User login
Fish oil: ‘No net benefit’ for depression prevention?
Fish oil supplementation does not help prevent depression or boost mood, new research suggests.
The VITAL-DEP study included more than 18,000 participants. Among adults aged 50 years or older free of clinically relevant depressive symptoms at baseline, long-term use of marine omega-3 fatty acid (omega-3) supplements did not reduce risk for depression or clinically relevant depressive symptoms — or make a difference in the quality of mood.
“While a small increase in risk of depression was inside the statistical margin of significance, there was no harmful or beneficial effect of omega-3 on the overall course of mood during the roughly 5 to 7 years of follow-up,” lead author Olivia I. Okereke, MD, Massachusetts General Hospital and Harvard Medical School, Boston, told Medscape Medical News.
“The takeaway from our study is that there is no net benefit of long-term use of daily omega-3 fish oil supplements for preventing depression or boosting mood,” Okereke said.
The findings were published online Dec. 21 in JAMA.
Assessing general population risk
For many years, experts have recommended omega-3 supplements for reduction in depression recurrence in some high-risk patients, Okereke noted.
“However, there are no guidelines related to the use of omega-3 supplements for preventing depression in the general population. Therefore, we undertook this study to provide clarity in the issue,” she said.
The VITAL-DEP study enrolled 18,353 older adults (mean age, 67.5 years; 49% women). Of these, 16,657 were at risk for incident depression, defined as having no previous history of depression; and 1696 were at risk for recurrent depression, defined as having a history of depression but not having undergone treatment for depression within the past 2 years.
Roughly half the participants were randomly assigned to receive marine omega-3 fatty acids (1 g/d of fish oil, including 465 mg of eicosapentaenoic acid [EPA] and 375 mg of docosahexaenoic acid [DHA]) and the other half to matching placebo for an average of 5.3 years.
“Because of the large sample size and long follow-up, we were able to test the effects of daily omega-3 fish oil supplements on universal prevention of depression in the adult population,” Okereke said.
No significant benefit
Results showed risk for depression or clinically relevant depressive symptoms (total of incident and recurrent cases) was not significantly different between the omega-3 group and the placebo group.
The omega-3 group had 651 depression or clinically relevant depressive symptom events (13.9 per 1000 person-years), and the placebo group had 583 depression or clinically relevant depressive symptom events (12.3 per 1000 person-years). The hazard ratio was 1.13 (95% CI, 1.01 - 1.26; P = .03).
There were also no significant between-group differences in longitudinal mood scores. The mean difference in change in 8-item Patient Health Questionnaire (PHQ-8) score was 0.03 points (95% CI, −0.01 to 0.07; P = .19).
“Patients, physicians, and other clinicians should understand that there are still many reasons for some people, under the guidance of their health care providers, to take omega-3 fish oil supplements,” Okereke noted.
“These supplements increasingly have been found to have benefits for cardiac disease prevention and treatment of inflammatory conditions, in addition to being used for management of existing depressive disorders in some high-risk patients,” she said.
“However, the results of our study indicate there is no reason for adults in the general population to be taking daily omega-3 fish oil supplements solely for the purpose of preventing depression or for maintaining a positive mood,” she added.
Okereke noted, however, that the VITAL-DEP study used 1 g/day of omega-3 fatty acids and there may be a greater benefit from taking higher doses, such as 4 g/day.
Cautionary notes
Commenting on the study for Medscape Medical News, Kuan-Pin Su, MD, PhD, chief of the Department of General Psychiatry, China Medical University, Taichung, Taiwan, highlighted some of the limitations cited by the investigators.
First, depression or depressive symptoms were defined using self-rating scales, which are “convenient to screen for depressive disorders, but a high score obtained on a self-rating scale does not necessarily indicate the presence of depressive psychopathology,” said Su, who was not involved with the research.
He also noted that use of 465 mg of EPA and 375 mg of DHA in VITAL-DEP “might be too low” to have an impact.
Finally, Su said it is “very important to also address the potential for type I error, which makes the secondary and subgroup analyses less reliable.”
VITAL-DEP was supported by a grant from the National Institute of Mental Health. Pronova BioPharma donated the fish oil and matching placebo. Okereke reported receiving royalties from Springer Publishing. Su is a founding committee member of the International Society for Nutritional Psychiatry Research, the board director of the International Society for the Study of Fatty Acids, and an associate editor of the journal Brain, Behavior, and Immunity.
A version of this article first appeared on Medscape.com.
Fish oil supplementation does not help prevent depression or boost mood, new research suggests.
The VITAL-DEP study included more than 18,000 participants. Among adults aged 50 years or older free of clinically relevant depressive symptoms at baseline, long-term use of marine omega-3 fatty acid (omega-3) supplements did not reduce risk for depression or clinically relevant depressive symptoms — or make a difference in the quality of mood.
“While a small increase in risk of depression was inside the statistical margin of significance, there was no harmful or beneficial effect of omega-3 on the overall course of mood during the roughly 5 to 7 years of follow-up,” lead author Olivia I. Okereke, MD, Massachusetts General Hospital and Harvard Medical School, Boston, told Medscape Medical News.
“The takeaway from our study is that there is no net benefit of long-term use of daily omega-3 fish oil supplements for preventing depression or boosting mood,” Okereke said.
The findings were published online Dec. 21 in JAMA.
Assessing general population risk
For many years, experts have recommended omega-3 supplements for reduction in depression recurrence in some high-risk patients, Okereke noted.
“However, there are no guidelines related to the use of omega-3 supplements for preventing depression in the general population. Therefore, we undertook this study to provide clarity in the issue,” she said.
The VITAL-DEP study enrolled 18,353 older adults (mean age, 67.5 years; 49% women). Of these, 16,657 were at risk for incident depression, defined as having no previous history of depression; and 1696 were at risk for recurrent depression, defined as having a history of depression but not having undergone treatment for depression within the past 2 years.
Roughly half the participants were randomly assigned to receive marine omega-3 fatty acids (1 g/d of fish oil, including 465 mg of eicosapentaenoic acid [EPA] and 375 mg of docosahexaenoic acid [DHA]) and the other half to matching placebo for an average of 5.3 years.
“Because of the large sample size and long follow-up, we were able to test the effects of daily omega-3 fish oil supplements on universal prevention of depression in the adult population,” Okereke said.
No significant benefit
Results showed risk for depression or clinically relevant depressive symptoms (total of incident and recurrent cases) was not significantly different between the omega-3 group and the placebo group.
The omega-3 group had 651 depression or clinically relevant depressive symptom events (13.9 per 1000 person-years), and the placebo group had 583 depression or clinically relevant depressive symptom events (12.3 per 1000 person-years). The hazard ratio was 1.13 (95% CI, 1.01 - 1.26; P = .03).
There were also no significant between-group differences in longitudinal mood scores. The mean difference in change in 8-item Patient Health Questionnaire (PHQ-8) score was 0.03 points (95% CI, −0.01 to 0.07; P = .19).
“Patients, physicians, and other clinicians should understand that there are still many reasons for some people, under the guidance of their health care providers, to take omega-3 fish oil supplements,” Okereke noted.
“These supplements increasingly have been found to have benefits for cardiac disease prevention and treatment of inflammatory conditions, in addition to being used for management of existing depressive disorders in some high-risk patients,” she said.
“However, the results of our study indicate there is no reason for adults in the general population to be taking daily omega-3 fish oil supplements solely for the purpose of preventing depression or for maintaining a positive mood,” she added.
Okereke noted, however, that the VITAL-DEP study used 1 g/day of omega-3 fatty acids and there may be a greater benefit from taking higher doses, such as 4 g/day.
Cautionary notes
Commenting on the study for Medscape Medical News, Kuan-Pin Su, MD, PhD, chief of the Department of General Psychiatry, China Medical University, Taichung, Taiwan, highlighted some of the limitations cited by the investigators.
First, depression or depressive symptoms were defined using self-rating scales, which are “convenient to screen for depressive disorders, but a high score obtained on a self-rating scale does not necessarily indicate the presence of depressive psychopathology,” said Su, who was not involved with the research.
He also noted that use of 465 mg of EPA and 375 mg of DHA in VITAL-DEP “might be too low” to have an impact.
Finally, Su said it is “very important to also address the potential for type I error, which makes the secondary and subgroup analyses less reliable.”
VITAL-DEP was supported by a grant from the National Institute of Mental Health. Pronova BioPharma donated the fish oil and matching placebo. Okereke reported receiving royalties from Springer Publishing. Su is a founding committee member of the International Society for Nutritional Psychiatry Research, the board director of the International Society for the Study of Fatty Acids, and an associate editor of the journal Brain, Behavior, and Immunity.
A version of this article first appeared on Medscape.com.
Fish oil supplementation does not help prevent depression or boost mood, new research suggests.
The VITAL-DEP study included more than 18,000 participants. Among adults aged 50 years or older free of clinically relevant depressive symptoms at baseline, long-term use of marine omega-3 fatty acid (omega-3) supplements did not reduce risk for depression or clinically relevant depressive symptoms — or make a difference in the quality of mood.
“While a small increase in risk of depression was inside the statistical margin of significance, there was no harmful or beneficial effect of omega-3 on the overall course of mood during the roughly 5 to 7 years of follow-up,” lead author Olivia I. Okereke, MD, Massachusetts General Hospital and Harvard Medical School, Boston, told Medscape Medical News.
“The takeaway from our study is that there is no net benefit of long-term use of daily omega-3 fish oil supplements for preventing depression or boosting mood,” Okereke said.
The findings were published online Dec. 21 in JAMA.
Assessing general population risk
For many years, experts have recommended omega-3 supplements for reduction in depression recurrence in some high-risk patients, Okereke noted.
“However, there are no guidelines related to the use of omega-3 supplements for preventing depression in the general population. Therefore, we undertook this study to provide clarity in the issue,” she said.
The VITAL-DEP study enrolled 18,353 older adults (mean age, 67.5 years; 49% women). Of these, 16,657 were at risk for incident depression, defined as having no previous history of depression; and 1696 were at risk for recurrent depression, defined as having a history of depression but not having undergone treatment for depression within the past 2 years.
Roughly half the participants were randomly assigned to receive marine omega-3 fatty acids (1 g/d of fish oil, including 465 mg of eicosapentaenoic acid [EPA] and 375 mg of docosahexaenoic acid [DHA]) and the other half to matching placebo for an average of 5.3 years.
“Because of the large sample size and long follow-up, we were able to test the effects of daily omega-3 fish oil supplements on universal prevention of depression in the adult population,” Okereke said.
No significant benefit
Results showed risk for depression or clinically relevant depressive symptoms (total of incident and recurrent cases) was not significantly different between the omega-3 group and the placebo group.
The omega-3 group had 651 depression or clinically relevant depressive symptom events (13.9 per 1000 person-years), and the placebo group had 583 depression or clinically relevant depressive symptom events (12.3 per 1000 person-years). The hazard ratio was 1.13 (95% CI, 1.01 - 1.26; P = .03).
There were also no significant between-group differences in longitudinal mood scores. The mean difference in change in 8-item Patient Health Questionnaire (PHQ-8) score was 0.03 points (95% CI, −0.01 to 0.07; P = .19).
“Patients, physicians, and other clinicians should understand that there are still many reasons for some people, under the guidance of their health care providers, to take omega-3 fish oil supplements,” Okereke noted.
“These supplements increasingly have been found to have benefits for cardiac disease prevention and treatment of inflammatory conditions, in addition to being used for management of existing depressive disorders in some high-risk patients,” she said.
“However, the results of our study indicate there is no reason for adults in the general population to be taking daily omega-3 fish oil supplements solely for the purpose of preventing depression or for maintaining a positive mood,” she added.
Okereke noted, however, that the VITAL-DEP study used 1 g/day of omega-3 fatty acids and there may be a greater benefit from taking higher doses, such as 4 g/day.
