User login
Occipital Scalp Nodule in a Newborn
The Diagnosis: Subcutaneous Fat Necrosis
Histopathology revealed lobular panniculitis with lymphohistiocytic inflammation, lipid crystals, and calcifications in our patient (Figure). Subcutaneous fat necrosis (SCFN) was diagnosed based on these characteristic histopathologic findings. No further treatment was pursued.

Subcutaneous fat necrosis is a rare, self-limiting panniculitis that typically resolves within several weeks to months without scarring. It manifests as red or violaceous subcutaneous nodules or plaques most commonly on the buttocks, trunk, proximal arms and legs, and cheeks.1 Histopathology reveals lobular panniculitis with dense granulomatous infiltrates of histiocytes, eosinophils, and multinucleated giant cells with needle-shaped crystals. Focal areas of fat necrosis with calcification also can be seen.2
The epidemiology of SCFN is unknown. Most cases occur in healthy full-term to postterm neonates who experience hypoxia, other prenatal stressors, or therapeutic hypothermia for the treatment of hypoxic-ischemic encephalopathy.3 Although the etiology is unclear, certain inciting factors such as local tissue hypoxia, cold exposure, meconium aspiration, maternal diabetes, preeclampsia, and mechanical pressure have been proposed. Our patient underwent hypothermic cooling protocol, and it has been suggested that the increased saturated to unsaturated fat concentration in the skin of newborns increases the melting point, thus predisposing them to fat crystalization.4 Cases of SCFN involving the scalp are rare; therefore, any newborns receiving hypothermic therapy for hypoxic-ischemic encephalopathy should have a thorough skin examination with possible biopsy of lesions that are characteristic of SCFN, such as red or violaceous subcutaneous nodules or plaques, for specific disease identification.
The main complication of SCFN is hypercalcemia, which occurs in approximately 50% of cases. Other serum abnormalities include hyperglycemia, hypertriglyceridemia, and thrombocytopenia, though these findings are not as well associated.4 Patients with associated hypercalcemia may be asymptomatic, as in our patient, but other presentations include irritability, weakness, anorexia, vomiting, renal failure, failure to thrive, and encephalopathy. Nephrocalcinosis is a common complication of severe hypercalcemia; however, there is little evidence of associated major renal dysfunction.5 The exact mechanism of hypercalcemia is poorly understood. A widely accepted theory postulates that a granulomatous inflammatory infiltrate upregulates 1-α-hydroxylase activity, which enzymatically converts 25-hydroxyvitamin D to its active form, 1,25-dihydroxycholecalciferol, which increases bone resorption and calcium absorption through the gastrointestinal tract and renal systems. Treatments for hypercalcemia include hyperhydration, calcium-wasting diuretics, and low calcium intake.6 Furthermore, calcium levels should be obtained at the time of diagnosis and 30, 45, and 60 days after the lesions resolve.4
Subcutaneous fat necrosis needs to be differentiated from the more severe panniculitis, sclerema neonatorum (SN), which typically affects critically ill, preterm, and small-for-gestational-age newborns. It is associated with a high mortality rate and is characterized by skin and subadjacent tissue structures. The process typically begins in the thighs, buttocks, or trunk and spreads diffusely, sparing the fat-free palms, soles, and genitalia.7 Although our patient was born preterm, the physical characteristics of the nodule and the lack of severe illness placed SN lower on our differential. Histopathologic differences between SCFN and SN involve the extent of tissue fibrosis and presence of inflammatory cells. Sclerema neonatorum typically manifests with thickened connective tissue with a sparse inflammatory infiltrate, including lymphocytes, histiocytes, and multinucleated giant cells.7 Conversely, SCFN manifests with fat necrosis with an extensive inflammatory infiltrate. It is important to be able to distinguish between these 2 conditions, as both have vastly different prognoses.
Cold panniculitis, sometimes called “popsicle panniculitis,” is a phenomenon in which cold contact with the skin causes eruption of firm, erythematous, indurated plaques at the site of exposure. This self-limiting condition typically appears hours to days after cold exposure and spontaneously resolves in a few weeks.8 Therapeutic hypothermic protocol treatment involves using cooling devices to lower the body temperature for a short duration. The temperature typically is lowered to approximately 32 °C to 36 °C. These temperatures are not low enough to induce cold panniculitis, which is more commonly seen in facial ice applications when managing supraventricular tachycardia in neonates.
Cephalohematoma is a birthing injury that causes blood accumulation within the subperiosteal space. During parturition, the compressive and sheering forces on the calvarium rupture the vessels passing through the periosteum, causing blood to pool slowly into the subperiostium; thus, a cephalohematoma usually manifests later at 1 to 3 days of life as localized head swelling.9 The bleeding typically does not cross suture lines and is primarily found in the occipital or parietal regions. The incidence has been reported to be 0.4% to 2.5% of all live births.10 Although the location of the nodule in our patient was in the occipital region, imaging and biopsy results did not show hemorrhagic findings consistent with cephalohematoma. Management of cephalohematoma mainly is observational, as the mass slowly regresses and the accumulated blood gradually is reabsorbed.
Fungal scalp infections (tinea capitis) are common in the pediatric population. The peak incidence of this infection has been reported in children aged 3 to 7 years, with Trichophyton tonsurans and Microsporum canis as the usual causative organisms.11 Clinical features of tinea capitis include scaly patches with hair loss, hair loss with black pigmented dots at the follicular openings, diffuse scalp scaling with subtle hair loss, and cervical lymphadenopathy.12 Although less common, tinea capitis can progress to a more severe form known as a kerion, which is characterized by a tender plaque with pustules and crusting. A kerion can result in permanent scarring and alopecia if left untreated.12 In our patient, a nodule with scaling and faint erythema was observed, but no black pigmented dots at the follicular orifices were present. Therefore, a potassium hydroxide wet mount preparation used to diagnose tinea capitis was unnecessary. Systemic oral antifungal therapy such as fluconazole or terbinafine is the standard treatment for tinea capitis.
- Coondoo A, Lahiry R, Choudhury A, et al. Tender skin nodules in a newborn. Indian J Dermatol. 2013;58:328. doi:10.4103/0019-5154.113983
- Mitra S, Dove J, Somisetty SK. Subcutaneous fat necrosis in newbornan unusual case and review of literature. Eur J Pediatr. 2011;170:1107- 1110. doi:10.1007/s00431-011-1405-x
- Velasquez JH, Mendez MD. Newborn subcutaneous fat necrosis. In: StatPearls. StatPearls Publishing; 2022.
- Stefanko NS, Drolet BA. Subcutaneous fat necrosis of the newborn and associated hypercalcemia: a systematic review of the literature. Pediatr Dermatol. 2019;36:24-30. doi:10.1111/pde.13640
- Shumer DE, Thaker V, Taylor GA, et al. Severe hypercalcaemia due to subcutaneous fat necrosis: presentation, management and complications. Arch Dis Child Fetal Neonatal Ed. 2014;99:F419-F421. doi:10.1136/ archdischild-2014-306069
- Farooque A, Moss C, Zehnder D, et al. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in subcutaneous fat necrosis. Br J Dermatol. 2009;160:423-425. doi:10.1111/j.1365-2133.2008.08844.x
- Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnoses and management. J Perinatol. 2008;28:453-460. doi:10.1038/jp.2008.33
- Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489, vii. doi:10.1016 /j.det.2008.05.015
- Raines DA, Krawiec C, Jain S. Cephalohematoma. In: StatPearls. StatPearls Publishing; 2023.
