User login
Early SAVR tops watchful waiting in severe, asymptomatic aortic stenosis: AVATAR
Better to intervene early with a new valve in patients with severe aortic stenosis (AS) who are asymptomatic, even during exercise, than to wait for the disease to progress and symptoms to emerge before operating, suggests a small, randomized trial that challenges the guidelines.

Of the trial’s 157 patients, all with negative results on stress tests and normal left ventricular (LV) function despite severe AS, those assigned to early surgical aortic valve replacement (SAVR), compared with standard watchful waiting, showed a better-than-50% drop in risk for death or major adverse cardiac events (MACE) over 2-3 years. The benefit appeared driven by fewer hospitalizations for heart failure (HF) and deaths in the early-surgery group.
The findings “advocate for early surgery once aortic stenosis becomes significant and regardless of symptom status,” Marko Banovic, MD, PhD, said during his presentation at the American Heart Association scientific sessions.
Dr. Banovic, from the University of Belgrade Medical School in Serbia, is coprincipal investigator on the trial, called AVATAR (Aortic Valve Replacement vs. Conservative Treatment in Asymptomatic Severe Aortic Stenosis). He is also lead author on the study’s publication in Circulation, timed to coincide with his AHA presentation.
“The AVATAR findings provide additional evidence to help clinicians in guiding their decision when seeing a patient with significant aortic stenosis, normal left ventricular function, overall low surgical risk, and without significant comorbidities,” Dr. Banovic told this news organization.
European and North American Guidelines favor watchful waiting for asymptomatic patients with severe aortic stenosis, with surgery upon development of symptoms or LV dysfunction, observed Victoria Delgado, MD, PhD, Leiden (the Netherlands) University Medical Center, an invited discussant for the AVATAR presentation.
AVATAR does suggest that “early surgery in truly asymptomatic patients with severe aortic stenosis and preserved ejection fraction seems to provide better outcomes as compared to the conservative treatment,” she said. “But I think that the long-term follow-up for potential events, such as valve durability or endocarditis, is still needed.”
The trial has strengths, compared with the recent RECOVERY trial, which also concluded in favor of early SAVR over watchful waiting in patients described as asymptomatic with severe aortic stenosis. Dr. Delgado and other observers, however, have pointed out limitations of that trial, including questions about whether the patients were truly asymptomatic – stress testing wasn›t routinely performed.
In AVATAR, all patients were negative at stress testing, which required them to reach their estimated maximum heart rate, Dr. Banovic noted. As he and his colleagues write, the trial expands on RECOVERY “by providing evidence of the benefit of early surgery in a setting representative of a dilemma in decision making, in truly asymptomatic patients with severe but not critical aortic stenosis and normal LV function.”
A role for TAVR?
Guidelines in general “can be very conservative and lag behind evidence a bit,” Patricia A. Pellikka, MD, Mayo Clinic, Rochester, Minn., who is not associated with AVATAR, said in an interview.
“I think when we see patients clinically, we can advise them that if they don’t have symptoms and they do have severe aortic stenosis,” she said, “they’re likely going to get symptoms within a reasonably short period of time, according to our retrospective databases, and that doing the intervention early may yield better long-term outcomes.”
The results of AVATAR, in which valve replacement consisted only of SAVR, “probably could be extrapolated” to transcatheter aortic valve replacement (TAVR), Dr. Pellikka observed. “Certainly, TAVR is the procedure that patients come asking for. It’s attractive to avoid a major surgery, and it seems very plausible that TAVR would have yielded similar results if that had been a therapy in this trial.”
In practice, patient age and functional status would figure heavily in deciding whether early valve replacement, and which procedure, is appropriate, Dr. Banovic said in an interview. Importantly, the trial’s patients were at low surgical risk and free of major chronic diseases or other important health concerns.
“Frailty and older age are known risk factors for suboptimal recovery” after SAVR, Dr. Banovic said when interviewed. Therefore, frail patients, who were not many in AVATAR, might be “more suitable for TAVR than SAVR, based on the TAVR-vs.-SAVR results in symptomatic AS patients,” he said.
“One might extrapolate experience from AVATAR trial to TAVR, which may lower the bar for TAVR indications,” but that would require more supporting evidence, Dr. Banovic said.
Confirmed asymptomatic
AVATAR, conducted at nine centers in seven countries in the European Union, randomly assigned 157 adults with severe AS by echocardiography and a LV ejection fraction (LVEF) greater than 50% to early SAVR or conservative management. They averaged 67 years in age, and 43% were women.
The trial excluded anyone with dyspnea, syncope, presyncope, angina, or LV dysfunction and anyone with a history of atrial fibrillation or significant cardiac, renal, or lung disease. The cohort’s average Society of Thoracic Surgeons Predicted Risk of Mortality (STS-PROM) score was 1.7%.
The 78 patients in the early-surgery group “were expected” to have the procedure within 8 weeks of randomization, the published report states; the median time was 55 days. Six of them ultimately did not have the surgery. There was only one periprocedural death, for an operative mortality of 1.4%.
The 79 patients assigned to conservative care were later referred for surgery if they developed symptoms, their LVEF dropped below 50%, or they showed a 0.3-m/sec jump in peak aortic jet velocity at follow-up echocardiography. That occurred with 25 patients a median of 400 days after randomization.
The rate of the primary endpoint – death from any cause, acute myocardial infarction, stroke, or unplanned HF hospitalization – was 16.6% in the early-surgery group and 32.9% for those managed conservatively over a median of 32 months. The hazard ratio by intention-to-treat analysis was 0.46 (95% confidence interval, 0.23-0.90; P = .02). The HR for death from any cause or HF hospitalization was 0.40 (95% CI, 0.19-0.84; P = .013). Any differences in the individual endpoints of death, first HF hospitalizations, thromboembolic complications, or major bleeding were not significant.
If early aortic valve replacement is better for patients like those in AVATAR, some sort of screening for previously unknown severe aortic stenosis may seem attractive for selected populations. “Echocardiography would be the screening test for aortic stenosis, but it’s fairly expensive and therefore has never been advocated as a test to screen everyone,” Dr. Pellikka observed.
“But things are changing,” given innovations such as point-of-care ultrasonography and machine learning, she noted. “Artificial intelligence is progressing in its application to echocardiography, and it’s conceivable that in the future, there might be some abbreviated or screening type of test. But I don’t think we’re quite there yet.”
Dr. Banovic had no conflicts; disclosures for the other authors are in the report. Dr. Delgado disclosed speaker fees from Edwards Lifesciences, Abbott Vascular, Medtronic, Merck, Novartis, and GE Healthcare and unrestricted research grants to her institution from Abbott Vascular, Bayer, Biotronik, Bioventrix, Boston Scientific, Edwards Lifesciences, GE Healthcare, Ionis, and Medtronic. Dr. Pellikka disclosed receiving a research grant from Ultromics and having unspecified modest relationships with GE Healthcare, Lantheus, and OxThera.
A version of this article first appeared on Medscape.com.
Better to intervene early with a new valve in patients with severe aortic stenosis (AS) who are asymptomatic, even during exercise, than to wait for the disease to progress and symptoms to emerge before operating, suggests a small, randomized trial that challenges the guidelines.
Of the trial’s 157 patients, all with negative results on stress tests and normal left ventricular (LV) function despite severe AS, those assigned to early surgical aortic valve replacement (SAVR), compared with standard watchful waiting, showed a better-than-50% drop in risk for death or major adverse cardiac events (MACE) over 2-3 years. The benefit appeared driven by fewer hospitalizations for heart failure (HF) and deaths in the early-surgery group.
The findings “advocate for early surgery once aortic stenosis becomes significant and regardless of symptom status,” Marko Banovic, MD, PhD, said during his presentation at the American Heart Association scientific sessions.
Dr. Banovic, from the University of Belgrade Medical School in Serbia, is coprincipal investigator on the trial, called AVATAR (Aortic Valve Replacement vs. Conservative Treatment in Asymptomatic Severe Aortic Stenosis). He is also lead author on the study’s publication in Circulation, timed to coincide with his AHA presentation.
“The AVATAR findings provide additional evidence to help clinicians in guiding their decision when seeing a patient with significant aortic stenosis, normal left ventricular function, overall low surgical risk, and without significant comorbidities,” Dr. Banovic told this news organization.
European and North American Guidelines favor watchful waiting for asymptomatic patients with severe aortic stenosis, with surgery upon development of symptoms or LV dysfunction, observed Victoria Delgado, MD, PhD, Leiden (the Netherlands) University Medical Center, an invited discussant for the AVATAR presentation.
AVATAR does suggest that “early surgery in truly asymptomatic patients with severe aortic stenosis and preserved ejection fraction seems to provide better outcomes as compared to the conservative treatment,” she said. “But I think that the long-term follow-up for potential events, such as valve durability or endocarditis, is still needed.”
The trial has strengths, compared with the recent RECOVERY trial, which also concluded in favor of early SAVR over watchful waiting in patients described as asymptomatic with severe aortic stenosis. Dr. Delgado and other observers, however, have pointed out limitations of that trial, including questions about whether the patients were truly asymptomatic – stress testing wasn›t routinely performed.
In AVATAR, all patients were negative at stress testing, which required them to reach their estimated maximum heart rate, Dr. Banovic noted. As he and his colleagues write, the trial expands on RECOVERY “by providing evidence of the benefit of early surgery in a setting representative of a dilemma in decision making, in truly asymptomatic patients with severe but not critical aortic stenosis and normal LV function.”
A role for TAVR?
Guidelines in general “can be very conservative and lag behind evidence a bit,” Patricia A. Pellikka, MD, Mayo Clinic, Rochester, Minn., who is not associated with AVATAR, said in an interview.
“I think when we see patients clinically, we can advise them that if they don’t have symptoms and they do have severe aortic stenosis,” she said, “they’re likely going to get symptoms within a reasonably short period of time, according to our retrospective databases, and that doing the intervention early may yield better long-term outcomes.”
The results of AVATAR, in which valve replacement consisted only of SAVR, “probably could be extrapolated” to transcatheter aortic valve replacement (TAVR), Dr. Pellikka observed. “Certainly, TAVR is the procedure that patients come asking for. It’s attractive to avoid a major surgery, and it seems very plausible that TAVR would have yielded similar results if that had been a therapy in this trial.”
In practice, patient age and functional status would figure heavily in deciding whether early valve replacement, and which procedure, is appropriate, Dr. Banovic said in an interview. Importantly, the trial’s patients were at low surgical risk and free of major chronic diseases or other important health concerns.
“Frailty and older age are known risk factors for suboptimal recovery” after SAVR, Dr. Banovic said when interviewed. Therefore, frail patients, who were not many in AVATAR, might be “more suitable for TAVR than SAVR, based on the TAVR-vs.-SAVR results in symptomatic AS patients,” he said.
“One might extrapolate experience from AVATAR trial to TAVR, which may lower the bar for TAVR indications,” but that would require more supporting evidence, Dr. Banovic said.
Confirmed asymptomatic
AVATAR, conducted at nine centers in seven countries in the European Union, randomly assigned 157 adults with severe AS by echocardiography and a LV ejection fraction (LVEF) greater than 50% to early SAVR or conservative management. They averaged 67 years in age, and 43% were women.
The trial excluded anyone with dyspnea, syncope, presyncope, angina, or LV dysfunction and anyone with a history of atrial fibrillation or significant cardiac, renal, or lung disease. The cohort’s average Society of Thoracic Surgeons Predicted Risk of Mortality (STS-PROM) score was 1.7%.
The 78 patients in the early-surgery group “were expected” to have the procedure within 8 weeks of randomization, the published report states; the median time was 55 days. Six of them ultimately did not have the surgery. There was only one periprocedural death, for an operative mortality of 1.4%.
The 79 patients assigned to conservative care were later referred for surgery if they developed symptoms, their LVEF dropped below 50%, or they showed a 0.3-m/sec jump in peak aortic jet velocity at follow-up echocardiography. That occurred with 25 patients a median of 400 days after randomization.
The rate of the primary endpoint – death from any cause, acute myocardial infarction, stroke, or unplanned HF hospitalization – was 16.6% in the early-surgery group and 32.9% for those managed conservatively over a median of 32 months. The hazard ratio by intention-to-treat analysis was 0.46 (95% confidence interval, 0.23-0.90; P = .02). The HR for death from any cause or HF hospitalization was 0.40 (95% CI, 0.19-0.84; P = .013). Any differences in the individual endpoints of death, first HF hospitalizations, thromboembolic complications, or major bleeding were not significant.
If early aortic valve replacement is better for patients like those in AVATAR, some sort of screening for previously unknown severe aortic stenosis may seem attractive for selected populations. “Echocardiography would be the screening test for aortic stenosis, but it’s fairly expensive and therefore has never been advocated as a test to screen everyone,” Dr. Pellikka observed.
“But things are changing,” given innovations such as point-of-care ultrasonography and machine learning, she noted. “Artificial intelligence is progressing in its application to echocardiography, and it’s conceivable that in the future, there might be some abbreviated or screening type of test. But I don’t think we’re quite there yet.”
Dr. Banovic had no conflicts; disclosures for the other authors are in the report. Dr. Delgado disclosed speaker fees from Edwards Lifesciences, Abbott Vascular, Medtronic, Merck, Novartis, and GE Healthcare and unrestricted research grants to her institution from Abbott Vascular, Bayer, Biotronik, Bioventrix, Boston Scientific, Edwards Lifesciences, GE Healthcare, Ionis, and Medtronic. Dr. Pellikka disclosed receiving a research grant from Ultromics and having unspecified modest relationships with GE Healthcare, Lantheus, and OxThera.
A version of this article first appeared on Medscape.com.
Better to intervene early with a new valve in patients with severe aortic stenosis (AS) who are asymptomatic, even during exercise, than to wait for the disease to progress and symptoms to emerge before operating, suggests a small, randomized trial that challenges the guidelines.
Of the trial’s 157 patients, all with negative results on stress tests and normal left ventricular (LV) function despite severe AS, those assigned to early surgical aortic valve replacement (SAVR), compared with standard watchful waiting, showed a better-than-50% drop in risk for death or major adverse cardiac events (MACE) over 2-3 years. The benefit appeared driven by fewer hospitalizations for heart failure (HF) and deaths in the early-surgery group.
The findings “advocate for early surgery once aortic stenosis becomes significant and regardless of symptom status,” Marko Banovic, MD, PhD, said during his presentation at the American Heart Association scientific sessions.
Dr. Banovic, from the University of Belgrade Medical School in Serbia, is coprincipal investigator on the trial, called AVATAR (Aortic Valve Replacement vs. Conservative Treatment in Asymptomatic Severe Aortic Stenosis). He is also lead author on the study’s publication in Circulation, timed to coincide with his AHA presentation.
“The AVATAR findings provide additional evidence to help clinicians in guiding their decision when seeing a patient with significant aortic stenosis, normal left ventricular function, overall low surgical risk, and without significant comorbidities,” Dr. Banovic told this news organization.
European and North American Guidelines favor watchful waiting for asymptomatic patients with severe aortic stenosis, with surgery upon development of symptoms or LV dysfunction, observed Victoria Delgado, MD, PhD, Leiden (the Netherlands) University Medical Center, an invited discussant for the AVATAR presentation.
AVATAR does suggest that “early surgery in truly asymptomatic patients with severe aortic stenosis and preserved ejection fraction seems to provide better outcomes as compared to the conservative treatment,” she said. “But I think that the long-term follow-up for potential events, such as valve durability or endocarditis, is still needed.”
The trial has strengths, compared with the recent RECOVERY trial, which also concluded in favor of early SAVR over watchful waiting in patients described as asymptomatic with severe aortic stenosis. Dr. Delgado and other observers, however, have pointed out limitations of that trial, including questions about whether the patients were truly asymptomatic – stress testing wasn›t routinely performed.
In AVATAR, all patients were negative at stress testing, which required them to reach their estimated maximum heart rate, Dr. Banovic noted. As he and his colleagues write, the trial expands on RECOVERY “by providing evidence of the benefit of early surgery in a setting representative of a dilemma in decision making, in truly asymptomatic patients with severe but not critical aortic stenosis and normal LV function.”
A role for TAVR?
Guidelines in general “can be very conservative and lag behind evidence a bit,” Patricia A. Pellikka, MD, Mayo Clinic, Rochester, Minn., who is not associated with AVATAR, said in an interview.
“I think when we see patients clinically, we can advise them that if they don’t have symptoms and they do have severe aortic stenosis,” she said, “they’re likely going to get symptoms within a reasonably short period of time, according to our retrospective databases, and that doing the intervention early may yield better long-term outcomes.”
The results of AVATAR, in which valve replacement consisted only of SAVR, “probably could be extrapolated” to transcatheter aortic valve replacement (TAVR), Dr. Pellikka observed. “Certainly, TAVR is the procedure that patients come asking for. It’s attractive to avoid a major surgery, and it seems very plausible that TAVR would have yielded similar results if that had been a therapy in this trial.”
In practice, patient age and functional status would figure heavily in deciding whether early valve replacement, and which procedure, is appropriate, Dr. Banovic said in an interview. Importantly, the trial’s patients were at low surgical risk and free of major chronic diseases or other important health concerns.
“Frailty and older age are known risk factors for suboptimal recovery” after SAVR, Dr. Banovic said when interviewed. Therefore, frail patients, who were not many in AVATAR, might be “more suitable for TAVR than SAVR, based on the TAVR-vs.-SAVR results in symptomatic AS patients,” he said.
“One might extrapolate experience from AVATAR trial to TAVR, which may lower the bar for TAVR indications,” but that would require more supporting evidence, Dr. Banovic said.
Confirmed asymptomatic
AVATAR, conducted at nine centers in seven countries in the European Union, randomly assigned 157 adults with severe AS by echocardiography and a LV ejection fraction (LVEF) greater than 50% to early SAVR or conservative management. They averaged 67 years in age, and 43% were women.
The trial excluded anyone with dyspnea, syncope, presyncope, angina, or LV dysfunction and anyone with a history of atrial fibrillation or significant cardiac, renal, or lung disease. The cohort’s average Society of Thoracic Surgeons Predicted Risk of Mortality (STS-PROM) score was 1.7%.
The 78 patients in the early-surgery group “were expected” to have the procedure within 8 weeks of randomization, the published report states; the median time was 55 days. Six of them ultimately did not have the surgery. There was only one periprocedural death, for an operative mortality of 1.4%.
The 79 patients assigned to conservative care were later referred for surgery if they developed symptoms, their LVEF dropped below 50%, or they showed a 0.3-m/sec jump in peak aortic jet velocity at follow-up echocardiography. That occurred with 25 patients a median of 400 days after randomization.
The rate of the primary endpoint – death from any cause, acute myocardial infarction, stroke, or unplanned HF hospitalization – was 16.6% in the early-surgery group and 32.9% for those managed conservatively over a median of 32 months. The hazard ratio by intention-to-treat analysis was 0.46 (95% confidence interval, 0.23-0.90; P = .02). The HR for death from any cause or HF hospitalization was 0.40 (95% CI, 0.19-0.84; P = .013). Any differences in the individual endpoints of death, first HF hospitalizations, thromboembolic complications, or major bleeding were not significant.
If early aortic valve replacement is better for patients like those in AVATAR, some sort of screening for previously unknown severe aortic stenosis may seem attractive for selected populations. “Echocardiography would be the screening test for aortic stenosis, but it’s fairly expensive and therefore has never been advocated as a test to screen everyone,” Dr. Pellikka observed.
“But things are changing,” given innovations such as point-of-care ultrasonography and machine learning, she noted. “Artificial intelligence is progressing in its application to echocardiography, and it’s conceivable that in the future, there might be some abbreviated or screening type of test. But I don’t think we’re quite there yet.”
Dr. Banovic had no conflicts; disclosures for the other authors are in the report. Dr. Delgado disclosed speaker fees from Edwards Lifesciences, Abbott Vascular, Medtronic, Merck, Novartis, and GE Healthcare and unrestricted research grants to her institution from Abbott Vascular, Bayer, Biotronik, Bioventrix, Boston Scientific, Edwards Lifesciences, GE Healthcare, Ionis, and Medtronic. Dr. Pellikka disclosed receiving a research grant from Ultromics and having unspecified modest relationships with GE Healthcare, Lantheus, and OxThera.
A version of this article first appeared on Medscape.com.
FROM AHA 2021
Fully endovascular mitral valve replacement a limited success in feasibility study
It remains early days for transcatheter mitral-valve replacement (TMVR) as a minimally invasive way to treat severe, mitral regurgitation (MR), but it’s even earlier days for TMVR as an endovascular procedure. Most of the technique’s limited experience with a dedicated mitral prosthesis has involved transapical delivery.
But now a 15-patient study of transfemoral, transeptal TMVR – with a prosthesis designed for the mitral position and previously tested only transapically – has shown good 30-day results in that MR was essentially abolished with virtually no paravalvular leakage.
Nor were there adverse clinical events such as death, stroke, reintervention, or new need for a pacemaker in any of the high-surgical-risk patients with MR in this feasibility study of the transfemoral Intrepid TMVR System (Medtronic). Implantation failed, however, in one patient who then received a surgical valve via sternotomy.
The current cohort is part of a larger ongoing trial that will track whether patients implanted transfemorally with the Intrepid also show reverse remodeling and good clinical outcomes over at least a year. That study, called APOLLO, is one of several exploring dedicated TMVR valves from different companies, with names like SUMMIT, MISCEND, and TIARA-2.
Currently, TMVR is approved in the United States only using one device designed for the aortic position and only for treating failed surgical mitral bioprostheses in high-risk patients.
If the Intrepid transfemoral system has an Achilles’ heel, at least in the current iteration, it might be its 35 F catheter delivery system that requires surgical access to the femoral vein. Seven of the patients in the small series experienced major bleeding events, including six at the femoral access site, listed as major vascular complications.
Overall, the study’s patients “were extremely sick with a lot of comorbidity. A lot of them had atrial fibrillation, a lot of them were on anticoagulation to start with,” observed Firas Zahr, MD, Oregon Health & Science University, Portland, as part of his presentation of the study at Transcatheter Cardiovascular Therapeutics (TCT) 2021, held virtually as well as onsite in Orlando, Florida.
All had moderate-to-severe, usually primary MR; two thirds of the cohort had been in NYHA class III or IV at baseline, and 40% had been hospitalized for heart failure within the past year. Eight had a history of cardiovascular surgery, and eight had diabetes. Their mean Society of Thoracic Surgeons Predicted Risk of Mortality (STS-PROM) score was 4.7, Dr. Zahr reported.
“At 30 days, there was a significant improvement in their heart failure classification; the vast majority of the patients were [NYHA] class I and class II,” said Dr. Zahr, who is also lead author on the study’s Nov. 6 publication in JACC: Cardiovascular Interventions.
Observers of the study at TCT 2021 seemed enthusiastic about the study’s results but recognized that TMVR in its current form still has formidable limitations.
“This is clearly an exciting look into the future and very reassuring to a degree, aside from the complications, which are somewhat expected as we go with 30-plus French devices,” Rajiv Tayal, MD, MPH, said at a press conference on the Intrepid study held before Dr. Zahr’s formal presentation. Dr. Tayal is an interventional cardiologist with Valley Health System, Ridgewood, New Jersey, and New York Medical College, Valhalla.
“I think we’ve all learned that transapical [access] is just not a viable procedure for a lot of these patients, and so we’ve got to get to transfemoral,” Susheel K. Kodali, MD, interventional cardiologist at New York-Presbyterian/Columbia University Irving Medical Center, said at the same forum.