Cautionary notes
Commenting on the study for Medscape Medical News, Kuan-Pin Su, MD, PhD, chief of the Department of General Psychiatry, China Medical University, Taichung, Taiwan, highlighted some of the limitations cited by the investigators.
First, depression or depressive symptoms were defined using self-rating scales, which are “convenient to screen for depressive disorders, but a high score obtained on a self-rating scale does not necessarily indicate the presence of depressive psychopathology,” said Su, who was not involved with the research.
He also noted that use of 465 mg of EPA and 375 mg of DHA in VITAL-DEP “might be too low” to have an impact.
Finally, Su said it is “very important to also address the potential for type I error, which makes the secondary and subgroup analyses less reliable.”
VITAL-DEP was supported by a grant from the National Institute of Mental Health. Pronova BioPharma donated the fish oil and matching placebo. Okereke reported receiving royalties from Springer Publishing. Su is a founding committee member of the International Society for Nutritional Psychiatry Research, the board director of the International Society for the Study of Fatty Acids, and an associate editor of the journal Brain, Behavior, and Immunity.
A version of this article first appeared on Medscape.com.
FDA grants new indication to lumateperone (Caplyta) for bipolar depression
The Food and Drug Administration has expanded approval of lumateperone (Caplyta) to include treatment of adults with depressive episodes associated with bipolar I and II disorder, as monotherapy or adjunctive therapy with lithium or valproate.

This makes lumateperone the only FDA-approved drug for this indication.
“The efficacy, and favorable safety and tolerability profile, make Caplyta an important treatment option for the millions of patients living with bipolar I or II depression and represents a major development for these patients,” Roger McIntyre, MD, professor of psychiatry and pharmacology, University of Toronto, and head of the mood disorders psychopharmacology unit, said in a company news release.
Lumateperone was first approved by the FDA in 2019 for the treatment of adults with schizophrenia.
‘Positioned to launch immediately’
that showed treatment with lumateperone, alone or with lithium or valproate, significantly improved depressive symptoms for patients with major depressive episodes associated with bipolar I and bipolar II disorders.
In these studies, treatment with a 42-mg once-daily dose was associated with significantly greater improvement from baseline in Montgomery-Åsberg Depression Rating Scale score versus placebo.
Lumateperone also showed a statistically significant improvement in the key secondary endpoint relating to clinical global impression of bipolar disorder.
Somnolence/sedation, dizziness, nausea, and dry mouth were the most commonly reported adverse events associated with the medication. Minimal changes were observed in weight and vital signs and in results of metabolic or endocrine assessments. Incidence of extrapyramidal symptom–related events was low and was similar to those with placebo.
Sharon Mates, PhD, chairman and CEO of Intra-Cellular Therapies, noted in the same press release that the company is “positioned to launch immediately and are excited to offer Caplyta to the millions of patients living with bipolar depression.”
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has expanded approval of lumateperone (Caplyta) to include treatment of adults with depressive episodes associated with bipolar I and II disorder, as monotherapy or adjunctive therapy with lithium or valproate.
This makes lumateperone the only FDA-approved drug for this indication.
“The efficacy, and favorable safety and tolerability profile, make Caplyta an important treatment option for the millions of patients living with bipolar I or II depression and represents a major development for these patients,” Roger McIntyre, MD, professor of psychiatry and pharmacology, University of Toronto, and head of the mood disorders psychopharmacology unit, said in a company news release.
Lumateperone was first approved by the FDA in 2019 for the treatment of adults with schizophrenia.
‘Positioned to launch immediately’
that showed treatment with lumateperone, alone or with lithium or valproate, significantly improved depressive symptoms for patients with major depressive episodes associated with bipolar I and bipolar II disorders.
In these studies, treatment with a 42-mg once-daily dose was associated with significantly greater improvement from baseline in Montgomery-Åsberg Depression Rating Scale score versus placebo.
Lumateperone also showed a statistically significant improvement in the key secondary endpoint relating to clinical global impression of bipolar disorder.
Somnolence/sedation, dizziness, nausea, and dry mouth were the most commonly reported adverse events associated with the medication. Minimal changes were observed in weight and vital signs and in results of metabolic or endocrine assessments. Incidence of extrapyramidal symptom–related events was low and was similar to those with placebo.
Sharon Mates, PhD, chairman and CEO of Intra-Cellular Therapies, noted in the same press release that the company is “positioned to launch immediately and are excited to offer Caplyta to the millions of patients living with bipolar depression.”
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has expanded approval of lumateperone (Caplyta) to include treatment of adults with depressive episodes associated with bipolar I and II disorder, as monotherapy or adjunctive therapy with lithium or valproate.
This makes lumateperone the only FDA-approved drug for this indication.
“The efficacy, and favorable safety and tolerability profile, make Caplyta an important treatment option for the millions of patients living with bipolar I or II depression and represents a major development for these patients,” Roger McIntyre, MD, professor of psychiatry and pharmacology, University of Toronto, and head of the mood disorders psychopharmacology unit, said in a company news release.
Lumateperone was first approved by the FDA in 2019 for the treatment of adults with schizophrenia.
‘Positioned to launch immediately’
that showed treatment with lumateperone, alone or with lithium or valproate, significantly improved depressive symptoms for patients with major depressive episodes associated with bipolar I and bipolar II disorders.
In these studies, treatment with a 42-mg once-daily dose was associated with significantly greater improvement from baseline in Montgomery-Åsberg Depression Rating Scale score versus placebo.
Lumateperone also showed a statistically significant improvement in the key secondary endpoint relating to clinical global impression of bipolar disorder.
Somnolence/sedation, dizziness, nausea, and dry mouth were the most commonly reported adverse events associated with the medication. Minimal changes were observed in weight and vital signs and in results of metabolic or endocrine assessments. Incidence of extrapyramidal symptom–related events was low and was similar to those with placebo.
Sharon Mates, PhD, chairman and CEO of Intra-Cellular Therapies, noted in the same press release that the company is “positioned to launch immediately and are excited to offer Caplyta to the millions of patients living with bipolar depression.”
Full prescribing information is available online.
A version of this article first appeared on Medscape.com.
Class I recall of percutaneous thrombolytic device
Arrow International, a subsidiary of Teleflex, has recalled a total of 3,241 Arrow-Trerotola over-the-wire 7FR percutaneous thrombolytic device (PTD) kits because of the risk of the orange inner lumen of the catheter’s tip component separating from the basket.
The U.S. Food and Drug Administration has identified this as a Class I recall, the most serious type, because of the potential for serious injury or death.
The recalled kits include a rotatable catheter with an outer sheath and an inner cable with a self-expanding basket. The Arrow-Trerotola PTD catheter is used with the Arrow rotator drive unit to remove clots in patients with arteriovenous fistulas and synthetic dialysis grafts.
“If the orange inner lumen separates from the basket, it may fracture and detach and block the blood vessel(s),” the FDA says in the recall notice posted on the FDA website.
“If the orange inner lumen detaches from the basket, health consequences depend upon where the fractured tip component embolizes. If the embolization is local to the treatment target site, retrieval may be attempted, requiring an additional intervention and consequent delay of therapy,” the agency notes.
“In some cases, the embolization could be central or possibly even to the heart or pulmonary arteries. This may lead to serious adverse events such as vessel damage, need for additional medical procedures, or possibly death,” the agency says.
To date, there have been seven complaints and no injuries or deaths reported for this device.
The recalled devices were distributed in the United States between Nov. 1, 2019, and July 31, 2021. Product codes and lot numbers pertaining to the devices are listed on the FDA website.
Teleflex has sent an urgent field safety notice to customers requesting that they check inventory for affected product and remove and quarantine all recalled product.
Customers are also asked to complete the enclosed acknowledgement form and fax it to 1-855-419-8507 (attention: customer service) or e-mail the form to recalls@teleflex.com.
Customers with recalled product service will be contacted by a company representative with instructions for returning any recalled products.
Customers who have questions about this recall should contact Teleflex customer service by phone at 1-866-396-2111, by fax at 1-855-419-8507, or by email at Recalls@teleflex.com.
Health care providers can report adverse reactions or quality problems they experience using these devices to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
Arrow International, a subsidiary of Teleflex, has recalled a total of 3,241 Arrow-Trerotola over-the-wire 7FR percutaneous thrombolytic device (PTD) kits because of the risk of the orange inner lumen of the catheter’s tip component separating from the basket.
The U.S. Food and Drug Administration has identified this as a Class I recall, the most serious type, because of the potential for serious injury or death.
The recalled kits include a rotatable catheter with an outer sheath and an inner cable with a self-expanding basket. The Arrow-Trerotola PTD catheter is used with the Arrow rotator drive unit to remove clots in patients with arteriovenous fistulas and synthetic dialysis grafts.
“If the orange inner lumen separates from the basket, it may fracture and detach and block the blood vessel(s),” the FDA says in the recall notice posted on the FDA website.
“If the orange inner lumen detaches from the basket, health consequences depend upon where the fractured tip component embolizes. If the embolization is local to the treatment target site, retrieval may be attempted, requiring an additional intervention and consequent delay of therapy,” the agency notes.
“In some cases, the embolization could be central or possibly even to the heart or pulmonary arteries. This may lead to serious adverse events such as vessel damage, need for additional medical procedures, or possibly death,” the agency says.
To date, there have been seven complaints and no injuries or deaths reported for this device.
The recalled devices were distributed in the United States between Nov. 1, 2019, and July 31, 2021. Product codes and lot numbers pertaining to the devices are listed on the FDA website.
Teleflex has sent an urgent field safety notice to customers requesting that they check inventory for affected product and remove and quarantine all recalled product.
Customers are also asked to complete the enclosed acknowledgement form and fax it to 1-855-419-8507 (attention: customer service) or e-mail the form to recalls@teleflex.com.
Customers with recalled product service will be contacted by a company representative with instructions for returning any recalled products.
Customers who have questions about this recall should contact Teleflex customer service by phone at 1-866-396-2111, by fax at 1-855-419-8507, or by email at Recalls@teleflex.com.
Health care providers can report adverse reactions or quality problems they experience using these devices to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
Arrow International, a subsidiary of Teleflex, has recalled a total of 3,241 Arrow-Trerotola over-the-wire 7FR percutaneous thrombolytic device (PTD) kits because of the risk of the orange inner lumen of the catheter’s tip component separating from the basket.
The U.S. Food and Drug Administration has identified this as a Class I recall, the most serious type, because of the potential for serious injury or death.
The recalled kits include a rotatable catheter with an outer sheath and an inner cable with a self-expanding basket. The Arrow-Trerotola PTD catheter is used with the Arrow rotator drive unit to remove clots in patients with arteriovenous fistulas and synthetic dialysis grafts.
“If the orange inner lumen separates from the basket, it may fracture and detach and block the blood vessel(s),” the FDA says in the recall notice posted on the FDA website.
“If the orange inner lumen detaches from the basket, health consequences depend upon where the fractured tip component embolizes. If the embolization is local to the treatment target site, retrieval may be attempted, requiring an additional intervention and consequent delay of therapy,” the agency notes.
“In some cases, the embolization could be central or possibly even to the heart or pulmonary arteries. This may lead to serious adverse events such as vessel damage, need for additional medical procedures, or possibly death,” the agency says.
To date, there have been seven complaints and no injuries or deaths reported for this device.
The recalled devices were distributed in the United States between Nov. 1, 2019, and July 31, 2021. Product codes and lot numbers pertaining to the devices are listed on the FDA website.
Teleflex has sent an urgent field safety notice to customers requesting that they check inventory for affected product and remove and quarantine all recalled product.
Customers are also asked to complete the enclosed acknowledgement form and fax it to 1-855-419-8507 (attention: customer service) or e-mail the form to recalls@teleflex.com.
Customers with recalled product service will be contacted by a company representative with instructions for returning any recalled products.
Customers who have questions about this recall should contact Teleflex customer service by phone at 1-866-396-2111, by fax at 1-855-419-8507, or by email at Recalls@teleflex.com.
Health care providers can report adverse reactions or quality problems they experience using these devices to the FDA’s MedWatch program.
