User login
Vulvar syringoma
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
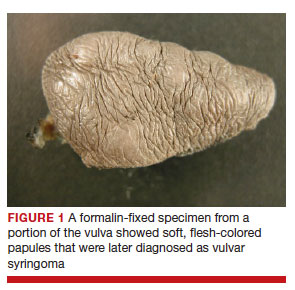
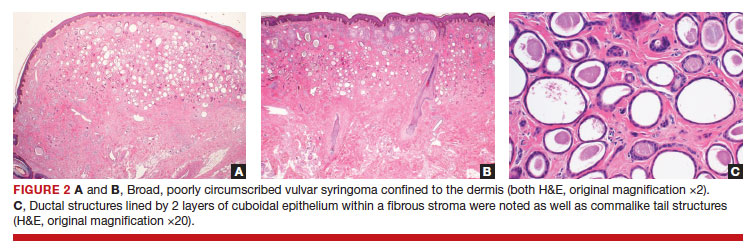
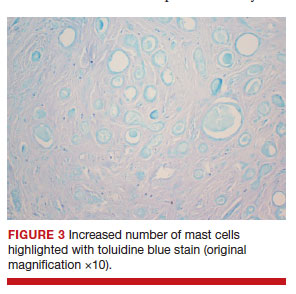
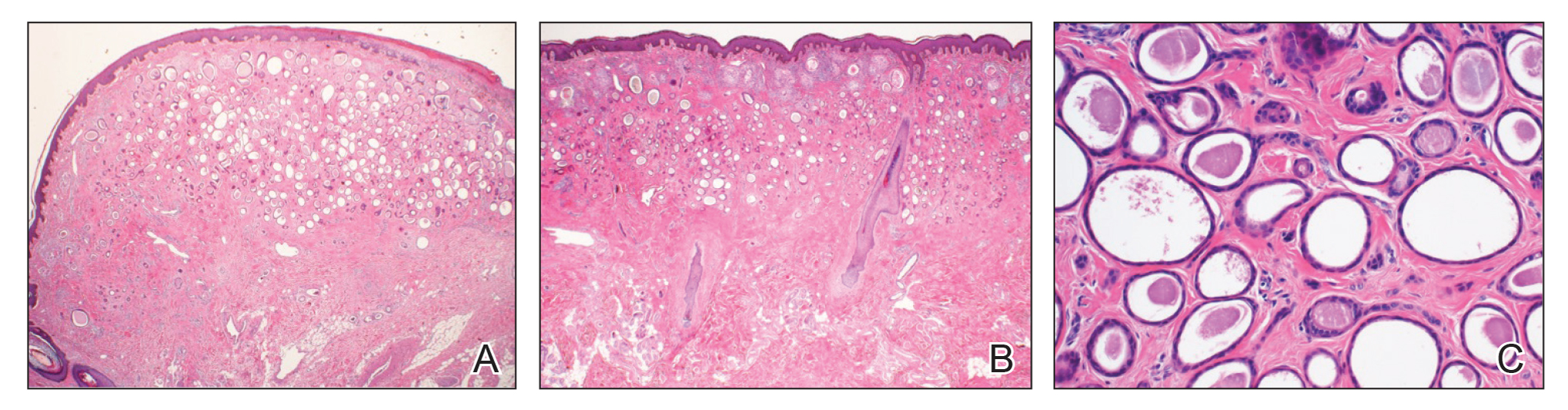
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (FIGURE 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (FIGURES 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (FIGURE 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (FIGURE 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulvar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (FIGURES 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (FIGURE 3).Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems. ●
- Ensure adequate depth of biopsy to assist in the histologic diagnosis of syringoma vs microcystic adnexal carcinoma.
- Vulvar syringomas also may contribute to notable pruritus and ultimately be the underlying etiology for secondary skin changes leading to a lichen simplex chronicus–like phenotype
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas DermoSifiliográficas. 2008; 99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995; 22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinomaaggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369372.
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (FIGURE 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (FIGURES 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (FIGURE 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (FIGURE 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulvar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (FIGURES 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (FIGURE 3).Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems. ●
- Ensure adequate depth of biopsy to assist in the histologic diagnosis of syringoma vs microcystic adnexal carcinoma.
