User login
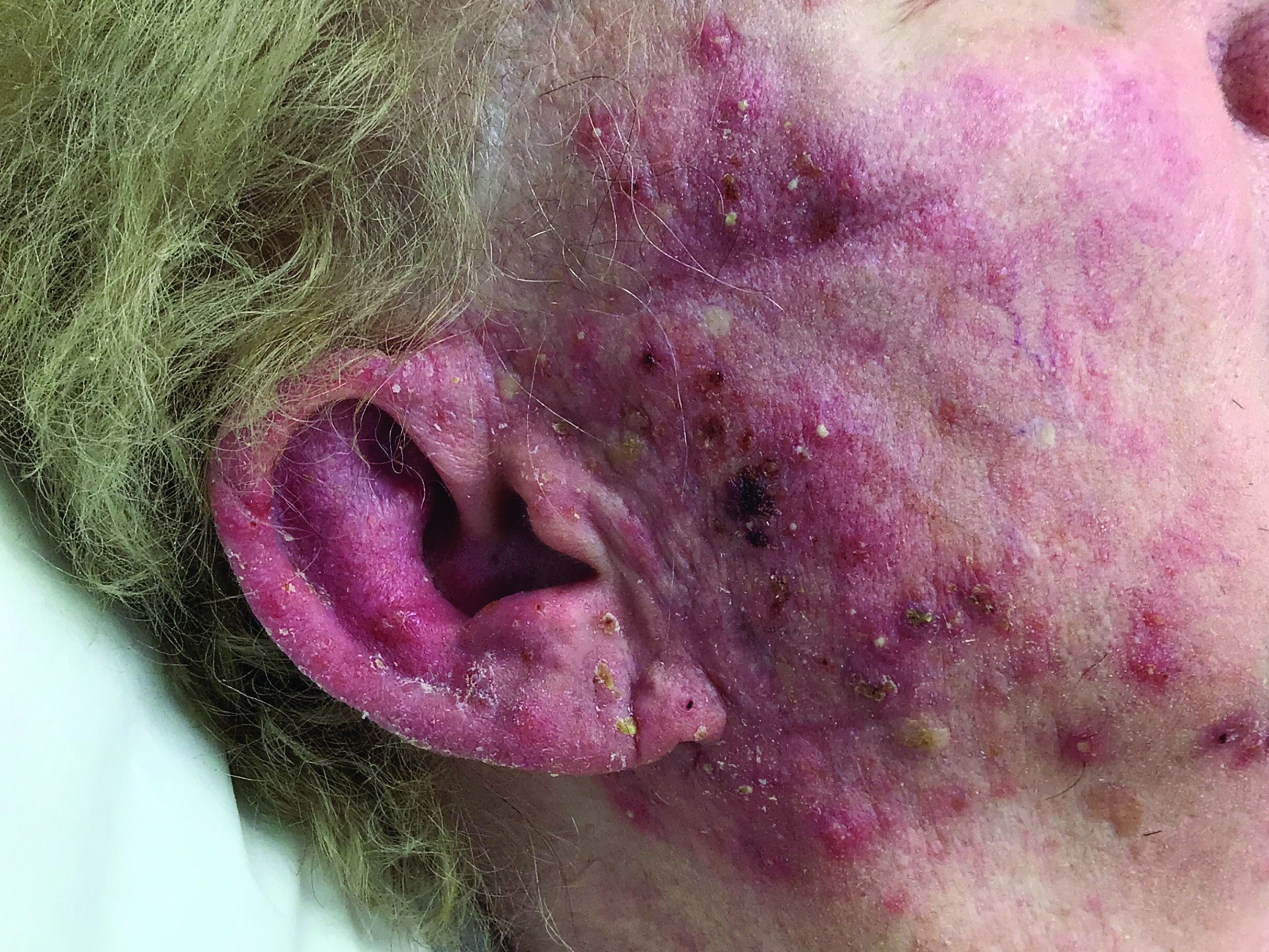
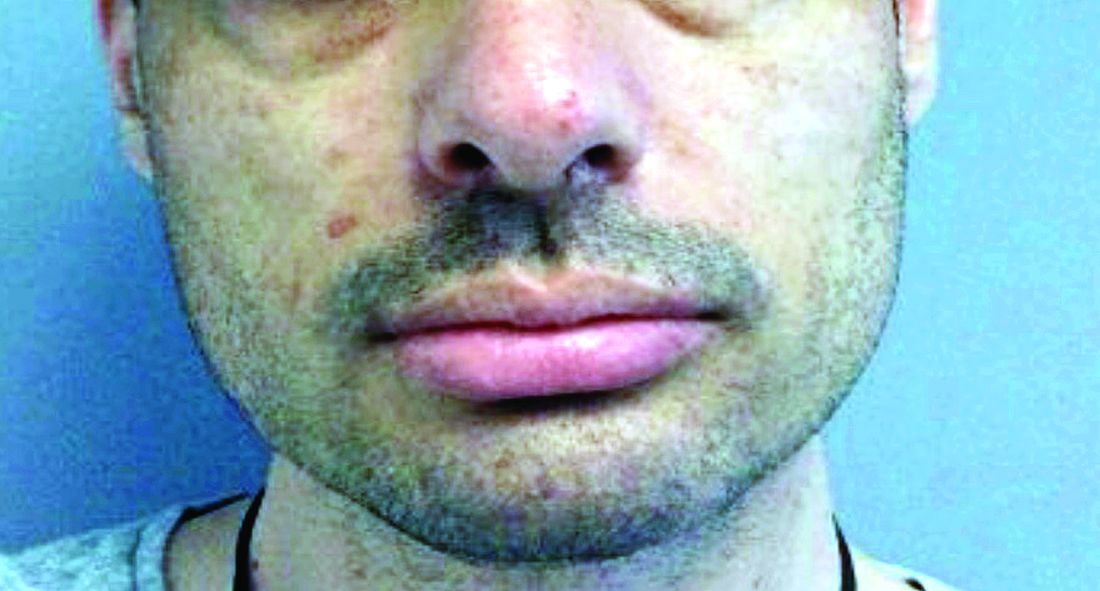
A woman with scaling, and painful, crusted, erythematous papules and pustules on her face
Biopsy for this patient revealed folliculitis with Demodex mites visualized on histology. Direct immunofluorescence was negative. A KOH preparation was performed and was positive for large numbers of Demodex. Bacterial cultures were negative. The patient was started on a course of submicrobial doxycycline and ivermectin and showed marked improvement 1 month following treatment.
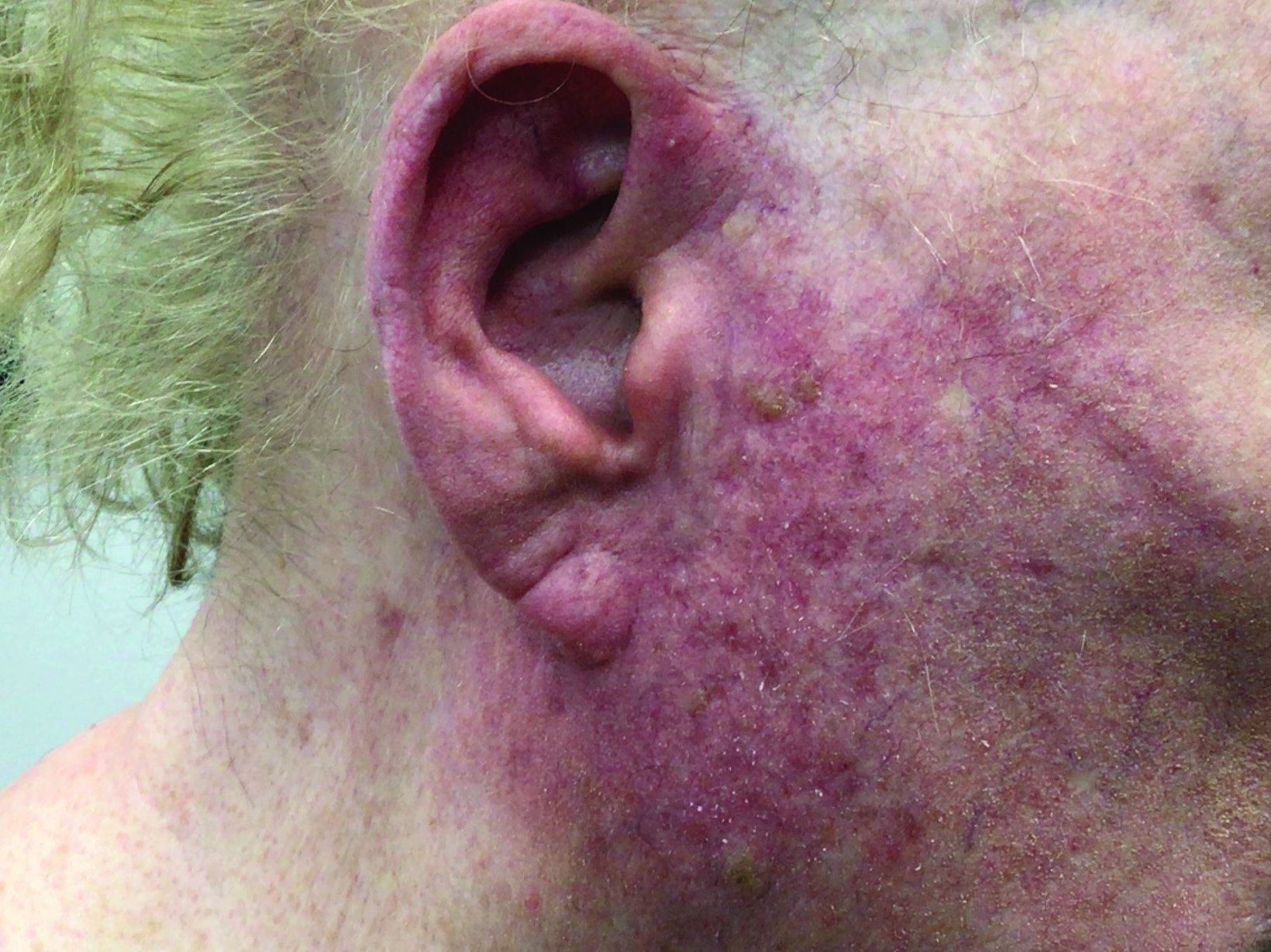
Demodex folliculorum and Demodex brevis (collectively referred to as Demodex) are microscopic parasitic mites that commonly live on human skin.1 Typically, the mite remains asymptomatic. However, in higher numbers, the infestation may cause dermatoses, called demodicosis. Lesions often present as itchy papules, pustules, and erythematous scaling on the face, ears, and scalp. Blepharitis may be present. Demodex folliculitis is more common in immunocompromised patients.2
Demodex may have a causative role in rosacea and present similarly, with a key difference being that Demodex-type rosacea is more scaly/dry and pustular than common rosacea.1 In Demodex folliculitis, bacterial cultures are often negative. A skin scraping for KOH will reveal increased mite colonization. The Demodex mite may also be seen in histologic slides.
Treatment of Demodex folliculitis includes crotamiton cream, permethrin cream, oral tetracyclines, topical or systemic metronidazole, and topical or oral ivermectin.
This case and photos were submitted by Susannah McClain, MD, Three Rivers Dermatology, Pittsburgh.
