a bigger difference [for] men than women,” reported Antti Palomäki, MD, PhD, of the center for rheumatology and clinical immunology at Turku (Finland) University.
The gain-of-function MUC5B variant, which encodes mucin 5B, was first linked to RA-associated interstitial lung disease (ILD) more than 3 years ago. At that time, it was already a known genetic risk factor for idiopathic pulmonary fibrosis in the general population. The new data confirm the association in a longitudinal analysis of a large biobank and suggest the association might have clinical utility.
“This is not ready for clinical practice at the moment. We do not yet know whether we can change therapy to reduce risk,” Dr. Palomäki said, adding “in the future we can look.”
One question that might be asked in clinical studies using MUC5B as a tool to assess and modify risk of ILD in patients with RA is whether one therapy is better than another in avoiding or delaying development of lung fibrosis. Dr. Palomäki noted that biologics, for example, might be a more favorable choice in patients with RA who are at high risk of developing ILD.
The association of the MUC5B variant with increased ILD incidence in patients with RA was drawn from a data set known as FinnGen, a biobank collection of epidemiologic cohorts and hospital samples with genotypes of about 10% of the Finnish population. Follow-up extends to 46 years in some of these individuals.
When 248,4000 individuals in this data set were evaluated, 5,534 had a diagnosis of RA. Of these, 178 (3.2%) developed ILD. About 20% of both those with and without RA were MUC5B variant carriers, meaning the remainder were not.
Sex and age factor into lifetime risk
In patients with RA, the lifetime rate of ILD among MUC5B variant carriers was 16.8% versus only 6.1% among noncarriers. This finding translated into a hazard ratio for ILD of 2.27 (95% confidence interval, 1.75–2.96) for variant carriers versus noncarriers.
The lifetime rate of ILD in patients with RA was greater in men versus women regardless of carrier status (18.5% vs. 8.5%). For women, the lifetime rate was lower for carriers, although the difference relative to female noncarriers was greater (14.5% vs. 4.7%).
ILD, whether in the general population or in patients with RA, is a disease of advancing age. When Dr. Palomäki showed a graph, the rise in ILD incidence did not start in any population, whether those with or without RA and regardless of carrier status, until about age 55. In those without RA and in noncarriers of the variant, ILD incidence remained low and began a discernible climb at around age 70.
In those who did not have RA but were positive for the variant, the rates rose more than twice as fast, particularly after age 70. In people who had RA but not the variant, the rate of ILD was greater than in patients who carried the variant without RA, starting the climb earlier and rising more steeply with age. In those with RA and the variant, the climb in ILD incidence rose rapidly after age 65 years even though the incidence remained fairly similar between all of these groups at age 60.
Putting the findings into context
The need to develop ways to prevent ILD in RA is urgent. ILD is one of the most common extraarticular manifestations of RA, developing in up to 60% of patients with RA in older age groups when evaluated with imaging, according to Dr. Palomäki. Although it develops into a clinically significant complication in only about 10% of these patients, ILD still is a significant cause of illness and death in elderly patients with RA.
In the 2018 study that first linked the MUC5B variant to RA-ILD, the investigators also found that the variant was associated with an increased likelihood of developing the usual interstitial pneumonia type of ILD on imaging. David Schwartz, MD, professor of medicine, pulmonary sciences, and critical care and chair of the department of medicine at the University of Colorado at Denver, Aurora, was a senior author of that study. He said these findings build on the 2018 study.
“While the gain-of-function MUC5B promoter variant is important in predicting who will develop RA-ILD, these findings also suggest that MUC5B may be involved in the etiology of RA-ILD, at least for those with the MUC5B variant,” he said.
“The study also raises the possibility that there are several subtypes of RA-ILD, and the subtype that is driven by MUC5B may respond differently to RA biologics or therapeutic agents to treat ILD,” he added.
In the discussion following the presentation by Dr. Palomäki, others agreed, with that statement including Dr. Palomäki. He expressed interest in clinical studies comparing different classes of RA therapies for their relative impact on the risk of developing ILD.Dr. Palomäki reported financial relationships with AbbVie, Merck, Pfizer, and Sanofi. Dr. Schwartz is the founder of Eleven P15, which is developing methods for early diagnosis and treatment of pulmonary fibrosis.
FROM THE EULAR 2021 CONGRESS
Nintedanib slows interstitial lung disease in RA patients
Subgroup analysis from INBUILD trial finds results similar to overall study cohort
In a new subgroup analysis of a previously published multinational trial, the preservation of lung function with nintedanib (Ofev) was about the same in patients with interstitial lung disease related to rheumatoid arthritis (RA-ILD) as it was in patients with other etiologies, according to data presented at the annual European Congress of Rheumatology.

“There was no significant heterogeneity across any of several characteristics we evaluated,” reported Clive Kelly, MBBS, of the Institute of Cellular Medicine at Newcastle University (England).
The INBUILD trial, which enrolled more than 600 patients in 15 countries with a range of fibrosing lung diseases, was published almost 2 years ago. On the primary endpoint of rate of decline in forced vital capacity (FVC), the medians were –80.8 mL per year among those randomized to nintedanib and –187.8 mL per year (P < .001) on placebo.
The INBUILD study provided evidence that fibrosing lung diseases have a common pathobiologic mechanism that can be slowed by targeting intracellular kinases. Nintedanib inhibits several growth factor receptors as well as nonreceptor tyrosine kinases, but its exact mechanism for slowing fibrosing lung diseases remains unclear. Initially approved for, nintedanib received approvals from the FDA for systemic sclerosis–associated ILD in 2019 and for chronic fibrosing ILD with progressive phenotypes in 2020 after being initially approved for the treatment of idiopathic pulmonary fibrosis in 2014.
When asked for comment, Paul F. Dellaripa, MD, an associate professor of medicine in the division of rheumatology, immunology, and allergy at Harvard Medical School, Boston, indicated these data are helpful in considering strategies for RA patients with ILD, but he encouraged collaboration between joint and lung specialists.
“Antifibrotic agents for patients with progressive ILD in autoimmune diseases like RA is a welcome addition to our care of this challenging complication,” said Dr. Dellaripa, who has published frequently on the diagnosis and treatment of lung diseases associated with RA. Yet, treatment must be individualized, he added.
“It will be incumbent for rheumatologists to incorporate lung health as a critical part of patient care and work closely with pulmonologists to consider when to institute antifibrotic therapy in patients with ILD,” he said.
Details of subanalysis
In the RA-ILD subpopulation of 89 patients, there was no further decline in FVC from 24 weeks after randomization to the end of 52 weeks for those on nintedanib, but the decline remained steady over the full course of follow-up among those in the placebo group. At 52 weeks, the decline in the placebo group reached –200 mL at the end of 52 weeks. As a result, the between-group relative reduction in FVC at 52 weeks of 116.7 mL favoring nintedanib over placebo (P < .037) slightly exceeded the 107-mL reduction (P < .001) observed in the overall INBUILD study population.