- Vulvar syringomas also may contribute to notable pruritus and ultimately be the underlying etiology for secondary skin changes leading to a lichen simplex chronicus–like phenotype
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (FIGURE 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (FIGURES 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (FIGURE 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (FIGURE 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulvar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (FIGURES 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (FIGURE 3).Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems. ●
- Ensure adequate depth of biopsy to assist in the histologic diagnosis of syringoma vs microcystic adnexal carcinoma.
- Vulvar syringomas also may contribute to notable pruritus and ultimately be the underlying etiology for secondary skin changes leading to a lichen simplex chronicus–like phenotype
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas DermoSifiliográficas. 2008; 99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995; 22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinomaaggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369372.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas DermoSifiliográficas. 2008; 99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995; 22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinomaaggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369372.
Vulvar Syringoma
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.

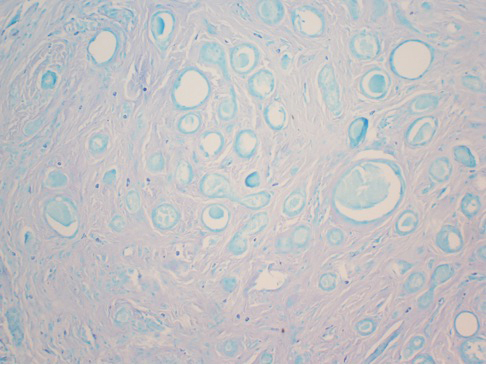
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
Practice Points
- Ensure adequate depth of biopsy to assist in the histologic diagnosis of syringoma vs microcystic adnexal carcinoma.
- Vulvar syringomas also may contribute to notable pruritus and ultimately be the underlying etiology for secondary skin changes leading to a lichen simplex chronicus–like phenotype.

Annular Atrophic Lichen Planus Responds to Hydroxychloroquine and Acitretin
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.


The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).

The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
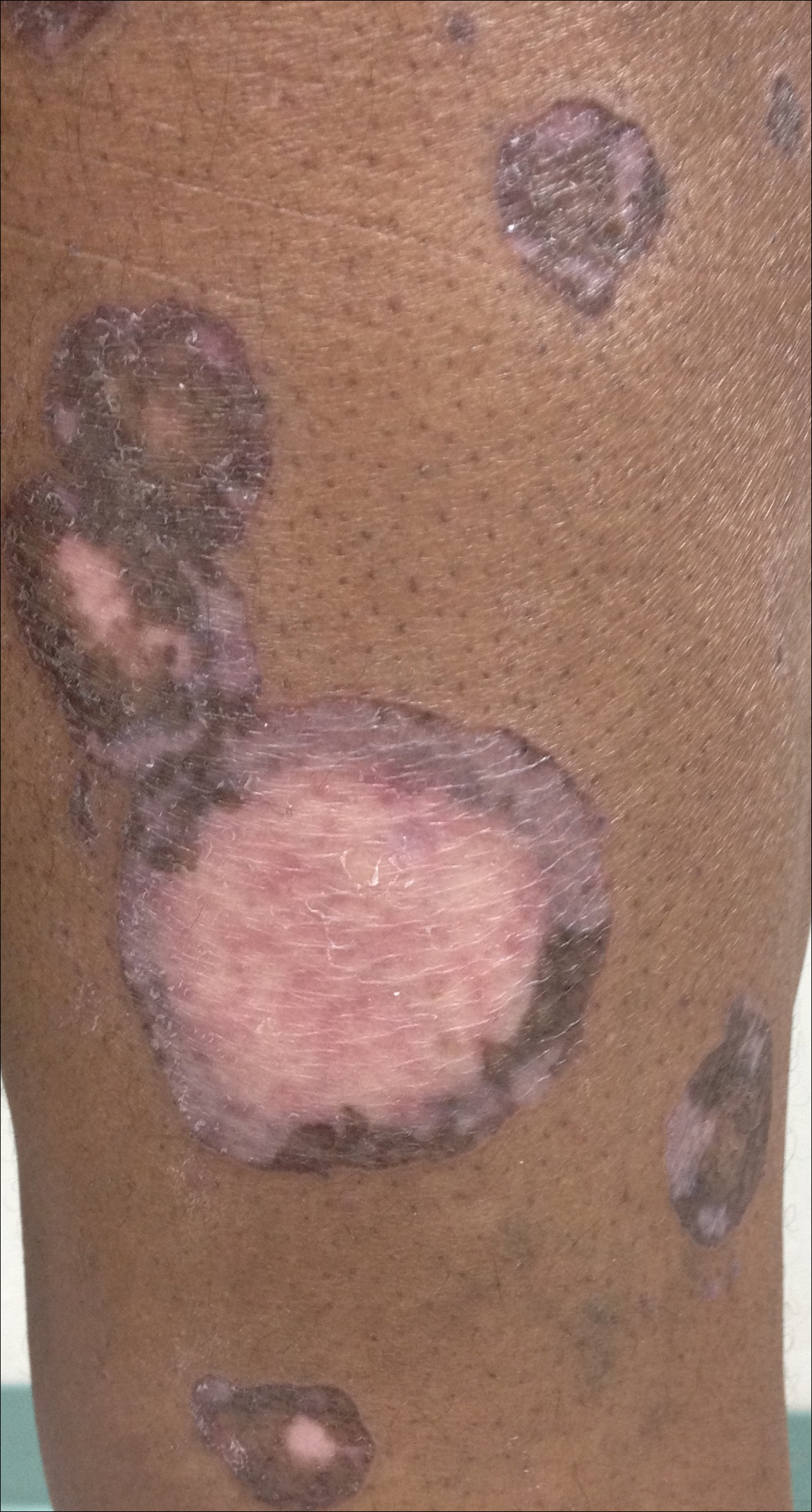
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).
The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).
The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
