User login
Marked BPN elevations found in a third of diastolic HF patients
VANCOUVER, B.C. – B-type natriuretic peptide elevations above 1,000 pg/mL may herald worse prognosis in patients with diastolic heart failure but normal renal function, according to a retrospective database analysis from the University of California, Davis.
Among 421 hospitalized patients with diastolic heart failure – heart failure (HF) with ejection fractions above 45% – investigators found that 117 (28%) patients had B-type natriuretic peptide (BNP) levels above 1,000 pg/mL.
Fifty of those patients (43%) had stage IV or V chronic kidney disease, compared with 7 (13%) of the 54 diastolic HF patients with BNPs below 100 pg/mL. The team also found a significant relationship between estimated glomerular filtration rate (eGFR) and BNP (R = 0.39). In most cases, BNP levels above 1,000 pg/mL "were likely due to reduced [renal] clearance and associated factors. The degree of renal dysfunction is related to the degree of BNP elevation," said senior investigator Dr. Saul Schaefer, professor at the university and also chief of cardiology at the Veterans Affairs Medical Center in Sacramento.

But 25 patients (21%) with BNPs above 1,000 pg/mL "actually had normal renal function; that is, an [eGFR] greater than 60 mL/min per 1.73 m2. It’s unclear why these patients would have such an elevated BNP. They were older [mean age 70.2 years vs. 67.7 years] and had slightly reduced ejection fractions in comparison to the overall group," he said at the 18th World Congress on Heart Disease.
"My guess is that these patients with high BNPs and normal renal function have more severe abnormalities and diastolic failure. It’s a potential marker of higher risk. We could potentially follow these patients more closely and treat them more aggressively," he said.
Before that happens, however, "we need to prospectively look at these patients and see if they can be distinguished from the lower-BNP patients, and track them to see whether their prognosis is adversely affected," Dr. Schaefer said.
Compared with diastolic HF patients in the study with BNPs below 100 pg/mL, the 117 with levels above 1,000 pg/mL tended to be older (68 vs. 61.4 years), have higher troponin levels (0.2 vs. 0 ng/mL), and have greater use of antihypertensive medications, suggesting that they had more chronic hypertension. There were no associations between markedly elevated BNP and gender, diabetes, or ischemic heart disease. Echo parameters did not predict BNP.
Dr. Schaefer said that he had no relevant disclosures.
VANCOUVER, B.C. – B-type natriuretic peptide elevations above 1,000 pg/mL may herald worse prognosis in patients with diastolic heart failure but normal renal function, according to a retrospective database analysis from the University of California, Davis.
Among 421 hospitalized patients with diastolic heart failure – heart failure (HF) with ejection fractions above 45% – investigators found that 117 (28%) patients had B-type natriuretic peptide (BNP) levels above 1,000 pg/mL.
Fifty of those patients (43%) had stage IV or V chronic kidney disease, compared with 7 (13%) of the 54 diastolic HF patients with BNPs below 100 pg/mL. The team also found a significant relationship between estimated glomerular filtration rate (eGFR) and BNP (R = 0.39). In most cases, BNP levels above 1,000 pg/mL "were likely due to reduced [renal] clearance and associated factors. The degree of renal dysfunction is related to the degree of BNP elevation," said senior investigator Dr. Saul Schaefer, professor at the university and also chief of cardiology at the Veterans Affairs Medical Center in Sacramento.
But 25 patients (21%) with BNPs above 1,000 pg/mL "actually had normal renal function; that is, an [eGFR] greater than 60 mL/min per 1.73 m2. It’s unclear why these patients would have such an elevated BNP. They were older [mean age 70.2 years vs. 67.7 years] and had slightly reduced ejection fractions in comparison to the overall group," he said at the 18th World Congress on Heart Disease.
"My guess is that these patients with high BNPs and normal renal function have more severe abnormalities and diastolic failure. It’s a potential marker of higher risk. We could potentially follow these patients more closely and treat them more aggressively," he said.
Before that happens, however, "we need to prospectively look at these patients and see if they can be distinguished from the lower-BNP patients, and track them to see whether their prognosis is adversely affected," Dr. Schaefer said.
Compared with diastolic HF patients in the study with BNPs below 100 pg/mL, the 117 with levels above 1,000 pg/mL tended to be older (68 vs. 61.4 years), have higher troponin levels (0.2 vs. 0 ng/mL), and have greater use of antihypertensive medications, suggesting that they had more chronic hypertension. There were no associations between markedly elevated BNP and gender, diabetes, or ischemic heart disease. Echo parameters did not predict BNP.
Dr. Schaefer said that he had no relevant disclosures.
VANCOUVER, B.C. – B-type natriuretic peptide elevations above 1,000 pg/mL may herald worse prognosis in patients with diastolic heart failure but normal renal function, according to a retrospective database analysis from the University of California, Davis.
Among 421 hospitalized patients with diastolic heart failure – heart failure (HF) with ejection fractions above 45% – investigators found that 117 (28%) patients had B-type natriuretic peptide (BNP) levels above 1,000 pg/mL.
Fifty of those patients (43%) had stage IV or V chronic kidney disease, compared with 7 (13%) of the 54 diastolic HF patients with BNPs below 100 pg/mL. The team also found a significant relationship between estimated glomerular filtration rate (eGFR) and BNP (R = 0.39). In most cases, BNP levels above 1,000 pg/mL "were likely due to reduced [renal] clearance and associated factors. The degree of renal dysfunction is related to the degree of BNP elevation," said senior investigator Dr. Saul Schaefer, professor at the university and also chief of cardiology at the Veterans Affairs Medical Center in Sacramento.
But 25 patients (21%) with BNPs above 1,000 pg/mL "actually had normal renal function; that is, an [eGFR] greater than 60 mL/min per 1.73 m2. It’s unclear why these patients would have such an elevated BNP. They were older [mean age 70.2 years vs. 67.7 years] and had slightly reduced ejection fractions in comparison to the overall group," he said at the 18th World Congress on Heart Disease.
"My guess is that these patients with high BNPs and normal renal function have more severe abnormalities and diastolic failure. It’s a potential marker of higher risk. We could potentially follow these patients more closely and treat them more aggressively," he said.
Before that happens, however, "we need to prospectively look at these patients and see if they can be distinguished from the lower-BNP patients, and track them to see whether their prognosis is adversely affected," Dr. Schaefer said.
Compared with diastolic HF patients in the study with BNPs below 100 pg/mL, the 117 with levels above 1,000 pg/mL tended to be older (68 vs. 61.4 years), have higher troponin levels (0.2 vs. 0 ng/mL), and have greater use of antihypertensive medications, suggesting that they had more chronic hypertension. There were no associations between markedly elevated BNP and gender, diabetes, or ischemic heart disease. Echo parameters did not predict BNP.
Dr. Schaefer said that he had no relevant disclosures.
AT THE 18TH WORLD CONGRESS ON HEART DISEASE
Major finding: Among 421 hospitalized patients with diastolic heart failure, 117 (28%) had BNP levels above 1,000 pg/mL.
Data Source: Record review.
Disclosures: Dr. Schaefer said that he had no relevant disclosures.

Advisers support FDA approval of wireless HF monitoring device
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
AT AN FDA ADVISORY PANEL MEETING
FDA panel narrowly backs expanding CRT devices’ indications
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
AT AN FDA ADVISORY COMMITTEE MEETING
Novel oral vasodilator approved to treat pulmonary hypertension
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
Full-dose beta-blockers still show heart failure benefit
AMSTERDAM – Patients with heart failure appeared to continue to benefit from full-dose beta-blocker therapy against a background of optimal, contemporary treatment, based on an analysis of data collected from an 1,800-patient trial of cardiac resynchronization therapy.
Dr. L. Brent Mitchell, who presented this finding at the annual congress of the European Society of Cardiology, acknowledged that assessing the efficacy of drugs in a study that did not randomize patients based on their use of those drugs had questionable validity and was vulnerable to unidentified confounding, but he also noted that data on the role of beta-blockers in contemporary heart failure management were unlikely to come from any other source.
"You need some tool to ask whether beta-blockers are still useful," said Dr. Mitchell, professor of cardiac sciences at the University of Calgary (Alta.). "The purpose of this analysis was to ask if beta-blockers at full dosages are still better than not using a full-dose beta-blocker despite all the other treatments" that patients received while in the study during 2003-2009. The analysis showed that those receiving full-dosage or nearly full-dosage beta-blocker regimens had a one-third cut in all-cause death or heart failure hospitalization, compared with patients receiving less than half the recommended dosage, after adjustment for many baseline differences between these two subgroups.
The analysis also showed that just half of the nearly 1,500 patients who received treatment with a beta-blocker in this cohort received an adequate dosage, defined as at least 50% of the level recommended by the European Society of Cardiology in heart failure treatment guidelines issued last year (Euro. Heart J. 2012;33:1787-847). Two-thirds of the patients on carvedilol received at least half of the recommended target dosage of 50 mg/day. In contrast, about a third of patients being treated with either bisoprolol or metoprolol received a dosage that was at least half the target dosage (10 mg/day for bisoprolol, 200 mg/day for metoprolol).
The analysis used data collected in the Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (RAFT), which enrolled 1,798 patients primarily with New York Heart Association class II heart failure at 34 centers worldwide. When the primary endpoint of RAFT was evaluated, treatment with cardiac resynchronization therapy (CRT) with defibrillator capability was more effective at preventing death or heart failure hospitalization in the study population than was an implanted defibrillator alone against a background of optimal medical treatment (N. Engl. J. Med. 2010;363:2385-95).
Dr. Mitchell and his associates focused on 1,474 patients who all received beta-blocker treatment while in RAFT. Like the entire RAFT population, 97% of these patients also received treatment with an ACE inhibitor or angiotensin receptor blocker, and 42% received treatment with spironolactone, and their average left ventricular ejection fraction was 23%.
