User login
What’s Eating You? Phlebotomine Sandflies and Leishmania Parasites
The genus Leishmania comprises protozoan parasites that cause approximately 2 million new cases of leishmaniasis each year across 98 countries.1 These protozoa are obligate intracellular parasites of phlebotomine sandfly species that transmit leishmaniasis and result in a considerable parasitic cause of fatalities globally, second only to malaria.2,3
Phlebotomine sandflies primarily live in tropical and subtropical regions and function as vectors for many pathogens in addition to Leishmania species, such as Bartonella species and arboviruses.3 In 2004, it was noted that the majority of leishmaniasis cases affected developing countries: 90% of visceral leishmaniasis cases occurred in Bangladesh, India, Nepal, Sudan, and Brazil, and 90% of cutaneous leishmaniasis cases occurred in Afghanistan, Algeria, Brazil, Iran, Peru, Saudi Arabia, and Syria.4 Of note, with recent environmental changes, phlebotomine sandflies have gradually migrated to more northerly latitudes, extending into Europe.5
Twenty Leishmania species and 30 sandfly species have been identified as causes of leishmaniasis.4Leishmania infection occurs when an infected sandfly bites a mammalian host and transmits the parasite’s flagellated form, known as a promastigote. Host inflammatory cells, such as monocytes and dendritic cells, phagocytize parasites that enter the skin. The interaction between parasites and dendritic cells become an important factor in the outcome of Leishmania infection in the host because dendritic cells promote development of CD4 and CD8 T lymphocytes with specificity to target Leishmania parasites and protect the host.1
The number of cases of leishmaniasis has increased worldwide, most likely due to changes in the environment and human behaviors such as urbanization, the creation of new settlements, and migration from rural to urban areas.3,5 Important risk factors in individual patients include malnutrition; low-quality housing and sanitation; a history of migration or travel; and immunosuppression, such as that caused by HIV co-infection.2,5
Case Report
An otherwise healthy 25-year-old Bangladeshi man presented to our community hospital for evaluation of a painful leg ulcer of 1 month’s duration. The patient had migrated from Bangladesh to Panama, then to Costa Rica, followed by Guatemala, Honduras, Mexico, and, last, Texas. In Texas, he was identified by the US Immigration and Customs Enforcement, transported to a detention facility, and transferred to this hospital shortly afterward.
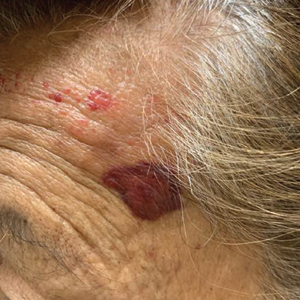
The patient reported that, during his extensive migration, he had lived in the jungle and reported what he described as mosquito bites on the legs. He subsequently developed a 3-cm ulcerated and crusted plaque with rolled borders on the right medial ankle (Figure 1). In addition, he had a palpable nodular cord on the medial leg from the ankle lesion to the mid thigh that was consistent with lymphocutaneous spread. Ultrasonography was negative for deep-vein thrombosis.

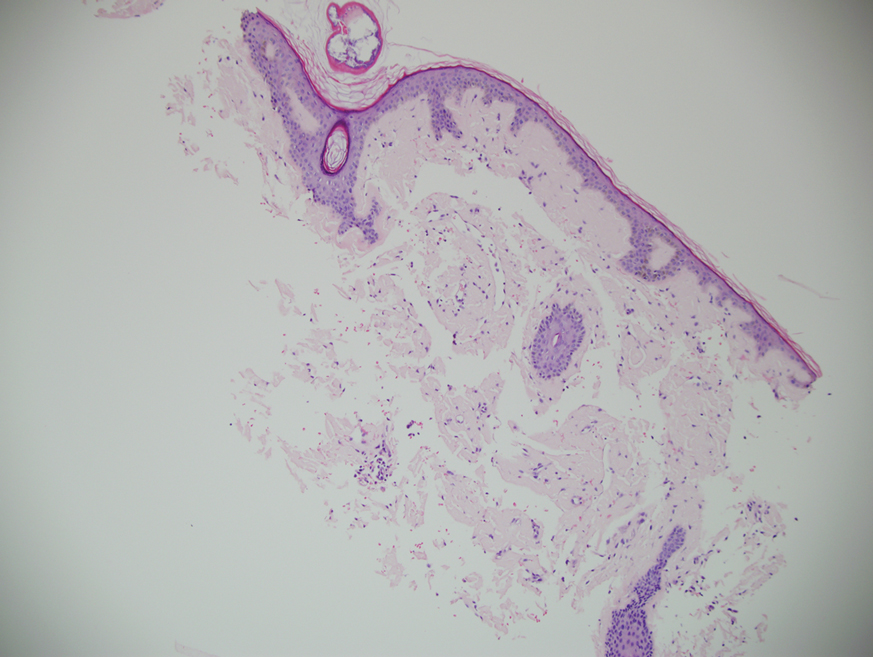
Because the patient’s recent migration from Central America was highly concerning for microbial infection, vancomycin and piperacillin-tazobactam were started empirically on admission. A punch biopsy from the right medial ankle was nondiagnostic, showing acute and chronic necrotizing inflammation along with numerous epithelioid histiocytes with a vaguely granulomatous appearance (Figure 2). A specimen from the right medial ankle that had already been taken by an astute border patrol medical provider was sent to the Centers for Disease Control and Prevention (CDC) for polymerase chain reaction analysis following admission and was found to be positive for Leishmania panamensis.
.")
Given the concern for mucocutaneous leishmaniasis with this particular species, otolaryngology was consulted; however, the patient did not demonstrate mucocutaneous disease. Because of the elevated risk for persistent disease with L panamensis, systemic therapy was indicated and administered: IV amphotericin B 200 mg on days 1 through 5 and again on day 10. Improvement in the ulcer was seen after the 10-day regimen was completed.