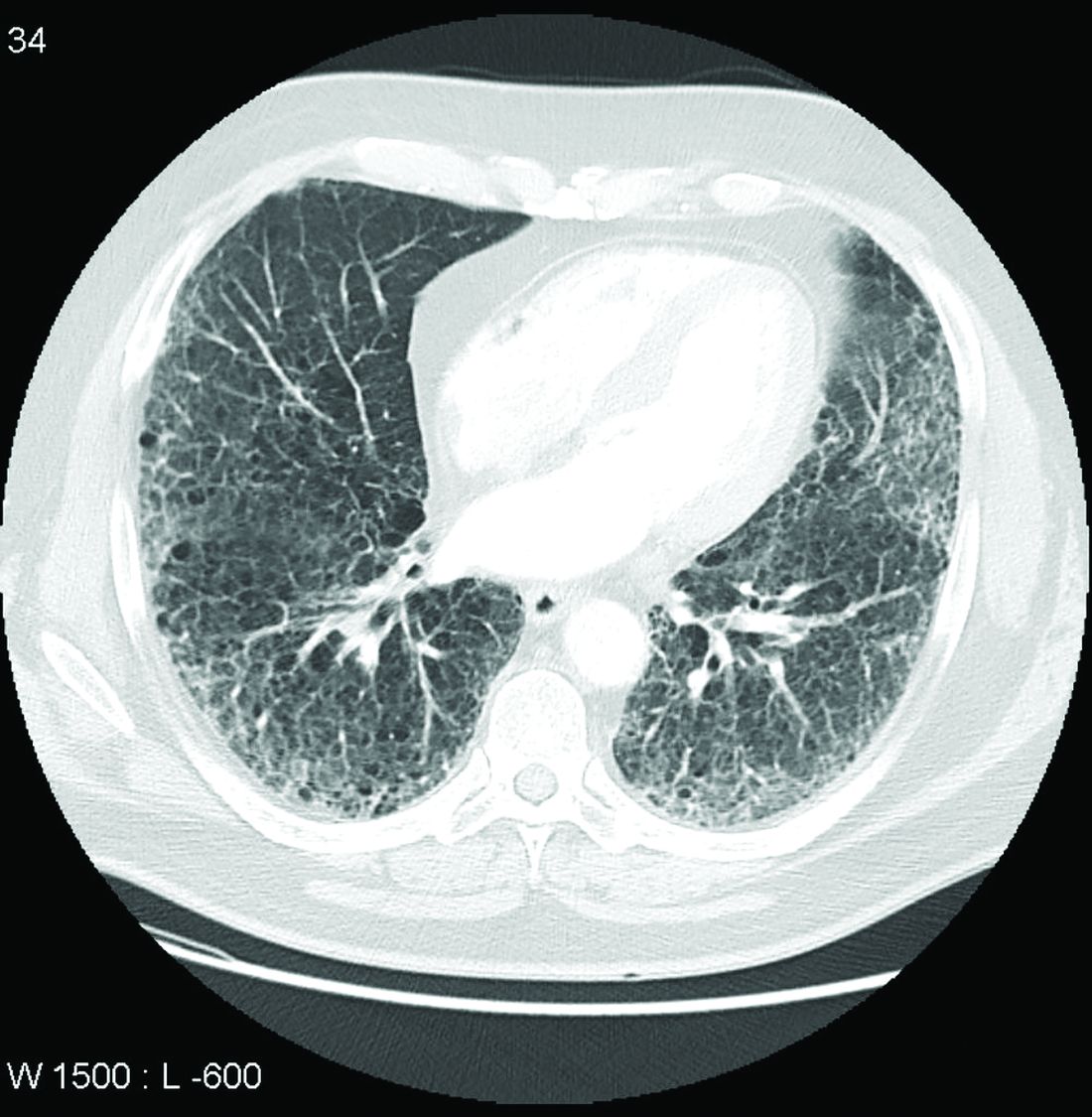
Among other subgroups the investigators evaluated, outcomes with nintedanib did not differ when patients were split into groups with higher or lower baseline levels of high-sensitivity C-reactive protein, regardless of whether the groups were defined by levels above and below 1 mg/L or 3 mg/L. The same was true for those who were taking nonbiologic disease-modifying antirheumatic drugs or glucocorticoids.
However, for these latter analyses, Dr. Kelly conceded that the differences were based on small numbers of patients and so cannot be considered conclusive.
The adverse event most closely associated with nintedanib in the RA-ILD population was diarrhea, just as in the overall study, and it was more than twice as frequent in the RA-ILD patients receiving the active therapy, compared with placebo (54.8% vs. 25.5%). Nausea was also more common (21.4% vs. 10.6%), and so was decreased appetite (11.9% vs. 2.1%) and weight reduction (9.5% vs. 2.1%).
Lung-related adverse events, such as bronchiolitis (21.4% vs. 17.0%) and dyspnea (11.9% vs. 10.6%), were only slightly more frequent in the nintedanib group. Nasopharyngitis (7.1% vs. 12.8%) was less common. Side effects leading to treatment discontinuation were higher on nintedanib (19.0% vs. 12.8%)
The RA-ILD subgroup represented 13.4% of those randomized in INBUILD. The mean time since diagnosis of RA was about 10 years. More than 60% were smokers or former smokers. At baseline, the mean FVC of predicted was 71%. More than 85% had a usual interstitial pneumonia (UIP) radiologic pattern.
Acute exacerbations and death were not evaluated in the RA-ILD subpopulation, but these were secondary endpoints in the published INBUILD study according to the presence or absence of a UIP-like fibrotic pattern. For the combined endpoint of acute exacerbation of ILD or death, the protection associated with nintedanib approached statistical significance for the population overall (odds ratio, 0.68; 95% confidence interval, 0.46-1.01) and reached significance for those with a UIP pattern (OR, 0.61; 95% CI, 0.38-0.98).
Nintedanib led to lower death rates at 52 weeks in the overall population (8.1% vs. 11.5% with placebo) and in the group with a UIP pattern (9.7% vs. 15.0% with placebo).
Dr. Kelly has financial relationships with multiple pharmaceutical companies, including Boehringer Ingelheim, which provided funding for INBUILD and this subpopulation analysis. Dr. Dellaripa reported financial relationships with Bristol-Myers Squibb and Genentech.
Subgroup analysis from INBUILD trial finds results similar to overall study cohort
Subgroup analysis from INBUILD trial finds results similar to overall study cohort
In a new subgroup analysis of a previously published multinational trial, the preservation of lung function with nintedanib (Ofev) was about the same in patients with interstitial lung disease related to rheumatoid arthritis (RA-ILD) as it was in patients with other etiologies, according to data presented at the annual European Congress of Rheumatology.
“There was no significant heterogeneity across any of several characteristics we evaluated,” reported Clive Kelly, MBBS, of the Institute of Cellular Medicine at Newcastle University (England).
The INBUILD trial, which enrolled more than 600 patients in 15 countries with a range of fibrosing lung diseases, was published almost 2 years ago. On the primary endpoint of rate of decline in forced vital capacity (FVC), the medians were –80.8 mL per year among those randomized to nintedanib and –187.8 mL per year (P < .001) on placebo.