- Chung HY, Chung JY, Lee DG, et al. Surgical treatment of ossified cephalhematoma. J Craniofac Surg. 2004;15:774-779. doi:10.1097/00001665- 200409000-00015
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68. doi:10.2174/1872 213x14666200106145624
- Kovitwanichkanont T, Chong A. Superficial fungal infections. Aust J Gen Pract. 2019;48:706-711. doi:10.31128/ajgp-05-19-4930
The Diagnosis: Subcutaneous Fat Necrosis
Histopathology revealed lobular panniculitis with lymphohistiocytic inflammation, lipid crystals, and calcifications in our patient (Figure). Subcutaneous fat necrosis (SCFN) was diagnosed based on these characteristic histopathologic findings. No further treatment was pursued.
Subcutaneous fat necrosis is a rare, self-limiting panniculitis that typically resolves within several weeks to months without scarring. It manifests as red or violaceous subcutaneous nodules or plaques most commonly on the buttocks, trunk, proximal arms and legs, and cheeks.1 Histopathology reveals lobular panniculitis with dense granulomatous infiltrates of histiocytes, eosinophils, and multinucleated giant cells with needle-shaped crystals. Focal areas of fat necrosis with calcification also can be seen.2
The epidemiology of SCFN is unknown. Most cases occur in healthy full-term to postterm neonates who experience hypoxia, other prenatal stressors, or therapeutic hypothermia for the treatment of hypoxic-ischemic encephalopathy.3 Although the etiology is unclear, certain inciting factors such as local tissue hypoxia, cold exposure, meconium aspiration, maternal diabetes, preeclampsia, and mechanical pressure have been proposed. Our patient underwent hypothermic cooling protocol, and it has been suggested that the increased saturated to unsaturated fat concentration in the skin of newborns increases the melting point, thus predisposing them to fat crystalization.4 Cases of SCFN involving the scalp are rare; therefore, any newborns receiving hypothermic therapy for hypoxic-ischemic encephalopathy should have a thorough skin examination with possible biopsy of lesions that are characteristic of SCFN, such as red or violaceous subcutaneous nodules or plaques, for specific disease identification.
The main complication of SCFN is hypercalcemia, which occurs in approximately 50% of cases. Other serum abnormalities include hyperglycemia, hypertriglyceridemia, and thrombocytopenia, though these findings are not as well associated.4 Patients with associated hypercalcemia may be asymptomatic, as in our patient, but other presentations include irritability, weakness, anorexia, vomiting, renal failure, failure to thrive, and encephalopathy. Nephrocalcinosis is a common complication of severe hypercalcemia; however, there is little evidence of associated major renal dysfunction.5 The exact mechanism of hypercalcemia is poorly understood. A widely accepted theory postulates that a granulomatous inflammatory infiltrate upregulates 1-α-hydroxylase activity, which enzymatically converts 25-hydroxyvitamin D to its active form, 1,25-dihydroxycholecalciferol, which increases bone resorption and calcium absorption through the gastrointestinal tract and renal systems. Treatments for hypercalcemia include hyperhydration, calcium-wasting diuretics, and low calcium intake.6 Furthermore, calcium levels should be obtained at the time of diagnosis and 30, 45, and 60 days after the lesions resolve.4
Subcutaneous fat necrosis needs to be differentiated from the more severe panniculitis, sclerema neonatorum (SN), which typically affects critically ill, preterm, and small-for-gestational-age newborns. It is associated with a high mortality rate and is characterized by skin and subadjacent tissue structures. The process typically begins in the thighs, buttocks, or trunk and spreads diffusely, sparing the fat-free palms, soles, and genitalia.7 Although our patient was born preterm, the physical characteristics of the nodule and the lack of severe illness placed SN lower on our differential. Histopathologic differences between SCFN and SN involve the extent of tissue fibrosis and presence of inflammatory cells. Sclerema neonatorum typically manifests with thickened connective tissue with a sparse inflammatory infiltrate, including lymphocytes, histiocytes, and multinucleated giant cells.7 Conversely, SCFN manifests with fat necrosis with an extensive inflammatory infiltrate. It is important to be able to distinguish between these 2 conditions, as both have vastly different prognoses.
Cold panniculitis, sometimes called “popsicle panniculitis,” is a phenomenon in which cold contact with the skin causes eruption of firm, erythematous, indurated plaques at the site of exposure. This self-limiting condition typically appears hours to days after cold exposure and spontaneously resolves in a few weeks.8 Therapeutic hypothermic protocol treatment involves using cooling devices to lower the body temperature for a short duration. The temperature typically is lowered to approximately 32 °C to 36 °C. These temperatures are not low enough to induce cold panniculitis, which is more commonly seen in facial ice applications when managing supraventricular tachycardia in neonates.
Cephalohematoma is a birthing injury that causes blood accumulation within the subperiosteal space. During parturition, the compressive and sheering forces on the calvarium rupture the vessels passing through the periosteum, causing blood to pool slowly into the subperiostium; thus, a cephalohematoma usually manifests later at 1 to 3 days of life as localized head swelling.9 The bleeding typically does not cross suture lines and is primarily found in the occipital or parietal regions. The incidence has been reported to be 0.4% to 2.5% of all live births.10 Although the location of the nodule in our patient was in the occipital region, imaging and biopsy results did not show hemorrhagic findings consistent with cephalohematoma. Management of cephalohematoma mainly is observational, as the mass slowly regresses and the accumulated blood gradually is reabsorbed.
Fungal scalp infections (tinea capitis) are common in the pediatric population. The peak incidence of this infection has been reported in children aged 3 to 7 years, with Trichophyton tonsurans and Microsporum canis as the usual causative organisms.11 Clinical features of tinea capitis include scaly patches with hair loss, hair loss with black pigmented dots at the follicular openings, diffuse scalp scaling with subtle hair loss, and cervical lymphadenopathy.12 Although less common, tinea capitis can progress to a more severe form known as a kerion, which is characterized by a tender plaque with pustules and crusting. A kerion can result in permanent scarring and alopecia if left untreated.12 In our patient, a nodule with scaling and faint erythema was observed, but no black pigmented dots at the follicular orifices were present. Therefore, a potassium hydroxide wet mount preparation used to diagnose tinea capitis was unnecessary. Systemic oral antifungal therapy such as fluconazole or terbinafine is the standard treatment for tinea capitis.
The Diagnosis: Subcutaneous Fat Necrosis
Histopathology revealed lobular panniculitis with lymphohistiocytic inflammation, lipid crystals, and calcifications in our patient (Figure). Subcutaneous fat necrosis (SCFN) was diagnosed based on these characteristic histopathologic findings. No further treatment was pursued.
Subcutaneous fat necrosis is a rare, self-limiting panniculitis that typically resolves within several weeks to months without scarring. It manifests as red or violaceous subcutaneous nodules or plaques most commonly on the buttocks, trunk, proximal arms and legs, and cheeks.1 Histopathology reveals lobular panniculitis with dense granulomatous infiltrates of histiocytes, eosinophils, and multinucleated giant cells with needle-shaped crystals. Focal areas of fat necrosis with calcification also can be seen.2
The epidemiology of SCFN is unknown. Most cases occur in healthy full-term to postterm neonates who experience hypoxia, other prenatal stressors, or therapeutic hypothermia for the treatment of hypoxic-ischemic encephalopathy.3 Although the etiology is unclear, certain inciting factors such as local tissue hypoxia, cold exposure, meconium aspiration, maternal diabetes, preeclampsia, and mechanical pressure have been proposed. Our patient underwent hypothermic cooling protocol, and it has been suggested that the increased saturated to unsaturated fat concentration in the skin of newborns increases the melting point, thus predisposing them to fat crystalization.4 Cases of SCFN involving the scalp are rare; therefore, any newborns receiving hypothermic therapy for hypoxic-ischemic encephalopathy should have a thorough skin examination with possible biopsy of lesions that are characteristic of SCFN, such as red or violaceous subcutaneous nodules or plaques, for specific disease identification.