Comment
Leishmaniasis can be broadly classified by geographic region or clinical presentation. Under the geographic region system, leishmaniasis can be categorized as Old World or New World. Old World leishmaniasis primarily is transmitted by Phlebotomus sandflies and carries the parasites Leishmania major and Leishmania tropica, among others. New World leishmaniasis is caused by Lutzomyia sandflies, which carry Leishmania mexicana, Leishmania braziliensis, Leishmania amazonensis, and others.6
Our patient presented with cutaneous leishmaniasis, one of 4 primary clinical disease forms of leishmaniasis; the other 3 forms under this classification system are diffuse cutaneous, mucocutaneous, and visceral leishmaniasis, also known as kala-azar.3,6 Cutaneous leishmaniasis is limited to the skin, particularly the face and extremities. This form is more common with Old World vectors, with most cases occurring in Peru, Brazil, and the Middle East. In Old World cutaneous leishmaniasis, the disease begins with a solitary nodule at the site of the bite that ulcerates and can continue to spread in a sporotrichoid pattern. This cutaneous form tends to heal slowly over months to years with residual scarring. New World cutaneous leishmaniasis can present with a variety of clinical manifestations, including ulcerative, sarcoidlike, miliary, and nodular lesions.6,7
The diffuse form of cutaneous leishmaniasis begins in a similar manner to the Old World cutaneous form: a single nodule spreads widely over the body, especially the nose, and covers the patient’s skin with keloidal or verrucous lesions that do not ulcerate. These nodules contain large groupings of Leishmania-filled foamy macrophages. Often, patients with diffuse cutaneous leishmaniasis are immunosuppressed and are unable to develop an immune response to leishmanin and other skin antigens.6,7
Mucocutaneous leishmaniasis predominantly is caused by the New World species L braziliensis but also has been attributed to L amazonensis, L panamensis, and L guyanensis. This form manifests as mucosal lesions that can develop simultaneously with cutaneous lesions but more commonly appear months to years after resolution of the skin infection. Patients often present with ulceration of the lip, nose, and oropharynx, and destruction of the nasopharynx can result in severe consequences such as obstruction of the airway and perforation of the nasal septum (also known as espundia).6,7
The most severe presentation of leishmaniasis is the visceral form (kala-azar), which presents with parasitic infection of the liver, spleen, and bone marrow. Most commonly caused by Leishmania donovani, Leishmania infantum, and Leishmania chagasi, this form has a long incubation period spanning months to years before presenting with diarrhea, hepatomegaly, splenomegaly, darkening of the skin (in Hindi, kala-azar means “black fever”), pancytopenia, lymphadenopathy, nephritis, and intestinal hemorrhage, among other severe manifestations. Visceral leishmaniasis has a poor prognosis: patients succumb to disease within 2 years if not treated.6,7
Diagnosis—Diagnosing leishmaniasis starts with a complete personal and medical history, paying close attention to travel and exposures. Diagnosis is most successfully performed by polymerase chain reaction analysis, which is both highly sensitive and specific but also can be determined by culture using Novy-McNeal-Nicolle medium or by light microscopy. Histologic findings include the marquee sign, which describes an array of amastigotes (promastigotes that have developed into the intracellular tissue-stage form) with kinetoplasts surrounding the periphery of parasitized histiocytes. Giemsa staining can be helpful in identifying organisms.2,6,7
The diagnosis in our case was challenging, as none of the above findings were seen in our patient. The specimen taken by the border patrol medical provider was negative on Gram, Giemsa, and Grocott-Gömöri methenamine silver staining; no amastigotes were identified. Another diagnostic modality (not performed in our patient) is the Montenegro delayed skin-reaction test, which often is positive in patients with cutaneous leishmaniasis but also yields a positive result in patients who have been cured of Leishmania infection.6
An important consideration in the diagnostic workup of leishmaniasis is that collaboration with the CDC can be helpful, such as in our case, as they provide clear guidance for specimen collection and processing.2
Treatment—Treating leishmaniasis is challenging and complex. Even the initial decision to treat depends on several factors, including the form of infection. Most visceral and mucocutaneous infections should be treated due to both the lack of self-resolution of these forms and the higher risk for a potentially life-threatening disease course; in contrast, cutaneous forms require further consideration before initiating treatment. Some indicators for treating cutaneous leishmaniasis include widespread infection, intention to decrease scarring, and lesions with the potential to cause further complications (eg, on the face or ears or close to joints).6-8
The treatment of choice for cutaneous and mucocutaneous leishmaniasis is pentavalent antimony; however, this drug can only be obtained in the United States for investigational use, requiring approval by the CDC. A 20-day intravenous or intramuscular course of 20 mg/kg per day typically is used for cutaneous cases; a 28-day course typically is used for mucosal forms.
Amphotericin B is not only the treatment of choice for visceral leishmaniasis but also is an important alternative therapy for patients with mucosal leishmaniasis or who are co-infected with HIV. Patients with visceral infection also should receive supportive care for any concomitant afflictions, such as malnutrition or other infections. Although different regimens have been described, the US Food and Drug Administration has created outlines of specific intravenous infusion schedules for liposomal amphotericin B in immunocompetent and immunosuppressed patients.8 Liposomal amphotericin B also has a more favorable toxicity profile than conventional amphotericin B deoxycholate, which is otherwise effective in combating visceral leishmaniasis.6-8
Other treatments that have been attempted include pentamidine, miltefosine, thermotherapy, oral itraconazole and fluconazole, rifampicin, metronidazole and cotrimoxazole, dapsone, photodynamic therapy, thermotherapy, topical paromomycin formulations, intralesional pentavalent antimony, and laser cryotherapy. Notable among these other agents is miltefosine, a US Food and Drug Administration–approved oral medication for adults and adolescents (used off-label for patients younger than 12 years) with cutaneous leishmaniasis caused by L braziliensis, L panamensis, or L guyanensis. Other oral options mentioned include the so-called azole antifungal medications, which historically have produced variable results. From the CDC’s reports, ketoconazole was moderately effective in Guatemala and Panama,8 whereas itraconazole did not demonstrate efficacy in Colombia, and the efficacy of fluconazole was inconsistent in different countries.8 When considering one of the local (as opposed to oral and parenteral) therapies mentioned, the extent of cutaneous findings as well as the risk of mucosal spread should be factored in.6-8
Understandably, a number of considerations can come into play in determining the appropriate treatment modality, including body region affected, clinical form, severity, and Leishmania species.6-8 Our case is of particular interest because it demonstrates the complexities behind the diagnosis and treatment of cutaneous leishmaniasis, with careful consideration geared toward the species; for example, because our patient was infected with L panamensis, which is known to cause mucocutaneous disease, the infectious disease service decided to pursue systemic therapy with amphotericin B rather than topical treatment.
Prevention—Vector control is the primary means of preventing leishmaniasis under 2 umbrellas: environmental management and synthetic insecticides. The goal of environmental management is to eliminate the phlebotomine sandfly habitat; this was the primary method of vector control until 1940. Until that time, tree stumps were removed, indoor cracks and crevices were filled to prevent sandfly emergence, and areas around animal shelters were cleaned. These methods were highly dependent on community awareness and involvement; today, they can be combined with synthetic insecticides to offer maximum protection.
