User login
Beta-blockers for hypertension: Are they going out of style?
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
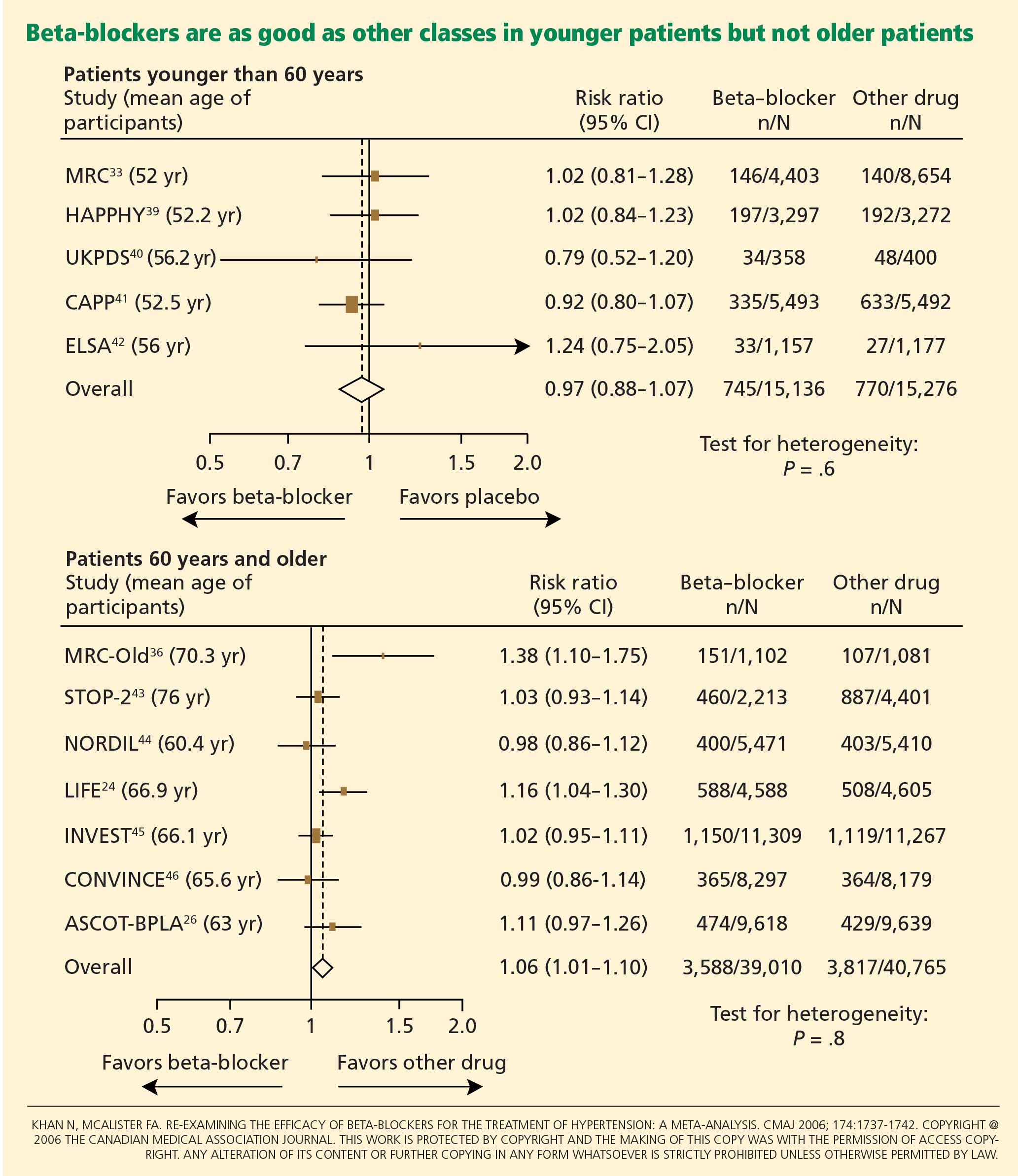
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
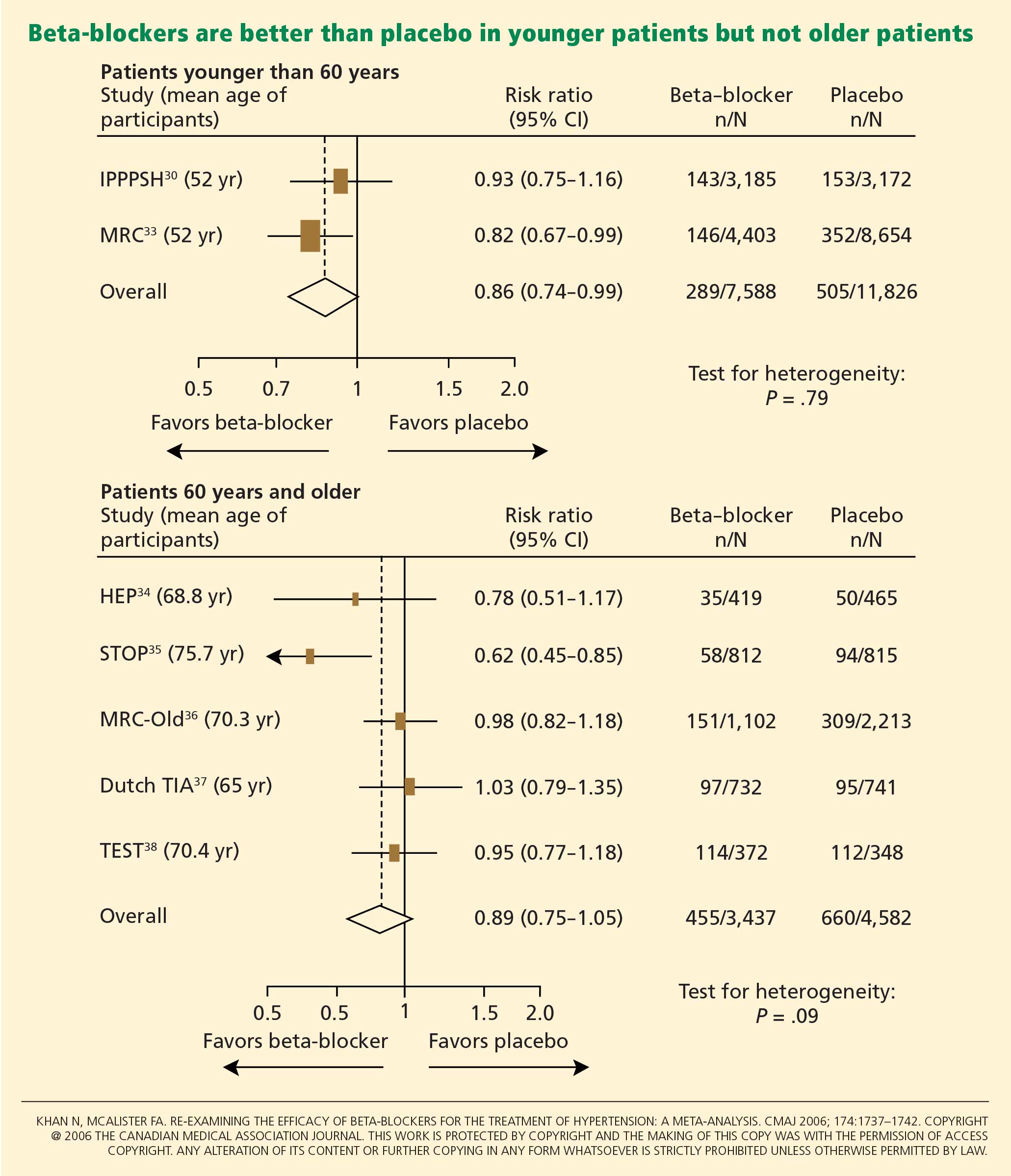
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
KEY POINTS
- No evidence exists that beta-blockers prevent first episodes of cardiovascular events in patients with hypertension, and in some trials, outcomes were worse with beta-blockers than with antihypertensive drugs of other classes.
- Younger hypertensive patients have hemodynamic characteristics that would seem to be amenable to beta-blocker therapy. However, most clinical trials of beta blockers did not stratify patients by age.
- Most trials of the antihypertensive effects of beta-blockers used atenolol (Tenormin), which is not an ideal representative of this class of drugs.
- Newer beta-blockers with vasodilatory properties may overcome the adverse effect of increased peripheral vascular resistance that occurs with older agents such as atenolol.
Pregabalin for fibromyalgia: Some relief but no cure
Pregabalin (Lyrica) is a novel analogue of the neurotransmitter gamma aminobutyric acid (GABA) with analgesic, anticonvulsant, and anxiolytic activity. Its approval by the US Food and Drug Administration (FDA) in 2007 for the treatment of fibromyalgia made it the first drug approved for this indication. Until then, management of fibromyalgia entailed drugs to treat pain, sleep, fatigue, and psychological disorders, and a strong emphasis on exercise and physical therapy.
Those who still question the validity of fibromyalgia as a diagnosis object to drug companies “benefiting” from the sale of such drugs.1 But many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it. A key question remains: How will pregabalin fit into the treatment of this often-challenging disease?
FROM FIBROSITIS TO FIBROMYALGIA
Fibromyalgia is a syndrome characterized by widespread pain. Chronic muscular pain is a common problem, but fibromyalgia is distinguished from other pain disorders by additional findings, such as consistent areas of tenderness (tender points), nonrestorative sleep, severe fatigue, and frequent psychological comorbidities such as depression and anxiety.
Fibromyalgia was originally termed “fibrositis” in 1904 by Sir William Gowers, who described it as a painful condition of the fibrous tissue, which he believed was due to inflammation in the muscles.2,3 For several decades, research was dedicated to looking for pathology in the muscle tissue, which was thought to be the major source of pain for most patients with fibromyalgia.
In the mid-1970s, Dr. H. Moldofsky, a noted sleep researcher, reported on abnormalities of the alpha-delta component of nonrapid-eye-movement sleep in these patients. He subsequently collaborated with Dr. Hugh A. Smythe, who helped define the fibromyalgia tender points. Fibrositis was subsequently renamed fibromyalgia syndrome, since it was agreed that there was no true inflammation in muscles or fibrous tissue.
In 1990, the American College of Rheumatology published “classification criteria” for the disease.4 The criteria include two main features:
- A history of widespread pain (“widespread” being defined as in the axial distribution, in both the left and right sides of the body, and above and below the waist), which must be present for 3 months or more, and
- Tenderness in at least 11 of 18 specified points that is elicited when a pressure of 4 kg (the amount of pressure required to blanch a thumbnail) is applied in steady increments starting at 1 kg.
Although pain is subjective and therefore difficult to assess, the classification criteria did make it easier to study the disease in a uniform way and led to an explosion of research in this field.
FUNCTIONAL ABNORMALITIES NI THE CENTRAL NERVOUS SYSTEM
Research to date points to the pain in fibromyalgia as being mediated by changes in the central nervous system rather than in the musculoskeletal system, as was initially thought.
In the dorsal horn of the spinal cord, nociceptive (pain-sensing) neurons from the periphery synapse with the second-order neurons that carry the pain signal to the brain. In fibromyalgia, several processes seem to amplify the signal.
Central sensitization is defined as enhanced excitability of neurons in the dorsal horn. Its features include augmented spontaneous neuronal activity, enlarged receptive field areas, and enhanced responses generated by large- and small-caliber primary afferent fibers. It can result from prolonged or strong activity in the dorsal horn neurons, and it leads to the spread of hyperactivity across multiple spinal segments.5–7
While much of the evidence for central sensitization in fibromyalgia is from animal studies, the phenomenon has also been studied in humans. Desmeules et al8 found that, compared with people without fibromyalgia, those with fibromyalgia had significantly lower thresholds of pain as assessed subjectively and measured objectively using the nociceptive flexion R-III reflex, which the authors described as “a specific physiologic correlate for the objective evaluation of central nociceptive pathways.”
Wind-up. Prolonged stimulation of C fibers in the dorsal horn can result in the phenomenon of wind-up, which refers to the temporal summation of second pain.
A painful stimulus evokes two pain signals. The first signal is brief and travels rapidly to the spinal cord via myelinated fibers (A fibers). The second signal, which is related to chronic pain and is described as dull, aching, or burning, travels more slowly to the dorsal horn via unmyelinated fibers (C fibers), the synapses of which use the neurotransmitter glutamate. Temporal summation is a phenomenon observed in experiments in which a series of painful stimuli are applied at regular intervals of about 2 seconds; although each stimulus is identical in intensity, subjects perceive them as increasing in intensity. The reason: during this repetitive stimulation, N-methyl-d-aspartate (NMDA) receptors become activated, leading to the removal of a magnesium block within the receptor. This results in an influx of calcium into the neuron and activation of protein kinase C, nitric oxide synthase, and cyclooxygenase. Ultimately, the firing rates of the nociceptive neurons are increased and the peripheral pain signal is strongly amplified.6
Wind-up has been shown to lead to characteristics of central sensitization related to C-fiber activity in animals.7 Staud et al9 studied wind-up in patients with and without fibromyalgia using series of repetitive thermal stimulation to produce temporal summation. Though wind-up was evoked in both groups, differences were observed both in the magnitude of sensory response to the first stimulus within a series and in the amount of temporal summation within a series.
Elevated excitatory neurotransmitters. In 1994, Russell et al10 showed that the concentration of substance P, an excitatory neurotransmitter, was three times higher in the cerebrospinal fluid of people with fibromyalgia than in normal controls.
Harris and colleagues11 reported that glutamate, another excitatory neurotransmitter, is elevated within the brain in people with fibromyalgia. They further showed that the levels of glutamate within the insula of the brain are directly associated with the levels of both experimental pressure-evoked pain thresholds and clinical pain ratings in fibromyalgia patients.
Evidence from imaging studies. Other objective evidence of central sensitization in fibromyalgia patients comes from studies using novel imaging.
Gracely et al12 performed functional magnetic resonance imaging (MRI) in people with and without fibromyalgia while applying pressure to their thumbs with a thumbscrew-type device. At equal levels of pressure, the people with fibromyalgia said the pressure hurt more, and specific areas of their brains lit up more on functional MRI. When the experimenters increased the pressure in the people without fibromyalgia until this group subjectively rated the pain as high as the fibromyalgia patients rated the lower level of pressure, their brains lit up to a similar degree in the same areas. These findings provide objective evidence of significantly lower pain thresholds in patients with fibromyalgia than in healthy controls, and they support the theory of central augmentation of pain sensitivity in fibromyalgia.
Staud et al13 also used functional MRI and found greater brain activity associated with temporal summation in fibromyalgia patients compared with controls. (In this experiment, the painful stimulus consisted of heat pulses to the foot.)
Drugs other than pregabalin that modulate the dorsal horn activity of the pain pathway include opioids, tramadol (Ultram), gabapentin (Neurontin), GABA agonists such as baclofen (Lioresal), antidepressants, alpha-2 adrenergic agonists (phenylephrine), and 5-HT3 antagonists such as ondansetron (Zofran), but none has been consistently effective for fibromyalgia.5,14
PREGABALIN
Pregabalin is an alpha-2-delta ligand similar to GABA, but it does not act on GABA receptors. Rather, it binds with high affinity to the alpha-2-delta subunit of voltage-gated presynaptic calcium channels, resulting in reduction of calcium flow through the channels, which subsequently inhibits the release of neurotransmitters including glutamate, norepinephrine, and substance P.15–17 Animal studies suggest that the decrease in the levels of these excitatory neurotransmitters is the mechanism of action of pregabalin, resulting in its analgesic, anticonvulsant, and anxiolytic benefit.15 Another potential mechanism of pregabalin is enhancement of slow-wave sleep, demonstrated in one study in healthy human subjects.18
Besides fibromyalgia, pregabalin is also approved for the treatment of diabetic peripheral neuropathy, postherpetic neuralgia, generalized anxiety disorder, and social anxiety disorder, and as adjunctive therapy for partial-onset seizure in adults.
Pharmacokinetics
Pregabalin is quickly absorbed, primarily in the proximal colon (bioavailability > 90%), and has highly predictable and linear pharmacokinetics.15 Food consumption does not affect its absorption or elimination but can delay its peak plasma concentration, which occurs at 1.5 hours. Its elimination half-life is approximately 6 hours.15 Because it does not bind to plasma proteins, it freely crosses the blood-brain barrier. The drug reaches its steady-state concentration within 2 days of starting therapy.
Its clearance is not affected by the sex or race of the patient, but its total clearance may be lower in the elderly because of age-related loss of renal function. Patients on hemodialysis may require a supplemental dose after dialysis because hemodialysis removes the pregabalin.
The drug is not metabolized by the P450 system in the liver, so it interacts only minimally with drugs that do use the P450 system. However, its clearance may be decreased when it is used concomitantly with drugs that can reduce the glomerular filtration rate, such as nonsteroidal anti-inflammatory drugs, aminoglycosides, and cyclosporine.15
Efficacy
The efficacy of pregabalin in fibromyalgia was evaluated in several recent trials.19
Crofford et al16 assessed pregabalin’s effects on pain, sleep, fatigue, and health-related quality of life. Some 529 patients with fibromyalgia were randomized in a double-blind fashion to four treatment groups: placebo, and pregabalin 150 mg/day, 300 mg/day, and 450 mg/day. The baseline mean pain scores (a 0-to-10 scale derived from daily diary ratings) were 6.9 in the placebo group, 6.9 for the pregabalin 150 mg/day group, 7.3 for the pregabalin 300 mg/day group, and 7.0 for the pregabalin 450 mg/day group.
The pain scores declined in all groups, but at 8 weeks, the mean score had declined 0.93 points more in the group receiving pregabalin 450 mg/day than in the placebo group (P ≤ .001). The scores in the groups taking pregabalin 150 mg/day and 300 mg/day were not significantly different from those in the placebo group. Significantly more patients in the 450-mg/day group (29%, vs 13% in the placebo group) had at least 50% improvement in pain at the end of the study. Patients in both the 300-mg/day group and the 450-mg/day had statistically significant improvement in their quality of sleep, in fatigue, and on the Patient Global Impression of Change (PGIC) scale.
Arnold et al20 conducted a trial with 750 patients in which three doses of pregabalin were compared with placebo: 300 mg/day, 450 mg/day, and 600 mg/day. The primary end point was also the change in pain score from baseline (using the 0-to-10 scale derived from a daily pain diary). The mean baseline pain score was 6.7.
At 14 weeks, the mean pain score was lower than at baseline in all the groups, but it had declined 0.71 more in the pregabalin 300-mg/day group than in the placebo group, 0.98 points more in the 450-mg/day group, and 1.0 points more in the 600-mg/day group. All three pregabalin groups also showed significant improvement on the PGIC scale, and patients in the 450-mg/day and 600-mg/day groups showed statistically significant improvement in the Fibromyalgia Impact Questionnaire (FIQ) score. All three pregabalin treatment groups also had significantly better patient-reported sleep outcomes than in the placebo group, both in measures of overall sleep and quality of sleep. With the exception of a significant improvement of anxiety on 600 mg/day, there was no significant difference between the treatment and placebo groups in the secondary outcomes of depression and anxiety symptoms and fatigue.
Duan et al21 presented a pooled analysis of this and a similarly designed double-blind, placebo-controlled trial (the results of which were not available individually) at the 71st annual meeting of the American College of Rheumatology in November 2007. The analysis included 1,493 patients with a mean baseline pain score of 6.9. Compared with the mean pain score in the placebo group, those in the pregabalin groups had declined more by the end of the study: 0.55 points more with 300 mg/day, 0.71 points more with 450 mg/day, and 0.82 points more with 600 mg/day. This pooled analysis also showed significant improvement in PGIC score with all pregabalin doses and in the FIQ score with 450 mg/day and 600 mg/day.
The FREEDOM trial22 (Fibromyalgia Relapse Evaluation and Efficacy for Durability of Meaningful Relief) evaluated the durability of effect of pregabalin in reducing pain and symptoms associated with fibromyalgia in 1,051 patients who initially responded to the drug.
The patients received 6 weeks of open-label treatment with pregabalin and then 26 weeks of double-blind treatment (dose adjustment was allowed based on efficacy and tolerability for the first 3 weeks). The time to loss of therapeutic response was significantly longer with pregabalin than with placebo. Loss of therapeutic response was defined as worsening of pain for two consecutive visits or worsening of fibromyalgia symptoms requiring alternative therapy.
By the end of the double-blind phase, 61% of those in the placebo group had loss of therapeutic response compared with only 32% in the pregabalin group. The time to worsening of the FIQ score was also significantly longer in the pregabalin group than in the placebo group.
Adverse effects: Dizziness, sleepiness, weight gain
Dizziness and sleepiness were the most common adverse events in these studies.
In the 8-week study by Crofford et al,16 dizziness was dose-related, occurring in 10.7% of those receiving placebo (one patient withdrew because of dizziness), 22.7% of those receiving 150 mg/day (two patients withdrew), 31.3% of those receiving 300 mg/day (four patients withdrew), and 49.2% of those receiving 450 mg/day (five patients withdrew). Somnolence was also dose-related, occurring in 4.6% in the placebo group, 15.9% in the 150-mg/day group (two patients withdrew due to somnolence), 27.6% in the 300-mg/day group (three withdrew), and 28.0% in the 450-mg/day group (five withdrew).
The 14-week study by Arnold et al20 also showed higher frequencies of adverse events with higher doses. The rates of dizziness were 7.6% with placebo, 27.9% with pregabalin 300 mg/day, 37.4% with 450 mg/day, and 42.0% with 600 mg/day. The rates of somnolence were 3.8% with placebo, 12.6% with 300 mg/day of pregabalin, 19.5% with 450 mg/day, and 21.8% with 600 mg/day. Dizziness and somnolence were also the most common adverse effects that led to discontinuation of pregabalin, with rates of 4% and 3%, respectively.
The open-label phase of the FREEDOM trial showed rates of 36% for dizziness and 22% for somnolence among pregabalin-treated patients.
Weight gain and peripheral edema were also common adverse effects in these studies.22 Definitions of weight gain varied, and edema was not accompanied by evidence of cardiac or renal dysfunction.
Less common side effects seen more frequently in the treated groups included dry mouth, blurred vision, and difficulty with concentration and attention. The package insert also warns of angioedema, hypersensitivity reaction, mild asymptomatic creatine kinase elevation, decreased platelet count (without bleeding), and prolongation of the PR interval on electrocardiography.
Pregabalin is a schedule V controlled substance; in clinical studies, abrupt or rapid discontinuation of the drug led to insomnia, nausea, headache, or diarrhea in some patients, suggesting symptoms of dependence. In clinical studies involving a total of more than 5,500 patients, 4% of patients on pregabalin and 1% of patients on placebo reported euphoria as an adverse effect,19 suggesting possible potential for abuse.
Dosing
As a result of the above studies, the recommended starting dose of pregabalin for fibromyalgia is 150 mg/day in two or three divided doses, gradually increased to 300 mg/day within 1 week based on tolerability and efficacy. The dose may be increased to a maximum of 450 mg/day. The 600-mg dose was found to have no significant additional benefit, but it did have more adverse effects and therefore is not recommended. It is important to note that in these studies multiple medications for pain and insomnia were prohibited, so data on drug interactions with pregabalin are limited.
Few achieve complete remission, but most patients feel better
Several studies of the natural history of fibromyalgia have shown that very few patients experience complete remission of the disease, even after many years. Therefore, one should try to set up realistic expectations for patients, with the goal of achieving functional improvement in activities of daily living and a return to one’s predisease state.
In the longest follow-up study, 39 patients in Boston, MA, were prospectively followed for over 10 years. No patient achieved complete remission: all of them reported some fibromyalgia-related symptoms at the end of the study.23 However, 66% of them felt a little to a lot better than when first diagnosed, 55% felt well or very well, and only 7% felt poorly.
Other studies have also shown complete remission to be rare.24,25 A 5-year follow-up study investigating fibromyalgia patients’ perceptions of their symptoms and its impact on everyday life activities demonstrated that the social consequences of fibromyalgia’s symptoms are severe and constant over time.26
Evidence of favorable outcomes was reported in one study in which 47% of patients reported moderate to marked improvement in overall fibromyalgia status upon 3-year follow-up,27 and in another study, in which remission was objectively identified in 24.2% of patients 2 years after diagnosis.28
OTHER THERAPIES
Although there have been many studies of pharmacologic therapies for fibromyalgia to date, the trials had significant limitations, such as short duration, inadequate sample size, nonstandardized measures of efficacy, question of regression to the mean, and inadequate blinding, resulting in insufficient evidence to recommend one drug over another.
Tricyclic antidepressants. Two meta-analyses and a clinical review have supported the efficacy of tricyclic antidepressants in improving symptoms in fibromyalgia patients.29–31
Selective serotonin reuptake inhibitors (SSRIs) have not been well studied, and the small size and methodologic shortcomings of these studies make it difficult to draw conclusions about the efficacy of SSRIs in reducing pain in fibromyalgia patients.30,31
Duloxetine (Cymbalta) and milnacipran (Savella) are serotonin and norepinephrine reuptake inhibitors.32–34 A randomized, double-blind placebo-controlled trial evaluated duloxetine in 520 fibromyalgia patients with and without major depressive disorder. Pain scores improved significantly over 6 months in duloxetine-treated patients at doses of 60 and 120 mg/day.33 Duloxetine became the second drug approved for the treatment of fibromyalgia in 2007, and milnacipran became the third in 2009.
WHAT ROLE FOR PREGABALIN?
Pregabalin may reduce pain in some patients with fibromyalgia. However, the presenting symptoms can vary significantly, and symptoms can vary even in individual patients over time. Therefore, in most patients with fibromyalgia, a multidisciplinary approach is used to treat pain, sleep disturbance, and fatigue, along with comorbidities such as neurally mediated hypotension and psychiatric disorders. Because treatment of fibromyalgia often involves multiple drugs in addition to exercise and behavioral therapies, future studies should examine combinations of drugs and the use of drugs in conjunction with nondrug treatments.
Pregabalin advances our knowledge of fibromyalgia through improving the understanding of central sensitization and how brain neurotransmitters control central pain perceptions. Drug treatment must still be part of the comprehensive management of this disease. Physician and patient education about the current understanding of the disease is paramount in setting realistic goals for treatment.14 Future strategies to manage fibromyalgia will be based on the pathophysiology of this complex condition.
- Berenson A. Drug approved. Is disease real? New York Times, January 14, 2008. http://www.nytimes.com/2008/01/14/health/14pain.html. Accessed February 2, 2009.
- White KP, Harth M. Classification, epidemiology, and natural history of fibromyalgia. Curr Pain Headache Rep 2001; 5:320–329.
- Bennett RM. Fibromyalgia: present to future. Curr Pain Headache Rep 2004; 8:379–384.
- Wolfe F, Smythe HA, Yunus MF, et al. The American College of Rheumatolgy 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum 1990; 33:160–172.
- Bennett RM. The rational management of fibromyalgia patients. Rheum Dis Clin North Am 2002; 28:181–199.
- Staud R. Evidence of involvement of central neural mechanisms in generating fibromyalgia pain. Curr Rheumatol Rep 2002; 4:299–305.
- Li J, Simone DA, Larson AA. Windup leads to characteristics of central sensitization. Pain 1999; 79:75–82.
- Desmeules JA, Cedraschi C, Rapiti E, et al. Neurophysiologic evidence for a central sensitization in patients with fibromyalgia. Arthritis Rheum 2003; 48:1420–1429.
- Staud R, Vierck CJ, Cannon RL, Mauderli AP, Price DD. Abnormal sensitization and temporal summation of pain (wind-up) in patients with fibromyalgia syndrome. Pain 2001; 91:165–175.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Harris RE, Sundgren PC, Pang Y, et al. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum 2008; 58:903–907.
- Gracely RH, Petzke F, Wolf JM, Clauw DJ. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum 2002; 46:1333–1343.
- Staud R, Craggs JG, Perlstein WM, Robinson ME, Price DD. Brain activity associated with slow temporal summation of C-fiber evoked pain in fibromyalgia patients and healthy controls. Eur J Pain 2008; 12:1078–1089.
- Baker K, Barkhuizen A. Pharmacologic treatment of fibromyalgia. Curr Pain Headache Rep 2005; 9:301–306.
- Tassone DM, Boyce E, Guyer J, Nuzum D. Pregabalin: a novel gamma-aminobutyric acid analogue in the treatment of neuropathic pain, partial-onset seizures, and anxiety disorders. Clin Ther 2007; 29:26–48.
- Crofford LJ, Rowbotham MC, Mease PJ, et al, and the Pregabalin 1008-105 Study Group. Pregabalin for the treatment of fibromyalgia syndrome. results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2005; 52:1264–1273.