The INBUILD study provided evidence that fibrosing lung diseases have a common pathobiologic mechanism that can be slowed by targeting intracellular kinases. Nintedanib inhibits several growth factor receptors as well as nonreceptor tyrosine kinases, but its exact mechanism for slowing fibrosing lung diseases remains unclear. Initially approved for, nintedanib received approvals from the FDA for systemic sclerosis–associated ILD in 2019 and for chronic fibrosing ILD with progressive phenotypes in 2020 after being initially approved for the treatment of idiopathic pulmonary fibrosis in 2014.
When asked for comment, Paul F. Dellaripa, MD, an associate professor of medicine in the division of rheumatology, immunology, and allergy at Harvard Medical School, Boston, indicated these data are helpful in considering strategies for RA patients with ILD, but he encouraged collaboration between joint and lung specialists.
“Antifibrotic agents for patients with progressive ILD in autoimmune diseases like RA is a welcome addition to our care of this challenging complication,” said Dr. Dellaripa, who has published frequently on the diagnosis and treatment of lung diseases associated with RA. Yet, treatment must be individualized, he added.
“It will be incumbent for rheumatologists to incorporate lung health as a critical part of patient care and work closely with pulmonologists to consider when to institute antifibrotic therapy in patients with ILD,” he said.
Details of subanalysis
In the RA-ILD subpopulation of 89 patients, there was no further decline in FVC from 24 weeks after randomization to the end of 52 weeks for those on nintedanib, but the decline remained steady over the full course of follow-up among those in the placebo group. At 52 weeks, the decline in the placebo group reached –200 mL at the end of 52 weeks. As a result, the between-group relative reduction in FVC at 52 weeks of 116.7 mL favoring nintedanib over placebo (P < .037) slightly exceeded the 107-mL reduction (P < .001) observed in the overall INBUILD study population.
Among other subgroups the investigators evaluated, outcomes with nintedanib did not differ when patients were split into groups with higher or lower baseline levels of high-sensitivity C-reactive protein, regardless of whether the groups were defined by levels above and below 1 mg/L or 3 mg/L. The same was true for those who were taking nonbiologic disease-modifying antirheumatic drugs or glucocorticoids.
However, for these latter analyses, Dr. Kelly conceded that the differences were based on small numbers of patients and so cannot be considered conclusive.
The adverse event most closely associated with nintedanib in the RA-ILD population was diarrhea, just as in the overall study, and it was more than twice as frequent in the RA-ILD patients receiving the active therapy, compared with placebo (54.8% vs. 25.5%). Nausea was also more common (21.4% vs. 10.6%), and so was decreased appetite (11.9% vs. 2.1%) and weight reduction (9.5% vs. 2.1%).
Lung-related adverse events, such as bronchiolitis (21.4% vs. 17.0%) and dyspnea (11.9% vs. 10.6%), were only slightly more frequent in the nintedanib group. Nasopharyngitis (7.1% vs. 12.8%) was less common. Side effects leading to treatment discontinuation were higher on nintedanib (19.0% vs. 12.8%)
The RA-ILD subgroup represented 13.4% of those randomized in INBUILD. The mean time since diagnosis of RA was about 10 years. More than 60% were smokers or former smokers. At baseline, the mean FVC of predicted was 71%. More than 85% had a usual interstitial pneumonia (UIP) radiologic pattern.
Acute exacerbations and death were not evaluated in the RA-ILD subpopulation, but these were secondary endpoints in the published INBUILD study according to the presence or absence of a UIP-like fibrotic pattern. For the combined endpoint of acute exacerbation of ILD or death, the protection associated with nintedanib approached statistical significance for the population overall (odds ratio, 0.68; 95% confidence interval, 0.46-1.01) and reached significance for those with a UIP pattern (OR, 0.61; 95% CI, 0.38-0.98).
Nintedanib led to lower death rates at 52 weeks in the overall population (8.1% vs. 11.5% with placebo) and in the group with a UIP pattern (9.7% vs. 15.0% with placebo).
Dr. Kelly has financial relationships with multiple pharmaceutical companies, including Boehringer Ingelheim, which provided funding for INBUILD and this subpopulation analysis. Dr. Dellaripa reported financial relationships with Bristol-Myers Squibb and Genentech.
In a new subgroup analysis of a previously published multinational trial, the preservation of lung function with nintedanib (Ofev) was about the same in patients with interstitial lung disease related to rheumatoid arthritis (RA-ILD) as it was in patients with other etiologies, according to data presented at the annual European Congress of Rheumatology.
“There was no significant heterogeneity across any of several characteristics we evaluated,” reported Clive Kelly, MBBS, of the Institute of Cellular Medicine at Newcastle University (England).
The INBUILD trial, which enrolled more than 600 patients in 15 countries with a range of fibrosing lung diseases, was published almost 2 years ago. On the primary endpoint of rate of decline in forced vital capacity (FVC), the medians were –80.8 mL per year among those randomized to nintedanib and –187.8 mL per year (P < .001) on placebo.
The INBUILD study provided evidence that fibrosing lung diseases have a common pathobiologic mechanism that can be slowed by targeting intracellular kinases. Nintedanib inhibits several growth factor receptors as well as nonreceptor tyrosine kinases, but its exact mechanism for slowing fibrosing lung diseases remains unclear. Initially approved for, nintedanib received approvals from the FDA for systemic sclerosis–associated ILD in 2019 and for chronic fibrosing ILD with progressive phenotypes in 2020 after being initially approved for the treatment of idiopathic pulmonary fibrosis in 2014.
When asked for comment, Paul F. Dellaripa, MD, an associate professor of medicine in the division of rheumatology, immunology, and allergy at Harvard Medical School, Boston, indicated these data are helpful in considering strategies for RA patients with ILD, but he encouraged collaboration between joint and lung specialists.
“Antifibrotic agents for patients with progressive ILD in autoimmune diseases like RA is a welcome addition to our care of this challenging complication,” said Dr. Dellaripa, who has published frequently on the diagnosis and treatment of lung diseases associated with RA. Yet, treatment must be individualized, he added.
“It will be incumbent for rheumatologists to incorporate lung health as a critical part of patient care and work closely with pulmonologists to consider when to institute antifibrotic therapy in patients with ILD,” he said.
Details of subanalysis
In the RA-ILD subpopulation of 89 patients, there was no further decline in FVC from 24 weeks after randomization to the end of 52 weeks for those on nintedanib, but the decline remained steady over the full course of follow-up among those in the placebo group. At 52 weeks, the decline in the placebo group reached –200 mL at the end of 52 weeks. As a result, the between-group relative reduction in FVC at 52 weeks of 116.7 mL favoring nintedanib over placebo (P < .037) slightly exceeded the 107-mL reduction (P < .001) observed in the overall INBUILD study population.