A version of this article first appeared on Medscape.com.
FDA approves new myasthenia gravis drug
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
FDA updates risks, cautions for clotting-bleeding disorder on Janssen COVID-19 vaccine
Updated Janssen/Johnson & Johnson COVID-19 vaccine fact sheets for health care professionals and the general public now include a contraindication to its use in persons with a history of thrombosis with thrombocytopenia after receiving it “or any other adenovirus-vectored COVID-19 vaccine,” the U.S. Food and Drug Administration has announced.

Thrombosis with thrombocytopenia syndrome (TTS) – thrombocytopenia and increased bleeding risk along with documented thrombosis – after administration of the Janssen Ad26.COV2.S vaccine remains rare. But over all age groups, about one in seven cases have been fatal, said the agency.
the provider fact sheet states.
Although TTS associated with the Janssen COVID-19 vaccine has been reported in men and women aged 18 and older, the highest reported rate has been for women aged 30-49, the agency states. The rate in that group has been about 1 case per 100,000 doses administered.
Symptoms of TTS may occur 1-2 weeks after administration of the Janssen COVID-19 vaccine, the FDA says, based on data from the Vaccine Adverse Events Reporting System (VAERS).
Its clinical course shares features with autoimmune heparin-induced thrombocytopenia. In individuals with suspected TTS following receipt of the Janssen COVID-19 vaccine, the agency cautions, the use of heparin “may be harmful and alternative treatments may be needed. Consultation with hematology specialists is strongly recommended.”
The apparent excess risk of TTS remains under investigation, but “the FDA continues to find that the known and potential benefits of the Janssen COVID-19 vaccine outweigh its known and potential risks in individuals 18 years of age and older,” the agency states.
A version of this article first appeared on Medscape.com.
Updated Janssen/Johnson & Johnson COVID-19 vaccine fact sheets for health care professionals and the general public now include a contraindication to its use in persons with a history of thrombosis with thrombocytopenia after receiving it “or any other adenovirus-vectored COVID-19 vaccine,” the U.S. Food and Drug Administration has announced.
Thrombosis with thrombocytopenia syndrome (TTS) – thrombocytopenia and increased bleeding risk along with documented thrombosis – after administration of the Janssen Ad26.COV2.S vaccine remains rare. But over all age groups, about one in seven cases have been fatal, said the agency.
the provider fact sheet states.
Although TTS associated with the Janssen COVID-19 vaccine has been reported in men and women aged 18 and older, the highest reported rate has been for women aged 30-49, the agency states. The rate in that group has been about 1 case per 100,000 doses administered.
Symptoms of TTS may occur 1-2 weeks after administration of the Janssen COVID-19 vaccine, the FDA says, based on data from the Vaccine Adverse Events Reporting System (VAERS).
Its clinical course shares features with autoimmune heparin-induced thrombocytopenia. In individuals with suspected TTS following receipt of the Janssen COVID-19 vaccine, the agency cautions, the use of heparin “may be harmful and alternative treatments may be needed. Consultation with hematology specialists is strongly recommended.”
The apparent excess risk of TTS remains under investigation, but “the FDA continues to find that the known and potential benefits of the Janssen COVID-19 vaccine outweigh its known and potential risks in individuals 18 years of age and older,” the agency states.
A version of this article first appeared on Medscape.com.
Updated Janssen/Johnson & Johnson COVID-19 vaccine fact sheets for health care professionals and the general public now include a contraindication to its use in persons with a history of thrombosis with thrombocytopenia after receiving it “or any other adenovirus-vectored COVID-19 vaccine,” the U.S. Food and Drug Administration has announced.
Thrombosis with thrombocytopenia syndrome (TTS) – thrombocytopenia and increased bleeding risk along with documented thrombosis – after administration of the Janssen Ad26.COV2.S vaccine remains rare. But over all age groups, about one in seven cases have been fatal, said the agency.
the provider fact sheet states.
Although TTS associated with the Janssen COVID-19 vaccine has been reported in men and women aged 18 and older, the highest reported rate has been for women aged 30-49, the agency states. The rate in that group has been about 1 case per 100,000 doses administered.
Symptoms of TTS may occur 1-2 weeks after administration of the Janssen COVID-19 vaccine, the FDA says, based on data from the Vaccine Adverse Events Reporting System (VAERS).
Its clinical course shares features with autoimmune heparin-induced thrombocytopenia. In individuals with suspected TTS following receipt of the Janssen COVID-19 vaccine, the agency cautions, the use of heparin “may be harmful and alternative treatments may be needed. Consultation with hematology specialists is strongly recommended.”
The apparent excess risk of TTS remains under investigation, but “the FDA continues to find that the known and potential benefits of the Janssen COVID-19 vaccine outweigh its known and potential risks in individuals 18 years of age and older,” the agency states.
A version of this article first appeared on Medscape.com.
A pandemic silver lining? Dramatic drop in teen drug use
Illicit drug use among U.S. teenagers dropped sharply in 2021, likely because of stay-at-home orders and other restrictions on social activities due to the COVID-19 pandemic.
The latest findings, from the Monitoring the Future survey, represent the largest 1-year decrease in overall illicit drug use reported since the survey began in 1975.
“We have never seen such dramatic decreases in drug use among teens in just a 1-year period,” Nora Volkow, MD, director of the National Institute on Drug Abuse (NIDA), said in a news release.
“These data are unprecedented and highlight one unexpected potential consequence of the COVID-19 pandemic, which caused seismic shifts in the day-to-day lives of adolescents,” said Dr. Volkow.
The annual Monitoring the Future survey is conducted by researchers at the University of Michigan, Ann Arbor, and funded by NIDA, to assess drug and alcohol use and related attitudes among adolescent students across the United States.
This year’s self-reported survey included 32,260 students in grades 8, 10, and 12 across 319 public and private schools.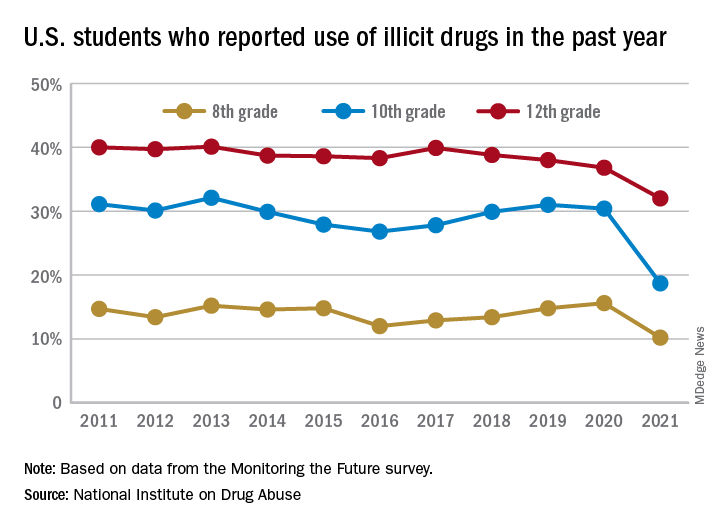
Compared with 2020, the percentage of students reporting any illicit drug use (other than marijuana) in 2021 decreased significantly for 8th graders (down 5.4%), 10th graders (down 11.7%), and 12th graders (down 4.8%).
For alcohol, about 47% of 12th graders and 29% of 10th graders said they drank alcohol in 2021, down significantly from 55% and 41%, respectively, in 2020. The percentage of 8th graders who said they drank alcohol remained stable (17% in 2021 and 20% in 2020).
For teen vaping, about 27% of 12th graders and 20% of 10th graders said they had vaped nicotine in 2021, down significantly from nearly 35% and 31%, respectively, in 2020. Fewer 8th graders also vaped nicotine in 2021 compared with 2020 (12% vs. 17%).
For marijuana, use dropped significantly for all three grades in 2021 compared with 2020. About 31% of 12th graders and 17% of 10th graders said they used marijuana in 2021, down from 35% and 28% in 2020. Among 8th graders, 7% used marijuana in 2021, down from 11% in 2020.
The latest survey also shows significant declines in use of a range of other drugs for many of the age cohorts, including cocaine, hallucinogens, and nonmedical use of amphetamines, tranquilizers, and prescription opioids.
“We knew that this year’s data would illuminate how the COVID-19 pandemic may have impacted substance use among young people, and in the coming years, we will find out whether those impacts are long-lasting as we continue tracking the drug use patterns of these unique cohorts of adolescents,” Richard A. Miech, PhD, who heads the Monitoring the Future study at the University of Michigan, said in the news release.
“Moving forward, it will be crucial to identify the pivotal elements of this past year that contributed to decreased drug use – whether related to drug availability, family involvement, differences in peer pressure, or other factors – and harness them to inform future prevention efforts,” Dr. Volkow added.
In 2021, students across all age groups reported moderate increases in feelings of boredom, anxiety, depression, loneliness, worry, difficulty sleeping, and other negative mental health indicators since the beginning of the pandemic.
A version of this article first appeared on Medscape.com.
Illicit drug use among U.S. teenagers dropped sharply in 2021, likely because of stay-at-home orders and other restrictions on social activities due to the COVID-19 pandemic.
The latest findings, from the Monitoring the Future survey, represent the largest 1-year decrease in overall illicit drug use reported since the survey began in 1975.
“We have never seen such dramatic decreases in drug use among teens in just a 1-year period,” Nora Volkow, MD, director of the National Institute on Drug Abuse (NIDA), said in a news release.
“These data are unprecedented and highlight one unexpected potential consequence of the COVID-19 pandemic, which caused seismic shifts in the day-to-day lives of adolescents,” said Dr. Volkow.
The annual Monitoring the Future survey is conducted by researchers at the University of Michigan, Ann Arbor, and funded by NIDA, to assess drug and alcohol use and related attitudes among adolescent students across the United States.
This year’s self-reported survey included 32,260 students in grades 8, 10, and 12 across 319 public and private schools.
Compared with 2020, the percentage of students reporting any illicit drug use (other than marijuana) in 2021 decreased significantly for 8th graders (down 5.4%), 10th graders (down 11.7%), and 12th graders (down 4.8%).
For alcohol, about 47% of 12th graders and 29% of 10th graders said they drank alcohol in 2021, down significantly from 55% and 41%, respectively, in 2020. The percentage of 8th graders who said they drank alcohol remained stable (17% in 2021 and 20% in 2020).
For teen vaping, about 27% of 12th graders and 20% of 10th graders said they had vaped nicotine in 2021, down significantly from nearly 35% and 31%, respectively, in 2020. Fewer 8th graders also vaped nicotine in 2021 compared with 2020 (12% vs. 17%).
For marijuana, use dropped significantly for all three grades in 2021 compared with 2020. About 31% of 12th graders and 17% of 10th graders said they used marijuana in 2021, down from 35% and 28% in 2020. Among 8th graders, 7% used marijuana in 2021, down from 11% in 2020.
The latest survey also shows significant declines in use of a range of other drugs for many of the age cohorts, including cocaine, hallucinogens, and nonmedical use of amphetamines, tranquilizers, and prescription opioids.
“We knew that this year’s data would illuminate how the COVID-19 pandemic may have impacted substance use among young people, and in the coming years, we will find out whether those impacts are long-lasting as we continue tracking the drug use patterns of these unique cohorts of adolescents,” Richard A. Miech, PhD, who heads the Monitoring the Future study at the University of Michigan, said in the news release.
“Moving forward, it will be crucial to identify the pivotal elements of this past year that contributed to decreased drug use – whether related to drug availability, family involvement, differences in peer pressure, or other factors – and harness them to inform future prevention efforts,” Dr. Volkow added.
In 2021, students across all age groups reported moderate increases in feelings of boredom, anxiety, depression, loneliness, worry, difficulty sleeping, and other negative mental health indicators since the beginning of the pandemic.
A version of this article first appeared on Medscape.com.
Illicit drug use among U.S. teenagers dropped sharply in 2021, likely because of stay-at-home orders and other restrictions on social activities due to the COVID-19 pandemic.