Synthetic insecticides include indoor sprays, treated nets, repellents, and impregnated dog collars, all of which control sandflies. However, the use of these insecticides in endemic areas, such as India, has driven development of insecticide resistance in many sandfly vector species.3
As of 2020, 5 vaccines against Leishmania have been created. Two are approved–one in Brazil and one in Uzbekistan–for human use as immunotherapy, while the other 3 have been developed to immunize dogs in Brazil. However, the effectiveness of these vaccines is under debate. First, one of the vaccines used as immunotherapy for cutaneous leishmaniasis must be used in combination with conventional chemotherapy; second, long-term effects of the canine vaccine are unknown.1 A preventive vaccine for humans is under development.1,3
Final Thoughts
Leishmaniasis remains a notable parasitic disease that is increasing in prevalence worldwide. Clinicians should be aware of this disease because early detection and treatment are essential to control infection.3 Health care providers in the United States should be especially aware of this condition among patients who have a history of travel or migration; those in Texas should recognize the current endemic status of leishmaniasis there.4,6
- Coutinho De Oliveira B, Duthie MS, Alves Pereira VR. Vaccines for leishmaniasis and the implications of their development for American tegumentary leishmaniasis. Hum Vaccin Immunother. 2020;16:919-930. doi:10.1080/21645515.2019.1678998
- Chan CX, Simmons BJ, Call JE, et al. Cutaneous leishmaniasis successfully treated with miltefosine. Cutis. 2020;106:206-209. doi:10.12788/cutis.0086
- Balaska S, Fotakis EA, Chaskopoulou A, et al. Chemical control and insecticide resistance status of sand fly vectors worldwide. PLoS Negl Trop Dis. 2021;15:E0009586. doi:10.1371/journal.pntd.0009586
- Desjeux P. Leishmaniasis. Nat Rev Microbiol. 2004;2:692. doi:10.1038/nrmicro981
- Michelutti A, Toniolo F, Bertola M, et al. Occurrence of Phlebotomine sand flies (Diptera: Psychodidae) in the northeastern plain of Italy. Parasit Vectors. 2021;14:164. doi:10.1186/s13071-021-04652-2
- Alkihan A, Hocker TLH. Infectious diseases: parasites and other creatures: protozoa. In: Alikhan A, Hocker TLH, eds. Review of Dermatology. Elsevier; 2024:329-331.
- Dinulos JGH. Infestations and bites. In: Habif TP, ed. Clinical Dermatology. Elsevier; 2016:630-634.
- Centers for Disease Control and Prevention. Leishmaniasis: resources for health professionals. US Department of Health and Human Services. March 20, 2023. Accessed October 5, 2023. https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#:~:text=Liposomal%20amphotericin%20B%20is%20FDA,treatment%20of%20choice%20for%20U.S
The genus Leishmania comprises protozoan parasites that cause approximately 2 million new cases of leishmaniasis each year across 98 countries.1 These protozoa are obligate intracellular parasites of phlebotomine sandfly species that transmit leishmaniasis and result in a considerable parasitic cause of fatalities globally, second only to malaria.2,3
Phlebotomine sandflies primarily live in tropical and subtropical regions and function as vectors for many pathogens in addition to Leishmania species, such as Bartonella species and arboviruses.3 In 2004, it was noted that the majority of leishmaniasis cases affected developing countries: 90% of visceral leishmaniasis cases occurred in Bangladesh, India, Nepal, Sudan, and Brazil, and 90% of cutaneous leishmaniasis cases occurred in Afghanistan, Algeria, Brazil, Iran, Peru, Saudi Arabia, and Syria.4 Of note, with recent environmental changes, phlebotomine sandflies have gradually migrated to more northerly latitudes, extending into Europe.5
Twenty Leishmania species and 30 sandfly species have been identified as causes of leishmaniasis.4Leishmania infection occurs when an infected sandfly bites a mammalian host and transmits the parasite’s flagellated form, known as a promastigote. Host inflammatory cells, such as monocytes and dendritic cells, phagocytize parasites that enter the skin. The interaction between parasites and dendritic cells become an important factor in the outcome of Leishmania infection in the host because dendritic cells promote development of CD4 and CD8 T lymphocytes with specificity to target Leishmania parasites and protect the host.1
The number of cases of leishmaniasis has increased worldwide, most likely due to changes in the environment and human behaviors such as urbanization, the creation of new settlements, and migration from rural to urban areas.3,5 Important risk factors in individual patients include malnutrition; low-quality housing and sanitation; a history of migration or travel; and immunosuppression, such as that caused by HIV co-infection.2,5
Case Report
An otherwise healthy 25-year-old Bangladeshi man presented to our community hospital for evaluation of a painful leg ulcer of 1 month’s duration. The patient had migrated from Bangladesh to Panama, then to Costa Rica, followed by Guatemala, Honduras, Mexico, and, last, Texas. In Texas, he was identified by the US Immigration and Customs Enforcement, transported to a detention facility, and transferred to this hospital shortly afterward.
The patient reported that, during his extensive migration, he had lived in the jungle and reported what he described as mosquito bites on the legs. He subsequently developed a 3-cm ulcerated and crusted plaque with rolled borders on the right medial ankle (Figure 1). In addition, he had a palpable nodular cord on the medial leg from the ankle lesion to the mid thigh that was consistent with lymphocutaneous spread. Ultrasonography was negative for deep-vein thrombosis.
Because the patient’s recent migration from Central America was highly concerning for microbial infection, vancomycin and piperacillin-tazobactam were started empirically on admission. A punch biopsy from the right medial ankle was nondiagnostic, showing acute and chronic necrotizing inflammation along with numerous epithelioid histiocytes with a vaguely granulomatous appearance (Figure 2). A specimen from the right medial ankle that had already been taken by an astute border patrol medical provider was sent to the Centers for Disease Control and Prevention (CDC) for polymerase chain reaction analysis following admission and was found to be positive for Leishmania panamensis.
Given the concern for mucocutaneous leishmaniasis with this particular species, otolaryngology was consulted; however, the patient did not demonstrate mucocutaneous disease. Because of the elevated risk for persistent disease with L panamensis, systemic therapy was indicated and administered: IV amphotericin B 200 mg on days 1 through 5 and again on day 10. Improvement in the ulcer was seen after the 10-day regimen was completed.