References
1. Rather PA and Hassan I. Indian J Dermatol. 2014 Jan;59(1):60-6.
2. Bachmeyer C and Moreno-Sabater A. CMAJ. 2017 Jun 26;189(25):E865.
Biopsy for this patient revealed folliculitis with Demodex mites visualized on histology. Direct immunofluorescence was negative. A KOH preparation was performed and was positive for large numbers of Demodex. Bacterial cultures were negative. The patient was started on a course of submicrobial doxycycline and ivermectin and showed marked improvement 1 month following treatment.
Demodex folliculorum and Demodex brevis (collectively referred to as Demodex) are microscopic parasitic mites that commonly live on human skin.1 Typically, the mite remains asymptomatic. However, in higher numbers, the infestation may cause dermatoses, called demodicosis. Lesions often present as itchy papules, pustules, and erythematous scaling on the face, ears, and scalp. Blepharitis may be present. Demodex folliculitis is more common in immunocompromised patients.2
Demodex may have a causative role in rosacea and present similarly, with a key difference being that Demodex-type rosacea is more scaly/dry and pustular than common rosacea.1 In Demodex folliculitis, bacterial cultures are often negative. A skin scraping for KOH will reveal increased mite colonization. The Demodex mite may also be seen in histologic slides.
Treatment of Demodex folliculitis includes crotamiton cream, permethrin cream, oral tetracyclines, topical or systemic metronidazole, and topical or oral ivermectin.
This case and photos were submitted by Susannah McClain, MD, Three Rivers Dermatology, Pittsburgh.
References
1. Rather PA and Hassan I. Indian J Dermatol. 2014 Jan;59(1):60-6.
2. Bachmeyer C and Moreno-Sabater A. CMAJ. 2017 Jun 26;189(25):E865.
Biopsy for this patient revealed folliculitis with Demodex mites visualized on histology. Direct immunofluorescence was negative. A KOH preparation was performed and was positive for large numbers of Demodex. Bacterial cultures were negative. The patient was started on a course of submicrobial doxycycline and ivermectin and showed marked improvement 1 month following treatment.
Demodex folliculorum and Demodex brevis (collectively referred to as Demodex) are microscopic parasitic mites that commonly live on human skin.1 Typically, the mite remains asymptomatic. However, in higher numbers, the infestation may cause dermatoses, called demodicosis. Lesions often present as itchy papules, pustules, and erythematous scaling on the face, ears, and scalp. Blepharitis may be present. Demodex folliculitis is more common in immunocompromised patients.2
Demodex may have a causative role in rosacea and present similarly, with a key difference being that Demodex-type rosacea is more scaly/dry and pustular than common rosacea.1 In Demodex folliculitis, bacterial cultures are often negative. A skin scraping for KOH will reveal increased mite colonization. The Demodex mite may also be seen in histologic slides.
Treatment of Demodex folliculitis includes crotamiton cream, permethrin cream, oral tetracyclines, topical or systemic metronidazole, and topical or oral ivermectin.
This case and photos were submitted by Susannah McClain, MD, Three Rivers Dermatology, Pittsburgh.
References
1. Rather PA and Hassan I. Indian J Dermatol. 2014 Jan;59(1):60-6.
2. Bachmeyer C and Moreno-Sabater A. CMAJ. 2017 Jun 26;189(25):E865.
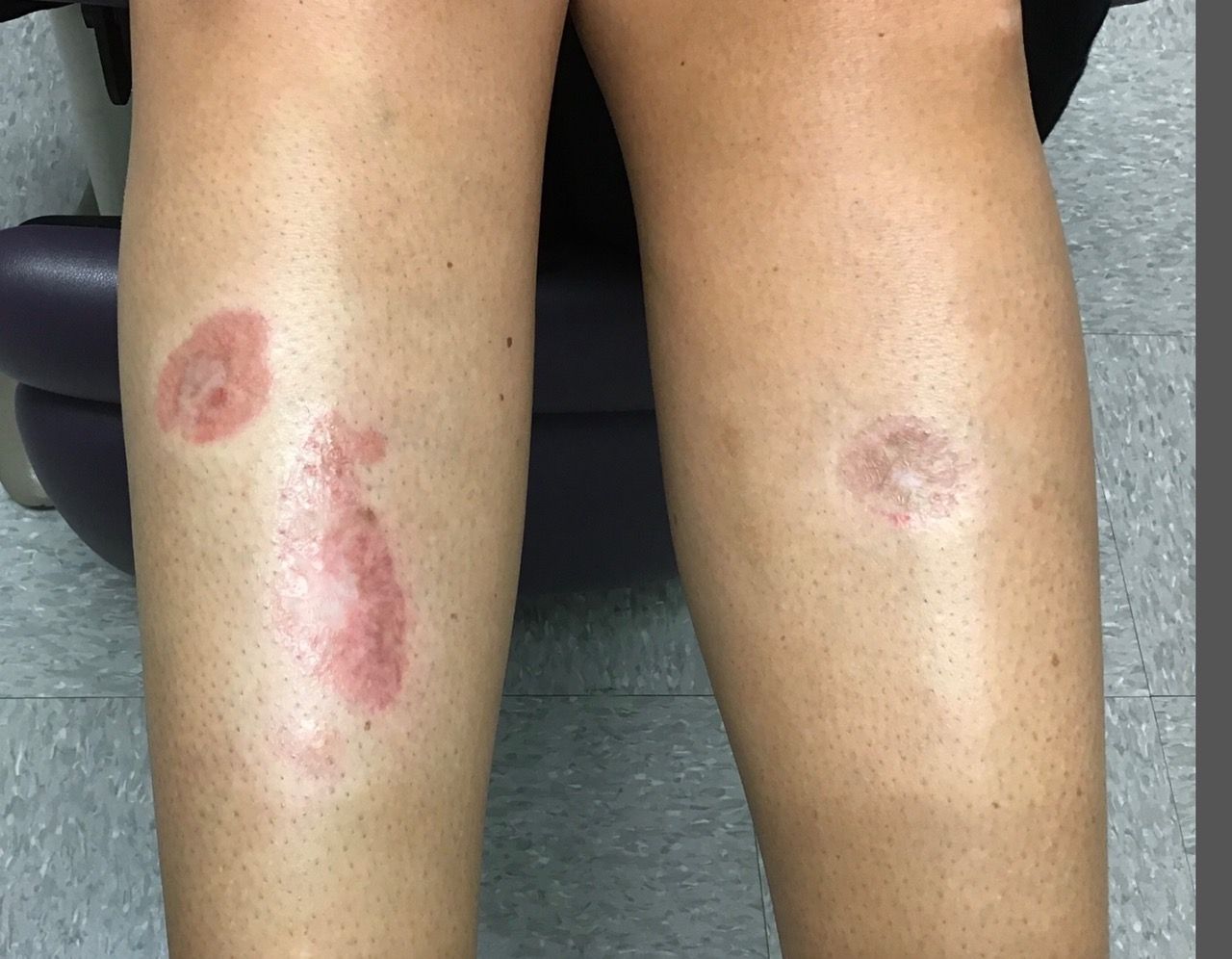
A woman with a history of diabetes, and plaques on both shins
. Women are often more affected than men. Patients often present in their 30s and 40s. The cause of NLD is unknown. Twenty percent of patients with NLD will have glucose intolerance or a family history of diabetes.1 The percentage of patients with NLD who have diabetes varies in reports from 11% to 65%.2 NLD may progress despite the diabetes treatment. Only 0.03% of patient with diabetes will have NLD.3

Lesions most commonly occur on the extremities, with shins being affected in most cases. They vary from asymptomatic to painful. Typically, lesions begin as small, firm erythematous papules that evolve into shiny, well-defined plaques. In older plaques, the center will often appear yellow, depressed, and atrophic, with telangiectasias. The periphery appears pink to violaceous to brown. Ulceration may be present, particularly after trauma, and there may be decreased sensation in the plaques. NLD is clinically distinct from diabetic dermopathy, which appear as brown macules, often in older patients with diabetes.
Ideally, biopsy should be taken at the edge of a lesion. Histologically, the epidermis appears normal or atrophic. A diffuse palisaded and interstitial granulomatous dermatitis consisting of histiocytes, multinucleated giant cells, lymphocytes, and plasma cells is seen in the dermis. Granulomas are often oriented parallel to the epidermis. There is no mucin at the center of the granulomas (as seen in granuloma annulare). Inflammation may extend into the subcutaneous fat. Asteroid bodies (as seen in sarcoid) are absent.
Unfortunately, treatment of NLD is often unsuccessful. Treatment includes potent topical corticosteroids for early lesions and intralesional triamcinolone to the leading edge of lesions. Care should be taken to avoid injecting centrally where atrophy and ulceration may result. Systemic steroids may be helpful in some cases, but can elevate glucose levels. Other reported medical treatments include pentoxifylline, cyclosporine, and niacinamide. Some lesions may spontaneously resolve. Ulcerations may require surgical excision with grafting.
This case and photo are provided by Dr. Bilu Martin, who is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. James WD et al. Andrews’ Diseases of the Skin: Clinical Dermatology. Philadelphia: Saunders Elsevier, 2006.
2. Hashemi D et al. JAMA Dermatol. 2019 Apr 1;155(4):455-9.
3. Bolognia JL et al. Dermatology. St. Louis, Mo.: Mosby Elsevier, 2008.
. Women are often more affected than men. Patients often present in their 30s and 40s. The cause of NLD is unknown. Twenty percent of patients with NLD will have glucose intolerance or a family history of diabetes.1 The percentage of patients with NLD who have diabetes varies in reports from 11% to 65%.2 NLD may progress despite the diabetes treatment. Only 0.03% of patient with diabetes will have NLD.3
Lesions most commonly occur on the extremities, with shins being affected in most cases. They vary from asymptomatic to painful. Typically, lesions begin as small, firm erythematous papules that evolve into shiny, well-defined plaques. In older plaques, the center will often appear yellow, depressed, and atrophic, with telangiectasias. The periphery appears pink to violaceous to brown. Ulceration may be present, particularly after trauma, and there may be decreased sensation in the plaques. NLD is clinically distinct from diabetic dermopathy, which appear as brown macules, often in older patients with diabetes.
Ideally, biopsy should be taken at the edge of a lesion. Histologically, the epidermis appears normal or atrophic. A diffuse palisaded and interstitial granulomatous dermatitis consisting of histiocytes, multinucleated giant cells, lymphocytes, and plasma cells is seen in the dermis. Granulomas are often oriented parallel to the epidermis. There is no mucin at the center of the granulomas (as seen in granuloma annulare). Inflammation may extend into the subcutaneous fat. Asteroid bodies (as seen in sarcoid) are absent.
Unfortunately, treatment of NLD is often unsuccessful. Treatment includes potent topical corticosteroids for early lesions and intralesional triamcinolone to the leading edge of lesions. Care should be taken to avoid injecting centrally where atrophy and ulceration may result. Systemic steroids may be helpful in some cases, but can elevate glucose levels. Other reported medical treatments include pentoxifylline, cyclosporine, and niacinamide. Some lesions may spontaneously resolve. Ulcerations may require surgical excision with grafting.
This case and photo are provided by Dr. Bilu Martin, who is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. James WD et al. Andrews’ Diseases of the Skin: Clinical Dermatology. Philadelphia: Saunders Elsevier, 2006.