Autosomal-Dominant Familial Angiolipomatosis
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
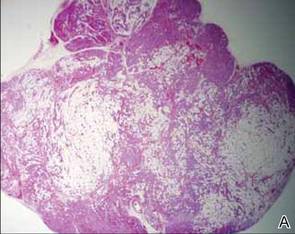
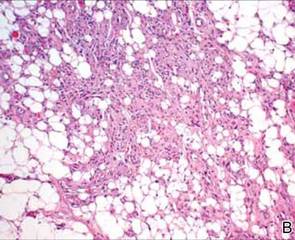
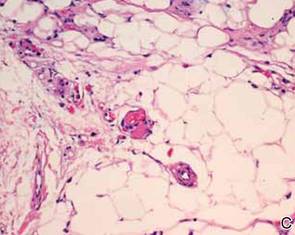
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
Angiolipomas are benign subcutaneous tumors that usually present on the arms, legs, and trunk in young men. Angiolipomas typically range in size from 1 to 4 cm in diameter, and multiple lesions often are present. Tenderness or mild pain may be elicited with palpation, particularly during the initial growth period. Grossly they appear as yellow, firm, circumscribed tumors. Histologic examination generally is characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.
Angiolipomas most often occur sporadically, but in a minority of cases a family history can be identified. Although the exact incidence of familial cases has not been identified in the literature, it is estimated to be 5% to 10%.1 This rare condition has been classified as familial angiolipomatosis, which may be inherited in either an autosomal-recessive or autosomal-dominant fashion, the former being far more prevalent.2 We report the case of a 31-year-old man with multiple angiolipomas who served as a proband for an evaluation of familial angiolipomatosis transmitted in an autosomal-dominant fashion among several male family members.
Case Report
A 31-year-old man presented with a history of fatty tumors on the bilateral upper extremities. The patient’s medical history was remarkable for allergy to dogs and cats, as confirmed by positive skin testing, which was treated with hydroxyzine and albuterol. Physical examination was unremarkable, except for the subcutaneous nodules on both arms and forearms. Laboratory results from a complete blood cell count and a comprehensive metabolic panel including total cholesterol, triglycerides, and high-density lipoproteins were all within reference range. A family history revealed that the patient’s brother, father, and 3 paternal uncles had a history of similar fatty tumors, as well as 2 of his paternal grandmother’s brothers (Figure 1). At the time of presentation, clinical examination revealed multiple tumors distributed on the upper and lower left arm as well as on the posterior and anterior aspect of the right forearm and upper arm. The patient did not report antecedent trauma to these areas.
