User login
Research and Reviews for the Practicing Oncologist
Game changers in pediatric cancer
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
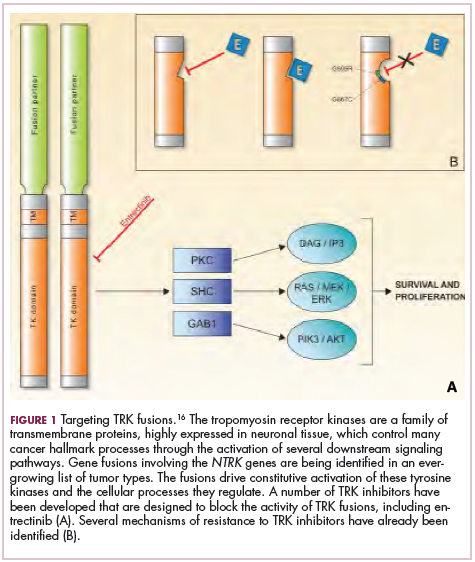
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
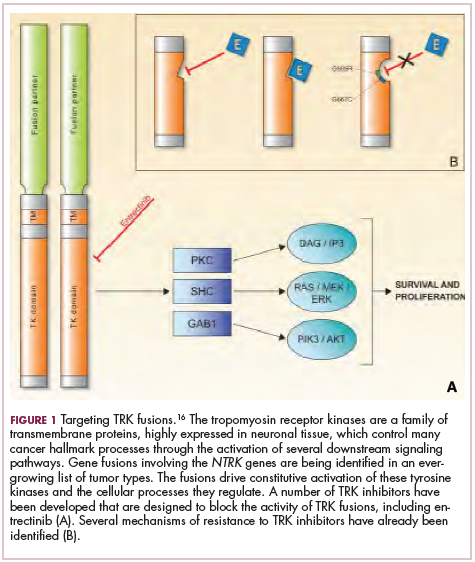
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.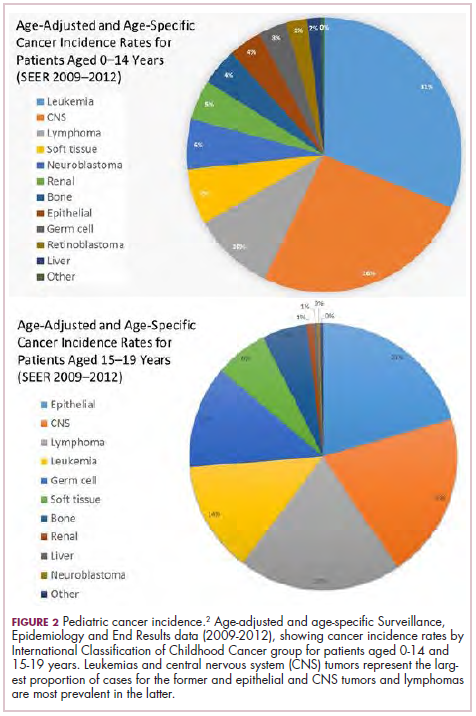
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).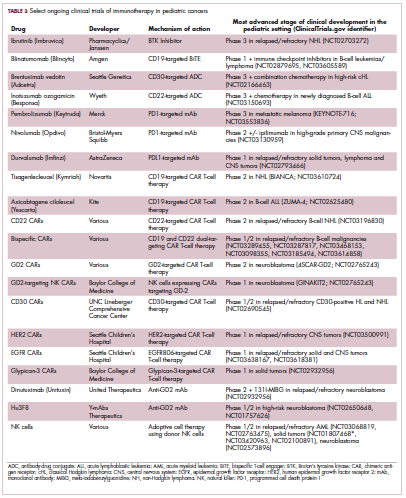
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
Although there have been significant improvements in patient outcomes for some forms of pediatric cancer, progress has been painfully slow for others. An increasing understanding of pediatric cancers is highlighting the unique molecular drivers and challenging the assumption that drugs developed in adults can be applied to children and young adults. Here, we discuss game-changing therapeutic advances and a shifting view of childhood cancers.
Unique genomic background
Although pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, they are the second leading cause of death in children aged 1 to 14 years. There are many different histological tumor types under the umbrella of childhood cancers, of which the most common are leukemias, central nervous system tumors, and lymphomas (Figure 1).1,2
Significant progress has been made in the treatment of certain pediatric cancers in recent decades, exemplified by pediatric acute lymphoblastic leukemia (ALL), which has been transformed from a virtually incurable cancer to one in which 5-year survival rates now reach up to 90%. In other forms of pediatric cancer, however, survival rates have stagnated and little progress has been made in the development of effective new therapies.3
Because of their rarity, pediatric cancers are difficult to study and adequate enrollment of children in clinical trials can be challenging. Pharmaceutical companies are often hesitant to test drugs in the pediatric population in patients who often cannot advocate for themselves. As a result, the activity of drugs developed in adult patients has often been inferred in pediatric patients with the same tumor type or molecular aberrations. However, as researchers have gathered more information about pediatric cancers, there has been increasing recognition of their unique attributes and the need for dedicated clinical trials in this patient population.
Pediatric cancers tend to be found in the developing mesodermic tissue, whereas adult cancers are more prevalent in the epithelial tissues. Genome sequencing studies have revealed a much lower mutational burden in pediatric cancers and the mechanisms of oncogenesis are also quite different; adult tumors can develop from a series of acquired gene mutations, but pediatric tumors tend to develop from a single catastrophic event.4,5
Even the same type of cancer in a pediatric and adult patient can be quite different, with very different underlying molecular mechanisms. In a recent genomic analysis of different types of pediatric cancer by researchers at St Jude’s Children’s Research Hospital, less than half of the identified mutated genes were found to be similar to those found in adult patients.6
A ‘magic bullet’?
Chromosomal rearrangements are common in pediatric cancers. This type of molecular abnormality can result in a fusion of 2 different genes when the chromosome breaks apart and the pieces join back together in a muddled order. If the genetic code fuses in a manner that is “readable” by the cell, then it can drive aberrant activation of one or both genes.7 Gene fusions often involve kinase enzymes that are essential players in cell signaling pathways regulating hallmark cancer processes, such as unchecked cell proliferation. The fusion drives the constitutive activation of the kinase and, thus, these downstream signaling pathways.
One of the first chromosomal rearrangements linked to cancer, BCR-ABL1 – more commonly known as the Philadelphia chromosome – results in aberrant activation of the ABL1 kinase. It is present in nearly all cases of chronic myeloid leukemia (CML) and 3% to 5% of patients with ALL, and thus became the central focus of targeted drug development. Imatinib was initially approved by the US Food and Drug Administration (FDA) in 2001 for the treatment of adult patients with CML and had such a significant impact on the treatment landscape that it made the cover of Time magazine as a “magic bullet” in the war on cancer.8
Approval was expanded into pediatric patients in 2006 and for pediatric patients with ALL in 2013. However, as with the use of most kinase inhibitors, tumors can evolve under the selective pressure of treatment, developing additional molecular abnormalities that drive resistance.9
Next-generation multikinase inhibitors that more potently inhibit the BCR-ABL1 fusion protein have been developed to provide additional treatment options for patients who become resistant to imatinib. Dasatinib and nilotinib are among several drugs that have recently been approved for pediatric cancer therapy (Table 1). Both therapies were approved to treat children with Philadelphia chromosome-positive CML in the chronic phase in either the front- or second-line setting after failure of imatinib.
The approval of dasatinib was based on data from 97 patients across 2 trials, 51 of whom were newly diagnosed and 46 previously treated with imatinib. Most of the patients were treated with dasatinib 60 mg/m2 once daily. After 2 years of follow-up, more than 95% of newly diagnosed patients and 82.6% of relapsed/refractory patients had complete cytogenetic response.10
Nilotinib was approved on the basis of findings from 2 clinical trials including 69 patients – 1 trial involving patients who were refractory to or relapsed after dasatinib and imatinib treatment, and 1 that included both relapsed/refractory and newly diagnosed patients. Patients received nilotinib 230 mg/m2 twice daily, rounded to the nearest 50-mg dose, in 28-day cycles. By cycle 12, the cumulative major molecular response rate (MMR) was 47.7% in patients with relapsed/refractory disease, and 64% in newly diagnosed patients.11 Clinical trials of both drugs in the pediatric setting are ongoing.
Other prominent gene fusions
Gene fusions involving the anaplastic lymphoma kinase (ALK) occur in patients with non–small-cell lung cancer and ALK inhibitors have provided an effective new treatment option for patients whose tumors display this abnormality.
ALK fusions are also a prominent feature of several kinds of pediatric cancers and ALK inhibitors offer promise in this setting.7,12 An NPM-ALK fusion is found in 90% of pediatric anaplastic large cell lymphoma (ALCL) cases,13 whereas a variety of ALK fusions are found in up to half of patients with inflammatory myofibroblastic tumor (IMT), a rare form of soft tissue sarcoma.14 ALK inhibitors are being tested in a variety of clinical trials in pediatric patients (Table 2).
The results of a small phase 1 study of crizotinib in pediatric patients with ALK-positive ALCL (n = 26) or IMT (n = 14) were recently published. ALCL patients received crizotinib at a dose of 165 mg/m2, while IMT patients were given 100, 165, or 280 mg/m2. For the latter, the results were presented as a pooled cohort since safety and efficacy data were similar across dose levels. The overall response rate (ORR) was 83% for patients with ALCL and 86% for those with IMT. Grade 3/4 adverse events occurred in 83% and 71% of patients, respectively, and most commonly involved reduced neutrophil count.15
Most recently and perhaps most promisingly, fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) gene have generated significant buzz. There are 3 NTRK genes, NTRK1, 2, and 3, which encode the TRKA, TRKB, and TRKC proteins, respectively.
To date, 22 different partner genes have been identified that can fuse with the NTRK genes and, as with other kinase fusions, drive constitutive activation of the receptor proteins and downstream oncogenic signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway (Figure 2).
NTRK fusions are being identified in an ever-growing number of cancer types, but are typically found in a small percentage of patients. However, in certain rare pediatric tumors, including congenital infantile fibrosarcoma and papillary thyroid cancer, they are found at much higher frequencies.
TRK inhibitors have been developed to target the fusion proteins and, given the spread of NTRK fusions across different types of cancers, they offer the most substantial promise as the next tumor agnostic cancer therapy – to treat patients based on the shared presence of a molecular aberration, irrespective of the type of cancer.16
The ongoing SCOUT trial is evaluating larotrectinib (LOXO-101) in pediatric patients. Among 24 patients (17 with NTRK fusions and 7 without) with infantile fibrosarcoma (47%), soft tissue sarcoma (41%) or papillary thyroid cancer (12%), the ORR was 93%, including complete response (CR) in 13% of patients.17
Preliminary results from an ongoing phase 1/2 study of entrectinib in pediatric patients with extracranial solid tumors were also recently presented at the annual meeting of the American Society for Clinical Oncology (ASCO). Among 15 evaluable patients enrolled to date, 3 have NTRK fusions and all experienced an objective response, with 1 (a patient with IMT) ongoing at 10 months.18
CAR T cells transformative in ALL
A variety of different types of immunotherapy have been tested in patients with pediatric cancers. In general, immunotherapy has proved less effective than in adult cancers, possibly because of the lower tumor mutation burden in pediatric cancers, which means there are likely fewer cancer antigens to provoke an anti-tumor immune response.
There are notable exceptions among the disappointments, however, and most exciting is the development of chimeric antigen receptor (CAR) T cells. CAR T cells fall into a category of immunotherapy known as adoptive cell therapy (ACT), in which immune cells are harvested from a patient and grown outside the body to increase their numbers before being reinfused into the patient.
In the case of CAR T-cell therapy, the cells are genetically engineered to express a CAR that endows them with tumor-targeting capabilities. To date, the development of CAR T cells has focused on the use of the CD19 antigen as a target, which is highly expressed on a variety of B-cell malignancies, including several of the most common forms of pediatric cancer. ASCO shined the spotlight on CAR T-cell therapy this year, naming it the Advance of the Year for 2018, saying that the treatment is “poised to transform childhood ALL.”19
Two CD19-targeted CAR T-cell therapies – tisagenlecleucel and axicabtagene ciloleucel – were brought to market in 2017. Only tisagenlecleucel is approved in the pediatric ALL population, however, having been awarded approval for the treatment of patients aged up to 25 years whose disease is refractory to or relapsed after receiving at least 2 prior therapies. In the pivotal trial, complete responses were observed in more than 60% of patients.20 Clinical trials of both CAR T-cell therapies in pediatric ALL and non-Hodgkin lymphoma are ongoing (Table 3).
CD19 has also proven to be a promising target for other forms of immunotherapy, including a new type of antibody known as a bispecific T-cell engager (BiTE). In 2014, blinatumomab became the first BiTE to receive regulatory approval, for the treatment of adult patients with relapsed/refractory ALL. Blinatumomab also targets the CD3 protein on T cells and helps to bring cancer cells and cytotoxic immune cells into close enough proximity that an immunological synapse can be formed between the two, facilitating tumor cell killing.21
In 2016, the approved indication was expanded into the pediatric population based on the results of a phase 1/2 study in which the safety and efficacy of blinatumomab were evaluated in 93 pediatric patients with relapsed/refractory ALL. Among the 70 patients who received the recommended dose of 5µg/m2 a day for the first 7 days, followed by 15µg/m2 a day thereafter, 51% achieved complete remission within the first 2 cycles, 52% of whom achieved minimal residual disease (MRD).22 Most recently, the FDA expanded the indication for blinatumomab to include patients (both adults and children) who are in remission, but MRD positive.23Despite the dramatic responses, many patients relapse after treatment with CD19-targeted CAR T cells, and researchers have uncovered numerous mechanisms of resistance. Among them is the loss of the CD19 antigen on the surface of target cells, such that a CD19-positive tumor becomes CD19-negative after treatment, driving relapse.24-26Several strategies for overcoming CD19-negative relapse are already being investigated, including the development of CD22-targeted CAR T cells and bispecific CAR T cells that target both CD19 and CD22. The results of a first-in-human trial of anti-CD22 CAR T-cell therapy were recently published. Among 21 pediatric and adult patients with relapsed/refractory B-cell ALL who were treated with either 3 x 105 cells/kg, 1 x 106 cells/kg, or 3 x 106 cells/kg, complete responses were observed in 57%.27
Results from 15 pediatric patients enrolled in a trial evaluating CD22-targeted CAR T cells as salvage therapy for those who relapse after CD19-targeted CAR T cell therapy were presented at the recent Congress of the European Hematology Association in Stockholm, Sweden. Patients who had undergone a stem cell transplant received the CAR T cells at a dose of 0.9 x 105 cell/kg and those who had not undergone a transplant received a dose of 8.2 x 105 cells/kg. At 30 days after CAR T cell infusion, the CR rate was 80% and the treatment was well tolerated.28
More immunotherapy approvals
The immune checkpoint inhibitors, which work by blocking inhibitory receptors on the surface of T cells, have also had recent approvals in pediatric patient populations. Pembrolizumab and nivolumab, inhibitors of the programmed cell death receptor 1 (PD-1) protein, have both been approved for use in adult and pediatric patients (older than 12 years) with relapsed/refractory metastatic colorectal cancer (and other solid tumors in the case of pembrolizumab) that display defects in the mismatch repair pathway that fixes damaged DNA or in patients that have high levels of microsatellite instability. Both deficient mismatch repair and microsatellite instability–high can indicate a high mutation burden in a tumor, which may predict increased sensitivity to immunotherapy.29
The approval in pediatric patients in both of those instances, however, was not based on data in pediatric patient populations but extrapolated from adult patients. Pembrolizumab is also approved for the treatment of adults and pediatric patients with classical Hodgkin lymphoma (cHL) after 3 or more previous treatments, but once again efficacy in the pediatric population was inferred from clinical trials performed in adults. Most recently, pembrolizumab was approved for the treatment of adult and pediatric patients with relapsed or refractory primary mediastinal large B-cell lymphoma.30Ipilimumab, which targets a different T cell receptor – cytotoxic T lymphocyte antigen-4 (CTLA-4) – has been approved for the treatment of pediatric patients aged 12 years and older with metastatic melanoma. This expanded indication, following on from its approval in adult patients in 2011, was based on data from 2 trials in which objective responses were observed in 2 out of 17 patients, including 1 partial response that lasted 16 months.31Finally, antibody-drug conjugates (ADC), in which tumor antigen-targeting monoclonal antibodies are conjugated to cytotoxic payloads to combine the specificity of an antibody with the cell-killing potency of chemotherapy, have also generated some recent successes in pediatric cancers.
Gemtuzumab ozogamicin is an ADC that targets the CD33 protein, which is highly expressed on 85%-90% of cases of acute myeloid leukemia (AML). In 2000, it was the first ADC to be brought to market in the United States, but it was subsequently voluntarily withdrawn by the manufacturer in 2010 after confirmatory trials failed to show a survival benefit.
Recently, a meta-analysis of gemtuzumab ozogamicin trials suggested that the drug likely does improve long-term overall survival (OS) and reduce the risk of relapse and researchers developed an intermittent dosing schedule to help mitigate toxicity.32 This new dosing regimen received FDA approval in 2017 for the treatment of pediatric patients aged 2 years and older on the basis of 2 clinical trials.
In the MyloFrance-1 trial, 57 patients were administered 3 mg/m2 gemtuzumab ozogamicin on days 1, 4, and 7 followed by cytarabine consolidation therapy and demonstrated a 26% CR rate and median recurrence-free survival of 11.6 months. In the phase 3 AML-19 trial, 237 patients received gemtuzumab ozogamicin at a dose of 6 mg/m2 on day 1 and 3 mg/m2 on day 8 or best supportive care. Gemtuzumab ozogamicin improved OS from 3.6 to 4.9 months.33,34
Inotuzumab ozogamicin is a CD22-targeting ADC that has been FDA approved for the treatment of adult patients with relapsed/refractory B-cell precursor ALL since last year. The therapy has been available to pediatric patients through a compassionate access program, but it has not been extensively evaluated in this population. The results of a retrospective analysis of pediatric patients who received at least 1 dose of inotuzumab ozogamicin were presented at ASCO in 2017. Among 29 patients with heavily pretreated disease the CR rate was 62%, 72% of whom achieved MRD negativity.35
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
1. American Cancer Society. Key statistics for childhood cancers. https://www.cancer.org/cancer/cancer-in-children/key-statistics.html. Last revised September 10, 2018. Accessed September 16, 2018.
2. NHI/National Cancer Institute website. Unusual cancers of childhood treatment (PDQ) - Health Professional Version. https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Last updated August 28, 2018. Accessed September 8, 2018.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
4. Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer. 2014;14(4):277-289.
5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
6. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371.
7. Dupain C, Harttrampf AC, Urbinati G, Geoerger B, Massaad-Massade L. Relevance of fusion genes in pediatric cancers: toward precision medicine. Molec Ther Nucleic Acids. 2017;6:315-326.
8. Lemonick MD, Park A. New hope for cancer. http://content.time.com/time/world/article/0,8599,2047900-2,00.html. Published May 28, 2001. Last accessed September 13, 2018.
9. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. https://www.hindawi.com/journals/cherp/2014/357027/. Published May 19, 2014. Accessed September 16, 2018.
10. Gore L, Kearns PR, Martino MLd, et al. Dasatinib in pediatric patients with chronic myeloid leukemia in chronic phase: results from a phase II trial. J Clin Oncol. 2018;36(13):1330-1338.
11. Novartis press release. Novartis drug Tasigna approved by FDA to treat children with rare form of leukemia. 2018; https://www.novartis.com/news/media-releases/novartis-drug-tasignar-approved-fda-treat-children-rare-form-leukemia. Released March 22, 2018. Accessed September 16, 2018.
12. Takita J. The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci. 2017;108(10):1913-1920.
13. Turner SD, Lamant L, Kenner L, Brugieres L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol. 2016;173(4):560-572.
14. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 fusions and rare novel RET gene rearrangement. Am J Surg Pathol. 2015;39(7):957-967.
15. Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a children's oncology group study. J Clin Oncol. 2017;35(28):3215-3221.
16. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5070277/. Published online March 18, 2016. Accessed September 16, 2018.
17. [Behind paywall.] Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705-714.
18. Desai AV, Brodeur GM, Foster J, et al. Phase 1 study of entrectinib (RXDX-101), a TRK, ROS1, and ALK inhibitor, in children, adolescents, and young adults with recurrent or refractory solid tumors. J Clin Oncol. 2018;36(suppl;):abstr 10536.
19. Heymach J, Krilov L, Alberg A, et al. Clinical cancer advances 2018: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2018;36(10):1020-1044.
20. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. NEJM. 2018;378(5):439-448.
21. Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
22. Stackelberg Av, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
23. Gokbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
24. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195.
25. Fousek K, Watanabe J, George A, et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J Clin Oncol. 2018;36(5_suppl):121-121.
26. Mejstríková E, Hrusak O, Borowitz MJ, et al. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017;7(12):659.
27. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
28. Pan J, Deng B, Liu S, et al. Efficacy and safety of CD22-directed CAR T-cell therapy in 15 pediatric refractory or relapsed b acute lymphoblastic leukemia patients. Paper presented at 23rd Congress of the European Hematology Association 2018; Stockholm, Sweden.
29. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35.
30. Drugs.com. Keytruda approval history. 2018; https://www.drugs.com/history/keytruda.html. Last update information not given. Accessed September 16, 2018.
31. Bristol Myers Squibb press release. US Food and Drug Administration expands approval of Yervoy (ipilimumab) to include pediatric patients 12 years and older with unresectable or metastatic melanoma. https://news.bms.com/press-release/corporatefinancial-news/us-food-and-drug-administration-expands-approval-yervoy-ipilim. Released July 24, 2017. Accessed September 16, 2018.
32. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
33. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972-979.
34. Taksin AL, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66-71.
35. Bhojwani D, Sposto R, Shah N, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). J Clin Oncol. 2017;35(15_suppl):10512-10512.
Prolonged survival in adenocarcinoma of unknown primary treated with chemoradiotherapy
Cancer of unknown primary (CUP) represents 3% to 5% of all cancer malignancies in the world.1 Since 2003, CUP has been divided into 2 subsets – favorable (20% of the cases) and unfavorable (80% of the cases) – based on histopathologic and clinical manifestations.2 The impact of locoregional therapies, such as surgery and radiation, in addition to systemic chemotherapy in adenocarcinomas of unknown primary is not well described in the literature.
Case presentation and summary
The patient was frustrated by the lack of diagnosis and extensive work-up and decided to travel to Bangladesh for several months. Upon her return in May 2015, the patient underwent dilation and curettage at an outside tertiary care center because of her persistently elevated beta-hCG levels (>500 mIU/mL; reference range for nonpregnant woman, <5 mIU/mL) that found no products of conception and excluded a malignant process. Endoscopy and colonoscopy at that time failed to reveal a primary tumor.
She was then referred to our institution. Her level of beta-hCG remained elevated, and another transvaginal ultrasound was performed but failed to reveal any masses or evidence of pregnancy. Mammogram and a breast ultrasound showed left breast lesions. Biopsy of the breast lesions was performed, and the pathology demonstrated fibrocystic changes.
The results of a PET-CT scan in August 2015 showed a lobulated abdominal mass of 5.7 x 3.7 cm, consisting of multiple periportal necrotic lymph nodes with a standardized uptake value (SUV) of 14 (Figure 1A) and a 2.0-cm hypermetabolic retroperitoneal lymph node at the aortic bifurcation level with an SUV of 8.6. The SUV is a ratio of activity per unit volume of a region of interest to the activity per unit whole body volume. An SUV of 2.5 or higher is generally considered to be indicative of malignant tissue. We conducted a detailed review of the lymph node pathologic specimen. Immunohistochemical (IHC) studies were positive for CK7, CDX2, and EMA; focally positive for PR and mammaglobin; and negative for CK20, ER, TTF-1, and WT-1. Nonspecific staining was seen with BRST2, and there was no staining with GATA3. IHC stain for HER2-NEU was equivocal. Molecular analysis did not detect BRAF, KRAS, NRAS, and PIK3CA mutations, but did find a CTNNB1 mutation. The IHC pattern suggested pancreatobiliary origin of the tumor.3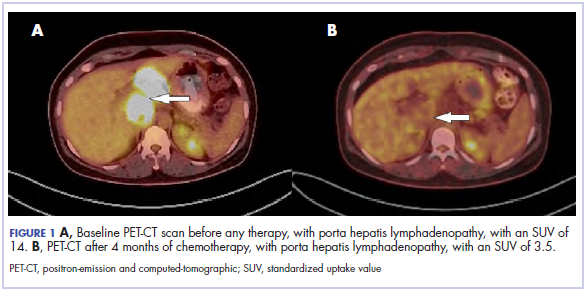
Although serum tumor marker pattern of elevated beta-hCG, AFP, and LDH can be seen in germ cell tumors, the pathology evaluation did not favor a germ cell tumor. No site of origin was evident on radiographic evaluation, and the patient was diagnosed with CUP. Based on tumor metastatic distribution and the elevated beta-hCG level,4 we suspected that an undetected pancreatic primary was possible, and we therefore chose the folinic acid, fluorouracil, irinotecan, oxaliplatin (FOLFIRINOX) chemotherapy regimen for its evidence in prolonging survival in metastatic pancreatic cancer.5 At the initiation of treatment, the patient’s elevated tumor markers were beta-hCG 953.6 mIU/mL (reference for nonpregnant woman, <5 mIU/mL) and AFP 1,800.7 ng/mL (reference range, 0.0-9.0 ng/mL). The patient began FOLFIRINOX chemotherapy in August 2015 and after 1 month of treatment, her beta-hCG and AFP levels declined notably to 1.7 mIU/mL and 11.2 ng/mL, respectively. She completed a total of 8 cycles of FOLFIRINOX in November 2015. After completion of chemotherapy, the PET-CT scan showed a decrease in fluoro-D-glucose (FDG) uptake in the porta hepatis and retroperitoneal lymph nodes (Figure 1B). SUV in the porta hepatis lymph nodes declined from 14 to 3.5. The patient’s case was presented to our institution’s multidisciplinary tumor board, and the members deemed the risk of possible lymph node dissection surgery would outweigh the benefit. It was recommended that we proceed with radiotherapy to the residual lymph node stations.
During December 2015 through February 2016, the patient underwent a course of consolidative chemoradiation therapy to the intra-abdominal lymph nodes to a dose of 5,400 cGy in 30 fractions, with concurrent capecitabine as radiosensitizer, using intensity-modulated radiation therapy. During both chemotherapy and CRT, the patient experienced nausea, vomiting, fatigue, and anorexia, which were treated with antiemetics. She completed therapy without major complications and recovered completely from the adverse effects.
Five weeks after completion of chemoradiation, a restaging PET-CT scan showed a persistent small FDG uptake in the periportal region (SUV, 4.2). After CRT, tumor markers beta-hCG and AFP declined to less than 1.2 mIU/mL and less than 2.0 ng/mL, respectively.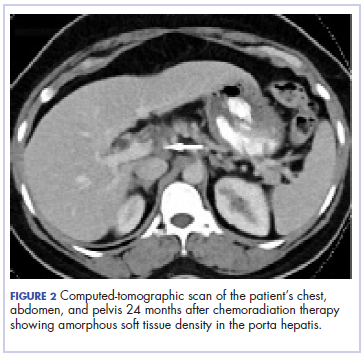
Discussion
CUP is divided into favorable and unfavorable subsets.1 The favorable subset includes women with adenocarcinoma involving axillary lymph nodes, women with papillary adenocarcinoma of peritoneal cavity, and adenocarcinoma with a colon profile. The unfavorable subset includes moderate to poorly differentiated adenocarcinomas (64%) and undifferentiated tumors (36%). It involves the liver in 40% to 50% of the cases, followed by lymph nodes (35%), lungs (31%), bones (28%), and the brain (15%).1,2,6 Although data suggest that CUP with lymph-node–only metastases generally fall into an unfavorable prognosis group, our patient’s survival and progression-free survival have been especially prolonged.
The combined platinum–paclitaxel-based regimens are the treatment of choice in this unfavorable subset of CUP,7,8 with patients showing 16% to 38% response rates and median overall survival times of 6.5 to 13 months.7 Platinum–gemcitabine combinations can also be used as an alternative first-line regimen, with an overall response rate of 55% and a median survival of 8 months.9 The addition of the targeted agents bevacizumab and erlotinib to the carboplatin–paclitaxel combination, followed by bevacizumab and erlotinib maintenance, has been shown to yield a median survival of 12.6 months but was not meaningfully superior to historical studies with chemotherapy alone.10
We chose the FOLFIRINOX regimen for our patient. Conroy and colleagues reported a notably improved survival of 11.1 months with that combination chemotherapy in patients with metastatic pancreatic cancer compared with 6.8 months with gemcitabine alone.5 Given the possible pancreatobiliary site of tumor origin on IHC, the lymph node pattern of spread, and the patient’s young age and robust performance status, we felt that this multiagent systemic therapy would offer the best chance of prolonged survival. FOLFIRINOX includes a platinum agent, oxaliplatin, and platinum agents are recommended to be included in chemotherapy combinations for CUP.9,10 Although there is no data to suggest the superiority of a triplet regimen over a doublet regimen in a CUP, a triplet chemotherapy regimen may be considered in select cases.
There have been only a few reports showing the effectiveness of radiotherapy in the treatment of adenocarcinomas of unknown primary outside of the head and neck. Kubisch and colleagues have reported a case of a woman with hepatic adenocarcinoma of unknown primary that was treated with chemotherapy and surgery. Upon recurrence, the patient was then treated with selective internal radiation therapy (SIRT). She was still alive 3 years after diagnosis, and there had been no tumor relapse 21 months after SIRT.11 Shiota and colleagues have reported a case of a mediastinal lymph node CUP that was treated with docetaxel and cisplatin with concurrent thoracic radiation therapy.12 The patient remained free of symptoms without regrowth of the primary site 22 months after disease onset, and exploration of the body with enhanced and PET-CT scan showed no further abnormalities.
Other reports suggest that locoregional therapy such as surgery and radiation may be of benefit to select patients with CUP. A retrospective study by Löffler and colleagues reported that patients with a limited local involvement who received radical surgery had a median overall survival of 52.7 months compared with those who received radiation (median overall survival, 19.4 months) and those who received chemotherapy alone (median overall survival, 16 months).13 A case of a metastatic undifferentiated CUP also reported a long-term (>5 years), disease-free survivor after pancreaticoduodenectomy and systemic adjuvant chemotherapy.14
Our case further demonstrates that a multidisciplinary approach to CUP may lead to excellent clinical outcomes. Chemotherapy followed by chemoradiation in our patient increased local tumor control and survival.
Adenocarcinomas of unknown primary cases should involve management by a multidisciplinary team. Clinical trials incorporating locoregional therapies for CUP in addition to systemic therapy are warranted.
1. Pavlidis N, Khaled H, Gaafar R. A mini review on cancer of unknown primary site: a clinical puzzle for the oncologists. J Adv Res. 2015;6(3):375-382.
2. Pavlidis N, Briasoulis E, Hainsworth J, Greco FA. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer. 2003;39(14):1990-2005.
3. Oien KA. Pathologic evaluation of unknown primary cancer. Semin Oncol. 2009;36(1):8-37.
4. Louhimo J, Alfthan H, Stenman UH, Hagland C. Serum HCG beta and CA 72-4 are stronger prognostic factors than CEA, CA 19-9 and CA 242 in pancreatic cancer. Oncology. 2004;66(2):126-131.
5. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817-1825.
6. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379:1428-1435.
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017;35(3):199-206.
8. Amela EY, Lauridant-Philippin G, Cousin S, Ryckewaert T, Adenis A, Penel N. Management of 'unfavourable' carcinoma of unknown primary site: synthesis of recent literature. Crit Rev Oncol Hematol. 2012;84(2):213-223.
9. Culine S, Lortholary A, Voigt J-J, et al. Cisplatin in combination with either gemcitabine or irinotecan in carcinomas of unknown primary site: results of a randomized phase II study--trial for the French study group on carcinomas of unknown primary (GEFCAPI 01). J Clin Oncol. 2003;21(18):3479-3482.
10. Hainsworth JD, Spigel DR, Thompson DS, et al. Paclitaxel/carboplatin plus bevacizumab/erlotinib in the first-line treatment of patients with carcinoma of unknown primary site. Oncologist. 2009;14(12):1189-1197.
11. Kubisch CH, Beigel F, Ihrler S, Goke B, Reiser MF, Hoffmann RT. Oesophageal ulceration after selective internal radiation therapy in a patient with carcinoma of unknown primary. Z Gastroenterol. 2010;48(5):546-550.
12. Shiota Y, Imai S, Sasaki N, et al. A case of mediastinal lymph node carcinoma of unknown primary site treated with docetaxel and cisplatin with concurrent thoracic radiation therapy. Acta Med Okayama. 2011;65(6):407-411.
13. Löffler H, Puthenparambil J, Hielscher T, Neben K, Krämer A. Patients with cancer of unknown primary: a retrospective analysis of 223 patients with adenocarcinoma or undifferentiated carcinoma. Dtsch Arztebl Int. 111(27-28):481-487.
14. Nakagawa Y, Todoroki T, Morishita Y, et al. A long-term survivor after pancreaticoduodenectomy for metastatic undifferentiated carcinoma of an unknown primary. Hepatogastroenterology. 2008;55(86-87):1557-1561.
15. Rodríguez-López JL, Toro-Bahamonde AM, Santiago-Méndez RJ, González-Cancel IF, Vélez-Cortés HA. An unusual case of colorectal adenocarcinoma presenting as an anterior mediastinal mass. Clin Colorectal Cancer. 2018;17(1):e115-e119.
Cancer of unknown primary (CUP) represents 3% to 5% of all cancer malignancies in the world.1 Since 2003, CUP has been divided into 2 subsets – favorable (20% of the cases) and unfavorable (80% of the cases) – based on histopathologic and clinical manifestations.2 The impact of locoregional therapies, such as surgery and radiation, in addition to systemic chemotherapy in adenocarcinomas of unknown primary is not well described in the literature.
Case presentation and summary
The patient was frustrated by the lack of diagnosis and extensive work-up and decided to travel to Bangladesh for several months. Upon her return in May 2015, the patient underwent dilation and curettage at an outside tertiary care center because of her persistently elevated beta-hCG levels (>500 mIU/mL; reference range for nonpregnant woman, <5 mIU/mL) that found no products of conception and excluded a malignant process. Endoscopy and colonoscopy at that time failed to reveal a primary tumor.
She was then referred to our institution. Her level of beta-hCG remained elevated, and another transvaginal ultrasound was performed but failed to reveal any masses or evidence of pregnancy. Mammogram and a breast ultrasound showed left breast lesions. Biopsy of the breast lesions was performed, and the pathology demonstrated fibrocystic changes.
The results of a PET-CT scan in August 2015 showed a lobulated abdominal mass of 5.7 x 3.7 cm, consisting of multiple periportal necrotic lymph nodes with a standardized uptake value (SUV) of 14 (Figure 1A) and a 2.0-cm hypermetabolic retroperitoneal lymph node at the aortic bifurcation level with an SUV of 8.6. The SUV is a ratio of activity per unit volume of a region of interest to the activity per unit whole body volume. An SUV of 2.5 or higher is generally considered to be indicative of malignant tissue. We conducted a detailed review of the lymph node pathologic specimen. Immunohistochemical (IHC) studies were positive for CK7, CDX2, and EMA; focally positive for PR and mammaglobin; and negative for CK20, ER, TTF-1, and WT-1. Nonspecific staining was seen with BRST2, and there was no staining with GATA3. IHC stain for HER2-NEU was equivocal. Molecular analysis did not detect BRAF, KRAS, NRAS, and PIK3CA mutations, but did find a CTNNB1 mutation. The IHC pattern suggested pancreatobiliary origin of the tumor.3
Although serum tumor marker pattern of elevated beta-hCG, AFP, and LDH can be seen in germ cell tumors, the pathology evaluation did not favor a germ cell tumor. No site of origin was evident on radiographic evaluation, and the patient was diagnosed with CUP. Based on tumor metastatic distribution and the elevated beta-hCG level,4 we suspected that an undetected pancreatic primary was possible, and we therefore chose the folinic acid, fluorouracil, irinotecan, oxaliplatin (FOLFIRINOX) chemotherapy regimen for its evidence in prolonging survival in metastatic pancreatic cancer.5 At the initiation of treatment, the patient’s elevated tumor markers were beta-hCG 953.6 mIU/mL (reference for nonpregnant woman, <5 mIU/mL) and AFP 1,800.7 ng/mL (reference range, 0.0-9.0 ng/mL). The patient began FOLFIRINOX chemotherapy in August 2015 and after 1 month of treatment, her beta-hCG and AFP levels declined notably to 1.7 mIU/mL and 11.2 ng/mL, respectively. She completed a total of 8 cycles of FOLFIRINOX in November 2015. After completion of chemotherapy, the PET-CT scan showed a decrease in fluoro-D-glucose (FDG) uptake in the porta hepatis and retroperitoneal lymph nodes (Figure 1B). SUV in the porta hepatis lymph nodes declined from 14 to 3.5. The patient’s case was presented to our institution’s multidisciplinary tumor board, and the members deemed the risk of possible lymph node dissection surgery would outweigh the benefit. It was recommended that we proceed with radiotherapy to the residual lymph node stations.
During December 2015 through February 2016, the patient underwent a course of consolidative chemoradiation therapy to the intra-abdominal lymph nodes to a dose of 5,400 cGy in 30 fractions, with concurrent capecitabine as radiosensitizer, using intensity-modulated radiation therapy. During both chemotherapy and CRT, the patient experienced nausea, vomiting, fatigue, and anorexia, which were treated with antiemetics. She completed therapy without major complications and recovered completely from the adverse effects.
