User login
CC-220 shows efficacy, safety concerns in phase II SLE trial
PORTLAND – The investigational agent CC-220 showed some efficacy but important safety signals in a 12-week, phase II, dose-escalation study of 42 patients with systemic lupus erythematosus (SLE).
In all, 14% of patients stopped treatment because of adverse effects, including five patients who were receiving CC-220 and one patient on placebo, Victoria Werth, MD, of the University of Pennsylvania, Philadelphia, said during a poster presentation at the annual meeting of the Society for Investigative Dermatology. Higher doses of CC-220 were associated with neutropenia, pneumonia, and dermatitis, she reported.
About 79% of patients completed the study, and none stopped because of lack of efficacy, Dr. Werth said. The most common treatment-emergent adverse effects were mild to moderate nausea and diarrhea. However, four patients experienced serious adverse events, including two patients in the placebo group and two patients who developed pneumonia at the highest (0.6 mg per day) CC-220 dose. In the two highest-dose groups, two patients also developed grade 3 neutropenia, one patient developed grade 1 neutropenia, and there were two cases each of dermatitis and urticaria.
CC-220 appeared to improve scores on the CLASI, SELENA-SENDAI, and PGA, Dr. Werth said. Average drops from baseline on the CLASI ranged from 0 (placebo group) to 7 points (0.3 mg every other day), and were 5 points at 0.3 mg daily, 4 points at alternating doses of 0.3 mg and 0.6 mg, and 6 points at the highest dose of 0.6 mg daily. Among patients whose baseline CLASI score was at least 10, mean decreases ranged between 0 (placebo) and 26 points (0.3 mg daily), and average decreases for the other groups were 9 points (0.3 mg every other day and 0.3 mg alternating with 0.6 mg) or 13 points (0.6 mg daily).
Compared with the placebo group, a greater proportion of CC-220 recipients also had at least a 4-point drop on SELENA-SENDAI and tended to improve more on the tender joint count and the swollen joint count, said Dr. Werth. The average drop in mean PGA score ranged between 0 (placebo group) and 0.9 (0.3 mg per day) and did not show a dose-response trend. The relatively small number of patients in this study and some variability in baseline SLE disease activity made it difficult to draw conclusions about dose-response relationships, Dr. Werth noted. However, there were signs of an overall dose-response trend when pooling mean changes in CLASI, tender and swollen joint counts, SELENA-SLEDAI, and PGA scores.
In terms of pharmacodynamics, doses of at least 0.3 mg CC-220 per day were associated with marked decreases in circulating plasmacytoid dendritic cells and B-cell subsets, which began as early as day 29, Dr. Werth said. Cereblon is part of the cullin-ring finger ligase-4 complex, she explained. Previous work has shown that CC-220 binds with high affinity to cereblon, inducing ubiquitination and increased breakdown of the transcription factors IKZF1 and IKZF3. The end result is an altered immune response.
This study includes an optional 2-year extension phase at the three highest doses of CC-220. Celgene also plans a phase IIb proof-of-concept study in a “broader SLE population,” Dr. Werth said.
Celgene funded the study. Dr. Werth disclosed grant or research support from Celgene, Janssen, Biogen, Roche, and Corbus Pharmaceuticals, and consulting or advisory fees from Celgene, Janssen, Genentech, and several other pharmaceutical companies.
PORTLAND – The investigational agent CC-220 showed some efficacy but important safety signals in a 12-week, phase II, dose-escalation study of 42 patients with systemic lupus erythematosus (SLE).
In all, 14% of patients stopped treatment because of adverse effects, including five patients who were receiving CC-220 and one patient on placebo, Victoria Werth, MD, of the University of Pennsylvania, Philadelphia, said during a poster presentation at the annual meeting of the Society for Investigative Dermatology. Higher doses of CC-220 were associated with neutropenia, pneumonia, and dermatitis, she reported.
About 79% of patients completed the study, and none stopped because of lack of efficacy, Dr. Werth said. The most common treatment-emergent adverse effects were mild to moderate nausea and diarrhea. However, four patients experienced serious adverse events, including two patients in the placebo group and two patients who developed pneumonia at the highest (0.6 mg per day) CC-220 dose. In the two highest-dose groups, two patients also developed grade 3 neutropenia, one patient developed grade 1 neutropenia, and there were two cases each of dermatitis and urticaria.
CC-220 appeared to improve scores on the CLASI, SELENA-SENDAI, and PGA, Dr. Werth said. Average drops from baseline on the CLASI ranged from 0 (placebo group) to 7 points (0.3 mg every other day), and were 5 points at 0.3 mg daily, 4 points at alternating doses of 0.3 mg and 0.6 mg, and 6 points at the highest dose of 0.6 mg daily. Among patients whose baseline CLASI score was at least 10, mean decreases ranged between 0 (placebo) and 26 points (0.3 mg daily), and average decreases for the other groups were 9 points (0.3 mg every other day and 0.3 mg alternating with 0.6 mg) or 13 points (0.6 mg daily).
Compared with the placebo group, a greater proportion of CC-220 recipients also had at least a 4-point drop on SELENA-SENDAI and tended to improve more on the tender joint count and the swollen joint count, said Dr. Werth. The average drop in mean PGA score ranged between 0 (placebo group) and 0.9 (0.3 mg per day) and did not show a dose-response trend. The relatively small number of patients in this study and some variability in baseline SLE disease activity made it difficult to draw conclusions about dose-response relationships, Dr. Werth noted. However, there were signs of an overall dose-response trend when pooling mean changes in CLASI, tender and swollen joint counts, SELENA-SLEDAI, and PGA scores.
In terms of pharmacodynamics, doses of at least 0.3 mg CC-220 per day were associated with marked decreases in circulating plasmacytoid dendritic cells and B-cell subsets, which began as early as day 29, Dr. Werth said. Cereblon is part of the cullin-ring finger ligase-4 complex, she explained. Previous work has shown that CC-220 binds with high affinity to cereblon, inducing ubiquitination and increased breakdown of the transcription factors IKZF1 and IKZF3. The end result is an altered immune response.
This study includes an optional 2-year extension phase at the three highest doses of CC-220. Celgene also plans a phase IIb proof-of-concept study in a “broader SLE population,” Dr. Werth said.
Celgene funded the study. Dr. Werth disclosed grant or research support from Celgene, Janssen, Biogen, Roche, and Corbus Pharmaceuticals, and consulting or advisory fees from Celgene, Janssen, Genentech, and several other pharmaceutical companies.
PORTLAND – The investigational agent CC-220 showed some efficacy but important safety signals in a 12-week, phase II, dose-escalation study of 42 patients with systemic lupus erythematosus (SLE).
In all, 14% of patients stopped treatment because of adverse effects, including five patients who were receiving CC-220 and one patient on placebo, Victoria Werth, MD, of the University of Pennsylvania, Philadelphia, said during a poster presentation at the annual meeting of the Society for Investigative Dermatology. Higher doses of CC-220 were associated with neutropenia, pneumonia, and dermatitis, she reported.
About 79% of patients completed the study, and none stopped because of lack of efficacy, Dr. Werth said. The most common treatment-emergent adverse effects were mild to moderate nausea and diarrhea. However, four patients experienced serious adverse events, including two patients in the placebo group and two patients who developed pneumonia at the highest (0.6 mg per day) CC-220 dose. In the two highest-dose groups, two patients also developed grade 3 neutropenia, one patient developed grade 1 neutropenia, and there were two cases each of dermatitis and urticaria.
CC-220 appeared to improve scores on the CLASI, SELENA-SENDAI, and PGA, Dr. Werth said. Average drops from baseline on the CLASI ranged from 0 (placebo group) to 7 points (0.3 mg every other day), and were 5 points at 0.3 mg daily, 4 points at alternating doses of 0.3 mg and 0.6 mg, and 6 points at the highest dose of 0.6 mg daily. Among patients whose baseline CLASI score was at least 10, mean decreases ranged between 0 (placebo) and 26 points (0.3 mg daily), and average decreases for the other groups were 9 points (0.3 mg every other day and 0.3 mg alternating with 0.6 mg) or 13 points (0.6 mg daily).
Compared with the placebo group, a greater proportion of CC-220 recipients also had at least a 4-point drop on SELENA-SENDAI and tended to improve more on the tender joint count and the swollen joint count, said Dr. Werth. The average drop in mean PGA score ranged between 0 (placebo group) and 0.9 (0.3 mg per day) and did not show a dose-response trend. The relatively small number of patients in this study and some variability in baseline SLE disease activity made it difficult to draw conclusions about dose-response relationships, Dr. Werth noted. However, there were signs of an overall dose-response trend when pooling mean changes in CLASI, tender and swollen joint counts, SELENA-SLEDAI, and PGA scores.
In terms of pharmacodynamics, doses of at least 0.3 mg CC-220 per day were associated with marked decreases in circulating plasmacytoid dendritic cells and B-cell subsets, which began as early as day 29, Dr. Werth said. Cereblon is part of the cullin-ring finger ligase-4 complex, she explained. Previous work has shown that CC-220 binds with high affinity to cereblon, inducing ubiquitination and increased breakdown of the transcription factors IKZF1 and IKZF3. The end result is an altered immune response.
This study includes an optional 2-year extension phase at the three highest doses of CC-220. Celgene also plans a phase IIb proof-of-concept study in a “broader SLE population,” Dr. Werth said.
Celgene funded the study. Dr. Werth disclosed grant or research support from Celgene, Janssen, Biogen, Roche, and Corbus Pharmaceuticals, and consulting or advisory fees from Celgene, Janssen, Genentech, and several other pharmaceutical companies.
AT SID 2017
Key clinical point:
Major finding: Among patients whose baseline CLASI score was at least 10, mean decreases ranged between 0 (placebo), and 26 points (0.3 mg daily).
Data source: A 12-week, phase II, placebo-controlled dose-escalation study of 42 patients with systemic lupus erythematosus.
Disclosures: Celgene funded the study. Dr. Werth disclosed grant or research support from Celgene, Janssen, Biogen, Roche, and Corbus Pharmaceuticals, and consulting or advisory fees from Celgene, Janssen, Genentech, and several other pharmaceutical companies.
SLE linked to subsequent risk of malignant melanoma
PORTLAND, ORE. – A diagnosis of systemic lupus erythematosus (SLE) significantly increases the risk of a subsequent diagnosis of malignant melanoma, according to the results of a large, first-in-kind, single-center longitudinal analysis of electronic medical records.
This finding expands the list of known associations between SLE and cancer, and highlights the need for careful surveillance of this population, Solomiya Grushchak, of the department of dermatology at Northwestern University, Chicago, and her associates, reported in a poster presented at the annual meeting of the Society for Investigative Dermatology.
SLE is increasingly being treated with immune checkpoint inhibitors, which can aggressively disrupt immune reactivity and trigger uncontrolled cellular responses in patients with SLE, Ms. Grushchak noted. “The findings in this large population warrant further exploration of the association between malignant melanoma and SLE to promote optimal patient management, especially in light of recent advances [in the use of] checkpoint inhibitors,” she added.
Past work has linked SLE with several other malignancies, including nonmelanoma skin cancers, non-Hodgkin and Hodgkin lymphomas, and cancers of the larynx, lungs, liver, vulva, vagina, and thyroid gland. Even when patients are not receiving checkpoint inhibitors, SLE causes chronic inflammation and is known to increase cellular dysplasia, which can ultimately trigger uncontrolled proliferation of tumor cells. In 2015, a meta-analysis showed that SLE was associated with a decreased risk of melanoma, but no studies had conclusively evaluated this relationship (PLoS One. 2015;10[4]:e0122964).
