User login
Sunscreen ingredients found in bloodstream, but health impact unknown
for sunscreens, in a phase 1 randomized controlled study of 24 healthy volunteers.

In the open-label study, 24 people (mean age 35.5 years) were randomized to one of four commercially available sunscreens (two sprays, one lotion, or one cream formulation); 2 mg of sunscreen per 1 cm2 was applied to 75% of their body surface four times a day for 4 days (described as “maximal use conditions consistent with current sunscreen labeling”), and 30 blood samples were collected over 7 days.
The primary outcome was the maximum plasma concentration of avobenzone, from days 1-7; secondary outcomes were maximum plasma concentrations of sunscreen ingredients oxybenzone, octocrylene, and ecamsule over the same period of time.
All but one participant completed the study. “All four sunscreen active ingredients tested resulted in exposures exceeding 0.5 ng/mL,” reported Murali Matta, PhD, of the FDA’s Center for Drug Evaluation and Research, and coauthors. “The clinical effect of plasma concentrations exceeding 0.5 ng/mL is unknown, necessitating further research,” they added.
According to the study, FDA sunscreen guidance and the proposed rule for over-the-counter sunscreen monograph, nonclinical toxicology studies, such as carcinogenicity and reproductive studies, “may be waived if results of an adequately conducted human pharmacokinetic maximal usage trial show a steady state blood level less than 0.5 ng/mL and an adequately conducted toxicology assessment does not reveal any potential safety concerns.”
The results of this study “do not indicate that individuals should refrain from the use of sunscreen,” the authors concluded, adding that the “systemic absorption of sunscreen ingredients supports the need for further studies to determine the clinical significance of these findings.” The study was published in JAMA.
In an accompanying editorial, former FDA commissioner Robert Califf, MD, professor of cardiology, Duke University, Durham, N.C., and JAMA Dermatology Editor Kanade Shinkai, MD, PhD, of the department of dermatology, University of California, San Francisco, noted that “the demonstration of systemic absorption well above the FDA guideline does not mean these ingredients are unsafe” (JAMA. 2019 May 6. doi: 10.1001/jama.2019.5528). But, they added, the results “raise many important questions about sunscreen and the process by which the sunscreen industry, clinicians, specialty organizations, and regulatory agencies evaluate the benefits and risks of this topical OTC medication. First and foremost, it is essential to determine whether systemic absorption of sunscreen poses risks to human health. Second, the effects of different sunscreen formulations, clinical characteristics (that is, skin type, age, presence of skin diseases that disrupt the skin barrier), physical activity level, and exposure to sun and water on systemic sunscreen levels require further study.”
In a statement, former American Academy of Dermatology President Darrel Rigel, MD, of the department of dermatology, New York University, said that he was concerned that the results were misleading. “We have always known that there is a very small amount of absorption of sunscreens in the bloodstream,” and there are no data that this is a problem, he said, adding: “Tens of millions of people use sunscreens in the U.S. every summer weekend for many years with no incidence. Daily use of a broad-spectrum SPF of at least 30 is the best way to protect yourself from skin cancer. For many people, the chemical formulations cited in the study are the only ones that feel cosmetically elegant enough to wear. Consumers should continue to use their preferred formulation if it means they will actually wear it.”
SOURCE: JAMA. 2019 May 6. doi: 10.1001/jama.2019.5586
It comes as no real surprise that in the wake of the recent FDA proposed rule on sunscreen, which is currently in the “open season” phase for public input, a pilot study supporting said proposal emerges from this very group. We certainly need a watchdog – one that protects us from potentially harmful things in this world. The study presented validates this role. However, let’s not misconstrue what is presented here. In fact, I credit the authors with highlighting a key point in the last sentence of their abstract: These data do not suggest that individuals should refrain from using sunscreen. This paper serves a purpose, which is to support the recommendation to evaluate the potential of these ingredients to penetrate, permeate, and absorb into the systemic circulation. And yes, these data certainly suggest specific filters and formulations can enable systemic absorption, but these findings cannot be correlated to toxicity or pathology.

Let’s critically evaluate what was investigated. The experimental protocol was not exactly realistic, rather, representative of optimal use (2mg/cm2, covering 75% body surface area, four times a day – let’s be real, who actually does that?). The number of those evaluated was low (six per group) and did not account for all skin type and external environments that do play a role in barrier integrity.
While the clinical relevance is unclear, let’s turn to what is not unclear: Ultraviolet radiation causes skin cancer, plain and simple. Therefore, a comprehensive sun protective regimen that includes sunscreen, sun avoidance, and protective clothing is central to prevention. If one is still concerned, there are always mineral sunscreens, zinc and titanium, which the FDA has deemed safe (“generally recognized as safe and effective” or GRASE).
Adam Friedman, MD, is professor and interim chief of dermatology, and director of the supportive oncodermatology clinic, at George Washington University, Washington. He is an advisor and consultant to Aveeno and LaRoche Posay.
It comes as no real surprise that in the wake of the recent FDA proposed rule on sunscreen, which is currently in the “open season” phase for public input, a pilot study supporting said proposal emerges from this very group. We certainly need a watchdog – one that protects us from potentially harmful things in this world. The study presented validates this role. However, let’s not misconstrue what is presented here. In fact, I credit the authors with highlighting a key point in the last sentence of their abstract: These data do not suggest that individuals should refrain from using sunscreen. This paper serves a purpose, which is to support the recommendation to evaluate the potential of these ingredients to penetrate, permeate, and absorb into the systemic circulation. And yes, these data certainly suggest specific filters and formulations can enable systemic absorption, but these findings cannot be correlated to toxicity or pathology.
Let’s critically evaluate what was investigated. The experimental protocol was not exactly realistic, rather, representative of optimal use (2mg/cm2, covering 75% body surface area, four times a day – let’s be real, who actually does that?). The number of those evaluated was low (six per group) and did not account for all skin type and external environments that do play a role in barrier integrity.
While the clinical relevance is unclear, let’s turn to what is not unclear: Ultraviolet radiation causes skin cancer, plain and simple. Therefore, a comprehensive sun protective regimen that includes sunscreen, sun avoidance, and protective clothing is central to prevention. If one is still concerned, there are always mineral sunscreens, zinc and titanium, which the FDA has deemed safe (“generally recognized as safe and effective” or GRASE).
Adam Friedman, MD, is professor and interim chief of dermatology, and director of the supportive oncodermatology clinic, at George Washington University, Washington. He is an advisor and consultant to Aveeno and LaRoche Posay.
It comes as no real surprise that in the wake of the recent FDA proposed rule on sunscreen, which is currently in the “open season” phase for public input, a pilot study supporting said proposal emerges from this very group. We certainly need a watchdog – one that protects us from potentially harmful things in this world. The study presented validates this role. However, let’s not misconstrue what is presented here. In fact, I credit the authors with highlighting a key point in the last sentence of their abstract: These data do not suggest that individuals should refrain from using sunscreen. This paper serves a purpose, which is to support the recommendation to evaluate the potential of these ingredients to penetrate, permeate, and absorb into the systemic circulation. And yes, these data certainly suggest specific filters and formulations can enable systemic absorption, but these findings cannot be correlated to toxicity or pathology.
Let’s critically evaluate what was investigated. The experimental protocol was not exactly realistic, rather, representative of optimal use (2mg/cm2, covering 75% body surface area, four times a day – let’s be real, who actually does that?). The number of those evaluated was low (six per group) and did not account for all skin type and external environments that do play a role in barrier integrity.