2. Hashemi D et al. JAMA Dermatol. 2019 Apr 1;155(4):455-9.
3. Bolognia JL et al. Dermatology. St. Louis, Mo.: Mosby Elsevier, 2008.
. Women are often more affected than men. Patients often present in their 30s and 40s. The cause of NLD is unknown. Twenty percent of patients with NLD will have glucose intolerance or a family history of diabetes.1 The percentage of patients with NLD who have diabetes varies in reports from 11% to 65%.2 NLD may progress despite the diabetes treatment. Only 0.03% of patient with diabetes will have NLD.3
Lesions most commonly occur on the extremities, with shins being affected in most cases. They vary from asymptomatic to painful. Typically, lesions begin as small, firm erythematous papules that evolve into shiny, well-defined plaques. In older plaques, the center will often appear yellow, depressed, and atrophic, with telangiectasias. The periphery appears pink to violaceous to brown. Ulceration may be present, particularly after trauma, and there may be decreased sensation in the plaques. NLD is clinically distinct from diabetic dermopathy, which appear as brown macules, often in older patients with diabetes.
Ideally, biopsy should be taken at the edge of a lesion. Histologically, the epidermis appears normal or atrophic. A diffuse palisaded and interstitial granulomatous dermatitis consisting of histiocytes, multinucleated giant cells, lymphocytes, and plasma cells is seen in the dermis. Granulomas are often oriented parallel to the epidermis. There is no mucin at the center of the granulomas (as seen in granuloma annulare). Inflammation may extend into the subcutaneous fat. Asteroid bodies (as seen in sarcoid) are absent.
Unfortunately, treatment of NLD is often unsuccessful. Treatment includes potent topical corticosteroids for early lesions and intralesional triamcinolone to the leading edge of lesions. Care should be taken to avoid injecting centrally where atrophy and ulceration may result. Systemic steroids may be helpful in some cases, but can elevate glucose levels. Other reported medical treatments include pentoxifylline, cyclosporine, and niacinamide. Some lesions may spontaneously resolve. Ulcerations may require surgical excision with grafting.
This case and photo are provided by Dr. Bilu Martin, who is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. James WD et al. Andrews’ Diseases of the Skin: Clinical Dermatology. Philadelphia: Saunders Elsevier, 2006.
2. Hashemi D et al. JAMA Dermatol. 2019 Apr 1;155(4):455-9.
3. Bolognia JL et al. Dermatology. St. Louis, Mo.: Mosby Elsevier, 2008.
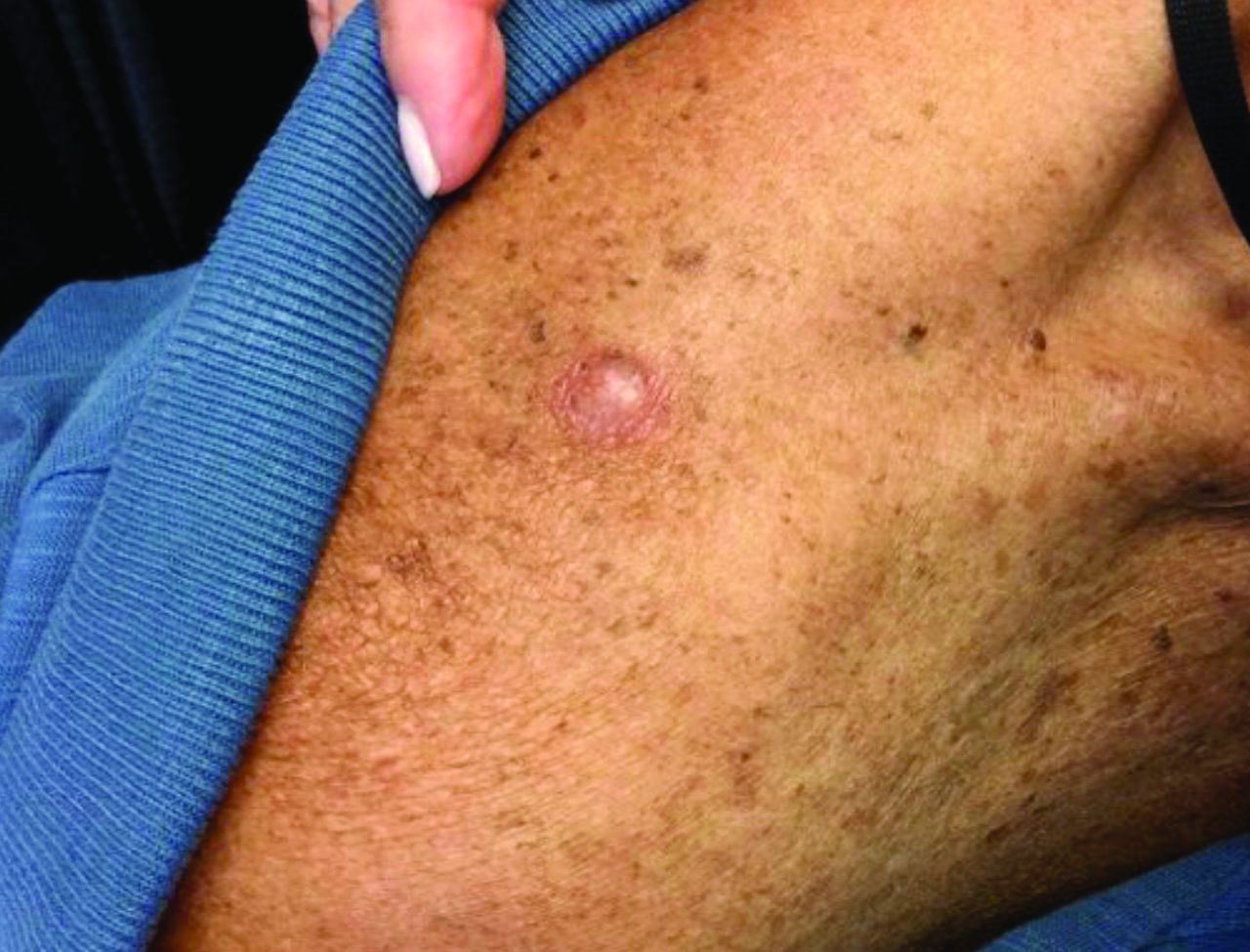
An 80-year-old patient presents with an asymptomatic firm pink plaque on his shoulder
Melanoma is a type of skin cancer that arises from melanocytes. According to the American Cancer Society, about 106,110 new melanomas will be diagnosed in the United States in 2021.The risk for developing melanoma increases with age. There are multiple clinical forms of cutaneous melanoma. The four main types are superficial spreading melanoma, nodular melanoma, melanoma in situ (lentigo maligna), and acral lentiginous melanoma. Rare variants include amelanotic melanoma, nevoid melanoma, spitzoid melanoma, and desmoplastic melanoma (DM). Melanoma can also rarely affect parts of the eye and mucosa.
, according to the Memorial Sloan Kettering Cancer Center. It typically presents as a subtle pigmented, pink, red, or skin colored patch, papule or plaque on sun-exposed skin (head and neck most frequently). Chronic UV exposure has been linked to DM. It may be mistaken for a scar or dermatofibroma. DM tends to grow locally and has less risk for nodal metastasis.1
Histologic diagnosis may be challenging. Two histologic variants in desmoplastic melanoma have been described: pure and mixed, depending on the degree of desmoplasia and cellularity present in the tumor.1 Pure DM tends to have a less aggressive course. Melanocytes can appear spindled in a fibrotic stroma. Patchy lymphocyte aggregates may be seen. Perineural invasion is more common in desmoplastic melanoma. Histologically, the differential includes spindle cell carcinoma and sarcoma. Immunostaining is helpful in differentiation.
Our patient had no lymphadenopathy on physical examination. Biopsy revealed a desmoplastic melanoma, 3.6 mm in depth, no ulceration, no regression, mitotic rate 1/mm2. He was referred to surgical oncology. The patient underwent wide excision. Sentinel lymph node biopsy was deferred.
It is imperative for dermatologists to be cognizant of this challenging subtype of melanoma when evaluating patients.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Chen L et al. J Am Acad Dermatol. 2013 May;68(5):825-33.
Melanoma is a type of skin cancer that arises from melanocytes. According to the American Cancer Society, about 106,110 new melanomas will be diagnosed in the United States in 2021.The risk for developing melanoma increases with age. There are multiple clinical forms of cutaneous melanoma. The four main types are superficial spreading melanoma, nodular melanoma, melanoma in situ (lentigo maligna), and acral lentiginous melanoma. Rare variants include amelanotic melanoma, nevoid melanoma, spitzoid melanoma, and desmoplastic melanoma (DM). Melanoma can also rarely affect parts of the eye and mucosa.
, according to the Memorial Sloan Kettering Cancer Center. It typically presents as a subtle pigmented, pink, red, or skin colored patch, papule or plaque on sun-exposed skin (head and neck most frequently). Chronic UV exposure has been linked to DM. It may be mistaken for a scar or dermatofibroma. DM tends to grow locally and has less risk for nodal metastasis.1
Histologic diagnosis may be challenging. Two histologic variants in desmoplastic melanoma have been described: pure and mixed, depending on the degree of desmoplasia and cellularity present in the tumor.1 Pure DM tends to have a less aggressive course. Melanocytes can appear spindled in a fibrotic stroma. Patchy lymphocyte aggregates may be seen. Perineural invasion is more common in desmoplastic melanoma. Histologically, the differential includes spindle cell carcinoma and sarcoma. Immunostaining is helpful in differentiation.
Our patient had no lymphadenopathy on physical examination. Biopsy revealed a desmoplastic melanoma, 3.6 mm in depth, no ulceration, no regression, mitotic rate 1/mm2. He was referred to surgical oncology. The patient underwent wide excision. Sentinel lymph node biopsy was deferred.
It is imperative for dermatologists to be cognizant of this challenging subtype of melanoma when evaluating patients.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Chen L et al. J Am Acad Dermatol. 2013 May;68(5):825-33.
Melanoma is a type of skin cancer that arises from melanocytes. According to the American Cancer Society, about 106,110 new melanomas will be diagnosed in the United States in 2021.The risk for developing melanoma increases with age. There are multiple clinical forms of cutaneous melanoma. The four main types are superficial spreading melanoma, nodular melanoma, melanoma in situ (lentigo maligna), and acral lentiginous melanoma. Rare variants include amelanotic melanoma, nevoid melanoma, spitzoid melanoma, and desmoplastic melanoma (DM). Melanoma can also rarely affect parts of the eye and mucosa.
, according to the Memorial Sloan Kettering Cancer Center. It typically presents as a subtle pigmented, pink, red, or skin colored patch, papule or plaque on sun-exposed skin (head and neck most frequently). Chronic UV exposure has been linked to DM. It may be mistaken for a scar or dermatofibroma. DM tends to grow locally and has less risk for nodal metastasis.1
Histologic diagnosis may be challenging. Two histologic variants in desmoplastic melanoma have been described: pure and mixed, depending on the degree of desmoplasia and cellularity present in the tumor.1 Pure DM tends to have a less aggressive course. Melanocytes can appear spindled in a fibrotic stroma. Patchy lymphocyte aggregates may be seen. Perineural invasion is more common in desmoplastic melanoma. Histologically, the differential includes spindle cell carcinoma and sarcoma. Immunostaining is helpful in differentiation.