Five weeks after completion of chemoradiation, a restaging PET-CT scan showed a persistent small FDG uptake in the periportal region (SUV, 4.2). After CRT, tumor markers beta-hCG and AFP declined to less than 1.2 mIU/mL and less than 2.0 ng/mL, respectively.
Discussion
CUP is divided into favorable and unfavorable subsets.1 The favorable subset includes women with adenocarcinoma involving axillary lymph nodes, women with papillary adenocarcinoma of peritoneal cavity, and adenocarcinoma with a colon profile. The unfavorable subset includes moderate to poorly differentiated adenocarcinomas (64%) and undifferentiated tumors (36%). It involves the liver in 40% to 50% of the cases, followed by lymph nodes (35%), lungs (31%), bones (28%), and the brain (15%).1,2,6 Although data suggest that CUP with lymph-node–only metastases generally fall into an unfavorable prognosis group, our patient’s survival and progression-free survival have been especially prolonged.
The combined platinum–paclitaxel-based regimens are the treatment of choice in this unfavorable subset of CUP,7,8 with patients showing 16% to 38% response rates and median overall survival times of 6.5 to 13 months.7 Platinum–gemcitabine combinations can also be used as an alternative first-line regimen, with an overall response rate of 55% and a median survival of 8 months.9 The addition of the targeted agents bevacizumab and erlotinib to the carboplatin–paclitaxel combination, followed by bevacizumab and erlotinib maintenance, has been shown to yield a median survival of 12.6 months but was not meaningfully superior to historical studies with chemotherapy alone.10
We chose the FOLFIRINOX regimen for our patient. Conroy and colleagues reported a notably improved survival of 11.1 months with that combination chemotherapy in patients with metastatic pancreatic cancer compared with 6.8 months with gemcitabine alone.5 Given the possible pancreatobiliary site of tumor origin on IHC, the lymph node pattern of spread, and the patient’s young age and robust performance status, we felt that this multiagent systemic therapy would offer the best chance of prolonged survival. FOLFIRINOX includes a platinum agent, oxaliplatin, and platinum agents are recommended to be included in chemotherapy combinations for CUP.9,10 Although there is no data to suggest the superiority of a triplet regimen over a doublet regimen in a CUP, a triplet chemotherapy regimen may be considered in select cases.
There have been only a few reports showing the effectiveness of radiotherapy in the treatment of adenocarcinomas of unknown primary outside of the head and neck. Kubisch and colleagues have reported a case of a woman with hepatic adenocarcinoma of unknown primary that was treated with chemotherapy and surgery. Upon recurrence, the patient was then treated with selective internal radiation therapy (SIRT). She was still alive 3 years after diagnosis, and there had been no tumor relapse 21 months after SIRT.11 Shiota and colleagues have reported a case of a mediastinal lymph node CUP that was treated with docetaxel and cisplatin with concurrent thoracic radiation therapy.12 The patient remained free of symptoms without regrowth of the primary site 22 months after disease onset, and exploration of the body with enhanced and PET-CT scan showed no further abnormalities.
Other reports suggest that locoregional therapy such as surgery and radiation may be of benefit to select patients with CUP. A retrospective study by Löffler and colleagues reported that patients with a limited local involvement who received radical surgery had a median overall survival of 52.7 months compared with those who received radiation (median overall survival, 19.4 months) and those who received chemotherapy alone (median overall survival, 16 months).13 A case of a metastatic undifferentiated CUP also reported a long-term (>5 years), disease-free survivor after pancreaticoduodenectomy and systemic adjuvant chemotherapy.14
Our case further demonstrates that a multidisciplinary approach to CUP may lead to excellent clinical outcomes. Chemotherapy followed by chemoradiation in our patient increased local tumor control and survival.
Adenocarcinomas of unknown primary cases should involve management by a multidisciplinary team. Clinical trials incorporating locoregional therapies for CUP in addition to systemic therapy are warranted.
Cancer of unknown primary (CUP) represents 3% to 5% of all cancer malignancies in the world.1 Since 2003, CUP has been divided into 2 subsets – favorable (20% of the cases) and unfavorable (80% of the cases) – based on histopathologic and clinical manifestations.2 The impact of locoregional therapies, such as surgery and radiation, in addition to systemic chemotherapy in adenocarcinomas of unknown primary is not well described in the literature.
Case presentation and summary
The patient was frustrated by the lack of diagnosis and extensive work-up and decided to travel to Bangladesh for several months. Upon her return in May 2015, the patient underwent dilation and curettage at an outside tertiary care center because of her persistently elevated beta-hCG levels (>500 mIU/mL; reference range for nonpregnant woman, <5 mIU/mL) that found no products of conception and excluded a malignant process. Endoscopy and colonoscopy at that time failed to reveal a primary tumor.
She was then referred to our institution. Her level of beta-hCG remained elevated, and another transvaginal ultrasound was performed but failed to reveal any masses or evidence of pregnancy. Mammogram and a breast ultrasound showed left breast lesions. Biopsy of the breast lesions was performed, and the pathology demonstrated fibrocystic changes.
The results of a PET-CT scan in August 2015 showed a lobulated abdominal mass of 5.7 x 3.7 cm, consisting of multiple periportal necrotic lymph nodes with a standardized uptake value (SUV) of 14 (Figure 1A) and a 2.0-cm hypermetabolic retroperitoneal lymph node at the aortic bifurcation level with an SUV of 8.6. The SUV is a ratio of activity per unit volume of a region of interest to the activity per unit whole body volume. An SUV of 2.5 or higher is generally considered to be indicative of malignant tissue. We conducted a detailed review of the lymph node pathologic specimen. Immunohistochemical (IHC) studies were positive for CK7, CDX2, and EMA; focally positive for PR and mammaglobin; and negative for CK20, ER, TTF-1, and WT-1. Nonspecific staining was seen with BRST2, and there was no staining with GATA3. IHC stain for HER2-NEU was equivocal. Molecular analysis did not detect BRAF, KRAS, NRAS, and PIK3CA mutations, but did find a CTNNB1 mutation. The IHC pattern suggested pancreatobiliary origin of the tumor.3
Although serum tumor marker pattern of elevated beta-hCG, AFP, and LDH can be seen in germ cell tumors, the pathology evaluation did not favor a germ cell tumor. No site of origin was evident on radiographic evaluation, and the patient was diagnosed with CUP. Based on tumor metastatic distribution and the elevated beta-hCG level,4 we suspected that an undetected pancreatic primary was possible, and we therefore chose the folinic acid, fluorouracil, irinotecan, oxaliplatin (FOLFIRINOX) chemotherapy regimen for its evidence in prolonging survival in metastatic pancreatic cancer.5 At the initiation of treatment, the patient’s elevated tumor markers were beta-hCG 953.6 mIU/mL (reference for nonpregnant woman, <5 mIU/mL) and AFP 1,800.7 ng/mL (reference range, 0.0-9.0 ng/mL). The patient began FOLFIRINOX chemotherapy in August 2015 and after 1 month of treatment, her beta-hCG and AFP levels declined notably to 1.7 mIU/mL and 11.2 ng/mL, respectively. She completed a total of 8 cycles of FOLFIRINOX in November 2015. After completion of chemotherapy, the PET-CT scan showed a decrease in fluoro-D-glucose (FDG) uptake in the porta hepatis and retroperitoneal lymph nodes (Figure 1B). SUV in the porta hepatis lymph nodes declined from 14 to 3.5. The patient’s case was presented to our institution’s multidisciplinary tumor board, and the members deemed the risk of possible lymph node dissection surgery would outweigh the benefit. It was recommended that we proceed with radiotherapy to the residual lymph node stations.
During December 2015 through February 2016, the patient underwent a course of consolidative chemoradiation therapy to the intra-abdominal lymph nodes to a dose of 5,400 cGy in 30 fractions, with concurrent capecitabine as radiosensitizer, using intensity-modulated radiation therapy. During both chemotherapy and CRT, the patient experienced nausea, vomiting, fatigue, and anorexia, which were treated with antiemetics. She completed therapy without major complications and recovered completely from the adverse effects.
Five weeks after completion of chemoradiation, a restaging PET-CT scan showed a persistent small FDG uptake in the periportal region (SUV, 4.2). After CRT, tumor markers beta-hCG and AFP declined to less than 1.2 mIU/mL and less than 2.0 ng/mL, respectively.
Discussion
CUP is divided into favorable and unfavorable subsets.1 The favorable subset includes women with adenocarcinoma involving axillary lymph nodes, women with papillary adenocarcinoma of peritoneal cavity, and adenocarcinoma with a colon profile. The unfavorable subset includes moderate to poorly differentiated adenocarcinomas (64%) and undifferentiated tumors (36%). It involves the liver in 40% to 50% of the cases, followed by lymph nodes (35%), lungs (31%), bones (28%), and the brain (15%).1,2,6 Although data suggest that CUP with lymph-node–only metastases generally fall into an unfavorable prognosis group, our patient’s survival and progression-free survival have been especially prolonged.
The combined platinum–paclitaxel-based regimens are the treatment of choice in this unfavorable subset of CUP,7,8 with patients showing 16% to 38% response rates and median overall survival times of 6.5 to 13 months.7 Platinum–gemcitabine combinations can also be used as an alternative first-line regimen, with an overall response rate of 55% and a median survival of 8 months.9 The addition of the targeted agents bevacizumab and erlotinib to the carboplatin–paclitaxel combination, followed by bevacizumab and erlotinib maintenance, has been shown to yield a median survival of 12.6 months but was not meaningfully superior to historical studies with chemotherapy alone.10
We chose the FOLFIRINOX regimen for our patient. Conroy and colleagues reported a notably improved survival of 11.1 months with that combination chemotherapy in patients with metastatic pancreatic cancer compared with 6.8 months with gemcitabine alone.5 Given the possible pancreatobiliary site of tumor origin on IHC, the lymph node pattern of spread, and the patient’s young age and robust performance status, we felt that this multiagent systemic therapy would offer the best chance of prolonged survival. FOLFIRINOX includes a platinum agent, oxaliplatin, and platinum agents are recommended to be included in chemotherapy combinations for CUP.9,10 Although there is no data to suggest the superiority of a triplet regimen over a doublet regimen in a CUP, a triplet chemotherapy regimen may be considered in select cases.
There have been only a few reports showing the effectiveness of radiotherapy in the treatment of adenocarcinomas of unknown primary outside of the head and neck. Kubisch and colleagues have reported a case of a woman with hepatic adenocarcinoma of unknown primary that was treated with chemotherapy and surgery. Upon recurrence, the patient was then treated with selective internal radiation therapy (SIRT). She was still alive 3 years after diagnosis, and there had been no tumor relapse 21 months after SIRT.11 Shiota and colleagues have reported a case of a mediastinal lymph node CUP that was treated with docetaxel and cisplatin with concurrent thoracic radiation therapy.12 The patient remained free of symptoms without regrowth of the primary site 22 months after disease onset, and exploration of the body with enhanced and PET-CT scan showed no further abnormalities.
Other reports suggest that locoregional therapy such as surgery and radiation may be of benefit to select patients with CUP. A retrospective study by Löffler and colleagues reported that patients with a limited local involvement who received radical surgery had a median overall survival of 52.7 months compared with those who received radiation (median overall survival, 19.4 months) and those who received chemotherapy alone (median overall survival, 16 months).13 A case of a metastatic undifferentiated CUP also reported a long-term (>5 years), disease-free survivor after pancreaticoduodenectomy and systemic adjuvant chemotherapy.14
Our case further demonstrates that a multidisciplinary approach to CUP may lead to excellent clinical outcomes. Chemotherapy followed by chemoradiation in our patient increased local tumor control and survival.
Adenocarcinomas of unknown primary cases should involve management by a multidisciplinary team. Clinical trials incorporating locoregional therapies for CUP in addition to systemic therapy are warranted.
1. Pavlidis N, Khaled H, Gaafar R. A mini review on cancer of unknown primary site: a clinical puzzle for the oncologists. J Adv Res. 2015;6(3):375-382.
2. Pavlidis N, Briasoulis E, Hainsworth J, Greco FA. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer. 2003;39(14):1990-2005.
3. Oien KA. Pathologic evaluation of unknown primary cancer. Semin Oncol. 2009;36(1):8-37.
4. Louhimo J, Alfthan H, Stenman UH, Hagland C. Serum HCG beta and CA 72-4 are stronger prognostic factors than CEA, CA 19-9 and CA 242 in pancreatic cancer. Oncology. 2004;66(2):126-131.
5. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817-1825.
6. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379:1428-1435.
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017;35(3):199-206.
8. Amela EY, Lauridant-Philippin G, Cousin S, Ryckewaert T, Adenis A, Penel N. Management of 'unfavourable' carcinoma of unknown primary site: synthesis of recent literature. Crit Rev Oncol Hematol. 2012;84(2):213-223.
9. Culine S, Lortholary A, Voigt J-J, et al. Cisplatin in combination with either gemcitabine or irinotecan in carcinomas of unknown primary site: results of a randomized phase II study--trial for the French study group on carcinomas of unknown primary (GEFCAPI 01). J Clin Oncol. 2003;21(18):3479-3482.
10. Hainsworth JD, Spigel DR, Thompson DS, et al. Paclitaxel/carboplatin plus bevacizumab/erlotinib in the first-line treatment of patients with carcinoma of unknown primary site. Oncologist. 2009;14(12):1189-1197.
11. Kubisch CH, Beigel F, Ihrler S, Goke B, Reiser MF, Hoffmann RT. Oesophageal ulceration after selective internal radiation therapy in a patient with carcinoma of unknown primary. Z Gastroenterol. 2010;48(5):546-550.
12. Shiota Y, Imai S, Sasaki N, et al. A case of mediastinal lymph node carcinoma of unknown primary site treated with docetaxel and cisplatin with concurrent thoracic radiation therapy. Acta Med Okayama. 2011;65(6):407-411.
13. Löffler H, Puthenparambil J, Hielscher T, Neben K, Krämer A. Patients with cancer of unknown primary: a retrospective analysis of 223 patients with adenocarcinoma or undifferentiated carcinoma. Dtsch Arztebl Int. 111(27-28):481-487.
14. Nakagawa Y, Todoroki T, Morishita Y, et al. A long-term survivor after pancreaticoduodenectomy for metastatic undifferentiated carcinoma of an unknown primary. Hepatogastroenterology. 2008;55(86-87):1557-1561.
15. Rodríguez-López JL, Toro-Bahamonde AM, Santiago-Méndez RJ, González-Cancel IF, Vélez-Cortés HA. An unusual case of colorectal adenocarcinoma presenting as an anterior mediastinal mass. Clin Colorectal Cancer. 2018;17(1):e115-e119.
1. Pavlidis N, Khaled H, Gaafar R. A mini review on cancer of unknown primary site: a clinical puzzle for the oncologists. J Adv Res. 2015;6(3):375-382.
2. Pavlidis N, Briasoulis E, Hainsworth J, Greco FA. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer. 2003;39(14):1990-2005.
3. Oien KA. Pathologic evaluation of unknown primary cancer. Semin Oncol. 2009;36(1):8-37.
4. Louhimo J, Alfthan H, Stenman UH, Hagland C. Serum HCG beta and CA 72-4 are stronger prognostic factors than CEA, CA 19-9 and CA 242 in pancreatic cancer. Oncology. 2004;66(2):126-131.
5. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817-1825.
6. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379:1428-1435.
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017;35(3):199-206.
8. Amela EY, Lauridant-Philippin G, Cousin S, Ryckewaert T, Adenis A, Penel N. Management of 'unfavourable' carcinoma of unknown primary site: synthesis of recent literature. Crit Rev Oncol Hematol. 2012;84(2):213-223.
9. Culine S, Lortholary A, Voigt J-J, et al. Cisplatin in combination with either gemcitabine or irinotecan in carcinomas of unknown primary site: results of a randomized phase II study--trial for the French study group on carcinomas of unknown primary (GEFCAPI 01). J Clin Oncol. 2003;21(18):3479-3482.
10. Hainsworth JD, Spigel DR, Thompson DS, et al. Paclitaxel/carboplatin plus bevacizumab/erlotinib in the first-line treatment of patients with carcinoma of unknown primary site. Oncologist. 2009;14(12):1189-1197.
11. Kubisch CH, Beigel F, Ihrler S, Goke B, Reiser MF, Hoffmann RT. Oesophageal ulceration after selective internal radiation therapy in a patient with carcinoma of unknown primary. Z Gastroenterol. 2010;48(5):546-550.
12. Shiota Y, Imai S, Sasaki N, et al. A case of mediastinal lymph node carcinoma of unknown primary site treated with docetaxel and cisplatin with concurrent thoracic radiation therapy. Acta Med Okayama. 2011;65(6):407-411.
13. Löffler H, Puthenparambil J, Hielscher T, Neben K, Krämer A. Patients with cancer of unknown primary: a retrospective analysis of 223 patients with adenocarcinoma or undifferentiated carcinoma. Dtsch Arztebl Int. 111(27-28):481-487.
14. Nakagawa Y, Todoroki T, Morishita Y, et al. A long-term survivor after pancreaticoduodenectomy for metastatic undifferentiated carcinoma of an unknown primary. Hepatogastroenterology. 2008;55(86-87):1557-1561.
15. Rodríguez-López JL, Toro-Bahamonde AM, Santiago-Méndez RJ, González-Cancel IF, Vélez-Cortés HA. An unusual case of colorectal adenocarcinoma presenting as an anterior mediastinal mass. Clin Colorectal Cancer. 2018;17(1):e115-e119.
Primary renal synovial sarcoma – a diagnostic dilemma
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).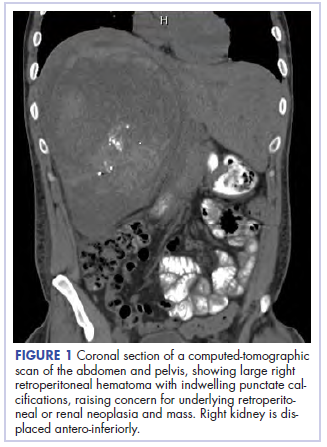
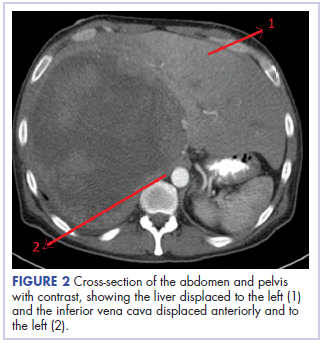
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).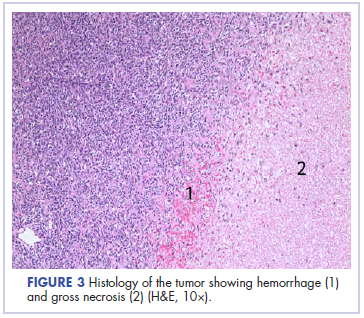
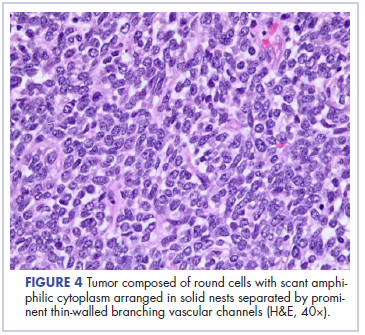
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
Marriage predicts for survival in patients with stage III non–small-cell lung cancer
Non–small-cell lung cancer (NSCLC) remains the leading cause of cancer death in the United States, where 29% of patients will present with stage III disease.1,2 Ongoing research efforts seek to improve these outcomes using novel systemic therapy options or modern radiation techniques. However, there have also been recent studies showing the importance of marital and/or partner status on clinical outcomes.3-7 For example, in a large Surveillance, Epidemiology, and End Results (SEER) analysis of 734,889 patients diagnosed with several types of cancer (including lung cancer), patients identified as married were less likely to present with metastatic disease, more likely to receive definitive therapy, and had superior cancer-related mortality even after adjusting for other variables such as cancer stage and treatment when compared with single patients.3 Population-based assessments are important in relaying information about trends and general outcomes based on marital status, but because they are large, they often lack patient-specific information such as nutrition, immunologic status, and variability in treatment paradigms, all of which can independently have an impact on overall survival (OS) in stage III NSCLC.8-10 In addition, population analyses have typically included patients of all cancer stages and hence involved a multitude of treatment approaches ranging from curative to palliative. There are limited well-annotated institutional data on the association of marital status on nonmetastatic, locally advanced (LA-NSCLC) in the setting of National Comprehensive Cancer Network-guided, standard-of-care definitive treatment.
The objective of this analysis is to evaluate the effect of marital status on OS and freedom from recurrence (FFR) in patients with stage III NSCLC who were treated at a National Cancer Institute–designated cancer center with curative intent from 2000 through 2013. We performed a detailed multivariate analysis (MVA) of patient-, disease-, and treatment-specific factors, including the interaction with racial, nutritional, and immunologic status, which to our knowledge has not been previously reported, to comprehensively evaluate the benefit of marital status in patients with LA-NSCLC.
Methods
Patient population and treatment
From January 2000 through December 2013, 355 patients diagnosed with clinical stage III NSCLC (American Joint Committee on Cancer 7th edition) were definitively treated at the University of Maryland in Baltimore, Maryland. Their clinical data were retrospectively analyzed under internal review board approval (GCC 1175, Thoracic Oncology Database). All of the patients were evaluated before treatment by a multidisciplinary team consisting of thoracic surgeons and medical and radiation oncologists. Before treatment, the patients underwent standard work-up, which included systemic imaging with positron-emission (PET), computed-tomographic (CT), PET–CT, and/or bone scan, brain imaging consisting of magnetic-resonance imaging or CT with contrast, and routine blood. Patients had documentation of mediastinal disease by either imaging, mediastinoscopy, or endobronchial ultrasound biopsy.
Definitive therapy was administered using the backbone of chemoradiation therapy (CRT) with (trimodality) or without (bimodality) surgical resection. Concurrent CRT was typically administered with weekly carboplatin–paclitaxel (areas under the curve [AUCs], 2 and 50 mg/m2, respectively) and was generally followed with 2 cycles of consolidative treatment with definitive doses of carboplatin–paclitaxel (AUCs, 5-6 and 200-225 mg/m2, respectively) as tolerated. The entire cohort was also assessed for possible trimodality therapy at the time of initial diagnosis, and patients who were potential surgical candidates were reassessed for mediastinal nodal clearance following repeat radiographic staging after full-dose CRT. Patients who experienced pathologic mediastinal clearance of disease underwent resection followed by consolidative chemotherapy. Unless there was evidence of disease progression, patients who did not have mediastinal lymph node clearance or who were found not to be a surgical candidate proceeded directly to consolidative chemotherapy. The details of patient selection for trimodality therapy and the oncological outcomes have been previously reported.10 For follow-up, patients were normally followed with serial CT or PET–CT scans as clinically indicated every 3 months for the first year, 4 to 6 months for the next 2 to 5 years, and then yearly thereafter.
For the analysis, patients were categorized as being either married or single based on self-reporting. As a surrogate for nutrition status, patients were stratified into 4 pretreatment body mass index (BMI) cohorts based on the following World Health Organization criteria: underweight, <18.5 kg/m2; normal weight, 18.5 to <25 kg/m2; overweight, 25 to <30 kg/m2; and obesity, ≥30 kg/m2. Pretreatment albumin was also evaluated as a continuous variable. For assessment of immunological status, neutrophil-to-lymphocyte ratio (NLR) was calculated at the time of diagnosis by dividing the absolute neutrophil count by the absolute lymphocyte count.
Statistics
We used the Pearson chi-square test to compare categorical variables. OS was calculated from the date of diagnosis (by biopsy of either primary tumor or mediastinal nodes) to the time of death or date of last follow-up. Patients were only censored if they were lost to follow-up. FFR was determined by the date of diagnosis to the time of first failure, with either distant or locoregional disease progression. For this analysis, patients were censored at the time of their last follow-up or death. The Kaplan-Meier product limit method was used to estimate OS and FFR, and we applied the log-rank test to compare outcomes between the 2 cohorts.
We conducted the multivariate analyses using Cox regression with forward model selection. Variables analyzed included age (<60 vs ≥60 years), sex, race (black vs nonblack), median household income, insurance status (Yes vs No), Eastern Cooperative Oncology Group Performance Status (ECOG PS) (range: 0-3; 0 = fully active and 3 = capable of limited self-care, confined to bed/chair >50% of day) at time of diagnosis (0 vs ≥1), pre-CRT BMI, smoking (pack-years), chronic obstructive pulmonary disorder (Yes vs No), Charlson Comorbidity Index score (≤6 vs >7; range, 3-15; this score takes into consideration age, cardiovascular disease, malignancy, and other chronic conditions to calculate 1-year mortality), histology, calculated pretreatment NLR (as a continuous variable), pretreatment albumin (as a continuous variable), T stage, N stage, overall stage (IIIA vs IIIB), radiation technique (3D-CRT vs intensity-modulated radiation therapy [IMRT]), date of diagnosis (divided into quartiles based on proportion diagnosed by years: 2000-2002, 2003-2005, 2006-2009, 2010-2013), use of trimodality therapy, and consolidation chemotherapy. SPSS software (version 23.0) was used for statistical analysis (IBM Corp, Armonk, NY).
Results
Treatment cohorts
Table 1 compares and summarizes patient demographics, disease, and treatment characteristics for married (n = 185; 52.1%) and nonmarried (n = 170; 47.9%) patients. Married patients were more likely to self-identify as being white (P < .0001), reside in zip codes with a higher household median income (P < .0001), have an ECOG PS of 0 (P = .001), have a higher distribution of pretreatment albumin levels (P = .009), and undergo trimodality therapy (P = .001), and they were twice as likely to have insurance (P = .029). Both cohorts were evenly distributed in terms of T stage, N stage, and overall staging. There was no difference in pretreatment NLR or pretreatment BMI between married and single patients. Concurrent CRT was used in more than 85% of patients in both groups, with approximately two-thirds also receiving consolidation chemotherapy (Table 1). Median delivered radiation dose was 64.8 Gy (range, 10.8-81.6 Gy). There was no statistically significant difference in radiation dose delivered to either group, with nearly 90% of the cohort receiving ≥60 Gy.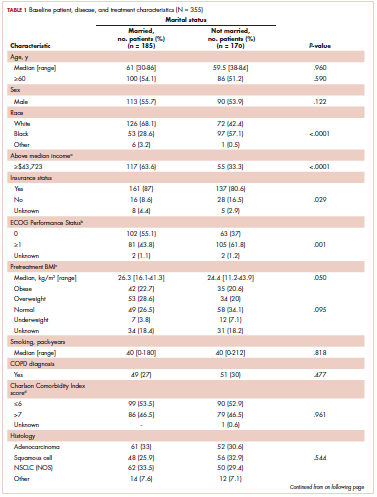
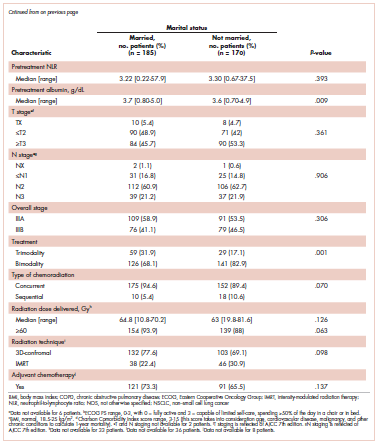
OS and FFR
With a median follow-up of 15 months for all patients and 89 months for surviving patients (range, 1-184 months), married patients had improved OS when compared with the single cohort, with a median survival of 29.6 and 18.4 months, respectively (unadjusted hazard ratio [HR] of married vs nonmarried, .640; 95% confidence interval [CI], 0.502-0.816; P < .0001; Figure 1A). The estimated 2- and 5-year OS for married and single patients were 56% and 31% and 38.6% and 15%, respectively. When stratified by stage, married patients with stage IIIB disease (median survival, 25 months; Figure 1B) had a similar survival to unmarried patients with stage IIIA disease (median survival, 24 months; Figure 1B).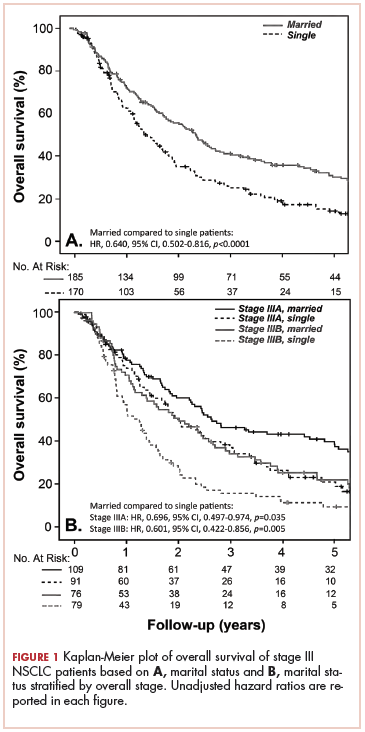
In stage IIIA patients, marital status was associated with an unadjusted HR of .696 (95% CI, 0.497-0.974; P = .035), with a larger OS benefit seen in the IIIB group (unadjusted HR, .601; 95% CI, 0.422-0.856; P = .005).
Survival as it pertains to marital status was further stratified by sex (Figure 2A) and race (Figure 2B). Married men had an improved estimated median survival of 30 months when compared with single men, whose median survival was 16 months (unadjusted HR, .541; 95% CI, 0.392-0.746; P < .0001). On the other hand, marital status had no statistically significant effect on OS when comparing married women with their single counterparts (unadjusted HR, .717; 95% CI, 0.491-1.048; P = .085; Figure 2A), with an overall median survival of approximately 28 months for the entire female cohort. Stratification by race also showed similar results, with married nonblack patients demonstrating better OS when compared with single nonblack patients (HR, .586; 95% CI, 0.420-0.820; P = .002; Figure 2B), with a median survival of 29 and 17 months, respectively. Black patients also had a similar improvement in survival when comparing the married (median survival, 30 months) and nonmarried groups (median survival, 19.6 months; unadjusted HR, .676; 95% CI, 0.457-1.000; P = .050; Figure 2B).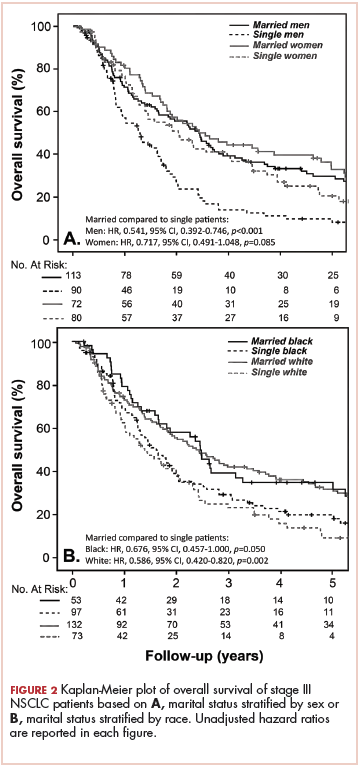
FFR did not differ between the 2 groups, with a median time to failure of 17 and 15 months for married and nonmarried patients, respectively (unadjusted HR, .799; 95% CI, 0.607-1.051; P = .108; Figure 3). Estimated 2- and 5-year FFR for married and nonmarried patients were 39.4% and 27% and 31.5% and 18.5%, respectively (Figure 3).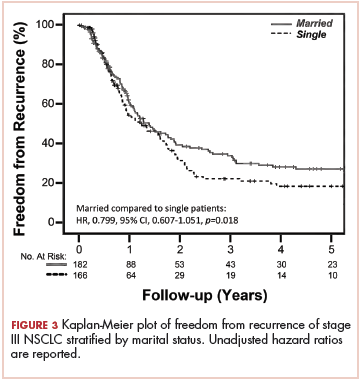
Clinical predictors of survival
On MVA, factors that were independent predictors for OS are summarized in Table 2. Risk of death was reduced by approximately 65% and 45% in patients who underwent trimodality treatment (P < .0001) or were able to undergo consolidative chemotherapy (P = .004) when compared with those who were treated definitively with bimodality treatment or did not undergo systemic doses of adjuvant chemotherapy, respectively. Having insurance (P = .048) and use of IMRT over 3D-CRT (P = .008) was associated with a reduction of mortality by about half in this cohort. Both gender (improved OS with female sex; P = .004) and marital status (improved OS with marriage; P = .006) were associated with a decreased the risk of death by 40% (Table 2). By contrast, a higher NLR resulted
Discussion
Our study continues to support the notion that marital status is an independent indicator of survival in stage III NSCLC (adjusted HR, .59; 95% CI, 0.404-0.859; P = .006). The benefit of marriage in this population seems to be better than that reported in the SEER analysis for all stages, wherein the HR for death of married patients compared with their single counterparts was .85 (95% CI, 0.83-0.87). In their analysis, the investigators hypothesized that this survival advantage could partially be explained by better access to health care and adherence to therapy, as was supported by the higher likelihood of married patients presenting with localized disease and receiving definitive treatment.3 Another population-based study using the Florida Cancer Data System identified 161,228 lung cancer patients (NSCLC and small-cell lung histology included), and on MVA, marital status remained an important prognostic indicator for OS when compared with never-married patients (HR, .86; P = .001).6 In addition to typically including patients with all stages of diseases, population-based studies often include patients who receive a heterogeneous combination of treatment modalities, possibly confounding the analysis. Furthermore, large population analyses typically do not report on patient-specific variables such as nutrition (ie, BMI and albumin) or immunologic status (ie, NLR), both of which have been shown to be independent predictors of survival in LA-NSCLC.8,9
In contrast, some other studies have failed to demonstrate an OS advantage with marital status in patients with NSCLC. For example, in a meta-analysis that evaluated the influence of race, gender, and marital status on 1,365 nonoperative NSCLC patients who were enrolled in 9 Radiation Therapy Oncology Group (RTOG) trials, the investigators did not find marital status to be independently predictive of survival.11 In addition, for the 5,898 patients who were prospectively enrolled in a Mayo Clinic Lung Cancer Cohort (MCLCC), marital status was also found not to be prognostic for NSCLC outcomes when all stages of the disease were analyzed together.4 There are some possible confounding factors in these studies. Patients recruited for clinical trials tend to be healthier with a better performance status and have a support system (including close monitoring by the study team) when compared with the general population diagnosed with lung cancer. About 70% to 76% of the patients in both the RTOG and MCLCC studies were married, which is significantly higher than both the national average (51%) and our group (52.1%). Like other population-based studies, the MCLCC included patients with all stages getting a variety of treatments. Although no overall impact on survival was noted, the investigators noted that single, divorced, and widowed patients were more likely to not receive cancer therapy(P < .0001). The marital status also influenced the choice of therapy, with subgroup analysis revealing inferior outcomes in widowed and divorced patients with stage IA, IIB, or IIIB disease. The authors also recognized an inherent referral bias from patients, with support system being typically seen at the Mayo clinics, which may have played an additional role. All of the patients in our analysis were appropriately staged and received curative-intent treatment by a team of physicians using essentially identical therapeutic strategies, thus minimizing some of these confounding factors. This allowed us to explore the impact of marital status while a patient was undergoing stage-appropriate treatment. We demonstrated a strong association with marital status and survival that even overcame the effects of stage (IIIA vs IIIB) on clinical outcomes (Figure 1B).
Furthermore, our analysis allowed us to explore the interaction of race and marital status more definitively because the demographics of the patients in the RTOG and MCLCC included 14% and less than 3% of patients identified as being nonwhite, respectively, in contrast to our analysis in which 41% of the patients self-identified as black.12 In our black population, marital status was associated with an observable improvement in OS, similar to our nonblack, predominantly white (97%) cohort (Figure 2B). Also, the results of our analysis may be a more accurate representation of the general population living in large urban or semiurban settings and further implies that an intact social support system could have a greater influence on clinical outcomes.
The current analysis is unique when compared with previous published studies in that beyond conventional demographic and treatment-related factors, we have comprehensively explored potential mechanisms that may explain the survival advantage seen in married patients by evaluating additional factors, such as functional status (ECOG and Charlson’s scores), nutritional status (BMI and albumin), immunologic characteristics (NLR), and other social factors (race, income, insurance status). Although married patients were more likely to have a higher BMI and albumin at diagnosis, when controlling for these factors in the multivariable analysis, marital status remained strongly prognostic (Table 2), suggesting that nutrition alone does not fully account for the observed survival advantage demonstrated. A similar conclusion can be drawn about immunologic status. NLR has previously been shown to be prognostic in a number of cancers,13-16 including in our own cohort.8 Although immune status remains an important predictor for OS in our locally advanced NSCLC population, when we take NLR into consideration in our analysis, marital status continues to be a strong indicator for survival (Table 2). In terms of other variables analyzed, insurance status was a significant predictor of OS in the MVA, though functional status and other social factors including race were not significant.
We also explored cancer control outcomes in the form of FFR. Married patients had an observable, although not statistically significant, improvement in FFR when compared with the single cohort (Figure 2). In our study, married patients were more likely to undergo trimodality therapy (Table 1), which has likely translated to the improvement of FFR seen in our group. In this case, marriage may serve as a surrogate for availability of a support system to undergo aggressive, potentially toxic treatment.3,17,18 Even in the setting of bimodality therapy, the RTOG 0617 study noted about 17.5% treatment interruptions because of adverse effects or illness, with more than 30% of patients experiencing grade 3 or more esophagitis, irrespective of radiation technique.19 In these scenarios, in addition to receiving better attention to nutrition and care, significant others often provide emotional and social support that, in turn, can lead to better compliance. Social supports and socio-demographic factors are especially critical in patient populations in which access to health care is challenging.
