User login
Orally dissolving buprenorphine tied to severe tooth decay, FDA warns
Orally dissolving medications containing buprenorphine are linked to severe dental problems, including total tooth loss, the U.S. Food and Drug Administration warns in a safety communication.
The oral side effects of these medications, which are used to treat opioid use disorder (OUD) and pain, include cavities/tooth decay, including rampant caries; dental abscesses/infection; tooth erosion; fillings falling out; and, in some cases, total tooth loss.

Multiple cases have been reported even in patients with no history of dental problems.
The FDA is adding a warning about the risk of dental problems to the prescribing information and the patient medication guide for all buprenorphine-containing medicines dissolved in the mouth.
The FDA emphasizes, however, that buprenorphine remains “an important treatment option for OUD and pain, and the benefits of these medicines clearly outweigh the risks.”
More than 300 reported cases
Buprenorphine was approved in 2002 as a sublingual tablet, and in 2015 as a film to be placed inside the cheek to treat pain. Both delivery methods have been associated with dental problems.
Since buprenorphine was approved, the FDA has identified 305 cases of dental problems associated with orally dissolving buprenorphine, including 131 classified as serious.
There may be other cases, the FDA says, as this represents only cases reported to the FDA or published in the medical literature.
, but those as young as 18 years old were also affected.
Most cases occurred in patients using the medicines for OUD; however, 28 cases of dental problems occurred in patients using it to treat pain.
In 26 cases, patients had no prior history of dental problems. Some dental problems developed as soon as 2 weeks after treatment began; the median time to diagnosis was about 2 years after starting treatment.
Among all 305 cases reported, 113 involved two or more teeth.
The most common treatment for the dental problems was tooth extraction/removal, which was reported in 71 cases. Other cases required root canals, dental surgery, and other procedures such as crowns and implants.
Recommendations
The FDA says health care providers should counsel patients that severe and extensive tooth decay, tooth loss, and tooth fracture have been reported with the use of transmucosal buprenorphine-containing medicines and emphasize the importance of visiting their dentist to closely monitor their teeth.
Patients should be counseled to continue taking buprenorphine medications as prescribed and not stop suddenly without first talking to their health care provider, as this could lead to serious consequences, including relapse, misuse or abuse of other opioids, overdose, and death.
Patients are also being advised to take extra steps to help lessen the risk of serious dental problems.
Patients should also be educated on strategies to maintain or improve oral health while taking transmucosal buprenorphine medicines.
Counsel them that after the medicine is completely dissolved, the patient should take a large sip of water, swish it gently around the teeth and gums, swallow, and wait at least 1 hour before brushing their teeth, as the FDA advises. This will allow time for the mouth to gradually return to oral homeostasis and avoid any mechanical damage that may occur due to brushing.
The FDA also advises that patients tell their provider about any history of tooth problems, including cavities, and schedule a dentist visit soon after starting the medicine.
Dental problems related to transmucosal buprenorphine-containing medicines should be reported to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
Orally dissolving medications containing buprenorphine are linked to severe dental problems, including total tooth loss, the U.S. Food and Drug Administration warns in a safety communication.
The oral side effects of these medications, which are used to treat opioid use disorder (OUD) and pain, include cavities/tooth decay, including rampant caries; dental abscesses/infection; tooth erosion; fillings falling out; and, in some cases, total tooth loss.
Multiple cases have been reported even in patients with no history of dental problems.
The FDA is adding a warning about the risk of dental problems to the prescribing information and the patient medication guide for all buprenorphine-containing medicines dissolved in the mouth.
The FDA emphasizes, however, that buprenorphine remains “an important treatment option for OUD and pain, and the benefits of these medicines clearly outweigh the risks.”
More than 300 reported cases
Buprenorphine was approved in 2002 as a sublingual tablet, and in 2015 as a film to be placed inside the cheek to treat pain. Both delivery methods have been associated with dental problems.
Since buprenorphine was approved, the FDA has identified 305 cases of dental problems associated with orally dissolving buprenorphine, including 131 classified as serious.
There may be other cases, the FDA says, as this represents only cases reported to the FDA or published in the medical literature.
, but those as young as 18 years old were also affected.
Most cases occurred in patients using the medicines for OUD; however, 28 cases of dental problems occurred in patients using it to treat pain.
In 26 cases, patients had no prior history of dental problems. Some dental problems developed as soon as 2 weeks after treatment began; the median time to diagnosis was about 2 years after starting treatment.
Among all 305 cases reported, 113 involved two or more teeth.
The most common treatment for the dental problems was tooth extraction/removal, which was reported in 71 cases. Other cases required root canals, dental surgery, and other procedures such as crowns and implants.
Recommendations
The FDA says health care providers should counsel patients that severe and extensive tooth decay, tooth loss, and tooth fracture have been reported with the use of transmucosal buprenorphine-containing medicines and emphasize the importance of visiting their dentist to closely monitor their teeth.
Patients should be counseled to continue taking buprenorphine medications as prescribed and not stop suddenly without first talking to their health care provider, as this could lead to serious consequences, including relapse, misuse or abuse of other opioids, overdose, and death.
Patients are also being advised to take extra steps to help lessen the risk of serious dental problems.
Patients should also be educated on strategies to maintain or improve oral health while taking transmucosal buprenorphine medicines.
Counsel them that after the medicine is completely dissolved, the patient should take a large sip of water, swish it gently around the teeth and gums, swallow, and wait at least 1 hour before brushing their teeth, as the FDA advises. This will allow time for the mouth to gradually return to oral homeostasis and avoid any mechanical damage that may occur due to brushing.
The FDA also advises that patients tell their provider about any history of tooth problems, including cavities, and schedule a dentist visit soon after starting the medicine.
Dental problems related to transmucosal buprenorphine-containing medicines should be reported to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
Orally dissolving medications containing buprenorphine are linked to severe dental problems, including total tooth loss, the U.S. Food and Drug Administration warns in a safety communication.
The oral side effects of these medications, which are used to treat opioid use disorder (OUD) and pain, include cavities/tooth decay, including rampant caries; dental abscesses/infection; tooth erosion; fillings falling out; and, in some cases, total tooth loss.
Multiple cases have been reported even in patients with no history of dental problems.
The FDA is adding a warning about the risk of dental problems to the prescribing information and the patient medication guide for all buprenorphine-containing medicines dissolved in the mouth.
The FDA emphasizes, however, that buprenorphine remains “an important treatment option for OUD and pain, and the benefits of these medicines clearly outweigh the risks.”
More than 300 reported cases
Buprenorphine was approved in 2002 as a sublingual tablet, and in 2015 as a film to be placed inside the cheek to treat pain. Both delivery methods have been associated with dental problems.
Since buprenorphine was approved, the FDA has identified 305 cases of dental problems associated with orally dissolving buprenorphine, including 131 classified as serious.
There may be other cases, the FDA says, as this represents only cases reported to the FDA or published in the medical literature.
, but those as young as 18 years old were also affected.
Most cases occurred in patients using the medicines for OUD; however, 28 cases of dental problems occurred in patients using it to treat pain.
In 26 cases, patients had no prior history of dental problems. Some dental problems developed as soon as 2 weeks after treatment began; the median time to diagnosis was about 2 years after starting treatment.
Among all 305 cases reported, 113 involved two or more teeth.
The most common treatment for the dental problems was tooth extraction/removal, which was reported in 71 cases. Other cases required root canals, dental surgery, and other procedures such as crowns and implants.
Recommendations
The FDA says health care providers should counsel patients that severe and extensive tooth decay, tooth loss, and tooth fracture have been reported with the use of transmucosal buprenorphine-containing medicines and emphasize the importance of visiting their dentist to closely monitor their teeth.
Patients should be counseled to continue taking buprenorphine medications as prescribed and not stop suddenly without first talking to their health care provider, as this could lead to serious consequences, including relapse, misuse or abuse of other opioids, overdose, and death.
Patients are also being advised to take extra steps to help lessen the risk of serious dental problems.
Patients should also be educated on strategies to maintain or improve oral health while taking transmucosal buprenorphine medicines.
Counsel them that after the medicine is completely dissolved, the patient should take a large sip of water, swish it gently around the teeth and gums, swallow, and wait at least 1 hour before brushing their teeth, as the FDA advises. This will allow time for the mouth to gradually return to oral homeostasis and avoid any mechanical damage that may occur due to brushing.
The FDA also advises that patients tell their provider about any history of tooth problems, including cavities, and schedule a dentist visit soon after starting the medicine.
Dental problems related to transmucosal buprenorphine-containing medicines should be reported to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
Ranking seven COVID-19 antigen tests by ease of use: Report
Some COVID-19 rapid antigen home test kits are much easier to use than others, according to an analysis by ECRI, an independent, nonprofit patient safety organization.
None of the tests were rated as “excellent” in terms of usability and some had “noteworthy” usability concerns, the company said.
If a test is hard to use, “chances are that you may miss a step or not follow the right order, or contaminate the testing area and that can definitely influence the accuracy of the test and lead to a wrong test result,” Marcus Schabacker, MD, PhD, president and CEO of ECRI, told this news organization.
To gauge usability, ECRI used the “industry-standard” system usability scale (SUS), which rates products on a scale of 0 to 100 with 100 being the easiest to use.
More than 30 points separated the top and bottom tests analyzed. The top performer was On/Go, followed by CareStart and Flowflex.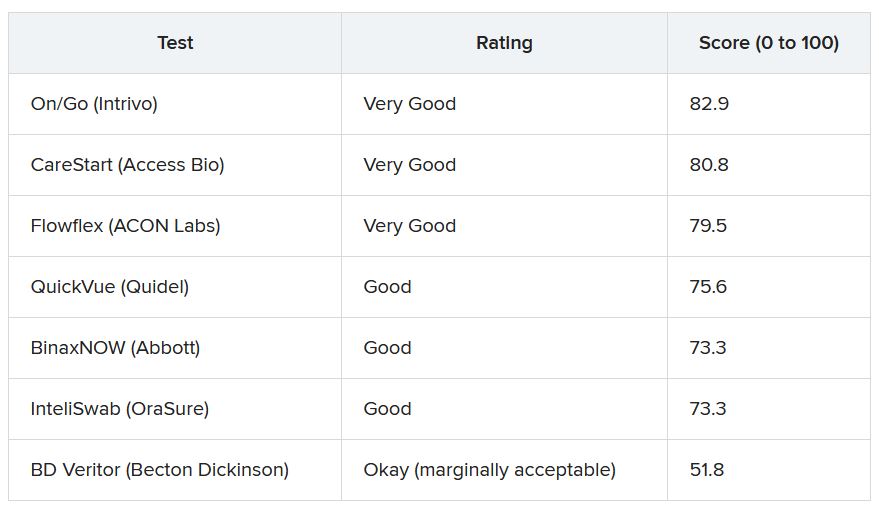
ECRI analysts found that some tests require particularly fine motor skills or have instructions with extremely small font size that may make it hard for older adults or people with complex health conditions to use the tests correctly.