Our patient had no lymphadenopathy on physical examination. Biopsy revealed a desmoplastic melanoma, 3.6 mm in depth, no ulceration, no regression, mitotic rate 1/mm2. He was referred to surgical oncology. The patient underwent wide excision. Sentinel lymph node biopsy was deferred.
It is imperative for dermatologists to be cognizant of this challenging subtype of melanoma when evaluating patients.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Chen L et al. J Am Acad Dermatol. 2013 May;68(5):825-33.
A 35-year-old male who takes antiseizure medications, has asymptomatic lesions on his nose and cheeks, present since birth
although up to 75% of cases may be caused by a spontaneous mutation. It is caused by mutations in the TSC1 gene on chromosome 9q34–encoding hamartin or the TSC2 gene on chromosome I6pl3–encoding tuberin. Patients present at birth and males and females are affected equally.
There are multiple skin findings in TS that may herald the diagnosis. The earliest findings are hypopigmented macules, found in 85% of patients. They may be in an ash-leaf shape or confetti pattern. Adenoma sebaceum, or angiofibromas, are present on the forehead, nose, and cheeks, and often present in childhood. Periungual angiofibromas called Koenen tumors tend to occur at puberty. Connective-tissue nevi called Shagreen plaques, or collagenomas, may be present, which is what our patient exhibits on his back. The lumbosacral region is the most common area for these to appear in the first decade of life.
TS can affect other organ systems in the body. Seizures, neuropsychiatric diseases, and mental deficiency are common. Cortical tumors, gliomas, and astrocytomas may develop in the brain. Congenital retinal hamartomas (phakomas) occur. Renal cysts and angiomyolipomas may occur in the kidneys. In the lungs, patients may develop lymphangiomyomatosis. Rhabdomyomas can occur in the heart in infancy and may regress spontaneously over time. Bony changes such as cysts and sclerosis may occur.
Treatment and monitoring of TS requires a multidisciplinary approach with neurology, pulmonology, cardiology, ophthalmology, orthopedics, and dermatology. Cosmetic treatment for angiofibromas includes CO2 laser, shaving, and dermabrasion. Topical rapamycin use has been described in the literature to improve the appearance of angiofibromas. Our patient has been using rapamycin 1% cream for more than 5 years and has had a substantial reduction in the size and number of angiofibromas.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References:
Spitz J. Genodermatoses. A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins, 2005.
James W et al. Andrews’ Diseases of the Skin. Philadelphia: Saunders, 2006.
Bolognia JL et al. Dermatology. London: Mosby Elsevier, 2008.
although up to 75% of cases may be caused by a spontaneous mutation. It is caused by mutations in the TSC1 gene on chromosome 9q34–encoding hamartin or the TSC2 gene on chromosome I6pl3–encoding tuberin. Patients present at birth and males and females are affected equally.
There are multiple skin findings in TS that may herald the diagnosis. The earliest findings are hypopigmented macules, found in 85% of patients. They may be in an ash-leaf shape or confetti pattern. Adenoma sebaceum, or angiofibromas, are present on the forehead, nose, and cheeks, and often present in childhood. Periungual angiofibromas called Koenen tumors tend to occur at puberty. Connective-tissue nevi called Shagreen plaques, or collagenomas, may be present, which is what our patient exhibits on his back. The lumbosacral region is the most common area for these to appear in the first decade of life.
TS can affect other organ systems in the body. Seizures, neuropsychiatric diseases, and mental deficiency are common. Cortical tumors, gliomas, and astrocytomas may develop in the brain. Congenital retinal hamartomas (phakomas) occur. Renal cysts and angiomyolipomas may occur in the kidneys. In the lungs, patients may develop lymphangiomyomatosis. Rhabdomyomas can occur in the heart in infancy and may regress spontaneously over time. Bony changes such as cysts and sclerosis may occur.
Treatment and monitoring of TS requires a multidisciplinary approach with neurology, pulmonology, cardiology, ophthalmology, orthopedics, and dermatology. Cosmetic treatment for angiofibromas includes CO2 laser, shaving, and dermabrasion. Topical rapamycin use has been described in the literature to improve the appearance of angiofibromas. Our patient has been using rapamycin 1% cream for more than 5 years and has had a substantial reduction in the size and number of angiofibromas.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References:
Spitz J. Genodermatoses. A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins, 2005.
James W et al. Andrews’ Diseases of the Skin. Philadelphia: Saunders, 2006.
Bolognia JL et al. Dermatology. London: Mosby Elsevier, 2008.
although up to 75% of cases may be caused by a spontaneous mutation. It is caused by mutations in the TSC1 gene on chromosome 9q34–encoding hamartin or the TSC2 gene on chromosome I6pl3–encoding tuberin. Patients present at birth and males and females are affected equally.
There are multiple skin findings in TS that may herald the diagnosis. The earliest findings are hypopigmented macules, found in 85% of patients. They may be in an ash-leaf shape or confetti pattern. Adenoma sebaceum, or angiofibromas, are present on the forehead, nose, and cheeks, and often present in childhood. Periungual angiofibromas called Koenen tumors tend to occur at puberty. Connective-tissue nevi called Shagreen plaques, or collagenomas, may be present, which is what our patient exhibits on his back. The lumbosacral region is the most common area for these to appear in the first decade of life.
TS can affect other organ systems in the body. Seizures, neuropsychiatric diseases, and mental deficiency are common. Cortical tumors, gliomas, and astrocytomas may develop in the brain. Congenital retinal hamartomas (phakomas) occur. Renal cysts and angiomyolipomas may occur in the kidneys. In the lungs, patients may develop lymphangiomyomatosis. Rhabdomyomas can occur in the heart in infancy and may regress spontaneously over time. Bony changes such as cysts and sclerosis may occur.
Treatment and monitoring of TS requires a multidisciplinary approach with neurology, pulmonology, cardiology, ophthalmology, orthopedics, and dermatology. Cosmetic treatment for angiofibromas includes CO2 laser, shaving, and dermabrasion. Topical rapamycin use has been described in the literature to improve the appearance of angiofibromas. Our patient has been using rapamycin 1% cream for more than 5 years and has had a substantial reduction in the size and number of angiofibromas.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References:
Spitz J. Genodermatoses. A Clinical Guide to Genetic Skin Disorders. Philadelphia: Lippincott Williams & Wilkins, 2005.
James W et al. Andrews’ Diseases of the Skin. Philadelphia: Saunders, 2006.
Bolognia JL et al. Dermatology. London: Mosby Elsevier, 2008.
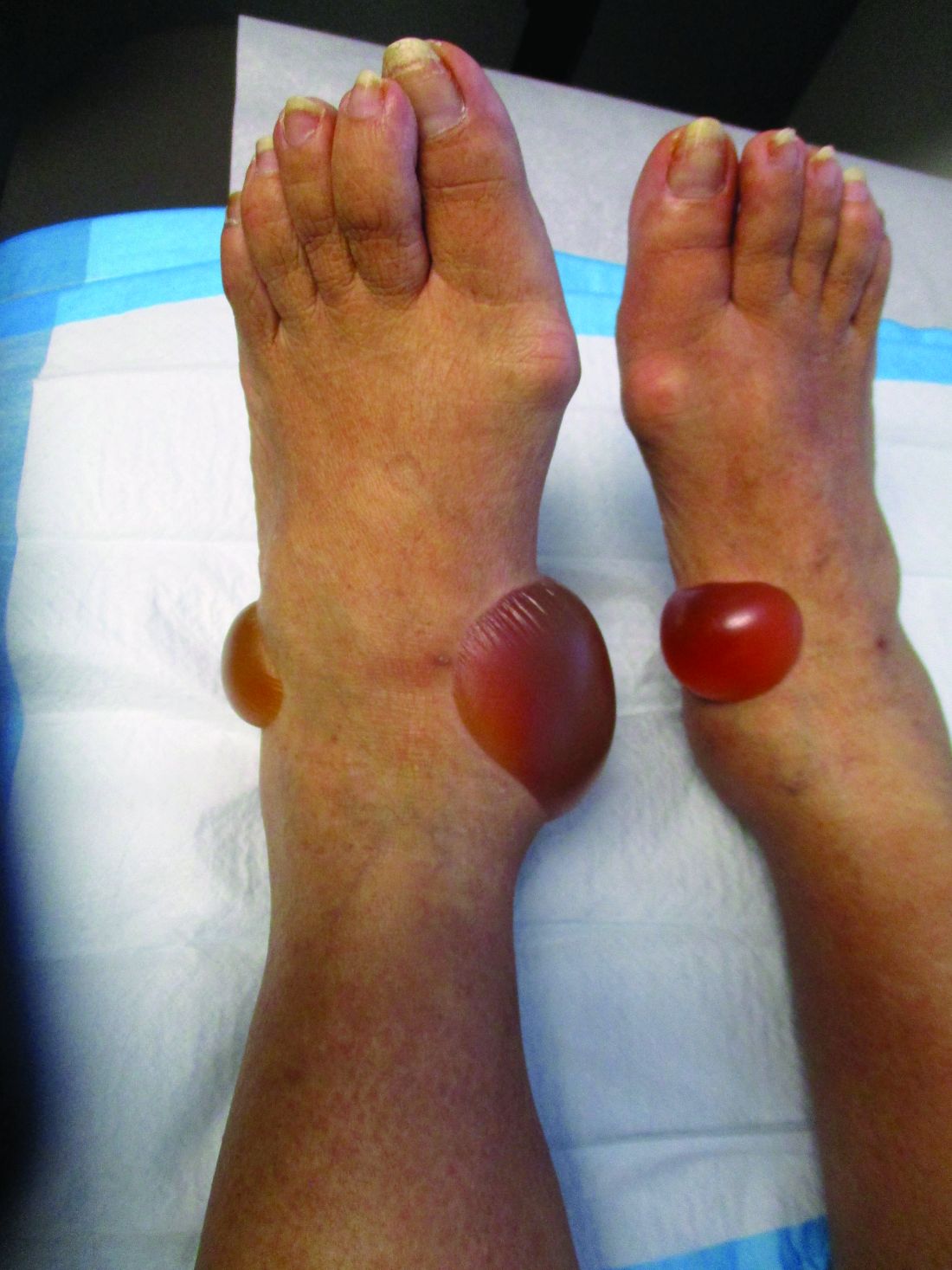
A 67-year-old White woman presented with 2 weeks of bullae on her lower feet
Bullous arthropod assault
Insect-bite reactions are commonly seen in dermatology practice. Most often, they present as pruritic papules. Vesicles and bullae can be seen as well but are less common. Flea bites are the most likely to cause blisters.1 Lesions may be grouped or in a linear pattern. Children tend to have more severe reactions than adults. Body temperature and odor may make some people more susceptible than others to bites. Of note, patients with chronic lymphocytic leukemia tend to have more severe, bullous reactions.2 The differential diagnosis includes bullous pemphigoid, bullous impetigo, bullous tinea, bullous fixed drug, and bullous diabeticorum.

In general, bullous arthropod reactions begin as intraepidermal vesicles that can progress to subepidermal blisters. Eosinophils can be present. Flame figures are often seen in patients with chronic lymphocytic leukemia.3 Histopathology in this patient revealed a subepidermal vesicular dermatitis with minimal inflammation. Periodic acid–Schiff (PAS) stain was negative. Direct immunofluorescence was negative for IgG, C3, IgA, IgM, and fibrinogen. Of note, systemic steroids may alter histologic and immunologic findings.