Despite the compelling outcomes presented, our study suffers from the common limitations of retrospective analyses. Marital status, in this setting, most likely correlates with improved socioeconomic status and greater support, which have resulted in improved survival. Furthermore, although patients were self-classified as married or single, our data were not able to capture whether patients were single but lived with another adult or had other types of social support. However, even if there was a proportion of the unmarried cohort that had an alternate support system, separating them out is likely to further expand the differences. Quantifying the amount of social, emotional, or even spiritual support was not possible to accomplish in our analysis, though we know that all 3 can play a role in cancer outcomes.20,21 Further prospective studies would have to be done to completely understand how marital status can influence clinical decisions. Understanding whether marital status is a proxy for social provisions may help to identify populations at risk for inferior outcomes. These at-risk patients may benefit from targeted clinical interventions, such as closer physician follow-up, more aggressive supportive care, access to support groups, or nurse navigator visits.
Conclusions
In patients with locally advanced NSCLC treated with curative-intent following uniform treatment algorithms, marital status was linked with improvement in survival even when adjusted for other key variables, with the second highest HR (after insurance status) among pretreatment demographic variables. Although marriage is an unmodifiable factor in itself, it is most likely a surrogate for better psychosocial support. The scale of these positive survival improvements emphasizes the need to institute targeted supportive care strategies to help advance overall outcomes in a tumor for which modern therapeutic approaches (novel systemic therapy and radiation) have yielded only modest improvement in outcomes yet come at the cost of considerable treatment-related toxicity.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
2. Goldstraw P, Chansky K, Crowley J, et al. The IASLC lung cancer staging project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11(1):39-51.
3. Aizer AA, Chen M-H, McCarthy EP, et al. Marital status and survival in patients with cancer. J Clin Oncol. 2013;31(31):3869-3876.
4. Jatoi A, Novotny P, Cassivi S, et al. Does marital status impact survival and quality of life in patients with non-small cell lung cancer? Observations from the mayo clinic lung cancer cohort. Oncologist. 2007;12(12):1456-1463.
5. Kravdal H, Syse A. Changes over time in the effect of marital status on cancer survival. BMC Public Health. 2011;11:804.
6. Tannenbaum SL, Zhao W, Koru-Sengul T, Miao F, Lee D, Byrne MM. Marital status and its effect on lung cancer survival. Springerplus. 2013;2:504.
7. Ellis L, Canchola AJ, Spiegel D, Ladabaum U, Haile R, Gomez SL. Racial and ethnic disparities in cancer survival: the contribution of tumor, sociodemographic, institutional, and neighborhood characteristics [published online October 16, 2017]. J Clin Oncol. 2018;36(1):25-33.
8. Scilla KA, Bentzen SM, Lam VK, et al. Neutrophil-lymphocyte ratio is a prognostic marker in patients with locally advanced (stage IIIA and IIIB) non-small cell lung cancer treated with combined modality therapy. Oncologist. 2017;22(6):737-742.
9. Lam VK, Bentzen SM, Mohindra P, et al. Obesity is associated with long-term improved survival in definitively treated locally advanced non-small cell lung cancer (NSCLC). Lung Cancer. 2017;104:52-57.
10. Vyfhuis MAL, Bhooshan N, Burrows WM, et al. Oncological outcomes from trimodality therapy receiving definitive doses of neoadjuvant chemoradiation (≥60 Gy) and factors influencing consideration for surgery in stage III non-small cell lung cancer. Adv Radiat Oncol. 2017;2(3):259-269.
11. Siddiqui F, Bae K, Langer CJ, et al. The influence of gender, race, and marital status on survival in lung cancer patients: analysis of radiation therapy oncology group trials. J Thorac Oncol. 2010;5(5):631-639.
12. Vyfhuis MAL, Bhooshan N, Molitoris J, et al. Clinical outcomes of black vs. non-black patients with locally advanced non–small cell lung cancer. Lung Cancer. 2017;114:44-49.
13. Beltran BE, Castro D, De La Cruz-Vargas JA, et al. The neutrophil-lymphocyte ratio is prognostic in patients with early stage aggressive peripheral T cell lymphoma [published online February 26, 2018]. Br J Haematol. doi:10.1111/bjh.15141.
14. Lee BM, Chung SY, Chang JS, Lee KJ, Seong J. The neutrophil-lymphocyte ratio and platelet-lymphocyte ratio are prognostic factors in patients with locally advanced pancreatic cancer treated with chemoradiotherapy. Gut Liver. 2018;12(3):342-352.
15. Najjar M, Agrawal S, Emond JC, Halazun KJ. Pretreatment neutrophil-lymphocyte ratio: useful prognostic biomarker in hepatocellular carcinoma. J Hepatocell Carcinoma. 2018;5:17-28.
16. Hu W, Yu J, Huang Y, Hu F, Zhang X, Wang Y. Lymphocyte-related inflammation and immune-based scores predict prognosis of chordoma patients after radical resection. Transl Oncol. 2018;11(2):444-449.
17. Mahal BA, Cooperberg MR, Aizer AA, et al. Who bears the greatest burden of aggressive treatment of indolent prostate cancer? Am J Med. 2015;128(6):609-616.
18. Inverso G, Mahal BA, Aizer AA, Donoff RB, Chau NG, Haddad RI. Marital status and head and neck cancer outcomes. Cancer. 2015;121(8):1273-1278.
19. Chun SG, Hu C, Choy H, et al. Impact of intensity-modulated radiation therapy technique for locally advanced non-small-cell lung cancer: a secondary analysis of the NRG oncology RTOG 0617 randomized clinical trial. J Clin Oncol. 2017;35(1):56-62.
20. Waite LJ, Lehrer EL. The benefits from marriage and religion in the United States: a comparative analysis. Popul Dev Rev. 2003;29(2):255-276.
21. Osborne C, Ostir GV, Du X, Peek MK, Goodwin JS. The influence of marital status on the stage at diagnosis, treatment, and survival of older women with breast cancer. Breast Cancer Res Treat. 2005;93(1):41-47.
Non–small-cell lung cancer (NSCLC) remains the leading cause of cancer death in the United States, where 29% of patients will present with stage III disease.1,2 Ongoing research efforts seek to improve these outcomes using novel systemic therapy options or modern radiation techniques. However, there have also been recent studies showing the importance of marital and/or partner status on clinical outcomes.3-7 For example, in a large Surveillance, Epidemiology, and End Results (SEER) analysis of 734,889 patients diagnosed with several types of cancer (including lung cancer), patients identified as married were less likely to present with metastatic disease, more likely to receive definitive therapy, and had superior cancer-related mortality even after adjusting for other variables such as cancer stage and treatment when compared with single patients.3 Population-based assessments are important in relaying information about trends and general outcomes based on marital status, but because they are large, they often lack patient-specific information such as nutrition, immunologic status, and variability in treatment paradigms, all of which can independently have an impact on overall survival (OS) in stage III NSCLC.8-10 In addition, population analyses have typically included patients of all cancer stages and hence involved a multitude of treatment approaches ranging from curative to palliative. There are limited well-annotated institutional data on the association of marital status on nonmetastatic, locally advanced (LA-NSCLC) in the setting of National Comprehensive Cancer Network-guided, standard-of-care definitive treatment.
The objective of this analysis is to evaluate the effect of marital status on OS and freedom from recurrence (FFR) in patients with stage III NSCLC who were treated at a National Cancer Institute–designated cancer center with curative intent from 2000 through 2013. We performed a detailed multivariate analysis (MVA) of patient-, disease-, and treatment-specific factors, including the interaction with racial, nutritional, and immunologic status, which to our knowledge has not been previously reported, to comprehensively evaluate the benefit of marital status in patients with LA-NSCLC.
Methods
Patient population and treatment
From January 2000 through December 2013, 355 patients diagnosed with clinical stage III NSCLC (American Joint Committee on Cancer 7th edition) were definitively treated at the University of Maryland in Baltimore, Maryland. Their clinical data were retrospectively analyzed under internal review board approval (GCC 1175, Thoracic Oncology Database). All of the patients were evaluated before treatment by a multidisciplinary team consisting of thoracic surgeons and medical and radiation oncologists. Before treatment, the patients underwent standard work-up, which included systemic imaging with positron-emission (PET), computed-tomographic (CT), PET–CT, and/or bone scan, brain imaging consisting of magnetic-resonance imaging or CT with contrast, and routine blood. Patients had documentation of mediastinal disease by either imaging, mediastinoscopy, or endobronchial ultrasound biopsy.
Definitive therapy was administered using the backbone of chemoradiation therapy (CRT) with (trimodality) or without (bimodality) surgical resection. Concurrent CRT was typically administered with weekly carboplatin–paclitaxel (areas under the curve [AUCs], 2 and 50 mg/m2, respectively) and was generally followed with 2 cycles of consolidative treatment with definitive doses of carboplatin–paclitaxel (AUCs, 5-6 and 200-225 mg/m2, respectively) as tolerated. The entire cohort was also assessed for possible trimodality therapy at the time of initial diagnosis, and patients who were potential surgical candidates were reassessed for mediastinal nodal clearance following repeat radiographic staging after full-dose CRT. Patients who experienced pathologic mediastinal clearance of disease underwent resection followed by consolidative chemotherapy. Unless there was evidence of disease progression, patients who did not have mediastinal lymph node clearance or who were found not to be a surgical candidate proceeded directly to consolidative chemotherapy. The details of patient selection for trimodality therapy and the oncological outcomes have been previously reported.10 For follow-up, patients were normally followed with serial CT or PET–CT scans as clinically indicated every 3 months for the first year, 4 to 6 months for the next 2 to 5 years, and then yearly thereafter.
For the analysis, patients were categorized as being either married or single based on self-reporting. As a surrogate for nutrition status, patients were stratified into 4 pretreatment body mass index (BMI) cohorts based on the following World Health Organization criteria: underweight, <18.5 kg/m2; normal weight, 18.5 to <25 kg/m2; overweight, 25 to <30 kg/m2; and obesity, ≥30 kg/m2. Pretreatment albumin was also evaluated as a continuous variable. For assessment of immunological status, neutrophil-to-lymphocyte ratio (NLR) was calculated at the time of diagnosis by dividing the absolute neutrophil count by the absolute lymphocyte count.
Statistics
We used the Pearson chi-square test to compare categorical variables. OS was calculated from the date of diagnosis (by biopsy of either primary tumor or mediastinal nodes) to the time of death or date of last follow-up. Patients were only censored if they were lost to follow-up. FFR was determined by the date of diagnosis to the time of first failure, with either distant or locoregional disease progression. For this analysis, patients were censored at the time of their last follow-up or death. The Kaplan-Meier product limit method was used to estimate OS and FFR, and we applied the log-rank test to compare outcomes between the 2 cohorts.
We conducted the multivariate analyses using Cox regression with forward model selection. Variables analyzed included age (<60 vs ≥60 years), sex, race (black vs nonblack), median household income, insurance status (Yes vs No), Eastern Cooperative Oncology Group Performance Status (ECOG PS) (range: 0-3; 0 = fully active and 3 = capable of limited self-care, confined to bed/chair >50% of day) at time of diagnosis (0 vs ≥1), pre-CRT BMI, smoking (pack-years), chronic obstructive pulmonary disorder (Yes vs No), Charlson Comorbidity Index score (≤6 vs >7; range, 3-15; this score takes into consideration age, cardiovascular disease, malignancy, and other chronic conditions to calculate 1-year mortality), histology, calculated pretreatment NLR (as a continuous variable), pretreatment albumin (as a continuous variable), T stage, N stage, overall stage (IIIA vs IIIB), radiation technique (3D-CRT vs intensity-modulated radiation therapy [IMRT]), date of diagnosis (divided into quartiles based on proportion diagnosed by years: 2000-2002, 2003-2005, 2006-2009, 2010-2013), use of trimodality therapy, and consolidation chemotherapy. SPSS software (version 23.0) was used for statistical analysis (IBM Corp, Armonk, NY).
Results
Treatment cohorts
Table 1 compares and summarizes patient demographics, disease, and treatment characteristics for married (n = 185; 52.1%) and nonmarried (n = 170; 47.9%) patients. Married patients were more likely to self-identify as being white (P < .0001), reside in zip codes with a higher household median income (P < .0001), have an ECOG PS of 0 (P = .001), have a higher distribution of pretreatment albumin levels (P = .009), and undergo trimodality therapy (P = .001), and they were twice as likely to have insurance (P = .029). Both cohorts were evenly distributed in terms of T stage, N stage, and overall staging. There was no difference in pretreatment NLR or pretreatment BMI between married and single patients. Concurrent CRT was used in more than 85% of patients in both groups, with approximately two-thirds also receiving consolidation chemotherapy (Table 1). Median delivered radiation dose was 64.8 Gy (range, 10.8-81.6 Gy). There was no statistically significant difference in radiation dose delivered to either group, with nearly 90% of the cohort receiving ≥60 Gy.
OS and FFR
With a median follow-up of 15 months for all patients and 89 months for surviving patients (range, 1-184 months), married patients had improved OS when compared with the single cohort, with a median survival of 29.6 and 18.4 months, respectively (unadjusted hazard ratio [HR] of married vs nonmarried, .640; 95% confidence interval [CI], 0.502-0.816; P < .0001; Figure 1A). The estimated 2- and 5-year OS for married and single patients were 56% and 31% and 38.6% and 15%, respectively. When stratified by stage, married patients with stage IIIB disease (median survival, 25 months; Figure 1B) had a similar survival to unmarried patients with stage IIIA disease (median survival, 24 months; Figure 1B).
In stage IIIA patients, marital status was associated with an unadjusted HR of .696 (95% CI, 0.497-0.974; P = .035), with a larger OS benefit seen in the IIIB group (unadjusted HR, .601; 95% CI, 0.422-0.856; P = .005).
Survival as it pertains to marital status was further stratified by sex (Figure 2A) and race (Figure 2B). Married men had an improved estimated median survival of 30 months when compared with single men, whose median survival was 16 months (unadjusted HR, .541; 95% CI, 0.392-0.746; P < .0001). On the other hand, marital status had no statistically significant effect on OS when comparing married women with their single counterparts (unadjusted HR, .717; 95% CI, 0.491-1.048; P = .085; Figure 2A), with an overall median survival of approximately 28 months for the entire female cohort. Stratification by race also showed similar results, with married nonblack patients demonstrating better OS when compared with single nonblack patients (HR, .586; 95% CI, 0.420-0.820; P = .002; Figure 2B), with a median survival of 29 and 17 months, respectively. Black patients also had a similar improvement in survival when comparing the married (median survival, 30 months) and nonmarried groups (median survival, 19.6 months; unadjusted HR, .676; 95% CI, 0.457-1.000; P = .050; Figure 2B).
FFR did not differ between the 2 groups, with a median time to failure of 17 and 15 months for married and nonmarried patients, respectively (unadjusted HR, .799; 95% CI, 0.607-1.051; P = .108; Figure 3). Estimated 2- and 5-year FFR for married and nonmarried patients were 39.4% and 27% and 31.5% and 18.5%, respectively (Figure 3).
Clinical predictors of survival
On MVA, factors that were independent predictors for OS are summarized in Table 2. Risk of death was reduced by approximately 65% and 45% in patients who underwent trimodality treatment (P < .0001) or were able to undergo consolidative chemotherapy (P = .004) when compared with those who were treated definitively with bimodality treatment or did not undergo systemic doses of adjuvant chemotherapy, respectively. Having insurance (P = .048) and use of IMRT over 3D-CRT (P = .008) was associated with a reduction of mortality by about half in this cohort. Both gender (improved OS with female sex; P = .004) and marital status (improved OS with marriage; P = .006) were associated with a decreased the risk of death by 40% (Table 2). By contrast, a higher NLR resulted
Discussion
Our study continues to support the notion that marital status is an independent indicator of survival in stage III NSCLC (adjusted HR, .59; 95% CI, 0.404-0.859; P = .006). The benefit of marriage in this population seems to be better than that reported in the SEER analysis for all stages, wherein the HR for death of married patients compared with their single counterparts was .85 (95% CI, 0.83-0.87). In their analysis, the investigators hypothesized that this survival advantage could partially be explained by better access to health care and adherence to therapy, as was supported by the higher likelihood of married patients presenting with localized disease and receiving definitive treatment.3 Another population-based study using the Florida Cancer Data System identified 161,228 lung cancer patients (NSCLC and small-cell lung histology included), and on MVA, marital status remained an important prognostic indicator for OS when compared with never-married patients (HR, .86; P = .001).6 In addition to typically including patients with all stages of diseases, population-based studies often include patients who receive a heterogeneous combination of treatment modalities, possibly confounding the analysis. Furthermore, large population analyses typically do not report on patient-specific variables such as nutrition (ie, BMI and albumin) or immunologic status (ie, NLR), both of which have been shown to be independent predictors of survival in LA-NSCLC.8,9
In contrast, some other studies have failed to demonstrate an OS advantage with marital status in patients with NSCLC. For example, in a meta-analysis that evaluated the influence of race, gender, and marital status on 1,365 nonoperative NSCLC patients who were enrolled in 9 Radiation Therapy Oncology Group (RTOG) trials, the investigators did not find marital status to be independently predictive of survival.11 In addition, for the 5,898 patients who were prospectively enrolled in a Mayo Clinic Lung Cancer Cohort (MCLCC), marital status was also found not to be prognostic for NSCLC outcomes when all stages of the disease were analyzed together.4 There are some possible confounding factors in these studies. Patients recruited for clinical trials tend to be healthier with a better performance status and have a support system (including close monitoring by the study team) when compared with the general population diagnosed with lung cancer. About 70% to 76% of the patients in both the RTOG and MCLCC studies were married, which is significantly higher than both the national average (51%) and our group (52.1%). Like other population-based studies, the MCLCC included patients with all stages getting a variety of treatments. Although no overall impact on survival was noted, the investigators noted that single, divorced, and widowed patients were more likely to not receive cancer therapy(P < .0001). The marital status also influenced the choice of therapy, with subgroup analysis revealing inferior outcomes in widowed and divorced patients with stage IA, IIB, or IIIB disease. The authors also recognized an inherent referral bias from patients, with support system being typically seen at the Mayo clinics, which may have played an additional role. All of the patients in our analysis were appropriately staged and received curative-intent treatment by a team of physicians using essentially identical therapeutic strategies, thus minimizing some of these confounding factors. This allowed us to explore the impact of marital status while a patient was undergoing stage-appropriate treatment. We demonstrated a strong association with marital status and survival that even overcame the effects of stage (IIIA vs IIIB) on clinical outcomes (Figure 1B).
Furthermore, our analysis allowed us to explore the interaction of race and marital status more definitively because the demographics of the patients in the RTOG and MCLCC included 14% and less than 3% of patients identified as being nonwhite, respectively, in contrast to our analysis in which 41% of the patients self-identified as black.12 In our black population, marital status was associated with an observable improvement in OS, similar to our nonblack, predominantly white (97%) cohort (Figure 2B). Also, the results of our analysis may be a more accurate representation of the general population living in large urban or semiurban settings and further implies that an intact social support system could have a greater influence on clinical outcomes.
The current analysis is unique when compared with previous published studies in that beyond conventional demographic and treatment-related factors, we have comprehensively explored potential mechanisms that may explain the survival advantage seen in married patients by evaluating additional factors, such as functional status (ECOG and Charlson’s scores), nutritional status (BMI and albumin), immunologic characteristics (NLR), and other social factors (race, income, insurance status). Although married patients were more likely to have a higher BMI and albumin at diagnosis, when controlling for these factors in the multivariable analysis, marital status remained strongly prognostic (Table 2), suggesting that nutrition alone does not fully account for the observed survival advantage demonstrated. A similar conclusion can be drawn about immunologic status. NLR has previously been shown to be prognostic in a number of cancers,13-16 including in our own cohort.8 Although immune status remains an important predictor for OS in our locally advanced NSCLC population, when we take NLR into consideration in our analysis, marital status continues to be a strong indicator for survival (Table 2). In terms of other variables analyzed, insurance status was a significant predictor of OS in the MVA, though functional status and other social factors including race were not significant.
We also explored cancer control outcomes in the form of FFR. Married patients had an observable, although not statistically significant, improvement in FFR when compared with the single cohort (Figure 2). In our study, married patients were more likely to undergo trimodality therapy (Table 1), which has likely translated to the improvement of FFR seen in our group. In this case, marriage may serve as a surrogate for availability of a support system to undergo aggressive, potentially toxic treatment.3,17,18 Even in the setting of bimodality therapy, the RTOG 0617 study noted about 17.5% treatment interruptions because of adverse effects or illness, with more than 30% of patients experiencing grade 3 or more esophagitis, irrespective of radiation technique.19 In these scenarios, in addition to receiving better attention to nutrition and care, significant others often provide emotional and social support that, in turn, can lead to better compliance. Social supports and socio-demographic factors are especially critical in patient populations in which access to health care is challenging.
Despite the compelling outcomes presented, our study suffers from the common limitations of retrospective analyses. Marital status, in this setting, most likely correlates with improved socioeconomic status and greater support, which have resulted in improved survival. Furthermore, although patients were self-classified as married or single, our data were not able to capture whether patients were single but lived with another adult or had other types of social support. However, even if there was a proportion of the unmarried cohort that had an alternate support system, separating them out is likely to further expand the differences. Quantifying the amount of social, emotional, or even spiritual support was not possible to accomplish in our analysis, though we know that all 3 can play a role in cancer outcomes.20,21 Further prospective studies would have to be done to completely understand how marital status can influence clinical decisions. Understanding whether marital status is a proxy for social provisions may help to identify populations at risk for inferior outcomes. These at-risk patients may benefit from targeted clinical interventions, such as closer physician follow-up, more aggressive supportive care, access to support groups, or nurse navigator visits.
Conclusions
In patients with locally advanced NSCLC treated with curative-intent following uniform treatment algorithms, marital status was linked with improvement in survival even when adjusted for other key variables, with the second highest HR (after insurance status) among pretreatment demographic variables. Although marriage is an unmodifiable factor in itself, it is most likely a surrogate for better psychosocial support. The scale of these positive survival improvements emphasizes the need to institute targeted supportive care strategies to help advance overall outcomes in a tumor for which modern therapeutic approaches (novel systemic therapy and radiation) have yielded only modest improvement in outcomes yet come at the cost of considerable treatment-related toxicity.
Non–small-cell lung cancer (NSCLC) remains the leading cause of cancer death in the United States, where 29% of patients will present with stage III disease.1,2 Ongoing research efforts seek to improve these outcomes using novel systemic therapy options or modern radiation techniques. However, there have also been recent studies showing the importance of marital and/or partner status on clinical outcomes.3-7 For example, in a large Surveillance, Epidemiology, and End Results (SEER) analysis of 734,889 patients diagnosed with several types of cancer (including lung cancer), patients identified as married were less likely to present with metastatic disease, more likely to receive definitive therapy, and had superior cancer-related mortality even after adjusting for other variables such as cancer stage and treatment when compared with single patients.3 Population-based assessments are important in relaying information about trends and general outcomes based on marital status, but because they are large, they often lack patient-specific information such as nutrition, immunologic status, and variability in treatment paradigms, all of which can independently have an impact on overall survival (OS) in stage III NSCLC.8-10 In addition, population analyses have typically included patients of all cancer stages and hence involved a multitude of treatment approaches ranging from curative to palliative. There are limited well-annotated institutional data on the association of marital status on nonmetastatic, locally advanced (LA-NSCLC) in the setting of National Comprehensive Cancer Network-guided, standard-of-care definitive treatment.
The objective of this analysis is to evaluate the effect of marital status on OS and freedom from recurrence (FFR) in patients with stage III NSCLC who were treated at a National Cancer Institute–designated cancer center with curative intent from 2000 through 2013. We performed a detailed multivariate analysis (MVA) of patient-, disease-, and treatment-specific factors, including the interaction with racial, nutritional, and immunologic status, which to our knowledge has not been previously reported, to comprehensively evaluate the benefit of marital status in patients with LA-NSCLC.
Methods
Patient population and treatment
From January 2000 through December 2013, 355 patients diagnosed with clinical stage III NSCLC (American Joint Committee on Cancer 7th edition) were definitively treated at the University of Maryland in Baltimore, Maryland. Their clinical data were retrospectively analyzed under internal review board approval (GCC 1175, Thoracic Oncology Database). All of the patients were evaluated before treatment by a multidisciplinary team consisting of thoracic surgeons and medical and radiation oncologists. Before treatment, the patients underwent standard work-up, which included systemic imaging with positron-emission (PET), computed-tomographic (CT), PET–CT, and/or bone scan, brain imaging consisting of magnetic-resonance imaging or CT with contrast, and routine blood. Patients had documentation of mediastinal disease by either imaging, mediastinoscopy, or endobronchial ultrasound biopsy.
Definitive therapy was administered using the backbone of chemoradiation therapy (CRT) with (trimodality) or without (bimodality) surgical resection. Concurrent CRT was typically administered with weekly carboplatin–paclitaxel (areas under the curve [AUCs], 2 and 50 mg/m2, respectively) and was generally followed with 2 cycles of consolidative treatment with definitive doses of carboplatin–paclitaxel (AUCs, 5-6 and 200-225 mg/m2, respectively) as tolerated. The entire cohort was also assessed for possible trimodality therapy at the time of initial diagnosis, and patients who were potential surgical candidates were reassessed for mediastinal nodal clearance following repeat radiographic staging after full-dose CRT. Patients who experienced pathologic mediastinal clearance of disease underwent resection followed by consolidative chemotherapy. Unless there was evidence of disease progression, patients who did not have mediastinal lymph node clearance or who were found not to be a surgical candidate proceeded directly to consolidative chemotherapy. The details of patient selection for trimodality therapy and the oncological outcomes have been previously reported.10 For follow-up, patients were normally followed with serial CT or PET–CT scans as clinically indicated every 3 months for the first year, 4 to 6 months for the next 2 to 5 years, and then yearly thereafter.
For the analysis, patients were categorized as being either married or single based on self-reporting. As a surrogate for nutrition status, patients were stratified into 4 pretreatment body mass index (BMI) cohorts based on the following World Health Organization criteria: underweight, <18.5 kg/m2; normal weight, 18.5 to <25 kg/m2; overweight, 25 to <30 kg/m2; and obesity, ≥30 kg/m2. Pretreatment albumin was also evaluated as a continuous variable. For assessment of immunological status, neutrophil-to-lymphocyte ratio (NLR) was calculated at the time of diagnosis by dividing the absolute neutrophil count by the absolute lymphocyte count.
Statistics
We used the Pearson chi-square test to compare categorical variables. OS was calculated from the date of diagnosis (by biopsy of either primary tumor or mediastinal nodes) to the time of death or date of last follow-up. Patients were only censored if they were lost to follow-up. FFR was determined by the date of diagnosis to the time of first failure, with either distant or locoregional disease progression. For this analysis, patients were censored at the time of their last follow-up or death. The Kaplan-Meier product limit method was used to estimate OS and FFR, and we applied the log-rank test to compare outcomes between the 2 cohorts.
We conducted the multivariate analyses using Cox regression with forward model selection. Variables analyzed included age (<60 vs ≥60 years), sex, race (black vs nonblack), median household income, insurance status (Yes vs No), Eastern Cooperative Oncology Group Performance Status (ECOG PS) (range: 0-3; 0 = fully active and 3 = capable of limited self-care, confined to bed/chair >50% of day) at time of diagnosis (0 vs ≥1), pre-CRT BMI, smoking (pack-years), chronic obstructive pulmonary disorder (Yes vs No), Charlson Comorbidity Index score (≤6 vs >7; range, 3-15; this score takes into consideration age, cardiovascular disease, malignancy, and other chronic conditions to calculate 1-year mortality), histology, calculated pretreatment NLR (as a continuous variable), pretreatment albumin (as a continuous variable), T stage, N stage, overall stage (IIIA vs IIIB), radiation technique (3D-CRT vs intensity-modulated radiation therapy [IMRT]), date of diagnosis (divided into quartiles based on proportion diagnosed by years: 2000-2002, 2003-2005, 2006-2009, 2010-2013), use of trimodality therapy, and consolidation chemotherapy. SPSS software (version 23.0) was used for statistical analysis (IBM Corp, Armonk, NY).
Results
Treatment cohorts
Table 1 compares and summarizes patient demographics, disease, and treatment characteristics for married (n = 185; 52.1%) and nonmarried (n = 170; 47.9%) patients. Married patients were more likely to self-identify as being white (P < .0001), reside in zip codes with a higher household median income (P < .0001), have an ECOG PS of 0 (P = .001), have a higher distribution of pretreatment albumin levels (P = .009), and undergo trimodality therapy (P = .001), and they were twice as likely to have insurance (P = .029). Both cohorts were evenly distributed in terms of T stage, N stage, and overall staging. There was no difference in pretreatment NLR or pretreatment BMI between married and single patients. Concurrent CRT was used in more than 85% of patients in both groups, with approximately two-thirds also receiving consolidation chemotherapy (Table 1). Median delivered radiation dose was 64.8 Gy (range, 10.8-81.6 Gy). There was no statistically significant difference in radiation dose delivered to either group, with nearly 90% of the cohort receiving ≥60 Gy.
OS and FFR
With a median follow-up of 15 months for all patients and 89 months for surviving patients (range, 1-184 months), married patients had improved OS when compared with the single cohort, with a median survival of 29.6 and 18.4 months, respectively (unadjusted hazard ratio [HR] of married vs nonmarried, .640; 95% confidence interval [CI], 0.502-0.816; P < .0001; Figure 1A). The estimated 2- and 5-year OS for married and single patients were 56% and 31% and 38.6% and 15%, respectively. When stratified by stage, married patients with stage IIIB disease (median survival, 25 months; Figure 1B) had a similar survival to unmarried patients with stage IIIA disease (median survival, 24 months; Figure 1B).
In stage IIIA patients, marital status was associated with an unadjusted HR of .696 (95% CI, 0.497-0.974; P = .035), with a larger OS benefit seen in the IIIB group (unadjusted HR, .601; 95% CI, 0.422-0.856; P = .005).
Survival as it pertains to marital status was further stratified by sex (Figure 2A) and race (Figure 2B). Married men had an improved estimated median survival of 30 months when compared with single men, whose median survival was 16 months (unadjusted HR, .541; 95% CI, 0.392-0.746; P < .0001). On the other hand, marital status had no statistically significant effect on OS when comparing married women with their single counterparts (unadjusted HR, .717; 95% CI, 0.491-1.048; P = .085; Figure 2A), with an overall median survival of approximately 28 months for the entire female cohort. Stratification by race also showed similar results, with married nonblack patients demonstrating better OS when compared with single nonblack patients (HR, .586; 95% CI, 0.420-0.820; P = .002; Figure 2B), with a median survival of 29 and 17 months, respectively. Black patients also had a similar improvement in survival when comparing the married (median survival, 30 months) and nonmarried groups (median survival, 19.6 months; unadjusted HR, .676; 95% CI, 0.457-1.000; P = .050; Figure 2B).
FFR did not differ between the 2 groups, with a median time to failure of 17 and 15 months for married and nonmarried patients, respectively (unadjusted HR, .799; 95% CI, 0.607-1.051; P = .108; Figure 3). Estimated 2- and 5-year FFR for married and nonmarried patients were 39.4% and 27% and 31.5% and 18.5%, respectively (Figure 3).
Clinical predictors of survival
On MVA, factors that were independent predictors for OS are summarized in Table 2. Risk of death was reduced by approximately 65% and 45% in patients who underwent trimodality treatment (P < .0001) or were able to undergo consolidative chemotherapy (P = .004) when compared with those who were treated definitively with bimodality treatment or did not undergo systemic doses of adjuvant chemotherapy, respectively. Having insurance (P = .048) and use of IMRT over 3D-CRT (P = .008) was associated with a reduction of mortality by about half in this cohort. Both gender (improved OS with female sex; P = .004) and marital status (improved OS with marriage; P = .006) were associated with a decreased the risk of death by 40% (Table 2). By contrast, a higher NLR resulted
Discussion
Our study continues to support the notion that marital status is an independent indicator of survival in stage III NSCLC (adjusted HR, .59; 95% CI, 0.404-0.859; P = .006). The benefit of marriage in this population seems to be better than that reported in the SEER analysis for all stages, wherein the HR for death of married patients compared with their single counterparts was .85 (95% CI, 0.83-0.87). In their analysis, the investigators hypothesized that this survival advantage could partially be explained by better access to health care and adherence to therapy, as was supported by the higher likelihood of married patients presenting with localized disease and receiving definitive treatment.3 Another population-based study using the Florida Cancer Data System identified 161,228 lung cancer patients (NSCLC and small-cell lung histology included), and on MVA, marital status remained an important prognostic indicator for OS when compared with never-married patients (HR, .86; P = .001).6 In addition to typically including patients with all stages of diseases, population-based studies often include patients who receive a heterogeneous combination of treatment modalities, possibly confounding the analysis. Furthermore, large population analyses typically do not report on patient-specific variables such as nutrition (ie, BMI and albumin) or immunologic status (ie, NLR), both of which have been shown to be independent predictors of survival in LA-NSCLC.8,9
In contrast, some other studies have failed to demonstrate an OS advantage with marital status in patients with NSCLC. For example, in a meta-analysis that evaluated the influence of race, gender, and marital status on 1,365 nonoperative NSCLC patients who were enrolled in 9 Radiation Therapy Oncology Group (RTOG) trials, the investigators did not find marital status to be independently predictive of survival.11 In addition, for the 5,898 patients who were prospectively enrolled in a Mayo Clinic Lung Cancer Cohort (MCLCC), marital status was also found not to be prognostic for NSCLC outcomes when all stages of the disease were analyzed together.4 There are some possible confounding factors in these studies. Patients recruited for clinical trials tend to be healthier with a better performance status and have a support system (including close monitoring by the study team) when compared with the general population diagnosed with lung cancer. About 70% to 76% of the patients in both the RTOG and MCLCC studies were married, which is significantly higher than both the national average (51%) and our group (52.1%). Like other population-based studies, the MCLCC included patients with all stages getting a variety of treatments. Although no overall impact on survival was noted, the investigators noted that single, divorced, and widowed patients were more likely to not receive cancer therapy(P < .0001). The marital status also influenced the choice of therapy, with subgroup analysis revealing inferior outcomes in widowed and divorced patients with stage IA, IIB, or IIIB disease. The authors also recognized an inherent referral bias from patients, with support system being typically seen at the Mayo clinics, which may have played an additional role. All of the patients in our analysis were appropriately staged and received curative-intent treatment by a team of physicians using essentially identical therapeutic strategies, thus minimizing some of these confounding factors. This allowed us to explore the impact of marital status while a patient was undergoing stage-appropriate treatment. We demonstrated a strong association with marital status and survival that even overcame the effects of stage (IIIA vs IIIB) on clinical outcomes (Figure 1B).
Furthermore, our analysis allowed us to explore the interaction of race and marital status more definitively because the demographics of the patients in the RTOG and MCLCC included 14% and less than 3% of patients identified as being nonwhite, respectively, in contrast to our analysis in which 41% of the patients self-identified as black.12 In our black population, marital status was associated with an observable improvement in OS, similar to our nonblack, predominantly white (97%) cohort (Figure 2B). Also, the results of our analysis may be a more accurate representation of the general population living in large urban or semiurban settings and further implies that an intact social support system could have a greater influence on clinical outcomes.
The current analysis is unique when compared with previous published studies in that beyond conventional demographic and treatment-related factors, we have comprehensively explored potential mechanisms that may explain the survival advantage seen in married patients by evaluating additional factors, such as functional status (ECOG and Charlson’s scores), nutritional status (BMI and albumin), immunologic characteristics (NLR), and other social factors (race, income, insurance status). Although married patients were more likely to have a higher BMI and albumin at diagnosis, when controlling for these factors in the multivariable analysis, marital status remained strongly prognostic (Table 2), suggesting that nutrition alone does not fully account for the observed survival advantage demonstrated. A similar conclusion can be drawn about immunologic status. NLR has previously been shown to be prognostic in a number of cancers,13-16 including in our own cohort.8 Although immune status remains an important predictor for OS in our locally advanced NSCLC population, when we take NLR into consideration in our analysis, marital status continues to be a strong indicator for survival (Table 2). In terms of other variables analyzed, insurance status was a significant predictor of OS in the MVA, though functional status and other social factors including race were not significant.
We also explored cancer control outcomes in the form of FFR. Married patients had an observable, although not statistically significant, improvement in FFR when compared with the single cohort (Figure 2). In our study, married patients were more likely to undergo trimodality therapy (Table 1), which has likely translated to the improvement of FFR seen in our group. In this case, marriage may serve as a surrogate for availability of a support system to undergo aggressive, potentially toxic treatment.3,17,18 Even in the setting of bimodality therapy, the RTOG 0617 study noted about 17.5% treatment interruptions because of adverse effects or illness, with more than 30% of patients experiencing grade 3 or more esophagitis, irrespective of radiation technique.19 In these scenarios, in addition to receiving better attention to nutrition and care, significant others often provide emotional and social support that, in turn, can lead to better compliance. Social supports and socio-demographic factors are especially critical in patient populations in which access to health care is challenging.
Despite the compelling outcomes presented, our study suffers from the common limitations of retrospective analyses. Marital status, in this setting, most likely correlates with improved socioeconomic status and greater support, which have resulted in improved survival. Furthermore, although patients were self-classified as married or single, our data were not able to capture whether patients were single but lived with another adult or had other types of social support. However, even if there was a proportion of the unmarried cohort that had an alternate support system, separating them out is likely to further expand the differences. Quantifying the amount of social, emotional, or even spiritual support was not possible to accomplish in our analysis, though we know that all 3 can play a role in cancer outcomes.20,21 Further prospective studies would have to be done to completely understand how marital status can influence clinical decisions. Understanding whether marital status is a proxy for social provisions may help to identify populations at risk for inferior outcomes. These at-risk patients may benefit from targeted clinical interventions, such as closer physician follow-up, more aggressive supportive care, access to support groups, or nurse navigator visits.
