User login
Biomarkers: Their potential in the diagnosis and treatment of heart failure
The growth in recognition and clinical adoption of blood and urine biomarkers over the last 20 years has been a major advance in the diagnosis and prognosis of heart failure (HF). While there have been numerous research studies and prospective clinical trials on this topic, healthcare providers often face limited availability of biomarker testing and a relative paucity of data to guide individual patient management. This is especially true since many guideline-directed medical therapies have long-established clinical indications and target populations, predating the clinical availability of biomarkers testing. This article addresses the salient insights gained from broad clinical use of biomarkers, as well as from clinical studies that helped define their appropriate use and lay the foundations of the major changes presented in the recently published clinical guidelines for the management of HF.
WHAT MAKES A BIOMARKER CLINICALLY USEFUL?
To appreciate the appropriate use of any clinical tool, clinicians need to first understand its indications and limitations and how they are defined. There are four major criteria regarding the clinical utility of a biomarker.
First, we have to establish what we are measuring, particularly with accurate and reproducible methods, with rapid turnaround, and at a reasonable cost. Second, we have to determine why we need the biomarker: ie, we need to determine if its measurement provides valuable new information to the clinician, if there is a strong and consistent association between the marker and the disease or outcome, and if this has been validated in a way that is generalizable. Third, we have to determine when measuring the biomarker would help clinical management, whether it is superior to existing tests, and whether there is evidence that it improves outcomes. Last, and perhaps most commonly overlooked, is practicality: ie, how can measuring the biomarkers be incorporated into the clinical workflow?
Not all biomarkers need to fulfill all these criteria in order to be useful, and the usefulness of a biomarker may differ from one patient population to another, from one clinician to another, or from one clinical scenario to another.1 Many clinical biomarkers are applied based on their ability to indicate a specific diagnosis or treatment (eg, glycated hemoglobin), and some have been used to determine the limits of therapy (eg, creatinine or liver function tests to detect end-organ damage). Nevertheless, the overarching goal is to establish the clinical role of a biomarker to provide the opportunity to gain additional insight into a disease state beyond that provided by a standard clinical assessment, and to determine if using the biomarker favorably alters the clinical course.
WHICH BIOMARKERS DO WE ALREADY ROUTINELY MEASURE?
Traditionally, the management of HF requires meticulous monitoring for adverse effects of drug therapy (eg, electrolyte and renal abnormalities with diuretics or drugs targeting the renin-angiotensin-aldosterone system). Although no specific clinical studies have been conducted to support their routine use, electrolytes (sodium, potassium, chloride, bicarbonate) and renal function measurements (blood urea nitrogen [BUN], creatinine) are often repeated periodically in the longitudinal care of patients with HF.2 Diagnostic tests for hemochromatosis, human immunodeficiency virus, rheumatologic disease, amyloidosis, and pheochromocytoma are reasonable in patients presenting with HF in whom there is a clinical suspicion of these diseases.2
For risk stratification, biomarkers that reflect renal insufficiency (particularly sodium, BUN, creatinine, and the estimated glomerular filtration rate [eGFR]) are powerful prognosticators.3 Newer renal markers of glomerular function (such as cystatin C)4,5 or of acute kidney injury (such as neutrophil gelatinase-associated lipocalin)6,7 have been proposed, although their clinical utility beyond prognostication remains to be determined. In fact, head- to-head comparisons have revealed that BUN appeared to be superior to most other renal biomarkers in stratifying short-term and long-term risk.8
Liver function, blood cell count, and thyroid function profiles are checked on some occasions to determine underlying end-organ dysfunction.2 Interestingly, several common laboratory values have consistently been associated with more advanced disease states or with a higher risk of future adverse events. These include serum uric acid (likely reflecting oxidative stress and nucleotide catabolism),9 anemia or red cell distribution width (likely reflecting iron deficiency or hematopoietic insufficiency),10 lymphocytopenia (likely reflecting immune dysfunction), and total bilirubin (likely reflection of hepatobiliary congestion).11
Some biomarkers have been incorporated into risk-stratification in patients with HF.2 However, drugs targeting these biomarkers have yet to be shown to improve clinical outcomes in prospective clinical trials. Several recent examples in chronic systolic HF include allopurinol for elevated uric acid levels12 and darbepoetin alfa for anemia (low hemoglobin).13 Thus, improving the biomarker level with specific treatment may not translate to improved clinical outcomes.
GUIDELINE RECOMMENDATIONS FOR CARDIAC BIOMARKERS IN HEART FAILURE
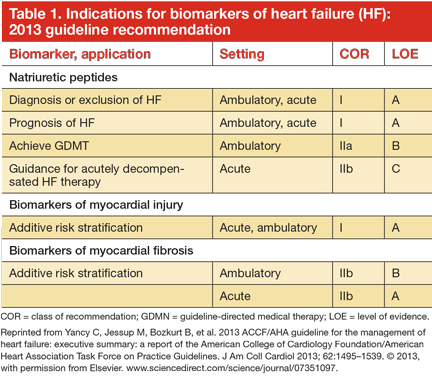
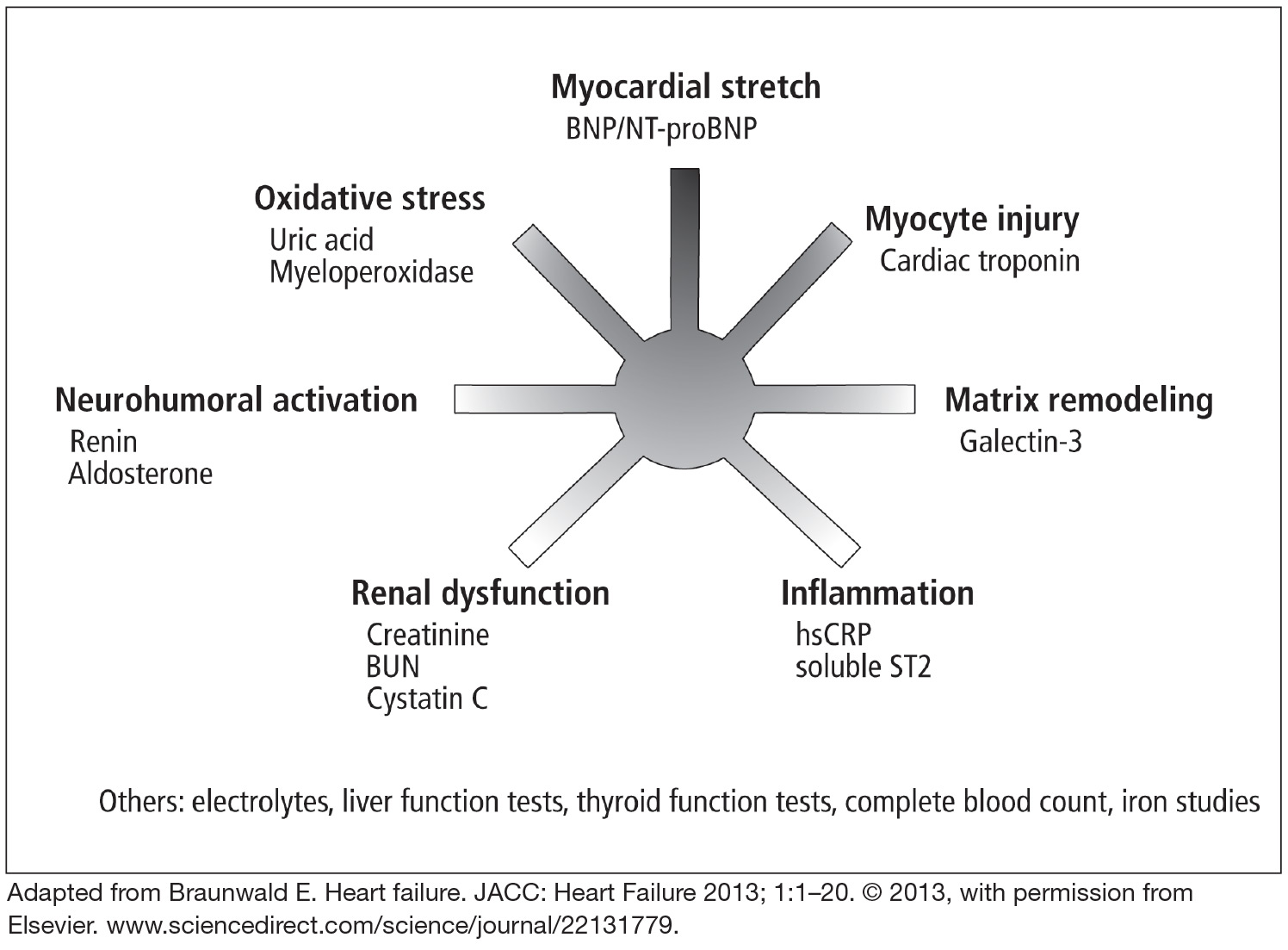
Clinical guidelines from several countries on the management of HF have expanded the role of biomarker testing in patients with HF.2,14–16 Table 1 shows the recommendations for biomarker testing in HF from the most recent joint guidelines of the American College of Cardiology and the American Heart Association. These recommendations will form the basis of the following discussion of clinically available biomarkers of HF that reflect distinct pathophysiologic processes and that have been cleared by the US Food and Drug Administration (Figure 1).
Biomarkers of myocardial stress: Natriuretic peptides
Natriuretic peptides are primary counterregulatory hormones produced in response to myocardial stress. Natriuretic peptide receptors stimulated by B-type (also “brain”) natriuretic peptide (BNP) lead to an increase in natriuresis, vasodilation, and opposing effects of other overactive neurohormonal systems. The contemporary understanding of how natriuretic peptides are being produced and metabolized is beyond the scope of this review, but generally it is now recognized that natriuretic peptide levels vary widely among patients with the same degree of symptoms or echocardiographic features.17
Of the several types of natriuretic peptide detectable by immunoassay, the two main types available for clinical use in the United States are BNP and amino acid N-terminal pro-BNP (NT-proBNP). Although there is no direct conversion available (NT-proBNP levels are five to eight times higher than BNP levels), their levels are often concordant and both are influenced by factors such as age, body mass index, and renal function. Specifically, natriuretic peptide levels in morbidly obese patients range 30% to 40% lower than levels in patients who are not morbidly obese.18
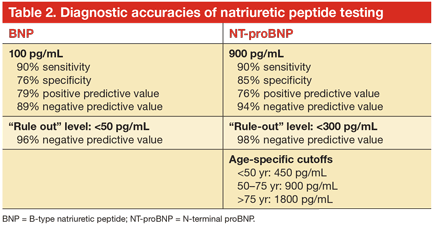
Studies over the past 10 years of natriuretic peptides in the diagnosis of HF have shown that levels are invariably elevated in underlying HF, while stable (and especially low) levels often track with clinical stability. In the latest clinical guidelines, natriuretic peptide testing has gained the highest level of recommendation for clinical use for any biomarker in HF, especially in the setting of clinical uncertainty (class 1 recommendation, level of evidence A).2,16 Two common clinical scenarios are represented in this indication. When patients present with signs and symptoms suspicious of HF (shortness of breath, fluid retention, peripheral edema, evidence of central congestion), natriuretic peptide testing provides confirmation of an underlying cardiac cause of these symptoms when elevated. Conversely, when there are alternative explanations or if the presentation is subtle and there is some degree of uncertainty, testing natriuretic peptide levels helps establish the diagnosis of HF when levels are higher than the cut-off values, and levels below the cut-off have a high negative predictive value (Table 2).19,20

Meanwhile, for patients with established HF, a deviation from “stable” natriuretic peptide levels (particularly an increase of more than 30%) may represent evolving destabilization that may warrant an intensification of therapy, whereas an unchanged or reduced level may be taken as objective evidence of clinical stability or favorable response to medical therapy. Table 3 outlines the latest Canadian guidelines that offer a practical approach as ongoing studies attempt to clarify the benefits of these strategies.15
The consistent association between elevated natriuretic peptide levels and worse prognosis21 has led to the promise that intensification of medical therapy in those with elevated natriuretic peptide levels can lead to better outcomes. Nevertheless, the rise in natriuretic peptide levels requires interpretation in the clinical context, as not all factors affecting the levels can be relieved by intensifying medical therapy (eg, age, renal insufficiency).