The main complication of SCFN is hypercalcemia, which occurs in approximately 50% of cases. Other serum abnormalities include hyperglycemia, hypertriglyceridemia, and thrombocytopenia, though these findings are not as well associated.4 Patients with associated hypercalcemia may be asymptomatic, as in our patient, but other presentations include irritability, weakness, anorexia, vomiting, renal failure, failure to thrive, and encephalopathy. Nephrocalcinosis is a common complication of severe hypercalcemia; however, there is little evidence of associated major renal dysfunction.5 The exact mechanism of hypercalcemia is poorly understood. A widely accepted theory postulates that a granulomatous inflammatory infiltrate upregulates 1-α-hydroxylase activity, which enzymatically converts 25-hydroxyvitamin D to its active form, 1,25-dihydroxycholecalciferol, which increases bone resorption and calcium absorption through the gastrointestinal tract and renal systems. Treatments for hypercalcemia include hyperhydration, calcium-wasting diuretics, and low calcium intake.6 Furthermore, calcium levels should be obtained at the time of diagnosis and 30, 45, and 60 days after the lesions resolve.4
Subcutaneous fat necrosis needs to be differentiated from the more severe panniculitis, sclerema neonatorum (SN), which typically affects critically ill, preterm, and small-for-gestational-age newborns. It is associated with a high mortality rate and is characterized by skin and subadjacent tissue structures. The process typically begins in the thighs, buttocks, or trunk and spreads diffusely, sparing the fat-free palms, soles, and genitalia.7 Although our patient was born preterm, the physical characteristics of the nodule and the lack of severe illness placed SN lower on our differential. Histopathologic differences between SCFN and SN involve the extent of tissue fibrosis and presence of inflammatory cells. Sclerema neonatorum typically manifests with thickened connective tissue with a sparse inflammatory infiltrate, including lymphocytes, histiocytes, and multinucleated giant cells.7 Conversely, SCFN manifests with fat necrosis with an extensive inflammatory infiltrate. It is important to be able to distinguish between these 2 conditions, as both have vastly different prognoses.
Cold panniculitis, sometimes called “popsicle panniculitis,” is a phenomenon in which cold contact with the skin causes eruption of firm, erythematous, indurated plaques at the site of exposure. This self-limiting condition typically appears hours to days after cold exposure and spontaneously resolves in a few weeks.8 Therapeutic hypothermic protocol treatment involves using cooling devices to lower the body temperature for a short duration. The temperature typically is lowered to approximately 32 °C to 36 °C. These temperatures are not low enough to induce cold panniculitis, which is more commonly seen in facial ice applications when managing supraventricular tachycardia in neonates.
Cephalohematoma is a birthing injury that causes blood accumulation within the subperiosteal space. During parturition, the compressive and sheering forces on the calvarium rupture the vessels passing through the periosteum, causing blood to pool slowly into the subperiostium; thus, a cephalohematoma usually manifests later at 1 to 3 days of life as localized head swelling.9 The bleeding typically does not cross suture lines and is primarily found in the occipital or parietal regions. The incidence has been reported to be 0.4% to 2.5% of all live births.10 Although the location of the nodule in our patient was in the occipital region, imaging and biopsy results did not show hemorrhagic findings consistent with cephalohematoma. Management of cephalohematoma mainly is observational, as the mass slowly regresses and the accumulated blood gradually is reabsorbed.
Fungal scalp infections (tinea capitis) are common in the pediatric population. The peak incidence of this infection has been reported in children aged 3 to 7 years, with Trichophyton tonsurans and Microsporum canis as the usual causative organisms.11 Clinical features of tinea capitis include scaly patches with hair loss, hair loss with black pigmented dots at the follicular openings, diffuse scalp scaling with subtle hair loss, and cervical lymphadenopathy.12 Although less common, tinea capitis can progress to a more severe form known as a kerion, which is characterized by a tender plaque with pustules and crusting. A kerion can result in permanent scarring and alopecia if left untreated.12 In our patient, a nodule with scaling and faint erythema was observed, but no black pigmented dots at the follicular orifices were present. Therefore, a potassium hydroxide wet mount preparation used to diagnose tinea capitis was unnecessary. Systemic oral antifungal therapy such as fluconazole or terbinafine is the standard treatment for tinea capitis.
- Coondoo A, Lahiry R, Choudhury A, et al. Tender skin nodules in a newborn. Indian J Dermatol. 2013;58:328. doi:10.4103/0019-5154.113983
- Mitra S, Dove J, Somisetty SK. Subcutaneous fat necrosis in newbornan unusual case and review of literature. Eur J Pediatr. 2011;170:1107- 1110. doi:10.1007/s00431-011-1405-x
- Velasquez JH, Mendez MD. Newborn subcutaneous fat necrosis. In: StatPearls. StatPearls Publishing; 2022.
- Stefanko NS, Drolet BA. Subcutaneous fat necrosis of the newborn and associated hypercalcemia: a systematic review of the literature. Pediatr Dermatol. 2019;36:24-30. doi:10.1111/pde.13640
- Shumer DE, Thaker V, Taylor GA, et al. Severe hypercalcaemia due to subcutaneous fat necrosis: presentation, management and complications. Arch Dis Child Fetal Neonatal Ed. 2014;99:F419-F421. doi:10.1136/ archdischild-2014-306069
- Farooque A, Moss C, Zehnder D, et al. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in subcutaneous fat necrosis. Br J Dermatol. 2009;160:423-425. doi:10.1111/j.1365-2133.2008.08844.x
- Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnoses and management. J Perinatol. 2008;28:453-460. doi:10.1038/jp.2008.33
- Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489, vii. doi:10.1016 /j.det.2008.05.015
- Raines DA, Krawiec C, Jain S. Cephalohematoma. In: StatPearls. StatPearls Publishing; 2023.
- Chung HY, Chung JY, Lee DG, et al. Surgical treatment of ossified cephalhematoma. J Craniofac Surg. 2004;15:774-779. doi:10.1097/00001665- 200409000-00015
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68. doi:10.2174/1872 213x14666200106145624
- Kovitwanichkanont T, Chong A. Superficial fungal infections. Aust J Gen Pract. 2019;48:706-711. doi:10.31128/ajgp-05-19-4930
- Coondoo A, Lahiry R, Choudhury A, et al. Tender skin nodules in a newborn. Indian J Dermatol. 2013;58:328. doi:10.4103/0019-5154.113983
- Mitra S, Dove J, Somisetty SK. Subcutaneous fat necrosis in newbornan unusual case and review of literature. Eur J Pediatr. 2011;170:1107- 1110. doi:10.1007/s00431-011-1405-x
- Velasquez JH, Mendez MD. Newborn subcutaneous fat necrosis. In: StatPearls. StatPearls Publishing; 2022.
- Stefanko NS, Drolet BA. Subcutaneous fat necrosis of the newborn and associated hypercalcemia: a systematic review of the literature. Pediatr Dermatol. 2019;36:24-30. doi:10.1111/pde.13640
- Shumer DE, Thaker V, Taylor GA, et al. Severe hypercalcaemia due to subcutaneous fat necrosis: presentation, management and complications. Arch Dis Child Fetal Neonatal Ed. 2014;99:F419-F421. doi:10.1136/ archdischild-2014-306069
- Farooque A, Moss C, Zehnder D, et al. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in subcutaneous fat necrosis. Br J Dermatol. 2009;160:423-425. doi:10.1111/j.1365-2133.2008.08844.x
- Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnoses and management. J Perinatol. 2008;28:453-460. doi:10.1038/jp.2008.33
- Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489, vii. doi:10.1016 /j.det.2008.05.015
- Raines DA, Krawiec C, Jain S. Cephalohematoma. In: StatPearls. StatPearls Publishing; 2023.
