User login
Research and Reviews for the Practicing Oncologist
Addressing the rarity and complexities of sarcomas
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
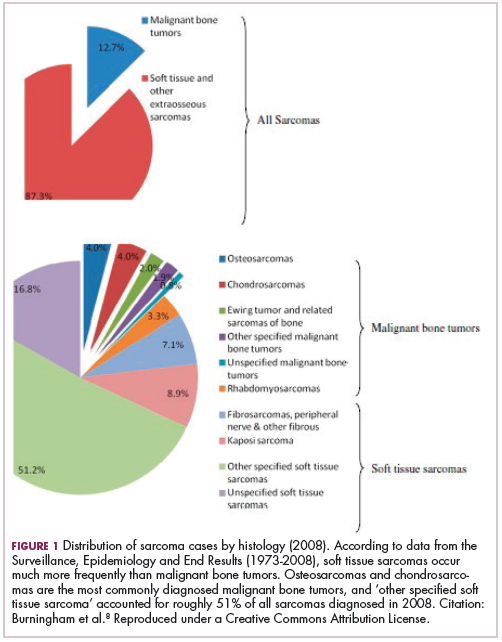
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3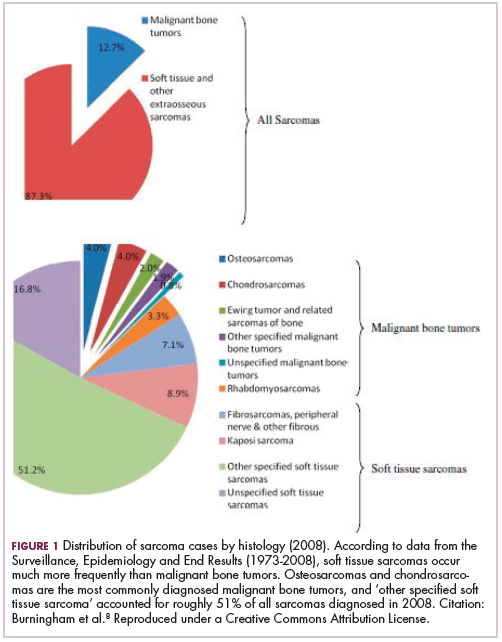
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).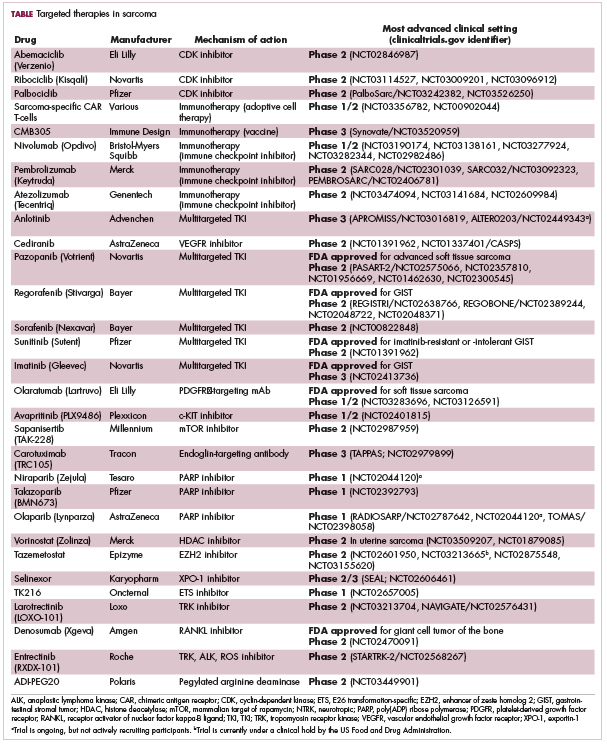
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33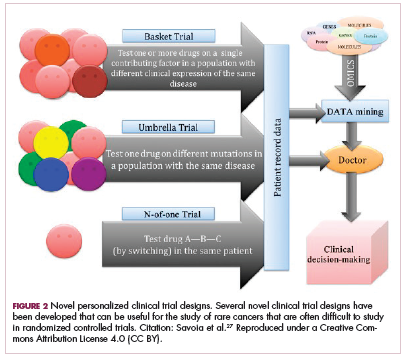
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
Extramedullary plasmacytoma of the thyroid, refractory to radiation therapy and treated with bortezomib
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).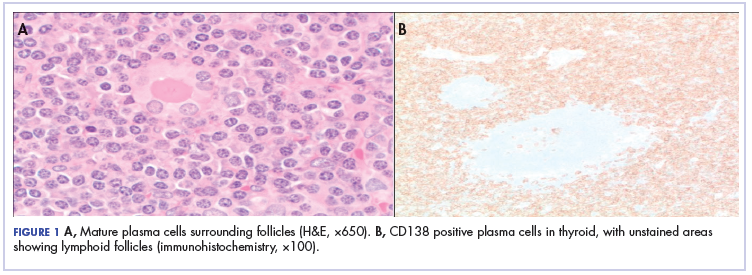
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).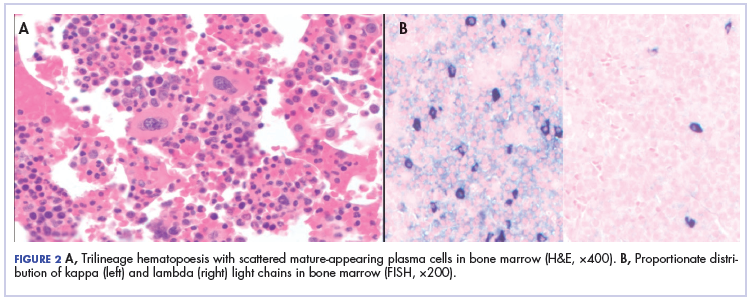
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
Salivary ductal adenocarcinoma with complete response to androgen blockade
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
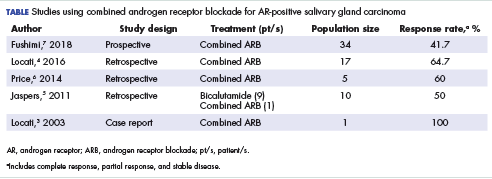
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.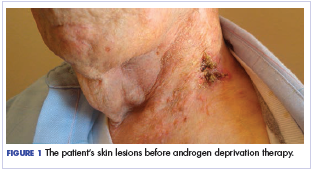
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.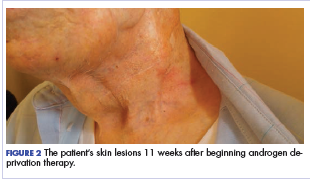
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
Characteristics of urgent palliative cancer care consultations encountered by radiation oncologists
Palliative radiation therapy (PRT) plays a major role in the management of incurable cancers. Study findings have demonstrated the efficacy of using PRT in treating tumor-related bone pain,1 brain metastases and related symptoms,2 thoracic disease-causing hemoptysis or obstruction,3 gastrointestinal involvement causing bleeding and/or obstruction,4 and genitourinary and/or gynecologic involvement causing bleeding.5,6
PRT accounts for between 30% and 50% of courses of radiotherapy delivered.3 These courses of RT typically require urgent evaluation since patients are seen because of new and/or progressive symptoms that give cause for concern. The urgency of presentation requires radiation oncologists and the departments receiving these patients to be equipped to manage these cases efficiently and effectively. Furthermore, the types of cases seen, including PRT indications and related symptoms requiring management, inform the training of radiation oncology physicians as well as nursing and other clinical staff. Finally, characterizing the types of urgent PRT cases that are seen can also guide research and quality improvement endeavors for advanced cancer care in radiation oncology settings.
There is currently a paucity of data characterizing the types and frequencies of urgent PRT indications in patients who present to radiation oncology departments, as well as a lack of data detailing the related symptoms radiation oncology clinicians are managing. The aim of this study was to characterize the types and frequencies of urgent PRT consultations and the related symptoms that radiation oncologists are managing as part of patient care.
Methods
Based on national palliative care practice and national oncology care practice guidelines,7,8 we designed a survey to categorize the cancer-related palliative care issues seen by radiation oncologists. Physical symptoms, psychosocial issues, cultural consideration, spiritual needs, care coordination, advanced-care planning, goals of care, and ethical and legal issues comprised the 8 palliative care domains that we evaluated. A survey was developed and critically evaluated by 3 investigators (MK, VL, TB). Each palliative care domain was ranked by clinicians by its relevance (5-point Likert scale [range, 1-5]: 1, Not Relevant, to 5, Extremely Relevant) to the patient’s care point in radiation oncology. In addition, 31 palliative care subissues related to the primary domains were identified by clinicians based on their presence (Yes, No, Not Assessed). Clinicians were also asked whether the consulted patient’s metastatic cancer diagnosis was established (longer than 1 month) or new (within the last 1 month). In addition, clinicians noted whether the patient was returning to active oncologic care (eg, chemotherapy) or to no further anticancer therapies (eg, hospice care) after RT consultation and intervention (if deemed necessary).
The survey’s face and content validity, ease of completion, and time of completion was assessed by a panel of 7 clinicians with expertise in medical oncology, radiation oncology, palliative care, and/or survey construction. The survey was then sent in a sequential manner to 1 member of the panel at a time after incorporating each panel member’s initial comments. After each panel member’s review, the survey was edited until 2 consecutive panel members had no further suggestions for improvement.
After receiving approval from the institutional review boards of participating radiation oncology centers, we electronically surveyed radiation oncology clinicians who were conducting PRT consultations. From May 19 to September 26, 2014, all consultations were evaluated prospectively for consideration of PRT performed by a dedicated PRT service at 3 centers (a large academic cancer center and 2 participating clinicians at affiliated regional hospitals). The consultations for patients aged 18 years or older with incurable, metastatic cancers were considered eligible. The consulting clinician was e-mailed a survey for completion within 5 business days immediately after each PRT consult. Three reminders to complete the survey were sent during the 5–business-day interval. Over the entire study period, 162 consecutive patients were identified, resulting in 162 surveys being sent to 15 radiation oncology clinicians, including nurse practitioners, resident physicians, and attending physicians. Each clinician received a $25 gift card for participating, regardless of the number of surveys completed. In total, 140 of the 162 surveys were returned, resulting in a response rate of 86%.
The investigators then collected patient demographics (age, gender, race, marital status) and disease characteristics (primary cancer type, Eastern Cooperative Oncology Group Performance Status, reasons for urgent RT consult, physical symptoms requiring management at presentation, patient’s place in illness trajectory, and RT recommendation) pertaining to each completed survey from the electronic medical record. Urgent consultations were defined as any patients who needed to be seen on the same day or within a few days of the consult request.
The descriptive statistics of all these data were calculated in terms of frequencies and percentage of categorical variables. Chi-squared statistics, Fisher exact test, and nonparametric rank sum tests were applied to various categories to determine any statistically significant differences between groups.
Results
In total, 162 patients were seen in consultation for PRT during the 19-week enrollment period, or an average of 8.7 consults a week. Of that total, surveys for 140 patients were returned (Table).
The median patient age was 63 years (range, 29-89 years). A sizeable minority (20%) was 50 years or younger. The most common cancer diagnosis was lung cancer (28%), followed by breast (13%) and prostate (10%) cancers, melanoma (10%), and sarcoma (7%). Other diagnoses accounted for the remaining 32%.
Timing of PRT consult in cancer trajectory
The points in the advanced cancer illness trajectory at which patients were seen for PRT evaluation are shown in Figure 1. Most patients (63%) were seen for a PRT evaluation at the time of an established diagnosis (>1 month after diagnosis of metastatic cancer) and were continuing to further cancer therapies. An additional 19% of patients with an established diagnosis proceeded to hospice or end-of-life care after the PRT evaluation. A notable minority of patients (18%) were seen for a PRT evaluation at the time of a new diagnosis (<1 month of diagnosis of metastatic cancer), and of those, 17% went on to receive anticancer therapy after the PRT evaluation and 1% proceeded to hospice or end-of-life care.
Characteristics of PRT consults and symptoms at presentation
The primary reasons for urgent consultation for PRT are shown in Figure 2. Cancer-related pain (57%), brain metastases (29%), and malignant spinal cord or cauda equina compression (13%) were the predominant reasons for consults. Notable minorities were seen for tumor-related dyspnea (10%), bleeding (8%), and bone fractures (4%).