“If you have a tremor from Parkinson’s, for example, or anything which won’t allow you to handle small items, you will have difficulties to do that test by yourself. That is the No. 1 concern we have,” Dr. Schabacker said.
“The second concern is readability, as all of these tests have relatively small instructions. One of them actually has doesn’t even have instructions – you have to download an app,” he noted.
Given demand and supply issues, Dr. Schabacker acknowledged that consumers might not have a choice in which test to use and may have to rely on whatever is available.
These tests are a “hot commodity right now,” he said. “If you have a choice, people should use the ones which are easiest to use, which is the On/Go, the CareStart, or the Flowflex.”
A version of this article first appeared on Medscape.com.
Some COVID-19 rapid antigen home test kits are much easier to use than others, according to an analysis by ECRI, an independent, nonprofit patient safety organization.
None of the tests were rated as “excellent” in terms of usability and some had “noteworthy” usability concerns, the company said.
If a test is hard to use, “chances are that you may miss a step or not follow the right order, or contaminate the testing area and that can definitely influence the accuracy of the test and lead to a wrong test result,” Marcus Schabacker, MD, PhD, president and CEO of ECRI, told this news organization.
To gauge usability, ECRI used the “industry-standard” system usability scale (SUS), which rates products on a scale of 0 to 100 with 100 being the easiest to use.
More than 30 points separated the top and bottom tests analyzed. The top performer was On/Go, followed by CareStart and Flowflex.
ECRI analysts found that some tests require particularly fine motor skills or have instructions with extremely small font size that may make it hard for older adults or people with complex health conditions to use the tests correctly.
“If you have a tremor from Parkinson’s, for example, or anything which won’t allow you to handle small items, you will have difficulties to do that test by yourself. That is the No. 1 concern we have,” Dr. Schabacker said.
“The second concern is readability, as all of these tests have relatively small instructions. One of them actually has doesn’t even have instructions – you have to download an app,” he noted.
Given demand and supply issues, Dr. Schabacker acknowledged that consumers might not have a choice in which test to use and may have to rely on whatever is available.
These tests are a “hot commodity right now,” he said. “If you have a choice, people should use the ones which are easiest to use, which is the On/Go, the CareStart, or the Flowflex.”
A version of this article first appeared on Medscape.com.
Some COVID-19 rapid antigen home test kits are much easier to use than others, according to an analysis by ECRI, an independent, nonprofit patient safety organization.
None of the tests were rated as “excellent” in terms of usability and some had “noteworthy” usability concerns, the company said.
If a test is hard to use, “chances are that you may miss a step or not follow the right order, or contaminate the testing area and that can definitely influence the accuracy of the test and lead to a wrong test result,” Marcus Schabacker, MD, PhD, president and CEO of ECRI, told this news organization.
To gauge usability, ECRI used the “industry-standard” system usability scale (SUS), which rates products on a scale of 0 to 100 with 100 being the easiest to use.
More than 30 points separated the top and bottom tests analyzed. The top performer was On/Go, followed by CareStart and Flowflex.
ECRI analysts found that some tests require particularly fine motor skills or have instructions with extremely small font size that may make it hard for older adults or people with complex health conditions to use the tests correctly.
“If you have a tremor from Parkinson’s, for example, or anything which won’t allow you to handle small items, you will have difficulties to do that test by yourself. That is the No. 1 concern we have,” Dr. Schabacker said.
“The second concern is readability, as all of these tests have relatively small instructions. One of them actually has doesn’t even have instructions – you have to download an app,” he noted.
Given demand and supply issues, Dr. Schabacker acknowledged that consumers might not have a choice in which test to use and may have to rely on whatever is available.
These tests are a “hot commodity right now,” he said. “If you have a choice, people should use the ones which are easiest to use, which is the On/Go, the CareStart, or the Flowflex.”
A version of this article first appeared on Medscape.com.
Statin therapy seems safe in pregnancy
Statins may be safe when used during pregnancy, with no increase in risk for fetal anomalies, although there may be a higher risk for low birth weight and preterm labor, results of a large study from Taiwan suggest.
The Food and Drug Administration relaxed its warning on statins in July 2021, removing the drug’s blanket contraindication in all pregnant women.
Removal of the broadly worded contraindication should “enable health care professionals and patients to make individual decisions about benefit and risk, especially for those at very high risk of heart attack or stroke,” the FDA said in their announcement.
“Our findings suggested that statins may be used during pregnancy with no increase in the rate of congenital anomalies,” wrote Jui-Chun Chang, MD, from Taichung Veterans General Hospital, Taiwan, and colleagues in the new study, published online Dec. 30, 2021, in JAMA Network Open.
“For pregnant women at low risk, statins should be used carefully after assessing the risks of low birth weight and preterm birth,” they said. “For women with dyslipidemia or high-risk cardiovascular disease, as well as those who use statins before conception, statins may be continuously used with no increased risks of neonatal adverse effects.”
The study included more than 1.4 million pregnant women aged 18 years and older who gave birth to their first child between 2004 and 2014.
A total of 469 women (mean age, 32.6 years; mean gestational age, 38.4 weeks) who used statins during pregnancy were compared with 4,690 matched controls who had no statin exposure during pregnancy.
After controlling for maternal comorbidities and age, women who used statins during pregnancy were more apt to have low-birth-weight babies weighing less than 2,500 g (risk ratio, 1.51; 95% confidence interval, 1.05-2.16) and to deliver preterm (RR, 1.99; 95% CI, 1.46-2.71).
The statin-exposed babies were also more likely to have a lower 1-minute Apgar score (RR, 1.83; 95% CI, 1.04-3.20). Importantly, however, there was no increase in risk for fetal anomalies in the statin-exposed infants, the researchers said.
In addition, for women who used statins for more than 3 months prior to pregnancy, maintaining statin use during pregnancy did not increase the risk for adverse neonatal outcomes, including congenital anomalies, low birth weight, preterm birth, very low birth weight, low Apgar scores, and fetal distress.
The researchers called for further studies to confirm their observations.
Funding for the study was provided by Taichung Veterans General Hospital. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Statins may be safe when used during pregnancy, with no increase in risk for fetal anomalies, although there may be a higher risk for low birth weight and preterm labor, results of a large study from Taiwan suggest.
The Food and Drug Administration relaxed its warning on statins in July 2021, removing the drug’s blanket contraindication in all pregnant women.
Removal of the broadly worded contraindication should “enable health care professionals and patients to make individual decisions about benefit and risk, especially for those at very high risk of heart attack or stroke,” the FDA said in their announcement.
“Our findings suggested that statins may be used during pregnancy with no increase in the rate of congenital anomalies,” wrote Jui-Chun Chang, MD, from Taichung Veterans General Hospital, Taiwan, and colleagues in the new study, published online Dec. 30, 2021, in JAMA Network Open.
“For pregnant women at low risk, statins should be used carefully after assessing the risks of low birth weight and preterm birth,” they said. “For women with dyslipidemia or high-risk cardiovascular disease, as well as those who use statins before conception, statins may be continuously used with no increased risks of neonatal adverse effects.”
The study included more than 1.4 million pregnant women aged 18 years and older who gave birth to their first child between 2004 and 2014.
A total of 469 women (mean age, 32.6 years; mean gestational age, 38.4 weeks) who used statins during pregnancy were compared with 4,690 matched controls who had no statin exposure during pregnancy.
After controlling for maternal comorbidities and age, women who used statins during pregnancy were more apt to have low-birth-weight babies weighing less than 2,500 g (risk ratio, 1.51; 95% confidence interval, 1.05-2.16) and to deliver preterm (RR, 1.99; 95% CI, 1.46-2.71).
The statin-exposed babies were also more likely to have a lower 1-minute Apgar score (RR, 1.83; 95% CI, 1.04-3.20). Importantly, however, there was no increase in risk for fetal anomalies in the statin-exposed infants, the researchers said.
In addition, for women who used statins for more than 3 months prior to pregnancy, maintaining statin use during pregnancy did not increase the risk for adverse neonatal outcomes, including congenital anomalies, low birth weight, preterm birth, very low birth weight, low Apgar scores, and fetal distress.
The researchers called for further studies to confirm their observations.
Funding for the study was provided by Taichung Veterans General Hospital. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Statins may be safe when used during pregnancy, with no increase in risk for fetal anomalies, although there may be a higher risk for low birth weight and preterm labor, results of a large study from Taiwan suggest.
The Food and Drug Administration relaxed its warning on statins in July 2021, removing the drug’s blanket contraindication in all pregnant women.
Removal of the broadly worded contraindication should “enable health care professionals and patients to make individual decisions about benefit and risk, especially for those at very high risk of heart attack or stroke,” the FDA said in their announcement.
“Our findings suggested that statins may be used during pregnancy with no increase in the rate of congenital anomalies,” wrote Jui-Chun Chang, MD, from Taichung Veterans General Hospital, Taiwan, and colleagues in the new study, published online Dec. 30, 2021, in JAMA Network Open.
“For pregnant women at low risk, statins should be used carefully after assessing the risks of low birth weight and preterm birth,” they said. “For women with dyslipidemia or high-risk cardiovascular disease, as well as those who use statins before conception, statins may be continuously used with no increased risks of neonatal adverse effects.”
The study included more than 1.4 million pregnant women aged 18 years and older who gave birth to their first child between 2004 and 2014.
A total of 469 women (mean age, 32.6 years; mean gestational age, 38.4 weeks) who used statins during pregnancy were compared with 4,690 matched controls who had no statin exposure during pregnancy.
After controlling for maternal comorbidities and age, women who used statins during pregnancy were more apt to have low-birth-weight babies weighing less than 2,500 g (risk ratio, 1.51; 95% confidence interval, 1.05-2.16) and to deliver preterm (RR, 1.99; 95% CI, 1.46-2.71).
The statin-exposed babies were also more likely to have a lower 1-minute Apgar score (RR, 1.83; 95% CI, 1.04-3.20). Importantly, however, there was no increase in risk for fetal anomalies in the statin-exposed infants, the researchers said.
In addition, for women who used statins for more than 3 months prior to pregnancy, maintaining statin use during pregnancy did not increase the risk for adverse neonatal outcomes, including congenital anomalies, low birth weight, preterm birth, very low birth weight, low Apgar scores, and fetal distress.
The researchers called for further studies to confirm their observations.