Bullous pemphigoid is an autoimmune blistering disorder where patients develop widespread tense bullae. Histopathology revealed a subepidermal blister with numerous eosinophils. Direct immunofluorescence study of perilesional skin showed linear IgG and C3 deposits at the basal membrane level. Systemic steroids, tetracyclines, and immunosuppressive medications are a mainstay of treatment. In bullous impetigo, the toxin of Staphylococcus aureus causes blister formation. It is treated with antistaphylococcal antibiotics. Bullous tinea reveals hyphae with PAS staining. Topical or systemic antifungals are used for treatment.
In severe cases, systemic steroids can be used as well. Bacterial culture was negative in this patient. The patient was treated with 1 week of oral prednisone prior to biopsy and topical betamethasone ointment. Her lesions subsequently resolved with no recurrence.
This case and photo were submitted by Brooke Resh Sateesh, MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1-3. “Dermatology” 2nd ed. (Maryland Heights, Mo.: Mosby, 2008).
Bullous arthropod assault
Insect-bite reactions are commonly seen in dermatology practice. Most often, they present as pruritic papules. Vesicles and bullae can be seen as well but are less common. Flea bites are the most likely to cause blisters.1 Lesions may be grouped or in a linear pattern. Children tend to have more severe reactions than adults. Body temperature and odor may make some people more susceptible than others to bites. Of note, patients with chronic lymphocytic leukemia tend to have more severe, bullous reactions.2 The differential diagnosis includes bullous pemphigoid, bullous impetigo, bullous tinea, bullous fixed drug, and bullous diabeticorum.
In general, bullous arthropod reactions begin as intraepidermal vesicles that can progress to subepidermal blisters. Eosinophils can be present. Flame figures are often seen in patients with chronic lymphocytic leukemia.3 Histopathology in this patient revealed a subepidermal vesicular dermatitis with minimal inflammation. Periodic acid–Schiff (PAS) stain was negative. Direct immunofluorescence was negative for IgG, C3, IgA, IgM, and fibrinogen. Of note, systemic steroids may alter histologic and immunologic findings.
Bullous pemphigoid is an autoimmune blistering disorder where patients develop widespread tense bullae. Histopathology revealed a subepidermal blister with numerous eosinophils. Direct immunofluorescence study of perilesional skin showed linear IgG and C3 deposits at the basal membrane level. Systemic steroids, tetracyclines, and immunosuppressive medications are a mainstay of treatment. In bullous impetigo, the toxin of Staphylococcus aureus causes blister formation. It is treated with antistaphylococcal antibiotics. Bullous tinea reveals hyphae with PAS staining. Topical or systemic antifungals are used for treatment.
In severe cases, systemic steroids can be used as well. Bacterial culture was negative in this patient. The patient was treated with 1 week of oral prednisone prior to biopsy and topical betamethasone ointment. Her lesions subsequently resolved with no recurrence.
This case and photo were submitted by Brooke Resh Sateesh, MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1-3. “Dermatology” 2nd ed. (Maryland Heights, Mo.: Mosby, 2008).
Bullous arthropod assault
Insect-bite reactions are commonly seen in dermatology practice. Most often, they present as pruritic papules. Vesicles and bullae can be seen as well but are less common. Flea bites are the most likely to cause blisters.1 Lesions may be grouped or in a linear pattern. Children tend to have more severe reactions than adults. Body temperature and odor may make some people more susceptible than others to bites. Of note, patients with chronic lymphocytic leukemia tend to have more severe, bullous reactions.2 The differential diagnosis includes bullous pemphigoid, bullous impetigo, bullous tinea, bullous fixed drug, and bullous diabeticorum.