- Chung HY, Chung JY, Lee DG, et al. Surgical treatment of ossified cephalhematoma. J Craniofac Surg. 2004;15:774-779. doi:10.1097/00001665- 200409000-00015
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68. doi:10.2174/1872 213x14666200106145624
- Kovitwanichkanont T, Chong A. Superficial fungal infections. Aust J Gen Pract. 2019;48:706-711. doi:10.31128/ajgp-05-19-4930
A 4-week-old male infant was referred to dermatology for evaluation of a nodule on the occipital protuberance of 2 weeks’ duration. The patient was born at 36 weeks and 6 days’ gestation via an emergency cesarean delivery due to fetal distress. He later was found to have hypoxic-ischemic encephalopathy, pulmonary hypertension, and hypertrophic cardiomyopathy. He underwent therapeutic hypothermia protocol treatment starting at less than 6 hours after birth. At the current presentation, physical examination showed a 2.5-cm, erythematous, firm, mobile nodule on the occipital scalp with some overlying crusting and minimal surrounding erythema. No other cutaneous features or lesions were present. Initial laboratory findings were remarkable for hypercalcemia at 11 mg/dL (reference range, 8.5-10.5 mg/dL). Magnetic resonance imaging showed a faint abnormality in the subcutaneous tissue in this region without a noted connection to the underlying brain/meningeal matter. A punch biopsy was performed.


Acquired Acrodermatitis Enteropathica in an Infant
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.

Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.

The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.

Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.
Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.
The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.
Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.
Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.
The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.
Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
Practice Points
- Although clinically characterized by the triad of acral and periorificial dermatitis, alopecia, and diarrhea, most cases of acrodermatitis enteropathica (AE) present with only partial features of this syndrome.
- Low levels of zinc-dependent enzymes such as alkaline phosphatase may support the diagnosis of AE.

Exaggerated Facial Lines on the Forehead and Cheeks
The Diagnosis: Pachydermoperiostosis
Histopathology of the forehead punch biopsy demonstrated sebaceous hyperplasia with an occupation rate of greater than 40%, increased mucin, elastic fiber degeneration, and fibrosis. Pachydermia is graded from 0 to 3 depending on the degree of these changes; our patient met criteria for grade 3 pachydermia (Figure 1). Radiography revealed diffuse cortical thickening of the long bones that was most marked in the left femur (Figure 2); however, no other findings were demonstrative of Paget disease.
. B, Colloidal iron stain demonstrated increased mucin (original magnification ×4). C, Verhoeff-van Gieson stain showed elastic fiber")
Pachydermoperiostosis (PDP)(also known as Touraine-Solente-Golé syndrome or primary hypertrophic osteoarthropathy) is a rare genetic condition that affects both the dermatologic and skeletal systems. Clinical features of the disease include progressive thickening and furrowing of the skin on the scalp and face (known as pachydermia), digital clubbing, and periostosis. Other potential cutaneous features include seborrhea, acne, hyperhidrosis of the palms and soles, cutis verticis gyrata, eczema, and a burning sensation of the hands and feet. Myelofibrosis and gastrointestinal abnormalities also have been reported.1

The disease typically affects males (7:1 ratio); also, men typically display a more severe phenotype of the disease.2 It most commonly begins during puberty and follows a generally progressive course of 5 to 20 years before eventually stabilizing. Both autosomal-dominant with incomplete penetrance and recessive inheritance versions of PDP can occur. Prostaglandin E2 (PGE2) has been implicated in the pathogenesis of PDP; PGE2 is important in the inflammatory response and may evolve from disrupted protein degradation pathways.3 Sasaki et al4 additionally reported that the severity of pachydermia clinically and histologically appeared to correlate with the serum PGE2 levels in affected patients. Prostaglandin E2 causes a vasodilatory effect, perhaps explaining the clubbing observed in PDP, and also modifies the activity of osteoblasts and osteoclasts, causing the bone remodeling observed in the disease.4
In our patient, the initial differential diagnosis included PDP, as well as lepromatous leprosy, acromegaly, Paget disease of the bone, amyloidosis, scleromyxedema, and cutaneous T-cell lymphoma. However, the time course of the disease, lack of numerous symmetric thickened plaques and madarosis, and pathology argued against lepromatous leprosy. Acromegaly was ruled out due to lack of macroglossia as well as laboratory analysis within reference range including IGF-1 levels and thyroid function tests. Biopsy findings ultimately ruled out amyloidosis and cutaneous T-cell lymphoma. The bone scan revealed diffuse cortical thickening consistent with PDP, and there were no other radiologic findings suggestive of Paget disease. Pachydermoperiostosis is diagnosed using the Borochowitz criteria, which entails that 2 of the following 4 fulfillment criteria must be met: familial transmission, pachydermia, digital clubbing, and/or bony involvement with evidence of radiologic alterations or pain. Our patient met all 4 criteria. The clinical manifestations of PDP are variable with respect to skin and bone changes. The various clinical expressions include the complete form (ie, pachydermia, cutis verticis gyrata, periostosis), the incomplete form (ie, absence of cutis verticis gyrata), and forme fruste (ie, pachydermia with minimal or absent periostosis).5
Management for PDP involves surgical correction for cosmesis as well as for functional concerns if present. Symptoms secondary to periostosis should be managed with symptomatic treatment such as nonsteroidal antiinflammatory drugs. Patients managed with etoricoxib, a COX-2–selective nonsteroidal anti-inflammatory drug, have had normalized inflammatory markers that resulted in the lessening of forehead skin folds. Oral aescin has been shown to relieve joint pain due to its antiedematous effect.6 Our patient received treatment with nonsteroidal anti-inflammatory drugs for symptomatic management of the associated joint pain but unfortunately was lost to follow-up.
- Castori M, Sinibaldi L, Mingarelli R, et al. Pachydermoperiostosis: an update. Clin Genet. 2005;68:477-486.
- Reginato AJ, Shipachasse V, Guerrero R. Familial idiopathic hypertrophic osteoarthropathy and cranial suture defects in children. Skel Radiol. 1982;8:105-109.
- Coggins KG, Coffman TM, Koller BH. The Hippocratic finger points the blame at PGE2. Nat Genet. 2008;40:691-692.
- Sasaki T, Niizeki H, Shimizu A, et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J Dermatol Sci. 2012;68:36-44.
- Bhaskaranand K, Shetty RR, Bhat AK. Pachydermoperiostosis: three case reports. J Orthop Surg (Hong Kong). 2001;9:61-66.
- Zhang H, Yang B. Successful treatment of pachydermoperiostosis patients with etoricoxib, aescin, and arthroscopic synovectomy: two case reports. Medicine (Baltimore). 2017;96:E8865.
The Diagnosis: Pachydermoperiostosis
Histopathology of the forehead punch biopsy demonstrated sebaceous hyperplasia with an occupation rate of greater than 40%, increased mucin, elastic fiber degeneration, and fibrosis. Pachydermia is graded from 0 to 3 depending on the degree of these changes; our patient met criteria for grade 3 pachydermia (Figure 1). Radiography revealed diffuse cortical thickening of the long bones that was most marked in the left femur (Figure 2); however, no other findings were demonstrative of Paget disease.