Funding for the study was provided by Taichung Veterans General Hospital. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA NETWORK OPEN
Novel antidepressant shows promise as add-on therapy
as add-on therapy
Adjunctive treatment with the novel oral medication REL-1017 (esmethadone) is effective in adults with major depressive disorder (MDD) who have failed other antidepressants, new research suggests.
REL-1017, from Relmada Therapeutics, is a novel N-methyl-D-aspartate receptor (NMDAR) channel blocker that preferentially targets hyperactive channels while maintaining physiologic glutamatergic neurotransmission.
Results from a phase 2a study showed rapid “therapeutic efficacy,” with a statistical difference by day 4, and the improvement was “robust,” with an effect size of 0.7 to 1. The positive outcome was also sustained for at least 1 week after treatment discontinuation, coinvestigator Paolo L. Manfredi, MD, chief scientific officer, Relmada Therapeutics, noted.
“Considering that the available traditional antidepressants have an average effect size around 0.3, this novel, potential rapid-acting antidepressant … holds great promise for millions of patients suffering from depression,” Dr. Manfredi told this news organization.
These results were obtained with a “very-well-tolerated once-daily oral NMDAR antagonist, without the dissociative effects seen with ketamine,” he added.
The findings were published online in the American Journal of Psychiatry.
‘Clear need’ for better therapies
It is estimated that more than half of patients with MDD fail to respond adequately following their first standard antidepressant treatment. In addition, responses are often delayed by 4-8 weeks after starting an antidepressant.
Therefore, there is a “clear need” to develop drugs for MDD that act quickly and with improved efficacy, the investigators note.
The phase 2a study of REL-1017 enrolled 62 adult patients (45% women) aged 18-65 years with moderate to severe MDD and no significant psychiatric comorbidity. All had failed to benefit from one to three standard antidepressant treatments in their current major depressive episode.
The researchers evaluated two doses of REL-1017 (25 mg and 50 mg once daily) vs. placebo given as adjunctive treatment. The assigned treatment lasted 7 days.
The primary study objectives were safety and tolerability. Results showed no serious adverse events (AEs), and no patients experienced treatment-emergent AEs that led to the stopping of treatment.
In addition, patients receiving the active drug experienced mild or moderate transient AEs comparable to placebo, with no opioid, dissociative, or psychotomimetic symptoms, or withdrawal effects when treatment ended.
The most common AEs reported were headache, constipation, nausea, and sleepiness.
Significant efficacy
The primary efficacy endpoint was the Montgomery–Åsberg Depression Scale (MADRS) score.
MADRS scores showed improvement on day 4 of treatment in both REL-1017 groups, and the improvement continued through day 7 (last dose) and day 14 (7 days after the last dose), with P ≤ .0308 and effect sizes ranging from 0.7 to 1.0.
Mean change from baseline in MADRS scores showed more improvement at the end of the dosing period for both dosing groups (–16.8 with 25 mg and –16.6 with 50 mg) vs. –8.8 with placebo.
Results of the other efficacy endpoints of Symptoms of Depression Questionnaire (SDQ) score and Clinical Global Impressions severity scale (CGI-S) and improvement scale (CGI-I) scores were similar to that of the MADRS.
Remission rates (defined as a MADRS score ≤10) on day 14, the last day of efficacy assessment, were 5% with placebo vs. 31% (P = .035) with REL-1017 25 mg and 39% (P = .01) with REL-1017 50 mg.
The number needed to treat to achieve remission on day 14 was four with the 25-mg dose and three with the 50-mg dose.
Phase 3 trials to confirm the efficacy and safety of REL-1017 are in progress, with topline results expected later this year, the investigators report.
The study was funded by Relmada Therapeutics. Dr. Manfredi has received personal fees from and/or held stock ownership in Relmada. Disclosures for the other investigators are fully listed in the original article.
A version of this article first appeared on Medscape.com.
Adjunctive treatment with the novel oral medication REL-1017 (esmethadone) is effective in adults with major depressive disorder (MDD) who have failed other antidepressants, new research suggests.
REL-1017, from Relmada Therapeutics, is a novel N-methyl-D-aspartate receptor (NMDAR) channel blocker that preferentially targets hyperactive channels while maintaining physiologic glutamatergic neurotransmission.
Results from a phase 2a study showed rapid “therapeutic efficacy,” with a statistical difference by day 4, and the improvement was “robust,” with an effect size of 0.7 to 1. The positive outcome was also sustained for at least 1 week after treatment discontinuation, coinvestigator Paolo L. Manfredi, MD, chief scientific officer, Relmada Therapeutics, noted.
“Considering that the available traditional antidepressants have an average effect size around 0.3, this novel, potential rapid-acting antidepressant … holds great promise for millions of patients suffering from depression,” Dr. Manfredi told this news organization.
These results were obtained with a “very-well-tolerated once-daily oral NMDAR antagonist, without the dissociative effects seen with ketamine,” he added.
The findings were published online in the American Journal of Psychiatry.
‘Clear need’ for better therapies
It is estimated that more than half of patients with MDD fail to respond adequately following their first standard antidepressant treatment. In addition, responses are often delayed by 4-8 weeks after starting an antidepressant.
Therefore, there is a “clear need” to develop drugs for MDD that act quickly and with improved efficacy, the investigators note.
The phase 2a study of REL-1017 enrolled 62 adult patients (45% women) aged 18-65 years with moderate to severe MDD and no significant psychiatric comorbidity. All had failed to benefit from one to three standard antidepressant treatments in their current major depressive episode.
The researchers evaluated two doses of REL-1017 (25 mg and 50 mg once daily) vs. placebo given as adjunctive treatment. The assigned treatment lasted 7 days.
The primary study objectives were safety and tolerability. Results showed no serious adverse events (AEs), and no patients experienced treatment-emergent AEs that led to the stopping of treatment.
In addition, patients receiving the active drug experienced mild or moderate transient AEs comparable to placebo, with no opioid, dissociative, or psychotomimetic symptoms, or withdrawal effects when treatment ended.
The most common AEs reported were headache, constipation, nausea, and sleepiness.
Significant efficacy
The primary efficacy endpoint was the Montgomery–Åsberg Depression Scale (MADRS) score.
MADRS scores showed improvement on day 4 of treatment in both REL-1017 groups, and the improvement continued through day 7 (last dose) and day 14 (7 days after the last dose), with P ≤ .0308 and effect sizes ranging from 0.7 to 1.0.
Mean change from baseline in MADRS scores showed more improvement at the end of the dosing period for both dosing groups (–16.8 with 25 mg and –16.6 with 50 mg) vs. –8.8 with placebo.
Results of the other efficacy endpoints of Symptoms of Depression Questionnaire (SDQ) score and Clinical Global Impressions severity scale (CGI-S) and improvement scale (CGI-I) scores were similar to that of the MADRS.
Remission rates (defined as a MADRS score ≤10) on day 14, the last day of efficacy assessment, were 5% with placebo vs. 31% (P = .035) with REL-1017 25 mg and 39% (P = .01) with REL-1017 50 mg.
The number needed to treat to achieve remission on day 14 was four with the 25-mg dose and three with the 50-mg dose.
Phase 3 trials to confirm the efficacy and safety of REL-1017 are in progress, with topline results expected later this year, the investigators report.
The study was funded by Relmada Therapeutics. Dr. Manfredi has received personal fees from and/or held stock ownership in Relmada. Disclosures for the other investigators are fully listed in the original article.
A version of this article first appeared on Medscape.com.
Adjunctive treatment with the novel oral medication REL-1017 (esmethadone) is effective in adults with major depressive disorder (MDD) who have failed other antidepressants, new research suggests.
REL-1017, from Relmada Therapeutics, is a novel N-methyl-D-aspartate receptor (NMDAR) channel blocker that preferentially targets hyperactive channels while maintaining physiologic glutamatergic neurotransmission.
Results from a phase 2a study showed rapid “therapeutic efficacy,” with a statistical difference by day 4, and the improvement was “robust,” with an effect size of 0.7 to 1. The positive outcome was also sustained for at least 1 week after treatment discontinuation, coinvestigator Paolo L. Manfredi, MD, chief scientific officer, Relmada Therapeutics, noted.
“Considering that the available traditional antidepressants have an average effect size around 0.3, this novel, potential rapid-acting antidepressant … holds great promise for millions of patients suffering from depression,” Dr. Manfredi told this news organization.
These results were obtained with a “very-well-tolerated once-daily oral NMDAR antagonist, without the dissociative effects seen with ketamine,” he added.
The findings were published online in the American Journal of Psychiatry.
‘Clear need’ for better therapies
It is estimated that more than half of patients with MDD fail to respond adequately following their first standard antidepressant treatment. In addition, responses are often delayed by 4-8 weeks after starting an antidepressant.
Therefore, there is a “clear need” to develop drugs for MDD that act quickly and with improved efficacy, the investigators note.
The phase 2a study of REL-1017 enrolled 62 adult patients (45% women) aged 18-65 years with moderate to severe MDD and no significant psychiatric comorbidity. All had failed to benefit from one to three standard antidepressant treatments in their current major depressive episode.
The researchers evaluated two doses of REL-1017 (25 mg and 50 mg once daily) vs. placebo given as adjunctive treatment. The assigned treatment lasted 7 days.
The primary study objectives were safety and tolerability. Results showed no serious adverse events (AEs), and no patients experienced treatment-emergent AEs that led to the stopping of treatment.
In addition, patients receiving the active drug experienced mild or moderate transient AEs comparable to placebo, with no opioid, dissociative, or psychotomimetic symptoms, or withdrawal effects when treatment ended.
The most common AEs reported were headache, constipation, nausea, and sleepiness.
Significant efficacy
The primary efficacy endpoint was the Montgomery–Åsberg Depression Scale (MADRS) score.
MADRS scores showed improvement on day 4 of treatment in both REL-1017 groups, and the improvement continued through day 7 (last dose) and day 14 (7 days after the last dose), with P ≤ .0308 and effect sizes ranging from 0.7 to 1.0.
Mean change from baseline in MADRS scores showed more improvement at the end of the dosing period for both dosing groups (–16.8 with 25 mg and –16.6 with 50 mg) vs. –8.8 with placebo.
Results of the other efficacy endpoints of Symptoms of Depression Questionnaire (SDQ) score and Clinical Global Impressions severity scale (CGI-S) and improvement scale (CGI-I) scores were similar to that of the MADRS.
Remission rates (defined as a MADRS score ≤10) on day 14, the last day of efficacy assessment, were 5% with placebo vs. 31% (P = .035) with REL-1017 25 mg and 39% (P = .01) with REL-1017 50 mg.
The number needed to treat to achieve remission on day 14 was four with the 25-mg dose and three with the 50-mg dose.