- Stahl SM. Anticonvulsants and the relief of chronic pain: pregabalin and gabapentin as alpha(2)delta ligands at voltage-gated calcium channels. J Clin Psychiatry 2004; 65:596–597.
- Hindmarch I, Dawson J, Stanley N. A double-blind study in healthy volunteers to assess the effects of sleep on pregabalin compared with alprazolam and placebo. Sleep 2005; 28:187–193.
- Pfizer Executive Summary. Lyrica (pregabalin) capsules c-v. July 2007. www.fda.gov/OHRMS/DOCKETS/ac/08/briefing/2008-4372b1-02-Pfizer.pdf. Accessed February 2, 2009.
- Arnold LM, Russell IJ, Diri EW, et al. A 14-week, randomized, double-blinded, placebo-controlled mono-therapy trial of pregabalin in patients with fibomyalgia. J Pain 2008; 9:792–805.
- Duan WR, Florian H, Young JP, Martin S, Haig G, Barrett JA. Pregabalin monotherapy for management of fibromyalgia: analysis of two double-blind, randomized, placebo-controlled trials (poster presentation). American College of Rheumatology Annual Scientific Meeting, Boston, MA, November 6–7, 2007.
- Crofford LJ, Mease PJ, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain 2008; 136:419–431.
- Kennedy M, Felson DT. A prospective long-term study of fibromyalgia syndrome. Arthritis Rheum 1996; 39:682–685.
- Bengtsson A, Backman E. Long-term follow-up of fibro-myalgia patients [abstract]. Scand J Rheumatolology 1992; 21(suppl 94):9.
- Ledingham J, Doherty S, Doherty M. Primary fibromyalgia syndrome—an outcome study. Br J Rheumatol 1993; 32:139–142.
- Henrikkson CM. Longterm effects of fibromyalgia on everyday life: a study of 56 patients. Scand J Rheumatol 1994; 23:36–41.
- Fitzcharles MA, Costa DD, Pöyhiä R. A study of standard care in fibromyalgia syndrome: a favorable outcome. J Rheumatol 2003; 30:154–159.
- Granges G, Zilko P, Littlejohn GO. Fibromyalgia syndrome: assessment of the severity of the condition 2 years after the diagnosis. J Rheumatol 1994; 21:523–529.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia: a meta-analysis and review. Psychosomatics 2000; 41:104–113.
- O’Malley PG, Balden E, Tomkins G, Santoro J, Kroenke K, Jackson JL. Treatment of fibromyalgia with anti-depressants: a meta-analysis. J Gen Intern Med 2000; 15:659–666.
- Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum 2004; 50:2974–2984.
- Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: Results from a 6-month, randomized, double-blind, placebo-controlled fixed-dose trial. Pain 2008; 136:432–444.
- Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther 2008; 30:1988–2004.
Pregabalin (Lyrica) is a novel analogue of the neurotransmitter gamma aminobutyric acid (GABA) with analgesic, anticonvulsant, and anxiolytic activity. Its approval by the US Food and Drug Administration (FDA) in 2007 for the treatment of fibromyalgia made it the first drug approved for this indication. Until then, management of fibromyalgia entailed drugs to treat pain, sleep, fatigue, and psychological disorders, and a strong emphasis on exercise and physical therapy.
Those who still question the validity of fibromyalgia as a diagnosis object to drug companies “benefiting” from the sale of such drugs.1 But many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it. A key question remains: How will pregabalin fit into the treatment of this often-challenging disease?
FROM FIBROSITIS TO FIBROMYALGIA
Fibromyalgia is a syndrome characterized by widespread pain. Chronic muscular pain is a common problem, but fibromyalgia is distinguished from other pain disorders by additional findings, such as consistent areas of tenderness (tender points), nonrestorative sleep, severe fatigue, and frequent psychological comorbidities such as depression and anxiety.
Fibromyalgia was originally termed “fibrositis” in 1904 by Sir William Gowers, who described it as a painful condition of the fibrous tissue, which he believed was due to inflammation in the muscles.2,3 For several decades, research was dedicated to looking for pathology in the muscle tissue, which was thought to be the major source of pain for most patients with fibromyalgia.
In the mid-1970s, Dr. H. Moldofsky, a noted sleep researcher, reported on abnormalities of the alpha-delta component of nonrapid-eye-movement sleep in these patients. He subsequently collaborated with Dr. Hugh A. Smythe, who helped define the fibromyalgia tender points. Fibrositis was subsequently renamed fibromyalgia syndrome, since it was agreed that there was no true inflammation in muscles or fibrous tissue.
In 1990, the American College of Rheumatology published “classification criteria” for the disease.4 The criteria include two main features:
- A history of widespread pain (“widespread” being defined as in the axial distribution, in both the left and right sides of the body, and above and below the waist), which must be present for 3 months or more, and
- Tenderness in at least 11 of 18 specified points that is elicited when a pressure of 4 kg (the amount of pressure required to blanch a thumbnail) is applied in steady increments starting at 1 kg.
Although pain is subjective and therefore difficult to assess, the classification criteria did make it easier to study the disease in a uniform way and led to an explosion of research in this field.
FUNCTIONAL ABNORMALITIES NI THE CENTRAL NERVOUS SYSTEM
Research to date points to the pain in fibromyalgia as being mediated by changes in the central nervous system rather than in the musculoskeletal system, as was initially thought.
In the dorsal horn of the spinal cord, nociceptive (pain-sensing) neurons from the periphery synapse with the second-order neurons that carry the pain signal to the brain. In fibromyalgia, several processes seem to amplify the signal.
Central sensitization is defined as enhanced excitability of neurons in the dorsal horn. Its features include augmented spontaneous neuronal activity, enlarged receptive field areas, and enhanced responses generated by large- and small-caliber primary afferent fibers. It can result from prolonged or strong activity in the dorsal horn neurons, and it leads to the spread of hyperactivity across multiple spinal segments.5–7
While much of the evidence for central sensitization in fibromyalgia is from animal studies, the phenomenon has also been studied in humans. Desmeules et al8 found that, compared with people without fibromyalgia, those with fibromyalgia had significantly lower thresholds of pain as assessed subjectively and measured objectively using the nociceptive flexion R-III reflex, which the authors described as “a specific physiologic correlate for the objective evaluation of central nociceptive pathways.”
Wind-up. Prolonged stimulation of C fibers in the dorsal horn can result in the phenomenon of wind-up, which refers to the temporal summation of second pain.
A painful stimulus evokes two pain signals. The first signal is brief and travels rapidly to the spinal cord via myelinated fibers (A fibers). The second signal, which is related to chronic pain and is described as dull, aching, or burning, travels more slowly to the dorsal horn via unmyelinated fibers (C fibers), the synapses of which use the neurotransmitter glutamate. Temporal summation is a phenomenon observed in experiments in which a series of painful stimuli are applied at regular intervals of about 2 seconds; although each stimulus is identical in intensity, subjects perceive them as increasing in intensity. The reason: during this repetitive stimulation, N-methyl-d-aspartate (NMDA) receptors become activated, leading to the removal of a magnesium block within the receptor. This results in an influx of calcium into the neuron and activation of protein kinase C, nitric oxide synthase, and cyclooxygenase. Ultimately, the firing rates of the nociceptive neurons are increased and the peripheral pain signal is strongly amplified.6
Wind-up has been shown to lead to characteristics of central sensitization related to C-fiber activity in animals.7 Staud et al9 studied wind-up in patients with and without fibromyalgia using series of repetitive thermal stimulation to produce temporal summation. Though wind-up was evoked in both groups, differences were observed both in the magnitude of sensory response to the first stimulus within a series and in the amount of temporal summation within a series.
Elevated excitatory neurotransmitters. In 1994, Russell et al10 showed that the concentration of substance P, an excitatory neurotransmitter, was three times higher in the cerebrospinal fluid of people with fibromyalgia than in normal controls.
Harris and colleagues11 reported that glutamate, another excitatory neurotransmitter, is elevated within the brain in people with fibromyalgia. They further showed that the levels of glutamate within the insula of the brain are directly associated with the levels of both experimental pressure-evoked pain thresholds and clinical pain ratings in fibromyalgia patients.
Evidence from imaging studies. Other objective evidence of central sensitization in fibromyalgia patients comes from studies using novel imaging.
Gracely et al12 performed functional magnetic resonance imaging (MRI) in people with and without fibromyalgia while applying pressure to their thumbs with a thumbscrew-type device. At equal levels of pressure, the people with fibromyalgia said the pressure hurt more, and specific areas of their brains lit up more on functional MRI. When the experimenters increased the pressure in the people without fibromyalgia until this group subjectively rated the pain as high as the fibromyalgia patients rated the lower level of pressure, their brains lit up to a similar degree in the same areas. These findings provide objective evidence of significantly lower pain thresholds in patients with fibromyalgia than in healthy controls, and they support the theory of central augmentation of pain sensitivity in fibromyalgia.
Staud et al13 also used functional MRI and found greater brain activity associated with temporal summation in fibromyalgia patients compared with controls. (In this experiment, the painful stimulus consisted of heat pulses to the foot.)
Drugs other than pregabalin that modulate the dorsal horn activity of the pain pathway include opioids, tramadol (Ultram), gabapentin (Neurontin), GABA agonists such as baclofen (Lioresal), antidepressants, alpha-2 adrenergic agonists (phenylephrine), and 5-HT3 antagonists such as ondansetron (Zofran), but none has been consistently effective for fibromyalgia.5,14
PREGABALIN
Pregabalin is an alpha-2-delta ligand similar to GABA, but it does not act on GABA receptors. Rather, it binds with high affinity to the alpha-2-delta subunit of voltage-gated presynaptic calcium channels, resulting in reduction of calcium flow through the channels, which subsequently inhibits the release of neurotransmitters including glutamate, norepinephrine, and substance P.15–17 Animal studies suggest that the decrease in the levels of these excitatory neurotransmitters is the mechanism of action of pregabalin, resulting in its analgesic, anticonvulsant, and anxiolytic benefit.15 Another potential mechanism of pregabalin is enhancement of slow-wave sleep, demonstrated in one study in healthy human subjects.18
Besides fibromyalgia, pregabalin is also approved for the treatment of diabetic peripheral neuropathy, postherpetic neuralgia, generalized anxiety disorder, and social anxiety disorder, and as adjunctive therapy for partial-onset seizure in adults.
Pharmacokinetics
Pregabalin is quickly absorbed, primarily in the proximal colon (bioavailability > 90%), and has highly predictable and linear pharmacokinetics.15 Food consumption does not affect its absorption or elimination but can delay its peak plasma concentration, which occurs at 1.5 hours. Its elimination half-life is approximately 6 hours.15 Because it does not bind to plasma proteins, it freely crosses the blood-brain barrier. The drug reaches its steady-state concentration within 2 days of starting therapy.
Its clearance is not affected by the sex or race of the patient, but its total clearance may be lower in the elderly because of age-related loss of renal function. Patients on hemodialysis may require a supplemental dose after dialysis because hemodialysis removes the pregabalin.
The drug is not metabolized by the P450 system in the liver, so it interacts only minimally with drugs that do use the P450 system. However, its clearance may be decreased when it is used concomitantly with drugs that can reduce the glomerular filtration rate, such as nonsteroidal anti-inflammatory drugs, aminoglycosides, and cyclosporine.15
Efficacy
The efficacy of pregabalin in fibromyalgia was evaluated in several recent trials.19
Crofford et al16 assessed pregabalin’s effects on pain, sleep, fatigue, and health-related quality of life. Some 529 patients with fibromyalgia were randomized in a double-blind fashion to four treatment groups: placebo, and pregabalin 150 mg/day, 300 mg/day, and 450 mg/day. The baseline mean pain scores (a 0-to-10 scale derived from daily diary ratings) were 6.9 in the placebo group, 6.9 for the pregabalin 150 mg/day group, 7.3 for the pregabalin 300 mg/day group, and 7.0 for the pregabalin 450 mg/day group.
The pain scores declined in all groups, but at 8 weeks, the mean score had declined 0.93 points more in the group receiving pregabalin 450 mg/day than in the placebo group (P ≤ .001). The scores in the groups taking pregabalin 150 mg/day and 300 mg/day were not significantly different from those in the placebo group. Significantly more patients in the 450-mg/day group (29%, vs 13% in the placebo group) had at least 50% improvement in pain at the end of the study. Patients in both the 300-mg/day group and the 450-mg/day had statistically significant improvement in their quality of sleep, in fatigue, and on the Patient Global Impression of Change (PGIC) scale.
Arnold et al20 conducted a trial with 750 patients in which three doses of pregabalin were compared with placebo: 300 mg/day, 450 mg/day, and 600 mg/day. The primary end point was also the change in pain score from baseline (using the 0-to-10 scale derived from a daily pain diary). The mean baseline pain score was 6.7.
At 14 weeks, the mean pain score was lower than at baseline in all the groups, but it had declined 0.71 more in the pregabalin 300-mg/day group than in the placebo group, 0.98 points more in the 450-mg/day group, and 1.0 points more in the 600-mg/day group. All three pregabalin groups also showed significant improvement on the PGIC scale, and patients in the 450-mg/day and 600-mg/day groups showed statistically significant improvement in the Fibromyalgia Impact Questionnaire (FIQ) score. All three pregabalin treatment groups also had significantly better patient-reported sleep outcomes than in the placebo group, both in measures of overall sleep and quality of sleep. With the exception of a significant improvement of anxiety on 600 mg/day, there was no significant difference between the treatment and placebo groups in the secondary outcomes of depression and anxiety symptoms and fatigue.
Duan et al21 presented a pooled analysis of this and a similarly designed double-blind, placebo-controlled trial (the results of which were not available individually) at the 71st annual meeting of the American College of Rheumatology in November 2007. The analysis included 1,493 patients with a mean baseline pain score of 6.9. Compared with the mean pain score in the placebo group, those in the pregabalin groups had declined more by the end of the study: 0.55 points more with 300 mg/day, 0.71 points more with 450 mg/day, and 0.82 points more with 600 mg/day. This pooled analysis also showed significant improvement in PGIC score with all pregabalin doses and in the FIQ score with 450 mg/day and 600 mg/day.
The FREEDOM trial22 (Fibromyalgia Relapse Evaluation and Efficacy for Durability of Meaningful Relief) evaluated the durability of effect of pregabalin in reducing pain and symptoms associated with fibromyalgia in 1,051 patients who initially responded to the drug.
The patients received 6 weeks of open-label treatment with pregabalin and then 26 weeks of double-blind treatment (dose adjustment was allowed based on efficacy and tolerability for the first 3 weeks). The time to loss of therapeutic response was significantly longer with pregabalin than with placebo. Loss of therapeutic response was defined as worsening of pain for two consecutive visits or worsening of fibromyalgia symptoms requiring alternative therapy.
By the end of the double-blind phase, 61% of those in the placebo group had loss of therapeutic response compared with only 32% in the pregabalin group. The time to worsening of the FIQ score was also significantly longer in the pregabalin group than in the placebo group.
Adverse effects: Dizziness, sleepiness, weight gain
Dizziness and sleepiness were the most common adverse events in these studies.
In the 8-week study by Crofford et al,16 dizziness was dose-related, occurring in 10.7% of those receiving placebo (one patient withdrew because of dizziness), 22.7% of those receiving 150 mg/day (two patients withdrew), 31.3% of those receiving 300 mg/day (four patients withdrew), and 49.2% of those receiving 450 mg/day (five patients withdrew). Somnolence was also dose-related, occurring in 4.6% in the placebo group, 15.9% in the 150-mg/day group (two patients withdrew due to somnolence), 27.6% in the 300-mg/day group (three withdrew), and 28.0% in the 450-mg/day group (five withdrew).
The 14-week study by Arnold et al20 also showed higher frequencies of adverse events with higher doses. The rates of dizziness were 7.6% with placebo, 27.9% with pregabalin 300 mg/day, 37.4% with 450 mg/day, and 42.0% with 600 mg/day. The rates of somnolence were 3.8% with placebo, 12.6% with 300 mg/day of pregabalin, 19.5% with 450 mg/day, and 21.8% with 600 mg/day. Dizziness and somnolence were also the most common adverse effects that led to discontinuation of pregabalin, with rates of 4% and 3%, respectively.
The open-label phase of the FREEDOM trial showed rates of 36% for dizziness and 22% for somnolence among pregabalin-treated patients.
Weight gain and peripheral edema were also common adverse effects in these studies.22 Definitions of weight gain varied, and edema was not accompanied by evidence of cardiac or renal dysfunction.
Less common side effects seen more frequently in the treated groups included dry mouth, blurred vision, and difficulty with concentration and attention. The package insert also warns of angioedema, hypersensitivity reaction, mild asymptomatic creatine kinase elevation, decreased platelet count (without bleeding), and prolongation of the PR interval on electrocardiography.
Pregabalin is a schedule V controlled substance; in clinical studies, abrupt or rapid discontinuation of the drug led to insomnia, nausea, headache, or diarrhea in some patients, suggesting symptoms of dependence. In clinical studies involving a total of more than 5,500 patients, 4% of patients on pregabalin and 1% of patients on placebo reported euphoria as an adverse effect,19 suggesting possible potential for abuse.
Dosing
As a result of the above studies, the recommended starting dose of pregabalin for fibromyalgia is 150 mg/day in two or three divided doses, gradually increased to 300 mg/day within 1 week based on tolerability and efficacy. The dose may be increased to a maximum of 450 mg/day. The 600-mg dose was found to have no significant additional benefit, but it did have more adverse effects and therefore is not recommended. It is important to note that in these studies multiple medications for pain and insomnia were prohibited, so data on drug interactions with pregabalin are limited.
Few achieve complete remission, but most patients feel better
Several studies of the natural history of fibromyalgia have shown that very few patients experience complete remission of the disease, even after many years. Therefore, one should try to set up realistic expectations for patients, with the goal of achieving functional improvement in activities of daily living and a return to one’s predisease state.
In the longest follow-up study, 39 patients in Boston, MA, were prospectively followed for over 10 years. No patient achieved complete remission: all of them reported some fibromyalgia-related symptoms at the end of the study.23 However, 66% of them felt a little to a lot better than when first diagnosed, 55% felt well or very well, and only 7% felt poorly.
Other studies have also shown complete remission to be rare.24,25 A 5-year follow-up study investigating fibromyalgia patients’ perceptions of their symptoms and its impact on everyday life activities demonstrated that the social consequences of fibromyalgia’s symptoms are severe and constant over time.26
Evidence of favorable outcomes was reported in one study in which 47% of patients reported moderate to marked improvement in overall fibromyalgia status upon 3-year follow-up,27 and in another study, in which remission was objectively identified in 24.2% of patients 2 years after diagnosis.28
OTHER THERAPIES
Although there have been many studies of pharmacologic therapies for fibromyalgia to date, the trials had significant limitations, such as short duration, inadequate sample size, nonstandardized measures of efficacy, question of regression to the mean, and inadequate blinding, resulting in insufficient evidence to recommend one drug over another.
Tricyclic antidepressants. Two meta-analyses and a clinical review have supported the efficacy of tricyclic antidepressants in improving symptoms in fibromyalgia patients.29–31
Selective serotonin reuptake inhibitors (SSRIs) have not been well studied, and the small size and methodologic shortcomings of these studies make it difficult to draw conclusions about the efficacy of SSRIs in reducing pain in fibromyalgia patients.30,31
Duloxetine (Cymbalta) and milnacipran (Savella) are serotonin and norepinephrine reuptake inhibitors.32–34 A randomized, double-blind placebo-controlled trial evaluated duloxetine in 520 fibromyalgia patients with and without major depressive disorder. Pain scores improved significantly over 6 months in duloxetine-treated patients at doses of 60 and 120 mg/day.33 Duloxetine became the second drug approved for the treatment of fibromyalgia in 2007, and milnacipran became the third in 2009.
WHAT ROLE FOR PREGABALIN?
Pregabalin may reduce pain in some patients with fibromyalgia. However, the presenting symptoms can vary significantly, and symptoms can vary even in individual patients over time. Therefore, in most patients with fibromyalgia, a multidisciplinary approach is used to treat pain, sleep disturbance, and fatigue, along with comorbidities such as neurally mediated hypotension and psychiatric disorders. Because treatment of fibromyalgia often involves multiple drugs in addition to exercise and behavioral therapies, future studies should examine combinations of drugs and the use of drugs in conjunction with nondrug treatments.
Pregabalin advances our knowledge of fibromyalgia through improving the understanding of central sensitization and how brain neurotransmitters control central pain perceptions. Drug treatment must still be part of the comprehensive management of this disease. Physician and patient education about the current understanding of the disease is paramount in setting realistic goals for treatment.14 Future strategies to manage fibromyalgia will be based on the pathophysiology of this complex condition.
Pregabalin (Lyrica) is a novel analogue of the neurotransmitter gamma aminobutyric acid (GABA) with analgesic, anticonvulsant, and anxiolytic activity. Its approval by the US Food and Drug Administration (FDA) in 2007 for the treatment of fibromyalgia made it the first drug approved for this indication. Until then, management of fibromyalgia entailed drugs to treat pain, sleep, fatigue, and psychological disorders, and a strong emphasis on exercise and physical therapy.
Those who still question the validity of fibromyalgia as a diagnosis object to drug companies “benefiting” from the sale of such drugs.1 But many hail pregabalin as an important advance in our understanding of the pathogenesis of fibromyalgia and how to treat it. A key question remains: How will pregabalin fit into the treatment of this often-challenging disease?
FROM FIBROSITIS TO FIBROMYALGIA
Fibromyalgia is a syndrome characterized by widespread pain. Chronic muscular pain is a common problem, but fibromyalgia is distinguished from other pain disorders by additional findings, such as consistent areas of tenderness (tender points), nonrestorative sleep, severe fatigue, and frequent psychological comorbidities such as depression and anxiety.
Fibromyalgia was originally termed “fibrositis” in 1904 by Sir William Gowers, who described it as a painful condition of the fibrous tissue, which he believed was due to inflammation in the muscles.2,3 For several decades, research was dedicated to looking for pathology in the muscle tissue, which was thought to be the major source of pain for most patients with fibromyalgia.
In the mid-1970s, Dr. H. Moldofsky, a noted sleep researcher, reported on abnormalities of the alpha-delta component of nonrapid-eye-movement sleep in these patients. He subsequently collaborated with Dr. Hugh A. Smythe, who helped define the fibromyalgia tender points. Fibrositis was subsequently renamed fibromyalgia syndrome, since it was agreed that there was no true inflammation in muscles or fibrous tissue.
In 1990, the American College of Rheumatology published “classification criteria” for the disease.4 The criteria include two main features:
- A history of widespread pain (“widespread” being defined as in the axial distribution, in both the left and right sides of the body, and above and below the waist), which must be present for 3 months or more, and
- Tenderness in at least 11 of 18 specified points that is elicited when a pressure of 4 kg (the amount of pressure required to blanch a thumbnail) is applied in steady increments starting at 1 kg.
Although pain is subjective and therefore difficult to assess, the classification criteria did make it easier to study the disease in a uniform way and led to an explosion of research in this field.
FUNCTIONAL ABNORMALITIES NI THE CENTRAL NERVOUS SYSTEM
Research to date points to the pain in fibromyalgia as being mediated by changes in the central nervous system rather than in the musculoskeletal system, as was initially thought.
In the dorsal horn of the spinal cord, nociceptive (pain-sensing) neurons from the periphery synapse with the second-order neurons that carry the pain signal to the brain. In fibromyalgia, several processes seem to amplify the signal.
Central sensitization is defined as enhanced excitability of neurons in the dorsal horn. Its features include augmented spontaneous neuronal activity, enlarged receptive field areas, and enhanced responses generated by large- and small-caliber primary afferent fibers. It can result from prolonged or strong activity in the dorsal horn neurons, and it leads to the spread of hyperactivity across multiple spinal segments.5–7
While much of the evidence for central sensitization in fibromyalgia is from animal studies, the phenomenon has also been studied in humans. Desmeules et al8 found that, compared with people without fibromyalgia, those with fibromyalgia had significantly lower thresholds of pain as assessed subjectively and measured objectively using the nociceptive flexion R-III reflex, which the authors described as “a specific physiologic correlate for the objective evaluation of central nociceptive pathways.”
Wind-up. Prolonged stimulation of C fibers in the dorsal horn can result in the phenomenon of wind-up, which refers to the temporal summation of second pain.
A painful stimulus evokes two pain signals. The first signal is brief and travels rapidly to the spinal cord via myelinated fibers (A fibers). The second signal, which is related to chronic pain and is described as dull, aching, or burning, travels more slowly to the dorsal horn via unmyelinated fibers (C fibers), the synapses of which use the neurotransmitter glutamate. Temporal summation is a phenomenon observed in experiments in which a series of painful stimuli are applied at regular intervals of about 2 seconds; although each stimulus is identical in intensity, subjects perceive them as increasing in intensity. The reason: during this repetitive stimulation, N-methyl-d-aspartate (NMDA) receptors become activated, leading to the removal of a magnesium block within the receptor. This results in an influx of calcium into the neuron and activation of protein kinase C, nitric oxide synthase, and cyclooxygenase. Ultimately, the firing rates of the nociceptive neurons are increased and the peripheral pain signal is strongly amplified.6
Wind-up has been shown to lead to characteristics of central sensitization related to C-fiber activity in animals.7 Staud et al9 studied wind-up in patients with and without fibromyalgia using series of repetitive thermal stimulation to produce temporal summation. Though wind-up was evoked in both groups, differences were observed both in the magnitude of sensory response to the first stimulus within a series and in the amount of temporal summation within a series.
Elevated excitatory neurotransmitters. In 1994, Russell et al10 showed that the concentration of substance P, an excitatory neurotransmitter, was three times higher in the cerebrospinal fluid of people with fibromyalgia than in normal controls.
Harris and colleagues11 reported that glutamate, another excitatory neurotransmitter, is elevated within the brain in people with fibromyalgia. They further showed that the levels of glutamate within the insula of the brain are directly associated with the levels of both experimental pressure-evoked pain thresholds and clinical pain ratings in fibromyalgia patients.
Evidence from imaging studies. Other objective evidence of central sensitization in fibromyalgia patients comes from studies using novel imaging.
Gracely et al12 performed functional magnetic resonance imaging (MRI) in people with and without fibromyalgia while applying pressure to their thumbs with a thumbscrew-type device. At equal levels of pressure, the people with fibromyalgia said the pressure hurt more, and specific areas of their brains lit up more on functional MRI. When the experimenters increased the pressure in the people without fibromyalgia until this group subjectively rated the pain as high as the fibromyalgia patients rated the lower level of pressure, their brains lit up to a similar degree in the same areas. These findings provide objective evidence of significantly lower pain thresholds in patients with fibromyalgia than in healthy controls, and they support the theory of central augmentation of pain sensitivity in fibromyalgia.
Staud et al13 also used functional MRI and found greater brain activity associated with temporal summation in fibromyalgia patients compared with controls. (In this experiment, the painful stimulus consisted of heat pulses to the foot.)
Drugs other than pregabalin that modulate the dorsal horn activity of the pain pathway include opioids, tramadol (Ultram), gabapentin (Neurontin), GABA agonists such as baclofen (Lioresal), antidepressants, alpha-2 adrenergic agonists (phenylephrine), and 5-HT3 antagonists such as ondansetron (Zofran), but none has been consistently effective for fibromyalgia.5,14
PREGABALIN
Pregabalin is an alpha-2-delta ligand similar to GABA, but it does not act on GABA receptors. Rather, it binds with high affinity to the alpha-2-delta subunit of voltage-gated presynaptic calcium channels, resulting in reduction of calcium flow through the channels, which subsequently inhibits the release of neurotransmitters including glutamate, norepinephrine, and substance P.15–17 Animal studies suggest that the decrease in the levels of these excitatory neurotransmitters is the mechanism of action of pregabalin, resulting in its analgesic, anticonvulsant, and anxiolytic benefit.15 Another potential mechanism of pregabalin is enhancement of slow-wave sleep, demonstrated in one study in healthy human subjects.18
Besides fibromyalgia, pregabalin is also approved for the treatment of diabetic peripheral neuropathy, postherpetic neuralgia, generalized anxiety disorder, and social anxiety disorder, and as adjunctive therapy for partial-onset seizure in adults.
Pharmacokinetics
Pregabalin is quickly absorbed, primarily in the proximal colon (bioavailability > 90%), and has highly predictable and linear pharmacokinetics.15 Food consumption does not affect its absorption or elimination but can delay its peak plasma concentration, which occurs at 1.5 hours. Its elimination half-life is approximately 6 hours.15 Because it does not bind to plasma proteins, it freely crosses the blood-brain barrier. The drug reaches its steady-state concentration within 2 days of starting therapy.
Its clearance is not affected by the sex or race of the patient, but its total clearance may be lower in the elderly because of age-related loss of renal function. Patients on hemodialysis may require a supplemental dose after dialysis because hemodialysis removes the pregabalin.