Pachydermoperiostosis (PDP)(also known as Touraine-Solente-Golé syndrome or primary hypertrophic osteoarthropathy) is a rare genetic condition that affects both the dermatologic and skeletal systems. Clinical features of the disease include progressive thickening and furrowing of the skin on the scalp and face (known as pachydermia), digital clubbing, and periostosis. Other potential cutaneous features include seborrhea, acne, hyperhidrosis of the palms and soles, cutis verticis gyrata, eczema, and a burning sensation of the hands and feet. Myelofibrosis and gastrointestinal abnormalities also have been reported.1
The disease typically affects males (7:1 ratio); also, men typically display a more severe phenotype of the disease.2 It most commonly begins during puberty and follows a generally progressive course of 5 to 20 years before eventually stabilizing. Both autosomal-dominant with incomplete penetrance and recessive inheritance versions of PDP can occur. Prostaglandin E2 (PGE2) has been implicated in the pathogenesis of PDP; PGE2 is important in the inflammatory response and may evolve from disrupted protein degradation pathways.3 Sasaki et al4 additionally reported that the severity of pachydermia clinically and histologically appeared to correlate with the serum PGE2 levels in affected patients. Prostaglandin E2 causes a vasodilatory effect, perhaps explaining the clubbing observed in PDP, and also modifies the activity of osteoblasts and osteoclasts, causing the bone remodeling observed in the disease.4
In our patient, the initial differential diagnosis included PDP, as well as lepromatous leprosy, acromegaly, Paget disease of the bone, amyloidosis, scleromyxedema, and cutaneous T-cell lymphoma. However, the time course of the disease, lack of numerous symmetric thickened plaques and madarosis, and pathology argued against lepromatous leprosy. Acromegaly was ruled out due to lack of macroglossia as well as laboratory analysis within reference range including IGF-1 levels and thyroid function tests. Biopsy findings ultimately ruled out amyloidosis and cutaneous T-cell lymphoma. The bone scan revealed diffuse cortical thickening consistent with PDP, and there were no other radiologic findings suggestive of Paget disease. Pachydermoperiostosis is diagnosed using the Borochowitz criteria, which entails that 2 of the following 4 fulfillment criteria must be met: familial transmission, pachydermia, digital clubbing, and/or bony involvement with evidence of radiologic alterations or pain. Our patient met all 4 criteria. The clinical manifestations of PDP are variable with respect to skin and bone changes. The various clinical expressions include the complete form (ie, pachydermia, cutis verticis gyrata, periostosis), the incomplete form (ie, absence of cutis verticis gyrata), and forme fruste (ie, pachydermia with minimal or absent periostosis).5
Management for PDP involves surgical correction for cosmesis as well as for functional concerns if present. Symptoms secondary to periostosis should be managed with symptomatic treatment such as nonsteroidal antiinflammatory drugs. Patients managed with etoricoxib, a COX-2–selective nonsteroidal anti-inflammatory drug, have had normalized inflammatory markers that resulted in the lessening of forehead skin folds. Oral aescin has been shown to relieve joint pain due to its antiedematous effect.6 Our patient received treatment with nonsteroidal anti-inflammatory drugs for symptomatic management of the associated joint pain but unfortunately was lost to follow-up.
The Diagnosis: Pachydermoperiostosis
Histopathology of the forehead punch biopsy demonstrated sebaceous hyperplasia with an occupation rate of greater than 40%, increased mucin, elastic fiber degeneration, and fibrosis. Pachydermia is graded from 0 to 3 depending on the degree of these changes; our patient met criteria for grade 3 pachydermia (Figure 1). Radiography revealed diffuse cortical thickening of the long bones that was most marked in the left femur (Figure 2); however, no other findings were demonstrative of Paget disease.
Pachydermoperiostosis (PDP)(also known as Touraine-Solente-Golé syndrome or primary hypertrophic osteoarthropathy) is a rare genetic condition that affects both the dermatologic and skeletal systems. Clinical features of the disease include progressive thickening and furrowing of the skin on the scalp and face (known as pachydermia), digital clubbing, and periostosis. Other potential cutaneous features include seborrhea, acne, hyperhidrosis of the palms and soles, cutis verticis gyrata, eczema, and a burning sensation of the hands and feet. Myelofibrosis and gastrointestinal abnormalities also have been reported.1
The disease typically affects males (7:1 ratio); also, men typically display a more severe phenotype of the disease.2 It most commonly begins during puberty and follows a generally progressive course of 5 to 20 years before eventually stabilizing. Both autosomal-dominant with incomplete penetrance and recessive inheritance versions of PDP can occur. Prostaglandin E2 (PGE2) has been implicated in the pathogenesis of PDP; PGE2 is important in the inflammatory response and may evolve from disrupted protein degradation pathways.3 Sasaki et al4 additionally reported that the severity of pachydermia clinically and histologically appeared to correlate with the serum PGE2 levels in affected patients. Prostaglandin E2 causes a vasodilatory effect, perhaps explaining the clubbing observed in PDP, and also modifies the activity of osteoblasts and osteoclasts, causing the bone remodeling observed in the disease.4
In our patient, the initial differential diagnosis included PDP, as well as lepromatous leprosy, acromegaly, Paget disease of the bone, amyloidosis, scleromyxedema, and cutaneous T-cell lymphoma. However, the time course of the disease, lack of numerous symmetric thickened plaques and madarosis, and pathology argued against lepromatous leprosy. Acromegaly was ruled out due to lack of macroglossia as well as laboratory analysis within reference range including IGF-1 levels and thyroid function tests. Biopsy findings ultimately ruled out amyloidosis and cutaneous T-cell lymphoma. The bone scan revealed diffuse cortical thickening consistent with PDP, and there were no other radiologic findings suggestive of Paget disease. Pachydermoperiostosis is diagnosed using the Borochowitz criteria, which entails that 2 of the following 4 fulfillment criteria must be met: familial transmission, pachydermia, digital clubbing, and/or bony involvement with evidence of radiologic alterations or pain. Our patient met all 4 criteria. The clinical manifestations of PDP are variable with respect to skin and bone changes. The various clinical expressions include the complete form (ie, pachydermia, cutis verticis gyrata, periostosis), the incomplete form (ie, absence of cutis verticis gyrata), and forme fruste (ie, pachydermia with minimal or absent periostosis).5
Management for PDP involves surgical correction for cosmesis as well as for functional concerns if present. Symptoms secondary to periostosis should be managed with symptomatic treatment such as nonsteroidal antiinflammatory drugs. Patients managed with etoricoxib, a COX-2–selective nonsteroidal anti-inflammatory drug, have had normalized inflammatory markers that resulted in the lessening of forehead skin folds. Oral aescin has been shown to relieve joint pain due to its antiedematous effect.6 Our patient received treatment with nonsteroidal anti-inflammatory drugs for symptomatic management of the associated joint pain but unfortunately was lost to follow-up.
- Castori M, Sinibaldi L, Mingarelli R, et al. Pachydermoperiostosis: an update. Clin Genet. 2005;68:477-486.
- Reginato AJ, Shipachasse V, Guerrero R. Familial idiopathic hypertrophic osteoarthropathy and cranial suture defects in children. Skel Radiol. 1982;8:105-109.
- Coggins KG, Coffman TM, Koller BH. The Hippocratic finger points the blame at PGE2. Nat Genet. 2008;40:691-692.
- Sasaki T, Niizeki H, Shimizu A, et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J Dermatol Sci. 2012;68:36-44.
- Bhaskaranand K, Shetty RR, Bhat AK. Pachydermoperiostosis: three case reports. J Orthop Surg (Hong Kong). 2001;9:61-66.
- Zhang H, Yang B. Successful treatment of pachydermoperiostosis patients with etoricoxib, aescin, and arthroscopic synovectomy: two case reports. Medicine (Baltimore). 2017;96:E8865.
- Castori M, Sinibaldi L, Mingarelli R, et al. Pachydermoperiostosis: an update. Clin Genet. 2005;68:477-486.
- Reginato AJ, Shipachasse V, Guerrero R. Familial idiopathic hypertrophic osteoarthropathy and cranial suture defects in children. Skel Radiol. 1982;8:105-109.