The patients in this group who were undertreated with beta-blockers were significantly older and had a significantly greater prevalence of New York Heart Association class III heart failure and heart failure with an ischemic etiology compared with those who received at least 50% of the target dosage.
In a regression model that adjusted for all measured baseline differences, patients treated with at least half of the recommended dosage had an incidence of mortality or heart failure hospitalization during an average 40 months of follow-up that was 33% below the rate among patients on lower beta-blocker dosages, a statistically significant difference. This 33% relative risk reduction with adequate beta-blocker treatment is "indistinguishable" from the risk reductions seen with placebo in the major beta-blocker efficacy trials in heart failure patients during the 1990s, Dr. Mitchell noted. "This suggests that beta-blocker treatment still has potency," he said.
Other significant, independent predictors of the primary outcome in this model were prior coronary artery bypass, which boosted the risk by 63%; a history of ischemic heart disease, which raised the risk by 39%; having peripheral vascular disease, which increased the risk by 36%; and receiving CRT, which cut the risk by 33%. In addition, adverse outcomes rose 10% for each reduction of 5 mL/min per 1.73 m2 in estimated glomerular filtration rate.
The impact of underdosing was roughly similar across all three beta-blockers, bisoprolol, carvedilol, and metoprolol. The adverse effect of underdosing was mitigated to some extent when patients also received CRT.
RAFT was partially funded by Medtronic of Canada. Dr. Mitchell said he had no relevant financial disclosures.
On Twitter @mitchelzoler
A post hoc analysis of the nonrandomized interventions that patients receive during randomized clinical trials is generally discouraged. Patients treated with lower dosages of hemodynamically active drugs like beta-blockers are often sicker and have a worse prognosis. This problem can be seen in the baseline characteristics of the two subgroups in this analysis, which revealed many significant differences between the two. Performing statistical compensations to counterbalance these differences is very uncertain.
In addition, a few years ago, my associates and I ran the Systolic Heart Failure Treatment With the If inhibitor Ivabradine Trial (SHIFT) (Lancet 2010;376:875-85) to assess the efficacy and safety of ivabradine to lower heart rate and improve outcomes in optimally treated heart failure patients. In SHIFT we saw a relationship similar to the one seen in RAFT by the current investigators: Patients on lower dosages of beta-blockers were sicker and at higher risk than patients on higher beta-blocker dosages. In our analyses, adjusting for differences in heart rate eliminated the link between beta-blocker dosage and outcomes. We found that the entire treatment effect could be explained by differences in patient heart rate.
Dr. Karl Swedberg is a professor of medicine at the University of Gothenburg (Sweden). He was lead investigator for the SHIFT trial, which was funded by Servier, the company that markets ivabradine (Procoralan). He made these comments as designated discussant for Dr. Mitchell’s report.
A post hoc analysis of the nonrandomized interventions that patients receive during randomized clinical trials is generally discouraged. Patients treated with lower dosages of hemodynamically active drugs like beta-blockers are often sicker and have a worse prognosis. This problem can be seen in the baseline characteristics of the two subgroups in this analysis, which revealed many significant differences between the two. Performing statistical compensations to counterbalance these differences is very uncertain.
In addition, a few years ago, my associates and I ran the Systolic Heart Failure Treatment With the If inhibitor Ivabradine Trial (SHIFT) (Lancet 2010;376:875-85) to assess the efficacy and safety of ivabradine to lower heart rate and improve outcomes in optimally treated heart failure patients. In SHIFT we saw a relationship similar to the one seen in RAFT by the current investigators: Patients on lower dosages of beta-blockers were sicker and at higher risk than patients on higher beta-blocker dosages. In our analyses, adjusting for differences in heart rate eliminated the link between beta-blocker dosage and outcomes. We found that the entire treatment effect could be explained by differences in patient heart rate.
Dr. Karl Swedberg is a professor of medicine at the University of Gothenburg (Sweden). He was lead investigator for the SHIFT trial, which was funded by Servier, the company that markets ivabradine (Procoralan). He made these comments as designated discussant for Dr. Mitchell’s report.
A post hoc analysis of the nonrandomized interventions that patients receive during randomized clinical trials is generally discouraged. Patients treated with lower dosages of hemodynamically active drugs like beta-blockers are often sicker and have a worse prognosis. This problem can be seen in the baseline characteristics of the two subgroups in this analysis, which revealed many significant differences between the two. Performing statistical compensations to counterbalance these differences is very uncertain.
In addition, a few years ago, my associates and I ran the Systolic Heart Failure Treatment With the If inhibitor Ivabradine Trial (SHIFT) (Lancet 2010;376:875-85) to assess the efficacy and safety of ivabradine to lower heart rate and improve outcomes in optimally treated heart failure patients. In SHIFT we saw a relationship similar to the one seen in RAFT by the current investigators: Patients on lower dosages of beta-blockers were sicker and at higher risk than patients on higher beta-blocker dosages. In our analyses, adjusting for differences in heart rate eliminated the link between beta-blocker dosage and outcomes. We found that the entire treatment effect could be explained by differences in patient heart rate.
Dr. Karl Swedberg is a professor of medicine at the University of Gothenburg (Sweden). He was lead investigator for the SHIFT trial, which was funded by Servier, the company that markets ivabradine (Procoralan). He made these comments as designated discussant for Dr. Mitchell’s report.
AMSTERDAM – Patients with heart failure appeared to continue to benefit from full-dose beta-blocker therapy against a background of optimal, contemporary treatment, based on an analysis of data collected from an 1,800-patient trial of cardiac resynchronization therapy.
Dr. L. Brent Mitchell, who presented this finding at the annual congress of the European Society of Cardiology, acknowledged that assessing the efficacy of drugs in a study that did not randomize patients based on their use of those drugs had questionable validity and was vulnerable to unidentified confounding, but he also noted that data on the role of beta-blockers in contemporary heart failure management were unlikely to come from any other source.
"You need some tool to ask whether beta-blockers are still useful," said Dr. Mitchell, professor of cardiac sciences at the University of Calgary (Alta.). "The purpose of this analysis was to ask if beta-blockers at full dosages are still better than not using a full-dose beta-blocker despite all the other treatments" that patients received while in the study during 2003-2009. The analysis showed that those receiving full-dosage or nearly full-dosage beta-blocker regimens had a one-third cut in all-cause death or heart failure hospitalization, compared with patients receiving less than half the recommended dosage, after adjustment for many baseline differences between these two subgroups.
The analysis also showed that just half of the nearly 1,500 patients who received treatment with a beta-blocker in this cohort received an adequate dosage, defined as at least 50% of the level recommended by the European Society of Cardiology in heart failure treatment guidelines issued last year (Euro. Heart J. 2012;33:1787-847). Two-thirds of the patients on carvedilol received at least half of the recommended target dosage of 50 mg/day. In contrast, about a third of patients being treated with either bisoprolol or metoprolol received a dosage that was at least half the target dosage (10 mg/day for bisoprolol, 200 mg/day for metoprolol).
The analysis used data collected in the Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (RAFT), which enrolled 1,798 patients primarily with New York Heart Association class II heart failure at 34 centers worldwide. When the primary endpoint of RAFT was evaluated, treatment with cardiac resynchronization therapy (CRT) with defibrillator capability was more effective at preventing death or heart failure hospitalization in the study population than was an implanted defibrillator alone against a background of optimal medical treatment (N. Engl. J. Med. 2010;363:2385-95).
Dr. Mitchell and his associates focused on 1,474 patients who all received beta-blocker treatment while in RAFT. Like the entire RAFT population, 97% of these patients also received treatment with an ACE inhibitor or angiotensin receptor blocker, and 42% received treatment with spironolactone, and their average left ventricular ejection fraction was 23%.
The patients in this group who were undertreated with beta-blockers were significantly older and had a significantly greater prevalence of New York Heart Association class III heart failure and heart failure with an ischemic etiology compared with those who received at least 50% of the target dosage.
In a regression model that adjusted for all measured baseline differences, patients treated with at least half of the recommended dosage had an incidence of mortality or heart failure hospitalization during an average 40 months of follow-up that was 33% below the rate among patients on lower beta-blocker dosages, a statistically significant difference. This 33% relative risk reduction with adequate beta-blocker treatment is "indistinguishable" from the risk reductions seen with placebo in the major beta-blocker efficacy trials in heart failure patients during the 1990s, Dr. Mitchell noted. "This suggests that beta-blocker treatment still has potency," he said.
Other significant, independent predictors of the primary outcome in this model were prior coronary artery bypass, which boosted the risk by 63%; a history of ischemic heart disease, which raised the risk by 39%; having peripheral vascular disease, which increased the risk by 36%; and receiving CRT, which cut the risk by 33%. In addition, adverse outcomes rose 10% for each reduction of 5 mL/min per 1.73 m2 in estimated glomerular filtration rate.
The impact of underdosing was roughly similar across all three beta-blockers, bisoprolol, carvedilol, and metoprolol. The adverse effect of underdosing was mitigated to some extent when patients also received CRT.
RAFT was partially funded by Medtronic of Canada. Dr. Mitchell said he had no relevant financial disclosures.
On Twitter @mitchelzoler
AMSTERDAM – Patients with heart failure appeared to continue to benefit from full-dose beta-blocker therapy against a background of optimal, contemporary treatment, based on an analysis of data collected from an 1,800-patient trial of cardiac resynchronization therapy.
Dr. L. Brent Mitchell, who presented this finding at the annual congress of the European Society of Cardiology, acknowledged that assessing the efficacy of drugs in a study that did not randomize patients based on their use of those drugs had questionable validity and was vulnerable to unidentified confounding, but he also noted that data on the role of beta-blockers in contemporary heart failure management were unlikely to come from any other source.
"You need some tool to ask whether beta-blockers are still useful," said Dr. Mitchell, professor of cardiac sciences at the University of Calgary (Alta.). "The purpose of this analysis was to ask if beta-blockers at full dosages are still better than not using a full-dose beta-blocker despite all the other treatments" that patients received while in the study during 2003-2009. The analysis showed that those receiving full-dosage or nearly full-dosage beta-blocker regimens had a one-third cut in all-cause death or heart failure hospitalization, compared with patients receiving less than half the recommended dosage, after adjustment for many baseline differences between these two subgroups.