The latest findings, from the Monitoring the Future survey, represent the largest 1-year decrease in overall illicit drug use reported since the survey began in 1975.
“We have never seen such dramatic decreases in drug use among teens in just a 1-year period,” Nora Volkow, MD, director of the National Institute on Drug Abuse (NIDA), said in a news release.
“These data are unprecedented and highlight one unexpected potential consequence of the COVID-19 pandemic, which caused seismic shifts in the day-to-day lives of adolescents,” said Dr. Volkow.
The annual Monitoring the Future survey is conducted by researchers at the University of Michigan, Ann Arbor, and funded by NIDA, to assess drug and alcohol use and related attitudes among adolescent students across the United States.
This year’s self-reported survey included 32,260 students in grades 8, 10, and 12 across 319 public and private schools.
Compared with 2020, the percentage of students reporting any illicit drug use (other than marijuana) in 2021 decreased significantly for 8th graders (down 5.4%), 10th graders (down 11.7%), and 12th graders (down 4.8%).
For alcohol, about 47% of 12th graders and 29% of 10th graders said they drank alcohol in 2021, down significantly from 55% and 41%, respectively, in 2020. The percentage of 8th graders who said they drank alcohol remained stable (17% in 2021 and 20% in 2020).
For teen vaping, about 27% of 12th graders and 20% of 10th graders said they had vaped nicotine in 2021, down significantly from nearly 35% and 31%, respectively, in 2020. Fewer 8th graders also vaped nicotine in 2021 compared with 2020 (12% vs. 17%).
For marijuana, use dropped significantly for all three grades in 2021 compared with 2020. About 31% of 12th graders and 17% of 10th graders said they used marijuana in 2021, down from 35% and 28% in 2020. Among 8th graders, 7% used marijuana in 2021, down from 11% in 2020.
The latest survey also shows significant declines in use of a range of other drugs for many of the age cohorts, including cocaine, hallucinogens, and nonmedical use of amphetamines, tranquilizers, and prescription opioids.
“We knew that this year’s data would illuminate how the COVID-19 pandemic may have impacted substance use among young people, and in the coming years, we will find out whether those impacts are long-lasting as we continue tracking the drug use patterns of these unique cohorts of adolescents,” Richard A. Miech, PhD, who heads the Monitoring the Future study at the University of Michigan, said in the news release.
“Moving forward, it will be crucial to identify the pivotal elements of this past year that contributed to decreased drug use – whether related to drug availability, family involvement, differences in peer pressure, or other factors – and harness them to inform future prevention efforts,” Dr. Volkow added.
In 2021, students across all age groups reported moderate increases in feelings of boredom, anxiety, depression, loneliness, worry, difficulty sleeping, and other negative mental health indicators since the beginning of the pandemic.
A version of this article first appeared on Medscape.com.
Cold snare polypectomy works for larger colorectal polyps too
Large colorectal polyps up to 15 mm in size can be safely and effectively removed with cold snare polypectomy (CSP), with a low incomplete resection rate, according to a single-center, observational study from China.
Multiple current guidelines recommend CSP for removing diminutive (≤5 mm) and small (6-9 mm) polyps, citing clinical data demonstrating that such an approach leads to high complete resection rates and a good safety profile.
However, the use of CSP for removing larger colorectal polyps (≥10 mm) remains controversial, with only limited safety and efficacy data.
Yuqi He, MD, from the department of gastroenterology at the Seventh Medical Center of Chinese PLA General Hospital in Beijing, and colleagues evaluated 440 neoplastic polyps removed by CSP from 261 patients (mean age, 56.6 years; 166 men).
Indications for colonoscopy were screening (53%), diagnostic (17%), polyp surveillance (24%), and other (6%).
Of the 440 polyps, 353 (80%) were small (5-9 mm) and 87 (20%) were large (10-15 mm); 379 (86%) were adenomas, 59 (13%) were sessile serrated lesions (SSL), and 2 (<1%) were high-grade dysplasia.
For all polyps (5-15 mm), the incomplete resection rate (primary outcome) was 2.27%. Incomplete resection was more common for large polyps (3.45%) compared with small polyps (1.98%), but the difference was not statistically significant (P = .411).
In univariate analysis, factors associated with incomplete resection were SSL, piecemeal resection, and prolonged resection time.
In multivariate regression analysis, independent risk factors for incomplete resection were SSL (odds ratio, 6.45; 95% CI, 1.48-28.03; P = .013) and prolonged resection time (OR, 7.39; 95% CI, 1.48-36.96; P = .015).
Immediate bleeding was more common with resection of large polyps (6.9% vs. 1.42%, P = .003).
There were no recurrences on follow-up colonoscopy in 37 cases with large polyps, further supporting the efficacy of CSP for this size group, the researchers say.
Their study was published online Nov. 16 in Clinical Gastroenterology and Hepatology.
Important study, lingering questions, experts say
Reached for comment, Rajesh N. Keswani, MD, of Northwestern University Feinberg School of Medicine, Chicago, said, “It is standard practice to remove polyps <10 mm using cold techniques. We generally advocate for use of a cold snare for polyps 3-9 mm in size; either large cold forceps or a cold snare can be used for polyps 1-2 mm in size,” a recommendation also conveyed in the American Gastroenterological Association’s recent clinical practice update.
“It is still unclear whether cold polypectomy techniques are appropriate for larger lesions,” Dr. Keswani, who wasn’t involved in the study, told this news organization.
“For larger polyps that require piecemeal technique (generally >2 cm), there are multiple small studies showing that resection using cold snare piecemeal technique may be safe and effective, at minimum for serrated lesions. There has been enough interest in this approach that is being formally studied in a large multicenter trial,” he noted.
“Based on the safety and efficacy data thus far for cold snare polypectomy in larger serrated lesions, it would be difficult to think that it would not be a safe choice for serrated lesions 10-20 mm,” Dr. Keswani commented.
However, the data remain “unclear for adenomatous lesions 10-20 mm in size. Unfortunately, in this study only 87 polyps 10-15 mm [in size] were removed, and a proportion were serrated and a proportion were adenomas,” he said in an interview.
“Thus, I don’t think this study is able to answer one of our key remaining polypectomy questions, which is whether cold snare techniques are the optimal approach for adenomas >10 mm in size,” said Dr. Keswani.
Also weighing in was Emre Gorgun, MD, with the Cleveland Clinic, who said it’s “an important study in the sense it raises awareness to the role of cold snaring techniques, especially since it is less expensive, quicker, and safe.”
“Late complications related to possibly using energy might be also eliminated. However, results for expanding the practice to larger polyps up to 15 mm should be taken cautiously into consideration,” Dr. Gorgun told this news organization.
“The threat of bleeding in high-risk patients, for example with a history of anticoagulation use, were not reported. Other patient-related factors can increase these risks as well. Additionally, the long-term follow-up is not reported,” commented Dr. Gorgun, who was not associated with the research.
“This study triggers the idea of using cold snaring in larger polyps; however, the results should be further reproduced and verified for long-term consequences and safety,” he concluded.
The authors have disclosed no relevant financial relationships. Dr. Keswani is a consultant for Boston Scientific and Neptune Medical and receives research support from Virgo. Dr. Gorgun is a consultant for Boston Scientific. Support for the study was provided b y grants from Project of Army Special Care and Beijing Municipal Science and Technology Commission.
A version of this article first appeared on Medscape.com.
Large colorectal polyps up to 15 mm in size can be safely and effectively removed with cold snare polypectomy (CSP), with a low incomplete resection rate, according to a single-center, observational study from China.
Multiple current guidelines recommend CSP for removing diminutive (≤5 mm) and small (6-9 mm) polyps, citing clinical data demonstrating that such an approach leads to high complete resection rates and a good safety profile.
However, the use of CSP for removing larger colorectal polyps (≥10 mm) remains controversial, with only limited safety and efficacy data.
Yuqi He, MD, from the department of gastroenterology at the Seventh Medical Center of Chinese PLA General Hospital in Beijing, and colleagues evaluated 440 neoplastic polyps removed by CSP from 261 patients (mean age, 56.6 years; 166 men).
Indications for colonoscopy were screening (53%), diagnostic (17%), polyp surveillance (24%), and other (6%).
Of the 440 polyps, 353 (80%) were small (5-9 mm) and 87 (20%) were large (10-15 mm); 379 (86%) were adenomas, 59 (13%) were sessile serrated lesions (SSL), and 2 (<1%) were high-grade dysplasia.
For all polyps (5-15 mm), the incomplete resection rate (primary outcome) was 2.27%. Incomplete resection was more common for large polyps (3.45%) compared with small polyps (1.98%), but the difference was not statistically significant (P = .411).
In univariate analysis, factors associated with incomplete resection were SSL, piecemeal resection, and prolonged resection time.
In multivariate regression analysis, independent risk factors for incomplete resection were SSL (odds ratio, 6.45; 95% CI, 1.48-28.03; P = .013) and prolonged resection time (OR, 7.39; 95% CI, 1.48-36.96; P = .015).
Immediate bleeding was more common with resection of large polyps (6.9% vs. 1.42%, P = .003).
There were no recurrences on follow-up colonoscopy in 37 cases with large polyps, further supporting the efficacy of CSP for this size group, the researchers say.
Their study was published online Nov. 16 in Clinical Gastroenterology and Hepatology.
Important study, lingering questions, experts say
Reached for comment, Rajesh N. Keswani, MD, of Northwestern University Feinberg School of Medicine, Chicago, said, “It is standard practice to remove polyps <10 mm using cold techniques. We generally advocate for use of a cold snare for polyps 3-9 mm in size; either large cold forceps or a cold snare can be used for polyps 1-2 mm in size,” a recommendation also conveyed in the American Gastroenterological Association’s recent clinical practice update.
“It is still unclear whether cold polypectomy techniques are appropriate for larger lesions,” Dr. Keswani, who wasn’t involved in the study, told this news organization.
“For larger polyps that require piecemeal technique (generally >2 cm), there are multiple small studies showing that resection using cold snare piecemeal technique may be safe and effective, at minimum for serrated lesions. There has been enough interest in this approach that is being formally studied in a large multicenter trial,” he noted.
“Based on the safety and efficacy data thus far for cold snare polypectomy in larger serrated lesions, it would be difficult to think that it would not be a safe choice for serrated lesions 10-20 mm,” Dr. Keswani commented.
However, the data remain “unclear for adenomatous lesions 10-20 mm in size. Unfortunately, in this study only 87 polyps 10-15 mm [in size] were removed, and a proportion were serrated and a proportion were adenomas,” he said in an interview.
“Thus, I don’t think this study is able to answer one of our key remaining polypectomy questions, which is whether cold snare techniques are the optimal approach for adenomas >10 mm in size,” said Dr. Keswani.
Also weighing in was Emre Gorgun, MD, with the Cleveland Clinic, who said it’s “an important study in the sense it raises awareness to the role of cold snaring techniques, especially since it is less expensive, quicker, and safe.”
“Late complications related to possibly using energy might be also eliminated. However, results for expanding the practice to larger polyps up to 15 mm should be taken cautiously into consideration,” Dr. Gorgun told this news organization.
“The threat of bleeding in high-risk patients, for example with a history of anticoagulation use, were not reported. Other patient-related factors can increase these risks as well. Additionally, the long-term follow-up is not reported,” commented Dr. Gorgun, who was not associated with the research.
“This study triggers the idea of using cold snaring in larger polyps; however, the results should be further reproduced and verified for long-term consequences and safety,” he concluded.
The authors have disclosed no relevant financial relationships. Dr. Keswani is a consultant for Boston Scientific and Neptune Medical and receives research support from Virgo. Dr. Gorgun is a consultant for Boston Scientific. Support for the study was provided b y grants from Project of Army Special Care and Beijing Municipal Science and Technology Commission.
A version of this article first appeared on Medscape.com.