In general, bullous arthropod reactions begin as intraepidermal vesicles that can progress to subepidermal blisters. Eosinophils can be present. Flame figures are often seen in patients with chronic lymphocytic leukemia.3 Histopathology in this patient revealed a subepidermal vesicular dermatitis with minimal inflammation. Periodic acid–Schiff (PAS) stain was negative. Direct immunofluorescence was negative for IgG, C3, IgA, IgM, and fibrinogen. Of note, systemic steroids may alter histologic and immunologic findings.
Bullous pemphigoid is an autoimmune blistering disorder where patients develop widespread tense bullae. Histopathology revealed a subepidermal blister with numerous eosinophils. Direct immunofluorescence study of perilesional skin showed linear IgG and C3 deposits at the basal membrane level. Systemic steroids, tetracyclines, and immunosuppressive medications are a mainstay of treatment. In bullous impetigo, the toxin of Staphylococcus aureus causes blister formation. It is treated with antistaphylococcal antibiotics. Bullous tinea reveals hyphae with PAS staining. Topical or systemic antifungals are used for treatment.
In severe cases, systemic steroids can be used as well. Bacterial culture was negative in this patient. The patient was treated with 1 week of oral prednisone prior to biopsy and topical betamethasone ointment. Her lesions subsequently resolved with no recurrence.
This case and photo were submitted by Brooke Resh Sateesh, MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1-3. “Dermatology” 2nd ed. (Maryland Heights, Mo.: Mosby, 2008).
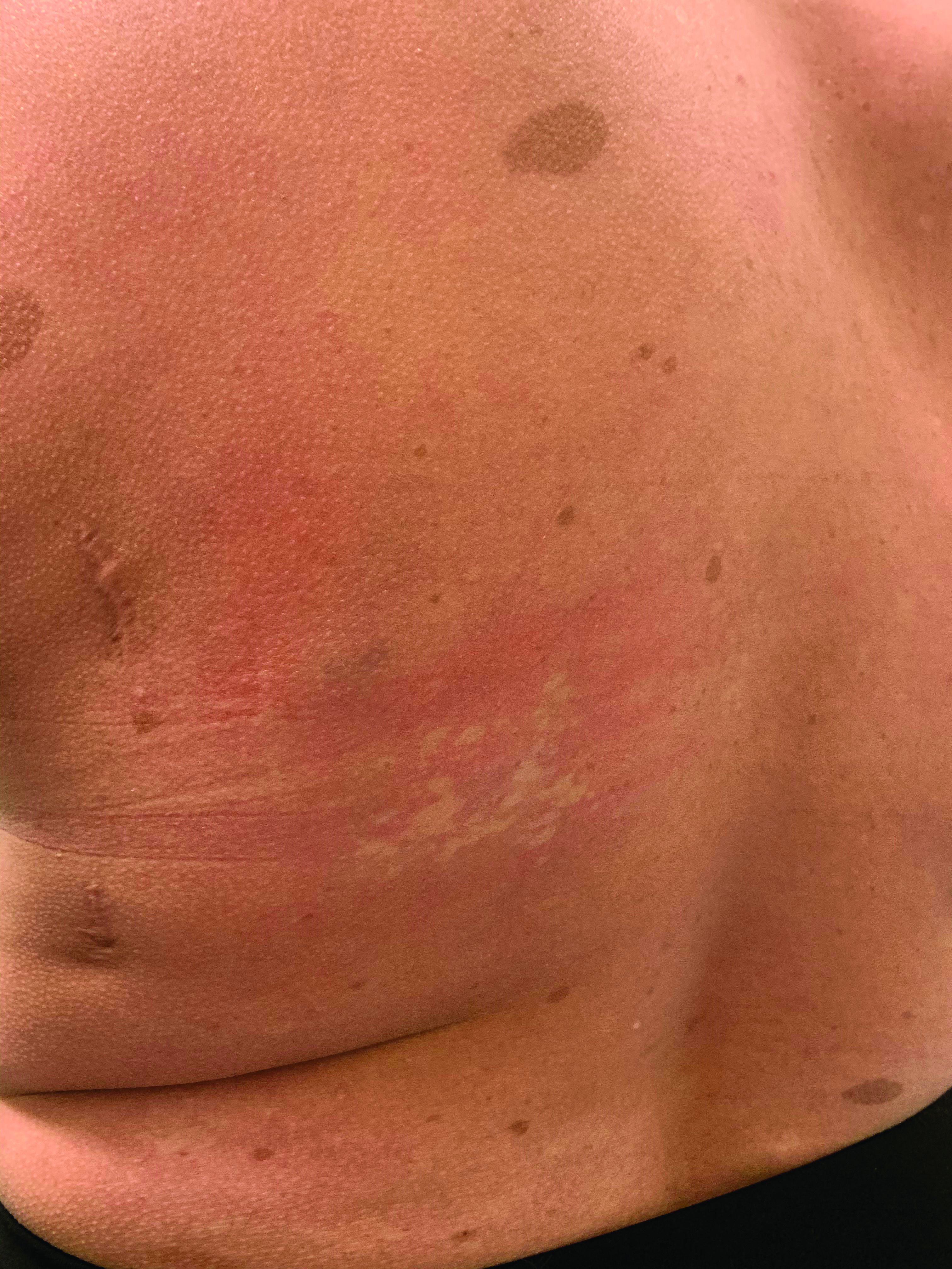
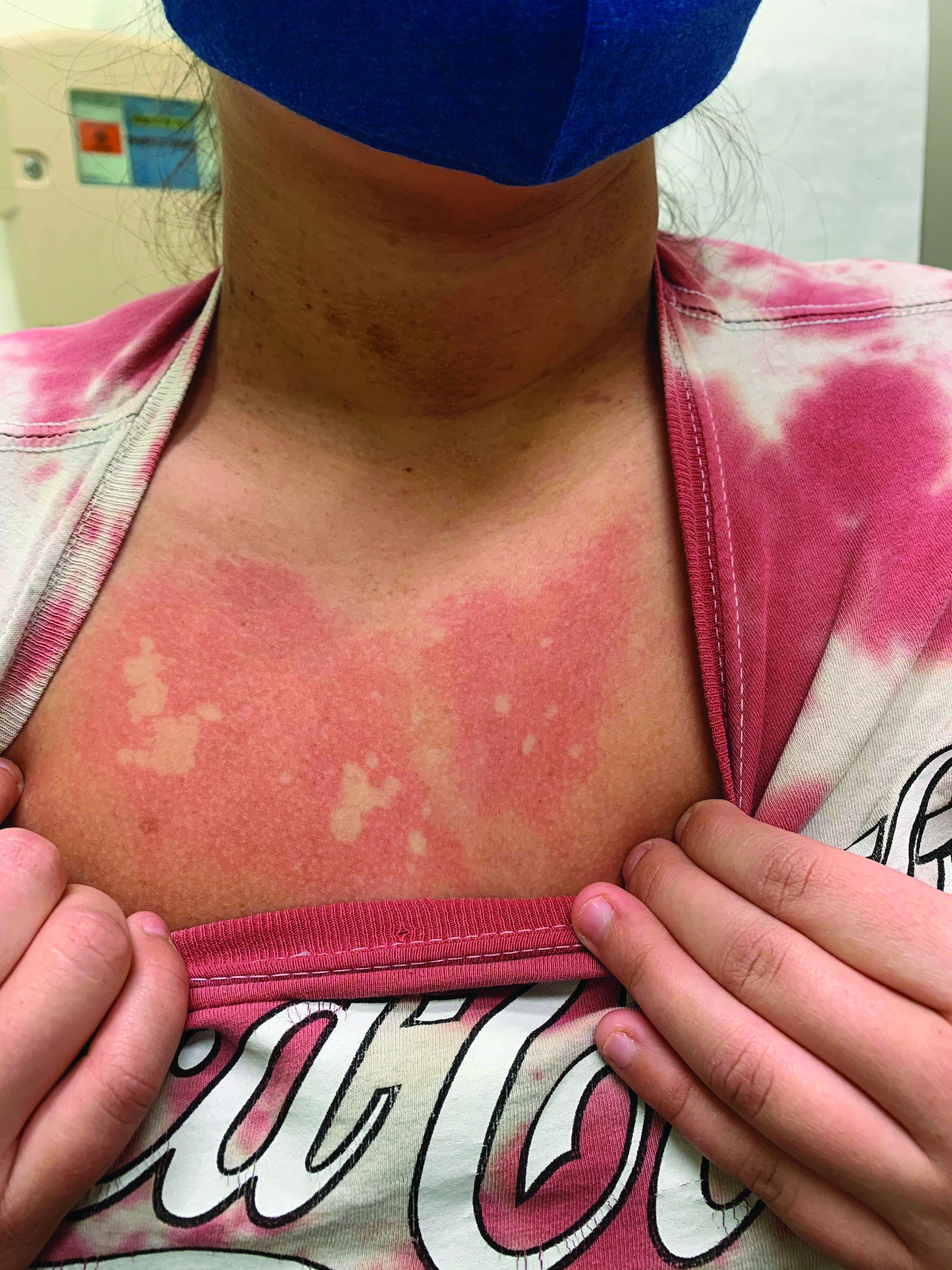
A girl presents with blotchy, slightly itchy spots on her chest, back
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1

Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1
Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1
Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
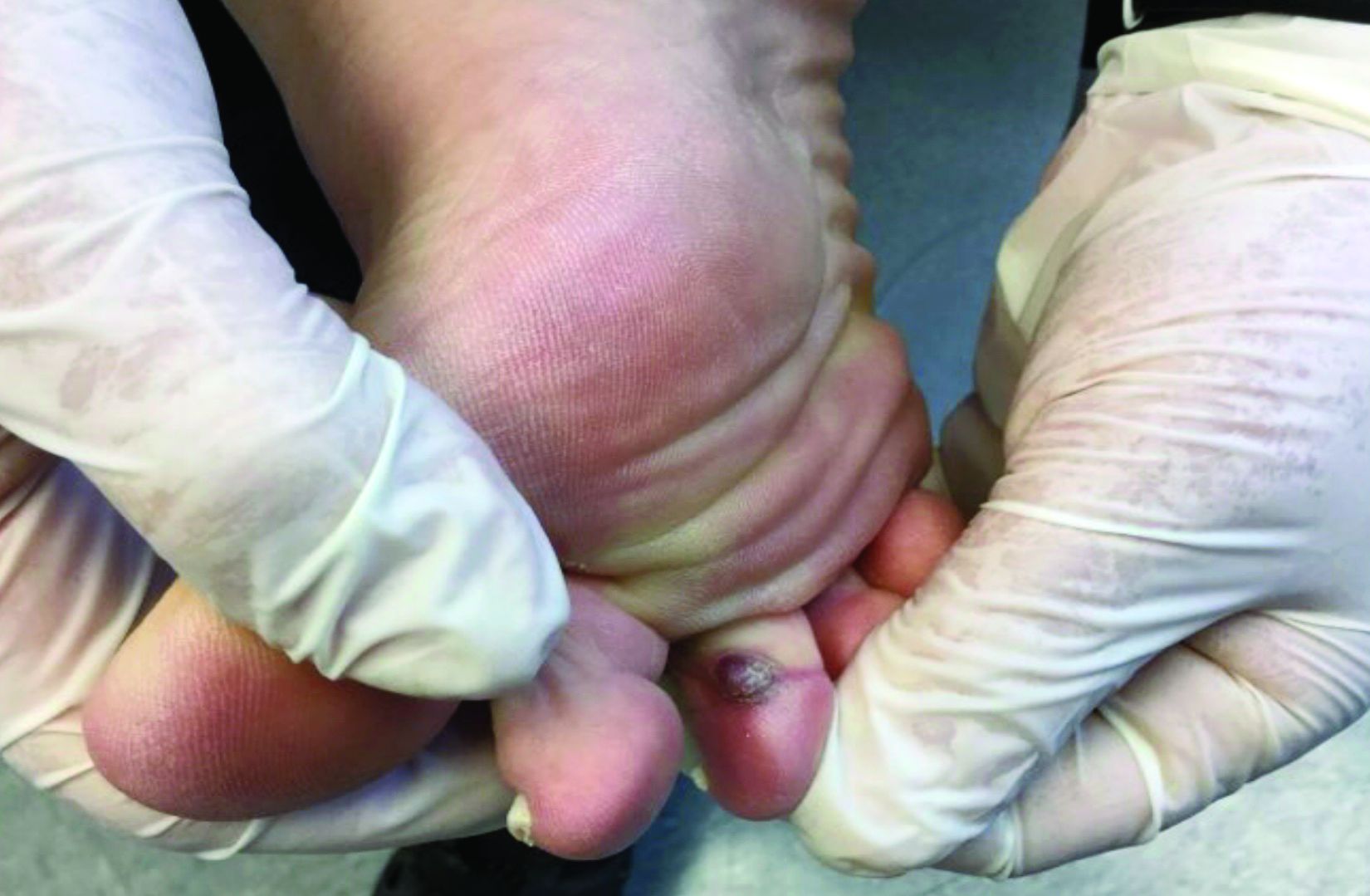
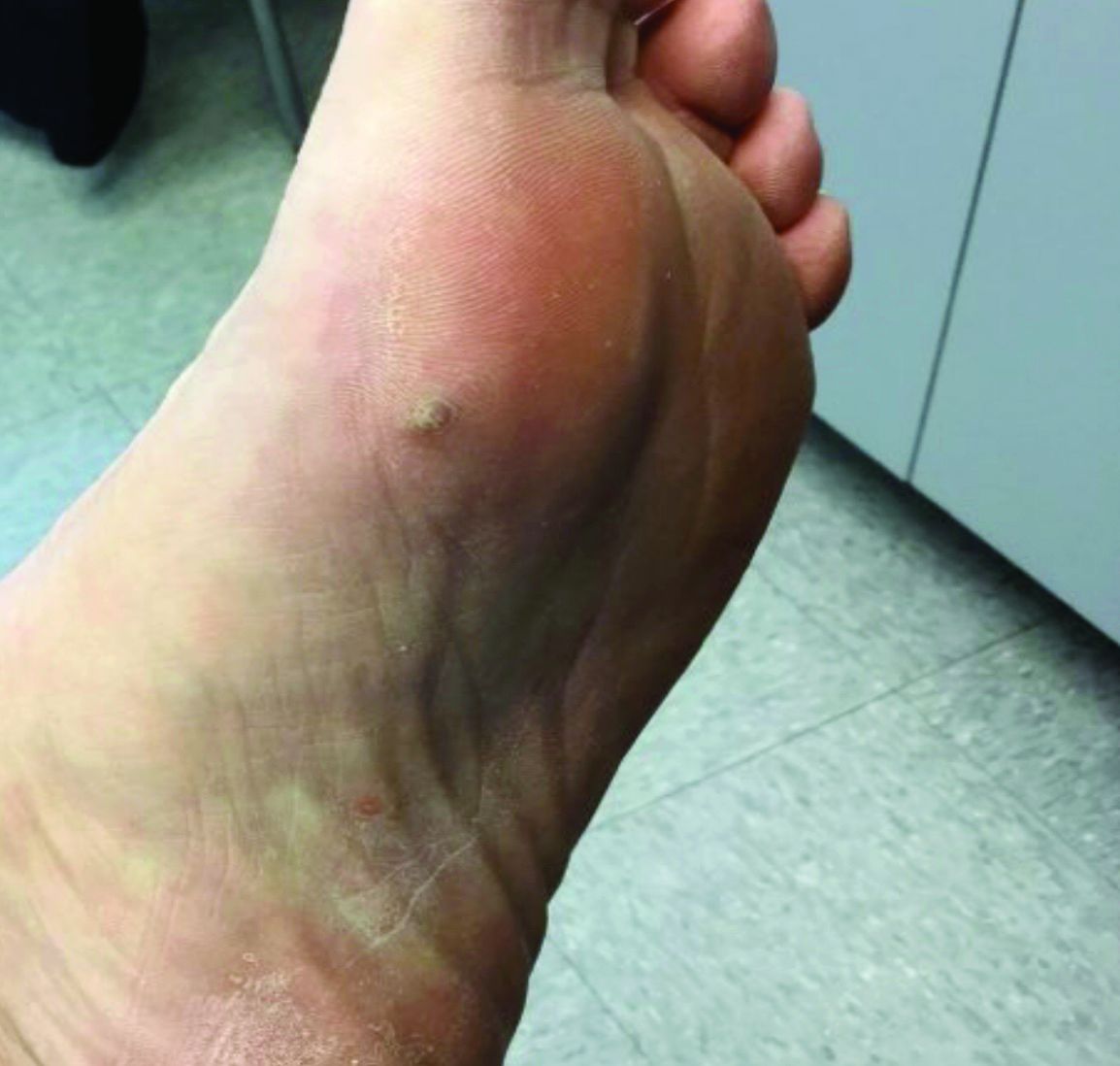
A 70-year-old presented with a 3-week history of asymptomatic violaceous papules on his feet
and named the condition multiple benign pigmented hemorrhagic sarcoma. The disease emerged again at the onset of the AIDS epidemic among homosexual men. There are five variants: HIV/AIDS–related KS, classic KS, African cutaneous KS, African lymphadenopathic KS, and immunosuppression-associated KS (from immunosuppressive therapy or malignancies such as lymphoma).
KS is caused by human herpes virus type 8 (HHV-8). Patients with KS have an increased risk of developing other malignancies such as lymphomas, leukemia, and myeloma. This patient exhibited classic KS.
The various forms of KS may appear different clinically. The lesions may appear as erythematous macules, small violaceous papules, large plaques, or ulcerated nodules. In classic KS, violaceous to bluish-black macules evolve to papules or plaques. Lesions are generally asymptomatic. The most common locations are the toes and soles, although other areas may be affected. Any mucocutaneous surface can be involved. The most common areas of internal involvement are the gastrointestinal system and lymphatics.
Histology reveals angular vessels lined by atypical cells. An associated inflammatory infiltrate containing plasma cells may be present in the upper dermis and perivascular areas. Nodules and plaques reveal a spindle cell neoplasm pattern. Lesions will stain positive for HHV-8.
In patients with HIV/AIDS–related KS, highly active antiretroviral therapy is the most important and beneficial treatment. Since the introduction of HAART, the incidence of KS has greatly decreased. However, there are a proportion of HIV/AIDS–associated Kaposi’s sarcoma patients with well-controlled HIV and undetectable viral loads who require further treatment.
Lesions may spontaneously resolve on their own. Other treatment methods include: cryotherapy, topical alitretinoin (9-cis-retinoic acid), intralesional interferon-alpha or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin or paclitaxel. Small lesions that are asymptomatic may be monitored.
This patient had no internal involvement and responded well to cryotherapy.