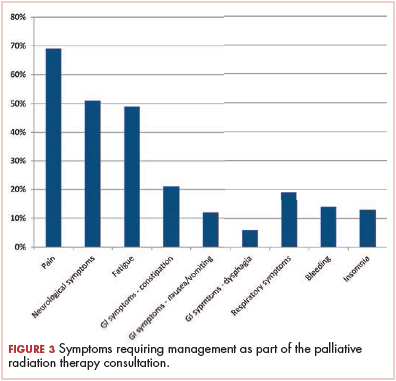
PRT recommendations and targets
Recommendations regarding PRT are shown in Figure 4A. Of the total 140 patients, 18 (13%) were not recommended for RT. Of the 122 patients for whom PRT was recommended, 11 (9%) received RT at more than 1 site.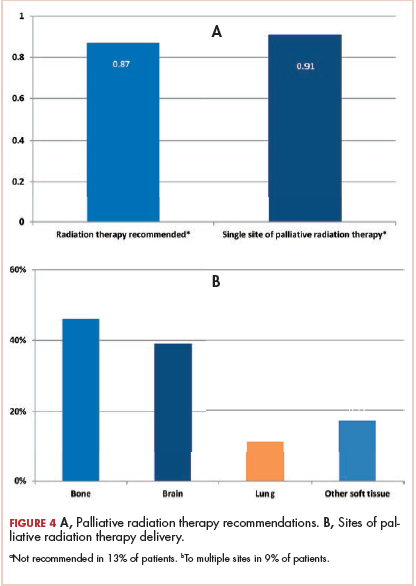
Discussion and conclusions
The present study provides a descriptive overview of urgent metastatic cancer patient presentations to radiation oncology clinicians through a comprehensive evaluation of 140 consults for PRT. The most common reasons for urgent evaluation were cancer-related pain (57%), but brain metastases (29%), spinal cord compression (13%), and respiratory symptoms (10%) were also common. Other less-common indications included cancer-related dysphagia, bleeding, and poststabilization management of bone fractures. The most common symptoms requiring management by radiation oncology clinicians were pain (69%), neurologic symptoms (51%), and fatigue (49%). The study also provides a comprehensive characterization of the timeframe of PRT consultation and the treatment recommendations in this cohort. Though most PRT consults occurred at the time of an established metastatic cancer diagnosis and before further anticancer therapies, sizeable minorities occurred at the time of a new diagnosis of metastatic cancer (18%) and before comfort-focused, end-of-life care and no further anticancer therapies (20%). Most patients (87%) were recommended PRT, and of those recommended RT, 11% received RT to more than 1 site. The most common PRT sites were to bone (46%), followed by brain (29%), nonlung soft-tissue sites (17%), and lung (8%). This comprehensive description of the day-to-day urgent, advanced cancer care issues seen and managed in radiation oncology practice can help guide PRT clinical structures, education, research, and quality improvement measures in clinical practice.
Our study provides an insight into urgent symptoms encountered by radiation oncology practitioners during their routine practice. Cancer-related pain remains the most common symptom requiring management. Given the frequency with which pain management is needed among PRT patients, this study highlights the need for radiation oncologists to be well trained in symptom management, particularly as the pain response to RT can often take several days. However, studies suggest that cancer-related pain is not frequently managed by radiation oncologists.9 For example, findings from an Italian study showed that the involvement of radiation oncologists in cancer pain management remains minimal compared with other medical professionals; during the treatment course, only half of the radiation oncologists implemented specific treatment for breakthrough pain.10 A nationwide survey in the United States implicated a number of barriers to adequate pain management, including poor assessment by the physician, reluctance in prescribing opioid analgesics, perceived excessive regulation, and patient reluctance to report pain.11 Notably, in a survey of the Radiation Therapy Oncology Group study physicians, 83% believed cancer patients with pain were undermedicated, and 40% reported that pain relief in their own practice setting was suboptimal. Furthermore, in the treatment plan, adjuvants and prophylactic side effect management were frequently not used properly.12 Education of radiation oncologists in pain assessment and management is key to overcome these barriers and to ensure adequate pain management and quality of life for patients in radiation oncology.
The next most common reason for which patients presented for palliative radiation oncology consultation was for central nervous system (CNS) metastatic disease, including brain metastases and spinal cord compression. Correspondingly, the next most common issue requiring management was neurologic symptoms. Management of CNS disease is becoming increasingly complex, and it benefits from multidisciplinary evaluation to guide optimal and personalized care for each patient, including medical oncology, radiation oncology, neurosurgery and/or orthopedic spine surgery, and palliative medicine. Treatment options include supportive care or corticosteroids alone, surgical resection, whole-brain RT, and/or radiosurgery or stereotactic RT alone. These treatment options are considered on the basis of global patient factors, such as prognosis, together with metastatic-site–specific factors, such as site-related symptoms and the number of metastatic diseases or the burden of the disease.13 For example, the use of the diagnosis-specific Graded Prognostic Assessment index to predict life expectancy can help tailor management of brain metastases based on performance status, age, number of brain metastases, extracranial metastases, and cancer type. Highlighting the complexity of this common PRT presentation, Tsao and colleagues showed that there was a lack of uniform agreement among radiation oncologists for common management issues in patients with brain metastatic disease.14
For metastatic spinal cord or cauda equina compression and the associated neurologic symptoms, initiation of immediate corticosteroids and implementation of local therapy within 24 hours of presentation is paramount,15 highlighting the need for rapid, comprehensive care decision-making for these patients. Treatment options that must be weighed include the potential benefit of upfront decompressive surgery, as supported by a randomized controlled trial by Patchell and colleagues16 for patients who are surgical candidates with true cord or cauda compression and have at least 1 neurologic symptom, a prognosis of ≥3 months, paraplegia of no longer than 48 hours, and no previous RT to the site or brain metastases. Compared with the RT alone, patients receiving surgery before RT had improved ambulatory status and overall survival. Hence, neurosurgical or orthopedic consultation should be standard in the evaluation of metastatic spinal cord or cauda equina compression patients. However, patients frequently do not meet these criteria, and corticosteroids and RT alone are considered. In addition to playing a role in surgical decision-making, prognosis also has a key role in decision-making about the RT fractionation. Short-course RT (8 Gy × 1) is as effectual as longer-course regimens (3 Gy × 10) in terms of motor function.17,18 However, more dose-intense or longer-course regimens, such as 3 Gy × 10, have been shown to have more durability beyond about 6 months and are therefore considered for intermediate to good prognosis.18 The common urgent presentation of CNS metastatic disease to radiation oncology clinics together with the complexity of management and urgency of care decision-making point to the need for dedicated structures of care for these patients in radiation oncology settings. For example, dedicated PRT programs, such as the Rapid Response Radiotherapy Program in Toronto and the Supportive and Palliative Radiation Oncology service in Boston, have demonstrated improved quality of care for patients being urgently evaluated for PRT.19
Following management of pain and neurologic symptoms, clinicians were faced with managing fatigue in nearly half of the patients (49%). The prevalence of fatigue among cancer patients and its impact on quality of life20 highlight the need for this key symptom to be addressed throughout the continuum of cancer care. National Comprehensive Cancer Network guidelines provide a comprehensive framework for addressing cancer-related fatigue.7 However, cancer-related fatigue is a largely underreported, underestimated, and thus undertreated problem.20 In a nationwide survey of members of the American Society for Radiation Oncology, radiation oncologists reported being significantly less confident in managing fatigue compared with managing other common symptoms.21 Furthermore, in a national survey of radiation oncology trainees, 67% of respondents indicated that they were not at all minimally or somewhat confident in their ability to manage fatigue symptoms. The frequency of this symptom together with the demonstrated need for improved education in fatigue management point to a need for radiation oncology palliative educational structures to include dedicated emphasis on managing fatigue in addition to other commonly encountered symptoms, such as pain.
Patients evaluated for PRT are seen across the trajectory of their metastatic cancer diagnosis. In our study, patients presented at all stages in their advanced cancers. These include patients seen at the time of initial diagnosis of cancer as well as those seen near the end of life when end-of-life care planning was underway. The broad spectrum of timing of PRT care underscores that radiation oncologists must be prepared to handle generalist palliative care issues encountered throughout the trajectory of advanced cancer care and hence need comprehensive education in generalist palliative care competencies. These include symptom management, end-of-life care coordination, and communication or goals-of-care discussions. Notably, a recent national survey of radiation oncology residents indicated that most residents, 77% on average, perceived their educational training as suboptimal across domains of generalist palliative care competencies needed in oncology practice.22 Furthermore, a majority (81%) desired greater palliative care education within training.
The most common sites treated in this study were bone, brain, and lung sites. These data provide guidance to both education and research initiatives aiming to advance PRT curriculum and care structures within departments. For example, a same-day simulation and radiation treatment program developed at Princess Margaret Hospital Palliative Radiation Oncology Program (Ontario, Canada) aids in providing streamlined care for patients with bone metastases, the most common presentation for PRT.23 Furthermore, education and research in the application of PRT techniques to bone, brain, and thoracic disease cover the majority of PRT presentations. It is notable, however, that 17% were other soft tissue body sites.
Limitations
There are a few limitations to this study. First, this is a survey-based study conducted at a single academic center within an urban setting and surrounding community regions, which affects its generalizability. Second, this study presents perspectives of radiation oncology practitioners evaluating patients and does not directly reflect patient perceptions or report of symptoms. Third, the data provided by this study are solely descriptive in nature. However, this can guide hypothesis-driven research regarding the evaluation and management of urgent palliative care issues encountered by radiation oncology clinicians and suggest educational objectives to address the needs of these patients.
Conclusions
Radiation oncologists are involved throughout the trajectory of care for advanced cancer patients. Furthermore, they manage a variety of urgent oncologic issues, most commonly metastases causing pain, brain metastases, and spinal cord or cauda equina compression. Radiation oncologists also manage many cancer-related symptoms, mostly pain, neurologic symptoms, fatigue, and gastrointestinal symptoms. These findings point toward the need for palliative care to be well integrated into radiation oncology training curricula and the need for dedicated care structures that enable rapid and multidisciplinary palliative oncology care within radiation oncology departments.
1. Chow E, Harris K, Fan G, Tsao M, Sze WM. Palliative radiotherapy trials for bone metastases: a systematic review. J Clin Oncol. 2007;25(11):1423-1436.
2. van Oorschot B, Rades D, Schulze W, Beckmann G, Feyer P. Palliative radiotherapy--new approaches. Semin Oncol. 2011;38(3):443-449.
3. Simone CB II, Jones JA. Palliative care for patients with locally advanced and metastatic non-small cell lung cancer. Ann Palliat Med. 2013;2(4):178-188.
4. Cihoric N, Crowe S, Eychmüller S, Aebersold DM, Ghadjar P. Clinically significant bleeding in incurable cancer patients: effectiveness of hemostatic radiotherapy. Radiat Oncol. 2012;7:132.
5. Duchesne GM, Bolger JJ, Griffiths GO, et al. A randomized trial of hypofractionated schedules of palliative radiotherapy in the management of bladder carcinoma: results of medical research council trial BA09. Int J Radiat Oncol Biol Phys. 2000;47(2):379-388.
6. Onsrud M, Hagen B, Strickert T. 10-Gy single-fraction pelvic irradiation for palliation and life prolongation in patients with cancer of the cervix and corpus uteri. Gynecol Oncol. 2001;82(1):167-171.
7. NCCN Guidelines(R) Updates. J Natl Compr Canc Netw. 2013;11(9):xxxii-xxxvi.
8. Colby WH, Dahlin C, Lantos J, Carney J, Christopher M. The National Consensus Project for Quality Palliative Care Clinical Practice Guidelines Domain 8: ethical and legal aspects of care. HEC Forum. 2010;22(2):117-131.
9. Stockler MR, Wilcken NR. Why is management of cancer pain still a problem? J Clin Oncol. 2012;30(16):1907-1908.
10. Caravatta L, Ramella S, Melano A, et al. Breakthrough pain management in patients undergoing radiotherapy: a national survey on behalf of the Palliative and Supportive Care Study Group. Tumori. 2015;101(6):603-608.
11. Breuer B, Fleishman SB, Cruciani RA, Portenoy RK. Medical oncologists’ attitudes and practice in cancer pain management: a national survey. J Clin Oncol. 2011;29(36):4769-4775.