Large colorectal polyps up to 15 mm in size can be safely and effectively removed with cold snare polypectomy (CSP), with a low incomplete resection rate, according to a single-center, observational study from China.
Multiple current guidelines recommend CSP for removing diminutive (≤5 mm) and small (6-9 mm) polyps, citing clinical data demonstrating that such an approach leads to high complete resection rates and a good safety profile.
However, the use of CSP for removing larger colorectal polyps (≥10 mm) remains controversial, with only limited safety and efficacy data.
Yuqi He, MD, from the department of gastroenterology at the Seventh Medical Center of Chinese PLA General Hospital in Beijing, and colleagues evaluated 440 neoplastic polyps removed by CSP from 261 patients (mean age, 56.6 years; 166 men).
Indications for colonoscopy were screening (53%), diagnostic (17%), polyp surveillance (24%), and other (6%).
Of the 440 polyps, 353 (80%) were small (5-9 mm) and 87 (20%) were large (10-15 mm); 379 (86%) were adenomas, 59 (13%) were sessile serrated lesions (SSL), and 2 (<1%) were high-grade dysplasia.
For all polyps (5-15 mm), the incomplete resection rate (primary outcome) was 2.27%. Incomplete resection was more common for large polyps (3.45%) compared with small polyps (1.98%), but the difference was not statistically significant (P = .411).
In univariate analysis, factors associated with incomplete resection were SSL, piecemeal resection, and prolonged resection time.
In multivariate regression analysis, independent risk factors for incomplete resection were SSL (odds ratio, 6.45; 95% CI, 1.48-28.03; P = .013) and prolonged resection time (OR, 7.39; 95% CI, 1.48-36.96; P = .015).
Immediate bleeding was more common with resection of large polyps (6.9% vs. 1.42%, P = .003).
There were no recurrences on follow-up colonoscopy in 37 cases with large polyps, further supporting the efficacy of CSP for this size group, the researchers say.
Their study was published online Nov. 16 in Clinical Gastroenterology and Hepatology.
Important study, lingering questions, experts say
Reached for comment, Rajesh N. Keswani, MD, of Northwestern University Feinberg School of Medicine, Chicago, said, “It is standard practice to remove polyps <10 mm using cold techniques. We generally advocate for use of a cold snare for polyps 3-9 mm in size; either large cold forceps or a cold snare can be used for polyps 1-2 mm in size,” a recommendation also conveyed in the American Gastroenterological Association’s recent clinical practice update.
“It is still unclear whether cold polypectomy techniques are appropriate for larger lesions,” Dr. Keswani, who wasn’t involved in the study, told this news organization.
“For larger polyps that require piecemeal technique (generally >2 cm), there are multiple small studies showing that resection using cold snare piecemeal technique may be safe and effective, at minimum for serrated lesions. There has been enough interest in this approach that is being formally studied in a large multicenter trial,” he noted.
“Based on the safety and efficacy data thus far for cold snare polypectomy in larger serrated lesions, it would be difficult to think that it would not be a safe choice for serrated lesions 10-20 mm,” Dr. Keswani commented.
However, the data remain “unclear for adenomatous lesions 10-20 mm in size. Unfortunately, in this study only 87 polyps 10-15 mm [in size] were removed, and a proportion were serrated and a proportion were adenomas,” he said in an interview.
“Thus, I don’t think this study is able to answer one of our key remaining polypectomy questions, which is whether cold snare techniques are the optimal approach for adenomas >10 mm in size,” said Dr. Keswani.
Also weighing in was Emre Gorgun, MD, with the Cleveland Clinic, who said it’s “an important study in the sense it raises awareness to the role of cold snaring techniques, especially since it is less expensive, quicker, and safe.”
“Late complications related to possibly using energy might be also eliminated. However, results for expanding the practice to larger polyps up to 15 mm should be taken cautiously into consideration,” Dr. Gorgun told this news organization.
“The threat of bleeding in high-risk patients, for example with a history of anticoagulation use, were not reported. Other patient-related factors can increase these risks as well. Additionally, the long-term follow-up is not reported,” commented Dr. Gorgun, who was not associated with the research.
“This study triggers the idea of using cold snaring in larger polyps; however, the results should be further reproduced and verified for long-term consequences and safety,” he concluded.
The authors have disclosed no relevant financial relationships. Dr. Keswani is a consultant for Boston Scientific and Neptune Medical and receives research support from Virgo. Dr. Gorgun is a consultant for Boston Scientific. Support for the study was provided b y grants from Project of Army Special Care and Beijing Municipal Science and Technology Commission.
A version of this article first appeared on Medscape.com.
More evidence ties some antipsychotics to increased breast cancer risk
New research provides more evidence that antipsychotics that raise prolactin levels are tied to a significantly increased risk for breast cancer.
The relative risk for breast cancer was 62% higher in women who took category 1 antipsychotic medications associated with high prolactin levels. These include haloperidol (Haldol), paliperidone (Invega), and risperidone (Risperdal). Additionally, the risk was 54% higher in those taking category 2 antipsychotics that have mid-range effects on prolactin. These include iloperidone (Fanapt), lurasidone (Latuda), and olanzapine (Zyprexa).
In contrast, category 3 antipsychotics which have a lesser effect on prolactin levels were not associated with any increase in breast cancer risk. These drugs include aripiprazole (Abilify), asenapine (Saphris), brexpiprazole (Rexulti), cariprazine (Vraylar), clozapine (multiple brands), quetiapine (Seroquel), and ziprasidone (Geodon).
While the “absolute” breast cancer risk for these drugs is unclear, “we can make the case that high circulating prolactin levels are associated with breast cancer risk. This follows what is already known about prolactin from prior studies, notably the nurses’ health studies,” Tahir Rahman, MD, associate professor of psychiatry, Washington University School of Medicine, St. Louis, told this news organization.
“We don’t want to alarm patients taking antipsychotic drugs for life-threatening mental health problems, but we also think it is time for doctors to track prolactin levels and vigilantly monitor their patients who are being treated with antipsychotics,” Dr. Rahman added in a news release.
The study was published online Dec. 3 in the Journal of Clinical Psychopharmacology.
Test prolactin levels
Using administrative claims data, the researchers evaluated breast cancer risk in women aged 18-64 exposed to antipsychotic medications compared with anticonvulsants and/or lithium.
They identified 914 cases of invasive breast cancer among 540,737 women.
Roughly 52% of the study population filled at least one prescription for a category 3 antipsychotic agent, whereas 15% filled at least one prescription for a category 1 agent; 49% of women filled at least one prescription for an anticonvulsant medication during the study period.
Exposure to all antipsychotics was independently associated with a 35% increased risk for breast cancer (adjusted hazard ratio, 1.35; 95% CI, 1.14-1.61), the study team found.
Compared with anticonvulsants or lithium, the risk for breast cancer was significantly increased for high prolactin (category 1) antipsychotics (adjusted hazard ratio, 1.62; 95% CI, 1.30-2.03) and for mid-prolactin (category 2) drugs (aHR 1.54; 95% CI, 1.19-1.99), with no increased risk for category 3 antipsychotics.
“Our research is obviously of interest for preventing breast cancer in antipsychotic-treated patients. Checking a blood prolactin level is cheap and easy [and a high level is] fairly simple to mitigate,” said Dr. Rahman.
A matter of debate
Reached for comment, Christoph Correll, MD, professor of psychiatry and molecular medicine, Zucker School of Medicine at Hofstra/Northwell, Hempstead, New York, said, “The potential elevation of breast cancer risk depending on the dose and time of treatment with antipsychotic medications with varying degrees of prolactin-raising properties has been a topic of research and matter of debate.”
This new study “adds another data point indicating that antipsychotics that are associated on average with a higher prolactin-raising effect than other antipsychotics may increase the risk of breast cancer in women to some degree,” said Dr. Correll, who was not involved with the study.
However, he cautioned that “naturalistic data are always vulnerable to residual confounding, for example, unmeasured effects that could also at least partially explain the results, and the follow-up time of only 4 years (maximum 6 years) in this study was relatively short.
“Nevertheless, given availability of many different antipsychotics with varying degrees of prolactin-raising potential, in women requiring antipsychotic treatment, less prolactin-raising antipsychotics may be preferable,” Dr. Correll said.
“In women receiving prolactin-raising antipsychotics for medium- and longer-term maintenance therapy, prolactin levels should be monitored,” he added.
When an elevated prolactin level is detected, this should be addressed “either via dose reduction, a switch to an alternative antipsychotic that does not raise prolactin levels significantly, or the addition of a partial or full D2 agonist when the prolactin-raising antipsychotic should be continued based on individualized risk assessment,” Dr. Correll advised.
This work was supported by an award from the Alvin J. Siteman Cancer Center; the National Cancer Institute and the National Center for Advancing Translational Sciences of the National Institutes of Health; the Taylor Family Institute for Innovative Psychiatric Research; and the Center for Brain Research in Mood Disorders. The authors have disclosed no relevant financial relationships. Dr. Correll has received royalties from UpToDate and is a stock option holder of LB Pharma.
A version of this article first appeared on Medscape.com.
New research provides more evidence that antipsychotics that raise prolactin levels are tied to a significantly increased risk for breast cancer.
The relative risk for breast cancer was 62% higher in women who took category 1 antipsychotic medications associated with high prolactin levels. These include haloperidol (Haldol), paliperidone (Invega), and risperidone (Risperdal). Additionally, the risk was 54% higher in those taking category 2 antipsychotics that have mid-range effects on prolactin. These include iloperidone (Fanapt), lurasidone (Latuda), and olanzapine (Zyprexa).
In contrast, category 3 antipsychotics which have a lesser effect on prolactin levels were not associated with any increase in breast cancer risk. These drugs include aripiprazole (Abilify), asenapine (Saphris), brexpiprazole (Rexulti), cariprazine (Vraylar), clozapine (multiple brands), quetiapine (Seroquel), and ziprasidone (Geodon).
While the “absolute” breast cancer risk for these drugs is unclear, “we can make the case that high circulating prolactin levels are associated with breast cancer risk. This follows what is already known about prolactin from prior studies, notably the nurses’ health studies,” Tahir Rahman, MD, associate professor of psychiatry, Washington University School of Medicine, St. Louis, told this news organization.
“We don’t want to alarm patients taking antipsychotic drugs for life-threatening mental health problems, but we also think it is time for doctors to track prolactin levels and vigilantly monitor their patients who are being treated with antipsychotics,” Dr. Rahman added in a news release.
The study was published online Dec. 3 in the Journal of Clinical Psychopharmacology.
Test prolactin levels
Using administrative claims data, the researchers evaluated breast cancer risk in women aged 18-64 exposed to antipsychotic medications compared with anticonvulsants and/or lithium.
They identified 914 cases of invasive breast cancer among 540,737 women.
Roughly 52% of the study population filled at least one prescription for a category 3 antipsychotic agent, whereas 15% filled at least one prescription for a category 1 agent; 49% of women filled at least one prescription for an anticonvulsant medication during the study period.
Exposure to all antipsychotics was independently associated with a 35% increased risk for breast cancer (adjusted hazard ratio, 1.35; 95% CI, 1.14-1.61), the study team found.
Compared with anticonvulsants or lithium, the risk for breast cancer was significantly increased for high prolactin (category 1) antipsychotics (adjusted hazard ratio, 1.62; 95% CI, 1.30-2.03) and for mid-prolactin (category 2) drugs (aHR 1.54; 95% CI, 1.19-1.99), with no increased risk for category 3 antipsychotics.
“Our research is obviously of interest for preventing breast cancer in antipsychotic-treated patients. Checking a blood prolactin level is cheap and easy [and a high level is] fairly simple to mitigate,” said Dr. Rahman.
A matter of debate
Reached for comment, Christoph Correll, MD, professor of psychiatry and molecular medicine, Zucker School of Medicine at Hofstra/Northwell, Hempstead, New York, said, “The potential elevation of breast cancer risk depending on the dose and time of treatment with antipsychotic medications with varying degrees of prolactin-raising properties has been a topic of research and matter of debate.”
