To the Editor:
Pityriasis rubra pilaris (PRP) is a rare inflammatory dermatosis of unknown etiology characterized by erythematosquamous salmon-colored plaques with well-demarcated islands of unaffected skin and hyperkeratotic follicles.1 In the United States, an incidence of 1 in 3500to 5000 patients presenting to dermatology clinics has been reported.2 Pityriasis rubra pilaris has several subtypes and variability in presentation that can make accurate and timely diagnosis challenging.3-5 Herein, we present a case of PRP with complex diagnostic and therapeutic challenges.
A 22-year-old woman presented with symmetrical, well-demarcated, hyperkeratotic, erythematous plaques with a carnauba wax–like appearance on the palms (Figure 1), soles, elbows, and trunk covering approximately 5% of the body surface area. Two weeks prior to presentation, she experienced an upper respiratory tract infection without any treatment and subsequently developed redness on the palms, which became very hard and scaly. The redness then spread to the elbows, soles, and trunk. She reported itching as well as pain in areas of fissuring. Hand mobility became restricted due to thick scale.
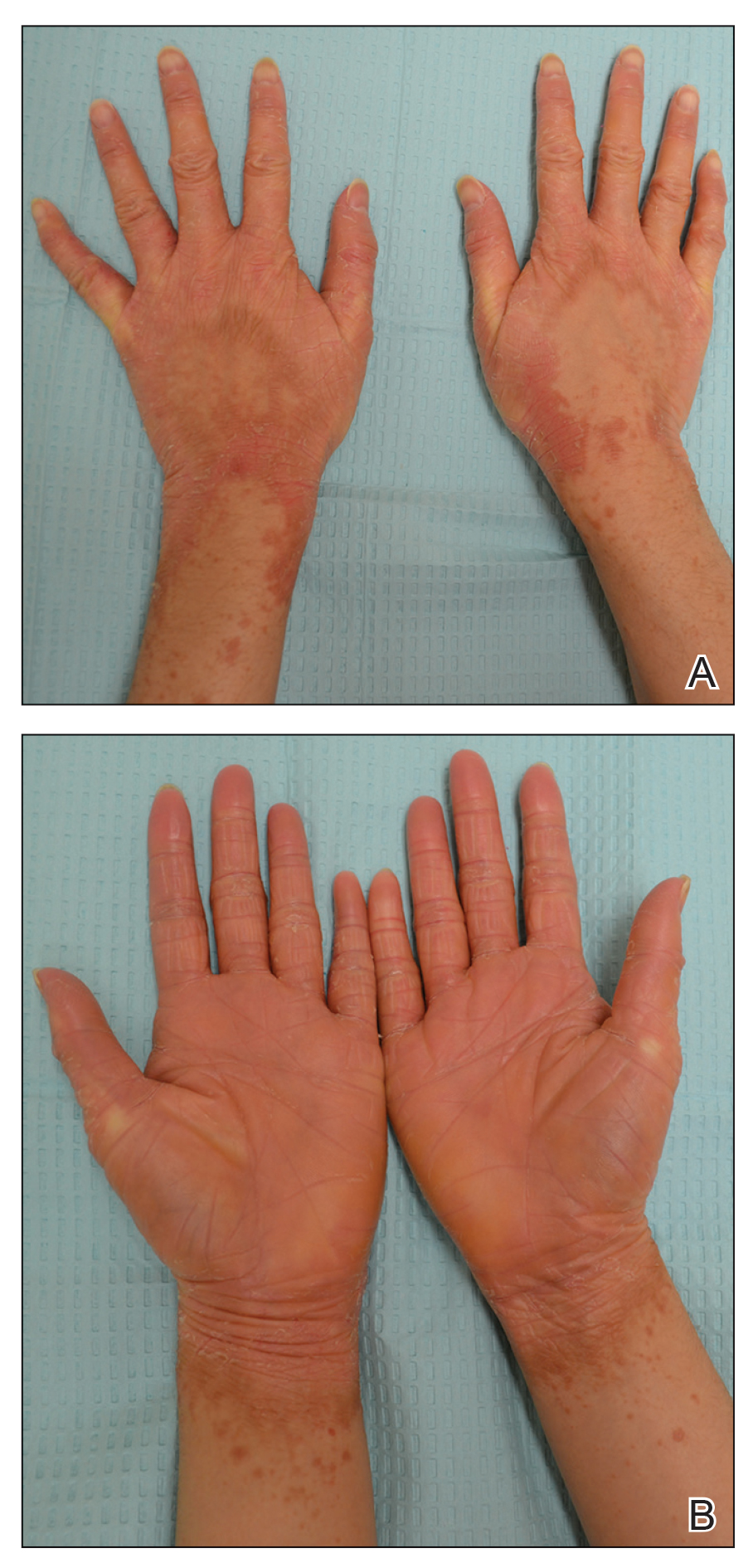
FIGURE 1. A and B, Pityriasis rubra pilaris on the hands before treatment.