While the clinical relevance is unclear, let’s turn to what is not unclear: Ultraviolet radiation causes skin cancer, plain and simple. Therefore, a comprehensive sun protective regimen that includes sunscreen, sun avoidance, and protective clothing is central to prevention. If one is still concerned, there are always mineral sunscreens, zinc and titanium, which the FDA has deemed safe (“generally recognized as safe and effective” or GRASE).
Adam Friedman, MD, is professor and interim chief of dermatology, and director of the supportive oncodermatology clinic, at George Washington University, Washington. He is an advisor and consultant to Aveeno and LaRoche Posay.
for sunscreens, in a phase 1 randomized controlled study of 24 healthy volunteers.
In the open-label study, 24 people (mean age 35.5 years) were randomized to one of four commercially available sunscreens (two sprays, one lotion, or one cream formulation); 2 mg of sunscreen per 1 cm2 was applied to 75% of their body surface four times a day for 4 days (described as “maximal use conditions consistent with current sunscreen labeling”), and 30 blood samples were collected over 7 days.
The primary outcome was the maximum plasma concentration of avobenzone, from days 1-7; secondary outcomes were maximum plasma concentrations of sunscreen ingredients oxybenzone, octocrylene, and ecamsule over the same period of time.
All but one participant completed the study. “All four sunscreen active ingredients tested resulted in exposures exceeding 0.5 ng/mL,” reported Murali Matta, PhD, of the FDA’s Center for Drug Evaluation and Research, and coauthors. “The clinical effect of plasma concentrations exceeding 0.5 ng/mL is unknown, necessitating further research,” they added.
According to the study, FDA sunscreen guidance and the proposed rule for over-the-counter sunscreen monograph, nonclinical toxicology studies, such as carcinogenicity and reproductive studies, “may be waived if results of an adequately conducted human pharmacokinetic maximal usage trial show a steady state blood level less than 0.5 ng/mL and an adequately conducted toxicology assessment does not reveal any potential safety concerns.”
The results of this study “do not indicate that individuals should refrain from the use of sunscreen,” the authors concluded, adding that the “systemic absorption of sunscreen ingredients supports the need for further studies to determine the clinical significance of these findings.” The study was published in JAMA.
In an accompanying editorial, former FDA commissioner Robert Califf, MD, professor of cardiology, Duke University, Durham, N.C., and JAMA Dermatology Editor Kanade Shinkai, MD, PhD, of the department of dermatology, University of California, San Francisco, noted that “the demonstration of systemic absorption well above the FDA guideline does not mean these ingredients are unsafe” (JAMA. 2019 May 6. doi: 10.1001/jama.2019.5528). But, they added, the results “raise many important questions about sunscreen and the process by which the sunscreen industry, clinicians, specialty organizations, and regulatory agencies evaluate the benefits and risks of this topical OTC medication. First and foremost, it is essential to determine whether systemic absorption of sunscreen poses risks to human health. Second, the effects of different sunscreen formulations, clinical characteristics (that is, skin type, age, presence of skin diseases that disrupt the skin barrier), physical activity level, and exposure to sun and water on systemic sunscreen levels require further study.”
In a statement, former American Academy of Dermatology President Darrel Rigel, MD, of the department of dermatology, New York University, said that he was concerned that the results were misleading. “We have always known that there is a very small amount of absorption of sunscreens in the bloodstream,” and there are no data that this is a problem, he said, adding: “Tens of millions of people use sunscreens in the U.S. every summer weekend for many years with no incidence. Daily use of a broad-spectrum SPF of at least 30 is the best way to protect yourself from skin cancer. For many people, the chemical formulations cited in the study are the only ones that feel cosmetically elegant enough to wear. Consumers should continue to use their preferred formulation if it means they will actually wear it.”
SOURCE: JAMA. 2019 May 6. doi: 10.1001/jama.2019.5586
for sunscreens, in a phase 1 randomized controlled study of 24 healthy volunteers.
In the open-label study, 24 people (mean age 35.5 years) were randomized to one of four commercially available sunscreens (two sprays, one lotion, or one cream formulation); 2 mg of sunscreen per 1 cm2 was applied to 75% of their body surface four times a day for 4 days (described as “maximal use conditions consistent with current sunscreen labeling”), and 30 blood samples were collected over 7 days.
The primary outcome was the maximum plasma concentration of avobenzone, from days 1-7; secondary outcomes were maximum plasma concentrations of sunscreen ingredients oxybenzone, octocrylene, and ecamsule over the same period of time.
All but one participant completed the study. “All four sunscreen active ingredients tested resulted in exposures exceeding 0.5 ng/mL,” reported Murali Matta, PhD, of the FDA’s Center for Drug Evaluation and Research, and coauthors. “The clinical effect of plasma concentrations exceeding 0.5 ng/mL is unknown, necessitating further research,” they added.
According to the study, FDA sunscreen guidance and the proposed rule for over-the-counter sunscreen monograph, nonclinical toxicology studies, such as carcinogenicity and reproductive studies, “may be waived if results of an adequately conducted human pharmacokinetic maximal usage trial show a steady state blood level less than 0.5 ng/mL and an adequately conducted toxicology assessment does not reveal any potential safety concerns.”
The results of this study “do not indicate that individuals should refrain from the use of sunscreen,” the authors concluded, adding that the “systemic absorption of sunscreen ingredients supports the need for further studies to determine the clinical significance of these findings.” The study was published in JAMA.
In an accompanying editorial, former FDA commissioner Robert Califf, MD, professor of cardiology, Duke University, Durham, N.C., and JAMA Dermatology Editor Kanade Shinkai, MD, PhD, of the department of dermatology, University of California, San Francisco, noted that “the demonstration of systemic absorption well above the FDA guideline does not mean these ingredients are unsafe” (JAMA. 2019 May 6. doi: 10.1001/jama.2019.5528). But, they added, the results “raise many important questions about sunscreen and the process by which the sunscreen industry, clinicians, specialty organizations, and regulatory agencies evaluate the benefits and risks of this topical OTC medication. First and foremost, it is essential to determine whether systemic absorption of sunscreen poses risks to human health. Second, the effects of different sunscreen formulations, clinical characteristics (that is, skin type, age, presence of skin diseases that disrupt the skin barrier), physical activity level, and exposure to sun and water on systemic sunscreen levels require further study.”
In a statement, former American Academy of Dermatology President Darrel Rigel, MD, of the department of dermatology, New York University, said that he was concerned that the results were misleading. “We have always known that there is a very small amount of absorption of sunscreens in the bloodstream,” and there are no data that this is a problem, he said, adding: “Tens of millions of people use sunscreens in the U.S. every summer weekend for many years with no incidence. Daily use of a broad-spectrum SPF of at least 30 is the best way to protect yourself from skin cancer. For many people, the chemical formulations cited in the study are the only ones that feel cosmetically elegant enough to wear. Consumers should continue to use their preferred formulation if it means they will actually wear it.”
SOURCE: JAMA. 2019 May 6. doi: 10.1001/jama.2019.5586
FROM JAMA
The 39th ASLMS meeting is now underway
WASHINGTON – At the annual meeting of the American Academy of Dermatology, the taking place March 27-31, 2019, in Denver.

“ASLMS is always an amazing meeting, and it’s a unique meeting,” said past president Mathew Avram, MD, director of the Dermatology Laser & Cosmetic Center at Massachusetts General Hospital, Boston. “At its core, it’s a scientific meeting ... you can take things back to your practice that change the practice of medicine.”
Current ASLMS president Eric Bernstein, MD, of Main Line Center for Laser Surgery, Ardmore, Pa., pointed out that, in addition to doctors and other health care practitioners, other available and accessible attendees include the engineers who build the lasers. And this year, injectables are being incorporated into the program.