This new study “adds another data point indicating that antipsychotics that are associated on average with a higher prolactin-raising effect than other antipsychotics may increase the risk of breast cancer in women to some degree,” said Dr. Correll, who was not involved with the study.
However, he cautioned that “naturalistic data are always vulnerable to residual confounding, for example, unmeasured effects that could also at least partially explain the results, and the follow-up time of only 4 years (maximum 6 years) in this study was relatively short.
“Nevertheless, given availability of many different antipsychotics with varying degrees of prolactin-raising potential, in women requiring antipsychotic treatment, less prolactin-raising antipsychotics may be preferable,” Dr. Correll said.
“In women receiving prolactin-raising antipsychotics for medium- and longer-term maintenance therapy, prolactin levels should be monitored,” he added.
When an elevated prolactin level is detected, this should be addressed “either via dose reduction, a switch to an alternative antipsychotic that does not raise prolactin levels significantly, or the addition of a partial or full D2 agonist when the prolactin-raising antipsychotic should be continued based on individualized risk assessment,” Dr. Correll advised.
This work was supported by an award from the Alvin J. Siteman Cancer Center; the National Cancer Institute and the National Center for Advancing Translational Sciences of the National Institutes of Health; the Taylor Family Institute for Innovative Psychiatric Research; and the Center for Brain Research in Mood Disorders. The authors have disclosed no relevant financial relationships. Dr. Correll has received royalties from UpToDate and is a stock option holder of LB Pharma.
A version of this article first appeared on Medscape.com.
New research provides more evidence that antipsychotics that raise prolactin levels are tied to a significantly increased risk for breast cancer.
The relative risk for breast cancer was 62% higher in women who took category 1 antipsychotic medications associated with high prolactin levels. These include haloperidol (Haldol), paliperidone (Invega), and risperidone (Risperdal). Additionally, the risk was 54% higher in those taking category 2 antipsychotics that have mid-range effects on prolactin. These include iloperidone (Fanapt), lurasidone (Latuda), and olanzapine (Zyprexa).
In contrast, category 3 antipsychotics which have a lesser effect on prolactin levels were not associated with any increase in breast cancer risk. These drugs include aripiprazole (Abilify), asenapine (Saphris), brexpiprazole (Rexulti), cariprazine (Vraylar), clozapine (multiple brands), quetiapine (Seroquel), and ziprasidone (Geodon).
While the “absolute” breast cancer risk for these drugs is unclear, “we can make the case that high circulating prolactin levels are associated with breast cancer risk. This follows what is already known about prolactin from prior studies, notably the nurses’ health studies,” Tahir Rahman, MD, associate professor of psychiatry, Washington University School of Medicine, St. Louis, told this news organization.
“We don’t want to alarm patients taking antipsychotic drugs for life-threatening mental health problems, but we also think it is time for doctors to track prolactin levels and vigilantly monitor their patients who are being treated with antipsychotics,” Dr. Rahman added in a news release.
The study was published online Dec. 3 in the Journal of Clinical Psychopharmacology.
Test prolactin levels
Using administrative claims data, the researchers evaluated breast cancer risk in women aged 18-64 exposed to antipsychotic medications compared with anticonvulsants and/or lithium.
They identified 914 cases of invasive breast cancer among 540,737 women.
Roughly 52% of the study population filled at least one prescription for a category 3 antipsychotic agent, whereas 15% filled at least one prescription for a category 1 agent; 49% of women filled at least one prescription for an anticonvulsant medication during the study period.
Exposure to all antipsychotics was independently associated with a 35% increased risk for breast cancer (adjusted hazard ratio, 1.35; 95% CI, 1.14-1.61), the study team found.
Compared with anticonvulsants or lithium, the risk for breast cancer was significantly increased for high prolactin (category 1) antipsychotics (adjusted hazard ratio, 1.62; 95% CI, 1.30-2.03) and for mid-prolactin (category 2) drugs (aHR 1.54; 95% CI, 1.19-1.99), with no increased risk for category 3 antipsychotics.
“Our research is obviously of interest for preventing breast cancer in antipsychotic-treated patients. Checking a blood prolactin level is cheap and easy [and a high level is] fairly simple to mitigate,” said Dr. Rahman.
A matter of debate
Reached for comment, Christoph Correll, MD, professor of psychiatry and molecular medicine, Zucker School of Medicine at Hofstra/Northwell, Hempstead, New York, said, “The potential elevation of breast cancer risk depending on the dose and time of treatment with antipsychotic medications with varying degrees of prolactin-raising properties has been a topic of research and matter of debate.”
This new study “adds another data point indicating that antipsychotics that are associated on average with a higher prolactin-raising effect than other antipsychotics may increase the risk of breast cancer in women to some degree,” said Dr. Correll, who was not involved with the study.
However, he cautioned that “naturalistic data are always vulnerable to residual confounding, for example, unmeasured effects that could also at least partially explain the results, and the follow-up time of only 4 years (maximum 6 years) in this study was relatively short.
“Nevertheless, given availability of many different antipsychotics with varying degrees of prolactin-raising potential, in women requiring antipsychotic treatment, less prolactin-raising antipsychotics may be preferable,” Dr. Correll said.
“In women receiving prolactin-raising antipsychotics for medium- and longer-term maintenance therapy, prolactin levels should be monitored,” he added.
When an elevated prolactin level is detected, this should be addressed “either via dose reduction, a switch to an alternative antipsychotic that does not raise prolactin levels significantly, or the addition of a partial or full D2 agonist when the prolactin-raising antipsychotic should be continued based on individualized risk assessment,” Dr. Correll advised.
This work was supported by an award from the Alvin J. Siteman Cancer Center; the National Cancer Institute and the National Center for Advancing Translational Sciences of the National Institutes of Health; the Taylor Family Institute for Innovative Psychiatric Research; and the Center for Brain Research in Mood Disorders. The authors have disclosed no relevant financial relationships. Dr. Correll has received royalties from UpToDate and is a stock option holder of LB Pharma.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF CLINICAL PSYCHOPHARMACOLOGY
Updated ACG GERD guideline addresses increased scrutiny of PPI therapy
For the first time since 2013, the American College of Gastroenterology has issued updated evidence-based recommendations and practical guidance on the evaluation and management of gastroesophageal reflux disease (GERD), including pharmacologic, lifestyle, surgical, and endoscopic management.
Over the past 8 years, understanding of the varied presentations of GERD, enhancements in diagnostic testing, and approach to patient management have evolved, and there has been closer scrutiny of proton pump inhibitor (PPI) therapy and its potential side effects, the guideline authors said.
While PPIs remain the “medical treatment of choice” for GERD, multiple studies have raised questions about adverse events, they noted.
“We now know a lot more about PPI adverse events in the sense that we have another 8 years of experience” since the 2013 guideline, first author Philip O. Katz, MD, professor of medicine and director of motility laboratories at Weill Cornell Medicine, New York, added in an interview.
This update emphasizes the importance of making an accurate diagnosis and recommends PPI therapy “when patients really have GERD and being careful to use the lowest effective dose,” Dr. Katz said.
The guideline was published online Nov. 22, 2021, in the American Journal of Gastroenterology.
Benefits outweigh risks
The guideline suggests telling patients that PPIs are the most effective medical treatment for GERD.
Some studies have identified an association between the long-term use of PPIs and the development of several adverse conditions, including intestinal infections, pneumonia, stomach cancer, osteoporosis-related bone fractures, chronic kidney disease, deficiencies of certain vitamins and minerals, heart attacks, strokes, dementia, and early death.
Clinicians should emphasize, however, that these studies have flaws, are not considered definitive, and do not establish a cause-and-effect relationship between PPIs and the adverse conditions.
They should also emphasize to patients that high-quality studies have found that PPIs do not significantly raise the risk of any of these conditions except intestinal infections.
Patients should be told that, for the treatment of GERD, “gastroenterologists generally agree that the well-established benefits of PPIs far outweigh their theoretical risks.”
“Everything in this guideline makes sense,” Scott Gabbard, MD, gastroenterologist and section head, Center for Neurogastroenterology and Motility, Cleveland Clinic, who wasn’t involved in the guideline development, said in an interview.
“A PPI trial for anyone with typical GERD symptoms and having those who respond taper to the lowest effective dose is still the first line for anyone with GERD,” Dr. Gabbard said.
Making the diagnosis
As there is currently no gold standard for the diagnosis of GERD, diagnosis is based on a combination of symptoms, endoscopic evaluation of esophageal mucosa, reflux monitoring, and response to therapeutic intervention, the guideline says.
For patients with classic symptoms of heartburn and regurgitation with no alarm symptoms, the authors recommend an 8-week trial of empiric once-daily PPIs before a meal. If the patient responds, the guideline recommends attempting to discontinue the medication.
The guideline recommends diagnostic endoscopy after PPIs are stopped for 2-4 weeks in patients whose classic symptoms fail to respond adequately to the 8-week empiric PPI trial or in those whose symptoms return when PPIs are discontinued.
For patients with chest pain but no heartburn who have undergone an adequate evaluation to exclude heart disease, the guideline advises objective testing for GERD (endoscopy and/or reflux monitoring).
The use of barium swallow solely as a diagnostic test for GERD is not recommended.
Endoscopy should be the first test for evaluating patients presenting with dysphagia or other alarm symptoms, such as weight loss and gastrointestinal bleeding, as well as for patients with risk factors for Barrett’s esophagus.
For patients in whom the diagnosis of GERD is suspected but unclear and endoscopy fails to show objective evidence of GERD, the guidelines advise reflux monitoring off therapy to establish the diagnosis.
The guideline recommends against reflux monitoring off therapy solely as a diagnostic test for GERD in patients with known endoscopic evidence of Los Angeles grade C or D reflux esophagitis or in patients with long-segment Barrett’s esophagus.
High-resolution manometry solely as a diagnostic test for GERD is also not recommended.
Medical management of GERD
Recommendations for medical management of GERD include weight loss in patients who are overweight or obese, avoidance of meals within 2-3 hours of bedtime, avoidance of tobacco products and “trigger foods,” and elevation of the head of the bed for nighttime symptoms.
Treatment with a PPI is recommended over histamine2-receptor antagonists for healing and maintenance of healing of eosinophilic esophagitis. Taking a PPI 30-60 minutes prior to a meal rather than at bedtime is recommended.
“Use of the lowest effective PPI dose is recommended and logical but must be individualized,” the guideline states.
There is “conceptual rationale” for a trial of switching PPIs for patients who don’t respond to one PPI. However, switching more than once to another PPI “cannot be supported,” the guideline says.
Dr. Gabbard said the advice about switching PPIs in nonresponders is particularly helpful.
“In clinical practice, I see patients who try one PPI, and if it doesn’t work, their doctor puts them on another PPI, then another and another, until they get through five PPIs and gotten nowhere,” he said in an interview.
“This new guideline is very helpful in saying, if a patient has GERD symptoms that do not respond to a PPI, you can do one switch. But, if that doesn’t work, have a low threshold to perform pH testing to determine if the patient truly has reflux or not,” Dr. Gabbard said.
“Some studies have suggested that up to 75% of PPI nonresponders actually don’t have reflux. They have functional heartburn, which is not reflux and is treated without PPIs,” he noted.
One area of controversy relates to abrupt PPI discontinuation and potential rebound acid hypersecretion, resulting in increased reflux symptoms. While this has been found in healthy control patients, strong evidence for an increase in symptoms after abrupt PPI withdrawal is lacking.
The guideline makes “no definitive recommendation as to whether weaning or stopping PPIs cold turkey is a better approach, due to a lack of evidence,” Dr. Katz said in an interview.
For patients with GERD without erosive esophagitis or Barrett’s esophagus and whose symptoms resolve with PPI therapy, the guideline says an attempt should be made to discontinue PPI therapy or to switch to on-demand therapy in which a PPI is taken only when symptoms occur and is stopped when they are relieved.