Several prospective, randomized controlled trials have tested this hypothesis, with favorable yet mixed results. Most studies have utilized a BNP measurement less than 100 pg/mL or an NT-proBNP measurement less than 1,000 pg/mL as a therapeutic target. In a recent prospective study that utilized the NT-proBNP threshold, only about half of patients were able to reach the target of less than 1,000 pg/mL.22 Often overlooked is the fact that in the same study, the inability to reach less than 5,000 pg/mL within 3 months after discharge clearly identified advanced, “nonresponsive” HF refractory to medical therapy and with a poor prognosis.23 This is an important point when assessing the clinical utility of biomarkers, as incremental prognostic values may not guarantee the feasibility or ultimate benefit of intensifying drug therapy according to specific biomarker targets. Until we have more insight into whether a care pathway guided by NT-proBNP measurements can lead to a consistent reduction in rates of hospitalization and mortality in HF, it is reasonable to target those with elevated natriuretic peptide levels by reevaluating their treatment regimen to achieve optimal dosing of guideline-directed medical therapy (Class 2a recommendation, level of evidence B).2 Also, the usefulness of BNP and NT-proBNP in guiding therapy for acutely decompensated HF is not well established (Class 2b recommendation, level of evidence C).2
Biomarkers of myocardial injury: Cardiac troponin
Whereas detecting circulating cardiac troponin is helpful in the diagnosis of acute coronary syndrome, the role of cardiac troponin levels in HF is primarily for risk stratification (Class 1 recommendation, level of evidence A in both acute and chronic HF).2 In patients hospitalized with acute decompensated HF, those with elevated troponin I or troponin T at the time of admission had lower systolic blood pressures, lower ejection fractions, and higher rate of in-hospital mortality.24,25 In chronic HF, elevations in both standard and high-sensitivity cardiac troponin levels were associated with increases in all-cause mortality,26 and rise in serial measurements appeared to correlate with an increased risk of future cardiovascular events.27 And with regard to cardiotoxicity, an increase in cardiac troponin over time (either after chemotherapy or with amyloidosis) is indicative of progressive cardiac dysfunction.28,29
Nevertheless, how to adjust medical therapy according to a rise in cardiac troponin levels remains unclear, as levels of cardiac troponin beyond the setting of acute coronary syndrome have appeared not to fluctuate significantly over time and do not seem to be related to underlying coronary events. Newer-generation cardiac troponin assays have yet to provide incremental value compared with standard clinical troponin assays despite their higher sensitivities.26
One common and underappreciated clinical application that combines both diagnostic and prognostic properties of both natriuretic peptide and cardiac troponin testing is the concept of HF staging. This is particularly relevant when there is a progressive change in clinical status (eg, need for hospitalization, change in signs or symptoms) or when a new therapy is started that may promote adverse effects. For example, a patient with pre-existing HF hospitalized with atypical symptoms and deemed not to have HF could be found to have subclinical myocardial necrosis as detected by low concentration of cardiac troponin or higher-than-baseline natriuretic peptide levels in the absence of hypervolemia. Careful assessment of the potential triggers of fluctuations from previous stable levels of cardiac biomarkers is also warranted (eg, atrial fibrillation, dietary indiscretion, infection, and ischemia). Indeed, these may represent objective rather than subjective changes in clinical manifestation of HF, which may warrant a reassessment of disease severity (eg, objective testing for functional capacity or hemodynamics, or even referral for consideration of advanced HF therapeutic options).
Biomarkers of inflammation and fibrosis: Soluble ST2 and galectin-3
Inflammation has long been associated with HF, and clinically available markers of inflammation such as high-sensitivity C-reactive protein (CRP)30,31 and myeloperoxidase32 have consistently tracked with prognosis. The search for a stable biomarker of inflammation has been challenging because inflammation is a dynamic process and because of the lack of treatment options for heightened inflammation.
A promising new protein biomarker, ST2 (suppression of tumorigenicity-2), has been identified in a soluble form (sST2) that binds to interleukin 33 (IL-33) to antagonize the maladaptive response of the myocardium to overload states.33 The levels of sST2 inversely correlate with the ejection fraction and have a positive association with increasing New York Heart Association class, worsening symptoms, and indicators of HF severity, such as norepinephrine levels, diastolic filling pressures, CRP, and natriuretic peptide levels.34 Unlike natriuretic peptides, levels of sST2 are not significantly affected by age, sex, body mass index, and valve disease,34 although recent observations have challenged its cardiac associations.35 In patients with chronic HF, elevated levels of sST2 (especially >35 ng/mL) have been associated with poorer clinical outcomes36 and increased risk of sudden cardiac death in HF.37 In addition, persistently elevated sST2 levels consistently confer poor long-term prognosis. Several studies have also demonstrated the prognostic value of elevated sST2 in predicting long-term risk of death in acute HF, either at baseline38,39 or on serial testing.40
Another new biomarker, galectin-3, has been implicated in fibrosis and in structural and pathophysiologic changes seen in HF.41 Studies have shown that higher levels of galectin-3 in patients with acute HF and chronic HF were associated with more severe cardiac fibrosis and with an increase in left ventricular remodeling.42–44 Serial measurements also confer prognostic information.45 However, many of these studies did not fully account for renal dysfunction as a major confounder, and the relationship between circulating galectin-3 and estimated GFR is strong.46,47 Meanwhile, head-to-head comparisons among galectin-3 and other clinically available biomarkers also revealed that the prognostic value of galectin-3 can be attenuated in the presence of sST2 and NT-proBNP.48,49 Furthermore, careful evaluation of diastolic parameters only showed a modest relationship with galectin-3 levels, especially in those with HF with preserved ejection fraction.50,51
In animal infarction models, disruption of the galectin-3 and IL-33/ST2 pathway with pharmacologic therapy such as mineralocorticoid receptor antagonists may attenuate cardiac remodeling.52,53 It is conceivable that these biomarkers may have mechanistic links with therapeutic benefits. However, the practical uses of galectin-3 and sST2 are still debated (Class 2b recommendation by the latest guidelines2) despite strong statistical associations between biomarker levels and adverse outcomes. The majority of biomarker substudies from clinical trials have suggested that improvements following drug or device therapy were largely confined to patients with lower rather than higher biomarker levels.54,55 Furthermore, validation studies have challenged the incremental prognostic value of these markers when natriuretic peptide levels are available.54,56–58 Thus, more clinical experience and research are warranted, and current clinical applications may be restricted to patient subsets.
BIOMARKERS IN EARLY STAGES OF HEART FAILURE
The potential benefit of biomarker testing may reside in the earlier end of the HF spectrum, especially in patients at risk of but not yet diagnosed with HF (so-called stage A). In the HealthABC study, the future risk of HF in elderly patients can be predicted with a combination of clinical risk factors (age, sex, left ventricular hypertrophy, systolic blood pressure, heart rate, smoking), as well as biochemical risk factors such as albumin, creatinine, and glucose.59 Patients with elevated natriuretic peptide levels are more likely to have underlying cardiac abnormalities and to have poorer long-term outcomes.60 In a recent prospective, randomized controlled trial, participants with a BNP-guided transition to HF therapies (when BNP >50 pg/mL) had a lower incidence of HF than participants without knowledge of BNP levels.61 Elevated levels of clinically available biomarkers of inflammation, such as myeloperoxidase,29,62 ceruloplasmin,63 and CRP,64 have also been associated with an increased risk of future HF. These findings support the notion that biomarkers, especially when combined with clinical risk factors, can serve as indicators of HF vulnerability. If independently confirmed, this will be an important therapeutic approach to the prevention of HF.
PRACTICAL CONSIDERATIONS
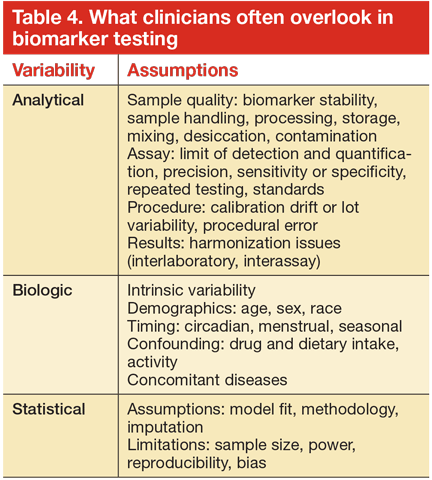
An important perspective often overlooked concerns the variability of a biomarker level as it is utilized in clinical practice (Table 4). In general, point-of-care assays are often more variable than the same tests done in clinical laboratories. Sample collection, handling, and processing also introduce a degree of variability. The biologic variability of specific measurements can significantly affect the precision of the measurement. In the case of HF, the biologic variability (as measured in stable patients over time) of natriuretic peptides and galectin-3 are significantly higher than those observed in cardiac troponins or sST2 (> 130% vs approximately 30%).65 Nevertheless because of their relative cardiac specificity, natriuretic peptides have maintained their clinical utility.
- Morrow DA, de Lemos JA. Benchmarks for the assessment of novel cardiovascular biomarkers. Circulation 2007; 115:949–952.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013; 128:e240–327.
- Halkar M, Tang WH. Incorporating common biomarkers into the clinical management of heart failure. Curr Heart Fail Rep 2013; 10:450–457.
- Dupont M, Wu Y, Hazen SL, Tang WH. Cystatin C identifies patients with stable chronic heart failure at increased risk for adverse cardiovascular events. Circ Heart Fail 2012; 5:602–609.
- Tang WH, Van Lente F, Shrestha K, et al. Impact of myocardial function on cystatin C measurements in chronic systolic heart failure. J Card Fail 2008; 14:394– 399.
- Dupont M, Shrestha K, Singh D, et al. Lack of significant renal tubular injury despite acute kidney injury in acute decompensated heart failure. Eur J Heart Fail 2012; 14:597–604.
- Verbrugge FH, Dupont M, Shao Z, et al. Novel urinary biomarkers in detecting acute kidney injury, persistent renal impairment, and all-cause mortality following decongestive therapy in acute decompensated heart failure. J Card Fail 2013; 19:621–628.
- Tang WH, Dupont M, Hernandez AF, et al. Comparative assessment of short-term adverse events in acute heart failure with cystatin C and other estimates of renal function: results from the ASCEND-HF trial. JACC Heart Fail 2015; 3:40–49.
- Vaduganathan M, Greene SJ, Ambrosy AP, et al. Relation of serum uric acid levels and outcomes among patients hospitalized for worsening heart failure with reduced ejection fraction (from the efficacy of vasopressin antagonism in heart failure outcome study with tolvaptan trial). Am J Cardiol 2014; 114:1713–1721.
- Felker GM, Allen LA, Pocock SJ, et al. Red cell distribution width as a novel prognostic marker in heart failure: data from the CHARM Program and the Duke Databank. J Am Coll Cardiol 2007; 50:40–47.
- Wu AH, Levy WC, Welch KB, et al. Association between bilirubin and mode of death in severe systolic heart failure. Am J Cardiol 2013; 111:1192–1197.
- Givertz MM, Anstrom KJ, Redfield MM, et al. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the Xanthine Oxidase Inhibition for Hyperuricemic Heart Failure Patients (EXACT-HF) study. Circulation 2015; 131:1763–1771.