MDedge reporter Doug Brunk will be reporting from the meeting.
WASHINGTON – At the annual meeting of the American Academy of Dermatology, the taking place March 27-31, 2019, in Denver.
“ASLMS is always an amazing meeting, and it’s a unique meeting,” said past president Mathew Avram, MD, director of the Dermatology Laser & Cosmetic Center at Massachusetts General Hospital, Boston. “At its core, it’s a scientific meeting ... you can take things back to your practice that change the practice of medicine.”
Current ASLMS president Eric Bernstein, MD, of Main Line Center for Laser Surgery, Ardmore, Pa., pointed out that, in addition to doctors and other health care practitioners, other available and accessible attendees include the engineers who build the lasers. And this year, injectables are being incorporated into the program.
MDedge reporter Doug Brunk will be reporting from the meeting.
WASHINGTON – At the annual meeting of the American Academy of Dermatology, the taking place March 27-31, 2019, in Denver.
“ASLMS is always an amazing meeting, and it’s a unique meeting,” said past president Mathew Avram, MD, director of the Dermatology Laser & Cosmetic Center at Massachusetts General Hospital, Boston. “At its core, it’s a scientific meeting ... you can take things back to your practice that change the practice of medicine.”
Current ASLMS president Eric Bernstein, MD, of Main Line Center for Laser Surgery, Ardmore, Pa., pointed out that, in addition to doctors and other health care practitioners, other available and accessible attendees include the engineers who build the lasers. And this year, injectables are being incorporated into the program.
MDedge reporter Doug Brunk will be reporting from the meeting.
REPORTING FROM AAD 2019

VIDEO: Immunomodulators for inflammatory skin diseases
WASHINGTON – During a session at the annual meeting of the American Academy of Dermatology, Adam Friedman, MD, presented on off-label use of immunomodulators for inflammatory skin diseases, the highlights of which he shared with fellow George Washington University dermatologist, A. Yasmine Kirkorian, MD, in an interview following the session.

Dr. Friedman, professor and interim chair of dermatology at George Washington University, Washington,
For example, as reflected in PubMed searches, low-dose naltrexone, which has to be compounded, is being used for such diseases as Hailey-Hailey and lichen planopilaris, said Dr. Friedman, who is using it for his mast cell activation syndrome patients. During the interview, he also describes his treatment approach for urticaria.
In his final remarks, Dr. Friedman encourages colleagues to “get creative,” publish, and talk about their experiences with off-label treatments in dermatology, citing the example of an article that mentioned using pioglitazone for lichen planopilaris. This article stimulated interest in using the type 2 diabetes agent pioglitazone to treat this skin disease, he notes.
Dr. Friedman and Dr. Kirkorian, a pediatric dermatologist at George Washington University and interim chief of pediatric dermatology at Children’s National in Washington had no relevant disclosures.
WASHINGTON – During a session at the annual meeting of the American Academy of Dermatology, Adam Friedman, MD, presented on off-label use of immunomodulators for inflammatory skin diseases, the highlights of which he shared with fellow George Washington University dermatologist, A. Yasmine Kirkorian, MD, in an interview following the session.
Dr. Friedman, professor and interim chair of dermatology at George Washington University, Washington,
For example, as reflected in PubMed searches, low-dose naltrexone, which has to be compounded, is being used for such diseases as Hailey-Hailey and lichen planopilaris, said Dr. Friedman, who is using it for his mast cell activation syndrome patients. During the interview, he also describes his treatment approach for urticaria.
In his final remarks, Dr. Friedman encourages colleagues to “get creative,” publish, and talk about their experiences with off-label treatments in dermatology, citing the example of an article that mentioned using pioglitazone for lichen planopilaris. This article stimulated interest in using the type 2 diabetes agent pioglitazone to treat this skin disease, he notes.
Dr. Friedman and Dr. Kirkorian, a pediatric dermatologist at George Washington University and interim chief of pediatric dermatology at Children’s National in Washington had no relevant disclosures.
WASHINGTON – During a session at the annual meeting of the American Academy of Dermatology, Adam Friedman, MD, presented on off-label use of immunomodulators for inflammatory skin diseases, the highlights of which he shared with fellow George Washington University dermatologist, A. Yasmine Kirkorian, MD, in an interview following the session.
Dr. Friedman, professor and interim chair of dermatology at George Washington University, Washington,
For example, as reflected in PubMed searches, low-dose naltrexone, which has to be compounded, is being used for such diseases as Hailey-Hailey and lichen planopilaris, said Dr. Friedman, who is using it for his mast cell activation syndrome patients. During the interview, he also describes his treatment approach for urticaria.
In his final remarks, Dr. Friedman encourages colleagues to “get creative,” publish, and talk about their experiences with off-label treatments in dermatology, citing the example of an article that mentioned using pioglitazone for lichen planopilaris. This article stimulated interest in using the type 2 diabetes agent pioglitazone to treat this skin disease, he notes.
Dr. Friedman and Dr. Kirkorian, a pediatric dermatologist at George Washington University and interim chief of pediatric dermatology at Children’s National in Washington had no relevant disclosures.

Food allergies and atopic dermatitis: What is the evidence?
WASHINGTON – The and brings up an interesting discussion, Peter Lio, MD, said in a video interview at the annual meeting of the American Academy of Dermatology.
Patients are often convinced that food is the main driver of their AD, or parents believe it is a trigger in their children with AD, according to Dr. Lio of the departments of dermatology and pediatrics at Northwestern University, Chicago.

There is an increased risk of true food allergy in patients with AD, which “seems to get worse with increasing severity of the disease,” he pointed out. However, he added, many patients believe that food allergy is what is driving their eczema, “and that’s the part we don’t really think bears out” in clinical trials.
In the interview, Dr. Lio reviewed some of the clinical trial data and discussed other issues, including foods that seem to have an inflammatory effect in the body, the concepts of “transcutaneous sensitization” in children with AD and the “leaky gut,” and why he tends to recommend probiotics for patients with AD.
Dr. Lio spoke on diet and AD during a session titled “Dietary Triggers and Modifications of Common Dermatologic Conditions – An Evidence Based Approach,” at the meeting, He had no relevant disclosures.
WASHINGTON – The and brings up an interesting discussion, Peter Lio, MD, said in a video interview at the annual meeting of the American Academy of Dermatology.
Patients are often convinced that food is the main driver of their AD, or parents believe it is a trigger in their children with AD, according to Dr. Lio of the departments of dermatology and pediatrics at Northwestern University, Chicago.
There is an increased risk of true food allergy in patients with AD, which “seems to get worse with increasing severity of the disease,” he pointed out. However, he added, many patients believe that food allergy is what is driving their eczema, “and that’s the part we don’t really think bears out” in clinical trials.
In the interview, Dr. Lio reviewed some of the clinical trial data and discussed other issues, including foods that seem to have an inflammatory effect in the body, the concepts of “transcutaneous sensitization” in children with AD and the “leaky gut,” and why he tends to recommend probiotics for patients with AD.
Dr. Lio spoke on diet and AD during a session titled “Dietary Triggers and Modifications of Common Dermatologic Conditions – An Evidence Based Approach,” at the meeting, He had no relevant disclosures.
WASHINGTON – The and brings up an interesting discussion, Peter Lio, MD, said in a video interview at the annual meeting of the American Academy of Dermatology.
Patients are often convinced that food is the main driver of their AD, or parents believe it is a trigger in their children with AD, according to Dr. Lio of the departments of dermatology and pediatrics at Northwestern University, Chicago.