Conclusions
In patients with locally advanced NSCLC treated with curative-intent following uniform treatment algorithms, marital status was linked with improvement in survival even when adjusted for other key variables, with the second highest HR (after insurance status) among pretreatment demographic variables. Although marriage is an unmodifiable factor in itself, it is most likely a surrogate for better psychosocial support. The scale of these positive survival improvements emphasizes the need to institute targeted supportive care strategies to help advance overall outcomes in a tumor for which modern therapeutic approaches (novel systemic therapy and radiation) have yielded only modest improvement in outcomes yet come at the cost of considerable treatment-related toxicity.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
2. Goldstraw P, Chansky K, Crowley J, et al. The IASLC lung cancer staging project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11(1):39-51.
3. Aizer AA, Chen M-H, McCarthy EP, et al. Marital status and survival in patients with cancer. J Clin Oncol. 2013;31(31):3869-3876.
4. Jatoi A, Novotny P, Cassivi S, et al. Does marital status impact survival and quality of life in patients with non-small cell lung cancer? Observations from the mayo clinic lung cancer cohort. Oncologist. 2007;12(12):1456-1463.
5. Kravdal H, Syse A. Changes over time in the effect of marital status on cancer survival. BMC Public Health. 2011;11:804.
6. Tannenbaum SL, Zhao W, Koru-Sengul T, Miao F, Lee D, Byrne MM. Marital status and its effect on lung cancer survival. Springerplus. 2013;2:504.
7. Ellis L, Canchola AJ, Spiegel D, Ladabaum U, Haile R, Gomez SL. Racial and ethnic disparities in cancer survival: the contribution of tumor, sociodemographic, institutional, and neighborhood characteristics [published online October 16, 2017]. J Clin Oncol. 2018;36(1):25-33.
8. Scilla KA, Bentzen SM, Lam VK, et al. Neutrophil-lymphocyte ratio is a prognostic marker in patients with locally advanced (stage IIIA and IIIB) non-small cell lung cancer treated with combined modality therapy. Oncologist. 2017;22(6):737-742.
9. Lam VK, Bentzen SM, Mohindra P, et al. Obesity is associated with long-term improved survival in definitively treated locally advanced non-small cell lung cancer (NSCLC). Lung Cancer. 2017;104:52-57.
10. Vyfhuis MAL, Bhooshan N, Burrows WM, et al. Oncological outcomes from trimodality therapy receiving definitive doses of neoadjuvant chemoradiation (≥60 Gy) and factors influencing consideration for surgery in stage III non-small cell lung cancer. Adv Radiat Oncol. 2017;2(3):259-269.
11. Siddiqui F, Bae K, Langer CJ, et al. The influence of gender, race, and marital status on survival in lung cancer patients: analysis of radiation therapy oncology group trials. J Thorac Oncol. 2010;5(5):631-639.
12. Vyfhuis MAL, Bhooshan N, Molitoris J, et al. Clinical outcomes of black vs. non-black patients with locally advanced non–small cell lung cancer. Lung Cancer. 2017;114:44-49.
13. Beltran BE, Castro D, De La Cruz-Vargas JA, et al. The neutrophil-lymphocyte ratio is prognostic in patients with early stage aggressive peripheral T cell lymphoma [published online February 26, 2018]. Br J Haematol. doi:10.1111/bjh.15141.
14. Lee BM, Chung SY, Chang JS, Lee KJ, Seong J. The neutrophil-lymphocyte ratio and platelet-lymphocyte ratio are prognostic factors in patients with locally advanced pancreatic cancer treated with chemoradiotherapy. Gut Liver. 2018;12(3):342-352.
15. Najjar M, Agrawal S, Emond JC, Halazun KJ. Pretreatment neutrophil-lymphocyte ratio: useful prognostic biomarker in hepatocellular carcinoma. J Hepatocell Carcinoma. 2018;5:17-28.
16. Hu W, Yu J, Huang Y, Hu F, Zhang X, Wang Y. Lymphocyte-related inflammation and immune-based scores predict prognosis of chordoma patients after radical resection. Transl Oncol. 2018;11(2):444-449.
17. Mahal BA, Cooperberg MR, Aizer AA, et al. Who bears the greatest burden of aggressive treatment of indolent prostate cancer? Am J Med. 2015;128(6):609-616.
18. Inverso G, Mahal BA, Aizer AA, Donoff RB, Chau NG, Haddad RI. Marital status and head and neck cancer outcomes. Cancer. 2015;121(8):1273-1278.
19. Chun SG, Hu C, Choy H, et al. Impact of intensity-modulated radiation therapy technique for locally advanced non-small-cell lung cancer: a secondary analysis of the NRG oncology RTOG 0617 randomized clinical trial. J Clin Oncol. 2017;35(1):56-62.
20. Waite LJ, Lehrer EL. The benefits from marriage and religion in the United States: a comparative analysis. Popul Dev Rev. 2003;29(2):255-276.
21. Osborne C, Ostir GV, Du X, Peek MK, Goodwin JS. The influence of marital status on the stage at diagnosis, treatment, and survival of older women with breast cancer. Breast Cancer Res Treat. 2005;93(1):41-47.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
2. Goldstraw P, Chansky K, Crowley J, et al. The IASLC lung cancer staging project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11(1):39-51.
3. Aizer AA, Chen M-H, McCarthy EP, et al. Marital status and survival in patients with cancer. J Clin Oncol. 2013;31(31):3869-3876.
4. Jatoi A, Novotny P, Cassivi S, et al. Does marital status impact survival and quality of life in patients with non-small cell lung cancer? Observations from the mayo clinic lung cancer cohort. Oncologist. 2007;12(12):1456-1463.
5. Kravdal H, Syse A. Changes over time in the effect of marital status on cancer survival. BMC Public Health. 2011;11:804.
6. Tannenbaum SL, Zhao W, Koru-Sengul T, Miao F, Lee D, Byrne MM. Marital status and its effect on lung cancer survival. Springerplus. 2013;2:504.
7. Ellis L, Canchola AJ, Spiegel D, Ladabaum U, Haile R, Gomez SL. Racial and ethnic disparities in cancer survival: the contribution of tumor, sociodemographic, institutional, and neighborhood characteristics [published online October 16, 2017]. J Clin Oncol. 2018;36(1):25-33.
8. Scilla KA, Bentzen SM, Lam VK, et al. Neutrophil-lymphocyte ratio is a prognostic marker in patients with locally advanced (stage IIIA and IIIB) non-small cell lung cancer treated with combined modality therapy. Oncologist. 2017;22(6):737-742.
9. Lam VK, Bentzen SM, Mohindra P, et al. Obesity is associated with long-term improved survival in definitively treated locally advanced non-small cell lung cancer (NSCLC). Lung Cancer. 2017;104:52-57.
10. Vyfhuis MAL, Bhooshan N, Burrows WM, et al. Oncological outcomes from trimodality therapy receiving definitive doses of neoadjuvant chemoradiation (≥60 Gy) and factors influencing consideration for surgery in stage III non-small cell lung cancer. Adv Radiat Oncol. 2017;2(3):259-269.
11. Siddiqui F, Bae K, Langer CJ, et al. The influence of gender, race, and marital status on survival in lung cancer patients: analysis of radiation therapy oncology group trials. J Thorac Oncol. 2010;5(5):631-639.
12. Vyfhuis MAL, Bhooshan N, Molitoris J, et al. Clinical outcomes of black vs. non-black patients with locally advanced non–small cell lung cancer. Lung Cancer. 2017;114:44-49.
13. Beltran BE, Castro D, De La Cruz-Vargas JA, et al. The neutrophil-lymphocyte ratio is prognostic in patients with early stage aggressive peripheral T cell lymphoma [published online February 26, 2018]. Br J Haematol. doi:10.1111/bjh.15141.
14. Lee BM, Chung SY, Chang JS, Lee KJ, Seong J. The neutrophil-lymphocyte ratio and platelet-lymphocyte ratio are prognostic factors in patients with locally advanced pancreatic cancer treated with chemoradiotherapy. Gut Liver. 2018;12(3):342-352.
15. Najjar M, Agrawal S, Emond JC, Halazun KJ. Pretreatment neutrophil-lymphocyte ratio: useful prognostic biomarker in hepatocellular carcinoma. J Hepatocell Carcinoma. 2018;5:17-28.
16. Hu W, Yu J, Huang Y, Hu F, Zhang X, Wang Y. Lymphocyte-related inflammation and immune-based scores predict prognosis of chordoma patients after radical resection. Transl Oncol. 2018;11(2):444-449.
17. Mahal BA, Cooperberg MR, Aizer AA, et al. Who bears the greatest burden of aggressive treatment of indolent prostate cancer? Am J Med. 2015;128(6):609-616.
18. Inverso G, Mahal BA, Aizer AA, Donoff RB, Chau NG, Haddad RI. Marital status and head and neck cancer outcomes. Cancer. 2015;121(8):1273-1278.
19. Chun SG, Hu C, Choy H, et al. Impact of intensity-modulated radiation therapy technique for locally advanced non-small-cell lung cancer: a secondary analysis of the NRG oncology RTOG 0617 randomized clinical trial. J Clin Oncol. 2017;35(1):56-62.
20. Waite LJ, Lehrer EL. The benefits from marriage and religion in the United States: a comparative analysis. Popul Dev Rev. 2003;29(2):255-276.
21. Osborne C, Ostir GV, Du X, Peek MK, Goodwin JS. The influence of marital status on the stage at diagnosis, treatment, and survival of older women with breast cancer. Breast Cancer Res Treat. 2005;93(1):41-47.
Effect of time of admission to treatment initiation on outcomes of patients with acute myeloid leukemia: a tertiary care referral center experience
Acute myeloid leukemia (AML) is the most common acute leukemia in adults in the United States.1 In 2018, the estimated annual incidence of AML is 19,520 (32.4% of all new leukemia cases), with 10,670 projected deaths (43.8% of all leukemia deaths).1 New molecularly targeted treatments are increasingly being used in treating AML, and some of them have shown improved health outcomes. In general, age, white blood cell (WBC) count at presentation, cytogenetics, and molecular characteristics are the major determinants of prognosis and treatment outcome. Studies analyzing the Surveillance Epidemiology and End Results database have also shown racial differences in outcomes.2 It is well known to the oncology community that patients with similar characteristics may respond differently to treatment and that outcome is not uniformly related to the well-defined clinical and laboratory characteristics. Issues related to health care disparities and access to health care are also known to affect the outcome in patients with cancer.3-9
AML is generally considered by the medical community as a time-sensitive condition. Treatment of patients with AML usually consists of induction chemotherapy followed by consolidation treatment with consideration for stem cell transplant. The duration of time from admission to treatment (TAT) of AML with induction chemotherapy is dependent on multiple factors. These may include the assessment of comorbid conditions and the availability of molecular studies at the time of treatment, which can be time consuming. The effect of treatment delays after AML diagnosis has been investigated, but with conflicting results. One study showed that time from diagnosis to treatment initiation affects survival in younger patients, and another showed it has no effect on survival regardless of patient age.10,11 We describe here the results of a retrospective analysis evaluating the impact of TAT and day of admission on outcomes of patients with AML who received treatment at a tertiary care referral center.
Methods and materials
We did a retrospective medical record review of all newly diagnosed AML patients at the Oklahoma University Health Sciences Center (OUHSC). Our sample was composed of 154 adult patients. Our inclusion criteria were an age of 18 years or older with complete insurance data, a diagnosis of AML, and having received treatment at our institution from January 2000 through June 2015. Data were obtained on laboratory values at diagnosis, pathology data including cytogenetics, molecular data, and bone marrow biopsies. Data on patient characteristics such as age, race and/or ethnicity, and comorbidities were obtained from the electronic medical records. Treatment data on type and dose of chemotherapy during induction, subsequent treatment phases, and number of treatments to achieve complete response (CR) as well as response data of CR achievement, relapse, date of CR, date of relapse, stem cell transplantation data, date of death, and date of last follow-up visit were recorded retrospectively from the electronic medical record. The study was approved by the OUHSC Institutional Review Board.
Statistical analysis
TAT was analyzed categorically (0-4 days vs >4 days), and day of admission was analyzed categorically (Monday to Thursday vs Friday to Sunday). Descriptive statistics were calculated overall and by TAT group. The chi-square test was used to compare the association between our covariates and TAT. Kaplan-Meier estimates (with a log-rank test) were used to assess the unadjusted effect of TAT with overall survival (OS) and event-free survival (EFS). Median OS and EFS and 95% confidence intervals (CIs) were also calculated. We used the Cox proportional hazards regression modeling to evaluate the relationship between OS and TAT. The initial model was built by including covariates, with P < .25 for the association between the covariates with OS. TAT was maintained in the final model because it was the primary variable of interest, whereas age and risk group were also included in the final model because those covariates are known prognostic risk factors in AML. Among the set of variables screened in, all 2-way interactions were assessed using P < .05. No significant interactions were found. Backward elimination was then performed. During the backward elimination, confounding was deemed to have been present if the measure of association of significant variables in the model changed by more than 20% and the P-value of the confounding variable was less than .30. Variables with P-values of less than .05 or deemed a confounder would then be retained. A similar modeling approach was used to examine EFS and CR. To evaluate the association between CR with potential predictors, binary logistic regression was used, whereby day of admission and time to treatment were explored unadjusted and then adjusted for age, WBC count, risk group, and undergoing allogeneic stem cell transplant (AlloSCT). SAS version 9.4 (SAS Institute Inc, Cary, North Carolina) was used for all analyses. A final alpha of 0.05 was used unless otherwise noted.
Results
Baseline characteristics are presented in Table 1. Treatment was initiated within 4 days for 71% (109/154) of patients. Most patients in our study were younger than 60 years (70%), male (64%), and white (77%). Most patients were admitted to the hospital for treatment between Monday and Thursday (75%). A higher proportion of patients in the 0-4 days TAT group were <60 years of age compared with patients in the >4 days TAT group (P = .0427). A higher proportion of patients in the 0-4 days TAT group had a WBC count of ≥50 x 103 μ/L compared with patients in the >4 days TAT group (27% vs 9%, respectively; P = .0148). A higher proportion of patients were admitted Friday to Sunday in the TAT >4 days group. Insured and uninsured patients were equally distributed between the 2 groups (P = .0014). Cytogenetic and/or molecular risk was not statistically different between the 0-4 days and >4 days TAT groups (unfavorable risk, 25% vs 23%, respectively; P = .6214). A higher proportion of patients received 7 + 3 induction chemotherapy (7 days cytarabine and 3 days anthracycline) in the TAT 0-4 days group compared with the >4 days TAT group (84% vs 69%, respectively; P = .0448). The most common intensive chemotherapy regimen used was 7 + 3 (80%). The rest of the patients (20%) received high-dose cytarabine clofarabine-based chemotherapy, hypomethylating agents, or other treatments. The proportion of patients who received an AlloSCT did not differ between the 0-4 days and >4 days TAT groups (24% vs 20%, respectively; P = .5655).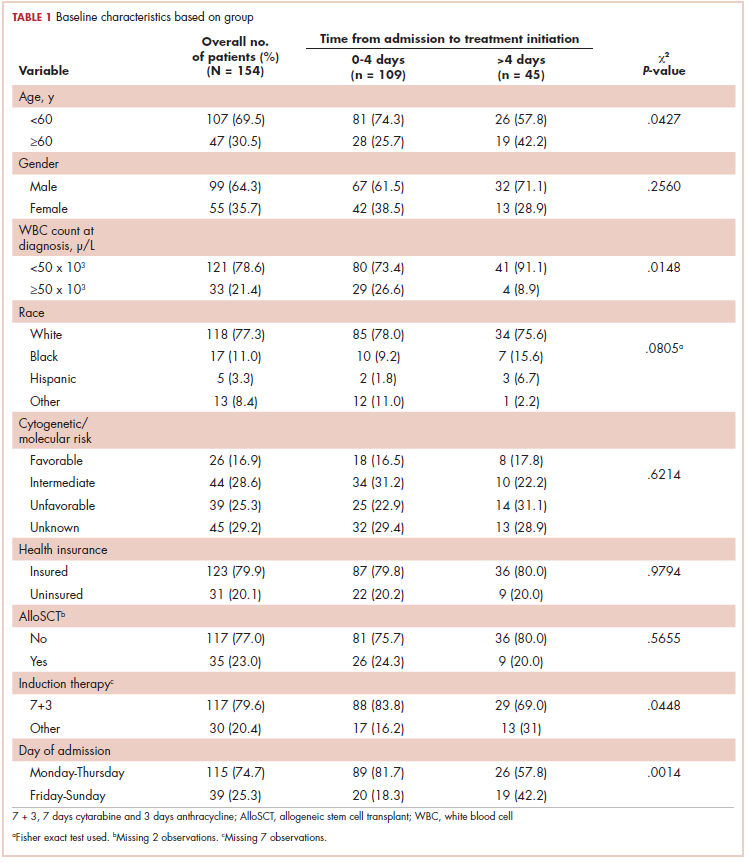
The median OS for all patients was 10.9 months (95% CI, 8.3-15.1), and the median EFS was 9.1 months (95% CI, 7.4-13.8). Median follow-up time was 8.6 months (95% CI, 6.7-11). We found a significant association between TAT and both OS and EFS without any adjustment (Table 2).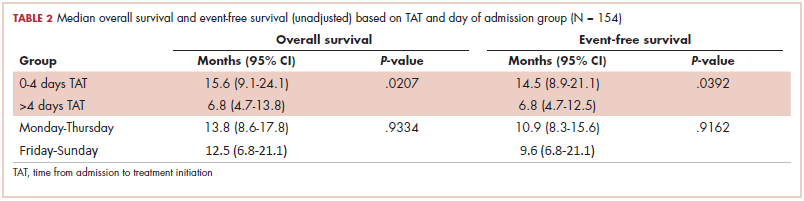
The median OS for the TAT 0-4 days group was 15.6 months, and for the TAT >4 days group, it was 6.8 months (P = .0207; Figure 1). The median EFS for the TAT 0-4 days group was 14.5 months, and for the TAT >4 days group, it was 6.8 months (P = .0240; Figure 2).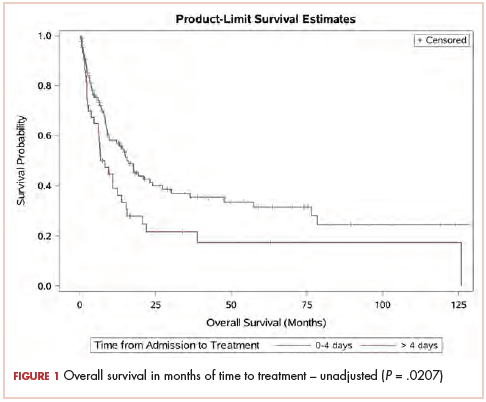
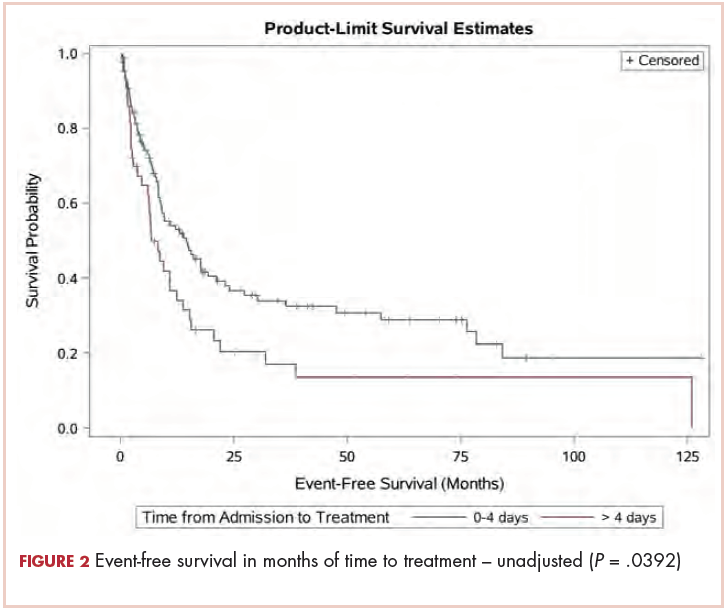
We found no association between the day of admission to hospital (Monday-Thursday vs Friday-Sunday) and either OS or EFS. After adjusting for age, WBC count, molecular risk status, and undergoing AlloSCT, the OS was shorter for those who received treatment >4 days after admission compared with those who received treatment within 0 to 4 days, with a hazard ratio (HR) of 1.59 (95% CI, 1.02-2.49; P = .0427; Table 3).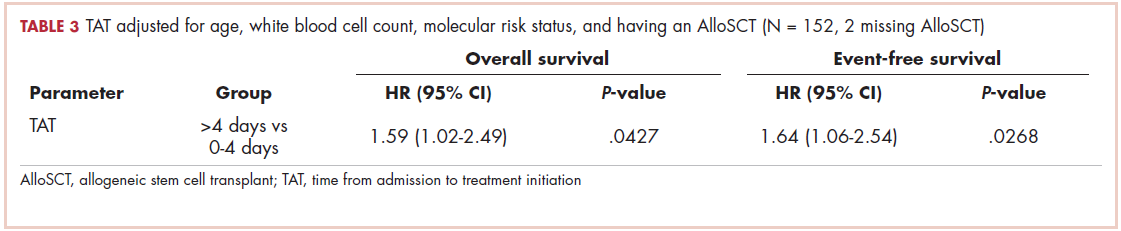
There was no association between day of admission with OS in the multivariable analysis. Similarly, after adjusting for age, WBC count, molecular risk status, and undergoing AlloSCT, EFS was shorter in patients who received treatment >4 days after admission compared with those who received treatment within 0 to 4 days (HR, 1.64; 95% CI, 1.06-2.54; P = .0268). There was no association between day of admission with EFS in the multivariable model. Although there was a trend for a higher CR rate with earlier treatment, this was not statistically significant (Table 4).
Discussion
Treatment outcomes for patients with AML are known to be affected by several patient- and disease-related factors. Patient-related factors can include age, performance status, comorbidities, and availability of a stem cell donor. Examples of disease-related factors include molecular alterations and site of disease involvement. Little is known about whether the timing of treatment initiation affects patient outcomes. Short-term treatment delays after the diagnosis of leukemia are not uncommon. Generally, patients are treated with anthracycline-based induction chemotherapy, but the response rate and survival are particularly poor in the older age group.12 Moreover, increasing comorbidities with aging are expected to lead to lower treatment tolerability.13 Therefore, elderly patients are particularly prone to treatment delays while providers await the results of the molecular studies to guide the use of less intensive targeted therapies.10 Other reasons for treatment delays may also include transfers between hospitals, suspected or documented infections, and evaluation of chronic illnesses. Our analysis also indicates that admission to the hospital on the weekend contributes to a delay in therapy compared with admission on a weekday.
We found a decreased OS and EFS in patients who received treatment >4 days after admission to the hospital compared with patients who received treatment within 0 to 4 days of admission. This association was statistically significant in a bivariate analysis as well as in a multivariable analysis with adjustment for age, WBC count on presentation, molecular risk group, and undergoing AlloSCT. A previous large retrospective study showed that the time from diagnosis to treatment initiation predicts survival in younger, but not older, patients with AML.10 This remained true after adjusting for age, performance status, WBC count, and the type of AML in a multivariable analysis. In our study, the declines in overall survival and event-free survival were evident after a delay of more than 4 days.
Another retrospective study that included 599 newly diagnosed AML patients, with a median time from diagnosis to treatment of 8 days, did not show any impact of treatment delay on overall survival, early death, or response rate.11 These differences in the effect of treatment delay on outcomes could be related to the differences in baseline characteristics of patients in these studies. Our study had a higher proportion of patients younger than 60 years, for example. We hypothesize that treatment delays, especially in patients with a high WBC count on presentation, might lead to further organ compromise and poorer outcomes with chemotherapy.
In our study, a higher proportion of patients were admitted over the weekend in the >4 days TAT group, but when we analyzed the day of admission to hospital separately, it was not associated with OS or EFS. Admission over the weekend was also not associated with clinical outcomes including 30-day mortality in a larger study that included 422 patients treated at a large teaching referral hospital.14
Limitations of our study include a small sample size and a short median follow-up time. Most of our patients were young and white, which may not be representative of the general population.
In conclusion, we found that treatment delays are associated with inferior outcomes in AML patients. It remains to be elucidated whether the benefit gained from using targeted and less-intensive chemotherapy, especially in elderly patients, outweighs the potential harm from delaying treatment. Additional studies are needed to confirm our findings in different settings and patient populations.
Acknowledgment
Statistical support was provided by the Stephenson Cancer Center Biostatistics and Research Design Shared Resource.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
2. Patel MI, Ma Y, Mitchell B, Rhoads KF. How do differences in treatment impact racial and ethnic disparities in acute myeloid leukemia? Cancer Epidemiol Biomarkers Prev. 2015;24(2):344-349.
3. Weber JJ, Kachare SD, Vohra NA, Fitzgerald TF, Wong JH. Regional disparities in breast cancer outcomes and the process of care. Am Surg. 2014;80(7):669-674.
4. Shippee TP, Kozhimannil KB, Rowan K, Virnig BA. Health insurance coverage and racial disparities in breast reconstruction after mastectomy. Womens Health Issues. 2014;24(3):e261-e269.
5. Dickens C, Joffe M, Jacobson J, et al. Stage at breast cancer diagnosis and distance from diagnostic hospital in a periurban setting: a South African public hospital case series of over 1,000 women. Int J Cancer. 2014;135(9):2173-2182.
6. Nguyen-Pham S, Leung J, McLaughlin D. Disparities in breast cancer stage at diagnosis in urban and rural adult women: a systematic review and meta-analysis. Ann Epidemiol. 2014;24(3):228-235.
7. Gong G, Belasco E, Hargrave KA, Lyford CP, Philips BU Jr. Determinants of delayed detection of cancers in Texas counties in the United States of America. Int J Equity Health. 2012;11:29.
8. Erhunmwunsee L, Joshi MB, Conlon DH, Harpole DH Jr. Neighborhood‐level socioeconomic determinants impact outcomes in nonsmall cell lung cancer patients in the Southeastern United States. Cancer. 2012;118(20):5117-5123.
9. Steele CB, Pisu M, Richardson LC. Urban/rural patterns in receipt of treatment for non–small cell lung cancer among black and white Medicare beneficiaries, 2000-2003. J Natl Med Assoc. 2011;103(8):711-718.
10. Sekeres MA, Elson P, Kalaycio ME, et al. Time from diagnosis to treatment initiation predicts survival in younger, but not older, acute myeloid leukemia patients. Blood. 2009;113(1):28-36.
11. Bertoli S, Bérard E, Huguet F, et al. Time from diagnosis to intensive chemotherapy initiation does not adversely impact the outcome of patients with acute myeloid leukemia. Blood. 2013:121(14):2618-2626.
12. Shah A, Andersson TM, Rachet B, Björkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971-2006: a population-based study. Br J Haematol. 2013;162(4):509-516.
13. Mohammadi M, Cao Y, Glimelius I, Bottai M, Eloranta S, Smedby KE. The impact of comorbid disease history on all-cause and cancer-specific mortality in myeloid leukemia and myeloma – a Swedish population-based study. BMC Cancer. 2015;15:850.
14. Bejanyan N, Fu AZ, Lazaryan A, et al. Impact of weekend admissions on quality of care and outcomes in patients with acute myeloid leukemia. Cancer. 2010;116(15):3614-3620.
Acute myeloid leukemia (AML) is the most common acute leukemia in adults in the United States.1 In 2018, the estimated annual incidence of AML is 19,520 (32.4% of all new leukemia cases), with 10,670 projected deaths (43.8% of all leukemia deaths).1 New molecularly targeted treatments are increasingly being used in treating AML, and some of them have shown improved health outcomes. In general, age, white blood cell (WBC) count at presentation, cytogenetics, and molecular characteristics are the major determinants of prognosis and treatment outcome. Studies analyzing the Surveillance Epidemiology and End Results database have also shown racial differences in outcomes.2 It is well known to the oncology community that patients with similar characteristics may respond differently to treatment and that outcome is not uniformly related to the well-defined clinical and laboratory characteristics. Issues related to health care disparities and access to health care are also known to affect the outcome in patients with cancer.3-9
AML is generally considered by the medical community as a time-sensitive condition. Treatment of patients with AML usually consists of induction chemotherapy followed by consolidation treatment with consideration for stem cell transplant. The duration of time from admission to treatment (TAT) of AML with induction chemotherapy is dependent on multiple factors. These may include the assessment of comorbid conditions and the availability of molecular studies at the time of treatment, which can be time consuming. The effect of treatment delays after AML diagnosis has been investigated, but with conflicting results. One study showed that time from diagnosis to treatment initiation affects survival in younger patients, and another showed it has no effect on survival regardless of patient age.10,11 We describe here the results of a retrospective analysis evaluating the impact of TAT and day of admission on outcomes of patients with AML who received treatment at a tertiary care referral center.
Methods and materials
We did a retrospective medical record review of all newly diagnosed AML patients at the Oklahoma University Health Sciences Center (OUHSC). Our sample was composed of 154 adult patients. Our inclusion criteria were an age of 18 years or older with complete insurance data, a diagnosis of AML, and having received treatment at our institution from January 2000 through June 2015. Data were obtained on laboratory values at diagnosis, pathology data including cytogenetics, molecular data, and bone marrow biopsies. Data on patient characteristics such as age, race and/or ethnicity, and comorbidities were obtained from the electronic medical records. Treatment data on type and dose of chemotherapy during induction, subsequent treatment phases, and number of treatments to achieve complete response (CR) as well as response data of CR achievement, relapse, date of CR, date of relapse, stem cell transplantation data, date of death, and date of last follow-up visit were recorded retrospectively from the electronic medical record. The study was approved by the OUHSC Institutional Review Board.
Statistical analysis
TAT was analyzed categorically (0-4 days vs >4 days), and day of admission was analyzed categorically (Monday to Thursday vs Friday to Sunday). Descriptive statistics were calculated overall and by TAT group. The chi-square test was used to compare the association between our covariates and TAT. Kaplan-Meier estimates (with a log-rank test) were used to assess the unadjusted effect of TAT with overall survival (OS) and event-free survival (EFS). Median OS and EFS and 95% confidence intervals (CIs) were also calculated. We used the Cox proportional hazards regression modeling to evaluate the relationship between OS and TAT. The initial model was built by including covariates, with P < .25 for the association between the covariates with OS. TAT was maintained in the final model because it was the primary variable of interest, whereas age and risk group were also included in the final model because those covariates are known prognostic risk factors in AML. Among the set of variables screened in, all 2-way interactions were assessed using P < .05. No significant interactions were found. Backward elimination was then performed. During the backward elimination, confounding was deemed to have been present if the measure of association of significant variables in the model changed by more than 20% and the P-value of the confounding variable was less than .30. Variables with P-values of less than .05 or deemed a confounder would then be retained. A similar modeling approach was used to examine EFS and CR. To evaluate the association between CR with potential predictors, binary logistic regression was used, whereby day of admission and time to treatment were explored unadjusted and then adjusted for age, WBC count, risk group, and undergoing allogeneic stem cell transplant (AlloSCT). SAS version 9.4 (SAS Institute Inc, Cary, North Carolina) was used for all analyses. A final alpha of 0.05 was used unless otherwise noted.
Results
Baseline characteristics are presented in Table 1. Treatment was initiated within 4 days for 71% (109/154) of patients. Most patients in our study were younger than 60 years (70%), male (64%), and white (77%). Most patients were admitted to the hospital for treatment between Monday and Thursday (75%). A higher proportion of patients in the 0-4 days TAT group were <60 years of age compared with patients in the >4 days TAT group (P = .0427). A higher proportion of patients in the 0-4 days TAT group had a WBC count of ≥50 x 103 μ/L compared with patients in the >4 days TAT group (27% vs 9%, respectively; P = .0148). A higher proportion of patients were admitted Friday to Sunday in the TAT >4 days group. Insured and uninsured patients were equally distributed between the 2 groups (P = .0014). Cytogenetic and/or molecular risk was not statistically different between the 0-4 days and >4 days TAT groups (unfavorable risk, 25% vs 23%, respectively; P = .6214). A higher proportion of patients received 7 + 3 induction chemotherapy (7 days cytarabine and 3 days anthracycline) in the TAT 0-4 days group compared with the >4 days TAT group (84% vs 69%, respectively; P = .0448). The most common intensive chemotherapy regimen used was 7 + 3 (80%). The rest of the patients (20%) received high-dose cytarabine clofarabine-based chemotherapy, hypomethylating agents, or other treatments. The proportion of patients who received an AlloSCT did not differ between the 0-4 days and >4 days TAT groups (24% vs 20%, respectively; P = .5655).
The median OS for all patients was 10.9 months (95% CI, 8.3-15.1), and the median EFS was 9.1 months (95% CI, 7.4-13.8). Median follow-up time was 8.6 months (95% CI, 6.7-11). We found a significant association between TAT and both OS and EFS without any adjustment (Table 2).
The median OS for the TAT 0-4 days group was 15.6 months, and for the TAT >4 days group, it was 6.8 months (P = .0207; Figure 1). The median EFS for the TAT 0-4 days group was 14.5 months, and for the TAT >4 days group, it was 6.8 months (P = .0240; Figure 2).
We found no association between the day of admission to hospital (Monday-Thursday vs Friday-Sunday) and either OS or EFS. After adjusting for age, WBC count, molecular risk status, and undergoing AlloSCT, the OS was shorter for those who received treatment >4 days after admission compared with those who received treatment within 0 to 4 days, with a hazard ratio (HR) of 1.59 (95% CI, 1.02-2.49; P = .0427; Table 3).
There was no association between day of admission with OS in the multivariable analysis. Similarly, after adjusting for age, WBC count, molecular risk status, and undergoing AlloSCT, EFS was shorter in patients who received treatment >4 days after admission compared with those who received treatment within 0 to 4 days (HR, 1.64; 95% CI, 1.06-2.54; P = .0268). There was no association between day of admission with EFS in the multivariable model. Although there was a trend for a higher CR rate with earlier treatment, this was not statistically significant (Table 4).
Discussion
Treatment outcomes for patients with AML are known to be affected by several patient- and disease-related factors. Patient-related factors can include age, performance status, comorbidities, and availability of a stem cell donor. Examples of disease-related factors include molecular alterations and site of disease involvement. Little is known about whether the timing of treatment initiation affects patient outcomes. Short-term treatment delays after the diagnosis of leukemia are not uncommon. Generally, patients are treated with anthracycline-based induction chemotherapy, but the response rate and survival are particularly poor in the older age group.12 Moreover, increasing comorbidities with aging are expected to lead to lower treatment tolerability.13 Therefore, elderly patients are particularly prone to treatment delays while providers await the results of the molecular studies to guide the use of less intensive targeted therapies.10 Other reasons for treatment delays may also include transfers between hospitals, suspected or documented infections, and evaluation of chronic illnesses. Our analysis also indicates that admission to the hospital on the weekend contributes to a delay in therapy compared with admission on a weekday.
We found a decreased OS and EFS in patients who received treatment >4 days after admission to the hospital compared with patients who received treatment within 0 to 4 days of admission. This association was statistically significant in a bivariate analysis as well as in a multivariable analysis with adjustment for age, WBC count on presentation, molecular risk group, and undergoing AlloSCT. A previous large retrospective study showed that the time from diagnosis to treatment initiation predicts survival in younger, but not older, patients with AML.10 This remained true after adjusting for age, performance status, WBC count, and the type of AML in a multivariable analysis. In our study, the declines in overall survival and event-free survival were evident after a delay of more than 4 days.
Another retrospective study that included 599 newly diagnosed AML patients, with a median time from diagnosis to treatment of 8 days, did not show any impact of treatment delay on overall survival, early death, or response rate.11 These differences in the effect of treatment delay on outcomes could be related to the differences in baseline characteristics of patients in these studies. Our study had a higher proportion of patients younger than 60 years, for example. We hypothesize that treatment delays, especially in patients with a high WBC count on presentation, might lead to further organ compromise and poorer outcomes with chemotherapy.
In our study, a higher proportion of patients were admitted over the weekend in the >4 days TAT group, but when we analyzed the day of admission to hospital separately, it was not associated with OS or EFS. Admission over the weekend was also not associated with clinical outcomes including 30-day mortality in a larger study that included 422 patients treated at a large teaching referral hospital.14
Limitations of our study include a small sample size and a short median follow-up time. Most of our patients were young and white, which may not be representative of the general population.
In conclusion, we found that treatment delays are associated with inferior outcomes in AML patients. It remains to be elucidated whether the benefit gained from using targeted and less-intensive chemotherapy, especially in elderly patients, outweighs the potential harm from delaying treatment. Additional studies are needed to confirm our findings in different settings and patient populations.
Acknowledgment
Statistical support was provided by the Stephenson Cancer Center Biostatistics and Research Design Shared Resource.