The drug is not metabolized by the P450 system in the liver, so it interacts only minimally with drugs that do use the P450 system. However, its clearance may be decreased when it is used concomitantly with drugs that can reduce the glomerular filtration rate, such as nonsteroidal anti-inflammatory drugs, aminoglycosides, and cyclosporine.15
Efficacy
The efficacy of pregabalin in fibromyalgia was evaluated in several recent trials.19
Crofford et al16 assessed pregabalin’s effects on pain, sleep, fatigue, and health-related quality of life. Some 529 patients with fibromyalgia were randomized in a double-blind fashion to four treatment groups: placebo, and pregabalin 150 mg/day, 300 mg/day, and 450 mg/day. The baseline mean pain scores (a 0-to-10 scale derived from daily diary ratings) were 6.9 in the placebo group, 6.9 for the pregabalin 150 mg/day group, 7.3 for the pregabalin 300 mg/day group, and 7.0 for the pregabalin 450 mg/day group.
The pain scores declined in all groups, but at 8 weeks, the mean score had declined 0.93 points more in the group receiving pregabalin 450 mg/day than in the placebo group (P ≤ .001). The scores in the groups taking pregabalin 150 mg/day and 300 mg/day were not significantly different from those in the placebo group. Significantly more patients in the 450-mg/day group (29%, vs 13% in the placebo group) had at least 50% improvement in pain at the end of the study. Patients in both the 300-mg/day group and the 450-mg/day had statistically significant improvement in their quality of sleep, in fatigue, and on the Patient Global Impression of Change (PGIC) scale.
Arnold et al20 conducted a trial with 750 patients in which three doses of pregabalin were compared with placebo: 300 mg/day, 450 mg/day, and 600 mg/day. The primary end point was also the change in pain score from baseline (using the 0-to-10 scale derived from a daily pain diary). The mean baseline pain score was 6.7.
At 14 weeks, the mean pain score was lower than at baseline in all the groups, but it had declined 0.71 more in the pregabalin 300-mg/day group than in the placebo group, 0.98 points more in the 450-mg/day group, and 1.0 points more in the 600-mg/day group. All three pregabalin groups also showed significant improvement on the PGIC scale, and patients in the 450-mg/day and 600-mg/day groups showed statistically significant improvement in the Fibromyalgia Impact Questionnaire (FIQ) score. All three pregabalin treatment groups also had significantly better patient-reported sleep outcomes than in the placebo group, both in measures of overall sleep and quality of sleep. With the exception of a significant improvement of anxiety on 600 mg/day, there was no significant difference between the treatment and placebo groups in the secondary outcomes of depression and anxiety symptoms and fatigue.
Duan et al21 presented a pooled analysis of this and a similarly designed double-blind, placebo-controlled trial (the results of which were not available individually) at the 71st annual meeting of the American College of Rheumatology in November 2007. The analysis included 1,493 patients with a mean baseline pain score of 6.9. Compared with the mean pain score in the placebo group, those in the pregabalin groups had declined more by the end of the study: 0.55 points more with 300 mg/day, 0.71 points more with 450 mg/day, and 0.82 points more with 600 mg/day. This pooled analysis also showed significant improvement in PGIC score with all pregabalin doses and in the FIQ score with 450 mg/day and 600 mg/day.
The FREEDOM trial22 (Fibromyalgia Relapse Evaluation and Efficacy for Durability of Meaningful Relief) evaluated the durability of effect of pregabalin in reducing pain and symptoms associated with fibromyalgia in 1,051 patients who initially responded to the drug.
The patients received 6 weeks of open-label treatment with pregabalin and then 26 weeks of double-blind treatment (dose adjustment was allowed based on efficacy and tolerability for the first 3 weeks). The time to loss of therapeutic response was significantly longer with pregabalin than with placebo. Loss of therapeutic response was defined as worsening of pain for two consecutive visits or worsening of fibromyalgia symptoms requiring alternative therapy.
By the end of the double-blind phase, 61% of those in the placebo group had loss of therapeutic response compared with only 32% in the pregabalin group. The time to worsening of the FIQ score was also significantly longer in the pregabalin group than in the placebo group.
Adverse effects: Dizziness, sleepiness, weight gain
Dizziness and sleepiness were the most common adverse events in these studies.
In the 8-week study by Crofford et al,16 dizziness was dose-related, occurring in 10.7% of those receiving placebo (one patient withdrew because of dizziness), 22.7% of those receiving 150 mg/day (two patients withdrew), 31.3% of those receiving 300 mg/day (four patients withdrew), and 49.2% of those receiving 450 mg/day (five patients withdrew). Somnolence was also dose-related, occurring in 4.6% in the placebo group, 15.9% in the 150-mg/day group (two patients withdrew due to somnolence), 27.6% in the 300-mg/day group (three withdrew), and 28.0% in the 450-mg/day group (five withdrew).
The 14-week study by Arnold et al20 also showed higher frequencies of adverse events with higher doses. The rates of dizziness were 7.6% with placebo, 27.9% with pregabalin 300 mg/day, 37.4% with 450 mg/day, and 42.0% with 600 mg/day. The rates of somnolence were 3.8% with placebo, 12.6% with 300 mg/day of pregabalin, 19.5% with 450 mg/day, and 21.8% with 600 mg/day. Dizziness and somnolence were also the most common adverse effects that led to discontinuation of pregabalin, with rates of 4% and 3%, respectively.
The open-label phase of the FREEDOM trial showed rates of 36% for dizziness and 22% for somnolence among pregabalin-treated patients.
Weight gain and peripheral edema were also common adverse effects in these studies.22 Definitions of weight gain varied, and edema was not accompanied by evidence of cardiac or renal dysfunction.
Less common side effects seen more frequently in the treated groups included dry mouth, blurred vision, and difficulty with concentration and attention. The package insert also warns of angioedema, hypersensitivity reaction, mild asymptomatic creatine kinase elevation, decreased platelet count (without bleeding), and prolongation of the PR interval on electrocardiography.
Pregabalin is a schedule V controlled substance; in clinical studies, abrupt or rapid discontinuation of the drug led to insomnia, nausea, headache, or diarrhea in some patients, suggesting symptoms of dependence. In clinical studies involving a total of more than 5,500 patients, 4% of patients on pregabalin and 1% of patients on placebo reported euphoria as an adverse effect,19 suggesting possible potential for abuse.
Dosing
As a result of the above studies, the recommended starting dose of pregabalin for fibromyalgia is 150 mg/day in two or three divided doses, gradually increased to 300 mg/day within 1 week based on tolerability and efficacy. The dose may be increased to a maximum of 450 mg/day. The 600-mg dose was found to have no significant additional benefit, but it did have more adverse effects and therefore is not recommended. It is important to note that in these studies multiple medications for pain and insomnia were prohibited, so data on drug interactions with pregabalin are limited.
Few achieve complete remission, but most patients feel better
Several studies of the natural history of fibromyalgia have shown that very few patients experience complete remission of the disease, even after many years. Therefore, one should try to set up realistic expectations for patients, with the goal of achieving functional improvement in activities of daily living and a return to one’s predisease state.
In the longest follow-up study, 39 patients in Boston, MA, were prospectively followed for over 10 years. No patient achieved complete remission: all of them reported some fibromyalgia-related symptoms at the end of the study.23 However, 66% of them felt a little to a lot better than when first diagnosed, 55% felt well or very well, and only 7% felt poorly.
Other studies have also shown complete remission to be rare.24,25 A 5-year follow-up study investigating fibromyalgia patients’ perceptions of their symptoms and its impact on everyday life activities demonstrated that the social consequences of fibromyalgia’s symptoms are severe and constant over time.26
Evidence of favorable outcomes was reported in one study in which 47% of patients reported moderate to marked improvement in overall fibromyalgia status upon 3-year follow-up,27 and in another study, in which remission was objectively identified in 24.2% of patients 2 years after diagnosis.28
OTHER THERAPIES
Although there have been many studies of pharmacologic therapies for fibromyalgia to date, the trials had significant limitations, such as short duration, inadequate sample size, nonstandardized measures of efficacy, question of regression to the mean, and inadequate blinding, resulting in insufficient evidence to recommend one drug over another.
Tricyclic antidepressants. Two meta-analyses and a clinical review have supported the efficacy of tricyclic antidepressants in improving symptoms in fibromyalgia patients.29–31
Selective serotonin reuptake inhibitors (SSRIs) have not been well studied, and the small size and methodologic shortcomings of these studies make it difficult to draw conclusions about the efficacy of SSRIs in reducing pain in fibromyalgia patients.30,31
Duloxetine (Cymbalta) and milnacipran (Savella) are serotonin and norepinephrine reuptake inhibitors.32–34 A randomized, double-blind placebo-controlled trial evaluated duloxetine in 520 fibromyalgia patients with and without major depressive disorder. Pain scores improved significantly over 6 months in duloxetine-treated patients at doses of 60 and 120 mg/day.33 Duloxetine became the second drug approved for the treatment of fibromyalgia in 2007, and milnacipran became the third in 2009.
WHAT ROLE FOR PREGABALIN?
Pregabalin may reduce pain in some patients with fibromyalgia. However, the presenting symptoms can vary significantly, and symptoms can vary even in individual patients over time. Therefore, in most patients with fibromyalgia, a multidisciplinary approach is used to treat pain, sleep disturbance, and fatigue, along with comorbidities such as neurally mediated hypotension and psychiatric disorders. Because treatment of fibromyalgia often involves multiple drugs in addition to exercise and behavioral therapies, future studies should examine combinations of drugs and the use of drugs in conjunction with nondrug treatments.
Pregabalin advances our knowledge of fibromyalgia through improving the understanding of central sensitization and how brain neurotransmitters control central pain perceptions. Drug treatment must still be part of the comprehensive management of this disease. Physician and patient education about the current understanding of the disease is paramount in setting realistic goals for treatment.14 Future strategies to manage fibromyalgia will be based on the pathophysiology of this complex condition.
- Berenson A. Drug approved. Is disease real? New York Times, January 14, 2008. http://www.nytimes.com/2008/01/14/health/14pain.html. Accessed February 2, 2009.
- White KP, Harth M. Classification, epidemiology, and natural history of fibromyalgia. Curr Pain Headache Rep 2001; 5:320–329.
- Bennett RM. Fibromyalgia: present to future. Curr Pain Headache Rep 2004; 8:379–384.
- Wolfe F, Smythe HA, Yunus MF, et al. The American College of Rheumatolgy 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum 1990; 33:160–172.
- Bennett RM. The rational management of fibromyalgia patients. Rheum Dis Clin North Am 2002; 28:181–199.
- Staud R. Evidence of involvement of central neural mechanisms in generating fibromyalgia pain. Curr Rheumatol Rep 2002; 4:299–305.
- Li J, Simone DA, Larson AA. Windup leads to characteristics of central sensitization. Pain 1999; 79:75–82.
- Desmeules JA, Cedraschi C, Rapiti E, et al. Neurophysiologic evidence for a central sensitization in patients with fibromyalgia. Arthritis Rheum 2003; 48:1420–1429.
- Staud R, Vierck CJ, Cannon RL, Mauderli AP, Price DD. Abnormal sensitization and temporal summation of pain (wind-up) in patients with fibromyalgia syndrome. Pain 2001; 91:165–175.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Harris RE, Sundgren PC, Pang Y, et al. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum 2008; 58:903–907.
- Gracely RH, Petzke F, Wolf JM, Clauw DJ. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum 2002; 46:1333–1343.
- Staud R, Craggs JG, Perlstein WM, Robinson ME, Price DD. Brain activity associated with slow temporal summation of C-fiber evoked pain in fibromyalgia patients and healthy controls. Eur J Pain 2008; 12:1078–1089.
- Baker K, Barkhuizen A. Pharmacologic treatment of fibromyalgia. Curr Pain Headache Rep 2005; 9:301–306.
- Tassone DM, Boyce E, Guyer J, Nuzum D. Pregabalin: a novel gamma-aminobutyric acid analogue in the treatment of neuropathic pain, partial-onset seizures, and anxiety disorders. Clin Ther 2007; 29:26–48.
- Crofford LJ, Rowbotham MC, Mease PJ, et al, and the Pregabalin 1008-105 Study Group. Pregabalin for the treatment of fibromyalgia syndrome. results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2005; 52:1264–1273.
- Stahl SM. Anticonvulsants and the relief of chronic pain: pregabalin and gabapentin as alpha(2)delta ligands at voltage-gated calcium channels. J Clin Psychiatry 2004; 65:596–597.
- Hindmarch I, Dawson J, Stanley N. A double-blind study in healthy volunteers to assess the effects of sleep on pregabalin compared with alprazolam and placebo. Sleep 2005; 28:187–193.
- Pfizer Executive Summary. Lyrica (pregabalin) capsules c-v. July 2007. www.fda.gov/OHRMS/DOCKETS/ac/08/briefing/2008-4372b1-02-Pfizer.pdf. Accessed February 2, 2009.
- Arnold LM, Russell IJ, Diri EW, et al. A 14-week, randomized, double-blinded, placebo-controlled mono-therapy trial of pregabalin in patients with fibomyalgia. J Pain 2008; 9:792–805.
- Duan WR, Florian H, Young JP, Martin S, Haig G, Barrett JA. Pregabalin monotherapy for management of fibromyalgia: analysis of two double-blind, randomized, placebo-controlled trials (poster presentation). American College of Rheumatology Annual Scientific Meeting, Boston, MA, November 6–7, 2007.
- Crofford LJ, Mease PJ, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain 2008; 136:419–431.
- Kennedy M, Felson DT. A prospective long-term study of fibromyalgia syndrome. Arthritis Rheum 1996; 39:682–685.
- Bengtsson A, Backman E. Long-term follow-up of fibro-myalgia patients [abstract]. Scand J Rheumatolology 1992; 21(suppl 94):9.
- Ledingham J, Doherty S, Doherty M. Primary fibromyalgia syndrome—an outcome study. Br J Rheumatol 1993; 32:139–142.
- Henrikkson CM. Longterm effects of fibromyalgia on everyday life: a study of 56 patients. Scand J Rheumatol 1994; 23:36–41.
- Fitzcharles MA, Costa DD, Pöyhiä R. A study of standard care in fibromyalgia syndrome: a favorable outcome. J Rheumatol 2003; 30:154–159.
- Granges G, Zilko P, Littlejohn GO. Fibromyalgia syndrome: assessment of the severity of the condition 2 years after the diagnosis. J Rheumatol 1994; 21:523–529.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia: a meta-analysis and review. Psychosomatics 2000; 41:104–113.
- O’Malley PG, Balden E, Tomkins G, Santoro J, Kroenke K, Jackson JL. Treatment of fibromyalgia with anti-depressants: a meta-analysis. J Gen Intern Med 2000; 15:659–666.
- Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum 2004; 50:2974–2984.
- Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: Results from a 6-month, randomized, double-blind, placebo-controlled fixed-dose trial. Pain 2008; 136:432–444.
- Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther 2008; 30:1988–2004.
- Berenson A. Drug approved. Is disease real? New York Times, January 14, 2008. http://www.nytimes.com/2008/01/14/health/14pain.html. Accessed February 2, 2009.
- White KP, Harth M. Classification, epidemiology, and natural history of fibromyalgia. Curr Pain Headache Rep 2001; 5:320–329.
- Bennett RM. Fibromyalgia: present to future. Curr Pain Headache Rep 2004; 8:379–384.
- Wolfe F, Smythe HA, Yunus MF, et al. The American College of Rheumatolgy 1990 Criteria for the Classification of Fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum 1990; 33:160–172.
- Bennett RM. The rational management of fibromyalgia patients. Rheum Dis Clin North Am 2002; 28:181–199.
- Staud R. Evidence of involvement of central neural mechanisms in generating fibromyalgia pain. Curr Rheumatol Rep 2002; 4:299–305.
- Li J, Simone DA, Larson AA. Windup leads to characteristics of central sensitization. Pain 1999; 79:75–82.
- Desmeules JA, Cedraschi C, Rapiti E, et al. Neurophysiologic evidence for a central sensitization in patients with fibromyalgia. Arthritis Rheum 2003; 48:1420–1429.
- Staud R, Vierck CJ, Cannon RL, Mauderli AP, Price DD. Abnormal sensitization and temporal summation of pain (wind-up) in patients with fibromyalgia syndrome. Pain 2001; 91:165–175.
- Russell IJ, Orr MD, Littman B, et al. Elevated cerebrospinal fluid levels of substance P in patients with fibromyalgia syndrome. Arthritis Rheum 1994; 37:1593–1601.
- Harris RE, Sundgren PC, Pang Y, et al. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum 2008; 58:903–907.
- Gracely RH, Petzke F, Wolf JM, Clauw DJ. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum 2002; 46:1333–1343.
- Staud R, Craggs JG, Perlstein WM, Robinson ME, Price DD. Brain activity associated with slow temporal summation of C-fiber evoked pain in fibromyalgia patients and healthy controls. Eur J Pain 2008; 12:1078–1089.
- Baker K, Barkhuizen A. Pharmacologic treatment of fibromyalgia. Curr Pain Headache Rep 2005; 9:301–306.
- Tassone DM, Boyce E, Guyer J, Nuzum D. Pregabalin: a novel gamma-aminobutyric acid analogue in the treatment of neuropathic pain, partial-onset seizures, and anxiety disorders. Clin Ther 2007; 29:26–48.
- Crofford LJ, Rowbotham MC, Mease PJ, et al, and the Pregabalin 1008-105 Study Group. Pregabalin for the treatment of fibromyalgia syndrome. results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2005; 52:1264–1273.
- Stahl SM. Anticonvulsants and the relief of chronic pain: pregabalin and gabapentin as alpha(2)delta ligands at voltage-gated calcium channels. J Clin Psychiatry 2004; 65:596–597.
- Hindmarch I, Dawson J, Stanley N. A double-blind study in healthy volunteers to assess the effects of sleep on pregabalin compared with alprazolam and placebo. Sleep 2005; 28:187–193.
- Pfizer Executive Summary. Lyrica (pregabalin) capsules c-v. July 2007. www.fda.gov/OHRMS/DOCKETS/ac/08/briefing/2008-4372b1-02-Pfizer.pdf. Accessed February 2, 2009.
- Arnold LM, Russell IJ, Diri EW, et al. A 14-week, randomized, double-blinded, placebo-controlled mono-therapy trial of pregabalin in patients with fibomyalgia. J Pain 2008; 9:792–805.
- Duan WR, Florian H, Young JP, Martin S, Haig G, Barrett JA. Pregabalin monotherapy for management of fibromyalgia: analysis of two double-blind, randomized, placebo-controlled trials (poster presentation). American College of Rheumatology Annual Scientific Meeting, Boston, MA, November 6–7, 2007.
- Crofford LJ, Mease PJ, Simpson SL, et al. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain 2008; 136:419–431.
- Kennedy M, Felson DT. A prospective long-term study of fibromyalgia syndrome. Arthritis Rheum 1996; 39:682–685.
- Bengtsson A, Backman E. Long-term follow-up of fibro-myalgia patients [abstract]. Scand J Rheumatolology 1992; 21(suppl 94):9.
- Ledingham J, Doherty S, Doherty M. Primary fibromyalgia syndrome—an outcome study. Br J Rheumatol 1993; 32:139–142.
- Henrikkson CM. Longterm effects of fibromyalgia on everyday life: a study of 56 patients. Scand J Rheumatol 1994; 23:36–41.
- Fitzcharles MA, Costa DD, Pöyhiä R. A study of standard care in fibromyalgia syndrome: a favorable outcome. J Rheumatol 2003; 30:154–159.
- Granges G, Zilko P, Littlejohn GO. Fibromyalgia syndrome: assessment of the severity of the condition 2 years after the diagnosis. J Rheumatol 1994; 21:523–529.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia: a meta-analysis and review. Psychosomatics 2000; 41:104–113.
- O’Malley PG, Balden E, Tomkins G, Santoro J, Kroenke K, Jackson JL. Treatment of fibromyalgia with anti-depressants: a meta-analysis. J Gen Intern Med 2000; 15:659–666.
- Arnold LM, Lu Y, Crofford LJ, et al. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum 2004; 50:2974–2984.
- Russell IJ, Mease PJ, Smith TR, et al. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: Results from a 6-month, randomized, double-blind, placebo-controlled fixed-dose trial. Pain 2008; 136:432–444.
- Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther 2008; 30:1988–2004.
KEY POINTS
- Several lines of evidence point to functional abnormalities in the central nervous system as being responsible for fibromyalgia.
- Clinical trials found pregabalin superior to placebo. Nevertheless, patients need to have reasonable expectations of its possible benefit.
- In most patients with fibromyalgia, a multidisciplinary approach is used to treat pain, sleep disturbance, and fatigue, along with comorbidities such as neurally mediated hypotension and psychiatric disorders.
- Research with pregabalin enhances our understanding of fibromyalgia and may point the way to future treatments.
Perioperative statins: More than lipid-lowering?
Soon, the checklist for internists seeing patients about to undergo surgery may include prescribing one of the lipid-lowering hydroxymethylglutaryl-CoA reductase inhibitors, also called statins.
Statins? Not long ago, we were debating whether patients who take statins should stop taking them before surgery, based on the manufacturers’ recommendations.1 The discussion, however, has changed to whether patients who have never received a statin should be started on one before surgery to provide immediate prophylaxis against cardiac morbidity, and how much harm long-term statin users face if these drugs are withheld perioperatively.
The evidence is still very preliminary and based mostly on studies in animals and retrospective studies in people. However, an expanding body of indirect evidence suggests that these drugs are beneficial in this situation.
In this review, we discuss the mechanisms by which statins may protect the heart in the short term, drawing on data from animal and human studies of acute myocardial infarction, and we review the current (albeit limited) data from the perioperative setting.
FEW INTERVENTIONS DECREASE RISK
Each year, approximately 50,000 patients suffer a perioperative cardiovascular event; the incidence of myocardial infarction during or after noncardiac surgery is 2% to 3%.2 The primary goal of preoperative cardiovascular risk assessment is to predict and avert these events.
But short of canceling surgery, few interventions have been found to reduce a patient’s risk. For example, a landmark study in 2004 cast doubt on the efficacy of preoperative coronary revascularization.3 Similarly, although early studies of beta-blockers were promising4,5 and although most internists prescribe these drugs before surgery, more recent studies have cast doubt on their efficacy, particularly in patients at low risk undergoing intermediate-risk (rather than vascular) surgery.6–8
This changing clinical landscape has prompted a search for new strategies for perioperative risk-reduction. Several recent studies have placed statins in the spotlight.
POTENTIAL MECHANISMS OF SHORT-TERM BENEFIT
Statins have been proven to save lives when used long-term, but how could this class of drugs, designed to prevent the accumulation of arterial plaques by lowering low-density lipoprotein cholesterol (LDL-C) levels, have any short-term impact on operative outcomes? Although LDL-C reduction is the principal mechanism of action of statins, not all of the benefit can be ascribed to this mechanism.9 The answer may lie in their “pleiotropic” effects—ie, actions other than LDL-C reduction.
The more immediate pleiotropic effects of statins in the proinflammatory and prothrombotic environment of the perioperative period are thought to include improved endothelial function (both antithrombotic function and vasomotor function in response to ischemic stress), enhanced stability of atherosclerotic plaques, decreased oxidative stress, and decreased vascular inflammation.10–12
EVIDENCE FROM ANIMAL STUDIES
Experiments in animals suggest that statins, given shortly before or after a cardiovascular event, confer benefit before any changes in LDL-C are measurable.
Lefer et al13 found that simvastatin (Zocor), given 18 hours before an ischemic episode in rats, blunted the inflammatory response in cardiac reperfusion injury. Not only was reperfusion injury significantly less in the hearts of the rats that received simvastatin than in the saline control group, but the simvastatin-treated hearts also expressed fewer neutrophil adhesion molecules such as P-selectin, and they had more basal release of nitric oxide, the potent endothelial-derived vasodilator with antithrombotic, anti-inflammatory, and antiproliferative effects.14 These results suggest that statins may improve endothelial function acutely, particularly during ischemic stress.
Osborne et al15 fed rabbits a cholesterol-rich diet plus either lovastatin (Mevacor) or placebo. After 2 weeks, the rabbits underwent either surgery to induce a myocardial infarction or a sham procedure. Regardless of the pretreatment, biopsies of the aorta did not reveal any atherosclerosis; yet the lovastatin-treated rabbits sustained less myocardial ischemic damage and they had more endothelium-mediated vasodilatation.
Statin therapy also may improve cerebral ischemia outcomes in animal models.14,16
Sironi et al16 induced strokes in rats by occluding the middle cerebral artery. The rats received either simvastatin or vehicle for 3 days before the stroke or immediately afterwards. Even though simvastatin did not have enough time to affect the total cholesterol level, rats treated with simvastatin had smaller infarcts (as measured by magnetic resonance imaging) and produced more nitric oxide.
Comment. Taken together, these studies offer tantalizing evidence that statins have short-term, beneficial nonlipid effects and may reduce not only the likelihood of an ischemic event, but—should one occur—the degree of tissue damage that ensues.
EFFECTS OF STATINS IN ACUTE CORONARY SYNDROME
The National Registry of Myocardial Infarction17 is a prospective, observational database of all patients with acute myocardial infarction admitted to 1,230 participating hospitals throughout the United States. In an analysis from this cohort, patients were divided into four groups: those receiving statins before and after admission, those receiving statins only before admission, those receiving statins only after admission, and those who never received statins.
Compared with those who never received statins, fewer patients who received them both before and after admission died while in the hospital (unadjusted odds ratio 0.23, 95% confidence interval [CI] 0.22–0.25), and the odds ratio for those who received statins for the first time was 0.31 (95% CI 0.29–0.33). Patients who stopped receiving a statin on admission were more likely to die than were patients who never received statins (odds ratio 1.09, 95% CI 1.03–1.15). These trends held true even when adjustments were made for potential confounding factors.
Comment. Unmeasured confounding factors (such as the inability to take pills due to altered mental status or the different practice styles of the providers who chose to discontinue statins) might have affected the results. Nevertheless, these results suggest that the protective effects of statins stop almost immediately when these drugs are discontinued, and that there may even be an adverse “rebound” effect when patients who have been taking these drugs for a long time stop taking them temporarily.
The Platelet Receptor Inhibition in Ischemic Syndrome Management trial,18 in a subgroup analysis, had nearly identical findings. In the main part of this trial, patients with coronary artery disease and chest pain at rest or accelerating pain in the last 24 hours were randomized to receive tirofiban (Aggrastat) or heparin. Complete data on statin use were available for 1,616 (50%) of the 3,232 patients in this trial, and the rate of the primary end point (death, myocardial infarction, or recurrent ischemia) was analyzed on the basis of statin therapy in this subgroup.
Comment. Together, these data lead to the conclusion that, when admitted for either acute myocardial infarction or acute coronary syndrome, patients already receiving statins should not have them stopped, and those who had not been receiving statins should receive them immediately. The safety of these medications in the acute setting appears excellent: in the Myocardial Ischemia Reduction With Acute Cholesterol Lowering (MIRACL)12 and the Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT)11 trials, fewer than 5% of statin-treated patients had transient elevations in transaminase levels, and no cases of rhabdomyolysis were reported.
PERIOPERATIVE STATIN STUDIES
The data on perioperative statin use are mostly observational and retrospective and fall into essentially four surgical categories: coronary artery bypass grafting (CABG), carotid endarterectomy,19,20 noncardiac vascular surgery, and major noncardiac surgery. Two meta-analyses have also evaluated the data.21,22 The only randomized controlled trial (performed by Durazzo et al23) was small and was carried out at a single center in vascular surgery patients, and the event rate was low.
Current recommendations from the National Cholesterol Education Program (NCEP)24 say that patients who need CABG, have peripheral arterial disease, have an abdominal aortic aneurysm, or have cerebrovascular disease should already be on a statin to achieve an LDL-C goal level of less than 100 mg/dL, with an optional goal of less than 70 mg/dL, independent of surgery.
Since not all patients who should be on statins are actually on them, questions arise:
- Is it important (and safe) to start statin treatment preoperatively?
- Will patients with cardiovascular risk factors but without known cardiovascular disease benefit from statins perioperatively?
Noncardiac vascular surgery
Multiple retrospective studies have evaluated the effect of statins in patients undergoing major noncardiac vascular surgery.25–32
Kertai et al25 evaluated 570 patients in Holland who underwent elective open surgery for infrarenal abdominal aortic aneurysms between 1991 and 2001, looking for an association between statin use and the incidence of perioperative death from myocardial infarction. Only 162 of the 570 patients had been on long-term statin therapy before the surgery. The use of statins was only one of many known baseline characteristics that were significantly different between the two groups, including age, body mass index, known coronary artery disease, and use of angiotensin-converting enzyme inhibitors and beta-blockers. In univariate analysis, statins appeared to be protective: 6 (3.7%) of the patients in the statin group died of a myocardial infarction, compared with 45 (11%) of those in the nostatin group. A multivariate analysis yielded similar findings, with an odds ratio of 0.24 (95% CI 0.11–0.54).