Among other subgroups the investigators evaluated, outcomes with nintedanib did not differ when patients were split into groups with higher or lower baseline levels of high-sensitivity C-reactive protein, regardless of whether the groups were defined by levels above and below 1 mg/L or 3 mg/L. The same was true for those who were taking nonbiologic disease-modifying antirheumatic drugs or glucocorticoids.
However, for these latter analyses, Dr. Kelly conceded that the differences were based on small numbers of patients and so cannot be considered conclusive.
The adverse event most closely associated with nintedanib in the RA-ILD population was diarrhea, just as in the overall study, and it was more than twice as frequent in the RA-ILD patients receiving the active therapy, compared with placebo (54.8% vs. 25.5%). Nausea was also more common (21.4% vs. 10.6%), and so was decreased appetite (11.9% vs. 2.1%) and weight reduction (9.5% vs. 2.1%).
Lung-related adverse events, such as bronchiolitis (21.4% vs. 17.0%) and dyspnea (11.9% vs. 10.6%), were only slightly more frequent in the nintedanib group. Nasopharyngitis (7.1% vs. 12.8%) was less common. Side effects leading to treatment discontinuation were higher on nintedanib (19.0% vs. 12.8%)
The RA-ILD subgroup represented 13.4% of those randomized in INBUILD. The mean time since diagnosis of RA was about 10 years. More than 60% were smokers or former smokers. At baseline, the mean FVC of predicted was 71%. More than 85% had a usual interstitial pneumonia (UIP) radiologic pattern.
Acute exacerbations and death were not evaluated in the RA-ILD subpopulation, but these were secondary endpoints in the published INBUILD study according to the presence or absence of a UIP-like fibrotic pattern. For the combined endpoint of acute exacerbation of ILD or death, the protection associated with nintedanib approached statistical significance for the population overall (odds ratio, 0.68; 95% confidence interval, 0.46-1.01) and reached significance for those with a UIP pattern (OR, 0.61; 95% CI, 0.38-0.98).
Nintedanib led to lower death rates at 52 weeks in the overall population (8.1% vs. 11.5% with placebo) and in the group with a UIP pattern (9.7% vs. 15.0% with placebo).
Dr. Kelly has financial relationships with multiple pharmaceutical companies, including Boehringer Ingelheim, which provided funding for INBUILD and this subpopulation analysis. Dr. Dellaripa reported financial relationships with Bristol-Myers Squibb and Genentech.
FROM THE EULAR 2021 CONGRESS
BEAT-LUPUS: Belimumab after rituximab delays severe flares
Using belimumab after rituximab to treat patients with systemic lupus erythematosus (SLE) refractory to conventional therapy not only significantly decreased levels of serum IgG anti-dsDNA antibody levels but also prolonged the time before severe flares of disease occurred in the phase 2b BEAT-LUPUS (Belimumab after B cell depletion in SLE) study.

The trial’s primary outcome of serum IgG anti-dsDNA antibody levels showed a decline from a geometric mean of 162 IU/mL at baseline to 69 IU/mL at 24 weeks and 47 IU/mL at 1 year in patients treated with belimumab (Benlysta) after rituximab (Rituxan and biosimilars). These reductions were significantly lower than the values seen in the placebo after rituximab arm (a respective 121 IU/mL, 99 IU/mL, and 103 IU/mL; P < .001).
Just 3 patients who received belimumab versus 10 who received placebo after rituximab experienced a severe BILAG (British Isles Lupus Assessment Group) index A flare by the end of the study at 52 weeks. The hazard ratio (HR) for the flare reduction was 0.27 (P = .03), indicating a 73% reduction.
The use of belimumab rather than a placebo also led to a small reduction in total serum IgG, and significantly suppressed B-cell repopulation (P = .03).
These results need confirming in a larger, phase 3 trial, the trial’s principal investigator, Michael Ehrenstein, PhD, said at the annual European Congress of Rheumatology. They are “clearly encouraging” and “support the hypothesis that BAFF [B-cell–activating factor] can drive flares after rituximab,” he said.
Although B-cell depletion with rituximab is recommended by national and international guidelines to treat some patients with SLE who are refractory to conventional therapy, its use is not licensed.
“Certainly, rituximab is a controversial drug in lupus,” Dr. Ehrenstein, a consultant rheumatologist based at University College London, said in an interview. Although there is real-world evidence from registries and open-label studies suggesting that it is widely used and effective in some patients, the randomized, controlled trials conducted with rituximab about 10 years ago failed to meet their primary endpoints.
“A lot has been written about why that was, but probably the biggest reason was the high dose of steroids in both groups,” Dr. Ehrenstein said. To try to avoid muddying the waters of the BEAT-LUPUS trial findings, the maximum dose of prednisolone allowed to be used as background therapy was 20 mg/day. The trial’s investigators were also encouraged to reduce the baseline steroid dose to at least 50% by the trial’s 6-month halfway point.
“We tried to reflect what was going on in the U.K.,” Dr. Ehrenstein said, noting that the inspiration for the trial was a patient who had received sequential rituximab treatment. Although she got better with each cycle of rituximab, when her disease flared it would be worse than the time before, with increasingly higher anti-dsDNA levels recorded. The reason for this seemed to be because of increasing BAFF levels, and so the hypothesis was that if rituximab was associated with increased BAFF levels, then co-targeting BAFF with belimumab should be able to prevent those flares from happening.
The BEAT-LUPUS trial has been a huge collaborative effort and was conducted across 16 U.K. centers. From initial funding to the data analysis, it has taken 6 years to complete and was made possible by a unique partnership between Versus Arthritis, University College London Hospitals Biomedical Research Center, the National Institute for Health Research UK Musculoskeletal Translational Research Collaboration, and GlaxoSmithKline (GSK). GSK provided belimumab free of charge, as well as additional funding, but had no role in the design of the study and will not have any role going forward.
From an initial 172 patients assessed for eligibility, 52 patients were finally enrolled into the trial and received rituximab as two infusions given 2 weeks apart. Patients were then randomized in a double-blind manner to receive either belimumab (n = 26) or placebo (n = 26) 4-8 weeks after their first dose of rituximab. The intention-to-treat population consisted of 43 patients.
The use of belimumab after rituximab did not increase the risk for infection – serious or otherwise – or adverse effects, Dr. Ehrenstein reported. Serious adverse events were reported in six (23%) patients in each arm, and serious infections were seen in two (8%) of the belimumab- and four (15%) of the placebo-treated patients.
“I think the take-home message is that it seems that belimumab can reduce the number of severe flares that occur after rituximab therapy,” Dr. Ehrenstein said. “It’s promising, but not definitive,” he added. The next step is of course to publish these data and to perform a phase 3 trial.
In the discussion time following the presentation, session moderator Xavier Mariette, MD, PhD, of Bicêtre Hospital, Paris-Saclay University, asked why not give belimumab first before rituximab if using belimumab afterward works?
“Our strategy was driven by the observation that BAFF levels surged after rituximab, and therefore it’s logical to give the belimumab to block that BAFF surge,” he answered.