Phase 3 trials to confirm the efficacy and safety of REL-1017 are in progress, with topline results expected later this year, the investigators report.
The study was funded by Relmada Therapeutics. Dr. Manfredi has received personal fees from and/or held stock ownership in Relmada. Disclosures for the other investigators are fully listed in the original article.
A version of this article first appeared on Medscape.com.
as add-on therapy
as add-on therapy
AAN updates treatment guidance on painful diabetic neuropathy
Painful diabetic neuropathy is very common and can greatly affect an individual’s quality of life, guideline author Brian Callaghan, MD, University of Michigan, Ann Arbor, noted in a news release.
“This guideline aims to help neurologists and other doctors provide the highest quality patient care based on the latest evidence,” Dr. Callaghan said.
The recommendations update the 2011 AAN guideline on the treatment of painful diabetic neuropathy. The new guidance was published online Dec. 27, 2021, in Neurology and has been endorsed by the American Association of Neuromuscular & Electrodiagnostic Medicine.
Multiple options
To update the guideline, an expert panel reviewed data from more than 100 randomized controlled trials published from January 2008 to April 2020.
The panel noted that more than 16% of individuals with diabetes experience painful diabetic neuropathy, but it often goes unrecognized and untreated. The guideline recommends clinicians assess patients with diabetes for peripheral neuropathic pain and its effect on their function and quality of life.
Before prescribing treatment, health providers should determine if the patient also has mood or sleep problems as both can influence pain perception.
The guideline recommends offering one of four classes of oral medications found to be effective for neuropathic pain: tricyclic antidepressants such as amitriptyline, nortriptyline, or imipramine; serotonin norepinephrine reuptake inhibitors such as duloxetine, venlafaxine, or desvenlafaxine; gabapentinoids such as gabapentin or pregabalin; and/or sodium channel blockers such as carbamazepine, oxcarbazepine, lamotrigine, or lacosamide.
All four classes of medications have “comparable effect sizes just above or just below our cutoff for a medium effect size” (standardized median difference, 0.5), the panel noted.
In addition, “new studies on sodium channel blockers published since the last guideline have resulted in these drugs now being recommended and considered as effective at providing pain relief as the other drug classes recommended in this guideline,” said Dr. Callaghan.
When an initial medication fails to provide meaningful improvement in pain, or produces significant side effects, a trial of another medication from a different class is recommended.
Pain reduction, not elimination
Opioids are not recommended for painful diabetic neuropathy. Not only do they come with risks, there is also no strong evidence they are effective for painful diabetic neuropathy in the long term, the panel wrote. Tramadol and tapentadol are also not recommended for the treatment of painful diabetic neuropathy.
“Current evidence suggests that the risks of the use of opioids for painful diabetic neuropathy therapy outweigh the benefits, so they should not be prescribed,” Dr. Callaghan said.
For patients interested in trying topical, nontraditional, or nondrug interventions to reduce pain, the guideline recommends a number of options including capsaicin, glyceryl trinitrate spray, and Citrullus colocynthis. Ginkgo biloba, exercise, mindfulness, cognitive-behavioral therapy, and tai chi are also suggested.
“It is important to note that the recommended drugs and topical treatments in this guideline may not eliminate pain, but they have been shown to reduce pain,” Dr. Callaghan said. “The good news is there are many treatment options for painful diabetic neuropathy, so a treatment plan can be tailored specifically to each person living with this condition.”
Along with the updated guideline, the AAN has also published a new Polyneuropathy Quality Measurement Set to assist neurologists and other health care providers in treating patients with painful diabetic neuropathy.
The updated guideline was developed with financial support from the AAN.
A version of this article first appeared on Medscape.com.
Painful diabetic neuropathy is very common and can greatly affect an individual’s quality of life, guideline author Brian Callaghan, MD, University of Michigan, Ann Arbor, noted in a news release.
“This guideline aims to help neurologists and other doctors provide the highest quality patient care based on the latest evidence,” Dr. Callaghan said.
The recommendations update the 2011 AAN guideline on the treatment of painful diabetic neuropathy. The new guidance was published online Dec. 27, 2021, in Neurology and has been endorsed by the American Association of Neuromuscular & Electrodiagnostic Medicine.
Multiple options
To update the guideline, an expert panel reviewed data from more than 100 randomized controlled trials published from January 2008 to April 2020.
The panel noted that more than 16% of individuals with diabetes experience painful diabetic neuropathy, but it often goes unrecognized and untreated. The guideline recommends clinicians assess patients with diabetes for peripheral neuropathic pain and its effect on their function and quality of life.
Before prescribing treatment, health providers should determine if the patient also has mood or sleep problems as both can influence pain perception.
The guideline recommends offering one of four classes of oral medications found to be effective for neuropathic pain: tricyclic antidepressants such as amitriptyline, nortriptyline, or imipramine; serotonin norepinephrine reuptake inhibitors such as duloxetine, venlafaxine, or desvenlafaxine; gabapentinoids such as gabapentin or pregabalin; and/or sodium channel blockers such as carbamazepine, oxcarbazepine, lamotrigine, or lacosamide.
All four classes of medications have “comparable effect sizes just above or just below our cutoff for a medium effect size” (standardized median difference, 0.5), the panel noted.
In addition, “new studies on sodium channel blockers published since the last guideline have resulted in these drugs now being recommended and considered as effective at providing pain relief as the other drug classes recommended in this guideline,” said Dr. Callaghan.
When an initial medication fails to provide meaningful improvement in pain, or produces significant side effects, a trial of another medication from a different class is recommended.
Pain reduction, not elimination
Opioids are not recommended for painful diabetic neuropathy. Not only do they come with risks, there is also no strong evidence they are effective for painful diabetic neuropathy in the long term, the panel wrote. Tramadol and tapentadol are also not recommended for the treatment of painful diabetic neuropathy.
“Current evidence suggests that the risks of the use of opioids for painful diabetic neuropathy therapy outweigh the benefits, so they should not be prescribed,” Dr. Callaghan said.
For patients interested in trying topical, nontraditional, or nondrug interventions to reduce pain, the guideline recommends a number of options including capsaicin, glyceryl trinitrate spray, and Citrullus colocynthis. Ginkgo biloba, exercise, mindfulness, cognitive-behavioral therapy, and tai chi are also suggested.
“It is important to note that the recommended drugs and topical treatments in this guideline may not eliminate pain, but they have been shown to reduce pain,” Dr. Callaghan said. “The good news is there are many treatment options for painful diabetic neuropathy, so a treatment plan can be tailored specifically to each person living with this condition.”
Along with the updated guideline, the AAN has also published a new Polyneuropathy Quality Measurement Set to assist neurologists and other health care providers in treating patients with painful diabetic neuropathy.
The updated guideline was developed with financial support from the AAN.
A version of this article first appeared on Medscape.com.
Painful diabetic neuropathy is very common and can greatly affect an individual’s quality of life, guideline author Brian Callaghan, MD, University of Michigan, Ann Arbor, noted in a news release.
“This guideline aims to help neurologists and other doctors provide the highest quality patient care based on the latest evidence,” Dr. Callaghan said.
The recommendations update the 2011 AAN guideline on the treatment of painful diabetic neuropathy. The new guidance was published online Dec. 27, 2021, in Neurology and has been endorsed by the American Association of Neuromuscular & Electrodiagnostic Medicine.
Multiple options
To update the guideline, an expert panel reviewed data from more than 100 randomized controlled trials published from January 2008 to April 2020.
The panel noted that more than 16% of individuals with diabetes experience painful diabetic neuropathy, but it often goes unrecognized and untreated. The guideline recommends clinicians assess patients with diabetes for peripheral neuropathic pain and its effect on their function and quality of life.
Before prescribing treatment, health providers should determine if the patient also has mood or sleep problems as both can influence pain perception.
The guideline recommends offering one of four classes of oral medications found to be effective for neuropathic pain: tricyclic antidepressants such as amitriptyline, nortriptyline, or imipramine; serotonin norepinephrine reuptake inhibitors such as duloxetine, venlafaxine, or desvenlafaxine; gabapentinoids such as gabapentin or pregabalin; and/or sodium channel blockers such as carbamazepine, oxcarbazepine, lamotrigine, or lacosamide.
All four classes of medications have “comparable effect sizes just above or just below our cutoff for a medium effect size” (standardized median difference, 0.5), the panel noted.
In addition, “new studies on sodium channel blockers published since the last guideline have resulted in these drugs now being recommended and considered as effective at providing pain relief as the other drug classes recommended in this guideline,” said Dr. Callaghan.
When an initial medication fails to provide meaningful improvement in pain, or produces significant side effects, a trial of another medication from a different class is recommended.
Pain reduction, not elimination
Opioids are not recommended for painful diabetic neuropathy. Not only do they come with risks, there is also no strong evidence they are effective for painful diabetic neuropathy in the long term, the panel wrote. Tramadol and tapentadol are also not recommended for the treatment of painful diabetic neuropathy.
“Current evidence suggests that the risks of the use of opioids for painful diabetic neuropathy therapy outweigh the benefits, so they should not be prescribed,” Dr. Callaghan said.
For patients interested in trying topical, nontraditional, or nondrug interventions to reduce pain, the guideline recommends a number of options including capsaicin, glyceryl trinitrate spray, and Citrullus colocynthis. Ginkgo biloba, exercise, mindfulness, cognitive-behavioral therapy, and tai chi are also suggested.
“It is important to note that the recommended drugs and topical treatments in this guideline may not eliminate pain, but they have been shown to reduce pain,” Dr. Callaghan said. “The good news is there are many treatment options for painful diabetic neuropathy, so a treatment plan can be tailored specifically to each person living with this condition.”
Along with the updated guideline, the AAN has also published a new Polyneuropathy Quality Measurement Set to assist neurologists and other health care providers in treating patients with painful diabetic neuropathy.
The updated guideline was developed with financial support from the AAN.
A version of this article first appeared on Medscape.com.
FROM NEUROLOGY
Effect of vitamin D supplementation in early psychosis
Low vitamin D is common in patients with first-episode psychosis (FEP), but supplementation does not appear to improve mental or physical symptoms, new data show.
“Previous work, our own and others, has shown that people with psychosis, even soon after their first diagnosis, have low vitamin D levels, but it was not known whether supplementing with vitamin D in people with early psychosis would improve health outcomes,” study investigator Fiona Gaughran, MD, with the Institute of Psychiatry, Psychology & Neuroscience, King’s College London, told this news organization.
“While we did not demonstrate a benefit of supplementation over 6 months, these very high rates of vitamin deficiency and insufficiency may have longer-term negative health impacts which we have not measured, so raising awareness of the need to optimize vitamin D in people with psychosis is important,” said Dr. Gaughran.