12. Cleeland CS, Janjan NA, Scott CB, Seiferheld WF, Curran WJ. Cancer pain management by radiotherapists: a survey of radiation therapy oncology group physicians. Int J Radiat Oncol Biol Phys. 2000;47(1):203-208.
13. Tsao MN, Rades D, Wirth A, et al. Radiotherapeutic and surgical management for newly diagnosed brain metastasis(es): an American Society for Radiation Oncology evidence-based guideline. Pract Radiat Oncol. 2012;2(3):210-225.
14. Tsao MN, Rades D, Wirth A, et al. International practice survey on the management of brain metastases: third international consensus workshop on palliative radiotherapy and symptom control. Clin Oncol (R Coll Radiol). 2012;24(6):e81-e92.
15. Tang V, Harvey D, Park Dorsay J, Jiang S, Rathbone MP. Prognostic indicators in metastatic spinal cord compression: using functional independence measure and Tokuhashi scale to optimize rehabilitation planning. Spinal Cord. 2007;45(10):671-677.
16. Patchell RA, Tibbs PA, Regine WF, et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomised trial. Lancet. 2005;366(9486):643-648.
17. Rades D, Stalpers LJ, Veninga T, et al. Evaluation of five radiation schedules and prognostic factors for metastatic spinal cord compression. J Clin Oncol. 2005;23(15):3366-3375.
18. Rades D, Stalpers LJ, Hulshof MC, et al. Comparison of 1 x 8 Gy and 10 x 3 Gy for functional outcome in patients with metastatic spinal cord compression. Int J Radiat Oncol Biol Phys. 2005;62(2):514-518.
19. Dennis K, Linden K, Balboni T, Chow E. Rapid access palliative radiation therapy programs: an efficient model of care. Future Oncol. 2015;11(17):2417-2426.
20. Kapoor A, Singhal MK, Bagri PK, Narayan S, Beniwal S, Kumar HS. Cancer related fatigue: a ubiquitous problem yet so under reported, under recognized and under treated. South Asian J Cancer. 2015;4(1):21-23.
21. Wei RL, Mattes MD, Yu J, et al. Attitudes of radiation oncologists toward palliative and supportive care in the United States: report on national membership survey by the American Society for Radiation Oncology (ASTRO). Pract Radiat Oncol. 2017;7(2):113-119.
22. Krishnan M, Racsa M, Jones J, et al. Radiation oncology resident palliative education. Pract Radiat Oncol. 2017;7(6):e439-e448.
23. McDonald R, Chow E, Lam H, Rowbottom L, Soliman H. International patterns of practice in radiotherapy for bone metastases: a review of the literature. J Bone Oncol. 2014;3(3-4):96-102.
Palliative radiation therapy (PRT) plays a major role in the management of incurable cancers. Study findings have demonstrated the efficacy of using PRT in treating tumor-related bone pain,1 brain metastases and related symptoms,2 thoracic disease-causing hemoptysis or obstruction,3 gastrointestinal involvement causing bleeding and/or obstruction,4 and genitourinary and/or gynecologic involvement causing bleeding.5,6
PRT accounts for between 30% and 50% of courses of radiotherapy delivered.3 These courses of RT typically require urgent evaluation since patients are seen because of new and/or progressive symptoms that give cause for concern. The urgency of presentation requires radiation oncologists and the departments receiving these patients to be equipped to manage these cases efficiently and effectively. Furthermore, the types of cases seen, including PRT indications and related symptoms requiring management, inform the training of radiation oncology physicians as well as nursing and other clinical staff. Finally, characterizing the types of urgent PRT cases that are seen can also guide research and quality improvement endeavors for advanced cancer care in radiation oncology settings.
There is currently a paucity of data characterizing the types and frequencies of urgent PRT indications in patients who present to radiation oncology departments, as well as a lack of data detailing the related symptoms radiation oncology clinicians are managing. The aim of this study was to characterize the types and frequencies of urgent PRT consultations and the related symptoms that radiation oncologists are managing as part of patient care.
Methods
Based on national palliative care practice and national oncology care practice guidelines,7,8 we designed a survey to categorize the cancer-related palliative care issues seen by radiation oncologists. Physical symptoms, psychosocial issues, cultural consideration, spiritual needs, care coordination, advanced-care planning, goals of care, and ethical and legal issues comprised the 8 palliative care domains that we evaluated. A survey was developed and critically evaluated by 3 investigators (MK, VL, TB). Each palliative care domain was ranked by clinicians by its relevance (5-point Likert scale [range, 1-5]: 1, Not Relevant, to 5, Extremely Relevant) to the patient’s care point in radiation oncology. In addition, 31 palliative care subissues related to the primary domains were identified by clinicians based on their presence (Yes, No, Not Assessed). Clinicians were also asked whether the consulted patient’s metastatic cancer diagnosis was established (longer than 1 month) or new (within the last 1 month). In addition, clinicians noted whether the patient was returning to active oncologic care (eg, chemotherapy) or to no further anticancer therapies (eg, hospice care) after RT consultation and intervention (if deemed necessary).
The survey’s face and content validity, ease of completion, and time of completion was assessed by a panel of 7 clinicians with expertise in medical oncology, radiation oncology, palliative care, and/or survey construction. The survey was then sent in a sequential manner to 1 member of the panel at a time after incorporating each panel member’s initial comments. After each panel member’s review, the survey was edited until 2 consecutive panel members had no further suggestions for improvement.
After receiving approval from the institutional review boards of participating radiation oncology centers, we electronically surveyed radiation oncology clinicians who were conducting PRT consultations. From May 19 to September 26, 2014, all consultations were evaluated prospectively for consideration of PRT performed by a dedicated PRT service at 3 centers (a large academic cancer center and 2 participating clinicians at affiliated regional hospitals). The consultations for patients aged 18 years or older with incurable, metastatic cancers were considered eligible. The consulting clinician was e-mailed a survey for completion within 5 business days immediately after each PRT consult. Three reminders to complete the survey were sent during the 5–business-day interval. Over the entire study period, 162 consecutive patients were identified, resulting in 162 surveys being sent to 15 radiation oncology clinicians, including nurse practitioners, resident physicians, and attending physicians. Each clinician received a $25 gift card for participating, regardless of the number of surveys completed. In total, 140 of the 162 surveys were returned, resulting in a response rate of 86%.
The investigators then collected patient demographics (age, gender, race, marital status) and disease characteristics (primary cancer type, Eastern Cooperative Oncology Group Performance Status, reasons for urgent RT consult, physical symptoms requiring management at presentation, patient’s place in illness trajectory, and RT recommendation) pertaining to each completed survey from the electronic medical record. Urgent consultations were defined as any patients who needed to be seen on the same day or within a few days of the consult request.
The descriptive statistics of all these data were calculated in terms of frequencies and percentage of categorical variables. Chi-squared statistics, Fisher exact test, and nonparametric rank sum tests were applied to various categories to determine any statistically significant differences between groups.
Results
In total, 162 patients were seen in consultation for PRT during the 19-week enrollment period, or an average of 8.7 consults a week. Of that total, surveys for 140 patients were returned (Table).
The median patient age was 63 years (range, 29-89 years). A sizeable minority (20%) was 50 years or younger. The most common cancer diagnosis was lung cancer (28%), followed by breast (13%) and prostate (10%) cancers, melanoma (10%), and sarcoma (7%). Other diagnoses accounted for the remaining 32%.
Timing of PRT consult in cancer trajectory
The points in the advanced cancer illness trajectory at which patients were seen for PRT evaluation are shown in Figure 1. Most patients (63%) were seen for a PRT evaluation at the time of an established diagnosis (>1 month after diagnosis of metastatic cancer) and were continuing to further cancer therapies. An additional 19% of patients with an established diagnosis proceeded to hospice or end-of-life care after the PRT evaluation. A notable minority of patients (18%) were seen for a PRT evaluation at the time of a new diagnosis (<1 month of diagnosis of metastatic cancer), and of those, 17% went on to receive anticancer therapy after the PRT evaluation and 1% proceeded to hospice or end-of-life care.
Characteristics of PRT consults and symptoms at presentation
The primary reasons for urgent consultation for PRT are shown in Figure 2. Cancer-related pain (57%), brain metastases (29%), and malignant spinal cord or cauda equina compression (13%) were the predominant reasons for consults. Notable minorities were seen for tumor-related dyspnea (10%), bleeding (8%), and bone fractures (4%).
PRT recommendations and targets
Recommendations regarding PRT are shown in Figure 4A. Of the total 140 patients, 18 (13%) were not recommended for RT. Of the 122 patients for whom PRT was recommended, 11 (9%) received RT at more than 1 site.
Discussion and conclusions
The present study provides a descriptive overview of urgent metastatic cancer patient presentations to radiation oncology clinicians through a comprehensive evaluation of 140 consults for PRT. The most common reasons for urgent evaluation were cancer-related pain (57%), but brain metastases (29%), spinal cord compression (13%), and respiratory symptoms (10%) were also common. Other less-common indications included cancer-related dysphagia, bleeding, and poststabilization management of bone fractures. The most common symptoms requiring management by radiation oncology clinicians were pain (69%), neurologic symptoms (51%), and fatigue (49%). The study also provides a comprehensive characterization of the timeframe of PRT consultation and the treatment recommendations in this cohort. Though most PRT consults occurred at the time of an established metastatic cancer diagnosis and before further anticancer therapies, sizeable minorities occurred at the time of a new diagnosis of metastatic cancer (18%) and before comfort-focused, end-of-life care and no further anticancer therapies (20%). Most patients (87%) were recommended PRT, and of those recommended RT, 11% received RT to more than 1 site. The most common PRT sites were to bone (46%), followed by brain (29%), nonlung soft-tissue sites (17%), and lung (8%). This comprehensive description of the day-to-day urgent, advanced cancer care issues seen and managed in radiation oncology practice can help guide PRT clinical structures, education, research, and quality improvement measures in clinical practice.
Our study provides an insight into urgent symptoms encountered by radiation oncology practitioners during their routine practice. Cancer-related pain remains the most common symptom requiring management. Given the frequency with which pain management is needed among PRT patients, this study highlights the need for radiation oncologists to be well trained in symptom management, particularly as the pain response to RT can often take several days. However, studies suggest that cancer-related pain is not frequently managed by radiation oncologists.9 For example, findings from an Italian study showed that the involvement of radiation oncologists in cancer pain management remains minimal compared with other medical professionals; during the treatment course, only half of the radiation oncologists implemented specific treatment for breakthrough pain.10 A nationwide survey in the United States implicated a number of barriers to adequate pain management, including poor assessment by the physician, reluctance in prescribing opioid analgesics, perceived excessive regulation, and patient reluctance to report pain.11 Notably, in a survey of the Radiation Therapy Oncology Group study physicians, 83% believed cancer patients with pain were undermedicated, and 40% reported that pain relief in their own practice setting was suboptimal. Furthermore, in the treatment plan, adjuvants and prophylactic side effect management were frequently not used properly.12 Education of radiation oncologists in pain assessment and management is key to overcome these barriers and to ensure adequate pain management and quality of life for patients in radiation oncology.