There is an increased risk of true food allergy in patients with AD, which “seems to get worse with increasing severity of the disease,” he pointed out. However, he added, many patients believe that food allergy is what is driving their eczema, “and that’s the part we don’t really think bears out” in clinical trials.
In the interview, Dr. Lio reviewed some of the clinical trial data and discussed other issues, including foods that seem to have an inflammatory effect in the body, the concepts of “transcutaneous sensitization” in children with AD and the “leaky gut,” and why he tends to recommend probiotics for patients with AD.
Dr. Lio spoke on diet and AD during a session titled “Dietary Triggers and Modifications of Common Dermatologic Conditions – An Evidence Based Approach,” at the meeting, He had no relevant disclosures.
REPORTING FROM AAD 2019
FDA approves new treatment for hereditary transthyretin-mediated amyloidosis
, the second treatment approved in 2 months for the rare genetic disorder.

Inotersen is a transthyretin-directed antisense oligonucleotide that inhibits production of the transthyretin (TTR) protein (amyloid), according to the FDA statement announcing the approval on Oct. 5. It is administered in a subcutaneous injection once a week and will be marketed as Tegsedi. In August, the FDA approved the first treatment for hATTR, patisiran (Onpattro).
The FDA statement described hATTR as a “rare, debilitating, and often fatal genetic disease characterized by the buildup of abnormal amyloid protein in peripheral nerves, the heart, and other organs,” with signs and symptoms that include effects on “sensation (pain and temperature), autonomic function (blood pressure changes and bowel and digestive problems), and muscle strength (weakness and immobility in the arms, legs, hands, and feet),” caused by amyloid deposition in the peripheral nervous system.
The mechanism-of-action section of the prescribing information states that inotersen “causes degradation of mutant and wild-type TTR mRNA through binding to the TTR mRNA, which results in a reduction of serum TTR protein and TTR protein deposits in tissues.”
A statement issued by the manufacturer, Ionis Pharmaceuticals, said that approval was based on a phase 3 study of patients with hATTR amyloidosis and polyneuropathy symptoms, which found significantly greater benefits among those treated with inotersen, compared with placebo, using primary endpoints that measured quality of life related to neuropathy and a measure of neuropathic disease progression.
Contraindications to inotersen include a history of acute glomerulonephritis caused by inotersen, history of a hypersensitivity reaction to the agent, and a platelet count below 100 × 109/L; the label includes a boxed warning about thrombocytopenia and glomerulonephritis associated with treatment. The drug also is being marketed with a risk evaluation and mitigation strategy (REMS).
Patisiran, the treatment approved in August, results in “degradation of mutant and wild-type TTR mRNA through RNA interference, which results in a reduction of serum TTR protein and TTR protein deposits in tissues,” according to its prescribing information.
, the second treatment approved in 2 months for the rare genetic disorder.
Inotersen is a transthyretin-directed antisense oligonucleotide that inhibits production of the transthyretin (TTR) protein (amyloid), according to the FDA statement announcing the approval on Oct. 5. It is administered in a subcutaneous injection once a week and will be marketed as Tegsedi. In August, the FDA approved the first treatment for hATTR, patisiran (Onpattro).
The FDA statement described hATTR as a “rare, debilitating, and often fatal genetic disease characterized by the buildup of abnormal amyloid protein in peripheral nerves, the heart, and other organs,” with signs and symptoms that include effects on “sensation (pain and temperature), autonomic function (blood pressure changes and bowel and digestive problems), and muscle strength (weakness and immobility in the arms, legs, hands, and feet),” caused by amyloid deposition in the peripheral nervous system.
The mechanism-of-action section of the prescribing information states that inotersen “causes degradation of mutant and wild-type TTR mRNA through binding to the TTR mRNA, which results in a reduction of serum TTR protein and TTR protein deposits in tissues.”
A statement issued by the manufacturer, Ionis Pharmaceuticals, said that approval was based on a phase 3 study of patients with hATTR amyloidosis and polyneuropathy symptoms, which found significantly greater benefits among those treated with inotersen, compared with placebo, using primary endpoints that measured quality of life related to neuropathy and a measure of neuropathic disease progression.
Contraindications to inotersen include a history of acute glomerulonephritis caused by inotersen, history of a hypersensitivity reaction to the agent, and a platelet count below 100 × 109/L; the label includes a boxed warning about thrombocytopenia and glomerulonephritis associated with treatment. The drug also is being marketed with a risk evaluation and mitigation strategy (REMS).
Patisiran, the treatment approved in August, results in “degradation of mutant and wild-type TTR mRNA through RNA interference, which results in a reduction of serum TTR protein and TTR protein deposits in tissues,” according to its prescribing information.
, the second treatment approved in 2 months for the rare genetic disorder.
Inotersen is a transthyretin-directed antisense oligonucleotide that inhibits production of the transthyretin (TTR) protein (amyloid), according to the FDA statement announcing the approval on Oct. 5. It is administered in a subcutaneous injection once a week and will be marketed as Tegsedi. In August, the FDA approved the first treatment for hATTR, patisiran (Onpattro).
The FDA statement described hATTR as a “rare, debilitating, and often fatal genetic disease characterized by the buildup of abnormal amyloid protein in peripheral nerves, the heart, and other organs,” with signs and symptoms that include effects on “sensation (pain and temperature), autonomic function (blood pressure changes and bowel and digestive problems), and muscle strength (weakness and immobility in the arms, legs, hands, and feet),” caused by amyloid deposition in the peripheral nervous system.
The mechanism-of-action section of the prescribing information states that inotersen “causes degradation of mutant and wild-type TTR mRNA through binding to the TTR mRNA, which results in a reduction of serum TTR protein and TTR protein deposits in tissues.”
A statement issued by the manufacturer, Ionis Pharmaceuticals, said that approval was based on a phase 3 study of patients with hATTR amyloidosis and polyneuropathy symptoms, which found significantly greater benefits among those treated with inotersen, compared with placebo, using primary endpoints that measured quality of life related to neuropathy and a measure of neuropathic disease progression.
Contraindications to inotersen include a history of acute glomerulonephritis caused by inotersen, history of a hypersensitivity reaction to the agent, and a platelet count below 100 × 109/L; the label includes a boxed warning about thrombocytopenia and glomerulonephritis associated with treatment. The drug also is being marketed with a risk evaluation and mitigation strategy (REMS).
Patisiran, the treatment approved in August, results in “degradation of mutant and wild-type TTR mRNA through RNA interference, which results in a reduction of serum TTR protein and TTR protein deposits in tissues,” according to its prescribing information.
FDA expands approval of 9-valent HPV vaccine
The , men and women aged 27-45 years, the Food and Drug Administration announced on Oct. 5.
The vaccine (Gardasil 9) was previously approved for those aged 9-26 years.
The approval “represents an important opportunity to help prevent HPV-related diseases and cancers in a broader age range,” Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research, said in the FDA statement announcing the approval.
“The Centers for Disease Control and Prevention has stated that HPV vaccination prior to becoming infected with the HPV types covered by the vaccine has the potential to prevent more than 90 percent of these cancers, or 31,200 cases every year, from ever developing,” he added.
Gardasil 9, approved in 2014, covers the four HPV types included in the original Gardasil vaccine approved in 2006, plus five additional HPV types.
The approval is based on the results of a study and follow-up of about 3,200 women aged 27-45 years, followed for an average of 3.5 years, which found that the vaccine was 88% percent effective “in the prevention of a combined endpoint of persistent infection, genital warts, vulvar and vaginal precancerous lesions, cervical precancerous lesions, and cervical cancer related to HPV types covered by the vaccine,” according to the FDA. The vaccine’s effectiveness in men in this age group is “inferred” from these results and from data on Gardasil in men aged 16-26 years, as well as “immunogenicity data from a clinical trial in which 150 men, 27 through 45 years of age, received a 3-dose regimen of Gardasil over 6 months,” the FDA statement noted.