Acute myeloid leukemia (AML) is the most common acute leukemia in adults in the United States.1 In 2018, the estimated annual incidence of AML is 19,520 (32.4% of all new leukemia cases), with 10,670 projected deaths (43.8% of all leukemia deaths).1 New molecularly targeted treatments are increasingly being used in treating AML, and some of them have shown improved health outcomes. In general, age, white blood cell (WBC) count at presentation, cytogenetics, and molecular characteristics are the major determinants of prognosis and treatment outcome. Studies analyzing the Surveillance Epidemiology and End Results database have also shown racial differences in outcomes.2 It is well known to the oncology community that patients with similar characteristics may respond differently to treatment and that outcome is not uniformly related to the well-defined clinical and laboratory characteristics. Issues related to health care disparities and access to health care are also known to affect the outcome in patients with cancer.3-9
AML is generally considered by the medical community as a time-sensitive condition. Treatment of patients with AML usually consists of induction chemotherapy followed by consolidation treatment with consideration for stem cell transplant. The duration of time from admission to treatment (TAT) of AML with induction chemotherapy is dependent on multiple factors. These may include the assessment of comorbid conditions and the availability of molecular studies at the time of treatment, which can be time consuming. The effect of treatment delays after AML diagnosis has been investigated, but with conflicting results. One study showed that time from diagnosis to treatment initiation affects survival in younger patients, and another showed it has no effect on survival regardless of patient age.10,11 We describe here the results of a retrospective analysis evaluating the impact of TAT and day of admission on outcomes of patients with AML who received treatment at a tertiary care referral center.
Methods and materials
We did a retrospective medical record review of all newly diagnosed AML patients at the Oklahoma University Health Sciences Center (OUHSC). Our sample was composed of 154 adult patients. Our inclusion criteria were an age of 18 years or older with complete insurance data, a diagnosis of AML, and having received treatment at our institution from January 2000 through June 2015. Data were obtained on laboratory values at diagnosis, pathology data including cytogenetics, molecular data, and bone marrow biopsies. Data on patient characteristics such as age, race and/or ethnicity, and comorbidities were obtained from the electronic medical records. Treatment data on type and dose of chemotherapy during induction, subsequent treatment phases, and number of treatments to achieve complete response (CR) as well as response data of CR achievement, relapse, date of CR, date of relapse, stem cell transplantation data, date of death, and date of last follow-up visit were recorded retrospectively from the electronic medical record. The study was approved by the OUHSC Institutional Review Board.
Statistical analysis
TAT was analyzed categorically (0-4 days vs >4 days), and day of admission was analyzed categorically (Monday to Thursday vs Friday to Sunday). Descriptive statistics were calculated overall and by TAT group. The chi-square test was used to compare the association between our covariates and TAT. Kaplan-Meier estimates (with a log-rank test) were used to assess the unadjusted effect of TAT with overall survival (OS) and event-free survival (EFS). Median OS and EFS and 95% confidence intervals (CIs) were also calculated. We used the Cox proportional hazards regression modeling to evaluate the relationship between OS and TAT. The initial model was built by including covariates, with P < .25 for the association between the covariates with OS. TAT was maintained in the final model because it was the primary variable of interest, whereas age and risk group were also included in the final model because those covariates are known prognostic risk factors in AML. Among the set of variables screened in, all 2-way interactions were assessed using P < .05. No significant interactions were found. Backward elimination was then performed. During the backward elimination, confounding was deemed to have been present if the measure of association of significant variables in the model changed by more than 20% and the P-value of the confounding variable was less than .30. Variables with P-values of less than .05 or deemed a confounder would then be retained. A similar modeling approach was used to examine EFS and CR. To evaluate the association between CR with potential predictors, binary logistic regression was used, whereby day of admission and time to treatment were explored unadjusted and then adjusted for age, WBC count, risk group, and undergoing allogeneic stem cell transplant (AlloSCT). SAS version 9.4 (SAS Institute Inc, Cary, North Carolina) was used for all analyses. A final alpha of 0.05 was used unless otherwise noted.
Results
Baseline characteristics are presented in Table 1. Treatment was initiated within 4 days for 71% (109/154) of patients. Most patients in our study were younger than 60 years (70%), male (64%), and white (77%). Most patients were admitted to the hospital for treatment between Monday and Thursday (75%). A higher proportion of patients in the 0-4 days TAT group were <60 years of age compared with patients in the >4 days TAT group (P = .0427). A higher proportion of patients in the 0-4 days TAT group had a WBC count of ≥50 x 103 μ/L compared with patients in the >4 days TAT group (27% vs 9%, respectively; P = .0148). A higher proportion of patients were admitted Friday to Sunday in the TAT >4 days group. Insured and uninsured patients were equally distributed between the 2 groups (P = .0014). Cytogenetic and/or molecular risk was not statistically different between the 0-4 days and >4 days TAT groups (unfavorable risk, 25% vs 23%, respectively; P = .6214). A higher proportion of patients received 7 + 3 induction chemotherapy (7 days cytarabine and 3 days anthracycline) in the TAT 0-4 days group compared with the >4 days TAT group (84% vs 69%, respectively; P = .0448). The most common intensive chemotherapy regimen used was 7 + 3 (80%). The rest of the patients (20%) received high-dose cytarabine clofarabine-based chemotherapy, hypomethylating agents, or other treatments. The proportion of patients who received an AlloSCT did not differ between the 0-4 days and >4 days TAT groups (24% vs 20%, respectively; P = .5655).
The median OS for all patients was 10.9 months (95% CI, 8.3-15.1), and the median EFS was 9.1 months (95% CI, 7.4-13.8). Median follow-up time was 8.6 months (95% CI, 6.7-11). We found a significant association between TAT and both OS and EFS without any adjustment (Table 2).
The median OS for the TAT 0-4 days group was 15.6 months, and for the TAT >4 days group, it was 6.8 months (P = .0207; Figure 1). The median EFS for the TAT 0-4 days group was 14.5 months, and for the TAT >4 days group, it was 6.8 months (P = .0240; Figure 2).
We found no association between the day of admission to hospital (Monday-Thursday vs Friday-Sunday) and either OS or EFS. After adjusting for age, WBC count, molecular risk status, and undergoing AlloSCT, the OS was shorter for those who received treatment >4 days after admission compared with those who received treatment within 0 to 4 days, with a hazard ratio (HR) of 1.59 (95% CI, 1.02-2.49; P = .0427; Table 3).
There was no association between day of admission with OS in the multivariable analysis. Similarly, after adjusting for age, WBC count, molecular risk status, and undergoing AlloSCT, EFS was shorter in patients who received treatment >4 days after admission compared with those who received treatment within 0 to 4 days (HR, 1.64; 95% CI, 1.06-2.54; P = .0268). There was no association between day of admission with EFS in the multivariable model. Although there was a trend for a higher CR rate with earlier treatment, this was not statistically significant (Table 4).
Discussion
Treatment outcomes for patients with AML are known to be affected by several patient- and disease-related factors. Patient-related factors can include age, performance status, comorbidities, and availability of a stem cell donor. Examples of disease-related factors include molecular alterations and site of disease involvement. Little is known about whether the timing of treatment initiation affects patient outcomes. Short-term treatment delays after the diagnosis of leukemia are not uncommon. Generally, patients are treated with anthracycline-based induction chemotherapy, but the response rate and survival are particularly poor in the older age group.12 Moreover, increasing comorbidities with aging are expected to lead to lower treatment tolerability.13 Therefore, elderly patients are particularly prone to treatment delays while providers await the results of the molecular studies to guide the use of less intensive targeted therapies.10 Other reasons for treatment delays may also include transfers between hospitals, suspected or documented infections, and evaluation of chronic illnesses. Our analysis also indicates that admission to the hospital on the weekend contributes to a delay in therapy compared with admission on a weekday.
We found a decreased OS and EFS in patients who received treatment >4 days after admission to the hospital compared with patients who received treatment within 0 to 4 days of admission. This association was statistically significant in a bivariate analysis as well as in a multivariable analysis with adjustment for age, WBC count on presentation, molecular risk group, and undergoing AlloSCT. A previous large retrospective study showed that the time from diagnosis to treatment initiation predicts survival in younger, but not older, patients with AML.10 This remained true after adjusting for age, performance status, WBC count, and the type of AML in a multivariable analysis. In our study, the declines in overall survival and event-free survival were evident after a delay of more than 4 days.
Another retrospective study that included 599 newly diagnosed AML patients, with a median time from diagnosis to treatment of 8 days, did not show any impact of treatment delay on overall survival, early death, or response rate.11 These differences in the effect of treatment delay on outcomes could be related to the differences in baseline characteristics of patients in these studies. Our study had a higher proportion of patients younger than 60 years, for example. We hypothesize that treatment delays, especially in patients with a high WBC count on presentation, might lead to further organ compromise and poorer outcomes with chemotherapy.
In our study, a higher proportion of patients were admitted over the weekend in the >4 days TAT group, but when we analyzed the day of admission to hospital separately, it was not associated with OS or EFS. Admission over the weekend was also not associated with clinical outcomes including 30-day mortality in a larger study that included 422 patients treated at a large teaching referral hospital.14
Limitations of our study include a small sample size and a short median follow-up time. Most of our patients were young and white, which may not be representative of the general population.
In conclusion, we found that treatment delays are associated with inferior outcomes in AML patients. It remains to be elucidated whether the benefit gained from using targeted and less-intensive chemotherapy, especially in elderly patients, outweighs the potential harm from delaying treatment. Additional studies are needed to confirm our findings in different settings and patient populations.
Acknowledgment
Statistical support was provided by the Stephenson Cancer Center Biostatistics and Research Design Shared Resource.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
2. Patel MI, Ma Y, Mitchell B, Rhoads KF. How do differences in treatment impact racial and ethnic disparities in acute myeloid leukemia? Cancer Epidemiol Biomarkers Prev. 2015;24(2):344-349.
3. Weber JJ, Kachare SD, Vohra NA, Fitzgerald TF, Wong JH. Regional disparities in breast cancer outcomes and the process of care. Am Surg. 2014;80(7):669-674.
4. Shippee TP, Kozhimannil KB, Rowan K, Virnig BA. Health insurance coverage and racial disparities in breast reconstruction after mastectomy. Womens Health Issues. 2014;24(3):e261-e269.
5. Dickens C, Joffe M, Jacobson J, et al. Stage at breast cancer diagnosis and distance from diagnostic hospital in a periurban setting: a South African public hospital case series of over 1,000 women. Int J Cancer. 2014;135(9):2173-2182.
6. Nguyen-Pham S, Leung J, McLaughlin D. Disparities in breast cancer stage at diagnosis in urban and rural adult women: a systematic review and meta-analysis. Ann Epidemiol. 2014;24(3):228-235.
7. Gong G, Belasco E, Hargrave KA, Lyford CP, Philips BU Jr. Determinants of delayed detection of cancers in Texas counties in the United States of America. Int J Equity Health. 2012;11:29.
8. Erhunmwunsee L, Joshi MB, Conlon DH, Harpole DH Jr. Neighborhood‐level socioeconomic determinants impact outcomes in nonsmall cell lung cancer patients in the Southeastern United States. Cancer. 2012;118(20):5117-5123.
9. Steele CB, Pisu M, Richardson LC. Urban/rural patterns in receipt of treatment for non–small cell lung cancer among black and white Medicare beneficiaries, 2000-2003. J Natl Med Assoc. 2011;103(8):711-718.
10. Sekeres MA, Elson P, Kalaycio ME, et al. Time from diagnosis to treatment initiation predicts survival in younger, but not older, acute myeloid leukemia patients. Blood. 2009;113(1):28-36.
11. Bertoli S, Bérard E, Huguet F, et al. Time from diagnosis to intensive chemotherapy initiation does not adversely impact the outcome of patients with acute myeloid leukemia. Blood. 2013:121(14):2618-2626.
12. Shah A, Andersson TM, Rachet B, Björkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971-2006: a population-based study. Br J Haematol. 2013;162(4):509-516.
13. Mohammadi M, Cao Y, Glimelius I, Bottai M, Eloranta S, Smedby KE. The impact of comorbid disease history on all-cause and cancer-specific mortality in myeloid leukemia and myeloma – a Swedish population-based study. BMC Cancer. 2015;15:850.
14. Bejanyan N, Fu AZ, Lazaryan A, et al. Impact of weekend admissions on quality of care and outcomes in patients with acute myeloid leukemia. Cancer. 2010;116(15):3614-3620.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
2. Patel MI, Ma Y, Mitchell B, Rhoads KF. How do differences in treatment impact racial and ethnic disparities in acute myeloid leukemia? Cancer Epidemiol Biomarkers Prev. 2015;24(2):344-349.
3. Weber JJ, Kachare SD, Vohra NA, Fitzgerald TF, Wong JH. Regional disparities in breast cancer outcomes and the process of care. Am Surg. 2014;80(7):669-674.
4. Shippee TP, Kozhimannil KB, Rowan K, Virnig BA. Health insurance coverage and racial disparities in breast reconstruction after mastectomy. Womens Health Issues. 2014;24(3):e261-e269.
5. Dickens C, Joffe M, Jacobson J, et al. Stage at breast cancer diagnosis and distance from diagnostic hospital in a periurban setting: a South African public hospital case series of over 1,000 women. Int J Cancer. 2014;135(9):2173-2182.
6. Nguyen-Pham S, Leung J, McLaughlin D. Disparities in breast cancer stage at diagnosis in urban and rural adult women: a systematic review and meta-analysis. Ann Epidemiol. 2014;24(3):228-235.
7. Gong G, Belasco E, Hargrave KA, Lyford CP, Philips BU Jr. Determinants of delayed detection of cancers in Texas counties in the United States of America. Int J Equity Health. 2012;11:29.
8. Erhunmwunsee L, Joshi MB, Conlon DH, Harpole DH Jr. Neighborhood‐level socioeconomic determinants impact outcomes in nonsmall cell lung cancer patients in the Southeastern United States. Cancer. 2012;118(20):5117-5123.
9. Steele CB, Pisu M, Richardson LC. Urban/rural patterns in receipt of treatment for non–small cell lung cancer among black and white Medicare beneficiaries, 2000-2003. J Natl Med Assoc. 2011;103(8):711-718.
10. Sekeres MA, Elson P, Kalaycio ME, et al. Time from diagnosis to treatment initiation predicts survival in younger, but not older, acute myeloid leukemia patients. Blood. 2009;113(1):28-36.
11. Bertoli S, Bérard E, Huguet F, et al. Time from diagnosis to intensive chemotherapy initiation does not adversely impact the outcome of patients with acute myeloid leukemia. Blood. 2013:121(14):2618-2626.
12. Shah A, Andersson TM, Rachet B, Björkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971-2006: a population-based study. Br J Haematol. 2013;162(4):509-516.
13. Mohammadi M, Cao Y, Glimelius I, Bottai M, Eloranta S, Smedby KE. The impact of comorbid disease history on all-cause and cancer-specific mortality in myeloid leukemia and myeloma – a Swedish population-based study. BMC Cancer. 2015;15:850.
14. Bejanyan N, Fu AZ, Lazaryan A, et al. Impact of weekend admissions on quality of care and outcomes in patients with acute myeloid leukemia. Cancer. 2010;116(15):3614-3620.
Venetoclax approved to treat CLL patients regardless of genotype
The approval of Bcl-2 inhibitor venetoclax was expanded by the US Food and Drug Administration in June 2018 to include the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL), regardless of their genotype, who have received at least 1 prior therapy.1 It was previously approved in 2016 for the treatment of patients who had a chromosome 17p deletion, which leads to loss of the tumor-suppressor gene TP53.
Approval was based on the positive results of the phase 3, randomized, multicenter, open-label MURANO trial in which 389 patients were randomized 1:1 to receive a combination of venetoclax and the CD20-targeting monoclonal antibody rituximab (venetoclax–rituximab) or bendamustine in combination with rituximab (bendamustine–rituximab).
Eligible patients were 18 years of age or older, had been diagnosed with relapsed/refractory CLL that required treatment, had received 1-3 prior therapies (including at least 1 chemotherapy regimen), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (on a 5-point scale, with 5 indicating the greatest level of disability), and had adequate bone marrow, renal, and hepatic function.
Patients who had received prior bendamustine treatment were eligible for the trial provided they had experienced a duration of response of 24 months or longer. However, patients with transformed CLL, central nervous system involvement, prior treatment with allogeneic or autologous stem cell transplant, major organ dysfunction, other active malignancy, or who were pregnant or breastfeeding, were excluded from the study.
Patients in the venetoclax arm received a 5-week ramp-up schedule, followed by a dose of 400 mg once daily for 24 months. Rituximab treatment started at the end of the venetoclax ramp-up period and was administered at a dose of 375 mg/m2 intravenously on cycle 1 day 1 and 500 mg/m2 on day 1 of cycles 2-6. In the control arm, patients received 6 cycles with the same rituxima
The primary endpoint was progression-free survival (PFS), as assessed by an independent review committee over a median follow-up of 23 months. Median PFS was significantly improved in the venetoclax arm (not yet reached versus 18.1 months in the bendamustine arm [HR, 0.19; P < .001]). In addition, objective response rate (ORR) and event-free survival (EFS) also favored the venetoclax arm; ORR was 92% compared with 72%, respectively, and 2-year EFS was 84.9% compared with 34.8%. There was also a trend toward improved 24-month overall survival (OS) rate (91.9% vs 86.6%), however this did not achieve statistical significance, nor did median OS.
The most common adverse events (AEs) in patients treated with venetoclax were neutropenia, diarrhea, upper-respiratory tract infection, fatigue, cough, and nausea. Grade 3/4 neutropenia occurred in 64% of patients, and serious AEs in 46% of patients. Serious infections occurred in 21% of patients, most commonly pneumonia. Ten deaths in the venetoclax arm were attributed to treatment, compared with 11 deaths in the bendamustine arm.2
The prescribing information details warnings and precautions relating to the risk of tumor lysis syndrome, which is increased in patients with higher tumor burden, reduced renal function, or in receipt of strong or moderate CYP3A inhibitors or P-gp inhibitors during the ramp-up stage. Patients should receive appropriate preventive strategies, including hydration and antihyperuricemics, blood chemistry should be monitored and abnormalities managed promptly, and dosing should be interrupted or adjusted as necessary.
Other warnings relate to neutropenia (complete blood counts should be monitored throughout treatment and venetoclax treatment interrupted or dose reduced for severe neutropenia, alongside possible use of supportive measures), immunization (live vaccines should not be administered before or during treatment or after treatment until B-cell recovery, and patients should be advised of the potentially reduced efficacy of vaccines), and embryofetal toxicity (patients should be advised of the risks and the need for effective contraception during and after treatment). Venetoclax is marketed as Venclexta by Genentech.3
1. US Food and Drug Administration website. FDA approves venetoclax for CLL or SLL, with or without 17p deletion, after one prior therapy. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm610308.htm. Last updated June 8, 2018. Accessed July 29, 2018.
2. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107-1120.
3. Venclexta (venetoclax tablets) for oral use. Prescribing information. Genentech USA, Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. Last updated June 2018. Accessed July 29, 2018.
The approval of Bcl-2 inhibitor venetoclax was expanded by the US Food and Drug Administration in June 2018 to include the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL), regardless of their genotype, who have received at least 1 prior therapy.1 It was previously approved in 2016 for the treatment of patients who had a chromosome 17p deletion, which leads to loss of the tumor-suppressor gene TP53.
Approval was based on the positive results of the phase 3, randomized, multicenter, open-label MURANO trial in which 389 patients were randomized 1:1 to receive a combination of venetoclax and the CD20-targeting monoclonal antibody rituximab (venetoclax–rituximab) or bendamustine in combination with rituximab (bendamustine–rituximab).
Eligible patients were 18 years of age or older, had been diagnosed with relapsed/refractory CLL that required treatment, had received 1-3 prior therapies (including at least 1 chemotherapy regimen), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (on a 5-point scale, with 5 indicating the greatest level of disability), and had adequate bone marrow, renal, and hepatic function.
Patients who had received prior bendamustine treatment were eligible for the trial provided they had experienced a duration of response of 24 months or longer. However, patients with transformed CLL, central nervous system involvement, prior treatment with allogeneic or autologous stem cell transplant, major organ dysfunction, other active malignancy, or who were pregnant or breastfeeding, were excluded from the study.
Patients in the venetoclax arm received a 5-week ramp-up schedule, followed by a dose of 400 mg once daily for 24 months. Rituximab treatment started at the end of the venetoclax ramp-up period and was administered at a dose of 375 mg/m2 intravenously on cycle 1 day 1 and 500 mg/m2 on day 1 of cycles 2-6. In the control arm, patients received 6 cycles with the same rituxima
The primary endpoint was progression-free survival (PFS), as assessed by an independent review committee over a median follow-up of 23 months. Median PFS was significantly improved in the venetoclax arm (not yet reached versus 18.1 months in the bendamustine arm [HR, 0.19; P < .001]). In addition, objective response rate (ORR) and event-free survival (EFS) also favored the venetoclax arm; ORR was 92% compared with 72%, respectively, and 2-year EFS was 84.9% compared with 34.8%. There was also a trend toward improved 24-month overall survival (OS) rate (91.9% vs 86.6%), however this did not achieve statistical significance, nor did median OS.
The most common adverse events (AEs) in patients treated with venetoclax were neutropenia, diarrhea, upper-respiratory tract infection, fatigue, cough, and nausea. Grade 3/4 neutropenia occurred in 64% of patients, and serious AEs in 46% of patients. Serious infections occurred in 21% of patients, most commonly pneumonia. Ten deaths in the venetoclax arm were attributed to treatment, compared with 11 deaths in the bendamustine arm.2
The prescribing information details warnings and precautions relating to the risk of tumor lysis syndrome, which is increased in patients with higher tumor burden, reduced renal function, or in receipt of strong or moderate CYP3A inhibitors or P-gp inhibitors during the ramp-up stage. Patients should receive appropriate preventive strategies, including hydration and antihyperuricemics, blood chemistry should be monitored and abnormalities managed promptly, and dosing should be interrupted or adjusted as necessary.
Other warnings relate to neutropenia (complete blood counts should be monitored throughout treatment and venetoclax treatment interrupted or dose reduced for severe neutropenia, alongside possible use of supportive measures), immunization (live vaccines should not be administered before or during treatment or after treatment until B-cell recovery, and patients should be advised of the potentially reduced efficacy of vaccines), and embryofetal toxicity (patients should be advised of the risks and the need for effective contraception during and after treatment). Venetoclax is marketed as Venclexta by Genentech.3
The approval of Bcl-2 inhibitor venetoclax was expanded by the US Food and Drug Administration in June 2018 to include the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL), regardless of their genotype, who have received at least 1 prior therapy.1 It was previously approved in 2016 for the treatment of patients who had a chromosome 17p deletion, which leads to loss of the tumor-suppressor gene TP53.
Approval was based on the positive results of the phase 3, randomized, multicenter, open-label MURANO trial in which 389 patients were randomized 1:1 to receive a combination of venetoclax and the CD20-targeting monoclonal antibody rituximab (venetoclax–rituximab) or bendamustine in combination with rituximab (bendamustine–rituximab).
Eligible patients were 18 years of age or older, had been diagnosed with relapsed/refractory CLL that required treatment, had received 1-3 prior therapies (including at least 1 chemotherapy regimen), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (on a 5-point scale, with 5 indicating the greatest level of disability), and had adequate bone marrow, renal, and hepatic function.
Patients who had received prior bendamustine treatment were eligible for the trial provided they had experienced a duration of response of 24 months or longer. However, patients with transformed CLL, central nervous system involvement, prior treatment with allogeneic or autologous stem cell transplant, major organ dysfunction, other active malignancy, or who were pregnant or breastfeeding, were excluded from the study.
Patients in the venetoclax arm received a 5-week ramp-up schedule, followed by a dose of 400 mg once daily for 24 months. Rituximab treatment started at the end of the venetoclax ramp-up period and was administered at a dose of 375 mg/m2 intravenously on cycle 1 day 1 and 500 mg/m2 on day 1 of cycles 2-6. In the control arm, patients received 6 cycles with the same rituxima
The primary endpoint was progression-free survival (PFS), as assessed by an independent review committee over a median follow-up of 23 months. Median PFS was significantly improved in the venetoclax arm (not yet reached versus 18.1 months in the bendamustine arm [HR, 0.19; P < .001]). In addition, objective response rate (ORR) and event-free survival (EFS) also favored the venetoclax arm; ORR was 92% compared with 72%, respectively, and 2-year EFS was 84.9% compared with 34.8%. There was also a trend toward improved 24-month overall survival (OS) rate (91.9% vs 86.6%), however this did not achieve statistical significance, nor did median OS.
The most common adverse events (AEs) in patients treated with venetoclax were neutropenia, diarrhea, upper-respiratory tract infection, fatigue, cough, and nausea. Grade 3/4 neutropenia occurred in 64% of patients, and serious AEs in 46% of patients. Serious infections occurred in 21% of patients, most commonly pneumonia. Ten deaths in the venetoclax arm were attributed to treatment, compared with 11 deaths in the bendamustine arm.2
The prescribing information details warnings and precautions relating to the risk of tumor lysis syndrome, which is increased in patients with higher tumor burden, reduced renal function, or in receipt of strong or moderate CYP3A inhibitors or P-gp inhibitors during the ramp-up stage. Patients should receive appropriate preventive strategies, including hydration and antihyperuricemics, blood chemistry should be monitored and abnormalities managed promptly, and dosing should be interrupted or adjusted as necessary.
Other warnings relate to neutropenia (complete blood counts should be monitored throughout treatment and venetoclax treatment interrupted or dose reduced for severe neutropenia, alongside possible use of supportive measures), immunization (live vaccines should not be administered before or during treatment or after treatment until B-cell recovery, and patients should be advised of the potentially reduced efficacy of vaccines), and embryofetal toxicity (patients should be advised of the risks and the need for effective contraception during and after treatment). Venetoclax is marketed as Venclexta by Genentech.3
1. US Food and Drug Administration website. FDA approves venetoclax for CLL or SLL, with or without 17p deletion, after one prior therapy. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm610308.htm. Last updated June 8, 2018. Accessed July 29, 2018.
2. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107-1120.
3. Venclexta (venetoclax tablets) for oral use. Prescribing information. Genentech USA, Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. Last updated June 2018. Accessed July 29, 2018.
1. US Food and Drug Administration website. FDA approves venetoclax for CLL or SLL, with or without 17p deletion, after one prior therapy. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm610308.htm. Last updated June 8, 2018. Accessed July 29, 2018.
2. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107-1120.
3. Venclexta (venetoclax tablets) for oral use. Prescribing information. Genentech USA, Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. Last updated June 2018. Accessed July 29, 2018.
Nivolumab and ipilimumab combination promises new standard of care for advanced RCC
In April 2018, the US Food and Drug Administration expanded the approval of the combination of nivolumab and ipilimumab into a new indication, following a previous approval in patients with metastatic melanoma. The double immune checkpoint inhibitor combination was approved on the basis of the phase 3 CheckMate-214 study for the treatment of patients with intermediate- or poor-risk, previously untreated advanced renal cell carcinoma (RCC).1
Nivolumab monotherapy is already approved in the second-line setting for the treatment of advanced RCC, and the demonstration of significantly improved overall survival (OS) in this study suggests that the combination should supplant sunitinib in the front-line setting in the treatment of this type of cancer.
A total of 1,096 patients at 175 sites in 28 countries were randomized 1:1 to receive nivolumab (3 mg/kg) and ipilimumab (1 mg/kg) intravenously every 3 weeks for 4 doses in an induction phase, followed by nivolumab monotherapy (3 mg/kg) every 2 weeks in a maintenance phase or sunitinib (50 mg) orally daily for 4 weeks of each 6-week cycle.
Eligible patients were 18 years or older, had previously untreated advanced RCC with a clear-cell component, had measurable disease according to Response Evaluation Criteria in Solid Tumors (version 1.1), and had a Karnofsky performance status of at least 70 (on a scale from 0 to 100, with lower scores indicating greater disability). Patients with central nervous system metastases or autoimmune disease who were being treated with glucocorticoids and immunosuppressants were excluded from the study.
Around three-quarters of patients with advanced RCC have intermediate- or poor-risk disease and experience worse outcomes than patients with favorable-risk disease. Patients in CheckMate-214 were stratified according to International Metastatic Renal Cell Carcinoma Database Consortium risk score as favorable (score of 0), intermediate (score of 1 or 2) or poor risk (score of 3-6), according to the number of risk factors present.
Risk factors included a Karnofsky performance score of 70, time from initial diagnosis to randomization of <1 year, a hemoglobin level below the lower limit of normal, a corrected serum calcium concentration of >10 mg/dL, or an absolute neutrophil count or platelet count above the upper limit of normal. Patients were also stratified according to geographic region (United States versus Canada and Europe versus the rest of the world).
The coprimary endpoints were objective response rate (ORR), progression-free survival (PFS), and OS in a subset of 847 intermediate- and poor-risk patients. Over a median follow-up of 25.2 months, there was a statistically significant improvement in OS and ORR in patients treated with nivolumab and ipilimumab (mPFS not reached; ORR, 41.6%), compared with sunitinib (OS, 25.9 months; ORR, 26.5%), with P <.001 for both. The immunotherapy combination was favored across subgroups.
The most common adverse events (AEs) in patients treated with nivolumab and ipilimumab included fatigue, rash, diarrhea, musculoskeletal pain, pruritus, nausea, cough, pyrexia, arthralgia, and decreased appetite. The combination was associated with fewer grade 3/4 AEs (63% vs 46% for sunitinib), but a higher rate of treatment discontinuations because of AEs (31% vs 21%, respectively). There were 8 deaths in the combination arm, and 4 in the sunitinib arm that were reported to be treatment related.2
The warnings and precautions related to nivolumab–ipilimumab combination therapy outlined in the prescribing information include mostly immune-mediated AEs, such as immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, nephritis and renal dysfunction, skin adverse reactions, and encephalitis. There are also warnings relating to the risk of infusion reactions and the potential for embryofetal toxicity.
Patients should be monitored for hyperglycemia and for changes in liver, thyroid, renal, and neurologic function. Treatment with nivolumab and ipilimumab should be withheld for moderate and permanently discontinued for severe or life-threatening immune-mediated pneumonitis, colitis, and hepatitis, as well as transaminase or total bilirubin elevation. It should also be withheld for moderate or severe hypophysitis and serum creatinine elevation, moderate adrenal insufficiency and severe hyperglycemia, and permanently discontinued for life-threatening hypophysitis and serum creatinine elevation, severe or life-threatening adrenal insufficiency, and life-threatening hyperglycemia.
New-onset moderate to severe neurologic signs or symptoms warrant treatment being withheld, and immune-mediated encephalitis should lead to treatment discontinuation. For mild or moderate infusion reactions, the infusion rate can be slowed or interrupted, and infusions should be discontinued in the event of severe or life-threatening infusion reactions. Patients should be advised of the potential for fetal harm and the need for effective contraception during and after treatment. Ipilimumab and nivolumab are marketed as Yervoy and Opdivo, respectively, by Bristol-Myers Squibb.3,4
1. US Food and Drug Administration website. FDA approves nivolumab plus ipilimumab combination for intermediate or poor-risk advanced renal cell carcinoma. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm604685.htm. Last updated April 16, 2018. Accessed July 25, 2018.
2. Motzer RJ, Tannir NM, McDermott O, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277-1290.
3. Opdivo (nivolumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf. Revised April 2018.
4. Yervoy (ipilimumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. July 2018. https://packageinserts.bms.com/pi/pi_yervoy.pdf. Accessed September, 2018.
In April 2018, the US Food and Drug Administration expanded the approval of the combination of nivolumab and ipilimumab into a new indication, following a previous approval in patients with metastatic melanoma. The double immune checkpoint inhibitor combination was approved on the basis of the phase 3 CheckMate-214 study for the treatment of patients with intermediate- or poor-risk, previously untreated advanced renal cell carcinoma (RCC).1
Nivolumab monotherapy is already approved in the second-line setting for the treatment of advanced RCC, and the demonstration of significantly improved overall survival (OS) in this study suggests that the combination should supplant sunitinib in the front-line setting in the treatment of this type of cancer.
A total of 1,096 patients at 175 sites in 28 countries were randomized 1:1 to receive nivolumab (3 mg/kg) and ipilimumab (1 mg/kg) intravenously every 3 weeks for 4 doses in an induction phase, followed by nivolumab monotherapy (3 mg/kg) every 2 weeks in a maintenance phase or sunitinib (50 mg) orally daily for 4 weeks of each 6-week cycle.
Eligible patients were 18 years or older, had previously untreated advanced RCC with a clear-cell component, had measurable disease according to Response Evaluation Criteria in Solid Tumors (version 1.1), and had a Karnofsky performance status of at least 70 (on a scale from 0 to 100, with lower scores indicating greater disability). Patients with central nervous system metastases or autoimmune disease who were being treated with glucocorticoids and immunosuppressants were excluded from the study.
Around three-quarters of patients with advanced RCC have intermediate- or poor-risk disease and experience worse outcomes than patients with favorable-risk disease. Patients in CheckMate-214 were stratified according to International Metastatic Renal Cell Carcinoma Database Consortium risk score as favorable (score of 0), intermediate (score of 1 or 2) or poor risk (score of 3-6), according to the number of risk factors present.
Risk factors included a Karnofsky performance score of 70, time from initial diagnosis to randomization of <1 year, a hemoglobin level below the lower limit of normal, a corrected serum calcium concentration of >10 mg/dL, or an absolute neutrophil count or platelet count above the upper limit of normal. Patients were also stratified according to geographic region (United States versus Canada and Europe versus the rest of the world).
The coprimary endpoints were objective response rate (ORR), progression-free survival (PFS), and OS in a subset of 847 intermediate- and poor-risk patients. Over a median follow-up of 25.2 months, there was a statistically significant improvement in OS and ORR in patients treated with nivolumab and ipilimumab (mPFS not reached; ORR, 41.6%), compared with sunitinib (OS, 25.9 months; ORR, 26.5%), with P <.001 for both. The immunotherapy combination was favored across subgroups.
The most common adverse events (AEs) in patients treated with nivolumab and ipilimumab included fatigue, rash, diarrhea, musculoskeletal pain, pruritus, nausea, cough, pyrexia, arthralgia, and decreased appetite. The combination was associated with fewer grade 3/4 AEs (63% vs 46% for sunitinib), but a higher rate of treatment discontinuations because of AEs (31% vs 21%, respectively). There were 8 deaths in the combination arm, and 4 in the sunitinib arm that were reported to be treatment related.2
The warnings and precautions related to nivolumab–ipilimumab combination therapy outlined in the prescribing information include mostly immune-mediated AEs, such as immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, nephritis and renal dysfunction, skin adverse reactions, and encephalitis. There are also warnings relating to the risk of infusion reactions and the potential for embryofetal toxicity.
Patients should be monitored for hyperglycemia and for changes in liver, thyroid, renal, and neurologic function. Treatment with nivolumab and ipilimumab should be withheld for moderate and permanently discontinued for severe or life-threatening immune-mediated pneumonitis, colitis, and hepatitis, as well as transaminase or total bilirubin elevation. It should also be withheld for moderate or severe hypophysitis and serum creatinine elevation, moderate adrenal insufficiency and severe hyperglycemia, and permanently discontinued for life-threatening hypophysitis and serum creatinine elevation, severe or life-threatening adrenal insufficiency, and life-threatening hyperglycemia.
New-onset moderate to severe neurologic signs or symptoms warrant treatment being withheld, and immune-mediated encephalitis should lead to treatment discontinuation. For mild or moderate infusion reactions, the infusion rate can be slowed or interrupted, and infusions should be discontinued in the event of severe or life-threatening infusion reactions. Patients should be advised of the potential for fetal harm and the need for effective contraception during and after treatment. Ipilimumab and nivolumab are marketed as Yervoy and Opdivo, respectively, by Bristol-Myers Squibb.3,4
In April 2018, the US Food and Drug Administration expanded the approval of the combination of nivolumab and ipilimumab into a new indication, following a previous approval in patients with metastatic melanoma. The double immune checkpoint inhibitor combination was approved on the basis of the phase 3 CheckMate-214 study for the treatment of patients with intermediate- or poor-risk, previously untreated advanced renal cell carcinoma (RCC).1
Nivolumab monotherapy is already approved in the second-line setting for the treatment of advanced RCC, and the demonstration of significantly improved overall survival (OS) in this study suggests that the combination should supplant sunitinib in the front-line setting in the treatment of this type of cancer.
A total of 1,096 patients at 175 sites in 28 countries were randomized 1:1 to receive nivolumab (3 mg/kg) and ipilimumab (1 mg/kg) intravenously every 3 weeks for 4 doses in an induction phase, followed by nivolumab monotherapy (3 mg/kg) every 2 weeks in a maintenance phase or sunitinib (50 mg) orally daily for 4 weeks of each 6-week cycle.
Eligible patients were 18 years or older, had previously untreated advanced RCC with a clear-cell component, had measurable disease according to Response Evaluation Criteria in Solid Tumors (version 1.1), and had a Karnofsky performance status of at least 70 (on a scale from 0 to 100, with lower scores indicating greater disability). Patients with central nervous system metastases or autoimmune disease who were being treated with glucocorticoids and immunosuppressants were excluded from the study.
Around three-quarters of patients with advanced RCC have intermediate- or poor-risk disease and experience worse outcomes than patients with favorable-risk disease. Patients in CheckMate-214 were stratified according to International Metastatic Renal Cell Carcinoma Database Consortium risk score as favorable (score of 0), intermediate (score of 1 or 2) or poor risk (score of 3-6), according to the number of risk factors present.
Risk factors included a Karnofsky performance score of 70, time from initial diagnosis to randomization of <1 year, a hemoglobin level below the lower limit of normal, a corrected serum calcium concentration of >10 mg/dL, or an absolute neutrophil count or platelet count above the upper limit of normal. Patients were also stratified according to geographic region (United States versus Canada and Europe versus the rest of the world).
The coprimary endpoints were objective response rate (ORR), progression-free survival (PFS), and OS in a subset of 847 intermediate- and poor-risk patients. Over a median follow-up of 25.2 months, there was a statistically significant improvement in OS and ORR in patients treated with nivolumab and ipilimumab (mPFS not reached; ORR, 41.6%), compared with sunitinib (OS, 25.9 months; ORR, 26.5%), with P <.001 for both. The immunotherapy combination was favored across subgroups.