The next most common reason for which patients presented for palliative radiation oncology consultation was for central nervous system (CNS) metastatic disease, including brain metastases and spinal cord compression. Correspondingly, the next most common issue requiring management was neurologic symptoms. Management of CNS disease is becoming increasingly complex, and it benefits from multidisciplinary evaluation to guide optimal and personalized care for each patient, including medical oncology, radiation oncology, neurosurgery and/or orthopedic spine surgery, and palliative medicine. Treatment options include supportive care or corticosteroids alone, surgical resection, whole-brain RT, and/or radiosurgery or stereotactic RT alone. These treatment options are considered on the basis of global patient factors, such as prognosis, together with metastatic-site–specific factors, such as site-related symptoms and the number of metastatic diseases or the burden of the disease.13 For example, the use of the diagnosis-specific Graded Prognostic Assessment index to predict life expectancy can help tailor management of brain metastases based on performance status, age, number of brain metastases, extracranial metastases, and cancer type. Highlighting the complexity of this common PRT presentation, Tsao and colleagues showed that there was a lack of uniform agreement among radiation oncologists for common management issues in patients with brain metastatic disease.14
For metastatic spinal cord or cauda equina compression and the associated neurologic symptoms, initiation of immediate corticosteroids and implementation of local therapy within 24 hours of presentation is paramount,15 highlighting the need for rapid, comprehensive care decision-making for these patients. Treatment options that must be weighed include the potential benefit of upfront decompressive surgery, as supported by a randomized controlled trial by Patchell and colleagues16 for patients who are surgical candidates with true cord or cauda compression and have at least 1 neurologic symptom, a prognosis of ≥3 months, paraplegia of no longer than 48 hours, and no previous RT to the site or brain metastases. Compared with the RT alone, patients receiving surgery before RT had improved ambulatory status and overall survival. Hence, neurosurgical or orthopedic consultation should be standard in the evaluation of metastatic spinal cord or cauda equina compression patients. However, patients frequently do not meet these criteria, and corticosteroids and RT alone are considered. In addition to playing a role in surgical decision-making, prognosis also has a key role in decision-making about the RT fractionation. Short-course RT (8 Gy × 1) is as effectual as longer-course regimens (3 Gy × 10) in terms of motor function.17,18 However, more dose-intense or longer-course regimens, such as 3 Gy × 10, have been shown to have more durability beyond about 6 months and are therefore considered for intermediate to good prognosis.18 The common urgent presentation of CNS metastatic disease to radiation oncology clinics together with the complexity of management and urgency of care decision-making point to the need for dedicated structures of care for these patients in radiation oncology settings. For example, dedicated PRT programs, such as the Rapid Response Radiotherapy Program in Toronto and the Supportive and Palliative Radiation Oncology service in Boston, have demonstrated improved quality of care for patients being urgently evaluated for PRT.19
Following management of pain and neurologic symptoms, clinicians were faced with managing fatigue in nearly half of the patients (49%). The prevalence of fatigue among cancer patients and its impact on quality of life20 highlight the need for this key symptom to be addressed throughout the continuum of cancer care. National Comprehensive Cancer Network guidelines provide a comprehensive framework for addressing cancer-related fatigue.7 However, cancer-related fatigue is a largely underreported, underestimated, and thus undertreated problem.20 In a nationwide survey of members of the American Society for Radiation Oncology, radiation oncologists reported being significantly less confident in managing fatigue compared with managing other common symptoms.21 Furthermore, in a national survey of radiation oncology trainees, 67% of respondents indicated that they were not at all minimally or somewhat confident in their ability to manage fatigue symptoms. The frequency of this symptom together with the demonstrated need for improved education in fatigue management point to a need for radiation oncology palliative educational structures to include dedicated emphasis on managing fatigue in addition to other commonly encountered symptoms, such as pain.
Patients evaluated for PRT are seen across the trajectory of their metastatic cancer diagnosis. In our study, patients presented at all stages in their advanced cancers. These include patients seen at the time of initial diagnosis of cancer as well as those seen near the end of life when end-of-life care planning was underway. The broad spectrum of timing of PRT care underscores that radiation oncologists must be prepared to handle generalist palliative care issues encountered throughout the trajectory of advanced cancer care and hence need comprehensive education in generalist palliative care competencies. These include symptom management, end-of-life care coordination, and communication or goals-of-care discussions. Notably, a recent national survey of radiation oncology residents indicated that most residents, 77% on average, perceived their educational training as suboptimal across domains of generalist palliative care competencies needed in oncology practice.22 Furthermore, a majority (81%) desired greater palliative care education within training.
The most common sites treated in this study were bone, brain, and lung sites. These data provide guidance to both education and research initiatives aiming to advance PRT curriculum and care structures within departments. For example, a same-day simulation and radiation treatment program developed at Princess Margaret Hospital Palliative Radiation Oncology Program (Ontario, Canada) aids in providing streamlined care for patients with bone metastases, the most common presentation for PRT.23 Furthermore, education and research in the application of PRT techniques to bone, brain, and thoracic disease cover the majority of PRT presentations. It is notable, however, that 17% were other soft tissue body sites.
Limitations
There are a few limitations to this study. First, this is a survey-based study conducted at a single academic center within an urban setting and surrounding community regions, which affects its generalizability. Second, this study presents perspectives of radiation oncology practitioners evaluating patients and does not directly reflect patient perceptions or report of symptoms. Third, the data provided by this study are solely descriptive in nature. However, this can guide hypothesis-driven research regarding the evaluation and management of urgent palliative care issues encountered by radiation oncology clinicians and suggest educational objectives to address the needs of these patients.
Conclusions
Radiation oncologists are involved throughout the trajectory of care for advanced cancer patients. Furthermore, they manage a variety of urgent oncologic issues, most commonly metastases causing pain, brain metastases, and spinal cord or cauda equina compression. Radiation oncologists also manage many cancer-related symptoms, mostly pain, neurologic symptoms, fatigue, and gastrointestinal symptoms. These findings point toward the need for palliative care to be well integrated into radiation oncology training curricula and the need for dedicated care structures that enable rapid and multidisciplinary palliative oncology care within radiation oncology departments.
Palliative radiation therapy (PRT) plays a major role in the management of incurable cancers. Study findings have demonstrated the efficacy of using PRT in treating tumor-related bone pain,1 brain metastases and related symptoms,2 thoracic disease-causing hemoptysis or obstruction,3 gastrointestinal involvement causing bleeding and/or obstruction,4 and genitourinary and/or gynecologic involvement causing bleeding.5,6
PRT accounts for between 30% and 50% of courses of radiotherapy delivered.3 These courses of RT typically require urgent evaluation since patients are seen because of new and/or progressive symptoms that give cause for concern. The urgency of presentation requires radiation oncologists and the departments receiving these patients to be equipped to manage these cases efficiently and effectively. Furthermore, the types of cases seen, including PRT indications and related symptoms requiring management, inform the training of radiation oncology physicians as well as nursing and other clinical staff. Finally, characterizing the types of urgent PRT cases that are seen can also guide research and quality improvement endeavors for advanced cancer care in radiation oncology settings.
There is currently a paucity of data characterizing the types and frequencies of urgent PRT indications in patients who present to radiation oncology departments, as well as a lack of data detailing the related symptoms radiation oncology clinicians are managing. The aim of this study was to characterize the types and frequencies of urgent PRT consultations and the related symptoms that radiation oncologists are managing as part of patient care.
Methods
Based on national palliative care practice and national oncology care practice guidelines,7,8 we designed a survey to categorize the cancer-related palliative care issues seen by radiation oncologists. Physical symptoms, psychosocial issues, cultural consideration, spiritual needs, care coordination, advanced-care planning, goals of care, and ethical and legal issues comprised the 8 palliative care domains that we evaluated. A survey was developed and critically evaluated by 3 investigators (MK, VL, TB). Each palliative care domain was ranked by clinicians by its relevance (5-point Likert scale [range, 1-5]: 1, Not Relevant, to 5, Extremely Relevant) to the patient’s care point in radiation oncology. In addition, 31 palliative care subissues related to the primary domains were identified by clinicians based on their presence (Yes, No, Not Assessed). Clinicians were also asked whether the consulted patient’s metastatic cancer diagnosis was established (longer than 1 month) or new (within the last 1 month). In addition, clinicians noted whether the patient was returning to active oncologic care (eg, chemotherapy) or to no further anticancer therapies (eg, hospice care) after RT consultation and intervention (if deemed necessary).
The survey’s face and content validity, ease of completion, and time of completion was assessed by a panel of 7 clinicians with expertise in medical oncology, radiation oncology, palliative care, and/or survey construction. The survey was then sent in a sequential manner to 1 member of the panel at a time after incorporating each panel member’s initial comments. After each panel member’s review, the survey was edited until 2 consecutive panel members had no further suggestions for improvement.
After receiving approval from the institutional review boards of participating radiation oncology centers, we electronically surveyed radiation oncology clinicians who were conducting PRT consultations. From May 19 to September 26, 2014, all consultations were evaluated prospectively for consideration of PRT performed by a dedicated PRT service at 3 centers (a large academic cancer center and 2 participating clinicians at affiliated regional hospitals). The consultations for patients aged 18 years or older with incurable, metastatic cancers were considered eligible. The consulting clinician was e-mailed a survey for completion within 5 business days immediately after each PRT consult. Three reminders to complete the survey were sent during the 5–business-day interval. Over the entire study period, 162 consecutive patients were identified, resulting in 162 surveys being sent to 15 radiation oncology clinicians, including nurse practitioners, resident physicians, and attending physicians. Each clinician received a $25 gift card for participating, regardless of the number of surveys completed. In total, 140 of the 162 surveys were returned, resulting in a response rate of 86%.
The investigators then collected patient demographics (age, gender, race, marital status) and disease characteristics (primary cancer type, Eastern Cooperative Oncology Group Performance Status, reasons for urgent RT consult, physical symptoms requiring management at presentation, patient’s place in illness trajectory, and RT recommendation) pertaining to each completed survey from the electronic medical record. Urgent consultations were defined as any patients who needed to be seen on the same day or within a few days of the consult request.
The descriptive statistics of all these data were calculated in terms of frequencies and percentage of categorical variables. Chi-squared statistics, Fisher exact test, and nonparametric rank sum tests were applied to various categories to determine any statistically significant differences between groups.
Results
In total, 162 patients were seen in consultation for PRT during the 19-week enrollment period, or an average of 8.7 consults a week. Of that total, surveys for 140 patients were returned (Table).
The median patient age was 63 years (range, 29-89 years). A sizeable minority (20%) was 50 years or younger. The most common cancer diagnosis was lung cancer (28%), followed by breast (13%) and prostate (10%) cancers, melanoma (10%), and sarcoma (7%). Other diagnoses accounted for the remaining 32%.
Timing of PRT consult in cancer trajectory
The points in the advanced cancer illness trajectory at which patients were seen for PRT evaluation are shown in Figure 1. Most patients (63%) were seen for a PRT evaluation at the time of an established diagnosis (>1 month after diagnosis of metastatic cancer) and were continuing to further cancer therapies. An additional 19% of patients with an established diagnosis proceeded to hospice or end-of-life care after the PRT evaluation. A notable minority of patients (18%) were seen for a PRT evaluation at the time of a new diagnosis (<1 month of diagnosis of metastatic cancer), and of those, 17% went on to receive anticancer therapy after the PRT evaluation and 1% proceeded to hospice or end-of-life care.
Characteristics of PRT consults and symptoms at presentation
The primary reasons for urgent consultation for PRT are shown in Figure 2. Cancer-related pain (57%), brain metastases (29%), and malignant spinal cord or cauda equina compression (13%) were the predominant reasons for consults. Notable minorities were seen for tumor-related dyspnea (10%), bleeding (8%), and bone fractures (4%).
PRT recommendations and targets
Recommendations regarding PRT are shown in Figure 4A. Of the total 140 patients, 18 (13%) were not recommended for RT. Of the 122 patients for whom PRT was recommended, 11 (9%) received RT at more than 1 site.
Discussion and conclusions
The present study provides a descriptive overview of urgent metastatic cancer patient presentations to radiation oncology clinicians through a comprehensive evaluation of 140 consults for PRT. The most common reasons for urgent evaluation were cancer-related pain (57%), but brain metastases (29%), spinal cord compression (13%), and respiratory symptoms (10%) were also common. Other less-common indications included cancer-related dysphagia, bleeding, and poststabilization management of bone fractures. The most common symptoms requiring management by radiation oncology clinicians were pain (69%), neurologic symptoms (51%), and fatigue (49%). The study also provides a comprehensive characterization of the timeframe of PRT consultation and the treatment recommendations in this cohort. Though most PRT consults occurred at the time of an established metastatic cancer diagnosis and before further anticancer therapies, sizeable minorities occurred at the time of a new diagnosis of metastatic cancer (18%) and before comfort-focused, end-of-life care and no further anticancer therapies (20%). Most patients (87%) were recommended PRT, and of those recommended RT, 11% received RT to more than 1 site. The most common PRT sites were to bone (46%), followed by brain (29%), nonlung soft-tissue sites (17%), and lung (8%). This comprehensive description of the day-to-day urgent, advanced cancer care issues seen and managed in radiation oncology practice can help guide PRT clinical structures, education, research, and quality improvement measures in clinical practice.