- Swedberg K, Young JB, Anand IS, et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med 2013; 368:1210–1219.
- McMurray JJ, Adamopoulos S, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 2012; 33:1787–1847.
- Moe GW, Ezekowitz JA, O’Meara E, et al. The 2014 Canadian Cardiovascular Society Heart Failure Management Guidelines Focus Update: anemia, biomarkers, and recent therapeutic trial implications. Can J Cardiol 2015; 31:3–16.
- Tang WH, Francis GS, Morrow DA, et al. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical utilization of cardiac biomarker testing in heart failure. Circulation 2007; 116:e99–109.
- Maisel AS, Daniels LB. Breathing not properly 10 years later: what we have learned and what we still need to learn. J Am Coll Cardiol 2012; 60:277–282.
- Mehra MR, Uber PA, Park MH, et al. Obesity and suppressed B-type natriuretic peptide levels in heart failure. J Am Coll Cardiol 2004; 43:1590–1595.
- Januzzi JL, Jr, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP Investigation of Dyspnea in the Emergency Department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Maisel AS, Krishnaswamy P, Nowak RM, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Fonarow GC, Peacock WF, Phillips CO, Givertz MM, Lopatin M. Admission B-type natriuretic peptide levels and in-hospital mortality in acute decompensated heart failure. J Am Coll Cardiol 2007; 49:1943–1950.
- Januzzi JL, Jr, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B- type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1889.
- Gaggin HK, Truong QA, Rehman SU, et al. Characterization and prediction of natriuretic peptide ‘nonresponse’ during heart failure management: results from the ProBNP Outpatient Tailored Chronic Heart Failure (PROTECT) and the NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death (BATTLESCARRED) study. Congest Heart Fail 2013; 19:135–142.
- Peacock WF 4th, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008; 358:2117–2126.
- Felker GM, Hasselblad V, Tang WH, et al. Troponin I in acute decompen- sated heart failure: insights from the ASCEND-HF study. Eur J Heart Fail 2012; 14:1257–1264.
- Grodin JL, Neale S, Wu Y, Hazen SL, Tang WH. Prognostic comparison of different sensitivity cardiac troponin assays in stable heart failure. Am J Med 2015; 128:276–282.
- Masson S, Anand I, Favero C, et al. Serial measurement of cardiac troponin T using a highly sensitive assay in patients with chronic heart failure: data from 2 large randomized clinical trials. Circulation 2012; 125:280–288.
- Cardinale D, Sandri MT, Colombo A, et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation 2004; 109:2749–2754.
- Ky B, Putt M, Sawaya H, et al. Early increases in multiple biomarkers predict subsequent cardiotoxicity in breast cancer patients treated with doxorubicin, taxanes, and trastuzumab. J Am Coll Cardiol 2014; 63:809–816.
- Kalogeropoulos AP, Tang WH, Hsu A, et al. High-sensitivity C-reactive protein in acute heart failure: insights from the ASCEND-HF trial. J Card Fail 2014; 20:319–326.
- Tang WH, Shrestha K, Van Lente F, et al. Usefulness of C-reactive protein and left ventricular diastolic performance for prognosis in patients with left ventricular systolic heart failure. Am J Cardiol 2008; 101:370–373.
- Tang WH, Wu Y, Nicholls SJ, Hazen SL. Plasma myeloperoxidase predicts incident cardiovascular risks in stable patients undergoing medical management for coronary artery disease. Clin Chem 2011; 57:33–39.
- Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest 2007; 117:1538–1549.
- Rehman SU, Mueller T, Januzzi JL, Jr. Characteristics of the novel interleukin family biomarker ST2 in patients with acute heart failure. J Am Coll Cardiol 2008; 52:1458–1465.
- Kaye DM, Mariani JA, van Empel V, Maeder MT. Determinants and implications of elevated soluble ST2 levels in heart failure. Int J Cardiol 2014; 176:1242–1243.
- Ky B, French B, McCloskey K, et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ Heart Fail 2011; 4:180–187.
- Ahmad T, Fiuzat M, Neely B, et al. Biomarkers of myocardial stress and fibrosis as predictors of mode of death in patients with chronic heart failure. JACC Heart Fail 2014; 2:260–268.
- Pascual-Figal DA, Manzano-Fernandez S, Boronat M, et al. Soluble ST2, high-sensitivity troponin T- and N-terminal pro-B-type natriuretic peptide: complementary role for risk stratification in acutely decompensated heart failure. Eur J Heart Fail 2011; 13:718–725.
- Januzzi JL, Jr, Peacock WF, Maisel AS, et al. Measurement of the interleukin family member ST2 in patients with acute dyspnea: results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) study. J Am Coll Cardiol 2007; 50:607-613.
- Boisot S, Beede J, Isakson S, et al. Serial sampling of ST2 predicts 90-day mortality following destabilized heart failure. J Card Fail 2008; 14:732–738.
- Sharma UC, Pokharel S, van Brakel TJ, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004; 110:3121–3128.
- van Kimmenade RR, Januzzi JL, Jr., Ellinor PT, et al. Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure. J Am Coll Cardiol 2006; 48:1217–1224.
- Lok DJ, Van Der Meer P, de la Porte PW, et al. Prognostic value of galectin-3, a novel marker of fibrosis, in patients with chronic heart failure: data from the DEAL-HF study. Clin Res Cardiol 2010; 99:323–328.
- Shah RV, Chen-Tournoux AA, Picard MH, van Kimmenade RR, Januzzi JL. Ga- lectin-3, cardiac structure and function, and long-term mortality in patients with acutely decompensated heart failure. Eur J Heart Fail 2010; 12:826–832.
- van der Velde AR, Gullestad L, Ueland T, et al. Prognostic value of changes in galectin-3 levels over time in patients with heart failure: data from CORONA and COACH. Circ Heart Fail 2013; 6:219–226.
- Tang WH, Shrestha K, Shao Z, et al. Usefulness of plasma galectin-3 levels in systolic heart failure to predict renal insufficiency and survival. Am J Cardiol 2011; 108:385–390.
- Gopal DM, Kommineni M, Ayalon N, et al. Relationship of plasma galectin-3 to renal function in patients with heart failure: effects of clinical status, pathophysiology of heart failure, and presence or absence of heart failure. J Am Heart Assoc 2012; 1:e000760.
- Bayes-Genis A, de Antonio M, Vila J, et al. Head-to-head comparison of 2 myocardial fibrosis biomarkers for long-term heart failure risk stratification: ST2 versus galectin-3. J Am Coll Cardiol 2014; 63:158–166.
- Felker GM, Fiuzat M, Shaw LK, et al. Galectin-3 in ambulatory patients with heart failure: results from the HF-ACTION study. Circ Heart Fail 2012; 5:72–78.
- Edelmann F, Holzendorf V, Wachter R, et al. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail 2015; 17:214–223.
- AbouEzzeddine OF, Haines P, Stevens S, et al. Galectin-3 in heart failure with preserved ejection fraction. A RELAX trial substudy (Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure). JACC Heart failure 2015; 3:245–252.
- Lax A, Sanchez-Mas J, Asensio-Lopez MC, et al. Mineralocorticoid receptor antagonists modulate galectin-3 and interleukin-33/ST2 signaling in left ventricular systolic dysfunction after acute myocardial infarction. JACC Heart failure 2015; 3:50–58.
- Calvier L, Martinez-Martinez E, Miana M, et al. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart failure 2015; 3:59–67.
- Anand IS, Rector TS, Kuskowski M, Adourian A, Muntendam P, Cohn JN. Baseline and serial measurements of galectin-3 in patients with heart failure: relationship to prognosis and effect of treatment with valsartan in the Val-HeFT. Eur J Heart Fail 2013; 15:511–518.
- Gullestad L, Ueland T, Kjekshus J, et al. Galectin-3 predicts response to statin therapy in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CO- RONA). Eur Heart J 2012; 33:2290–2296.
- Felker GM, Fiuzat M, Thompson V, et al. Soluble ST2 in ambulatory patients with heart failure: association with functional capacity and long-term outcomes. Circ Heart Fail 2013; 6:1172–1179.
- Anand IS, Rector TS, Kuskowski M, Snider J, Cohn JN. Prognostic value of soluble ST2 in the Valsartan Heart Failure Trial. Circ Heart Fail 2014; 7:418–426.
- Gullestad L, Ueland T, Kjekshus J, et al. The predictive value of galectin-3 for mortality and cardiovascular events in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA). Am Heart J 2012; 164:878–883.
- Butler J, Kalogeropoulos A, Georgiopoulou V, et al. Incident heart failure prediction in the elderly: the health ABC heart failure score. Circ Heart Fail 2008; 1:125–133.
- McKie PM, Rodeheffer RJ, Cataliotti A, et al. Amino-terminal pro-B-type natriuretic peptide and B-type natriuretic peptide: biomarkers for mortality in a large community-based cohort free of heart failure. Hypertension 2006; 47:874–880.
- Ledwidge M, Gallagher J, Conlon C, et al. Natriuretic peptide-based screening and collaborative care for heart failure: the STOP-HF randomized trial. JAMA 2013; 310:66–74.
- Tang WH, Katz R, Brennan ML, et al. Usefulness of myeloperoxidase levels in healthy elderly subjects to predict risk of developing heart failure. Am J Cardiol 2009; 103:1269–1274.
- Dadu RT, Dodge R, Nambi V, et al. Ceruloplasmin and heart failure in the Atherosclerosis Risk in Communities study. Circ Heart Fail 2013; 6:936–943.
- Kalogeropoulos A, Georgiopoulou V, Psaty BM, et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol 2010; 55:2129–2137.
- Wu AH, Wians F, Jaffe A. Biological variation of galectin-3 and soluble ST2 for chronic heart failure: implication on interpretation of test results. Am Heart J 2013; 165:995–999.
The growth in recognition and clinical adoption of blood and urine biomarkers over the last 20 years has been a major advance in the diagnosis and prognosis of heart failure (HF). While there have been numerous research studies and prospective clinical trials on this topic, healthcare providers often face limited availability of biomarker testing and a relative paucity of data to guide individual patient management. This is especially true since many guideline-directed medical therapies have long-established clinical indications and target populations, predating the clinical availability of biomarkers testing. This article addresses the salient insights gained from broad clinical use of biomarkers, as well as from clinical studies that helped define their appropriate use and lay the foundations of the major changes presented in the recently published clinical guidelines for the management of HF.
WHAT MAKES A BIOMARKER CLINICALLY USEFUL?
To appreciate the appropriate use of any clinical tool, clinicians need to first understand its indications and limitations and how they are defined. There are four major criteria regarding the clinical utility of a biomarker.
First, we have to establish what we are measuring, particularly with accurate and reproducible methods, with rapid turnaround, and at a reasonable cost. Second, we have to determine why we need the biomarker: ie, we need to determine if its measurement provides valuable new information to the clinician, if there is a strong and consistent association between the marker and the disease or outcome, and if this has been validated in a way that is generalizable. Third, we have to determine when measuring the biomarker would help clinical management, whether it is superior to existing tests, and whether there is evidence that it improves outcomes. Last, and perhaps most commonly overlooked, is practicality: ie, how can measuring the biomarkers be incorporated into the clinical workflow?
Not all biomarkers need to fulfill all these criteria in order to be useful, and the usefulness of a biomarker may differ from one patient population to another, from one clinician to another, or from one clinical scenario to another.1 Many clinical biomarkers are applied based on their ability to indicate a specific diagnosis or treatment (eg, glycated hemoglobin), and some have been used to determine the limits of therapy (eg, creatinine or liver function tests to detect end-organ damage). Nevertheless, the overarching goal is to establish the clinical role of a biomarker to provide the opportunity to gain additional insight into a disease state beyond that provided by a standard clinical assessment, and to determine if using the biomarker favorably alters the clinical course.