The analysis also showed that just half of the nearly 1,500 patients who received treatment with a beta-blocker in this cohort received an adequate dosage, defined as at least 50% of the level recommended by the European Society of Cardiology in heart failure treatment guidelines issued last year (Euro. Heart J. 2012;33:1787-847). Two-thirds of the patients on carvedilol received at least half of the recommended target dosage of 50 mg/day. In contrast, about a third of patients being treated with either bisoprolol or metoprolol received a dosage that was at least half the target dosage (10 mg/day for bisoprolol, 200 mg/day for metoprolol).
The analysis used data collected in the Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (RAFT), which enrolled 1,798 patients primarily with New York Heart Association class II heart failure at 34 centers worldwide. When the primary endpoint of RAFT was evaluated, treatment with cardiac resynchronization therapy (CRT) with defibrillator capability was more effective at preventing death or heart failure hospitalization in the study population than was an implanted defibrillator alone against a background of optimal medical treatment (N. Engl. J. Med. 2010;363:2385-95).
Dr. Mitchell and his associates focused on 1,474 patients who all received beta-blocker treatment while in RAFT. Like the entire RAFT population, 97% of these patients also received treatment with an ACE inhibitor or angiotensin receptor blocker, and 42% received treatment with spironolactone, and their average left ventricular ejection fraction was 23%.
The patients in this group who were undertreated with beta-blockers were significantly older and had a significantly greater prevalence of New York Heart Association class III heart failure and heart failure with an ischemic etiology compared with those who received at least 50% of the target dosage.
In a regression model that adjusted for all measured baseline differences, patients treated with at least half of the recommended dosage had an incidence of mortality or heart failure hospitalization during an average 40 months of follow-up that was 33% below the rate among patients on lower beta-blocker dosages, a statistically significant difference. This 33% relative risk reduction with adequate beta-blocker treatment is "indistinguishable" from the risk reductions seen with placebo in the major beta-blocker efficacy trials in heart failure patients during the 1990s, Dr. Mitchell noted. "This suggests that beta-blocker treatment still has potency," he said.
Other significant, independent predictors of the primary outcome in this model were prior coronary artery bypass, which boosted the risk by 63%; a history of ischemic heart disease, which raised the risk by 39%; having peripheral vascular disease, which increased the risk by 36%; and receiving CRT, which cut the risk by 33%. In addition, adverse outcomes rose 10% for each reduction of 5 mL/min per 1.73 m2 in estimated glomerular filtration rate.
The impact of underdosing was roughly similar across all three beta-blockers, bisoprolol, carvedilol, and metoprolol. The adverse effect of underdosing was mitigated to some extent when patients also received CRT.
RAFT was partially funded by Medtronic of Canada. Dr. Mitchell said he had no relevant financial disclosures.
On Twitter @mitchelzoler
AT THE ESC CONGRESS 2013
Major finding: Heart failure patients on recommended beta-blocker dosages had 33% fewer deaths or hospitalizations than did patients on lower dosages.
Data source: A post hoc subgroup analysis of results from RAFT, with 1,798 patients with heart failure at 34 worldwide sites.
Disclosures: RAFT was partially funded by Medtronic of Canada. Dr. Mitchell said he had no relevant financial disclosures.
HFSA13: Health reform, mechanical support devices take center stage
At the 2013 Heart Failure Society of America, held in Orlando, hotly debated topics will include multidisciplinary approaches to treating heart failure patients, health reform, better selection of mechanical support devices for heart failure patients, and heart failure readmissions.
Dr. David DeNofrio, the scientific program's co-chair, and director of heart failure and cardiac transplantation at Tufts Medical Center, Boston, discusses the issues in an interview with IMNG Medical Media.
At the 2013 Heart Failure Society of America, held in Orlando, hotly debated topics will include multidisciplinary approaches to treating heart failure patients, health reform, better selection of mechanical support devices for heart failure patients, and heart failure readmissions.
Dr. David DeNofrio, the scientific program's co-chair, and director of heart failure and cardiac transplantation at Tufts Medical Center, Boston, discusses the issues in an interview with IMNG Medical Media.
At the 2013 Heart Failure Society of America, held in Orlando, hotly debated topics will include multidisciplinary approaches to treating heart failure patients, health reform, better selection of mechanical support devices for heart failure patients, and heart failure readmissions.
Dr. David DeNofrio, the scientific program's co-chair, and director of heart failure and cardiac transplantation at Tufts Medical Center, Boston, discusses the issues in an interview with IMNG Medical Media.

Losartan shown effective in Marfan syndrome
AMSTERDAM – Daily losartan significantly slowed the aortic root dilatation rate in adults with Marfan syndrome in a 3-year randomized clinical trial.
"I think we can be positive about this treatment. We can now recommend losartan in clinical practice," Dr. Maarten Groenink said at the annual congress of the European Society of Cardiology.
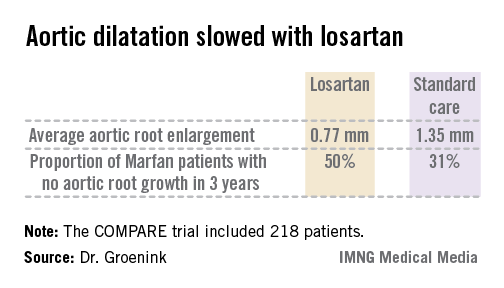
The COMPARE (Cozaar in Marfan Patients Reduces Aortic Enlargement) trial included 218 patients at all four university Marfan centers in the Netherlands. Patients were randomized to oral losartan at a target dose of 100 mg/day or no losartan in addition to standard-of-care treatment with beta-blockers. Roughly half of the patients in the losartan group were unable to tolerate the full dose of losartan in addition to a beta-blocker; those patients were maintained on losartan at 50 mg/day. Aortic root diameter was measured by MRI at enrollment and after 3 years of prospective follow-up. The aortic dilatation rate was significantly lower in the losartan group than in controls both in the patients with a native root and in those who had undergone aortic root replacement surgery, reported Dr. Groenink, a cardiologist at the Academic Medical Center, Amsterdam.

There were no aortic dissections in the losartan group and two in the control arm. Elective aortic replacement surgery was performed in a similar number of patients in both groups.
Blood pressure was lower in the losartan group, yet blood pressure didn’t correlate with the aortic dilatation rate. Dr. Groenink speculated that losartan’s chief mechanism of benefit in Marfan syndrome is its ability to curb overexpression of transforming growth factor-beta, which weakens the structure of the media layer of the aortic wall.
Dr. Groenink said it’s unknown whether losartan’s benefits are specific to that drug or are a class effect obtainable with other angiotensin II receptor antagonists, though he suspects it’s a class effect.
Ongoing clinical trials are evaluating losartan in children and adolescents with Marfan syndrome, he said, adding that there is a solid rationale for beginning treatment as early in life as possible.
"I believe the adverse effects on the aortic wall in Marfan syndrome are caused by the fibrillin defect but also by wear and tear due to cyclic stress by the beating heart. So you can hypothesize that the earlier you start treatment, the better the results," he explained.
Marfan syndrome is a genetic connective tissue disorder affecting multiple organ systems. The prognosis is mainly determined by the aortic complications, including dilatation, aneurysm formation, and possible acute dissection. Affected individuals tend to be tall, long-limbed, and have distinctively long, thin fingers. The prevalence of Marfan syndrome has been estimated at 1 in 5,000, but Dr. Groenink suspects the syndrome may actually be more common than that.
Simultaneous with Dr. Groenink’s presentation at the ESC, the COMPARE results were published online (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht334]).
The COMPARE trial was funded by the Dutch Heart Association. Dr. Groenink reported having no relevant financial interests.
*CORRECTION 11/14/13: The first version of this story had Dr. Groenink's name misspelled.
 |
Bruce Jancin/IMNG Medical Media
|
COMPARE is a very important study whose results are going to mean a paradigm shift for the management of Marfan syndrome.
It is intriguing to consider that the benefits of losartan might possibly also extend to patients with thoracic aortic disease in general, a worthy topic for future investigation.
Dr. John Gordon Harold is with Cedars-Sinai Heart Institute, Los Angeles, and president of the American College of Cardiology. He had no relevant financial disclosures.
|
Bruce Jancin/IMNG Medical Media
|
COMPARE is a very important study whose results are going to mean a paradigm shift for the management of Marfan syndrome.
It is intriguing to consider that the benefits of losartan might possibly also extend to patients with thoracic aortic disease in general, a worthy topic for future investigation.
Dr. John Gordon Harold is with Cedars-Sinai Heart Institute, Los Angeles, and president of the American College of Cardiology. He had no relevant financial disclosures.
|
Bruce Jancin/IMNG Medical Media
|
COMPARE is a very important study whose results are going to mean a paradigm shift for the management of Marfan syndrome.
It is intriguing to consider that the benefits of losartan might possibly also extend to patients with thoracic aortic disease in general, a worthy topic for future investigation.
Dr. John Gordon Harold is with Cedars-Sinai Heart Institute, Los Angeles, and president of the American College of Cardiology. He had no relevant financial disclosures.
AMSTERDAM – Daily losartan significantly slowed the aortic root dilatation rate in adults with Marfan syndrome in a 3-year randomized clinical trial.
"I think we can be positive about this treatment. We can now recommend losartan in clinical practice," Dr. Maarten Groenink said at the annual congress of the European Society of Cardiology.