Based on safety data in about 13,000 men and women, injection-site pain, swelling, redness, and headaches are the most common adverse reactions associated with Gardasil 9, the statement said. Gardasil 9 is manufactured by Merck.
The , men and women aged 27-45 years, the Food and Drug Administration announced on Oct. 5.
The vaccine (Gardasil 9) was previously approved for those aged 9-26 years.
The approval “represents an important opportunity to help prevent HPV-related diseases and cancers in a broader age range,” Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research, said in the FDA statement announcing the approval.
“The Centers for Disease Control and Prevention has stated that HPV vaccination prior to becoming infected with the HPV types covered by the vaccine has the potential to prevent more than 90 percent of these cancers, or 31,200 cases every year, from ever developing,” he added.
Gardasil 9, approved in 2014, covers the four HPV types included in the original Gardasil vaccine approved in 2006, plus five additional HPV types.
The approval is based on the results of a study and follow-up of about 3,200 women aged 27-45 years, followed for an average of 3.5 years, which found that the vaccine was 88% percent effective “in the prevention of a combined endpoint of persistent infection, genital warts, vulvar and vaginal precancerous lesions, cervical precancerous lesions, and cervical cancer related to HPV types covered by the vaccine,” according to the FDA. The vaccine’s effectiveness in men in this age group is “inferred” from these results and from data on Gardasil in men aged 16-26 years, as well as “immunogenicity data from a clinical trial in which 150 men, 27 through 45 years of age, received a 3-dose regimen of Gardasil over 6 months,” the FDA statement noted.
Based on safety data in about 13,000 men and women, injection-site pain, swelling, redness, and headaches are the most common adverse reactions associated with Gardasil 9, the statement said. Gardasil 9 is manufactured by Merck.
The , men and women aged 27-45 years, the Food and Drug Administration announced on Oct. 5.
The vaccine (Gardasil 9) was previously approved for those aged 9-26 years.
The approval “represents an important opportunity to help prevent HPV-related diseases and cancers in a broader age range,” Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research, said in the FDA statement announcing the approval.
“The Centers for Disease Control and Prevention has stated that HPV vaccination prior to becoming infected with the HPV types covered by the vaccine has the potential to prevent more than 90 percent of these cancers, or 31,200 cases every year, from ever developing,” he added.
Gardasil 9, approved in 2014, covers the four HPV types included in the original Gardasil vaccine approved in 2006, plus five additional HPV types.
The approval is based on the results of a study and follow-up of about 3,200 women aged 27-45 years, followed for an average of 3.5 years, which found that the vaccine was 88% percent effective “in the prevention of a combined endpoint of persistent infection, genital warts, vulvar and vaginal precancerous lesions, cervical precancerous lesions, and cervical cancer related to HPV types covered by the vaccine,” according to the FDA. The vaccine’s effectiveness in men in this age group is “inferred” from these results and from data on Gardasil in men aged 16-26 years, as well as “immunogenicity data from a clinical trial in which 150 men, 27 through 45 years of age, received a 3-dose regimen of Gardasil over 6 months,” the FDA statement noted.
Based on safety data in about 13,000 men and women, injection-site pain, swelling, redness, and headaches are the most common adverse reactions associated with Gardasil 9, the statement said. Gardasil 9 is manufactured by Merck.
Bertilimumab granted orphan drug status for bullous pemphigoid
The , according to an announcement issued by the company that is developing the treatment.
The company, Immune Pharmaceuticals, plans to launch a pivotal phase 2/3 study in 2019, the company said in an Aug. 20 press release. Two phase 2 studies of bertilimumab are currently underway, one in patients with bullous pemphigoid, and another in patients with ulcerative colitis. Bertilimumab is “a first-in-class, fully human monoclonal antibody that targets and lowers levels of eotaxin-1, a chemokine that plays a role in immune responses and attracts eosinophils to the site of inflammation,” the statement said.“By neutralizing eotaxin-1, bertilimumab may prevent the migration of eosinophils and other cells, thus helping to relieve associated inflammatory conditions.”
The FDA’s Orphan Drug Designation program grants orphan status to drugs and biologics, which are “defined as those intended for the safe and effective treatment, diagnosis or prevention of rare diseases/disorders that affect fewer than 200,000 people in the United States, or that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug,” according to the agency.
The , according to an announcement issued by the company that is developing the treatment.
The company, Immune Pharmaceuticals, plans to launch a pivotal phase 2/3 study in 2019, the company said in an Aug. 20 press release. Two phase 2 studies of bertilimumab are currently underway, one in patients with bullous pemphigoid, and another in patients with ulcerative colitis. Bertilimumab is “a first-in-class, fully human monoclonal antibody that targets and lowers levels of eotaxin-1, a chemokine that plays a role in immune responses and attracts eosinophils to the site of inflammation,” the statement said.“By neutralizing eotaxin-1, bertilimumab may prevent the migration of eosinophils and other cells, thus helping to relieve associated inflammatory conditions.”
The FDA’s Orphan Drug Designation program grants orphan status to drugs and biologics, which are “defined as those intended for the safe and effective treatment, diagnosis or prevention of rare diseases/disorders that affect fewer than 200,000 people in the United States, or that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug,” according to the agency.
The , according to an announcement issued by the company that is developing the treatment.
The company, Immune Pharmaceuticals, plans to launch a pivotal phase 2/3 study in 2019, the company said in an Aug. 20 press release. Two phase 2 studies of bertilimumab are currently underway, one in patients with bullous pemphigoid, and another in patients with ulcerative colitis. Bertilimumab is “a first-in-class, fully human monoclonal antibody that targets and lowers levels of eotaxin-1, a chemokine that plays a role in immune responses and attracts eosinophils to the site of inflammation,” the statement said.“By neutralizing eotaxin-1, bertilimumab may prevent the migration of eosinophils and other cells, thus helping to relieve associated inflammatory conditions.”
The FDA’s Orphan Drug Designation program grants orphan status to drugs and biologics, which are “defined as those intended for the safe and effective treatment, diagnosis or prevention of rare diseases/disorders that affect fewer than 200,000 people in the United States, or that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug,” according to the agency.
CUBE-C initiative aims to educate about atopic dermatitis
WASHINGTON – The National Eczema Association (NEA) has established the Coalition United for Better Eczema Care (CUBE-C) to provide practitioners with a resource for “trustworthy, up-to-date, state of the art” information on atopic dermatitis, with the goal of improving health outcomes, according to Julie Block, president and chief executive officer of the NEA.
In an interview at a symposium presented by CUBE-C, Ms. Block provided more information on CUBE-C, including how and why it started and what it can offer to dermatologists, as well as primary care physicians, who care for patients with atopic dermatitis. She said that the NEA convened dermatologists, allergists, immunologists, psychologists, nurse practitioners, physician assistants, and patients “to design a curriculum that provided an entire picture of the patient experience, so that we could go out and educate not only on the basics of eczema and atopic dermatitis for a variety of practitioners ... but also for the specialists who are now going to be engaging in new innovations and new therapies for their patients.”