“Certainly, there are ideas that belimumab releases mature B cells into the circulation and rituximab can target that,” he added. That strategy is under investigation in the BLISS-BELIEVE trial, which should also report by the end of this year. It’s a much larger, phase 3 trial, involving nearly 300 patients and is sponsored by GSK.
“Clearly, this is a combination treatment [but] whether you give one before the other is uncertain,” Dr. Ehrenstein observed.
Another member of the viewing audience asked whether it would have been a fairer comparison if another dose of rituximab had been given to patients at week 24 instead of no treatment. Dr. Ehrenstein noted that it was a “good point” to make, but the investigators mainly wanted to answer whether giving belimumab after rituximab would target BAFF and thereby drop serum anti-dsDNA antibody levels. He said that a full trial of rituximab for patients with SLE, perhaps adding this extra dose, needs to be conducted.
Dr. Ehrenstein disclosed receiving research funding and educational grants from GSK and participating in advisory panels for the company.
Using belimumab after rituximab to treat patients with systemic lupus erythematosus (SLE) refractory to conventional therapy not only significantly decreased levels of serum IgG anti-dsDNA antibody levels but also prolonged the time before severe flares of disease occurred in the phase 2b BEAT-LUPUS (Belimumab after B cell depletion in SLE) study.
The trial’s primary outcome of serum IgG anti-dsDNA antibody levels showed a decline from a geometric mean of 162 IU/mL at baseline to 69 IU/mL at 24 weeks and 47 IU/mL at 1 year in patients treated with belimumab (Benlysta) after rituximab (Rituxan and biosimilars). These reductions were significantly lower than the values seen in the placebo after rituximab arm (a respective 121 IU/mL, 99 IU/mL, and 103 IU/mL; P < .001).
Just 3 patients who received belimumab versus 10 who received placebo after rituximab experienced a severe BILAG (British Isles Lupus Assessment Group) index A flare by the end of the study at 52 weeks. The hazard ratio (HR) for the flare reduction was 0.27 (P = .03), indicating a 73% reduction.
The use of belimumab rather than a placebo also led to a small reduction in total serum IgG, and significantly suppressed B-cell repopulation (P = .03).
These results need confirming in a larger, phase 3 trial, the trial’s principal investigator, Michael Ehrenstein, PhD, said at the annual European Congress of Rheumatology. They are “clearly encouraging” and “support the hypothesis that BAFF [B-cell–activating factor] can drive flares after rituximab,” he said.
Although B-cell depletion with rituximab is recommended by national and international guidelines to treat some patients with SLE who are refractory to conventional therapy, its use is not licensed.
“Certainly, rituximab is a controversial drug in lupus,” Dr. Ehrenstein, a consultant rheumatologist based at University College London, said in an interview. Although there is real-world evidence from registries and open-label studies suggesting that it is widely used and effective in some patients, the randomized, controlled trials conducted with rituximab about 10 years ago failed to meet their primary endpoints.
“A lot has been written about why that was, but probably the biggest reason was the high dose of steroids in both groups,” Dr. Ehrenstein said. To try to avoid muddying the waters of the BEAT-LUPUS trial findings, the maximum dose of prednisolone allowed to be used as background therapy was 20 mg/day. The trial’s investigators were also encouraged to reduce the baseline steroid dose to at least 50% by the trial’s 6-month halfway point.
“We tried to reflect what was going on in the U.K.,” Dr. Ehrenstein said, noting that the inspiration for the trial was a patient who had received sequential rituximab treatment. Although she got better with each cycle of rituximab, when her disease flared it would be worse than the time before, with increasingly higher anti-dsDNA levels recorded. The reason for this seemed to be because of increasing BAFF levels, and so the hypothesis was that if rituximab was associated with increased BAFF levels, then co-targeting BAFF with belimumab should be able to prevent those flares from happening.
The BEAT-LUPUS trial has been a huge collaborative effort and was conducted across 16 U.K. centers. From initial funding to the data analysis, it has taken 6 years to complete and was made possible by a unique partnership between Versus Arthritis, University College London Hospitals Biomedical Research Center, the National Institute for Health Research UK Musculoskeletal Translational Research Collaboration, and GlaxoSmithKline (GSK). GSK provided belimumab free of charge, as well as additional funding, but had no role in the design of the study and will not have any role going forward.
From an initial 172 patients assessed for eligibility, 52 patients were finally enrolled into the trial and received rituximab as two infusions given 2 weeks apart. Patients were then randomized in a double-blind manner to receive either belimumab (n = 26) or placebo (n = 26) 4-8 weeks after their first dose of rituximab. The intention-to-treat population consisted of 43 patients.
The use of belimumab after rituximab did not increase the risk for infection – serious or otherwise – or adverse effects, Dr. Ehrenstein reported. Serious adverse events were reported in six (23%) patients in each arm, and serious infections were seen in two (8%) of the belimumab- and four (15%) of the placebo-treated patients.
“I think the take-home message is that it seems that belimumab can reduce the number of severe flares that occur after rituximab therapy,” Dr. Ehrenstein said. “It’s promising, but not definitive,” he added. The next step is of course to publish these data and to perform a phase 3 trial.
In the discussion time following the presentation, session moderator Xavier Mariette, MD, PhD, of Bicêtre Hospital, Paris-Saclay University, asked why not give belimumab first before rituximab if using belimumab afterward works?
“Our strategy was driven by the observation that BAFF levels surged after rituximab, and therefore it’s logical to give the belimumab to block that BAFF surge,” he answered.
“Certainly, there are ideas that belimumab releases mature B cells into the circulation and rituximab can target that,” he added. That strategy is under investigation in the BLISS-BELIEVE trial, which should also report by the end of this year. It’s a much larger, phase 3 trial, involving nearly 300 patients and is sponsored by GSK.
“Clearly, this is a combination treatment [but] whether you give one before the other is uncertain,” Dr. Ehrenstein observed.
Another member of the viewing audience asked whether it would have been a fairer comparison if another dose of rituximab had been given to patients at week 24 instead of no treatment. Dr. Ehrenstein noted that it was a “good point” to make, but the investigators mainly wanted to answer whether giving belimumab after rituximab would target BAFF and thereby drop serum anti-dsDNA antibody levels. He said that a full trial of rituximab for patients with SLE, perhaps adding this extra dose, needs to be conducted.
Dr. Ehrenstein disclosed receiving research funding and educational grants from GSK and participating in advisory panels for the company.
Using belimumab after rituximab to treat patients with systemic lupus erythematosus (SLE) refractory to conventional therapy not only significantly decreased levels of serum IgG anti-dsDNA antibody levels but also prolonged the time before severe flares of disease occurred in the phase 2b BEAT-LUPUS (Belimumab after B cell depletion in SLE) study.
The trial’s primary outcome of serum IgG anti-dsDNA antibody levels showed a decline from a geometric mean of 162 IU/mL at baseline to 69 IU/mL at 24 weeks and 47 IU/mL at 1 year in patients treated with belimumab (Benlysta) after rituximab (Rituxan and biosimilars). These reductions were significantly lower than the values seen in the placebo after rituximab arm (a respective 121 IU/mL, 99 IU/mL, and 103 IU/mL; P < .001).