Our study provides an insight into urgent symptoms encountered by radiation oncology practitioners during their routine practice. Cancer-related pain remains the most common symptom requiring management. Given the frequency with which pain management is needed among PRT patients, this study highlights the need for radiation oncologists to be well trained in symptom management, particularly as the pain response to RT can often take several days. However, studies suggest that cancer-related pain is not frequently managed by radiation oncologists.9 For example, findings from an Italian study showed that the involvement of radiation oncologists in cancer pain management remains minimal compared with other medical professionals; during the treatment course, only half of the radiation oncologists implemented specific treatment for breakthrough pain.10 A nationwide survey in the United States implicated a number of barriers to adequate pain management, including poor assessment by the physician, reluctance in prescribing opioid analgesics, perceived excessive regulation, and patient reluctance to report pain.11 Notably, in a survey of the Radiation Therapy Oncology Group study physicians, 83% believed cancer patients with pain were undermedicated, and 40% reported that pain relief in their own practice setting was suboptimal. Furthermore, in the treatment plan, adjuvants and prophylactic side effect management were frequently not used properly.12 Education of radiation oncologists in pain assessment and management is key to overcome these barriers and to ensure adequate pain management and quality of life for patients in radiation oncology.
The next most common reason for which patients presented for palliative radiation oncology consultation was for central nervous system (CNS) metastatic disease, including brain metastases and spinal cord compression. Correspondingly, the next most common issue requiring management was neurologic symptoms. Management of CNS disease is becoming increasingly complex, and it benefits from multidisciplinary evaluation to guide optimal and personalized care for each patient, including medical oncology, radiation oncology, neurosurgery and/or orthopedic spine surgery, and palliative medicine. Treatment options include supportive care or corticosteroids alone, surgical resection, whole-brain RT, and/or radiosurgery or stereotactic RT alone. These treatment options are considered on the basis of global patient factors, such as prognosis, together with metastatic-site–specific factors, such as site-related symptoms and the number of metastatic diseases or the burden of the disease.13 For example, the use of the diagnosis-specific Graded Prognostic Assessment index to predict life expectancy can help tailor management of brain metastases based on performance status, age, number of brain metastases, extracranial metastases, and cancer type. Highlighting the complexity of this common PRT presentation, Tsao and colleagues showed that there was a lack of uniform agreement among radiation oncologists for common management issues in patients with brain metastatic disease.14
For metastatic spinal cord or cauda equina compression and the associated neurologic symptoms, initiation of immediate corticosteroids and implementation of local therapy within 24 hours of presentation is paramount,15 highlighting the need for rapid, comprehensive care decision-making for these patients. Treatment options that must be weighed include the potential benefit of upfront decompressive surgery, as supported by a randomized controlled trial by Patchell and colleagues16 for patients who are surgical candidates with true cord or cauda compression and have at least 1 neurologic symptom, a prognosis of ≥3 months, paraplegia of no longer than 48 hours, and no previous RT to the site or brain metastases. Compared with the RT alone, patients receiving surgery before RT had improved ambulatory status and overall survival. Hence, neurosurgical or orthopedic consultation should be standard in the evaluation of metastatic spinal cord or cauda equina compression patients. However, patients frequently do not meet these criteria, and corticosteroids and RT alone are considered. In addition to playing a role in surgical decision-making, prognosis also has a key role in decision-making about the RT fractionation. Short-course RT (8 Gy × 1) is as effectual as longer-course regimens (3 Gy × 10) in terms of motor function.17,18 However, more dose-intense or longer-course regimens, such as 3 Gy × 10, have been shown to have more durability beyond about 6 months and are therefore considered for intermediate to good prognosis.18 The common urgent presentation of CNS metastatic disease to radiation oncology clinics together with the complexity of management and urgency of care decision-making point to the need for dedicated structures of care for these patients in radiation oncology settings. For example, dedicated PRT programs, such as the Rapid Response Radiotherapy Program in Toronto and the Supportive and Palliative Radiation Oncology service in Boston, have demonstrated improved quality of care for patients being urgently evaluated for PRT.19
Following management of pain and neurologic symptoms, clinicians were faced with managing fatigue in nearly half of the patients (49%). The prevalence of fatigue among cancer patients and its impact on quality of life20 highlight the need for this key symptom to be addressed throughout the continuum of cancer care. National Comprehensive Cancer Network guidelines provide a comprehensive framework for addressing cancer-related fatigue.7 However, cancer-related fatigue is a largely underreported, underestimated, and thus undertreated problem.20 In a nationwide survey of members of the American Society for Radiation Oncology, radiation oncologists reported being significantly less confident in managing fatigue compared with managing other common symptoms.21 Furthermore, in a national survey of radiation oncology trainees, 67% of respondents indicated that they were not at all minimally or somewhat confident in their ability to manage fatigue symptoms. The frequency of this symptom together with the demonstrated need for improved education in fatigue management point to a need for radiation oncology palliative educational structures to include dedicated emphasis on managing fatigue in addition to other commonly encountered symptoms, such as pain.
Patients evaluated for PRT are seen across the trajectory of their metastatic cancer diagnosis. In our study, patients presented at all stages in their advanced cancers. These include patients seen at the time of initial diagnosis of cancer as well as those seen near the end of life when end-of-life care planning was underway. The broad spectrum of timing of PRT care underscores that radiation oncologists must be prepared to handle generalist palliative care issues encountered throughout the trajectory of advanced cancer care and hence need comprehensive education in generalist palliative care competencies. These include symptom management, end-of-life care coordination, and communication or goals-of-care discussions. Notably, a recent national survey of radiation oncology residents indicated that most residents, 77% on average, perceived their educational training as suboptimal across domains of generalist palliative care competencies needed in oncology practice.22 Furthermore, a majority (81%) desired greater palliative care education within training.
The most common sites treated in this study were bone, brain, and lung sites. These data provide guidance to both education and research initiatives aiming to advance PRT curriculum and care structures within departments. For example, a same-day simulation and radiation treatment program developed at Princess Margaret Hospital Palliative Radiation Oncology Program (Ontario, Canada) aids in providing streamlined care for patients with bone metastases, the most common presentation for PRT.23 Furthermore, education and research in the application of PRT techniques to bone, brain, and thoracic disease cover the majority of PRT presentations. It is notable, however, that 17% were other soft tissue body sites.
Limitations
There are a few limitations to this study. First, this is a survey-based study conducted at a single academic center within an urban setting and surrounding community regions, which affects its generalizability. Second, this study presents perspectives of radiation oncology practitioners evaluating patients and does not directly reflect patient perceptions or report of symptoms. Third, the data provided by this study are solely descriptive in nature. However, this can guide hypothesis-driven research regarding the evaluation and management of urgent palliative care issues encountered by radiation oncology clinicians and suggest educational objectives to address the needs of these patients.
Conclusions
Radiation oncologists are involved throughout the trajectory of care for advanced cancer patients. Furthermore, they manage a variety of urgent oncologic issues, most commonly metastases causing pain, brain metastases, and spinal cord or cauda equina compression. Radiation oncologists also manage many cancer-related symptoms, mostly pain, neurologic symptoms, fatigue, and gastrointestinal symptoms. These findings point toward the need for palliative care to be well integrated into radiation oncology training curricula and the need for dedicated care structures that enable rapid and multidisciplinary palliative oncology care within radiation oncology departments.
1. Chow E, Harris K, Fan G, Tsao M, Sze WM. Palliative radiotherapy trials for bone metastases: a systematic review. J Clin Oncol. 2007;25(11):1423-1436.
2. van Oorschot B, Rades D, Schulze W, Beckmann G, Feyer P. Palliative radiotherapy--new approaches. Semin Oncol. 2011;38(3):443-449.
3. Simone CB II, Jones JA. Palliative care for patients with locally advanced and metastatic non-small cell lung cancer. Ann Palliat Med. 2013;2(4):178-188.
4. Cihoric N, Crowe S, Eychmüller S, Aebersold DM, Ghadjar P. Clinically significant bleeding in incurable cancer patients: effectiveness of hemostatic radiotherapy. Radiat Oncol. 2012;7:132.
5. Duchesne GM, Bolger JJ, Griffiths GO, et al. A randomized trial of hypofractionated schedules of palliative radiotherapy in the management of bladder carcinoma: results of medical research council trial BA09. Int J Radiat Oncol Biol Phys. 2000;47(2):379-388.
6. Onsrud M, Hagen B, Strickert T. 10-Gy single-fraction pelvic irradiation for palliation and life prolongation in patients with cancer of the cervix and corpus uteri. Gynecol Oncol. 2001;82(1):167-171.
7. NCCN Guidelines(R) Updates. J Natl Compr Canc Netw. 2013;11(9):xxxii-xxxvi.
8. Colby WH, Dahlin C, Lantos J, Carney J, Christopher M. The National Consensus Project for Quality Palliative Care Clinical Practice Guidelines Domain 8: ethical and legal aspects of care. HEC Forum. 2010;22(2):117-131.
9. Stockler MR, Wilcken NR. Why is management of cancer pain still a problem? J Clin Oncol. 2012;30(16):1907-1908.
10. Caravatta L, Ramella S, Melano A, et al. Breakthrough pain management in patients undergoing radiotherapy: a national survey on behalf of the Palliative and Supportive Care Study Group. Tumori. 2015;101(6):603-608.
11. Breuer B, Fleishman SB, Cruciani RA, Portenoy RK. Medical oncologists’ attitudes and practice in cancer pain management: a national survey. J Clin Oncol. 2011;29(36):4769-4775.
12. Cleeland CS, Janjan NA, Scott CB, Seiferheld WF, Curran WJ. Cancer pain management by radiotherapists: a survey of radiation therapy oncology group physicians. Int J Radiat Oncol Biol Phys. 2000;47(1):203-208.
13. Tsao MN, Rades D, Wirth A, et al. Radiotherapeutic and surgical management for newly diagnosed brain metastasis(es): an American Society for Radiation Oncology evidence-based guideline. Pract Radiat Oncol. 2012;2(3):210-225.
14. Tsao MN, Rades D, Wirth A, et al. International practice survey on the management of brain metastases: third international consensus workshop on palliative radiotherapy and symptom control. Clin Oncol (R Coll Radiol). 2012;24(6):e81-e92.
15. Tang V, Harvey D, Park Dorsay J, Jiang S, Rathbone MP. Prognostic indicators in metastatic spinal cord compression: using functional independence measure and Tokuhashi scale to optimize rehabilitation planning. Spinal Cord. 2007;45(10):671-677.
16. Patchell RA, Tibbs PA, Regine WF, et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomised trial. Lancet. 2005;366(9486):643-648.
17. Rades D, Stalpers LJ, Veninga T, et al. Evaluation of five radiation schedules and prognostic factors for metastatic spinal cord compression. J Clin Oncol. 2005;23(15):3366-3375.
18. Rades D, Stalpers LJ, Hulshof MC, et al. Comparison of 1 x 8 Gy and 10 x 3 Gy for functional outcome in patients with metastatic spinal cord compression. Int J Radiat Oncol Biol Phys. 2005;62(2):514-518.
19. Dennis K, Linden K, Balboni T, Chow E. Rapid access palliative radiation therapy programs: an efficient model of care. Future Oncol. 2015;11(17):2417-2426.
20. Kapoor A, Singhal MK, Bagri PK, Narayan S, Beniwal S, Kumar HS. Cancer related fatigue: a ubiquitous problem yet so under reported, under recognized and under treated. South Asian J Cancer. 2015;4(1):21-23.
21. Wei RL, Mattes MD, Yu J, et al. Attitudes of radiation oncologists toward palliative and supportive care in the United States: report on national membership survey by the American Society for Radiation Oncology (ASTRO). Pract Radiat Oncol. 2017;7(2):113-119.
22. Krishnan M, Racsa M, Jones J, et al. Radiation oncology resident palliative education. Pract Radiat Oncol. 2017;7(6):e439-e448.
23. McDonald R, Chow E, Lam H, Rowbottom L, Soliman H. International patterns of practice in radiotherapy for bone metastases: a review of the literature. J Bone Oncol. 2014;3(3-4):96-102.
1. Chow E, Harris K, Fan G, Tsao M, Sze WM. Palliative radiotherapy trials for bone metastases: a systematic review. J Clin Oncol. 2007;25(11):1423-1436.
2. van Oorschot B, Rades D, Schulze W, Beckmann G, Feyer P. Palliative radiotherapy--new approaches. Semin Oncol. 2011;38(3):443-449.
3. Simone CB II, Jones JA. Palliative care for patients with locally advanced and metastatic non-small cell lung cancer. Ann Palliat Med. 2013;2(4):178-188.