The COMPARE (Cozaar in Marfan Patients Reduces Aortic Enlargement) trial included 218 patients at all four university Marfan centers in the Netherlands. Patients were randomized to oral losartan at a target dose of 100 mg/day or no losartan in addition to standard-of-care treatment with beta-blockers. Roughly half of the patients in the losartan group were unable to tolerate the full dose of losartan in addition to a beta-blocker; those patients were maintained on losartan at 50 mg/day. Aortic root diameter was measured by MRI at enrollment and after 3 years of prospective follow-up. The aortic dilatation rate was significantly lower in the losartan group than in controls both in the patients with a native root and in those who had undergone aortic root replacement surgery, reported Dr. Groenink, a cardiologist at the Academic Medical Center, Amsterdam.
There were no aortic dissections in the losartan group and two in the control arm. Elective aortic replacement surgery was performed in a similar number of patients in both groups.
Blood pressure was lower in the losartan group, yet blood pressure didn’t correlate with the aortic dilatation rate. Dr. Groenink speculated that losartan’s chief mechanism of benefit in Marfan syndrome is its ability to curb overexpression of transforming growth factor-beta, which weakens the structure of the media layer of the aortic wall.
Dr. Groenink said it’s unknown whether losartan’s benefits are specific to that drug or are a class effect obtainable with other angiotensin II receptor antagonists, though he suspects it’s a class effect.
Ongoing clinical trials are evaluating losartan in children and adolescents with Marfan syndrome, he said, adding that there is a solid rationale for beginning treatment as early in life as possible.
"I believe the adverse effects on the aortic wall in Marfan syndrome are caused by the fibrillin defect but also by wear and tear due to cyclic stress by the beating heart. So you can hypothesize that the earlier you start treatment, the better the results," he explained.
Marfan syndrome is a genetic connective tissue disorder affecting multiple organ systems. The prognosis is mainly determined by the aortic complications, including dilatation, aneurysm formation, and possible acute dissection. Affected individuals tend to be tall, long-limbed, and have distinctively long, thin fingers. The prevalence of Marfan syndrome has been estimated at 1 in 5,000, but Dr. Groenink suspects the syndrome may actually be more common than that.
Simultaneous with Dr. Groenink’s presentation at the ESC, the COMPARE results were published online (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht334]).
The COMPARE trial was funded by the Dutch Heart Association. Dr. Groenink reported having no relevant financial interests.
*CORRECTION 11/14/13: The first version of this story had Dr. Groenink's name misspelled.
AMSTERDAM – Daily losartan significantly slowed the aortic root dilatation rate in adults with Marfan syndrome in a 3-year randomized clinical trial.
"I think we can be positive about this treatment. We can now recommend losartan in clinical practice," Dr. Maarten Groenink said at the annual congress of the European Society of Cardiology.
The COMPARE (Cozaar in Marfan Patients Reduces Aortic Enlargement) trial included 218 patients at all four university Marfan centers in the Netherlands. Patients were randomized to oral losartan at a target dose of 100 mg/day or no losartan in addition to standard-of-care treatment with beta-blockers. Roughly half of the patients in the losartan group were unable to tolerate the full dose of losartan in addition to a beta-blocker; those patients were maintained on losartan at 50 mg/day. Aortic root diameter was measured by MRI at enrollment and after 3 years of prospective follow-up. The aortic dilatation rate was significantly lower in the losartan group than in controls both in the patients with a native root and in those who had undergone aortic root replacement surgery, reported Dr. Groenink, a cardiologist at the Academic Medical Center, Amsterdam.
There were no aortic dissections in the losartan group and two in the control arm. Elective aortic replacement surgery was performed in a similar number of patients in both groups.
Blood pressure was lower in the losartan group, yet blood pressure didn’t correlate with the aortic dilatation rate. Dr. Groenink speculated that losartan’s chief mechanism of benefit in Marfan syndrome is its ability to curb overexpression of transforming growth factor-beta, which weakens the structure of the media layer of the aortic wall.
Dr. Groenink said it’s unknown whether losartan’s benefits are specific to that drug or are a class effect obtainable with other angiotensin II receptor antagonists, though he suspects it’s a class effect.
Ongoing clinical trials are evaluating losartan in children and adolescents with Marfan syndrome, he said, adding that there is a solid rationale for beginning treatment as early in life as possible.
"I believe the adverse effects on the aortic wall in Marfan syndrome are caused by the fibrillin defect but also by wear and tear due to cyclic stress by the beating heart. So you can hypothesize that the earlier you start treatment, the better the results," he explained.
Marfan syndrome is a genetic connective tissue disorder affecting multiple organ systems. The prognosis is mainly determined by the aortic complications, including dilatation, aneurysm formation, and possible acute dissection. Affected individuals tend to be tall, long-limbed, and have distinctively long, thin fingers. The prevalence of Marfan syndrome has been estimated at 1 in 5,000, but Dr. Groenink suspects the syndrome may actually be more common than that.
Simultaneous with Dr. Groenink’s presentation at the ESC, the COMPARE results were published online (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht334]).
The COMPARE trial was funded by the Dutch Heart Association. Dr. Groenink reported having no relevant financial interests.
*CORRECTION 11/14/13: The first version of this story had Dr. Groenink's name misspelled.
AT THE ESC CONGRESS 2013
Major finding: The rate of aortic root enlargement during 3 years of prospective follow-up was 0.77 mm in losartan-treated patients with Marfan syndrome, significantly less than the 1.35 mm in patients on standard-of-care treatment with no losartan.
Data source: The COMPARE trial was a randomized, prospective, open-label multicenter study in which 218 patients with Marfan syndrome were randomized to losartan at a target dose of 100 mg or to no losartan and followed for 3 years with the aortic root dilatation rate as measured by MRI the primary endpoint.
Disclosures: The COMPARE trial was supported by the Dutch Heart Association. Dr. Groenink reported having no financial conflicts.
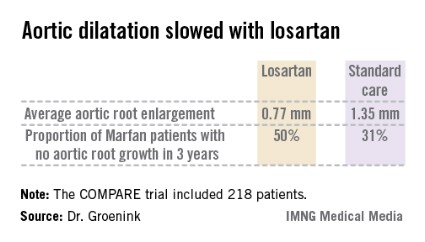
Psoriasis linked to increased heart failure risk
AMSTERDAM – Psoriasis proved to be independently associated with an increased risk for new-onset heart failure in the first nationwide study to look at a possible relationship.
As word of the newly identified psoriasis/heart failure link spreads, it’s likely cardiologists will receive a growing number of referrals from dermatologists and primary care physicians for evaluation of possible heart failure in psoriasis patients who develop shortness of breath or other symptoms suggestive of ventricular dysfunction, Dr. Usman Khalid said at the annual congress of the European Society of Cardiology.

"Our results underlie the importance of considering the psoriatic population as a high-risk patient group in terms of cardiovascular risk. We encourage early screening for cardiovascular risk factors in psoriasis patients," he said.
Psoriasis is known to be associated with an increased cardiovascular event rate, presumably due to shared systemic inflammatory pathways, but the association between the dermatologic disease and heart failure, specifically, hadn’t been looked at in depth prior to Dr. Khalid’s presentation of a Danish study involving all 5.85 million Danish adults, who were followed from 1997 through 2009.
Among the Danish population without known heart failure at baseline were 57,049 individuals with mild psoriasis – identified by their use of prescription topical agents – and 11,638 others with severe psoriasis. The incidence of new-onset heart failure during follow-up was 2.27 cases per 1,000 person-years in subjects without psoriasis and a significantly greater 4.02 per 1,000 person-years in those with mild psoriasis and 4.50 per 1,000 person-years in individuals with severe psoriasis, reported Dr. Khalid of the University of Copenhagen.
In a multivariate regression analysis adjusted for demographic and socioeconomic factors, comorbid conditions, and use of cardiovascular medications, the likelihood of developing new-onset heart failure during follow-up was 64% greater among individuals with mild psoriasis and 85% greater in those with severe psoriasis than in psoriasis-free subjects.
Because of limitations in the registry database, it is not possible to determine the proportion of new-onset heart failure among psoriasis patients that involved systolic as opposed to diastolic dysfunction, or ischemic versus nonischemic etiology, Dr. Khalid said in response to audience questions.
This study was supported by a research grant from Leo Pharma. Dr. Khalid reported having no financial conflicts of interest.
AMSTERDAM – Psoriasis proved to be independently associated with an increased risk for new-onset heart failure in the first nationwide study to look at a possible relationship.
As word of the newly identified psoriasis/heart failure link spreads, it’s likely cardiologists will receive a growing number of referrals from dermatologists and primary care physicians for evaluation of possible heart failure in psoriasis patients who develop shortness of breath or other symptoms suggestive of ventricular dysfunction, Dr. Usman Khalid said at the annual congress of the European Society of Cardiology.
"Our results underlie the importance of considering the psoriatic population as a high-risk patient group in terms of cardiovascular risk. We encourage early screening for cardiovascular risk factors in psoriasis patients," he said.
Psoriasis is known to be associated with an increased cardiovascular event rate, presumably due to shared systemic inflammatory pathways, but the association between the dermatologic disease and heart failure, specifically, hadn’t been looked at in depth prior to Dr. Khalid’s presentation of a Danish study involving all 5.85 million Danish adults, who were followed from 1997 through 2009.
Among the Danish population without known heart failure at baseline were 57,049 individuals with mild psoriasis – identified by their use of prescription topical agents – and 11,638 others with severe psoriasis. The incidence of new-onset heart failure during follow-up was 2.27 cases per 1,000 person-years in subjects without psoriasis and a significantly greater 4.02 per 1,000 person-years in those with mild psoriasis and 4.50 per 1,000 person-years in individuals with severe psoriasis, reported Dr. Khalid of the University of Copenhagen.
In a multivariate regression analysis adjusted for demographic and socioeconomic factors, comorbid conditions, and use of cardiovascular medications, the likelihood of developing new-onset heart failure during follow-up was 64% greater among individuals with mild psoriasis and 85% greater in those with severe psoriasis than in psoriasis-free subjects.
Because of limitations in the registry database, it is not possible to determine the proportion of new-onset heart failure among psoriasis patients that involved systolic as opposed to diastolic dysfunction, or ischemic versus nonischemic etiology, Dr. Khalid said in response to audience questions.