Just 3 patients who received belimumab versus 10 who received placebo after rituximab experienced a severe BILAG (British Isles Lupus Assessment Group) index A flare by the end of the study at 52 weeks. The hazard ratio (HR) for the flare reduction was 0.27 (P = .03), indicating a 73% reduction.
The use of belimumab rather than a placebo also led to a small reduction in total serum IgG, and significantly suppressed B-cell repopulation (P = .03).
These results need confirming in a larger, phase 3 trial, the trial’s principal investigator, Michael Ehrenstein, PhD, said at the annual European Congress of Rheumatology. They are “clearly encouraging” and “support the hypothesis that BAFF [B-cell–activating factor] can drive flares after rituximab,” he said.
Although B-cell depletion with rituximab is recommended by national and international guidelines to treat some patients with SLE who are refractory to conventional therapy, its use is not licensed.
“Certainly, rituximab is a controversial drug in lupus,” Dr. Ehrenstein, a consultant rheumatologist based at University College London, said in an interview. Although there is real-world evidence from registries and open-label studies suggesting that it is widely used and effective in some patients, the randomized, controlled trials conducted with rituximab about 10 years ago failed to meet their primary endpoints.
“A lot has been written about why that was, but probably the biggest reason was the high dose of steroids in both groups,” Dr. Ehrenstein said. To try to avoid muddying the waters of the BEAT-LUPUS trial findings, the maximum dose of prednisolone allowed to be used as background therapy was 20 mg/day. The trial’s investigators were also encouraged to reduce the baseline steroid dose to at least 50% by the trial’s 6-month halfway point.
“We tried to reflect what was going on in the U.K.,” Dr. Ehrenstein said, noting that the inspiration for the trial was a patient who had received sequential rituximab treatment. Although she got better with each cycle of rituximab, when her disease flared it would be worse than the time before, with increasingly higher anti-dsDNA levels recorded. The reason for this seemed to be because of increasing BAFF levels, and so the hypothesis was that if rituximab was associated with increased BAFF levels, then co-targeting BAFF with belimumab should be able to prevent those flares from happening.
The BEAT-LUPUS trial has been a huge collaborative effort and was conducted across 16 U.K. centers. From initial funding to the data analysis, it has taken 6 years to complete and was made possible by a unique partnership between Versus Arthritis, University College London Hospitals Biomedical Research Center, the National Institute for Health Research UK Musculoskeletal Translational Research Collaboration, and GlaxoSmithKline (GSK). GSK provided belimumab free of charge, as well as additional funding, but had no role in the design of the study and will not have any role going forward.
From an initial 172 patients assessed for eligibility, 52 patients were finally enrolled into the trial and received rituximab as two infusions given 2 weeks apart. Patients were then randomized in a double-blind manner to receive either belimumab (n = 26) or placebo (n = 26) 4-8 weeks after their first dose of rituximab. The intention-to-treat population consisted of 43 patients.
The use of belimumab after rituximab did not increase the risk for infection – serious or otherwise – or adverse effects, Dr. Ehrenstein reported. Serious adverse events were reported in six (23%) patients in each arm, and serious infections were seen in two (8%) of the belimumab- and four (15%) of the placebo-treated patients.
“I think the take-home message is that it seems that belimumab can reduce the number of severe flares that occur after rituximab therapy,” Dr. Ehrenstein said. “It’s promising, but not definitive,” he added. The next step is of course to publish these data and to perform a phase 3 trial.
In the discussion time following the presentation, session moderator Xavier Mariette, MD, PhD, of Bicêtre Hospital, Paris-Saclay University, asked why not give belimumab first before rituximab if using belimumab afterward works?
“Our strategy was driven by the observation that BAFF levels surged after rituximab, and therefore it’s logical to give the belimumab to block that BAFF surge,” he answered.
“Certainly, there are ideas that belimumab releases mature B cells into the circulation and rituximab can target that,” he added. That strategy is under investigation in the BLISS-BELIEVE trial, which should also report by the end of this year. It’s a much larger, phase 3 trial, involving nearly 300 patients and is sponsored by GSK.
“Clearly, this is a combination treatment [but] whether you give one before the other is uncertain,” Dr. Ehrenstein observed.
Another member of the viewing audience asked whether it would have been a fairer comparison if another dose of rituximab had been given to patients at week 24 instead of no treatment. Dr. Ehrenstein noted that it was a “good point” to make, but the investigators mainly wanted to answer whether giving belimumab after rituximab would target BAFF and thereby drop serum anti-dsDNA antibody levels. He said that a full trial of rituximab for patients with SLE, perhaps adding this extra dose, needs to be conducted.
Dr. Ehrenstein disclosed receiving research funding and educational grants from GSK and participating in advisory panels for the company.
FROM THE EULAR 2021 CONGRESS
EULAR COVID-19 recommendations set for update
The European Alliance of Associations for Rheumatology has started the process of updating their recommendations on how to manage patients with rheumatic and musculoskeletal diseases (RMDs) in the context of the SARS-CoV-2 pandemic.

So far, the first part of the systematic literature review has been performed and the conclusions that have been drawn appear to back up the recommendations that have already been made. It’s “hard to say” if there will need to be changes, said Robert B.M. Landewé, MD, PhD, at the annual European Congress of Rheumatology, as the next phase will be for the task force members to meet and discuss the implications of the literature research.“I think there will only be minor modifications and a few novel recommendations, but that is personal opinion,” speculated Dr. Landewé, who is professor of rheumatology at the Amsterdam Medical Center, University of Amsterdam.
The recommendations, which were developed a little over a year ago and published in Annals of the Rheumatic Diseases, set out provisional guidance covering four themes: infection prevention, managing patients when social distancing measures are in effect, managing patients with RMDs who develop COVID-19, and the prevention of infections other than SARS-CoV-2.
Emphasis on quality of evidence
According to EULAR’s standard operating procedures “updates should only be done if the evolving evidence mandates to do so,” and be based on “rational arguments,” Dr. Landewé said. “The last year was a bit unprecedented in that regard as we didn’t have those rational arguments before we designed our first set of recommendations and, as you can expect, that is totally due to the character of the pandemic.”
So much has been published on COVID-19 since then it was time to reappraise the situation. The task force behind the recommendations met in January 2021 to discuss the results of the literature search that was centered around five main research questions.
- Do patients with RMDs face more risk of contracting SARS-CoV-2 than the general population?
- If patients contract the virus, do they have a worse prognosis?
- Are antirheumatic medications associated with a worse outcome in people with RMDs?
- Should patients continue their antirheumatic medications?
- What evidence informs the use of vaccination against SARS-CoV-2 in patients with RMDs?
The latter research question is pending discussion since there were no studies to review at the time as the various vaccines had only just started to be widely available.