WHICH BIOMARKERS DO WE ALREADY ROUTINELY MEASURE?
Traditionally, the management of HF requires meticulous monitoring for adverse effects of drug therapy (eg, electrolyte and renal abnormalities with diuretics or drugs targeting the renin-angiotensin-aldosterone system). Although no specific clinical studies have been conducted to support their routine use, electrolytes (sodium, potassium, chloride, bicarbonate) and renal function measurements (blood urea nitrogen [BUN], creatinine) are often repeated periodically in the longitudinal care of patients with HF.2 Diagnostic tests for hemochromatosis, human immunodeficiency virus, rheumatologic disease, amyloidosis, and pheochromocytoma are reasonable in patients presenting with HF in whom there is a clinical suspicion of these diseases.2
For risk stratification, biomarkers that reflect renal insufficiency (particularly sodium, BUN, creatinine, and the estimated glomerular filtration rate [eGFR]) are powerful prognosticators.3 Newer renal markers of glomerular function (such as cystatin C)4,5 or of acute kidney injury (such as neutrophil gelatinase-associated lipocalin)6,7 have been proposed, although their clinical utility beyond prognostication remains to be determined. In fact, head- to-head comparisons have revealed that BUN appeared to be superior to most other renal biomarkers in stratifying short-term and long-term risk.8
Liver function, blood cell count, and thyroid function profiles are checked on some occasions to determine underlying end-organ dysfunction.2 Interestingly, several common laboratory values have consistently been associated with more advanced disease states or with a higher risk of future adverse events. These include serum uric acid (likely reflecting oxidative stress and nucleotide catabolism),9 anemia or red cell distribution width (likely reflecting iron deficiency or hematopoietic insufficiency),10 lymphocytopenia (likely reflecting immune dysfunction), and total bilirubin (likely reflection of hepatobiliary congestion).11
Some biomarkers have been incorporated into risk-stratification in patients with HF.2 However, drugs targeting these biomarkers have yet to be shown to improve clinical outcomes in prospective clinical trials. Several recent examples in chronic systolic HF include allopurinol for elevated uric acid levels12 and darbepoetin alfa for anemia (low hemoglobin).13 Thus, improving the biomarker level with specific treatment may not translate to improved clinical outcomes.
GUIDELINE RECOMMENDATIONS FOR CARDIAC BIOMARKERS IN HEART FAILURE
Clinical guidelines from several countries on the management of HF have expanded the role of biomarker testing in patients with HF.2,14–16 Table 1 shows the recommendations for biomarker testing in HF from the most recent joint guidelines of the American College of Cardiology and the American Heart Association. These recommendations will form the basis of the following discussion of clinically available biomarkers of HF that reflect distinct pathophysiologic processes and that have been cleared by the US Food and Drug Administration (Figure 1).
Biomarkers of myocardial stress: Natriuretic peptides
Natriuretic peptides are primary counterregulatory hormones produced in response to myocardial stress. Natriuretic peptide receptors stimulated by B-type (also “brain”) natriuretic peptide (BNP) lead to an increase in natriuresis, vasodilation, and opposing effects of other overactive neurohormonal systems. The contemporary understanding of how natriuretic peptides are being produced and metabolized is beyond the scope of this review, but generally it is now recognized that natriuretic peptide levels vary widely among patients with the same degree of symptoms or echocardiographic features.17
Of the several types of natriuretic peptide detectable by immunoassay, the two main types available for clinical use in the United States are BNP and amino acid N-terminal pro-BNP (NT-proBNP). Although there is no direct conversion available (NT-proBNP levels are five to eight times higher than BNP levels), their levels are often concordant and both are influenced by factors such as age, body mass index, and renal function. Specifically, natriuretic peptide levels in morbidly obese patients range 30% to 40% lower than levels in patients who are not morbidly obese.18
Studies over the past 10 years of natriuretic peptides in the diagnosis of HF have shown that levels are invariably elevated in underlying HF, while stable (and especially low) levels often track with clinical stability. In the latest clinical guidelines, natriuretic peptide testing has gained the highest level of recommendation for clinical use for any biomarker in HF, especially in the setting of clinical uncertainty (class 1 recommendation, level of evidence A).2,16 Two common clinical scenarios are represented in this indication. When patients present with signs and symptoms suspicious of HF (shortness of breath, fluid retention, peripheral edema, evidence of central congestion), natriuretic peptide testing provides confirmation of an underlying cardiac cause of these symptoms when elevated. Conversely, when there are alternative explanations or if the presentation is subtle and there is some degree of uncertainty, testing natriuretic peptide levels helps establish the diagnosis of HF when levels are higher than the cut-off values, and levels below the cut-off have a high negative predictive value (Table 2).19,20
Meanwhile, for patients with established HF, a deviation from “stable” natriuretic peptide levels (particularly an increase of more than 30%) may represent evolving destabilization that may warrant an intensification of therapy, whereas an unchanged or reduced level may be taken as objective evidence of clinical stability or favorable response to medical therapy. Table 3 outlines the latest Canadian guidelines that offer a practical approach as ongoing studies attempt to clarify the benefits of these strategies.15
The consistent association between elevated natriuretic peptide levels and worse prognosis21 has led to the promise that intensification of medical therapy in those with elevated natriuretic peptide levels can lead to better outcomes. Nevertheless, the rise in natriuretic peptide levels requires interpretation in the clinical context, as not all factors affecting the levels can be relieved by intensifying medical therapy (eg, age, renal insufficiency).
Several prospective, randomized controlled trials have tested this hypothesis, with favorable yet mixed results. Most studies have utilized a BNP measurement less than 100 pg/mL or an NT-proBNP measurement less than 1,000 pg/mL as a therapeutic target. In a recent prospective study that utilized the NT-proBNP threshold, only about half of patients were able to reach the target of less than 1,000 pg/mL.22 Often overlooked is the fact that in the same study, the inability to reach less than 5,000 pg/mL within 3 months after discharge clearly identified advanced, “nonresponsive” HF refractory to medical therapy and with a poor prognosis.23 This is an important point when assessing the clinical utility of biomarkers, as incremental prognostic values may not guarantee the feasibility or ultimate benefit of intensifying drug therapy according to specific biomarker targets. Until we have more insight into whether a care pathway guided by NT-proBNP measurements can lead to a consistent reduction in rates of hospitalization and mortality in HF, it is reasonable to target those with elevated natriuretic peptide levels by reevaluating their treatment regimen to achieve optimal dosing of guideline-directed medical therapy (Class 2a recommendation, level of evidence B).2 Also, the usefulness of BNP and NT-proBNP in guiding therapy for acutely decompensated HF is not well established (Class 2b recommendation, level of evidence C).2
Biomarkers of myocardial injury: Cardiac troponin
Whereas detecting circulating cardiac troponin is helpful in the diagnosis of acute coronary syndrome, the role of cardiac troponin levels in HF is primarily for risk stratification (Class 1 recommendation, level of evidence A in both acute and chronic HF).2 In patients hospitalized with acute decompensated HF, those with elevated troponin I or troponin T at the time of admission had lower systolic blood pressures, lower ejection fractions, and higher rate of in-hospital mortality.24,25 In chronic HF, elevations in both standard and high-sensitivity cardiac troponin levels were associated with increases in all-cause mortality,26 and rise in serial measurements appeared to correlate with an increased risk of future cardiovascular events.27 And with regard to cardiotoxicity, an increase in cardiac troponin over time (either after chemotherapy or with amyloidosis) is indicative of progressive cardiac dysfunction.28,29
Nevertheless, how to adjust medical therapy according to a rise in cardiac troponin levels remains unclear, as levels of cardiac troponin beyond the setting of acute coronary syndrome have appeared not to fluctuate significantly over time and do not seem to be related to underlying coronary events. Newer-generation cardiac troponin assays have yet to provide incremental value compared with standard clinical troponin assays despite their higher sensitivities.26
One common and underappreciated clinical application that combines both diagnostic and prognostic properties of both natriuretic peptide and cardiac troponin testing is the concept of HF staging. This is particularly relevant when there is a progressive change in clinical status (eg, need for hospitalization, change in signs or symptoms) or when a new therapy is started that may promote adverse effects. For example, a patient with pre-existing HF hospitalized with atypical symptoms and deemed not to have HF could be found to have subclinical myocardial necrosis as detected by low concentration of cardiac troponin or higher-than-baseline natriuretic peptide levels in the absence of hypervolemia. Careful assessment of the potential triggers of fluctuations from previous stable levels of cardiac biomarkers is also warranted (eg, atrial fibrillation, dietary indiscretion, infection, and ischemia). Indeed, these may represent objective rather than subjective changes in clinical manifestation of HF, which may warrant a reassessment of disease severity (eg, objective testing for functional capacity or hemodynamics, or even referral for consideration of advanced HF therapeutic options).
Biomarkers of inflammation and fibrosis: Soluble ST2 and galectin-3
Inflammation has long been associated with HF, and clinically available markers of inflammation such as high-sensitivity C-reactive protein (CRP)30,31 and myeloperoxidase32 have consistently tracked with prognosis. The search for a stable biomarker of inflammation has been challenging because inflammation is a dynamic process and because of the lack of treatment options for heightened inflammation.
A promising new protein biomarker, ST2 (suppression of tumorigenicity-2), has been identified in a soluble form (sST2) that binds to interleukin 33 (IL-33) to antagonize the maladaptive response of the myocardium to overload states.33 The levels of sST2 inversely correlate with the ejection fraction and have a positive association with increasing New York Heart Association class, worsening symptoms, and indicators of HF severity, such as norepinephrine levels, diastolic filling pressures, CRP, and natriuretic peptide levels.34 Unlike natriuretic peptides, levels of sST2 are not significantly affected by age, sex, body mass index, and valve disease,34 although recent observations have challenged its cardiac associations.35 In patients with chronic HF, elevated levels of sST2 (especially >35 ng/mL) have been associated with poorer clinical outcomes36 and increased risk of sudden cardiac death in HF.37 In addition, persistently elevated sST2 levels consistently confer poor long-term prognosis. Several studies have also demonstrated the prognostic value of elevated sST2 in predicting long-term risk of death in acute HF, either at baseline38,39 or on serial testing.40
Another new biomarker, galectin-3, has been implicated in fibrosis and in structural and pathophysiologic changes seen in HF.41 Studies have shown that higher levels of galectin-3 in patients with acute HF and chronic HF were associated with more severe cardiac fibrosis and with an increase in left ventricular remodeling.42–44 Serial measurements also confer prognostic information.45 However, many of these studies did not fully account for renal dysfunction as a major confounder, and the relationship between circulating galectin-3 and estimated GFR is strong.46,47 Meanwhile, head-to-head comparisons among galectin-3 and other clinically available biomarkers also revealed that the prognostic value of galectin-3 can be attenuated in the presence of sST2 and NT-proBNP.48,49 Furthermore, careful evaluation of diastolic parameters only showed a modest relationship with galectin-3 levels, especially in those with HF with preserved ejection fraction.50,51
In animal infarction models, disruption of the galectin-3 and IL-33/ST2 pathway with pharmacologic therapy such as mineralocorticoid receptor antagonists may attenuate cardiac remodeling.52,53 It is conceivable that these biomarkers may have mechanistic links with therapeutic benefits. However, the practical uses of galectin-3 and sST2 are still debated (Class 2b recommendation by the latest guidelines2) despite strong statistical associations between biomarker levels and adverse outcomes. The majority of biomarker substudies from clinical trials have suggested that improvements following drug or device therapy were largely confined to patients with lower rather than higher biomarker levels.54,55 Furthermore, validation studies have challenged the incremental prognostic value of these markers when natriuretic peptide levels are available.54,56–58 Thus, more clinical experience and research are warranted, and current clinical applications may be restricted to patient subsets.