She was joined by Adam Friedman, MD, professor of dermatology and residency program director at George Washington University, Washington, where the symposium, a resident’s boot camp, was held. The boot camp was somewhat unique in that it was geared more towards trainees; typically, the CUBE-C program is a CME program for practitioners, but this reflects the flexibility of the program, which can be tailored to the audience, Dr. Friedman pointed out. “The hope is that programs like these pop up all over the place ... anywhere you have a critical mass of individuals who want to learn about this,” where planners can choose from a menu of topics provided by CUBE-C – which include therapeutics, infections, pathogenesis, and access to care – and “easily formulate a conference like we held here today for the right audience.”
Topics covered at the George Washington University symposium included the impact of climate on the prevalence of childhood eczema, the diagnosis and differential diagnosis in children, infections in atopic dermatitis patients, and itch treatment.
More information on CUBE-C is available on the NEA website.
The symposium was supported by an educational grant from Sanofi Genzyme, Regeneron Pharmaceuticals, and Pfizer. Dr. Friedman reported serving as a speaker for Regeneron, Pfizer, and other companies. He also consults and serves on the advisory board for Pfizer and multiple other companies developing and marketing atopic dermatitis therapies and products.
WASHINGTON – The National Eczema Association (NEA) has established the Coalition United for Better Eczema Care (CUBE-C) to provide practitioners with a resource for “trustworthy, up-to-date, state of the art” information on atopic dermatitis, with the goal of improving health outcomes, according to Julie Block, president and chief executive officer of the NEA.
In an interview at a symposium presented by CUBE-C, Ms. Block provided more information on CUBE-C, including how and why it started and what it can offer to dermatologists, as well as primary care physicians, who care for patients with atopic dermatitis. She said that the NEA convened dermatologists, allergists, immunologists, psychologists, nurse practitioners, physician assistants, and patients “to design a curriculum that provided an entire picture of the patient experience, so that we could go out and educate not only on the basics of eczema and atopic dermatitis for a variety of practitioners ... but also for the specialists who are now going to be engaging in new innovations and new therapies for their patients.”
She was joined by Adam Friedman, MD, professor of dermatology and residency program director at George Washington University, Washington, where the symposium, a resident’s boot camp, was held. The boot camp was somewhat unique in that it was geared more towards trainees; typically, the CUBE-C program is a CME program for practitioners, but this reflects the flexibility of the program, which can be tailored to the audience, Dr. Friedman pointed out. “The hope is that programs like these pop up all over the place ... anywhere you have a critical mass of individuals who want to learn about this,” where planners can choose from a menu of topics provided by CUBE-C – which include therapeutics, infections, pathogenesis, and access to care – and “easily formulate a conference like we held here today for the right audience.”
Topics covered at the George Washington University symposium included the impact of climate on the prevalence of childhood eczema, the diagnosis and differential diagnosis in children, infections in atopic dermatitis patients, and itch treatment.
More information on CUBE-C is available on the NEA website.
The symposium was supported by an educational grant from Sanofi Genzyme, Regeneron Pharmaceuticals, and Pfizer. Dr. Friedman reported serving as a speaker for Regeneron, Pfizer, and other companies. He also consults and serves on the advisory board for Pfizer and multiple other companies developing and marketing atopic dermatitis therapies and products.
WASHINGTON – The National Eczema Association (NEA) has established the Coalition United for Better Eczema Care (CUBE-C) to provide practitioners with a resource for “trustworthy, up-to-date, state of the art” information on atopic dermatitis, with the goal of improving health outcomes, according to Julie Block, president and chief executive officer of the NEA.
In an interview at a symposium presented by CUBE-C, Ms. Block provided more information on CUBE-C, including how and why it started and what it can offer to dermatologists, as well as primary care physicians, who care for patients with atopic dermatitis. She said that the NEA convened dermatologists, allergists, immunologists, psychologists, nurse practitioners, physician assistants, and patients “to design a curriculum that provided an entire picture of the patient experience, so that we could go out and educate not only on the basics of eczema and atopic dermatitis for a variety of practitioners ... but also for the specialists who are now going to be engaging in new innovations and new therapies for their patients.”
She was joined by Adam Friedman, MD, professor of dermatology and residency program director at George Washington University, Washington, where the symposium, a resident’s boot camp, was held. The boot camp was somewhat unique in that it was geared more towards trainees; typically, the CUBE-C program is a CME program for practitioners, but this reflects the flexibility of the program, which can be tailored to the audience, Dr. Friedman pointed out. “The hope is that programs like these pop up all over the place ... anywhere you have a critical mass of individuals who want to learn about this,” where planners can choose from a menu of topics provided by CUBE-C – which include therapeutics, infections, pathogenesis, and access to care – and “easily formulate a conference like we held here today for the right audience.”
Topics covered at the George Washington University symposium included the impact of climate on the prevalence of childhood eczema, the diagnosis and differential diagnosis in children, infections in atopic dermatitis patients, and itch treatment.
More information on CUBE-C is available on the NEA website.
The symposium was supported by an educational grant from Sanofi Genzyme, Regeneron Pharmaceuticals, and Pfizer. Dr. Friedman reported serving as a speaker for Regeneron, Pfizer, and other companies. He also consults and serves on the advisory board for Pfizer and multiple other companies developing and marketing atopic dermatitis therapies and products.

SPOTme addresses unmet need for skin cancer screening
Almost half of the individuals diagnosed with melanoma in a free skin cancer screening program otherwise would not have gone to a doctor to have their skin examined, according to an analysis of the American Academy of Dermatology’s national skin cancer screening program, during 1986-2014.
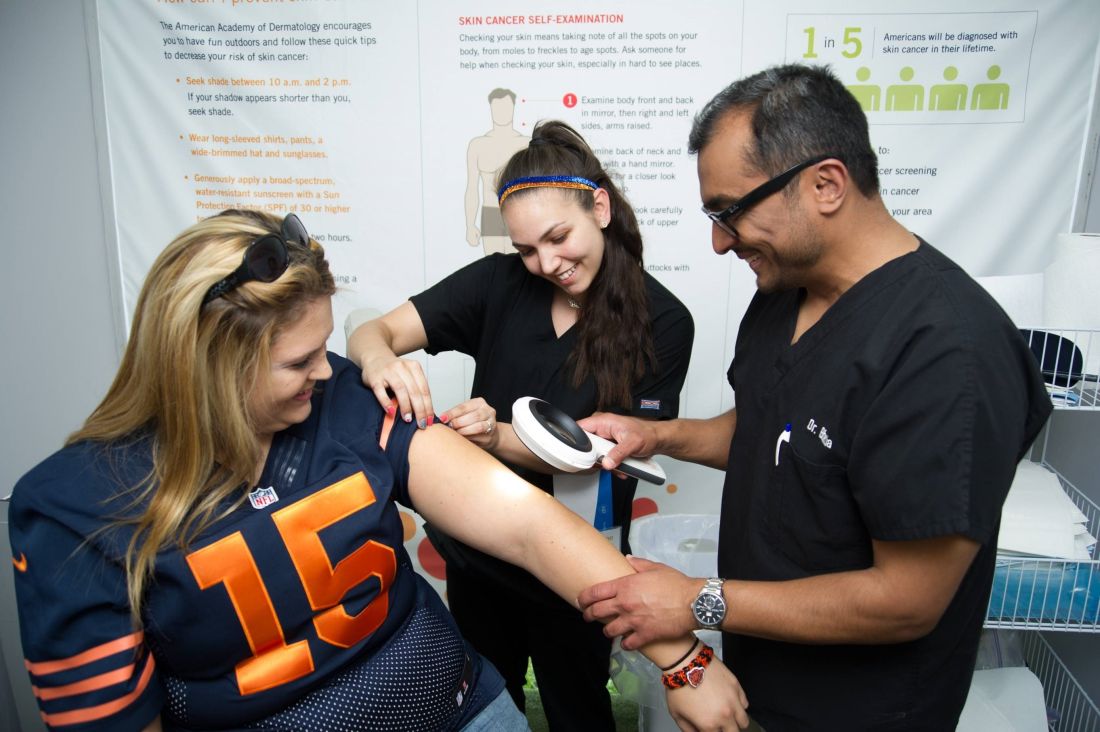
The SPOTme program, a national skin cancer screening and education program conducted by volunteer dermatologists, was launched in 1985. More than 2 million free screenings have been provided by the program in a “predominantly high-risk population, rendering important clinical diagnoses for hundreds of thousands of participants,” according to first authors Jean-Phillip Okhovat, MD, of Beth Israel Deaconess Medical Center, and Derek Beaulieu, MD, of Tufts University, both in Boston, and their colleagues.