This study was supported by a research grant from Leo Pharma. Dr. Khalid reported having no financial conflicts of interest.
AMSTERDAM – Psoriasis proved to be independently associated with an increased risk for new-onset heart failure in the first nationwide study to look at a possible relationship.
As word of the newly identified psoriasis/heart failure link spreads, it’s likely cardiologists will receive a growing number of referrals from dermatologists and primary care physicians for evaluation of possible heart failure in psoriasis patients who develop shortness of breath or other symptoms suggestive of ventricular dysfunction, Dr. Usman Khalid said at the annual congress of the European Society of Cardiology.
"Our results underlie the importance of considering the psoriatic population as a high-risk patient group in terms of cardiovascular risk. We encourage early screening for cardiovascular risk factors in psoriasis patients," he said.
Psoriasis is known to be associated with an increased cardiovascular event rate, presumably due to shared systemic inflammatory pathways, but the association between the dermatologic disease and heart failure, specifically, hadn’t been looked at in depth prior to Dr. Khalid’s presentation of a Danish study involving all 5.85 million Danish adults, who were followed from 1997 through 2009.
Among the Danish population without known heart failure at baseline were 57,049 individuals with mild psoriasis – identified by their use of prescription topical agents – and 11,638 others with severe psoriasis. The incidence of new-onset heart failure during follow-up was 2.27 cases per 1,000 person-years in subjects without psoriasis and a significantly greater 4.02 per 1,000 person-years in those with mild psoriasis and 4.50 per 1,000 person-years in individuals with severe psoriasis, reported Dr. Khalid of the University of Copenhagen.
In a multivariate regression analysis adjusted for demographic and socioeconomic factors, comorbid conditions, and use of cardiovascular medications, the likelihood of developing new-onset heart failure during follow-up was 64% greater among individuals with mild psoriasis and 85% greater in those with severe psoriasis than in psoriasis-free subjects.
Because of limitations in the registry database, it is not possible to determine the proportion of new-onset heart failure among psoriasis patients that involved systolic as opposed to diastolic dysfunction, or ischemic versus nonischemic etiology, Dr. Khalid said in response to audience questions.
This study was supported by a research grant from Leo Pharma. Dr. Khalid reported having no financial conflicts of interest.
AT THE ESC CONGRESS 2013
Major finding: Danish patients with mild psoriasis developed new-onset heart failure at the rate of 4.02 cases per 1,000 person-years of follow-up, while those with severe psoriasis did so at a pace of 4.50 cases per 1,000 person-years, both significantly higher rates than the 2.27 per 1,000 person-years in the general psoriasis-free population.
Data source: This was a Danish nationwide registry study that included all 5.85 million Danish adults. They were followed during 1997-2009 for development of heart failure.
Disclosures: The study was supported by a research grant from Leo Pharma. The presenter reported having no financial conflicts of interest.

CRT appears deadly in short-QRS patients
AMSTERDAM – Patients with severe heart failure and a narrow QRS duration who received cardiac resynchronization therapy had more than twice the rate of cardiovascular deaths as patients who did not undergo active CRT treatment in a multinational randomized study with 809 patients.
The study’s enrollment criteria focused on patients with a QRS duration of less than 130 msec, and in this group active CRT produced no benefit, compared with the CRT function turned off in control patients for the study’s primary efficacy endpoint of death from any cause or first hospitalization for worsening heart failure.

In addition, during an average 19 months’ follow-up, patients in the CRT-on group had an all-cause death rate nearly five percentage points above the controls, an 80% relatively increased hazard, and a cardiovascular death rate also five percentage points above the controls, a greater than two-fold relative hazard, Dr. Johannes Holzmeister said at the annual congress of the European Society of Cardiology.
Both increased hazard rates were statistically significant and were a "surprising, unexpected finding," said Dr. Holzmeister, a cardiologist at the University of Zurich.
"This is the final nail in the coffin for CRT in patients with only slightly-prolonged QRS," commented Dr. Douglas P. Zipes, a professor and electrophysiologist at Indiana University in Indianapolis.
Existing recommendations from the American College of Cardiology and American Heart Association (J. Am. Coll. Cardiol. 2013 [doi:10.1016/j.jacc2013.05.019]) restrict unequivocal endorsement of CRT to heart failure patients with a QRS duration of at least 150 msec and say that CRT can be useful for patients with New York Heart Association class II, III, or IV heart failure, left bundle branch block (LBBB), and a QRS duration of 120-149 msec. Recommendations from the European Society of Cardiology are more permissive, calling CRT recommended for patients in sinus rhythm with a QRS duration of at least 120 msec, LBBB morphology on their QRS wave, and an ejection fraction of at least 35% who are expected to remain in good functional status for more than 1 year (Eur. Heart J. 2012;33:1787-847).
The ESC recommendations also say that CRT should be considered in patients with a QRS duration of at least 150 msec, irrespective of their QRS morphology.
It seems like many patients with shorter QRS durations have been receiving CRT treatment. During his report at the meeting, Dr. Holzmeister cited data from a 2008-2009 European-wide survey of CRT patients by the European Heart Rhythm Association, which showed that, 4-5 years ago, 9% of patients in Europe who received a CRT device had a QRS duration of less than 120 msec and another 10% of the CRT recipients had durations of 120-129 msec (Eur. Heart J. 2009;30:2450-60).
Substantial numbers of patients continue to receive CRT treatment with a QRS duration of 120-129 msec, a treatment that the data indicate should be a "last resort," said Dr. John G.F. Cleland, professor of medicine and a heart failure specialist at the University of Hull (England).
Although existing society recommendations sanction CRT for selected heart failure patients with a QRS duration of 120-149 msec, many experts have become convinced that the field needs a new set of QRS criteria.
"I think the cutoff for CRT implantation should be 150 msec," said Dr. Gordon F. Tomaselli, professor of cardiology and an electrophysiologist at Johns Hopkins University in Baltimore. "Patients with a QRS duration of less than 150 msec, particularly without left bundle branch block, should probably not get CRTs unless some special circumstances are present," he said in an interview.
"This reinforces the conclusion that we should not use CRT devices in heart failure patients with a QRS duration of less than 130 msec," commented Dr. Stefan D. Anker, professor and cardiologist at Charitè Hospital in Berlin and designated discussant for Dr. Holzmeister’s talk at the meeting. Dr. Anker called CRT use in patients with QRS durations of 130-149 msec uncertain and in need of more evidence for efficacy and safety from trials. "What is certain is that in patients with a QRS of 150 msec or greater there is a strong mortality reduction with CRT," he said.
The Echocardiography Guided Cardiac Resynchronization Therapy (EchoCRT) study randomized 809 patients with a QRS duration of less than 130 msec and left ventricular ejection fraction of 35% or less, and also required that patients meet criteria for having left-ventricular dyssynchrony detected by echocardiography. All patients received a CRT device, which was turned on in 404 patients. The study began in August 2008, and enrollment stopped on March 13, 2013, because of futility and a potential for harm.
At that time, cardiovascular deaths had occurred in 9% of patients with their CRT turned on and in 4% of those with the device turned off. Concurrent with Dr. Holzmeister’s report at the meeting, an article with the results was published online (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1306687]).
The EchoCRT trial was sponsored by Biotronik and GE Healthcare. Dr. Holzmeister said that he has been a consultant to Biotronik. Dr. Zipes said that he has been a consultant to Health System Networks and consults for and is a co-owner of Insight Telehealth. Dr. Anker said that he has received fees and honoraria from St. Jude, Abbott Vascular, and several other drug and device companies. Dr. Tomaselli said that he had no disclosures.
On Twitter @mitchelzoler
The results from EchoCRT fit well with the results of a meta-analysis that my associates and I recently ran on data from 3,782 heart failure patients treated with CRT in five trials sponsored by Medtronic. The results showed that QRS duration was a powerful predictor of the effects of CRT on morbidity and mortality.
The meta-analysis results confirmed the benefit of CRT in patients with mild, moderate, or severe heart failure symptoms, in sinus rhythm, and with a QRS duration of at least 140 msec, and in these patients CRT is standard of care. The results also showed that the benefits from CRT diminish as the QRS duration goes below 140 msec. Patients with a QRS duration of 130-139 msec are in a gray zone. If they need a defibrillator, then making it a CRT device makes sense, but if no device implant is planned then continued attempts at medical treatment are probably better than going to CRT.
 |
|
I would avoid CRT in patients with a QRS of less than 130 msec, and now the EchoCRT results show evidence of harm in these patients. A lot of patients with a QRS duration of 120-129 msec have been receiving CRT devices when the chances of benefit are small. This might be a treatment of last resort, but only when you have exhausted all the other treatment alternatives.
The meta-analysis also showed that QRS duration was the only independent predictor of CRT outcomes (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht290]). QRS morphology – whether or not there is a left bundle branch block – was not a significant factor relative to QRS duration. The EchoCRT results also showed that echocardiography used to diagnose left-ventricular dyssynchrony failed to identify a subgroup of patients with a narrow QRS duration who benefited from CRT.
Dr. John G.F. Cleland is professor of medicine at the University of Hull (England). He has been a consultant to Biotronik, St. Jude, and Medtronic. He made these comments in an interview.
The results from EchoCRT fit well with the results of a meta-analysis that my associates and I recently ran on data from 3,782 heart failure patients treated with CRT in five trials sponsored by Medtronic. The results showed that QRS duration was a powerful predictor of the effects of CRT on morbidity and mortality.
The meta-analysis results confirmed the benefit of CRT in patients with mild, moderate, or severe heart failure symptoms, in sinus rhythm, and with a QRS duration of at least 140 msec, and in these patients CRT is standard of care. The results also showed that the benefits from CRT diminish as the QRS duration goes below 140 msec. Patients with a QRS duration of 130-139 msec are in a gray zone. If they need a defibrillator, then making it a CRT device makes sense, but if no device implant is planned then continued attempts at medical treatment are probably better than going to CRT.
|
|
I would avoid CRT in patients with a QRS of less than 130 msec, and now the EchoCRT results show evidence of harm in these patients. A lot of patients with a QRS duration of 120-129 msec have been receiving CRT devices when the chances of benefit are small. This might be a treatment of last resort, but only when you have exhausted all the other treatment alternatives.