For patients with Los Angeles grade C or D esophagitis, the recommendation is for maintenance PPI therapy indefinitely or antireflux surgery.
Dr. Gabbard said it’s “nice to have in writing from the ACG that patients with erosive esophagitis or Barrett’s esophagus – those who truly need a PPI – should be on indefinite PPI therapy, because the benefit of a PPI far outweighs the theoretical risks.”
The research had no financial support. Dr. Katz has served as consultant for Phathom Pharma and Medtronic, has received research support from Diversatek and royalties from UpToDate, and serves on the Medscape Gastroenterology advisory board. Dr. Gabbard disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For the first time since 2013, the American College of Gastroenterology has issued updated evidence-based recommendations and practical guidance on the evaluation and management of gastroesophageal reflux disease (GERD), including pharmacologic, lifestyle, surgical, and endoscopic management.
Over the past 8 years, understanding of the varied presentations of GERD, enhancements in diagnostic testing, and approach to patient management have evolved, and there has been closer scrutiny of proton pump inhibitor (PPI) therapy and its potential side effects, the guideline authors said.
While PPIs remain the “medical treatment of choice” for GERD, multiple studies have raised questions about adverse events, they noted.
“We now know a lot more about PPI adverse events in the sense that we have another 8 years of experience” since the 2013 guideline, first author Philip O. Katz, MD, professor of medicine and director of motility laboratories at Weill Cornell Medicine, New York, added in an interview.
This update emphasizes the importance of making an accurate diagnosis and recommends PPI therapy “when patients really have GERD and being careful to use the lowest effective dose,” Dr. Katz said.
The guideline was published online Nov. 22, 2021, in the American Journal of Gastroenterology.
Benefits outweigh risks
The guideline suggests telling patients that PPIs are the most effective medical treatment for GERD.
Some studies have identified an association between the long-term use of PPIs and the development of several adverse conditions, including intestinal infections, pneumonia, stomach cancer, osteoporosis-related bone fractures, chronic kidney disease, deficiencies of certain vitamins and minerals, heart attacks, strokes, dementia, and early death.
Clinicians should emphasize, however, that these studies have flaws, are not considered definitive, and do not establish a cause-and-effect relationship between PPIs and the adverse conditions.
They should also emphasize to patients that high-quality studies have found that PPIs do not significantly raise the risk of any of these conditions except intestinal infections.
Patients should be told that, for the treatment of GERD, “gastroenterologists generally agree that the well-established benefits of PPIs far outweigh their theoretical risks.”
“Everything in this guideline makes sense,” Scott Gabbard, MD, gastroenterologist and section head, Center for Neurogastroenterology and Motility, Cleveland Clinic, who wasn’t involved in the guideline development, said in an interview.
“A PPI trial for anyone with typical GERD symptoms and having those who respond taper to the lowest effective dose is still the first line for anyone with GERD,” Dr. Gabbard said.
Making the diagnosis
As there is currently no gold standard for the diagnosis of GERD, diagnosis is based on a combination of symptoms, endoscopic evaluation of esophageal mucosa, reflux monitoring, and response to therapeutic intervention, the guideline says.
For patients with classic symptoms of heartburn and regurgitation with no alarm symptoms, the authors recommend an 8-week trial of empiric once-daily PPIs before a meal. If the patient responds, the guideline recommends attempting to discontinue the medication.
The guideline recommends diagnostic endoscopy after PPIs are stopped for 2-4 weeks in patients whose classic symptoms fail to respond adequately to the 8-week empiric PPI trial or in those whose symptoms return when PPIs are discontinued.
For patients with chest pain but no heartburn who have undergone an adequate evaluation to exclude heart disease, the guideline advises objective testing for GERD (endoscopy and/or reflux monitoring).
The use of barium swallow solely as a diagnostic test for GERD is not recommended.
Endoscopy should be the first test for evaluating patients presenting with dysphagia or other alarm symptoms, such as weight loss and gastrointestinal bleeding, as well as for patients with risk factors for Barrett’s esophagus.
For patients in whom the diagnosis of GERD is suspected but unclear and endoscopy fails to show objective evidence of GERD, the guidelines advise reflux monitoring off therapy to establish the diagnosis.
The guideline recommends against reflux monitoring off therapy solely as a diagnostic test for GERD in patients with known endoscopic evidence of Los Angeles grade C or D reflux esophagitis or in patients with long-segment Barrett’s esophagus.
High-resolution manometry solely as a diagnostic test for GERD is also not recommended.
Medical management of GERD
Recommendations for medical management of GERD include weight loss in patients who are overweight or obese, avoidance of meals within 2-3 hours of bedtime, avoidance of tobacco products and “trigger foods,” and elevation of the head of the bed for nighttime symptoms.
Treatment with a PPI is recommended over histamine2-receptor antagonists for healing and maintenance of healing of eosinophilic esophagitis. Taking a PPI 30-60 minutes prior to a meal rather than at bedtime is recommended.
“Use of the lowest effective PPI dose is recommended and logical but must be individualized,” the guideline states.
There is “conceptual rationale” for a trial of switching PPIs for patients who don’t respond to one PPI. However, switching more than once to another PPI “cannot be supported,” the guideline says.
Dr. Gabbard said the advice about switching PPIs in nonresponders is particularly helpful.
“In clinical practice, I see patients who try one PPI, and if it doesn’t work, their doctor puts them on another PPI, then another and another, until they get through five PPIs and gotten nowhere,” he said in an interview.
“This new guideline is very helpful in saying, if a patient has GERD symptoms that do not respond to a PPI, you can do one switch. But, if that doesn’t work, have a low threshold to perform pH testing to determine if the patient truly has reflux or not,” Dr. Gabbard said.
“Some studies have suggested that up to 75% of PPI nonresponders actually don’t have reflux. They have functional heartburn, which is not reflux and is treated without PPIs,” he noted.
One area of controversy relates to abrupt PPI discontinuation and potential rebound acid hypersecretion, resulting in increased reflux symptoms. While this has been found in healthy control patients, strong evidence for an increase in symptoms after abrupt PPI withdrawal is lacking.
The guideline makes “no definitive recommendation as to whether weaning or stopping PPIs cold turkey is a better approach, due to a lack of evidence,” Dr. Katz said in an interview.
For patients with GERD without erosive esophagitis or Barrett’s esophagus and whose symptoms resolve with PPI therapy, the guideline says an attempt should be made to discontinue PPI therapy or to switch to on-demand therapy in which a PPI is taken only when symptoms occur and is stopped when they are relieved.
For patients with Los Angeles grade C or D esophagitis, the recommendation is for maintenance PPI therapy indefinitely or antireflux surgery.
Dr. Gabbard said it’s “nice to have in writing from the ACG that patients with erosive esophagitis or Barrett’s esophagus – those who truly need a PPI – should be on indefinite PPI therapy, because the benefit of a PPI far outweighs the theoretical risks.”
The research had no financial support. Dr. Katz has served as consultant for Phathom Pharma and Medtronic, has received research support from Diversatek and royalties from UpToDate, and serves on the Medscape Gastroenterology advisory board. Dr. Gabbard disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
For the first time since 2013, the American College of Gastroenterology has issued updated evidence-based recommendations and practical guidance on the evaluation and management of gastroesophageal reflux disease (GERD), including pharmacologic, lifestyle, surgical, and endoscopic management.
Over the past 8 years, understanding of the varied presentations of GERD, enhancements in diagnostic testing, and approach to patient management have evolved, and there has been closer scrutiny of proton pump inhibitor (PPI) therapy and its potential side effects, the guideline authors said.
While PPIs remain the “medical treatment of choice” for GERD, multiple studies have raised questions about adverse events, they noted.
“We now know a lot more about PPI adverse events in the sense that we have another 8 years of experience” since the 2013 guideline, first author Philip O. Katz, MD, professor of medicine and director of motility laboratories at Weill Cornell Medicine, New York, added in an interview.
This update emphasizes the importance of making an accurate diagnosis and recommends PPI therapy “when patients really have GERD and being careful to use the lowest effective dose,” Dr. Katz said.
The guideline was published online Nov. 22, 2021, in the American Journal of Gastroenterology.
Benefits outweigh risks
The guideline suggests telling patients that PPIs are the most effective medical treatment for GERD.
Some studies have identified an association between the long-term use of PPIs and the development of several adverse conditions, including intestinal infections, pneumonia, stomach cancer, osteoporosis-related bone fractures, chronic kidney disease, deficiencies of certain vitamins and minerals, heart attacks, strokes, dementia, and early death.
Clinicians should emphasize, however, that these studies have flaws, are not considered definitive, and do not establish a cause-and-effect relationship between PPIs and the adverse conditions.
They should also emphasize to patients that high-quality studies have found that PPIs do not significantly raise the risk of any of these conditions except intestinal infections.
Patients should be told that, for the treatment of GERD, “gastroenterologists generally agree that the well-established benefits of PPIs far outweigh their theoretical risks.”
“Everything in this guideline makes sense,” Scott Gabbard, MD, gastroenterologist and section head, Center for Neurogastroenterology and Motility, Cleveland Clinic, who wasn’t involved in the guideline development, said in an interview.
“A PPI trial for anyone with typical GERD symptoms and having those who respond taper to the lowest effective dose is still the first line for anyone with GERD,” Dr. Gabbard said.
Making the diagnosis
As there is currently no gold standard for the diagnosis of GERD, diagnosis is based on a combination of symptoms, endoscopic evaluation of esophageal mucosa, reflux monitoring, and response to therapeutic intervention, the guideline says.
For patients with classic symptoms of heartburn and regurgitation with no alarm symptoms, the authors recommend an 8-week trial of empiric once-daily PPIs before a meal. If the patient responds, the guideline recommends attempting to discontinue the medication.
The guideline recommends diagnostic endoscopy after PPIs are stopped for 2-4 weeks in patients whose classic symptoms fail to respond adequately to the 8-week empiric PPI trial or in those whose symptoms return when PPIs are discontinued.
For patients with chest pain but no heartburn who have undergone an adequate evaluation to exclude heart disease, the guideline advises objective testing for GERD (endoscopy and/or reflux monitoring).
The use of barium swallow solely as a diagnostic test for GERD is not recommended.
Endoscopy should be the first test for evaluating patients presenting with dysphagia or other alarm symptoms, such as weight loss and gastrointestinal bleeding, as well as for patients with risk factors for Barrett’s esophagus.
For patients in whom the diagnosis of GERD is suspected but unclear and endoscopy fails to show objective evidence of GERD, the guidelines advise reflux monitoring off therapy to establish the diagnosis.
The guideline recommends against reflux monitoring off therapy solely as a diagnostic test for GERD in patients with known endoscopic evidence of Los Angeles grade C or D reflux esophagitis or in patients with long-segment Barrett’s esophagus.
High-resolution manometry solely as a diagnostic test for GERD is also not recommended.
Medical management of GERD
Recommendations for medical management of GERD include weight loss in patients who are overweight or obese, avoidance of meals within 2-3 hours of bedtime, avoidance of tobacco products and “trigger foods,” and elevation of the head of the bed for nighttime symptoms.
Treatment with a PPI is recommended over histamine2-receptor antagonists for healing and maintenance of healing of eosinophilic esophagitis. Taking a PPI 30-60 minutes prior to a meal rather than at bedtime is recommended.
“Use of the lowest effective PPI dose is recommended and logical but must be individualized,” the guideline states.
There is “conceptual rationale” for a trial of switching PPIs for patients who don’t respond to one PPI. However, switching more than once to another PPI “cannot be supported,” the guideline says.
Dr. Gabbard said the advice about switching PPIs in nonresponders is particularly helpful.
“In clinical practice, I see patients who try one PPI, and if it doesn’t work, their doctor puts them on another PPI, then another and another, until they get through five PPIs and gotten nowhere,” he said in an interview.
“This new guideline is very helpful in saying, if a patient has GERD symptoms that do not respond to a PPI, you can do one switch. But, if that doesn’t work, have a low threshold to perform pH testing to determine if the patient truly has reflux or not,” Dr. Gabbard said.