Comment
Leishmaniasis can be broadly classified by geographic region or clinical presentation. Under the geographic region system, leishmaniasis can be categorized as Old World or New World. Old World leishmaniasis primarily is transmitted by Phlebotomus sandflies and carries the parasites Leishmania major and Leishmania tropica, among others. New World leishmaniasis is caused by Lutzomyia sandflies, which carry Leishmania mexicana, Leishmania braziliensis, Leishmania amazonensis, and others.6
Our patient presented with cutaneous leishmaniasis, one of 4 primary clinical disease forms of leishmaniasis; the other 3 forms under this classification system are diffuse cutaneous, mucocutaneous, and visceral leishmaniasis, also known as kala-azar.3,6 Cutaneous leishmaniasis is limited to the skin, particularly the face and extremities. This form is more common with Old World vectors, with most cases occurring in Peru, Brazil, and the Middle East. In Old World cutaneous leishmaniasis, the disease begins with a solitary nodule at the site of the bite that ulcerates and can continue to spread in a sporotrichoid pattern. This cutaneous form tends to heal slowly over months to years with residual scarring. New World cutaneous leishmaniasis can present with a variety of clinical manifestations, including ulcerative, sarcoidlike, miliary, and nodular lesions.6,7
The diffuse form of cutaneous leishmaniasis begins in a similar manner to the Old World cutaneous form: a single nodule spreads widely over the body, especially the nose, and covers the patient’s skin with keloidal or verrucous lesions that do not ulcerate. These nodules contain large groupings of Leishmania-filled foamy macrophages. Often, patients with diffuse cutaneous leishmaniasis are immunosuppressed and are unable to develop an immune response to leishmanin and other skin antigens.6,7
Mucocutaneous leishmaniasis predominantly is caused by the New World species L braziliensis but also has been attributed to L amazonensis, L panamensis, and L guyanensis. This form manifests as mucosal lesions that can develop simultaneously with cutaneous lesions but more commonly appear months to years after resolution of the skin infection. Patients often present with ulceration of the lip, nose, and oropharynx, and destruction of the nasopharynx can result in severe consequences such as obstruction of the airway and perforation of the nasal septum (also known as espundia).6,7
The most severe presentation of leishmaniasis is the visceral form (kala-azar), which presents with parasitic infection of the liver, spleen, and bone marrow. Most commonly caused by Leishmania donovani, Leishmania infantum, and Leishmania chagasi, this form has a long incubation period spanning months to years before presenting with diarrhea, hepatomegaly, splenomegaly, darkening of the skin (in Hindi, kala-azar means “black fever”), pancytopenia, lymphadenopathy, nephritis, and intestinal hemorrhage, among other severe manifestations. Visceral leishmaniasis has a poor prognosis: patients succumb to disease within 2 years if not treated.6,7
Diagnosis—Diagnosing leishmaniasis starts with a complete personal and medical history, paying close attention to travel and exposures. Diagnosis is most successfully performed by polymerase chain reaction analysis, which is both highly sensitive and specific but also can be determined by culture using Novy-McNeal-Nicolle medium or by light microscopy. Histologic findings include the marquee sign, which describes an array of amastigotes (promastigotes that have developed into the intracellular tissue-stage form) with kinetoplasts surrounding the periphery of parasitized histiocytes. Giemsa staining can be helpful in identifying organisms.2,6,7
The diagnosis in our case was challenging, as none of the above findings were seen in our patient. The specimen taken by the border patrol medical provider was negative on Gram, Giemsa, and Grocott-Gömöri methenamine silver staining; no amastigotes were identified. Another diagnostic modality (not performed in our patient) is the Montenegro delayed skin-reaction test, which often is positive in patients with cutaneous leishmaniasis but also yields a positive result in patients who have been cured of Leishmania infection.6
An important consideration in the diagnostic workup of leishmaniasis is that collaboration with the CDC can be helpful, such as in our case, as they provide clear guidance for specimen collection and processing.2
Treatment—Treating leishmaniasis is challenging and complex. Even the initial decision to treat depends on several factors, including the form of infection. Most visceral and mucocutaneous infections should be treated due to both the lack of self-resolution of these forms and the higher risk for a potentially life-threatening disease course; in contrast, cutaneous forms require further consideration before initiating treatment. Some indicators for treating cutaneous leishmaniasis include widespread infection, intention to decrease scarring, and lesions with the potential to cause further complications (eg, on the face or ears or close to joints).6-8
The treatment of choice for cutaneous and mucocutaneous leishmaniasis is pentavalent antimony; however, this drug can only be obtained in the United States for investigational use, requiring approval by the CDC. A 20-day intravenous or intramuscular course of 20 mg/kg per day typically is used for cutaneous cases; a 28-day course typically is used for mucosal forms.
Amphotericin B is not only the treatment of choice for visceral leishmaniasis but also is an important alternative therapy for patients with mucosal leishmaniasis or who are co-infected with HIV. Patients with visceral infection also should receive supportive care for any concomitant afflictions, such as malnutrition or other infections. Although different regimens have been described, the US Food and Drug Administration has created outlines of specific intravenous infusion schedules for liposomal amphotericin B in immunocompetent and immunosuppressed patients.8 Liposomal amphotericin B also has a more favorable toxicity profile than conventional amphotericin B deoxycholate, which is otherwise effective in combating visceral leishmaniasis.6-8
Other treatments that have been attempted include pentamidine, miltefosine, thermotherapy, oral itraconazole and fluconazole, rifampicin, metronidazole and cotrimoxazole, dapsone, photodynamic therapy, thermotherapy, topical paromomycin formulations, intralesional pentavalent antimony, and laser cryotherapy. Notable among these other agents is miltefosine, a US Food and Drug Administration–approved oral medication for adults and adolescents (used off-label for patients younger than 12 years) with cutaneous leishmaniasis caused by L braziliensis, L panamensis, or L guyanensis. Other oral options mentioned include the so-called azole antifungal medications, which historically have produced variable results. From the CDC’s reports, ketoconazole was moderately effective in Guatemala and Panama,8 whereas itraconazole did not demonstrate efficacy in Colombia, and the efficacy of fluconazole was inconsistent in different countries.8 When considering one of the local (as opposed to oral and parenteral) therapies mentioned, the extent of cutaneous findings as well as the risk of mucosal spread should be factored in.6-8
Understandably, a number of considerations can come into play in determining the appropriate treatment modality, including body region affected, clinical form, severity, and Leishmania species.6-8 Our case is of particular interest because it demonstrates the complexities behind the diagnosis and treatment of cutaneous leishmaniasis, with careful consideration geared toward the species; for example, because our patient was infected with L panamensis, which is known to cause mucocutaneous disease, the infectious disease service decided to pursue systemic therapy with amphotericin B rather than topical treatment.
Prevention—Vector control is the primary means of preventing leishmaniasis under 2 umbrellas: environmental management and synthetic insecticides. The goal of environmental management is to eliminate the phlebotomine sandfly habitat; this was the primary method of vector control until 1940. Until that time, tree stumps were removed, indoor cracks and crevices were filled to prevent sandfly emergence, and areas around animal shelters were cleaned. These methods were highly dependent on community awareness and involvement; today, they can be combined with synthetic insecticides to offer maximum protection.