This case and photo were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
and named the condition multiple benign pigmented hemorrhagic sarcoma. The disease emerged again at the onset of the AIDS epidemic among homosexual men. There are five variants: HIV/AIDS–related KS, classic KS, African cutaneous KS, African lymphadenopathic KS, and immunosuppression-associated KS (from immunosuppressive therapy or malignancies such as lymphoma).
KS is caused by human herpes virus type 8 (HHV-8). Patients with KS have an increased risk of developing other malignancies such as lymphomas, leukemia, and myeloma. This patient exhibited classic KS.
The various forms of KS may appear different clinically. The lesions may appear as erythematous macules, small violaceous papules, large plaques, or ulcerated nodules. In classic KS, violaceous to bluish-black macules evolve to papules or plaques. Lesions are generally asymptomatic. The most common locations are the toes and soles, although other areas may be affected. Any mucocutaneous surface can be involved. The most common areas of internal involvement are the gastrointestinal system and lymphatics.
Histology reveals angular vessels lined by atypical cells. An associated inflammatory infiltrate containing plasma cells may be present in the upper dermis and perivascular areas. Nodules and plaques reveal a spindle cell neoplasm pattern. Lesions will stain positive for HHV-8.
In patients with HIV/AIDS–related KS, highly active antiretroviral therapy is the most important and beneficial treatment. Since the introduction of HAART, the incidence of KS has greatly decreased. However, there are a proportion of HIV/AIDS–associated Kaposi’s sarcoma patients with well-controlled HIV and undetectable viral loads who require further treatment.
Lesions may spontaneously resolve on their own. Other treatment methods include: cryotherapy, topical alitretinoin (9-cis-retinoic acid), intralesional interferon-alpha or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin or paclitaxel. Small lesions that are asymptomatic may be monitored.
This patient had no internal involvement and responded well to cryotherapy.
This case and photo were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
and named the condition multiple benign pigmented hemorrhagic sarcoma. The disease emerged again at the onset of the AIDS epidemic among homosexual men. There are five variants: HIV/AIDS–related KS, classic KS, African cutaneous KS, African lymphadenopathic KS, and immunosuppression-associated KS (from immunosuppressive therapy or malignancies such as lymphoma).
KS is caused by human herpes virus type 8 (HHV-8). Patients with KS have an increased risk of developing other malignancies such as lymphomas, leukemia, and myeloma. This patient exhibited classic KS.
The various forms of KS may appear different clinically. The lesions may appear as erythematous macules, small violaceous papules, large plaques, or ulcerated nodules. In classic KS, violaceous to bluish-black macules evolve to papules or plaques. Lesions are generally asymptomatic. The most common locations are the toes and soles, although other areas may be affected. Any mucocutaneous surface can be involved. The most common areas of internal involvement are the gastrointestinal system and lymphatics.
Histology reveals angular vessels lined by atypical cells. An associated inflammatory infiltrate containing plasma cells may be present in the upper dermis and perivascular areas. Nodules and plaques reveal a spindle cell neoplasm pattern. Lesions will stain positive for HHV-8.
In patients with HIV/AIDS–related KS, highly active antiretroviral therapy is the most important and beneficial treatment. Since the introduction of HAART, the incidence of KS has greatly decreased. However, there are a proportion of HIV/AIDS–associated Kaposi’s sarcoma patients with well-controlled HIV and undetectable viral loads who require further treatment.
Lesions may spontaneously resolve on their own. Other treatment methods include: cryotherapy, topical alitretinoin (9-cis-retinoic acid), intralesional interferon-alpha or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin or paclitaxel. Small lesions that are asymptomatic may be monitored.
This patient had no internal involvement and responded well to cryotherapy.
This case and photo were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
An 11-year-old female with a 3-year history of alopecia
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.

Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
An 11-year-old female is seen in clinic with a 3-year history of alopecia. The patient recently immigrated to the United States from Afghanistan. Prior to immigrating, she was evaluated for "scarring alopecia" and had been treated with oral and topical steroids as well as oral and topical antifungals. When active, she had itching and tenderness. She is not actively losing any hair at this time, but she has not regrown any of her hair. The patient has no family members with alopecia. She reports some burning pain and itching of her scalp, and denies any muscle pain or weakness or sun sensitivity.
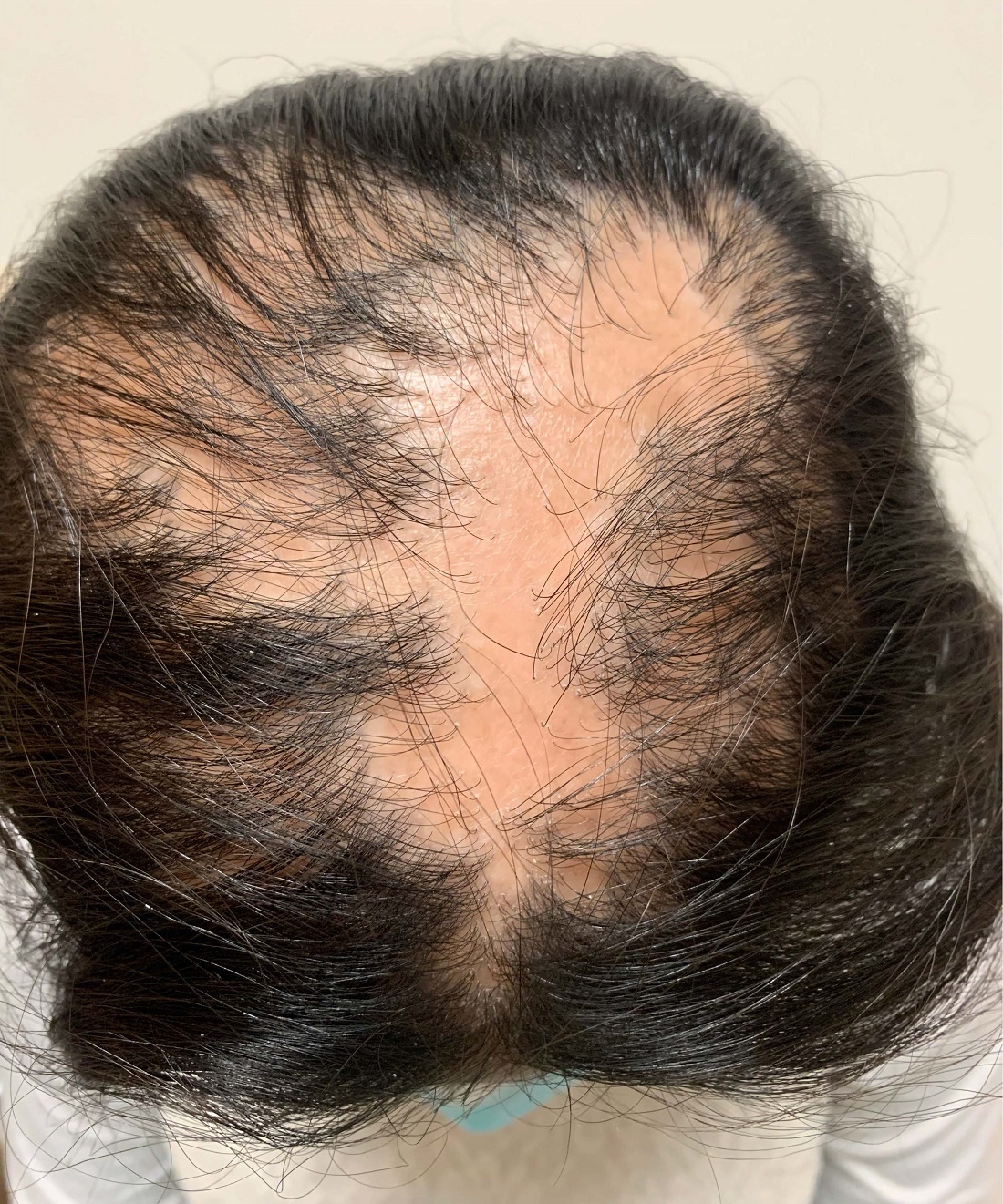
On physical exam, you see 50% loss of hair on the superior scalp with preservation of the anterior hair line. Patches of hair can be seen throughout, with segments of smooth-skinned alopecia, without pustules. There is a loss of the follicle pattern in scarred areas, and magnification or "dermoscopy" shows perifollicular erythema and scaling at the border of the affected scalp. Labs are all within normal limits. Bacterial and fungal cultures of the scalp do not grow organisms.
A 4-year-old presented to our pediatric dermatology clinic for evaluation of asymptomatic "brown spots."
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1

In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.

Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.
Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.
Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
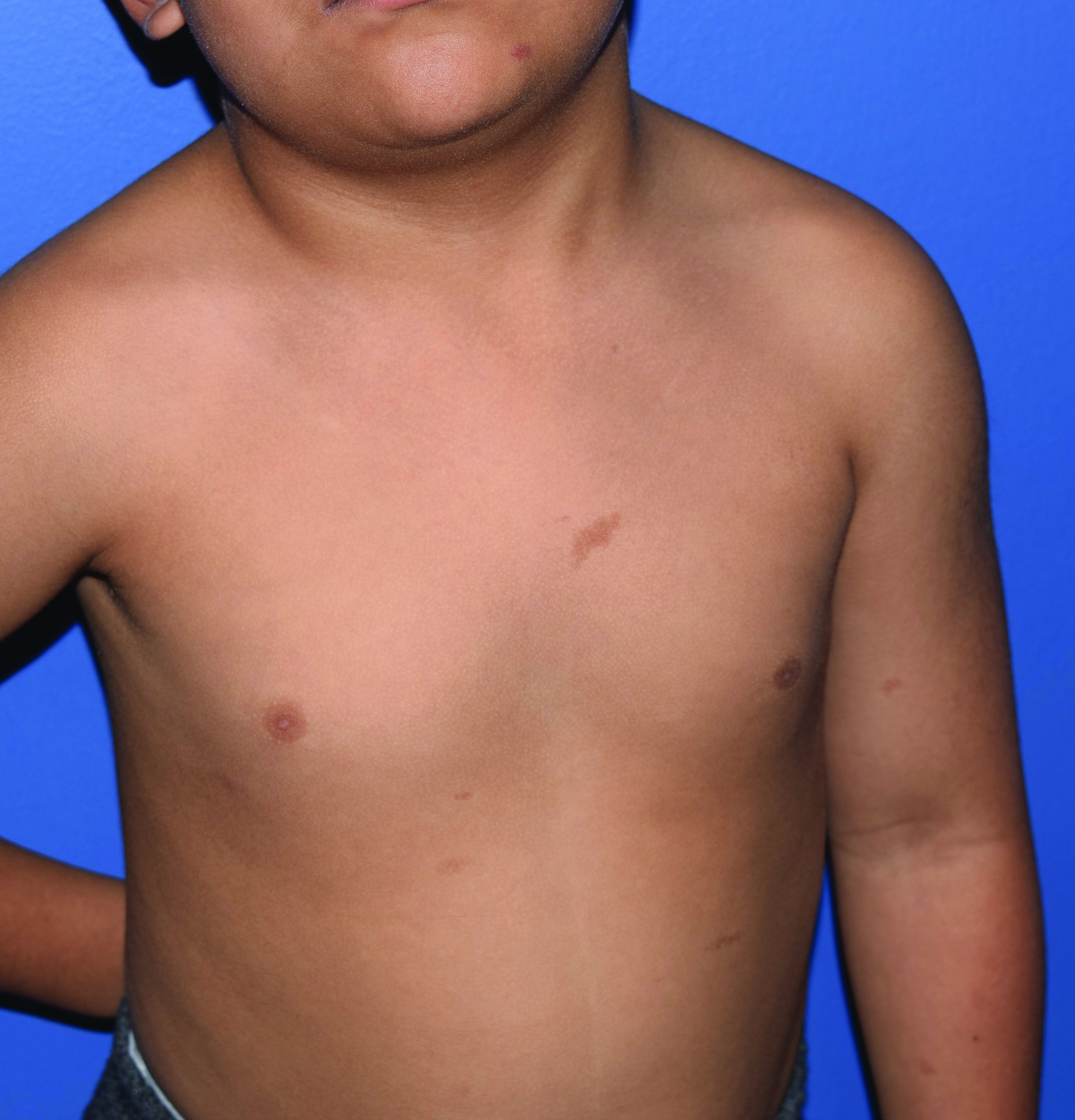
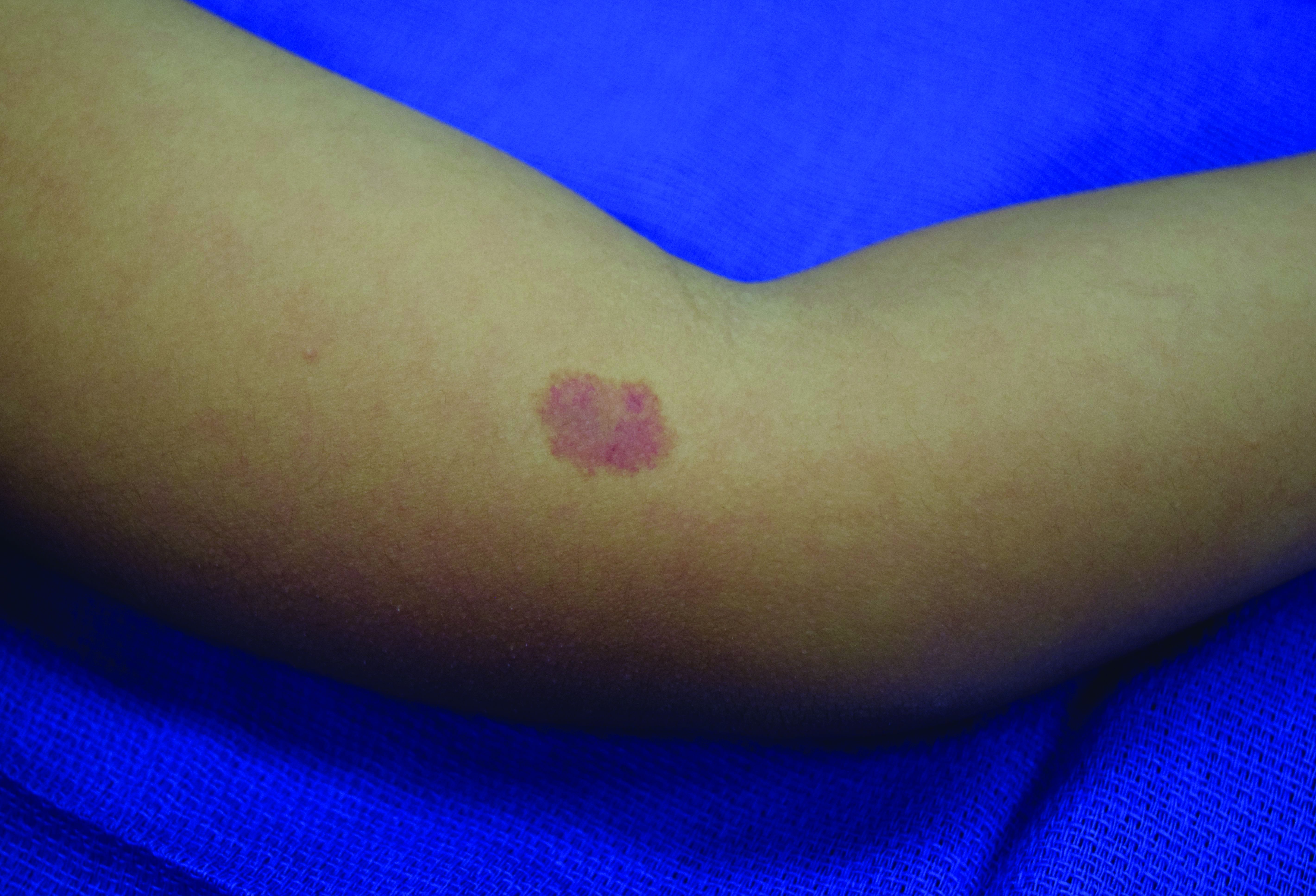
A teen presents with a severe, tender rash on the extremities
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.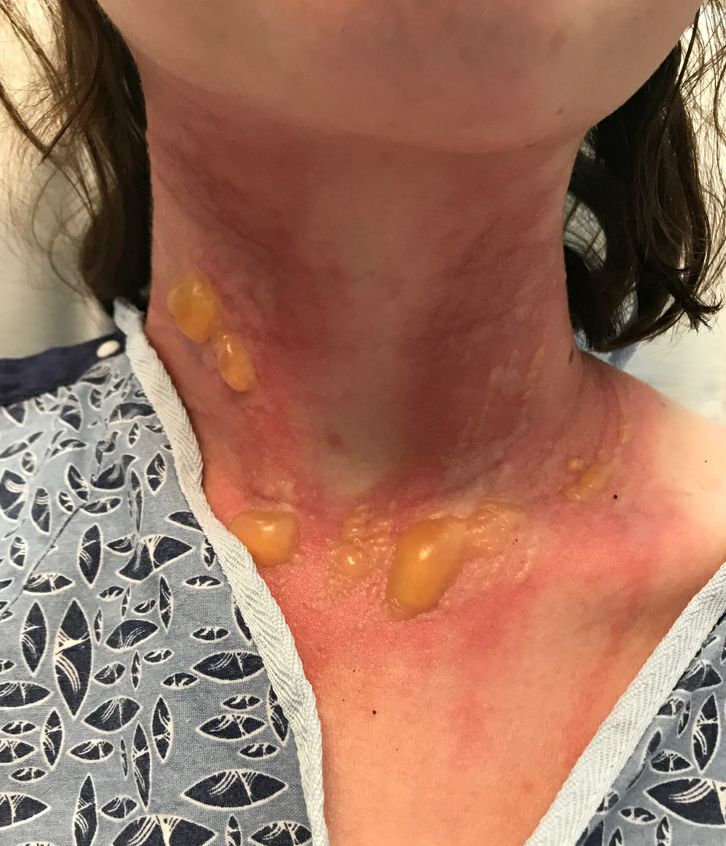
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.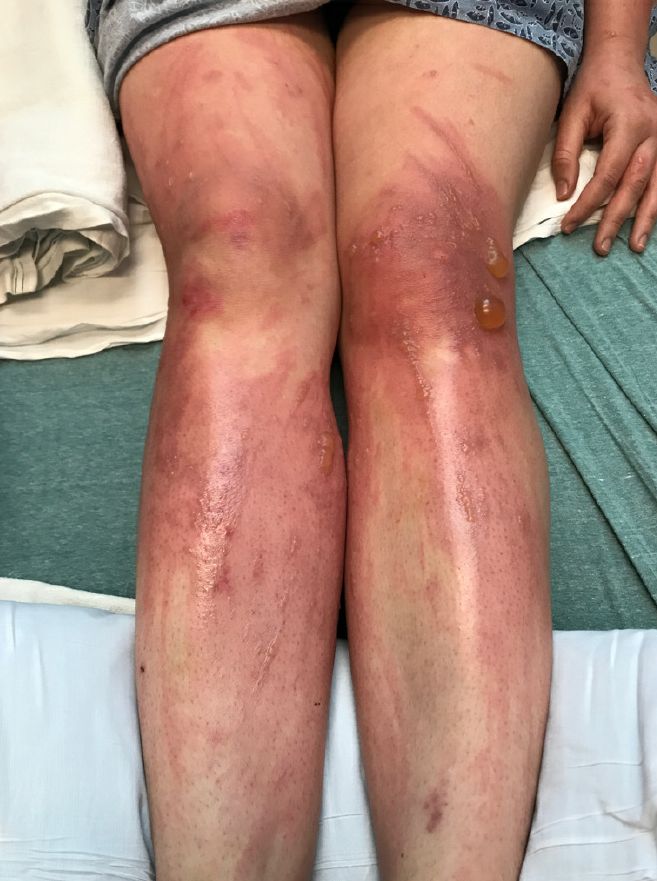
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
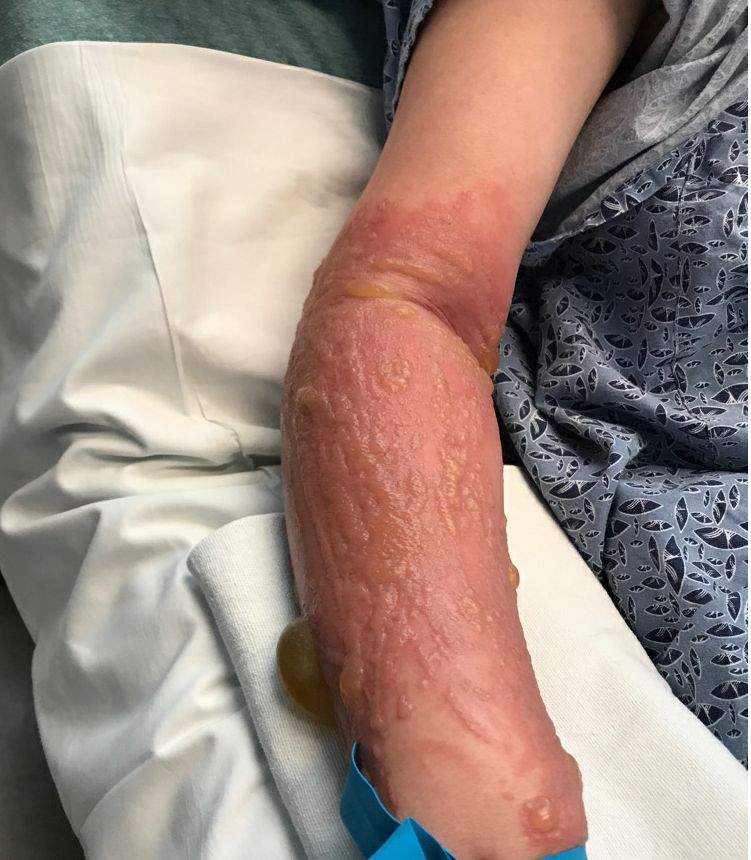
She started taking lithium for depression and anxiety 3 weeks prior to her developing the rash. She denies taking any other medications, supplements, or recreational drugs.
She denied any prior history of photosensitivity, no history of mouth ulcers, joint pain, muscle weakness, hair loss, or any other symptoms.
Besides her brother, there are no other affected family members, and no history of immune bullous disorders or other skin conditions.
On physical exam, the girl appears in a lot of pain and is uncomfortable. The skin is red and hot, and there are tense bullae on the neck, arms, and legs. There are no ocular or mucosal lesions.