The meta-analysis also showed that QRS duration was the only independent predictor of CRT outcomes (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht290]). QRS morphology – whether or not there is a left bundle branch block – was not a significant factor relative to QRS duration. The EchoCRT results also showed that echocardiography used to diagnose left-ventricular dyssynchrony failed to identify a subgroup of patients with a narrow QRS duration who benefited from CRT.
Dr. John G.F. Cleland is professor of medicine at the University of Hull (England). He has been a consultant to Biotronik, St. Jude, and Medtronic. He made these comments in an interview.
The results from EchoCRT fit well with the results of a meta-analysis that my associates and I recently ran on data from 3,782 heart failure patients treated with CRT in five trials sponsored by Medtronic. The results showed that QRS duration was a powerful predictor of the effects of CRT on morbidity and mortality.
The meta-analysis results confirmed the benefit of CRT in patients with mild, moderate, or severe heart failure symptoms, in sinus rhythm, and with a QRS duration of at least 140 msec, and in these patients CRT is standard of care. The results also showed that the benefits from CRT diminish as the QRS duration goes below 140 msec. Patients with a QRS duration of 130-139 msec are in a gray zone. If they need a defibrillator, then making it a CRT device makes sense, but if no device implant is planned then continued attempts at medical treatment are probably better than going to CRT.
|
|
I would avoid CRT in patients with a QRS of less than 130 msec, and now the EchoCRT results show evidence of harm in these patients. A lot of patients with a QRS duration of 120-129 msec have been receiving CRT devices when the chances of benefit are small. This might be a treatment of last resort, but only when you have exhausted all the other treatment alternatives.
The meta-analysis also showed that QRS duration was the only independent predictor of CRT outcomes (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht290]). QRS morphology – whether or not there is a left bundle branch block – was not a significant factor relative to QRS duration. The EchoCRT results also showed that echocardiography used to diagnose left-ventricular dyssynchrony failed to identify a subgroup of patients with a narrow QRS duration who benefited from CRT.
Dr. John G.F. Cleland is professor of medicine at the University of Hull (England). He has been a consultant to Biotronik, St. Jude, and Medtronic. He made these comments in an interview.
AMSTERDAM – Patients with severe heart failure and a narrow QRS duration who received cardiac resynchronization therapy had more than twice the rate of cardiovascular deaths as patients who did not undergo active CRT treatment in a multinational randomized study with 809 patients.
The study’s enrollment criteria focused on patients with a QRS duration of less than 130 msec, and in this group active CRT produced no benefit, compared with the CRT function turned off in control patients for the study’s primary efficacy endpoint of death from any cause or first hospitalization for worsening heart failure.
In addition, during an average 19 months’ follow-up, patients in the CRT-on group had an all-cause death rate nearly five percentage points above the controls, an 80% relatively increased hazard, and a cardiovascular death rate also five percentage points above the controls, a greater than two-fold relative hazard, Dr. Johannes Holzmeister said at the annual congress of the European Society of Cardiology.
Both increased hazard rates were statistically significant and were a "surprising, unexpected finding," said Dr. Holzmeister, a cardiologist at the University of Zurich.
"This is the final nail in the coffin for CRT in patients with only slightly-prolonged QRS," commented Dr. Douglas P. Zipes, a professor and electrophysiologist at Indiana University in Indianapolis.
Existing recommendations from the American College of Cardiology and American Heart Association (J. Am. Coll. Cardiol. 2013 [doi:10.1016/j.jacc2013.05.019]) restrict unequivocal endorsement of CRT to heart failure patients with a QRS duration of at least 150 msec and say that CRT can be useful for patients with New York Heart Association class II, III, or IV heart failure, left bundle branch block (LBBB), and a QRS duration of 120-149 msec. Recommendations from the European Society of Cardiology are more permissive, calling CRT recommended for patients in sinus rhythm with a QRS duration of at least 120 msec, LBBB morphology on their QRS wave, and an ejection fraction of at least 35% who are expected to remain in good functional status for more than 1 year (Eur. Heart J. 2012;33:1787-847).
The ESC recommendations also say that CRT should be considered in patients with a QRS duration of at least 150 msec, irrespective of their QRS morphology.
It seems like many patients with shorter QRS durations have been receiving CRT treatment. During his report at the meeting, Dr. Holzmeister cited data from a 2008-2009 European-wide survey of CRT patients by the European Heart Rhythm Association, which showed that, 4-5 years ago, 9% of patients in Europe who received a CRT device had a QRS duration of less than 120 msec and another 10% of the CRT recipients had durations of 120-129 msec (Eur. Heart J. 2009;30:2450-60).
Substantial numbers of patients continue to receive CRT treatment with a QRS duration of 120-129 msec, a treatment that the data indicate should be a "last resort," said Dr. John G.F. Cleland, professor of medicine and a heart failure specialist at the University of Hull (England).
Although existing society recommendations sanction CRT for selected heart failure patients with a QRS duration of 120-149 msec, many experts have become convinced that the field needs a new set of QRS criteria.
"I think the cutoff for CRT implantation should be 150 msec," said Dr. Gordon F. Tomaselli, professor of cardiology and an electrophysiologist at Johns Hopkins University in Baltimore. "Patients with a QRS duration of less than 150 msec, particularly without left bundle branch block, should probably not get CRTs unless some special circumstances are present," he said in an interview.
"This reinforces the conclusion that we should not use CRT devices in heart failure patients with a QRS duration of less than 130 msec," commented Dr. Stefan D. Anker, professor and cardiologist at Charitè Hospital in Berlin and designated discussant for Dr. Holzmeister’s talk at the meeting. Dr. Anker called CRT use in patients with QRS durations of 130-149 msec uncertain and in need of more evidence for efficacy and safety from trials. "What is certain is that in patients with a QRS of 150 msec or greater there is a strong mortality reduction with CRT," he said.
The Echocardiography Guided Cardiac Resynchronization Therapy (EchoCRT) study randomized 809 patients with a QRS duration of less than 130 msec and left ventricular ejection fraction of 35% or less, and also required that patients meet criteria for having left-ventricular dyssynchrony detected by echocardiography. All patients received a CRT device, which was turned on in 404 patients. The study began in August 2008, and enrollment stopped on March 13, 2013, because of futility and a potential for harm.
At that time, cardiovascular deaths had occurred in 9% of patients with their CRT turned on and in 4% of those with the device turned off. Concurrent with Dr. Holzmeister’s report at the meeting, an article with the results was published online (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1306687]).
The EchoCRT trial was sponsored by Biotronik and GE Healthcare. Dr. Holzmeister said that he has been a consultant to Biotronik. Dr. Zipes said that he has been a consultant to Health System Networks and consults for and is a co-owner of Insight Telehealth. Dr. Anker said that he has received fees and honoraria from St. Jude, Abbott Vascular, and several other drug and device companies. Dr. Tomaselli said that he had no disclosures.
On Twitter @mitchelzoler
AMSTERDAM – Patients with severe heart failure and a narrow QRS duration who received cardiac resynchronization therapy had more than twice the rate of cardiovascular deaths as patients who did not undergo active CRT treatment in a multinational randomized study with 809 patients.
The study’s enrollment criteria focused on patients with a QRS duration of less than 130 msec, and in this group active CRT produced no benefit, compared with the CRT function turned off in control patients for the study’s primary efficacy endpoint of death from any cause or first hospitalization for worsening heart failure.
In addition, during an average 19 months’ follow-up, patients in the CRT-on group had an all-cause death rate nearly five percentage points above the controls, an 80% relatively increased hazard, and a cardiovascular death rate also five percentage points above the controls, a greater than two-fold relative hazard, Dr. Johannes Holzmeister said at the annual congress of the European Society of Cardiology.
Both increased hazard rates were statistically significant and were a "surprising, unexpected finding," said Dr. Holzmeister, a cardiologist at the University of Zurich.
"This is the final nail in the coffin for CRT in patients with only slightly-prolonged QRS," commented Dr. Douglas P. Zipes, a professor and electrophysiologist at Indiana University in Indianapolis.
Existing recommendations from the American College of Cardiology and American Heart Association (J. Am. Coll. Cardiol. 2013 [doi:10.1016/j.jacc2013.05.019]) restrict unequivocal endorsement of CRT to heart failure patients with a QRS duration of at least 150 msec and say that CRT can be useful for patients with New York Heart Association class II, III, or IV heart failure, left bundle branch block (LBBB), and a QRS duration of 120-149 msec. Recommendations from the European Society of Cardiology are more permissive, calling CRT recommended for patients in sinus rhythm with a QRS duration of at least 120 msec, LBBB morphology on their QRS wave, and an ejection fraction of at least 35% who are expected to remain in good functional status for more than 1 year (Eur. Heart J. 2012;33:1787-847).
The ESC recommendations also say that CRT should be considered in patients with a QRS duration of at least 150 msec, irrespective of their QRS morphology.
It seems like many patients with shorter QRS durations have been receiving CRT treatment. During his report at the meeting, Dr. Holzmeister cited data from a 2008-2009 European-wide survey of CRT patients by the European Heart Rhythm Association, which showed that, 4-5 years ago, 9% of patients in Europe who received a CRT device had a QRS duration of less than 120 msec and another 10% of the CRT recipients had durations of 120-129 msec (Eur. Heart J. 2009;30:2450-60).
Substantial numbers of patients continue to receive CRT treatment with a QRS duration of 120-129 msec, a treatment that the data indicate should be a "last resort," said Dr. John G.F. Cleland, professor of medicine and a heart failure specialist at the University of Hull (England).