Synthetic insecticides include indoor sprays, treated nets, repellents, and impregnated dog collars, all of which control sandflies. However, the use of these insecticides in endemic areas, such as India, has driven development of insecticide resistance in many sandfly vector species.3
As of 2020, 5 vaccines against Leishmania have been created. Two are approved–one in Brazil and one in Uzbekistan–for human use as immunotherapy, while the other 3 have been developed to immunize dogs in Brazil. However, the effectiveness of these vaccines is under debate. First, one of the vaccines used as immunotherapy for cutaneous leishmaniasis must be used in combination with conventional chemotherapy; second, long-term effects of the canine vaccine are unknown.1 A preventive vaccine for humans is under development.1,3
Final Thoughts
Leishmaniasis remains a notable parasitic disease that is increasing in prevalence worldwide. Clinicians should be aware of this disease because early detection and treatment are essential to control infection.3 Health care providers in the United States should be especially aware of this condition among patients who have a history of travel or migration; those in Texas should recognize the current endemic status of leishmaniasis there.4,6
The genus Leishmania comprises protozoan parasites that cause approximately 2 million new cases of leishmaniasis each year across 98 countries.1 These protozoa are obligate intracellular parasites of phlebotomine sandfly species that transmit leishmaniasis and result in a considerable parasitic cause of fatalities globally, second only to malaria.2,3
Phlebotomine sandflies primarily live in tropical and subtropical regions and function as vectors for many pathogens in addition to Leishmania species, such as Bartonella species and arboviruses.3 In 2004, it was noted that the majority of leishmaniasis cases affected developing countries: 90% of visceral leishmaniasis cases occurred in Bangladesh, India, Nepal, Sudan, and Brazil, and 90% of cutaneous leishmaniasis cases occurred in Afghanistan, Algeria, Brazil, Iran, Peru, Saudi Arabia, and Syria.4 Of note, with recent environmental changes, phlebotomine sandflies have gradually migrated to more northerly latitudes, extending into Europe.5
Twenty Leishmania species and 30 sandfly species have been identified as causes of leishmaniasis.4Leishmania infection occurs when an infected sandfly bites a mammalian host and transmits the parasite’s flagellated form, known as a promastigote. Host inflammatory cells, such as monocytes and dendritic cells, phagocytize parasites that enter the skin. The interaction between parasites and dendritic cells become an important factor in the outcome of Leishmania infection in the host because dendritic cells promote development of CD4 and CD8 T lymphocytes with specificity to target Leishmania parasites and protect the host.1
The number of cases of leishmaniasis has increased worldwide, most likely due to changes in the environment and human behaviors such as urbanization, the creation of new settlements, and migration from rural to urban areas.3,5 Important risk factors in individual patients include malnutrition; low-quality housing and sanitation; a history of migration or travel; and immunosuppression, such as that caused by HIV co-infection.2,5
Case Report
An otherwise healthy 25-year-old Bangladeshi man presented to our community hospital for evaluation of a painful leg ulcer of 1 month’s duration. The patient had migrated from Bangladesh to Panama, then to Costa Rica, followed by Guatemala, Honduras, Mexico, and, last, Texas. In Texas, he was identified by the US Immigration and Customs Enforcement, transported to a detention facility, and transferred to this hospital shortly afterward.
The patient reported that, during his extensive migration, he had lived in the jungle and reported what he described as mosquito bites on the legs. He subsequently developed a 3-cm ulcerated and crusted plaque with rolled borders on the right medial ankle (Figure 1). In addition, he had a palpable nodular cord on the medial leg from the ankle lesion to the mid thigh that was consistent with lymphocutaneous spread. Ultrasonography was negative for deep-vein thrombosis.
Because the patient’s recent migration from Central America was highly concerning for microbial infection, vancomycin and piperacillin-tazobactam were started empirically on admission. A punch biopsy from the right medial ankle was nondiagnostic, showing acute and chronic necrotizing inflammation along with numerous epithelioid histiocytes with a vaguely granulomatous appearance (Figure 2). A specimen from the right medial ankle that had already been taken by an astute border patrol medical provider was sent to the Centers for Disease Control and Prevention (CDC) for polymerase chain reaction analysis following admission and was found to be positive for Leishmania panamensis.
Given the concern for mucocutaneous leishmaniasis with this particular species, otolaryngology was consulted; however, the patient did not demonstrate mucocutaneous disease. Because of the elevated risk for persistent disease with L panamensis, systemic therapy was indicated and administered: IV amphotericin B 200 mg on days 1 through 5 and again on day 10. Improvement in the ulcer was seen after the 10-day regimen was completed.
Comment
Leishmaniasis can be broadly classified by geographic region or clinical presentation. Under the geographic region system, leishmaniasis can be categorized as Old World or New World. Old World leishmaniasis primarily is transmitted by Phlebotomus sandflies and carries the parasites Leishmania major and Leishmania tropica, among others. New World leishmaniasis is caused by Lutzomyia sandflies, which carry Leishmania mexicana, Leishmania braziliensis, Leishmania amazonensis, and others.6
Our patient presented with cutaneous leishmaniasis, one of 4 primary clinical disease forms of leishmaniasis; the other 3 forms under this classification system are diffuse cutaneous, mucocutaneous, and visceral leishmaniasis, also known as kala-azar.3,6 Cutaneous leishmaniasis is limited to the skin, particularly the face and extremities. This form is more common with Old World vectors, with most cases occurring in Peru, Brazil, and the Middle East. In Old World cutaneous leishmaniasis, the disease begins with a solitary nodule at the site of the bite that ulcerates and can continue to spread in a sporotrichoid pattern. This cutaneous form tends to heal slowly over months to years with residual scarring. New World cutaneous leishmaniasis can present with a variety of clinical manifestations, including ulcerative, sarcoidlike, miliary, and nodular lesions.6,7
The diffuse form of cutaneous leishmaniasis begins in a similar manner to the Old World cutaneous form: a single nodule spreads widely over the body, especially the nose, and covers the patient’s skin with keloidal or verrucous lesions that do not ulcerate. These nodules contain large groupings of Leishmania-filled foamy macrophages. Often, patients with diffuse cutaneous leishmaniasis are immunosuppressed and are unable to develop an immune response to leishmanin and other skin antigens.6,7
Mucocutaneous leishmaniasis predominantly is caused by the New World species L braziliensis but also has been attributed to L amazonensis, L panamensis, and L guyanensis. This form manifests as mucosal lesions that can develop simultaneously with cutaneous lesions but more commonly appear months to years after resolution of the skin infection. Patients often present with ulceration of the lip, nose, and oropharynx, and destruction of the nasopharynx can result in severe consequences such as obstruction of the airway and perforation of the nasal septum (also known as espundia).6,7
The most severe presentation of leishmaniasis is the visceral form (kala-azar), which presents with parasitic infection of the liver, spleen, and bone marrow. Most commonly caused by Leishmania donovani, Leishmania infantum, and Leishmania chagasi, this form has a long incubation period spanning months to years before presenting with diarrhea, hepatomegaly, splenomegaly, darkening of the skin (in Hindi, kala-azar means “black fever”), pancytopenia, lymphadenopathy, nephritis, and intestinal hemorrhage, among other severe manifestations. Visceral leishmaniasis has a poor prognosis: patients succumb to disease within 2 years if not treated.6,7
Diagnosis—Diagnosing leishmaniasis starts with a complete personal and medical history, paying close attention to travel and exposures. Diagnosis is most successfully performed by polymerase chain reaction analysis, which is both highly sensitive and specific but also can be determined by culture using Novy-McNeal-Nicolle medium or by light microscopy. Histologic findings include the marquee sign, which describes an array of amastigotes (promastigotes that have developed into the intracellular tissue-stage form) with kinetoplasts surrounding the periphery of parasitized histiocytes. Giemsa staining can be helpful in identifying organisms.2,6,7
The diagnosis in our case was challenging, as none of the above findings were seen in our patient. The specimen taken by the border patrol medical provider was negative on Gram, Giemsa, and Grocott-Gömöri methenamine silver staining; no amastigotes were identified. Another diagnostic modality (not performed in our patient) is the Montenegro delayed skin-reaction test, which often is positive in patients with cutaneous leishmaniasis but also yields a positive result in patients who have been cured of Leishmania infection.6
An important consideration in the diagnostic workup of leishmaniasis is that collaboration with the CDC can be helpful, such as in our case, as they provide clear guidance for specimen collection and processing.2
Treatment—Treating leishmaniasis is challenging and complex. Even the initial decision to treat depends on several factors, including the form of infection. Most visceral and mucocutaneous infections should be treated due to both the lack of self-resolution of these forms and the higher risk for a potentially life-threatening disease course; in contrast, cutaneous forms require further consideration before initiating treatment. Some indicators for treating cutaneous leishmaniasis include widespread infection, intention to decrease scarring, and lesions with the potential to cause further complications (eg, on the face or ears or close to joints).6-8
The treatment of choice for cutaneous and mucocutaneous leishmaniasis is pentavalent antimony; however, this drug can only be obtained in the United States for investigational use, requiring approval by the CDC. A 20-day intravenous or intramuscular course of 20 mg/kg per day typically is used for cutaneous cases; a 28-day course typically is used for mucosal forms.