The results of the randomized clinical trial were published online Dec. 28 in JAMA Network Open.
Thoughtful approach, negative result
Participants included 149 adults within 3 years of a first presentation with a functional psychotic disorder. The cohort’s mean age was 28 years, 60% were men, 44% were Black or of other racial and ethnic minority groups, and 56% were White.
Seventy-five participants were randomly assigned to receive 120,000 IU of cholecalciferol or matching placebo administered by the researchers in monthly doses with an oral syringe.
“We chose a dose of 120,000 IU monthly (equivalent to 4,000 IU daily) which was expected to safely increase vitamin D levels. The regimen was discussed with experts with lived experience, and took into account that a daily preparation would add to the significant medication load that people with psychosis already carry,” said Dr. Gaughran.
Vitamin D supplementation as administered in this study was safe and led to a significant increase in 25-hydroxyvitamin D concentrations.
However, at 6 months (mean difference, 3.57; 95% confidence interval, –1.11 to 8.25; P = .13).
There was also no apparent benefit of vitamin D supplementation on any secondary outcome, including the PANSS subscores of global function and depression or cardiometabolic risk factors.
“With respect to clinical practice, we cannot now recommend monthly treatments with 120,000 IU of cholecalciferol in FEP,” the investigators note.
The prevalence of vitamin D insufficiency and deficiency was high in the population – 74.6% overall and 93.4% among ethnic minorities.
“Thus, the sample was well suited to detecting any potential benefits that may have arisen from correcting this. However, even in this subgroup, there was no evidence to support the guiding hypothesis” that vitamin D supplementation would improve outcomes in patients with early psychosis, the researchers note.
They suggest that future studies examine the association of vitamin D with brain-related outcomes based on periods of treatment longer than 6 months and administered as daily rather than bolus treatments.
“Future public health strategies should acknowledge the high prevalence of vitamin D insufficiency and deficiency in people with psychosis and consider any reasonable adjustments which may be needed to address this over and above general population guidance,” said Dr. Gaughran.
The study was funded by the Stanley Medical Research Institute and received support from the National Institute for Health Research Maudsley Biomedical Research Centre, King’s College London, and the NIHR Applied Research Collaboration South London. Dr. Gaughran reported receiving speaking honoraria from Otsuka Lundbeck outside the submitted work. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.
Low vitamin D is common in patients with first-episode psychosis (FEP), but supplementation does not appear to improve mental or physical symptoms, new data show.
“Previous work, our own and others, has shown that people with psychosis, even soon after their first diagnosis, have low vitamin D levels, but it was not known whether supplementing with vitamin D in people with early psychosis would improve health outcomes,” study investigator Fiona Gaughran, MD, with the Institute of Psychiatry, Psychology & Neuroscience, King’s College London, told this news organization.
“While we did not demonstrate a benefit of supplementation over 6 months, these very high rates of vitamin deficiency and insufficiency may have longer-term negative health impacts which we have not measured, so raising awareness of the need to optimize vitamin D in people with psychosis is important,” said Dr. Gaughran.
The results of the randomized clinical trial were published online Dec. 28 in JAMA Network Open.
Thoughtful approach, negative result
Participants included 149 adults within 3 years of a first presentation with a functional psychotic disorder. The cohort’s mean age was 28 years, 60% were men, 44% were Black or of other racial and ethnic minority groups, and 56% were White.
Seventy-five participants were randomly assigned to receive 120,000 IU of cholecalciferol or matching placebo administered by the researchers in monthly doses with an oral syringe.
“We chose a dose of 120,000 IU monthly (equivalent to 4,000 IU daily) which was expected to safely increase vitamin D levels. The regimen was discussed with experts with lived experience, and took into account that a daily preparation would add to the significant medication load that people with psychosis already carry,” said Dr. Gaughran.
Vitamin D supplementation as administered in this study was safe and led to a significant increase in 25-hydroxyvitamin D concentrations.
However, at 6 months (mean difference, 3.57; 95% confidence interval, –1.11 to 8.25; P = .13).
There was also no apparent benefit of vitamin D supplementation on any secondary outcome, including the PANSS subscores of global function and depression or cardiometabolic risk factors.
“With respect to clinical practice, we cannot now recommend monthly treatments with 120,000 IU of cholecalciferol in FEP,” the investigators note.
The prevalence of vitamin D insufficiency and deficiency was high in the population – 74.6% overall and 93.4% among ethnic minorities.
“Thus, the sample was well suited to detecting any potential benefits that may have arisen from correcting this. However, even in this subgroup, there was no evidence to support the guiding hypothesis” that vitamin D supplementation would improve outcomes in patients with early psychosis, the researchers note.
They suggest that future studies examine the association of vitamin D with brain-related outcomes based on periods of treatment longer than 6 months and administered as daily rather than bolus treatments.
“Future public health strategies should acknowledge the high prevalence of vitamin D insufficiency and deficiency in people with psychosis and consider any reasonable adjustments which may be needed to address this over and above general population guidance,” said Dr. Gaughran.
The study was funded by the Stanley Medical Research Institute and received support from the National Institute for Health Research Maudsley Biomedical Research Centre, King’s College London, and the NIHR Applied Research Collaboration South London. Dr. Gaughran reported receiving speaking honoraria from Otsuka Lundbeck outside the submitted work. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.
Low vitamin D is common in patients with first-episode psychosis (FEP), but supplementation does not appear to improve mental or physical symptoms, new data show.
“Previous work, our own and others, has shown that people with psychosis, even soon after their first diagnosis, have low vitamin D levels, but it was not known whether supplementing with vitamin D in people with early psychosis would improve health outcomes,” study investigator Fiona Gaughran, MD, with the Institute of Psychiatry, Psychology & Neuroscience, King’s College London, told this news organization.
“While we did not demonstrate a benefit of supplementation over 6 months, these very high rates of vitamin deficiency and insufficiency may have longer-term negative health impacts which we have not measured, so raising awareness of the need to optimize vitamin D in people with psychosis is important,” said Dr. Gaughran.
The results of the randomized clinical trial were published online Dec. 28 in JAMA Network Open.
Thoughtful approach, negative result
Participants included 149 adults within 3 years of a first presentation with a functional psychotic disorder. The cohort’s mean age was 28 years, 60% were men, 44% were Black or of other racial and ethnic minority groups, and 56% were White.
Seventy-five participants were randomly assigned to receive 120,000 IU of cholecalciferol or matching placebo administered by the researchers in monthly doses with an oral syringe.
“We chose a dose of 120,000 IU monthly (equivalent to 4,000 IU daily) which was expected to safely increase vitamin D levels. The regimen was discussed with experts with lived experience, and took into account that a daily preparation would add to the significant medication load that people with psychosis already carry,” said Dr. Gaughran.
Vitamin D supplementation as administered in this study was safe and led to a significant increase in 25-hydroxyvitamin D concentrations.
However, at 6 months (mean difference, 3.57; 95% confidence interval, –1.11 to 8.25; P = .13).
There was also no apparent benefit of vitamin D supplementation on any secondary outcome, including the PANSS subscores of global function and depression or cardiometabolic risk factors.
“With respect to clinical practice, we cannot now recommend monthly treatments with 120,000 IU of cholecalciferol in FEP,” the investigators note.
The prevalence of vitamin D insufficiency and deficiency was high in the population – 74.6% overall and 93.4% among ethnic minorities.
“Thus, the sample was well suited to detecting any potential benefits that may have arisen from correcting this. However, even in this subgroup, there was no evidence to support the guiding hypothesis” that vitamin D supplementation would improve outcomes in patients with early psychosis, the researchers note.
They suggest that future studies examine the association of vitamin D with brain-related outcomes based on periods of treatment longer than 6 months and administered as daily rather than bolus treatments.
“Future public health strategies should acknowledge the high prevalence of vitamin D insufficiency and deficiency in people with psychosis and consider any reasonable adjustments which may be needed to address this over and above general population guidance,” said Dr. Gaughran.
The study was funded by the Stanley Medical Research Institute and received support from the National Institute for Health Research Maudsley Biomedical Research Centre, King’s College London, and the NIHR Applied Research Collaboration South London. Dr. Gaughran reported receiving speaking honoraria from Otsuka Lundbeck outside the submitted work. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.
FROM JAMA NETWORK OPEN
Cardiac device interrogation after death ‘richly informative’
Interrogating the cardiac implantable electronic device (CIED) after death can yield important information about critical device malfunction, premortem abnormalities, and the mechanism and timing of death, a new study suggests.
Postmortem CIED interrogation is “richly informative” in assisting both cardiac and forensic investigations and “should be considered for select patients with CIEDs undergoing autopsy,” say Elizabeth Paratz, MBBS, department of cardiology, Baker Heart and Diabetes Institute, Prahran, Australia, and colleagues.
Their study results were published online in JACC: Clinical Electrophysiology.
Cause of death revealed in half of cases
They reviewed CIED interrogations in 260 deceased individuals undergoing medicolegal investigation of sudden death (162 patients) or unexplained death (98 patients) by the Victorian Institute of Forensic Medicine between 2005 and 2020.
Roughly two-thirds were male (68.8%) and their median age was 72.8 years; 202 patients had pacemakers, 56 had defibrillators, and 2 had loop recorders. The cause of death was cardiac in 79.6% of cases.
Postmortem CIED interrogation was successful in 98.5% cases and directly informed cause of death in 131 cases (50.4%), with fatal ventricular arrhythmias identified in 121 patients.
CIED interrogation assisted in determining the cause of death in 63.6% of cases of sudden death and 28.6% of nonsudden death cases.
In 20 cases (7.7%), CIED interrogation uncovered potential device malfunction. Issues included failure to appropriately treat ventricular arrhythmias in 13 cases; lead issues in 3 cases, including 2 cases resulting in failure to treat ventricular arrhythmias; as well as battery depletion in 6 cases.
In 72 patients (27.7%), the device recorded abnormalities in the 30 days before death. These abnormalities included nonsustained ventricular tachycardia in 26 cases, rapid atrial fibrillation in 17, elective replacement indicator or end-of-life status in 22, intrathoracic impedance alarms or lead issues in 3 each, and therapy delivered in 1 instance.
“In several cases, the absence of an arrhythmia carried medicolegal implications: For example, in eight fatal motor vehicle accident cases, only one patient had a ventricular arrhythmia documented on their CIED,” Dr. Paratz and colleagues report.
And in six cases in which the patient was found dead after a prolonged period, CIED interrogation determined time of death. And in one case, CIED interrogation was the primary means of identifying the patient.
Still, postmortem CIED interrogation remains uncommon, the study team notes.