Ward et al27 performed a very similar retrospective study, with similar findings. In 446 patients who underwent surgery for infrarenal abdominal aortic aneurysm, statin therapy was associated with a significantly lower incidence of the combined end point of death, myocardial infarction, stroke, and major peripheral vascular complications, with an adjusted odds ratio of 0.36 (95% CI 0.14–0.93).
Poldermans et al26 noted similar findings in a case-control study of noncardiac vascular surgery patients. Statin users had a much lower perioperative risk of death than did nonusers, with an adjusted odds ratio of 0.22 (95% CI 0.10–0.47).
O’Neil-Callahan et al,28 in a cohort study, found that statin users had fewer perioperative cardiac complications, with an adjusted odds ratio of 0.49 (95% CI 0.28–0.84, P = .009).
Dogma of withdrawing statins before major surgery is challenged
Le Manach et al33 reviewed the outcomes for all patients of a single hospital in Paris who underwent nonemergency infrarenal aortic procedures between January 2001 and December 2004. In January 2004, the hospital instituted guidelines to ensure that patients on statins continue taking them up to the evening before surgery and that statins be restarted on the first postoperative day (via nasogastric tube if necessary). Before 2004, there had been no specific guidelines, and patients on statins did not receive them for a median of 4 days postoperatively. Types of procedures were similar during the two time periods, as were the rates of beta-blocker use, preoperative revascularization, venous thromboembolism prophylaxis, and perioperative blood pressure control. After surgery, topononin I levels were measured in all patients as surveillance for cardiac events, and were defined as elevated when greater than 0.2 ng/mL.
Compared with patients not on statins at all, those treated with statins continuously throughout the perioperative period (after January 2004) had a lower rate of elevated troponin (relative risk 0.38). In contrast, those who had their statins transiently discontinued perioperatively (prior to 2004) had troponin elevations more often than those who had never been treated (relative risk 2.1). This suggested an over fivefold risk reduction (P < .001) conferred by not discontinuing statins in the immediate postoperative period. This finding was maintained after multivariate adjustment: statin withdrawal was associated with a 2.9-fold (95% CI 1.6–5.5) increase in the risk of cardiac enzyme elevations postoperatively. No fewer deaths were noted, but the study was not powered to detect a mortality difference.
Comment. Although secular trends cannot be entirely discounted as contributing to these findings, the prompt increase in cardiac events after just 4 days of statin withdrawal adds to the growing body of evidence suggesting that statin discontinuation can have harmful acute effects. It also brings up the question: Can starting statins benefit patients in the same time period?
Should statins be started before vascular surgery?
Schouten et al32 evaluated the effects of newly started or continued statin treatment in patients undergoing major elective vascular surgery. Patients were screened before surgery and started on statins if they were not already receiving them and their total cholesterol levels were elevated; new users received the medication for about 40 days before surgery. Of the 981 screened patients, 44 (5%) were newly started on statins and 182 (19%) were continued on their therapy. Perioperative death or myocardial infarction occurred in 22 (8.8%) of the statin users and 111 (14.7%) of the nonusers, a statistically significant difference. Temporary discontinuation (median 1 day) of statins in this study due to the inability to take an oral medication did not appear to affect the likelihood of a myocardial infarction.
Durazzo et al23 performed a single-center, randomized, prospective, placebo-controlled, double-blind clinical trial of atorvastatin (Lipitor) 20 mg daily vs placebo in 100 patients undergoing noncardiac arterial vascular surgery. Patients were excluded if they had previously used medications to treat dyslipidemia, recently had a cardiovascular event, or had contraindications to statin treatment such as a baseline creatinine level greater than 2.0 mg/dL or severe hepatic disease. The intervention group received atorvastatin starting at least 2 weeks before surgery for a total of 45 days. Patients were then continued or started on a statin after surgery if their LDL-C level was greater than 100 mg/dL. Beta-blocker use was recommended “on the basis of current guidelines.”
One month after surgery, the LDL-C level was statistically significantly lower in the atorvastatin group. Since most patients did not continue or start statin therapy after the 45-day treatment period, the LDL-C levels were not statistically different at 3 and 6 months after surgery.
At 6 months, the rate of the primary end point (death from cardiovascular causes, nonfatal acute myocardial infarction, ischemic stroke, or unstable angina) was 26.0% in the placebo group and 8.0% in the atorvastatin group, a statistically significant difference. Three patients in the atorvastatin group had cardiac events in the first 10 days after surgery, compared with 11 patients in the placebo group. Thirteen of the 17 total cardiac events took place within 10 days after surgery.
One of the atorvastatin patients developed rhabdomyolysis and elevated aminotransferase levels.
Major noncardiac surgery
Lindenauer et al2 performed a retrospective cohort study of surgical patients who were at least 18 years old and survived beyond the second hospital day. Patients were divided into a group receiving any form of lipid-lowering treatment (of whom more than 90% were taking statins) and a group that had never never received a lipid-lowering drug or only started one on the third day of the hospitalization or later. The period of study was from January 1, 2000, to December 31, 2001.
In all, 780,591 patients from 329 hospitals throughout the United States were included, of whom only 77,082 (9.9%) received lipid-lowering therapy. Eight percent of the patients underwent vascular surgery. Not surprisingly, the treated patients were more likely to have a history of hypertension, diabetes, ischemic heart disease, or hyperlipidemia. They also were more likely to have a vascular procedure performed, to have two or more cardiac risk factors (high-risk surgery, ischemic heart disease, congestive heart failure, cerebrovascular disease, renal insufficiency, or diabetes mellitus), and to be treated with beta-blockers and angiotensin-converting enzyme inhibitors, but they were less likely to have high-risk and emergency surgery performed.
The primary end point, perioperative death, occurred in 2.13% of the treated patients and 3.05% of the nontreated group. Compared with the rate in a propensity-matched cohort, the odds ratio adjusted for unbalanced covariates was 0.62 (95% CI 0.58–0.67) in favor of lipid treatment. Stratification by cardiac risk index revealed a number needed to treat of 186 for those with no risk factors, 60 for those with two risk factors, and 30 for those with four or more risk factors.
Unfortunately, this analysis was not able to take into account whether and for how long patients were receiving lipid-lowering therapy before hospitalization. It therefore does not answer the questions of whether starting lipid-lowering therapy before surgery is beneficial or whether stopping it is harmful. It also does not shed light on whether perioperative lipid-lowering increases the risk of rhabdomyolysis or liver disease.
Carotid endarterectomy
Two recent retrospective cohort studies evaluated the outcomes in patients undergoing carotid endarterectomy.19,20
Kennedy et al19 found that patients on a statin at the time of admission who had symptomatic carotid disease had lower rates of inhospital death (adjusted odds ratio 0.24, 95% CI 0.06–0.91) and ischemic stroke or death (adjusted odds ratio 0.55, 95% CI 0.31–0.97). However, cardiac outcomes among these symptomatic patients were not significantly improved (odds ratio 0.82, 95% CI 0.45–1.50), nor was there benefit for asymptomatic patients, raising the possibility that the positive findings were due to chance or that patients at lower baseline risk for vascular events may have less benefit.
McGirt et al20 performed a similar study; they did not, however, distinguish whether patients had symptomatic vs asymptomatic carotid disease. The 30-day risk of perioperative stroke was lower in patients treated with a statin, with an odds ratio of 0.41 (95% CI 0.18–0.93); the odds ratio for death was 0.21 (95% CI 0.05–0.96). Cardiac outcomes were not significantly affected.
Coronary artery bypass graft surgery
According to the NCEP recommendations, nearly all patients undergoing CABG should already be on a statin before surgery since they all have known coronary artery disease. Multiple observational studies have offered confirmatory evidence that statins are beneficial in this setting.34–38
Liakopoulos et al39 evaluated whether the anti-inflammatory effects of statins may, in part, account for their beneficial effect in the perioperative period. The authors prospectively matched 18 patients who were taking statins and were referred for elective CABG with 18 patients who were not prescribed statins previously. The only major measured baseline characteristic that differed between the two groups was a statistically significantly lower LDL-C level in the statin group. The operative characteristics did not differ, and cytokine levels at baseline were similar.
Tumor necrosis factor alpha levels increased significantly in the control group but did not change significantly in the statin group. Interleukin 8 increased in both groups by a similar amount. Interleukin 6 (the major inducer of C-reactive protein) increased from baseline in both groups but did not increase nearly as much in the statin group as in the control group; the intergroup difference was statistically significant. The anti-inflammatory cytokine interleukin 10 increased minimally from baseline in the control group, while the statin group’s levels increased significantly above baseline and those of the control group.
Christenson40 also found that inflammatory markers were improved with pre-CABG statin treatment in a small randomized trial in which patients received simvastatin 20 mg 4 weeks prior to CABG surgery vs no statin. Interestingly, far fewer statin-treated patients developed thrombocytosis (platelet count > 400 × 109/L) than did control patients (3% vs 81%, P < .0001).
RISKS OF PERIOPERATIVE STATINS
The risks associated with statin therapy in general appear low, but specific perioperative risks have not been well studied.
Baigent et al,41 in a meta-analysis of randomized trials of nonperioperative statin therapy, found that rhabdomyolysis occurred in 9 (0.023%) of 39,884 patients receiving statins vs 6 (0.015%) of the 39,817 controls, with a number needed to harm of 12,500. Moreover, the rates of nonvascular death and cancer did not increase. It is plausible that the risk is somewhat greater in the perioperative setting but is likely not enough to outweigh the potential benefits, especially since the risk of ischemic vascular events is particularly high then.
Some of the perioperative studies cited above specifically addressed potential risks. For example, in the study by Schouten et al,32 mild creatine kinase elevations were more common in the statin-treated group, but the incidence of moderate and severe creatine kinase elevations did not differ significantly. No case of rhabdomyolysis occurred, and length of surgery was the only predictor of myopathy. MIRACL and PROVE-IT revealed similar safety profiles; aminotransferase levels normalized when statins were stopped, and no cases of rhabdomyolysis occurred.11,12 In the vascular surgery study by Durazzo et al,23 1 (2%) of the 50 atorvastatin-treated patients developed both rhabdomyolysis and elevated aminotransferase levels that prompted discontinuation of the statin.
Overall, the observational studies do not indicate that statin continuation or treatment is harmful in perioperative patients. However, these studies did not specifically evaluate patients with acute insults from surgery such as sepsis, renal failure, or hepatitis. It is unknown what effect statin therapy would have in those patients and whether statins should be selectively discontinued in patients who develop major hepatic, musculoskeletal, or renal complications after surgery.
OUR RECOMMENDATIONS
Before CABG or vascular surgery
Given the NCEP recommendations, existing primary and secondary prevention studies, observational studies of CABG and noncardiac vascular surgery patients, and the one randomized trial of vascular surgery patients, data support the use of statins in nearly all patients undergoing cardiac or vascular surgery. We advocate starting statins in the perioperative period to take advantage of their rapid-acting pleiotropic effects, and continuing them long-term to take advantage of their lipid-lowering effects. This recommendation is in line with the recently released American College of Cardiology/American Heart Association (ACC/AHA) 2007 perioperative guidelines that state “for patients undergoing vascular surgery with or without clinical risk factors, statin use is reasonable.”42
Although the ideal time to start statins is not certain, the study by Durazzo et al23 suggests that they should be started at least 2 weeks before surgery if possible. Moreover, patients already taking statins should definitely not have their statins discontinued if at all possible.
Before major nonvascular surgery
For patients undergoing major nonvascular (intermediate-risk) surgery, physicians should first ascertain if the patient has an indication for statin therapy based on current nonsurgical lipid level recommendations. However, even if there is no clear indication for statin therapy based on NCEP guidelines, we endorse the recently released ACC/AHA perioperative guidelines that state that statin therapy can be considered in patients with a risk factor who are undergoing intermediate-risk procedures. Moreover, we wholeheartedly support the ACC/AHA’s strongest recommendation that patients who are already receiving statins and are undergoing noncardiac surgery should not have their statins discontinued.
When to discontinue statins?
The risk of harm overall appears to be minimal and certainly less than the likelihood of benefit. It is reasonable to observe patients postoperatively for adverse clinical events that may increase the risk of perioperative statin treatment, such as acute renal failure, hepatic failure, or sepsis, but whether statins should be stopped in patients with these complications remains unknown; we advocate individualizing the decision.
More studies needed
We need more data on whether moderate-risk patients undergoing moderate-risk surgery benefit from perioperative statin therapy, when therapy should be started, whether therapy should be started on the day of surgery if it was not started earlier, which statin and what doses are optimal, how long therapy should be continued, and what degree of risk is associated with perioperative statin therapy.
Fortunately, important data should be forthcoming in the next few years: the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE-IV) study43 is a 4-year two-by-two factorial placebo-controlled study evaluating the use of fluvastatin (Lescol) and bisoprolol (Zebeta, a beta-blocker) separately and together in patients who are older than 40 years, are undergoing elective noncardiac surgery, have an estimated risk of cardiovascular death of more than 1%, have not used statins previously, and do not have elevated cholesterol.
- Grant PJ, Kedia N. Should statins be discontinued preoperatively? IMPACT consults. Proceedings of the 2nd Annual Cleveland Clinic Perioperative Medicine Summit. Cleve Clin J Med 2006; 73 Electronic suppl 1:S9–S10.
- Lindenauer PK, Pekow P, Wang K, Gutierrez B, Benjamin EM. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- McFalls EO, Ward HB, Moritz TE, et al. Coronary-artery revascularization before elective major vascular surgery. N Engl J Med 2004; 351:2795–2804.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Ito MK, Talbert RL, Tsimikas S. Statin-associated pleiotropy: possible beneficial effects beyond cholesterol reduction. Pharmacotherapy 2006; 26:85S–97S.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999; 100:178–184.
- Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci 2004; 27:283–289.
- Osborne JA, Lento PH, Siegfried MR, Stahl GL, Fusman B, Lefer AM. Cardiovascular effects of acute hypercholesterolemia in rabbits. Reversal with lovastatin treatment. J Clin Invest 1989; 83:465–473.
- Sironi L, Cimino M, Guerrini U, et al. Treatment with statins after induction of focal ischemia in rats reduces the extent of brain damage. Arterioscler Thromb Vasc Biol 2003; 23:322–327.
- Fonarow GC, Wright RS, Spencer FA, et al. Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. Am J Cardiol 2005; 96:611–616.
- Heeschen C, Hamm CW, Laufs U, Snapinn S, Bohm M, White HD. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation 2002; 105:1446–1452.
- Kennedy J, Quan H, Buchan AM, Ghali WA, Feasby TE. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- McGirt MJ, Perler BA, Brooke BS, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors reduce the risk of perioperative stroke and mortality after carotid endarterectomy. J Vasc Surg 2005; 42:829–836.
- Hindler K, Shaw AD, Samuels J, Fulton S, Collard CD, Riedel B. Improved postoperative outcomes associated with preoperative statin therapy. Anesthesiology 2006; 105:1260–1272.
- Kapoor AS, Kanji H, Buckingham J, Devereaux PJ, McAlister FA. Strength of evidence for perioperative use of statins to reduce cardiovascular risk: systematic review of controlled studies. BMJ 2006; 333:1149.
- Durazzo AE, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Kertai MD, Boersma E, Westerhout CM, et al. A combination of statins and beta-blockers is independently associated with a reduction in the incidence of perioperative mortality and nonfatal myocardial infarction in patients undergoing abdominal aortic aneurysm surgery. Eur J Vasc Endovasc Surg 2004; 28:343–352.
- Poldermans D, Bax JJ, Kertai MD, et al. Statins are associated with a reduced incidence of perioperative mortality in patients undergoing major noncardiac vascular surgery. Circulation 2003; 107:1848–1851.
- Ward RP, Leeper NJ, Kirkpatrick JN, Lang RM, Sorrentino MJ, Williams KA. The effect of preoperative statin therapy on cardiovascular outcomes in patients undergoing infrainguinal vascular surgery. Int J Cardiol 2005; 104:264–268.
- O’Neil-Callahan K, Katsimaglis G, Tepper MR, et al. Statins decrease perioperative cardiac complications in patients undergoing non-cardiac vascular surgery: the Statins for Risk Reduction in Surgery (StaRRS) study. J Am Coll Cardiol 2005; 45:336–342.
- Abbruzzese TA, Havens J, Belkin M, et al. Statin therapy is associated with improved patency of autogenous infrainguinal bypass grafts. J Vasc Surg 2004; 39:1178–1185.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Landesberg G, Mosseri M, Wolf YG, et al. Preoperative thallium scanning, selective coronary revascularization, and long-term survival after major vascular surgery. Circulation 2003; 108:177–183.
- Schouten O, Kertai MD, Bax JJ, et al. Safety of perioperative statin use in high-risk patients undergoing major vascular surgery. Am J Cardiol 2005; 95:658–660.
- Le Manach Y, Godet G, Coriat P, et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg 2007; 104:1326–1333.
- Ali IS, Buth KJ. Preoperative statin use and outcomes following cardiac surgery. Int J Cardiol 2005; 103:12–18.
- Clark LL, Ikonomidis JS, Crawford FA, et al. Preoperative statin treatment is associated with reduced postoperative mortality and morbidity in patients undergoing cardiac surgery: an 8-year retrospective cohort study. J Thorac Cardiovasc Surg 2006; 131:679–685.
- Pan W, Pintar T, Anton J, Lee VV, Vaughn WK, Collard CD. Statins are associated with a reduced incidence of perioperative mortality after coronary artery bypass graft surgery. Circulation 2004; 110(suppl 2):II45–II49.
- Pascual DA, Arribas JM, Tornel PL, et al. Preoperative statin therapy and troponin T predict early complications of coronary artery surgery. Ann Thorac Surg 2006; 81:78–83.
- Dotani MI, Elnicki DM, Jain AC, Gibson CM. Effect of preoperative statin therapy and cardiac outcomes after coronary artery bypass grafting. Am J Cardiol 2000; 86:1128–1130.
- Liakopoulos OJ, Dorge H, Schmitto JD, Nagorsnik U, Grabedunkel J, Schoendube FA. Effects of preoperative statin therapy on cytokines after cardiac surgery. Thorac Cardiovasc Surg 2006; 54:250–254.
- Christenson JT. Preoperative lipid-control with simvastatin reduces the risk of postoperative thrombocytosis and thrombotic complications following CABG. Eur J Cardiothorac Surg 1999; 15:394–399.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 Guidelines on Perioperative Cardiovascular Evaluation and Care for Noncardiac Surgery. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:e418–e499.
- Schouten O, Poldermans D, Visser L, et al. Fluvastatin and bisoprolol for the reduction of perioperative cardiac mortality and morbidity in high-risk patients undergoing non-cardiac surgery: rationale and design of the DECREASE-IV study. Am Heart J 2004; 148:1047–1052.
- Amar D, Zhang H, Heerdt PM, Park B, Fleisher M, Thaler HT. Statin use is associated with a reduction in atrial fibrillation after noncardiac thoracic surgery independent of C-reactive protein. Chest 2005; 128:3421–3427.
Soon, the checklist for internists seeing patients about to undergo surgery may include prescribing one of the lipid-lowering hydroxymethylglutaryl-CoA reductase inhibitors, also called statins.
Statins? Not long ago, we were debating whether patients who take statins should stop taking them before surgery, based on the manufacturers’ recommendations.1 The discussion, however, has changed to whether patients who have never received a statin should be started on one before surgery to provide immediate prophylaxis against cardiac morbidity, and how much harm long-term statin users face if these drugs are withheld perioperatively.
The evidence is still very preliminary and based mostly on studies in animals and retrospective studies in people. However, an expanding body of indirect evidence suggests that these drugs are beneficial in this situation.
In this review, we discuss the mechanisms by which statins may protect the heart in the short term, drawing on data from animal and human studies of acute myocardial infarction, and we review the current (albeit limited) data from the perioperative setting.
FEW INTERVENTIONS DECREASE RISK
Each year, approximately 50,000 patients suffer a perioperative cardiovascular event; the incidence of myocardial infarction during or after noncardiac surgery is 2% to 3%.2 The primary goal of preoperative cardiovascular risk assessment is to predict and avert these events.
But short of canceling surgery, few interventions have been found to reduce a patient’s risk. For example, a landmark study in 2004 cast doubt on the efficacy of preoperative coronary revascularization.3 Similarly, although early studies of beta-blockers were promising4,5 and although most internists prescribe these drugs before surgery, more recent studies have cast doubt on their efficacy, particularly in patients at low risk undergoing intermediate-risk (rather than vascular) surgery.6–8
This changing clinical landscape has prompted a search for new strategies for perioperative risk-reduction. Several recent studies have placed statins in the spotlight.
POTENTIAL MECHANISMS OF SHORT-TERM BENEFIT
Statins have been proven to save lives when used long-term, but how could this class of drugs, designed to prevent the accumulation of arterial plaques by lowering low-density lipoprotein cholesterol (LDL-C) levels, have any short-term impact on operative outcomes? Although LDL-C reduction is the principal mechanism of action of statins, not all of the benefit can be ascribed to this mechanism.9 The answer may lie in their “pleiotropic” effects—ie, actions other than LDL-C reduction.
The more immediate pleiotropic effects of statins in the proinflammatory and prothrombotic environment of the perioperative period are thought to include improved endothelial function (both antithrombotic function and vasomotor function in response to ischemic stress), enhanced stability of atherosclerotic plaques, decreased oxidative stress, and decreased vascular inflammation.10–12
EVIDENCE FROM ANIMAL STUDIES
Experiments in animals suggest that statins, given shortly before or after a cardiovascular event, confer benefit before any changes in LDL-C are measurable.
Lefer et al13 found that simvastatin (Zocor), given 18 hours before an ischemic episode in rats, blunted the inflammatory response in cardiac reperfusion injury. Not only was reperfusion injury significantly less in the hearts of the rats that received simvastatin than in the saline control group, but the simvastatin-treated hearts also expressed fewer neutrophil adhesion molecules such as P-selectin, and they had more basal release of nitric oxide, the potent endothelial-derived vasodilator with antithrombotic, anti-inflammatory, and antiproliferative effects.14 These results suggest that statins may improve endothelial function acutely, particularly during ischemic stress.
Osborne et al15 fed rabbits a cholesterol-rich diet plus either lovastatin (Mevacor) or placebo. After 2 weeks, the rabbits underwent either surgery to induce a myocardial infarction or a sham procedure. Regardless of the pretreatment, biopsies of the aorta did not reveal any atherosclerosis; yet the lovastatin-treated rabbits sustained less myocardial ischemic damage and they had more endothelium-mediated vasodilatation.
Statin therapy also may improve cerebral ischemia outcomes in animal models.14,16
Sironi et al16 induced strokes in rats by occluding the middle cerebral artery. The rats received either simvastatin or vehicle for 3 days before the stroke or immediately afterwards. Even though simvastatin did not have enough time to affect the total cholesterol level, rats treated with simvastatin had smaller infarcts (as measured by magnetic resonance imaging) and produced more nitric oxide.
Comment. Taken together, these studies offer tantalizing evidence that statins have short-term, beneficial nonlipid effects and may reduce not only the likelihood of an ischemic event, but—should one occur—the degree of tissue damage that ensues.
EFFECTS OF STATINS IN ACUTE CORONARY SYNDROME
The National Registry of Myocardial Infarction17 is a prospective, observational database of all patients with acute myocardial infarction admitted to 1,230 participating hospitals throughout the United States. In an analysis from this cohort, patients were divided into four groups: those receiving statins before and after admission, those receiving statins only before admission, those receiving statins only after admission, and those who never received statins.
Compared with those who never received statins, fewer patients who received them both before and after admission died while in the hospital (unadjusted odds ratio 0.23, 95% confidence interval [CI] 0.22–0.25), and the odds ratio for those who received statins for the first time was 0.31 (95% CI 0.29–0.33). Patients who stopped receiving a statin on admission were more likely to die than were patients who never received statins (odds ratio 1.09, 95% CI 1.03–1.15). These trends held true even when adjustments were made for potential confounding factors.
Comment. Unmeasured confounding factors (such as the inability to take pills due to altered mental status or the different practice styles of the providers who chose to discontinue statins) might have affected the results. Nevertheless, these results suggest that the protective effects of statins stop almost immediately when these drugs are discontinued, and that there may even be an adverse “rebound” effect when patients who have been taking these drugs for a long time stop taking them temporarily.
The Platelet Receptor Inhibition in Ischemic Syndrome Management trial,18 in a subgroup analysis, had nearly identical findings. In the main part of this trial, patients with coronary artery disease and chest pain at rest or accelerating pain in the last 24 hours were randomized to receive tirofiban (Aggrastat) or heparin. Complete data on statin use were available for 1,616 (50%) of the 3,232 patients in this trial, and the rate of the primary end point (death, myocardial infarction, or recurrent ischemia) was analyzed on the basis of statin therapy in this subgroup.
Comment. Together, these data lead to the conclusion that, when admitted for either acute myocardial infarction or acute coronary syndrome, patients already receiving statins should not have them stopped, and those who had not been receiving statins should receive them immediately. The safety of these medications in the acute setting appears excellent: in the Myocardial Ischemia Reduction With Acute Cholesterol Lowering (MIRACL)12 and the Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT)11 trials, fewer than 5% of statin-treated patients had transient elevations in transaminase levels, and no cases of rhabdomyolysis were reported.
PERIOPERATIVE STATIN STUDIES
The data on perioperative statin use are mostly observational and retrospective and fall into essentially four surgical categories: coronary artery bypass grafting (CABG), carotid endarterectomy,19,20 noncardiac vascular surgery, and major noncardiac surgery. Two meta-analyses have also evaluated the data.21,22 The only randomized controlled trial (performed by Durazzo et al23) was small and was carried out at a single center in vascular surgery patients, and the event rate was low.
Current recommendations from the National Cholesterol Education Program (NCEP)24 say that patients who need CABG, have peripheral arterial disease, have an abdominal aortic aneurysm, or have cerebrovascular disease should already be on a statin to achieve an LDL-C goal level of less than 100 mg/dL, with an optional goal of less than 70 mg/dL, independent of surgery.
Since not all patients who should be on statins are actually on them, questions arise:
- Is it important (and safe) to start statin treatment preoperatively?
- Will patients with cardiovascular risk factors but without known cardiovascular disease benefit from statins perioperatively?
Noncardiac vascular surgery
Multiple retrospective studies have evaluated the effect of statins in patients undergoing major noncardiac vascular surgery.25–32
Kertai et al25 evaluated 570 patients in Holland who underwent elective open surgery for infrarenal abdominal aortic aneurysms between 1991 and 2001, looking for an association between statin use and the incidence of perioperative death from myocardial infarction. Only 162 of the 570 patients had been on long-term statin therapy before the surgery. The use of statins was only one of many known baseline characteristics that were significantly different between the two groups, including age, body mass index, known coronary artery disease, and use of angiotensin-converting enzyme inhibitors and beta-blockers. In univariate analysis, statins appeared to be protective: 6 (3.7%) of the patients in the statin group died of a myocardial infarction, compared with 45 (11%) of those in the nostatin group. A multivariate analysis yielded similar findings, with an odds ratio of 0.24 (95% CI 0.11–0.54).
Ward et al27 performed a very similar retrospective study, with similar findings. In 446 patients who underwent surgery for infrarenal abdominal aortic aneurysm, statin therapy was associated with a significantly lower incidence of the combined end point of death, myocardial infarction, stroke, and major peripheral vascular complications, with an adjusted odds ratio of 0.36 (95% CI 0.14–0.93).
Poldermans et al26 noted similar findings in a case-control study of noncardiac vascular surgery patients. Statin users had a much lower perioperative risk of death than did nonusers, with an adjusted odds ratio of 0.22 (95% CI 0.10–0.47).
O’Neil-Callahan et al,28 in a cohort study, found that statin users had fewer perioperative cardiac complications, with an adjusted odds ratio of 0.49 (95% CI 0.28–0.84, P = .009).
Dogma of withdrawing statins before major surgery is challenged
Le Manach et al33 reviewed the outcomes for all patients of a single hospital in Paris who underwent nonemergency infrarenal aortic procedures between January 2001 and December 2004. In January 2004, the hospital instituted guidelines to ensure that patients on statins continue taking them up to the evening before surgery and that statins be restarted on the first postoperative day (via nasogastric tube if necessary). Before 2004, there had been no specific guidelines, and patients on statins did not receive them for a median of 4 days postoperatively. Types of procedures were similar during the two time periods, as were the rates of beta-blocker use, preoperative revascularization, venous thromboembolism prophylaxis, and perioperative blood pressure control. After surgery, topononin I levels were measured in all patients as surveillance for cardiac events, and were defined as elevated when greater than 0.2 ng/mL.