- Coggins KG, Coffman TM, Koller BH. The Hippocratic finger points the blame at PGE2. Nat Genet. 2008;40:691-692.
- Sasaki T, Niizeki H, Shimizu A, et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J Dermatol Sci. 2012;68:36-44.
- Bhaskaranand K, Shetty RR, Bhat AK. Pachydermoperiostosis: three case reports. J Orthop Surg (Hong Kong). 2001;9:61-66.
- Zhang H, Yang B. Successful treatment of pachydermoperiostosis patients with etoricoxib, aescin, and arthroscopic synovectomy: two case reports. Medicine (Baltimore). 2017;96:E8865.
A 36-year-old man presented to the emergency department with an olecranon fracture after falling from a tree. The patient had a medical history of type 2 diabetes mellitus and a surgical history of facial cosmetic surgery. He underwent internal fixation with orthopedic surgery for the olecranon fracture, and dermatology subsequently was consulted due to diffuse skin changes on the face. He reported that these dermatologic changes began around 17 years of age and had progressed to the current presentation. He denied itching, burning, pain, or contact with armadillos. A family history revealed the patient’s brother also had a similar appearance. Physical examination revealed exaggerated facial lines on the forehead (top) and cheeks. Digital clubbing and skin thickening were noted on the hands (bottom) and feet; examination of the back revealed multiple hypopigmented patches. Observation of the scalp showed multiple symmetric ridges and grooves with sparse overlying hair consistent with cutis verticis gyrata. A punch biopsy of the forehead was obtained as well as bone radiography taken previously by the primary team.


Generalized rash follows ankle ulceration
A 31-year-old incarcerated man sought care for one crusted ulcer and one adjacent open ulcer with granulation tissue on his left malleolus. The ulcers were caused by chronic venous insufficiency—the result of previous trauma to the ankle. Concerned that the ulcers would become infected, the physician prescribed one double-strength tablet twice a day of trimethoprim-sulfamethoxazole (TMP-SMX). The patient took 2 doses of the antibiotic and one dose of naproxen.
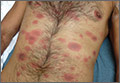
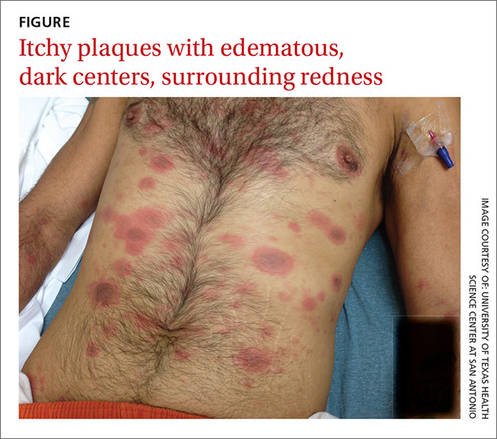
When the patient awoke the next morning, he had a generalized skin eruption on his chin, trunk, buttocks, glans penis, and extremities (FIGURE). The rash began as red edematous plaques that became itchy and painful with dark, violaceous dusky centers surrounded by redness. The patient was treated with topical hydrocortisone 2.5% twice a day and oral diphenhydramine 25 mg followed by 50 mg, but the rash didn’t improve.
The patient was transported to the local emergency department where physicians noted that the patient had about 30 to 40 well-demarcated papules and plaques of various sizes that were haphazardly located over the patient’s chin, chest, back, upper and lower extremities, and genitalia. There was one lesion on the chest with central vesiculation. There were no lesions on the mucous membranes of his eyes, ears, nose, mouth, or anus.
The patient, whose vital signs were within normal limits, was empirically treated with one dose of methylprednisolone (125 mg intravenous [IV]) and started on IV piperacillin-tazobactam and vancomycin. Lab work revealed no elevation in his white blood cell count, creatinine, liver function enzymes, or C-reactive protein.
The patient subsequently revealed that he’d had a similar experience a year earlier after being treated with TMP-SMX for cellulitis. He noted that during the previous episode, the lesions were located on the exact same areas of his glans penis and chin.
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Disseminated fixed-drug eruption
The diagnosis was based on the morphologic characteristics of the eruption and the patient’s history of similar lesions that appeared in the exact same initial locations (chin and glans penis) following previous treatment with TMP-SMX.
A fixed-drug eruption is an adverse cutaneous reaction to a drug that is defined by a dusky red or violaceous macule, which evolves into a patch, and eventually, an edematous plaque. Fixed-drug eruptions are typically solitary, but may be generalized (as was the case with our patient).
The pathophysiology of the disease involves resident intra-epidermal CD8+ T-cells resembling effector memory T-cells. These T-cells are increased in number at the dermoepidermal junction of normal appearing skin; their aberrant activation leads to an inflammatory response, stimulating tissue destruction and formation of the classic fixed-drug lesion.1
The diagnosis is usually made based on a history of similar lesions recurring at the same location in response to a specific drug2 and the classic physical exam findings of well-demarcated, edematous, and violaceous plaques. To confirm a fixed-drug eruption in the case of clinical equipoise, a skin biopsy may be performed.
Classic histologic findings of a fixed-drug eruption include:
- band-like lichenoid lymphocytic infiltrates with vacuolar changes at the dermoepidermal junction,
- mixed cellular infiltrates, including eosinophils, throughout the dermis and occasional superficial and deep mixed cellular perivascular infiltrates, and
- abundant melanophages suggesting pigment incontinence.
There are several reports of similar TMP-SMX–induced generalized fixed-drug eruptions in the literature.3 One study of 64 cases of fixed-drug eruption found that TMP-SMX was the most common offender, causing 75% of fixed-drug eruption cases; naproxen sodium came in second with 12.5%.3 Other common culprits include the antipyretic metamizole and other pyrazolone derivatives such as tetracycline, metronidazole, ciprofloxacin, and phenytoin sodium.4 There is evidence supporting a correlation between the offending drug and the subsequent site of reaction; TMP-SMX is associated with mucosal junction and genital involvement.4,5 This finding may aid physicians in the investigation of provoking agents.
Distinguish fixed-drug eruptions from serious bullous diseases
Fixed-drug eruptions occasionally exhibit bullae and erosions and must be differentiated from more serious generalized bullous diseases, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). The differential diagnosis also includes erythema multiforme, early bullous drug eruption, and bullous arthropod assault, which may leave similar hyperpigmented patches. Fixed-drug eruptions can be distinguished by the lack of simultaneous involvement of 2 mucosal surfaces, lack of generalized desquamation, and normal vital signs and lab values, including white blood cell count and erythrocyte sedimentation rate/C-reactive protein.
A subset of fixed-drug eruption, generalized bullous fixed-drug eruption (which has been defined as blistering on >10% of the body’s surface area at 3 different anatomic sites), may be particularly hard to distinguish from SJS and TEN. Generalized bullous fixed-drug eruption generally has a shorter latency period than SJS or TEN (usually <3 days compared to 7-10 days) and has less mucosal involvement.6
Symptomatic therapy includes antihistamines, glucocorticoid ointment
Management of a disseminated fixed-drug eruption requires a thorough history to identify the causative agent (including over-the-counter drugs, herbals, topicals, and eye drops). Most patients are asymptomatic, but some (like our patient) are symptomatic and experience generalized pruritus, cutaneous burning, and/or pain. Symptomatic therapy includes oral antihistamines and potent topical glucocorticoid ointment for non-eroded lesions. Additionally, if not medically contraindicated, oral steroids may be used for generalized or extremely painful mucosal lesions at a dose of 0.5 mg/kg daily for 3 to 5 days. Be advised, however, that these therapies are based on case report level data.2
Local wound care of eroded lesions includes keeping the site moist with a bland emollient and bandaging. The inciting agent must be added to the patient’s allergy list and avoided in the future. In equivocal cases, it is prudent to admit the patient for observation to ensure that the eruption is not a nascent SJS or TEN eruption.