Therefore, Ms. Grushchak and her associates analyzed medical records from 2,351 patients from the urban Midwest with SLE diagnosed by a dermatologist or rheumatologist between 2000 and 2016. The data source was the Northwestern Enterprise Data Warehouse, which integrates clinical and research information from more than 50 health data systems used by the Northwestern University Feinberg School of Medicine and its health care partners. To avoid detection bias, the researchers constructed a comparison group from the same database of 1,676 patients diagnosed with systemic sclerosis.
Ten patients (0.4%) with a diagnostic code for SLE were later diagnosed with malignant melanoma, compared with one patient with systemic sclerosis (0.06%), the investigators reported. A Fisher’s exact test confirmed a statistically significant difference between these rates (P = .03). Among the 10 SLE patients with melanoma, 7 were white, 2 were black, and 1 was of Asian ancestry. Nine were females, and one was male. The patient with systemic sclerosis and melanoma was a white male.
The study had several limitations. The investigators did not report how much time elapsed between the diagnoses of SLE and melanoma, or the rates or cumulative exposure to checkpoint inhibitors.
The National Institutes of Health provides support to the Northwestern Enterprise Data Warehouse. The investigators had no relevant financial conflicts.
PORTLAND, ORE. – A diagnosis of systemic lupus erythematosus (SLE) significantly increases the risk of a subsequent diagnosis of malignant melanoma, according to the results of a large, first-in-kind, single-center longitudinal analysis of electronic medical records.
This finding expands the list of known associations between SLE and cancer, and highlights the need for careful surveillance of this population, Solomiya Grushchak, of the department of dermatology at Northwestern University, Chicago, and her associates, reported in a poster presented at the annual meeting of the Society for Investigative Dermatology.
SLE is increasingly being treated with immune checkpoint inhibitors, which can aggressively disrupt immune reactivity and trigger uncontrolled cellular responses in patients with SLE, Ms. Grushchak noted. “The findings in this large population warrant further exploration of the association between malignant melanoma and SLE to promote optimal patient management, especially in light of recent advances [in the use of] checkpoint inhibitors,” she added.
Past work has linked SLE with several other malignancies, including nonmelanoma skin cancers, non-Hodgkin and Hodgkin lymphomas, and cancers of the larynx, lungs, liver, vulva, vagina, and thyroid gland. Even when patients are not receiving checkpoint inhibitors, SLE causes chronic inflammation and is known to increase cellular dysplasia, which can ultimately trigger uncontrolled proliferation of tumor cells. In 2015, a meta-analysis showed that SLE was associated with a decreased risk of melanoma, but no studies had conclusively evaluated this relationship (PLoS One. 2015;10[4]:e0122964).
Therefore, Ms. Grushchak and her associates analyzed medical records from 2,351 patients from the urban Midwest with SLE diagnosed by a dermatologist or rheumatologist between 2000 and 2016. The data source was the Northwestern Enterprise Data Warehouse, which integrates clinical and research information from more than 50 health data systems used by the Northwestern University Feinberg School of Medicine and its health care partners. To avoid detection bias, the researchers constructed a comparison group from the same database of 1,676 patients diagnosed with systemic sclerosis.
Ten patients (0.4%) with a diagnostic code for SLE were later diagnosed with malignant melanoma, compared with one patient with systemic sclerosis (0.06%), the investigators reported. A Fisher’s exact test confirmed a statistically significant difference between these rates (P = .03). Among the 10 SLE patients with melanoma, 7 were white, 2 were black, and 1 was of Asian ancestry. Nine were females, and one was male. The patient with systemic sclerosis and melanoma was a white male.
The study had several limitations. The investigators did not report how much time elapsed between the diagnoses of SLE and melanoma, or the rates or cumulative exposure to checkpoint inhibitors.
The National Institutes of Health provides support to the Northwestern Enterprise Data Warehouse. The investigators had no relevant financial conflicts.
PORTLAND, ORE. – A diagnosis of systemic lupus erythematosus (SLE) significantly increases the risk of a subsequent diagnosis of malignant melanoma, according to the results of a large, first-in-kind, single-center longitudinal analysis of electronic medical records.
This finding expands the list of known associations between SLE and cancer, and highlights the need for careful surveillance of this population, Solomiya Grushchak, of the department of dermatology at Northwestern University, Chicago, and her associates, reported in a poster presented at the annual meeting of the Society for Investigative Dermatology.
SLE is increasingly being treated with immune checkpoint inhibitors, which can aggressively disrupt immune reactivity and trigger uncontrolled cellular responses in patients with SLE, Ms. Grushchak noted. “The findings in this large population warrant further exploration of the association between malignant melanoma and SLE to promote optimal patient management, especially in light of recent advances [in the use of] checkpoint inhibitors,” she added.
Past work has linked SLE with several other malignancies, including nonmelanoma skin cancers, non-Hodgkin and Hodgkin lymphomas, and cancers of the larynx, lungs, liver, vulva, vagina, and thyroid gland. Even when patients are not receiving checkpoint inhibitors, SLE causes chronic inflammation and is known to increase cellular dysplasia, which can ultimately trigger uncontrolled proliferation of tumor cells. In 2015, a meta-analysis showed that SLE was associated with a decreased risk of melanoma, but no studies had conclusively evaluated this relationship (PLoS One. 2015;10[4]:e0122964).
Therefore, Ms. Grushchak and her associates analyzed medical records from 2,351 patients from the urban Midwest with SLE diagnosed by a dermatologist or rheumatologist between 2000 and 2016. The data source was the Northwestern Enterprise Data Warehouse, which integrates clinical and research information from more than 50 health data systems used by the Northwestern University Feinberg School of Medicine and its health care partners. To avoid detection bias, the researchers constructed a comparison group from the same database of 1,676 patients diagnosed with systemic sclerosis.
Ten patients (0.4%) with a diagnostic code for SLE were later diagnosed with malignant melanoma, compared with one patient with systemic sclerosis (0.06%), the investigators reported. A Fisher’s exact test confirmed a statistically significant difference between these rates (P = .03). Among the 10 SLE patients with melanoma, 7 were white, 2 were black, and 1 was of Asian ancestry. Nine were females, and one was male. The patient with systemic sclerosis and melanoma was a white male.
The study had several limitations. The investigators did not report how much time elapsed between the diagnoses of SLE and melanoma, or the rates or cumulative exposure to checkpoint inhibitors.
The National Institutes of Health provides support to the Northwestern Enterprise Data Warehouse. The investigators had no relevant financial conflicts.
AT SID 2017
Key clinical point: Compared with controls, patients with systemic lupus erythematosus (SLE) were at significantly increased risk of later being diagnosed with malignant melanoma.
Major finding: Ten patients with SLE (0.4%) were later diagnosed with malignant melanoma, compared with one patient with systemic sclerosis (0.06%), a statistically significant difference (P = .03).
Data source: Electronic medical record reviews of 2,351 patients with SLE and 1,676 patients with systemic sclerosis (controls) between 2000 and 2016.
Disclosures: The National Institutes of Health provides support to the Northwestern Enterprise Data Warehouse. The investigators had no relevant financial conflicts.
High-dose oral vitamin D3 significantly reduced effects of sunburn
PORTLAND, ORE. – When given within an hour, a single dose of at least 100,000 IU vitamin D3 rapidly attenuates sunburn, according to the results of a randomized, double-blind, placebo-controlled pilot study of 25 healthy adults.
This is the first in vivo study to evaluate whether vitamin D3 can modulate acute inflammation in target tissues, Jeffrey F. Scott, MD, wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology. The findings “have broad implications for the role of vitamin D in skin homeostasis, and suggest that oral vitamin D may be clinically therapeutic for its immunomodulatory properties,” he and his coauthors concluded.
Higher doses of vitamin D3 also produced significant decreases in skin levels of tumor necrosis factor–alpha (TNF-alpha) (P = .02) and inducible nitric oxide synthase (iNOS) (P = .04) compared with placebo, reported Dr. Scott, a resident in dermatology at University Hospitals Cleveland Medical Center.
Notably, 48 hours after sunburn, hematoxylin and eosin histology of punch biopsies showed that participants who received 200,000 IU vitamin D3 had the least structural damage to the skin, while placebo recipients had the most damage. Expression profiling also linked vitamin D3 treatment with upregulation of genes associated with skin barrier repair.
Studies continue to document diverse biologic effects of vitamin D, including “modulation of immune response, inflammatory disease, cardiovascular health, and carcinogenesis,” the researchers wrote. Vitamin D3 also has been shown to suppress inflammatory mediators and induce autophagy, they added. Previously, their group showed that oral D3 induced similar protective effects in a mouse model of chemical skin injury. Treatment inhibited proinflammatory cytokines and chemokines within the skin, including iNOS and TNF-alpha.
The current study also included a control phase in which participants underwent experimental sunburn on the right arm without any treatment. Forty-eight hours later, punch biopsies revealed high levels of iNOS and TNF-alpha, with increased expression of proinflammatory genes, the researchers wrote.
During the subsequent experimental phase, seven participants were assigned to receive placebo, and six were assigned to receive 50,000 IU, 100,000 IU, or 200,000 IU of vitamin D3. After treatment, participants with the highest serum D3 levels had significantly decreased skin redness compared with participants with lower serum D3 levels (P less than .05). Higher vitamin D3 serum levels were also associated with significant (P less than .05) upregulation of skin barrier repair genes and of arginase-1, a cytosolic enzyme that helps mediate anti-inflammatory activity.
“Arginase-1 may be a clinically useful tissue biomarker for monitoring the immunomodulatory effects of vitamin D3 in humans,” the researchers concluded.
The National Institute of Arthritis Musculoskeletal and Skin Diseases and the National Institutes of Health supported the work. Dr. Scott had no relevant financial disclosures.
PORTLAND, ORE. – When given within an hour, a single dose of at least 100,000 IU vitamin D3 rapidly attenuates sunburn, according to the results of a randomized, double-blind, placebo-controlled pilot study of 25 healthy adults.
This is the first in vivo study to evaluate whether vitamin D3 can modulate acute inflammation in target tissues, Jeffrey F. Scott, MD, wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology. The findings “have broad implications for the role of vitamin D in skin homeostasis, and suggest that oral vitamin D may be clinically therapeutic for its immunomodulatory properties,” he and his coauthors concluded.
Higher doses of vitamin D3 also produced significant decreases in skin levels of tumor necrosis factor–alpha (TNF-alpha) (P = .02) and inducible nitric oxide synthase (iNOS) (P = .04) compared with placebo, reported Dr. Scott, a resident in dermatology at University Hospitals Cleveland Medical Center.
Notably, 48 hours after sunburn, hematoxylin and eosin histology of punch biopsies showed that participants who received 200,000 IU vitamin D3 had the least structural damage to the skin, while placebo recipients had the most damage. Expression profiling also linked vitamin D3 treatment with upregulation of genes associated with skin barrier repair.
Studies continue to document diverse biologic effects of vitamin D, including “modulation of immune response, inflammatory disease, cardiovascular health, and carcinogenesis,” the researchers wrote. Vitamin D3 also has been shown to suppress inflammatory mediators and induce autophagy, they added. Previously, their group showed that oral D3 induced similar protective effects in a mouse model of chemical skin injury. Treatment inhibited proinflammatory cytokines and chemokines within the skin, including iNOS and TNF-alpha.
The current study also included a control phase in which participants underwent experimental sunburn on the right arm without any treatment. Forty-eight hours later, punch biopsies revealed high levels of iNOS and TNF-alpha, with increased expression of proinflammatory genes, the researchers wrote.