The patient’s medical history was notable for suspected psoriasis 9 years prior, but there were no records or biopsy reports that could be obtained to confirm the diagnosis. She also reported a similar skin condition in her father, which also was diagnosed as psoriasis, but this diagnosis could not be verified.
Although the morphology of the lesions was most consistent with localized PRP, atypical psoriasis, palmoplantar keratoderma (PPK), and erythroderma progressive symmetrica (EPS) also were considered given the personal and family history of suspected psoriasis. A biopsy could not be obtained due to an insurance issue. She was started on clobetasol cream 0.05% and ointment. At 2-week follow-up, her condition remained unchanged. Empiric systemic treatment was discussed, which would potentially work for diagnoses of both PRP and psoriasis. Due to the history of psoriasis and level of discomfort, cyclosporine 300 mg once daily was started to gain rapid control of the disease. Methotrexate also was considered due to its efficacy and economic considerations but was not selected due to patient concerns about the medication.
After 10 weeks of cyclosporine treatment, our patient showed some improvement of the skin with decreased scale and flattening of plaques but not complete resolution. At this point, a biopsy was able to be obtained with prior authorization. A 4-mm punch biopsy of the right flank demonstrated a psoriasiform and papillated epidermis with multifocally capped, compact parakeratosis and minimal lymphocytic infiltrate consistent with PRP. Although EPS also was on the histologic differential, clinical history was more consistent with a diagnosis of PRP. There was some minimal improvement with cyclosporine, but with the diagnosis of PRP confirmed, a systemic retinoid became the treatment of choice. Although acitretin is the preferred treatment for PRP, given that pregnancy would be contraindicated during and for 3 years following acitretin therapy, a trial of isotretinoin 40 mg once daily was started due to its shorter half-life compared to acitretin and was continued for 3 months (Figure 2).6,7