During surgical evaluation several months later, the subcutaneous nodules were preliminarily diagnosed by the surgeon as lipomas. Following surgical excision of all 5 lesions, gross examination revealed tan-yellow, circumscribed, soft-tissue nodules measuring 0.6 to 2.1 cm. Histologic examination revealed circumscribed nodules surrounded by a thin fibrous capsule. The lesions were composed of mature fat cells and benign vessels arranged in lobules of various sizes divided by fibrous septa. The vascular component ranged from 10% to approximately 50% of the lesion and was predominantly composed of capillary-sized vessels with scattered intraluminal fibrin thrombi (Figure 2). The histologic findings were considered a classic presentation of angiolipoma. Unfortunately, the patient was not able to provide pathology results pertaining to the lesions of his relatives, which he referred to as fatty tumors. At follow-up 13 months after excision, the patient developed new lesions and was planning to return for further excisions.
Comment
|
Angiolipomas are benign mesenchymal neoplasms composed of adipose tissue and blood vessels. They usually present subcutaneously but have been documented in other areas including the spinal region in rare instances.3 The most common locations include the forearms, upper arms, and trunk.4 Our case demonstrates a classic presentation of angiolipomatosis manifesting as multiple subcutaneous nodules on the upper arms of a young man. Although lipomas were clinically suspected, histologic examination revealed that the lesions were in fact angiolipomas.
Angiolipomas account for approximately 17% of all fatty tumors and are characterized by mature adipose tissue with an admixture of capillaries that often contain fibrin thrombi.4 Histologic variants of angiolipomas including cellular angiolipomas and angiomyxolipomas rarely are encountered.5-7 Cellular angiolipomas are composed almost entirely of small vessels (>95% of the lesion).5,6 In addition to the classic presentation, cellular angiolipomas also have been documented in unusual locations. Kahng et al8 reported a 73-year-old woman with abnormal mammographic findings who was found to have a cellular angiolipoma of the breast. Cellular angiolipoma with lymph node involvement was reported in a 67-year-old man with adenocarcinoma of the prostate who underwent a radical retropubic prostatectomy.9 Due to their prominent vascular component, cellular angiolipomas must be differentiated from spindle cell lipomas, Kaposi sarcoma, and other vascular tumors. Kaposi sarcomas usually have slitlike vascular spaces, contain globules in the cytoplasm of some cells that are positive on periodic acid–Schiff staining, display immunoreactivity for human herpesvirus 8, and lack microthrombi. Angiomyxolipomas also are rare. This variant of angiolipomas contains mature adipose tissue, extensive myxoid stroma, and numerous blood vessels.7 The differential diagnosis for angiomyxolipomas includes myxoid liposarcomas and other adipocytic lesions (eg, myxolipomas, myxoid spindle cell lipomas).
Angiolipomas most often occur sporadically; however, family history has been identified in a minority of cases. This rare finding has been classified as familial angiolipomatosis (Online Mendelian Inheritance in Man [OMIM] 206550), which can be inherited in either anautosomal-recessive or very rarely in an autosomal-dominant fashion.2 Our patient had numerous relatives with a history of similar lesions, which supported the diagnosis of familial angiolipomatosis in an autosomal-dominant inheritance pattern (Figure 1). Patients with autosomal-dominant familial angiolipomatosis also have been described to have other coincidental medical conditions, such as polycystic kidney disease.10
The clinical presentation of familial angiolipomatosis includes multiple subcutaneous tumors and a family history of similar lesions that are not associated with malignant transformation. Subcutaneous tumors and a family history with autosomal-dominant inheritance also can be seen in neurofibromatosis type I, which is associated with various benign and malignant neoplasms (eg, meningiomas, gliomas, pheochromocytomas). Therefore, in familial cases of multiple subcutaneous tumors transmitted in an autosomal-dominant pattern, histologic examination is essential to establish the correct diagnosis. Goodman and Baskin11 reported a patient with familial angiolipomatosis who