Compared with patients not on statins at all, those treated with statins continuously throughout the perioperative period (after January 2004) had a lower rate of elevated troponin (relative risk 0.38). In contrast, those who had their statins transiently discontinued perioperatively (prior to 2004) had troponin elevations more often than those who had never been treated (relative risk 2.1). This suggested an over fivefold risk reduction (P < .001) conferred by not discontinuing statins in the immediate postoperative period. This finding was maintained after multivariate adjustment: statin withdrawal was associated with a 2.9-fold (95% CI 1.6–5.5) increase in the risk of cardiac enzyme elevations postoperatively. No fewer deaths were noted, but the study was not powered to detect a mortality difference.
Comment. Although secular trends cannot be entirely discounted as contributing to these findings, the prompt increase in cardiac events after just 4 days of statin withdrawal adds to the growing body of evidence suggesting that statin discontinuation can have harmful acute effects. It also brings up the question: Can starting statins benefit patients in the same time period?
Should statins be started before vascular surgery?
Schouten et al32 evaluated the effects of newly started or continued statin treatment in patients undergoing major elective vascular surgery. Patients were screened before surgery and started on statins if they were not already receiving them and their total cholesterol levels were elevated; new users received the medication for about 40 days before surgery. Of the 981 screened patients, 44 (5%) were newly started on statins and 182 (19%) were continued on their therapy. Perioperative death or myocardial infarction occurred in 22 (8.8%) of the statin users and 111 (14.7%) of the nonusers, a statistically significant difference. Temporary discontinuation (median 1 day) of statins in this study due to the inability to take an oral medication did not appear to affect the likelihood of a myocardial infarction.
Durazzo et al23 performed a single-center, randomized, prospective, placebo-controlled, double-blind clinical trial of atorvastatin (Lipitor) 20 mg daily vs placebo in 100 patients undergoing noncardiac arterial vascular surgery. Patients were excluded if they had previously used medications to treat dyslipidemia, recently had a cardiovascular event, or had contraindications to statin treatment such as a baseline creatinine level greater than 2.0 mg/dL or severe hepatic disease. The intervention group received atorvastatin starting at least 2 weeks before surgery for a total of 45 days. Patients were then continued or started on a statin after surgery if their LDL-C level was greater than 100 mg/dL. Beta-blocker use was recommended “on the basis of current guidelines.”
One month after surgery, the LDL-C level was statistically significantly lower in the atorvastatin group. Since most patients did not continue or start statin therapy after the 45-day treatment period, the LDL-C levels were not statistically different at 3 and 6 months after surgery.
At 6 months, the rate of the primary end point (death from cardiovascular causes, nonfatal acute myocardial infarction, ischemic stroke, or unstable angina) was 26.0% in the placebo group and 8.0% in the atorvastatin group, a statistically significant difference. Three patients in the atorvastatin group had cardiac events in the first 10 days after surgery, compared with 11 patients in the placebo group. Thirteen of the 17 total cardiac events took place within 10 days after surgery.
One of the atorvastatin patients developed rhabdomyolysis and elevated aminotransferase levels.
Major noncardiac surgery
Lindenauer et al2 performed a retrospective cohort study of surgical patients who were at least 18 years old and survived beyond the second hospital day. Patients were divided into a group receiving any form of lipid-lowering treatment (of whom more than 90% were taking statins) and a group that had never never received a lipid-lowering drug or only started one on the third day of the hospitalization or later. The period of study was from January 1, 2000, to December 31, 2001.
In all, 780,591 patients from 329 hospitals throughout the United States were included, of whom only 77,082 (9.9%) received lipid-lowering therapy. Eight percent of the patients underwent vascular surgery. Not surprisingly, the treated patients were more likely to have a history of hypertension, diabetes, ischemic heart disease, or hyperlipidemia. They also were more likely to have a vascular procedure performed, to have two or more cardiac risk factors (high-risk surgery, ischemic heart disease, congestive heart failure, cerebrovascular disease, renal insufficiency, or diabetes mellitus), and to be treated with beta-blockers and angiotensin-converting enzyme inhibitors, but they were less likely to have high-risk and emergency surgery performed.
The primary end point, perioperative death, occurred in 2.13% of the treated patients and 3.05% of the nontreated group. Compared with the rate in a propensity-matched cohort, the odds ratio adjusted for unbalanced covariates was 0.62 (95% CI 0.58–0.67) in favor of lipid treatment. Stratification by cardiac risk index revealed a number needed to treat of 186 for those with no risk factors, 60 for those with two risk factors, and 30 for those with four or more risk factors.
Unfortunately, this analysis was not able to take into account whether and for how long patients were receiving lipid-lowering therapy before hospitalization. It therefore does not answer the questions of whether starting lipid-lowering therapy before surgery is beneficial or whether stopping it is harmful. It also does not shed light on whether perioperative lipid-lowering increases the risk of rhabdomyolysis or liver disease.
Carotid endarterectomy
Two recent retrospective cohort studies evaluated the outcomes in patients undergoing carotid endarterectomy.19,20
Kennedy et al19 found that patients on a statin at the time of admission who had symptomatic carotid disease had lower rates of inhospital death (adjusted odds ratio 0.24, 95% CI 0.06–0.91) and ischemic stroke or death (adjusted odds ratio 0.55, 95% CI 0.31–0.97). However, cardiac outcomes among these symptomatic patients were not significantly improved (odds ratio 0.82, 95% CI 0.45–1.50), nor was there benefit for asymptomatic patients, raising the possibility that the positive findings were due to chance or that patients at lower baseline risk for vascular events may have less benefit.
McGirt et al20 performed a similar study; they did not, however, distinguish whether patients had symptomatic vs asymptomatic carotid disease. The 30-day risk of perioperative stroke was lower in patients treated with a statin, with an odds ratio of 0.41 (95% CI 0.18–0.93); the odds ratio for death was 0.21 (95% CI 0.05–0.96). Cardiac outcomes were not significantly affected.
Coronary artery bypass graft surgery
According to the NCEP recommendations, nearly all patients undergoing CABG should already be on a statin before surgery since they all have known coronary artery disease. Multiple observational studies have offered confirmatory evidence that statins are beneficial in this setting.34–38
Liakopoulos et al39 evaluated whether the anti-inflammatory effects of statins may, in part, account for their beneficial effect in the perioperative period. The authors prospectively matched 18 patients who were taking statins and were referred for elective CABG with 18 patients who were not prescribed statins previously. The only major measured baseline characteristic that differed between the two groups was a statistically significantly lower LDL-C level in the statin group. The operative characteristics did not differ, and cytokine levels at baseline were similar.
Tumor necrosis factor alpha levels increased significantly in the control group but did not change significantly in the statin group. Interleukin 8 increased in both groups by a similar amount. Interleukin 6 (the major inducer of C-reactive protein) increased from baseline in both groups but did not increase nearly as much in the statin group as in the control group; the intergroup difference was statistically significant. The anti-inflammatory cytokine interleukin 10 increased minimally from baseline in the control group, while the statin group’s levels increased significantly above baseline and those of the control group.
Christenson40 also found that inflammatory markers were improved with pre-CABG statin treatment in a small randomized trial in which patients received simvastatin 20 mg 4 weeks prior to CABG surgery vs no statin. Interestingly, far fewer statin-treated patients developed thrombocytosis (platelet count > 400 × 109/L) than did control patients (3% vs 81%, P < .0001).
RISKS OF PERIOPERATIVE STATINS
The risks associated with statin therapy in general appear low, but specific perioperative risks have not been well studied.
Baigent et al,41 in a meta-analysis of randomized trials of nonperioperative statin therapy, found that rhabdomyolysis occurred in 9 (0.023%) of 39,884 patients receiving statins vs 6 (0.015%) of the 39,817 controls, with a number needed to harm of 12,500. Moreover, the rates of nonvascular death and cancer did not increase. It is plausible that the risk is somewhat greater in the perioperative setting but is likely not enough to outweigh the potential benefits, especially since the risk of ischemic vascular events is particularly high then.
Some of the perioperative studies cited above specifically addressed potential risks. For example, in the study by Schouten et al,32 mild creatine kinase elevations were more common in the statin-treated group, but the incidence of moderate and severe creatine kinase elevations did not differ significantly. No case of rhabdomyolysis occurred, and length of surgery was the only predictor of myopathy. MIRACL and PROVE-IT revealed similar safety profiles; aminotransferase levels normalized when statins were stopped, and no cases of rhabdomyolysis occurred.11,12 In the vascular surgery study by Durazzo et al,23 1 (2%) of the 50 atorvastatin-treated patients developed both rhabdomyolysis and elevated aminotransferase levels that prompted discontinuation of the statin.
Overall, the observational studies do not indicate that statin continuation or treatment is harmful in perioperative patients. However, these studies did not specifically evaluate patients with acute insults from surgery such as sepsis, renal failure, or hepatitis. It is unknown what effect statin therapy would have in those patients and whether statins should be selectively discontinued in patients who develop major hepatic, musculoskeletal, or renal complications after surgery.
OUR RECOMMENDATIONS
Before CABG or vascular surgery
Given the NCEP recommendations, existing primary and secondary prevention studies, observational studies of CABG and noncardiac vascular surgery patients, and the one randomized trial of vascular surgery patients, data support the use of statins in nearly all patients undergoing cardiac or vascular surgery. We advocate starting statins in the perioperative period to take advantage of their rapid-acting pleiotropic effects, and continuing them long-term to take advantage of their lipid-lowering effects. This recommendation is in line with the recently released American College of Cardiology/American Heart Association (ACC/AHA) 2007 perioperative guidelines that state “for patients undergoing vascular surgery with or without clinical risk factors, statin use is reasonable.”42
Although the ideal time to start statins is not certain, the study by Durazzo et al23 suggests that they should be started at least 2 weeks before surgery if possible. Moreover, patients already taking statins should definitely not have their statins discontinued if at all possible.
Before major nonvascular surgery
For patients undergoing major nonvascular (intermediate-risk) surgery, physicians should first ascertain if the patient has an indication for statin therapy based on current nonsurgical lipid level recommendations. However, even if there is no clear indication for statin therapy based on NCEP guidelines, we endorse the recently released ACC/AHA perioperative guidelines that state that statin therapy can be considered in patients with a risk factor who are undergoing intermediate-risk procedures. Moreover, we wholeheartedly support the ACC/AHA’s strongest recommendation that patients who are already receiving statins and are undergoing noncardiac surgery should not have their statins discontinued.
When to discontinue statins?
The risk of harm overall appears to be minimal and certainly less than the likelihood of benefit. It is reasonable to observe patients postoperatively for adverse clinical events that may increase the risk of perioperative statin treatment, such as acute renal failure, hepatic failure, or sepsis, but whether statins should be stopped in patients with these complications remains unknown; we advocate individualizing the decision.
More studies needed
We need more data on whether moderate-risk patients undergoing moderate-risk surgery benefit from perioperative statin therapy, when therapy should be started, whether therapy should be started on the day of surgery if it was not started earlier, which statin and what doses are optimal, how long therapy should be continued, and what degree of risk is associated with perioperative statin therapy.
Fortunately, important data should be forthcoming in the next few years: the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE-IV) study43 is a 4-year two-by-two factorial placebo-controlled study evaluating the use of fluvastatin (Lescol) and bisoprolol (Zebeta, a beta-blocker) separately and together in patients who are older than 40 years, are undergoing elective noncardiac surgery, have an estimated risk of cardiovascular death of more than 1%, have not used statins previously, and do not have elevated cholesterol.
Soon, the checklist for internists seeing patients about to undergo surgery may include prescribing one of the lipid-lowering hydroxymethylglutaryl-CoA reductase inhibitors, also called statins.
Statins? Not long ago, we were debating whether patients who take statins should stop taking them before surgery, based on the manufacturers’ recommendations.1 The discussion, however, has changed to whether patients who have never received a statin should be started on one before surgery to provide immediate prophylaxis against cardiac morbidity, and how much harm long-term statin users face if these drugs are withheld perioperatively.
The evidence is still very preliminary and based mostly on studies in animals and retrospective studies in people. However, an expanding body of indirect evidence suggests that these drugs are beneficial in this situation.
In this review, we discuss the mechanisms by which statins may protect the heart in the short term, drawing on data from animal and human studies of acute myocardial infarction, and we review the current (albeit limited) data from the perioperative setting.
FEW INTERVENTIONS DECREASE RISK
Each year, approximately 50,000 patients suffer a perioperative cardiovascular event; the incidence of myocardial infarction during or after noncardiac surgery is 2% to 3%.2 The primary goal of preoperative cardiovascular risk assessment is to predict and avert these events.
But short of canceling surgery, few interventions have been found to reduce a patient’s risk. For example, a landmark study in 2004 cast doubt on the efficacy of preoperative coronary revascularization.3 Similarly, although early studies of beta-blockers were promising4,5 and although most internists prescribe these drugs before surgery, more recent studies have cast doubt on their efficacy, particularly in patients at low risk undergoing intermediate-risk (rather than vascular) surgery.6–8
This changing clinical landscape has prompted a search for new strategies for perioperative risk-reduction. Several recent studies have placed statins in the spotlight.
POTENTIAL MECHANISMS OF SHORT-TERM BENEFIT
Statins have been proven to save lives when used long-term, but how could this class of drugs, designed to prevent the accumulation of arterial plaques by lowering low-density lipoprotein cholesterol (LDL-C) levels, have any short-term impact on operative outcomes? Although LDL-C reduction is the principal mechanism of action of statins, not all of the benefit can be ascribed to this mechanism.9 The answer may lie in their “pleiotropic” effects—ie, actions other than LDL-C reduction.
The more immediate pleiotropic effects of statins in the proinflammatory and prothrombotic environment of the perioperative period are thought to include improved endothelial function (both antithrombotic function and vasomotor function in response to ischemic stress), enhanced stability of atherosclerotic plaques, decreased oxidative stress, and decreased vascular inflammation.10–12
EVIDENCE FROM ANIMAL STUDIES
Experiments in animals suggest that statins, given shortly before or after a cardiovascular event, confer benefit before any changes in LDL-C are measurable.
Lefer et al13 found that simvastatin (Zocor), given 18 hours before an ischemic episode in rats, blunted the inflammatory response in cardiac reperfusion injury. Not only was reperfusion injury significantly less in the hearts of the rats that received simvastatin than in the saline control group, but the simvastatin-treated hearts also expressed fewer neutrophil adhesion molecules such as P-selectin, and they had more basal release of nitric oxide, the potent endothelial-derived vasodilator with antithrombotic, anti-inflammatory, and antiproliferative effects.14 These results suggest that statins may improve endothelial function acutely, particularly during ischemic stress.
Osborne et al15 fed rabbits a cholesterol-rich diet plus either lovastatin (Mevacor) or placebo. After 2 weeks, the rabbits underwent either surgery to induce a myocardial infarction or a sham procedure. Regardless of the pretreatment, biopsies of the aorta did not reveal any atherosclerosis; yet the lovastatin-treated rabbits sustained less myocardial ischemic damage and they had more endothelium-mediated vasodilatation.
Statin therapy also may improve cerebral ischemia outcomes in animal models.14,16
Sironi et al16 induced strokes in rats by occluding the middle cerebral artery. The rats received either simvastatin or vehicle for 3 days before the stroke or immediately afterwards. Even though simvastatin did not have enough time to affect the total cholesterol level, rats treated with simvastatin had smaller infarcts (as measured by magnetic resonance imaging) and produced more nitric oxide.
Comment. Taken together, these studies offer tantalizing evidence that statins have short-term, beneficial nonlipid effects and may reduce not only the likelihood of an ischemic event, but—should one occur—the degree of tissue damage that ensues.
EFFECTS OF STATINS IN ACUTE CORONARY SYNDROME
The National Registry of Myocardial Infarction17 is a prospective, observational database of all patients with acute myocardial infarction admitted to 1,230 participating hospitals throughout the United States. In an analysis from this cohort, patients were divided into four groups: those receiving statins before and after admission, those receiving statins only before admission, those receiving statins only after admission, and those who never received statins.
Compared with those who never received statins, fewer patients who received them both before and after admission died while in the hospital (unadjusted odds ratio 0.23, 95% confidence interval [CI] 0.22–0.25), and the odds ratio for those who received statins for the first time was 0.31 (95% CI 0.29–0.33). Patients who stopped receiving a statin on admission were more likely to die than were patients who never received statins (odds ratio 1.09, 95% CI 1.03–1.15). These trends held true even when adjustments were made for potential confounding factors.
Comment. Unmeasured confounding factors (such as the inability to take pills due to altered mental status or the different practice styles of the providers who chose to discontinue statins) might have affected the results. Nevertheless, these results suggest that the protective effects of statins stop almost immediately when these drugs are discontinued, and that there may even be an adverse “rebound” effect when patients who have been taking these drugs for a long time stop taking them temporarily.
The Platelet Receptor Inhibition in Ischemic Syndrome Management trial,18 in a subgroup analysis, had nearly identical findings. In the main part of this trial, patients with coronary artery disease and chest pain at rest or accelerating pain in the last 24 hours were randomized to receive tirofiban (Aggrastat) or heparin. Complete data on statin use were available for 1,616 (50%) of the 3,232 patients in this trial, and the rate of the primary end point (death, myocardial infarction, or recurrent ischemia) was analyzed on the basis of statin therapy in this subgroup.
Comment. Together, these data lead to the conclusion that, when admitted for either acute myocardial infarction or acute coronary syndrome, patients already receiving statins should not have them stopped, and those who had not been receiving statins should receive them immediately. The safety of these medications in the acute setting appears excellent: in the Myocardial Ischemia Reduction With Acute Cholesterol Lowering (MIRACL)12 and the Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT)11 trials, fewer than 5% of statin-treated patients had transient elevations in transaminase levels, and no cases of rhabdomyolysis were reported.
PERIOPERATIVE STATIN STUDIES
The data on perioperative statin use are mostly observational and retrospective and fall into essentially four surgical categories: coronary artery bypass grafting (CABG), carotid endarterectomy,19,20 noncardiac vascular surgery, and major noncardiac surgery. Two meta-analyses have also evaluated the data.21,22 The only randomized controlled trial (performed by Durazzo et al23) was small and was carried out at a single center in vascular surgery patients, and the event rate was low.
Current recommendations from the National Cholesterol Education Program (NCEP)24 say that patients who need CABG, have peripheral arterial disease, have an abdominal aortic aneurysm, or have cerebrovascular disease should already be on a statin to achieve an LDL-C goal level of less than 100 mg/dL, with an optional goal of less than 70 mg/dL, independent of surgery.
Since not all patients who should be on statins are actually on them, questions arise:
- Is it important (and safe) to start statin treatment preoperatively?
- Will patients with cardiovascular risk factors but without known cardiovascular disease benefit from statins perioperatively?
Noncardiac vascular surgery
Multiple retrospective studies have evaluated the effect of statins in patients undergoing major noncardiac vascular surgery.25–32
Kertai et al25 evaluated 570 patients in Holland who underwent elective open surgery for infrarenal abdominal aortic aneurysms between 1991 and 2001, looking for an association between statin use and the incidence of perioperative death from myocardial infarction. Only 162 of the 570 patients had been on long-term statin therapy before the surgery. The use of statins was only one of many known baseline characteristics that were significantly different between the two groups, including age, body mass index, known coronary artery disease, and use of angiotensin-converting enzyme inhibitors and beta-blockers. In univariate analysis, statins appeared to be protective: 6 (3.7%) of the patients in the statin group died of a myocardial infarction, compared with 45 (11%) of those in the nostatin group. A multivariate analysis yielded similar findings, with an odds ratio of 0.24 (95% CI 0.11–0.54).
Ward et al27 performed a very similar retrospective study, with similar findings. In 446 patients who underwent surgery for infrarenal abdominal aortic aneurysm, statin therapy was associated with a significantly lower incidence of the combined end point of death, myocardial infarction, stroke, and major peripheral vascular complications, with an adjusted odds ratio of 0.36 (95% CI 0.14–0.93).
Poldermans et al26 noted similar findings in a case-control study of noncardiac vascular surgery patients. Statin users had a much lower perioperative risk of death than did nonusers, with an adjusted odds ratio of 0.22 (95% CI 0.10–0.47).
O’Neil-Callahan et al,28 in a cohort study, found that statin users had fewer perioperative cardiac complications, with an adjusted odds ratio of 0.49 (95% CI 0.28–0.84, P = .009).
Dogma of withdrawing statins before major surgery is challenged
Le Manach et al33 reviewed the outcomes for all patients of a single hospital in Paris who underwent nonemergency infrarenal aortic procedures between January 2001 and December 2004. In January 2004, the hospital instituted guidelines to ensure that patients on statins continue taking them up to the evening before surgery and that statins be restarted on the first postoperative day (via nasogastric tube if necessary). Before 2004, there had been no specific guidelines, and patients on statins did not receive them for a median of 4 days postoperatively. Types of procedures were similar during the two time periods, as were the rates of beta-blocker use, preoperative revascularization, venous thromboembolism prophylaxis, and perioperative blood pressure control. After surgery, topononin I levels were measured in all patients as surveillance for cardiac events, and were defined as elevated when greater than 0.2 ng/mL.
Compared with patients not on statins at all, those treated with statins continuously throughout the perioperative period (after January 2004) had a lower rate of elevated troponin (relative risk 0.38). In contrast, those who had their statins transiently discontinued perioperatively (prior to 2004) had troponin elevations more often than those who had never been treated (relative risk 2.1). This suggested an over fivefold risk reduction (P < .001) conferred by not discontinuing statins in the immediate postoperative period. This finding was maintained after multivariate adjustment: statin withdrawal was associated with a 2.9-fold (95% CI 1.6–5.5) increase in the risk of cardiac enzyme elevations postoperatively. No fewer deaths were noted, but the study was not powered to detect a mortality difference.
Comment. Although secular trends cannot be entirely discounted as contributing to these findings, the prompt increase in cardiac events after just 4 days of statin withdrawal adds to the growing body of evidence suggesting that statin discontinuation can have harmful acute effects. It also brings up the question: Can starting statins benefit patients in the same time period?
Should statins be started before vascular surgery?
Schouten et al32 evaluated the effects of newly started or continued statin treatment in patients undergoing major elective vascular surgery. Patients were screened before surgery and started on statins if they were not already receiving them and their total cholesterol levels were elevated; new users received the medication for about 40 days before surgery. Of the 981 screened patients, 44 (5%) were newly started on statins and 182 (19%) were continued on their therapy. Perioperative death or myocardial infarction occurred in 22 (8.8%) of the statin users and 111 (14.7%) of the nonusers, a statistically significant difference. Temporary discontinuation (median 1 day) of statins in this study due to the inability to take an oral medication did not appear to affect the likelihood of a myocardial infarction.
Durazzo et al23 performed a single-center, randomized, prospective, placebo-controlled, double-blind clinical trial of atorvastatin (Lipitor) 20 mg daily vs placebo in 100 patients undergoing noncardiac arterial vascular surgery. Patients were excluded if they had previously used medications to treat dyslipidemia, recently had a cardiovascular event, or had contraindications to statin treatment such as a baseline creatinine level greater than 2.0 mg/dL or severe hepatic disease. The intervention group received atorvastatin starting at least 2 weeks before surgery for a total of 45 days. Patients were then continued or started on a statin after surgery if their LDL-C level was greater than 100 mg/dL. Beta-blocker use was recommended “on the basis of current guidelines.”
One month after surgery, the LDL-C level was statistically significantly lower in the atorvastatin group. Since most patients did not continue or start statin therapy after the 45-day treatment period, the LDL-C levels were not statistically different at 3 and 6 months after surgery.
At 6 months, the rate of the primary end point (death from cardiovascular causes, nonfatal acute myocardial infarction, ischemic stroke, or unstable angina) was 26.0% in the placebo group and 8.0% in the atorvastatin group, a statistically significant difference. Three patients in the atorvastatin group had cardiac events in the first 10 days after surgery, compared with 11 patients in the placebo group. Thirteen of the 17 total cardiac events took place within 10 days after surgery.
One of the atorvastatin patients developed rhabdomyolysis and elevated aminotransferase levels.
Major noncardiac surgery
Lindenauer et al2 performed a retrospective cohort study of surgical patients who were at least 18 years old and survived beyond the second hospital day. Patients were divided into a group receiving any form of lipid-lowering treatment (of whom more than 90% were taking statins) and a group that had never never received a lipid-lowering drug or only started one on the third day of the hospitalization or later. The period of study was from January 1, 2000, to December 31, 2001.
In all, 780,591 patients from 329 hospitals throughout the United States were included, of whom only 77,082 (9.9%) received lipid-lowering therapy. Eight percent of the patients underwent vascular surgery. Not surprisingly, the treated patients were more likely to have a history of hypertension, diabetes, ischemic heart disease, or hyperlipidemia. They also were more likely to have a vascular procedure performed, to have two or more cardiac risk factors (high-risk surgery, ischemic heart disease, congestive heart failure, cerebrovascular disease, renal insufficiency, or diabetes mellitus), and to be treated with beta-blockers and angiotensin-converting enzyme inhibitors, but they were less likely to have high-risk and emergency surgery performed.
The primary end point, perioperative death, occurred in 2.13% of the treated patients and 3.05% of the nontreated group. Compared with the rate in a propensity-matched cohort, the odds ratio adjusted for unbalanced covariates was 0.62 (95% CI 0.58–0.67) in favor of lipid treatment. Stratification by cardiac risk index revealed a number needed to treat of 186 for those with no risk factors, 60 for those with two risk factors, and 30 for those with four or more risk factors.
Unfortunately, this analysis was not able to take into account whether and for how long patients were receiving lipid-lowering therapy before hospitalization. It therefore does not answer the questions of whether starting lipid-lowering therapy before surgery is beneficial or whether stopping it is harmful. It also does not shed light on whether perioperative lipid-lowering increases the risk of rhabdomyolysis or liver disease.
Carotid endarterectomy
Two recent retrospective cohort studies evaluated the outcomes in patients undergoing carotid endarterectomy.19,20
Kennedy et al19 found that patients on a statin at the time of admission who had symptomatic carotid disease had lower rates of inhospital death (adjusted odds ratio 0.24, 95% CI 0.06–0.91) and ischemic stroke or death (adjusted odds ratio 0.55, 95% CI 0.31–0.97). However, cardiac outcomes among these symptomatic patients were not significantly improved (odds ratio 0.82, 95% CI 0.45–1.50), nor was there benefit for asymptomatic patients, raising the possibility that the positive findings were due to chance or that patients at lower baseline risk for vascular events may have less benefit.
McGirt et al20 performed a similar study; they did not, however, distinguish whether patients had symptomatic vs asymptomatic carotid disease. The 30-day risk of perioperative stroke was lower in patients treated with a statin, with an odds ratio of 0.41 (95% CI 0.18–0.93); the odds ratio for death was 0.21 (95% CI 0.05–0.96). Cardiac outcomes were not significantly affected.
Coronary artery bypass graft surgery
According to the NCEP recommendations, nearly all patients undergoing CABG should already be on a statin before surgery since they all have known coronary artery disease. Multiple observational studies have offered confirmatory evidence that statins are beneficial in this setting.34–38
Liakopoulos et al39 evaluated whether the anti-inflammatory effects of statins may, in part, account for their beneficial effect in the perioperative period. The authors prospectively matched 18 patients who were taking statins and were referred for elective CABG with 18 patients who were not prescribed statins previously. The only major measured baseline characteristic that differed between the two groups was a statistically significantly lower LDL-C level in the statin group. The operative characteristics did not differ, and cytokine levels at baseline were similar.
Tumor necrosis factor alpha levels increased significantly in the control group but did not change significantly in the statin group. Interleukin 8 increased in both groups by a similar amount. Interleukin 6 (the major inducer of C-reactive protein) increased from baseline in both groups but did not increase nearly as much in the statin group as in the control group; the intergroup difference was statistically significant. The anti-inflammatory cytokine interleukin 10 increased minimally from baseline in the control group, while the statin group’s levels increased significantly above baseline and those of the control group.
Christenson40 also found that inflammatory markers were improved with pre-CABG statin treatment in a small randomized trial in which patients received simvastatin 20 mg 4 weeks prior to CABG surgery vs no statin. Interestingly, far fewer statin-treated patients developed thrombocytosis (platelet count > 400 × 109/L) than did control patients (3% vs 81%, P < .0001).
RISKS OF PERIOPERATIVE STATINS
The risks associated with statin therapy in general appear low, but specific perioperative risks have not been well studied.