“We put a lot of emphasis on the quality of evidence,” Dr. Landewé said. In addition to making sure that patients did indeed have COVID-19 and checking that hospitalization and death records were caused by the disease, the task force team also looked to see if there was a control group being used. An extensive risk of bias assessment was undertaken, the results of which are pending.
Of 6,665 records identified during the literature search, just 113 full-text articles were assessed for eligibility. Of those, 60% were rejected as they did not pass the quality assessment, leaving 49 articles for consideration. The majority of these looked at the incidence of COVID-19, with others focusing on risk factors or both.
Literature search findings on main research questions
Dr. Landewé observed that the task force concluded that “current literature provides no evidence that patients with RMDs face more risk of contracting SARS-CoV-2 than individuals without RMDs.” They also concluded that patients with RMDs who do contract COVID-19 do not have a worse prognosis either, even though there have been a few studies suggesting a higher rate of hospitalization.
Both findings are reassuring as they fit with the existing recommendation to follow the same preventive and control measures in patients with RMDs as for the general population, but the task force is yet to determine if that recommendation should be amended.
There did not appear to be any hard evidence of any unique demographic feature or comorbidity that puts people with RMDs at more risk for severe COVID-19 than the general population. Think older age, male gender, high bodyweight, cardiovascular disease, diabetes, and chronic lung disease, Dr. Landewé said.
He noted, however, that there were some single-center reports suggesting that moderate or high levels of disease activity could put people with RMDs at greater risk for COVID-related death, “which is an intriguing finding in the context of discontinuing antirheumatic medication.” That is likely something the task force will be discussing when they decide how to update their recommendations.
The type of RMD may also be important, but again only single-center evidence to show that there might be an increased hospitalization risk in patients with autoinflammatory disease or risk for severe COVID-19 in those with certain connective tissue diseases. “These associations were not consistently found in other studies,” so it’s an open question how the task force decides to incorporate this into the updated guidance.
As for antirheumatic medications, conclusions from the literature review suggest that there doesn’t appear to be an increased or decreased risk for severe COVID-19 among users of NSAIDs or antimalarials.
That’s not the case for glucocorticoids. There appears to be an increased risk for hospitalization and COVID-19–related death, notably among those using higher (>10 mg) daily doses. “This is, so to say, the elephant in the room,” Dr. Landewé said. The current recommendation states that chronic users of glucocorticoids should continue their treatment. “The reports of additional risk could be due to glucocorticoids or to biases such as confounding by indication. So, the conclusion that we draw [is] not completely clear.”
In response to a question, he clarified this a little further: “We think ‘glucocorticoid use’ is a determinant of worse health, as is the case in many RMDs. Be aware that finding a positive association between [glucocorticoid] use and bad outcome does not mean that if you reduce [glucocorticoids], your patient will have a better outcome.”
The jury is also out on rituximab, which has been reported to increase the risk of severe COVID-19 and COVID-related death in two studies. There are also equivocal data on whether not using disease-modifying antirheumatic drugs increases the risk for these worse outcomes.
Asked about the absence of a recommendation on the use of the interleukin-6 inhibitor tocilizumab, Dr. Landewé responded: “We are caught up by evolving evidence. That is a generic problem in a dynamic field of COVID-19, I am afraid. What you recommend today is sometimes ‘old history’ tomorrow.”
Dr. Landewé had no relevant disclosures to make.
The European Alliance of Associations for Rheumatology has started the process of updating their recommendations on how to manage patients with rheumatic and musculoskeletal diseases (RMDs) in the context of the SARS-CoV-2 pandemic.
So far, the first part of the systematic literature review has been performed and the conclusions that have been drawn appear to back up the recommendations that have already been made. It’s “hard to say” if there will need to be changes, said Robert B.M. Landewé, MD, PhD, at the annual European Congress of Rheumatology, as the next phase will be for the task force members to meet and discuss the implications of the literature research.“I think there will only be minor modifications and a few novel recommendations, but that is personal opinion,” speculated Dr. Landewé, who is professor of rheumatology at the Amsterdam Medical Center, University of Amsterdam.
The recommendations, which were developed a little over a year ago and published in Annals of the Rheumatic Diseases, set out provisional guidance covering four themes: infection prevention, managing patients when social distancing measures are in effect, managing patients with RMDs who develop COVID-19, and the prevention of infections other than SARS-CoV-2.
Emphasis on quality of evidence
According to EULAR’s standard operating procedures “updates should only be done if the evolving evidence mandates to do so,” and be based on “rational arguments,” Dr. Landewé said. “The last year was a bit unprecedented in that regard as we didn’t have those rational arguments before we designed our first set of recommendations and, as you can expect, that is totally due to the character of the pandemic.”
So much has been published on COVID-19 since then it was time to reappraise the situation. The task force behind the recommendations met in January 2021 to discuss the results of the literature search that was centered around five main research questions.
- Do patients with RMDs face more risk of contracting SARS-CoV-2 than the general population?
- If patients contract the virus, do they have a worse prognosis?
- Are antirheumatic medications associated with a worse outcome in people with RMDs?
- Should patients continue their antirheumatic medications?
- What evidence informs the use of vaccination against SARS-CoV-2 in patients with RMDs?
The latter research question is pending discussion since there were no studies to review at the time as the various vaccines had only just started to be widely available.
“We put a lot of emphasis on the quality of evidence,” Dr. Landewé said. In addition to making sure that patients did indeed have COVID-19 and checking that hospitalization and death records were caused by the disease, the task force team also looked to see if there was a control group being used. An extensive risk of bias assessment was undertaken, the results of which are pending.
Of 6,665 records identified during the literature search, just 113 full-text articles were assessed for eligibility. Of those, 60% were rejected as they did not pass the quality assessment, leaving 49 articles for consideration. The majority of these looked at the incidence of COVID-19, with others focusing on risk factors or both.
Literature search findings on main research questions
Dr. Landewé observed that the task force concluded that “current literature provides no evidence that patients with RMDs face more risk of contracting SARS-CoV-2 than individuals without RMDs.” They also concluded that patients with RMDs who do contract COVID-19 do not have a worse prognosis either, even though there have been a few studies suggesting a higher rate of hospitalization.
Both findings are reassuring as they fit with the existing recommendation to follow the same preventive and control measures in patients with RMDs as for the general population, but the task force is yet to determine if that recommendation should be amended.
There did not appear to be any hard evidence of any unique demographic feature or comorbidity that puts people with RMDs at more risk for severe COVID-19 than the general population. Think older age, male gender, high bodyweight, cardiovascular disease, diabetes, and chronic lung disease, Dr. Landewé said.
He noted, however, that there were some single-center reports suggesting that moderate or high levels of disease activity could put people with RMDs at greater risk for COVID-related death, “which is an intriguing finding in the context of discontinuing antirheumatic medication.” That is likely something the task force will be discussing when they decide how to update their recommendations.
The type of RMD may also be important, but again only single-center evidence to show that there might be an increased hospitalization risk in patients with autoinflammatory disease or risk for severe COVID-19 in those with certain connective tissue diseases. “These associations were not consistently found in other studies,” so it’s an open question how the task force decides to incorporate this into the updated guidance.