During the subsequent experimental phase, seven participants were assigned to receive placebo, and six were assigned to receive 50,000 IU, 100,000 IU, or 200,000 IU of vitamin D3. After treatment, participants with the highest serum D3 levels had significantly decreased skin redness compared with participants with lower serum D3 levels (P less than .05). Higher vitamin D3 serum levels were also associated with significant (P less than .05) upregulation of skin barrier repair genes and of arginase-1, a cytosolic enzyme that helps mediate anti-inflammatory activity.
“Arginase-1 may be a clinically useful tissue biomarker for monitoring the immunomodulatory effects of vitamin D3 in humans,” the researchers concluded.
The National Institute of Arthritis Musculoskeletal and Skin Diseases and the National Institutes of Health supported the work. Dr. Scott had no relevant financial disclosures.
PORTLAND, ORE. – When given within an hour, a single dose of at least 100,000 IU vitamin D3 rapidly attenuates sunburn, according to the results of a randomized, double-blind, placebo-controlled pilot study of 25 healthy adults.
This is the first in vivo study to evaluate whether vitamin D3 can modulate acute inflammation in target tissues, Jeffrey F. Scott, MD, wrote in a poster presented at the annual meeting of the Society for Investigative Dermatology. The findings “have broad implications for the role of vitamin D in skin homeostasis, and suggest that oral vitamin D may be clinically therapeutic for its immunomodulatory properties,” he and his coauthors concluded.
Higher doses of vitamin D3 also produced significant decreases in skin levels of tumor necrosis factor–alpha (TNF-alpha) (P = .02) and inducible nitric oxide synthase (iNOS) (P = .04) compared with placebo, reported Dr. Scott, a resident in dermatology at University Hospitals Cleveland Medical Center.
Notably, 48 hours after sunburn, hematoxylin and eosin histology of punch biopsies showed that participants who received 200,000 IU vitamin D3 had the least structural damage to the skin, while placebo recipients had the most damage. Expression profiling also linked vitamin D3 treatment with upregulation of genes associated with skin barrier repair.
Studies continue to document diverse biologic effects of vitamin D, including “modulation of immune response, inflammatory disease, cardiovascular health, and carcinogenesis,” the researchers wrote. Vitamin D3 also has been shown to suppress inflammatory mediators and induce autophagy, they added. Previously, their group showed that oral D3 induced similar protective effects in a mouse model of chemical skin injury. Treatment inhibited proinflammatory cytokines and chemokines within the skin, including iNOS and TNF-alpha.
The current study also included a control phase in which participants underwent experimental sunburn on the right arm without any treatment. Forty-eight hours later, punch biopsies revealed high levels of iNOS and TNF-alpha, with increased expression of proinflammatory genes, the researchers wrote.
During the subsequent experimental phase, seven participants were assigned to receive placebo, and six were assigned to receive 50,000 IU, 100,000 IU, or 200,000 IU of vitamin D3. After treatment, participants with the highest serum D3 levels had significantly decreased skin redness compared with participants with lower serum D3 levels (P less than .05). Higher vitamin D3 serum levels were also associated with significant (P less than .05) upregulation of skin barrier repair genes and of arginase-1, a cytosolic enzyme that helps mediate anti-inflammatory activity.
“Arginase-1 may be a clinically useful tissue biomarker for monitoring the immunomodulatory effects of vitamin D3 in humans,” the researchers concluded.
The National Institute of Arthritis Musculoskeletal and Skin Diseases and the National Institutes of Health supported the work. Dr. Scott had no relevant financial disclosures.
Key clinical point: A single dose of at least 100,000 IU vitamin D3 rapidly attenuated experimental sunburn.
Major finding: Twenty-four hours after sunburn, recipients of 100,000 IU or 200,000 IU D3 had a marked, sustained reduction in skin redness compared with the placebo and 50,000 IU groups. Higher doses of D3 produced significant decreases in skin levels of tumor necrosis factor–alpha and inducible nitric oxide synthase, compared with placebo.
Data source: A double-blind, randomized, placebo-controlled pilot study of 25 healthy adults.
Disclosures: The National Institute of Arthritis Musculoskeletal and Skin Diseases and the National Institutes of Health supported the work. Dr. Scott had no relevant financial conflicts.

Cutaneous manifestations can signify severe systemic disease in ANCA-associated vasculitis
PORTLAND, ORE. – Clinicians who treat or diagnose ANCA-associated vasculitis should watch for a variety of skin lesions, which can signify severe systemic manifestations of disease, according to the results of a cross-sectional study of 1,184 patients from 130 centers worldwide.
Among patients with granulomatosis with polyangiitis (GPA) or eosinophilic granulomatosis with polyangiitis (EGPA), the presence of skin lesions approximately doubled the likelihood of renal, pulmonary, neurologic, or other severe systemic manifestations of ANCA-associated vasculitis (hazard ratios, 2.0; P less than .03).
This cohort is part of the Diagnostic and Classification Criteria in Vasculitis Study (DCVAS), which aims to develop classification and diagnostic criteria for primary systemic vasculitis. Fully 35% of patients had cutaneous manifestations of ANCA-associated vasculitis, including 47% of those with EGPA, 34% of those with GPA, and 28% of those with microscopic polyangiitis (MPA).
Petechiae/purpura were the most common cutaneous manifestations of all three subtypes, affecting 15% of the overall cohort, 21% of patients with EGPA, 16% of those with GPA, and 9% of those with MPA (P less than .01 for differences among groups). Petechiae/purpura did not more accurately predict systemic disease than other cutaneous findings, and skin lesions were not significantly associated with severe systemic disease in patients with MPA (HR, 0.63; 95% confidence interval, 0.35-1.14; P = .13), the investigators reported.
Besides petechiae/purpura, patients with EGPA most often presented with allergic and nonspecific cutaneous manifestations, such as pruritus (13% of patients), urticaria (8%), and maculopapular rash (8%), they said. In contrast, patients with GPA most often had painful skin lesions (10%) or maculopapular rash (7%), while those with MPA were more likely to have livedo reticularis or racemosa (7%).
Study participants tended to be in their mid-50s to mid-60s at diagnosis, about 48% were male, and most were Northern European, Southern European, or American whites, while 28% of those with MPA were Han Chinese, of another Chinese ethnicity, or Japanese.
“This study demonstrates that skin lesions are quite common and varied in granulomatosis with polyangiitis, microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis,” the investigators concluded.
Funders included the American College of Rheumatology, the European League Against Rheumatism, the Vasculitis Foundation, and the Dermatology Foundation. Dr. Micheletti had no conflicts of interest.
PORTLAND, ORE. – Clinicians who treat or diagnose ANCA-associated vasculitis should watch for a variety of skin lesions, which can signify severe systemic manifestations of disease, according to the results of a cross-sectional study of 1,184 patients from 130 centers worldwide.
Among patients with granulomatosis with polyangiitis (GPA) or eosinophilic granulomatosis with polyangiitis (EGPA), the presence of skin lesions approximately doubled the likelihood of renal, pulmonary, neurologic, or other severe systemic manifestations of ANCA-associated vasculitis (hazard ratios, 2.0; P less than .03).
This cohort is part of the Diagnostic and Classification Criteria in Vasculitis Study (DCVAS), which aims to develop classification and diagnostic criteria for primary systemic vasculitis. Fully 35% of patients had cutaneous manifestations of ANCA-associated vasculitis, including 47% of those with EGPA, 34% of those with GPA, and 28% of those with microscopic polyangiitis (MPA).
Petechiae/purpura were the most common cutaneous manifestations of all three subtypes, affecting 15% of the overall cohort, 21% of patients with EGPA, 16% of those with GPA, and 9% of those with MPA (P less than .01 for differences among groups). Petechiae/purpura did not more accurately predict systemic disease than other cutaneous findings, and skin lesions were not significantly associated with severe systemic disease in patients with MPA (HR, 0.63; 95% confidence interval, 0.35-1.14; P = .13), the investigators reported.
Besides petechiae/purpura, patients with EGPA most often presented with allergic and nonspecific cutaneous manifestations, such as pruritus (13% of patients), urticaria (8%), and maculopapular rash (8%), they said. In contrast, patients with GPA most often had painful skin lesions (10%) or maculopapular rash (7%), while those with MPA were more likely to have livedo reticularis or racemosa (7%).
Study participants tended to be in their mid-50s to mid-60s at diagnosis, about 48% were male, and most were Northern European, Southern European, or American whites, while 28% of those with MPA were Han Chinese, of another Chinese ethnicity, or Japanese.
“This study demonstrates that skin lesions are quite common and varied in granulomatosis with polyangiitis, microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis,” the investigators concluded.
Funders included the American College of Rheumatology, the European League Against Rheumatism, the Vasculitis Foundation, and the Dermatology Foundation. Dr. Micheletti had no conflicts of interest.
PORTLAND, ORE. – Clinicians who treat or diagnose ANCA-associated vasculitis should watch for a variety of skin lesions, which can signify severe systemic manifestations of disease, according to the results of a cross-sectional study of 1,184 patients from 130 centers worldwide.
Among patients with granulomatosis with polyangiitis (GPA) or eosinophilic granulomatosis with polyangiitis (EGPA), the presence of skin lesions approximately doubled the likelihood of renal, pulmonary, neurologic, or other severe systemic manifestations of ANCA-associated vasculitis (hazard ratios, 2.0; P less than .03).
This cohort is part of the Diagnostic and Classification Criteria in Vasculitis Study (DCVAS), which aims to develop classification and diagnostic criteria for primary systemic vasculitis. Fully 35% of patients had cutaneous manifestations of ANCA-associated vasculitis, including 47% of those with EGPA, 34% of those with GPA, and 28% of those with microscopic polyangiitis (MPA).
Petechiae/purpura were the most common cutaneous manifestations of all three subtypes, affecting 15% of the overall cohort, 21% of patients with EGPA, 16% of those with GPA, and 9% of those with MPA (P less than .01 for differences among groups). Petechiae/purpura did not more accurately predict systemic disease than other cutaneous findings, and skin lesions were not significantly associated with severe systemic disease in patients with MPA (HR, 0.63; 95% confidence interval, 0.35-1.14; P = .13), the investigators reported.
Besides petechiae/purpura, patients with EGPA most often presented with allergic and nonspecific cutaneous manifestations, such as pruritus (13% of patients), urticaria (8%), and maculopapular rash (8%), they said. In contrast, patients with GPA most often had painful skin lesions (10%) or maculopapular rash (7%), while those with MPA were more likely to have livedo reticularis or racemosa (7%).
Study participants tended to be in their mid-50s to mid-60s at diagnosis, about 48% were male, and most were Northern European, Southern European, or American whites, while 28% of those with MPA were Han Chinese, of another Chinese ethnicity, or Japanese.
“This study demonstrates that skin lesions are quite common and varied in granulomatosis with polyangiitis, microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis,” the investigators concluded.
Funders included the American College of Rheumatology, the European League Against Rheumatism, the Vasculitis Foundation, and the Dermatology Foundation. Dr. Micheletti had no conflicts of interest.
AT SID 2017
Key clinical point: Skin lesions can be a red flag for severe systemic disease in patients with ANCA-associated vasculitis.
Major finding: Among patients with granulomatosis with polyangiitis or eosinophilic granulomatosis with polyangiitis, the presence of skin lesions approximately doubled the likelihood of renal, pulmonary, neurologic, or other severe systemic manifestations of ANCA-associated vasculitis (HR, 2.0, P less than .03). The hazard ratio was not elevated in patients with microscopic polyangiitis.
Data source: A cross-sectional study of 1,184 patients with ANCA-associated vasculitis from 130 centers worldwide.
Disclosures: Funders included the American College of Rheumatology, the European League Against Rheumatism, the Vasculitis Foundation, and the Dermatology Foundation. Dr. Micheletti had no conflicts of interest.