The analysis was published online in the Journal of the American Academy of Dermatology on July 26.
Their study analyzed data on almost 2 million people screened through the program from 1986-2014. About 62% were women; 90% were white, about 2% were black, and almost 4% were Hispanic. Almost 80% had no regular dermatologist, almost 73% had not been screened previously, almost 45% had never had a skin cancer check, and 9% were uninsured. Almost 31% reported a mole that had recently change in size, color, or shape; almost 34% said they had a family history of skin cancer, and about 14% said they had a personal history of skin cancer.
Participants were asked about demographics and risk factors, although some questions changed from year to year (for example, in 2009 and 2010, participants were asked about melanoma risk factors, and from 1992 through 2010, participants were asked about their access to dermatologic care).
During 1991-2014 (which did not include data for 1995, 1996, and 2000, which were not available), the screening program resulted in 20,628 clinical melanoma diagnoses, 156,087 clinical dysplastic nevi diagnoses, 32,893 clinical squamous cell carcinoma diagnoses, and 129,848 clinical basal cell carcinoma diagnoses.
Of those clinically diagnosed with melanoma during 1992-2010, 83% said they did not have a regular dermatologist, 77% said they had not been screened previously, and 47% said they would not have seen a doctor for a skin exam if the SPOTme program had not been available.
Of those screened in 2009 and 2010 , 72% were considered at high risk for melanoma (older than age 65 years, having a history of sunburns, a family history of skin cancer, and/or more than 50 moles or unusual moles).
Among the other findings was that from 1992 to 2010, about 12% of those with a clinical melanoma diagnosis were not insured, which increased over time, from almost 11% during 1992-1999 to almost 16% during 2007-2010.
The “consistently high rates” of multiple skin cancer risk factors among those newly screened in the study are consistent with previously reported data, “suggesting that there is an untapped pool of at-risk Americans who have yet to be screened for skin cancer,” the authors wrote. “While the SPOTme program cannot be expected to meet the demands of this larger at-risk population, increased publicity and educational campaigns led by the AAD and assistance to primary care physicians in triaging of patients who should be seen by dermatologists could decrease the number of Americans who need to be screened,” they added.
Limitations of the study included the inability to confirm the clinical diagnoses with histopathology, and no data from the providers were available.
The authors had no disclosures. SPOTme, part of the AAD’s SPOT Skin Cancer initiative, is supported by a grant from Bristol-Myers Squibb.
SOURCE: Okhovat JP et al. J Am Acad Dermatol. https://doi./org/10.1016.j.jaad.2018.05.1242.
Almost half of the individuals diagnosed with melanoma in a free skin cancer screening program otherwise would not have gone to a doctor to have their skin examined, according to an analysis of the American Academy of Dermatology’s national skin cancer screening program, during 1986-2014.
The SPOTme program, a national skin cancer screening and education program conducted by volunteer dermatologists, was launched in 1985. More than 2 million free screenings have been provided by the program in a “predominantly high-risk population, rendering important clinical diagnoses for hundreds of thousands of participants,” according to first authors Jean-Phillip Okhovat, MD, of Beth Israel Deaconess Medical Center, and Derek Beaulieu, MD, of Tufts University, both in Boston, and their colleagues.
The analysis was published online in the Journal of the American Academy of Dermatology on July 26.
Their study analyzed data on almost 2 million people screened through the program from 1986-2014. About 62% were women; 90% were white, about 2% were black, and almost 4% were Hispanic. Almost 80% had no regular dermatologist, almost 73% had not been screened previously, almost 45% had never had a skin cancer check, and 9% were uninsured. Almost 31% reported a mole that had recently change in size, color, or shape; almost 34% said they had a family history of skin cancer, and about 14% said they had a personal history of skin cancer.
Participants were asked about demographics and risk factors, although some questions changed from year to year (for example, in 2009 and 2010, participants were asked about melanoma risk factors, and from 1992 through 2010, participants were asked about their access to dermatologic care).
During 1991-2014 (which did not include data for 1995, 1996, and 2000, which were not available), the screening program resulted in 20,628 clinical melanoma diagnoses, 156,087 clinical dysplastic nevi diagnoses, 32,893 clinical squamous cell carcinoma diagnoses, and 129,848 clinical basal cell carcinoma diagnoses.
Of those clinically diagnosed with melanoma during 1992-2010, 83% said they did not have a regular dermatologist, 77% said they had not been screened previously, and 47% said they would not have seen a doctor for a skin exam if the SPOTme program had not been available.
Of those screened in 2009 and 2010 , 72% were considered at high risk for melanoma (older than age 65 years, having a history of sunburns, a family history of skin cancer, and/or more than 50 moles or unusual moles).
Among the other findings was that from 1992 to 2010, about 12% of those with a clinical melanoma diagnosis were not insured, which increased over time, from almost 11% during 1992-1999 to almost 16% during 2007-2010.
The “consistently high rates” of multiple skin cancer risk factors among those newly screened in the study are consistent with previously reported data, “suggesting that there is an untapped pool of at-risk Americans who have yet to be screened for skin cancer,” the authors wrote. “While the SPOTme program cannot be expected to meet the demands of this larger at-risk population, increased publicity and educational campaigns led by the AAD and assistance to primary care physicians in triaging of patients who should be seen by dermatologists could decrease the number of Americans who need to be screened,” they added.
Limitations of the study included the inability to confirm the clinical diagnoses with histopathology, and no data from the providers were available.
The authors had no disclosures. SPOTme, part of the AAD’s SPOT Skin Cancer initiative, is supported by a grant from Bristol-Myers Squibb.
SOURCE: Okhovat JP et al. J Am Acad Dermatol. https://doi./org/10.1016.j.jaad.2018.05.1242.
Almost half of the individuals diagnosed with melanoma in a free skin cancer screening program otherwise would not have gone to a doctor to have their skin examined, according to an analysis of the American Academy of Dermatology’s national skin cancer screening program, during 1986-2014.
The SPOTme program, a national skin cancer screening and education program conducted by volunteer dermatologists, was launched in 1985. More than 2 million free screenings have been provided by the program in a “predominantly high-risk population, rendering important clinical diagnoses for hundreds of thousands of participants,” according to first authors Jean-Phillip Okhovat, MD, of Beth Israel Deaconess Medical Center, and Derek Beaulieu, MD, of Tufts University, both in Boston, and their colleagues.
The analysis was published online in the Journal of the American Academy of Dermatology on July 26.
Their study analyzed data on almost 2 million people screened through the program from 1986-2014. About 62% were women; 90% were white, about 2% were black, and almost 4% were Hispanic. Almost 80% had no regular dermatologist, almost 73% had not been screened previously, almost 45% had never had a skin cancer check, and 9% were uninsured. Almost 31% reported a mole that had recently change in size, color, or shape; almost 34% said they had a family history of skin cancer, and about 14% said they had a personal history of skin cancer.