A 35 F device “is going to be too big,” he said. However, “it is the first step to iterate to a smaller device.” Dr. Kodali said his center contributed a patient to the study, and he is listed as a coauthor on the publication.
The delivery system’s large profile is only part of the vascular complication issue. Not only did the procedure require surgical cutdown for venous access, but “we were fairly aggressive in anticoagulating these patients with the fear of thrombus formation,” Dr. Zahr said in the discussion following his presentation.
“A postprocedure anticoagulation regimen is recommended within the protocol, but ultimate therapy was left to the discretion of the treating site physician,” the published report states, noting that all 14 patients with successful TMVR were discharged on warfarin. They included 12 who were also put on a single antiplatelet and one given dual antiplatelet therapy on top of the oral anticoagulant.
“One thing that we learned is that we probably should standardize our approach to perioperative anticoagulation,” Dr. Zahr observed. Also, a 29 F sheath for the system is in the works, “and we’re hoping that with smaller sheath size, and hopefully going even to percutaneous, might have an impact on lowering the vascular complications.”
Explanations for the “higher-than-expected vascular complication rate” remains somewhat unclear, agreed an editorial accompanying the study’s publication, “but may include a learning curve with the system, the large introducer sheath, the need for surgical cutdown, and postprocedural anticoagulation.”
For trans-septal TMVR to become a default approach, “venous access will need to be achieved percutaneously and vascular complications need to be infrequent,” contends the editorial, with lead author Mohamad Alkhouli, MD, Mayo Clinic, Rochester, Minn.
“These data provide a glimpse into the future of TMVR. The excellent short-term safety and effectiveness of this still very early-stage procedure represent a major step forward in the field,” they write.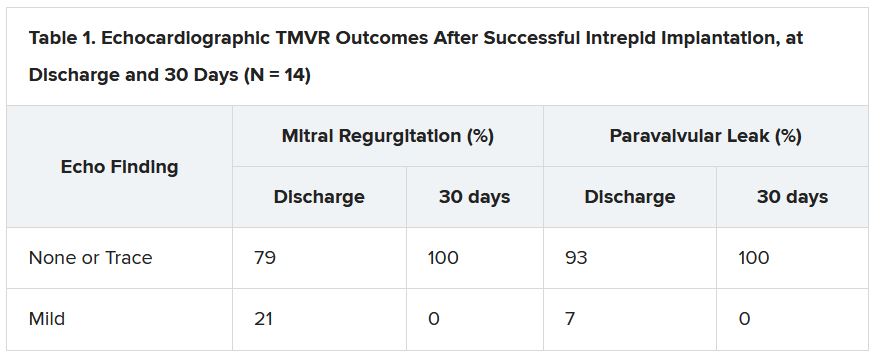
“The main question that the Intrepid early feasibility data raise is whether transfemoral, trans-septal TMVR will evolve to become the preferred strategy over transapical TMVR,” as occurred with transcatheter aortic-valve replacement (TAVR), the editorial states. “The answer is likely yes, but a few matters specific to trans-septal route will need be addressed first.”
Among those matters: The 35 F catheter leaves behind a considerable atrial septal defect (ASD). At operator discretion in this series, 11 patients received an ASD closure device.
None of the remaining four patients “developed significant heart failure or right ventricular dysfunction,” Dr. Zahr observed. “So, it seems like those patients who had their ASD left open tolerated it fairly well, at least until 30 days.”
But “we still need to learn what to do with those ASDs,” he said. “What is an acceptable residual shunt and what is an acceptable ASD size is to be determined.”
In general, the editorial notes, “the TMVR population has a high prevalence of cardiomyopathy, and a large residual iatrogenic ASD may lead to worsening volume overload and heart failure decompensation in some patients.”
Insertion of a closure device has its own issues, it continues. “Closure of the ASD might impede future access to the left atrium, which could impact life-long management of this high-risk population. A large septal occluder may hinder potentially needed procedures such as paravalvular leak closure, left atrial appendage closure, or pulmonary vein isolation.”
Patients like those in the current series, Dr. Kodali observed, will face “a lifetime of management challenges, and you want to make sure you don’t take away other options.”
The study was funded by Medtronic. Dr. Zahr reported institutional grant support from Edwards Lifesciences and Medtronic. Dr. Kodali disclosed consultant fees from Admedus and Dura Biotech; equity in Dura Biotech, Microinterventional Devices, Thubrika Aortic Valve, Supira, Admedus, TriFlo, and Anona; and institutional grant support from Edwards Lifesciences, Medtronic, Abbott Vascular, Boston Scientific, and JenaValve. The editorial writers have disclosed no relevant financial relationships. Dr. Tayal disclosed consultant fees or honoraria from or serving on a speakers bureau for Abiomed, Edwards Lifesciences, Abbott Vascular, and Shockwave Medical.
A version of this article first appeared on Medscape.com.
It remains early days for transcatheter mitral-valve replacement (TMVR) as a minimally invasive way to treat severe, mitral regurgitation (MR), but it’s even earlier days for TMVR as an endovascular procedure. Most of the technique’s limited experience with a dedicated mitral prosthesis has involved transapical delivery.
But now a 15-patient study of transfemoral, transeptal TMVR – with a prosthesis designed for the mitral position and previously tested only transapically – has shown good 30-day results in that MR was essentially abolished with virtually no paravalvular leakage.
Nor were there adverse clinical events such as death, stroke, reintervention, or new need for a pacemaker in any of the high-surgical-risk patients with MR in this feasibility study of the transfemoral Intrepid TMVR System (Medtronic). Implantation failed, however, in one patient who then received a surgical valve via sternotomy.
The current cohort is part of a larger ongoing trial that will track whether patients implanted transfemorally with the Intrepid also show reverse remodeling and good clinical outcomes over at least a year. That study, called APOLLO, is one of several exploring dedicated TMVR valves from different companies, with names like SUMMIT, MISCEND, and TIARA-2.
Currently, TMVR is approved in the United States only using one device designed for the aortic position and only for treating failed surgical mitral bioprostheses in high-risk patients.
If the Intrepid transfemoral system has an Achilles’ heel, at least in the current iteration, it might be its 35 F catheter delivery system that requires surgical access to the femoral vein. Seven of the patients in the small series experienced major bleeding events, including six at the femoral access site, listed as major vascular complications.
Overall, the study’s patients “were extremely sick with a lot of comorbidity. A lot of them had atrial fibrillation, a lot of them were on anticoagulation to start with,” observed Firas Zahr, MD, Oregon Health & Science University, Portland, as part of his presentation of the study at Transcatheter Cardiovascular Therapeutics (TCT) 2021, held virtually as well as onsite in Orlando, Florida.
All had moderate-to-severe, usually primary MR; two thirds of the cohort had been in NYHA class III or IV at baseline, and 40% had been hospitalized for heart failure within the past year. Eight had a history of cardiovascular surgery, and eight had diabetes. Their mean Society of Thoracic Surgeons Predicted Risk of Mortality (STS-PROM) score was 4.7, Dr. Zahr reported.
“At 30 days, there was a significant improvement in their heart failure classification; the vast majority of the patients were [NYHA] class I and class II,” said Dr. Zahr, who is also lead author on the study’s Nov. 6 publication in JACC: Cardiovascular Interventions.
Observers of the study at TCT 2021 seemed enthusiastic about the study’s results but recognized that TMVR in its current form still has formidable limitations.
“This is clearly an exciting look into the future and very reassuring to a degree, aside from the complications, which are somewhat expected as we go with 30-plus French devices,” Rajiv Tayal, MD, MPH, said at a press conference on the Intrepid study held before Dr. Zahr’s formal presentation. Dr. Tayal is an interventional cardiologist with Valley Health System, Ridgewood, New Jersey, and New York Medical College, Valhalla.
“I think we’ve all learned that transapical [access] is just not a viable procedure for a lot of these patients, and so we’ve got to get to transfemoral,” Susheel K. Kodali, MD, interventional cardiologist at New York-Presbyterian/Columbia University Irving Medical Center, said at the same forum.
A 35 F device “is going to be too big,” he said. However, “it is the first step to iterate to a smaller device.” Dr. Kodali said his center contributed a patient to the study, and he is listed as a coauthor on the publication.
The delivery system’s large profile is only part of the vascular complication issue. Not only did the procedure require surgical cutdown for venous access, but “we were fairly aggressive in anticoagulating these patients with the fear of thrombus formation,” Dr. Zahr said in the discussion following his presentation.
“A postprocedure anticoagulation regimen is recommended within the protocol, but ultimate therapy was left to the discretion of the treating site physician,” the published report states, noting that all 14 patients with successful TMVR were discharged on warfarin. They included 12 who were also put on a single antiplatelet and one given dual antiplatelet therapy on top of the oral anticoagulant.
“One thing that we learned is that we probably should standardize our approach to perioperative anticoagulation,” Dr. Zahr observed. Also, a 29 F sheath for the system is in the works, “and we’re hoping that with smaller sheath size, and hopefully going even to percutaneous, might have an impact on lowering the vascular complications.”
Explanations for the “higher-than-expected vascular complication rate” remains somewhat unclear, agreed an editorial accompanying the study’s publication, “but may include a learning curve with the system, the large introducer sheath, the need for surgical cutdown, and postprocedural anticoagulation.”
For trans-septal TMVR to become a default approach, “venous access will need to be achieved percutaneously and vascular complications need to be infrequent,” contends the editorial, with lead author Mohamad Alkhouli, MD, Mayo Clinic, Rochester, Minn.
“These data provide a glimpse into the future of TMVR. The excellent short-term safety and effectiveness of this still very early-stage procedure represent a major step forward in the field,” they write.
“The main question that the Intrepid early feasibility data raise is whether transfemoral, trans-septal TMVR will evolve to become the preferred strategy over transapical TMVR,” as occurred with transcatheter aortic-valve replacement (TAVR), the editorial states. “The answer is likely yes, but a few matters specific to trans-septal route will need be addressed first.”
Among those matters: The 35 F catheter leaves behind a considerable atrial septal defect (ASD). At operator discretion in this series, 11 patients received an ASD closure device.
None of the remaining four patients “developed significant heart failure or right ventricular dysfunction,” Dr. Zahr observed. “So, it seems like those patients who had their ASD left open tolerated it fairly well, at least until 30 days.”
But “we still need to learn what to do with those ASDs,” he said. “What is an acceptable residual shunt and what is an acceptable ASD size is to be determined.”
In general, the editorial notes, “the TMVR population has a high prevalence of cardiomyopathy, and a large residual iatrogenic ASD may lead to worsening volume overload and heart failure decompensation in some patients.”
Insertion of a closure device has its own issues, it continues. “Closure of the ASD might impede future access to the left atrium, which could impact life-long management of this high-risk population. A large septal occluder may hinder potentially needed procedures such as paravalvular leak closure, left atrial appendage closure, or pulmonary vein isolation.”
Patients like those in the current series, Dr. Kodali observed, will face “a lifetime of management challenges, and you want to make sure you don’t take away other options.”
The study was funded by Medtronic. Dr. Zahr reported institutional grant support from Edwards Lifesciences and Medtronic. Dr. Kodali disclosed consultant fees from Admedus and Dura Biotech; equity in Dura Biotech, Microinterventional Devices, Thubrika Aortic Valve, Supira, Admedus, TriFlo, and Anona; and institutional grant support from Edwards Lifesciences, Medtronic, Abbott Vascular, Boston Scientific, and JenaValve. The editorial writers have disclosed no relevant financial relationships. Dr. Tayal disclosed consultant fees or honoraria from or serving on a speakers bureau for Abiomed, Edwards Lifesciences, Abbott Vascular, and Shockwave Medical.
A version of this article first appeared on Medscape.com.
It remains early days for transcatheter mitral-valve replacement (TMVR) as a minimally invasive way to treat severe, mitral regurgitation (MR), but it’s even earlier days for TMVR as an endovascular procedure. Most of the technique’s limited experience with a dedicated mitral prosthesis has involved transapical delivery.
But now a 15-patient study of transfemoral, transeptal TMVR – with a prosthesis designed for the mitral position and previously tested only transapically – has shown good 30-day results in that MR was essentially abolished with virtually no paravalvular leakage.
Nor were there adverse clinical events such as death, stroke, reintervention, or new need for a pacemaker in any of the high-surgical-risk patients with MR in this feasibility study of the transfemoral Intrepid TMVR System (Medtronic). Implantation failed, however, in one patient who then received a surgical valve via sternotomy.
The current cohort is part of a larger ongoing trial that will track whether patients implanted transfemorally with the Intrepid also show reverse remodeling and good clinical outcomes over at least a year. That study, called APOLLO, is one of several exploring dedicated TMVR valves from different companies, with names like SUMMIT, MISCEND, and TIARA-2.
Currently, TMVR is approved in the United States only using one device designed for the aortic position and only for treating failed surgical mitral bioprostheses in high-risk patients.
If the Intrepid transfemoral system has an Achilles’ heel, at least in the current iteration, it might be its 35 F catheter delivery system that requires surgical access to the femoral vein. Seven of the patients in the small series experienced major bleeding events, including six at the femoral access site, listed as major vascular complications.
Overall, the study’s patients “were extremely sick with a lot of comorbidity. A lot of them had atrial fibrillation, a lot of them were on anticoagulation to start with,” observed Firas Zahr, MD, Oregon Health & Science University, Portland, as part of his presentation of the study at Transcatheter Cardiovascular Therapeutics (TCT) 2021, held virtually as well as onsite in Orlando, Florida.
All had moderate-to-severe, usually primary MR; two thirds of the cohort had been in NYHA class III or IV at baseline, and 40% had been hospitalized for heart failure within the past year. Eight had a history of cardiovascular surgery, and eight had diabetes. Their mean Society of Thoracic Surgeons Predicted Risk of Mortality (STS-PROM) score was 4.7, Dr. Zahr reported.
“At 30 days, there was a significant improvement in their heart failure classification; the vast majority of the patients were [NYHA] class I and class II,” said Dr. Zahr, who is also lead author on the study’s Nov. 6 publication in JACC: Cardiovascular Interventions.
Observers of the study at TCT 2021 seemed enthusiastic about the study’s results but recognized that TMVR in its current form still has formidable limitations.
“This is clearly an exciting look into the future and very reassuring to a degree, aside from the complications, which are somewhat expected as we go with 30-plus French devices,” Rajiv Tayal, MD, MPH, said at a press conference on the Intrepid study held before Dr. Zahr’s formal presentation. Dr. Tayal is an interventional cardiologist with Valley Health System, Ridgewood, New Jersey, and New York Medical College, Valhalla.
“I think we’ve all learned that transapical [access] is just not a viable procedure for a lot of these patients, and so we’ve got to get to transfemoral,” Susheel K. Kodali, MD, interventional cardiologist at New York-Presbyterian/Columbia University Irving Medical Center, said at the same forum.
A 35 F device “is going to be too big,” he said. However, “it is the first step to iterate to a smaller device.” Dr. Kodali said his center contributed a patient to the study, and he is listed as a coauthor on the publication.
The delivery system’s large profile is only part of the vascular complication issue. Not only did the procedure require surgical cutdown for venous access, but “we were fairly aggressive in anticoagulating these patients with the fear of thrombus formation,” Dr. Zahr said in the discussion following his presentation.
“A postprocedure anticoagulation regimen is recommended within the protocol, but ultimate therapy was left to the discretion of the treating site physician,” the published report states, noting that all 14 patients with successful TMVR were discharged on warfarin. They included 12 who were also put on a single antiplatelet and one given dual antiplatelet therapy on top of the oral anticoagulant.
“One thing that we learned is that we probably should standardize our approach to perioperative anticoagulation,” Dr. Zahr observed. Also, a 29 F sheath for the system is in the works, “and we’re hoping that with smaller sheath size, and hopefully going even to percutaneous, might have an impact on lowering the vascular complications.”
Explanations for the “higher-than-expected vascular complication rate” remains somewhat unclear, agreed an editorial accompanying the study’s publication, “but may include a learning curve with the system, the large introducer sheath, the need for surgical cutdown, and postprocedural anticoagulation.”
For trans-septal TMVR to become a default approach, “venous access will need to be achieved percutaneously and vascular complications need to be infrequent,” contends the editorial, with lead author Mohamad Alkhouli, MD, Mayo Clinic, Rochester, Minn.
“These data provide a glimpse into the future of TMVR. The excellent short-term safety and effectiveness of this still very early-stage procedure represent a major step forward in the field,” they write.
“The main question that the Intrepid early feasibility data raise is whether transfemoral, trans-septal TMVR will evolve to become the preferred strategy over transapical TMVR,” as occurred with transcatheter aortic-valve replacement (TAVR), the editorial states. “The answer is likely yes, but a few matters specific to trans-septal route will need be addressed first.”
Among those matters: The 35 F catheter leaves behind a considerable atrial septal defect (ASD). At operator discretion in this series, 11 patients received an ASD closure device.
None of the remaining four patients “developed significant heart failure or right ventricular dysfunction,” Dr. Zahr observed. “So, it seems like those patients who had their ASD left open tolerated it fairly well, at least until 30 days.”
But “we still need to learn what to do with those ASDs,” he said. “What is an acceptable residual shunt and what is an acceptable ASD size is to be determined.”
In general, the editorial notes, “the TMVR population has a high prevalence of cardiomyopathy, and a large residual iatrogenic ASD may lead to worsening volume overload and heart failure decompensation in some patients.”
Insertion of a closure device has its own issues, it continues. “Closure of the ASD might impede future access to the left atrium, which could impact life-long management of this high-risk population. A large septal occluder may hinder potentially needed procedures such as paravalvular leak closure, left atrial appendage closure, or pulmonary vein isolation.”
Patients like those in the current series, Dr. Kodali observed, will face “a lifetime of management challenges, and you want to make sure you don’t take away other options.”
The study was funded by Medtronic. Dr. Zahr reported institutional grant support from Edwards Lifesciences and Medtronic. Dr. Kodali disclosed consultant fees from Admedus and Dura Biotech; equity in Dura Biotech, Microinterventional Devices, Thubrika Aortic Valve, Supira, Admedus, TriFlo, and Anona; and institutional grant support from Edwards Lifesciences, Medtronic, Abbott Vascular, Boston Scientific, and JenaValve. The editorial writers have disclosed no relevant financial relationships. Dr. Tayal disclosed consultant fees or honoraria from or serving on a speakers bureau for Abiomed, Edwards Lifesciences, Abbott Vascular, and Shockwave Medical.
A version of this article first appeared on Medscape.com.
FDA panel slams Endologix response to stent-graft safety issues
The Food and Drug Administration has long kept a watchful eye over successive iterations of endovascular stent graphs in the Endologix AFX line, designed for repair of abdominal aortic aneurysms (AAA). For years, the devices, first approved in 2011, have drawn safety alerts and recalls , stemming from what the agency says was a “higher than expected” risk for potentially injurious or fatal type III endoleaks.

As part of the latest review process, Endologix recently showed regulators data from a rare randomized trial of the AAA endovascular aneurysm repair (EVAR) technique. The company said the recent postmarket study LEOPARD suggested the type III endoleaks – blood seeping around or through the device into the aneurysm – are no more common with the current AFX2 system than with other available AAA stent-grafts.
Technical upgrades to its AFX line of EVAR devices in recent years have largely resolved the safety issues identified in previous models, the company argued.
But the company’s case was unconvincing for a majority of the FDA Circulatory System Devices Advisory Panel that assembled virtually on Nov. 2. A number of panelists questioned the earnestness with which Endologix worked to rectify the safety alert and recall issues. Many also decried the real-world relevance of the randomized trial presented as evidence, with its follow-up time of only a few years.
The panel that included more than a dozen clinicians – mostly surgeons or interventional cardiologists or radiologists – were not instructed to formally vote on the issues. But it ultimately advised the FDA that more exacting studies with longer follow-ups appear needed to show that the device’s benefits in routine use outweigh its risks, especially for type III endoleaks.
“There isn’t a tremendous amount of confidence” that Endologix had enacted sufficient risk-mitigation measures in the wake of the safety alerts and recalls, chair Richard A. Lange, MD, MBA, Foster School of Medicine and Texas Tech University Health Sciences Center, El Paso, said when summarizing the panel’s take on the day’s proceedings.
Although the stent-graft’s safety seemed improved with recent design changes, the panel wasn’t convinced the upgrades could take the credit, or even that they were aimed specifically at preventing endoleaks, Dr. Lange said. “Nobody feels assurance that the problem has been solved.”
“I believe that the type-three endoleaks pose a challenge to patients, and I have not seen enough data to assure me with a degree of certainty that that problem no longer persists,” said panelist Joaquin E. Cigarroa, MD, a cardiologist at Oregon Health & Science University, Portland. His take on the LEOPARD trial, moreover, is that it “does not refute that there is an issue, given the duration of follow-up.”
On the other hand, a majority of the panel agreed that, currently, the AFX2’s benefits would likely outweigh risks for patients in narrowly defined high-risk anatomic or clinical scenarios and those with no other endovascular or surgical option.
“I do believe that there are patient subsets where the Endologix graft can play an important and vital role,” surgeon Keith B. Allen, MD, St. Luke’s Mid America Heart & Vascular Institute, Kansas City, Missouri, offered from the panel.
“In patients that don’t have aneurysmal disease but have distal bifurcation proximal iliac disease, it can be a very nice graft to use and solves a problem,” he said. “To remove that graft completely from the market, I believe, would deny a subset of patients.”
But for aortic aneurysms in routine practice, Dr. Allen said, “I think there are some red flags with it.”
Joining the day’s proceedings as a public commenter, surgeon Mark Conrad, MD, St. Elizabeth’s Hospital, Boston, agreed that “there’s not one commercial device out there that is able to handle every anatomy.”
Having options for patients is important, he said, because “the biggest problems we run into are when somebody only uses one graft, and they try to make that fit everything.”
Another public commenter offered a similar take. “I think we haven’t done a great job in the vascular surgery community really honing in on the detailed nuances that separate one device from another,” said Naiem Nassiri, MD, Yale New Haven Hospital Heart & Vascular Center, Connecticut.
The Endologix device, he said, “serves a very specific role under certain anatomic configurations and limitations, and really, truly fills a gap” left by other available grafts. It suits a very specific niche, “and I think it needs to be explored further for that.”
Endologix representatives who advise clinicians could play a better role in familiarizing operators with the EVAR system’s strengths and limitations, proposed several panelists, including Minhaj S. Khaja, MD, MBA, interventional radiologist at UVA Health and the University of Virginia, Charlottesville.
“There definitely needs to be more education of the clinical reps as well as the physicians implanting these devices,” he said, regarding the type III leaks, patient selection issues, appropriate imaging follow-up, “and the potential for increased reintervention.”
All public commenters, Dr. Lange observed, had been invited to disclose potential conflicts of interest, but it was not mandatory and none did so during the public forum. Disclosures of potential conflicts for the panelists are available on the FDA site.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has long kept a watchful eye over successive iterations of endovascular stent graphs in the Endologix AFX line, designed for repair of abdominal aortic aneurysms (AAA). For years, the devices, first approved in 2011, have drawn safety alerts and recalls , stemming from what the agency says was a “higher than expected” risk for potentially injurious or fatal type III endoleaks.
As part of the latest review process, Endologix recently showed regulators data from a rare randomized trial of the AAA endovascular aneurysm repair (EVAR) technique. The company said the recent postmarket study LEOPARD suggested the type III endoleaks – blood seeping around or through the device into the aneurysm – are no more common with the current AFX2 system than with other available AAA stent-grafts.