Merkel cell carcinoma most likely to recur within 2 years of diagnosis
PORTLAND, ORE. – The first 2 years after diagnosis are crucial when conducting surveillance for recurrence of Merkel cell carcinoma (MCC), Aubriana McEvoy said at the annual meeting of the Society for Investigative Dermatology.
Regardless of stage at diagnosis, the risk of recurrence peaked at about 1 year and leveled off by about year 2 in a retrospective cohort study, according to Ms. McEvoy, a medical student at the University of Washington, Seattle, who conducted the study with colleagues under the mentorship of Paul Nghiem, MD, PhD, professor and head of the division of dermatology. The study also inversely linked primary MCC stage with subsequent recurrence-free survival, highlighted the role of imaging for surveillance of patients who have advanced primary disease, and linked distant metastatic recurrence with significantly worse survival, compared with local or nodal recurrence.
“Patients with Merkel cell carcinoma always ask about recurrence,” Ms. McEvoy said. “Now, for the first time, we have the data to answer their questions.”
Surveillance of MCC is increasingly important, she said: The “treatment landscape is evolving quickly, and immunotherapies such as pembrolizumab can have a good response rate, especially in the setting of lower burden of disease.” But follow-up is costly on several fronts, making it crucial to aim for “enough” and not “too much” surveillance, she added.
“Imaging often costs thousands of dollars, and that’s only one piece of the pie. There’s also the cost of office visits, time spent by the patient and their family, and the emotional investment and uncertainty a patient goes through every time they have to come for a follow-up visit and scan,” Ms. McEvoy said.
Comprehensive, stage-specific guidelines can help clinicians and patients balance the benefits and costs of surveillance, but are lacking in MCC because no published study has characterized recurrence by stage, she said. To fill this gap, she and her associates analyzed 10 years of longitudinal MCC surveillance data on 468 patients who underwent pathologic staging and were followed at the Nghiem laboratory.
The risk of recurrence was highest within the first 2 years after diagnosis, regardless of whether patients had local (pathologic stage I–II) or nodal (stage III) MCC. However, the probability of recurrence-free survival correlated inversely with pathologic stage of primary MCC (P = .003). Median recurrence-free survival time was not reached by the 186 patients with local disease and small (2-cm maximum dimension) primary lesions, or by 135 patients with clinically occult nodal disease.
In contrast, median recurrence-free survival was about 6 years among 84 patients with local disease and lesions measuring more than 2 cm; was less than 2 years among 35 patients with clinically apparent, pathologically confirmed nodal disease or in-transit metastases; and was less than 1 year among patients with distant metastatic disease.
The researchers also investigated the risk of distant metastatic recurrence to confirm which patients need most intensive follow-up. Among 138 individuals with available data, 40% of stage I primary MCC patients developed a distant metastatic recurrence, as did 60% of patients with stage IIA or stage IIB primary MCC. And 80% of recurrences among patients with stage IIIA or stage IIIB primary disease were distant metastases. “I think it’s safe to say that stage III patients should receive appropriate, if not vigilant, surveillance,” Ms. McEvoy said. The site of recurrence also was significantly (P less than .001) tied to the risk of subsequent death from recurrent MCC; median survival time was not reached when recurrence was local or nodal, but was less than 2 years when it was distant or metastatic.
Early in 2018, the American Joint Committee on Cancer will update its MCC staging system to distinguish clinical versus pathologic staging. “This is important, because pathologic staging remains the gold standard, providing a much more in-depth view of the patient’s disease,” Ms. McEvoy commented. Ideally, clinicians would use more information to help predict the prognosis of MCC, including sex and immune and viral status, she noted. “But we hope these data provide information for more consistency across the country, so we can catch recurrences earlier, and avoid unnecessary visits and imaging scans for lower-risk patients.”
The study was supported by the National Institutes of Health, the Seattle Cancer Care Alliance, the University of Washington, and the Institute of Translational Health Sciences. Ms. McEvoy had no conflicts of interest.
PORTLAND, ORE. – The first 2 years after diagnosis are crucial when conducting surveillance for recurrence of Merkel cell carcinoma (MCC), Aubriana McEvoy said at the annual meeting of the Society for Investigative Dermatology.
Regardless of stage at diagnosis, the risk of recurrence peaked at about 1 year and leveled off by about year 2 in a retrospective cohort study, according to Ms. McEvoy, a medical student at the University of Washington, Seattle, who conducted the study with colleagues under the mentorship of Paul Nghiem, MD, PhD, professor and head of the division of dermatology. The study also inversely linked primary MCC stage with subsequent recurrence-free survival, highlighted the role of imaging for surveillance of patients who have advanced primary disease, and linked distant metastatic recurrence with significantly worse survival, compared with local or nodal recurrence.
“Patients with Merkel cell carcinoma always ask about recurrence,” Ms. McEvoy said. “Now, for the first time, we have the data to answer their questions.”
Surveillance of MCC is increasingly important, she said: The “treatment landscape is evolving quickly, and immunotherapies such as pembrolizumab can have a good response rate, especially in the setting of lower burden of disease.” But follow-up is costly on several fronts, making it crucial to aim for “enough” and not “too much” surveillance, she added.
“Imaging often costs thousands of dollars, and that’s only one piece of the pie. There’s also the cost of office visits, time spent by the patient and their family, and the emotional investment and uncertainty a patient goes through every time they have to come for a follow-up visit and scan,” Ms. McEvoy said.
Comprehensive, stage-specific guidelines can help clinicians and patients balance the benefits and costs of surveillance, but are lacking in MCC because no published study has characterized recurrence by stage, she said. To fill this gap, she and her associates analyzed 10 years of longitudinal MCC surveillance data on 468 patients who underwent pathologic staging and were followed at the Nghiem laboratory.
The risk of recurrence was highest within the first 2 years after diagnosis, regardless of whether patients had local (pathologic stage I–II) or nodal (stage III) MCC. However, the probability of recurrence-free survival correlated inversely with pathologic stage of primary MCC (P = .003). Median recurrence-free survival time was not reached by the 186 patients with local disease and small (2-cm maximum dimension) primary lesions, or by 135 patients with clinically occult nodal disease.
In contrast, median recurrence-free survival was about 6 years among 84 patients with local disease and lesions measuring more than 2 cm; was less than 2 years among 35 patients with clinically apparent, pathologically confirmed nodal disease or in-transit metastases; and was less than 1 year among patients with distant metastatic disease.
The researchers also investigated the risk of distant metastatic recurrence to confirm which patients need most intensive follow-up. Among 138 individuals with available data, 40% of stage I primary MCC patients developed a distant metastatic recurrence, as did 60% of patients with stage IIA or stage IIB primary MCC. And 80% of recurrences among patients with stage IIIA or stage IIIB primary disease were distant metastases. “I think it’s safe to say that stage III patients should receive appropriate, if not vigilant, surveillance,” Ms. McEvoy said. The site of recurrence also was significantly (P less than .001) tied to the risk of subsequent death from recurrent MCC; median survival time was not reached when recurrence was local or nodal, but was less than 2 years when it was distant or metastatic.
Early in 2018, the American Joint Committee on Cancer will update its MCC staging system to distinguish clinical versus pathologic staging. “This is important, because pathologic staging remains the gold standard, providing a much more in-depth view of the patient’s disease,” Ms. McEvoy commented. Ideally, clinicians would use more information to help predict the prognosis of MCC, including sex and immune and viral status, she noted. “But we hope these data provide information for more consistency across the country, so we can catch recurrences earlier, and avoid unnecessary visits and imaging scans for lower-risk patients.”
The study was supported by the National Institutes of Health, the Seattle Cancer Care Alliance, the University of Washington, and the Institute of Translational Health Sciences. Ms. McEvoy had no conflicts of interest.
PORTLAND, ORE. – The first 2 years after diagnosis are crucial when conducting surveillance for recurrence of Merkel cell carcinoma (MCC), Aubriana McEvoy said at the annual meeting of the Society for Investigative Dermatology.
Regardless of stage at diagnosis, the risk of recurrence peaked at about 1 year and leveled off by about year 2 in a retrospective cohort study, according to Ms. McEvoy, a medical student at the University of Washington, Seattle, who conducted the study with colleagues under the mentorship of Paul Nghiem, MD, PhD, professor and head of the division of dermatology. The study also inversely linked primary MCC stage with subsequent recurrence-free survival, highlighted the role of imaging for surveillance of patients who have advanced primary disease, and linked distant metastatic recurrence with significantly worse survival, compared with local or nodal recurrence.
“Patients with Merkel cell carcinoma always ask about recurrence,” Ms. McEvoy said. “Now, for the first time, we have the data to answer their questions.”
Surveillance of MCC is increasingly important, she said: The “treatment landscape is evolving quickly, and immunotherapies such as pembrolizumab can have a good response rate, especially in the setting of lower burden of disease.” But follow-up is costly on several fronts, making it crucial to aim for “enough” and not “too much” surveillance, she added.
“Imaging often costs thousands of dollars, and that’s only one piece of the pie. There’s also the cost of office visits, time spent by the patient and their family, and the emotional investment and uncertainty a patient goes through every time they have to come for a follow-up visit and scan,” Ms. McEvoy said.
Comprehensive, stage-specific guidelines can help clinicians and patients balance the benefits and costs of surveillance, but are lacking in MCC because no published study has characterized recurrence by stage, she said. To fill this gap, she and her associates analyzed 10 years of longitudinal MCC surveillance data on 468 patients who underwent pathologic staging and were followed at the Nghiem laboratory.
The risk of recurrence was highest within the first 2 years after diagnosis, regardless of whether patients had local (pathologic stage I–II) or nodal (stage III) MCC. However, the probability of recurrence-free survival correlated inversely with pathologic stage of primary MCC (P = .003). Median recurrence-free survival time was not reached by the 186 patients with local disease and small (2-cm maximum dimension) primary lesions, or by 135 patients with clinically occult nodal disease.
In contrast, median recurrence-free survival was about 6 years among 84 patients with local disease and lesions measuring more than 2 cm; was less than 2 years among 35 patients with clinically apparent, pathologically confirmed nodal disease or in-transit metastases; and was less than 1 year among patients with distant metastatic disease.
The researchers also investigated the risk of distant metastatic recurrence to confirm which patients need most intensive follow-up. Among 138 individuals with available data, 40% of stage I primary MCC patients developed a distant metastatic recurrence, as did 60% of patients with stage IIA or stage IIB primary MCC. And 80% of recurrences among patients with stage IIIA or stage IIIB primary disease were distant metastases. “I think it’s safe to say that stage III patients should receive appropriate, if not vigilant, surveillance,” Ms. McEvoy said. The site of recurrence also was significantly (P less than .001) tied to the risk of subsequent death from recurrent MCC; median survival time was not reached when recurrence was local or nodal, but was less than 2 years when it was distant or metastatic.
Early in 2018, the American Joint Committee on Cancer will update its MCC staging system to distinguish clinical versus pathologic staging. “This is important, because pathologic staging remains the gold standard, providing a much more in-depth view of the patient’s disease,” Ms. McEvoy commented. Ideally, clinicians would use more information to help predict the prognosis of MCC, including sex and immune and viral status, she noted. “But we hope these data provide information for more consistency across the country, so we can catch recurrences earlier, and avoid unnecessary visits and imaging scans for lower-risk patients.”
The study was supported by the National Institutes of Health, the Seattle Cancer Care Alliance, the University of Washington, and the Institute of Translational Health Sciences. Ms. McEvoy had no conflicts of interest.
AT SID 2017
Key clinical point:
Major finding: The risk of recurrence peaked about 1 year after diagnosis and leveled off at about year 2, regardless of whether patients had local (pathologic stage I–II) or nodal (stage III) disease.
Data source: A retrospective cohort study of 544 patients with Merkel cell carcinoma (468 with pathologic stage disease).