Although existing society recommendations sanction CRT for selected heart failure patients with a QRS duration of 120-149 msec, many experts have become convinced that the field needs a new set of QRS criteria.
"I think the cutoff for CRT implantation should be 150 msec," said Dr. Gordon F. Tomaselli, professor of cardiology and an electrophysiologist at Johns Hopkins University in Baltimore. "Patients with a QRS duration of less than 150 msec, particularly without left bundle branch block, should probably not get CRTs unless some special circumstances are present," he said in an interview.
"This reinforces the conclusion that we should not use CRT devices in heart failure patients with a QRS duration of less than 130 msec," commented Dr. Stefan D. Anker, professor and cardiologist at Charitè Hospital in Berlin and designated discussant for Dr. Holzmeister’s talk at the meeting. Dr. Anker called CRT use in patients with QRS durations of 130-149 msec uncertain and in need of more evidence for efficacy and safety from trials. "What is certain is that in patients with a QRS of 150 msec or greater there is a strong mortality reduction with CRT," he said.
The Echocardiography Guided Cardiac Resynchronization Therapy (EchoCRT) study randomized 809 patients with a QRS duration of less than 130 msec and left ventricular ejection fraction of 35% or less, and also required that patients meet criteria for having left-ventricular dyssynchrony detected by echocardiography. All patients received a CRT device, which was turned on in 404 patients. The study began in August 2008, and enrollment stopped on March 13, 2013, because of futility and a potential for harm.
At that time, cardiovascular deaths had occurred in 9% of patients with their CRT turned on and in 4% of those with the device turned off. Concurrent with Dr. Holzmeister’s report at the meeting, an article with the results was published online (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1306687]).
The EchoCRT trial was sponsored by Biotronik and GE Healthcare. Dr. Holzmeister said that he has been a consultant to Biotronik. Dr. Zipes said that he has been a consultant to Health System Networks and consults for and is a co-owner of Insight Telehealth. Dr. Anker said that he has received fees and honoraria from St. Jude, Abbott Vascular, and several other drug and device companies. Dr. Tomaselli said that he had no disclosures.
On Twitter @mitchelzoler
AT THE ESC CONGRESS 2013
Major finding: Cardiovascular death occurred in 9% of patients on active cardiac resynchronization therapy and in 4% of control patients.
Data source: Data came from the EchoCRT trial, a multicenter, randomized trial with 809 patients with NYHA class III or IV heart failure and a QRS duration of less than 130 msec.
Disclosures: EchoCRT was sponsored by Biotronik and GE Healthcare. Dr. Holzmeister has been a consultant to Biotronik. Dr. Zipes has been a consultant to Health System Networks and consults for and is a co-owner of Insight Telehealth. Dr. Anker has received fees and honoraria from St. Jude, Abbott Vascular, and several other drug and device companies. Dr. Tomaselli said that he had no disclosures. Dr. Cleland said that he has been a consultant to Biotronik, Medtronic, and St. Jude.

New heart failure inotrope could be ‘Holy Grail’
AMSTERDAM – The novel intravenous inotropic agent omecamtiv mecarbil achieved greater reduction in dyspnea scores and less likelihood of worsening heart failure at the highest dose studied, compared with placebo, in patients with acute decompensated heart failure in a phase II, dose-ranging study.
The drug also demonstrated a strong dose-dependent increase in systolic ejection time, no evidence of a proarrhythmic effect, and a greater reduction in heart rate with a larger increase in blood pressure than in placebo-treated controls, Dr. John R. Teerlink reported at the annual congress of the European Society of Cardiology.

In the overall population on omecamtiv mecarbil, however, which included the patients in the low- and intermediate-dose arms of this dose-ranging study, the dyspnea response rate wasn’t better than with placebo. Hence, the drug’s future is uncertain.
In a press release following Dr. Teerlink’s presentation, Amgen, cosponsor of the phase II ATOMIC-AHF (Acute Treatment With Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure) trial, said that no decision has been made as to whether to move omecamtiv mecarbil into phase III clinical trials. Dr. Teerlink thinks the drug has earned a shot.
"I personally believe this drug has continued to demonstrate its promise all along the way. It could replace milrinone and dobutamine in the hospital to improve cardiac performance," predicted Dr. Teerlink, professor of medicine at the University of California, San Francisco, and director of the heart failure program at the San Francisco Veterans Affairs Medical Center.
Omecamtiv mecarbil is a novel selective cardiac myosin activator. It increases the entry rate of myosin into the force-producing state with actin. It boosts cardiac output through prolongation of the duration of systole and increased stroke volume, with no change in myocardial contractility and no increases in myocardial oxygen demand or myocyte calcium.
"It’s essentially producing more hands pulling on the rope," the cardiologist explained.
Discussant Theresa A. McDonagh said omecamtiv mecarbil has drawn great interest within the heart failure field because current inotropes are recognized as "very inadequate."
"It is truly a new inotrope, a drug that acts on the sarcomere itself. It does seem to be free of the nemesis of other inotropes: increased heart rate, greater myocardial oxygen consumption, arrhythmia production, and increased mortality in heart failure. It appears to be the Holy Grail, and may be capable of replacing dobutamine and milrinone," said Dr. McDonagh, professor of heart failure at King’s College London.
"I think this [ATOMIC-AHF trial] is a very promising start for omecamtiv mecarbil in acute heart failure," she added.
ATOMIC-AHF included 613 patients with a left ventricular ejection fraction of 40% or less who were hospitalized for acute heart failure with dyspnea at rest or with minimal exertion despite intravenous diuretic therapy. The study was conducted at 106 sites on three continents. Participants were randomized to 48 hours of intravenous placebo or omecamtiv mecarbil in three different ascending dose regimens.
The primary efficacy endpoint, dyspnea at 6, 24, and 48 hours across the three doses, compared with the pooled placebo group, was not met.
However, patients in the highest-dose omecamtiv mecarbil study arm had a 51% rate of significant improvement in dyspnea on a self-rated 7-point Likert scale at 48 hours, a significantly better response than the 37% rate in matched placebo-treated controls (P = .03). This comparison was a prespecified secondary endpoint in the study.
In addition, patients in the middle- and high-dose omecamtiv mecarbil arms had roughly a 45% reduction in the 7-day incidence of worsening heart failure, compared with the 17% rate in controls.
Not only was there no evidence of a proarrhythmic effect with omecamtiv mecarbil, the drug was associated with a 50% reduction in the incidence of supraventricular tachyarrhythmias, compared with placebo, he continued.
A modest increase in troponin I levels was noted in the omecamtiv mecarbil–treated group, but it was unrelated to plasma drug concentrations, which Dr. Teerlink found reassuring. Nevertheless, Dr. McDonagh said the small increases in troponin "should herald some caution amid the enthusiasm here."
The drug’s mechanism of action, which results in greater systolic ejection time, always raises the concern that it might also shorten diastole and compromise coronary perfusion. However, the observed reduction in heart rate makes that seem less likely, she added.
The ATOMIC-AHF study population was narrowly defined and of a profile commonly seen in clinical practice. They had an estimated glomerular filtration rate of about 50 mL/min, and they presented with dyspnea but didn’t have an acute coronary syndrome.
"This study population is an area of great unmet need in heart failure, with mortality rates of about 10% during their hospitalization and 30% at 1 year following discharge," according to Dr. McDonagh.
Nevertheless, it will be important in phase III testing to see how the drug performs in a broader group of heart failure patients, including those with lower estimated glomerular filtration rates, she added.
In an interview, Dr. Teerlink said that before a decision is reached regarding advancement of omecamtiv mecarbil to phase III studies, investigators and the study sponsors first want to see the data from an ongoing phase II dose-finding study of an oral formulation of the drug in chronic heart failure patients. This study, COSMIC-HF (Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure), is still enrolling patients.
"We’ll get the combined data from ATOMIC-AHF and COSMIC-HF and then decide what phase III should look like. There are a lot of options here: IV [intravenous] alone, IV transitioning to chronic oral therapy, or even oral alone," he explained.
"One of the greatest challenges of the ATOMIC-AHF trial has been managing expectations," according to Dr. Teerlink. "Omecamtiv mecarbil is probably one of the most interesting new chemical entities in cardiovascular medicine now, and consequently expectations are very high in the scientific and lay communities."
The studies are sponsored by Amgen and Cytokinetics. Dr. Teerlink has received research grants and/or consulting fees from those companies as well as a handful of others. Dr. McDonagh reported no financial conflicts.
AMSTERDAM – The novel intravenous inotropic agent omecamtiv mecarbil achieved greater reduction in dyspnea scores and less likelihood of worsening heart failure at the highest dose studied, compared with placebo, in patients with acute decompensated heart failure in a phase II, dose-ranging study.
The drug also demonstrated a strong dose-dependent increase in systolic ejection time, no evidence of a proarrhythmic effect, and a greater reduction in heart rate with a larger increase in blood pressure than in placebo-treated controls, Dr. John R. Teerlink reported at the annual congress of the European Society of Cardiology.
In the overall population on omecamtiv mecarbil, however, which included the patients in the low- and intermediate-dose arms of this dose-ranging study, the dyspnea response rate wasn’t better than with placebo. Hence, the drug’s future is uncertain.
In a press release following Dr. Teerlink’s presentation, Amgen, cosponsor of the phase II ATOMIC-AHF (Acute Treatment With Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure) trial, said that no decision has been made as to whether to move omecamtiv mecarbil into phase III clinical trials. Dr. Teerlink thinks the drug has earned a shot.
"I personally believe this drug has continued to demonstrate its promise all along the way. It could replace milrinone and dobutamine in the hospital to improve cardiac performance," predicted Dr. Teerlink, professor of medicine at the University of California, San Francisco, and director of the heart failure program at the San Francisco Veterans Affairs Medical Center.
Omecamtiv mecarbil is a novel selective cardiac myosin activator. It increases the entry rate of myosin into the force-producing state with actin. It boosts cardiac output through prolongation of the duration of systole and increased stroke volume, with no change in myocardial contractility and no increases in myocardial oxygen demand or myocyte calcium.