Technical upgrades to its AFX line of EVAR devices in recent years have largely resolved the safety issues identified in previous models, the company argued.
But the company’s case was unconvincing for a majority of the FDA Circulatory System Devices Advisory Panel that assembled virtually on Nov. 2. A number of panelists questioned the earnestness with which Endologix worked to rectify the safety alert and recall issues. Many also decried the real-world relevance of the randomized trial presented as evidence, with its follow-up time of only a few years.
The panel that included more than a dozen clinicians – mostly surgeons or interventional cardiologists or radiologists – were not instructed to formally vote on the issues. But it ultimately advised the FDA that more exacting studies with longer follow-ups appear needed to show that the device’s benefits in routine use outweigh its risks, especially for type III endoleaks.
“There isn’t a tremendous amount of confidence” that Endologix had enacted sufficient risk-mitigation measures in the wake of the safety alerts and recalls, chair Richard A. Lange, MD, MBA, Foster School of Medicine and Texas Tech University Health Sciences Center, El Paso, said when summarizing the panel’s take on the day’s proceedings.
Although the stent-graft’s safety seemed improved with recent design changes, the panel wasn’t convinced the upgrades could take the credit, or even that they were aimed specifically at preventing endoleaks, Dr. Lange said. “Nobody feels assurance that the problem has been solved.”
“I believe that the type-three endoleaks pose a challenge to patients, and I have not seen enough data to assure me with a degree of certainty that that problem no longer persists,” said panelist Joaquin E. Cigarroa, MD, a cardiologist at Oregon Health & Science University, Portland. His take on the LEOPARD trial, moreover, is that it “does not refute that there is an issue, given the duration of follow-up.”
On the other hand, a majority of the panel agreed that, currently, the AFX2’s benefits would likely outweigh risks for patients in narrowly defined high-risk anatomic or clinical scenarios and those with no other endovascular or surgical option.
“I do believe that there are patient subsets where the Endologix graft can play an important and vital role,” surgeon Keith B. Allen, MD, St. Luke’s Mid America Heart & Vascular Institute, Kansas City, Missouri, offered from the panel.
“In patients that don’t have aneurysmal disease but have distal bifurcation proximal iliac disease, it can be a very nice graft to use and solves a problem,” he said. “To remove that graft completely from the market, I believe, would deny a subset of patients.”
But for aortic aneurysms in routine practice, Dr. Allen said, “I think there are some red flags with it.”
Joining the day’s proceedings as a public commenter, surgeon Mark Conrad, MD, St. Elizabeth’s Hospital, Boston, agreed that “there’s not one commercial device out there that is able to handle every anatomy.”
Having options for patients is important, he said, because “the biggest problems we run into are when somebody only uses one graft, and they try to make that fit everything.”
Another public commenter offered a similar take. “I think we haven’t done a great job in the vascular surgery community really honing in on the detailed nuances that separate one device from another,” said Naiem Nassiri, MD, Yale New Haven Hospital Heart & Vascular Center, Connecticut.
The Endologix device, he said, “serves a very specific role under certain anatomic configurations and limitations, and really, truly fills a gap” left by other available grafts. It suits a very specific niche, “and I think it needs to be explored further for that.”
Endologix representatives who advise clinicians could play a better role in familiarizing operators with the EVAR system’s strengths and limitations, proposed several panelists, including Minhaj S. Khaja, MD, MBA, interventional radiologist at UVA Health and the University of Virginia, Charlottesville.
“There definitely needs to be more education of the clinical reps as well as the physicians implanting these devices,” he said, regarding the type III leaks, patient selection issues, appropriate imaging follow-up, “and the potential for increased reintervention.”
All public commenters, Dr. Lange observed, had been invited to disclose potential conflicts of interest, but it was not mandatory and none did so during the public forum. Disclosures of potential conflicts for the panelists are available on the FDA site.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has long kept a watchful eye over successive iterations of endovascular stent graphs in the Endologix AFX line, designed for repair of abdominal aortic aneurysms (AAA). For years, the devices, first approved in 2011, have drawn safety alerts and recalls , stemming from what the agency says was a “higher than expected” risk for potentially injurious or fatal type III endoleaks.
As part of the latest review process, Endologix recently showed regulators data from a rare randomized trial of the AAA endovascular aneurysm repair (EVAR) technique. The company said the recent postmarket study LEOPARD suggested the type III endoleaks – blood seeping around or through the device into the aneurysm – are no more common with the current AFX2 system than with other available AAA stent-grafts.
Technical upgrades to its AFX line of EVAR devices in recent years have largely resolved the safety issues identified in previous models, the company argued.
But the company’s case was unconvincing for a majority of the FDA Circulatory System Devices Advisory Panel that assembled virtually on Nov. 2. A number of panelists questioned the earnestness with which Endologix worked to rectify the safety alert and recall issues. Many also decried the real-world relevance of the randomized trial presented as evidence, with its follow-up time of only a few years.
The panel that included more than a dozen clinicians – mostly surgeons or interventional cardiologists or radiologists – were not instructed to formally vote on the issues. But it ultimately advised the FDA that more exacting studies with longer follow-ups appear needed to show that the device’s benefits in routine use outweigh its risks, especially for type III endoleaks.
“There isn’t a tremendous amount of confidence” that Endologix had enacted sufficient risk-mitigation measures in the wake of the safety alerts and recalls, chair Richard A. Lange, MD, MBA, Foster School of Medicine and Texas Tech University Health Sciences Center, El Paso, said when summarizing the panel’s take on the day’s proceedings.
Although the stent-graft’s safety seemed improved with recent design changes, the panel wasn’t convinced the upgrades could take the credit, or even that they were aimed specifically at preventing endoleaks, Dr. Lange said. “Nobody feels assurance that the problem has been solved.”
“I believe that the type-three endoleaks pose a challenge to patients, and I have not seen enough data to assure me with a degree of certainty that that problem no longer persists,” said panelist Joaquin E. Cigarroa, MD, a cardiologist at Oregon Health & Science University, Portland. His take on the LEOPARD trial, moreover, is that it “does not refute that there is an issue, given the duration of follow-up.”
On the other hand, a majority of the panel agreed that, currently, the AFX2’s benefits would likely outweigh risks for patients in narrowly defined high-risk anatomic or clinical scenarios and those with no other endovascular or surgical option.
“I do believe that there are patient subsets where the Endologix graft can play an important and vital role,” surgeon Keith B. Allen, MD, St. Luke’s Mid America Heart & Vascular Institute, Kansas City, Missouri, offered from the panel.
“In patients that don’t have aneurysmal disease but have distal bifurcation proximal iliac disease, it can be a very nice graft to use and solves a problem,” he said. “To remove that graft completely from the market, I believe, would deny a subset of patients.”
But for aortic aneurysms in routine practice, Dr. Allen said, “I think there are some red flags with it.”
Joining the day’s proceedings as a public commenter, surgeon Mark Conrad, MD, St. Elizabeth’s Hospital, Boston, agreed that “there’s not one commercial device out there that is able to handle every anatomy.”
Having options for patients is important, he said, because “the biggest problems we run into are when somebody only uses one graft, and they try to make that fit everything.”
Another public commenter offered a similar take. “I think we haven’t done a great job in the vascular surgery community really honing in on the detailed nuances that separate one device from another,” said Naiem Nassiri, MD, Yale New Haven Hospital Heart & Vascular Center, Connecticut.
The Endologix device, he said, “serves a very specific role under certain anatomic configurations and limitations, and really, truly fills a gap” left by other available grafts. It suits a very specific niche, “and I think it needs to be explored further for that.”
Endologix representatives who advise clinicians could play a better role in familiarizing operators with the EVAR system’s strengths and limitations, proposed several panelists, including Minhaj S. Khaja, MD, MBA, interventional radiologist at UVA Health and the University of Virginia, Charlottesville.
“There definitely needs to be more education of the clinical reps as well as the physicians implanting these devices,” he said, regarding the type III leaks, patient selection issues, appropriate imaging follow-up, “and the potential for increased reintervention.”
All public commenters, Dr. Lange observed, had been invited to disclose potential conflicts of interest, but it was not mandatory and none did so during the public forum. Disclosures of potential conflicts for the panelists are available on the FDA site.
A version of this article first appeared on Medscape.com.
FFR-guided PCI falls short vs. surgery in multivessel disease: FAME 3
Coronary stenting guided by fractional flow reserve (FFR) readings, considered to reflect the targeted lesion’s functional impact, was no match for coronary bypass surgery (CABG) in patients with multivessel disease (MVD) in a major international randomized trial.

Indeed, FFR-guided percutaneous coronary intervention (PCI) using one of the latest drug-eluting stents (DES) seemed to perform poorly in the trial, compared with surgery, apparently upping the risk for clinical events by 50% over 1 year.
Designed statistically for noninferiority, the third Fractional Flow Reserve Versus Angiography for Multivessel Evaluation (FAME 3) trial, with 1,500 randomized patients, showed that FFR-guided PCI was “not noninferior” to CABG. Of those randomized to PCI, 10.6% met the 1-year primary endpoint of major adverse cardiac or cerebrovascular events (MACCE), compared with only 6.9% of patients assigned to CABG.
The trial enrolled only patients with three-vessel coronary disease with no left-main coronary artery involvement, who were declared by their institution’s multidisciplinary heart team to be appropriate for either form of revascularization.
One of the roles of FFR for PCI guidance is to identify significant lesions “that are underrecognized by the angiogram,” which is less likely to happen in patients with very complex coronary anatomy, study chair William F. Fearon, MD, Stanford (Calif.) University, said in an interview.
“That’s what we saw in a subgroup analysis based on SYNTAX score,” an index of lesion complexity. “In patients with very high SYNTAX scores, CABG outperformed FFR-guided PCI. But if you look at patients with low SYNTAX scores, actually, FFR-guided PCI outperformed CABG for 1-year MACCE.”
Dr. Fearon is lead author on the study’s Nov. 4, 2021, publication in the New England Journal of Medicine, its release timed to coincide with his presentation of the trial at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
He noted that FAME-3 “wasn’t designed or powered to test for superiority,” so its results do not imply CABG is superior to FFR-PCI in patients with MVD, and remains “inconclusive” on that question.
“I think what this study does is provide both the physician and patients more contemporary data and information on options and expected outcomes in multivessel disease. So if you are a patient who has less complex disease, I think you can feel comfortable that you will get an equivalent result with FFR-guided PCI.” But, at least based on FAME-3, Dr. Fearon said, CABG provides better outcomes in patients with more complex disease.
“I think there are still patients that look at trade-offs. Some patients will accept a higher event rate in order to avoid a long recovery, and vice versa.” So the trial may allow patients and physicians to make more informed decisions, he said.
A main message of FAME-3 “is that we’re getting very good results with three-vessel PCI, but better results with surgery,” Ran Kornowski, MD, Rabin Medical Center, Petah Tikva, Israel, and Tel Aviv University, said as a discussant following Dr. Fearon’s presentation of the trial. The subanalysis by SYNTAX score, he agreed, probably could be used as part of shared decision-making with patients.
Not all that surprising
“It’s a well-designed study, with a lot of patients,” said surgeon Frank W. Sellke, MD, of Rhode Island Hospital, Miriam Hospital, and Brown University, all in Providence.
“I don’t think it’s all that surprising,” he said in an interview. “It’s very consistent with what other studies have shown, that for three-vessel disease, surgery tends to have the edge,” even when pitted against FFR-guided PCI.
Indeed, pressure-wire FFR-PCI has a spotty history, even as an alternative to standard angiography-based PCI. For example, it has performed well in registry and other cohort studies but showed no advantage in the all-comers RIPCORD-2 trial or in the setting of complete revascularization PCI for acute MI in FLOWER-MI. And it emitted an increased-mortality signal in the prematurely halted FUTURE trial.
In FAME-3, “the 1-year follow-up was the best chance for FFR-PCI to be noninferior to CABG. The CABG advantage is only going to get better with time if prior experience and pathobiology is true,” Sanjay Kaul, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
Overall, “the quality and quantity of evidence is insufficient to support FFR-guided PCI” in patients with complex coronary artery disease (CAD), he said. “I would also argue that the evidence for FFR-guided PCI for simple CAD is also not high quality.”
Dr. Kaul also blasted the claim that FFR-PCI was seen to perform better against CABG in patients with low SYNTAX scores. “In general, one cannot use a positive subgroup in a null or negative trial, as is the case with FAME-3, to ‘rescue’ the treatment intervention.” Such a positive subgroup finding, he said, “would at best be deemed hypothesis-generating and not hypothesis validating.”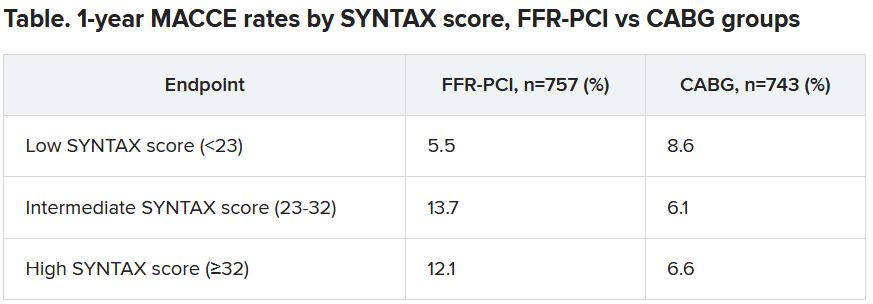
Dr. Fearon agreed that the subgroup analysis by SYNTAX score, though prespecified, was only hypothesis generating. “But I think that other studies have shown the same thing – that in less complex disease, the two strategies appear to perform in a similar fashion.”
The FAME-3 trial’s 1,500 patients were randomly assigned at 48 centers to undergo standard CABG or FFR-guided PCI with Resolute Integrity (Medtronic) zotarolimus-eluting DES. Lesions with a pressure-wire FFR of 0.80 or less were stented and those with higher FFR readings were deferred.
The 1-year hazard ratio for the primary endpoint—a composite of death from any cause, MI, stroke, or repeat revascularization – was 1.5 (95% confidence interval, 1.1-2.2) with a noninferiority P value of .35 for the comparison of FFR-PCI versus CABG.
FFR-guided PCI fared significantly better than CABG for some safety endpoints, including major bleeding (1.6% vs 3.8%, P < .01), arrhythmia including atrial fibrillation (2.4% vs. 14.1%, P < .001), acute kidney injury (0.1% vs 0.9%, P < .04), and 30-day rehospitalization (5.5% vs 10.2%, P < .001).
Did the primary endpoint favor CABG?
At a media briefing prior to Dr. Fearon’s TCT 2021 presentation of the trail, Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, proposed that the inclusion of repeat revascularization in the trial’s composite primary endpoint tilted the outcome in favor of CABG. “To me, the FAME-3 results are predictable because repeat revascularization is in the equation.”

It’s well recognized that the endpoint is less likely after CABG than PCI. The latter treats focal lesions that are a limited part of a coronary artery in which CAD is still likely progressing. CABG, on the other hand, can bypass longer segments of diseased artery.
Indeed, as Dr. Fearon reported, the rates of death, MI, or stroke excluding repeat revascularization were 7.3% with FFR-PCI and 5.2% for CABG, for an HR of 1.4 (95% CI, 0.9-2.1).
Dr. Mehran also proposed that intravascular-ultrasound (IVUS) guidance, had it been part of the trial, could potentially have boosted the performance of FFR-PCI.
Repeat revascularization, Dr. Kaul agreed, “should not have been included” in the trial’s primary endpoint. It had been added “to amplify events and to minimize sample size. Not including revascularization would render the sample size prohibitive. There is always give and take in designing clinical trials.”
And he agreed that “IVUS-based PCI optimization would have further improved PCI outcomes.” However, “IVUS plus FFR adds to the procedural burden and limited resources available.” Dr. Fearon said when interviewed that the trial’s definition of procedural MI, a component of the primary endpoint, might potentially be seen as controversial. Procedural MIs in both the PCI and CABG groups were required to meet the standards of CABG-related type-5 MI according to the third and fourth Universal Definitions. The had also had to be accompanied by “a significant finding like new Q waves or a new wall-motion abnormality on echocardiography,” he said.
“That’s fairly strict. Because of that, we had a low rate of periprocedural MI and it was similar between the two groups, around 1.5% in both arms.”
FAME-3 was funded by Medtronic and Abbott Vascular. Dr. Kaul disclosed no relevant financial relationships. Dr. Kornowsky receives royalties from or holds intellectual property rights with CathWorks. Dr. Mehran disclosed financial ties to numerous pharmaceutical and device companies, and that she, her spouse, or her institution hold equity in Elixir Medical, Applied Therapeutics, and ControlRad.
A version of this article first appeared on Medscape.com.
Coronary stenting guided by fractional flow reserve (FFR) readings, considered to reflect the targeted lesion’s functional impact, was no match for coronary bypass surgery (CABG) in patients with multivessel disease (MVD) in a major international randomized trial.
Indeed, FFR-guided percutaneous coronary intervention (PCI) using one of the latest drug-eluting stents (DES) seemed to perform poorly in the trial, compared with surgery, apparently upping the risk for clinical events by 50% over 1 year.
Designed statistically for noninferiority, the third Fractional Flow Reserve Versus Angiography for Multivessel Evaluation (FAME 3) trial, with 1,500 randomized patients, showed that FFR-guided PCI was “not noninferior” to CABG. Of those randomized to PCI, 10.6% met the 1-year primary endpoint of major adverse cardiac or cerebrovascular events (MACCE), compared with only 6.9% of patients assigned to CABG.
The trial enrolled only patients with three-vessel coronary disease with no left-main coronary artery involvement, who were declared by their institution’s multidisciplinary heart team to be appropriate for either form of revascularization.
One of the roles of FFR for PCI guidance is to identify significant lesions “that are underrecognized by the angiogram,” which is less likely to happen in patients with very complex coronary anatomy, study chair William F. Fearon, MD, Stanford (Calif.) University, said in an interview.
“That’s what we saw in a subgroup analysis based on SYNTAX score,” an index of lesion complexity. “In patients with very high SYNTAX scores, CABG outperformed FFR-guided PCI. But if you look at patients with low SYNTAX scores, actually, FFR-guided PCI outperformed CABG for 1-year MACCE.”
Dr. Fearon is lead author on the study’s Nov. 4, 2021, publication in the New England Journal of Medicine, its release timed to coincide with his presentation of the trial at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
He noted that FAME-3 “wasn’t designed or powered to test for superiority,” so its results do not imply CABG is superior to FFR-PCI in patients with MVD, and remains “inconclusive” on that question.
“I think what this study does is provide both the physician and patients more contemporary data and information on options and expected outcomes in multivessel disease. So if you are a patient who has less complex disease, I think you can feel comfortable that you will get an equivalent result with FFR-guided PCI.” But, at least based on FAME-3, Dr. Fearon said, CABG provides better outcomes in patients with more complex disease.
“I think there are still patients that look at trade-offs. Some patients will accept a higher event rate in order to avoid a long recovery, and vice versa.” So the trial may allow patients and physicians to make more informed decisions, he said.
A main message of FAME-3 “is that we’re getting very good results with three-vessel PCI, but better results with surgery,” Ran Kornowski, MD, Rabin Medical Center, Petah Tikva, Israel, and Tel Aviv University, said as a discussant following Dr. Fearon’s presentation of the trial. The subanalysis by SYNTAX score, he agreed, probably could be used as part of shared decision-making with patients.
Not all that surprising
“It’s a well-designed study, with a lot of patients,” said surgeon Frank W. Sellke, MD, of Rhode Island Hospital, Miriam Hospital, and Brown University, all in Providence.
“I don’t think it’s all that surprising,” he said in an interview. “It’s very consistent with what other studies have shown, that for three-vessel disease, surgery tends to have the edge,” even when pitted against FFR-guided PCI.
Indeed, pressure-wire FFR-PCI has a spotty history, even as an alternative to standard angiography-based PCI. For example, it has performed well in registry and other cohort studies but showed no advantage in the all-comers RIPCORD-2 trial or in the setting of complete revascularization PCI for acute MI in FLOWER-MI. And it emitted an increased-mortality signal in the prematurely halted FUTURE trial.
In FAME-3, “the 1-year follow-up was the best chance for FFR-PCI to be noninferior to CABG. The CABG advantage is only going to get better with time if prior experience and pathobiology is true,” Sanjay Kaul, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
Overall, “the quality and quantity of evidence is insufficient to support FFR-guided PCI” in patients with complex coronary artery disease (CAD), he said. “I would also argue that the evidence for FFR-guided PCI for simple CAD is also not high quality.”
Dr. Kaul also blasted the claim that FFR-PCI was seen to perform better against CABG in patients with low SYNTAX scores. “In general, one cannot use a positive subgroup in a null or negative trial, as is the case with FAME-3, to ‘rescue’ the treatment intervention.” Such a positive subgroup finding, he said, “would at best be deemed hypothesis-generating and not hypothesis validating.”
Dr. Fearon agreed that the subgroup analysis by SYNTAX score, though prespecified, was only hypothesis generating. “But I think that other studies have shown the same thing – that in less complex disease, the two strategies appear to perform in a similar fashion.”
The FAME-3 trial’s 1,500 patients were randomly assigned at 48 centers to undergo standard CABG or FFR-guided PCI with Resolute Integrity (Medtronic) zotarolimus-eluting DES. Lesions with a pressure-wire FFR of 0.80 or less were stented and those with higher FFR readings were deferred.
The 1-year hazard ratio for the primary endpoint—a composite of death from any cause, MI, stroke, or repeat revascularization – was 1.5 (95% confidence interval, 1.1-2.2) with a noninferiority P value of .35 for the comparison of FFR-PCI versus CABG.
FFR-guided PCI fared significantly better than CABG for some safety endpoints, including major bleeding (1.6% vs 3.8%, P < .01), arrhythmia including atrial fibrillation (2.4% vs. 14.1%, P < .001), acute kidney injury (0.1% vs 0.9%, P < .04), and 30-day rehospitalization (5.5% vs 10.2%, P < .001).
Did the primary endpoint favor CABG?
At a media briefing prior to Dr. Fearon’s TCT 2021 presentation of the trail, Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, proposed that the inclusion of repeat revascularization in the trial’s composite primary endpoint tilted the outcome in favor of CABG. “To me, the FAME-3 results are predictable because repeat revascularization is in the equation.”
It’s well recognized that the endpoint is less likely after CABG than PCI. The latter treats focal lesions that are a limited part of a coronary artery in which CAD is still likely progressing. CABG, on the other hand, can bypass longer segments of diseased artery.
Indeed, as Dr. Fearon reported, the rates of death, MI, or stroke excluding repeat revascularization were 7.3% with FFR-PCI and 5.2% for CABG, for an HR of 1.4 (95% CI, 0.9-2.1).
Dr. Mehran also proposed that intravascular-ultrasound (IVUS) guidance, had it been part of the trial, could potentially have boosted the performance of FFR-PCI.
Repeat revascularization, Dr. Kaul agreed, “should not have been included” in the trial’s primary endpoint. It had been added “to amplify events and to minimize sample size. Not including revascularization would render the sample size prohibitive. There is always give and take in designing clinical trials.”
And he agreed that “IVUS-based PCI optimization would have further improved PCI outcomes.” However, “IVUS plus FFR adds to the procedural burden and limited resources available.” Dr. Fearon said when interviewed that the trial’s definition of procedural MI, a component of the primary endpoint, might potentially be seen as controversial. Procedural MIs in both the PCI and CABG groups were required to meet the standards of CABG-related type-5 MI according to the third and fourth Universal Definitions. The had also had to be accompanied by “a significant finding like new Q waves or a new wall-motion abnormality on echocardiography,” he said.