4. Cihoric N, Crowe S, Eychmüller S, Aebersold DM, Ghadjar P. Clinically significant bleeding in incurable cancer patients: effectiveness of hemostatic radiotherapy. Radiat Oncol. 2012;7:132.
5. Duchesne GM, Bolger JJ, Griffiths GO, et al. A randomized trial of hypofractionated schedules of palliative radiotherapy in the management of bladder carcinoma: results of medical research council trial BA09. Int J Radiat Oncol Biol Phys. 2000;47(2):379-388.
6. Onsrud M, Hagen B, Strickert T. 10-Gy single-fraction pelvic irradiation for palliation and life prolongation in patients with cancer of the cervix and corpus uteri. Gynecol Oncol. 2001;82(1):167-171.
7. NCCN Guidelines(R) Updates. J Natl Compr Canc Netw. 2013;11(9):xxxii-xxxvi.
8. Colby WH, Dahlin C, Lantos J, Carney J, Christopher M. The National Consensus Project for Quality Palliative Care Clinical Practice Guidelines Domain 8: ethical and legal aspects of care. HEC Forum. 2010;22(2):117-131.
9. Stockler MR, Wilcken NR. Why is management of cancer pain still a problem? J Clin Oncol. 2012;30(16):1907-1908.
10. Caravatta L, Ramella S, Melano A, et al. Breakthrough pain management in patients undergoing radiotherapy: a national survey on behalf of the Palliative and Supportive Care Study Group. Tumori. 2015;101(6):603-608.
11. Breuer B, Fleishman SB, Cruciani RA, Portenoy RK. Medical oncologists’ attitudes and practice in cancer pain management: a national survey. J Clin Oncol. 2011;29(36):4769-4775.
12. Cleeland CS, Janjan NA, Scott CB, Seiferheld WF, Curran WJ. Cancer pain management by radiotherapists: a survey of radiation therapy oncology group physicians. Int J Radiat Oncol Biol Phys. 2000;47(1):203-208.
13. Tsao MN, Rades D, Wirth A, et al. Radiotherapeutic and surgical management for newly diagnosed brain metastasis(es): an American Society for Radiation Oncology evidence-based guideline. Pract Radiat Oncol. 2012;2(3):210-225.
14. Tsao MN, Rades D, Wirth A, et al. International practice survey on the management of brain metastases: third international consensus workshop on palliative radiotherapy and symptom control. Clin Oncol (R Coll Radiol). 2012;24(6):e81-e92.
15. Tang V, Harvey D, Park Dorsay J, Jiang S, Rathbone MP. Prognostic indicators in metastatic spinal cord compression: using functional independence measure and Tokuhashi scale to optimize rehabilitation planning. Spinal Cord. 2007;45(10):671-677.
16. Patchell RA, Tibbs PA, Regine WF, et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomised trial. Lancet. 2005;366(9486):643-648.
17. Rades D, Stalpers LJ, Veninga T, et al. Evaluation of five radiation schedules and prognostic factors for metastatic spinal cord compression. J Clin Oncol. 2005;23(15):3366-3375.
18. Rades D, Stalpers LJ, Hulshof MC, et al. Comparison of 1 x 8 Gy and 10 x 3 Gy for functional outcome in patients with metastatic spinal cord compression. Int J Radiat Oncol Biol Phys. 2005;62(2):514-518.
19. Dennis K, Linden K, Balboni T, Chow E. Rapid access palliative radiation therapy programs: an efficient model of care. Future Oncol. 2015;11(17):2417-2426.
20. Kapoor A, Singhal MK, Bagri PK, Narayan S, Beniwal S, Kumar HS. Cancer related fatigue: a ubiquitous problem yet so under reported, under recognized and under treated. South Asian J Cancer. 2015;4(1):21-23.
21. Wei RL, Mattes MD, Yu J, et al. Attitudes of radiation oncologists toward palliative and supportive care in the United States: report on national membership survey by the American Society for Radiation Oncology (ASTRO). Pract Radiat Oncol. 2017;7(2):113-119.
22. Krishnan M, Racsa M, Jones J, et al. Radiation oncology resident palliative education. Pract Radiat Oncol. 2017;7(6):e439-e448.
23. McDonald R, Chow E, Lam H, Rowbottom L, Soliman H. International patterns of practice in radiotherapy for bone metastases: a review of the literature. J Bone Oncol. 2014;3(3-4):96-102.
Neratinib extends adjuvant treatment of patients with HER2-positive breast cancer
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
More biosimilars reach the market in efforts to improve access and cut costs
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
Finding your practice home base
As summer winds down and we begin to gear up to return to school or work, I was thinking about new and returning hem-onc residents, fellows, and young attendings and a question I routinely get from them: what should I do next in my career? I always answer by holding up 3 fingers and telling them that they can practice 1, at a university hospital; 2, at a university teaching affiliate; or 3, at a community hospital or practice with a little or no university affiliation. These days, trainees in hematology-oncology are often advised to be highly specialty-specific when they plan their long-term careers and to focus on a particular cancer or hematologic disorder. That is fine if you want to remain in an academic or university-based practice, but not if community practice is your preference. So, what are the differences among these 3 options?
Option 1, to remain in a university setting where you can be highly focused and specialized in a single narrowly defined area, could be satisfying, but keep in mind that the institution expects results! You will be carefully monitored for research output and teaching and administration commitments, and your interaction with patients could add up to less than 50% of your time. Publication and grant renewal will also play a role and therefore take up your time.
If you are considering option 2 – to work at a university teaching affiliate hospital – you need to bear in mind that you likely will see a patient population with a much broader range of diagnoses than would be the case with the first option. Patient care for option 2 will take up more than 50% of your time, so it might be a little more challenging to stay current, but perhaps more refreshing if you enjoy contact with patients. Teaching, research, and administration will surely be available, and publication and grant renewal will play as big or small a role as you want.
Option 3 would be to join a community hospital or practice where the primary focus is on patient care and the diagnoses will span the hematology and oncology spectrum. This type of practice can be very demanding of one’s time, but as rewarding as the other options, especially if you value contact with patients. With this option, one is more likely to practice as a generalist, perhaps with an emphasis in one of the hem-onc specialties, but able to treat a cluster of different types of cancer as well.
I always advise trainees to be sure they ask physicians practicing in each of these options to give examples of what their best and worst days are like so that they can get some idea of what the daily humdrum and challenges would encompass. What did I choose? I have always gone with option 2 and have been very happy in that setting.
In this issue…
More biosimilars head our way. Turning to the current issue of the journal, on page e181, Dr Jane de Lartigue discusses 2 new biosimilars recently approved by the United States Food and Drug Administration (FDA) – epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for chemotherapy-induced anemia (CIA), and pegfilgrastim-jmdb (Fulphila; Mylan and Biocon) for prevention of febrile neutropenia. As Dr de Lartigue notes, biosimilars are copies of FDA-approved biologic drugs that cannot be identical to the reference drug but demonstrate a high similarity to it. In this case, the reference drug for epoetin alfa-epbx is epoetin alfa (Epogen/Procrit, Amgen) and for pegfilgrastim-jmdb, it is pegfilgrastim (Neulasta, Amgen). As the reference drugs’ patents expire, biosimilars are being developed to increase competition in the marketplace in an effort to reduce costs and improve patient access to these therapies. Indeed, the FDA is working to streamline the biosimilar approval process to facilitate that access.
Reading this article got me thinking about something I often have to consider in the course of my work: transfusion versus erythropoiesis-stimulating agents (ESAs)? Recombinant erythropoietin drugs such as the biosimilar, epoetin alfa-epbx, and its reference drug are grouped together as ESAs, and have been used to treat CIA since the late 1980s. However, there were a few trials that used higher-dose ESA or set high hemoglobin targets, and their findings suggested that ESAs may shorten survival in patients with cancer or increase tumor growth, or both. The use of ESAs took a nosedive after the 2007 decision by the FDA’s Oncologic Drugs Advisory Committee to rein in their use for a hard start of ESA treatment at less than 10 g/dL hemoglobin, and not higher. Subsequent trials addressed the concerns about survival and tumor growth. A meta-analysis of 60 randomized, placebo-controlled trials of ESAs in CIA found that there was no difference in overall survival between the study and control groups.1 Likewise, findings from an FDA-mandated trial with epoetin alfa (Procrit) in patients with metastatic breast cancer have reported that there was no significant difference in overall survival between the study and control groups.2 The results of a second FDA-mandated trial with darbepoetin alfa (Aranesp, Amgen) in patients with metastatic lung cancer are expected to be released soon. The FDA lifted the ESA Risk Evaluation and Mitigation Strategy based on those findings. However, many practitioners, both young and old, continue to shy away from using ESAs because of the FDA black box warning that remains in place despite the latest data.3The use of transfusion ticked up reciprocally with the decline in ESA use, but perhaps we should re-evaluate the use of these agents in our practice, especially now that the less costly, equally safe and effective biosimilars are becoming available and we have the new survival data. Transfusions are time consuming and have side effects, including allergic reaction and infection risk, whereas ESAs are easily administered by injection, which patients might find preferable.
Malignancies in patients with HIV-AIDS. On page e188, Koppaka and colleagues report on a study in India of the patterns of malignancies in patients with HIV-AIDS. I began my career just as the first reports of what became known as HIV-AIDS emerged, and we were all mystified by what was killing these patients and the curious hematologic and oncologic problems they developed. Back then, the patients were profoundly immunosuppressed, and the immunosuppression cancers of non-Hodgkin lymphoma, usually higher grade, and Kaposi sarcoma were most prevalent and today are collectively labeled AIDS-defining malignancies (ADMs).
Fast forward to present day, and we have extremely effective antiretroviral therapies that have resulted in a significant reduction in mortality among HIV-infected individuals who are now living long enough to get what we call non–AIDS-defining malignancies (NADMs) such as anal or cervical cancers, hepatoma (hepatocellular carcinoma), Hodgkin lymphoma, and lung cancer. Of note is that these NADMs are all highly viral associated, with anal and cervical cancers linked to infection with the human papillomavirus; hepatoma linked to the hepatitis B/C viruses; Hodgkin lymphoma to the Epstein-Barr virus; and lung cancer, possibly also HPV. Fortunately, these days we can use standard-dose chemoradiation therapy for all HIV-related cancers because the patients’ immune systems are much better reconstituted and the modern-day antiretroviral therapies have much less drug–drug interaction thanks to the advent of the integrase inhibitors. The researchers give an excellent breakdown of the occurrence of these malignancies, as well as an analysis of the correlation between CD4 counts and the different malignancies.
Immunotherapy-related side effects in the ED. What happens when our patients who are on immunotherapy end up in the emergency department (ED) with therapy-related symptoms? And what can the treating oncologist do to help the ED physician achieve the best possible outcome for the patient? I spoke to Dr Maura Sammon, an ED physician, about some of the more common of these side effects – lung, gastrointestinal, rash, and endocrine-related problems – and she describes in detail how physicians in the ED would triage and treat the patient. Dr Sammon also emphasizes the importance of communication: first, between the treating oncologist and patient, about the differences between chemotherapy and immunotherapy; and second, between the ED physician and the treating oncologist as soon as possible after the patient has presented to ensure a good outcome. The interview is part of The JCSO Interview series. It is jam-packed with useful, how-to information, and you can read a transcript of it on page e216 of this issue, or you can listen to it online.4
We round off the issue with a selection of Case Reports (pp. e200-e209), an original report on the characteristics of urgent palliative cancer care consultations encountered by radiation oncologists (p. e193), and a New Therapies feature, also by Dr de Lartigue, focusing on the rarity and complexities of sarcomas (p. e210).
Those are my dog-day-of-summer thoughts as we head toward another Labor Day and a new academic year. Since we are all online now, we encourage you to listen to my bimonthly podcast of each issue on our website at www.jcso-online.com, and of course, follow us on Twitter (@jcs_onc) and Instagram (@jcsoncology) and like us on Facebook.
1. Glaspy J, Crawford J, Vansteenkiste J, et al. Erythropoiesis-stimulating agents in oncology: a study-level meta-analysis of survival and other safety outcomes. Br J Cancer. 2010;102(2):301-315.
2. Leyland-Jones B, Bondarenko I, Nemsadze G, et al. A randomized, open-label, multicenter, phase III study of epoetin alfa versus best standard of care in anemic patients with metastatic breast cancer receiving standard chemotherapy. J Clin Oncol. 2016;34:1197-1207.