Disclosures: The study was supported by the National Institutes of Health, the Seattle Cancer Care Alliance, the University of Washington, and the Institute of Translational Health Sciences. Ms. McEvoy had no conflicts of interest.
JAK inhibitors and alopecia: After positive early data, various trials now underway
PORTLAND, ORE. – Janus kinase inhibitors are relatively safe and can produce a full head of hair in patients with moderate to severe alopecia areata (AA), although patients tend to shed hair after stopping treatment, Julian Mackay-Wiggan, MD, said at the annual meeting of the Society for Investigative Dermatology.
“At this point, there are 17 publications in the literature, from clinical trials to case reports, looking at JAK [Janus kinase] inhibitors in patients with alopecia areata,” said Dr. Mackay-Wiggan of the department of dermatology, Columbia University, New York, where she specializes in hair disorders. “Pretty much all report very positive findings. It definitely appears that Janus kinase inhibitors can play a very significant role in treatment.”
In an open label, uncontrolled pilot study at Columbia, 9 of 12 (75%) patients with moderate to severe AA improved by at least 50% on the Severity of Alopecia Tool (SALT) after receiving 20 mg ruxolitinib twice daily for 3 to 6 months (JCI Insight. 2016 Sep 22;1[15]:e89790). Responses started with the first month, and all but one responder achieved at least 50% hair regrowth by week 12, said Dr. Mackay-Wiggan, who is also the director of the Dermatology Clinical Research Unit at Columbia.
By the end of treatment, seven of nine responders achieved more than 95% regrowth, one achieved 85% regrowth, and one achieved 55% regrowth. Importantly, none of these relatively healthy patients experienced serious adverse events on ruxolitinib, and none needed to stop treatment, although one patient experienced declining hemoglobin levels that resolved after dose modification.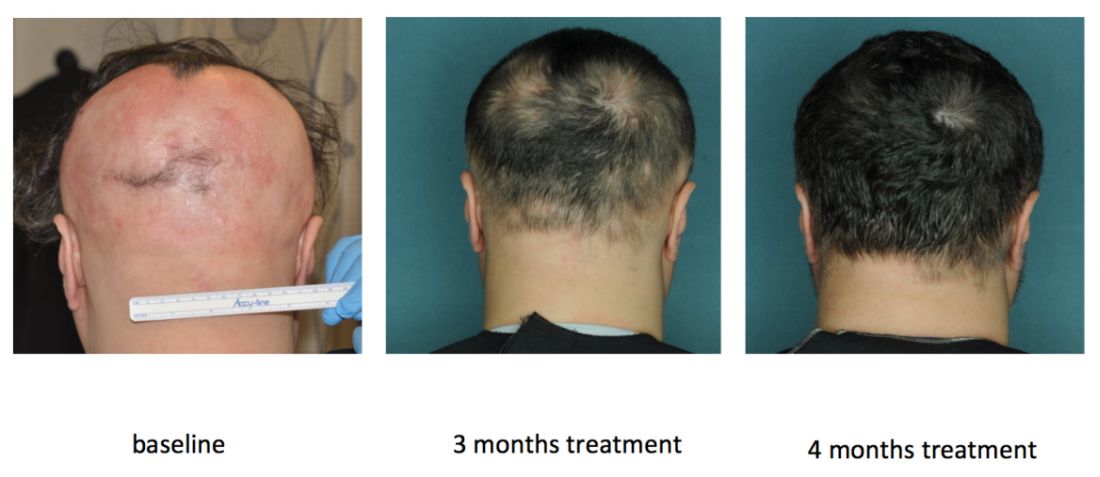
Columbia researchers are also conducting an uncontrolled, open label pilot trial of the JAK inhibitor tofacitinib (Xeljanz) in 12 patients, of whom seven have moderate to severe patchy AA and five have alopecia totalis or universalis. Tofacitinib is approved for treating rheumatoid arthritis at a dose of 5 mg twice daily, but patients have needed up to 10 mg twice daily to achieve hair regrowth, Dr. Mackay-Wiggan said. To date, 11 (92%) have achieved at least some hair regrowth, and 8 (67%) have achieved at least 50% regrowth. So far, there have been no serious adverse events over 6 to 16 months of treatment, although one patient stopped treatment after developing hypertension, a known adverse effect of tofacitinib.
In this study, heatmaps of RNA sequencing of CD8+ T cell populations clearly showed pathogenic signatures for AA and a “robust molecular response to treatment,” Dr. Mackay-Wiggan said. “These two signatures also overlapped statistically, producing 114 genes that may be targetable mediators of disease.” But as with ruxolitinib, regrowth started to decline as patients were taken off treatment.
Research indicates that inhibiting the JAK-STAT signaling pathway induces anagen and subsequent hair growth, but activating STAT 5 in the dermal papilla is also important to induce the growth phase of the hair follicle, according to Dr. Mackay-Wiggan. “Bottom line, it’s complicated,” she added. “The mode of delivery – topical versus systemic – may be important, and the timing of delivery may be crucial.”
Other studies point to a role for JAK inhibition in treating AA. In an uncontrolled, retrospective study of 90 adults with alopecia totalis, alopecia universalis, or moderate to severe AA, 58% had SALT scores of 50% or better after receiving 5 mg tofacitinib twice daily for 4 to 18 months. Patients with AA improved more than those with alopecia totalis or universalis. There were no severe adverse effects, although nearly a third of patients developed upper respiratory tract infections. In another uncontrolled study of 13 patients with AA, totalis, or universalis, 9 (70%) patients achieved full regrowth and there were no serious adverse effects, although patients experienced headaches, upper respiratory infections, and mild increases in liver transaminase levels.
JAK inhibition also has a potential role for treating some scarring alopecias, including lichen planopilaris and frontal fibrosing alopecia. These diseases are histologically “identical” and both exhibit perifollicular erythema, papules, and scale, all of which suggest active inflammation, Dr. Mackay-Wiggan said. Hair follicles from affected patients show immune markers such as interferon-inducible chemokines, cytotoxic T cell responses, and expression of major histocompatibility complexes I and II. “The important message here is that JAK/STAT signaling may play a significant role in other types of hair loss other than alopecia areata,” Dr. Mackay-Wiggan said. “These diseases may also be autoimmune diseases, and may also be treatable with JAK inhibitors.”
Studies continue to evaluate JAK inhibitors for treating alopecia and its variants. Investigators at Yale and Stanford are conducting three uncontrolled trials of oral or topical tofacitinib, while Incyte, the manufacturer of ruxolitinib, is sponsoring a multicenter, randomized, placebo-controlled trial of ruxolitinib phosphate cream for adults with AA, with topline results expected in May 2018. Concert Pharmaceuticals also is recruiting for a trial of a modified, investigational form of ruxolitinib called CTP-543 for treating moderate to severe AA. “Many more trials are in development,” Dr. Mackay-Wiggan noted.
The ruxolitinib pilot study was funded by the Locks of Love Foundation, the Alopecia Areata Initiative, NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases, and by an Irving Institute for Clinical and Translational Research/Columbia University Medical Center Clinical and Translational Science Award. The ongoing tofacitinib pilot study is sponsored by Dr. Mackay-Wiggan, Locks of Love, and Columbia University.
Dr. Mackay-Wiggan also acknowledged support from the Alopecia Areata Initiative – the Gates Foundation, the National Alopecia Areata Registry, and the National Alopecia Areata Foundation. She had no other relevant financial disclosures.
PORTLAND, ORE. – Janus kinase inhibitors are relatively safe and can produce a full head of hair in patients with moderate to severe alopecia areata (AA), although patients tend to shed hair after stopping treatment, Julian Mackay-Wiggan, MD, said at the annual meeting of the Society for Investigative Dermatology.
“At this point, there are 17 publications in the literature, from clinical trials to case reports, looking at JAK [Janus kinase] inhibitors in patients with alopecia areata,” said Dr. Mackay-Wiggan of the department of dermatology, Columbia University, New York, where she specializes in hair disorders. “Pretty much all report very positive findings. It definitely appears that Janus kinase inhibitors can play a very significant role in treatment.”
In an open label, uncontrolled pilot study at Columbia, 9 of 12 (75%) patients with moderate to severe AA improved by at least 50% on the Severity of Alopecia Tool (SALT) after receiving 20 mg ruxolitinib twice daily for 3 to 6 months (JCI Insight. 2016 Sep 22;1[15]:e89790). Responses started with the first month, and all but one responder achieved at least 50% hair regrowth by week 12, said Dr. Mackay-Wiggan, who is also the director of the Dermatology Clinical Research Unit at Columbia.
By the end of treatment, seven of nine responders achieved more than 95% regrowth, one achieved 85% regrowth, and one achieved 55% regrowth. Importantly, none of these relatively healthy patients experienced serious adverse events on ruxolitinib, and none needed to stop treatment, although one patient experienced declining hemoglobin levels that resolved after dose modification.
Columbia researchers are also conducting an uncontrolled, open label pilot trial of the JAK inhibitor tofacitinib (Xeljanz) in 12 patients, of whom seven have moderate to severe patchy AA and five have alopecia totalis or universalis. Tofacitinib is approved for treating rheumatoid arthritis at a dose of 5 mg twice daily, but patients have needed up to 10 mg twice daily to achieve hair regrowth, Dr. Mackay-Wiggan said. To date, 11 (92%) have achieved at least some hair regrowth, and 8 (67%) have achieved at least 50% regrowth. So far, there have been no serious adverse events over 6 to 16 months of treatment, although one patient stopped treatment after developing hypertension, a known adverse effect of tofacitinib.
In this study, heatmaps of RNA sequencing of CD8+ T cell populations clearly showed pathogenic signatures for AA and a “robust molecular response to treatment,” Dr. Mackay-Wiggan said. “These two signatures also overlapped statistically, producing 114 genes that may be targetable mediators of disease.” But as with ruxolitinib, regrowth started to decline as patients were taken off treatment.
Research indicates that inhibiting the JAK-STAT signaling pathway induces anagen and subsequent hair growth, but activating STAT 5 in the dermal papilla is also important to induce the growth phase of the hair follicle, according to Dr. Mackay-Wiggan. “Bottom line, it’s complicated,” she added. “The mode of delivery – topical versus systemic – may be important, and the timing of delivery may be crucial.”
Other studies point to a role for JAK inhibition in treating AA. In an uncontrolled, retrospective study of 90 adults with alopecia totalis, alopecia universalis, or moderate to severe AA, 58% had SALT scores of 50% or better after receiving 5 mg tofacitinib twice daily for 4 to 18 months. Patients with AA improved more than those with alopecia totalis or universalis. There were no severe adverse effects, although nearly a third of patients developed upper respiratory tract infections. In another uncontrolled study of 13 patients with AA, totalis, or universalis, 9 (70%) patients achieved full regrowth and there were no serious adverse effects, although patients experienced headaches, upper respiratory infections, and mild increases in liver transaminase levels.
JAK inhibition also has a potential role for treating some scarring alopecias, including lichen planopilaris and frontal fibrosing alopecia. These diseases are histologically “identical” and both exhibit perifollicular erythema, papules, and scale, all of which suggest active inflammation, Dr. Mackay-Wiggan said. Hair follicles from affected patients show immune markers such as interferon-inducible chemokines, cytotoxic T cell responses, and expression of major histocompatibility complexes I and II. “The important message here is that JAK/STAT signaling may play a significant role in other types of hair loss other than alopecia areata,” Dr. Mackay-Wiggan said. “These diseases may also be autoimmune diseases, and may also be treatable with JAK inhibitors.”