“That’s fairly strict. Because of that, we had a low rate of periprocedural MI and it was similar between the two groups, around 1.5% in both arms.”
FAME-3 was funded by Medtronic and Abbott Vascular. Dr. Kaul disclosed no relevant financial relationships. Dr. Kornowsky receives royalties from or holds intellectual property rights with CathWorks. Dr. Mehran disclosed financial ties to numerous pharmaceutical and device companies, and that she, her spouse, or her institution hold equity in Elixir Medical, Applied Therapeutics, and ControlRad.
A version of this article first appeared on Medscape.com.
Coronary stenting guided by fractional flow reserve (FFR) readings, considered to reflect the targeted lesion’s functional impact, was no match for coronary bypass surgery (CABG) in patients with multivessel disease (MVD) in a major international randomized trial.
Indeed, FFR-guided percutaneous coronary intervention (PCI) using one of the latest drug-eluting stents (DES) seemed to perform poorly in the trial, compared with surgery, apparently upping the risk for clinical events by 50% over 1 year.
Designed statistically for noninferiority, the third Fractional Flow Reserve Versus Angiography for Multivessel Evaluation (FAME 3) trial, with 1,500 randomized patients, showed that FFR-guided PCI was “not noninferior” to CABG. Of those randomized to PCI, 10.6% met the 1-year primary endpoint of major adverse cardiac or cerebrovascular events (MACCE), compared with only 6.9% of patients assigned to CABG.
The trial enrolled only patients with three-vessel coronary disease with no left-main coronary artery involvement, who were declared by their institution’s multidisciplinary heart team to be appropriate for either form of revascularization.
One of the roles of FFR for PCI guidance is to identify significant lesions “that are underrecognized by the angiogram,” which is less likely to happen in patients with very complex coronary anatomy, study chair William F. Fearon, MD, Stanford (Calif.) University, said in an interview.
“That’s what we saw in a subgroup analysis based on SYNTAX score,” an index of lesion complexity. “In patients with very high SYNTAX scores, CABG outperformed FFR-guided PCI. But if you look at patients with low SYNTAX scores, actually, FFR-guided PCI outperformed CABG for 1-year MACCE.”
Dr. Fearon is lead author on the study’s Nov. 4, 2021, publication in the New England Journal of Medicine, its release timed to coincide with his presentation of the trial at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
He noted that FAME-3 “wasn’t designed or powered to test for superiority,” so its results do not imply CABG is superior to FFR-PCI in patients with MVD, and remains “inconclusive” on that question.
“I think what this study does is provide both the physician and patients more contemporary data and information on options and expected outcomes in multivessel disease. So if you are a patient who has less complex disease, I think you can feel comfortable that you will get an equivalent result with FFR-guided PCI.” But, at least based on FAME-3, Dr. Fearon said, CABG provides better outcomes in patients with more complex disease.
“I think there are still patients that look at trade-offs. Some patients will accept a higher event rate in order to avoid a long recovery, and vice versa.” So the trial may allow patients and physicians to make more informed decisions, he said.
A main message of FAME-3 “is that we’re getting very good results with three-vessel PCI, but better results with surgery,” Ran Kornowski, MD, Rabin Medical Center, Petah Tikva, Israel, and Tel Aviv University, said as a discussant following Dr. Fearon’s presentation of the trial. The subanalysis by SYNTAX score, he agreed, probably could be used as part of shared decision-making with patients.
Not all that surprising
“It’s a well-designed study, with a lot of patients,” said surgeon Frank W. Sellke, MD, of Rhode Island Hospital, Miriam Hospital, and Brown University, all in Providence.
“I don’t think it’s all that surprising,” he said in an interview. “It’s very consistent with what other studies have shown, that for three-vessel disease, surgery tends to have the edge,” even when pitted against FFR-guided PCI.
Indeed, pressure-wire FFR-PCI has a spotty history, even as an alternative to standard angiography-based PCI. For example, it has performed well in registry and other cohort studies but showed no advantage in the all-comers RIPCORD-2 trial or in the setting of complete revascularization PCI for acute MI in FLOWER-MI. And it emitted an increased-mortality signal in the prematurely halted FUTURE trial.
In FAME-3, “the 1-year follow-up was the best chance for FFR-PCI to be noninferior to CABG. The CABG advantage is only going to get better with time if prior experience and pathobiology is true,” Sanjay Kaul, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
Overall, “the quality and quantity of evidence is insufficient to support FFR-guided PCI” in patients with complex coronary artery disease (CAD), he said. “I would also argue that the evidence for FFR-guided PCI for simple CAD is also not high quality.”
Dr. Kaul also blasted the claim that FFR-PCI was seen to perform better against CABG in patients with low SYNTAX scores. “In general, one cannot use a positive subgroup in a null or negative trial, as is the case with FAME-3, to ‘rescue’ the treatment intervention.” Such a positive subgroup finding, he said, “would at best be deemed hypothesis-generating and not hypothesis validating.”
Dr. Fearon agreed that the subgroup analysis by SYNTAX score, though prespecified, was only hypothesis generating. “But I think that other studies have shown the same thing – that in less complex disease, the two strategies appear to perform in a similar fashion.”
The FAME-3 trial’s 1,500 patients were randomly assigned at 48 centers to undergo standard CABG or FFR-guided PCI with Resolute Integrity (Medtronic) zotarolimus-eluting DES. Lesions with a pressure-wire FFR of 0.80 or less were stented and those with higher FFR readings were deferred.
The 1-year hazard ratio for the primary endpoint—a composite of death from any cause, MI, stroke, or repeat revascularization – was 1.5 (95% confidence interval, 1.1-2.2) with a noninferiority P value of .35 for the comparison of FFR-PCI versus CABG.
FFR-guided PCI fared significantly better than CABG for some safety endpoints, including major bleeding (1.6% vs 3.8%, P < .01), arrhythmia including atrial fibrillation (2.4% vs. 14.1%, P < .001), acute kidney injury (0.1% vs 0.9%, P < .04), and 30-day rehospitalization (5.5% vs 10.2%, P < .001).
Did the primary endpoint favor CABG?
At a media briefing prior to Dr. Fearon’s TCT 2021 presentation of the trail, Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, proposed that the inclusion of repeat revascularization in the trial’s composite primary endpoint tilted the outcome in favor of CABG. “To me, the FAME-3 results are predictable because repeat revascularization is in the equation.”
It’s well recognized that the endpoint is less likely after CABG than PCI. The latter treats focal lesions that are a limited part of a coronary artery in which CAD is still likely progressing. CABG, on the other hand, can bypass longer segments of diseased artery.
Indeed, as Dr. Fearon reported, the rates of death, MI, or stroke excluding repeat revascularization were 7.3% with FFR-PCI and 5.2% for CABG, for an HR of 1.4 (95% CI, 0.9-2.1).
Dr. Mehran also proposed that intravascular-ultrasound (IVUS) guidance, had it been part of the trial, could potentially have boosted the performance of FFR-PCI.
Repeat revascularization, Dr. Kaul agreed, “should not have been included” in the trial’s primary endpoint. It had been added “to amplify events and to minimize sample size. Not including revascularization would render the sample size prohibitive. There is always give and take in designing clinical trials.”
And he agreed that “IVUS-based PCI optimization would have further improved PCI outcomes.” However, “IVUS plus FFR adds to the procedural burden and limited resources available.” Dr. Fearon said when interviewed that the trial’s definition of procedural MI, a component of the primary endpoint, might potentially be seen as controversial. Procedural MIs in both the PCI and CABG groups were required to meet the standards of CABG-related type-5 MI according to the third and fourth Universal Definitions. The had also had to be accompanied by “a significant finding like new Q waves or a new wall-motion abnormality on echocardiography,” he said.
“That’s fairly strict. Because of that, we had a low rate of periprocedural MI and it was similar between the two groups, around 1.5% in both arms.”
FAME-3 was funded by Medtronic and Abbott Vascular. Dr. Kaul disclosed no relevant financial relationships. Dr. Kornowsky receives royalties from or holds intellectual property rights with CathWorks. Dr. Mehran disclosed financial ties to numerous pharmaceutical and device companies, and that she, her spouse, or her institution hold equity in Elixir Medical, Applied Therapeutics, and ControlRad.
A version of this article first appeared on Medscape.com.
ASNC rejects new chest pain guideline it helped create
It was Oct. 28 when the two big North American cardiology societies issued a joint practice guideline on evaluating and managing chest pain that was endorsed by five other subspecialty groups. The next day, another group that had taken part in the document’s genesis explained why it wasn’t one of those five.
Although the American Society of Nuclear Cardiology (ASNC) was “actively engaged at every stage of the guideline-writing and review process,” the society “could not endorse the guideline,” the society announced in a statement released to clinicians and the media. The most prominent cited reason: It doesn’t adequately “support the principle of Patient First Imaging.”
The guideline was published in Circulation and the Journal of the American College of Cardiology, flagship journals of the American Heart Association and American College of Cardiology, respectively.
The document notes at least two clinicians represented ASNC as peer reviewers, and another was on the writing committee, but the organization does not appear in the list of societies endorsing the document.
“We believe that the document fails to provide unbiased guidance to health care professionals on the optimal evaluation of patients with chest pain,” contends an editorial ASNC board members have scheduled for the Jan. 10 issue of the Journal of Nuclear Medicine but is available now on an open-access preprint server.
“Despite the many important and helpful recommendations in the new guideline, there are several recommendations that we could not support,” it states.
“The ASNC board of directors reviewed the document twice during the endorsement process,” and the society “offered substantive comments after the first endorsement review, several of which were addressed,” Randall C. Thompson, MD, St. Luke’s Mid America Heart Institute and University of Missouri–Kansas City, said in an interview.
“However, some of the board’s concerns went unresolved. It was after the board’s second review, when the document had been declared finalized, that they voted not to endorse,” said Dr. Thompson, who is ASNC president.
“When we gather multiple organizations together to review and summarize the evidence, we work collaboratively to interpret the extensive catalog of peer-reviewed, published literature and create clinical practice recommendations,” Guideline Writing Committee Chair Martha Gulati, MD, University of Arizona, Phoenix, told this news organization in a prepared statement.
“The ASNC had a representative on the writing committee who is a coauthor on the paper and actively participated throughout the writing process the past 4 years,” she said. “The final guideline reflects the latest evidence-based recommendations for the evaluation and diagnosis of chest pain, as agreed by the seven endorsing organizations.”
The document does not clearly note that an ASNC representative was on the writing committee. However, ASNC confirmed that Renee Bullock-Palmer, MD, Deborah Heart and Lung Center, Browns Mills, N.J., is a fellow of the ASNC and had represented the group as one of the coauthors. Two “official reviewers” of the document, however, are listed as ASNC representatives.
Points of contention
“The decision about which test to order can be a nuanced one, and cardiac imaging tests tend to be complementary,” elaborates the editorial on the issue of patient-centered management.
Careful patient selection for different tests is important, “and physician and technical local expertise, availability, quality of equipment, and patient preference are extremely important factors to consider. There is not enough emphasis on this important point,” contend the authors. “This is an important limitation of the guideline.”
Other issues of concern include “lack of balance in the document’s presentation of the science on FFR-CT [fractional flow reserve assessment with computed tomography] and its inappropriately prominent endorsement,” the editorial states.
The U.S. Food and Drug Administration–recognized “limitations and contraindications” to FFR-CT tend to be glossed over in the document, Dr. Thompson said. And most ASNC board members were “concerned with the prominent location of the recommendations for FFR-CT in various tables – especially since there was minimal-to-no discussion of the fact that it is currently provided by only one company, that it is not widely available nor covered routinely by health insurance carriers, and [that] the accuracy in the most relevant population is disputed.”
In other concerns, the document “inadequately discusses the benefit” of combining coronary artery calcium (CAC) scores with functional testing, which ASNC said it supports. For example, adding CAC scores to myocardial perfusion imaging improves its diagnostic accuracy and prognostic power.
Functional vs. anatomic testing?
Moreover, “it is no longer appropriate to bundle all types of stress testing together. All stress imaging tests have their unique advantages and limitations.” Yet, “the concept of the dichotomy of functional testing versus anatomic testing is a common theme in the guideline in many important patient groups,” the editorial states. That could overemphasize CT angiography and thus “blur distinction between different types of functional tests.”
Such concerns about “imbalance” in the portrayals of the two kinds of tests were “amplified by the problem of health insurance companies and radiologic benefits managers inappropriately substituting a test that was ordered by a physician with a different test,” Dr. Thompson elaborated. “There is the impression that some of them ‘cherry-pick’ certain guidelines and that this practice is harmful to patients.”
The ASNC currently does not plan its own corresponding guideline, he said. But the editorial says that “over the coming weeks and months ASNC will offer a series of webinars and other programs that address specific patient populations and dilemmas.” Also, “we will enhance our focus on programs to address quality and efficiency to support a patient-first approach to imaging.”
The five subspecialty groups that have endorsed the document are the American Society of Echocardiography, American College of Chest Physicians, Society for Academic Emergency Medicine, Society of Cardiovascular Computed Tomography, and Society for Cardiovascular Magnetic Resonance.
Dr. Thompson has reported no relevant financial relationships. Statements of disclosure for the other editorial writers are listed in the publication.
A version of this article first appeared on Medscape.com.
It was Oct. 28 when the two big North American cardiology societies issued a joint practice guideline on evaluating and managing chest pain that was endorsed by five other subspecialty groups. The next day, another group that had taken part in the document’s genesis explained why it wasn’t one of those five.
Although the American Society of Nuclear Cardiology (ASNC) was “actively engaged at every stage of the guideline-writing and review process,” the society “could not endorse the guideline,” the society announced in a statement released to clinicians and the media. The most prominent cited reason: It doesn’t adequately “support the principle of Patient First Imaging.”
The guideline was published in Circulation and the Journal of the American College of Cardiology, flagship journals of the American Heart Association and American College of Cardiology, respectively.
The document notes at least two clinicians represented ASNC as peer reviewers, and another was on the writing committee, but the organization does not appear in the list of societies endorsing the document.
“We believe that the document fails to provide unbiased guidance to health care professionals on the optimal evaluation of patients with chest pain,” contends an editorial ASNC board members have scheduled for the Jan. 10 issue of the Journal of Nuclear Medicine but is available now on an open-access preprint server.
“Despite the many important and helpful recommendations in the new guideline, there are several recommendations that we could not support,” it states.
“The ASNC board of directors reviewed the document twice during the endorsement process,” and the society “offered substantive comments after the first endorsement review, several of which were addressed,” Randall C. Thompson, MD, St. Luke’s Mid America Heart Institute and University of Missouri–Kansas City, said in an interview.
“However, some of the board’s concerns went unresolved. It was after the board’s second review, when the document had been declared finalized, that they voted not to endorse,” said Dr. Thompson, who is ASNC president.
“When we gather multiple organizations together to review and summarize the evidence, we work collaboratively to interpret the extensive catalog of peer-reviewed, published literature and create clinical practice recommendations,” Guideline Writing Committee Chair Martha Gulati, MD, University of Arizona, Phoenix, told this news organization in a prepared statement.
“The ASNC had a representative on the writing committee who is a coauthor on the paper and actively participated throughout the writing process the past 4 years,” she said. “The final guideline reflects the latest evidence-based recommendations for the evaluation and diagnosis of chest pain, as agreed by the seven endorsing organizations.”
The document does not clearly note that an ASNC representative was on the writing committee. However, ASNC confirmed that Renee Bullock-Palmer, MD, Deborah Heart and Lung Center, Browns Mills, N.J., is a fellow of the ASNC and had represented the group as one of the coauthors. Two “official reviewers” of the document, however, are listed as ASNC representatives.
Points of contention
“The decision about which test to order can be a nuanced one, and cardiac imaging tests tend to be complementary,” elaborates the editorial on the issue of patient-centered management.
Careful patient selection for different tests is important, “and physician and technical local expertise, availability, quality of equipment, and patient preference are extremely important factors to consider. There is not enough emphasis on this important point,” contend the authors. “This is an important limitation of the guideline.”
Other issues of concern include “lack of balance in the document’s presentation of the science on FFR-CT [fractional flow reserve assessment with computed tomography] and its inappropriately prominent endorsement,” the editorial states.
The U.S. Food and Drug Administration–recognized “limitations and contraindications” to FFR-CT tend to be glossed over in the document, Dr. Thompson said. And most ASNC board members were “concerned with the prominent location of the recommendations for FFR-CT in various tables – especially since there was minimal-to-no discussion of the fact that it is currently provided by only one company, that it is not widely available nor covered routinely by health insurance carriers, and [that] the accuracy in the most relevant population is disputed.”
In other concerns, the document “inadequately discusses the benefit” of combining coronary artery calcium (CAC) scores with functional testing, which ASNC said it supports. For example, adding CAC scores to myocardial perfusion imaging improves its diagnostic accuracy and prognostic power.
Functional vs. anatomic testing?
Moreover, “it is no longer appropriate to bundle all types of stress testing together. All stress imaging tests have their unique advantages and limitations.” Yet, “the concept of the dichotomy of functional testing versus anatomic testing is a common theme in the guideline in many important patient groups,” the editorial states. That could overemphasize CT angiography and thus “blur distinction between different types of functional tests.”
Such concerns about “imbalance” in the portrayals of the two kinds of tests were “amplified by the problem of health insurance companies and radiologic benefits managers inappropriately substituting a test that was ordered by a physician with a different test,” Dr. Thompson elaborated. “There is the impression that some of them ‘cherry-pick’ certain guidelines and that this practice is harmful to patients.”
The ASNC currently does not plan its own corresponding guideline, he said. But the editorial says that “over the coming weeks and months ASNC will offer a series of webinars and other programs that address specific patient populations and dilemmas.” Also, “we will enhance our focus on programs to address quality and efficiency to support a patient-first approach to imaging.”
The five subspecialty groups that have endorsed the document are the American Society of Echocardiography, American College of Chest Physicians, Society for Academic Emergency Medicine, Society of Cardiovascular Computed Tomography, and Society for Cardiovascular Magnetic Resonance.
Dr. Thompson has reported no relevant financial relationships. Statements of disclosure for the other editorial writers are listed in the publication.
A version of this article first appeared on Medscape.com.
It was Oct. 28 when the two big North American cardiology societies issued a joint practice guideline on evaluating and managing chest pain that was endorsed by five other subspecialty groups. The next day, another group that had taken part in the document’s genesis explained why it wasn’t one of those five.
Although the American Society of Nuclear Cardiology (ASNC) was “actively engaged at every stage of the guideline-writing and review process,” the society “could not endorse the guideline,” the society announced in a statement released to clinicians and the media. The most prominent cited reason: It doesn’t adequately “support the principle of Patient First Imaging.”
The guideline was published in Circulation and the Journal of the American College of Cardiology, flagship journals of the American Heart Association and American College of Cardiology, respectively.
The document notes at least two clinicians represented ASNC as peer reviewers, and another was on the writing committee, but the organization does not appear in the list of societies endorsing the document.
“We believe that the document fails to provide unbiased guidance to health care professionals on the optimal evaluation of patients with chest pain,” contends an editorial ASNC board members have scheduled for the Jan. 10 issue of the Journal of Nuclear Medicine but is available now on an open-access preprint server.
“Despite the many important and helpful recommendations in the new guideline, there are several recommendations that we could not support,” it states.
“The ASNC board of directors reviewed the document twice during the endorsement process,” and the society “offered substantive comments after the first endorsement review, several of which were addressed,” Randall C. Thompson, MD, St. Luke’s Mid America Heart Institute and University of Missouri–Kansas City, said in an interview.
“However, some of the board’s concerns went unresolved. It was after the board’s second review, when the document had been declared finalized, that they voted not to endorse,” said Dr. Thompson, who is ASNC president.
“When we gather multiple organizations together to review and summarize the evidence, we work collaboratively to interpret the extensive catalog of peer-reviewed, published literature and create clinical practice recommendations,” Guideline Writing Committee Chair Martha Gulati, MD, University of Arizona, Phoenix, told this news organization in a prepared statement.
“The ASNC had a representative on the writing committee who is a coauthor on the paper and actively participated throughout the writing process the past 4 years,” she said. “The final guideline reflects the latest evidence-based recommendations for the evaluation and diagnosis of chest pain, as agreed by the seven endorsing organizations.”
The document does not clearly note that an ASNC representative was on the writing committee. However, ASNC confirmed that Renee Bullock-Palmer, MD, Deborah Heart and Lung Center, Browns Mills, N.J., is a fellow of the ASNC and had represented the group as one of the coauthors. Two “official reviewers” of the document, however, are listed as ASNC representatives.
Points of contention
“The decision about which test to order can be a nuanced one, and cardiac imaging tests tend to be complementary,” elaborates the editorial on the issue of patient-centered management.
Careful patient selection for different tests is important, “and physician and technical local expertise, availability, quality of equipment, and patient preference are extremely important factors to consider. There is not enough emphasis on this important point,” contend the authors. “This is an important limitation of the guideline.”
Other issues of concern include “lack of balance in the document’s presentation of the science on FFR-CT [fractional flow reserve assessment with computed tomography] and its inappropriately prominent endorsement,” the editorial states.
The U.S. Food and Drug Administration–recognized “limitations and contraindications” to FFR-CT tend to be glossed over in the document, Dr. Thompson said. And most ASNC board members were “concerned with the prominent location of the recommendations for FFR-CT in various tables – especially since there was minimal-to-no discussion of the fact that it is currently provided by only one company, that it is not widely available nor covered routinely by health insurance carriers, and [that] the accuracy in the most relevant population is disputed.”
In other concerns, the document “inadequately discusses the benefit” of combining coronary artery calcium (CAC) scores with functional testing, which ASNC said it supports. For example, adding CAC scores to myocardial perfusion imaging improves its diagnostic accuracy and prognostic power.
Functional vs. anatomic testing?
Moreover, “it is no longer appropriate to bundle all types of stress testing together. All stress imaging tests have their unique advantages and limitations.” Yet, “the concept of the dichotomy of functional testing versus anatomic testing is a common theme in the guideline in many important patient groups,” the editorial states. That could overemphasize CT angiography and thus “blur distinction between different types of functional tests.”
Such concerns about “imbalance” in the portrayals of the two kinds of tests were “amplified by the problem of health insurance companies and radiologic benefits managers inappropriately substituting a test that was ordered by a physician with a different test,” Dr. Thompson elaborated. “There is the impression that some of them ‘cherry-pick’ certain guidelines and that this practice is harmful to patients.”
The ASNC currently does not plan its own corresponding guideline, he said. But the editorial says that “over the coming weeks and months ASNC will offer a series of webinars and other programs that address specific patient populations and dilemmas.” Also, “we will enhance our focus on programs to address quality and efficiency to support a patient-first approach to imaging.”
The five subspecialty groups that have endorsed the document are the American Society of Echocardiography, American College of Chest Physicians, Society for Academic Emergency Medicine, Society of Cardiovascular Computed Tomography, and Society for Cardiovascular Magnetic Resonance.
Dr. Thompson has reported no relevant financial relationships. Statements of disclosure for the other editorial writers are listed in the publication.
A version of this article first appeared on Medscape.com.
New reports help nail down myocarditis risk with COVID-19 vaccine
Recent literature features new descriptions of myocarditis linked to the two available mRNA vaccines against SARS-CoV-2. They tell a story largely consistent with experience to date, and support what might be its most useful public health message: The associated myocarditis is usually mild and self-limiting, and is far less likely to occur than myocarditis or death in unvaccinated people with COVID-19.