Baigent et al,41 in a meta-analysis of randomized trials of nonperioperative statin therapy, found that rhabdomyolysis occurred in 9 (0.023%) of 39,884 patients receiving statins vs 6 (0.015%) of the 39,817 controls, with a number needed to harm of 12,500. Moreover, the rates of nonvascular death and cancer did not increase. It is plausible that the risk is somewhat greater in the perioperative setting but is likely not enough to outweigh the potential benefits, especially since the risk of ischemic vascular events is particularly high then.
Some of the perioperative studies cited above specifically addressed potential risks. For example, in the study by Schouten et al,32 mild creatine kinase elevations were more common in the statin-treated group, but the incidence of moderate and severe creatine kinase elevations did not differ significantly. No case of rhabdomyolysis occurred, and length of surgery was the only predictor of myopathy. MIRACL and PROVE-IT revealed similar safety profiles; aminotransferase levels normalized when statins were stopped, and no cases of rhabdomyolysis occurred.11,12 In the vascular surgery study by Durazzo et al,23 1 (2%) of the 50 atorvastatin-treated patients developed both rhabdomyolysis and elevated aminotransferase levels that prompted discontinuation of the statin.
Overall, the observational studies do not indicate that statin continuation or treatment is harmful in perioperative patients. However, these studies did not specifically evaluate patients with acute insults from surgery such as sepsis, renal failure, or hepatitis. It is unknown what effect statin therapy would have in those patients and whether statins should be selectively discontinued in patients who develop major hepatic, musculoskeletal, or renal complications after surgery.
OUR RECOMMENDATIONS
Before CABG or vascular surgery
Given the NCEP recommendations, existing primary and secondary prevention studies, observational studies of CABG and noncardiac vascular surgery patients, and the one randomized trial of vascular surgery patients, data support the use of statins in nearly all patients undergoing cardiac or vascular surgery. We advocate starting statins in the perioperative period to take advantage of their rapid-acting pleiotropic effects, and continuing them long-term to take advantage of their lipid-lowering effects. This recommendation is in line with the recently released American College of Cardiology/American Heart Association (ACC/AHA) 2007 perioperative guidelines that state “for patients undergoing vascular surgery with or without clinical risk factors, statin use is reasonable.”42
Although the ideal time to start statins is not certain, the study by Durazzo et al23 suggests that they should be started at least 2 weeks before surgery if possible. Moreover, patients already taking statins should definitely not have their statins discontinued if at all possible.
Before major nonvascular surgery
For patients undergoing major nonvascular (intermediate-risk) surgery, physicians should first ascertain if the patient has an indication for statin therapy based on current nonsurgical lipid level recommendations. However, even if there is no clear indication for statin therapy based on NCEP guidelines, we endorse the recently released ACC/AHA perioperative guidelines that state that statin therapy can be considered in patients with a risk factor who are undergoing intermediate-risk procedures. Moreover, we wholeheartedly support the ACC/AHA’s strongest recommendation that patients who are already receiving statins and are undergoing noncardiac surgery should not have their statins discontinued.
When to discontinue statins?
The risk of harm overall appears to be minimal and certainly less than the likelihood of benefit. It is reasonable to observe patients postoperatively for adverse clinical events that may increase the risk of perioperative statin treatment, such as acute renal failure, hepatic failure, or sepsis, but whether statins should be stopped in patients with these complications remains unknown; we advocate individualizing the decision.
More studies needed
We need more data on whether moderate-risk patients undergoing moderate-risk surgery benefit from perioperative statin therapy, when therapy should be started, whether therapy should be started on the day of surgery if it was not started earlier, which statin and what doses are optimal, how long therapy should be continued, and what degree of risk is associated with perioperative statin therapy.
Fortunately, important data should be forthcoming in the next few years: the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE-IV) study43 is a 4-year two-by-two factorial placebo-controlled study evaluating the use of fluvastatin (Lescol) and bisoprolol (Zebeta, a beta-blocker) separately and together in patients who are older than 40 years, are undergoing elective noncardiac surgery, have an estimated risk of cardiovascular death of more than 1%, have not used statins previously, and do not have elevated cholesterol.
- Grant PJ, Kedia N. Should statins be discontinued preoperatively? IMPACT consults. Proceedings of the 2nd Annual Cleveland Clinic Perioperative Medicine Summit. Cleve Clin J Med 2006; 73 Electronic suppl 1:S9–S10.
- Lindenauer PK, Pekow P, Wang K, Gutierrez B, Benjamin EM. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- McFalls EO, Ward HB, Moritz TE, et al. Coronary-artery revascularization before elective major vascular surgery. N Engl J Med 2004; 351:2795–2804.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Ito MK, Talbert RL, Tsimikas S. Statin-associated pleiotropy: possible beneficial effects beyond cholesterol reduction. Pharmacotherapy 2006; 26:85S–97S.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999; 100:178–184.
- Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci 2004; 27:283–289.
- Osborne JA, Lento PH, Siegfried MR, Stahl GL, Fusman B, Lefer AM. Cardiovascular effects of acute hypercholesterolemia in rabbits. Reversal with lovastatin treatment. J Clin Invest 1989; 83:465–473.
- Sironi L, Cimino M, Guerrini U, et al. Treatment with statins after induction of focal ischemia in rats reduces the extent of brain damage. Arterioscler Thromb Vasc Biol 2003; 23:322–327.
- Fonarow GC, Wright RS, Spencer FA, et al. Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. Am J Cardiol 2005; 96:611–616.
- Heeschen C, Hamm CW, Laufs U, Snapinn S, Bohm M, White HD. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation 2002; 105:1446–1452.
- Kennedy J, Quan H, Buchan AM, Ghali WA, Feasby TE. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- McGirt MJ, Perler BA, Brooke BS, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors reduce the risk of perioperative stroke and mortality after carotid endarterectomy. J Vasc Surg 2005; 42:829–836.
- Hindler K, Shaw AD, Samuels J, Fulton S, Collard CD, Riedel B. Improved postoperative outcomes associated with preoperative statin therapy. Anesthesiology 2006; 105:1260–1272.
- Kapoor AS, Kanji H, Buckingham J, Devereaux PJ, McAlister FA. Strength of evidence for perioperative use of statins to reduce cardiovascular risk: systematic review of controlled studies. BMJ 2006; 333:1149.
- Durazzo AE, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Kertai MD, Boersma E, Westerhout CM, et al. A combination of statins and beta-blockers is independently associated with a reduction in the incidence of perioperative mortality and nonfatal myocardial infarction in patients undergoing abdominal aortic aneurysm surgery. Eur J Vasc Endovasc Surg 2004; 28:343–352.
- Poldermans D, Bax JJ, Kertai MD, et al. Statins are associated with a reduced incidence of perioperative mortality in patients undergoing major noncardiac vascular surgery. Circulation 2003; 107:1848–1851.
- Ward RP, Leeper NJ, Kirkpatrick JN, Lang RM, Sorrentino MJ, Williams KA. The effect of preoperative statin therapy on cardiovascular outcomes in patients undergoing infrainguinal vascular surgery. Int J Cardiol 2005; 104:264–268.
- O’Neil-Callahan K, Katsimaglis G, Tepper MR, et al. Statins decrease perioperative cardiac complications in patients undergoing non-cardiac vascular surgery: the Statins for Risk Reduction in Surgery (StaRRS) study. J Am Coll Cardiol 2005; 45:336–342.
- Abbruzzese TA, Havens J, Belkin M, et al. Statin therapy is associated with improved patency of autogenous infrainguinal bypass grafts. J Vasc Surg 2004; 39:1178–1185.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Landesberg G, Mosseri M, Wolf YG, et al. Preoperative thallium scanning, selective coronary revascularization, and long-term survival after major vascular surgery. Circulation 2003; 108:177–183.
- Schouten O, Kertai MD, Bax JJ, et al. Safety of perioperative statin use in high-risk patients undergoing major vascular surgery. Am J Cardiol 2005; 95:658–660.
- Le Manach Y, Godet G, Coriat P, et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg 2007; 104:1326–1333.
- Ali IS, Buth KJ. Preoperative statin use and outcomes following cardiac surgery. Int J Cardiol 2005; 103:12–18.
- Clark LL, Ikonomidis JS, Crawford FA, et al. Preoperative statin treatment is associated with reduced postoperative mortality and morbidity in patients undergoing cardiac surgery: an 8-year retrospective cohort study. J Thorac Cardiovasc Surg 2006; 131:679–685.
- Pan W, Pintar T, Anton J, Lee VV, Vaughn WK, Collard CD. Statins are associated with a reduced incidence of perioperative mortality after coronary artery bypass graft surgery. Circulation 2004; 110(suppl 2):II45–II49.
- Pascual DA, Arribas JM, Tornel PL, et al. Preoperative statin therapy and troponin T predict early complications of coronary artery surgery. Ann Thorac Surg 2006; 81:78–83.
- Dotani MI, Elnicki DM, Jain AC, Gibson CM. Effect of preoperative statin therapy and cardiac outcomes after coronary artery bypass grafting. Am J Cardiol 2000; 86:1128–1130.
- Liakopoulos OJ, Dorge H, Schmitto JD, Nagorsnik U, Grabedunkel J, Schoendube FA. Effects of preoperative statin therapy on cytokines after cardiac surgery. Thorac Cardiovasc Surg 2006; 54:250–254.
- Christenson JT. Preoperative lipid-control with simvastatin reduces the risk of postoperative thrombocytosis and thrombotic complications following CABG. Eur J Cardiothorac Surg 1999; 15:394–399.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 Guidelines on Perioperative Cardiovascular Evaluation and Care for Noncardiac Surgery. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:e418–e499.
- Schouten O, Poldermans D, Visser L, et al. Fluvastatin and bisoprolol for the reduction of perioperative cardiac mortality and morbidity in high-risk patients undergoing non-cardiac surgery: rationale and design of the DECREASE-IV study. Am Heart J 2004; 148:1047–1052.
- Amar D, Zhang H, Heerdt PM, Park B, Fleisher M, Thaler HT. Statin use is associated with a reduction in atrial fibrillation after noncardiac thoracic surgery independent of C-reactive protein. Chest 2005; 128:3421–3427.
- Grant PJ, Kedia N. Should statins be discontinued preoperatively? IMPACT consults. Proceedings of the 2nd Annual Cleveland Clinic Perioperative Medicine Summit. Cleve Clin J Med 2006; 73 Electronic suppl 1:S9–S10.
- Lindenauer PK, Pekow P, Wang K, Gutierrez B, Benjamin EM. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- McFalls EO, Ward HB, Moritz TE, et al. Coronary-artery revascularization before elective major vascular surgery. N Engl J Med 2004; 351:2795–2804.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Ito MK, Talbert RL, Tsimikas S. Statin-associated pleiotropy: possible beneficial effects beyond cholesterol reduction. Pharmacotherapy 2006; 26:85S–97S.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999; 100:178–184.
- Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci 2004; 27:283–289.
- Osborne JA, Lento PH, Siegfried MR, Stahl GL, Fusman B, Lefer AM. Cardiovascular effects of acute hypercholesterolemia in rabbits. Reversal with lovastatin treatment. J Clin Invest 1989; 83:465–473.
- Sironi L, Cimino M, Guerrini U, et al. Treatment with statins after induction of focal ischemia in rats reduces the extent of brain damage. Arterioscler Thromb Vasc Biol 2003; 23:322–327.
- Fonarow GC, Wright RS, Spencer FA, et al. Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. Am J Cardiol 2005; 96:611–616.
- Heeschen C, Hamm CW, Laufs U, Snapinn S, Bohm M, White HD. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation 2002; 105:1446–1452.
- Kennedy J, Quan H, Buchan AM, Ghali WA, Feasby TE. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- McGirt MJ, Perler BA, Brooke BS, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors reduce the risk of perioperative stroke and mortality after carotid endarterectomy. J Vasc Surg 2005; 42:829–836.
- Hindler K, Shaw AD, Samuels J, Fulton S, Collard CD, Riedel B. Improved postoperative outcomes associated with preoperative statin therapy. Anesthesiology 2006; 105:1260–1272.
- Kapoor AS, Kanji H, Buckingham J, Devereaux PJ, McAlister FA. Strength of evidence for perioperative use of statins to reduce cardiovascular risk: systematic review of controlled studies. BMJ 2006; 333:1149.
- Durazzo AE, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Kertai MD, Boersma E, Westerhout CM, et al. A combination of statins and beta-blockers is independently associated with a reduction in the incidence of perioperative mortality and nonfatal myocardial infarction in patients undergoing abdominal aortic aneurysm surgery. Eur J Vasc Endovasc Surg 2004; 28:343–352.
- Poldermans D, Bax JJ, Kertai MD, et al. Statins are associated with a reduced incidence of perioperative mortality in patients undergoing major noncardiac vascular surgery. Circulation 2003; 107:1848–1851.
- Ward RP, Leeper NJ, Kirkpatrick JN, Lang RM, Sorrentino MJ, Williams KA. The effect of preoperative statin therapy on cardiovascular outcomes in patients undergoing infrainguinal vascular surgery. Int J Cardiol 2005; 104:264–268.
- O’Neil-Callahan K, Katsimaglis G, Tepper MR, et al. Statins decrease perioperative cardiac complications in patients undergoing non-cardiac vascular surgery: the Statins for Risk Reduction in Surgery (StaRRS) study. J Am Coll Cardiol 2005; 45:336–342.
- Abbruzzese TA, Havens J, Belkin M, et al. Statin therapy is associated with improved patency of autogenous infrainguinal bypass grafts. J Vasc Surg 2004; 39:1178–1185.
- Boersma E, Poldermans D, Bax JJ, et al. Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Landesberg G, Mosseri M, Wolf YG, et al. Preoperative thallium scanning, selective coronary revascularization, and long-term survival after major vascular surgery. Circulation 2003; 108:177–183.
- Schouten O, Kertai MD, Bax JJ, et al. Safety of perioperative statin use in high-risk patients undergoing major vascular surgery. Am J Cardiol 2005; 95:658–660.
- Le Manach Y, Godet G, Coriat P, et al. The impact of postoperative discontinuation or continuation of chronic statin therapy on cardiac outcome after major vascular surgery. Anesth Analg 2007; 104:1326–1333.
- Ali IS, Buth KJ. Preoperative statin use and outcomes following cardiac surgery. Int J Cardiol 2005; 103:12–18.
- Clark LL, Ikonomidis JS, Crawford FA, et al. Preoperative statin treatment is associated with reduced postoperative mortality and morbidity in patients undergoing cardiac surgery: an 8-year retrospective cohort study. J Thorac Cardiovasc Surg 2006; 131:679–685.
- Pan W, Pintar T, Anton J, Lee VV, Vaughn WK, Collard CD. Statins are associated with a reduced incidence of perioperative mortality after coronary artery bypass graft surgery. Circulation 2004; 110(suppl 2):II45–II49.
- Pascual DA, Arribas JM, Tornel PL, et al. Preoperative statin therapy and troponin T predict early complications of coronary artery surgery. Ann Thorac Surg 2006; 81:78–83.
- Dotani MI, Elnicki DM, Jain AC, Gibson CM. Effect of preoperative statin therapy and cardiac outcomes after coronary artery bypass grafting. Am J Cardiol 2000; 86:1128–1130.
- Liakopoulos OJ, Dorge H, Schmitto JD, Nagorsnik U, Grabedunkel J, Schoendube FA. Effects of preoperative statin therapy on cytokines after cardiac surgery. Thorac Cardiovasc Surg 2006; 54:250–254.
- Christenson JT. Preoperative lipid-control with simvastatin reduces the risk of postoperative thrombocytosis and thrombotic complications following CABG. Eur J Cardiothorac Surg 1999; 15:394–399.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 Guidelines on Perioperative Cardiovascular Evaluation and Care for Noncardiac Surgery. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation 2007; 116:e418–e499.
- Schouten O, Poldermans D, Visser L, et al. Fluvastatin and bisoprolol for the reduction of perioperative cardiac mortality and morbidity in high-risk patients undergoing non-cardiac surgery: rationale and design of the DECREASE-IV study. Am Heart J 2004; 148:1047–1052.
- Amar D, Zhang H, Heerdt PM, Park B, Fleisher M, Thaler HT. Statin use is associated with a reduction in atrial fibrillation after noncardiac thoracic surgery independent of C-reactive protein. Chest 2005; 128:3421–3427.
KEY POINTS
- Experiments in animals suggest that statins, given shortly before or after a cardiovascular event, confer benefit before any changes in lipids are measurable.
- Retrospective and prospective studies indicate that patients with either acute myocardial infarction or acute coronary syndrome who are already receiving statins should not have them stopped, and those who had not been receiving statins should receive them immediately.
- Most patients undergoing coronary artery bypass grafting or noncardiac vascular surgery should already be receiving a statin. These drugs can also be considered in patients undergoing intermediate-risk nonvascular surgery. Patients who have been receiving statins prior to surgery should not have them stopped for surgery.
Anemia of chronic kidney disease: When normalcy becomes undesirable
The last several years have seen increased debate over the appropriate hemoglobin target range when using erythropoiesis-stimulating agents (ESAs) to treat the anemia of chronic kidney disease and kidney failure. But several recent studies have raised alarms, and in November 2006 the US Food and Drug Administration (FDA) issued a new warning regarding the use of ESAs in renal disease.
For a perspective on the use of erythropoiesis-stimulating agents in cancer patients, see the related editorial.
This article will discuss the history of ESAs and the current guidelines for their use. ESAs are also indicated to treat anemia in patients undergoing cancer chemotherapy or surgery, but those uses will not be discussed in this article.
THE BENEFITS OF ESAs
The first ESA, Epogen, was approved by the FDA in 1989 to treat anemia associated with kidney disease.
Since then, ESAs have made a revolutionary change in the care of patients with kidney failure by allowing them to avoid blood transfusions, which were the norm, and by improving the quality of life, although the evidence for the latter is less compelling.1 The benefits of avoiding the use of blood products include a lower risk of reactions, lower cost, and avoiding sensitization of the human lymphocyte antigen (HLA) system in kidney transplant candidates.
To date, however, no randomized, placebo-controlled clinical trial with adequate power to detect a reduction in adverse clinical outcomes (hospitalizations, nonfatal cardiovascular events, or deaths) has assessed the effect of raising hemoglobin levels with ESAs in patients with chronic kidney disease or end-stage renal disease. Nevertheless, several small studies have shown ESAs to have favorable effects on surrogate end points, and an impressive amount of observational data have shown higher survival rates with higher hemoglobin levels.2–6
HOW HIGH SHOULD THE HEMOGLOBIN BE RAISED?
During ESA treatment, the FDA first approved a target hemoglobin range of 10 to 11 g/dL, and subsequently changed it to 10 to 12 g/dL in 1994. The National Kidney Foundation, in its 1997 practice guidelines, endorsed a target range of 11 to 12 g/dL.
The US Normal Hematocrit Study (1998) struck a sour note. In this study, 1,233 dialysis patients with cardiovascular disease were randomized to either a low hematocrit target (33%) or a normal hematocrit target (42%). The trial was stopped early when the investigators recognized that more patients in the normal-hematocrit group had died, that the difference was nearing statistical significance, and that continuing the study was unlikely to reveal a benefit in the normal-hematocrit group. Also of note, the incidence of vascular access thrombosis was higher in the normal-hematocrit group.8
In 2006 the National Kidney Foundation modified its 1997 guidelines, suggesting an upper hemoglobin boundary of 13 g/dL. But in early 2007 it retreated to a hemoglobin target range of 11–12 g/dL,9 after the simultaneous publication of two randomized controlled trials that found no improved outcomes with hemoglobin normalization, and some evidence of harm.10,11
The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial randomized predialysis patients to a hemoglobin goal of either 11.3 g/dL or 13.5 g/dL. The trial was terminated early because the likelihood of benefit with the high hemoglobin goal was low. In fact, the higher-hemoglobin group had a higher incidence of the primary end point, ie, the composite of death, stroke, myocardial infarction, and hospitalization for congestive heart failure. Death and hospitalization for congestive heart failure were the main drivers of the difference in the composite end point between the groups. Quality of life was no better with the higher goal than with the lower goal.10
The Cardiovascular Risk Reduction by Early Anemia Treatment With Epoetin Beta (CREATE) trial11 found that the risk of cardiovascular events in predialysis patients was no lower when anemia was completely corrected (target hemoglobin range 13.0–15.0 g/dL) than with a goal of 10.5 to 11.5 g/dL. Moreover, renal function declined faster in the higher-goal group than in the lower-goal group. However, this study did show higher quality-of-life scores in the group with the higher hemoglobin goal.11
AN FDA ALERT
On November 16, 2006, the FDA issued an alert and required that ESA product labeling include a new boxed warning with the following information12:
- Use the lowest dose of an ESA (Procrit, Epogen, or Aranesp) that will gradually raise the hemoglobin concentration to the lowest level sufficient to avoid the need for blood transfusion.
- ESAs should not be given to treat symptoms of anemia or poor quality of life.
- Maintain the hemoglobin level in the target range of 10 to 12 g/dL.
- Decrease the dose if the hemoglobin level increases by more than 1 g/dL in any 2-week period.
ANOTHER LOOK AT THE DATA
In post hoc analyses, data from the US Normal Hematocrit and CHOIR studies were analyzed on an “as-treated” basis instead of on an intention-to-treat basis as originally reported.13,14 Although the original studies found no survival advantage (and perhaps harm) with higher hemoglobin targets (ie, by intention-to-treat analysis), when the investigators looked at the actual hemoglobin levels achieved, they found that event rates were higher with low hemoglobin levels.
Such discordant findings highlight the importance of randomized experimental designs to avoid bias due to confounding factors (measured and unmeasured) linked to both hemoglobin level and outcome. To reconcile the above findings, we offer the following observations:
- In each treatment group, event rates were higher among those who responded poorly to ESAs (hyporesponders). This finding undermines the intuitive assumption that higher achieved hemoglobin levels were causing volume-related events (congestive heart failure or pulmonary edema) and thrombotic events. Of note, rapid changes in hemoglobin levels in either direction further increased the frequency of events among hyporesponders (which might be associated with the more aggressive algorithm needed in the higher target group).
- Within each treatment group, the difference in event rates is unlikely to be explained by the variation in hemoglobin within its narrow range. Rather, it was mostly due to a higher burden of disease among the hyporesponders. This problem—called targeting bias—is peculiar to therapies that are adjusted according to a target level, eg, of serum hemoglobin.15 Therefore, any association of mortality with achieved hemoglobin within the individual target hemoglobin group is more likely due to other factors such as patient comorbidities.
- Patients assigned to the higher hemoglobin targets received more than just higher doses of ESAs: they also got more of other interventions such as intravenous iron supplementation. Therefore, the results of the trials reflect not only the target level achieved but also the independent effects of the study drug, the co-interventions, and the treatment algorithm.
TAKE-HOME POINTS
Partial correction of the anemia associated with kidney disease reduces transfusion requirements, but normalizing the hemoglobin level does not confer survival benefit and may be harmful. In accordance with the FDA recommendations and the available evidence, we agree that the goal for treating anemia associated with kidney disease should be partial correction: the upper boundary of hemoglobin should be 12 g/dL. However, transient trespasses beyond the upper boundary in day-to-day clinical practice should not trigger a panic response in the health care provider (as seen with hyperkalemia, for instance). Rather, they should result in appropriate and timely treatment adjustments.
Further efforts should explore the merits of treatment algorithms that minimize rapid changes in hemoglobin levels, as well as dose limitation of ESAs and co-interventions among hyporesponders.
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Ma JZ, Ebben J, Xia H, Collins AJ. Hematocrit level and associated mortality in hemodialysis patients. J Am Soc Nephrol 1999; 10:610–619.
- Xue JL, St Peter WL, Ebben JP, Everson SE, Collins AJ. Anemia treatment in the pre-ESRD period and associated mortality in elderly patients. Am J Kidney Dis 2002; 40:1153–1161.
- Levin A, Thompson CR, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis 1999; 34:125–134.
- Gouva C, Nikolopoulos P, Ioannidis JP, Siamopoulos KC. Treating anemia early in renal failure patients slows the decline of renal function: a randomized controlled trial. Kidney Int 2004; 66:753–760.
- Ritz E, Laville M, Bilous RW, et al. Target level for hemoglobin correction in patients with diabetes and CKD: primary results of the Anemia Correction in Diabetes (ACORD) Study. Am J Kidney Dis 2007; 49:194–207.
- KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis 2006; 47 suppl 3:S11–S145.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis 2007; 50:471–530.
- Singh AK, Szczech L, Tang KL, et al; CHOIR investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- US Food and Drug Administration. www.fda.gov/cder/drug/InfoSheets/HCP/RHE2007HCP.htm. Accessed 2/5/08.
- US Food and Drug Administration Advisory Committee briefing document. www.fda.gov/ohrms/dockets/AC/07/briefing/2007-4315b1-01-FDA.pdf. Accessed 2/5/08.
- Macdougall IC, Ritz E. The Normal Haematocrit Trial in patients with cardiac disease: are we any the less confused about target haemoglobin? Nephrol Dial Transplant 1998; 13:3030–3033.
- Greene T, Daugirdas J, Depner T, et al. Association of achieved dialysis dose with mortality in the hemodialysis study: an example of “dose-targeting bias.” J Am Soc Nephrol 2005; 16:3371–3380.
The last several years have seen increased debate over the appropriate hemoglobin target range when using erythropoiesis-stimulating agents (ESAs) to treat the anemia of chronic kidney disease and kidney failure. But several recent studies have raised alarms, and in November 2006 the US Food and Drug Administration (FDA) issued a new warning regarding the use of ESAs in renal disease.
For a perspective on the use of erythropoiesis-stimulating agents in cancer patients, see the related editorial.
This article will discuss the history of ESAs and the current guidelines for their use. ESAs are also indicated to treat anemia in patients undergoing cancer chemotherapy or surgery, but those uses will not be discussed in this article.
THE BENEFITS OF ESAs
The first ESA, Epogen, was approved by the FDA in 1989 to treat anemia associated with kidney disease.
Since then, ESAs have made a revolutionary change in the care of patients with kidney failure by allowing them to avoid blood transfusions, which were the norm, and by improving the quality of life, although the evidence for the latter is less compelling.1 The benefits of avoiding the use of blood products include a lower risk of reactions, lower cost, and avoiding sensitization of the human lymphocyte antigen (HLA) system in kidney transplant candidates.
To date, however, no randomized, placebo-controlled clinical trial with adequate power to detect a reduction in adverse clinical outcomes (hospitalizations, nonfatal cardiovascular events, or deaths) has assessed the effect of raising hemoglobin levels with ESAs in patients with chronic kidney disease or end-stage renal disease. Nevertheless, several small studies have shown ESAs to have favorable effects on surrogate end points, and an impressive amount of observational data have shown higher survival rates with higher hemoglobin levels.2–6
HOW HIGH SHOULD THE HEMOGLOBIN BE RAISED?
During ESA treatment, the FDA first approved a target hemoglobin range of 10 to 11 g/dL, and subsequently changed it to 10 to 12 g/dL in 1994. The National Kidney Foundation, in its 1997 practice guidelines, endorsed a target range of 11 to 12 g/dL.
The US Normal Hematocrit Study (1998) struck a sour note. In this study, 1,233 dialysis patients with cardiovascular disease were randomized to either a low hematocrit target (33%) or a normal hematocrit target (42%). The trial was stopped early when the investigators recognized that more patients in the normal-hematocrit group had died, that the difference was nearing statistical significance, and that continuing the study was unlikely to reveal a benefit in the normal-hematocrit group. Also of note, the incidence of vascular access thrombosis was higher in the normal-hematocrit group.8
In 2006 the National Kidney Foundation modified its 1997 guidelines, suggesting an upper hemoglobin boundary of 13 g/dL. But in early 2007 it retreated to a hemoglobin target range of 11–12 g/dL,9 after the simultaneous publication of two randomized controlled trials that found no improved outcomes with hemoglobin normalization, and some evidence of harm.10,11
The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial randomized predialysis patients to a hemoglobin goal of either 11.3 g/dL or 13.5 g/dL. The trial was terminated early because the likelihood of benefit with the high hemoglobin goal was low. In fact, the higher-hemoglobin group had a higher incidence of the primary end point, ie, the composite of death, stroke, myocardial infarction, and hospitalization for congestive heart failure. Death and hospitalization for congestive heart failure were the main drivers of the difference in the composite end point between the groups. Quality of life was no better with the higher goal than with the lower goal.10
The Cardiovascular Risk Reduction by Early Anemia Treatment With Epoetin Beta (CREATE) trial11 found that the risk of cardiovascular events in predialysis patients was no lower when anemia was completely corrected (target hemoglobin range 13.0–15.0 g/dL) than with a goal of 10.5 to 11.5 g/dL. Moreover, renal function declined faster in the higher-goal group than in the lower-goal group. However, this study did show higher quality-of-life scores in the group with the higher hemoglobin goal.11
AN FDA ALERT
On November 16, 2006, the FDA issued an alert and required that ESA product labeling include a new boxed warning with the following information12:
- Use the lowest dose of an ESA (Procrit, Epogen, or Aranesp) that will gradually raise the hemoglobin concentration to the lowest level sufficient to avoid the need for blood transfusion.