Studies continue to evaluate JAK inhibitors for treating alopecia and its variants. Investigators at Yale and Stanford are conducting three uncontrolled trials of oral or topical tofacitinib, while Incyte, the manufacturer of ruxolitinib, is sponsoring a multicenter, randomized, placebo-controlled trial of ruxolitinib phosphate cream for adults with AA, with topline results expected in May 2018. Concert Pharmaceuticals also is recruiting for a trial of a modified, investigational form of ruxolitinib called CTP-543 for treating moderate to severe AA. “Many more trials are in development,” Dr. Mackay-Wiggan noted.
The ruxolitinib pilot study was funded by the Locks of Love Foundation, the Alopecia Areata Initiative, NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases, and by an Irving Institute for Clinical and Translational Research/Columbia University Medical Center Clinical and Translational Science Award. The ongoing tofacitinib pilot study is sponsored by Dr. Mackay-Wiggan, Locks of Love, and Columbia University.
Dr. Mackay-Wiggan also acknowledged support from the Alopecia Areata Initiative – the Gates Foundation, the National Alopecia Areata Registry, and the National Alopecia Areata Foundation. She had no other relevant financial disclosures.
PORTLAND, ORE. – Janus kinase inhibitors are relatively safe and can produce a full head of hair in patients with moderate to severe alopecia areata (AA), although patients tend to shed hair after stopping treatment, Julian Mackay-Wiggan, MD, said at the annual meeting of the Society for Investigative Dermatology.
“At this point, there are 17 publications in the literature, from clinical trials to case reports, looking at JAK [Janus kinase] inhibitors in patients with alopecia areata,” said Dr. Mackay-Wiggan of the department of dermatology, Columbia University, New York, where she specializes in hair disorders. “Pretty much all report very positive findings. It definitely appears that Janus kinase inhibitors can play a very significant role in treatment.”
In an open label, uncontrolled pilot study at Columbia, 9 of 12 (75%) patients with moderate to severe AA improved by at least 50% on the Severity of Alopecia Tool (SALT) after receiving 20 mg ruxolitinib twice daily for 3 to 6 months (JCI Insight. 2016 Sep 22;1[15]:e89790). Responses started with the first month, and all but one responder achieved at least 50% hair regrowth by week 12, said Dr. Mackay-Wiggan, who is also the director of the Dermatology Clinical Research Unit at Columbia.
By the end of treatment, seven of nine responders achieved more than 95% regrowth, one achieved 85% regrowth, and one achieved 55% regrowth. Importantly, none of these relatively healthy patients experienced serious adverse events on ruxolitinib, and none needed to stop treatment, although one patient experienced declining hemoglobin levels that resolved after dose modification.
Columbia researchers are also conducting an uncontrolled, open label pilot trial of the JAK inhibitor tofacitinib (Xeljanz) in 12 patients, of whom seven have moderate to severe patchy AA and five have alopecia totalis or universalis. Tofacitinib is approved for treating rheumatoid arthritis at a dose of 5 mg twice daily, but patients have needed up to 10 mg twice daily to achieve hair regrowth, Dr. Mackay-Wiggan said. To date, 11 (92%) have achieved at least some hair regrowth, and 8 (67%) have achieved at least 50% regrowth. So far, there have been no serious adverse events over 6 to 16 months of treatment, although one patient stopped treatment after developing hypertension, a known adverse effect of tofacitinib.
In this study, heatmaps of RNA sequencing of CD8+ T cell populations clearly showed pathogenic signatures for AA and a “robust molecular response to treatment,” Dr. Mackay-Wiggan said. “These two signatures also overlapped statistically, producing 114 genes that may be targetable mediators of disease.” But as with ruxolitinib, regrowth started to decline as patients were taken off treatment.
Research indicates that inhibiting the JAK-STAT signaling pathway induces anagen and subsequent hair growth, but activating STAT 5 in the dermal papilla is also important to induce the growth phase of the hair follicle, according to Dr. Mackay-Wiggan. “Bottom line, it’s complicated,” she added. “The mode of delivery – topical versus systemic – may be important, and the timing of delivery may be crucial.”
Other studies point to a role for JAK inhibition in treating AA. In an uncontrolled, retrospective study of 90 adults with alopecia totalis, alopecia universalis, or moderate to severe AA, 58% had SALT scores of 50% or better after receiving 5 mg tofacitinib twice daily for 4 to 18 months. Patients with AA improved more than those with alopecia totalis or universalis. There were no severe adverse effects, although nearly a third of patients developed upper respiratory tract infections. In another uncontrolled study of 13 patients with AA, totalis, or universalis, 9 (70%) patients achieved full regrowth and there were no serious adverse effects, although patients experienced headaches, upper respiratory infections, and mild increases in liver transaminase levels.
JAK inhibition also has a potential role for treating some scarring alopecias, including lichen planopilaris and frontal fibrosing alopecia. These diseases are histologically “identical” and both exhibit perifollicular erythema, papules, and scale, all of which suggest active inflammation, Dr. Mackay-Wiggan said. Hair follicles from affected patients show immune markers such as interferon-inducible chemokines, cytotoxic T cell responses, and expression of major histocompatibility complexes I and II. “The important message here is that JAK/STAT signaling may play a significant role in other types of hair loss other than alopecia areata,” Dr. Mackay-Wiggan said. “These diseases may also be autoimmune diseases, and may also be treatable with JAK inhibitors.”
Studies continue to evaluate JAK inhibitors for treating alopecia and its variants. Investigators at Yale and Stanford are conducting three uncontrolled trials of oral or topical tofacitinib, while Incyte, the manufacturer of ruxolitinib, is sponsoring a multicenter, randomized, placebo-controlled trial of ruxolitinib phosphate cream for adults with AA, with topline results expected in May 2018. Concert Pharmaceuticals also is recruiting for a trial of a modified, investigational form of ruxolitinib called CTP-543 for treating moderate to severe AA. “Many more trials are in development,” Dr. Mackay-Wiggan noted.
The ruxolitinib pilot study was funded by the Locks of Love Foundation, the Alopecia Areata Initiative, NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases, and by an Irving Institute for Clinical and Translational Research/Columbia University Medical Center Clinical and Translational Science Award. The ongoing tofacitinib pilot study is sponsored by Dr. Mackay-Wiggan, Locks of Love, and Columbia University.
Dr. Mackay-Wiggan also acknowledged support from the Alopecia Areata Initiative – the Gates Foundation, the National Alopecia Areata Registry, and the National Alopecia Areata Foundation. She had no other relevant financial disclosures.
AT SID 2017
5-Fluorouracil failed four separate measures of photoaging
PORTLAND – A standard course of topical 5-fluorouracil (5-FU) does not noticeably improve visual signs of facial photoaging, such as forehead lines and crow’s feet, according to the results of a blinded, controlled study of 281 elderly white men.
Four validated photonumeric measures revealed no statistically significant differences between the intervention and vehicle control arms at 6, 12, or 18 months’ follow-up, Kaveri Korgavkar, MD, said at the annual meeting of the Society for Investigative Dermatology. “This might be a true lack of impact, or current scales may not be sensitive enough to capture aspects of aging that are improved by 5-fluorouracil,” commented Dr. Korgavkar, who presented the findings on behalf of the VAKCCT (the Veterans Affairs Keratinocyte Carcinoma Chemoprevention Trial) work group.
The treatment and control groups resembled each other demographically and clinically at baseline. Participants averaged 71.5 years of age (standard deviation, 0.57 years), 97% were male, 99% were white, and all had clinically meaningful histories of sun damage with at least two keratinocyte carcinomas in the previous 2 years, including at least one lesion on the face or ears. Previously, the VAKCCT investigators reported positive results for 5-FU as a chemopreventive – for example, it was associated with about a 60% reduction in actinic keratoses, compared with placebo, and the effects persisted for up to 3 years.
However, none of the four photonumeric scales of photoaging uncovered significant differences between the treatment and control groups at 6, 12, or 18 months’ follow-up, Dr. Korgavkar reported. That finding belies the results of two other previous studies, but they were small and uncontrolled, she added. One study of 19 patients reported statistically significant improvements over time in wrinkling, hyperpigmentation, lentigines, and sallowness based on the Griffith’s scale, while a second prospective study of 32 patients reported significant improvements in visual signs of photoaging on the forearms, with a corresponding rise in levels of procollagen 1 and a decrease in dermal elastosis at 1 month.
Existing scales might more effectively capture some aspects of photoaging – such as wrinkles or crow’s feet – than others, Dr. Korgavkar said in an interview. Therefore, she and her associates are working to construct more sensitive and comprehensive visual scales of photoaging, she said.
The VAKCCT was sponsored by the VA Office of Research and Development. Dr. Korgavkar had no conflicts of interest.
PORTLAND – A standard course of topical 5-fluorouracil (5-FU) does not noticeably improve visual signs of facial photoaging, such as forehead lines and crow’s feet, according to the results of a blinded, controlled study of 281 elderly white men.
Four validated photonumeric measures revealed no statistically significant differences between the intervention and vehicle control arms at 6, 12, or 18 months’ follow-up, Kaveri Korgavkar, MD, said at the annual meeting of the Society for Investigative Dermatology. “This might be a true lack of impact, or current scales may not be sensitive enough to capture aspects of aging that are improved by 5-fluorouracil,” commented Dr. Korgavkar, who presented the findings on behalf of the VAKCCT (the Veterans Affairs Keratinocyte Carcinoma Chemoprevention Trial) work group.
The treatment and control groups resembled each other demographically and clinically at baseline. Participants averaged 71.5 years of age (standard deviation, 0.57 years), 97% were male, 99% were white, and all had clinically meaningful histories of sun damage with at least two keratinocyte carcinomas in the previous 2 years, including at least one lesion on the face or ears. Previously, the VAKCCT investigators reported positive results for 5-FU as a chemopreventive – for example, it was associated with about a 60% reduction in actinic keratoses, compared with placebo, and the effects persisted for up to 3 years.
However, none of the four photonumeric scales of photoaging uncovered significant differences between the treatment and control groups at 6, 12, or 18 months’ follow-up, Dr. Korgavkar reported. That finding belies the results of two other previous studies, but they were small and uncontrolled, she added. One study of 19 patients reported statistically significant improvements over time in wrinkling, hyperpigmentation, lentigines, and sallowness based on the Griffith’s scale, while a second prospective study of 32 patients reported significant improvements in visual signs of photoaging on the forearms, with a corresponding rise in levels of procollagen 1 and a decrease in dermal elastosis at 1 month.
Existing scales might more effectively capture some aspects of photoaging – such as wrinkles or crow’s feet – than others, Dr. Korgavkar said in an interview. Therefore, she and her associates are working to construct more sensitive and comprehensive visual scales of photoaging, she said.
The VAKCCT was sponsored by the VA Office of Research and Development. Dr. Korgavkar had no conflicts of interest.
PORTLAND – A standard course of topical 5-fluorouracil (5-FU) does not noticeably improve visual signs of facial photoaging, such as forehead lines and crow’s feet, according to the results of a blinded, controlled study of 281 elderly white men.
Four validated photonumeric measures revealed no statistically significant differences between the intervention and vehicle control arms at 6, 12, or 18 months’ follow-up, Kaveri Korgavkar, MD, said at the annual meeting of the Society for Investigative Dermatology. “This might be a true lack of impact, or current scales may not be sensitive enough to capture aspects of aging that are improved by 5-fluorouracil,” commented Dr. Korgavkar, who presented the findings on behalf of the VAKCCT (the Veterans Affairs Keratinocyte Carcinoma Chemoprevention Trial) work group.
The treatment and control groups resembled each other demographically and clinically at baseline. Participants averaged 71.5 years of age (standard deviation, 0.57 years), 97% were male, 99% were white, and all had clinically meaningful histories of sun damage with at least two keratinocyte carcinomas in the previous 2 years, including at least one lesion on the face or ears. Previously, the VAKCCT investigators reported positive results for 5-FU as a chemopreventive – for example, it was associated with about a 60% reduction in actinic keratoses, compared with placebo, and the effects persisted for up to 3 years.