In line with previous research, the new analyses suggest the myocarditis – with onset usually a few days to a week after injection – has an overall incidence that ranges from less than 1 to perhaps 3 per 100,000 people who received at least one of the full mRNA-vaccine regimen’s two injections. Also, as in earlier studies, the incidence climbed higher – sometimes sharply – in certain groups by age and sex, particularly in young men and older male teens.
The new studies “are confirmatory, in terms of the risk being low,” but underscore that clinicians still must be wary of myocarditis as a potential complication of the mRNA vaccines, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, told this news organization.
Dr. Bozkurt, a leading heart failure specialist and researcher, did not contribute to any of the new reports but does study the myocarditis of COVID-19 and was lead author on a recent review of the potential vaccine complication’s features and possible mechanisms.
In the new myocarditis reports, she observed, more than 90% of the cases were mild and “resolved on their own without a major adverse outcome.” Dr. Bozkurt emphasized the need for perspective regarding the risk. For example, the myocarditis associated with SARS-CoV-2 infection is not only more likely than the vaccine-related myocarditis, but it’s also usually far more severe.
Dr. Bozkurt pointed to a recent study in which the mRNA vaccines, compared with no vaccination, appeared to escalate the myocarditis risk by a factor of 3, whereas the risk for myocarditis in SARS-CoV-2 infection was increased 18 times.
In contrast, she observed, the new myocarditis cases reported this week feature a few that are novel or are at least very rare, including the case of a patient who developed cardiogenic shock and another with fulminant myocarditis who died.
The Centers for Disease Control and Prevention in May publicly described the apparent link between myocarditis and the two available mRNA vaccines against SARS-CoV-2: BNT162b2 (Pfizer-BioNTech) and mRNA-1273 (Moderna). The next month, the Food and Drug Administration added a warning about the risk to the labeling.
Less than 1 case per 100,000
Fifteen confirmed cases of myocarditis were identified among about 2.4 million members of Kaiser Permanente Southern California aged 18 or older who received at least one injection of the Pfizer or Moderna mRNA vaccines between December 2020 and July 2021, in a report published in JAMA Internal Medicine. The study counted cases up to 10 days after the first or second injection, of which there were 2 and 13, respectively.

All eight patients who received the Pfizer BNT162b2 vaccine and the eight given the Moderna mRNA-1273 vaccine were male with a median age of 25 years (interquartile range, 20-32 years).
“The main takeaway messages from our study are that the incidence of myocarditis after COVID-19 mRNA vaccinations is very low, that this condition is primarily observed in young men within a few days after the second dose, and that most patients recover quickly,” senior author Mingsum Lee, MD, PhD, Kaiser Permanente Los Angeles Medical Center, told this news organization.
“The incidence of vaccine-related myocarditis was significantly lower than rates of COVID-19 hospitalization during the same period and population area,” she added.
The group saw a per-million incidence of 0.8 and 5.8 myocarditis cases in the 10 days after first and second injections, respectively. That made for an incidence of 0.58 per 100,000, or 1 case per 172,414 fully vaccinated adults.
The group also considered a cohort of 1,577,741 unvaccinated people with a median age of 39 years (interquartile range, 28-53 years) during the same period. Of the 75 cases of myocarditis, 52% were in men, they reported.
Comparing the vaccinated and unvaccinated cohorts, they saw a 10-day vaccine-associated myocarditis incidence rate ratio of 0.38 (95% confidence interval, 0.05-1.40; P = .15) after the first dose, and 2.7 (95% CI, 1.4-4.8; P = .004) after the second dose.
In a comparison of the vaccinated group with itself using data from a 10-day period in the previous year, the corresponding myocarditis IRRs were 1.0 (P > .99) and 3.3 (P = .03), respectively.
Dr. Lee said none of the 15 patients required admission to an intensive care unit. “All patients with myocarditis responded well to treatment and felt better quickly,” she noted.
Myocarditis after an mRNA vaccine injection is rare and, Dr. Lee said emphatically, and “the benefits of the COVID-19 vaccine greatly outweigh the risks.”
Sex- and age-stratified rates
In a separate analysis of 5,442,696 people given a first dose of the Pfizer BNT162b2 vaccine and 5,125,635 given a second dose, there were 142 cases of myocarditis with onset 21 days after dose 1 and 30 days after dose 2. Of those cases, 136 were documented as “definite or probable” in an Israeli Ministry of Health database that covered up to the end of May 2021.
There were also 40 cases among vaccinated people seen after the 30-day window, which were considered not related to the vaccination, and 101 cases among unvaccinated people; of the latter, 29 had confirmed diagnoses of COVID-19.
Of the 136 people with definite or probable cases, the myocarditis was “generally mild” in 129 and usually resolved on its own, notes the report on the study, published in the New England Journal of Medicine, with lead author Dror Mevorach, MD, Hadassah-Hebrew University Medical Center, Jerusalem.
The estimated myocarditis incidence after a second such vaccine dose across the entire Israeli population, based on the current study, was about one per 26,000 males and one per 218,000 females, the group writes. Those figures compare with one case per 10,857 among “the general unvaccinated population.”
Again, the risk was concentrated among younger men and male adolescents. In an analysis limited to vaccinated people aged 16-19 years, myocarditis in the 21 days after a second mRNA injection was seen in about one of 6,637 males and one of 99,853 females, the group reported.
The standardized incidence ratio of 5.34 (95% CI, 4.48-6.40) after a second injection, across all groups, “was driven mostly by the diagnosis of myocarditis in younger male recipients.” Among that male subgroup, the ratios by age group were 13.60 (95% CI, 9.30-19.20) for 16-19 years, 8.53 (95% CI, 5.57-12.50) for 20-24 years, and 6.96 (95% CI, 4.25-10.75) for 25-29 years.
Among people who received a second injection, compared with unvaccinated people, the 30-day rate ratio was 2.35 (95% CI, 1.10-5.02). Again, the effect was concentrated in males aged 16-19 years. Among them, the myocarditis rate ratios in the 30 days after a second mRNA vaccine injection were 8.96 (95% CI, 4.50-17.83) for the 16-19 years group, 6.13 (95% CI, 3.16-11.88) for the 20-24 group, and 3.58 (95% CI, 1.82-7.01) for 25-29 years.
Most of the patients with myocarditis showed “significant clinical improvement,” with a mean hospitalization time of only 3-4 days, the report notes. Treatment consisted of nonsteroidal anti-inflammatory drugs “with or without colchicine for presumed pericardial inflammation.”
However, seven patients (4.9%) developed important complications, including left-ventricular dysfunction, ventricular arrhythmias, and heart failure. Among them was a 22-year-old patient who died of fulminant myocarditis within 24 hours of diagnosis, the group wrote.
From an Israeli health care organization
Published by the same journal as the study by Dr. Menvorach and associates, an analysis of a separate database showed largely consistent findings among patients in the largest of Israel’s four health care organizations charged by the government to administer health services.
The report, with authors led by Guy Witberg, MD, Rabin Medical Center, Petah Tikva, Israel, focused on members of the health care organization aged 16 years or older who had received at least one Pfizer mRNA vaccine dose by the end of May 2021.
The cohorts from the two separate reports surely overlap substantially, as the Ministry of Health analysis from Dr. Mevorach and colleagues derived from a nationwide database, and – as Dr. Witberg and associates wrote – the health care organization providing their data covers 52% of the Israeli population.
Of 2,558,421 vaccinated people in the analysis, of whom 94% received two doses, 54 developed confirmed myocarditis in the 42 days after the first dose. Their median age was 27 years (interquartile range, 21-35 years) and all but three (94%) were male. Of those 54 cases, 41 were considered mild and 12 intermediate in severity, and one was fulminant with the patient in cardiogenic shock, the group writes. In addition, nonsustained ventricular tachycardia and atrial fibrillation developed in 5% and 3% of cases, respectively.
The estimated myocarditis incidence in the 42 days after administration of at least one mRNA vaccine dose was 2.13 per 100,000 vaccinated people. In that group, Dr. Witberg and colleagues note, the corresponding incidences per 100,000 were 4.12 and 0.23 for males and females, respectively.
Also in the current report, incidences per 100,000 vaccinated people aged 16-29 years, by sex, included 5.49 (95% CI, 3.59-7.39) overall, and 10.69 (95% CI, 6.93-14.46) for males (the highest rate in the report).
There was only one case in a female aged 16-29 years, and two cases in females 30 years or older.
Of note, some authors of the current study are also authors on the high-profile report from Noam Barda, MD, and colleagues published in the New England Journal of Medicine, that used the same database to arrive at an mRNA-vaccine-related incidence of myocarditis of 2.7 per 100,000. Eligibility criteria and follow-up time were different in that report, as were case ascertainment criteria.
The myocarditis risk associated with the two mRNA vaccines is small compared with “the morbidity and mortality of COVID-19 infection, in which up to 28% of hospitalized patients showed signs of myocardial injury,” wrote Vinay Guduguntla, MD, University of California, San Francisco, and Mitchell H. Katz, MD, NYC Health + Hospitals, New York, in an editorial accompanying the report from Dr. Lee and associates.
“Randomized clinical trials show that COVID-19 mRNA vaccines represent a safe and effective method of preventing infection,” they stated. “The identification of rare myocarditis does not change clinical decision-making.”
Dr. Bozkurt, who is immediate past president of the Heart Failure Society of America, has disclosed consulting for Bayer and scPharmaceuticals and serving on a clinical events committee for a trial supported by Abbott Pharmaceuticals and on a data and safety monitoring board for a trial supported by Liva Nova Pharmaceuticals. Dr. Lee and the report’s other authors had no disclosures. Dr. Mevorach discloses consulting for Enlivex Therapeutics; disclosures for the other authors are available at NEJM.org. Dr. Witberg said he has no interests to disclose; disclosures for the other authors are available at NEJM.org. Dr. Guduguntla is an editorial fellow and Dr. Katz a deputy editor at JAMA Internal Medicine; neither had disclosures.
A version of this article first appeared on Medscape.com.
Recent literature features new descriptions of myocarditis linked to the two available mRNA vaccines against SARS-CoV-2. They tell a story largely consistent with experience to date, and support what might be its most useful public health message: The associated myocarditis is usually mild and self-limiting, and is far less likely to occur than myocarditis or death in unvaccinated people with COVID-19.
In line with previous research, the new analyses suggest the myocarditis – with onset usually a few days to a week after injection – has an overall incidence that ranges from less than 1 to perhaps 3 per 100,000 people who received at least one of the full mRNA-vaccine regimen’s two injections. Also, as in earlier studies, the incidence climbed higher – sometimes sharply – in certain groups by age and sex, particularly in young men and older male teens.
The new studies “are confirmatory, in terms of the risk being low,” but underscore that clinicians still must be wary of myocarditis as a potential complication of the mRNA vaccines, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, told this news organization.
Dr. Bozkurt, a leading heart failure specialist and researcher, did not contribute to any of the new reports but does study the myocarditis of COVID-19 and was lead author on a recent review of the potential vaccine complication’s features and possible mechanisms.
In the new myocarditis reports, she observed, more than 90% of the cases were mild and “resolved on their own without a major adverse outcome.” Dr. Bozkurt emphasized the need for perspective regarding the risk. For example, the myocarditis associated with SARS-CoV-2 infection is not only more likely than the vaccine-related myocarditis, but it’s also usually far more severe.
Dr. Bozkurt pointed to a recent study in which the mRNA vaccines, compared with no vaccination, appeared to escalate the myocarditis risk by a factor of 3, whereas the risk for myocarditis in SARS-CoV-2 infection was increased 18 times.
In contrast, she observed, the new myocarditis cases reported this week feature a few that are novel or are at least very rare, including the case of a patient who developed cardiogenic shock and another with fulminant myocarditis who died.
The Centers for Disease Control and Prevention in May publicly described the apparent link between myocarditis and the two available mRNA vaccines against SARS-CoV-2: BNT162b2 (Pfizer-BioNTech) and mRNA-1273 (Moderna). The next month, the Food and Drug Administration added a warning about the risk to the labeling.
Less than 1 case per 100,000
Fifteen confirmed cases of myocarditis were identified among about 2.4 million members of Kaiser Permanente Southern California aged 18 or older who received at least one injection of the Pfizer or Moderna mRNA vaccines between December 2020 and July 2021, in a report published in JAMA Internal Medicine. The study counted cases up to 10 days after the first or second injection, of which there were 2 and 13, respectively.
All eight patients who received the Pfizer BNT162b2 vaccine and the eight given the Moderna mRNA-1273 vaccine were male with a median age of 25 years (interquartile range, 20-32 years).
“The main takeaway messages from our study are that the incidence of myocarditis after COVID-19 mRNA vaccinations is very low, that this condition is primarily observed in young men within a few days after the second dose, and that most patients recover quickly,” senior author Mingsum Lee, MD, PhD, Kaiser Permanente Los Angeles Medical Center, told this news organization.
“The incidence of vaccine-related myocarditis was significantly lower than rates of COVID-19 hospitalization during the same period and population area,” she added.
The group saw a per-million incidence of 0.8 and 5.8 myocarditis cases in the 10 days after first and second injections, respectively. That made for an incidence of 0.58 per 100,000, or 1 case per 172,414 fully vaccinated adults.
The group also considered a cohort of 1,577,741 unvaccinated people with a median age of 39 years (interquartile range, 28-53 years) during the same period. Of the 75 cases of myocarditis, 52% were in men, they reported.
Comparing the vaccinated and unvaccinated cohorts, they saw a 10-day vaccine-associated myocarditis incidence rate ratio of 0.38 (95% confidence interval, 0.05-1.40; P = .15) after the first dose, and 2.7 (95% CI, 1.4-4.8; P = .004) after the second dose.
In a comparison of the vaccinated group with itself using data from a 10-day period in the previous year, the corresponding myocarditis IRRs were 1.0 (P > .99) and 3.3 (P = .03), respectively.
Dr. Lee said none of the 15 patients required admission to an intensive care unit. “All patients with myocarditis responded well to treatment and felt better quickly,” she noted.
Myocarditis after an mRNA vaccine injection is rare and, Dr. Lee said emphatically, and “the benefits of the COVID-19 vaccine greatly outweigh the risks.”
Sex- and age-stratified rates
In a separate analysis of 5,442,696 people given a first dose of the Pfizer BNT162b2 vaccine and 5,125,635 given a second dose, there were 142 cases of myocarditis with onset 21 days after dose 1 and 30 days after dose 2. Of those cases, 136 were documented as “definite or probable” in an Israeli Ministry of Health database that covered up to the end of May 2021.
There were also 40 cases among vaccinated people seen after the 30-day window, which were considered not related to the vaccination, and 101 cases among unvaccinated people; of the latter, 29 had confirmed diagnoses of COVID-19.
Of the 136 people with definite or probable cases, the myocarditis was “generally mild” in 129 and usually resolved on its own, notes the report on the study, published in the New England Journal of Medicine, with lead author Dror Mevorach, MD, Hadassah-Hebrew University Medical Center, Jerusalem.
The estimated myocarditis incidence after a second such vaccine dose across the entire Israeli population, based on the current study, was about one per 26,000 males and one per 218,000 females, the group writes. Those figures compare with one case per 10,857 among “the general unvaccinated population.”
Again, the risk was concentrated among younger men and male adolescents. In an analysis limited to vaccinated people aged 16-19 years, myocarditis in the 21 days after a second mRNA injection was seen in about one of 6,637 males and one of 99,853 females, the group reported.
The standardized incidence ratio of 5.34 (95% CI, 4.48-6.40) after a second injection, across all groups, “was driven mostly by the diagnosis of myocarditis in younger male recipients.” Among that male subgroup, the ratios by age group were 13.60 (95% CI, 9.30-19.20) for 16-19 years, 8.53 (95% CI, 5.57-12.50) for 20-24 years, and 6.96 (95% CI, 4.25-10.75) for 25-29 years.
Among people who received a second injection, compared with unvaccinated people, the 30-day rate ratio was 2.35 (95% CI, 1.10-5.02). Again, the effect was concentrated in males aged 16-19 years. Among them, the myocarditis rate ratios in the 30 days after a second mRNA vaccine injection were 8.96 (95% CI, 4.50-17.83) for the 16-19 years group, 6.13 (95% CI, 3.16-11.88) for the 20-24 group, and 3.58 (95% CI, 1.82-7.01) for 25-29 years.
Most of the patients with myocarditis showed “significant clinical improvement,” with a mean hospitalization time of only 3-4 days, the report notes. Treatment consisted of nonsteroidal anti-inflammatory drugs “with or without colchicine for presumed pericardial inflammation.”
However, seven patients (4.9%) developed important complications, including left-ventricular dysfunction, ventricular arrhythmias, and heart failure. Among them was a 22-year-old patient who died of fulminant myocarditis within 24 hours of diagnosis, the group wrote.
From an Israeli health care organization
Published by the same journal as the study by Dr. Menvorach and associates, an analysis of a separate database showed largely consistent findings among patients in the largest of Israel’s four health care organizations charged by the government to administer health services.
The report, with authors led by Guy Witberg, MD, Rabin Medical Center, Petah Tikva, Israel, focused on members of the health care organization aged 16 years or older who had received at least one Pfizer mRNA vaccine dose by the end of May 2021.
The cohorts from the two separate reports surely overlap substantially, as the Ministry of Health analysis from Dr. Mevorach and colleagues derived from a nationwide database, and – as Dr. Witberg and associates wrote – the health care organization providing their data covers 52% of the Israeli population.
Of 2,558,421 vaccinated people in the analysis, of whom 94% received two doses, 54 developed confirmed myocarditis in the 42 days after the first dose. Their median age was 27 years (interquartile range, 21-35 years) and all but three (94%) were male. Of those 54 cases, 41 were considered mild and 12 intermediate in severity, and one was fulminant with the patient in cardiogenic shock, the group writes. In addition, nonsustained ventricular tachycardia and atrial fibrillation developed in 5% and 3% of cases, respectively.
The estimated myocarditis incidence in the 42 days after administration of at least one mRNA vaccine dose was 2.13 per 100,000 vaccinated people. In that group, Dr. Witberg and colleagues note, the corresponding incidences per 100,000 were 4.12 and 0.23 for males and females, respectively.
Also in the current report, incidences per 100,000 vaccinated people aged 16-29 years, by sex, included 5.49 (95% CI, 3.59-7.39) overall, and 10.69 (95% CI, 6.93-14.46) for males (the highest rate in the report).
There was only one case in a female aged 16-29 years, and two cases in females 30 years or older.
Of note, some authors of the current study are also authors on the high-profile report from Noam Barda, MD, and colleagues published in the New England Journal of Medicine, that used the same database to arrive at an mRNA-vaccine-related incidence of myocarditis of 2.7 per 100,000. Eligibility criteria and follow-up time were different in that report, as were case ascertainment criteria.
The myocarditis risk associated with the two mRNA vaccines is small compared with “the morbidity and mortality of COVID-19 infection, in which up to 28% of hospitalized patients showed signs of myocardial injury,” wrote Vinay Guduguntla, MD, University of California, San Francisco, and Mitchell H. Katz, MD, NYC Health + Hospitals, New York, in an editorial accompanying the report from Dr. Lee and associates.
“Randomized clinical trials show that COVID-19 mRNA vaccines represent a safe and effective method of preventing infection,” they stated. “The identification of rare myocarditis does not change clinical decision-making.”
Dr. Bozkurt, who is immediate past president of the Heart Failure Society of America, has disclosed consulting for Bayer and scPharmaceuticals and serving on a clinical events committee for a trial supported by Abbott Pharmaceuticals and on a data and safety monitoring board for a trial supported by Liva Nova Pharmaceuticals. Dr. Lee and the report’s other authors had no disclosures. Dr. Mevorach discloses consulting for Enlivex Therapeutics; disclosures for the other authors are available at NEJM.org. Dr. Witberg said he has no interests to disclose; disclosures for the other authors are available at NEJM.org. Dr. Guduguntla is an editorial fellow and Dr. Katz a deputy editor at JAMA Internal Medicine; neither had disclosures.
A version of this article first appeared on Medscape.com.
Recent literature features new descriptions of myocarditis linked to the two available mRNA vaccines against SARS-CoV-2. They tell a story largely consistent with experience to date, and support what might be its most useful public health message: The associated myocarditis is usually mild and self-limiting, and is far less likely to occur than myocarditis or death in unvaccinated people with COVID-19.
In line with previous research, the new analyses suggest the myocarditis – with onset usually a few days to a week after injection – has an overall incidence that ranges from less than 1 to perhaps 3 per 100,000 people who received at least one of the full mRNA-vaccine regimen’s two injections. Also, as in earlier studies, the incidence climbed higher – sometimes sharply – in certain groups by age and sex, particularly in young men and older male teens.
The new studies “are confirmatory, in terms of the risk being low,” but underscore that clinicians still must be wary of myocarditis as a potential complication of the mRNA vaccines, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, told this news organization.
Dr. Bozkurt, a leading heart failure specialist and researcher, did not contribute to any of the new reports but does study the myocarditis of COVID-19 and was lead author on a recent review of the potential vaccine complication’s features and possible mechanisms.
In the new myocarditis reports, she observed, more than 90% of the cases were mild and “resolved on their own without a major adverse outcome.” Dr. Bozkurt emphasized the need for perspective regarding the risk. For example, the myocarditis associated with SARS-CoV-2 infection is not only more likely than the vaccine-related myocarditis, but it’s also usually far more severe.
Dr. Bozkurt pointed to a recent study in which the mRNA vaccines, compared with no vaccination, appeared to escalate the myocarditis risk by a factor of 3, whereas the risk for myocarditis in SARS-CoV-2 infection was increased 18 times.
In contrast, she observed, the new myocarditis cases reported this week feature a few that are novel or are at least very rare, including the case of a patient who developed cardiogenic shock and another with fulminant myocarditis who died.
The Centers for Disease Control and Prevention in May publicly described the apparent link between myocarditis and the two available mRNA vaccines against SARS-CoV-2: BNT162b2 (Pfizer-BioNTech) and mRNA-1273 (Moderna). The next month, the Food and Drug Administration added a warning about the risk to the labeling.
Less than 1 case per 100,000
Fifteen confirmed cases of myocarditis were identified among about 2.4 million members of Kaiser Permanente Southern California aged 18 or older who received at least one injection of the Pfizer or Moderna mRNA vaccines between December 2020 and July 2021, in a report published in JAMA Internal Medicine. The study counted cases up to 10 days after the first or second injection, of which there were 2 and 13, respectively.
All eight patients who received the Pfizer BNT162b2 vaccine and the eight given the Moderna mRNA-1273 vaccine were male with a median age of 25 years (interquartile range, 20-32 years).
“The main takeaway messages from our study are that the incidence of myocarditis after COVID-19 mRNA vaccinations is very low, that this condition is primarily observed in young men within a few days after the second dose, and that most patients recover quickly,” senior author Mingsum Lee, MD, PhD, Kaiser Permanente Los Angeles Medical Center, told this news organization.
“The incidence of vaccine-related myocarditis was significantly lower than rates of COVID-19 hospitalization during the same period and population area,” she added.
The group saw a per-million incidence of 0.8 and 5.8 myocarditis cases in the 10 days after first and second injections, respectively. That made for an incidence of 0.58 per 100,000, or 1 case per 172,414 fully vaccinated adults.