Participants were asked about demographics and risk factors, although some questions changed from year to year (for example, in 2009 and 2010, participants were asked about melanoma risk factors, and from 1992 through 2010, participants were asked about their access to dermatologic care).
During 1991-2014 (which did not include data for 1995, 1996, and 2000, which were not available), the screening program resulted in 20,628 clinical melanoma diagnoses, 156,087 clinical dysplastic nevi diagnoses, 32,893 clinical squamous cell carcinoma diagnoses, and 129,848 clinical basal cell carcinoma diagnoses.
Of those clinically diagnosed with melanoma during 1992-2010, 83% said they did not have a regular dermatologist, 77% said they had not been screened previously, and 47% said they would not have seen a doctor for a skin exam if the SPOTme program had not been available.
Of those screened in 2009 and 2010 , 72% were considered at high risk for melanoma (older than age 65 years, having a history of sunburns, a family history of skin cancer, and/or more than 50 moles or unusual moles).
Among the other findings was that from 1992 to 2010, about 12% of those with a clinical melanoma diagnosis were not insured, which increased over time, from almost 11% during 1992-1999 to almost 16% during 2007-2010.
The “consistently high rates” of multiple skin cancer risk factors among those newly screened in the study are consistent with previously reported data, “suggesting that there is an untapped pool of at-risk Americans who have yet to be screened for skin cancer,” the authors wrote. “While the SPOTme program cannot be expected to meet the demands of this larger at-risk population, increased publicity and educational campaigns led by the AAD and assistance to primary care physicians in triaging of patients who should be seen by dermatologists could decrease the number of Americans who need to be screened,” they added.
Limitations of the study included the inability to confirm the clinical diagnoses with histopathology, and no data from the providers were available.
The authors had no disclosures. SPOTme, part of the AAD’s SPOT Skin Cancer initiative, is supported by a grant from Bristol-Myers Squibb.
SOURCE: Okhovat JP et al. J Am Acad Dermatol. https://doi./org/10.1016.j.jaad.2018.05.1242.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Key clinical point: Free skin cancer screening programs help meet an unmet need for people at high risk for skin cancer.
Major finding: Of those who received a clinical diagnosis of melanoma during 1992-2010, 47% said they would not have seen a doctor for a skin exam if the free program had not been available.
Study details: The study analyzed data on almost 2 million people screened through the free SPOTme skin cancer screening program during 1986-2014.
Disclosures: The authors had no disclosures. SPOTme, part of the AAD’s SPOT Skin Cancer initiative, is supported by a grant from Bristol-Myers Squibb.
Source: Okhovat JP et al. J Am Acad Dermatol. https://doi./org/10.1016.j.jaad.2018.05.1242.
FDA approves rituximab for treating pemphigus vulgaris
Rituximab (Rituxan) has been approved for the treatment of adults with moderate to severe pemphigus vulgaris, the manufacturer announced on June 7.
Rituximab is the first biologic approved for treating pemphigus vulgaris, Genentech, a member of the Roche group, stated in a press release announcing the approval.
The prospective, multicenter, open-label, randomized trial, conducted in France, compared the rituximab product approved in the European Union, plus short-term corticosteroid therapy (1,000 mg rituximab administered intravenously at baseline and day 14, then 500 mg at 12 and 18 months, plus 0.5 mg/kg or 1.0 mg/kg per day of prednisone tapered over 3 or 6 months) to corticosteroid therapy alone (oral prednisone 1.0 or 1.5 mg/kg per day tapered over 12 or 18 months), in 90 patients newly diagnosed with moderate to severe pemphigus.
At 24 months, 89% of those in the rituximab group met the primary endpoint, complete remission off therapy at 24 months, compared with 34% of those on corticosteroids (P less than .0001), the investigators reported (Lancet. 2017 May 20;389[10083]:2031-40). Severe adverse events were more commonly reported in the prednisone-only group.
First approved in 1997, rituximab, an anti-CD20 monoclonal antibody, is approved for non-Hodgkin lymphoma, rheumatoid arthritis, chronic lymphocytic leukemia, and granulomatosis with polyangiitis. The prescribing information includes a boxed warning about the risks of fatal infusion reactions, severe mucocutaneous reactions, hepatitis B virus reactivation, and progressive multifocal leukoencephalopathy.
The study published in the Lancet was funded by Roche, the French Ministry of Health, and the French Society of Dermatology.
Rituximab (Rituxan) has been approved for the treatment of adults with moderate to severe pemphigus vulgaris, the manufacturer announced on June 7.
Rituximab is the first biologic approved for treating pemphigus vulgaris, Genentech, a member of the Roche group, stated in a press release announcing the approval.
The prospective, multicenter, open-label, randomized trial, conducted in France, compared the rituximab product approved in the European Union, plus short-term corticosteroid therapy (1,000 mg rituximab administered intravenously at baseline and day 14, then 500 mg at 12 and 18 months, plus 0.5 mg/kg or 1.0 mg/kg per day of prednisone tapered over 3 or 6 months) to corticosteroid therapy alone (oral prednisone 1.0 or 1.5 mg/kg per day tapered over 12 or 18 months), in 90 patients newly diagnosed with moderate to severe pemphigus.
At 24 months, 89% of those in the rituximab group met the primary endpoint, complete remission off therapy at 24 months, compared with 34% of those on corticosteroids (P less than .0001), the investigators reported (Lancet. 2017 May 20;389[10083]:2031-40). Severe adverse events were more commonly reported in the prednisone-only group.
First approved in 1997, rituximab, an anti-CD20 monoclonal antibody, is approved for non-Hodgkin lymphoma, rheumatoid arthritis, chronic lymphocytic leukemia, and granulomatosis with polyangiitis. The prescribing information includes a boxed warning about the risks of fatal infusion reactions, severe mucocutaneous reactions, hepatitis B virus reactivation, and progressive multifocal leukoencephalopathy.
The study published in the Lancet was funded by Roche, the French Ministry of Health, and the French Society of Dermatology.
Rituximab (Rituxan) has been approved for the treatment of adults with moderate to severe pemphigus vulgaris, the manufacturer announced on June 7.
Rituximab is the first biologic approved for treating pemphigus vulgaris, Genentech, a member of the Roche group, stated in a press release announcing the approval.
The prospective, multicenter, open-label, randomized trial, conducted in France, compared the rituximab product approved in the European Union, plus short-term corticosteroid therapy (1,000 mg rituximab administered intravenously at baseline and day 14, then 500 mg at 12 and 18 months, plus 0.5 mg/kg or 1.0 mg/kg per day of prednisone tapered over 3 or 6 months) to corticosteroid therapy alone (oral prednisone 1.0 or 1.5 mg/kg per day tapered over 12 or 18 months), in 90 patients newly diagnosed with moderate to severe pemphigus.
At 24 months, 89% of those in the rituximab group met the primary endpoint, complete remission off therapy at 24 months, compared with 34% of those on corticosteroids (P less than .0001), the investigators reported (Lancet. 2017 May 20;389[10083]:2031-40). Severe adverse events were more commonly reported in the prednisone-only group.
First approved in 1997, rituximab, an anti-CD20 monoclonal antibody, is approved for non-Hodgkin lymphoma, rheumatoid arthritis, chronic lymphocytic leukemia, and granulomatosis with polyangiitis. The prescribing information includes a boxed warning about the risks of fatal infusion reactions, severe mucocutaneous reactions, hepatitis B virus reactivation, and progressive multifocal leukoencephalopathy.
The study published in the Lancet was funded by Roche, the French Ministry of Health, and the French Society of Dermatology.