However, none of the four photonumeric scales of photoaging uncovered significant differences between the treatment and control groups at 6, 12, or 18 months’ follow-up, Dr. Korgavkar reported. That finding belies the results of two other previous studies, but they were small and uncontrolled, she added. One study of 19 patients reported statistically significant improvements over time in wrinkling, hyperpigmentation, lentigines, and sallowness based on the Griffith’s scale, while a second prospective study of 32 patients reported significant improvements in visual signs of photoaging on the forearms, with a corresponding rise in levels of procollagen 1 and a decrease in dermal elastosis at 1 month.
Existing scales might more effectively capture some aspects of photoaging – such as wrinkles or crow’s feet – than others, Dr. Korgavkar said in an interview. Therefore, she and her associates are working to construct more sensitive and comprehensive visual scales of photoaging, she said.
The VAKCCT was sponsored by the VA Office of Research and Development. Dr. Korgavkar had no conflicts of interest.
AT SID 2017
Key clinical point: A standard topical course of 5-fluorouracil did not noticeably improve visual signs of photoaging, such as forehead lines and crow’s feet.
Major finding: Four validated photonumeric measures of photoaging revealed no statistically significant differences between the intervention and the vehicle control at 6, 12, or 18 months’ follow-up.
Data source: An analysis of data from 281 participants in the Veterans Affairs Keratinocyte Carcinoma Chemoprevention trial (VAKCCT).
Disclosures: The VAKCCT was sponsored by the VA Office of Research and Development. Dr. Korgavkar had no conflicts of interest.
Acne, rosacea prescriptions cost more when prescribed by dermatologists
PORTLAND, ORE. – Dermatologists have plenty of room to improve when it comes to choosing cost-effective medications for acne or rosacea, based on the results of a large retrospective analysis of Medicare claims data.
Patients with acne or rosacea consistently paid more for topical retinoids, topical antibiotics, and oral tetracyclines when the prescriber was a dermatologist instead of a family or internal medicine physician, Myron Zhang reported at the annual meeting of the Society for Investigative Dermatology.
“There is a large potential for reducing expenditures on health care for patients with acne and rosacea by choosing more generics and less expensive options within drug classes,” said Mr. Zhang, a medical student at the Ohio State University, Columbus, who conducted the study with colleagues there and at Northwestern University in Chicago.
Treating acne and rosacea falls under the purview of both primary and specialty outpatient care, but, researchers had not broken down costs of prescriptions for these conditions by provider type. To help fill that gap, Mr. Zhang and his colleagues retrospectively analyzed all Medicare drug claims for topical retinoids, topical antibiotics, isotretinoin, and oral tetracycline-class antibiotics used to treat acne and rosacea in the United States in 2008 and 2010.
Medicare claims for these prescriptions added up to $65 million in 2008 and $74 million in 2004, Mr. Zhang said. Although most generics either dropped in cost or rose by small amounts in that 2-year span, many brand name prescriptions rose by between 30% and 70%. “Specialist prescriptions were associated with higher brand name usage, a greater variety of medications, and higher costs,” Mr. Zhang said.
Patients paid an average of about $2-$3 more for a topical retinoid, $3-$4 more for oral tetracycline, and $1 more for a topical antibiotic prescribed by a specialist instead of a primary care physician.
This finding might indicate that dermatologists are more comfortable prescribing a greater variety of medications for acne and rosacea, such as tazarotene, azelaic acid, and sulfacetamide, while primary care physicians might stick to a narrower range of medicines, Mr. Zhang said. However, the prescribing behavior of dermatologists could also reflect “external factors,” such as increased contact with representatives from pharmaceutical companies, he added.
“As always, prescription costs need to be weighed against patient preference, compliance, and quality of care,” he said. “More outcomes research is needed to understand the comparative efficacy of generic and brand name treatments.”
The study included both rosacea and acne so that the researchers could capture the total costs of medications such as tretinoin, which is used to treat both conditions. Approximately 80% of topical retinoid prescriptions were for tretinoin, about 50% of topical antibiotic prescriptions were for metronidazole, while about 27% were for clindamycin. About half of oral antibiotic prescriptions were for tetracycline, while about 40% were for minocycline.
Mr. Zhang reported having no conflicts of interest.
PORTLAND, ORE. – Dermatologists have plenty of room to improve when it comes to choosing cost-effective medications for acne or rosacea, based on the results of a large retrospective analysis of Medicare claims data.
Patients with acne or rosacea consistently paid more for topical retinoids, topical antibiotics, and oral tetracyclines when the prescriber was a dermatologist instead of a family or internal medicine physician, Myron Zhang reported at the annual meeting of the Society for Investigative Dermatology.
“There is a large potential for reducing expenditures on health care for patients with acne and rosacea by choosing more generics and less expensive options within drug classes,” said Mr. Zhang, a medical student at the Ohio State University, Columbus, who conducted the study with colleagues there and at Northwestern University in Chicago.
Treating acne and rosacea falls under the purview of both primary and specialty outpatient care, but, researchers had not broken down costs of prescriptions for these conditions by provider type. To help fill that gap, Mr. Zhang and his colleagues retrospectively analyzed all Medicare drug claims for topical retinoids, topical antibiotics, isotretinoin, and oral tetracycline-class antibiotics used to treat acne and rosacea in the United States in 2008 and 2010.
Medicare claims for these prescriptions added up to $65 million in 2008 and $74 million in 2004, Mr. Zhang said. Although most generics either dropped in cost or rose by small amounts in that 2-year span, many brand name prescriptions rose by between 30% and 70%. “Specialist prescriptions were associated with higher brand name usage, a greater variety of medications, and higher costs,” Mr. Zhang said.
Patients paid an average of about $2-$3 more for a topical retinoid, $3-$4 more for oral tetracycline, and $1 more for a topical antibiotic prescribed by a specialist instead of a primary care physician.
This finding might indicate that dermatologists are more comfortable prescribing a greater variety of medications for acne and rosacea, such as tazarotene, azelaic acid, and sulfacetamide, while primary care physicians might stick to a narrower range of medicines, Mr. Zhang said. However, the prescribing behavior of dermatologists could also reflect “external factors,” such as increased contact with representatives from pharmaceutical companies, he added.
“As always, prescription costs need to be weighed against patient preference, compliance, and quality of care,” he said. “More outcomes research is needed to understand the comparative efficacy of generic and brand name treatments.”
The study included both rosacea and acne so that the researchers could capture the total costs of medications such as tretinoin, which is used to treat both conditions. Approximately 80% of topical retinoid prescriptions were for tretinoin, about 50% of topical antibiotic prescriptions were for metronidazole, while about 27% were for clindamycin. About half of oral antibiotic prescriptions were for tetracycline, while about 40% were for minocycline.
Mr. Zhang reported having no conflicts of interest.
PORTLAND, ORE. – Dermatologists have plenty of room to improve when it comes to choosing cost-effective medications for acne or rosacea, based on the results of a large retrospective analysis of Medicare claims data.
Patients with acne or rosacea consistently paid more for topical retinoids, topical antibiotics, and oral tetracyclines when the prescriber was a dermatologist instead of a family or internal medicine physician, Myron Zhang reported at the annual meeting of the Society for Investigative Dermatology.
“There is a large potential for reducing expenditures on health care for patients with acne and rosacea by choosing more generics and less expensive options within drug classes,” said Mr. Zhang, a medical student at the Ohio State University, Columbus, who conducted the study with colleagues there and at Northwestern University in Chicago.
Treating acne and rosacea falls under the purview of both primary and specialty outpatient care, but, researchers had not broken down costs of prescriptions for these conditions by provider type. To help fill that gap, Mr. Zhang and his colleagues retrospectively analyzed all Medicare drug claims for topical retinoids, topical antibiotics, isotretinoin, and oral tetracycline-class antibiotics used to treat acne and rosacea in the United States in 2008 and 2010.
Medicare claims for these prescriptions added up to $65 million in 2008 and $74 million in 2004, Mr. Zhang said. Although most generics either dropped in cost or rose by small amounts in that 2-year span, many brand name prescriptions rose by between 30% and 70%. “Specialist prescriptions were associated with higher brand name usage, a greater variety of medications, and higher costs,” Mr. Zhang said.
Patients paid an average of about $2-$3 more for a topical retinoid, $3-$4 more for oral tetracycline, and $1 more for a topical antibiotic prescribed by a specialist instead of a primary care physician.
This finding might indicate that dermatologists are more comfortable prescribing a greater variety of medications for acne and rosacea, such as tazarotene, azelaic acid, and sulfacetamide, while primary care physicians might stick to a narrower range of medicines, Mr. Zhang said. However, the prescribing behavior of dermatologists could also reflect “external factors,” such as increased contact with representatives from pharmaceutical companies, he added.
“As always, prescription costs need to be weighed against patient preference, compliance, and quality of care,” he said. “More outcomes research is needed to understand the comparative efficacy of generic and brand name treatments.”
The study included both rosacea and acne so that the researchers could capture the total costs of medications such as tretinoin, which is used to treat both conditions. Approximately 80% of topical retinoid prescriptions were for tretinoin, about 50% of topical antibiotic prescriptions were for metronidazole, while about 27% were for clindamycin. About half of oral antibiotic prescriptions were for tetracycline, while about 40% were for minocycline.
Mr. Zhang reported having no conflicts of interest.
At SID 2017
Key clinical point:
Major finding: Patients paid an average of $2-$3 more for a topical retinoid, $3-$4 more for an oral tetracycline-class antibiotic, and $1 more for a topical antibiotic prescribed by a dermatologist, compared with prescriptions from a primary care physician.
Data source: A retrospective analysis of Medicare prescription claims for oral antibiotics, topical antibiotics, and topical retinoids for acne and rosacea in 2008 and 2010.
Disclosures: Mr. Zhang reported having no conflicts of interest.
Vitamin D3 supplementation beat placebo in atopic dermatitis trial
PORTLAND – Short-term daily oral supplementation with 5000 IU of vitamin D3 was safe and significantly outperformed placebo for lessening the signs and symptoms of atopic dermatitis (AD), according to the results of a randomized, double-blind trial of 65 patients with moderate to severe disease.
At week 12, average improvements on the Scoring Atopic Dermatitis (SCORAD) index (standard deviation, 11.1 points) were 21.2 points in the vitamin D3 group and 13.9 points (standard deviation, 12.3 points) in the placebo group (P = .02), Karen Sanchez-Armendariz, MD, reported at the annual meeting of the Society for Investigative Dermatology. “Vitamin D3 should be considered a valuable adjuvant in the treatment of AD, especially in patients with relapses,” she said.
Past studies have reported an inverse relationship between 25(OH)D (vitamin D) serum levels and the severity of AD, noted Dr. Sanchez-Armendariz, who is with the National Autonomous University of Mexico in Mexico City. This finding makes sense, as vitamin D3 induces the topical production of antimicrobial peptides, which have direct antimicrobial activity and induce a host response characterized by cytokine release, chemotaxis, inflammation, angiogenesis, and re-epithelialization, she added.
Consequently, 25(OH)D deficiency has been posited as a culprit in the pathophysiology of AD. However, scientific and endocrine societies have published differing recommendations about safe and optimal doses for vitamin D3 supplementation, while experts have noted that 5000 IU per day is needed to achieve an increase of 10 ng 25(OH)D per mL serum (J Am Board Fam Med. 2014 Jul-Aug;27[4]:495-509).