3. US Food and Drug Administration release. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Last updated April 13, 2017. Accessed August 20, 2018.
4. Henry D, Sammon M. Treating immunotherapy-related AEs in the emergency department [Audio]. https://www.mdedge.com/jcso/article/171966/patient-survivor-care/treating-immunotherapy-related-aes-emergency-department. Published August 6, 2018.
As summer winds down and we begin to gear up to return to school or work, I was thinking about new and returning hem-onc residents, fellows, and young attendings and a question I routinely get from them: what should I do next in my career? I always answer by holding up 3 fingers and telling them that they can practice 1, at a university hospital; 2, at a university teaching affiliate; or 3, at a community hospital or practice with a little or no university affiliation. These days, trainees in hematology-oncology are often advised to be highly specialty-specific when they plan their long-term careers and to focus on a particular cancer or hematologic disorder. That is fine if you want to remain in an academic or university-based practice, but not if community practice is your preference. So, what are the differences among these 3 options?
Option 1, to remain in a university setting where you can be highly focused and specialized in a single narrowly defined area, could be satisfying, but keep in mind that the institution expects results! You will be carefully monitored for research output and teaching and administration commitments, and your interaction with patients could add up to less than 50% of your time. Publication and grant renewal will also play a role and therefore take up your time.
If you are considering option 2 – to work at a university teaching affiliate hospital – you need to bear in mind that you likely will see a patient population with a much broader range of diagnoses than would be the case with the first option. Patient care for option 2 will take up more than 50% of your time, so it might be a little more challenging to stay current, but perhaps more refreshing if you enjoy contact with patients. Teaching, research, and administration will surely be available, and publication and grant renewal will play as big or small a role as you want.
Option 3 would be to join a community hospital or practice where the primary focus is on patient care and the diagnoses will span the hematology and oncology spectrum. This type of practice can be very demanding of one’s time, but as rewarding as the other options, especially if you value contact with patients. With this option, one is more likely to practice as a generalist, perhaps with an emphasis in one of the hem-onc specialties, but able to treat a cluster of different types of cancer as well.
I always advise trainees to be sure they ask physicians practicing in each of these options to give examples of what their best and worst days are like so that they can get some idea of what the daily humdrum and challenges would encompass. What did I choose? I have always gone with option 2 and have been very happy in that setting.
In this issue…
More biosimilars head our way. Turning to the current issue of the journal, on page e181, Dr Jane de Lartigue discusses 2 new biosimilars recently approved by the United States Food and Drug Administration (FDA) – epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for chemotherapy-induced anemia (CIA), and pegfilgrastim-jmdb (Fulphila; Mylan and Biocon) for prevention of febrile neutropenia. As Dr de Lartigue notes, biosimilars are copies of FDA-approved biologic drugs that cannot be identical to the reference drug but demonstrate a high similarity to it. In this case, the reference drug for epoetin alfa-epbx is epoetin alfa (Epogen/Procrit, Amgen) and for pegfilgrastim-jmdb, it is pegfilgrastim (Neulasta, Amgen). As the reference drugs’ patents expire, biosimilars are being developed to increase competition in the marketplace in an effort to reduce costs and improve patient access to these therapies. Indeed, the FDA is working to streamline the biosimilar approval process to facilitate that access.
Reading this article got me thinking about something I often have to consider in the course of my work: transfusion versus erythropoiesis-stimulating agents (ESAs)? Recombinant erythropoietin drugs such as the biosimilar, epoetin alfa-epbx, and its reference drug are grouped together as ESAs, and have been used to treat CIA since the late 1980s. However, there were a few trials that used higher-dose ESA or set high hemoglobin targets, and their findings suggested that ESAs may shorten survival in patients with cancer or increase tumor growth, or both. The use of ESAs took a nosedive after the 2007 decision by the FDA’s Oncologic Drugs Advisory Committee to rein in their use for a hard start of ESA treatment at less than 10 g/dL hemoglobin, and not higher. Subsequent trials addressed the concerns about survival and tumor growth. A meta-analysis of 60 randomized, placebo-controlled trials of ESAs in CIA found that there was no difference in overall survival between the study and control groups.1 Likewise, findings from an FDA-mandated trial with epoetin alfa (Procrit) in patients with metastatic breast cancer have reported that there was no significant difference in overall survival between the study and control groups.2 The results of a second FDA-mandated trial with darbepoetin alfa (Aranesp, Amgen) in patients with metastatic lung cancer are expected to be released soon. The FDA lifted the ESA Risk Evaluation and Mitigation Strategy based on those findings. However, many practitioners, both young and old, continue to shy away from using ESAs because of the FDA black box warning that remains in place despite the latest data.3The use of transfusion ticked up reciprocally with the decline in ESA use, but perhaps we should re-evaluate the use of these agents in our practice, especially now that the less costly, equally safe and effective biosimilars are becoming available and we have the new survival data. Transfusions are time consuming and have side effects, including allergic reaction and infection risk, whereas ESAs are easily administered by injection, which patients might find preferable.
Malignancies in patients with HIV-AIDS. On page e188, Koppaka and colleagues report on a study in India of the patterns of malignancies in patients with HIV-AIDS. I began my career just as the first reports of what became known as HIV-AIDS emerged, and we were all mystified by what was killing these patients and the curious hematologic and oncologic problems they developed. Back then, the patients were profoundly immunosuppressed, and the immunosuppression cancers of non-Hodgkin lymphoma, usually higher grade, and Kaposi sarcoma were most prevalent and today are collectively labeled AIDS-defining malignancies (ADMs).
Fast forward to present day, and we have extremely effective antiretroviral therapies that have resulted in a significant reduction in mortality among HIV-infected individuals who are now living long enough to get what we call non–AIDS-defining malignancies (NADMs) such as anal or cervical cancers, hepatoma (hepatocellular carcinoma), Hodgkin lymphoma, and lung cancer. Of note is that these NADMs are all highly viral associated, with anal and cervical cancers linked to infection with the human papillomavirus; hepatoma linked to the hepatitis B/C viruses; Hodgkin lymphoma to the Epstein-Barr virus; and lung cancer, possibly also HPV. Fortunately, these days we can use standard-dose chemoradiation therapy for all HIV-related cancers because the patients’ immune systems are much better reconstituted and the modern-day antiretroviral therapies have much less drug–drug interaction thanks to the advent of the integrase inhibitors. The researchers give an excellent breakdown of the occurrence of these malignancies, as well as an analysis of the correlation between CD4 counts and the different malignancies.
Immunotherapy-related side effects in the ED. What happens when our patients who are on immunotherapy end up in the emergency department (ED) with therapy-related symptoms? And what can the treating oncologist do to help the ED physician achieve the best possible outcome for the patient? I spoke to Dr Maura Sammon, an ED physician, about some of the more common of these side effects – lung, gastrointestinal, rash, and endocrine-related problems – and she describes in detail how physicians in the ED would triage and treat the patient. Dr Sammon also emphasizes the importance of communication: first, between the treating oncologist and patient, about the differences between chemotherapy and immunotherapy; and second, between the ED physician and the treating oncologist as soon as possible after the patient has presented to ensure a good outcome. The interview is part of The JCSO Interview series. It is jam-packed with useful, how-to information, and you can read a transcript of it on page e216 of this issue, or you can listen to it online.4
We round off the issue with a selection of Case Reports (pp. e200-e209), an original report on the characteristics of urgent palliative cancer care consultations encountered by radiation oncologists (p. e193), and a New Therapies feature, also by Dr de Lartigue, focusing on the rarity and complexities of sarcomas (p. e210).
Those are my dog-day-of-summer thoughts as we head toward another Labor Day and a new academic year. Since we are all online now, we encourage you to listen to my bimonthly podcast of each issue on our website at www.jcso-online.com, and of course, follow us on Twitter (@jcs_onc) and Instagram (@jcsoncology) and like us on Facebook.
As summer winds down and we begin to gear up to return to school or work, I was thinking about new and returning hem-onc residents, fellows, and young attendings and a question I routinely get from them: what should I do next in my career? I always answer by holding up 3 fingers and telling them that they can practice 1, at a university hospital; 2, at a university teaching affiliate; or 3, at a community hospital or practice with a little or no university affiliation. These days, trainees in hematology-oncology are often advised to be highly specialty-specific when they plan their long-term careers and to focus on a particular cancer or hematologic disorder. That is fine if you want to remain in an academic or university-based practice, but not if community practice is your preference. So, what are the differences among these 3 options?
Option 1, to remain in a university setting where you can be highly focused and specialized in a single narrowly defined area, could be satisfying, but keep in mind that the institution expects results! You will be carefully monitored for research output and teaching and administration commitments, and your interaction with patients could add up to less than 50% of your time. Publication and grant renewal will also play a role and therefore take up your time.
If you are considering option 2 – to work at a university teaching affiliate hospital – you need to bear in mind that you likely will see a patient population with a much broader range of diagnoses than would be the case with the first option. Patient care for option 2 will take up more than 50% of your time, so it might be a little more challenging to stay current, but perhaps more refreshing if you enjoy contact with patients. Teaching, research, and administration will surely be available, and publication and grant renewal will play as big or small a role as you want.
Option 3 would be to join a community hospital or practice where the primary focus is on patient care and the diagnoses will span the hematology and oncology spectrum. This type of practice can be very demanding of one’s time, but as rewarding as the other options, especially if you value contact with patients. With this option, one is more likely to practice as a generalist, perhaps with an emphasis in one of the hem-onc specialties, but able to treat a cluster of different types of cancer as well.
I always advise trainees to be sure they ask physicians practicing in each of these options to give examples of what their best and worst days are like so that they can get some idea of what the daily humdrum and challenges would encompass. What did I choose? I have always gone with option 2 and have been very happy in that setting.
In this issue…
More biosimilars head our way. Turning to the current issue of the journal, on page e181, Dr Jane de Lartigue discusses 2 new biosimilars recently approved by the United States Food and Drug Administration (FDA) – epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for chemotherapy-induced anemia (CIA), and pegfilgrastim-jmdb (Fulphila; Mylan and Biocon) for prevention of febrile neutropenia. As Dr de Lartigue notes, biosimilars are copies of FDA-approved biologic drugs that cannot be identical to the reference drug but demonstrate a high similarity to it. In this case, the reference drug for epoetin alfa-epbx is epoetin alfa (Epogen/Procrit, Amgen) and for pegfilgrastim-jmdb, it is pegfilgrastim (Neulasta, Amgen). As the reference drugs’ patents expire, biosimilars are being developed to increase competition in the marketplace in an effort to reduce costs and improve patient access to these therapies. Indeed, the FDA is working to streamline the biosimilar approval process to facilitate that access.
Reading this article got me thinking about something I often have to consider in the course of my work: transfusion versus erythropoiesis-stimulating agents (ESAs)? Recombinant erythropoietin drugs such as the biosimilar, epoetin alfa-epbx, and its reference drug are grouped together as ESAs, and have been used to treat CIA since the late 1980s. However, there were a few trials that used higher-dose ESA or set high hemoglobin targets, and their findings suggested that ESAs may shorten survival in patients with cancer or increase tumor growth, or both. The use of ESAs took a nosedive after the 2007 decision by the FDA’s Oncologic Drugs Advisory Committee to rein in their use for a hard start of ESA treatment at less than 10 g/dL hemoglobin, and not higher. Subsequent trials addressed the concerns about survival and tumor growth. A meta-analysis of 60 randomized, placebo-controlled trials of ESAs in CIA found that there was no difference in overall survival between the study and control groups.1 Likewise, findings from an FDA-mandated trial with epoetin alfa (Procrit) in patients with metastatic breast cancer have reported that there was no significant difference in overall survival between the study and control groups.2 The results of a second FDA-mandated trial with darbepoetin alfa (Aranesp, Amgen) in patients with metastatic lung cancer are expected to be released soon. The FDA lifted the ESA Risk Evaluation and Mitigation Strategy based on those findings. However, many practitioners, both young and old, continue to shy away from using ESAs because of the FDA black box warning that remains in place despite the latest data.3The use of transfusion ticked up reciprocally with the decline in ESA use, but perhaps we should re-evaluate the use of these agents in our practice, especially now that the less costly, equally safe and effective biosimilars are becoming available and we have the new survival data. Transfusions are time consuming and have side effects, including allergic reaction and infection risk, whereas ESAs are easily administered by injection, which patients might find preferable.
