Dr. Tsai is from the Department of Internal Medicine, Cedars-Sinai Medical Center, Los Angeles, California. Drs. Hu and Worswick are from the Division of Dermatology, David Geffen School of Medicine, University of California, Los Angeles. Dr. Zussman is from the Department of Dermatology, University of Utah, Salt Lake City.
The authors report no conflict of interest.
Correspondence: Scott Worswick, MD, 200 Medical Plaza at UCLA, Ste 450, Los Angeles, CA 90095 (sworswick@mednet.ucla.edu).
Relapsing polychondritis (RP) is a rare autoimmune disease of the cartilaginous structures of the body with many systemic manifestations including meningoencephalitis (ME). We present a case of a man with RP-associated ME that was responsive to steroid treatment. An updated literature review of 7 cases of RP-associated ME also is provided. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
Meningoencephalitis (ME) is a potentially rare complication of relapsing polychondritis (RP).
Treatment of ME due to RP can include high-dose steroids and biologics.
References
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
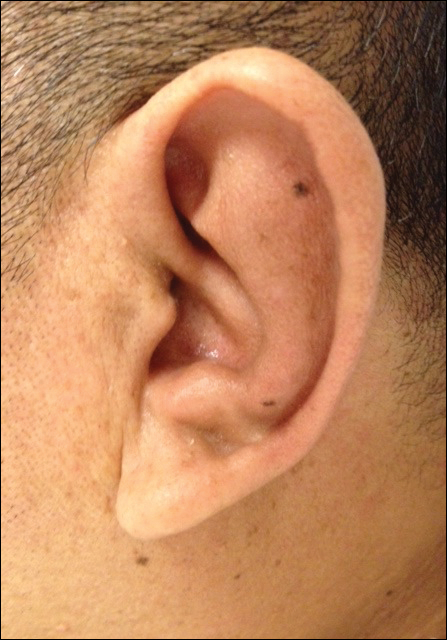
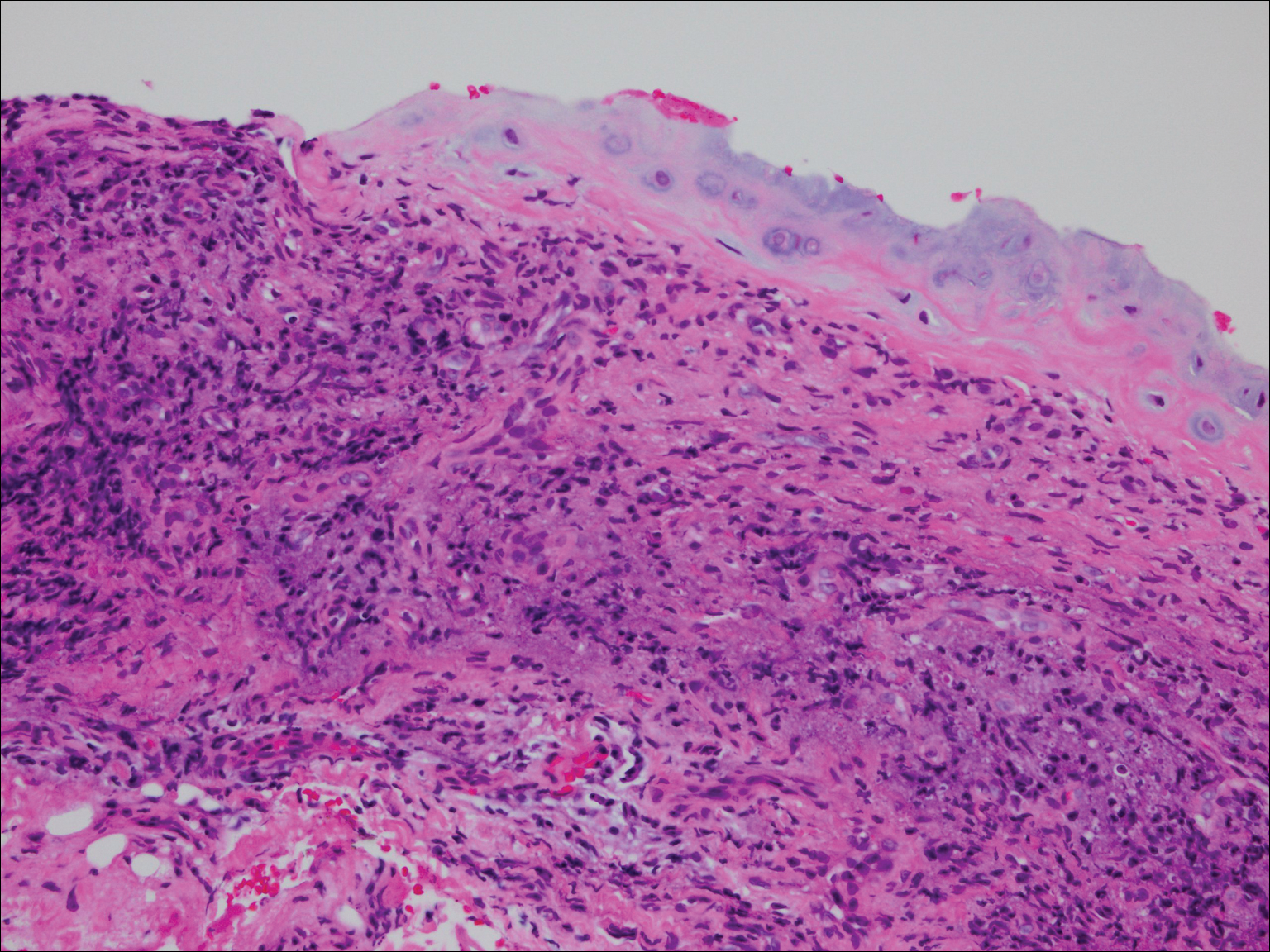
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
Figure 1. Auricular erythema and edema on the left ear with sparing of the lobule.
Figure 2. Histopathology revealed prominent neutrophilic inflammatory infiltrate with scattered lymphocytes and histiocytes. Degeneration of the cartilaginous tissue also was evident with pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes (H&E, original magnification ×40).
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.