Amphotericin B is not only the treatment of choice for visceral leishmaniasis but also is an important alternative therapy for patients with mucosal leishmaniasis or who are co-infected with HIV. Patients with visceral infection also should receive supportive care for any concomitant afflictions, such as malnutrition or other infections. Although different regimens have been described, the US Food and Drug Administration has created outlines of specific intravenous infusion schedules for liposomal amphotericin B in immunocompetent and immunosuppressed patients.8 Liposomal amphotericin B also has a more favorable toxicity profile than conventional amphotericin B deoxycholate, which is otherwise effective in combating visceral leishmaniasis.6-8
Other treatments that have been attempted include pentamidine, miltefosine, thermotherapy, oral itraconazole and fluconazole, rifampicin, metronidazole and cotrimoxazole, dapsone, photodynamic therapy, thermotherapy, topical paromomycin formulations, intralesional pentavalent antimony, and laser cryotherapy. Notable among these other agents is miltefosine, a US Food and Drug Administration–approved oral medication for adults and adolescents (used off-label for patients younger than 12 years) with cutaneous leishmaniasis caused by L braziliensis, L panamensis, or L guyanensis. Other oral options mentioned include the so-called azole antifungal medications, which historically have produced variable results. From the CDC’s reports, ketoconazole was moderately effective in Guatemala and Panama,8 whereas itraconazole did not demonstrate efficacy in Colombia, and the efficacy of fluconazole was inconsistent in different countries.8 When considering one of the local (as opposed to oral and parenteral) therapies mentioned, the extent of cutaneous findings as well as the risk of mucosal spread should be factored in.6-8
Understandably, a number of considerations can come into play in determining the appropriate treatment modality, including body region affected, clinical form, severity, and Leishmania species.6-8 Our case is of particular interest because it demonstrates the complexities behind the diagnosis and treatment of cutaneous leishmaniasis, with careful consideration geared toward the species; for example, because our patient was infected with L panamensis, which is known to cause mucocutaneous disease, the infectious disease service decided to pursue systemic therapy with amphotericin B rather than topical treatment.
Prevention—Vector control is the primary means of preventing leishmaniasis under 2 umbrellas: environmental management and synthetic insecticides. The goal of environmental management is to eliminate the phlebotomine sandfly habitat; this was the primary method of vector control until 1940. Until that time, tree stumps were removed, indoor cracks and crevices were filled to prevent sandfly emergence, and areas around animal shelters were cleaned. These methods were highly dependent on community awareness and involvement; today, they can be combined with synthetic insecticides to offer maximum protection.
Synthetic insecticides include indoor sprays, treated nets, repellents, and impregnated dog collars, all of which control sandflies. However, the use of these insecticides in endemic areas, such as India, has driven development of insecticide resistance in many sandfly vector species.3
As of 2020, 5 vaccines against Leishmania have been created. Two are approved–one in Brazil and one in Uzbekistan–for human use as immunotherapy, while the other 3 have been developed to immunize dogs in Brazil. However, the effectiveness of these vaccines is under debate. First, one of the vaccines used as immunotherapy for cutaneous leishmaniasis must be used in combination with conventional chemotherapy; second, long-term effects of the canine vaccine are unknown.1 A preventive vaccine for humans is under development.1,3
Final Thoughts
Leishmaniasis remains a notable parasitic disease that is increasing in prevalence worldwide. Clinicians should be aware of this disease because early detection and treatment are essential to control infection.3 Health care providers in the United States should be especially aware of this condition among patients who have a history of travel or migration; those in Texas should recognize the current endemic status of leishmaniasis there.4,6
- Coutinho De Oliveira B, Duthie MS, Alves Pereira VR. Vaccines for leishmaniasis and the implications of their development for American tegumentary leishmaniasis. Hum Vaccin Immunother. 2020;16:919-930. doi:10.1080/21645515.2019.1678998
- Chan CX, Simmons BJ, Call JE, et al. Cutaneous leishmaniasis successfully treated with miltefosine. Cutis. 2020;106:206-209. doi:10.12788/cutis.0086
- Balaska S, Fotakis EA, Chaskopoulou A, et al. Chemical control and insecticide resistance status of sand fly vectors worldwide. PLoS Negl Trop Dis. 2021;15:E0009586. doi:10.1371/journal.pntd.0009586
- Desjeux P. Leishmaniasis. Nat Rev Microbiol. 2004;2:692. doi:10.1038/nrmicro981
- Michelutti A, Toniolo F, Bertola M, et al. Occurrence of Phlebotomine sand flies (Diptera: Psychodidae) in the northeastern plain of Italy. Parasit Vectors. 2021;14:164. doi:10.1186/s13071-021-04652-2
- Alkihan A, Hocker TLH. Infectious diseases: parasites and other creatures: protozoa. In: Alikhan A, Hocker TLH, eds. Review of Dermatology. Elsevier; 2024:329-331.
- Dinulos JGH. Infestations and bites. In: Habif TP, ed. Clinical Dermatology. Elsevier; 2016:630-634.
- Centers for Disease Control and Prevention. Leishmaniasis: resources for health professionals. US Department of Health and Human Services. March 20, 2023. Accessed October 5, 2023. https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#:~:text=Liposomal%20amphotericin%20B%20is%20FDA,treatment%20of%20choice%20for%20U.S
- Coutinho De Oliveira B, Duthie MS, Alves Pereira VR. Vaccines for leishmaniasis and the implications of their development for American tegumentary leishmaniasis. Hum Vaccin Immunother. 2020;16:919-930. doi:10.1080/21645515.2019.1678998
- Chan CX, Simmons BJ, Call JE, et al. Cutaneous leishmaniasis successfully treated with miltefosine. Cutis. 2020;106:206-209. doi:10.12788/cutis.0086
- Balaska S, Fotakis EA, Chaskopoulou A, et al. Chemical control and insecticide resistance status of sand fly vectors worldwide. PLoS Negl Trop Dis. 2021;15:E0009586. doi:10.1371/journal.pntd.0009586
- Desjeux P. Leishmaniasis. Nat Rev Microbiol. 2004;2:692. doi:10.1038/nrmicro981
- Michelutti A, Toniolo F, Bertola M, et al. Occurrence of Phlebotomine sand flies (Diptera: Psychodidae) in the northeastern plain of Italy. Parasit Vectors. 2021;14:164. doi:10.1186/s13071-021-04652-2
- Alkihan A, Hocker TLH. Infectious diseases: parasites and other creatures: protozoa. In: Alikhan A, Hocker TLH, eds. Review of Dermatology. Elsevier; 2024:329-331.