Malignancies in patients with HIV-AIDS. On page e188, Koppaka and colleagues report on a study in India of the patterns of malignancies in patients with HIV-AIDS. I began my career just as the first reports of what became known as HIV-AIDS emerged, and we were all mystified by what was killing these patients and the curious hematologic and oncologic problems they developed. Back then, the patients were profoundly immunosuppressed, and the immunosuppression cancers of non-Hodgkin lymphoma, usually higher grade, and Kaposi sarcoma were most prevalent and today are collectively labeled AIDS-defining malignancies (ADMs).
Fast forward to present day, and we have extremely effective antiretroviral therapies that have resulted in a significant reduction in mortality among HIV-infected individuals who are now living long enough to get what we call non–AIDS-defining malignancies (NADMs) such as anal or cervical cancers, hepatoma (hepatocellular carcinoma), Hodgkin lymphoma, and lung cancer. Of note is that these NADMs are all highly viral associated, with anal and cervical cancers linked to infection with the human papillomavirus; hepatoma linked to the hepatitis B/C viruses; Hodgkin lymphoma to the Epstein-Barr virus; and lung cancer, possibly also HPV. Fortunately, these days we can use standard-dose chemoradiation therapy for all HIV-related cancers because the patients’ immune systems are much better reconstituted and the modern-day antiretroviral therapies have much less drug–drug interaction thanks to the advent of the integrase inhibitors. The researchers give an excellent breakdown of the occurrence of these malignancies, as well as an analysis of the correlation between CD4 counts and the different malignancies.
Immunotherapy-related side effects in the ED. What happens when our patients who are on immunotherapy end up in the emergency department (ED) with therapy-related symptoms? And what can the treating oncologist do to help the ED physician achieve the best possible outcome for the patient? I spoke to Dr Maura Sammon, an ED physician, about some of the more common of these side effects – lung, gastrointestinal, rash, and endocrine-related problems – and she describes in detail how physicians in the ED would triage and treat the patient. Dr Sammon also emphasizes the importance of communication: first, between the treating oncologist and patient, about the differences between chemotherapy and immunotherapy; and second, between the ED physician and the treating oncologist as soon as possible after the patient has presented to ensure a good outcome. The interview is part of The JCSO Interview series. It is jam-packed with useful, how-to information, and you can read a transcript of it on page e216 of this issue, or you can listen to it online.4
We round off the issue with a selection of Case Reports (pp. e200-e209), an original report on the characteristics of urgent palliative cancer care consultations encountered by radiation oncologists (p. e193), and a New Therapies feature, also by Dr de Lartigue, focusing on the rarity and complexities of sarcomas (p. e210).
Those are my dog-day-of-summer thoughts as we head toward another Labor Day and a new academic year. Since we are all online now, we encourage you to listen to my bimonthly podcast of each issue on our website at www.jcso-online.com, and of course, follow us on Twitter (@jcs_onc) and Instagram (@jcsoncology) and like us on Facebook.
1. Glaspy J, Crawford J, Vansteenkiste J, et al. Erythropoiesis-stimulating agents in oncology: a study-level meta-analysis of survival and other safety outcomes. Br J Cancer. 2010;102(2):301-315.
2. Leyland-Jones B, Bondarenko I, Nemsadze G, et al. A randomized, open-label, multicenter, phase III study of epoetin alfa versus best standard of care in anemic patients with metastatic breast cancer receiving standard chemotherapy. J Clin Oncol. 2016;34:1197-1207.
3. US Food and Drug Administration release. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Last updated April 13, 2017. Accessed August 20, 2018.
4. Henry D, Sammon M. Treating immunotherapy-related AEs in the emergency department [Audio]. https://www.mdedge.com/jcso/article/171966/patient-survivor-care/treating-immunotherapy-related-aes-emergency-department. Published August 6, 2018.
1. Glaspy J, Crawford J, Vansteenkiste J, et al. Erythropoiesis-stimulating agents in oncology: a study-level meta-analysis of survival and other safety outcomes. Br J Cancer. 2010;102(2):301-315.
2. Leyland-Jones B, Bondarenko I, Nemsadze G, et al. A randomized, open-label, multicenter, phase III study of epoetin alfa versus best standard of care in anemic patients with metastatic breast cancer receiving standard chemotherapy. J Clin Oncol. 2016;34:1197-1207.
3. US Food and Drug Administration release. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Last updated April 13, 2017. Accessed August 20, 2018.
4. Henry D, Sammon M. Treating immunotherapy-related AEs in the emergency department [Audio]. https://www.mdedge.com/jcso/article/171966/patient-survivor-care/treating-immunotherapy-related-aes-emergency-department. Published August 6, 2018.
Carcinoma of the colon in a child
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).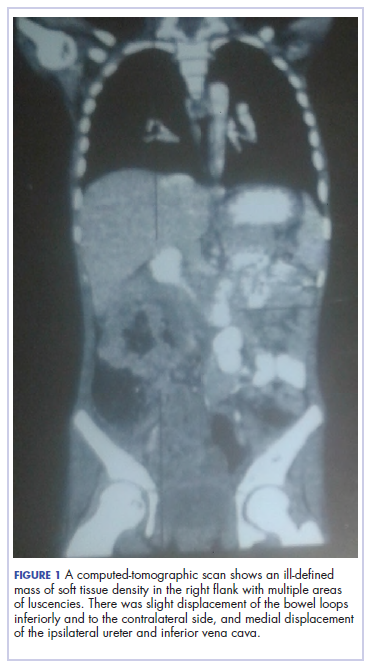
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).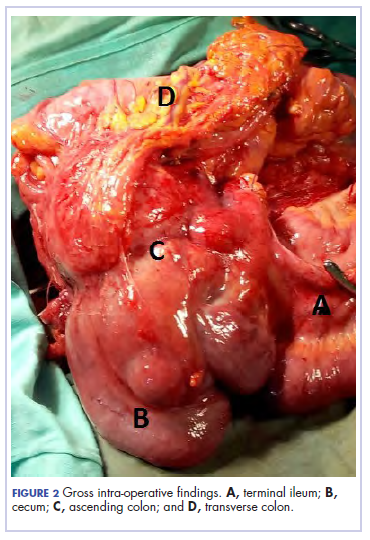
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).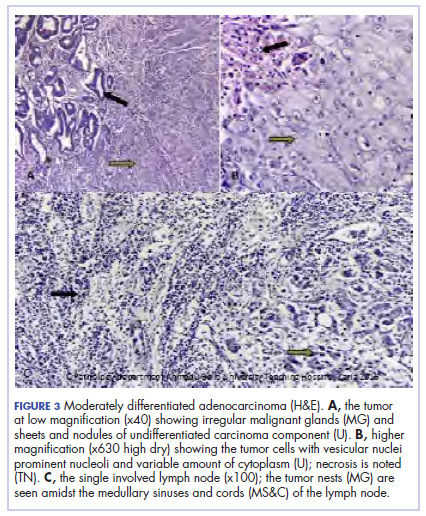
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.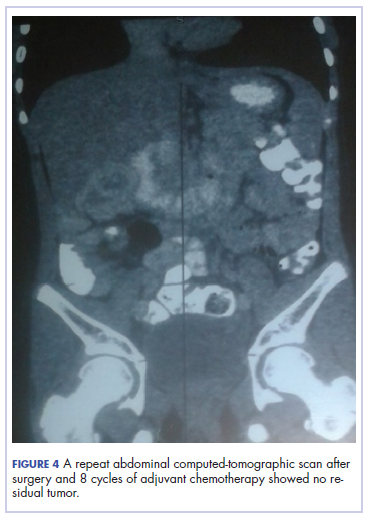
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
Dr David Henry's top ASCO selections in hematology and bone health
David Henry, MD, the Editor-in-Chief of The Journal of Community and Supportive Oncology, shares his top selections in hematology and bone health from this year's line-up of abstracts at the annual meeting of the American Society of Clinical Oncology in Chicago.
8004 Phase 2 study of venetoclax plus carfilzomib and dexamethasone in patients with relapsed refractory myeloma (Luciano J Costa et al). Of 17 patients evaluated after completing at least two cycles of therapy, 3 had complete responses, 2 had very good partial response, 3 had partial response, 3 experienced stable disease, and 2 had progressive disease (response data for 4 patients still to come). The study drug combination was well tolerated, and phase 3 trial is planned.
8008 FDA analysis of pembrolizumab trials in multiple myeloma: immune-related adverse events and response (Aviva C Krauss et al). An evaluation of the KEYNOTE-183 and KEYNOTE-185 trials of nearly 250 patients showed no significant myeloma response with pembrolizumab compared with the control arm of pomalidomide+dexamethasone.
10113 Prospsective phase II pilot study to evaluate the use of intravenous iron in the treatment of anemia in cancer patients (Youjin Kim et al). IV iron supplementation alone showed encouraging results in improving anemia, with hepcidin level possibly predicting response to IV iron and may be superior to the TSAT test.
500 Adjuvant denosumab in early breast cancer: disease free survival analysis of postmenopausal patients in the ABCSG-18 trial (Michael Gnant et al). In this double-blind placebo controlled trial, disease-free survival in the denosumab group was 89% at 5 years and 80% at 8 years, compared with 87% and 77%, respectively, for placebo.
David Henry, MD, the Editor-in-Chief of The Journal of Community and Supportive Oncology, shares his top selections in hematology and bone health from this year's line-up of abstracts at the annual meeting of the American Society of Clinical Oncology in Chicago.
8004 Phase 2 study of venetoclax plus carfilzomib and dexamethasone in patients with relapsed refractory myeloma (Luciano J Costa et al). Of 17 patients evaluated after completing at least two cycles of therapy, 3 had complete responses, 2 had very good partial response, 3 had partial response, 3 experienced stable disease, and 2 had progressive disease (response data for 4 patients still to come). The study drug combination was well tolerated, and phase 3 trial is planned.
8008 FDA analysis of pembrolizumab trials in multiple myeloma: immune-related adverse events and response (Aviva C Krauss et al). An evaluation of the KEYNOTE-183 and KEYNOTE-185 trials of nearly 250 patients showed no significant myeloma response with pembrolizumab compared with the control arm of pomalidomide+dexamethasone.
10113 Prospsective phase II pilot study to evaluate the use of intravenous iron in the treatment of anemia in cancer patients (Youjin Kim et al). IV iron supplementation alone showed encouraging results in improving anemia, with hepcidin level possibly predicting response to IV iron and may be superior to the TSAT test.
500 Adjuvant denosumab in early breast cancer: disease free survival analysis of postmenopausal patients in the ABCSG-18 trial (Michael Gnant et al). In this double-blind placebo controlled trial, disease-free survival in the denosumab group was 89% at 5 years and 80% at 8 years, compared with 87% and 77%, respectively, for placebo.
David Henry, MD, the Editor-in-Chief of The Journal of Community and Supportive Oncology, shares his top selections in hematology and bone health from this year's line-up of abstracts at the annual meeting of the American Society of Clinical Oncology in Chicago.
8004 Phase 2 study of venetoclax plus carfilzomib and dexamethasone in patients with relapsed refractory myeloma (Luciano J Costa et al). Of 17 patients evaluated after completing at least two cycles of therapy, 3 had complete responses, 2 had very good partial response, 3 had partial response, 3 experienced stable disease, and 2 had progressive disease (response data for 4 patients still to come). The study drug combination was well tolerated, and phase 3 trial is planned.
8008 FDA analysis of pembrolizumab trials in multiple myeloma: immune-related adverse events and response (Aviva C Krauss et al). An evaluation of the KEYNOTE-183 and KEYNOTE-185 trials of nearly 250 patients showed no significant myeloma response with pembrolizumab compared with the control arm of pomalidomide+dexamethasone.
10113 Prospsective phase II pilot study to evaluate the use of intravenous iron in the treatment of anemia in cancer patients (Youjin Kim et al). IV iron supplementation alone showed encouraging results in improving anemia, with hepcidin level possibly predicting response to IV iron and may be superior to the TSAT test.
500 Adjuvant denosumab in early breast cancer: disease free survival analysis of postmenopausal patients in the ABCSG-18 trial (Michael Gnant et al). In this double-blind placebo controlled trial, disease-free survival in the denosumab group was 89% at 5 years and 80% at 8 years, compared with 87% and 77%, respectively, for placebo.