- ESAs should not be given to treat symptoms of anemia or poor quality of life.
- Maintain the hemoglobin level in the target range of 10 to 12 g/dL.
- Decrease the dose if the hemoglobin level increases by more than 1 g/dL in any 2-week period.
ANOTHER LOOK AT THE DATA
In post hoc analyses, data from the US Normal Hematocrit and CHOIR studies were analyzed on an “as-treated” basis instead of on an intention-to-treat basis as originally reported.13,14 Although the original studies found no survival advantage (and perhaps harm) with higher hemoglobin targets (ie, by intention-to-treat analysis), when the investigators looked at the actual hemoglobin levels achieved, they found that event rates were higher with low hemoglobin levels.
Such discordant findings highlight the importance of randomized experimental designs to avoid bias due to confounding factors (measured and unmeasured) linked to both hemoglobin level and outcome. To reconcile the above findings, we offer the following observations:
- In each treatment group, event rates were higher among those who responded poorly to ESAs (hyporesponders). This finding undermines the intuitive assumption that higher achieved hemoglobin levels were causing volume-related events (congestive heart failure or pulmonary edema) and thrombotic events. Of note, rapid changes in hemoglobin levels in either direction further increased the frequency of events among hyporesponders (which might be associated with the more aggressive algorithm needed in the higher target group).
- Within each treatment group, the difference in event rates is unlikely to be explained by the variation in hemoglobin within its narrow range. Rather, it was mostly due to a higher burden of disease among the hyporesponders. This problem—called targeting bias—is peculiar to therapies that are adjusted according to a target level, eg, of serum hemoglobin.15 Therefore, any association of mortality with achieved hemoglobin within the individual target hemoglobin group is more likely due to other factors such as patient comorbidities.
- Patients assigned to the higher hemoglobin targets received more than just higher doses of ESAs: they also got more of other interventions such as intravenous iron supplementation. Therefore, the results of the trials reflect not only the target level achieved but also the independent effects of the study drug, the co-interventions, and the treatment algorithm.
TAKE-HOME POINTS
Partial correction of the anemia associated with kidney disease reduces transfusion requirements, but normalizing the hemoglobin level does not confer survival benefit and may be harmful. In accordance with the FDA recommendations and the available evidence, we agree that the goal for treating anemia associated with kidney disease should be partial correction: the upper boundary of hemoglobin should be 12 g/dL. However, transient trespasses beyond the upper boundary in day-to-day clinical practice should not trigger a panic response in the health care provider (as seen with hyperkalemia, for instance). Rather, they should result in appropriate and timely treatment adjustments.
Further efforts should explore the merits of treatment algorithms that minimize rapid changes in hemoglobin levels, as well as dose limitation of ESAs and co-interventions among hyporesponders.
The last several years have seen increased debate over the appropriate hemoglobin target range when using erythropoiesis-stimulating agents (ESAs) to treat the anemia of chronic kidney disease and kidney failure. But several recent studies have raised alarms, and in November 2006 the US Food and Drug Administration (FDA) issued a new warning regarding the use of ESAs in renal disease.
For a perspective on the use of erythropoiesis-stimulating agents in cancer patients, see the related editorial.
This article will discuss the history of ESAs and the current guidelines for their use. ESAs are also indicated to treat anemia in patients undergoing cancer chemotherapy or surgery, but those uses will not be discussed in this article.
THE BENEFITS OF ESAs
The first ESA, Epogen, was approved by the FDA in 1989 to treat anemia associated with kidney disease.
Since then, ESAs have made a revolutionary change in the care of patients with kidney failure by allowing them to avoid blood transfusions, which were the norm, and by improving the quality of life, although the evidence for the latter is less compelling.1 The benefits of avoiding the use of blood products include a lower risk of reactions, lower cost, and avoiding sensitization of the human lymphocyte antigen (HLA) system in kidney transplant candidates.
To date, however, no randomized, placebo-controlled clinical trial with adequate power to detect a reduction in adverse clinical outcomes (hospitalizations, nonfatal cardiovascular events, or deaths) has assessed the effect of raising hemoglobin levels with ESAs in patients with chronic kidney disease or end-stage renal disease. Nevertheless, several small studies have shown ESAs to have favorable effects on surrogate end points, and an impressive amount of observational data have shown higher survival rates with higher hemoglobin levels.2–6
HOW HIGH SHOULD THE HEMOGLOBIN BE RAISED?
During ESA treatment, the FDA first approved a target hemoglobin range of 10 to 11 g/dL, and subsequently changed it to 10 to 12 g/dL in 1994. The National Kidney Foundation, in its 1997 practice guidelines, endorsed a target range of 11 to 12 g/dL.
The US Normal Hematocrit Study (1998) struck a sour note. In this study, 1,233 dialysis patients with cardiovascular disease were randomized to either a low hematocrit target (33%) or a normal hematocrit target (42%). The trial was stopped early when the investigators recognized that more patients in the normal-hematocrit group had died, that the difference was nearing statistical significance, and that continuing the study was unlikely to reveal a benefit in the normal-hematocrit group. Also of note, the incidence of vascular access thrombosis was higher in the normal-hematocrit group.8
In 2006 the National Kidney Foundation modified its 1997 guidelines, suggesting an upper hemoglobin boundary of 13 g/dL. But in early 2007 it retreated to a hemoglobin target range of 11–12 g/dL,9 after the simultaneous publication of two randomized controlled trials that found no improved outcomes with hemoglobin normalization, and some evidence of harm.10,11
The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial randomized predialysis patients to a hemoglobin goal of either 11.3 g/dL or 13.5 g/dL. The trial was terminated early because the likelihood of benefit with the high hemoglobin goal was low. In fact, the higher-hemoglobin group had a higher incidence of the primary end point, ie, the composite of death, stroke, myocardial infarction, and hospitalization for congestive heart failure. Death and hospitalization for congestive heart failure were the main drivers of the difference in the composite end point between the groups. Quality of life was no better with the higher goal than with the lower goal.10
The Cardiovascular Risk Reduction by Early Anemia Treatment With Epoetin Beta (CREATE) trial11 found that the risk of cardiovascular events in predialysis patients was no lower when anemia was completely corrected (target hemoglobin range 13.0–15.0 g/dL) than with a goal of 10.5 to 11.5 g/dL. Moreover, renal function declined faster in the higher-goal group than in the lower-goal group. However, this study did show higher quality-of-life scores in the group with the higher hemoglobin goal.11
AN FDA ALERT
On November 16, 2006, the FDA issued an alert and required that ESA product labeling include a new boxed warning with the following information12:
- Use the lowest dose of an ESA (Procrit, Epogen, or Aranesp) that will gradually raise the hemoglobin concentration to the lowest level sufficient to avoid the need for blood transfusion.
- ESAs should not be given to treat symptoms of anemia or poor quality of life.
- Maintain the hemoglobin level in the target range of 10 to 12 g/dL.
- Decrease the dose if the hemoglobin level increases by more than 1 g/dL in any 2-week period.
ANOTHER LOOK AT THE DATA
In post hoc analyses, data from the US Normal Hematocrit and CHOIR studies were analyzed on an “as-treated” basis instead of on an intention-to-treat basis as originally reported.13,14 Although the original studies found no survival advantage (and perhaps harm) with higher hemoglobin targets (ie, by intention-to-treat analysis), when the investigators looked at the actual hemoglobin levels achieved, they found that event rates were higher with low hemoglobin levels.
Such discordant findings highlight the importance of randomized experimental designs to avoid bias due to confounding factors (measured and unmeasured) linked to both hemoglobin level and outcome. To reconcile the above findings, we offer the following observations:
- In each treatment group, event rates were higher among those who responded poorly to ESAs (hyporesponders). This finding undermines the intuitive assumption that higher achieved hemoglobin levels were causing volume-related events (congestive heart failure or pulmonary edema) and thrombotic events. Of note, rapid changes in hemoglobin levels in either direction further increased the frequency of events among hyporesponders (which might be associated with the more aggressive algorithm needed in the higher target group).
- Within each treatment group, the difference in event rates is unlikely to be explained by the variation in hemoglobin within its narrow range. Rather, it was mostly due to a higher burden of disease among the hyporesponders. This problem—called targeting bias—is peculiar to therapies that are adjusted according to a target level, eg, of serum hemoglobin.15 Therefore, any association of mortality with achieved hemoglobin within the individual target hemoglobin group is more likely due to other factors such as patient comorbidities.
- Patients assigned to the higher hemoglobin targets received more than just higher doses of ESAs: they also got more of other interventions such as intravenous iron supplementation. Therefore, the results of the trials reflect not only the target level achieved but also the independent effects of the study drug, the co-interventions, and the treatment algorithm.
TAKE-HOME POINTS
Partial correction of the anemia associated with kidney disease reduces transfusion requirements, but normalizing the hemoglobin level does not confer survival benefit and may be harmful. In accordance with the FDA recommendations and the available evidence, we agree that the goal for treating anemia associated with kidney disease should be partial correction: the upper boundary of hemoglobin should be 12 g/dL. However, transient trespasses beyond the upper boundary in day-to-day clinical practice should not trigger a panic response in the health care provider (as seen with hyperkalemia, for instance). Rather, they should result in appropriate and timely treatment adjustments.
Further efforts should explore the merits of treatment algorithms that minimize rapid changes in hemoglobin levels, as well as dose limitation of ESAs and co-interventions among hyporesponders.
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Ma JZ, Ebben J, Xia H, Collins AJ. Hematocrit level and associated mortality in hemodialysis patients. J Am Soc Nephrol 1999; 10:610–619.
- Xue JL, St Peter WL, Ebben JP, Everson SE, Collins AJ. Anemia treatment in the pre-ESRD period and associated mortality in elderly patients. Am J Kidney Dis 2002; 40:1153–1161.
- Levin A, Thompson CR, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis 1999; 34:125–134.
- Gouva C, Nikolopoulos P, Ioannidis JP, Siamopoulos KC. Treating anemia early in renal failure patients slows the decline of renal function: a randomized controlled trial. Kidney Int 2004; 66:753–760.
- Ritz E, Laville M, Bilous RW, et al. Target level for hemoglobin correction in patients with diabetes and CKD: primary results of the Anemia Correction in Diabetes (ACORD) Study. Am J Kidney Dis 2007; 49:194–207.
- KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis 2006; 47 suppl 3:S11–S145.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis 2007; 50:471–530.
- Singh AK, Szczech L, Tang KL, et al; CHOIR investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- US Food and Drug Administration. www.fda.gov/cder/drug/InfoSheets/HCP/RHE2007HCP.htm. Accessed 2/5/08.
- US Food and Drug Administration Advisory Committee briefing document. www.fda.gov/ohrms/dockets/AC/07/briefing/2007-4315b1-01-FDA.pdf. Accessed 2/5/08.
- Macdougall IC, Ritz E. The Normal Haematocrit Trial in patients with cardiac disease: are we any the less confused about target haemoglobin? Nephrol Dial Transplant 1998; 13:3030–3033.
- Greene T, Daugirdas J, Depner T, et al. Association of achieved dialysis dose with mortality in the hemodialysis study: an example of “dose-targeting bias.” J Am Soc Nephrol 2005; 16:3371–3380.
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Ma JZ, Ebben J, Xia H, Collins AJ. Hematocrit level and associated mortality in hemodialysis patients. J Am Soc Nephrol 1999; 10:610–619.
- Xue JL, St Peter WL, Ebben JP, Everson SE, Collins AJ. Anemia treatment in the pre-ESRD period and associated mortality in elderly patients. Am J Kidney Dis 2002; 40:1153–1161.
- Levin A, Thompson CR, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis 1999; 34:125–134.
- Gouva C, Nikolopoulos P, Ioannidis JP, Siamopoulos KC. Treating anemia early in renal failure patients slows the decline of renal function: a randomized controlled trial. Kidney Int 2004; 66:753–760.
- Ritz E, Laville M, Bilous RW, et al. Target level for hemoglobin correction in patients with diabetes and CKD: primary results of the Anemia Correction in Diabetes (ACORD) Study. Am J Kidney Dis 2007; 49:194–207.
- KDOQI clinical practice guidelines and clinical practice recommendations for anemia in chronic kidney disease. Am J Kidney Dis 2006; 47 suppl 3:S11–S145.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- KDOQI clinical practice guideline and clinical practice recommendations for anemia in chronic kidney disease: 2007 update of hemoglobin target. Am J Kidney Dis 2007; 50:471–530.
- Singh AK, Szczech L, Tang KL, et al; CHOIR investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- US Food and Drug Administration. www.fda.gov/cder/drug/InfoSheets/HCP/RHE2007HCP.htm. Accessed 2/5/08.
- US Food and Drug Administration Advisory Committee briefing document. www.fda.gov/ohrms/dockets/AC/07/briefing/2007-4315b1-01-FDA.pdf. Accessed 2/5/08.
- Macdougall IC, Ritz E. The Normal Haematocrit Trial in patients with cardiac disease: are we any the less confused about target haemoglobin? Nephrol Dial Transplant 1998; 13:3030–3033.
- Greene T, Daugirdas J, Depner T, et al. Association of achieved dialysis dose with mortality in the hemodialysis study: an example of “dose-targeting bias.” J Am Soc Nephrol 2005; 16:3371–3380.
KEY POINTS
- ESAs reduce the need for blood transfusions and possibly improve quality of life.
- It is unclear if higher hemoglobin levels per se actually caused the adverse events in these trials. Event rates were highest in patients who responded poorly to ESAs.
- We concur with the FDA’s recommendation that the hemoglobin level be raised to no higher than 12 g/dL with ESAs in patients with chronic kidney disease or renal failure.
- Transient excursions of the hemoglobin level above 12 g/dL should not be a cause for panic. Rather, the next ESA dose should be reduced.
What role will ‘gliptins’ play in glycemic control?
The “gliptins”—the nickname for dipeptidyl peptidase 4 (DPP-4) inhibitors—are one of the newest classes of drugs for the treatment of type 2 diabetes mellitus.
These drugs work by prolonging the action of gut hormones called incretins, which boost insulin levels. The greatest advantage of the gliptins appears to be their ability to stimulate insulin production with little risk of corresponding hypoglycemia.
Sitagliptin (Januvia), the first commercially available DPP-4 inhibitor, has been approved by the US Food and Drug Administration (FDA) and is currently in clinical use, and vildagliptin (Galvus) awaits FDA approval at the time of this writing. Other drugs of this class are in development.
However, because these drugs are so new, a number of questions remain about their use. In this article, we discuss the rationale behind gliptin drugs, the evidence to date on their use alone or in combination with current oral hypoglycemic drugs (and even with insulin), and when and how to use them in daily practice.
THE NEED FOR MORE EFFECTIVE DIABETES TREATMENT
As the number of patients with type 2 diabetes continues its steep and steady rise,1,2 much work has gone into studying treatment goals and how to achieve them. Although experts generally agree on glycemic goals,3 we currently fail to achieve those goals in close to two-thirds of patients: only 37% have a hemo-globin A1c (HbA1c) value at or below the goal of 7%, and the same number have levels exceeding 8%.4
Part of the problem is that treatment regimens are not adjusted in a timely fashion. In a prescribing database of almost 4,000 patients with type 2 diabetes,5 the mean time from the first HbA1c reading above 8% to an actual change in therapy was about 15 months for those taking metformin (Glucophage) alone, and 21 months for those taking a sulfonylurea alone. Another part of the problem is that, on average, patients with an HbA1c of 8.0% to 8.9% can expect only a 0.6% lowering with the addition of one agent.6 Clearly, we need new pharmacologic approaches and new management paradigms. One new approach is the use of gliptins.
HOW GLIPTINS WORK
Incretins promote insulin secretion
We have known for more than 20 years that insulin levels rise considerably higher in response to an oral glucose load than to an intravenous glucose infusion, even though the plasma glucose concentrations may be similar.7 This phenomenon involves a myriad of neural and nutritional factors, but the gut hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) appear to be key.
These peptides—called incretins—have a high degree of homology, and both promote insulin secretion. However, GLP-1, produced by the L cells of the ileum and colon, inhibits glucagon secretion and slows gastric emptying, whereas GIP, secreted from the K cells of the duodenum, has no effect on glucagon and little effect on gastric emptying. Both peptides appear to promote pancreatic beta cell growth and survival,8,9 an effect that in theory might allow us to slow the progressive loss of insulin secretory capacity in type 2 diabetes.
Furthermore, the effect of GLP-1 on insulin secretion depends on the plasma glucose concentration, with a greater insulin secretory effect at higher glucose levels and minimal effect at euglycemic levels.10 This phenomenon suggests that drugs that boost GLP-1 activity should not cause the troublesome hypoglycemia typically seen in patients taking insulin, insulin secretagogues, sulfonyl-ureas, or the meglitinides repaglinide (Prandin) or nateglinide (Starlix). Studies of combination treatment with metformin and the GLP-1 receptor agonist exenatide (Byetta) have shown little risk of hypoglycemia,11 offering evidence favoring this conjecture.
Inhibition of DPP-4 boosts incretin action
The challenge for creating treatments that take advantage of the beneficial effects of GLP-1 and GIP is that they have very short physiologic half-lives, ie, less than 10 minutes. GLP-1 and GIP both have two N-terminal amino acids that are quickly cleaved by DPP-4,12 an enzyme present in the circulation13 and on endothelial cells.14
Currently, there are two classes of drugs based on incretins. One class, the incretin mimetics or GLP-1 receptor agonists, includes drugs that mimic the effect of GLP-1 but are not so quickly degraded by DPP-4. Examples of these drugs are exenatide, which is currently FDA-approved, and liraglutide, which is not yet approved.
On the other hand, by inhibiting the cleaving action of DPP-4, the gliptins can prolong the half-life of endogenous GLP-1, increasing its physiologic effects.
Studies comparing gliptins with GLP-1 receptor agonists are only at the preclinical phase. Liraglutide showed an antiglycemic effect similar to that of vildagliptin in an animal model of glucose intolerance.15 This and other16,17 preclinical studies have shown evidence of improved beta cell growth and survival with DPP-4 inhibitor treatment, to an extent similar to that reported with thiazo-lidinediones, whereas sulfonylureas show no evidence either of increase in beta cells or of improved intrinsic beta cell secretory function in these models. Of course, animal studies can only be cautiously extrapolated to potential effects in humans, and it is uncertain whether such benefits will occur with the therapeutic use of DPP-4 inhibitors.
RANDOMIZED CLINICAL TRIALS OF SITAGLIPTIN
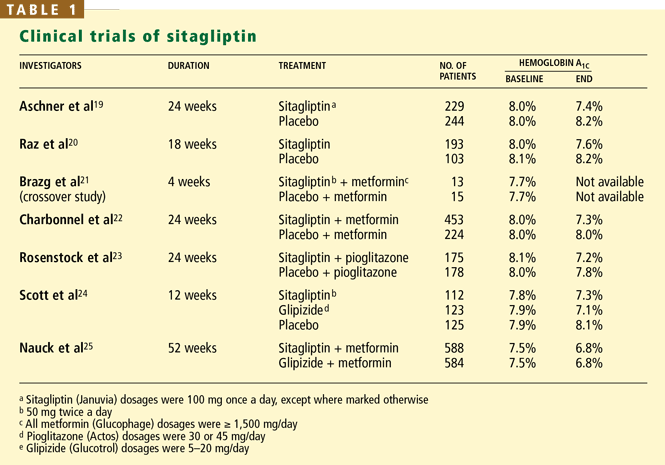
Sitagliptin is effective when used by itself,reducing a baseline HbA1c level of about 8% by 0.6% to 0.8%,19,20,24 and is similarly effective when combined with metformin21,22,25 or pioglitazone (Actos, a thiazolidinedione).23 It also decreases fasting blood glucose levels and improves other measures of glucose control.
A study comparing sitagliptin and the sul-fonylurea glipizide (Glucotrol) showed identical glucose-lowering over a 1-year period, with less hypoglycemia and weight gain with sitagliptin.25 Hypoglycemic episodes occurred in 32% of patients taking glipizide but in only 5% of those taking sitagliptin.
Studies noted several trends in laboratory values, though none was associated with clinical evidence of adverse outcome:
- White blood cell counts were noted to increase in three of the studies by 4.7% to 10%, owing to increases in neutro-phils19,20,22
- Alkaline phosphatase concentrations decreased in four studies19,20,22,23
- Uric acid levels increased in four studies.19,20,22,23
RENAL INSUFFICIENCY SLOWS SITAGLIPTIN CLEARANCE
Lower doses and periodic monitoring of renal function are recommended in patients taking sitagliptin who have some degree of renal insufficiency. Clearance of sitagliptin is delayed in patients with renal insufficiency (creatinine clearance < 50 mL/minute).
In a placebo-controlled study of sitagliptin safety, Scott et al26 found that the area under the sitagliptin concentration-time curve was 2.3 times greater in patients with moderate renal insufficiency (creatinine clearance rate 30–49.9 mL/minute), 3.8 times greater in those with severe renal insufficiency (15–29.9 mL/minute), and 4.5 times greater in those with end-stage renal disease (< 15 mL/minute).
The Januvia package insert27 recommends that the daily dose be decreased to 50 mg in patients with creatinine clearance rates of 30 to 49.9 mL/minute (serum creatinine > 1.7 mg/dL in men, > 1.5 mg/dL in women), and that the dose be decreased to 25 mg per day in those with creatinine clearance rates below 30 mL/minute (creatinine > 3.0/2.5 mg/dL).
CLINICAL TRIALS OF VILDAGLIPTIN BEGIN
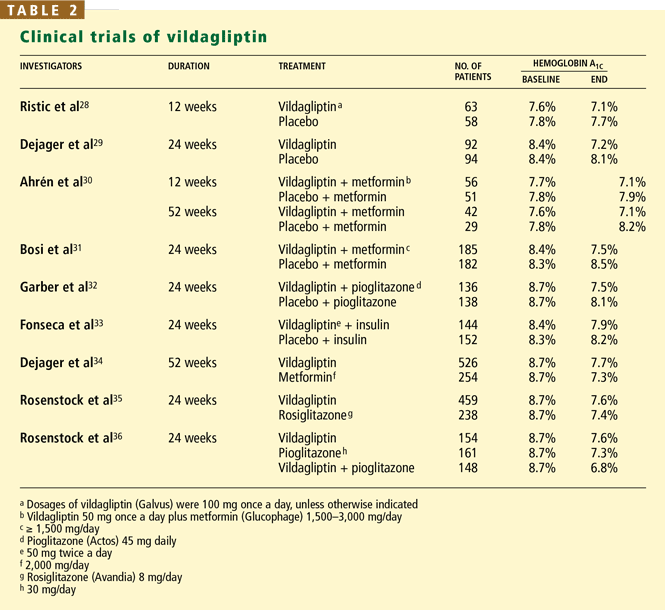
A study comparing vildagliptin against metformin34 showed less glucose-lowering over a 1-year period with vildagliptin, albeit with fewer gastrointestinal side effects, while comparisons with rosiglitazone (Avandia)35 and with pioglitazone36 showed similar glucose-lowering ability.
In a 24-week study,33 256 patients with type 2 diabetes who had a mean body mass index of 33 kg/m2 and who were taking more than 30 units of insulin daily (an average of 82 units) were randomized to additionally receive either vildagliptin 50 mg twice daily or placebo. The HbA1c decreased by 0.5% with vildagliptin and by 0.2% with placebo, from a baseline level of 8.5%. Of interest, 33 patients receiving vildagliptin had a hypo-glycemic episode (a total of 113 events), compared with 45 patients in the placebo group (185 events). None of the episodes in the vildagliptin group was classified as severe, whereas six episodes in the placebo group were classified as severe. This suggests that adding vildagliptin in patients taking insulin can improve glycemia without causing excessive hypoglycemia.
A weakness of the design of this study is that it did not include patients who were receiving an insulin sensitizer, an approach that is typically taken. Given this, it is understandable that overall glycemic control was relatively poor. More effort is needed to explore the use of gliptins with insulin.
WHAT ROLE FOR GLIPTINS?
The evidence from the studies reviewed in this article suggests that gliptins can play an important role in the treatment of type 2 diabetes. In certain patient groups such as the elderly, who cannot take either metformin or a thiazolidinedione and in whom concerns about hypoglycemia are greatest, thus precluding sulfonylurea therapy, gliptins may be the agents of choice. The trials reviewed here suggest that gliptins have glucose-lowering efficacy similar to that of these classes of agents. Gliptins are also effective when combined with metformin or a thiazolidinedione and, as discussed above, may prove to be useful in combination with insulin.
The eventual role of gliptins in the treatment of type 2 diabetes will depend on the answers to several questions. For example, do they preserve beta cell function and reverse the progression of diabetes? Do they affect insulin resistance? Do they have cardiovascular benefits beyond glucose-lowering? Also, since DPP-4 is widely distributed in the body, and since we do not yet know the effects of all the proteins cleaved by this enzyme, will this affect the long-term safety of these drugs?
For now, we can state with reasonable certainty that gliptins lower blood sugar levels to a degree similar to that of other oral hypo-glycemic therapies, with minimal risk of hypo-glycemia, with few immediate adverse effects, and without requiring dose titration. These characteristics suggest that gliptins should be considered useful agents in monotherapy and combination therapy for the treatment of type 2 diabetes.
- National Diabetes Surveillance System. www.cdc.gov/diabetes/statistics/prev/national/figpersons.htm. Last accessed February 28, 2008.
- Narayan KM, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden: US, 2005–2050. Diabetes Care 2006; 29:2114–2116.
- American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007; 30 suppl 1:S4–S41.
- Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA 2004; 291:335–342.
- Brown JB, Nichols GA, Perry A. The burden of treatment failure in type 2 diabetes. Diabetes Care 2004; 27:1535–1540.
- Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose-lowering efficacy: a meta-regression analysis. Diabetes Care 2006; 29:2137–2139.
- Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 2003; 26:2929–2940.
- Bloomgarden ZT. Gut hormones and related concepts. Diabetes Care 2006; 29:2319–2324.
- Nauck MA, Kleine N, Orskov C, et al. Normalization of fasting hyper-glycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-4 inhibitors. Diabetologia 2005; 48:612–615.
- Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140:5356–5363.
- Raun K, von Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long-acting glucagon-like peptide-1 analog, reduces body weight and food intake in obese candy-fed rats, whereas a dipeptidyl peptidase-IV inhibitor, vildagliptin, does not. Diabetes 2007; 56:8–15.
- Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase IV with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006; 55:1695–1704.
- Pospisilik JA, Martin J, Doty T, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes 2003; 52:741–750.
- Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91:4612–4619.
- Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Brazg R, Xu L, Dalla Man C, Cobelli C, Thomas K, Stein PP. Effect of adding sitagliptin, a dipeptidyl peptidase-4 inhibitor, to metformin on 24-h glycaemic control and beta-cell function in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:186–193.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Scott R, Wu M, Sanchez M, Stein P. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract 2007; 61:171–180.
- Nauck MA, Meininger JG, Sheng D, Terranella L, Stein PP. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Scott RS, Hartley P, Luo E, et al. Use of sitagliptin in patients with type 2 diabetes and renal insufficiency [abtract]. Diabetes 2006; 55 suppl 1:A462.
- Januvia prescribing information. www.merck.com/product/usa/pi_circulars/j/products_j.html. Last accessed February 28, 2008.
- Ristic S, Byiers S, Foley J, Holmes D. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: vildagliptin (LAF237) dose response. Diabetes Obes Metab 2005; 7:692–698.
- Dejager S, Baron M, Razac S, Foley JE, Dickinson S, Schweizer S. Effect of vildagliptin on drug-naïve patients with type 2 diabetes. Diabetologia 2006; 49 suppl 1:479–480.
- Ahrén B, Gomis R, Standl E, Mills D, Schweizer A. Twelve- and 52-week efficacy of the dipeptidyl peptidase iv inhibitor laf237 in metformin-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2874–2880.
- Bosi E, Camisasca RP, Collober C, Rochotte E, Garber AJ. Effects of vildagliptin on glucose control over 24 weeks in patients with type 2 diabetes inadequately controlled with metformin. Diabetes Care 2007; 30:890–895.
- Garber A, Schweizer A, Baron MA, Rochotte E, Dejager S. Vildagliptin in combination with pioglitazone improves glycaemic control in patients with type 2 diabetes failing thiazolidinedione monotherapy: a randomized, placebo-controlled study. Diabetes Obes Metab 2007; 9:166–174.