The most common adverse events (AEs) in patients treated with nivolumab and ipilimumab included fatigue, rash, diarrhea, musculoskeletal pain, pruritus, nausea, cough, pyrexia, arthralgia, and decreased appetite. The combination was associated with fewer grade 3/4 AEs (63% vs 46% for sunitinib), but a higher rate of treatment discontinuations because of AEs (31% vs 21%, respectively). There were 8 deaths in the combination arm, and 4 in the sunitinib arm that were reported to be treatment related.2
The warnings and precautions related to nivolumab–ipilimumab combination therapy outlined in the prescribing information include mostly immune-mediated AEs, such as immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, nephritis and renal dysfunction, skin adverse reactions, and encephalitis. There are also warnings relating to the risk of infusion reactions and the potential for embryofetal toxicity.
Patients should be monitored for hyperglycemia and for changes in liver, thyroid, renal, and neurologic function. Treatment with nivolumab and ipilimumab should be withheld for moderate and permanently discontinued for severe or life-threatening immune-mediated pneumonitis, colitis, and hepatitis, as well as transaminase or total bilirubin elevation. It should also be withheld for moderate or severe hypophysitis and serum creatinine elevation, moderate adrenal insufficiency and severe hyperglycemia, and permanently discontinued for life-threatening hypophysitis and serum creatinine elevation, severe or life-threatening adrenal insufficiency, and life-threatening hyperglycemia.
New-onset moderate to severe neurologic signs or symptoms warrant treatment being withheld, and immune-mediated encephalitis should lead to treatment discontinuation. For mild or moderate infusion reactions, the infusion rate can be slowed or interrupted, and infusions should be discontinued in the event of severe or life-threatening infusion reactions. Patients should be advised of the potential for fetal harm and the need for effective contraception during and after treatment. Ipilimumab and nivolumab are marketed as Yervoy and Opdivo, respectively, by Bristol-Myers Squibb.3,4
1. US Food and Drug Administration website. FDA approves nivolumab plus ipilimumab combination for intermediate or poor-risk advanced renal cell carcinoma. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm604685.htm. Last updated April 16, 2018. Accessed July 25, 2018.
2. Motzer RJ, Tannir NM, McDermott O, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277-1290.
3. Opdivo (nivolumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf. Revised April 2018.
4. Yervoy (ipilimumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. July 2018. https://packageinserts.bms.com/pi/pi_yervoy.pdf. Accessed September, 2018.
1. US Food and Drug Administration website. FDA approves nivolumab plus ipilimumab combination for intermediate or poor-risk advanced renal cell carcinoma. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm604685.htm. Last updated April 16, 2018. Accessed July 25, 2018.
2. Motzer RJ, Tannir NM, McDermott O, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277-1290.
3. Opdivo (nivolumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf. Revised April 2018.
4. Yervoy (ipilimumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. July 2018. https://packageinserts.bms.com/pi/pi_yervoy.pdf. Accessed September, 2018.
Finding that sweet spot where science, practice, and best-possible outcomes come together
The practice of oncology and the science driving it have undergone substantial change in recent years, so it was particularly exciting when this year’s Nobel Prize for Physiology or Medicine was awarded to James Allison and Tasuko Honjo for their discovery that the body’s immune system can be harnessed to fight cancer. The advent of immunotherapy has expanded our therapeutic options, especially for patients whose previous treatments have failed, and in some patients, improvement in overall survival and safety profiles have been encouraging. But we still have a way to go with immunotherapies: not all patients respond to them and they are a costly therapeutic option. In addition, while chemotherapy supresses the immune system, immune-checkpoint inhibitors can hyperactivate it, and patients can experience serious immune-related adverse events that can result in life-threatening toxicities. Among the many things we grapple with in our daily practice is pairing these new and thrilling findings with our patients on a case-by-case basis to ensure the best-possible outcomes at every level – clinical, psychosocial, financial.
In recent years, we have seen an uptick in the number of FDA approvals, and as our therapeutic options have expanded, we have been able to refine and microtarget our treatment approaches, with encouraging clinical and quality-of-life outcomes. Our approach to practice has changed as well – our care is more patient focused, and we work more as part of a team, rather than individually, to ensure that our patients’ clinical and supportive needs are met. We hope our content reflects these shifts. For example, on page e188, Ibrahimi and colleagues looked at the time from admission to treatment initiation (TAT) in patients who were newly diagnosed with acute myeloid leukemia to see if it had an impact on overall survival (OS) and event-free survival. They obtained retrospective data over 5 years, focusing on patients with a TAT of 0-4 days and those with a TAT of >4 days, and found that the median OS in the 0-4 days group was almost double that of the <4 days group (1.3 years and 0.57 years, respectively). Median event-free survival for the groups was 1.21 years and 0.57 years, respectively. Moreover, that association remained significant in a multivariate analysis adjusting for age, white blood cell count, molecular risk group, and undergoing allogeneic stem cell transplant.
Marriage and survival
Does marital status have a prognostic bearing on outcomes in patients with cancer? Vyfhuis and colleagues addressed that question in their study of patients with stage III non–small-cell lung cancer (NSCLC) who had been treated uniformly with curative intent (p. e194). Specifically, they looked at OS and freedom from recurrence and they adjusted for patient-, disease-, and treatment-specific factors, as well as the interaction with racial, nutritional, and immunologic status.
In all, 52% of patients in the study were married, and were more likely to self-identify as white; live in areas with a higher household median income; undergo surgery; and have insurance, an ECOG of 0, and higher pretreatment albumin. The authors report that on multivariate analysis, marital status remained an independent predictor of survival and was associated with a 40% decreased risk of death, further stratifying outcomes beyond gender and stage grouping. Freedom from recurrence was comparable between the married and not-married patients. These findings suggest that in a cancer such as NSCLC, for which survival is modest despite therapeutic advances and which is associated with considerable treatment-related toxicities, marital status might be an independent predictor for survival. The authors suggest that marriage is likely a surrogate for better psychosocial support, and that the survival improvements might justify investment in supportive care interventional strategies to help advance overall outcomes.
Cancer in children and AYAs
Two articles in this issue examine cancers in pediatric patients and in adolescents and young adults (AYAs), and by doing so, demonstrate the importance of having evidence-based research findings to help us refine and deliver better-quality, patient-focused care. On page e217, Sharon Worcester documents the growing efforts by researchers and clinicians to understand and address the disparities in survival outcomes between AYAs with cancer and their pediatric and adult counterparts.
It has been known for a while that some cancers are more common among AYAs compared with the other 2 populations, and others are less common. More recent findings suggest that the biology and molecular make-up of AYA cancers might also be different and therefore necessitate different therapeutic protocols, and that the social and psychological needs unique to this population also require specifically tailored supportive care. What about treatment setting for AYAs with cancer – would outcomes be better in a pediatric or adult care center? There is evidence that the pediatric setting might have some advantage, but a recent study from Canada suggests that the cost of care in that setting might be higher. Despite these encouraging findings, there are very few trials designed specifically for the AYA cancer population, and the “pediatric-versus-adult” question also applies to AYA participation in trials. Worcester’s comprehensive article weaves together these issues and offers insights and useful explanations from a number of experts who study or care for AYAs with cancers.
Pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, but they are the second leading cause of death in children aged 1 to 14 years and therefore warrant attention, writes Jane de Lartigue in an article on page e210. She echoes Worcester’s point that better understanding of cancers in this younger population has brought to light their unique molecular drivers and challenged the assumption that drugs developed for adults can be used in children and young adults. Dr de Lartigue drills down into the science behind the unique biology and molecular aberrations in pediatric cancers and provides a useful list of ongoing clinical trials of targeted therapies in this population. She notes that because of their rarity, pediatric cancers are difficult to study and adequate enrollment in trials is challenging, although that is changing with researchers’ greater awareness of the uniqueness of these cancers and need for age-specific trials.
Also included in this issue are Community Translation articles on the approval of an immunotherapy combination – nivolumab plus ipilimumab – for the treatment of advanced RCC (p. e182), and for venetoclax as a therapy for patients with chronic lymphocytic leukemia, regardless of genotype (p. e185); and 2 Case Reports, one describing a diagnostic dilemma relating to a patient eventually diagnosed with primary renal synovial sarcoma (p. e202), and another detailing prolonged survival in a patient with adenocarcinoma of unknown primary who was treated with chemoradiotherapy (p. e206).
The practice of oncology and the science driving it have undergone substantial change in recent years, so it was particularly exciting when this year’s Nobel Prize for Physiology or Medicine was awarded to James Allison and Tasuko Honjo for their discovery that the body’s immune system can be harnessed to fight cancer. The advent of immunotherapy has expanded our therapeutic options, especially for patients whose previous treatments have failed, and in some patients, improvement in overall survival and safety profiles have been encouraging. But we still have a way to go with immunotherapies: not all patients respond to them and they are a costly therapeutic option. In addition, while chemotherapy supresses the immune system, immune-checkpoint inhibitors can hyperactivate it, and patients can experience serious immune-related adverse events that can result in life-threatening toxicities. Among the many things we grapple with in our daily practice is pairing these new and thrilling findings with our patients on a case-by-case basis to ensure the best-possible outcomes at every level – clinical, psychosocial, financial.
In recent years, we have seen an uptick in the number of FDA approvals, and as our therapeutic options have expanded, we have been able to refine and microtarget our treatment approaches, with encouraging clinical and quality-of-life outcomes. Our approach to practice has changed as well – our care is more patient focused, and we work more as part of a team, rather than individually, to ensure that our patients’ clinical and supportive needs are met. We hope our content reflects these shifts. For example, on page e188, Ibrahimi and colleagues looked at the time from admission to treatment initiation (TAT) in patients who were newly diagnosed with acute myeloid leukemia to see if it had an impact on overall survival (OS) and event-free survival. They obtained retrospective data over 5 years, focusing on patients with a TAT of 0-4 days and those with a TAT of >4 days, and found that the median OS in the 0-4 days group was almost double that of the <4 days group (1.3 years and 0.57 years, respectively). Median event-free survival for the groups was 1.21 years and 0.57 years, respectively. Moreover, that association remained significant in a multivariate analysis adjusting for age, white blood cell count, molecular risk group, and undergoing allogeneic stem cell transplant.
Marriage and survival
Does marital status have a prognostic bearing on outcomes in patients with cancer? Vyfhuis and colleagues addressed that question in their study of patients with stage III non–small-cell lung cancer (NSCLC) who had been treated uniformly with curative intent (p. e194). Specifically, they looked at OS and freedom from recurrence and they adjusted for patient-, disease-, and treatment-specific factors, as well as the interaction with racial, nutritional, and immunologic status.
In all, 52% of patients in the study were married, and were more likely to self-identify as white; live in areas with a higher household median income; undergo surgery; and have insurance, an ECOG of 0, and higher pretreatment albumin. The authors report that on multivariate analysis, marital status remained an independent predictor of survival and was associated with a 40% decreased risk of death, further stratifying outcomes beyond gender and stage grouping. Freedom from recurrence was comparable between the married and not-married patients. These findings suggest that in a cancer such as NSCLC, for which survival is modest despite therapeutic advances and which is associated with considerable treatment-related toxicities, marital status might be an independent predictor for survival. The authors suggest that marriage is likely a surrogate for better psychosocial support, and that the survival improvements might justify investment in supportive care interventional strategies to help advance overall outcomes.
Cancer in children and AYAs
Two articles in this issue examine cancers in pediatric patients and in adolescents and young adults (AYAs), and by doing so, demonstrate the importance of having evidence-based research findings to help us refine and deliver better-quality, patient-focused care. On page e217, Sharon Worcester documents the growing efforts by researchers and clinicians to understand and address the disparities in survival outcomes between AYAs with cancer and their pediatric and adult counterparts.
It has been known for a while that some cancers are more common among AYAs compared with the other 2 populations, and others are less common. More recent findings suggest that the biology and molecular make-up of AYA cancers might also be different and therefore necessitate different therapeutic protocols, and that the social and psychological needs unique to this population also require specifically tailored supportive care. What about treatment setting for AYAs with cancer – would outcomes be better in a pediatric or adult care center? There is evidence that the pediatric setting might have some advantage, but a recent study from Canada suggests that the cost of care in that setting might be higher. Despite these encouraging findings, there are very few trials designed specifically for the AYA cancer population, and the “pediatric-versus-adult” question also applies to AYA participation in trials. Worcester’s comprehensive article weaves together these issues and offers insights and useful explanations from a number of experts who study or care for AYAs with cancers.
Pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, but they are the second leading cause of death in children aged 1 to 14 years and therefore warrant attention, writes Jane de Lartigue in an article on page e210. She echoes Worcester’s point that better understanding of cancers in this younger population has brought to light their unique molecular drivers and challenged the assumption that drugs developed for adults can be used in children and young adults. Dr de Lartigue drills down into the science behind the unique biology and molecular aberrations in pediatric cancers and provides a useful list of ongoing clinical trials of targeted therapies in this population. She notes that because of their rarity, pediatric cancers are difficult to study and adequate enrollment in trials is challenging, although that is changing with researchers’ greater awareness of the uniqueness of these cancers and need for age-specific trials.
Also included in this issue are Community Translation articles on the approval of an immunotherapy combination – nivolumab plus ipilimumab – for the treatment of advanced RCC (p. e182), and for venetoclax as a therapy for patients with chronic lymphocytic leukemia, regardless of genotype (p. e185); and 2 Case Reports, one describing a diagnostic dilemma relating to a patient eventually diagnosed with primary renal synovial sarcoma (p. e202), and another detailing prolonged survival in a patient with adenocarcinoma of unknown primary who was treated with chemoradiotherapy (p. e206).
The practice of oncology and the science driving it have undergone substantial change in recent years, so it was particularly exciting when this year’s Nobel Prize for Physiology or Medicine was awarded to James Allison and Tasuko Honjo for their discovery that the body’s immune system can be harnessed to fight cancer. The advent of immunotherapy has expanded our therapeutic options, especially for patients whose previous treatments have failed, and in some patients, improvement in overall survival and safety profiles have been encouraging. But we still have a way to go with immunotherapies: not all patients respond to them and they are a costly therapeutic option. In addition, while chemotherapy supresses the immune system, immune-checkpoint inhibitors can hyperactivate it, and patients can experience serious immune-related adverse events that can result in life-threatening toxicities. Among the many things we grapple with in our daily practice is pairing these new and thrilling findings with our patients on a case-by-case basis to ensure the best-possible outcomes at every level – clinical, psychosocial, financial.
In recent years, we have seen an uptick in the number of FDA approvals, and as our therapeutic options have expanded, we have been able to refine and microtarget our treatment approaches, with encouraging clinical and quality-of-life outcomes. Our approach to practice has changed as well – our care is more patient focused, and we work more as part of a team, rather than individually, to ensure that our patients’ clinical and supportive needs are met. We hope our content reflects these shifts. For example, on page e188, Ibrahimi and colleagues looked at the time from admission to treatment initiation (TAT) in patients who were newly diagnosed with acute myeloid leukemia to see if it had an impact on overall survival (OS) and event-free survival. They obtained retrospective data over 5 years, focusing on patients with a TAT of 0-4 days and those with a TAT of >4 days, and found that the median OS in the 0-4 days group was almost double that of the <4 days group (1.3 years and 0.57 years, respectively). Median event-free survival for the groups was 1.21 years and 0.57 years, respectively. Moreover, that association remained significant in a multivariate analysis adjusting for age, white blood cell count, molecular risk group, and undergoing allogeneic stem cell transplant.
Marriage and survival
Does marital status have a prognostic bearing on outcomes in patients with cancer? Vyfhuis and colleagues addressed that question in their study of patients with stage III non–small-cell lung cancer (NSCLC) who had been treated uniformly with curative intent (p. e194). Specifically, they looked at OS and freedom from recurrence and they adjusted for patient-, disease-, and treatment-specific factors, as well as the interaction with racial, nutritional, and immunologic status.
In all, 52% of patients in the study were married, and were more likely to self-identify as white; live in areas with a higher household median income; undergo surgery; and have insurance, an ECOG of 0, and higher pretreatment albumin. The authors report that on multivariate analysis, marital status remained an independent predictor of survival and was associated with a 40% decreased risk of death, further stratifying outcomes beyond gender and stage grouping. Freedom from recurrence was comparable between the married and not-married patients. These findings suggest that in a cancer such as NSCLC, for which survival is modest despite therapeutic advances and which is associated with considerable treatment-related toxicities, marital status might be an independent predictor for survival. The authors suggest that marriage is likely a surrogate for better psychosocial support, and that the survival improvements might justify investment in supportive care interventional strategies to help advance overall outcomes.
Cancer in children and AYAs
Two articles in this issue examine cancers in pediatric patients and in adolescents and young adults (AYAs), and by doing so, demonstrate the importance of having evidence-based research findings to help us refine and deliver better-quality, patient-focused care. On page e217, Sharon Worcester documents the growing efforts by researchers and clinicians to understand and address the disparities in survival outcomes between AYAs with cancer and their pediatric and adult counterparts.
It has been known for a while that some cancers are more common among AYAs compared with the other 2 populations, and others are less common. More recent findings suggest that the biology and molecular make-up of AYA cancers might also be different and therefore necessitate different therapeutic protocols, and that the social and psychological needs unique to this population also require specifically tailored supportive care. What about treatment setting for AYAs with cancer – would outcomes be better in a pediatric or adult care center? There is evidence that the pediatric setting might have some advantage, but a recent study from Canada suggests that the cost of care in that setting might be higher. Despite these encouraging findings, there are very few trials designed specifically for the AYA cancer population, and the “pediatric-versus-adult” question also applies to AYA participation in trials. Worcester’s comprehensive article weaves together these issues and offers insights and useful explanations from a number of experts who study or care for AYAs with cancers.
Pediatric cancers are rare, representing just 1% of all new cancers diagnosed annually in the United States, but they are the second leading cause of death in children aged 1 to 14 years and therefore warrant attention, writes Jane de Lartigue in an article on page e210. She echoes Worcester’s point that better understanding of cancers in this younger population has brought to light their unique molecular drivers and challenged the assumption that drugs developed for adults can be used in children and young adults. Dr de Lartigue drills down into the science behind the unique biology and molecular aberrations in pediatric cancers and provides a useful list of ongoing clinical trials of targeted therapies in this population. She notes that because of their rarity, pediatric cancers are difficult to study and adequate enrollment in trials is challenging, although that is changing with researchers’ greater awareness of the uniqueness of these cancers and need for age-specific trials.
Also included in this issue are Community Translation articles on the approval of an immunotherapy combination – nivolumab plus ipilimumab – for the treatment of advanced RCC (p. e182), and for venetoclax as a therapy for patients with chronic lymphocytic leukemia, regardless of genotype (p. e185); and 2 Case Reports, one describing a diagnostic dilemma relating to a patient eventually diagnosed with primary renal synovial sarcoma (p. e202), and another detailing prolonged survival in a patient with adenocarcinoma of unknown primary who was treated with chemoradiotherapy (p. e206).
Advances in precision medicine help refine – and redefine – cancer care
Among the groundbreaking findings presented at this year’s annual meeting of the American Society of Clinical Oncology were those showing that most women with HR-positive, HER2-negative, early-stage breast cancer who have an intermediate recurrence score can safely skip adjuvant chemotherapy, and that upfront pembrolizumab for patients with NSCLC expressing PD-L1 on at least 1% of tumor cells can not only significantly improve overall survival, but do so with less toxicity than standard chemotherapy.
TAILORx marks major advance for precision medicine in breast cancer
Key clinical point The majority of women with HR-positive, HER2-negative, node-negative early-stage breast cancer who have an intermediate recurrence score can safely skip adjuvant chemotherapy. Major finding In women with an Oncotype DX Recurrence Score in the midrange (11-25), invasive DFS with endocrine therapy alone was not inferior to that with chemotherapy plus endocrine therapy (HR, 1.08; P = .26). Study details A phase 3 trial in 10,273 women with HR-positive, HER2-negative, node-negative, early-stage breast cancer, with a noninferiority randomized component in the 6,711 women with a midrange recurrence score (TAILORx trial). Funding This study received funding primarily from the National Cancer Institute, National Institutes of Health. Additional support was provided by the Breast Cancer Research Foundation, Komen Foundation, and US Postal Service Breast Cancer Stamp. Disclosures Dr Sparano disclosed that he has a consulting or advisory role with Genentech/Roche, Novartis, AstraZeneca, Celgene, Lilly, Celldex, Pfizer, Prescient Therapeutics, Juno Therapeutics, and Merrimack; has stock or other ownership interests with MetaStat; and receives research funding (institutional) from Prescient Therapeutics, Deciphera, Genentech/Roche, Merck, Novartis, and Merrimack. Source Sparano et al. ASCO 2018 Abstract LBA1: https://meetinglibrary.asco.org/record/161490/abstract
Use of the 21-tumor gene expression assay (Oncotype DX Recurrence Score) allows nearly 70% of women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer to safely forgo adjuvant chemotherapy, sparing them adverse effects and preventing overtreatment, TAILORx trial results show.
The findings, which were reported in the plenary session at the meeting and simultaneously published in the New England Journal of Medicine (N Engl J Med. 2018; 379:111-121; [behind paywall]), mark a major advance in precision medicine.
“The rationale for the TAILORx precision medicine trial is that we are really trying to ‘thread the needle,’ “ lead study author Joseph A Sparano, MD, commented in a press briefing. Oncologists typically recommend adjuvant chemotherapy for the half of all breast cancers that are hormone receptor positive, HER2 negative, and node negative, even though its absolute benefit in reducing recurrences in this population is small. “This results in most patients being overtreated because endocrine therapy alone is adequate. But some are undertreated: They do not receive chemotherapy although they could have benefited from it,” he noted.
The recurrence score is known to be prognostic and predictive of benefit from adding chemotherapy to endocrine therapy, Dr Sparano said. “But there was a major gap: There was uncertain benefit for patients who had a midrange score, which is about two-thirds of all patients who are treated,” said Dr Sparano, the associate director for clinical research at Albert Einstein Cancer Center and Montefiore Health System in New York, and vice-chair of the ECOG-ACRIN Cancer Research Group.
The phase 3 TAILORx trial registered 10,273 women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer, making it the largest adjuvant breast cancer trial to date. Analyses focused on the 6,711 evaluable women with a midrange recurrence score (defined as 11 through 25 in the trial), who were randomized to receive endocrine therapy alone or adjuvant chemotherapy plus endocrine therapy, with a noninferiority design. Of note is that contemporary drugs and regimens were used.
Results at a median follow-up of 7.5 years showed that the trial met its primary endpoint: the risk of invasive disease-free survival (DFS) events (invasive disease recurrence, second primary cancer, or death) was not inferior for women given endocrine therapy alone compared with counterparts given chemotherapy plus endocrine therapy (hazard ratio [HR], 1.08; P = .26), Dr Sparano reported.
The groups were also on par, with absolute differences of no more than 1% between rates, with respect to a variety of other efficacy outcomes: freedom from distant recurrence and any recurrence, and overall survival (OS).
Findings were similar across most subgroups. But analyses suggested that women aged 50 years and younger and who had a recurrence score of 16-25 fared better when they received chemotherapy. “Though exploratory from a statistical perspective, this is a highly clinically relevant observation,” Dr Sparano said. “It suggests ... that chemotherapy should be spared with caution in this subgroup, after a careful discussion of potential benefits and risks in a shared decision process.”
In other findings, analyses of the trial’s nonrandomized groups confirmed excellent outcomes in women with a low recurrence score (0-10) who were given endocrine therapy alone, and at the other end of the spectrum, there was need for a more aggressive approach, including chemotherapy, in women with a high recurrence score (26-100).
Ultimately, application of the recurrence score allowed 69% of the trial population to skip chemotherapy: all of the women with a score of 0-10 (16% of the trial population), those older than 50 years with a score of 11-25 (45%), and those aged 50 years or younger with a score of 11-15 (8%).
An ongoing companion phase 3 trial, RxPONDER, is assessing the benefit of applying the recurrence score in women who are similar but ihave node-positive disease.
Study details
All of the women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer enrolled in TAILORx met National Comprehensive Cancer Network guidelines for receiving adjuvant chemotherapy. About 69% had an intermediate recurrence score (11-25) and were randomized. All of the 17% with a low recurrence score (0-10) were given only endocrine therapy, and all of the 14% with a high recurrence score (26-100) were given both adjuvant chemotherapy and endocrine therapy.
Of note, the recurrence scores used to define midrange were adjusted downward from those conventionally used to account for exclusion of patients with higher-risk HER2-positive disease and to minimize potential for undertreatment, Dr Sparano explained.
In the women with midrange scores who were randomized, the hazard ratio of 1.08 for invasive DFS with endocrine therapy alone compared with chemotherapy plus endocrine therapy fell well within the predefined hazard ratio for noninferiority (1.322). The 9-year rate of invasive DFS was 83.3% with endocrine therapy and 84.3% with chemotherapy plus endocrine therapy.
The groups had similar rates of freedom from distant recurrence (94.5% vs 95.0%; HR, 1.10; P = .48) and distant or locoregional recurrence (92.2% vs 92.9%; HR, 1.11; P = .33), and similar OSs (93.9% vs 93.8%; HR for death, 0.99; P = .89).
In exploratory analyses, there was an interaction of age and recurrence score (P = .004) whereby women aged 50 years or younger derived some benefit from chemotherapy if they had a recurrence score of 16-20 (9% fewer invasive DFS events, including 2% fewer distant recurrences) or a recurrence score 21-25 (6% fewer invasive DFS events, mainly distant recurrences). “This is information that could drive some younger women who have a recurrence score in this range to accept chemotherapy,” Dr Sparano said.
The 9-year rate of distant recurrence averaged 5% in women with midrange scores overall. It was 3% in those with a low recurrence score given endocrine therapy alone, but it was still 13% in those with a high recurrence score despite receiving both endocrine therapy and chemotherapy. The latter finding may “indicate the need to explore potentially more effective therapies in this setting,” he proposed.
Tailoring treatment: ‘not too much and not too little’
“These are important data because this is the most common form of breast cancer in the United States and other developed countries, and the most challenging decision we make with these patients is whether or not to recommend adjuvant chemotherapy with all its side effects and potential benefits,” said ASCO expert Harold Burstein, MD, PhD, FASCO. “The data show that the majority of women who have this test performed on their tumor can be told that they don’t need chemotherapy, and that can be said with tremendous confidence and reassurance.”
The recurrence score has been used for a decade, but the trial was necessary because the score was originally developed in patients who were receiving older chemotherapy regimens and older endocrine therapies, and because there have been few data to guide decision making in the large group of patients with midrange scores, he said. “Now we can say with confidence ... that the patients got contemporary chemo regimens and still saw no benefit from chemotherapy.
“This is not so much about de-escalation ... the goal of this study was not to just use less treatment but to tailor treatment. The investigators chose the title very aptly,” said Dr Burstein, a medical oncologist at the Dana-Farber Cancer Institute and associate professor of medicine at the Harvard Medical School, Boston.
“This is extraordinary data for breast cancer doctors and women who have breast cancer. It allows you to individualize treatment based on extraordinary science, which now has tremendous prospective validation,” he said. Overall, “women with breast cancer who are getting modern therapy are doing well, and this test shows us how to tailor that management so that they get exactly the right amount of treatment – not too much and not too little.”
— Susan London
First-line immunotherapy boosts survival in NSCLC patients
Key clinical point Many patients with previously untreated NSCLC could benefit from first-line therapy with the checkpoint inhibitor pembrolizumab. Major finding In all patients with expression of PD-L1 on 1% or more of tumor cells, OS was 16.7 months with pembrolizumab, compared with 12.1 months for chemotherapy. Study details Randomized phase 3 trial of 1,274 patients with advanced or metastatic NSCLC. Funding Merck, the maker of the study drug, funded the study. Disclosures Dr Lopes disclosed institutional research funding from Merck Sharp & Dohme, EMD Serono, and AstraZeneca. Dr Heymach disclosed stock/ownership in Bio-Tree and Cardinal Spine, a consulting or advisory role for Abbvie, ARIAD, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Calithera Biosciences, Genentech, Medivation, Novartis, Oncomed, and Synta, and institutional research funding from AstraZeneca. Dr Gandhi reported having no relevant disclosures. Source Lopes G et al. ASCO 2018, abstract LBA4. https://meetinglibrary.asco.org/record/165950/abstract
Pembrolizumab as first-line treatment of advanced non–small-cell lung cancer (NSCLC) offered longer OS with better tolerability compared with chemotherapy, results of the Keynote-042 phase 3 randomized trial show.
In 1,274 patients with advanced, previously untreated NSCLC with expression of the PD-L1 on 1% or more of tumor cells, median OS after a median follow-up of 12.8 months was 16.7 months for patients treated with pembrolizumab monotherapy, compared with 12.1 months for patients treated with either paclitaxel or pemetrexed plus carboplatin, reported lead author Gilberto Lopes, MD, of the Sylvester Comprehensive Cancer Center at the University of Miami.
The survival benefit for immunotherapy was even greater for patients with higher levels of PD-L1 expression: 20 versus 12.2 months for patients with PD-L1 expression of 50% or greater, and 17.7 versus 13 months for patients with PD-L1 expression of 20% or greater, Dr Lopes noted.
For all 3 PD-L1 expression groups, the median duration of response was 20.2 months, compared with 10.8-8.3 months for patients in the chemotherapy arm.
“These are responses that are unlike anything that we have seen with chemotherapy in the past for non–small-cell lung cancer,” Dr Lopes said at a briefing before his presentation. “In addition to that, and probably more importantly, patients had fewer adverse events [with pembrolizumab]. Overall, about 60% had any treatment-related adverse event with pembrolizumab, versus 90% with chemotherapy,” he added.
‘A true milestone’
ASCO expert John Heymach, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, said at the briefing that the study was “a true milestone for the field, because now, for the first time, we can say that in non–small-cell lung cancer patients receiving first-line therapy, the vast majority can receive immunotherapy with pembrolizumab instead of chemotherapy.”
He noted that an earlier study, Keynote-024, showed that pembrolizumab significantly improved progression-free survival in patients with tumors expressing PD-L1 on at least 50% of cells compared with standard platinum-based chemotherapy (10.3 vs 6 months).
“This more than doubles that population that can start immunotherapy as a first-line treatment, assuming the [Food and Drug Administration] modifies the label in accordance with this study,” he added.
The Keynote-042 investigators enrolled 1,274 patients with locally advanced or metastatic NSCLC, and randomly assigned them to receive either a maximum of 35 cycles of pembrolizumab 200 mg every 3 weeks, or the investigators’ choice of not more than 6 cycles of either paclitaxel–carboplatin or pemetrexed–carboplatin, with optional pemetrexed maintenance for patients with nonsquamous histologies only.
The randomization was stratified by region (Asia vs non-East Asia), Eastern Cooperative Oncology Group performance status 0 or 1, squamous versus nonsquamous histology, and PD-L1 expression, or TPS (tumor proportion score) greater than 50% versus 1%-49%.
As noted before, the primary endpoint of OS in all patients with a TPS of 1% or greater was met, with respective median OS in the pembrolizumab versus chemotherapy groups of 16.7 and 12.1 months, translating into an HR favoring pembrolizumab of 0.81 (P = .0018). Respective hazard ratios for the TPS 20% or greater and TPS 50% or greater groups were 0.77 (P = .0020) and 0.69 (P = .0003).
At 12.8 months of median follow-up, 13% of patients assigned to pembrolizumab were still on the drug, and 4.3% of patients were receiving maintenance pemetrexed.
Treatment-related adverse events of any grade occurred in 399 of 636 patients assigned to pembrolizumab (62.7%), compared with 553 of 615 patients assigned to chemotherapy (89.9%). Grade 3 or greater events occurred in 17.8% and 41% of patients, respectively. There were 13 deaths related to therapy in the pembrolizumab arm (2.0%), and 14 in the chemotherapy arm (2.3%). Adverse events leading to discontinuation were similar between the groups, at 9% and 9.4%, respectively.
There were more immune-mediated adverse events in the pembrolizumab arm than in the chemotherapy arm (27.8% vs 7.2%, respectively), and of those, grade 3 or higher events occurred in 8% and 1.5% of patients. There was 1 immune-mediated death, from pneumonitis, in the immunotherapy arm; there were no deaths related to immune-mediated side effects in the chemotherapy arm.
“I really view this as a ‘double whammy’ for patients,” Dr Heymach said at the briefing. “Often advances in survival for our lung cancer patients come at the cost of significant toxicities. Here, by contrast, not only are patients living longer and having a much higher likelihood of prolonged survival in years, often instead of months, but they’re also receiving a treatment that has substantially less toxicity across virtually all measures, and this really impacts the day-to-day life of these patients.”
Leena Gandhi, MD, PhD, of the Perlmutter Cancer Center at New York University, the invited discussant at the plenary, agreed that pembrolizumab improves survival, compared with chemotherapy patients with PD-L1 expression levels greater than 1%, but noted that most of the benefit – as also seen in Keynote-024 – was in those patients whose tumors had high levels of PD-L1 expression.
She emphasized that although PD-L1 is an imperfect biomarker, it should still be used to help select patients for therapy and it may be complementary with tumor mutational burden for more precise treatment selection.
“What we know, and what this study adds to, is that PD-L1 really does define a patient population that could receive benefit from pembrolizumab over chemotherapy. Patients with low or no PD-L1 expression likely should get some type of combination therapy,” she said. “This study extends what we’ve seen from other recent studies, which is that chemotherapy alone is no longer a first-line standard of care in non–small-cell lung cancer.”
— Neil Osterweil
Maintenance chemo improves survival in youth with rhabdomyosarcoma
Key clinical point 6 months of maintenance chemotherapy improves survival in youth with high-risk rhabdomyosarcoma. Major finding Patients given maintenance low-dose vinorelbine and cyclophosphamide had better 5-year OS compared with those not receiving any additional treatment (86.5% vs 73.7%; HR, 0.52). Study details A phase 3 randomized controlled trial in 371 patients aged 0-21 years with high-risk rhabdomyosarcoma who had had a complete response to standard intensive therapy. Funding The study received funding from Fondazione Città della Speranza, Italy. Disclosures Dr Bisogno disclosed that he has a consulting or advisory role with Clinigen Group, and receives travel, accommodations, and/or expenses from Jazz Pharmaceuticals. Source Bisogno et al. ASCO 2018, Abstract LBA2. https://meetinglibrary.asco.org/record/161695/abstract
Six months of maintenance chemotherapy prolongs OS in youth with high-risk rhabdomyosarcoma, finds a phase 3 randomized controlled trial of the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG).
Rhabdomyosarcoma is a rare but highly aggressive tumor, lead study author Gianni Bisogno, MD, PhD, a professor at the University Hospital of Padova, Italy, and chair of the EpSSG, noted in a press briefing at the meeting, where the findings were reported. In pediatric patients who achieve complete response to standard therapy, “we know that after 1 or 2 years, one-third of these children relapse, and most of them die,” he said.
The EpSSG trial, which took about 10 years to conduct, enrolled 371 patients aged 0-21 years with high-risk rhabdomyosarcoma who had had a complete response to standard intensive therapy. They were randomized evenly to stop treatment or to receive 6 months of maintenance treatment consisting of low-dose vinorelbine and cyclophosphamide.
Results reported in the meeting’s plenary session showed that giving maintenance chemotherapy improved the 5-year OS rate by an absolute 12.8%, which translated to a near halving of the risk of death. And the maintenance regimen used was generally well tolerated.
“At the end of this long, not-easy study, we concluded that maintenance chemotherapy is an effective and well tolerated treatment for children with high-risk rhabdomyosarcoma,” Dr Bisogno said.
There are three possibilities for its efficacy, he speculated. “It may be the duration, the type of drugs used, or the metronomic approach. Maybe altogether, these three different actions have a benefit to increase survival.
“Our group has decided this is the new standard treatment for patients. At least in Europe, we give standard intensive therapy and then we continue with 6 more months of low-dose chemotherapy,” Dr Bisogno concluded. “We think that this approach – a new way of using old drugs – can be of interest also for other pediatric tumors.”
The trial is noteworthy in that it shows “how to successfully conduct large and important trials in rare diseases,” said ASCO expert Warren Chow, MD.
The standard therapy for rhabdomyosarcomas is somewhat different in the United States, typically a regimen containing vincristine, actinomycin D, cyclophosphamide, and (more recently) irinotecan, he noted. “We have not been traditionally using maintenance chemo for any of the pediatric sarcomas, so this is a paradigm shift. These results will need to be tested with US-based protocols before becoming standard of care in the United States. Also, we will need to determine if these results are applicable to patients older than 21 years of age who are considered high risk based solely on their age.
“Even with these caveats, this is the first significant treatment advance in this rare cancer in more than 30 years,” concluded Dr Chow, a medical oncologist and clinical professor at City of Hope, Duarte, Calif. “No doubt, this trial was a home run.”
Study details
Patients enrolled in the EpSSG trial had had a complete response to the standard intensive therapy used in Europe: high-dose chemotherapy (ifosfamide, vincristine, and actinomycin D, with or without doxorubicin), radiation therapy, and surgery.
The maintenance chemotherapy consisted of a combination of low-dose intravenous vinorelbine given weekly and oral cyclophosphamide given daily. The 6-month duration was somewhat arbitrary, according to Dr Bisogno. “We had to start somewhere. So when we started, we decided to use 6 months because there was some evidence in the past for regimens that long. In our next European trial, we are going to test different kinds and durations of maintenance because this is very important.”