They point to a 2007 survey of Chicago morticians that found roughly 370 CIEDs were explanted per year prior to cremation, but only 4% of morticians had ever returned a CIED to the manufacturer for analysis.
“Encouraging postmortem interrogation of CIEDs may assist in postmarketing surveillance for critical faults, as well as in providing an electrophysiological appraisal of terminal rhythms and device responses in a variety of physiological scenarios,” the researchers say.
The study had no commercial funding. Dr. Paratz is supported by a National Health and Medical Research Council/National Heart Foundation cofunded Postgraduate Scholarship, Royal Australasian College of Physicians JJ Billings Scholarship, and PSA Insurance Cardiovascular Scholarship. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Interrogating the cardiac implantable electronic device (CIED) after death can yield important information about critical device malfunction, premortem abnormalities, and the mechanism and timing of death, a new study suggests.
Postmortem CIED interrogation is “richly informative” in assisting both cardiac and forensic investigations and “should be considered for select patients with CIEDs undergoing autopsy,” say Elizabeth Paratz, MBBS, department of cardiology, Baker Heart and Diabetes Institute, Prahran, Australia, and colleagues.
Their study results were published online in JACC: Clinical Electrophysiology.
Cause of death revealed in half of cases
They reviewed CIED interrogations in 260 deceased individuals undergoing medicolegal investigation of sudden death (162 patients) or unexplained death (98 patients) by the Victorian Institute of Forensic Medicine between 2005 and 2020.
Roughly two-thirds were male (68.8%) and their median age was 72.8 years; 202 patients had pacemakers, 56 had defibrillators, and 2 had loop recorders. The cause of death was cardiac in 79.6% of cases.
Postmortem CIED interrogation was successful in 98.5% cases and directly informed cause of death in 131 cases (50.4%), with fatal ventricular arrhythmias identified in 121 patients.
CIED interrogation assisted in determining the cause of death in 63.6% of cases of sudden death and 28.6% of nonsudden death cases.
In 20 cases (7.7%), CIED interrogation uncovered potential device malfunction. Issues included failure to appropriately treat ventricular arrhythmias in 13 cases; lead issues in 3 cases, including 2 cases resulting in failure to treat ventricular arrhythmias; as well as battery depletion in 6 cases.
In 72 patients (27.7%), the device recorded abnormalities in the 30 days before death. These abnormalities included nonsustained ventricular tachycardia in 26 cases, rapid atrial fibrillation in 17, elective replacement indicator or end-of-life status in 22, intrathoracic impedance alarms or lead issues in 3 each, and therapy delivered in 1 instance.
“In several cases, the absence of an arrhythmia carried medicolegal implications: For example, in eight fatal motor vehicle accident cases, only one patient had a ventricular arrhythmia documented on their CIED,” Dr. Paratz and colleagues report.
And in six cases in which the patient was found dead after a prolonged period, CIED interrogation determined time of death. And in one case, CIED interrogation was the primary means of identifying the patient.
Still, postmortem CIED interrogation remains uncommon, the study team notes.
They point to a 2007 survey of Chicago morticians that found roughly 370 CIEDs were explanted per year prior to cremation, but only 4% of morticians had ever returned a CIED to the manufacturer for analysis.
“Encouraging postmortem interrogation of CIEDs may assist in postmarketing surveillance for critical faults, as well as in providing an electrophysiological appraisal of terminal rhythms and device responses in a variety of physiological scenarios,” the researchers say.
The study had no commercial funding. Dr. Paratz is supported by a National Health and Medical Research Council/National Heart Foundation cofunded Postgraduate Scholarship, Royal Australasian College of Physicians JJ Billings Scholarship, and PSA Insurance Cardiovascular Scholarship. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Interrogating the cardiac implantable electronic device (CIED) after death can yield important information about critical device malfunction, premortem abnormalities, and the mechanism and timing of death, a new study suggests.
Postmortem CIED interrogation is “richly informative” in assisting both cardiac and forensic investigations and “should be considered for select patients with CIEDs undergoing autopsy,” say Elizabeth Paratz, MBBS, department of cardiology, Baker Heart and Diabetes Institute, Prahran, Australia, and colleagues.
Their study results were published online in JACC: Clinical Electrophysiology.
Cause of death revealed in half of cases
They reviewed CIED interrogations in 260 deceased individuals undergoing medicolegal investigation of sudden death (162 patients) or unexplained death (98 patients) by the Victorian Institute of Forensic Medicine between 2005 and 2020.
Roughly two-thirds were male (68.8%) and their median age was 72.8 years; 202 patients had pacemakers, 56 had defibrillators, and 2 had loop recorders. The cause of death was cardiac in 79.6% of cases.
Postmortem CIED interrogation was successful in 98.5% cases and directly informed cause of death in 131 cases (50.4%), with fatal ventricular arrhythmias identified in 121 patients.
CIED interrogation assisted in determining the cause of death in 63.6% of cases of sudden death and 28.6% of nonsudden death cases.
In 20 cases (7.7%), CIED interrogation uncovered potential device malfunction. Issues included failure to appropriately treat ventricular arrhythmias in 13 cases; lead issues in 3 cases, including 2 cases resulting in failure to treat ventricular arrhythmias; as well as battery depletion in 6 cases.
In 72 patients (27.7%), the device recorded abnormalities in the 30 days before death. These abnormalities included nonsustained ventricular tachycardia in 26 cases, rapid atrial fibrillation in 17, elective replacement indicator or end-of-life status in 22, intrathoracic impedance alarms or lead issues in 3 each, and therapy delivered in 1 instance.
“In several cases, the absence of an arrhythmia carried medicolegal implications: For example, in eight fatal motor vehicle accident cases, only one patient had a ventricular arrhythmia documented on their CIED,” Dr. Paratz and colleagues report.
And in six cases in which the patient was found dead after a prolonged period, CIED interrogation determined time of death. And in one case, CIED interrogation was the primary means of identifying the patient.
Still, postmortem CIED interrogation remains uncommon, the study team notes.
They point to a 2007 survey of Chicago morticians that found roughly 370 CIEDs were explanted per year prior to cremation, but only 4% of morticians had ever returned a CIED to the manufacturer for analysis.
“Encouraging postmortem interrogation of CIEDs may assist in postmarketing surveillance for critical faults, as well as in providing an electrophysiological appraisal of terminal rhythms and device responses in a variety of physiological scenarios,” the researchers say.
The study had no commercial funding. Dr. Paratz is supported by a National Health and Medical Research Council/National Heart Foundation cofunded Postgraduate Scholarship, Royal Australasian College of Physicians JJ Billings Scholarship, and PSA Insurance Cardiovascular Scholarship. The authors have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JACC: CLINICAL ELECTROPHYSIOLOGY
New understanding of suicide attempts emerges
even in the absence of a psychiatric disorder.
This finding suggests the genetic underpinnings of suicide attempts are partially shared and partially distinct from those of related psychiatric disorders, the investigators note.
“This study brings us a step closer to understanding the neurobiology of suicidality, with the ultimate goal of developing new treatments and prevention strategies,” Niamh Mullins, PhD, department of psychiatry, department of genetics and genomic sciences, Icahn School of Medicine at Mount Sinai in New York, said in an interview.
The study was published online in Biological Psychiatry.
Largest study to date
In the largest genetic association study of suicide attempt published to date, the researchers conducted a genome-wide association study (GWAS) of 29,782 suicide attempt cases and 519,961 controls in the International Suicide Genetics Consortium (ISGC).
Two loci reached genome-wide significance for suicide attempt – the major histocompatibility complex and an intergenic locus on chromosome 7, the latter of which remained associated with suicide attempt after conditioning on psychiatric disorders and was replicated in an independent cohort of over 14,000 veterans in the Million Veteran Program.
“This is the first replicated genetic locus that contributes more to suicide attempt than related psychiatric disorders,” Dr. Mullins said.
“The study found overlap in the genetic basis of suicide attempt and that of related psychiatric disorders, particularly major depression, but also with that of nonpsychiatric risk factors such as smoking, pain, risk-taking behavior, sleep disturbances, and poorer general health,” Dr. Mullins said.
“These genetic relationships between suicide attempt and nonpsychiatric risk factors were not a by-product of comorbid psychiatric illness, suggesting that there is some shared biological basis between suicide attempt and nonpsychiatric risk factors,” she added.
Dr. Mullins cautioned that the findings do not have any immediate impact on patient care.
“The ultimate goal of this research is to gain insight into the underlying biological pathways involved in suicide attempts or suicidal thoughts, providing potential avenues to treatments and prevention strategies,” she said.
“The study findings also point to the importance of studying the potential direct causal paths between these risk factors and suicide attempt in patients with and without psychiatric illness,” Douglas Ruderfer, PhD, of Vanderbilt University Medical Center, Nashville, Tenn., cofounder and cochair of the consortium and senior author of the paper, added in a news release.
A version of this article first appeared on Medscape.com.
even in the absence of a psychiatric disorder.
This finding suggests the genetic underpinnings of suicide attempts are partially shared and partially distinct from those of related psychiatric disorders, the investigators note.
“This study brings us a step closer to understanding the neurobiology of suicidality, with the ultimate goal of developing new treatments and prevention strategies,” Niamh Mullins, PhD, department of psychiatry, department of genetics and genomic sciences, Icahn School of Medicine at Mount Sinai in New York, said in an interview.
The study was published online in Biological Psychiatry.
Largest study to date
In the largest genetic association study of suicide attempt published to date, the researchers conducted a genome-wide association study (GWAS) of 29,782 suicide attempt cases and 519,961 controls in the International Suicide Genetics Consortium (ISGC).
Two loci reached genome-wide significance for suicide attempt – the major histocompatibility complex and an intergenic locus on chromosome 7, the latter of which remained associated with suicide attempt after conditioning on psychiatric disorders and was replicated in an independent cohort of over 14,000 veterans in the Million Veteran Program.
“This is the first replicated genetic locus that contributes more to suicide attempt than related psychiatric disorders,” Dr. Mullins said.
“The study found overlap in the genetic basis of suicide attempt and that of related psychiatric disorders, particularly major depression, but also with that of nonpsychiatric risk factors such as smoking, pain, risk-taking behavior, sleep disturbances, and poorer general health,” Dr. Mullins said.
“These genetic relationships between suicide attempt and nonpsychiatric risk factors were not a by-product of comorbid psychiatric illness, suggesting that there is some shared biological basis between suicide attempt and nonpsychiatric risk factors,” she added.
Dr. Mullins cautioned that the findings do not have any immediate impact on patient care.
“The ultimate goal of this research is to gain insight into the underlying biological pathways involved in suicide attempts or suicidal thoughts, providing potential avenues to treatments and prevention strategies,” she said.