The group also considered a cohort of 1,577,741 unvaccinated people with a median age of 39 years (interquartile range, 28-53 years) during the same period. Of the 75 cases of myocarditis, 52% were in men, they reported.
Comparing the vaccinated and unvaccinated cohorts, they saw a 10-day vaccine-associated myocarditis incidence rate ratio of 0.38 (95% confidence interval, 0.05-1.40; P = .15) after the first dose, and 2.7 (95% CI, 1.4-4.8; P = .004) after the second dose.
In a comparison of the vaccinated group with itself using data from a 10-day period in the previous year, the corresponding myocarditis IRRs were 1.0 (P > .99) and 3.3 (P = .03), respectively.
Dr. Lee said none of the 15 patients required admission to an intensive care unit. “All patients with myocarditis responded well to treatment and felt better quickly,” she noted.
Myocarditis after an mRNA vaccine injection is rare and, Dr. Lee said emphatically, and “the benefits of the COVID-19 vaccine greatly outweigh the risks.”
Sex- and age-stratified rates
In a separate analysis of 5,442,696 people given a first dose of the Pfizer BNT162b2 vaccine and 5,125,635 given a second dose, there were 142 cases of myocarditis with onset 21 days after dose 1 and 30 days after dose 2. Of those cases, 136 were documented as “definite or probable” in an Israeli Ministry of Health database that covered up to the end of May 2021.
There were also 40 cases among vaccinated people seen after the 30-day window, which were considered not related to the vaccination, and 101 cases among unvaccinated people; of the latter, 29 had confirmed diagnoses of COVID-19.
Of the 136 people with definite or probable cases, the myocarditis was “generally mild” in 129 and usually resolved on its own, notes the report on the study, published in the New England Journal of Medicine, with lead author Dror Mevorach, MD, Hadassah-Hebrew University Medical Center, Jerusalem.
The estimated myocarditis incidence after a second such vaccine dose across the entire Israeli population, based on the current study, was about one per 26,000 males and one per 218,000 females, the group writes. Those figures compare with one case per 10,857 among “the general unvaccinated population.”
Again, the risk was concentrated among younger men and male adolescents. In an analysis limited to vaccinated people aged 16-19 years, myocarditis in the 21 days after a second mRNA injection was seen in about one of 6,637 males and one of 99,853 females, the group reported.
The standardized incidence ratio of 5.34 (95% CI, 4.48-6.40) after a second injection, across all groups, “was driven mostly by the diagnosis of myocarditis in younger male recipients.” Among that male subgroup, the ratios by age group were 13.60 (95% CI, 9.30-19.20) for 16-19 years, 8.53 (95% CI, 5.57-12.50) for 20-24 years, and 6.96 (95% CI, 4.25-10.75) for 25-29 years.
Among people who received a second injection, compared with unvaccinated people, the 30-day rate ratio was 2.35 (95% CI, 1.10-5.02). Again, the effect was concentrated in males aged 16-19 years. Among them, the myocarditis rate ratios in the 30 days after a second mRNA vaccine injection were 8.96 (95% CI, 4.50-17.83) for the 16-19 years group, 6.13 (95% CI, 3.16-11.88) for the 20-24 group, and 3.58 (95% CI, 1.82-7.01) for 25-29 years.
Most of the patients with myocarditis showed “significant clinical improvement,” with a mean hospitalization time of only 3-4 days, the report notes. Treatment consisted of nonsteroidal anti-inflammatory drugs “with or without colchicine for presumed pericardial inflammation.”
However, seven patients (4.9%) developed important complications, including left-ventricular dysfunction, ventricular arrhythmias, and heart failure. Among them was a 22-year-old patient who died of fulminant myocarditis within 24 hours of diagnosis, the group wrote.
From an Israeli health care organization
Published by the same journal as the study by Dr. Menvorach and associates, an analysis of a separate database showed largely consistent findings among patients in the largest of Israel’s four health care organizations charged by the government to administer health services.
The report, with authors led by Guy Witberg, MD, Rabin Medical Center, Petah Tikva, Israel, focused on members of the health care organization aged 16 years or older who had received at least one Pfizer mRNA vaccine dose by the end of May 2021.
The cohorts from the two separate reports surely overlap substantially, as the Ministry of Health analysis from Dr. Mevorach and colleagues derived from a nationwide database, and – as Dr. Witberg and associates wrote – the health care organization providing their data covers 52% of the Israeli population.
Of 2,558,421 vaccinated people in the analysis, of whom 94% received two doses, 54 developed confirmed myocarditis in the 42 days after the first dose. Their median age was 27 years (interquartile range, 21-35 years) and all but three (94%) were male. Of those 54 cases, 41 were considered mild and 12 intermediate in severity, and one was fulminant with the patient in cardiogenic shock, the group writes. In addition, nonsustained ventricular tachycardia and atrial fibrillation developed in 5% and 3% of cases, respectively.
The estimated myocarditis incidence in the 42 days after administration of at least one mRNA vaccine dose was 2.13 per 100,000 vaccinated people. In that group, Dr. Witberg and colleagues note, the corresponding incidences per 100,000 were 4.12 and 0.23 for males and females, respectively.
Also in the current report, incidences per 100,000 vaccinated people aged 16-29 years, by sex, included 5.49 (95% CI, 3.59-7.39) overall, and 10.69 (95% CI, 6.93-14.46) for males (the highest rate in the report).
There was only one case in a female aged 16-29 years, and two cases in females 30 years or older.
Of note, some authors of the current study are also authors on the high-profile report from Noam Barda, MD, and colleagues published in the New England Journal of Medicine, that used the same database to arrive at an mRNA-vaccine-related incidence of myocarditis of 2.7 per 100,000. Eligibility criteria and follow-up time were different in that report, as were case ascertainment criteria.
The myocarditis risk associated with the two mRNA vaccines is small compared with “the morbidity and mortality of COVID-19 infection, in which up to 28% of hospitalized patients showed signs of myocardial injury,” wrote Vinay Guduguntla, MD, University of California, San Francisco, and Mitchell H. Katz, MD, NYC Health + Hospitals, New York, in an editorial accompanying the report from Dr. Lee and associates.
“Randomized clinical trials show that COVID-19 mRNA vaccines represent a safe and effective method of preventing infection,” they stated. “The identification of rare myocarditis does not change clinical decision-making.”
Dr. Bozkurt, who is immediate past president of the Heart Failure Society of America, has disclosed consulting for Bayer and scPharmaceuticals and serving on a clinical events committee for a trial supported by Abbott Pharmaceuticals and on a data and safety monitoring board for a trial supported by Liva Nova Pharmaceuticals. Dr. Lee and the report’s other authors had no disclosures. Dr. Mevorach discloses consulting for Enlivex Therapeutics; disclosures for the other authors are available at NEJM.org. Dr. Witberg said he has no interests to disclose; disclosures for the other authors are available at NEJM.org. Dr. Guduguntla is an editorial fellow and Dr. Katz a deputy editor at JAMA Internal Medicine; neither had disclosures.
A version of this article first appeared on Medscape.com.
Is AFib a stroke cause or innocent bystander? The debate continues
Discovery of substantial atrial fibrillation (AFib) is usually an indication to start oral anticoagulation (OAC) for stroke prevention, but it’s far from settled whether such AFib is actually a direct cause of thromboembolic stroke. And that has implications for whether patients with occasional bouts of the arrhythmia need to be on continuous OAC.
It’s possible that some with infrequent paroxysmal AFib can get away with OAC maintained only about as long as the arrhythmia persists, and then go off the drugs, say researchers based on their study, which, they caution, would need the support of prospective trials before such a strategy could be considered.
But importantly, in their patients who had been continuously monitored by their cardiac implantable electronic devices (CIEDs) prior to experiencing a stroke, the 30-day risk of that stroke more than tripled if their AFib burden on 1 day reached at least 5-6 hours. The risk jumped especially high within the first few days after accumulating that amount of AFib in a day, but then fell off sharply over the next few days.
Based on the study, “Your risk of stroke goes up acutely when you have an episode of AFib, and it decreases rapidly, back to baseline – certainly by 30 days and it looked like in our data by 5 days,” Daniel E. Singer, MD, of Massachusetts General Hospital, Boston, said in an interview.
Increasingly, he noted, “there’s a widespread belief that AFib is a risk marker, not a causal risk factor.” In that scenario, most embolic strokes are caused by thrombi formed as a result of an atrial myopathy, characterized by fibrosis and inflammation, that also happens to trigger AFib.
But said Dr. Singer, who is lead author on the analysis published online Sept. 29 in JAMA Cardiology.
Some studies have “shown that anticoagulants seem to lower stroke risk even in patients without atrial fib, and even from sources not likely to be coming from the atrium,” Mintu P. Turakhia, MD, of Stanford (Calif.) University, Palo Alto, said in an interview. Collectively they point to “atrial fibrillation as a cause of and a noncausal marker for stroke.”
For example, Dr. Turakhia pointed out in an editorial accompanying the current report that stroke in patients with CIEDs “may occur during prolonged periods of sinus rhythm.”
The current study, he said in an interview, doesn’t preclude atrial myopathy as one direct cause of stroke-associated thrombus, because probably both the myopathy and AFib can be culprits. Still, AFib itself it may bear more responsibility for strokes in patients with fewer competing risks for stroke.
In such patients at lower vascular risk, who may have a CHA2DS2-VASc score of only 1 or 2, for example, “AFib can become a more important cause” of ischemic stroke, Dr. Turakhia said. That’s when AFib is more likely to be temporally related to stroke as the likely culprit, the mechanism addressed by Dr. Singer and associates.
“I think we’re all trying to grapple with what the truth is,” Dr. Singer observed. Still, the current study was unusual for primarily looking at the temporal relationship between AFib and stroke, rather than stroke risk. “And once again, as we found in our earlier study, but now a much larger study, it’s a tight relationship.”
Based on the current results, he said, the risk is “high when you have AFib, and it decreases very rapidly after the AFib is over.” And, “it takes multiple hours of AFib to raise stroke risk.” Inclusion in the analysis required accumulation of at least 5.5 hours of AFib on at least 1 day in a month, the cut point at which stroke risk started to climb significantly in an earlier trial.
In the current analysis, however, the 30-day odds ratio for stroke was a nonsignificant 2.75 for an AFib burden of 6-23 hours in a day and jumped to a significant 5.0 for a burden in excess of 23 hours in a day. “That’s a lot of AFib” before the risk actually goes up, and supports AFib as causative, Dr. Singer said. If it were the myopathy itself triggering stroke in these particular patients, the risk would be ongoing and not subject to a threshold of AFib burden.
Implications for noncontinuous OAC
“The hope is that there are people who have very little AFib: They may have several hours, and then they have nothing for 6 months. Do they have to be anticoagulated or not?” Dr. Singer asked.
“If you believe the risk-marker story, you might say they have to be anticoagulated. But if you believe our results, you would certainly think there’s a good chance they don’t have to be anticoagulated,” he said.
“So it is logical to think, if you have the right people and continuous monitoring, that you could have time-delimited anticoagulation.” That is, patients might start right away on a direct OAC once reaching the AFib threshold in a day, Dr. Singer said, “going on and off anticoagulants in parallel with their episodes of AFib.”
The strategy wouldn’t be feasible in patients who often experience AFib, Dr. Singer noted, “but it might work for people who have infrequent paroxysmal AFib.” It certainly would first have to be tested in prospective trials, he said. Such trials would be more practical than ever to carry out given the growing availability of continuous AFib monitoring by wearables.
“We need a trial to make the case whether it’s safe or not,” Dr. Turakhia said of such a rhythm-guided approach to OAC for AFib. The population to start with, he said, would be patients with paroxysmal AFib and low CHA2DS2-VASc scores. “If you think CHA2DS2-VASc as an integrated score of vascular risk, such patients would have a lot fewer reasons to have strokes. And if they do have a stroke, it’s more reasonable to assume that it’s likely caused by atrial fib and not just a marker.”
Importantly, such a strategy could well be safer than continuous OAC for some patients – those at the lowest vascular risk and with the most occasional AFib and lowest AFib burden “who are otherwise doing fine,” Dr. Turakhia said. In such patients on continuous OAC, he proposed, the risks of bleeding and intracranial hemorrhage could potentially exceed the expected degree of protection from ischemic events.
Discordant periods of AFib burden
Dr. Singer and his colleagues linked a national electronic health record database with Medtronic CareLink records covering 10 years to identify 891 patients who experienced an ischemic stroke preceded by at least 120 days of continuous heart-rhythm monitoring.
The patients were then categorized by their pattern of AFib, if any, within each of two prestroke periods: the most recent 30 days, which was the test period, and the preceding 91-120 days, the control period.
The analysis then excluded any patients who reached an AFib-burden threshold of at least 5.5 hours on any day during both the test and control periods, and those who did not attain that threshold in either period.
“The ones who had AFib in both periods mostly had permanent AFib, and ones that didn’t have AFib in either period mostly were in sinus rhythm,” Dr. Singer said. It was “close to 100%” in both cases.
Those exclusions left 66 patients, 7.4% of the total, who reached the AFib-burden threshold on at least 1 day during either the test or control periods, but not both. They included 52 and 14 patients, respectively, with “discordant” periods, that is, at least that burden of AFib in a day during either the test or control period, but not both.
Comparing AFib burden at test versus control periods among patients for whom the two periods were discordant yielded an OR for stroke of 3.71 (95% confidence interval, 2.06-6.70).
Stroke risk levels were not evenly spread throughout the 24-hour periods that met the AFib-burden threshold or the 30 days preceding the patients’ strokes. The OR for stroke was 5.00 (95% CI, 2.62-9.55) during days 1-5 following the day in which the AFib-burden threshold was met. And it was 5.00 (95% CI, 2.08-12.01) over 30 days if the AFib burden exceeded 23 hours on any day of the test period.
The study’s case-crossover design, in which each patient served as their own control, is one of its advantages, Dr. Singer observed. Most patient features, including CHA2DS2-VASc score and comorbidities, did not change appreciably from earliest to the latest 30-day period, which strengthens the comparison of the two because “you don’t have to worry about long-term confounding.”
Dr. Singer was supported by the Eliot B. and Edith C. Shoolman fund of the Massachusetts General Hospital. He discloses receiving grants from Boehringer Ingelheim and Bristol-Myers Squibb; personal fees from Boehringer Ingelheim, Bristol-Myers Squibb, Fitbit, Johnson & Johnson, Merck, and Pfizer; and royalties from UpToDate.
Dr. Turakhia discloses personal fees from Medtronic, Abbott, Sanofi, Pfizer, Myokardia, Johnson & Johnson, Milestone Pharmaceuticals, InCarda Therapeutics, 100Plus, Forward Pharma, and AliveCor; and grants from Bristol-Myers Squibb, the American Heart Association, Apple, and Bayer.
A version of this article first appeared on Medscape.com.
Discovery of substantial atrial fibrillation (AFib) is usually an indication to start oral anticoagulation (OAC) for stroke prevention, but it’s far from settled whether such AFib is actually a direct cause of thromboembolic stroke. And that has implications for whether patients with occasional bouts of the arrhythmia need to be on continuous OAC.
It’s possible that some with infrequent paroxysmal AFib can get away with OAC maintained only about as long as the arrhythmia persists, and then go off the drugs, say researchers based on their study, which, they caution, would need the support of prospective trials before such a strategy could be considered.
But importantly, in their patients who had been continuously monitored by their cardiac implantable electronic devices (CIEDs) prior to experiencing a stroke, the 30-day risk of that stroke more than tripled if their AFib burden on 1 day reached at least 5-6 hours. The risk jumped especially high within the first few days after accumulating that amount of AFib in a day, but then fell off sharply over the next few days.
Based on the study, “Your risk of stroke goes up acutely when you have an episode of AFib, and it decreases rapidly, back to baseline – certainly by 30 days and it looked like in our data by 5 days,” Daniel E. Singer, MD, of Massachusetts General Hospital, Boston, said in an interview.
Increasingly, he noted, “there’s a widespread belief that AFib is a risk marker, not a causal risk factor.” In that scenario, most embolic strokes are caused by thrombi formed as a result of an atrial myopathy, characterized by fibrosis and inflammation, that also happens to trigger AFib.
But said Dr. Singer, who is lead author on the analysis published online Sept. 29 in JAMA Cardiology.
Some studies have “shown that anticoagulants seem to lower stroke risk even in patients without atrial fib, and even from sources not likely to be coming from the atrium,” Mintu P. Turakhia, MD, of Stanford (Calif.) University, Palo Alto, said in an interview. Collectively they point to “atrial fibrillation as a cause of and a noncausal marker for stroke.”
For example, Dr. Turakhia pointed out in an editorial accompanying the current report that stroke in patients with CIEDs “may occur during prolonged periods of sinus rhythm.”
The current study, he said in an interview, doesn’t preclude atrial myopathy as one direct cause of stroke-associated thrombus, because probably both the myopathy and AFib can be culprits. Still, AFib itself it may bear more responsibility for strokes in patients with fewer competing risks for stroke.
In such patients at lower vascular risk, who may have a CHA2DS2-VASc score of only 1 or 2, for example, “AFib can become a more important cause” of ischemic stroke, Dr. Turakhia said. That’s when AFib is more likely to be temporally related to stroke as the likely culprit, the mechanism addressed by Dr. Singer and associates.
“I think we’re all trying to grapple with what the truth is,” Dr. Singer observed. Still, the current study was unusual for primarily looking at the temporal relationship between AFib and stroke, rather than stroke risk. “And once again, as we found in our earlier study, but now a much larger study, it’s a tight relationship.”
Based on the current results, he said, the risk is “high when you have AFib, and it decreases very rapidly after the AFib is over.” And, “it takes multiple hours of AFib to raise stroke risk.” Inclusion in the analysis required accumulation of at least 5.5 hours of AFib on at least 1 day in a month, the cut point at which stroke risk started to climb significantly in an earlier trial.
In the current analysis, however, the 30-day odds ratio for stroke was a nonsignificant 2.75 for an AFib burden of 6-23 hours in a day and jumped to a significant 5.0 for a burden in excess of 23 hours in a day. “That’s a lot of AFib” before the risk actually goes up, and supports AFib as causative, Dr. Singer said. If it were the myopathy itself triggering stroke in these particular patients, the risk would be ongoing and not subject to a threshold of AFib burden.
Implications for noncontinuous OAC
“The hope is that there are people who have very little AFib: They may have several hours, and then they have nothing for 6 months. Do they have to be anticoagulated or not?” Dr. Singer asked.
“If you believe the risk-marker story, you might say they have to be anticoagulated. But if you believe our results, you would certainly think there’s a good chance they don’t have to be anticoagulated,” he said.
“So it is logical to think, if you have the right people and continuous monitoring, that you could have time-delimited anticoagulation.” That is, patients might start right away on a direct OAC once reaching the AFib threshold in a day, Dr. Singer said, “going on and off anticoagulants in parallel with their episodes of AFib.”
The strategy wouldn’t be feasible in patients who often experience AFib, Dr. Singer noted, “but it might work for people who have infrequent paroxysmal AFib.” It certainly would first have to be tested in prospective trials, he said. Such trials would be more practical than ever to carry out given the growing availability of continuous AFib monitoring by wearables.
“We need a trial to make the case whether it’s safe or not,” Dr. Turakhia said of such a rhythm-guided approach to OAC for AFib. The population to start with, he said, would be patients with paroxysmal AFib and low CHA2DS2-VASc scores. “If you think CHA2DS2-VASc as an integrated score of vascular risk, such patients would have a lot fewer reasons to have strokes. And if they do have a stroke, it’s more reasonable to assume that it’s likely caused by atrial fib and not just a marker.”
Importantly, such a strategy could well be safer than continuous OAC for some patients – those at the lowest vascular risk and with the most occasional AFib and lowest AFib burden “who are otherwise doing fine,” Dr. Turakhia said. In such patients on continuous OAC, he proposed, the risks of bleeding and intracranial hemorrhage could potentially exceed the expected degree of protection from ischemic events.
Discordant periods of AFib burden
Dr. Singer and his colleagues linked a national electronic health record database with Medtronic CareLink records covering 10 years to identify 891 patients who experienced an ischemic stroke preceded by at least 120 days of continuous heart-rhythm monitoring.
The patients were then categorized by their pattern of AFib, if any, within each of two prestroke periods: the most recent 30 days, which was the test period, and the preceding 91-120 days, the control period.
The analysis then excluded any patients who reached an AFib-burden threshold of at least 5.5 hours on any day during both the test and control periods, and those who did not attain that threshold in either period.
“The ones who had AFib in both periods mostly had permanent AFib, and ones that didn’t have AFib in either period mostly were in sinus rhythm,” Dr. Singer said. It was “close to 100%” in both cases.
Those exclusions left 66 patients, 7.4% of the total, who reached the AFib-burden threshold on at least 1 day during either the test or control periods, but not both. They included 52 and 14 patients, respectively, with “discordant” periods, that is, at least that burden of AFib in a day during either the test or control period, but not both.
Comparing AFib burden at test versus control periods among patients for whom the two periods were discordant yielded an OR for stroke of 3.71 (95% confidence interval, 2.06-6.70).
Stroke risk levels were not evenly spread throughout the 24-hour periods that met the AFib-burden threshold or the 30 days preceding the patients’ strokes. The OR for stroke was 5.00 (95% CI, 2.62-9.55) during days 1-5 following the day in which the AFib-burden threshold was met. And it was 5.00 (95% CI, 2.08-12.01) over 30 days if the AFib burden exceeded 23 hours on any day of the test period.
The study’s case-crossover design, in which each patient served as their own control, is one of its advantages, Dr. Singer observed. Most patient features, including CHA2DS2-VASc score and comorbidities, did not change appreciably from earliest to the latest 30-day period, which strengthens the comparison of the two because “you don’t have to worry about long-term confounding.”
Dr. Singer was supported by the Eliot B. and Edith C. Shoolman fund of the Massachusetts General Hospital. He discloses receiving grants from Boehringer Ingelheim and Bristol-Myers Squibb; personal fees from Boehringer Ingelheim, Bristol-Myers Squibb, Fitbit, Johnson & Johnson, Merck, and Pfizer; and royalties from UpToDate.
Dr. Turakhia discloses personal fees from Medtronic, Abbott, Sanofi, Pfizer, Myokardia, Johnson & Johnson, Milestone Pharmaceuticals, InCarda Therapeutics, 100Plus, Forward Pharma, and AliveCor; and grants from Bristol-Myers Squibb, the American Heart Association, Apple, and Bayer.
A version of this article first appeared on Medscape.com.
Discovery of substantial atrial fibrillation (AFib) is usually an indication to start oral anticoagulation (OAC) for stroke prevention, but it’s far from settled whether such AFib is actually a direct cause of thromboembolic stroke. And that has implications for whether patients with occasional bouts of the arrhythmia need to be on continuous OAC.
It’s possible that some with infrequent paroxysmal AFib can get away with OAC maintained only about as long as the arrhythmia persists, and then go off the drugs, say researchers based on their study, which, they caution, would need the support of prospective trials before such a strategy could be considered.
But importantly, in their patients who had been continuously monitored by their cardiac implantable electronic devices (CIEDs) prior to experiencing a stroke, the 30-day risk of that stroke more than tripled if their AFib burden on 1 day reached at least 5-6 hours. The risk jumped especially high within the first few days after accumulating that amount of AFib in a day, but then fell off sharply over the next few days.
Based on the study, “Your risk of stroke goes up acutely when you have an episode of AFib, and it decreases rapidly, back to baseline – certainly by 30 days and it looked like in our data by 5 days,” Daniel E. Singer, MD, of Massachusetts General Hospital, Boston, said in an interview.