BIOMARKERS IN EARLY STAGES OF HEART FAILURE
The potential benefit of biomarker testing may reside in the earlier end of the HF spectrum, especially in patients at risk of but not yet diagnosed with HF (so-called stage A). In the HealthABC study, the future risk of HF in elderly patients can be predicted with a combination of clinical risk factors (age, sex, left ventricular hypertrophy, systolic blood pressure, heart rate, smoking), as well as biochemical risk factors such as albumin, creatinine, and glucose.59 Patients with elevated natriuretic peptide levels are more likely to have underlying cardiac abnormalities and to have poorer long-term outcomes.60 In a recent prospective, randomized controlled trial, participants with a BNP-guided transition to HF therapies (when BNP >50 pg/mL) had a lower incidence of HF than participants without knowledge of BNP levels.61 Elevated levels of clinically available biomarkers of inflammation, such as myeloperoxidase,29,62 ceruloplasmin,63 and CRP,64 have also been associated with an increased risk of future HF. These findings support the notion that biomarkers, especially when combined with clinical risk factors, can serve as indicators of HF vulnerability. If independently confirmed, this will be an important therapeutic approach to the prevention of HF.
PRACTICAL CONSIDERATIONS
An important perspective often overlooked concerns the variability of a biomarker level as it is utilized in clinical practice (Table 4). In general, point-of-care assays are often more variable than the same tests done in clinical laboratories. Sample collection, handling, and processing also introduce a degree of variability. The biologic variability of specific measurements can significantly affect the precision of the measurement. In the case of HF, the biologic variability (as measured in stable patients over time) of natriuretic peptides and galectin-3 are significantly higher than those observed in cardiac troponins or sST2 (> 130% vs approximately 30%).65 Nevertheless because of their relative cardiac specificity, natriuretic peptides have maintained their clinical utility.
The growth in recognition and clinical adoption of blood and urine biomarkers over the last 20 years has been a major advance in the diagnosis and prognosis of heart failure (HF). While there have been numerous research studies and prospective clinical trials on this topic, healthcare providers often face limited availability of biomarker testing and a relative paucity of data to guide individual patient management. This is especially true since many guideline-directed medical therapies have long-established clinical indications and target populations, predating the clinical availability of biomarkers testing. This article addresses the salient insights gained from broad clinical use of biomarkers, as well as from clinical studies that helped define their appropriate use and lay the foundations of the major changes presented in the recently published clinical guidelines for the management of HF.
WHAT MAKES A BIOMARKER CLINICALLY USEFUL?
To appreciate the appropriate use of any clinical tool, clinicians need to first understand its indications and limitations and how they are defined. There are four major criteria regarding the clinical utility of a biomarker.
First, we have to establish what we are measuring, particularly with accurate and reproducible methods, with rapid turnaround, and at a reasonable cost. Second, we have to determine why we need the biomarker: ie, we need to determine if its measurement provides valuable new information to the clinician, if there is a strong and consistent association between the marker and the disease or outcome, and if this has been validated in a way that is generalizable. Third, we have to determine when measuring the biomarker would help clinical management, whether it is superior to existing tests, and whether there is evidence that it improves outcomes. Last, and perhaps most commonly overlooked, is practicality: ie, how can measuring the biomarkers be incorporated into the clinical workflow?
Not all biomarkers need to fulfill all these criteria in order to be useful, and the usefulness of a biomarker may differ from one patient population to another, from one clinician to another, or from one clinical scenario to another.1 Many clinical biomarkers are applied based on their ability to indicate a specific diagnosis or treatment (eg, glycated hemoglobin), and some have been used to determine the limits of therapy (eg, creatinine or liver function tests to detect end-organ damage). Nevertheless, the overarching goal is to establish the clinical role of a biomarker to provide the opportunity to gain additional insight into a disease state beyond that provided by a standard clinical assessment, and to determine if using the biomarker favorably alters the clinical course.
WHICH BIOMARKERS DO WE ALREADY ROUTINELY MEASURE?
Traditionally, the management of HF requires meticulous monitoring for adverse effects of drug therapy (eg, electrolyte and renal abnormalities with diuretics or drugs targeting the renin-angiotensin-aldosterone system). Although no specific clinical studies have been conducted to support their routine use, electrolytes (sodium, potassium, chloride, bicarbonate) and renal function measurements (blood urea nitrogen [BUN], creatinine) are often repeated periodically in the longitudinal care of patients with HF.2 Diagnostic tests for hemochromatosis, human immunodeficiency virus, rheumatologic disease, amyloidosis, and pheochromocytoma are reasonable in patients presenting with HF in whom there is a clinical suspicion of these diseases.2
For risk stratification, biomarkers that reflect renal insufficiency (particularly sodium, BUN, creatinine, and the estimated glomerular filtration rate [eGFR]) are powerful prognosticators.3 Newer renal markers of glomerular function (such as cystatin C)4,5 or of acute kidney injury (such as neutrophil gelatinase-associated lipocalin)6,7 have been proposed, although their clinical utility beyond prognostication remains to be determined. In fact, head- to-head comparisons have revealed that BUN appeared to be superior to most other renal biomarkers in stratifying short-term and long-term risk.8
Liver function, blood cell count, and thyroid function profiles are checked on some occasions to determine underlying end-organ dysfunction.2 Interestingly, several common laboratory values have consistently been associated with more advanced disease states or with a higher risk of future adverse events. These include serum uric acid (likely reflecting oxidative stress and nucleotide catabolism),9 anemia or red cell distribution width (likely reflecting iron deficiency or hematopoietic insufficiency),10 lymphocytopenia (likely reflecting immune dysfunction), and total bilirubin (likely reflection of hepatobiliary congestion).11
Some biomarkers have been incorporated into risk-stratification in patients with HF.2 However, drugs targeting these biomarkers have yet to be shown to improve clinical outcomes in prospective clinical trials. Several recent examples in chronic systolic HF include allopurinol for elevated uric acid levels12 and darbepoetin alfa for anemia (low hemoglobin).13 Thus, improving the biomarker level with specific treatment may not translate to improved clinical outcomes.
GUIDELINE RECOMMENDATIONS FOR CARDIAC BIOMARKERS IN HEART FAILURE
Clinical guidelines from several countries on the management of HF have expanded the role of biomarker testing in patients with HF.2,14–16 Table 1 shows the recommendations for biomarker testing in HF from the most recent joint guidelines of the American College of Cardiology and the American Heart Association. These recommendations will form the basis of the following discussion of clinically available biomarkers of HF that reflect distinct pathophysiologic processes and that have been cleared by the US Food and Drug Administration (Figure 1).
Biomarkers of myocardial stress: Natriuretic peptides
Natriuretic peptides are primary counterregulatory hormones produced in response to myocardial stress. Natriuretic peptide receptors stimulated by B-type (also “brain”) natriuretic peptide (BNP) lead to an increase in natriuresis, vasodilation, and opposing effects of other overactive neurohormonal systems. The contemporary understanding of how natriuretic peptides are being produced and metabolized is beyond the scope of this review, but generally it is now recognized that natriuretic peptide levels vary widely among patients with the same degree of symptoms or echocardiographic features.17
Of the several types of natriuretic peptide detectable by immunoassay, the two main types available for clinical use in the United States are BNP and amino acid N-terminal pro-BNP (NT-proBNP). Although there is no direct conversion available (NT-proBNP levels are five to eight times higher than BNP levels), their levels are often concordant and both are influenced by factors such as age, body mass index, and renal function. Specifically, natriuretic peptide levels in morbidly obese patients range 30% to 40% lower than levels in patients who are not morbidly obese.18
Studies over the past 10 years of natriuretic peptides in the diagnosis of HF have shown that levels are invariably elevated in underlying HF, while stable (and especially low) levels often track with clinical stability. In the latest clinical guidelines, natriuretic peptide testing has gained the highest level of recommendation for clinical use for any biomarker in HF, especially in the setting of clinical uncertainty (class 1 recommendation, level of evidence A).2,16 Two common clinical scenarios are represented in this indication. When patients present with signs and symptoms suspicious of HF (shortness of breath, fluid retention, peripheral edema, evidence of central congestion), natriuretic peptide testing provides confirmation of an underlying cardiac cause of these symptoms when elevated. Conversely, when there are alternative explanations or if the presentation is subtle and there is some degree of uncertainty, testing natriuretic peptide levels helps establish the diagnosis of HF when levels are higher than the cut-off values, and levels below the cut-off have a high negative predictive value (Table 2).19,20
Meanwhile, for patients with established HF, a deviation from “stable” natriuretic peptide levels (particularly an increase of more than 30%) may represent evolving destabilization that may warrant an intensification of therapy, whereas an unchanged or reduced level may be taken as objective evidence of clinical stability or favorable response to medical therapy. Table 3 outlines the latest Canadian guidelines that offer a practical approach as ongoing studies attempt to clarify the benefits of these strategies.15
The consistent association between elevated natriuretic peptide levels and worse prognosis21 has led to the promise that intensification of medical therapy in those with elevated natriuretic peptide levels can lead to better outcomes. Nevertheless, the rise in natriuretic peptide levels requires interpretation in the clinical context, as not all factors affecting the levels can be relieved by intensifying medical therapy (eg, age, renal insufficiency).
Several prospective, randomized controlled trials have tested this hypothesis, with favorable yet mixed results. Most studies have utilized a BNP measurement less than 100 pg/mL or an NT-proBNP measurement less than 1,000 pg/mL as a therapeutic target. In a recent prospective study that utilized the NT-proBNP threshold, only about half of patients were able to reach the target of less than 1,000 pg/mL.22 Often overlooked is the fact that in the same study, the inability to reach less than 5,000 pg/mL within 3 months after discharge clearly identified advanced, “nonresponsive” HF refractory to medical therapy and with a poor prognosis.23 This is an important point when assessing the clinical utility of biomarkers, as incremental prognostic values may not guarantee the feasibility or ultimate benefit of intensifying drug therapy according to specific biomarker targets. Until we have more insight into whether a care pathway guided by NT-proBNP measurements can lead to a consistent reduction in rates of hospitalization and mortality in HF, it is reasonable to target those with elevated natriuretic peptide levels by reevaluating their treatment regimen to achieve optimal dosing of guideline-directed medical therapy (Class 2a recommendation, level of evidence B).2 Also, the usefulness of BNP and NT-proBNP in guiding therapy for acutely decompensated HF is not well established (Class 2b recommendation, level of evidence C).2
Biomarkers of myocardial injury: Cardiac troponin
Whereas detecting circulating cardiac troponin is helpful in the diagnosis of acute coronary syndrome, the role of cardiac troponin levels in HF is primarily for risk stratification (Class 1 recommendation, level of evidence A in both acute and chronic HF).2 In patients hospitalized with acute decompensated HF, those with elevated troponin I or troponin T at the time of admission had lower systolic blood pressures, lower ejection fractions, and higher rate of in-hospital mortality.24,25 In chronic HF, elevations in both standard and high-sensitivity cardiac troponin levels were associated with increases in all-cause mortality,26 and rise in serial measurements appeared to correlate with an increased risk of future cardiovascular events.27 And with regard to cardiotoxicity, an increase in cardiac troponin over time (either after chemotherapy or with amyloidosis) is indicative of progressive cardiac dysfunction.28,29
Nevertheless, how to adjust medical therapy according to a rise in cardiac troponin levels remains unclear, as levels of cardiac troponin beyond the setting of acute coronary syndrome have appeared not to fluctuate significantly over time and do not seem to be related to underlying coronary events. Newer-generation cardiac troponin assays have yet to provide incremental value compared with standard clinical troponin assays despite their higher sensitivities.26
One common and underappreciated clinical application that combines both diagnostic and prognostic properties of both natriuretic peptide and cardiac troponin testing is the concept of HF staging. This is particularly relevant when there is a progressive change in clinical status (eg, need for hospitalization, change in signs or symptoms) or when a new therapy is started that may promote adverse effects. For example, a patient with pre-existing HF hospitalized with atypical symptoms and deemed not to have HF could be found to have subclinical myocardial necrosis as detected by low concentration of cardiac troponin or higher-than-baseline natriuretic peptide levels in the absence of hypervolemia. Careful assessment of the potential triggers of fluctuations from previous stable levels of cardiac biomarkers is also warranted (eg, atrial fibrillation, dietary indiscretion, infection, and ischemia). Indeed, these may represent objective rather than subjective changes in clinical manifestation of HF, which may warrant a reassessment of disease severity (eg, objective testing for functional capacity or hemodynamics, or even referral for consideration of advanced HF therapeutic options).