“Some studies have suggested that up to 75% of PPI nonresponders actually don’t have reflux. They have functional heartburn, which is not reflux and is treated without PPIs,” he noted.
One area of controversy relates to abrupt PPI discontinuation and potential rebound acid hypersecretion, resulting in increased reflux symptoms. While this has been found in healthy control patients, strong evidence for an increase in symptoms after abrupt PPI withdrawal is lacking.
The guideline makes “no definitive recommendation as to whether weaning or stopping PPIs cold turkey is a better approach, due to a lack of evidence,” Dr. Katz said in an interview.
For patients with GERD without erosive esophagitis or Barrett’s esophagus and whose symptoms resolve with PPI therapy, the guideline says an attempt should be made to discontinue PPI therapy or to switch to on-demand therapy in which a PPI is taken only when symptoms occur and is stopped when they are relieved.
For patients with Los Angeles grade C or D esophagitis, the recommendation is for maintenance PPI therapy indefinitely or antireflux surgery.
Dr. Gabbard said it’s “nice to have in writing from the ACG that patients with erosive esophagitis or Barrett’s esophagus – those who truly need a PPI – should be on indefinite PPI therapy, because the benefit of a PPI far outweighs the theoretical risks.”
The research had no financial support. Dr. Katz has served as consultant for Phathom Pharma and Medtronic, has received research support from Diversatek and royalties from UpToDate, and serves on the Medscape Gastroenterology advisory board. Dr. Gabbard disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE AMERICAN JOURNAL OF GASTROENTEROLOGY
New data on rare myocarditis after COVID-19 vaccination
Adolescents and adults younger than age 21 who develop myocarditis after mRNA COVID-19 vaccination frequently have abnormal findings on cardiac MRI (cMRI) but most have a mild clinical course with rapid resolution of symptoms, a new study concludes.

“This study supports what we’ve been seeing. People identified and treated early and appropriately for the rare complication of COVID-19 vaccine-related myocarditis typically experienced only mild cases and short recovery times,” American Heart Association President Donald M. Lloyd-Jones, MD, said in a podcast.
“Overwhelmingly, the data continue to indicate [that] the benefits of COVID-19 vaccine far outweigh any very rare risks of adverse events from the vaccine, including myocarditis,” Dr. Lloyd-Jones added.
The study was published online Dec. 6 in Circulation.
Using data from 26 pediatric medical centers across the United States and Canada, the researchers reviewed the medical records of 139 patients younger than 21 with suspected myocarditis within 1 month of receiving a COVID-19 vaccination.
They made the following key observations:
- Most patients were male (90.6%), White (66.2%) and with a median age of 15.8 years.
- Suspected myocarditis occurred in 136 patients (97.8%) following mRNA vaccine, with 131 (94.2%) following the Pfizer-BioNTech vaccine; 128 cases (91.4%) occurred after the second dose.
- Symptoms started a median of 2 days (range 0 to 22 days) following vaccination administration.
- Chest pain was the most common symptom (99.3%), with fever present in 30.9% of patients and shortness of breath in 27.3%.
- Patients were treated with nonsteroidal anti-inflammatory drugs (81.3%), intravenous immunoglobulin (21.6%), glucocorticoids (21.6%), colchicine (7.9%) or no anti-inflammatory therapies (8.6%).
- Twenty-six patients (18.7%) were admitted to the intensive care unit; 2 received inotropic/vasoactive support; none required extracorporeal membrane oxygenation or died.
- Median time spent in the hospital was 2 days.
- A total of 111 patients had elevated troponin I (8.12 ng/mL) and 28 had elevated troponin T (0.61 ng/mL).
- More than two-thirds (69.8%) had abnormal electrocardiograms and/or arrhythmias (7 with nonsustained ventricular tachycardia).
- Twenty-six patients (18.7%) had left ventricular ejection fraction (LVEF) less than 55% on echocardiogram; LVEF had returned to normal in the 25 who returned for follow-up.
- 75 of 97 patients (77.3%) who underwent cMRI at a median of 5 days from symptom onset had abnormal findings; 74 (76.3%) had late gadolinium enhancement, 54 (55.7%) had myocardial edema, and 49 (50.5%) met Lake Louise criteria for myocarditis.
“These data suggest that most cases of suspected COVID-19 vaccine–related myocarditis in people younger than 21 are mild and resolve quickly,” corresponding author Dongngan Truong, MD, Division of Pediatric Cardiology, University of Utah and Primary Children’s Hospital, Salt Lake City, said in a statement.
“We were very happy to see that type of recovery. However, we are awaiting further studies to better understand the long-term outcomes of patients who have had COVID-19 vaccination-related myocarditis. We also need to study the risk factors and mechanisms for this rare complication,” Dr. Truong added.
Dr. Lloyd-Jones said these findings support the AHA’s position that COVID-19 vaccines are “safe, highly effective, and fundamental to saving lives, protecting our families and communities against COVID-19, and ending the pandemic.”
The study received no funding. Dr. Truong consults for Pfizer on vaccine-associated myocarditis. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.
Adolescents and adults younger than age 21 who develop myocarditis after mRNA COVID-19 vaccination frequently have abnormal findings on cardiac MRI (cMRI) but most have a mild clinical course with rapid resolution of symptoms, a new study concludes.
“This study supports what we’ve been seeing. People identified and treated early and appropriately for the rare complication of COVID-19 vaccine-related myocarditis typically experienced only mild cases and short recovery times,” American Heart Association President Donald M. Lloyd-Jones, MD, said in a podcast.
“Overwhelmingly, the data continue to indicate [that] the benefits of COVID-19 vaccine far outweigh any very rare risks of adverse events from the vaccine, including myocarditis,” Dr. Lloyd-Jones added.
The study was published online Dec. 6 in Circulation.
Using data from 26 pediatric medical centers across the United States and Canada, the researchers reviewed the medical records of 139 patients younger than 21 with suspected myocarditis within 1 month of receiving a COVID-19 vaccination.
They made the following key observations:
- Most patients were male (90.6%), White (66.2%) and with a median age of 15.8 years.
- Suspected myocarditis occurred in 136 patients (97.8%) following mRNA vaccine, with 131 (94.2%) following the Pfizer-BioNTech vaccine; 128 cases (91.4%) occurred after the second dose.
- Symptoms started a median of 2 days (range 0 to 22 days) following vaccination administration.
- Chest pain was the most common symptom (99.3%), with fever present in 30.9% of patients and shortness of breath in 27.3%.
- Patients were treated with nonsteroidal anti-inflammatory drugs (81.3%), intravenous immunoglobulin (21.6%), glucocorticoids (21.6%), colchicine (7.9%) or no anti-inflammatory therapies (8.6%).
- Twenty-six patients (18.7%) were admitted to the intensive care unit; 2 received inotropic/vasoactive support; none required extracorporeal membrane oxygenation or died.
- Median time spent in the hospital was 2 days.
- A total of 111 patients had elevated troponin I (8.12 ng/mL) and 28 had elevated troponin T (0.61 ng/mL).
- More than two-thirds (69.8%) had abnormal electrocardiograms and/or arrhythmias (7 with nonsustained ventricular tachycardia).
- Twenty-six patients (18.7%) had left ventricular ejection fraction (LVEF) less than 55% on echocardiogram; LVEF had returned to normal in the 25 who returned for follow-up.
- 75 of 97 patients (77.3%) who underwent cMRI at a median of 5 days from symptom onset had abnormal findings; 74 (76.3%) had late gadolinium enhancement, 54 (55.7%) had myocardial edema, and 49 (50.5%) met Lake Louise criteria for myocarditis.
“These data suggest that most cases of suspected COVID-19 vaccine–related myocarditis in people younger than 21 are mild and resolve quickly,” corresponding author Dongngan Truong, MD, Division of Pediatric Cardiology, University of Utah and Primary Children’s Hospital, Salt Lake City, said in a statement.
“We were very happy to see that type of recovery. However, we are awaiting further studies to better understand the long-term outcomes of patients who have had COVID-19 vaccination-related myocarditis. We also need to study the risk factors and mechanisms for this rare complication,” Dr. Truong added.
Dr. Lloyd-Jones said these findings support the AHA’s position that COVID-19 vaccines are “safe, highly effective, and fundamental to saving lives, protecting our families and communities against COVID-19, and ending the pandemic.”
The study received no funding. Dr. Truong consults for Pfizer on vaccine-associated myocarditis. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.
Adolescents and adults younger than age 21 who develop myocarditis after mRNA COVID-19 vaccination frequently have abnormal findings on cardiac MRI (cMRI) but most have a mild clinical course with rapid resolution of symptoms, a new study concludes.
“This study supports what we’ve been seeing. People identified and treated early and appropriately for the rare complication of COVID-19 vaccine-related myocarditis typically experienced only mild cases and short recovery times,” American Heart Association President Donald M. Lloyd-Jones, MD, said in a podcast.
“Overwhelmingly, the data continue to indicate [that] the benefits of COVID-19 vaccine far outweigh any very rare risks of adverse events from the vaccine, including myocarditis,” Dr. Lloyd-Jones added.
The study was published online Dec. 6 in Circulation.
Using data from 26 pediatric medical centers across the United States and Canada, the researchers reviewed the medical records of 139 patients younger than 21 with suspected myocarditis within 1 month of receiving a COVID-19 vaccination.
They made the following key observations:
- Most patients were male (90.6%), White (66.2%) and with a median age of 15.8 years.
- Suspected myocarditis occurred in 136 patients (97.8%) following mRNA vaccine, with 131 (94.2%) following the Pfizer-BioNTech vaccine; 128 cases (91.4%) occurred after the second dose.
- Symptoms started a median of 2 days (range 0 to 22 days) following vaccination administration.
- Chest pain was the most common symptom (99.3%), with fever present in 30.9% of patients and shortness of breath in 27.3%.
- Patients were treated with nonsteroidal anti-inflammatory drugs (81.3%), intravenous immunoglobulin (21.6%), glucocorticoids (21.6%), colchicine (7.9%) or no anti-inflammatory therapies (8.6%).
- Twenty-six patients (18.7%) were admitted to the intensive care unit; 2 received inotropic/vasoactive support; none required extracorporeal membrane oxygenation or died.
- Median time spent in the hospital was 2 days.
- A total of 111 patients had elevated troponin I (8.12 ng/mL) and 28 had elevated troponin T (0.61 ng/mL).
- More than two-thirds (69.8%) had abnormal electrocardiograms and/or arrhythmias (7 with nonsustained ventricular tachycardia).
- Twenty-six patients (18.7%) had left ventricular ejection fraction (LVEF) less than 55% on echocardiogram; LVEF had returned to normal in the 25 who returned for follow-up.
- 75 of 97 patients (77.3%) who underwent cMRI at a median of 5 days from symptom onset had abnormal findings; 74 (76.3%) had late gadolinium enhancement, 54 (55.7%) had myocardial edema, and 49 (50.5%) met Lake Louise criteria for myocarditis.
“These data suggest that most cases of suspected COVID-19 vaccine–related myocarditis in people younger than 21 are mild and resolve quickly,” corresponding author Dongngan Truong, MD, Division of Pediatric Cardiology, University of Utah and Primary Children’s Hospital, Salt Lake City, said in a statement.
“We were very happy to see that type of recovery. However, we are awaiting further studies to better understand the long-term outcomes of patients who have had COVID-19 vaccination-related myocarditis. We also need to study the risk factors and mechanisms for this rare complication,” Dr. Truong added.
Dr. Lloyd-Jones said these findings support the AHA’s position that COVID-19 vaccines are “safe, highly effective, and fundamental to saving lives, protecting our families and communities against COVID-19, and ending the pandemic.”
The study received no funding. Dr. Truong consults for Pfizer on vaccine-associated myocarditis. A complete list of author disclosures is available with the original article.
A version of this article first appeared on Medscape.com.