As for antirheumatic medications, conclusions from the literature review suggest that there doesn’t appear to be an increased or decreased risk for severe COVID-19 among users of NSAIDs or antimalarials.
That’s not the case for glucocorticoids. There appears to be an increased risk for hospitalization and COVID-19–related death, notably among those using higher (>10 mg) daily doses. “This is, so to say, the elephant in the room,” Dr. Landewé said. The current recommendation states that chronic users of glucocorticoids should continue their treatment. “The reports of additional risk could be due to glucocorticoids or to biases such as confounding by indication. So, the conclusion that we draw [is] not completely clear.”
In response to a question, he clarified this a little further: “We think ‘glucocorticoid use’ is a determinant of worse health, as is the case in many RMDs. Be aware that finding a positive association between [glucocorticoid] use and bad outcome does not mean that if you reduce [glucocorticoids], your patient will have a better outcome.”
The jury is also out on rituximab, which has been reported to increase the risk of severe COVID-19 and COVID-related death in two studies. There are also equivocal data on whether not using disease-modifying antirheumatic drugs increases the risk for these worse outcomes.
Asked about the absence of a recommendation on the use of the interleukin-6 inhibitor tocilizumab, Dr. Landewé responded: “We are caught up by evolving evidence. That is a generic problem in a dynamic field of COVID-19, I am afraid. What you recommend today is sometimes ‘old history’ tomorrow.”
Dr. Landewé had no relevant disclosures to make.
The European Alliance of Associations for Rheumatology has started the process of updating their recommendations on how to manage patients with rheumatic and musculoskeletal diseases (RMDs) in the context of the SARS-CoV-2 pandemic.
So far, the first part of the systematic literature review has been performed and the conclusions that have been drawn appear to back up the recommendations that have already been made. It’s “hard to say” if there will need to be changes, said Robert B.M. Landewé, MD, PhD, at the annual European Congress of Rheumatology, as the next phase will be for the task force members to meet and discuss the implications of the literature research.“I think there will only be minor modifications and a few novel recommendations, but that is personal opinion,” speculated Dr. Landewé, who is professor of rheumatology at the Amsterdam Medical Center, University of Amsterdam.
The recommendations, which were developed a little over a year ago and published in Annals of the Rheumatic Diseases, set out provisional guidance covering four themes: infection prevention, managing patients when social distancing measures are in effect, managing patients with RMDs who develop COVID-19, and the prevention of infections other than SARS-CoV-2.
Emphasis on quality of evidence
According to EULAR’s standard operating procedures “updates should only be done if the evolving evidence mandates to do so,” and be based on “rational arguments,” Dr. Landewé said. “The last year was a bit unprecedented in that regard as we didn’t have those rational arguments before we designed our first set of recommendations and, as you can expect, that is totally due to the character of the pandemic.”
So much has been published on COVID-19 since then it was time to reappraise the situation. The task force behind the recommendations met in January 2021 to discuss the results of the literature search that was centered around five main research questions.
- Do patients with RMDs face more risk of contracting SARS-CoV-2 than the general population?
- If patients contract the virus, do they have a worse prognosis?
- Are antirheumatic medications associated with a worse outcome in people with RMDs?
- Should patients continue their antirheumatic medications?
- What evidence informs the use of vaccination against SARS-CoV-2 in patients with RMDs?
The latter research question is pending discussion since there were no studies to review at the time as the various vaccines had only just started to be widely available.
“We put a lot of emphasis on the quality of evidence,” Dr. Landewé said. In addition to making sure that patients did indeed have COVID-19 and checking that hospitalization and death records were caused by the disease, the task force team also looked to see if there was a control group being used. An extensive risk of bias assessment was undertaken, the results of which are pending.
Of 6,665 records identified during the literature search, just 113 full-text articles were assessed for eligibility. Of those, 60% were rejected as they did not pass the quality assessment, leaving 49 articles for consideration. The majority of these looked at the incidence of COVID-19, with others focusing on risk factors or both.
Literature search findings on main research questions
Dr. Landewé observed that the task force concluded that “current literature provides no evidence that patients with RMDs face more risk of contracting SARS-CoV-2 than individuals without RMDs.” They also concluded that patients with RMDs who do contract COVID-19 do not have a worse prognosis either, even though there have been a few studies suggesting a higher rate of hospitalization.
Both findings are reassuring as they fit with the existing recommendation to follow the same preventive and control measures in patients with RMDs as for the general population, but the task force is yet to determine if that recommendation should be amended.
There did not appear to be any hard evidence of any unique demographic feature or comorbidity that puts people with RMDs at more risk for severe COVID-19 than the general population. Think older age, male gender, high bodyweight, cardiovascular disease, diabetes, and chronic lung disease, Dr. Landewé said.
He noted, however, that there were some single-center reports suggesting that moderate or high levels of disease activity could put people with RMDs at greater risk for COVID-related death, “which is an intriguing finding in the context of discontinuing antirheumatic medication.” That is likely something the task force will be discussing when they decide how to update their recommendations.
The type of RMD may also be important, but again only single-center evidence to show that there might be an increased hospitalization risk in patients with autoinflammatory disease or risk for severe COVID-19 in those with certain connective tissue diseases. “These associations were not consistently found in other studies,” so it’s an open question how the task force decides to incorporate this into the updated guidance.
As for antirheumatic medications, conclusions from the literature review suggest that there doesn’t appear to be an increased or decreased risk for severe COVID-19 among users of NSAIDs or antimalarials.
That’s not the case for glucocorticoids. There appears to be an increased risk for hospitalization and COVID-19–related death, notably among those using higher (>10 mg) daily doses. “This is, so to say, the elephant in the room,” Dr. Landewé said. The current recommendation states that chronic users of glucocorticoids should continue their treatment. “The reports of additional risk could be due to glucocorticoids or to biases such as confounding by indication. So, the conclusion that we draw [is] not completely clear.”
In response to a question, he clarified this a little further: “We think ‘glucocorticoid use’ is a determinant of worse health, as is the case in many RMDs. Be aware that finding a positive association between [glucocorticoid] use and bad outcome does not mean that if you reduce [glucocorticoids], your patient will have a better outcome.”
The jury is also out on rituximab, which has been reported to increase the risk of severe COVID-19 and COVID-related death in two studies. There are also equivocal data on whether not using disease-modifying antirheumatic drugs increases the risk for these worse outcomes.
Asked about the absence of a recommendation on the use of the interleukin-6 inhibitor tocilizumab, Dr. Landewé responded: “We are caught up by evolving evidence. That is a generic problem in a dynamic field of COVID-19, I am afraid. What you recommend today is sometimes ‘old history’ tomorrow.”
Dr. Landewé had no relevant disclosures to make.
FROM THE EULAR 2021 CONGRESS
Intravenous immunoglobulin controls dermatomyositis in phase 3 trial
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
Nearly 50% achieve moderate improvement or better
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
FROM THE EULAR 2021 CONGRESS