The maintenance regimen was well tolerated compared with the regimen given during standard intensive therapy, with, for example, lower rates of grade 3 and 4 anemia (8.9% vs 48.9%), neutropenia (80.6% vs 91.6%), and thrombocytopenia (0.6% vs 26.0%), which translated to less of a need for transfusions, and a lower rate of grade 3 or 4 infection (29.4% vs 56.4%), Dr Bisogno reported. There were no cases of grade 3 or 4 cardiac, hepatobiliary/pancreatic, or renal toxicity.
Relative to peers who stopped treatment after standard intensive therapy, patients who received maintenance treatment tended to have better DFS (77.6% vs 69.8%; HR, 0.68; P = .0613) and had significantly better OS (86.5% vs 73.7%; HR, 0.52; P = .0111).
— Susan London
Among the groundbreaking findings presented at this year’s annual meeting of the American Society of Clinical Oncology were those showing that most women with HR-positive, HER2-negative, early-stage breast cancer who have an intermediate recurrence score can safely skip adjuvant chemotherapy, and that upfront pembrolizumab for patients with NSCLC expressing PD-L1 on at least 1% of tumor cells can not only significantly improve overall survival, but do so with less toxicity than standard chemotherapy.
TAILORx marks major advance for precision medicine in breast cancer
Key clinical point The majority of women with HR-positive, HER2-negative, node-negative early-stage breast cancer who have an intermediate recurrence score can safely skip adjuvant chemotherapy. Major finding In women with an Oncotype DX Recurrence Score in the midrange (11-25), invasive DFS with endocrine therapy alone was not inferior to that with chemotherapy plus endocrine therapy (HR, 1.08; P = .26). Study details A phase 3 trial in 10,273 women with HR-positive, HER2-negative, node-negative, early-stage breast cancer, with a noninferiority randomized component in the 6,711 women with a midrange recurrence score (TAILORx trial). Funding This study received funding primarily from the National Cancer Institute, National Institutes of Health. Additional support was provided by the Breast Cancer Research Foundation, Komen Foundation, and US Postal Service Breast Cancer Stamp. Disclosures Dr Sparano disclosed that he has a consulting or advisory role with Genentech/Roche, Novartis, AstraZeneca, Celgene, Lilly, Celldex, Pfizer, Prescient Therapeutics, Juno Therapeutics, and Merrimack; has stock or other ownership interests with MetaStat; and receives research funding (institutional) from Prescient Therapeutics, Deciphera, Genentech/Roche, Merck, Novartis, and Merrimack. Source Sparano et al. ASCO 2018 Abstract LBA1: https://meetinglibrary.asco.org/record/161490/abstract
Use of the 21-tumor gene expression assay (Oncotype DX Recurrence Score) allows nearly 70% of women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer to safely forgo adjuvant chemotherapy, sparing them adverse effects and preventing overtreatment, TAILORx trial results show.
The findings, which were reported in the plenary session at the meeting and simultaneously published in the New England Journal of Medicine (N Engl J Med. 2018; 379:111-121; [behind paywall]), mark a major advance in precision medicine.
“The rationale for the TAILORx precision medicine trial is that we are really trying to ‘thread the needle,’ “ lead study author Joseph A Sparano, MD, commented in a press briefing. Oncologists typically recommend adjuvant chemotherapy for the half of all breast cancers that are hormone receptor positive, HER2 negative, and node negative, even though its absolute benefit in reducing recurrences in this population is small. “This results in most patients being overtreated because endocrine therapy alone is adequate. But some are undertreated: They do not receive chemotherapy although they could have benefited from it,” he noted.
The recurrence score is known to be prognostic and predictive of benefit from adding chemotherapy to endocrine therapy, Dr Sparano said. “But there was a major gap: There was uncertain benefit for patients who had a midrange score, which is about two-thirds of all patients who are treated,” said Dr Sparano, the associate director for clinical research at Albert Einstein Cancer Center and Montefiore Health System in New York, and vice-chair of the ECOG-ACRIN Cancer Research Group.
The phase 3 TAILORx trial registered 10,273 women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer, making it the largest adjuvant breast cancer trial to date. Analyses focused on the 6,711 evaluable women with a midrange recurrence score (defined as 11 through 25 in the trial), who were randomized to receive endocrine therapy alone or adjuvant chemotherapy plus endocrine therapy, with a noninferiority design. Of note is that contemporary drugs and regimens were used.
Results at a median follow-up of 7.5 years showed that the trial met its primary endpoint: the risk of invasive disease-free survival (DFS) events (invasive disease recurrence, second primary cancer, or death) was not inferior for women given endocrine therapy alone compared with counterparts given chemotherapy plus endocrine therapy (hazard ratio [HR], 1.08; P = .26), Dr Sparano reported.
The groups were also on par, with absolute differences of no more than 1% between rates, with respect to a variety of other efficacy outcomes: freedom from distant recurrence and any recurrence, and overall survival (OS).
Findings were similar across most subgroups. But analyses suggested that women aged 50 years and younger and who had a recurrence score of 16-25 fared better when they received chemotherapy. “Though exploratory from a statistical perspective, this is a highly clinically relevant observation,” Dr Sparano said. “It suggests ... that chemotherapy should be spared with caution in this subgroup, after a careful discussion of potential benefits and risks in a shared decision process.”
In other findings, analyses of the trial’s nonrandomized groups confirmed excellent outcomes in women with a low recurrence score (0-10) who were given endocrine therapy alone, and at the other end of the spectrum, there was need for a more aggressive approach, including chemotherapy, in women with a high recurrence score (26-100).
Ultimately, application of the recurrence score allowed 69% of the trial population to skip chemotherapy: all of the women with a score of 0-10 (16% of the trial population), those older than 50 years with a score of 11-25 (45%), and those aged 50 years or younger with a score of 11-15 (8%).
An ongoing companion phase 3 trial, RxPONDER, is assessing the benefit of applying the recurrence score in women who are similar but ihave node-positive disease.
Study details
All of the women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer enrolled in TAILORx met National Comprehensive Cancer Network guidelines for receiving adjuvant chemotherapy. About 69% had an intermediate recurrence score (11-25) and were randomized. All of the 17% with a low recurrence score (0-10) were given only endocrine therapy, and all of the 14% with a high recurrence score (26-100) were given both adjuvant chemotherapy and endocrine therapy.
Of note, the recurrence scores used to define midrange were adjusted downward from those conventionally used to account for exclusion of patients with higher-risk HER2-positive disease and to minimize potential for undertreatment, Dr Sparano explained.
In the women with midrange scores who were randomized, the hazard ratio of 1.08 for invasive DFS with endocrine therapy alone compared with chemotherapy plus endocrine therapy fell well within the predefined hazard ratio for noninferiority (1.322). The 9-year rate of invasive DFS was 83.3% with endocrine therapy and 84.3% with chemotherapy plus endocrine therapy.
The groups had similar rates of freedom from distant recurrence (94.5% vs 95.0%; HR, 1.10; P = .48) and distant or locoregional recurrence (92.2% vs 92.9%; HR, 1.11; P = .33), and similar OSs (93.9% vs 93.8%; HR for death, 0.99; P = .89).
In exploratory analyses, there was an interaction of age and recurrence score (P = .004) whereby women aged 50 years or younger derived some benefit from chemotherapy if they had a recurrence score of 16-20 (9% fewer invasive DFS events, including 2% fewer distant recurrences) or a recurrence score 21-25 (6% fewer invasive DFS events, mainly distant recurrences). “This is information that could drive some younger women who have a recurrence score in this range to accept chemotherapy,” Dr Sparano said.
The 9-year rate of distant recurrence averaged 5% in women with midrange scores overall. It was 3% in those with a low recurrence score given endocrine therapy alone, but it was still 13% in those with a high recurrence score despite receiving both endocrine therapy and chemotherapy. The latter finding may “indicate the need to explore potentially more effective therapies in this setting,” he proposed.
Tailoring treatment: ‘not too much and not too little’
“These are important data because this is the most common form of breast cancer in the United States and other developed countries, and the most challenging decision we make with these patients is whether or not to recommend adjuvant chemotherapy with all its side effects and potential benefits,” said ASCO expert Harold Burstein, MD, PhD, FASCO. “The data show that the majority of women who have this test performed on their tumor can be told that they don’t need chemotherapy, and that can be said with tremendous confidence and reassurance.”
The recurrence score has been used for a decade, but the trial was necessary because the score was originally developed in patients who were receiving older chemotherapy regimens and older endocrine therapies, and because there have been few data to guide decision making in the large group of patients with midrange scores, he said. “Now we can say with confidence ... that the patients got contemporary chemo regimens and still saw no benefit from chemotherapy.
“This is not so much about de-escalation ... the goal of this study was not to just use less treatment but to tailor treatment. The investigators chose the title very aptly,” said Dr Burstein, a medical oncologist at the Dana-Farber Cancer Institute and associate professor of medicine at the Harvard Medical School, Boston.
“This is extraordinary data for breast cancer doctors and women who have breast cancer. It allows you to individualize treatment based on extraordinary science, which now has tremendous prospective validation,” he said. Overall, “women with breast cancer who are getting modern therapy are doing well, and this test shows us how to tailor that management so that they get exactly the right amount of treatment – not too much and not too little.”
— Susan London
First-line immunotherapy boosts survival in NSCLC patients
Key clinical point Many patients with previously untreated NSCLC could benefit from first-line therapy with the checkpoint inhibitor pembrolizumab. Major finding In all patients with expression of PD-L1 on 1% or more of tumor cells, OS was 16.7 months with pembrolizumab, compared with 12.1 months for chemotherapy. Study details Randomized phase 3 trial of 1,274 patients with advanced or metastatic NSCLC. Funding Merck, the maker of the study drug, funded the study. Disclosures Dr Lopes disclosed institutional research funding from Merck Sharp & Dohme, EMD Serono, and AstraZeneca. Dr Heymach disclosed stock/ownership in Bio-Tree and Cardinal Spine, a consulting or advisory role for Abbvie, ARIAD, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Calithera Biosciences, Genentech, Medivation, Novartis, Oncomed, and Synta, and institutional research funding from AstraZeneca. Dr Gandhi reported having no relevant disclosures. Source Lopes G et al. ASCO 2018, abstract LBA4. https://meetinglibrary.asco.org/record/165950/abstract
Pembrolizumab as first-line treatment of advanced non–small-cell lung cancer (NSCLC) offered longer OS with better tolerability compared with chemotherapy, results of the Keynote-042 phase 3 randomized trial show.
In 1,274 patients with advanced, previously untreated NSCLC with expression of the PD-L1 on 1% or more of tumor cells, median OS after a median follow-up of 12.8 months was 16.7 months for patients treated with pembrolizumab monotherapy, compared with 12.1 months for patients treated with either paclitaxel or pemetrexed plus carboplatin, reported lead author Gilberto Lopes, MD, of the Sylvester Comprehensive Cancer Center at the University of Miami.
The survival benefit for immunotherapy was even greater for patients with higher levels of PD-L1 expression: 20 versus 12.2 months for patients with PD-L1 expression of 50% or greater, and 17.7 versus 13 months for patients with PD-L1 expression of 20% or greater, Dr Lopes noted.
For all 3 PD-L1 expression groups, the median duration of response was 20.2 months, compared with 10.8-8.3 months for patients in the chemotherapy arm.
“These are responses that are unlike anything that we have seen with chemotherapy in the past for non–small-cell lung cancer,” Dr Lopes said at a briefing before his presentation. “In addition to that, and probably more importantly, patients had fewer adverse events [with pembrolizumab]. Overall, about 60% had any treatment-related adverse event with pembrolizumab, versus 90% with chemotherapy,” he added.
‘A true milestone’
ASCO expert John Heymach, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, said at the briefing that the study was “a true milestone for the field, because now, for the first time, we can say that in non–small-cell lung cancer patients receiving first-line therapy, the vast majority can receive immunotherapy with pembrolizumab instead of chemotherapy.”
He noted that an earlier study, Keynote-024, showed that pembrolizumab significantly improved progression-free survival in patients with tumors expressing PD-L1 on at least 50% of cells compared with standard platinum-based chemotherapy (10.3 vs 6 months).
“This more than doubles that population that can start immunotherapy as a first-line treatment, assuming the [Food and Drug Administration] modifies the label in accordance with this study,” he added.
The Keynote-042 investigators enrolled 1,274 patients with locally advanced or metastatic NSCLC, and randomly assigned them to receive either a maximum of 35 cycles of pembrolizumab 200 mg every 3 weeks, or the investigators’ choice of not more than 6 cycles of either paclitaxel–carboplatin or pemetrexed–carboplatin, with optional pemetrexed maintenance for patients with nonsquamous histologies only.
The randomization was stratified by region (Asia vs non-East Asia), Eastern Cooperative Oncology Group performance status 0 or 1, squamous versus nonsquamous histology, and PD-L1 expression, or TPS (tumor proportion score) greater than 50% versus 1%-49%.
As noted before, the primary endpoint of OS in all patients with a TPS of 1% or greater was met, with respective median OS in the pembrolizumab versus chemotherapy groups of 16.7 and 12.1 months, translating into an HR favoring pembrolizumab of 0.81 (P = .0018). Respective hazard ratios for the TPS 20% or greater and TPS 50% or greater groups were 0.77 (P = .0020) and 0.69 (P = .0003).
At 12.8 months of median follow-up, 13% of patients assigned to pembrolizumab were still on the drug, and 4.3% of patients were receiving maintenance pemetrexed.
Treatment-related adverse events of any grade occurred in 399 of 636 patients assigned to pembrolizumab (62.7%), compared with 553 of 615 patients assigned to chemotherapy (89.9%). Grade 3 or greater events occurred in 17.8% and 41% of patients, respectively. There were 13 deaths related to therapy in the pembrolizumab arm (2.0%), and 14 in the chemotherapy arm (2.3%). Adverse events leading to discontinuation were similar between the groups, at 9% and 9.4%, respectively.
There were more immune-mediated adverse events in the pembrolizumab arm than in the chemotherapy arm (27.8% vs 7.2%, respectively), and of those, grade 3 or higher events occurred in 8% and 1.5% of patients. There was 1 immune-mediated death, from pneumonitis, in the immunotherapy arm; there were no deaths related to immune-mediated side effects in the chemotherapy arm.
“I really view this as a ‘double whammy’ for patients,” Dr Heymach said at the briefing. “Often advances in survival for our lung cancer patients come at the cost of significant toxicities. Here, by contrast, not only are patients living longer and having a much higher likelihood of prolonged survival in years, often instead of months, but they’re also receiving a treatment that has substantially less toxicity across virtually all measures, and this really impacts the day-to-day life of these patients.”
Leena Gandhi, MD, PhD, of the Perlmutter Cancer Center at New York University, the invited discussant at the plenary, agreed that pembrolizumab improves survival, compared with chemotherapy patients with PD-L1 expression levels greater than 1%, but noted that most of the benefit – as also seen in Keynote-024 – was in those patients whose tumors had high levels of PD-L1 expression.
She emphasized that although PD-L1 is an imperfect biomarker, it should still be used to help select patients for therapy and it may be complementary with tumor mutational burden for more precise treatment selection.
“What we know, and what this study adds to, is that PD-L1 really does define a patient population that could receive benefit from pembrolizumab over chemotherapy. Patients with low or no PD-L1 expression likely should get some type of combination therapy,” she said. “This study extends what we’ve seen from other recent studies, which is that chemotherapy alone is no longer a first-line standard of care in non–small-cell lung cancer.”
— Neil Osterweil
Maintenance chemo improves survival in youth with rhabdomyosarcoma
Key clinical point 6 months of maintenance chemotherapy improves survival in youth with high-risk rhabdomyosarcoma. Major finding Patients given maintenance low-dose vinorelbine and cyclophosphamide had better 5-year OS compared with those not receiving any additional treatment (86.5% vs 73.7%; HR, 0.52). Study details A phase 3 randomized controlled trial in 371 patients aged 0-21 years with high-risk rhabdomyosarcoma who had had a complete response to standard intensive therapy. Funding The study received funding from Fondazione Città della Speranza, Italy. Disclosures Dr Bisogno disclosed that he has a consulting or advisory role with Clinigen Group, and receives travel, accommodations, and/or expenses from Jazz Pharmaceuticals. Source Bisogno et al. ASCO 2018, Abstract LBA2. https://meetinglibrary.asco.org/record/161695/abstract
Six months of maintenance chemotherapy prolongs OS in youth with high-risk rhabdomyosarcoma, finds a phase 3 randomized controlled trial of the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG).
Rhabdomyosarcoma is a rare but highly aggressive tumor, lead study author Gianni Bisogno, MD, PhD, a professor at the University Hospital of Padova, Italy, and chair of the EpSSG, noted in a press briefing at the meeting, where the findings were reported. In pediatric patients who achieve complete response to standard therapy, “we know that after 1 or 2 years, one-third of these children relapse, and most of them die,” he said.
The EpSSG trial, which took about 10 years to conduct, enrolled 371 patients aged 0-21 years with high-risk rhabdomyosarcoma who had had a complete response to standard intensive therapy. They were randomized evenly to stop treatment or to receive 6 months of maintenance treatment consisting of low-dose vinorelbine and cyclophosphamide.
Results reported in the meeting’s plenary session showed that giving maintenance chemotherapy improved the 5-year OS rate by an absolute 12.8%, which translated to a near halving of the risk of death. And the maintenance regimen used was generally well tolerated.
“At the end of this long, not-easy study, we concluded that maintenance chemotherapy is an effective and well tolerated treatment for children with high-risk rhabdomyosarcoma,” Dr Bisogno said.
There are three possibilities for its efficacy, he speculated. “It may be the duration, the type of drugs used, or the metronomic approach. Maybe altogether, these three different actions have a benefit to increase survival.
“Our group has decided this is the new standard treatment for patients. At least in Europe, we give standard intensive therapy and then we continue with 6 more months of low-dose chemotherapy,” Dr Bisogno concluded. “We think that this approach – a new way of using old drugs – can be of interest also for other pediatric tumors.”
The trial is noteworthy in that it shows “how to successfully conduct large and important trials in rare diseases,” said ASCO expert Warren Chow, MD.
The standard therapy for rhabdomyosarcomas is somewhat different in the United States, typically a regimen containing vincristine, actinomycin D, cyclophosphamide, and (more recently) irinotecan, he noted. “We have not been traditionally using maintenance chemo for any of the pediatric sarcomas, so this is a paradigm shift. These results will need to be tested with US-based protocols before becoming standard of care in the United States. Also, we will need to determine if these results are applicable to patients older than 21 years of age who are considered high risk based solely on their age.
“Even with these caveats, this is the first significant treatment advance in this rare cancer in more than 30 years,” concluded Dr Chow, a medical oncologist and clinical professor at City of Hope, Duarte, Calif. “No doubt, this trial was a home run.”
Study details
Patients enrolled in the EpSSG trial had had a complete response to the standard intensive therapy used in Europe: high-dose chemotherapy (ifosfamide, vincristine, and actinomycin D, with or without doxorubicin), radiation therapy, and surgery.
The maintenance chemotherapy consisted of a combination of low-dose intravenous vinorelbine given weekly and oral cyclophosphamide given daily. The 6-month duration was somewhat arbitrary, according to Dr Bisogno. “We had to start somewhere. So when we started, we decided to use 6 months because there was some evidence in the past for regimens that long. In our next European trial, we are going to test different kinds and durations of maintenance because this is very important.”
The maintenance regimen was well tolerated compared with the regimen given during standard intensive therapy, with, for example, lower rates of grade 3 and 4 anemia (8.9% vs 48.9%), neutropenia (80.6% vs 91.6%), and thrombocytopenia (0.6% vs 26.0%), which translated to less of a need for transfusions, and a lower rate of grade 3 or 4 infection (29.4% vs 56.4%), Dr Bisogno reported. There were no cases of grade 3 or 4 cardiac, hepatobiliary/pancreatic, or renal toxicity.
Relative to peers who stopped treatment after standard intensive therapy, patients who received maintenance treatment tended to have better DFS (77.6% vs 69.8%; HR, 0.68; P = .0613) and had significantly better OS (86.5% vs 73.7%; HR, 0.52; P = .0111).
— Susan London
Among the groundbreaking findings presented at this year’s annual meeting of the American Society of Clinical Oncology were those showing that most women with HR-positive, HER2-negative, early-stage breast cancer who have an intermediate recurrence score can safely skip adjuvant chemotherapy, and that upfront pembrolizumab for patients with NSCLC expressing PD-L1 on at least 1% of tumor cells can not only significantly improve overall survival, but do so with less toxicity than standard chemotherapy.
TAILORx marks major advance for precision medicine in breast cancer
Key clinical point The majority of women with HR-positive, HER2-negative, node-negative early-stage breast cancer who have an intermediate recurrence score can safely skip adjuvant chemotherapy. Major finding In women with an Oncotype DX Recurrence Score in the midrange (11-25), invasive DFS with endocrine therapy alone was not inferior to that with chemotherapy plus endocrine therapy (HR, 1.08; P = .26). Study details A phase 3 trial in 10,273 women with HR-positive, HER2-negative, node-negative, early-stage breast cancer, with a noninferiority randomized component in the 6,711 women with a midrange recurrence score (TAILORx trial). Funding This study received funding primarily from the National Cancer Institute, National Institutes of Health. Additional support was provided by the Breast Cancer Research Foundation, Komen Foundation, and US Postal Service Breast Cancer Stamp. Disclosures Dr Sparano disclosed that he has a consulting or advisory role with Genentech/Roche, Novartis, AstraZeneca, Celgene, Lilly, Celldex, Pfizer, Prescient Therapeutics, Juno Therapeutics, and Merrimack; has stock or other ownership interests with MetaStat; and receives research funding (institutional) from Prescient Therapeutics, Deciphera, Genentech/Roche, Merck, Novartis, and Merrimack. Source Sparano et al. ASCO 2018 Abstract LBA1: https://meetinglibrary.asco.org/record/161490/abstract
Use of the 21-tumor gene expression assay (Oncotype DX Recurrence Score) allows nearly 70% of women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer to safely forgo adjuvant chemotherapy, sparing them adverse effects and preventing overtreatment, TAILORx trial results show.
The findings, which were reported in the plenary session at the meeting and simultaneously published in the New England Journal of Medicine (N Engl J Med. 2018; 379:111-121; [behind paywall]), mark a major advance in precision medicine.
“The rationale for the TAILORx precision medicine trial is that we are really trying to ‘thread the needle,’ “ lead study author Joseph A Sparano, MD, commented in a press briefing. Oncologists typically recommend adjuvant chemotherapy for the half of all breast cancers that are hormone receptor positive, HER2 negative, and node negative, even though its absolute benefit in reducing recurrences in this population is small. “This results in most patients being overtreated because endocrine therapy alone is adequate. But some are undertreated: They do not receive chemotherapy although they could have benefited from it,” he noted.
The recurrence score is known to be prognostic and predictive of benefit from adding chemotherapy to endocrine therapy, Dr Sparano said. “But there was a major gap: There was uncertain benefit for patients who had a midrange score, which is about two-thirds of all patients who are treated,” said Dr Sparano, the associate director for clinical research at Albert Einstein Cancer Center and Montefiore Health System in New York, and vice-chair of the ECOG-ACRIN Cancer Research Group.
The phase 3 TAILORx trial registered 10,273 women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer, making it the largest adjuvant breast cancer trial to date. Analyses focused on the 6,711 evaluable women with a midrange recurrence score (defined as 11 through 25 in the trial), who were randomized to receive endocrine therapy alone or adjuvant chemotherapy plus endocrine therapy, with a noninferiority design. Of note is that contemporary drugs and regimens were used.
Results at a median follow-up of 7.5 years showed that the trial met its primary endpoint: the risk of invasive disease-free survival (DFS) events (invasive disease recurrence, second primary cancer, or death) was not inferior for women given endocrine therapy alone compared with counterparts given chemotherapy plus endocrine therapy (hazard ratio [HR], 1.08; P = .26), Dr Sparano reported.
The groups were also on par, with absolute differences of no more than 1% between rates, with respect to a variety of other efficacy outcomes: freedom from distant recurrence and any recurrence, and overall survival (OS).
Findings were similar across most subgroups. But analyses suggested that women aged 50 years and younger and who had a recurrence score of 16-25 fared better when they received chemotherapy. “Though exploratory from a statistical perspective, this is a highly clinically relevant observation,” Dr Sparano said. “It suggests ... that chemotherapy should be spared with caution in this subgroup, after a careful discussion of potential benefits and risks in a shared decision process.”
In other findings, analyses of the trial’s nonrandomized groups confirmed excellent outcomes in women with a low recurrence score (0-10) who were given endocrine therapy alone, and at the other end of the spectrum, there was need for a more aggressive approach, including chemotherapy, in women with a high recurrence score (26-100).
Ultimately, application of the recurrence score allowed 69% of the trial population to skip chemotherapy: all of the women with a score of 0-10 (16% of the trial population), those older than 50 years with a score of 11-25 (45%), and those aged 50 years or younger with a score of 11-15 (8%).
An ongoing companion phase 3 trial, RxPONDER, is assessing the benefit of applying the recurrence score in women who are similar but ihave node-positive disease.
Study details
All of the women with hormone receptor-positive, HER2-negative, node-negative, early-stage breast cancer enrolled in TAILORx met National Comprehensive Cancer Network guidelines for receiving adjuvant chemotherapy. About 69% had an intermediate recurrence score (11-25) and were randomized. All of the 17% with a low recurrence score (0-10) were given only endocrine therapy, and all of the 14% with a high recurrence score (26-100) were given both adjuvant chemotherapy and endocrine therapy.
Of note, the recurrence scores used to define midrange were adjusted downward from those conventionally used to account for exclusion of patients with higher-risk HER2-positive disease and to minimize potential for undertreatment, Dr Sparano explained.
In the women with midrange scores who were randomized, the hazard ratio of 1.08 for invasive DFS with endocrine therapy alone compared with chemotherapy plus endocrine therapy fell well within the predefined hazard ratio for noninferiority (1.322). The 9-year rate of invasive DFS was 83.3% with endocrine therapy and 84.3% with chemotherapy plus endocrine therapy.
The groups had similar rates of freedom from distant recurrence (94.5% vs 95.0%; HR, 1.10; P = .48) and distant or locoregional recurrence (92.2% vs 92.9%; HR, 1.11; P = .33), and similar OSs (93.9% vs 93.8%; HR for death, 0.99; P = .89).
In exploratory analyses, there was an interaction of age and recurrence score (P = .004) whereby women aged 50 years or younger derived some benefit from chemotherapy if they had a recurrence score of 16-20 (9% fewer invasive DFS events, including 2% fewer distant recurrences) or a recurrence score 21-25 (6% fewer invasive DFS events, mainly distant recurrences). “This is information that could drive some younger women who have a recurrence score in this range to accept chemotherapy,” Dr Sparano said.
The 9-year rate of distant recurrence averaged 5% in women with midrange scores overall. It was 3% in those with a low recurrence score given endocrine therapy alone, but it was still 13% in those with a high recurrence score despite receiving both endocrine therapy and chemotherapy. The latter finding may “indicate the need to explore potentially more effective therapies in this setting,” he proposed.
Tailoring treatment: ‘not too much and not too little’
“These are important data because this is the most common form of breast cancer in the United States and other developed countries, and the most challenging decision we make with these patients is whether or not to recommend adjuvant chemotherapy with all its side effects and potential benefits,” said ASCO expert Harold Burstein, MD, PhD, FASCO. “The data show that the majority of women who have this test performed on their tumor can be told that they don’t need chemotherapy, and that can be said with tremendous confidence and reassurance.”
The recurrence score has been used for a decade, but the trial was necessary because the score was originally developed in patients who were receiving older chemotherapy regimens and older endocrine therapies, and because there have been few data to guide decision making in the large group of patients with midrange scores, he said. “Now we can say with confidence ... that the patients got contemporary chemo regimens and still saw no benefit from chemotherapy.
“This is not so much about de-escalation ... the goal of this study was not to just use less treatment but to tailor treatment. The investigators chose the title very aptly,” said Dr Burstein, a medical oncologist at the Dana-Farber Cancer Institute and associate professor of medicine at the Harvard Medical School, Boston.
“This is extraordinary data for breast cancer doctors and women who have breast cancer. It allows you to individualize treatment based on extraordinary science, which now has tremendous prospective validation,” he said. Overall, “women with breast cancer who are getting modern therapy are doing well, and this test shows us how to tailor that management so that they get exactly the right amount of treatment – not too much and not too little.”
— Susan London
First-line immunotherapy boosts survival in NSCLC patients
Key clinical point Many patients with previously untreated NSCLC could benefit from first-line therapy with the checkpoint inhibitor pembrolizumab. Major finding In all patients with expression of PD-L1 on 1% or more of tumor cells, OS was 16.7 months with pembrolizumab, compared with 12.1 months for chemotherapy. Study details Randomized phase 3 trial of 1,274 patients with advanced or metastatic NSCLC. Funding Merck, the maker of the study drug, funded the study. Disclosures Dr Lopes disclosed institutional research funding from Merck Sharp & Dohme, EMD Serono, and AstraZeneca. Dr Heymach disclosed stock/ownership in Bio-Tree and Cardinal Spine, a consulting or advisory role for Abbvie, ARIAD, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Calithera Biosciences, Genentech, Medivation, Novartis, Oncomed, and Synta, and institutional research funding from AstraZeneca. Dr Gandhi reported having no relevant disclosures. Source Lopes G et al. ASCO 2018, abstract LBA4. https://meetinglibrary.asco.org/record/165950/abstract
Pembrolizumab as first-line treatment of advanced non–small-cell lung cancer (NSCLC) offered longer OS with better tolerability compared with chemotherapy, results of the Keynote-042 phase 3 randomized trial show.
In 1,274 patients with advanced, previously untreated NSCLC with expression of the PD-L1 on 1% or more of tumor cells, median OS after a median follow-up of 12.8 months was 16.7 months for patients treated with pembrolizumab monotherapy, compared with 12.1 months for patients treated with either paclitaxel or pemetrexed plus carboplatin, reported lead author Gilberto Lopes, MD, of the Sylvester Comprehensive Cancer Center at the University of Miami.
The survival benefit for immunotherapy was even greater for patients with higher levels of PD-L1 expression: 20 versus 12.2 months for patients with PD-L1 expression of 50% or greater, and 17.7 versus 13 months for patients with PD-L1 expression of 20% or greater, Dr Lopes noted.
For all 3 PD-L1 expression groups, the median duration of response was 20.2 months, compared with 10.8-8.3 months for patients in the chemotherapy arm.
“These are responses that are unlike anything that we have seen with chemotherapy in the past for non–small-cell lung cancer,” Dr Lopes said at a briefing before his presentation. “In addition to that, and probably more importantly, patients had fewer adverse events [with pembrolizumab]. Overall, about 60% had any treatment-related adverse event with pembrolizumab, versus 90% with chemotherapy,” he added.
‘A true milestone’
ASCO expert John Heymach, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston, said at the briefing that the study was “a true milestone for the field, because now, for the first time, we can say that in non–small-cell lung cancer patients receiving first-line therapy, the vast majority can receive immunotherapy with pembrolizumab instead of chemotherapy.”
He noted that an earlier study, Keynote-024, showed that pembrolizumab significantly improved progression-free survival in patients with tumors expressing PD-L1 on at least 50% of cells compared with standard platinum-based chemotherapy (10.3 vs 6 months).
“This more than doubles that population that can start immunotherapy as a first-line treatment, assuming the [Food and Drug Administration] modifies the label in accordance with this study,” he added.
The Keynote-042 investigators enrolled 1,274 patients with locally advanced or metastatic NSCLC, and randomly assigned them to receive either a maximum of 35 cycles of pembrolizumab 200 mg every 3 weeks, or the investigators’ choice of not more than 6 cycles of either paclitaxel–carboplatin or pemetrexed–carboplatin, with optional pemetrexed maintenance for patients with nonsquamous histologies only.
The randomization was stratified by region (Asia vs non-East Asia), Eastern Cooperative Oncology Group performance status 0 or 1, squamous versus nonsquamous histology, and PD-L1 expression, or TPS (tumor proportion score) greater than 50% versus 1%-49%.
As noted before, the primary endpoint of OS in all patients with a TPS of 1% or greater was met, with respective median OS in the pembrolizumab versus chemotherapy groups of 16.7 and 12.1 months, translating into an HR favoring pembrolizumab of 0.81 (P = .0018). Respective hazard ratios for the TPS 20% or greater and TPS 50% or greater groups were 0.77 (P = .0020) and 0.69 (P = .0003).
At 12.8 months of median follow-up, 13% of patients assigned to pembrolizumab were still on the drug, and 4.3% of patients were receiving maintenance pemetrexed.
Treatment-related adverse events of any grade occurred in 399 of 636 patients assigned to pembrolizumab (62.7%), compared with 553 of 615 patients assigned to chemotherapy (89.9%). Grade 3 or greater events occurred in 17.8% and 41% of patients, respectively. There were 13 deaths related to therapy in the pembrolizumab arm (2.0%), and 14 in the chemotherapy arm (2.3%). Adverse events leading to discontinuation were similar between the groups, at 9% and 9.4%, respectively.
There were more immune-mediated adverse events in the pembrolizumab arm than in the chemotherapy arm (27.8% vs 7.2%, respectively), and of those, grade 3 or higher events occurred in 8% and 1.5% of patients. There was 1 immune-mediated death, from pneumonitis, in the immunotherapy arm; there were no deaths related to immune-mediated side effects in the chemotherapy arm.
“I really view this as a ‘double whammy’ for patients,” Dr Heymach said at the briefing. “Often advances in survival for our lung cancer patients come at the cost of significant toxicities. Here, by contrast, not only are patients living longer and having a much higher likelihood of prolonged survival in years, often instead of months, but they’re also receiving a treatment that has substantially less toxicity across virtually all measures, and this really impacts the day-to-day life of these patients.”
Leena Gandhi, MD, PhD, of the Perlmutter Cancer Center at New York University, the invited discussant at the plenary, agreed that pembrolizumab improves survival, compared with chemotherapy patients with PD-L1 expression levels greater than 1%, but noted that most of the benefit – as also seen in Keynote-024 – was in those patients whose tumors had high levels of PD-L1 expression.
She emphasized that although PD-L1 is an imperfect biomarker, it should still be used to help select patients for therapy and it may be complementary with tumor mutational burden for more precise treatment selection.
“What we know, and what this study adds to, is that PD-L1 really does define a patient population that could receive benefit from pembrolizumab over chemotherapy. Patients with low or no PD-L1 expression likely should get some type of combination therapy,” she said. “This study extends what we’ve seen from other recent studies, which is that chemotherapy alone is no longer a first-line standard of care in non–small-cell lung cancer.”
— Neil Osterweil
Maintenance chemo improves survival in youth with rhabdomyosarcoma
Key clinical point 6 months of maintenance chemotherapy improves survival in youth with high-risk rhabdomyosarcoma. Major finding Patients given maintenance low-dose vinorelbine and cyclophosphamide had better 5-year OS compared with those not receiving any additional treatment (86.5% vs 73.7%; HR, 0.52). Study details A phase 3 randomized controlled trial in 371 patients aged 0-21 years with high-risk rhabdomyosarcoma who had had a complete response to standard intensive therapy. Funding The study received funding from Fondazione Città della Speranza, Italy. Disclosures Dr Bisogno disclosed that he has a consulting or advisory role with Clinigen Group, and receives travel, accommodations, and/or expenses from Jazz Pharmaceuticals. Source Bisogno et al. ASCO 2018, Abstract LBA2. https://meetinglibrary.asco.org/record/161695/abstract
Six months of maintenance chemotherapy prolongs OS in youth with high-risk rhabdomyosarcoma, finds a phase 3 randomized controlled trial of the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG).
Rhabdomyosarcoma is a rare but highly aggressive tumor, lead study author Gianni Bisogno, MD, PhD, a professor at the University Hospital of Padova, Italy, and chair of the EpSSG, noted in a press briefing at the meeting, where the findings were reported. In pediatric patients who achieve complete response to standard therapy, “we know that after 1 or 2 years, one-third of these children relapse, and most of them die,” he said.
The EpSSG trial, which took about 10 years to conduct, enrolled 371 patients aged 0-21 years with high-risk rhabdomyosarcoma who had had a complete response to standard intensive therapy. They were randomized evenly to stop treatment or to receive 6 months of maintenance treatment consisting of low-dose vinorelbine and cyclophosphamide.
Results reported in the meeting’s plenary session showed that giving maintenance chemotherapy improved the 5-year OS rate by an absolute 12.8%, which translated to a near halving of the risk of death. And the maintenance regimen used was generally well tolerated.
“At the end of this long, not-easy study, we concluded that maintenance chemotherapy is an effective and well tolerated treatment for children with high-risk rhabdomyosarcoma,” Dr Bisogno said.
There are three possibilities for its efficacy, he speculated. “It may be the duration, the type of drugs used, or the metronomic approach. Maybe altogether, these three different actions have a benefit to increase survival.
“Our group has decided this is the new standard treatment for patients. At least in Europe, we give standard intensive therapy and then we continue with 6 more months of low-dose chemotherapy,” Dr Bisogno concluded. “We think that this approach – a new way of using old drugs – can be of interest also for other pediatric tumors.”
The trial is noteworthy in that it shows “how to successfully conduct large and important trials in rare diseases,” said ASCO expert Warren Chow, MD.
The standard therapy for rhabdomyosarcomas is somewhat different in the United States, typically a regimen containing vincristine, actinomycin D, cyclophosphamide, and (more recently) irinotecan, he noted. “We have not been traditionally using maintenance chemo for any of the pediatric sarcomas, so this is a paradigm shift. These results will need to be tested with US-based protocols before becoming standard of care in the United States. Also, we will need to determine if these results are applicable to patients older than 21 years of age who are considered high risk based solely on their age.