initially was suspected to have neurofibromatosis. The patient also had a granular cell tumor, which occasionally can be seen in neurofibromatosis.11 Another diagnostic problem between familial angiolipomatosis and neurofibromatosis was described by Cina et al2 who documented a case of familial angiolipomatosis with Lisch nodules, which are common in neurofibromatosis but rarely are seen in patients without this condition.12 These reported parallels have prompted some investigators to suggest that similar pathogenetic mechanisms might be involved in both familial angiolipomatosis with an autosomal-dominant inheritance and neurofibromatosis type I.11 Karyotyping performed on angiolipomas has failed to reveal reproducible cytogenetic abnormalities,13 with the exception of 1 report that documented a patient in which 1 of 5 angiolipomas had a t(X;2) abnormality.14 Conversely, ordinary lipomas are associated with numerous karyotypic abnormalities.14
Angiolipomas are benign tumors, but patients with large or disfiguring angiolipomas may choose to undergo surgical excision. For neoplasms that deeply extend between muscles, tendons, and joint capsules, subtotal excision may be required to restore regular function; however, local recurrence with muscular hypotrophy and deformation of the bones near the affected joints may occur.15
Conclusion
We present the case of a 31-year-old man with a rare form of familial angiolipomatosis characterized by an autosomal-dominant inheritance pattern. Our case emphasizes the need to obtain a detailed family history to determine the inheritance pattern in patients with multiple lesions of angiolipoma. Pathology review is essential to differentiate other diseases such as neurofibromatosis, which may present in a similar fashion. We encourage reports of further cases of familial angiolipomatosis to document the inheritance patterns.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
1. Weedon D, Strutton G, Rubin AI. Weedon’s Skin Pathology. Edinburgh, Scotland: Churchill Livingstone/Elsevier; 2010.
2. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123:946-948.
3. Konya D, Ozgen S, Kurtkaya O, et al. Lumbar spinal angiolipoma: case report and review of the literature [published online ahead of print September 20, 2005]. Eur Spine J. 2006;15:1025-1028.
4. Howard WR, Helwig EB. Angiolipoma. Arch Dermatol. 1960;82:924-931.
5. Hunt SJ, Santa Cruz DJ, Barr RJ. Cellular angiolipoma. Am J Surg Pathol. 1990;14:75-81.
6. Kanik AB, Oh CH, Bhawan J. Cellular angiolipoma. Am J Dermatopathol. 1995;17:312-315.
7. Lee HW, Lee DK, Lee MW, et al. Two cases of angiomyxolipoma (vascular myxolipoma) of subcutaneous tissue. J Cutan Pathol. 2005;32:379-382.
8. Kahng HC, Chin NW, Opitz LM, et al. Cellular angiolipoma of the breast: immunohistochemical study and review of the literature. Breast J. 2002;8:47-49.
9. Kazakov DV, Hes O, Hora M, et al. Primary intranodal cellular angiolipoma. Int J Surg Pathol. 2005;13:99-101.
10. Kumar R, Pereira BJ, Sakhuja V, et al. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35:202-204.
11. Goodman JC, Baskin DS. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosis. 1989;2:326-31.
12. Cassiman C, Legius E, Spileers W, et al. Ophthalmological assessment of children with neurofibromatosis type 1 [published online ahead of print May 25, 2013]. Eur J Pediatr. 2013;172:1327-1333.
13. Sciot R, Akerman M, Dal Cin P, et al. Cytogenetic analysis of subcutaneous angiolipoma: further evidence supporting its difference from ordinary pure lipomas: a report of the CHAMP Study Group. Am J Surg Pathol. 1997;21:441-444.
14. Mandahl N, Höglund M, Mertens F, et al. Cytogenetic aberrations in 188 benign and borderline adipose tissue tumors. Genes Chromosomes Cancer. 1994;9:207-215.
15. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17:202-208.
Practice Points
- Dermatologists should be familiar with the clinical and histological features of angiolipomas along with their potential inheritance patterns.
- Familial angiolipomatosis is a rare condition characterized by multiple angiolipomas that has been described as having an autosomal-recessive transmission pattern. Autosomal-dominant inheritance also may occur, as illustrated in the current case report.
- Awareness of the autosomal-dominant form of this entity is important to prevent its misdiagnosis as
neurofibromatosis type I, which has a similar family history and clinical presentation.