For the study, therefore, Dr. Sanchez-Armendariz and her associates evaluated this dose as adjuvant therapy in patients with moderate to severe AD despite topical hydrocortisone aceponate, soap substitute, and emollient. Patients continued this usual care and received either a cellulose capsule placebo or 5000 IU oral vitamin D3 per day. Based on previously recommended cutoff values, the researchers considered serum 25(OH)D levels sufficient if they exceeded 30 ng per mL, insufficient if they were between 20 and 30 ng per mL, and deficient if they were less than 20 ng per mL (J Bone Miner Res. 2011 Mar;26[3]:455-7).
At baseline, the placebo and intervention groups resembled each other in terms of baseline levels of 25(OH)D, calcium, and phosphorus, pruritus, and scores on SCORAD and the six-area Total Body Severity Assessment. A total of 59% of patients had insufficient serum 25(OH)D, and 39% were deficient.
Patients in both groups improved on the Total Body Severity Assessment, but the extent of improvement did not statistically differ between groups, Dr. Sanchez-Armendariz reported. Supplementation did significantly increase serum 25(OH)D levels, which at week 12 averaged 58.3 ng per mL (SD, 18.5 ng per mL), compared with 22.2 (SD, 7.6 ng per mL) with placebo (P less than .001).
Interestingly, at week 12, all patients in the vitamin D3 group achieved serum 25(OH)D levels of at least 30 ng per mL, as did 41% of patients in the placebo arm. Patients in both groups who achieved this “sufficient” serum 25(OH)D level averaged a nearly 21-point improvement on the SCORAD (SD, 11.1 points), compared with 7.5 points (SD, 9.9 points) among patients who remained deficient, and 17.4 points (SD, 12.7 points) among patients who remained insufficient (P = .004 for difference among groups; P = .02 for difference between sufficient and insufficient groups).
An analysis of variance confirmed this finding, showing that, regardless of treatment group, patients whose final serum 25(OH)D level was at least 20 ng per mL scored an average of 20 points on the SCORAD, compared with 38 points for patients whose 25(OH)D level remained below 20 ng per mL. Patients whose final 25(OH)D level exceeded 20 ng per mL had fivefold greater odds of having a SCORAD below 25 than did patients with lower serum 25(OH)D levels (odds ratio, 5.1; 95% confidence interval, 1.2-22.6; P = .03).
The investigators identified no adverse effects of vitamin D3 supplementation, Dr. Sanchez-Armendariz said. “Attaining 25(OH)D levels above 20 ng per mL in addition to baseline therapy reduced the SCORAD of patients with moderate to severe atopic dermatitis,” she added. Clinicians should consider vitamin D3 deficiency and supplementation in patients with relapsing or refractory AD, she emphasized.
Dr. Sanchez-Armendariz reported no external funding sources and no conflicts of interest.
PORTLAND – Short-term daily oral supplementation with 5000 IU of vitamin D3 was safe and significantly outperformed placebo for lessening the signs and symptoms of atopic dermatitis (AD), according to the results of a randomized, double-blind trial of 65 patients with moderate to severe disease.
At week 12, average improvements on the Scoring Atopic Dermatitis (SCORAD) index (standard deviation, 11.1 points) were 21.2 points in the vitamin D3 group and 13.9 points (standard deviation, 12.3 points) in the placebo group (P = .02), Karen Sanchez-Armendariz, MD, reported at the annual meeting of the Society for Investigative Dermatology. “Vitamin D3 should be considered a valuable adjuvant in the treatment of AD, especially in patients with relapses,” she said.
Past studies have reported an inverse relationship between 25(OH)D (vitamin D) serum levels and the severity of AD, noted Dr. Sanchez-Armendariz, who is with the National Autonomous University of Mexico in Mexico City. This finding makes sense, as vitamin D3 induces the topical production of antimicrobial peptides, which have direct antimicrobial activity and induce a host response characterized by cytokine release, chemotaxis, inflammation, angiogenesis, and re-epithelialization, she added.
Consequently, 25(OH)D deficiency has been posited as a culprit in the pathophysiology of AD. However, scientific and endocrine societies have published differing recommendations about safe and optimal doses for vitamin D3 supplementation, while experts have noted that 5000 IU per day is needed to achieve an increase of 10 ng 25(OH)D per mL serum (J Am Board Fam Med. 2014 Jul-Aug;27[4]:495-509).
For the study, therefore, Dr. Sanchez-Armendariz and her associates evaluated this dose as adjuvant therapy in patients with moderate to severe AD despite topical hydrocortisone aceponate, soap substitute, and emollient. Patients continued this usual care and received either a cellulose capsule placebo or 5000 IU oral vitamin D3 per day. Based on previously recommended cutoff values, the researchers considered serum 25(OH)D levels sufficient if they exceeded 30 ng per mL, insufficient if they were between 20 and 30 ng per mL, and deficient if they were less than 20 ng per mL (J Bone Miner Res. 2011 Mar;26[3]:455-7).
At baseline, the placebo and intervention groups resembled each other in terms of baseline levels of 25(OH)D, calcium, and phosphorus, pruritus, and scores on SCORAD and the six-area Total Body Severity Assessment. A total of 59% of patients had insufficient serum 25(OH)D, and 39% were deficient.
Patients in both groups improved on the Total Body Severity Assessment, but the extent of improvement did not statistically differ between groups, Dr. Sanchez-Armendariz reported. Supplementation did significantly increase serum 25(OH)D levels, which at week 12 averaged 58.3 ng per mL (SD, 18.5 ng per mL), compared with 22.2 (SD, 7.6 ng per mL) with placebo (P less than .001).
Interestingly, at week 12, all patients in the vitamin D3 group achieved serum 25(OH)D levels of at least 30 ng per mL, as did 41% of patients in the placebo arm. Patients in both groups who achieved this “sufficient” serum 25(OH)D level averaged a nearly 21-point improvement on the SCORAD (SD, 11.1 points), compared with 7.5 points (SD, 9.9 points) among patients who remained deficient, and 17.4 points (SD, 12.7 points) among patients who remained insufficient (P = .004 for difference among groups; P = .02 for difference between sufficient and insufficient groups).
An analysis of variance confirmed this finding, showing that, regardless of treatment group, patients whose final serum 25(OH)D level was at least 20 ng per mL scored an average of 20 points on the SCORAD, compared with 38 points for patients whose 25(OH)D level remained below 20 ng per mL. Patients whose final 25(OH)D level exceeded 20 ng per mL had fivefold greater odds of having a SCORAD below 25 than did patients with lower serum 25(OH)D levels (odds ratio, 5.1; 95% confidence interval, 1.2-22.6; P = .03).
The investigators identified no adverse effects of vitamin D3 supplementation, Dr. Sanchez-Armendariz said. “Attaining 25(OH)D levels above 20 ng per mL in addition to baseline therapy reduced the SCORAD of patients with moderate to severe atopic dermatitis,” she added. Clinicians should consider vitamin D3 deficiency and supplementation in patients with relapsing or refractory AD, she emphasized.
Dr. Sanchez-Armendariz reported no external funding sources and no conflicts of interest.
PORTLAND – Short-term daily oral supplementation with 5000 IU of vitamin D3 was safe and significantly outperformed placebo for lessening the signs and symptoms of atopic dermatitis (AD), according to the results of a randomized, double-blind trial of 65 patients with moderate to severe disease.
At week 12, average improvements on the Scoring Atopic Dermatitis (SCORAD) index (standard deviation, 11.1 points) were 21.2 points in the vitamin D3 group and 13.9 points (standard deviation, 12.3 points) in the placebo group (P = .02), Karen Sanchez-Armendariz, MD, reported at the annual meeting of the Society for Investigative Dermatology. “Vitamin D3 should be considered a valuable adjuvant in the treatment of AD, especially in patients with relapses,” she said.
Past studies have reported an inverse relationship between 25(OH)D (vitamin D) serum levels and the severity of AD, noted Dr. Sanchez-Armendariz, who is with the National Autonomous University of Mexico in Mexico City. This finding makes sense, as vitamin D3 induces the topical production of antimicrobial peptides, which have direct antimicrobial activity and induce a host response characterized by cytokine release, chemotaxis, inflammation, angiogenesis, and re-epithelialization, she added.
Consequently, 25(OH)D deficiency has been posited as a culprit in the pathophysiology of AD. However, scientific and endocrine societies have published differing recommendations about safe and optimal doses for vitamin D3 supplementation, while experts have noted that 5000 IU per day is needed to achieve an increase of 10 ng 25(OH)D per mL serum (J Am Board Fam Med. 2014 Jul-Aug;27[4]:495-509).
For the study, therefore, Dr. Sanchez-Armendariz and her associates evaluated this dose as adjuvant therapy in patients with moderate to severe AD despite topical hydrocortisone aceponate, soap substitute, and emollient. Patients continued this usual care and received either a cellulose capsule placebo or 5000 IU oral vitamin D3 per day. Based on previously recommended cutoff values, the researchers considered serum 25(OH)D levels sufficient if they exceeded 30 ng per mL, insufficient if they were between 20 and 30 ng per mL, and deficient if they were less than 20 ng per mL (J Bone Miner Res. 2011 Mar;26[3]:455-7).
At baseline, the placebo and intervention groups resembled each other in terms of baseline levels of 25(OH)D, calcium, and phosphorus, pruritus, and scores on SCORAD and the six-area Total Body Severity Assessment. A total of 59% of patients had insufficient serum 25(OH)D, and 39% were deficient.
Patients in both groups improved on the Total Body Severity Assessment, but the extent of improvement did not statistically differ between groups, Dr. Sanchez-Armendariz reported. Supplementation did significantly increase serum 25(OH)D levels, which at week 12 averaged 58.3 ng per mL (SD, 18.5 ng per mL), compared with 22.2 (SD, 7.6 ng per mL) with placebo (P less than .001).
Interestingly, at week 12, all patients in the vitamin D3 group achieved serum 25(OH)D levels of at least 30 ng per mL, as did 41% of patients in the placebo arm. Patients in both groups who achieved this “sufficient” serum 25(OH)D level averaged a nearly 21-point improvement on the SCORAD (SD, 11.1 points), compared with 7.5 points (SD, 9.9 points) among patients who remained deficient, and 17.4 points (SD, 12.7 points) among patients who remained insufficient (P = .004 for difference among groups; P = .02 for difference between sufficient and insufficient groups).
An analysis of variance confirmed this finding, showing that, regardless of treatment group, patients whose final serum 25(OH)D level was at least 20 ng per mL scored an average of 20 points on the SCORAD, compared with 38 points for patients whose 25(OH)D level remained below 20 ng per mL. Patients whose final 25(OH)D level exceeded 20 ng per mL had fivefold greater odds of having a SCORAD below 25 than did patients with lower serum 25(OH)D levels (odds ratio, 5.1; 95% confidence interval, 1.2-22.6; P = .03).
The investigators identified no adverse effects of vitamin D3 supplementation, Dr. Sanchez-Armendariz said. “Attaining 25(OH)D levels above 20 ng per mL in addition to baseline therapy reduced the SCORAD of patients with moderate to severe atopic dermatitis,” she added. Clinicians should consider vitamin D3 deficiency and supplementation in patients with relapsing or refractory AD, she emphasized.
Dr. Sanchez-Armendariz reported no external funding sources and no conflicts of interest.
Key clinical point:
Major finding: At week 12, average improvements on the SCORAD index were 21.2 points with vitamin D3 and 13.9 points (SD, 12.3 points) with placebo (P = .02).
Data source: A double-blind, placebo-controlled, randomized trial of 65 patients with moderate to severe atopic dermatitis despite usual therapy with topical steroids, soap substitute, and topical emollient.
Disclosures: Dr. Sanchez-Armendariz did not acknowledge external funding sources. She reported having no conflicts of interest.