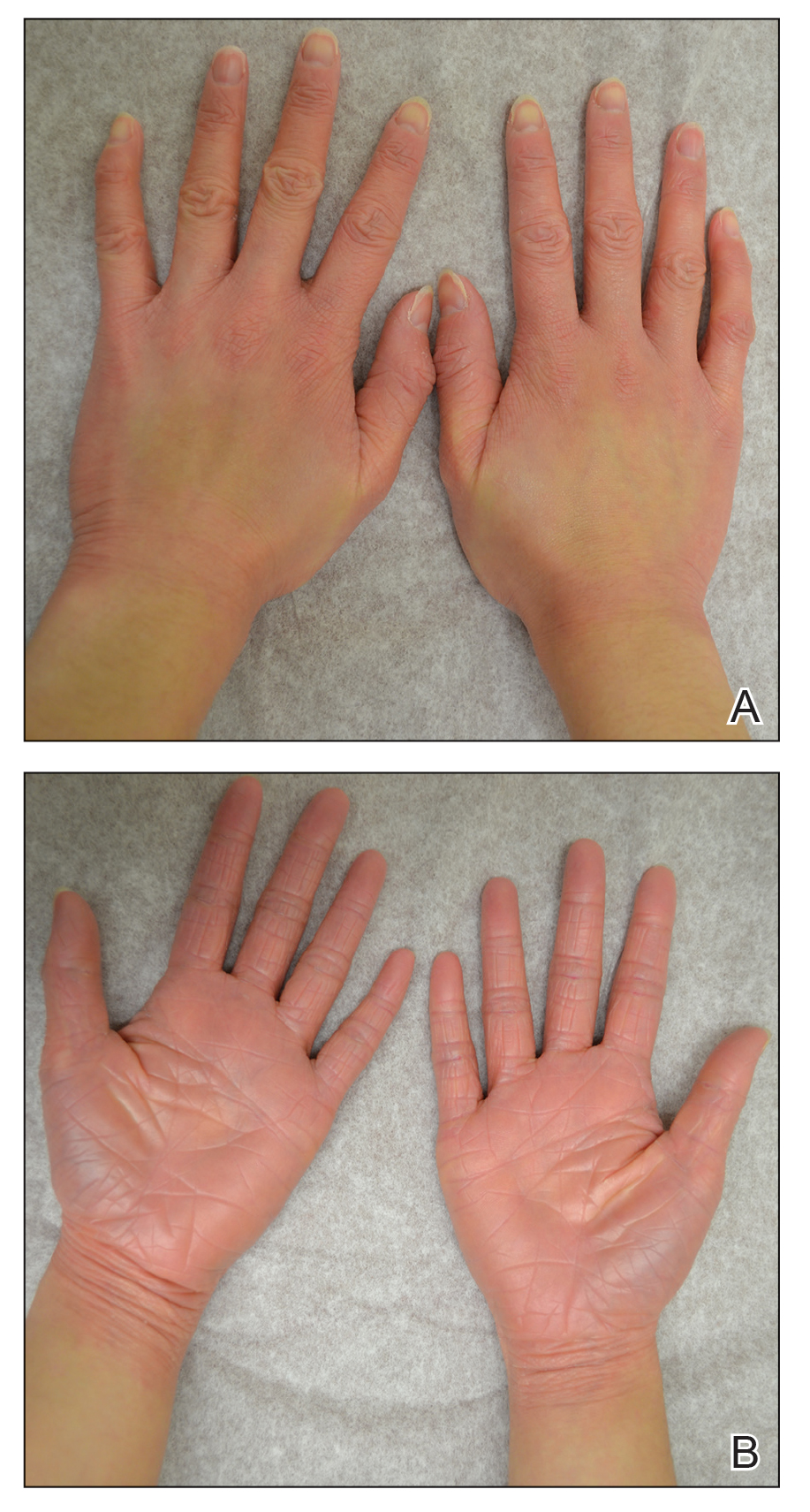
FIGURE 2. A and B, The hands after treatment with cyclosporine 300 mg daily for 10 weeks, followed by isotretinoin 40 mg daily for 3 months.
The diagnosis of PRP often can be challenging given the variety of clinical presentations. This case was an atypical presentation of PRP with several learning points, as our patient’s condition did not fit perfectly into any of the 6 types of PRP. The age of onset was atypical at 22 years old. Pityriasis rubra pilaris typically presents with a bimodal age distribution, appearing either in the first decade or the fifth to sixth decades of life.3,8 Her clinical presentation was atypical for adult-onset types I and II, which typically present with cephalocaudal progression or ichthyosiform dermatitis, respectively. Her presentation also was atypical for juvenile onset in types III, IV, and V, which tend to present in younger children and with different physical examination findings.3,8
The morphology of our patient’s lesions also was atypical for PRP, PPK, EPS, and psoriasis. The clinical presentation had features of these entities with erythema, fissuring, xerosis, carnauba wax–like appearance, symmetric scale, and well-demarcated plaques. Although these findings are not mutually exclusive, their combined presentation is atypical. Coupled with the ambiguous family history of similar skin disease in the patient’s father, the discussion of genodermatoses, particularly PPK, further confounded the diagnosis.4,9 When evaluating for PRP, especially with any family history of skin conditions, genodermatoses should be considered. Furthermore, our patient’s remote and unverifiable history of psoriasis serves as a cautionary reminder that prior diagnoses and medical history always should be reasonably scrutinized. Additionally, a drug-induced PRP eruption also should be considered. Although our patient received no medical treatment for the upper respiratory tract infection prior to the onset of PRP, there have been several reports of drug-induced PRP.10-12