Our patient was admitted to the observation unit overnight to monitor for the appearance of systemic symptoms and to assess the evolution of the rash for further mucosal involvement that could have indicated SJS. Upon reassessment the next day, his older lesions had evolved into vesiculated and necrotic areas as per the natural history of severe fixed-drug eruption.
He was prescribed prednisone 40 mg/d for 3 days to help with local inflammation, pain, and itching. TMP-SMX was added to his allergy list and he was given local wound care instructions. He was told to return if he developed any systemic symptoms.
CORRESPONDENCE
Jackie Bucher, MD, 7733 Louis Pasteur Drive Apt. 209, San Antonio, TX 78229; bucher@uthscsa.edu.
1. Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
2. Wolff K, Johnson RA. Dermatology and internal medicine: fixed drug eruption. In: Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York: McGraw-Hill; 2009:566-568.
3. Ozkaya-Bayazit E, Bayazit H, Ozarmagan G. Drug related clinical pattern in fixed drug eruption. Eur J Dermatol. 2000;10:288-291.
4. Sharma VK, Dhar S, Gill AN. Drug related involvement of specific sites in fixed eruptions: a statistical evaluation. J Dermatol. 1996;23:530-534.
5. Thankappan TP, Zachariah J. Drug-specific clinical pattern in fixed drug eruptions. Int J Dermatol. 1991;30:867-870.
6. Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.
A 31-year-old incarcerated man sought care for one crusted ulcer and one adjacent open ulcer with granulation tissue on his left malleolus. The ulcers were caused by chronic venous insufficiency—the result of previous trauma to the ankle. Concerned that the ulcers would become infected, the physician prescribed one double-strength tablet twice a day of trimethoprim-sulfamethoxazole (TMP-SMX). The patient took 2 doses of the antibiotic and one dose of naproxen.
When the patient awoke the next morning, he had a generalized skin eruption on his chin, trunk, buttocks, glans penis, and extremities (FIGURE). The rash began as red edematous plaques that became itchy and painful with dark, violaceous dusky centers surrounded by redness. The patient was treated with topical hydrocortisone 2.5% twice a day and oral diphenhydramine 25 mg followed by 50 mg, but the rash didn’t improve.
The patient was transported to the local emergency department where physicians noted that the patient had about 30 to 40 well-demarcated papules and plaques of various sizes that were haphazardly located over the patient’s chin, chest, back, upper and lower extremities, and genitalia. There was one lesion on the chest with central vesiculation. There were no lesions on the mucous membranes of his eyes, ears, nose, mouth, or anus.
The patient, whose vital signs were within normal limits, was empirically treated with one dose of methylprednisolone (125 mg intravenous [IV]) and started on IV piperacillin-tazobactam and vancomycin. Lab work revealed no elevation in his white blood cell count, creatinine, liver function enzymes, or C-reactive protein.
The patient subsequently revealed that he’d had a similar experience a year earlier after being treated with TMP-SMX for cellulitis. He noted that during the previous episode, the lesions were located on the exact same areas of his glans penis and chin.
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Disseminated fixed-drug eruption
The diagnosis was based on the morphologic characteristics of the eruption and the patient’s history of similar lesions that appeared in the exact same initial locations (chin and glans penis) following previous treatment with TMP-SMX.
A fixed-drug eruption is an adverse cutaneous reaction to a drug that is defined by a dusky red or violaceous macule, which evolves into a patch, and eventually, an edematous plaque. Fixed-drug eruptions are typically solitary, but may be generalized (as was the case with our patient).
The pathophysiology of the disease involves resident intra-epidermal CD8+ T-cells resembling effector memory T-cells. These T-cells are increased in number at the dermoepidermal junction of normal appearing skin; their aberrant activation leads to an inflammatory response, stimulating tissue destruction and formation of the classic fixed-drug lesion.1
The diagnosis is usually made based on a history of similar lesions recurring at the same location in response to a specific drug2 and the classic physical exam findings of well-demarcated, edematous, and violaceous plaques. To confirm a fixed-drug eruption in the case of clinical equipoise, a skin biopsy may be performed.
Classic histologic findings of a fixed-drug eruption include:
- band-like lichenoid lymphocytic infiltrates with vacuolar changes at the dermoepidermal junction,
- mixed cellular infiltrates, including eosinophils, throughout the dermis and occasional superficial and deep mixed cellular perivascular infiltrates, and
- abundant melanophages suggesting pigment incontinence.
There are several reports of similar TMP-SMX–induced generalized fixed-drug eruptions in the literature.3 One study of 64 cases of fixed-drug eruption found that TMP-SMX was the most common offender, causing 75% of fixed-drug eruption cases; naproxen sodium came in second with 12.5%.3 Other common culprits include the antipyretic metamizole and other pyrazolone derivatives such as tetracycline, metronidazole, ciprofloxacin, and phenytoin sodium.4 There is evidence supporting a correlation between the offending drug and the subsequent site of reaction; TMP-SMX is associated with mucosal junction and genital involvement.4,5 This finding may aid physicians in the investigation of provoking agents.
Distinguish fixed-drug eruptions from serious bullous diseases
Fixed-drug eruptions occasionally exhibit bullae and erosions and must be differentiated from more serious generalized bullous diseases, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). The differential diagnosis also includes erythema multiforme, early bullous drug eruption, and bullous arthropod assault, which may leave similar hyperpigmented patches. Fixed-drug eruptions can be distinguished by the lack of simultaneous involvement of 2 mucosal surfaces, lack of generalized desquamation, and normal vital signs and lab values, including white blood cell count and erythrocyte sedimentation rate/C-reactive protein.
A subset of fixed-drug eruption, generalized bullous fixed-drug eruption (which has been defined as blistering on >10% of the body’s surface area at 3 different anatomic sites), may be particularly hard to distinguish from SJS and TEN. Generalized bullous fixed-drug eruption generally has a shorter latency period than SJS or TEN (usually <3 days compared to 7-10 days) and has less mucosal involvement.6
Symptomatic therapy includes antihistamines, glucocorticoid ointment
Management of a disseminated fixed-drug eruption requires a thorough history to identify the causative agent (including over-the-counter drugs, herbals, topicals, and eye drops). Most patients are asymptomatic, but some (like our patient) are symptomatic and experience generalized pruritus, cutaneous burning, and/or pain. Symptomatic therapy includes oral antihistamines and potent topical glucocorticoid ointment for non-eroded lesions. Additionally, if not medically contraindicated, oral steroids may be used for generalized or extremely painful mucosal lesions at a dose of 0.5 mg/kg daily for 3 to 5 days. Be advised, however, that these therapies are based on case report level data.2
Local wound care of eroded lesions includes keeping the site moist with a bland emollient and bandaging. The inciting agent must be added to the patient’s allergy list and avoided in the future. In equivocal cases, it is prudent to admit the patient for observation to ensure that the eruption is not a nascent SJS or TEN eruption.
Our patient was admitted to the observation unit overnight to monitor for the appearance of systemic symptoms and to assess the evolution of the rash for further mucosal involvement that could have indicated SJS. Upon reassessment the next day, his older lesions had evolved into vesiculated and necrotic areas as per the natural history of severe fixed-drug eruption.
He was prescribed prednisone 40 mg/d for 3 days to help with local inflammation, pain, and itching. TMP-SMX was added to his allergy list and he was given local wound care instructions. He was told to return if he developed any systemic symptoms.