- Dinulos JGH. Infestations and bites. In: Habif TP, ed. Clinical Dermatology. Elsevier; 2016:630-634.
- Centers for Disease Control and Prevention. Leishmaniasis: resources for health professionals. US Department of Health and Human Services. March 20, 2023. Accessed October 5, 2023. https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#:~:text=Liposomal%20amphotericin%20B%20is%20FDA,treatment%20of%20choice%20for%20U.S
Practice Points
- The Phlebotomus and Lutzomyia genera of sandflies are vectors of Leishmania parasites, which can result in an array of clinical findings associated with leishmaniasis.
- Treatment options for leishmaniasis differ based on whether the infection is considered uncomplicated or complicated, which depends on the species of Leishmania; the number, size, and location of the lesion(s); and host immune status.
- All US practitioners should be aware of this pathogen, especially with regard to patients who have a history of travel to other countries. Health care professionals in states such as Texas and Oklahoma should be especially cognizant because these constitute one of the few areas in the United States where locally acquired cases of leishmaniasis have been reported.

Acquired Acrodermatitis Enteropathica in an Infant
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.

Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.

The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.

Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.
Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.
The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.
Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
Acrodermatitis enteropathica (AE) is a rare disorder of zinc metabolism that typically presents in infancy.1 Although it is clinically characterized by acral and periorificial dermatitis, alopecia, and diarrhea, only 20% of cases present with this triad.2 Zinc deficiency in AE can either be acquired or inborn (congenital). Acquired forms can occur from dietary inadequacy or malabsorption, whereas genetic causes are related to an autosomal-recessive disorder affecting zinc transporters.1 We report a case of a 3-month-old female infant with acquired AE who was successfully treated with zinc supplementation over the course of 3 weeks.
Case Report
A 3-month-old female infant presented to the emergency department with a rash of 2 weeks’ duration. She was born full term with no birth complications. The patient’s mother reported that the rash started on the cheeks, then enlarged and spread to the neck, back, and perineum. The patient also had been having diarrhea during this time. She previously had received mupirocin and cephalexin with no response to treatment. Maternal history was negative for lupus, and the mother’s diet consisted of a variety of foods but not many vegetables. The patient was exclusively breastfed, and there was no pertinent history of similar rashes occurring in other family members.
Physical examination revealed the patient had annular and polycyclic, hyperkeratotic, crusted papules and plaques on the cheeks, neck, back, and axillae, as well as the perineum/groin and perianal regions (Figure 1). The differential diagnosis at the time included neonatal lupus, zinc deficiency, and syphilis. Relevant laboratory testing and a shave biopsy of the left axilla were obtained.
Pertinent laboratory findings included a low zinc level (23 μg/dL [reference range, 26–141 μg/dL]), low alkaline phosphatase level (74 U/L [reference range, 94–486 U/L]), and thrombocytosis (826×109/L [reference range, 150–400×109/L). Results for antinuclear antibody and anti–Sjögren syndrome–related antigen A and B antibody testing were negative. A rapid plasma reagin test was nonreactive. Histologic examination revealed psoriasiform hyperplasia with overlying confluent parakeratosis, focal spongiosis, multiple dyskeratotic keratinocytes, and mitotic figures (Figure 2). Ballooning was evident in focal cells in the subcorneal region in addition to an accompanying lymphocytic infiltrate and occasional neutrophils.
The patient was given a 10-mg/mL suspension of elemental zinc and was advised to take 1 mL (10 mg) by mouth twice daily with food. This dosage equated to 3 mg/kg/d. On follow-up 3 weeks later, the skin began to clear (Figure 3). Follow-up laboratory testing showed an increase in zinc (114 μg/dL) and alkaline phosphatase levels (313 U/L). The patient was able to discontinue the zinc supplementation, and follow-up during the next year revealed no recurrence.
Comment
Etiology of AE—Acrodermatitis enteropathica was first identified in 1942 as an acral rash associated with diarrhea3; in 1973, Barnes and Moynahan4 discovered zinc deficiency as a causal agent for these findings. The causes of AE are further subclassified as either an acquired or inborn etiology. Congenital causes commonly are seen in infants within the first few months of life, whereas acquired forms are seen at any age. Acquired forms in infants can occur from failure of the mother to secrete zinc in breast milk, low maternal serum zinc levels, or other reasons causing low nutritional intake. A single mutation in the SLC30A2 gene has been found to markedly reduce zinc concentrations in breast milk, thus causing zinc deficiency in breastfed infants.5 Other acquired forms can be caused by malabsorption, sometimes after surgery such as intestinal bypass or from intravenous nutrition without sufficient zinc.1 The congenital form of AE is an autosomal-recessive disorder occurring from mutations in the SLC39A4 gene located on band 8q24.3. Affected individuals have a decreased ability to absorb zinc in the small intestine because of defects in zinc transporters ZIP and ZnT.6 Based on our patient’s laboratory findings and history, it is believed that the zinc deficiency was acquired, as the condition normalized with repletion and has not required any supplementation in the year of follow-up. In addition, the absence of a pertinent family history supported an acquired diagnosis, which has various etiologies, whereas the congenital form primarily is a genetic disease.
Management—Treatment of AE includes supplementation with oral elemental zinc; however, there are scant evidence-based recommendations on the exact dose of zinc to be given. Generally, the recommended amount is 3 mg/kg/d.8 For individuals with the congenital form of AE, lifelong zinc supplementation is additionally recommended.9 It is important to recognize this presentation because the patient can develop worsening irritability, severe diarrhea, nail dystrophy, hair loss, immune dysfunction, and numerous ophthalmic disorders if left untreated. Acute zinc toxicity due to excess administration is rare, with symptoms of nausea and vomiting occurring with dosages of 50 to 100 mg/d. Additionally, dosages of up to 70 mg twice weekly have been provided without any toxic effect.10 In our case, 3 mg/kg/d of oral zinc supplementation proved to be effective in resolving the patient’s symptoms of acquired zinc deficiency.