“Even with these caveats, this is the first significant treatment advance in this rare cancer in more than 30 years,” concluded Dr Chow, a medical oncologist and clinical professor at City of Hope, Duarte, Calif. “No doubt, this trial was a home run.”
Study details
Patients enrolled in the EpSSG trial had had a complete response to the standard intensive therapy used in Europe: high-dose chemotherapy (ifosfamide, vincristine, and actinomycin D, with or without doxorubicin), radiation therapy, and surgery.
The maintenance chemotherapy consisted of a combination of low-dose intravenous vinorelbine given weekly and oral cyclophosphamide given daily. The 6-month duration was somewhat arbitrary, according to Dr Bisogno. “We had to start somewhere. So when we started, we decided to use 6 months because there was some evidence in the past for regimens that long. In our next European trial, we are going to test different kinds and durations of maintenance because this is very important.”
The maintenance regimen was well tolerated compared with the regimen given during standard intensive therapy, with, for example, lower rates of grade 3 and 4 anemia (8.9% vs 48.9%), neutropenia (80.6% vs 91.6%), and thrombocytopenia (0.6% vs 26.0%), which translated to less of a need for transfusions, and a lower rate of grade 3 or 4 infection (29.4% vs 56.4%), Dr Bisogno reported. There were no cases of grade 3 or 4 cardiac, hepatobiliary/pancreatic, or renal toxicity.
Relative to peers who stopped treatment after standard intensive therapy, patients who received maintenance treatment tended to have better DFS (77.6% vs 69.8%; HR, 0.68; P = .0613) and had significantly better OS (86.5% vs 73.7%; HR, 0.52; P = .0111).
— Susan London
Immunotherapy-related adverse effects: how to identify and treat them in the emergency department
DR HENRY I am pleased to be talking with Dr Maura Sammon, an emergency department (ED) physician, about identifying and treating immunotherapy-related side effects in the ED. This is a hot topic in oncology, and I was very interested in having an ED physician talk about what happens when treating oncologists send their patients to the ED, because a physician may think it is chemotherapy when it is immunotherapy. Let’s start with the example of an oncology patient going to the ED with some symptoms, and the ED physician asks the patient what they’re being treated with. The patient may or may not say the right thing – that is, inform you whether they are being treated with chemotherapy or immunotherapy. How do you morph over into knowing that they are not getting chemotherapy?
DR SAMMON Yes, that’s a big problem in the ED. Patients come to the ED and say they’re being treated for cancer. They say they’re on chemotherapy, when they’re actually on immunotherapy, and it can really send the treatment team down the wrong path. I have a metaphor to explain this. They say that Great Britain and the United States are two nations separated by a common language. For example, when a British person talks about football, they mean something very different than when an American talks about football. If someone in Great Britain asks you to come play football, you might show up with shoulder pads and a helmet rather than shin guards, and you’re left without having the right tools to participate in the game.
How this sometimes plays out with immunotherapy, unfortunately, is that a patient will present to the ED and say they’re having a cough and that they’re on chemotherapy for melanoma. Usually, this patient would be worked up for being in a potentially immunosuppressed state. You might get a white blood cell count. You might get a chest X-ray. You might see what looks to you like a new infiltrate on this chest X-ray and then start going down the path of treating someone whom you think is immunosuppressed with pneumonia and giving them antibiotics rather than what could be life-saving steroids, as would be the case if the patient were on immunotherapy.
It’s a real problem, because you have one word that patients may use meaning two very different things. It can get you into trouble if you are treating someone for potentially infectious causes rather than immunotherapy-related adverse reactions, which are much more similar to graft-versus-host disease than in the case of traditional chemotherapy.
DR HENRY That’s a very good point. I think, as we in oncology use these immunotherapies/checkpoint inhibitors more often, you will see them more often in the ED. Let’s get right into that. You’ve identified this patient as not getting a traditional chemotherapy – hopefully, all our records on these patients are available. You’ve decided to follow onto what might be a side effect of the immunotherapy, so I’m going to name the side effects that always occur to me: lung, gastrointestinal (GI) – which could be loose bowels or liver function – rash, endocrine problems. Let’s start with lung symptoms. You see the patient is short of breath and you identified immunotherapy. What’s your next step?
DR SAMMON That’s a great example, because the problem is that you see these patients with cough or shortness of breath and pulmonary complications, and pulmonary complications of immunotherapy, while rare, are potentially life threatening if they’re not identified quickly.
You can start with a chest X-ray on these patients knowing, however, that for a good percentage of them you won’t see findings on their chest X-ray (Figure 1, from Sammon M, Tobin T. Identification and management of immune-related adverse events in the emergency setting. Presented at: Advances in Cancer Immunotherapy – Society for Immunotherapy of Cancer (SITC); August 4, 2017; Philadelphia, PA). You need to proceed to computed tomography (CT), because the issue is that you can have protean findings on the CT related to immunotherapy treatment/adverse reactions. You need to have a very high index of suspicion regardless of what abnormal findings you’re seeing on this CT and erring toward withholding the drugs, starting treatment, and being more aggressive with this type of finding.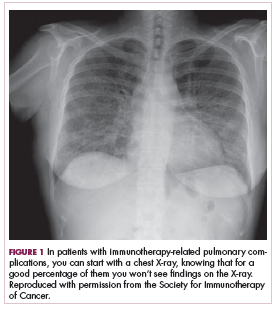
DR HENRY I’ve heard you talk at a conference about a patient with metastatic lung cancer, or some other tumor that may have existing disease in lung. The patient is aware of that, and the chart reflects that. Then you have this difficulty where the CT scan shows a pneumonitis, and it may not be tumor progression at all – it may be the drug. How do you work through that? Of course, your additional problem is you don’t have a whole lot of time. You must decide to whether you’re going to keep them in the ED, admit them, or send them home.
DR SAMMON Right. One of the first things is to get the oncologist involved at an early stage in treating this. We are all a team. We are all working together, and it’s very important to have that communication occur very early. I’m going to err on admitting those folks who have had symptoms and who have had findings on a chest CT, because they can progress. They can get much worse. I’m going to be getting the oncologist involved very early. We’re going to have the conversation about whether we should be starting steroids on them in the ED, getting them upstairs, and being aggressive in treating this.
DR HENRY So, time and therapy are very important.
DR SAMMON Yes.
DR HENRY You’ll get that steroid started as an antidote right away.
DR SAMMON Absolutely. For grade 2, you’re going to use methylprednisolone, 1 mg /kg daily. For anything higher than that – grade 3 or grade 4 – we’re going to start higher. We’re going to start at 2-4 mg/kg a day and get the patient upstairs and taper them slowly.
DR HENRY That’s worth empathizing. I, sadly, had a patient who ultimately did well, but had severe grade 4 pneumonitis. T
DR SAMMON Absolutely, but it’s very important to work together as a team to make these patients have good outcomes.
DR HENRY Agree. Let’s change over to the GI symptoms. The patient comes in and misidentifies him- or herself as having chemotherapy and diarrhea. We are used to causing nausea, vomiting, and diarrhea with some of our therapies. You realize, in talking to the oncologist, the patient is taking a checkpoint inhibitor. How would you approach the patient with diarrhea?
DR SAMMON First, I would talk to the patient. I would try to establish the baseline number of stools per day, because it’s not defined as a definite number of stools per day, it’s the number of stools above their baseline per day. If they’re having fewer than 4 stools above their baseline per day, I would send off some tests. We can send off a C diff (Clostridium difficile) test, we can send off a stool culture – all the parasites. Make sure the oncologist is going to be able to get these results and get them followed up, because these are results I’m not going to get back myself in the ED.
Then I’m going to talk to the patient about symptomatic treatment. I’m going to talk to them about oral hydration, a bland diet. Avoid using loperamide or any other antidiarrheal medicines, because that could decrease the frequency of stools but mask more severe symptoms that they may be having.
If I have a patient who is having more than 4 stools above their daily baseline and it’s been happening 4 to 6 stools a day for more than a week, I’m going to be sending those studies off, and I’m going to be having a conversation with the treating oncologist to find out if they want me to start the patient on steroids immediately, or if they want to wait for the test results to come back and have the steroids started as an outpatient.
These moderate patients can maybe wait a day until these test results come back. Those who are having more than 7 stools above baseline per day, peritoneal signs, ileus, or fever, are the patients you should worry about. You need to admit them for IV hydration. You need to do the stools, so you might need to keep them in the hospital until you find the results of the stool studies. You need to rule out perforation.
You may be starting steroids on these patients sooner rather than later. They’re going to be getting systemic corticosteroids at about 1-2 mg/kg of prednisone equivalent, assuming there is no perforation and their stool studies are negative. If they are unstable, though, they are really going to need high-dose corticosteroids. They are going to need methylprednisolone, 125 mg IV, to evaluate for their responsiveness. These folks really need to be treated as inpatients, and they need to have their oncologists involved early on with their treatment.
DR HENRY Yes. I couldn’t agree with you more. When I talk to the diarrhea side effect patients that we see, I tend to think it’s a curse. It’s volume. It’s calories. It’s electrolytes. The number of stools you’re mentioning, it is almost certainly going to need admission to rule out other causes. Then, if it’s the checkpoint inhibitor, the steroid antidotes.
Let’s move on to the rash. This is another organ system that can be affected by immunotherapy. What is your approach when you see a generalized body rash in a patient on one of these drugs who is sent to the ED?
DR SAMMON I am obviously going to be ruling out other causes first, but generally you’re going to see a maculopapular rash. It may be itchy. It may be burning. Patients will often describe it as just sort of having a tight sensation. I’m going to be looking at them a little bit like I look at a burn patient. What is their total body surface area that’s involved? If they’ve got less than 30% of their total body surface area involved, that’s considered a mild reaction.
For those folks, I’m not going to use systemic steroids, but I can give them some topical steroids, and I can give them some Benadryl, some diphenhydramine, and really treat them symptomatically as well as ensuring that they have early follow-up to make sure this isn’t progressing. Once we get between 30% and 50% body area, we’re talking about a moderate toxicity. If these patients are not improving rapidly with just withholding the drug, they need systemic corticosteroids.
We usually treat them at about 0.5-1 mg/kg body weight a day of prednisone equivalent. Just as with burn patients, these patients’ symptoms can become very severe. You can see signs of blistering, dermal ulceration, necrotic, bolus, hemorrhagic lesions (Figure 2, A-D).1 These folks can have very difficult-to-manage fluid balances, and they’re at very high risk for skin infections as well. They need to be treated as inpatients. If possible, you might want to consider sending these patients to a burn unit. They need systemic corticosteroids, 1-2 mg/kg per day, and they need careful monitoring for signs of dehydration, electrolytic abnormalities, and/or skin infections. They need excellent wound care.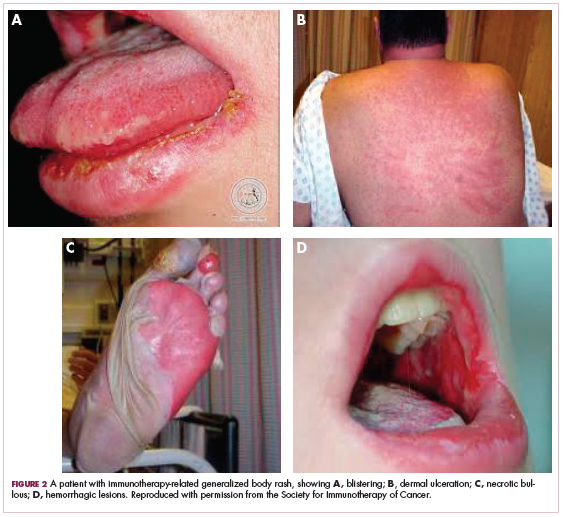
DR HENRY That’s very well put and always difficult, because there are so many causes of rash. That takes me to an area that has always been difficult for me, which is therapy-related endocrine problems. It’s interesting to note that these drugs can cause endocrine problems. I’ve heard you speak about the pituitary affecting vision, as well as thyroid or adrenal issues. Let’s start with how you’d approach vision difficulty in a patient on these drugs.
DR SAMMON The endocrinopathies that you can get with these checkpoint inhibitors really have a myriad of symptoms. Your patient may present saying that they’re feeling tired, that they’re feeling weak, or they may have a headache. If your patient is having actual pituitary enlargement, they can present with headaches, visual field defects, or cranial nerve defects. The reason for those symptoms is that the pituitary sits in the cavernous sinus, and you have various cranial nerves passing through that area as well as the optic chiasm just above the pituitary gland (Figure 3).1 Your patient may present with a bitemporal hemianopia. Or with diplopia. You are going to want to very quickly get either a CT scan or an MRI to find out if that is what’s going on (Figure 4).1 These folks need to be treated aggressively as well.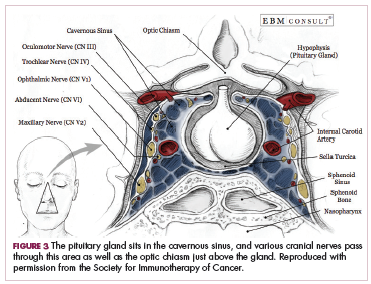
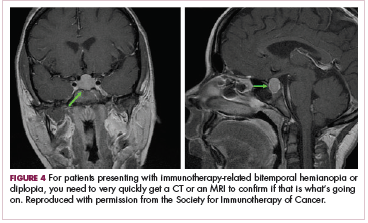
DR HENRY You’ll get your CT scan or your MRI and rule out an enlargement or a change to the visual field. I haven’t seen this yet, but certainly exciting when you see it to treat it. Would you get the radialis brevis involved, steroids involved? How would you manage that?
DR SAMMON It’s interesting, because you do want to use corticosteroids. One of the questions here is, which corticosteroid do you want to use? If you’re talking about someone who may have adrenal insufficiency, you may want to be able to do a stimulation test. In these patients, you may want to choose using dexamethasone, because you can still do the corticotropin stimulation test. However, if your patient is in frank shock because of what you think is an adrenal crisis, you’re going to want to use hydrocortisone. If a patient is truly hypotensive and unstable, the testing is at that point less important than the treatment.
DR HENRY Very interesting. We have covered what I would consider the major aspects of these fascinating drugs. We haven’t covered all of what they do when they work well, which hopefully we’re seeing more and more often, but we have covered very well what can happen when things go wrong in side effects. Anything else that you would like to add from the ED perspective or other side effects worth mentioning?
DR SAMMON The thing that I would most like to share with the oncology office is the importance of communicating with your patients that, when they’re on these drugs, they need to tell emergency physicians that they’re on immunotherapy, not chemotherapy. It might be helpful to give these patients a card stating that they’re on immunotherapy, not chemotherapy, and outlining some of the side effects that ED physicians should be looking out for in these patients.
DR HENRY That’s a great point. I’ve seen that some of the manufacturers have little cheat cards that the patient can carry naming the drug and the side effects, because not all ED doctors are aware of the side effects of these drugs.
DR SAMMON Absolutely. We love those cards.
DR HENRY Yes. I’ve also given some to the ED doctors at Pennsylvania Hospital, and they love it. I think we’ve covered everything in quite a bit of detail. Thank you, Dr Sammon, for sharing this information from the frontlines of the ED.
DR HENRY I am pleased to be talking with Dr Maura Sammon, an emergency department (ED) physician, about identifying and treating immunotherapy-related side effects in the ED. This is a hot topic in oncology, and I was very interested in having an ED physician talk about what happens when treating oncologists send their patients to the ED, because a physician may think it is chemotherapy when it is immunotherapy. Let’s start with the example of an oncology patient going to the ED with some symptoms, and the ED physician asks the patient what they’re being treated with. The patient may or may not say the right thing – that is, inform you whether they are being treated with chemotherapy or immunotherapy. How do you morph over into knowing that they are not getting chemotherapy?
DR SAMMON Yes, that’s a big problem in the ED. Patients come to the ED and say they’re being treated for cancer. They say they’re on chemotherapy, when they’re actually on immunotherapy, and it can really send the treatment team down the wrong path. I have a metaphor to explain this. They say that Great Britain and the United States are two nations separated by a common language. For example, when a British person talks about football, they mean something very different than when an American talks about football. If someone in Great Britain asks you to come play football, you might show up with shoulder pads and a helmet rather than shin guards, and you’re left without having the right tools to participate in the game.
How this sometimes plays out with immunotherapy, unfortunately, is that a patient will present to the ED and say they’re having a cough and that they’re on chemotherapy for melanoma. Usually, this patient would be worked up for being in a potentially immunosuppressed state. You might get a white blood cell count. You might get a chest X-ray. You might see what looks to you like a new infiltrate on this chest X-ray and then start going down the path of treating someone whom you think is immunosuppressed with pneumonia and giving them antibiotics rather than what could be life-saving steroids, as would be the case if the patient were on immunotherapy.
It’s a real problem, because you have one word that patients may use meaning two very different things. It can get you into trouble if you are treating someone for potentially infectious causes rather than immunotherapy-related adverse reactions, which are much more similar to graft-versus-host disease than in the case of traditional chemotherapy.
DR HENRY That’s a very good point. I think, as we in oncology use these immunotherapies/checkpoint inhibitors more often, you will see them more often in the ED. Let’s get right into that. You’ve identified this patient as not getting a traditional chemotherapy – hopefully, all our records on these patients are available. You’ve decided to follow onto what might be a side effect of the immunotherapy, so I’m going to name the side effects that always occur to me: lung, gastrointestinal (GI) – which could be loose bowels or liver function – rash, endocrine problems. Let’s start with lung symptoms. You see the patient is short of breath and you identified immunotherapy. What’s your next step?
DR SAMMON That’s a great example, because the problem is that you see these patients with cough or shortness of breath and pulmonary complications, and pulmonary complications of immunotherapy, while rare, are potentially life threatening if they’re not identified quickly.
You can start with a chest X-ray on these patients knowing, however, that for a good percentage of them you won’t see findings on their chest X-ray (Figure 1, from Sammon M, Tobin T. Identification and management of immune-related adverse events in the emergency setting. Presented at: Advances in Cancer Immunotherapy – Society for Immunotherapy of Cancer (SITC); August 4, 2017; Philadelphia, PA). You need to proceed to computed tomography (CT), because the issue is that you can have protean findings on the CT related to immunotherapy treatment/adverse reactions. You need to have a very high index of suspicion regardless of what abnormal findings you’re seeing on this CT and erring toward withholding the drugs, starting treatment, and being more aggressive with this type of finding.
DR HENRY I’ve heard you talk at a conference about a patient with metastatic lung cancer, or some other tumor that may have existing disease in lung. The patient is aware of that, and the chart reflects that. Then you have this difficulty where the CT scan shows a pneumonitis, and it may not be tumor progression at all – it may be the drug. How do you work through that? Of course, your additional problem is you don’t have a whole lot of time. You must decide to whether you’re going to keep them in the ED, admit them, or send them home.
DR SAMMON Right. One of the first things is to get the oncologist involved at an early stage in treating this. We are all a team. We are all working together, and it’s very important to have that communication occur very early. I’m going to err on admitting those folks who have had symptoms and who have had findings on a chest CT, because they can progress. They can get much worse. I’m going to be getting the oncologist involved very early. We’re going to have the conversation about whether we should be starting steroids on them in the ED, getting them upstairs, and being aggressive in treating this.
DR HENRY So, time and therapy are very important.
DR SAMMON Yes.
DR HENRY You’ll get that steroid started as an antidote right away.
DR SAMMON Absolutely. For grade 2, you’re going to use methylprednisolone, 1 mg /kg daily. For anything higher than that – grade 3 or grade 4 – we’re going to start higher. We’re going to start at 2-4 mg/kg a day and get the patient upstairs and taper them slowly.
DR HENRY That’s worth empathizing. I, sadly, had a patient who ultimately did well, but had severe grade 4 pneumonitis. T
DR SAMMON Absolutely, but it’s very important to work together as a team to make these patients have good outcomes.
DR HENRY Agree. Let’s change over to the GI symptoms. The patient comes in and misidentifies him- or herself as having chemotherapy and diarrhea. We are used to causing nausea, vomiting, and diarrhea with some of our therapies. You realize, in talking to the oncologist, the patient is taking a checkpoint inhibitor. How would you approach the patient with diarrhea?
DR SAMMON First, I would talk to the patient. I would try to establish the baseline number of stools per day, because it’s not defined as a definite number of stools per day, it’s the number of stools above their baseline per day. If they’re having fewer than 4 stools above their baseline per day, I would send off some tests. We can send off a C diff (Clostridium difficile) test, we can send off a stool culture – all the parasites. Make sure the oncologist is going to be able to get these results and get them followed up, because these are results I’m not going to get back myself in the ED.
Then I’m going to talk to the patient about symptomatic treatment. I’m going to talk to them about oral hydration, a bland diet. Avoid using loperamide or any other antidiarrheal medicines, because that could decrease the frequency of stools but mask more severe symptoms that they may be having.
If I have a patient who is having more than 4 stools above their daily baseline and it’s been happening 4 to 6 stools a day for more than a week, I’m going to be sending those studies off, and I’m going to be having a conversation with the treating oncologist to find out if they want me to start the patient on steroids immediately, or if they want to wait for the test results to come back and have the steroids started as an outpatient.
These moderate patients can maybe wait a day until these test results come back. Those who are having more than 7 stools above baseline per day, peritoneal signs, ileus, or fever, are the patients you should worry about. You need to admit them for IV hydration. You need to do the stools, so you might need to keep them in the hospital until you find the results of the stool studies. You need to rule out perforation.
You may be starting steroids on these patients sooner rather than later. They’re going to be getting systemic corticosteroids at about 1-2 mg/kg of prednisone equivalent, assuming there is no perforation and their stool studies are negative. If they are unstable, though, they are really going to need high-dose corticosteroids. They are going to need methylprednisolone, 125 mg IV, to evaluate for their responsiveness. These folks really need to be treated as inpatients, and they need to have their oncologists involved early on with their treatment.
DR HENRY Yes. I couldn’t agree with you more. When I talk to the diarrhea side effect patients that we see, I tend to think it’s a curse. It’s volume. It’s calories. It’s electrolytes. The number of stools you’re mentioning, it is almost certainly going to need admission to rule out other causes. Then, if it’s the checkpoint inhibitor, the steroid antidotes.
Let’s move on to the rash. This is another organ system that can be affected by immunotherapy. What is your approach when you see a generalized body rash in a patient on one of these drugs who is sent to the ED?
DR SAMMON I am obviously going to be ruling out other causes first, but generally you’re going to see a maculopapular rash. It may be itchy. It may be burning. Patients will often describe it as just sort of having a tight sensation. I’m going to be looking at them a little bit like I look at a burn patient. What is their total body surface area that’s involved? If they’ve got less than 30% of their total body surface area involved, that’s considered a mild reaction.
For those folks, I’m not going to use systemic steroids, but I can give them some topical steroids, and I can give them some Benadryl, some diphenhydramine, and really treat them symptomatically as well as ensuring that they have early follow-up to make sure this isn’t progressing. Once we get between 30% and 50% body area, we’re talking about a moderate toxicity. If these patients are not improving rapidly with just withholding the drug, they need systemic corticosteroids.
We usually treat them at about 0.5-1 mg/kg body weight a day of prednisone equivalent. Just as with burn patients, these patients’ symptoms can become very severe. You can see signs of blistering, dermal ulceration, necrotic, bolus, hemorrhagic lesions (Figure 2, A-D).1 These folks can have very difficult-to-manage fluid balances, and they’re at very high risk for skin infections as well. They need to be treated as inpatients. If possible, you might want to consider sending these patients to a burn unit. They need systemic corticosteroids, 1-2 mg/kg per day, and they need careful monitoring for signs of dehydration, electrolytic abnormalities, and/or skin infections. They need excellent wound care.
DR HENRY That’s very well put and always difficult, because there are so many causes of rash. That takes me to an area that has always been difficult for me, which is therapy-related endocrine problems. It’s interesting to note that these drugs can cause endocrine problems. I’ve heard you speak about the pituitary affecting vision, as well as thyroid or adrenal issues. Let’s start with how you’d approach vision difficulty in a patient on these drugs.
DR SAMMON The endocrinopathies that you can get with these checkpoint inhibitors really have a myriad of symptoms. Your patient may present saying that they’re feeling tired, that they’re feeling weak, or they may have a headache. If your patient is having actual pituitary enlargement, they can present with headaches, visual field defects, or cranial nerve defects. The reason for those symptoms is that the pituitary sits in the cavernous sinus, and you have various cranial nerves passing through that area as well as the optic chiasm just above the pituitary gland (Figure 3).1 Your patient may present with a bitemporal hemianopia. Or with diplopia. You are going to want to very quickly get either a CT scan or an MRI to find out if that is what’s going on (Figure 4).1 These folks need to be treated aggressively as well.
DR HENRY You’ll get your CT scan or your MRI and rule out an enlargement or a change to the visual field. I haven’t seen this yet, but certainly exciting when you see it to treat it. Would you get the radialis brevis involved, steroids involved? How would you manage that?
DR SAMMON It’s interesting, because you do want to use corticosteroids. One of the questions here is, which corticosteroid do you want to use? If you’re talking about someone who may have adrenal insufficiency, you may want to be able to do a stimulation test. In these patients, you may want to choose using dexamethasone, because you can still do the corticotropin stimulation test. However, if your patient is in frank shock because of what you think is an adrenal crisis, you’re going to want to use hydrocortisone. If a patient is truly hypotensive and unstable, the testing is at that point less important than the treatment.
DR HENRY Very interesting. We have covered what I would consider the major aspects of these fascinating drugs. We haven’t covered all of what they do when they work well, which hopefully we’re seeing more and more often, but we have covered very well what can happen when things go wrong in side effects. Anything else that you would like to add from the ED perspective or other side effects worth mentioning?
DR SAMMON The thing that I would most like to share with the oncology office is the importance of communicating with your patients that, when they’re on these drugs, they need to tell emergency physicians that they’re on immunotherapy, not chemotherapy. It might be helpful to give these patients a card stating that they’re on immunotherapy, not chemotherapy, and outlining some of the side effects that ED physicians should be looking out for in these patients.
DR HENRY That’s a great point. I’ve seen that some of the manufacturers have little cheat cards that the patient can carry naming the drug and the side effects, because not all ED doctors are aware of the side effects of these drugs.
DR SAMMON Absolutely. We love those cards.
DR HENRY Yes. I’ve also given some to the ED doctors at Pennsylvania Hospital, and they love it. I think we’ve covered everything in quite a bit of detail. Thank you, Dr Sammon, for sharing this information from the frontlines of the ED.
DR HENRY I am pleased to be talking with Dr Maura Sammon, an emergency department (ED) physician, about identifying and treating immunotherapy-related side effects in the ED. This is a hot topic in oncology, and I was very interested in having an ED physician talk about what happens when treating oncologists send their patients to the ED, because a physician may think it is chemotherapy when it is immunotherapy. Let’s start with the example of an oncology patient going to the ED with some symptoms, and the ED physician asks the patient what they’re being treated with. The patient may or may not say the right thing – that is, inform you whether they are being treated with chemotherapy or immunotherapy. How do you morph over into knowing that they are not getting chemotherapy?
DR SAMMON Yes, that’s a big problem in the ED. Patients come to the ED and say they’re being treated for cancer. They say they’re on chemotherapy, when they’re actually on immunotherapy, and it can really send the treatment team down the wrong path. I have a metaphor to explain this. They say that Great Britain and the United States are two nations separated by a common language. For example, when a British person talks about football, they mean something very different than when an American talks about football. If someone in Great Britain asks you to come play football, you might show up with shoulder pads and a helmet rather than shin guards, and you’re left without having the right tools to participate in the game.
How this sometimes plays out with immunotherapy, unfortunately, is that a patient will present to the ED and say they’re having a cough and that they’re on chemotherapy for melanoma. Usually, this patient would be worked up for being in a potentially immunosuppressed state. You might get a white blood cell count. You might get a chest X-ray. You might see what looks to you like a new infiltrate on this chest X-ray and then start going down the path of treating someone whom you think is immunosuppressed with pneumonia and giving them antibiotics rather than what could be life-saving steroids, as would be the case if the patient were on immunotherapy.
It’s a real problem, because you have one word that patients may use meaning two very different things. It can get you into trouble if you are treating someone for potentially infectious causes rather than immunotherapy-related adverse reactions, which are much more similar to graft-versus-host disease than in the case of traditional chemotherapy.
DR HENRY That’s a very good point. I think, as we in oncology use these immunotherapies/checkpoint inhibitors more often, you will see them more often in the ED. Let’s get right into that. You’ve identified this patient as not getting a traditional chemotherapy – hopefully, all our records on these patients are available. You’ve decided to follow onto what might be a side effect of the immunotherapy, so I’m going to name the side effects that always occur to me: lung, gastrointestinal (GI) – which could be loose bowels or liver function – rash, endocrine problems. Let’s start with lung symptoms. You see the patient is short of breath and you identified immunotherapy. What’s your next step?
DR SAMMON That’s a great example, because the problem is that you see these patients with cough or shortness of breath and pulmonary complications, and pulmonary complications of immunotherapy, while rare, are potentially life threatening if they’re not identified quickly.
You can start with a chest X-ray on these patients knowing, however, that for a good percentage of them you won’t see findings on their chest X-ray (Figure 1, from Sammon M, Tobin T. Identification and management of immune-related adverse events in the emergency setting. Presented at: Advances in Cancer Immunotherapy – Society for Immunotherapy of Cancer (SITC); August 4, 2017; Philadelphia, PA). You need to proceed to computed tomography (CT), because the issue is that you can have protean findings on the CT related to immunotherapy treatment/adverse reactions. You need to have a very high index of suspicion regardless of what abnormal findings you’re seeing on this CT and erring toward withholding the drugs, starting treatment, and being more aggressive with this type of finding.
DR HENRY I’ve heard you talk at a conference about a patient with metastatic lung cancer, or some other tumor that may have existing disease in lung. The patient is aware of that, and the chart reflects that. Then you have this difficulty where the CT scan shows a pneumonitis, and it may not be tumor progression at all – it may be the drug. How do you work through that? Of course, your additional problem is you don’t have a whole lot of time. You must decide to whether you’re going to keep them in the ED, admit them, or send them home.
DR SAMMON Right. One of the first things is to get the oncologist involved at an early stage in treating this. We are all a team. We are all working together, and it’s very important to have that communication occur very early. I’m going to err on admitting those folks who have had symptoms and who have had findings on a chest CT, because they can progress. They can get much worse. I’m going to be getting the oncologist involved very early. We’re going to have the conversation about whether we should be starting steroids on them in the ED, getting them upstairs, and being aggressive in treating this.
DR HENRY So, time and therapy are very important.
DR SAMMON Yes.
DR HENRY You’ll get that steroid started as an antidote right away.
DR SAMMON Absolutely. For grade 2, you’re going to use methylprednisolone, 1 mg /kg daily. For anything higher than that – grade 3 or grade 4 – we’re going to start higher. We’re going to start at 2-4 mg/kg a day and get the patient upstairs and taper them slowly.
DR HENRY That’s worth empathizing. I, sadly, had a patient who ultimately did well, but had severe grade 4 pneumonitis. T
DR SAMMON Absolutely, but it’s very important to work together as a team to make these patients have good outcomes.
DR HENRY Agree. Let’s change over to the GI symptoms. The patient comes in and misidentifies him- or herself as having chemotherapy and diarrhea. We are used to causing nausea, vomiting, and diarrhea with some of our therapies. You realize, in talking to the oncologist, the patient is taking a checkpoint inhibitor. How would you approach the patient with diarrhea?
DR SAMMON First, I would talk to the patient. I would try to establish the baseline number of stools per day, because it’s not defined as a definite number of stools per day, it’s the number of stools above their baseline per day. If they’re having fewer than 4 stools above their baseline per day, I would send off some tests. We can send off a C diff (Clostridium difficile) test, we can send off a stool culture – all the parasites. Make sure the oncologist is going to be able to get these results and get them followed up, because these are results I’m not going to get back myself in the ED.
Then I’m going to talk to the patient about symptomatic treatment. I’m going to talk to them about oral hydration, a bland diet. Avoid using loperamide or any other antidiarrheal medicines, because that could decrease the frequency of stools but mask more severe symptoms that they may be having.
If I have a patient who is having more than 4 stools above their daily baseline and it’s been happening 4 to 6 stools a day for more than a week, I’m going to be sending those studies off, and I’m going to be having a conversation with the treating oncologist to find out if they want me to start the patient on steroids immediately, or if they want to wait for the test results to come back and have the steroids started as an outpatient.
These moderate patients can maybe wait a day until these test results come back. Those who are having more than 7 stools above baseline per day, peritoneal signs, ileus, or fever, are the patients you should worry about. You need to admit them for IV hydration. You need to do the stools, so you might need to keep them in the hospital until you find the results of the stool studies. You need to rule out perforation.
You may be starting steroids on these patients sooner rather than later. They’re going to be getting systemic corticosteroids at about 1-2 mg/kg of prednisone equivalent, assuming there is no perforation and their stool studies are negative. If they are unstable, though, they are really going to need high-dose corticosteroids. They are going to need methylprednisolone, 125 mg IV, to evaluate for their responsiveness. These folks really need to be treated as inpatients, and they need to have their oncologists involved early on with their treatment.
DR HENRY Yes. I couldn’t agree with you more. When I talk to the diarrhea side effect patients that we see, I tend to think it’s a curse. It’s volume. It’s calories. It’s electrolytes. The number of stools you’re mentioning, it is almost certainly going to need admission to rule out other causes. Then, if it’s the checkpoint inhibitor, the steroid antidotes.
Let’s move on to the rash. This is another organ system that can be affected by immunotherapy. What is your approach when you see a generalized body rash in a patient on one of these drugs who is sent to the ED?
DR SAMMON I am obviously going to be ruling out other causes first, but generally you’re going to see a maculopapular rash. It may be itchy. It may be burning. Patients will often describe it as just sort of having a tight sensation. I’m going to be looking at them a little bit like I look at a burn patient. What is their total body surface area that’s involved? If they’ve got less than 30% of their total body surface area involved, that’s considered a mild reaction.
For those folks, I’m not going to use systemic steroids, but I can give them some topical steroids, and I can give them some Benadryl, some diphenhydramine, and really treat them symptomatically as well as ensuring that they have early follow-up to make sure this isn’t progressing. Once we get between 30% and 50% body area, we’re talking about a moderate toxicity. If these patients are not improving rapidly with just withholding the drug, they need systemic corticosteroids.
We usually treat them at about 0.5-1 mg/kg body weight a day of prednisone equivalent. Just as with burn patients, these patients’ symptoms can become very severe. You can see signs of blistering, dermal ulceration, necrotic, bolus, hemorrhagic lesions (Figure 2, A-D).1 These folks can have very difficult-to-manage fluid balances, and they’re at very high risk for skin infections as well. They need to be treated as inpatients. If possible, you might want to consider sending these patients to a burn unit. They need systemic corticosteroids, 1-2 mg/kg per day, and they need careful monitoring for signs of dehydration, electrolytic abnormalities, and/or skin infections. They need excellent wound care.
DR HENRY That’s very well put and always difficult, because there are so many causes of rash. That takes me to an area that has always been difficult for me, which is therapy-related endocrine problems. It’s interesting to note that these drugs can cause endocrine problems. I’ve heard you speak about the pituitary affecting vision, as well as thyroid or adrenal issues. Let’s start with how you’d approach vision difficulty in a patient on these drugs.
DR SAMMON The endocrinopathies that you can get with these checkpoint inhibitors really have a myriad of symptoms. Your patient may present saying that they’re feeling tired, that they’re feeling weak, or they may have a headache. If your patient is having actual pituitary enlargement, they can present with headaches, visual field defects, or cranial nerve defects. The reason for those symptoms is that the pituitary sits in the cavernous sinus, and you have various cranial nerves passing through that area as well as the optic chiasm just above the pituitary gland (Figure 3).1 Your patient may present with a bitemporal hemianopia. Or with diplopia. You are going to want to very quickly get either a CT scan or an MRI to find out if that is what’s going on (Figure 4).1 These folks need to be treated aggressively as well.
DR HENRY You’ll get your CT scan or your MRI and rule out an enlargement or a change to the visual field. I haven’t seen this yet, but certainly exciting when you see it to treat it. Would you get the radialis brevis involved, steroids involved? How would you manage that?
DR SAMMON It’s interesting, because you do want to use corticosteroids. One of the questions here is, which corticosteroid do you want to use? If you’re talking about someone who may have adrenal insufficiency, you may want to be able to do a stimulation test. In these patients, you may want to choose using dexamethasone, because you can still do the corticotropin stimulation test. However, if your patient is in frank shock because of what you think is an adrenal crisis, you’re going to want to use hydrocortisone. If a patient is truly hypotensive and unstable, the testing is at that point less important than the treatment.
DR HENRY Very interesting. We have covered what I would consider the major aspects of these fascinating drugs. We haven’t covered all of what they do when they work well, which hopefully we’re seeing more and more often, but we have covered very well what can happen when things go wrong in side effects. Anything else that you would like to add from the ED perspective or other side effects worth mentioning?
DR SAMMON The thing that I would most like to share with the oncology office is the importance of communicating with your patients that, when they’re on these drugs, they need to tell emergency physicians that they’re on immunotherapy, not chemotherapy. It might be helpful to give these patients a card stating that they’re on immunotherapy, not chemotherapy, and outlining some of the side effects that ED physicians should be looking out for in these patients.
DR HENRY That’s a great point. I’ve seen that some of the manufacturers have little cheat cards that the patient can carry naming the drug and the side effects, because not all ED doctors are aware of the side effects of these drugs.
DR SAMMON Absolutely. We love those cards.
DR HENRY Yes. I’ve also given some to the ED doctors at Pennsylvania Hospital, and they love it. I think we’ve covered everything in quite a bit of detail. Thank you, Dr Sammon, for sharing this information from the frontlines of the ED.