Increasingly, he noted, “there’s a widespread belief that AFib is a risk marker, not a causal risk factor.” In that scenario, most embolic strokes are caused by thrombi formed as a result of an atrial myopathy, characterized by fibrosis and inflammation, that also happens to trigger AFib.
But said Dr. Singer, who is lead author on the analysis published online Sept. 29 in JAMA Cardiology.
Some studies have “shown that anticoagulants seem to lower stroke risk even in patients without atrial fib, and even from sources not likely to be coming from the atrium,” Mintu P. Turakhia, MD, of Stanford (Calif.) University, Palo Alto, said in an interview. Collectively they point to “atrial fibrillation as a cause of and a noncausal marker for stroke.”
For example, Dr. Turakhia pointed out in an editorial accompanying the current report that stroke in patients with CIEDs “may occur during prolonged periods of sinus rhythm.”
The current study, he said in an interview, doesn’t preclude atrial myopathy as one direct cause of stroke-associated thrombus, because probably both the myopathy and AFib can be culprits. Still, AFib itself it may bear more responsibility for strokes in patients with fewer competing risks for stroke.
In such patients at lower vascular risk, who may have a CHA2DS2-VASc score of only 1 or 2, for example, “AFib can become a more important cause” of ischemic stroke, Dr. Turakhia said. That’s when AFib is more likely to be temporally related to stroke as the likely culprit, the mechanism addressed by Dr. Singer and associates.
“I think we’re all trying to grapple with what the truth is,” Dr. Singer observed. Still, the current study was unusual for primarily looking at the temporal relationship between AFib and stroke, rather than stroke risk. “And once again, as we found in our earlier study, but now a much larger study, it’s a tight relationship.”
Based on the current results, he said, the risk is “high when you have AFib, and it decreases very rapidly after the AFib is over.” And, “it takes multiple hours of AFib to raise stroke risk.” Inclusion in the analysis required accumulation of at least 5.5 hours of AFib on at least 1 day in a month, the cut point at which stroke risk started to climb significantly in an earlier trial.
In the current analysis, however, the 30-day odds ratio for stroke was a nonsignificant 2.75 for an AFib burden of 6-23 hours in a day and jumped to a significant 5.0 for a burden in excess of 23 hours in a day. “That’s a lot of AFib” before the risk actually goes up, and supports AFib as causative, Dr. Singer said. If it were the myopathy itself triggering stroke in these particular patients, the risk would be ongoing and not subject to a threshold of AFib burden.
Implications for noncontinuous OAC
“The hope is that there are people who have very little AFib: They may have several hours, and then they have nothing for 6 months. Do they have to be anticoagulated or not?” Dr. Singer asked.
“If you believe the risk-marker story, you might say they have to be anticoagulated. But if you believe our results, you would certainly think there’s a good chance they don’t have to be anticoagulated,” he said.
“So it is logical to think, if you have the right people and continuous monitoring, that you could have time-delimited anticoagulation.” That is, patients might start right away on a direct OAC once reaching the AFib threshold in a day, Dr. Singer said, “going on and off anticoagulants in parallel with their episodes of AFib.”
The strategy wouldn’t be feasible in patients who often experience AFib, Dr. Singer noted, “but it might work for people who have infrequent paroxysmal AFib.” It certainly would first have to be tested in prospective trials, he said. Such trials would be more practical than ever to carry out given the growing availability of continuous AFib monitoring by wearables.
“We need a trial to make the case whether it’s safe or not,” Dr. Turakhia said of such a rhythm-guided approach to OAC for AFib. The population to start with, he said, would be patients with paroxysmal AFib and low CHA2DS2-VASc scores. “If you think CHA2DS2-VASc as an integrated score of vascular risk, such patients would have a lot fewer reasons to have strokes. And if they do have a stroke, it’s more reasonable to assume that it’s likely caused by atrial fib and not just a marker.”
Importantly, such a strategy could well be safer than continuous OAC for some patients – those at the lowest vascular risk and with the most occasional AFib and lowest AFib burden “who are otherwise doing fine,” Dr. Turakhia said. In such patients on continuous OAC, he proposed, the risks of bleeding and intracranial hemorrhage could potentially exceed the expected degree of protection from ischemic events.
Discordant periods of AFib burden
Dr. Singer and his colleagues linked a national electronic health record database with Medtronic CareLink records covering 10 years to identify 891 patients who experienced an ischemic stroke preceded by at least 120 days of continuous heart-rhythm monitoring.
The patients were then categorized by their pattern of AFib, if any, within each of two prestroke periods: the most recent 30 days, which was the test period, and the preceding 91-120 days, the control period.
The analysis then excluded any patients who reached an AFib-burden threshold of at least 5.5 hours on any day during both the test and control periods, and those who did not attain that threshold in either period.
“The ones who had AFib in both periods mostly had permanent AFib, and ones that didn’t have AFib in either period mostly were in sinus rhythm,” Dr. Singer said. It was “close to 100%” in both cases.
Those exclusions left 66 patients, 7.4% of the total, who reached the AFib-burden threshold on at least 1 day during either the test or control periods, but not both. They included 52 and 14 patients, respectively, with “discordant” periods, that is, at least that burden of AFib in a day during either the test or control period, but not both.
Comparing AFib burden at test versus control periods among patients for whom the two periods were discordant yielded an OR for stroke of 3.71 (95% confidence interval, 2.06-6.70).
Stroke risk levels were not evenly spread throughout the 24-hour periods that met the AFib-burden threshold or the 30 days preceding the patients’ strokes. The OR for stroke was 5.00 (95% CI, 2.62-9.55) during days 1-5 following the day in which the AFib-burden threshold was met. And it was 5.00 (95% CI, 2.08-12.01) over 30 days if the AFib burden exceeded 23 hours on any day of the test period.
The study’s case-crossover design, in which each patient served as their own control, is one of its advantages, Dr. Singer observed. Most patient features, including CHA2DS2-VASc score and comorbidities, did not change appreciably from earliest to the latest 30-day period, which strengthens the comparison of the two because “you don’t have to worry about long-term confounding.”
Dr. Singer was supported by the Eliot B. and Edith C. Shoolman fund of the Massachusetts General Hospital. He discloses receiving grants from Boehringer Ingelheim and Bristol-Myers Squibb; personal fees from Boehringer Ingelheim, Bristol-Myers Squibb, Fitbit, Johnson & Johnson, Merck, and Pfizer; and royalties from UpToDate.
Dr. Turakhia discloses personal fees from Medtronic, Abbott, Sanofi, Pfizer, Myokardia, Johnson & Johnson, Milestone Pharmaceuticals, InCarda Therapeutics, 100Plus, Forward Pharma, and AliveCor; and grants from Bristol-Myers Squibb, the American Heart Association, Apple, and Bayer.
A version of this article first appeared on Medscape.com.
FDA approves Abbott’s Portico valve for TAVR
The Food and Drug Administration has approved the Portico with FlexNav (Abbott) transcatheter aortic valve replacement (TAVR) system for patients with “symptomatic, severe aortic stenosis who are at high or extreme risk for open-heart surgery,” the company has announced.

The approval indication is in line with the entry criteria of PORTICO IDE, the investigational device exemption trial from which the FDA largely made its decision.
With the self-expanding Portico valve, Abbott joins two other companies with TAVR valves on the U.S. market: Medtronic with the self-expanding Corevalve Evolut (Medtronic) line, and Edwards Lifesciences with its Sapien (Edwards Lifesciences) valves, both of which can be used in patients at low surgical risk.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the Portico with FlexNav (Abbott) transcatheter aortic valve replacement (TAVR) system for patients with “symptomatic, severe aortic stenosis who are at high or extreme risk for open-heart surgery,” the company has announced.
The approval indication is in line with the entry criteria of PORTICO IDE, the investigational device exemption trial from which the FDA largely made its decision.
With the self-expanding Portico valve, Abbott joins two other companies with TAVR valves on the U.S. market: Medtronic with the self-expanding Corevalve Evolut (Medtronic) line, and Edwards Lifesciences with its Sapien (Edwards Lifesciences) valves, both of which can be used in patients at low surgical risk.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the Portico with FlexNav (Abbott) transcatheter aortic valve replacement (TAVR) system for patients with “symptomatic, severe aortic stenosis who are at high or extreme risk for open-heart surgery,” the company has announced.
The approval indication is in line with the entry criteria of PORTICO IDE, the investigational device exemption trial from which the FDA largely made its decision.
With the self-expanding Portico valve, Abbott joins two other companies with TAVR valves on the U.S. market: Medtronic with the self-expanding Corevalve Evolut (Medtronic) line, and Edwards Lifesciences with its Sapien (Edwards Lifesciences) valves, both of which can be used in patients at low surgical risk.
A version of this article first appeared on Medscape.com.
Most muscle pain on statins not a drug effect: SAMSON in print
but by the expectation of such adverse effects, conclude researchers behind the randomized SAMSON trial, now fully published .
It’s common for patients to stop taking their statin because of muscle pain and their belief that the drug itself is to blame. That can sometimes be true, but the SAMSON trial, owing to its unusual design, makes a strong case that such symptoms are usually a nocebo effect.
That is, most statin-related muscle symptoms are likely “driven by the act of taking tablets rather than whether the tablets contain a statin,” concludes the report, which appears in the September 21 issue of the Journal of the American College of Cardiology, with lead authors James P. Howard, PhD, and Frances A. Wood, MPhil, Imperial College London.
SAMSON had been presented at the American Heart Association Scientific Sessions 2020 virtual meeting, covered at the time by this news organization, and simultaneously published in abbreviated form as correspondence in the New England Journal of Medicine.
“SAMSON suggests that the bulk of statin-related intolerable side effects arise from the taking of a tablet, not from statin therapy per se,” agrees an editorial accompanying the new publication.
“The study also demonstrates that the informal experimentation of stopping and restarting a statin to evaluate symptom resolution and reinduction without use of a placebo leads to nocebo symptoms misattributed to the statin,” writes Peter P. Toth, MD, PhD, Johns Hopkins University, Baltimore.
Statin intolerance, he continues, “warrants considerable further investigation, because it undermines standard of care for a very large number of patients worldwide,” leaving them vulnerable to atherosclerotic cardiovascular disease events. “Aches and pains are a fact of life; just because a patient has them does not mean they should be attributed to their statin.”
SAMSON assigned 35 men and 25 women to take atorvastatin 20 mg/day, its matching placebo, or neither pill each for 1 month in randomly alternating order for 12 months, with double-blinding, such that each of the three regimens was maintained for a total of 4 months.
The patients, 77% of whom were prescribed statins for primary prevention and all of whom had a history of stopping the drugs because of adverse effects, documented the severity of any perceived adverse effects on a smartphone app, with a “symptom score” ranging from 0 to 100.
The symptom score averaged 8.0 in months when no tablet was taken, but it was much higher in other months: 15.4 in placebo-pill months and 16.3 in months when atorvastatin was taken. The no-tablet score was significantly lower (P < .001) than either of the two other scores, which themselves were not significantly different from each other.
Eleven patients were unable to complete all 12 one-month segments of the trial, including five because of severe symptoms, but discontinuation was no more likely to occur in the atorvastatin group than in the placebo group.
The authors calculated an overall 0.90 “nocebo ratio” for the study, defined as the difference between symptom intensity on placebo and on no pill, divided by the difference between symptom intensity on atorvastatin and on no pill.
That means, the authors propose, that 90% of the symptom burden felt by patients receiving atorvastatin was also felt on the placebo pill and could be attributed to the nocebo effect.
“Prompt onset and offset of symptoms after starting and stopping tablets is often interpreted by patients and clinicians as evidence of causation. Our data indicate that this is true,” the authors write, but “the causation is from taking a tablet, rather than from the tablet being a statin.”
SAMSON was funded by the British Heart Foundation and supported by the National Institute for Health Research Imperial Biomedical Research Centre and the Imperial Clinical Trials Unit. Dr. Howard is supported by the Wellcome Trust. Dr. Wood declared no conflicts. Disclosures for the other authors are in the report. Dr. Toth discloses serving as a consultant to Amarin, Amgen, AstraZeneca, nio89, Kowa, Merck, Resverlogix, and Theravance; and serving on a speaker’s bureau for Amarin, Amgen, Esperion, and NovoNordisk.
A version of this article first appeared on Medscape.com.
but by the expectation of such adverse effects, conclude researchers behind the randomized SAMSON trial, now fully published .
It’s common for patients to stop taking their statin because of muscle pain and their belief that the drug itself is to blame. That can sometimes be true, but the SAMSON trial, owing to its unusual design, makes a strong case that such symptoms are usually a nocebo effect.
That is, most statin-related muscle symptoms are likely “driven by the act of taking tablets rather than whether the tablets contain a statin,” concludes the report, which appears in the September 21 issue of the Journal of the American College of Cardiology, with lead authors James P. Howard, PhD, and Frances A. Wood, MPhil, Imperial College London.
SAMSON had been presented at the American Heart Association Scientific Sessions 2020 virtual meeting, covered at the time by this news organization, and simultaneously published in abbreviated form as correspondence in the New England Journal of Medicine.
“SAMSON suggests that the bulk of statin-related intolerable side effects arise from the taking of a tablet, not from statin therapy per se,” agrees an editorial accompanying the new publication.
“The study also demonstrates that the informal experimentation of stopping and restarting a statin to evaluate symptom resolution and reinduction without use of a placebo leads to nocebo symptoms misattributed to the statin,” writes Peter P. Toth, MD, PhD, Johns Hopkins University, Baltimore.
Statin intolerance, he continues, “warrants considerable further investigation, because it undermines standard of care for a very large number of patients worldwide,” leaving them vulnerable to atherosclerotic cardiovascular disease events. “Aches and pains are a fact of life; just because a patient has them does not mean they should be attributed to their statin.”
SAMSON assigned 35 men and 25 women to take atorvastatin 20 mg/day, its matching placebo, or neither pill each for 1 month in randomly alternating order for 12 months, with double-blinding, such that each of the three regimens was maintained for a total of 4 months.
The patients, 77% of whom were prescribed statins for primary prevention and all of whom had a history of stopping the drugs because of adverse effects, documented the severity of any perceived adverse effects on a smartphone app, with a “symptom score” ranging from 0 to 100.
The symptom score averaged 8.0 in months when no tablet was taken, but it was much higher in other months: 15.4 in placebo-pill months and 16.3 in months when atorvastatin was taken. The no-tablet score was significantly lower (P < .001) than either of the two other scores, which themselves were not significantly different from each other.
Eleven patients were unable to complete all 12 one-month segments of the trial, including five because of severe symptoms, but discontinuation was no more likely to occur in the atorvastatin group than in the placebo group.
The authors calculated an overall 0.90 “nocebo ratio” for the study, defined as the difference between symptom intensity on placebo and on no pill, divided by the difference between symptom intensity on atorvastatin and on no pill.
That means, the authors propose, that 90% of the symptom burden felt by patients receiving atorvastatin was also felt on the placebo pill and could be attributed to the nocebo effect.
“Prompt onset and offset of symptoms after starting and stopping tablets is often interpreted by patients and clinicians as evidence of causation. Our data indicate that this is true,” the authors write, but “the causation is from taking a tablet, rather than from the tablet being a statin.”
SAMSON was funded by the British Heart Foundation and supported by the National Institute for Health Research Imperial Biomedical Research Centre and the Imperial Clinical Trials Unit. Dr. Howard is supported by the Wellcome Trust. Dr. Wood declared no conflicts. Disclosures for the other authors are in the report. Dr. Toth discloses serving as a consultant to Amarin, Amgen, AstraZeneca, nio89, Kowa, Merck, Resverlogix, and Theravance; and serving on a speaker’s bureau for Amarin, Amgen, Esperion, and NovoNordisk.
A version of this article first appeared on Medscape.com.
but by the expectation of such adverse effects, conclude researchers behind the randomized SAMSON trial, now fully published .
It’s common for patients to stop taking their statin because of muscle pain and their belief that the drug itself is to blame. That can sometimes be true, but the SAMSON trial, owing to its unusual design, makes a strong case that such symptoms are usually a nocebo effect.
That is, most statin-related muscle symptoms are likely “driven by the act of taking tablets rather than whether the tablets contain a statin,” concludes the report, which appears in the September 21 issue of the Journal of the American College of Cardiology, with lead authors James P. Howard, PhD, and Frances A. Wood, MPhil, Imperial College London.
SAMSON had been presented at the American Heart Association Scientific Sessions 2020 virtual meeting, covered at the time by this news organization, and simultaneously published in abbreviated form as correspondence in the New England Journal of Medicine.
“SAMSON suggests that the bulk of statin-related intolerable side effects arise from the taking of a tablet, not from statin therapy per se,” agrees an editorial accompanying the new publication.
“The study also demonstrates that the informal experimentation of stopping and restarting a statin to evaluate symptom resolution and reinduction without use of a placebo leads to nocebo symptoms misattributed to the statin,” writes Peter P. Toth, MD, PhD, Johns Hopkins University, Baltimore.
Statin intolerance, he continues, “warrants considerable further investigation, because it undermines standard of care for a very large number of patients worldwide,” leaving them vulnerable to atherosclerotic cardiovascular disease events. “Aches and pains are a fact of life; just because a patient has them does not mean they should be attributed to their statin.”
SAMSON assigned 35 men and 25 women to take atorvastatin 20 mg/day, its matching placebo, or neither pill each for 1 month in randomly alternating order for 12 months, with double-blinding, such that each of the three regimens was maintained for a total of 4 months.
The patients, 77% of whom were prescribed statins for primary prevention and all of whom had a history of stopping the drugs because of adverse effects, documented the severity of any perceived adverse effects on a smartphone app, with a “symptom score” ranging from 0 to 100.
The symptom score averaged 8.0 in months when no tablet was taken, but it was much higher in other months: 15.4 in placebo-pill months and 16.3 in months when atorvastatin was taken. The no-tablet score was significantly lower (P < .001) than either of the two other scores, which themselves were not significantly different from each other.
Eleven patients were unable to complete all 12 one-month segments of the trial, including five because of severe symptoms, but discontinuation was no more likely to occur in the atorvastatin group than in the placebo group.
The authors calculated an overall 0.90 “nocebo ratio” for the study, defined as the difference between symptom intensity on placebo and on no pill, divided by the difference between symptom intensity on atorvastatin and on no pill.
That means, the authors propose, that 90% of the symptom burden felt by patients receiving atorvastatin was also felt on the placebo pill and could be attributed to the nocebo effect.
“Prompt onset and offset of symptoms after starting and stopping tablets is often interpreted by patients and clinicians as evidence of causation. Our data indicate that this is true,” the authors write, but “the causation is from taking a tablet, rather than from the tablet being a statin.”
SAMSON was funded by the British Heart Foundation and supported by the National Institute for Health Research Imperial Biomedical Research Centre and the Imperial Clinical Trials Unit. Dr. Howard is supported by the Wellcome Trust. Dr. Wood declared no conflicts. Disclosures for the other authors are in the report. Dr. Toth discloses serving as a consultant to Amarin, Amgen, AstraZeneca, nio89, Kowa, Merck, Resverlogix, and Theravance; and serving on a speaker’s bureau for Amarin, Amgen, Esperion, and NovoNordisk.
A version of this article first appeared on Medscape.com.
Are ESC’s new heart failure guidelines already outdated?
The new guideline on management of heart failure (HF) from the European Society of Cardiology seemed to bear an asterisk or footnote even before its full unveiling in the early hours of ESC Congress 2021.

The document would offer little new in the arena of HF with preserved ejection fraction (HFpEF), so understandably the fast-approaching presentation of a major HFpEF trial – arguably the conference’s marquee event – would feel to some like the elephant in the room.
“I’d like to highlight this unfortunate timing of the guideline, because it’s an hour or 2 before we hear the full story from EMPEROR-Preserved, which I’m sure will change the guidelines,” Faiez Zannad, MD, PhD, University of Lorraine, Vandoeuvre-Les-Nancy, France, said wryly.
Anticipation of the trial’s full presentation was intense as the ESC congress got underway, in part because the top-line and incomplete message from EMPEROR-Preserved had already been released: Patients with HFpEF treated with the sodium-glucose cotransporter 2 inhibitor empagliflozin (Jardiance, Boehringer Ingelheim/Eli Lilly) showed a significant benefit for the primary endpoint of cardiovascular (CV) death or HF hospitalization.
Although empagliflozin is the first medication to achieve that status in a major HFpEF trial, conspicuously absent from the early announcement were the magnitude of “benefit” and any data. Still, the tantalizing top-line results mean that technically, at least, “we have a drug which is effective in reduced and preserved ejection fraction,” Dr. Zannad said.
But the new guideline, published online Aug. 27, 2021, in the European Heart Journal and comprehensively described that day at the congress, was never really expected to consider results from EMPEROR-Reduced. “These new indications do need to go through the regulatory authorities,” such as the European Medicines Agency and the U.S. Food and Drug Administration, observed Carlos Aguiar, MD, Hospital Santa Cruz, Carnaxide, Portugal.
“It does take some time for the whole process to be concluded and, finally, as physicians, being able to implement it in clinical practice,” Dr. Aguiar said as moderator of press briefing prior to the ESC congress.
The ESC guideline’s next iteration or update could well include an SGLT2 inhibitor recommendation that applies beyond the ejection fraction limits of HFrEF. Still, the document summarized that day reflects a number of pivotal concepts with profound treatment implications. Among them are the field’s latest paradigm for medical therapy of HFrEF and the increasingly accepted division of traditional HFpEF into two entities: HF with mildly reduced ejection fraction (HFmrEF); and HFpEF, with its left ventricular ejection fraction (LVEF) threshold raised to 50%.
In fact, HFmrEF in the new document is a drug-therapy indication that barely existed a few years ago but grew in prominence after secondary findings from trials like TOPCAT for spironolactone and PARAGON-HF for sacubitril-valsartan (Entresto, Novartis), an angiotensin-receptor/neprilysin inhibitor (ARNI). Still, the HFmrEF recommendations come with different class and level-of-evidence designations.
Those new guideline features and others in the realm of pharmacologic therapy were summarized by the document’s authors at the 2021 Heart Failure Association of the European Society of Cardiology (ESC-HFA) meeting, and covered at the time by this news organization
The ‘fantastic four’
One of the document’s central recommendations specifies which contemporary drug classes should be initiated, and when, in patients with HFrEF. An ACE inhibitor or ARNI, a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and an SGLT2 inhibitor collectively earned a class I recommendation, “given the importance of these key HFrEF therapies, some of which have been shown to improve outcomes within a month of initiation,” observed Roy S. Gardner, MBChB, MD.
An agent from each of the four classes is to be “commenced and up-titrated as quickly and as safely as possible, whilst using the lowest effective dose of loop diuretic to relieve congestion,” said Dr. Gardner, from Golden Jubilee National Hospital, Clydebank, Scotland, when presenting the full HFrEF portion of the guidelines.
The oral soluble guanylate-cyclase receptor stimulator vericiguat (Verquvo, Merck), which recently emerged from the VICTORIA trial as a modest success for patients with HFrEF and a previous HF hospitalization, gained a class IIb recommendation.
The document’s “simplified algorithm” for managing such patients overall and the advent of SGLT2 inhibitors are new twists in ESC guidelines for HF. But the way the four drug classes are started in patients is key and could take some practitioners time to get used to. There is no prespecified order of initiation.