CORRESPONDENCE
Jackie Bucher, MD, 7733 Louis Pasteur Drive Apt. 209, San Antonio, TX 78229; bucher@uthscsa.edu.
A 31-year-old incarcerated man sought care for one crusted ulcer and one adjacent open ulcer with granulation tissue on his left malleolus. The ulcers were caused by chronic venous insufficiency—the result of previous trauma to the ankle. Concerned that the ulcers would become infected, the physician prescribed one double-strength tablet twice a day of trimethoprim-sulfamethoxazole (TMP-SMX). The patient took 2 doses of the antibiotic and one dose of naproxen.
When the patient awoke the next morning, he had a generalized skin eruption on his chin, trunk, buttocks, glans penis, and extremities (FIGURE). The rash began as red edematous plaques that became itchy and painful with dark, violaceous dusky centers surrounded by redness. The patient was treated with topical hydrocortisone 2.5% twice a day and oral diphenhydramine 25 mg followed by 50 mg, but the rash didn’t improve.
The patient was transported to the local emergency department where physicians noted that the patient had about 30 to 40 well-demarcated papules and plaques of various sizes that were haphazardly located over the patient’s chin, chest, back, upper and lower extremities, and genitalia. There was one lesion on the chest with central vesiculation. There were no lesions on the mucous membranes of his eyes, ears, nose, mouth, or anus.
The patient, whose vital signs were within normal limits, was empirically treated with one dose of methylprednisolone (125 mg intravenous [IV]) and started on IV piperacillin-tazobactam and vancomycin. Lab work revealed no elevation in his white blood cell count, creatinine, liver function enzymes, or C-reactive protein.
The patient subsequently revealed that he’d had a similar experience a year earlier after being treated with TMP-SMX for cellulitis. He noted that during the previous episode, the lesions were located on the exact same areas of his glans penis and chin.
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Disseminated fixed-drug eruption
The diagnosis was based on the morphologic characteristics of the eruption and the patient’s history of similar lesions that appeared in the exact same initial locations (chin and glans penis) following previous treatment with TMP-SMX.
A fixed-drug eruption is an adverse cutaneous reaction to a drug that is defined by a dusky red or violaceous macule, which evolves into a patch, and eventually, an edematous plaque. Fixed-drug eruptions are typically solitary, but may be generalized (as was the case with our patient).
The pathophysiology of the disease involves resident intra-epidermal CD8+ T-cells resembling effector memory T-cells. These T-cells are increased in number at the dermoepidermal junction of normal appearing skin; their aberrant activation leads to an inflammatory response, stimulating tissue destruction and formation of the classic fixed-drug lesion.1
The diagnosis is usually made based on a history of similar lesions recurring at the same location in response to a specific drug2 and the classic physical exam findings of well-demarcated, edematous, and violaceous plaques. To confirm a fixed-drug eruption in the case of clinical equipoise, a skin biopsy may be performed.
Classic histologic findings of a fixed-drug eruption include:
- band-like lichenoid lymphocytic infiltrates with vacuolar changes at the dermoepidermal junction,
- mixed cellular infiltrates, including eosinophils, throughout the dermis and occasional superficial and deep mixed cellular perivascular infiltrates, and
- abundant melanophages suggesting pigment incontinence.
There are several reports of similar TMP-SMX–induced generalized fixed-drug eruptions in the literature.3 One study of 64 cases of fixed-drug eruption found that TMP-SMX was the most common offender, causing 75% of fixed-drug eruption cases; naproxen sodium came in second with 12.5%.3 Other common culprits include the antipyretic metamizole and other pyrazolone derivatives such as tetracycline, metronidazole, ciprofloxacin, and phenytoin sodium.4 There is evidence supporting a correlation between the offending drug and the subsequent site of reaction; TMP-SMX is associated with mucosal junction and genital involvement.4,5 This finding may aid physicians in the investigation of provoking agents.
Distinguish fixed-drug eruptions from serious bullous diseases
Fixed-drug eruptions occasionally exhibit bullae and erosions and must be differentiated from more serious generalized bullous diseases, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). The differential diagnosis also includes erythema multiforme, early bullous drug eruption, and bullous arthropod assault, which may leave similar hyperpigmented patches. Fixed-drug eruptions can be distinguished by the lack of simultaneous involvement of 2 mucosal surfaces, lack of generalized desquamation, and normal vital signs and lab values, including white blood cell count and erythrocyte sedimentation rate/C-reactive protein.
A subset of fixed-drug eruption, generalized bullous fixed-drug eruption (which has been defined as blistering on >10% of the body’s surface area at 3 different anatomic sites), may be particularly hard to distinguish from SJS and TEN. Generalized bullous fixed-drug eruption generally has a shorter latency period than SJS or TEN (usually <3 days compared to 7-10 days) and has less mucosal involvement.6
Symptomatic therapy includes antihistamines, glucocorticoid ointment
Management of a disseminated fixed-drug eruption requires a thorough history to identify the causative agent (including over-the-counter drugs, herbals, topicals, and eye drops). Most patients are asymptomatic, but some (like our patient) are symptomatic and experience generalized pruritus, cutaneous burning, and/or pain. Symptomatic therapy includes oral antihistamines and potent topical glucocorticoid ointment for non-eroded lesions. Additionally, if not medically contraindicated, oral steroids may be used for generalized or extremely painful mucosal lesions at a dose of 0.5 mg/kg daily for 3 to 5 days. Be advised, however, that these therapies are based on case report level data.2
Local wound care of eroded lesions includes keeping the site moist with a bland emollient and bandaging. The inciting agent must be added to the patient’s allergy list and avoided in the future. In equivocal cases, it is prudent to admit the patient for observation to ensure that the eruption is not a nascent SJS or TEN eruption.
Our patient was admitted to the observation unit overnight to monitor for the appearance of systemic symptoms and to assess the evolution of the rash for further mucosal involvement that could have indicated SJS. Upon reassessment the next day, his older lesions had evolved into vesiculated and necrotic areas as per the natural history of severe fixed-drug eruption.
He was prescribed prednisone 40 mg/d for 3 days to help with local inflammation, pain, and itching. TMP-SMX was added to his allergy list and he was given local wound care instructions. He was told to return if he developed any systemic symptoms.
CORRESPONDENCE
Jackie Bucher, MD, 7733 Louis Pasteur Drive Apt. 209, San Antonio, TX 78229; bucher@uthscsa.edu.
1. Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
2. Wolff K, Johnson RA. Dermatology and internal medicine: fixed drug eruption. In: Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York: McGraw-Hill; 2009:566-568.
3. Ozkaya-Bayazit E, Bayazit H, Ozarmagan G. Drug related clinical pattern in fixed drug eruption. Eur J Dermatol. 2000;10:288-291.
4. Sharma VK, Dhar S, Gill AN. Drug related involvement of specific sites in fixed eruptions: a statistical evaluation. J Dermatol. 1996;23:530-534.
5. Thankappan TP, Zachariah J. Drug-specific clinical pattern in fixed drug eruptions. Int J Dermatol. 1991;30:867-870.
6. Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.
1. Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
2. Wolff K, Johnson RA. Dermatology and internal medicine: fixed drug eruption. In: Wolff K, Johnson RA, Saavedra AP, eds. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York: McGraw-Hill; 2009:566-568.
3. Ozkaya-Bayazit E, Bayazit H, Ozarmagan G. Drug related clinical pattern in fixed drug eruption. Eur J Dermatol. 2000;10:288-291.
4. Sharma VK, Dhar S, Gill AN. Drug related involvement of specific sites in fixed eruptions: a statistical evaluation. J Dermatol. 1996;23:530-534.
5. Thankappan TP, Zachariah J. Drug-specific clinical pattern in fixed drug eruptions. Int J Dermatol. 1991;30:867-870.
6. Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.