Biomarkers of inflammation and fibrosis: Soluble ST2 and galectin-3
Inflammation has long been associated with HF, and clinically available markers of inflammation such as high-sensitivity C-reactive protein (CRP)30,31 and myeloperoxidase32 have consistently tracked with prognosis. The search for a stable biomarker of inflammation has been challenging because inflammation is a dynamic process and because of the lack of treatment options for heightened inflammation.
A promising new protein biomarker, ST2 (suppression of tumorigenicity-2), has been identified in a soluble form (sST2) that binds to interleukin 33 (IL-33) to antagonize the maladaptive response of the myocardium to overload states.33 The levels of sST2 inversely correlate with the ejection fraction and have a positive association with increasing New York Heart Association class, worsening symptoms, and indicators of HF severity, such as norepinephrine levels, diastolic filling pressures, CRP, and natriuretic peptide levels.34 Unlike natriuretic peptides, levels of sST2 are not significantly affected by age, sex, body mass index, and valve disease,34 although recent observations have challenged its cardiac associations.35 In patients with chronic HF, elevated levels of sST2 (especially >35 ng/mL) have been associated with poorer clinical outcomes36 and increased risk of sudden cardiac death in HF.37 In addition, persistently elevated sST2 levels consistently confer poor long-term prognosis. Several studies have also demonstrated the prognostic value of elevated sST2 in predicting long-term risk of death in acute HF, either at baseline38,39 or on serial testing.40
Another new biomarker, galectin-3, has been implicated in fibrosis and in structural and pathophysiologic changes seen in HF.41 Studies have shown that higher levels of galectin-3 in patients with acute HF and chronic HF were associated with more severe cardiac fibrosis and with an increase in left ventricular remodeling.42–44 Serial measurements also confer prognostic information.45 However, many of these studies did not fully account for renal dysfunction as a major confounder, and the relationship between circulating galectin-3 and estimated GFR is strong.46,47 Meanwhile, head-to-head comparisons among galectin-3 and other clinically available biomarkers also revealed that the prognostic value of galectin-3 can be attenuated in the presence of sST2 and NT-proBNP.48,49 Furthermore, careful evaluation of diastolic parameters only showed a modest relationship with galectin-3 levels, especially in those with HF with preserved ejection fraction.50,51
In animal infarction models, disruption of the galectin-3 and IL-33/ST2 pathway with pharmacologic therapy such as mineralocorticoid receptor antagonists may attenuate cardiac remodeling.52,53 It is conceivable that these biomarkers may have mechanistic links with therapeutic benefits. However, the practical uses of galectin-3 and sST2 are still debated (Class 2b recommendation by the latest guidelines2) despite strong statistical associations between biomarker levels and adverse outcomes. The majority of biomarker substudies from clinical trials have suggested that improvements following drug or device therapy were largely confined to patients with lower rather than higher biomarker levels.54,55 Furthermore, validation studies have challenged the incremental prognostic value of these markers when natriuretic peptide levels are available.54,56–58 Thus, more clinical experience and research are warranted, and current clinical applications may be restricted to patient subsets.
BIOMARKERS IN EARLY STAGES OF HEART FAILURE
The potential benefit of biomarker testing may reside in the earlier end of the HF spectrum, especially in patients at risk of but not yet diagnosed with HF (so-called stage A). In the HealthABC study, the future risk of HF in elderly patients can be predicted with a combination of clinical risk factors (age, sex, left ventricular hypertrophy, systolic blood pressure, heart rate, smoking), as well as biochemical risk factors such as albumin, creatinine, and glucose.59 Patients with elevated natriuretic peptide levels are more likely to have underlying cardiac abnormalities and to have poorer long-term outcomes.60 In a recent prospective, randomized controlled trial, participants with a BNP-guided transition to HF therapies (when BNP >50 pg/mL) had a lower incidence of HF than participants without knowledge of BNP levels.61 Elevated levels of clinically available biomarkers of inflammation, such as myeloperoxidase,29,62 ceruloplasmin,63 and CRP,64 have also been associated with an increased risk of future HF. These findings support the notion that biomarkers, especially when combined with clinical risk factors, can serve as indicators of HF vulnerability. If independently confirmed, this will be an important therapeutic approach to the prevention of HF.
PRACTICAL CONSIDERATIONS
An important perspective often overlooked concerns the variability of a biomarker level as it is utilized in clinical practice (Table 4). In general, point-of-care assays are often more variable than the same tests done in clinical laboratories. Sample collection, handling, and processing also introduce a degree of variability. The biologic variability of specific measurements can significantly affect the precision of the measurement. In the case of HF, the biologic variability (as measured in stable patients over time) of natriuretic peptides and galectin-3 are significantly higher than those observed in cardiac troponins or sST2 (> 130% vs approximately 30%).65 Nevertheless because of their relative cardiac specificity, natriuretic peptides have maintained their clinical utility.
- Morrow DA, de Lemos JA. Benchmarks for the assessment of novel cardiovascular biomarkers. Circulation 2007; 115:949–952.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013; 128:e240–327.
- Halkar M, Tang WH. Incorporating common biomarkers into the clinical management of heart failure. Curr Heart Fail Rep 2013; 10:450–457.
- Dupont M, Wu Y, Hazen SL, Tang WH. Cystatin C identifies patients with stable chronic heart failure at increased risk for adverse cardiovascular events. Circ Heart Fail 2012; 5:602–609.
- Tang WH, Van Lente F, Shrestha K, et al. Impact of myocardial function on cystatin C measurements in chronic systolic heart failure. J Card Fail 2008; 14:394– 399.
- Dupont M, Shrestha K, Singh D, et al. Lack of significant renal tubular injury despite acute kidney injury in acute decompensated heart failure. Eur J Heart Fail 2012; 14:597–604.
- Verbrugge FH, Dupont M, Shao Z, et al. Novel urinary biomarkers in detecting acute kidney injury, persistent renal impairment, and all-cause mortality following decongestive therapy in acute decompensated heart failure. J Card Fail 2013; 19:621–628.
- Tang WH, Dupont M, Hernandez AF, et al. Comparative assessment of short-term adverse events in acute heart failure with cystatin C and other estimates of renal function: results from the ASCEND-HF trial. JACC Heart Fail 2015; 3:40–49.
- Vaduganathan M, Greene SJ, Ambrosy AP, et al. Relation of serum uric acid levels and outcomes among patients hospitalized for worsening heart failure with reduced ejection fraction (from the efficacy of vasopressin antagonism in heart failure outcome study with tolvaptan trial). Am J Cardiol 2014; 114:1713–1721.
- Felker GM, Allen LA, Pocock SJ, et al. Red cell distribution width as a novel prognostic marker in heart failure: data from the CHARM Program and the Duke Databank. J Am Coll Cardiol 2007; 50:40–47.
- Wu AH, Levy WC, Welch KB, et al. Association between bilirubin and mode of death in severe systolic heart failure. Am J Cardiol 2013; 111:1192–1197.
- Givertz MM, Anstrom KJ, Redfield MM, et al. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the Xanthine Oxidase Inhibition for Hyperuricemic Heart Failure Patients (EXACT-HF) study. Circulation 2015; 131:1763–1771.
- Swedberg K, Young JB, Anand IS, et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med 2013; 368:1210–1219.
- McMurray JJ, Adamopoulos S, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 2012; 33:1787–1847.
- Moe GW, Ezekowitz JA, O’Meara E, et al. The 2014 Canadian Cardiovascular Society Heart Failure Management Guidelines Focus Update: anemia, biomarkers, and recent therapeutic trial implications. Can J Cardiol 2015; 31:3–16.
- Tang WH, Francis GS, Morrow DA, et al. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical utilization of cardiac biomarker testing in heart failure. Circulation 2007; 116:e99–109.
- Maisel AS, Daniels LB. Breathing not properly 10 years later: what we have learned and what we still need to learn. J Am Coll Cardiol 2012; 60:277–282.
- Mehra MR, Uber PA, Park MH, et al. Obesity and suppressed B-type natriuretic peptide levels in heart failure. J Am Coll Cardiol 2004; 43:1590–1595.
- Januzzi JL, Jr, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP Investigation of Dyspnea in the Emergency Department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Maisel AS, Krishnaswamy P, Nowak RM, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Fonarow GC, Peacock WF, Phillips CO, Givertz MM, Lopatin M. Admission B-type natriuretic peptide levels and in-hospital mortality in acute decompensated heart failure. J Am Coll Cardiol 2007; 49:1943–1950.
- Januzzi JL, Jr, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B- type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1889.
- Gaggin HK, Truong QA, Rehman SU, et al. Characterization and prediction of natriuretic peptide ‘nonresponse’ during heart failure management: results from the ProBNP Outpatient Tailored Chronic Heart Failure (PROTECT) and the NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death (BATTLESCARRED) study. Congest Heart Fail 2013; 19:135–142.
- Peacock WF 4th, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008; 358:2117–2126.
- Felker GM, Hasselblad V, Tang WH, et al. Troponin I in acute decompen- sated heart failure: insights from the ASCEND-HF study. Eur J Heart Fail 2012; 14:1257–1264.
- Grodin JL, Neale S, Wu Y, Hazen SL, Tang WH. Prognostic comparison of different sensitivity cardiac troponin assays in stable heart failure. Am J Med 2015; 128:276–282.
- Masson S, Anand I, Favero C, et al. Serial measurement of cardiac troponin T using a highly sensitive assay in patients with chronic heart failure: data from 2 large randomized clinical trials. Circulation 2012; 125:280–288.
- Cardinale D, Sandri MT, Colombo A, et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation 2004; 109:2749–2754.
- Ky B, Putt M, Sawaya H, et al. Early increases in multiple biomarkers predict subsequent cardiotoxicity in breast cancer patients treated with doxorubicin, taxanes, and trastuzumab. J Am Coll Cardiol 2014; 63:809–816.
- Kalogeropoulos AP, Tang WH, Hsu A, et al. High-sensitivity C-reactive protein in acute heart failure: insights from the ASCEND-HF trial. J Card Fail 2014; 20:319–326.
- Tang WH, Shrestha K, Van Lente F, et al. Usefulness of C-reactive protein and left ventricular diastolic performance for prognosis in patients with left ventricular systolic heart failure. Am J Cardiol 2008; 101:370–373.
- Tang WH, Wu Y, Nicholls SJ, Hazen SL. Plasma myeloperoxidase predicts incident cardiovascular risks in stable patients undergoing medical management for coronary artery disease. Clin Chem 2011; 57:33–39.
- Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest 2007; 117:1538–1549.
- Rehman SU, Mueller T, Januzzi JL, Jr. Characteristics of the novel interleukin family biomarker ST2 in patients with acute heart failure. J Am Coll Cardiol 2008; 52:1458–1465.