Differential Diagnosis—It is important to note that deficiencies of other nutrients may present as an AE-like eruption called acrodermatitis dysmetabolica (AD). Both diseases may present with the triad of dermatitis, alopecia, and diarrhea; however, AD is associated with inborn errors of metabolism. There have been cases that describe AD in patients with a zinc deficiency in conjunction with a deficiency of branched-chain amino acids.11,12 It is important to consider AD in the differential diagnosis of an AE eruption, especially in the context of a metabolic disorder, as it may affect the treatment plan. One case described the dermatitis of AD as not responding to zinc supplementation alone, while another described improvement after increasing an isoleucine supplementation dose.11,12
Other considerations in the differential diagnoses include AE-like conditions such as biotinidase deficiency, multiple carboxylase deficiency, and essential fatty acid deficiency. An AE-like condition may present with the triad of dermatitis, alopecia, and diarrhea. However, unlike in true AE, zinc and alkaline phosphatase levels tend to be normal in these conditions. Other features seen in AE-like conditions depend on the underlying cause but often include failure to thrive, neurologic defects, ophthalmic abnormalities, and metabolic abnormalities.13
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
- Acrodermatitis enteropathica. National Organization for Rare Disorders. Accessed October 16, 2022. https://rarediseases.org/rare-diseases/acrodermatitis-enteropathica/
- Perafán-Riveros C, França LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Danbolt N. Acrodermatitis enteropathica. Br J Dermatol. 1979;100:37-40.
- Barnes PM, Moynahan EJ. Zinc deficiency in acrodermatitis enteropathica: multiple dietary intolerance treated with synthetic diet. Proc R Soc Med. 1973;66:327-329.
- Lee S, Zhou Y, Gill DL, et al. A genetic variant in SLC30A2 causes breast dysfunction during lactation by inducing ER stress, oxidative stress and epithelial barrier defects. Sci Rep. 2018;8:3542.
- Kaur S, Sangwan A, Sahu P, et al. Clinical variants of acrodermatitis enteropathica and its co-relation with genetics. Indian J Paediatr Dermatol. 2016;17:35-37.
- Dela Rosa KM, James WD. Acrodermatitis enteropathica workup. Medscape. Updated June 4, 2021. Accessed October 16, 2022. https://emedicine.medscape.com/article/1102575-workup#showall
- Ngan V, Gangakhedkar A, Oakley A. Acrodermatitis enteropathica. DermNet. Accessed October 16, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica/
- Ranugha P, Sethi P, Veeranna S. Acrodermatitis enteropathica: the need for sustained high dose zinc supplementation. Dermatol Online J. 2018;24:13030/qt1w9002sr.
- Larson CP, Roy SK, Khan AI, et al. Zinc treatment to under-five children: applications to improve child survival and reduce burden of disease. J Health Popul Nutr. 2008;26:356-365.
- Samady JA, Schwartz RA, Shih LY, et al. Acrodermatitis enteropathica-like eruption in an infant with nonketotic hyperglycinemia. J Dermatol. 2000;27:604-608.
- Flores K, Chikowski R, Morrell DS. Acrodermatitis dysmetabolica in an infant with maple syrup urine disease. Clin Exp Dermatol. 2016;41:651-654.
- Jones L, Oakley A. Acrodermatitis enteropathica-like conditions. DermNet. Accessed August 30, 2022. https://dermnetnz.org/topics/acrodermatitis-enteropathica-like-conditions
Practice Points
- Although clinically characterized by the triad of acral and periorificial dermatitis, alopecia, and diarrhea, most cases of acrodermatitis enteropathica (AE) present with only partial features of this syndrome.
- Low levels of zinc-dependent enzymes such as alkaline phosphatase may support the diagnosis of AE.

Asymptomatic Hemorrhagic Lesions in an Anemic Woman
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
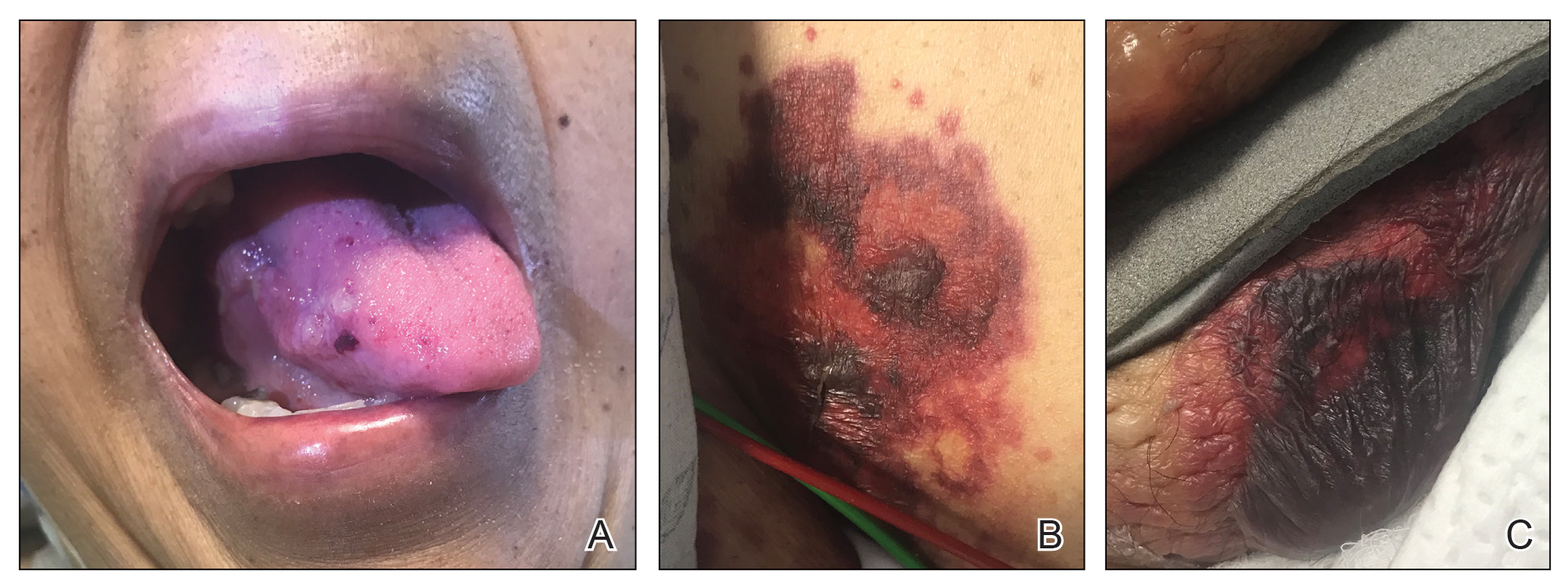
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
A 67-year-old woman with a medical history of type 2 diabetes mellitus, unspecified leukocytosis, and anemia presented to the dermatology clinic with asymptomatic hemorrhagic bullae on the face, chest, and tongue, as well as a large, tender, tense, hemorrhagic bulla on the groin of 3 to 4 months’ duration. A review of systems was negative for fever, chills, night sweats, malaise, shortness of breath, and dyspnea on exertion. A complete blood cell count showed mild leukocytosis, anemia, and thrombocytopenia. Her creatinine level was slightly elevated. Chest computed tomography showed early pulmonary fibrosis and coronary artery calcification. An echocardiogram showed diastolic dysfunction with moderate left ventricle thickening. A serum and urine electrophoresis demonstrated elevated free λ light chains with an M-spike. A punch biopsy was performed.