- Fonseca V, Schweizer A, Albrecht D, Baron MA, Chang I, Dejager S. Addition of vildagliptin to insulin improves glycaemic control in type 2 diabetes. Diabetologia 2007; 50:1148–1155.
- Dejager S, LeBeaut A, Couturier A, Schweizer A. Sustained reduction in HbA1c during one-year treatment with vildagliptin in patients with type 2 diabetes (T2DM) [abstract]. Diabetes 2006; 55 suppl 1:A29.
- Rosenstock J, Baron MA, Dejager S, Mills D, Schweizer A. Comparison of vildagliptin and rosiglitazone monotherapy in patients with type 2 diabetes. Diabetes Care 2007; 30:217–223.
- Rosenstock J, Baron MA, Camisasca R-P, Cressier F, Couturier A, Dejager S. Efficacy and tolerability of initial combination therapy with vildagliptin and pioglitazone compared with component monotherapy in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:175–185.
The “gliptins”—the nickname for dipeptidyl peptidase 4 (DPP-4) inhibitors—are one of the newest classes of drugs for the treatment of type 2 diabetes mellitus.
These drugs work by prolonging the action of gut hormones called incretins, which boost insulin levels. The greatest advantage of the gliptins appears to be their ability to stimulate insulin production with little risk of corresponding hypoglycemia.
Sitagliptin (Januvia), the first commercially available DPP-4 inhibitor, has been approved by the US Food and Drug Administration (FDA) and is currently in clinical use, and vildagliptin (Galvus) awaits FDA approval at the time of this writing. Other drugs of this class are in development.
However, because these drugs are so new, a number of questions remain about their use. In this article, we discuss the rationale behind gliptin drugs, the evidence to date on their use alone or in combination with current oral hypoglycemic drugs (and even with insulin), and when and how to use them in daily practice.
THE NEED FOR MORE EFFECTIVE DIABETES TREATMENT
As the number of patients with type 2 diabetes continues its steep and steady rise,1,2 much work has gone into studying treatment goals and how to achieve them. Although experts generally agree on glycemic goals,3 we currently fail to achieve those goals in close to two-thirds of patients: only 37% have a hemo-globin A1c (HbA1c) value at or below the goal of 7%, and the same number have levels exceeding 8%.4
Part of the problem is that treatment regimens are not adjusted in a timely fashion. In a prescribing database of almost 4,000 patients with type 2 diabetes,5 the mean time from the first HbA1c reading above 8% to an actual change in therapy was about 15 months for those taking metformin (Glucophage) alone, and 21 months for those taking a sulfonylurea alone. Another part of the problem is that, on average, patients with an HbA1c of 8.0% to 8.9% can expect only a 0.6% lowering with the addition of one agent.6 Clearly, we need new pharmacologic approaches and new management paradigms. One new approach is the use of gliptins.
HOW GLIPTINS WORK
Incretins promote insulin secretion
We have known for more than 20 years that insulin levels rise considerably higher in response to an oral glucose load than to an intravenous glucose infusion, even though the plasma glucose concentrations may be similar.7 This phenomenon involves a myriad of neural and nutritional factors, but the gut hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) appear to be key.
These peptides—called incretins—have a high degree of homology, and both promote insulin secretion. However, GLP-1, produced by the L cells of the ileum and colon, inhibits glucagon secretion and slows gastric emptying, whereas GIP, secreted from the K cells of the duodenum, has no effect on glucagon and little effect on gastric emptying. Both peptides appear to promote pancreatic beta cell growth and survival,8,9 an effect that in theory might allow us to slow the progressive loss of insulin secretory capacity in type 2 diabetes.
Furthermore, the effect of GLP-1 on insulin secretion depends on the plasma glucose concentration, with a greater insulin secretory effect at higher glucose levels and minimal effect at euglycemic levels.10 This phenomenon suggests that drugs that boost GLP-1 activity should not cause the troublesome hypoglycemia typically seen in patients taking insulin, insulin secretagogues, sulfonyl-ureas, or the meglitinides repaglinide (Prandin) or nateglinide (Starlix). Studies of combination treatment with metformin and the GLP-1 receptor agonist exenatide (Byetta) have shown little risk of hypoglycemia,11 offering evidence favoring this conjecture.
Inhibition of DPP-4 boosts incretin action
The challenge for creating treatments that take advantage of the beneficial effects of GLP-1 and GIP is that they have very short physiologic half-lives, ie, less than 10 minutes. GLP-1 and GIP both have two N-terminal amino acids that are quickly cleaved by DPP-4,12 an enzyme present in the circulation13 and on endothelial cells.14
Currently, there are two classes of drugs based on incretins. One class, the incretin mimetics or GLP-1 receptor agonists, includes drugs that mimic the effect of GLP-1 but are not so quickly degraded by DPP-4. Examples of these drugs are exenatide, which is currently FDA-approved, and liraglutide, which is not yet approved.
On the other hand, by inhibiting the cleaving action of DPP-4, the gliptins can prolong the half-life of endogenous GLP-1, increasing its physiologic effects.
Studies comparing gliptins with GLP-1 receptor agonists are only at the preclinical phase. Liraglutide showed an antiglycemic effect similar to that of vildagliptin in an animal model of glucose intolerance.15 This and other16,17 preclinical studies have shown evidence of improved beta cell growth and survival with DPP-4 inhibitor treatment, to an extent similar to that reported with thiazo-lidinediones, whereas sulfonylureas show no evidence either of increase in beta cells or of improved intrinsic beta cell secretory function in these models. Of course, animal studies can only be cautiously extrapolated to potential effects in humans, and it is uncertain whether such benefits will occur with the therapeutic use of DPP-4 inhibitors.
RANDOMIZED CLINICAL TRIALS OF SITAGLIPTIN
Sitagliptin is effective when used by itself,reducing a baseline HbA1c level of about 8% by 0.6% to 0.8%,19,20,24 and is similarly effective when combined with metformin21,22,25 or pioglitazone (Actos, a thiazolidinedione).23 It also decreases fasting blood glucose levels and improves other measures of glucose control.
A study comparing sitagliptin and the sul-fonylurea glipizide (Glucotrol) showed identical glucose-lowering over a 1-year period, with less hypoglycemia and weight gain with sitagliptin.25 Hypoglycemic episodes occurred in 32% of patients taking glipizide but in only 5% of those taking sitagliptin.
Studies noted several trends in laboratory values, though none was associated with clinical evidence of adverse outcome:
- White blood cell counts were noted to increase in three of the studies by 4.7% to 10%, owing to increases in neutro-phils19,20,22
- Alkaline phosphatase concentrations decreased in four studies19,20,22,23
- Uric acid levels increased in four studies.19,20,22,23
RENAL INSUFFICIENCY SLOWS SITAGLIPTIN CLEARANCE
Lower doses and periodic monitoring of renal function are recommended in patients taking sitagliptin who have some degree of renal insufficiency. Clearance of sitagliptin is delayed in patients with renal insufficiency (creatinine clearance < 50 mL/minute).
In a placebo-controlled study of sitagliptin safety, Scott et al26 found that the area under the sitagliptin concentration-time curve was 2.3 times greater in patients with moderate renal insufficiency (creatinine clearance rate 30–49.9 mL/minute), 3.8 times greater in those with severe renal insufficiency (15–29.9 mL/minute), and 4.5 times greater in those with end-stage renal disease (< 15 mL/minute).
The Januvia package insert27 recommends that the daily dose be decreased to 50 mg in patients with creatinine clearance rates of 30 to 49.9 mL/minute (serum creatinine > 1.7 mg/dL in men, > 1.5 mg/dL in women), and that the dose be decreased to 25 mg per day in those with creatinine clearance rates below 30 mL/minute (creatinine > 3.0/2.5 mg/dL).
CLINICAL TRIALS OF VILDAGLIPTIN BEGIN
A study comparing vildagliptin against metformin34 showed less glucose-lowering over a 1-year period with vildagliptin, albeit with fewer gastrointestinal side effects, while comparisons with rosiglitazone (Avandia)35 and with pioglitazone36 showed similar glucose-lowering ability.
In a 24-week study,33 256 patients with type 2 diabetes who had a mean body mass index of 33 kg/m2 and who were taking more than 30 units of insulin daily (an average of 82 units) were randomized to additionally receive either vildagliptin 50 mg twice daily or placebo. The HbA1c decreased by 0.5% with vildagliptin and by 0.2% with placebo, from a baseline level of 8.5%. Of interest, 33 patients receiving vildagliptin had a hypo-glycemic episode (a total of 113 events), compared with 45 patients in the placebo group (185 events). None of the episodes in the vildagliptin group was classified as severe, whereas six episodes in the placebo group were classified as severe. This suggests that adding vildagliptin in patients taking insulin can improve glycemia without causing excessive hypoglycemia.
A weakness of the design of this study is that it did not include patients who were receiving an insulin sensitizer, an approach that is typically taken. Given this, it is understandable that overall glycemic control was relatively poor. More effort is needed to explore the use of gliptins with insulin.
WHAT ROLE FOR GLIPTINS?
The evidence from the studies reviewed in this article suggests that gliptins can play an important role in the treatment of type 2 diabetes. In certain patient groups such as the elderly, who cannot take either metformin or a thiazolidinedione and in whom concerns about hypoglycemia are greatest, thus precluding sulfonylurea therapy, gliptins may be the agents of choice. The trials reviewed here suggest that gliptins have glucose-lowering efficacy similar to that of these classes of agents. Gliptins are also effective when combined with metformin or a thiazolidinedione and, as discussed above, may prove to be useful in combination with insulin.
The eventual role of gliptins in the treatment of type 2 diabetes will depend on the answers to several questions. For example, do they preserve beta cell function and reverse the progression of diabetes? Do they affect insulin resistance? Do they have cardiovascular benefits beyond glucose-lowering? Also, since DPP-4 is widely distributed in the body, and since we do not yet know the effects of all the proteins cleaved by this enzyme, will this affect the long-term safety of these drugs?
For now, we can state with reasonable certainty that gliptins lower blood sugar levels to a degree similar to that of other oral hypo-glycemic therapies, with minimal risk of hypo-glycemia, with few immediate adverse effects, and without requiring dose titration. These characteristics suggest that gliptins should be considered useful agents in monotherapy and combination therapy for the treatment of type 2 diabetes.
The “gliptins”—the nickname for dipeptidyl peptidase 4 (DPP-4) inhibitors—are one of the newest classes of drugs for the treatment of type 2 diabetes mellitus.
These drugs work by prolonging the action of gut hormones called incretins, which boost insulin levels. The greatest advantage of the gliptins appears to be their ability to stimulate insulin production with little risk of corresponding hypoglycemia.
Sitagliptin (Januvia), the first commercially available DPP-4 inhibitor, has been approved by the US Food and Drug Administration (FDA) and is currently in clinical use, and vildagliptin (Galvus) awaits FDA approval at the time of this writing. Other drugs of this class are in development.
However, because these drugs are so new, a number of questions remain about their use. In this article, we discuss the rationale behind gliptin drugs, the evidence to date on their use alone or in combination with current oral hypoglycemic drugs (and even with insulin), and when and how to use them in daily practice.
THE NEED FOR MORE EFFECTIVE DIABETES TREATMENT
As the number of patients with type 2 diabetes continues its steep and steady rise,1,2 much work has gone into studying treatment goals and how to achieve them. Although experts generally agree on glycemic goals,3 we currently fail to achieve those goals in close to two-thirds of patients: only 37% have a hemo-globin A1c (HbA1c) value at or below the goal of 7%, and the same number have levels exceeding 8%.4
Part of the problem is that treatment regimens are not adjusted in a timely fashion. In a prescribing database of almost 4,000 patients with type 2 diabetes,5 the mean time from the first HbA1c reading above 8% to an actual change in therapy was about 15 months for those taking metformin (Glucophage) alone, and 21 months for those taking a sulfonylurea alone. Another part of the problem is that, on average, patients with an HbA1c of 8.0% to 8.9% can expect only a 0.6% lowering with the addition of one agent.6 Clearly, we need new pharmacologic approaches and new management paradigms. One new approach is the use of gliptins.
HOW GLIPTINS WORK
Incretins promote insulin secretion
We have known for more than 20 years that insulin levels rise considerably higher in response to an oral glucose load than to an intravenous glucose infusion, even though the plasma glucose concentrations may be similar.7 This phenomenon involves a myriad of neural and nutritional factors, but the gut hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) appear to be key.
These peptides—called incretins—have a high degree of homology, and both promote insulin secretion. However, GLP-1, produced by the L cells of the ileum and colon, inhibits glucagon secretion and slows gastric emptying, whereas GIP, secreted from the K cells of the duodenum, has no effect on glucagon and little effect on gastric emptying. Both peptides appear to promote pancreatic beta cell growth and survival,8,9 an effect that in theory might allow us to slow the progressive loss of insulin secretory capacity in type 2 diabetes.
Furthermore, the effect of GLP-1 on insulin secretion depends on the plasma glucose concentration, with a greater insulin secretory effect at higher glucose levels and minimal effect at euglycemic levels.10 This phenomenon suggests that drugs that boost GLP-1 activity should not cause the troublesome hypoglycemia typically seen in patients taking insulin, insulin secretagogues, sulfonyl-ureas, or the meglitinides repaglinide (Prandin) or nateglinide (Starlix). Studies of combination treatment with metformin and the GLP-1 receptor agonist exenatide (Byetta) have shown little risk of hypoglycemia,11 offering evidence favoring this conjecture.
Inhibition of DPP-4 boosts incretin action
The challenge for creating treatments that take advantage of the beneficial effects of GLP-1 and GIP is that they have very short physiologic half-lives, ie, less than 10 minutes. GLP-1 and GIP both have two N-terminal amino acids that are quickly cleaved by DPP-4,12 an enzyme present in the circulation13 and on endothelial cells.14
Currently, there are two classes of drugs based on incretins. One class, the incretin mimetics or GLP-1 receptor agonists, includes drugs that mimic the effect of GLP-1 but are not so quickly degraded by DPP-4. Examples of these drugs are exenatide, which is currently FDA-approved, and liraglutide, which is not yet approved.
On the other hand, by inhibiting the cleaving action of DPP-4, the gliptins can prolong the half-life of endogenous GLP-1, increasing its physiologic effects.
Studies comparing gliptins with GLP-1 receptor agonists are only at the preclinical phase. Liraglutide showed an antiglycemic effect similar to that of vildagliptin in an animal model of glucose intolerance.15 This and other16,17 preclinical studies have shown evidence of improved beta cell growth and survival with DPP-4 inhibitor treatment, to an extent similar to that reported with thiazo-lidinediones, whereas sulfonylureas show no evidence either of increase in beta cells or of improved intrinsic beta cell secretory function in these models. Of course, animal studies can only be cautiously extrapolated to potential effects in humans, and it is uncertain whether such benefits will occur with the therapeutic use of DPP-4 inhibitors.
RANDOMIZED CLINICAL TRIALS OF SITAGLIPTIN
Sitagliptin is effective when used by itself,reducing a baseline HbA1c level of about 8% by 0.6% to 0.8%,19,20,24 and is similarly effective when combined with metformin21,22,25 or pioglitazone (Actos, a thiazolidinedione).23 It also decreases fasting blood glucose levels and improves other measures of glucose control.
A study comparing sitagliptin and the sul-fonylurea glipizide (Glucotrol) showed identical glucose-lowering over a 1-year period, with less hypoglycemia and weight gain with sitagliptin.25 Hypoglycemic episodes occurred in 32% of patients taking glipizide but in only 5% of those taking sitagliptin.
Studies noted several trends in laboratory values, though none was associated with clinical evidence of adverse outcome:
- White blood cell counts were noted to increase in three of the studies by 4.7% to 10%, owing to increases in neutro-phils19,20,22
- Alkaline phosphatase concentrations decreased in four studies19,20,22,23
- Uric acid levels increased in four studies.19,20,22,23
RENAL INSUFFICIENCY SLOWS SITAGLIPTIN CLEARANCE
Lower doses and periodic monitoring of renal function are recommended in patients taking sitagliptin who have some degree of renal insufficiency. Clearance of sitagliptin is delayed in patients with renal insufficiency (creatinine clearance < 50 mL/minute).
In a placebo-controlled study of sitagliptin safety, Scott et al26 found that the area under the sitagliptin concentration-time curve was 2.3 times greater in patients with moderate renal insufficiency (creatinine clearance rate 30–49.9 mL/minute), 3.8 times greater in those with severe renal insufficiency (15–29.9 mL/minute), and 4.5 times greater in those with end-stage renal disease (< 15 mL/minute).
The Januvia package insert27 recommends that the daily dose be decreased to 50 mg in patients with creatinine clearance rates of 30 to 49.9 mL/minute (serum creatinine > 1.7 mg/dL in men, > 1.5 mg/dL in women), and that the dose be decreased to 25 mg per day in those with creatinine clearance rates below 30 mL/minute (creatinine > 3.0/2.5 mg/dL).
CLINICAL TRIALS OF VILDAGLIPTIN BEGIN
A study comparing vildagliptin against metformin34 showed less glucose-lowering over a 1-year period with vildagliptin, albeit with fewer gastrointestinal side effects, while comparisons with rosiglitazone (Avandia)35 and with pioglitazone36 showed similar glucose-lowering ability.
In a 24-week study,33 256 patients with type 2 diabetes who had a mean body mass index of 33 kg/m2 and who were taking more than 30 units of insulin daily (an average of 82 units) were randomized to additionally receive either vildagliptin 50 mg twice daily or placebo. The HbA1c decreased by 0.5% with vildagliptin and by 0.2% with placebo, from a baseline level of 8.5%. Of interest, 33 patients receiving vildagliptin had a hypo-glycemic episode (a total of 113 events), compared with 45 patients in the placebo group (185 events). None of the episodes in the vildagliptin group was classified as severe, whereas six episodes in the placebo group were classified as severe. This suggests that adding vildagliptin in patients taking insulin can improve glycemia without causing excessive hypoglycemia.
A weakness of the design of this study is that it did not include patients who were receiving an insulin sensitizer, an approach that is typically taken. Given this, it is understandable that overall glycemic control was relatively poor. More effort is needed to explore the use of gliptins with insulin.
WHAT ROLE FOR GLIPTINS?
The evidence from the studies reviewed in this article suggests that gliptins can play an important role in the treatment of type 2 diabetes. In certain patient groups such as the elderly, who cannot take either metformin or a thiazolidinedione and in whom concerns about hypoglycemia are greatest, thus precluding sulfonylurea therapy, gliptins may be the agents of choice. The trials reviewed here suggest that gliptins have glucose-lowering efficacy similar to that of these classes of agents. Gliptins are also effective when combined with metformin or a thiazolidinedione and, as discussed above, may prove to be useful in combination with insulin.
The eventual role of gliptins in the treatment of type 2 diabetes will depend on the answers to several questions. For example, do they preserve beta cell function and reverse the progression of diabetes? Do they affect insulin resistance? Do they have cardiovascular benefits beyond glucose-lowering? Also, since DPP-4 is widely distributed in the body, and since we do not yet know the effects of all the proteins cleaved by this enzyme, will this affect the long-term safety of these drugs?
For now, we can state with reasonable certainty that gliptins lower blood sugar levels to a degree similar to that of other oral hypo-glycemic therapies, with minimal risk of hypo-glycemia, with few immediate adverse effects, and without requiring dose titration. These characteristics suggest that gliptins should be considered useful agents in monotherapy and combination therapy for the treatment of type 2 diabetes.
- National Diabetes Surveillance System. www.cdc.gov/diabetes/statistics/prev/national/figpersons.htm. Last accessed February 28, 2008.
- Narayan KM, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden: US, 2005–2050. Diabetes Care 2006; 29:2114–2116.
- American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007; 30 suppl 1:S4–S41.
- Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA 2004; 291:335–342.
- Brown JB, Nichols GA, Perry A. The burden of treatment failure in type 2 diabetes. Diabetes Care 2004; 27:1535–1540.
- Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose-lowering efficacy: a meta-regression analysis. Diabetes Care 2006; 29:2137–2139.
- Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 2003; 26:2929–2940.
- Bloomgarden ZT. Gut hormones and related concepts. Diabetes Care 2006; 29:2319–2324.
- Nauck MA, Kleine N, Orskov C, et al. Normalization of fasting hyper-glycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-4 inhibitors. Diabetologia 2005; 48:612–615.
- Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140:5356–5363.
- Raun K, von Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long-acting glucagon-like peptide-1 analog, reduces body weight and food intake in obese candy-fed rats, whereas a dipeptidyl peptidase-IV inhibitor, vildagliptin, does not. Diabetes 2007; 56:8–15.
- Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase IV with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006; 55:1695–1704.
- Pospisilik JA, Martin J, Doty T, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes 2003; 52:741–750.
- Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91:4612–4619.
- Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Brazg R, Xu L, Dalla Man C, Cobelli C, Thomas K, Stein PP. Effect of adding sitagliptin, a dipeptidyl peptidase-4 inhibitor, to metformin on 24-h glycaemic control and beta-cell function in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:186–193.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Scott R, Wu M, Sanchez M, Stein P. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract 2007; 61:171–180.
- Nauck MA, Meininger JG, Sheng D, Terranella L, Stein PP. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Scott RS, Hartley P, Luo E, et al. Use of sitagliptin in patients with type 2 diabetes and renal insufficiency [abtract]. Diabetes 2006; 55 suppl 1:A462.
- Januvia prescribing information. www.merck.com/product/usa/pi_circulars/j/products_j.html. Last accessed February 28, 2008.
- Ristic S, Byiers S, Foley J, Holmes D. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: vildagliptin (LAF237) dose response. Diabetes Obes Metab 2005; 7:692–698.
- Dejager S, Baron M, Razac S, Foley JE, Dickinson S, Schweizer S. Effect of vildagliptin on drug-naïve patients with type 2 diabetes. Diabetologia 2006; 49 suppl 1:479–480.
- Ahrén B, Gomis R, Standl E, Mills D, Schweizer A. Twelve- and 52-week efficacy of the dipeptidyl peptidase iv inhibitor laf237 in metformin-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2874–2880.
- Bosi E, Camisasca RP, Collober C, Rochotte E, Garber AJ. Effects of vildagliptin on glucose control over 24 weeks in patients with type 2 diabetes inadequately controlled with metformin. Diabetes Care 2007; 30:890–895.
- Garber A, Schweizer A, Baron MA, Rochotte E, Dejager S. Vildagliptin in combination with pioglitazone improves glycaemic control in patients with type 2 diabetes failing thiazolidinedione monotherapy: a randomized, placebo-controlled study. Diabetes Obes Metab 2007; 9:166–174.
- Fonseca V, Schweizer A, Albrecht D, Baron MA, Chang I, Dejager S. Addition of vildagliptin to insulin improves glycaemic control in type 2 diabetes. Diabetologia 2007; 50:1148–1155.
- Dejager S, LeBeaut A, Couturier A, Schweizer A. Sustained reduction in HbA1c during one-year treatment with vildagliptin in patients with type 2 diabetes (T2DM) [abstract]. Diabetes 2006; 55 suppl 1:A29.
- Rosenstock J, Baron MA, Dejager S, Mills D, Schweizer A. Comparison of vildagliptin and rosiglitazone monotherapy in patients with type 2 diabetes. Diabetes Care 2007; 30:217–223.
- Rosenstock J, Baron MA, Camisasca R-P, Cressier F, Couturier A, Dejager S. Efficacy and tolerability of initial combination therapy with vildagliptin and pioglitazone compared with component monotherapy in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:175–185.
- National Diabetes Surveillance System. www.cdc.gov/diabetes/statistics/prev/national/figpersons.htm. Last accessed February 28, 2008.
- Narayan KM, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden: US, 2005–2050. Diabetes Care 2006; 29:2114–2116.
- American Diabetes Association. Standards of medical care in diabetes—2007. Diabetes Care 2007; 30 suppl 1:S4–S41.
- Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA 2004; 291:335–342.
- Brown JB, Nichols GA, Perry A. The burden of treatment failure in type 2 diabetes. Diabetes Care 2004; 27:1535–1540.
- Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose-lowering efficacy: a meta-regression analysis. Diabetes Care 2006; 29:2137–2139.
- Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 2003; 26:2929–2940.
- Bloomgarden ZT. Gut hormones and related concepts. Diabetes Care 2006; 29:2319–2324.
- Nauck MA, Kleine N, Orskov C, et al. Normalization of fasting hyper-glycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Holst JJ, Deacon CF. Glucagon-like peptide-1 mediates the therapeutic actions of DPP-4 inhibitors. Diabetologia 2005; 48:612–615.
- Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7–36)amide is transformed to glucagon-like peptide-1-(9–36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140:5356–5363.
- Raun K, von Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long-acting glucagon-like peptide-1 analog, reduces body weight and food intake in obese candy-fed rats, whereas a dipeptidyl peptidase-IV inhibitor, vildagliptin, does not. Diabetes 2007; 56:8–15.
- Mu J, Woods J, Zhou YP, et al. Chronic inhibition of dipeptidyl peptidase IV with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes 2006; 55:1695–1704.
- Pospisilik JA, Martin J, Doty T, et al. Dipeptidyl peptidase IV inhibitor treatment stimulates beta-cell survival and islet neogenesis in streptozotocin-induced diabetic rats. Diabetes 2003; 52:741–750.
- Herman GA, Bergman A, Stevens C, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab 2006; 91:4612–4619.
- Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Brazg R, Xu L, Dalla Man C, Cobelli C, Thomas K, Stein PP. Effect of adding sitagliptin, a dipeptidyl peptidase-4 inhibitor, to metformin on 24-h glycaemic control and beta-cell function in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:186–193.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Scott R, Wu M, Sanchez M, Stein P. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int J Clin Pract 2007; 61:171–180.
- Nauck MA, Meininger JG, Sheng D, Terranella L, Stein PP. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Scott RS, Hartley P, Luo E, et al. Use of sitagliptin in patients with type 2 diabetes and renal insufficiency [abtract]. Diabetes 2006; 55 suppl 1:A462.
- Januvia prescribing information. www.merck.com/product/usa/pi_circulars/j/products_j.html. Last accessed February 28, 2008.
- Ristic S, Byiers S, Foley J, Holmes D. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: vildagliptin (LAF237) dose response. Diabetes Obes Metab 2005; 7:692–698.
- Dejager S, Baron M, Razac S, Foley JE, Dickinson S, Schweizer S. Effect of vildagliptin on drug-naïve patients with type 2 diabetes. Diabetologia 2006; 49 suppl 1:479–480.
- Ahrén B, Gomis R, Standl E, Mills D, Schweizer A. Twelve- and 52-week efficacy of the dipeptidyl peptidase iv inhibitor laf237 in metformin-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2874–2880.
- Bosi E, Camisasca RP, Collober C, Rochotte E, Garber AJ. Effects of vildagliptin on glucose control over 24 weeks in patients with type 2 diabetes inadequately controlled with metformin. Diabetes Care 2007; 30:890–895.
- Garber A, Schweizer A, Baron MA, Rochotte E, Dejager S. Vildagliptin in combination with pioglitazone improves glycaemic control in patients with type 2 diabetes failing thiazolidinedione monotherapy: a randomized, placebo-controlled study. Diabetes Obes Metab 2007; 9:166–174.
- Fonseca V, Schweizer A, Albrecht D, Baron MA, Chang I, Dejager S. Addition of vildagliptin to insulin improves glycaemic control in type 2 diabetes. Diabetologia 2007; 50:1148–1155.
- Dejager S, LeBeaut A, Couturier A, Schweizer A. Sustained reduction in HbA1c during one-year treatment with vildagliptin in patients with type 2 diabetes (T2DM) [abstract]. Diabetes 2006; 55 suppl 1:A29.
- Rosenstock J, Baron MA, Dejager S, Mills D, Schweizer A. Comparison of vildagliptin and rosiglitazone monotherapy in patients with type 2 diabetes. Diabetes Care 2007; 30:217–223.
- Rosenstock J, Baron MA, Camisasca R-P, Cressier F, Couturier A, Dejager S. Efficacy and tolerability of initial combination therapy with vildagliptin and pioglitazone compared with component monotherapy in patients with type 2 diabetes. Diabetes Obes Metab 2007; 9:175–185.
KEY POINTS
- Sitagliptin (Januvia) is now available, and vildagliptin (Galvus) is awaiting approval. Other gliptins are under development.
- The gliptins effectively lower blood glucose levels, do not require titration, are unlikely to cause hypoglycemia, do not cause weight gain or loss, and are well tolerated.
- Gliptins can be used alone or in combination with metformin (Glucophage) or a thiazolidinedione. Preliminary studies also show evidence of benefit when they are used in combination with insulin.
- Comparative studies suggest that gliptins lower blood glucose levels by about the same amount as other oral hypoglycemic agents.