- Kaye DM, Mariani JA, van Empel V, Maeder MT. Determinants and implications of elevated soluble ST2 levels in heart failure. Int J Cardiol 2014; 176:1242–1243.
- Ky B, French B, McCloskey K, et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ Heart Fail 2011; 4:180–187.
- Ahmad T, Fiuzat M, Neely B, et al. Biomarkers of myocardial stress and fibrosis as predictors of mode of death in patients with chronic heart failure. JACC Heart Fail 2014; 2:260–268.
- Pascual-Figal DA, Manzano-Fernandez S, Boronat M, et al. Soluble ST2, high-sensitivity troponin T- and N-terminal pro-B-type natriuretic peptide: complementary role for risk stratification in acutely decompensated heart failure. Eur J Heart Fail 2011; 13:718–725.
- Januzzi JL, Jr, Peacock WF, Maisel AS, et al. Measurement of the interleukin family member ST2 in patients with acute dyspnea: results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) study. J Am Coll Cardiol 2007; 50:607-613.
- Boisot S, Beede J, Isakson S, et al. Serial sampling of ST2 predicts 90-day mortality following destabilized heart failure. J Card Fail 2008; 14:732–738.
- Sharma UC, Pokharel S, van Brakel TJ, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004; 110:3121–3128.
- van Kimmenade RR, Januzzi JL, Jr., Ellinor PT, et al. Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure. J Am Coll Cardiol 2006; 48:1217–1224.
- Lok DJ, Van Der Meer P, de la Porte PW, et al. Prognostic value of galectin-3, a novel marker of fibrosis, in patients with chronic heart failure: data from the DEAL-HF study. Clin Res Cardiol 2010; 99:323–328.
- Shah RV, Chen-Tournoux AA, Picard MH, van Kimmenade RR, Januzzi JL. Ga- lectin-3, cardiac structure and function, and long-term mortality in patients with acutely decompensated heart failure. Eur J Heart Fail 2010; 12:826–832.
- van der Velde AR, Gullestad L, Ueland T, et al. Prognostic value of changes in galectin-3 levels over time in patients with heart failure: data from CORONA and COACH. Circ Heart Fail 2013; 6:219–226.
- Tang WH, Shrestha K, Shao Z, et al. Usefulness of plasma galectin-3 levels in systolic heart failure to predict renal insufficiency and survival. Am J Cardiol 2011; 108:385–390.
- Gopal DM, Kommineni M, Ayalon N, et al. Relationship of plasma galectin-3 to renal function in patients with heart failure: effects of clinical status, pathophysiology of heart failure, and presence or absence of heart failure. J Am Heart Assoc 2012; 1:e000760.
- Bayes-Genis A, de Antonio M, Vila J, et al. Head-to-head comparison of 2 myocardial fibrosis biomarkers for long-term heart failure risk stratification: ST2 versus galectin-3. J Am Coll Cardiol 2014; 63:158–166.
- Felker GM, Fiuzat M, Shaw LK, et al. Galectin-3 in ambulatory patients with heart failure: results from the HF-ACTION study. Circ Heart Fail 2012; 5:72–78.
- Edelmann F, Holzendorf V, Wachter R, et al. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail 2015; 17:214–223.
- AbouEzzeddine OF, Haines P, Stevens S, et al. Galectin-3 in heart failure with preserved ejection fraction. A RELAX trial substudy (Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure). JACC Heart failure 2015; 3:245–252.
- Lax A, Sanchez-Mas J, Asensio-Lopez MC, et al. Mineralocorticoid receptor antagonists modulate galectin-3 and interleukin-33/ST2 signaling in left ventricular systolic dysfunction after acute myocardial infarction. JACC Heart failure 2015; 3:50–58.
- Calvier L, Martinez-Martinez E, Miana M, et al. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart failure 2015; 3:59–67.
- Anand IS, Rector TS, Kuskowski M, Adourian A, Muntendam P, Cohn JN. Baseline and serial measurements of galectin-3 in patients with heart failure: relationship to prognosis and effect of treatment with valsartan in the Val-HeFT. Eur J Heart Fail 2013; 15:511–518.
- Gullestad L, Ueland T, Kjekshus J, et al. Galectin-3 predicts response to statin therapy in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CO- RONA). Eur Heart J 2012; 33:2290–2296.
- Felker GM, Fiuzat M, Thompson V, et al. Soluble ST2 in ambulatory patients with heart failure: association with functional capacity and long-term outcomes. Circ Heart Fail 2013; 6:1172–1179.
- Anand IS, Rector TS, Kuskowski M, Snider J, Cohn JN. Prognostic value of soluble ST2 in the Valsartan Heart Failure Trial. Circ Heart Fail 2014; 7:418–426.
- Gullestad L, Ueland T, Kjekshus J, et al. The predictive value of galectin-3 for mortality and cardiovascular events in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA). Am Heart J 2012; 164:878–883.
- Butler J, Kalogeropoulos A, Georgiopoulou V, et al. Incident heart failure prediction in the elderly: the health ABC heart failure score. Circ Heart Fail 2008; 1:125–133.
- McKie PM, Rodeheffer RJ, Cataliotti A, et al. Amino-terminal pro-B-type natriuretic peptide and B-type natriuretic peptide: biomarkers for mortality in a large community-based cohort free of heart failure. Hypertension 2006; 47:874–880.
- Ledwidge M, Gallagher J, Conlon C, et al. Natriuretic peptide-based screening and collaborative care for heart failure: the STOP-HF randomized trial. JAMA 2013; 310:66–74.
- Tang WH, Katz R, Brennan ML, et al. Usefulness of myeloperoxidase levels in healthy elderly subjects to predict risk of developing heart failure. Am J Cardiol 2009; 103:1269–1274.
- Dadu RT, Dodge R, Nambi V, et al. Ceruloplasmin and heart failure in the Atherosclerosis Risk in Communities study. Circ Heart Fail 2013; 6:936–943.
- Kalogeropoulos A, Georgiopoulou V, Psaty BM, et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol 2010; 55:2129–2137.
- Wu AH, Wians F, Jaffe A. Biological variation of galectin-3 and soluble ST2 for chronic heart failure: implication on interpretation of test results. Am Heart J 2013; 165:995–999.
- Morrow DA, de Lemos JA. Benchmarks for the assessment of novel cardiovascular biomarkers. Circulation 2007; 115:949–952.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013; 128:e240–327.
- Halkar M, Tang WH. Incorporating common biomarkers into the clinical management of heart failure. Curr Heart Fail Rep 2013; 10:450–457.
- Dupont M, Wu Y, Hazen SL, Tang WH. Cystatin C identifies patients with stable chronic heart failure at increased risk for adverse cardiovascular events. Circ Heart Fail 2012; 5:602–609.
- Tang WH, Van Lente F, Shrestha K, et al. Impact of myocardial function on cystatin C measurements in chronic systolic heart failure. J Card Fail 2008; 14:394– 399.
- Dupont M, Shrestha K, Singh D, et al. Lack of significant renal tubular injury despite acute kidney injury in acute decompensated heart failure. Eur J Heart Fail 2012; 14:597–604.
- Verbrugge FH, Dupont M, Shao Z, et al. Novel urinary biomarkers in detecting acute kidney injury, persistent renal impairment, and all-cause mortality following decongestive therapy in acute decompensated heart failure. J Card Fail 2013; 19:621–628.
- Tang WH, Dupont M, Hernandez AF, et al. Comparative assessment of short-term adverse events in acute heart failure with cystatin C and other estimates of renal function: results from the ASCEND-HF trial. JACC Heart Fail 2015; 3:40–49.
- Vaduganathan M, Greene SJ, Ambrosy AP, et al. Relation of serum uric acid levels and outcomes among patients hospitalized for worsening heart failure with reduced ejection fraction (from the efficacy of vasopressin antagonism in heart failure outcome study with tolvaptan trial). Am J Cardiol 2014; 114:1713–1721.
- Felker GM, Allen LA, Pocock SJ, et al. Red cell distribution width as a novel prognostic marker in heart failure: data from the CHARM Program and the Duke Databank. J Am Coll Cardiol 2007; 50:40–47.
- Wu AH, Levy WC, Welch KB, et al. Association between bilirubin and mode of death in severe systolic heart failure. Am J Cardiol 2013; 111:1192–1197.
- Givertz MM, Anstrom KJ, Redfield MM, et al. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the Xanthine Oxidase Inhibition for Hyperuricemic Heart Failure Patients (EXACT-HF) study. Circulation 2015; 131:1763–1771.
- Swedberg K, Young JB, Anand IS, et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med 2013; 368:1210–1219.
- McMurray JJ, Adamopoulos S, Anker SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 2012; 33:1787–1847.
- Moe GW, Ezekowitz JA, O’Meara E, et al. The 2014 Canadian Cardiovascular Society Heart Failure Management Guidelines Focus Update: anemia, biomarkers, and recent therapeutic trial implications. Can J Cardiol 2015; 31:3–16.
- Tang WH, Francis GS, Morrow DA, et al. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical utilization of cardiac biomarker testing in heart failure. Circulation 2007; 116:e99–109.
- Maisel AS, Daniels LB. Breathing not properly 10 years later: what we have learned and what we still need to learn. J Am Coll Cardiol 2012; 60:277–282.
- Mehra MR, Uber PA, Park MH, et al. Obesity and suppressed B-type natriuretic peptide levels in heart failure. J Am Coll Cardiol 2004; 43:1590–1595.
- Januzzi JL, Jr, Camargo CA, Anwaruddin S, et al. The N-terminal Pro-BNP Investigation of Dyspnea in the Emergency Department (PRIDE) study. Am J Cardiol 2005; 95:948–954.
- Maisel AS, Krishnaswamy P, Nowak RM, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002; 347:161–167.
- Fonarow GC, Peacock WF, Phillips CO, Givertz MM, Lopatin M. Admission B-type natriuretic peptide levels and in-hospital mortality in acute decompensated heart failure. J Am Coll Cardiol 2007; 49:1943–1950.
- Januzzi JL, Jr, Rehman SU, Mohammed AA, et al. Use of amino-terminal pro-B- type natriuretic peptide to guide outpatient therapy of patients with chronic left ventricular systolic dysfunction. J Am Coll Cardiol 2011; 58:1881–1889.
- Gaggin HK, Truong QA, Rehman SU, et al. Characterization and prediction of natriuretic peptide ‘nonresponse’ during heart failure management: results from the ProBNP Outpatient Tailored Chronic Heart Failure (PROTECT) and the NT-proBNP-Assisted Treatment to Lessen Serial Cardiac Readmissions and Death (BATTLESCARRED) study. Congest Heart Fail 2013; 19:135–142.
- Peacock WF 4th, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008; 358:2117–2126.
- Felker GM, Hasselblad V, Tang WH, et al. Troponin I in acute decompen- sated heart failure: insights from the ASCEND-HF study. Eur J Heart Fail 2012; 14:1257–1264.
- Grodin JL, Neale S, Wu Y, Hazen SL, Tang WH. Prognostic comparison of different sensitivity cardiac troponin assays in stable heart failure. Am J Med 2015; 128:276–282.
- Masson S, Anand I, Favero C, et al. Serial measurement of cardiac troponin T using a highly sensitive assay in patients with chronic heart failure: data from 2 large randomized clinical trials. Circulation 2012; 125:280–288.
- Cardinale D, Sandri MT, Colombo A, et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation 2004; 109:2749–2754.
- Ky B, Putt M, Sawaya H, et al. Early increases in multiple biomarkers predict subsequent cardiotoxicity in breast cancer patients treated with doxorubicin, taxanes, and trastuzumab. J Am Coll Cardiol 2014; 63:809–816.
- Kalogeropoulos AP, Tang WH, Hsu A, et al. High-sensitivity C-reactive protein in acute heart failure: insights from the ASCEND-HF trial. J Card Fail 2014; 20:319–326.