“We’ve left the door open for clinicians to evaluate the evidence to make sure these four drugs are started, and to tailor how to do it according to the patient,” based on clinical considerations such as blood pressure or renal function, said Theresa A. McDonagh, MD, King’s College London, cochair of the guideline task force.
“The SGLT2 inhibitor trials were done on top of therapy with ACE inhibitors or ARNI, beta-blockers, and MRAs, so some people no doubt will choose to follow a sequenced approach,” Dr. McDonagh said. Other practitioners will consider each patient and attempt to get all four started “as quickly and safely as possible based on the phenotype.”
Importantly, clinicians “should not wait for weeks, months, or years until you have the four drugs in the patient, but you should do this within weeks,” cautioned Johann Bauersachs, MD, Hannover (Germany) Medical School, a discussant for the guideline presentation who is listed as a reviewer on the document.
Although angiotensin-receptor blockers (ARBs) and ACE inhibitors are sometimes thought of as interchangeable, the new guideline does not give them the same weight. “The angiotensin-receptor blocker valsartan is a constituent of the ARNI,” Dr. McDonagh noted. “So, the place of ARBs in heart failure has been downgraded in HFrEF. They are really for those who are intolerant of an ACE inhibitor or an ARNI.”
In practice, ARBs are likely to be used as first-line therapy in some circumstances, observed Dr. Bauersachs. They are “the default option in, unfortunately, many low-income countries that may not afford sacubitril-valsartan. And I know that there are many of them.”
Tweaks to device recommendations
The new document contains several new wrinkles in the recommendations for HF device therapy, which should usually be considered only if still appropriate after at least 3 months of optimal medical therapy, Dr. Gardner said.
For example, use of an implantable cardioverter-defibrillator (ICD) has been demoted from its previous class I recommendation to class II, level of evidence A, in patients with nonischemic cardiomyopathy “in light of the data from the DANISH study,” Dr. Gardner said.
The 2016 DANISH trial was noteworthy for questioning the survival benefits of ICDs in patients with nonischemic cardiomyopathy, whether or not they were also receiving cardiac resynchronization therapy (CRT).
The new document also puts greater emphasis on a range of specific CRT patient-selection criteria. Beyond the conventional recommended standards of an LVEF of 35% or less, QRS of at least 150 ms, and left-bundle-branch block on optimal meds, consideration can be given to CRT if the QRS is only 130 ms or greater. “And where it’s appropriate to do so, an ICD could be an option,” Dr. Gardner said.
It also recommends CRT as a replacement for right ventricular pacing in patients with high-degree atrioventricular block. “And this, for the first time, includes patients with atrial fibrillation,” he said. “The previous indications for CRT were in individuals in sinus rhythm.”
The new document recommends that HF in any patient be classified as HFrEF, defined by an LVEF of ≤40%; HFmrEF, defined by an LVEF of 41%-49%; or HFpEF, defined by an LVEF of at least 50%. “Importantly, for all forms, the presence of the clinical syndrome of heart failure is a prerequisite,” observed Carolyn S.P. Lam, MBBS, PhD, Duke-NUS Graduate Medical School, Singapore, at the presentation.
In a critical update from previous guidelines, the term HF with “mid-range” ejection fraction was replaced by the term specifying “mildly reduced” ejection fraction, Dr. Lam noted. The shift retains the acronym but now reflects growing appreciation that HFmrEF patients can benefit from treatments also used in HFrEF, including ACE inhibitors, ARBs, beta-blockers, MRAs, and sacubitril-valsartan, she said.
Support for that relationship comes largely from post hoc subgroup analyses of trials that featured some patients with LVEF 40%-49%. That includes most HFpEF trials represented in the guideline document, but also EMPEROR-Preserved, which saw gains for the primary outcome across the entire range of LVEF above 40%.
The LVEF-based definitions are consistent with a recent HF classification proposal endorsed by the ESC and subspecialty societies in Europe, North America, Japan, India, Australia, New Zealand, and China.
The document doesn’t update recommendations for HFpEF, in which “no treatment has been shown to convincingly reduce mortality or morbidity,” Dr. Lam observed. Still, she noted, the guideline task force “acknowledges that treatment options for HFpEF are being revised even as the guidelines have been published.”
That could be a reference to empagliflozin in EMPEROR-Preserved, but it also refers to the strikingly broad wording of an expanded indication for sacubitril-valsartan in the United States – “to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure” – without specific restrictions on the basis of LVEF. The new indication was announced in early 2021, too late to be considered in the new guidelines.
Whither LVEF-based definitions?
During discussion after the guideline presentation, Dr. Zannad speculated on the future of HF classifications based on ventricular function, given trial evidence in recent years that some agents – notably spironolactone, sacubitril-valsartan, and now, apparently, empagliflozin – might be effective in HFpEF as well as HFrEF.
Will the field continue with “LVEF-centric” distinctions across the range of HF, or transition to “some definition in which drug therapies can be used independently across the full spectrum of ejection fraction?” Dr. Zannad posed.
“I think we need to wait and see what some of these trials with the SGLT2 inhibitors are going to show in heart failure with preserved ejection fraction,” Dr. McDonagh replied. “And I think that will be a step for the next guideline, completely redefining heart failure.”
A version of this article first appeared on Medscape.com.
The new guideline on management of heart failure (HF) from the European Society of Cardiology seemed to bear an asterisk or footnote even before its full unveiling in the early hours of ESC Congress 2021.
The document would offer little new in the arena of HF with preserved ejection fraction (HFpEF), so understandably the fast-approaching presentation of a major HFpEF trial – arguably the conference’s marquee event – would feel to some like the elephant in the room.
“I’d like to highlight this unfortunate timing of the guideline, because it’s an hour or 2 before we hear the full story from EMPEROR-Preserved, which I’m sure will change the guidelines,” Faiez Zannad, MD, PhD, University of Lorraine, Vandoeuvre-Les-Nancy, France, said wryly.
Anticipation of the trial’s full presentation was intense as the ESC congress got underway, in part because the top-line and incomplete message from EMPEROR-Preserved had already been released: Patients with HFpEF treated with the sodium-glucose cotransporter 2 inhibitor empagliflozin (Jardiance, Boehringer Ingelheim/Eli Lilly) showed a significant benefit for the primary endpoint of cardiovascular (CV) death or HF hospitalization.
Although empagliflozin is the first medication to achieve that status in a major HFpEF trial, conspicuously absent from the early announcement were the magnitude of “benefit” and any data. Still, the tantalizing top-line results mean that technically, at least, “we have a drug which is effective in reduced and preserved ejection fraction,” Dr. Zannad said.
But the new guideline, published online Aug. 27, 2021, in the European Heart Journal and comprehensively described that day at the congress, was never really expected to consider results from EMPEROR-Reduced. “These new indications do need to go through the regulatory authorities,” such as the European Medicines Agency and the U.S. Food and Drug Administration, observed Carlos Aguiar, MD, Hospital Santa Cruz, Carnaxide, Portugal.
“It does take some time for the whole process to be concluded and, finally, as physicians, being able to implement it in clinical practice,” Dr. Aguiar said as moderator of press briefing prior to the ESC congress.
The ESC guideline’s next iteration or update could well include an SGLT2 inhibitor recommendation that applies beyond the ejection fraction limits of HFrEF. Still, the document summarized that day reflects a number of pivotal concepts with profound treatment implications. Among them are the field’s latest paradigm for medical therapy of HFrEF and the increasingly accepted division of traditional HFpEF into two entities: HF with mildly reduced ejection fraction (HFmrEF); and HFpEF, with its left ventricular ejection fraction (LVEF) threshold raised to 50%.
In fact, HFmrEF in the new document is a drug-therapy indication that barely existed a few years ago but grew in prominence after secondary findings from trials like TOPCAT for spironolactone and PARAGON-HF for sacubitril-valsartan (Entresto, Novartis), an angiotensin-receptor/neprilysin inhibitor (ARNI). Still, the HFmrEF recommendations come with different class and level-of-evidence designations.
Those new guideline features and others in the realm of pharmacologic therapy were summarized by the document’s authors at the 2021 Heart Failure Association of the European Society of Cardiology (ESC-HFA) meeting, and covered at the time by this news organization
The ‘fantastic four’
One of the document’s central recommendations specifies which contemporary drug classes should be initiated, and when, in patients with HFrEF. An ACE inhibitor or ARNI, a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and an SGLT2 inhibitor collectively earned a class I recommendation, “given the importance of these key HFrEF therapies, some of which have been shown to improve outcomes within a month of initiation,” observed Roy S. Gardner, MBChB, MD.
An agent from each of the four classes is to be “commenced and up-titrated as quickly and as safely as possible, whilst using the lowest effective dose of loop diuretic to relieve congestion,” said Dr. Gardner, from Golden Jubilee National Hospital, Clydebank, Scotland, when presenting the full HFrEF portion of the guidelines.
The oral soluble guanylate-cyclase receptor stimulator vericiguat (Verquvo, Merck), which recently emerged from the VICTORIA trial as a modest success for patients with HFrEF and a previous HF hospitalization, gained a class IIb recommendation.
The document’s “simplified algorithm” for managing such patients overall and the advent of SGLT2 inhibitors are new twists in ESC guidelines for HF. But the way the four drug classes are started in patients is key and could take some practitioners time to get used to. There is no prespecified order of initiation.
“We’ve left the door open for clinicians to evaluate the evidence to make sure these four drugs are started, and to tailor how to do it according to the patient,” based on clinical considerations such as blood pressure or renal function, said Theresa A. McDonagh, MD, King’s College London, cochair of the guideline task force.
“The SGLT2 inhibitor trials were done on top of therapy with ACE inhibitors or ARNI, beta-blockers, and MRAs, so some people no doubt will choose to follow a sequenced approach,” Dr. McDonagh said. Other practitioners will consider each patient and attempt to get all four started “as quickly and safely as possible based on the phenotype.”
Importantly, clinicians “should not wait for weeks, months, or years until you have the four drugs in the patient, but you should do this within weeks,” cautioned Johann Bauersachs, MD, Hannover (Germany) Medical School, a discussant for the guideline presentation who is listed as a reviewer on the document.
Although angiotensin-receptor blockers (ARBs) and ACE inhibitors are sometimes thought of as interchangeable, the new guideline does not give them the same weight. “The angiotensin-receptor blocker valsartan is a constituent of the ARNI,” Dr. McDonagh noted. “So, the place of ARBs in heart failure has been downgraded in HFrEF. They are really for those who are intolerant of an ACE inhibitor or an ARNI.”
In practice, ARBs are likely to be used as first-line therapy in some circumstances, observed Dr. Bauersachs. They are “the default option in, unfortunately, many low-income countries that may not afford sacubitril-valsartan. And I know that there are many of them.”
Tweaks to device recommendations
The new document contains several new wrinkles in the recommendations for HF device therapy, which should usually be considered only if still appropriate after at least 3 months of optimal medical therapy, Dr. Gardner said.
For example, use of an implantable cardioverter-defibrillator (ICD) has been demoted from its previous class I recommendation to class II, level of evidence A, in patients with nonischemic cardiomyopathy “in light of the data from the DANISH study,” Dr. Gardner said.
The 2016 DANISH trial was noteworthy for questioning the survival benefits of ICDs in patients with nonischemic cardiomyopathy, whether or not they were also receiving cardiac resynchronization therapy (CRT).
The new document also puts greater emphasis on a range of specific CRT patient-selection criteria. Beyond the conventional recommended standards of an LVEF of 35% or less, QRS of at least 150 ms, and left-bundle-branch block on optimal meds, consideration can be given to CRT if the QRS is only 130 ms or greater. “And where it’s appropriate to do so, an ICD could be an option,” Dr. Gardner said.
It also recommends CRT as a replacement for right ventricular pacing in patients with high-degree atrioventricular block. “And this, for the first time, includes patients with atrial fibrillation,” he said. “The previous indications for CRT were in individuals in sinus rhythm.”
The new document recommends that HF in any patient be classified as HFrEF, defined by an LVEF of ≤40%; HFmrEF, defined by an LVEF of 41%-49%; or HFpEF, defined by an LVEF of at least 50%. “Importantly, for all forms, the presence of the clinical syndrome of heart failure is a prerequisite,” observed Carolyn S.P. Lam, MBBS, PhD, Duke-NUS Graduate Medical School, Singapore, at the presentation.
In a critical update from previous guidelines, the term HF with “mid-range” ejection fraction was replaced by the term specifying “mildly reduced” ejection fraction, Dr. Lam noted. The shift retains the acronym but now reflects growing appreciation that HFmrEF patients can benefit from treatments also used in HFrEF, including ACE inhibitors, ARBs, beta-blockers, MRAs, and sacubitril-valsartan, she said.
Support for that relationship comes largely from post hoc subgroup analyses of trials that featured some patients with LVEF 40%-49%. That includes most HFpEF trials represented in the guideline document, but also EMPEROR-Preserved, which saw gains for the primary outcome across the entire range of LVEF above 40%.
The LVEF-based definitions are consistent with a recent HF classification proposal endorsed by the ESC and subspecialty societies in Europe, North America, Japan, India, Australia, New Zealand, and China.
The document doesn’t update recommendations for HFpEF, in which “no treatment has been shown to convincingly reduce mortality or morbidity,” Dr. Lam observed. Still, she noted, the guideline task force “acknowledges that treatment options for HFpEF are being revised even as the guidelines have been published.”
That could be a reference to empagliflozin in EMPEROR-Preserved, but it also refers to the strikingly broad wording of an expanded indication for sacubitril-valsartan in the United States – “to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure” – without specific restrictions on the basis of LVEF. The new indication was announced in early 2021, too late to be considered in the new guidelines.
Whither LVEF-based definitions?
During discussion after the guideline presentation, Dr. Zannad speculated on the future of HF classifications based on ventricular function, given trial evidence in recent years that some agents – notably spironolactone, sacubitril-valsartan, and now, apparently, empagliflozin – might be effective in HFpEF as well as HFrEF.
Will the field continue with “LVEF-centric” distinctions across the range of HF, or transition to “some definition in which drug therapies can be used independently across the full spectrum of ejection fraction?” Dr. Zannad posed.
“I think we need to wait and see what some of these trials with the SGLT2 inhibitors are going to show in heart failure with preserved ejection fraction,” Dr. McDonagh replied. “And I think that will be a step for the next guideline, completely redefining heart failure.”
A version of this article first appeared on Medscape.com.
The new guideline on management of heart failure (HF) from the European Society of Cardiology seemed to bear an asterisk or footnote even before its full unveiling in the early hours of ESC Congress 2021.
The document would offer little new in the arena of HF with preserved ejection fraction (HFpEF), so understandably the fast-approaching presentation of a major HFpEF trial – arguably the conference’s marquee event – would feel to some like the elephant in the room.
“I’d like to highlight this unfortunate timing of the guideline, because it’s an hour or 2 before we hear the full story from EMPEROR-Preserved, which I’m sure will change the guidelines,” Faiez Zannad, MD, PhD, University of Lorraine, Vandoeuvre-Les-Nancy, France, said wryly.
Anticipation of the trial’s full presentation was intense as the ESC congress got underway, in part because the top-line and incomplete message from EMPEROR-Preserved had already been released: Patients with HFpEF treated with the sodium-glucose cotransporter 2 inhibitor empagliflozin (Jardiance, Boehringer Ingelheim/Eli Lilly) showed a significant benefit for the primary endpoint of cardiovascular (CV) death or HF hospitalization.
Although empagliflozin is the first medication to achieve that status in a major HFpEF trial, conspicuously absent from the early announcement were the magnitude of “benefit” and any data. Still, the tantalizing top-line results mean that technically, at least, “we have a drug which is effective in reduced and preserved ejection fraction,” Dr. Zannad said.
But the new guideline, published online Aug. 27, 2021, in the European Heart Journal and comprehensively described that day at the congress, was never really expected to consider results from EMPEROR-Reduced. “These new indications do need to go through the regulatory authorities,” such as the European Medicines Agency and the U.S. Food and Drug Administration, observed Carlos Aguiar, MD, Hospital Santa Cruz, Carnaxide, Portugal.
“It does take some time for the whole process to be concluded and, finally, as physicians, being able to implement it in clinical practice,” Dr. Aguiar said as moderator of press briefing prior to the ESC congress.
The ESC guideline’s next iteration or update could well include an SGLT2 inhibitor recommendation that applies beyond the ejection fraction limits of HFrEF. Still, the document summarized that day reflects a number of pivotal concepts with profound treatment implications. Among them are the field’s latest paradigm for medical therapy of HFrEF and the increasingly accepted division of traditional HFpEF into two entities: HF with mildly reduced ejection fraction (HFmrEF); and HFpEF, with its left ventricular ejection fraction (LVEF) threshold raised to 50%.
In fact, HFmrEF in the new document is a drug-therapy indication that barely existed a few years ago but grew in prominence after secondary findings from trials like TOPCAT for spironolactone and PARAGON-HF for sacubitril-valsartan (Entresto, Novartis), an angiotensin-receptor/neprilysin inhibitor (ARNI). Still, the HFmrEF recommendations come with different class and level-of-evidence designations.
Those new guideline features and others in the realm of pharmacologic therapy were summarized by the document’s authors at the 2021 Heart Failure Association of the European Society of Cardiology (ESC-HFA) meeting, and covered at the time by this news organization
The ‘fantastic four’
One of the document’s central recommendations specifies which contemporary drug classes should be initiated, and when, in patients with HFrEF. An ACE inhibitor or ARNI, a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and an SGLT2 inhibitor collectively earned a class I recommendation, “given the importance of these key HFrEF therapies, some of which have been shown to improve outcomes within a month of initiation,” observed Roy S. Gardner, MBChB, MD.
An agent from each of the four classes is to be “commenced and up-titrated as quickly and as safely as possible, whilst using the lowest effective dose of loop diuretic to relieve congestion,” said Dr. Gardner, from Golden Jubilee National Hospital, Clydebank, Scotland, when presenting the full HFrEF portion of the guidelines.
The oral soluble guanylate-cyclase receptor stimulator vericiguat (Verquvo, Merck), which recently emerged from the VICTORIA trial as a modest success for patients with HFrEF and a previous HF hospitalization, gained a class IIb recommendation.
The document’s “simplified algorithm” for managing such patients overall and the advent of SGLT2 inhibitors are new twists in ESC guidelines for HF. But the way the four drug classes are started in patients is key and could take some practitioners time to get used to. There is no prespecified order of initiation.
“We’ve left the door open for clinicians to evaluate the evidence to make sure these four drugs are started, and to tailor how to do it according to the patient,” based on clinical considerations such as blood pressure or renal function, said Theresa A. McDonagh, MD, King’s College London, cochair of the guideline task force.
“The SGLT2 inhibitor trials were done on top of therapy with ACE inhibitors or ARNI, beta-blockers, and MRAs, so some people no doubt will choose to follow a sequenced approach,” Dr. McDonagh said. Other practitioners will consider each patient and attempt to get all four started “as quickly and safely as possible based on the phenotype.”
Importantly, clinicians “should not wait for weeks, months, or years until you have the four drugs in the patient, but you should do this within weeks,” cautioned Johann Bauersachs, MD, Hannover (Germany) Medical School, a discussant for the guideline presentation who is listed as a reviewer on the document.
Although angiotensin-receptor blockers (ARBs) and ACE inhibitors are sometimes thought of as interchangeable, the new guideline does not give them the same weight. “The angiotensin-receptor blocker valsartan is a constituent of the ARNI,” Dr. McDonagh noted. “So, the place of ARBs in heart failure has been downgraded in HFrEF. They are really for those who are intolerant of an ACE inhibitor or an ARNI.”
In practice, ARBs are likely to be used as first-line therapy in some circumstances, observed Dr. Bauersachs. They are “the default option in, unfortunately, many low-income countries that may not afford sacubitril-valsartan. And I know that there are many of them.”
Tweaks to device recommendations
The new document contains several new wrinkles in the recommendations for HF device therapy, which should usually be considered only if still appropriate after at least 3 months of optimal medical therapy, Dr. Gardner said.
For example, use of an implantable cardioverter-defibrillator (ICD) has been demoted from its previous class I recommendation to class II, level of evidence A, in patients with nonischemic cardiomyopathy “in light of the data from the DANISH study,” Dr. Gardner said.
The 2016 DANISH trial was noteworthy for questioning the survival benefits of ICDs in patients with nonischemic cardiomyopathy, whether or not they were also receiving cardiac resynchronization therapy (CRT).
The new document also puts greater emphasis on a range of specific CRT patient-selection criteria. Beyond the conventional recommended standards of an LVEF of 35% or less, QRS of at least 150 ms, and left-bundle-branch block on optimal meds, consideration can be given to CRT if the QRS is only 130 ms or greater. “And where it’s appropriate to do so, an ICD could be an option,” Dr. Gardner said.
It also recommends CRT as a replacement for right ventricular pacing in patients with high-degree atrioventricular block. “And this, for the first time, includes patients with atrial fibrillation,” he said. “The previous indications for CRT were in individuals in sinus rhythm.”
The new document recommends that HF in any patient be classified as HFrEF, defined by an LVEF of ≤40%; HFmrEF, defined by an LVEF of 41%-49%; or HFpEF, defined by an LVEF of at least 50%. “Importantly, for all forms, the presence of the clinical syndrome of heart failure is a prerequisite,” observed Carolyn S.P. Lam, MBBS, PhD, Duke-NUS Graduate Medical School, Singapore, at the presentation.
In a critical update from previous guidelines, the term HF with “mid-range” ejection fraction was replaced by the term specifying “mildly reduced” ejection fraction, Dr. Lam noted. The shift retains the acronym but now reflects growing appreciation that HFmrEF patients can benefit from treatments also used in HFrEF, including ACE inhibitors, ARBs, beta-blockers, MRAs, and sacubitril-valsartan, she said.
Support for that relationship comes largely from post hoc subgroup analyses of trials that featured some patients with LVEF 40%-49%. That includes most HFpEF trials represented in the guideline document, but also EMPEROR-Preserved, which saw gains for the primary outcome across the entire range of LVEF above 40%.
The LVEF-based definitions are consistent with a recent HF classification proposal endorsed by the ESC and subspecialty societies in Europe, North America, Japan, India, Australia, New Zealand, and China.
The document doesn’t update recommendations for HFpEF, in which “no treatment has been shown to convincingly reduce mortality or morbidity,” Dr. Lam observed. Still, she noted, the guideline task force “acknowledges that treatment options for HFpEF are being revised even as the guidelines have been published.”
That could be a reference to empagliflozin in EMPEROR-Preserved, but it also refers to the strikingly broad wording of an expanded indication for sacubitril-valsartan in the United States – “to reduce the risk of cardiovascular death and hospitalization for heart failure in adult patients with chronic heart failure” – without specific restrictions on the basis of LVEF. The new indication was announced in early 2021, too late to be considered in the new guidelines.
Whither LVEF-based definitions?
During discussion after the guideline presentation, Dr. Zannad speculated on the future of HF classifications based on ventricular function, given trial evidence in recent years that some agents – notably spironolactone, sacubitril-valsartan, and now, apparently, empagliflozin – might be effective in HFpEF as well as HFrEF.
Will the field continue with “LVEF-centric” distinctions across the range of HF, or transition to “some definition in which drug therapies can be used independently across the full spectrum of ejection fraction?” Dr. Zannad posed.
“I think we need to wait and see what some of these trials with the SGLT2 inhibitors are going to show in heart failure with preserved ejection fraction,” Dr. McDonagh replied. “And I think that will be a step for the next guideline, completely redefining heart failure.”
A version of this article first appeared on Medscape.com.