“The study findings also point to the importance of studying the potential direct causal paths between these risk factors and suicide attempt in patients with and without psychiatric illness,” Douglas Ruderfer, PhD, of Vanderbilt University Medical Center, Nashville, Tenn., cofounder and cochair of the consortium and senior author of the paper, added in a news release.
A version of this article first appeared on Medscape.com.
even in the absence of a psychiatric disorder.
This finding suggests the genetic underpinnings of suicide attempts are partially shared and partially distinct from those of related psychiatric disorders, the investigators note.
“This study brings us a step closer to understanding the neurobiology of suicidality, with the ultimate goal of developing new treatments and prevention strategies,” Niamh Mullins, PhD, department of psychiatry, department of genetics and genomic sciences, Icahn School of Medicine at Mount Sinai in New York, said in an interview.
The study was published online in Biological Psychiatry.
Largest study to date
In the largest genetic association study of suicide attempt published to date, the researchers conducted a genome-wide association study (GWAS) of 29,782 suicide attempt cases and 519,961 controls in the International Suicide Genetics Consortium (ISGC).
Two loci reached genome-wide significance for suicide attempt – the major histocompatibility complex and an intergenic locus on chromosome 7, the latter of which remained associated with suicide attempt after conditioning on psychiatric disorders and was replicated in an independent cohort of over 14,000 veterans in the Million Veteran Program.
“This is the first replicated genetic locus that contributes more to suicide attempt than related psychiatric disorders,” Dr. Mullins said.
“The study found overlap in the genetic basis of suicide attempt and that of related psychiatric disorders, particularly major depression, but also with that of nonpsychiatric risk factors such as smoking, pain, risk-taking behavior, sleep disturbances, and poorer general health,” Dr. Mullins said.
“These genetic relationships between suicide attempt and nonpsychiatric risk factors were not a by-product of comorbid psychiatric illness, suggesting that there is some shared biological basis between suicide attempt and nonpsychiatric risk factors,” she added.
Dr. Mullins cautioned that the findings do not have any immediate impact on patient care.
“The ultimate goal of this research is to gain insight into the underlying biological pathways involved in suicide attempts or suicidal thoughts, providing potential avenues to treatments and prevention strategies,” she said.
“The study findings also point to the importance of studying the potential direct causal paths between these risk factors and suicide attempt in patients with and without psychiatric illness,” Douglas Ruderfer, PhD, of Vanderbilt University Medical Center, Nashville, Tenn., cofounder and cochair of the consortium and senior author of the paper, added in a news release.
A version of this article first appeared on Medscape.com.
FROM BIOLOGICAL PSYCHIATRY
COVID-19 antigen tests may be less sensitive to Omicron: FDA
Rapid antigen tests for COVID-19 might be less effective at detecting the Omicron variant that is spreading rapidly across the United States, according to the Food and Drug Administration.
Early data suggest that COVID-19 antigen tests “do detect the Omicron variant but may have reduced sensitivity,” the FDA said in a statement posted Dec. 28 on its website.
The FDA is working with the National Institutes of Health’s Rapid Acceleration of Diagnostics (RADx) initiative to assess the performance of antigen tests with patient samples that have the Omicron variant.
The potential for antigen tests to be less sensitive for the Omicron variant emerged in tests using patient samples containing live virus, “which represents the best way to evaluate true test performance in the short term,” the FDA said.
Initial laboratory tests using heat-activated (killed) virus samples found that antigen tests were able to detect the Omicron variant.
“It is important to note that these laboratory data are not a replacement for clinical study evaluations using patient samples with live virus, which are ongoing. The FDA and RADx are continuing to further evaluate the performance of antigen tests using patient samples with live virus,” the FDA said.
Testing still important
The agency continues to recommend use of antigen tests as directed in the authorized labeling and in accordance with the instructions included with the tests.
They note that antigen tests are generally less sensitive and less likely to pick up very early infections, compared with molecular tests.
The FDA continues to recommend that an individual with a negative antigen test who has symptoms or a high likelihood of infection because of exposure follow-up with a molecular test to determine if they have COVID-19.
An individual with a positive antigen test should self-isolate and seek follow-up care with a health care provider to determine the next steps.
The FDA, with partners and test developers, are continuing to evaluate test sensitivity, as well as the best timing and frequency of antigen testing.
The agency said that it will provide updated information and any needed recommendations when appropriate.
A version of this article first appeared on Medscape.com.
Rapid antigen tests for COVID-19 might be less effective at detecting the Omicron variant that is spreading rapidly across the United States, according to the Food and Drug Administration.
Early data suggest that COVID-19 antigen tests “do detect the Omicron variant but may have reduced sensitivity,” the FDA said in a statement posted Dec. 28 on its website.
The FDA is working with the National Institutes of Health’s Rapid Acceleration of Diagnostics (RADx) initiative to assess the performance of antigen tests with patient samples that have the Omicron variant.
The potential for antigen tests to be less sensitive for the Omicron variant emerged in tests using patient samples containing live virus, “which represents the best way to evaluate true test performance in the short term,” the FDA said.
Initial laboratory tests using heat-activated (killed) virus samples found that antigen tests were able to detect the Omicron variant.
“It is important to note that these laboratory data are not a replacement for clinical study evaluations using patient samples with live virus, which are ongoing. The FDA and RADx are continuing to further evaluate the performance of antigen tests using patient samples with live virus,” the FDA said.
Testing still important
The agency continues to recommend use of antigen tests as directed in the authorized labeling and in accordance with the instructions included with the tests.
They note that antigen tests are generally less sensitive and less likely to pick up very early infections, compared with molecular tests.
The FDA continues to recommend that an individual with a negative antigen test who has symptoms or a high likelihood of infection because of exposure follow-up with a molecular test to determine if they have COVID-19.
An individual with a positive antigen test should self-isolate and seek follow-up care with a health care provider to determine the next steps.
The FDA, with partners and test developers, are continuing to evaluate test sensitivity, as well as the best timing and frequency of antigen testing.
The agency said that it will provide updated information and any needed recommendations when appropriate.
A version of this article first appeared on Medscape.com.
Rapid antigen tests for COVID-19 might be less effective at detecting the Omicron variant that is spreading rapidly across the United States, according to the Food and Drug Administration.
Early data suggest that COVID-19 antigen tests “do detect the Omicron variant but may have reduced sensitivity,” the FDA said in a statement posted Dec. 28 on its website.
The FDA is working with the National Institutes of Health’s Rapid Acceleration of Diagnostics (RADx) initiative to assess the performance of antigen tests with patient samples that have the Omicron variant.
The potential for antigen tests to be less sensitive for the Omicron variant emerged in tests using patient samples containing live virus, “which represents the best way to evaluate true test performance in the short term,” the FDA said.
Initial laboratory tests using heat-activated (killed) virus samples found that antigen tests were able to detect the Omicron variant.
“It is important to note that these laboratory data are not a replacement for clinical study evaluations using patient samples with live virus, which are ongoing. The FDA and RADx are continuing to further evaluate the performance of antigen tests using patient samples with live virus,” the FDA said.
Testing still important
The agency continues to recommend use of antigen tests as directed in the authorized labeling and in accordance with the instructions included with the tests.
They note that antigen tests are generally less sensitive and less likely to pick up very early infections, compared with molecular tests.
The FDA continues to recommend that an individual with a negative antigen test who has symptoms or a high likelihood of infection because of exposure follow-up with a molecular test to determine if they have COVID-19.
An individual with a positive antigen test should self-isolate and seek follow-up care with a health care provider to determine the next steps.
The FDA, with partners and test developers, are continuing to evaluate test sensitivity, as well as the best timing and frequency of antigen testing.
The agency said that it will provide updated information and any needed recommendations when appropriate.
A version of this article first appeared on Medscape.com.
Formaldehyde exposure tied to cognitive impairment
Long-term exposure to formaldehyde on the job is linked to cognitive impairment down the road, new research suggests.
In a large observational study of adults aged 45-70 years, researchers found a 17% higher risk for cognitive problems in those with occupational formaldehyde exposure – and higher risks for those with longer duration of exposure.
“The effect of formaldehyde on the brain has been previously shown mainly in animal experiments, but very few studies have been done on humans,” lead author Noemie Letellier, PhD, Institute for Neurosciences of Montpellier, University of Montpellier (France), said in an interview.
“Our results show that being or having been occupationally exposed to formaldehyde is associated with cognitive impairment in a relatively young population,” Dr. Letellier said.
The findings were published online Dec. 22, 2021, in the journal Neurology.
Dose-effect relationship
The investigators assessed a representative sample of 75,322 adults in France (median age, 57.5 years; 53% women). All were part of the CONSTANCES cohort, an observational cohort with a focus on occupational and environmental factors.
A total of 6,026 participants (8%) were exposed to formaldehyde during their careers. Their occupations included nurses, caregivers, medical technicians, workers in the textile, chemistry and metal industries, carpenters, and cleaners.
The researchers calculated lifetime formaldehyde exposure using a French job-exposure matrix created to estimate a person’s exposure to potential health hazards in different occupations.
Individuals were divided into three equal groups according to their years of exposure to formaldehyde. “Low” was considered to be 6 or fewer years of exposure, “medium” was 7-21 years, and “high” was 22 or more years.
Participants were also split into three groups according to their cumulative exposure (total lifetime formaldehyde exposure based on the probability, intensity, and frequency of exposure).
Prevention efforts needed
After adjusting for age, sex, education and other confounders, participants exposed to formaldehyde were at higher risk for global cognitive impairment (adjusted relative risk, 1.17; 95% confidence interval, 1.1-1.2).
Longer duration of exposure and high cumulative lifetime exposure were associated with worse cognitive impairment, “with a dose-effect relationship for exposure duration,” the researchers reported.
Those exposed to formaldehyde for 22 years or more had a 21% higher risk of global cognitive impairment and workers with the highest cumulative exposure had a 19% higher risk of cognitive impairment, compared with workers with no exposure.
Although workers with recent exposure showed higher cognitive impairment, “time may not fully attenuate formaldehyde-associated cognitive deficits, especially in highly exposed but also in moderately exposed workers,” the researchers wrote.
They caution that their findings only show an association and does not prove that exposure to formaldehyde causes cognitive impairment.
Nonetheless, Dr. Letellier encourages health care providers to “be aware of lifetime occupational exposure to target prevention efforts to the identified occupational groups.” This especially includes the care sector where the most people are exposed to formaldehyde, such as nurses, caregivers, and medical technicians.
“Despite the restrictions on the use of formaldehyde due to the better knowledge of its toxicity, especially its carcinogenic effect, formaldehyde is still widely used in many sectors. These results encourage prevention efforts to further limit worker exposure to formaldehyde,” Dr. Letellier said.