- Tang WH, Shrestha K, Van Lente F, et al. Usefulness of C-reactive protein and left ventricular diastolic performance for prognosis in patients with left ventricular systolic heart failure. Am J Cardiol 2008; 101:370–373.
- Tang WH, Wu Y, Nicholls SJ, Hazen SL. Plasma myeloperoxidase predicts incident cardiovascular risks in stable patients undergoing medical management for coronary artery disease. Clin Chem 2011; 57:33–39.
- Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest 2007; 117:1538–1549.
- Rehman SU, Mueller T, Januzzi JL, Jr. Characteristics of the novel interleukin family biomarker ST2 in patients with acute heart failure. J Am Coll Cardiol 2008; 52:1458–1465.
- Kaye DM, Mariani JA, van Empel V, Maeder MT. Determinants and implications of elevated soluble ST2 levels in heart failure. Int J Cardiol 2014; 176:1242–1243.
- Ky B, French B, McCloskey K, et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ Heart Fail 2011; 4:180–187.
- Ahmad T, Fiuzat M, Neely B, et al. Biomarkers of myocardial stress and fibrosis as predictors of mode of death in patients with chronic heart failure. JACC Heart Fail 2014; 2:260–268.
- Pascual-Figal DA, Manzano-Fernandez S, Boronat M, et al. Soluble ST2, high-sensitivity troponin T- and N-terminal pro-B-type natriuretic peptide: complementary role for risk stratification in acutely decompensated heart failure. Eur J Heart Fail 2011; 13:718–725.
- Januzzi JL, Jr, Peacock WF, Maisel AS, et al. Measurement of the interleukin family member ST2 in patients with acute dyspnea: results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) study. J Am Coll Cardiol 2007; 50:607-613.
- Boisot S, Beede J, Isakson S, et al. Serial sampling of ST2 predicts 90-day mortality following destabilized heart failure. J Card Fail 2008; 14:732–738.
- Sharma UC, Pokharel S, van Brakel TJ, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004; 110:3121–3128.
- van Kimmenade RR, Januzzi JL, Jr., Ellinor PT, et al. Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure. J Am Coll Cardiol 2006; 48:1217–1224.
- Lok DJ, Van Der Meer P, de la Porte PW, et al. Prognostic value of galectin-3, a novel marker of fibrosis, in patients with chronic heart failure: data from the DEAL-HF study. Clin Res Cardiol 2010; 99:323–328.
- Shah RV, Chen-Tournoux AA, Picard MH, van Kimmenade RR, Januzzi JL. Ga- lectin-3, cardiac structure and function, and long-term mortality in patients with acutely decompensated heart failure. Eur J Heart Fail 2010; 12:826–832.
- van der Velde AR, Gullestad L, Ueland T, et al. Prognostic value of changes in galectin-3 levels over time in patients with heart failure: data from CORONA and COACH. Circ Heart Fail 2013; 6:219–226.
- Tang WH, Shrestha K, Shao Z, et al. Usefulness of plasma galectin-3 levels in systolic heart failure to predict renal insufficiency and survival. Am J Cardiol 2011; 108:385–390.
- Gopal DM, Kommineni M, Ayalon N, et al. Relationship of plasma galectin-3 to renal function in patients with heart failure: effects of clinical status, pathophysiology of heart failure, and presence or absence of heart failure. J Am Heart Assoc 2012; 1:e000760.
- Bayes-Genis A, de Antonio M, Vila J, et al. Head-to-head comparison of 2 myocardial fibrosis biomarkers for long-term heart failure risk stratification: ST2 versus galectin-3. J Am Coll Cardiol 2014; 63:158–166.
- Felker GM, Fiuzat M, Shaw LK, et al. Galectin-3 in ambulatory patients with heart failure: results from the HF-ACTION study. Circ Heart Fail 2012; 5:72–78.
- Edelmann F, Holzendorf V, Wachter R, et al. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail 2015; 17:214–223.
- AbouEzzeddine OF, Haines P, Stevens S, et al. Galectin-3 in heart failure with preserved ejection fraction. A RELAX trial substudy (Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure). JACC Heart failure 2015; 3:245–252.
- Lax A, Sanchez-Mas J, Asensio-Lopez MC, et al. Mineralocorticoid receptor antagonists modulate galectin-3 and interleukin-33/ST2 signaling in left ventricular systolic dysfunction after acute myocardial infarction. JACC Heart failure 2015; 3:50–58.
- Calvier L, Martinez-Martinez E, Miana M, et al. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart failure 2015; 3:59–67.
- Anand IS, Rector TS, Kuskowski M, Adourian A, Muntendam P, Cohn JN. Baseline and serial measurements of galectin-3 in patients with heart failure: relationship to prognosis and effect of treatment with valsartan in the Val-HeFT. Eur J Heart Fail 2013; 15:511–518.
- Gullestad L, Ueland T, Kjekshus J, et al. Galectin-3 predicts response to statin therapy in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CO- RONA). Eur Heart J 2012; 33:2290–2296.
- Felker GM, Fiuzat M, Thompson V, et al. Soluble ST2 in ambulatory patients with heart failure: association with functional capacity and long-term outcomes. Circ Heart Fail 2013; 6:1172–1179.
- Anand IS, Rector TS, Kuskowski M, Snider J, Cohn JN. Prognostic value of soluble ST2 in the Valsartan Heart Failure Trial. Circ Heart Fail 2014; 7:418–426.
- Gullestad L, Ueland T, Kjekshus J, et al. The predictive value of galectin-3 for mortality and cardiovascular events in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA). Am Heart J 2012; 164:878–883.
- Butler J, Kalogeropoulos A, Georgiopoulou V, et al. Incident heart failure prediction in the elderly: the health ABC heart failure score. Circ Heart Fail 2008; 1:125–133.
- McKie PM, Rodeheffer RJ, Cataliotti A, et al. Amino-terminal pro-B-type natriuretic peptide and B-type natriuretic peptide: biomarkers for mortality in a large community-based cohort free of heart failure. Hypertension 2006; 47:874–880.
- Ledwidge M, Gallagher J, Conlon C, et al. Natriuretic peptide-based screening and collaborative care for heart failure: the STOP-HF randomized trial. JAMA 2013; 310:66–74.
- Tang WH, Katz R, Brennan ML, et al. Usefulness of myeloperoxidase levels in healthy elderly subjects to predict risk of developing heart failure. Am J Cardiol 2009; 103:1269–1274.
- Dadu RT, Dodge R, Nambi V, et al. Ceruloplasmin and heart failure in the Atherosclerosis Risk in Communities study. Circ Heart Fail 2013; 6:936–943.
- Kalogeropoulos A, Georgiopoulou V, Psaty BM, et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol 2010; 55:2129–2137.
- Wu AH, Wians F, Jaffe A. Biological variation of galectin-3 and soluble ST2 for chronic heart failure: implication on interpretation of test results. Am Heart J 2013; 165:995–999.
KEY POINTS
- The usefulness of a biomarker may differ from one patient population to another, from one clinician to another, or from one clinical scenario to another.
- For risk stratification in heart failure (HF), biomarkers that reflect renal insufficiency are especially powerful prognosticators.
- In the latest clinical guidelines, natriuretic peptide testing has gained the highest level of recommendation for clinical use for any biomarker in HF.
- In general, point-of-care assays are often more variable than the same tests done in clinical laboratories; sample collection, handling, and processing also introduce variability.
The role of aldosterone receptor antagonists in the management of heart failure: An update
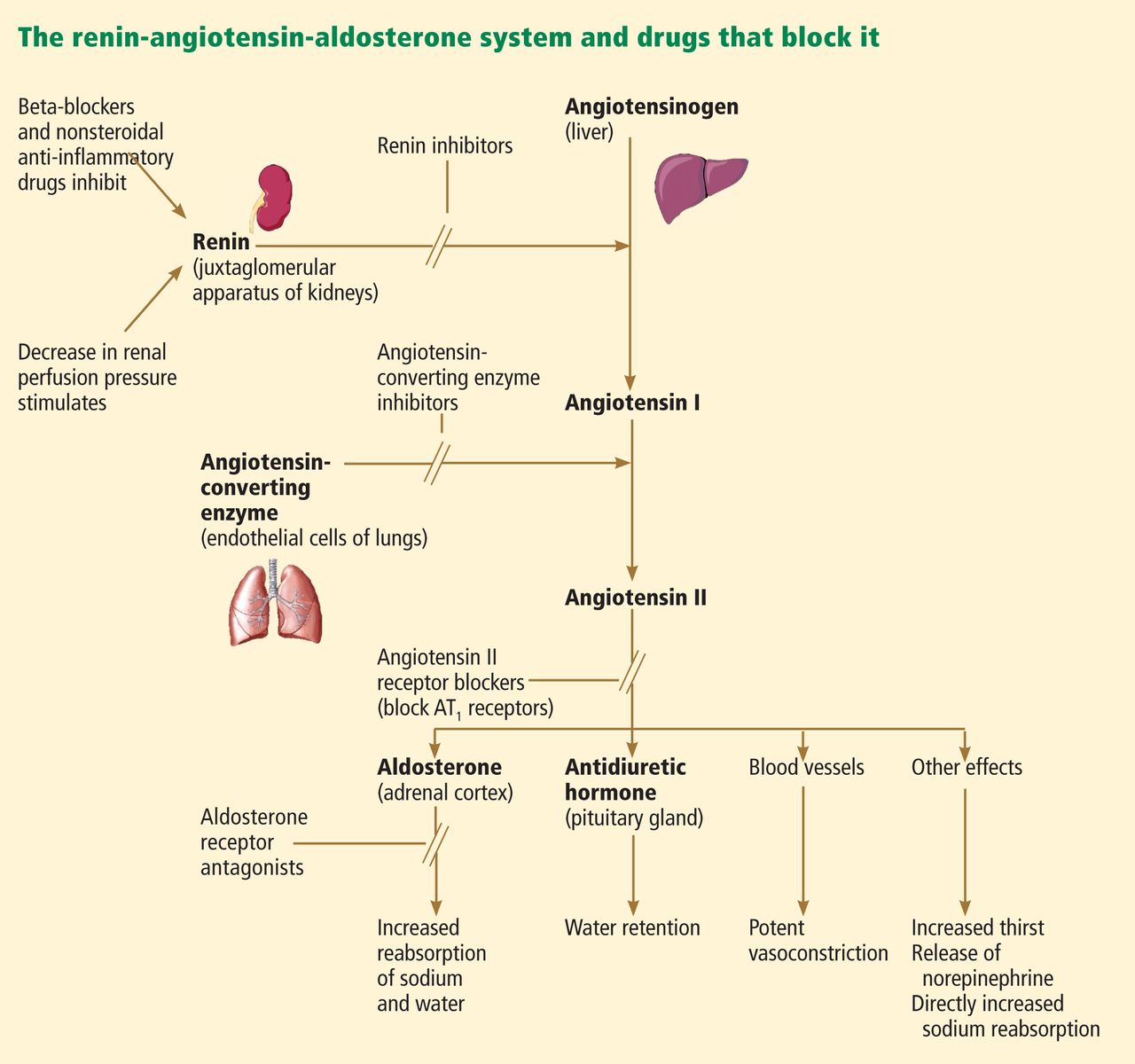
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23

Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
KEY POINTS
- Although caution is advised in starting ARAs, these drugs are commonly underused in heart failure.
- Aldosterone “escape” can blunt the effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. This is the rationale for also using ARAs.
- The major trials of ARAs in heart failure to date have been the Randomized Aldactone Evaluation Study (RALES), the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), and the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF).
- Close monitoring is essential when starting an ARA, as severe hyperkalemia and renal insufficiency can occur.