"It’s essentially producing more hands pulling on the rope," the cardiologist explained.
Discussant Theresa A. McDonagh said omecamtiv mecarbil has drawn great interest within the heart failure field because current inotropes are recognized as "very inadequate."
"It is truly a new inotrope, a drug that acts on the sarcomere itself. It does seem to be free of the nemesis of other inotropes: increased heart rate, greater myocardial oxygen consumption, arrhythmia production, and increased mortality in heart failure. It appears to be the Holy Grail, and may be capable of replacing dobutamine and milrinone," said Dr. McDonagh, professor of heart failure at King’s College London.
"I think this [ATOMIC-AHF trial] is a very promising start for omecamtiv mecarbil in acute heart failure," she added.
ATOMIC-AHF included 613 patients with a left ventricular ejection fraction of 40% or less who were hospitalized for acute heart failure with dyspnea at rest or with minimal exertion despite intravenous diuretic therapy. The study was conducted at 106 sites on three continents. Participants were randomized to 48 hours of intravenous placebo or omecamtiv mecarbil in three different ascending dose regimens.
The primary efficacy endpoint, dyspnea at 6, 24, and 48 hours across the three doses, compared with the pooled placebo group, was not met.
However, patients in the highest-dose omecamtiv mecarbil study arm had a 51% rate of significant improvement in dyspnea on a self-rated 7-point Likert scale at 48 hours, a significantly better response than the 37% rate in matched placebo-treated controls (P = .03). This comparison was a prespecified secondary endpoint in the study.
In addition, patients in the middle- and high-dose omecamtiv mecarbil arms had roughly a 45% reduction in the 7-day incidence of worsening heart failure, compared with the 17% rate in controls.
Not only was there no evidence of a proarrhythmic effect with omecamtiv mecarbil, the drug was associated with a 50% reduction in the incidence of supraventricular tachyarrhythmias, compared with placebo, he continued.
A modest increase in troponin I levels was noted in the omecamtiv mecarbil–treated group, but it was unrelated to plasma drug concentrations, which Dr. Teerlink found reassuring. Nevertheless, Dr. McDonagh said the small increases in troponin "should herald some caution amid the enthusiasm here."
The drug’s mechanism of action, which results in greater systolic ejection time, always raises the concern that it might also shorten diastole and compromise coronary perfusion. However, the observed reduction in heart rate makes that seem less likely, she added.
The ATOMIC-AHF study population was narrowly defined and of a profile commonly seen in clinical practice. They had an estimated glomerular filtration rate of about 50 mL/min, and they presented with dyspnea but didn’t have an acute coronary syndrome.
"This study population is an area of great unmet need in heart failure, with mortality rates of about 10% during their hospitalization and 30% at 1 year following discharge," according to Dr. McDonagh.
Nevertheless, it will be important in phase III testing to see how the drug performs in a broader group of heart failure patients, including those with lower estimated glomerular filtration rates, she added.
In an interview, Dr. Teerlink said that before a decision is reached regarding advancement of omecamtiv mecarbil to phase III studies, investigators and the study sponsors first want to see the data from an ongoing phase II dose-finding study of an oral formulation of the drug in chronic heart failure patients. This study, COSMIC-HF (Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure), is still enrolling patients.
"We’ll get the combined data from ATOMIC-AHF and COSMIC-HF and then decide what phase III should look like. There are a lot of options here: IV [intravenous] alone, IV transitioning to chronic oral therapy, or even oral alone," he explained.
"One of the greatest challenges of the ATOMIC-AHF trial has been managing expectations," according to Dr. Teerlink. "Omecamtiv mecarbil is probably one of the most interesting new chemical entities in cardiovascular medicine now, and consequently expectations are very high in the scientific and lay communities."
The studies are sponsored by Amgen and Cytokinetics. Dr. Teerlink has received research grants and/or consulting fees from those companies as well as a handful of others. Dr. McDonagh reported no financial conflicts.
AMSTERDAM – The novel intravenous inotropic agent omecamtiv mecarbil achieved greater reduction in dyspnea scores and less likelihood of worsening heart failure at the highest dose studied, compared with placebo, in patients with acute decompensated heart failure in a phase II, dose-ranging study.
The drug also demonstrated a strong dose-dependent increase in systolic ejection time, no evidence of a proarrhythmic effect, and a greater reduction in heart rate with a larger increase in blood pressure than in placebo-treated controls, Dr. John R. Teerlink reported at the annual congress of the European Society of Cardiology.
In the overall population on omecamtiv mecarbil, however, which included the patients in the low- and intermediate-dose arms of this dose-ranging study, the dyspnea response rate wasn’t better than with placebo. Hence, the drug’s future is uncertain.
In a press release following Dr. Teerlink’s presentation, Amgen, cosponsor of the phase II ATOMIC-AHF (Acute Treatment With Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure) trial, said that no decision has been made as to whether to move omecamtiv mecarbil into phase III clinical trials. Dr. Teerlink thinks the drug has earned a shot.
"I personally believe this drug has continued to demonstrate its promise all along the way. It could replace milrinone and dobutamine in the hospital to improve cardiac performance," predicted Dr. Teerlink, professor of medicine at the University of California, San Francisco, and director of the heart failure program at the San Francisco Veterans Affairs Medical Center.
Omecamtiv mecarbil is a novel selective cardiac myosin activator. It increases the entry rate of myosin into the force-producing state with actin. It boosts cardiac output through prolongation of the duration of systole and increased stroke volume, with no change in myocardial contractility and no increases in myocardial oxygen demand or myocyte calcium.
"It’s essentially producing more hands pulling on the rope," the cardiologist explained.
Discussant Theresa A. McDonagh said omecamtiv mecarbil has drawn great interest within the heart failure field because current inotropes are recognized as "very inadequate."
"It is truly a new inotrope, a drug that acts on the sarcomere itself. It does seem to be free of the nemesis of other inotropes: increased heart rate, greater myocardial oxygen consumption, arrhythmia production, and increased mortality in heart failure. It appears to be the Holy Grail, and may be capable of replacing dobutamine and milrinone," said Dr. McDonagh, professor of heart failure at King’s College London.
"I think this [ATOMIC-AHF trial] is a very promising start for omecamtiv mecarbil in acute heart failure," she added.
ATOMIC-AHF included 613 patients with a left ventricular ejection fraction of 40% or less who were hospitalized for acute heart failure with dyspnea at rest or with minimal exertion despite intravenous diuretic therapy. The study was conducted at 106 sites on three continents. Participants were randomized to 48 hours of intravenous placebo or omecamtiv mecarbil in three different ascending dose regimens.
The primary efficacy endpoint, dyspnea at 6, 24, and 48 hours across the three doses, compared with the pooled placebo group, was not met.
However, patients in the highest-dose omecamtiv mecarbil study arm had a 51% rate of significant improvement in dyspnea on a self-rated 7-point Likert scale at 48 hours, a significantly better response than the 37% rate in matched placebo-treated controls (P = .03). This comparison was a prespecified secondary endpoint in the study.
In addition, patients in the middle- and high-dose omecamtiv mecarbil arms had roughly a 45% reduction in the 7-day incidence of worsening heart failure, compared with the 17% rate in controls.
Not only was there no evidence of a proarrhythmic effect with omecamtiv mecarbil, the drug was associated with a 50% reduction in the incidence of supraventricular tachyarrhythmias, compared with placebo, he continued.
A modest increase in troponin I levels was noted in the omecamtiv mecarbil–treated group, but it was unrelated to plasma drug concentrations, which Dr. Teerlink found reassuring. Nevertheless, Dr. McDonagh said the small increases in troponin "should herald some caution amid the enthusiasm here."
The drug’s mechanism of action, which results in greater systolic ejection time, always raises the concern that it might also shorten diastole and compromise coronary perfusion. However, the observed reduction in heart rate makes that seem less likely, she added.
The ATOMIC-AHF study population was narrowly defined and of a profile commonly seen in clinical practice. They had an estimated glomerular filtration rate of about 50 mL/min, and they presented with dyspnea but didn’t have an acute coronary syndrome.
"This study population is an area of great unmet need in heart failure, with mortality rates of about 10% during their hospitalization and 30% at 1 year following discharge," according to Dr. McDonagh.
Nevertheless, it will be important in phase III testing to see how the drug performs in a broader group of heart failure patients, including those with lower estimated glomerular filtration rates, she added.
In an interview, Dr. Teerlink said that before a decision is reached regarding advancement of omecamtiv mecarbil to phase III studies, investigators and the study sponsors first want to see the data from an ongoing phase II dose-finding study of an oral formulation of the drug in chronic heart failure patients. This study, COSMIC-HF (Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure), is still enrolling patients.
"We’ll get the combined data from ATOMIC-AHF and COSMIC-HF and then decide what phase III should look like. There are a lot of options here: IV [intravenous] alone, IV transitioning to chronic oral therapy, or even oral alone," he explained.
"One of the greatest challenges of the ATOMIC-AHF trial has been managing expectations," according to Dr. Teerlink. "Omecamtiv mecarbil is probably one of the most interesting new chemical entities in cardiovascular medicine now, and consequently expectations are very high in the scientific and lay communities."
The studies are sponsored by Amgen and Cytokinetics. Dr. Teerlink has received research grants and/or consulting fees from those companies as well as a handful of others. Dr. McDonagh reported no financial conflicts.
AT THE ESC CONGRESS 2013
Major finding: Fifty-one percent of patients with acute heart failure assigned to the high-dose arm in a study of intravenous omecamtiv mecarbil showed significant improvement in dyspnea scores at 48 hours, compared with 37% of placebo-treated controls.
Data source: The ATOMIC-AHF trial was a 613-patient, phase II, randomized, placebo-controlled, double-blind, dose-finding study.
Disclosures: The study was sponsored by Amgen and Cytokinetics. The presenter has received research grants and consultant fees from the companies.