Relevant to health care workers
Commenting on the study, Shaheen E. Lakhan, MD, PhD, a neurologist in Newton, Mass., said in an interview that exposure to some degree of formaldehyde is found in every home and workplace, “from the floors to furniture.”
“If you have cigarette smoke in the environment, your exposure rises sharply. When limiting your exposure, it’s not only cancer that you are preventing, but also your brain health,” added Dr. Lakhan, who was not involved with the research.
He said the disturbances in cognitive function noted in the current study were “particularly relevant to health care workers, given the use of formaldehyde in sterilization, tissue pathology processing, and embalming.”
“Interestingly, with only past exposure, there seems to be some degree of cognitive recovery,” but it does not return to a level before any exposure when corrected for age and other factors, Dr. Lakhan said.
Some caveats should also be noted, he pointed out. The study included a French population, but regulators such as the U.S. Occupational Safety and Health Administration and the California Office of Environmental Health Hazard Assessment have strict standards on formaldehyde use in a variety of work settings.
On the flip side, given the COVID-19 pandemic, there has been greater use of chemical disinfectants in and out the workplace, some of which contain formaldehyde, Dr. Lakhan said.
In addition, he noted the study assessed data from 1950 to 2018, so prepandemic.
“A word of advice from a brain doc: Check with your employer on the level of occupational exposure to formaldehyde, heavy metals, and other toxic substances – and cross-reference with your local environmental standards,” Dr. Lakhan concluded.
The research was supported by a grant from the French Agency for Food, Environmental, and Occupational Health & Safety. The investigators and Dr. Lakhan disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Long-term exposure to formaldehyde on the job is linked to cognitive impairment down the road, new research suggests.
In a large observational study of adults aged 45-70 years, researchers found a 17% higher risk for cognitive problems in those with occupational formaldehyde exposure – and higher risks for those with longer duration of exposure.
“The effect of formaldehyde on the brain has been previously shown mainly in animal experiments, but very few studies have been done on humans,” lead author Noemie Letellier, PhD, Institute for Neurosciences of Montpellier, University of Montpellier (France), said in an interview.
“Our results show that being or having been occupationally exposed to formaldehyde is associated with cognitive impairment in a relatively young population,” Dr. Letellier said.
The findings were published online Dec. 22, 2021, in the journal Neurology.
Dose-effect relationship
The investigators assessed a representative sample of 75,322 adults in France (median age, 57.5 years; 53% women). All were part of the CONSTANCES cohort, an observational cohort with a focus on occupational and environmental factors.
A total of 6,026 participants (8%) were exposed to formaldehyde during their careers. Their occupations included nurses, caregivers, medical technicians, workers in the textile, chemistry and metal industries, carpenters, and cleaners.
The researchers calculated lifetime formaldehyde exposure using a French job-exposure matrix created to estimate a person’s exposure to potential health hazards in different occupations.
Individuals were divided into three equal groups according to their years of exposure to formaldehyde. “Low” was considered to be 6 or fewer years of exposure, “medium” was 7-21 years, and “high” was 22 or more years.
Participants were also split into three groups according to their cumulative exposure (total lifetime formaldehyde exposure based on the probability, intensity, and frequency of exposure).
Prevention efforts needed
After adjusting for age, sex, education and other confounders, participants exposed to formaldehyde were at higher risk for global cognitive impairment (adjusted relative risk, 1.17; 95% confidence interval, 1.1-1.2).
Longer duration of exposure and high cumulative lifetime exposure were associated with worse cognitive impairment, “with a dose-effect relationship for exposure duration,” the researchers reported.
Those exposed to formaldehyde for 22 years or more had a 21% higher risk of global cognitive impairment and workers with the highest cumulative exposure had a 19% higher risk of cognitive impairment, compared with workers with no exposure.
Although workers with recent exposure showed higher cognitive impairment, “time may not fully attenuate formaldehyde-associated cognitive deficits, especially in highly exposed but also in moderately exposed workers,” the researchers wrote.
They caution that their findings only show an association and does not prove that exposure to formaldehyde causes cognitive impairment.
Nonetheless, Dr. Letellier encourages health care providers to “be aware of lifetime occupational exposure to target prevention efforts to the identified occupational groups.” This especially includes the care sector where the most people are exposed to formaldehyde, such as nurses, caregivers, and medical technicians.
“Despite the restrictions on the use of formaldehyde due to the better knowledge of its toxicity, especially its carcinogenic effect, formaldehyde is still widely used in many sectors. These results encourage prevention efforts to further limit worker exposure to formaldehyde,” Dr. Letellier said.
Relevant to health care workers
Commenting on the study, Shaheen E. Lakhan, MD, PhD, a neurologist in Newton, Mass., said in an interview that exposure to some degree of formaldehyde is found in every home and workplace, “from the floors to furniture.”
“If you have cigarette smoke in the environment, your exposure rises sharply. When limiting your exposure, it’s not only cancer that you are preventing, but also your brain health,” added Dr. Lakhan, who was not involved with the research.
He said the disturbances in cognitive function noted in the current study were “particularly relevant to health care workers, given the use of formaldehyde in sterilization, tissue pathology processing, and embalming.”
“Interestingly, with only past exposure, there seems to be some degree of cognitive recovery,” but it does not return to a level before any exposure when corrected for age and other factors, Dr. Lakhan said.
Some caveats should also be noted, he pointed out. The study included a French population, but regulators such as the U.S. Occupational Safety and Health Administration and the California Office of Environmental Health Hazard Assessment have strict standards on formaldehyde use in a variety of work settings.
On the flip side, given the COVID-19 pandemic, there has been greater use of chemical disinfectants in and out the workplace, some of which contain formaldehyde, Dr. Lakhan said.
In addition, he noted the study assessed data from 1950 to 2018, so prepandemic.
“A word of advice from a brain doc: Check with your employer on the level of occupational exposure to formaldehyde, heavy metals, and other toxic substances – and cross-reference with your local environmental standards,” Dr. Lakhan concluded.
The research was supported by a grant from the French Agency for Food, Environmental, and Occupational Health & Safety. The investigators and Dr. Lakhan disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Long-term exposure to formaldehyde on the job is linked to cognitive impairment down the road, new research suggests.
In a large observational study of adults aged 45-70 years, researchers found a 17% higher risk for cognitive problems in those with occupational formaldehyde exposure – and higher risks for those with longer duration of exposure.
“The effect of formaldehyde on the brain has been previously shown mainly in animal experiments, but very few studies have been done on humans,” lead author Noemie Letellier, PhD, Institute for Neurosciences of Montpellier, University of Montpellier (France), said in an interview.
“Our results show that being or having been occupationally exposed to formaldehyde is associated with cognitive impairment in a relatively young population,” Dr. Letellier said.
The findings were published online Dec. 22, 2021, in the journal Neurology.
Dose-effect relationship
The investigators assessed a representative sample of 75,322 adults in France (median age, 57.5 years; 53% women). All were part of the CONSTANCES cohort, an observational cohort with a focus on occupational and environmental factors.
A total of 6,026 participants (8%) were exposed to formaldehyde during their careers. Their occupations included nurses, caregivers, medical technicians, workers in the textile, chemistry and metal industries, carpenters, and cleaners.
The researchers calculated lifetime formaldehyde exposure using a French job-exposure matrix created to estimate a person’s exposure to potential health hazards in different occupations.
Individuals were divided into three equal groups according to their years of exposure to formaldehyde. “Low” was considered to be 6 or fewer years of exposure, “medium” was 7-21 years, and “high” was 22 or more years.
Participants were also split into three groups according to their cumulative exposure (total lifetime formaldehyde exposure based on the probability, intensity, and frequency of exposure).
Prevention efforts needed
After adjusting for age, sex, education and other confounders, participants exposed to formaldehyde were at higher risk for global cognitive impairment (adjusted relative risk, 1.17; 95% confidence interval, 1.1-1.2).
Longer duration of exposure and high cumulative lifetime exposure were associated with worse cognitive impairment, “with a dose-effect relationship for exposure duration,” the researchers reported.
Those exposed to formaldehyde for 22 years or more had a 21% higher risk of global cognitive impairment and workers with the highest cumulative exposure had a 19% higher risk of cognitive impairment, compared with workers with no exposure.
Although workers with recent exposure showed higher cognitive impairment, “time may not fully attenuate formaldehyde-associated cognitive deficits, especially in highly exposed but also in moderately exposed workers,” the researchers wrote.
They caution that their findings only show an association and does not prove that exposure to formaldehyde causes cognitive impairment.
Nonetheless, Dr. Letellier encourages health care providers to “be aware of lifetime occupational exposure to target prevention efforts to the identified occupational groups.” This especially includes the care sector where the most people are exposed to formaldehyde, such as nurses, caregivers, and medical technicians.
“Despite the restrictions on the use of formaldehyde due to the better knowledge of its toxicity, especially its carcinogenic effect, formaldehyde is still widely used in many sectors. These results encourage prevention efforts to further limit worker exposure to formaldehyde,” Dr. Letellier said.
Relevant to health care workers
Commenting on the study, Shaheen E. Lakhan, MD, PhD, a neurologist in Newton, Mass., said in an interview that exposure to some degree of formaldehyde is found in every home and workplace, “from the floors to furniture.”
“If you have cigarette smoke in the environment, your exposure rises sharply. When limiting your exposure, it’s not only cancer that you are preventing, but also your brain health,” added Dr. Lakhan, who was not involved with the research.
He said the disturbances in cognitive function noted in the current study were “particularly relevant to health care workers, given the use of formaldehyde in sterilization, tissue pathology processing, and embalming.”
“Interestingly, with only past exposure, there seems to be some degree of cognitive recovery,” but it does not return to a level before any exposure when corrected for age and other factors, Dr. Lakhan said.
Some caveats should also be noted, he pointed out. The study included a French population, but regulators such as the U.S. Occupational Safety and Health Administration and the California Office of Environmental Health Hazard Assessment have strict standards on formaldehyde use in a variety of work settings.
On the flip side, given the COVID-19 pandemic, there has been greater use of chemical disinfectants in and out the workplace, some of which contain formaldehyde, Dr. Lakhan said.
In addition, he noted the study assessed data from 1950 to 2018, so prepandemic.
“A word of advice from a brain doc: Check with your employer on the level of occupational exposure to formaldehyde, heavy metals, and other toxic substances – and cross-reference with your local environmental standards,” Dr. Lakhan concluded.
The research was supported by a grant from the French Agency for Food, Environmental, and Occupational Health & Safety. The investigators and Dr. Lakhan disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM NEUROLOGY