User login
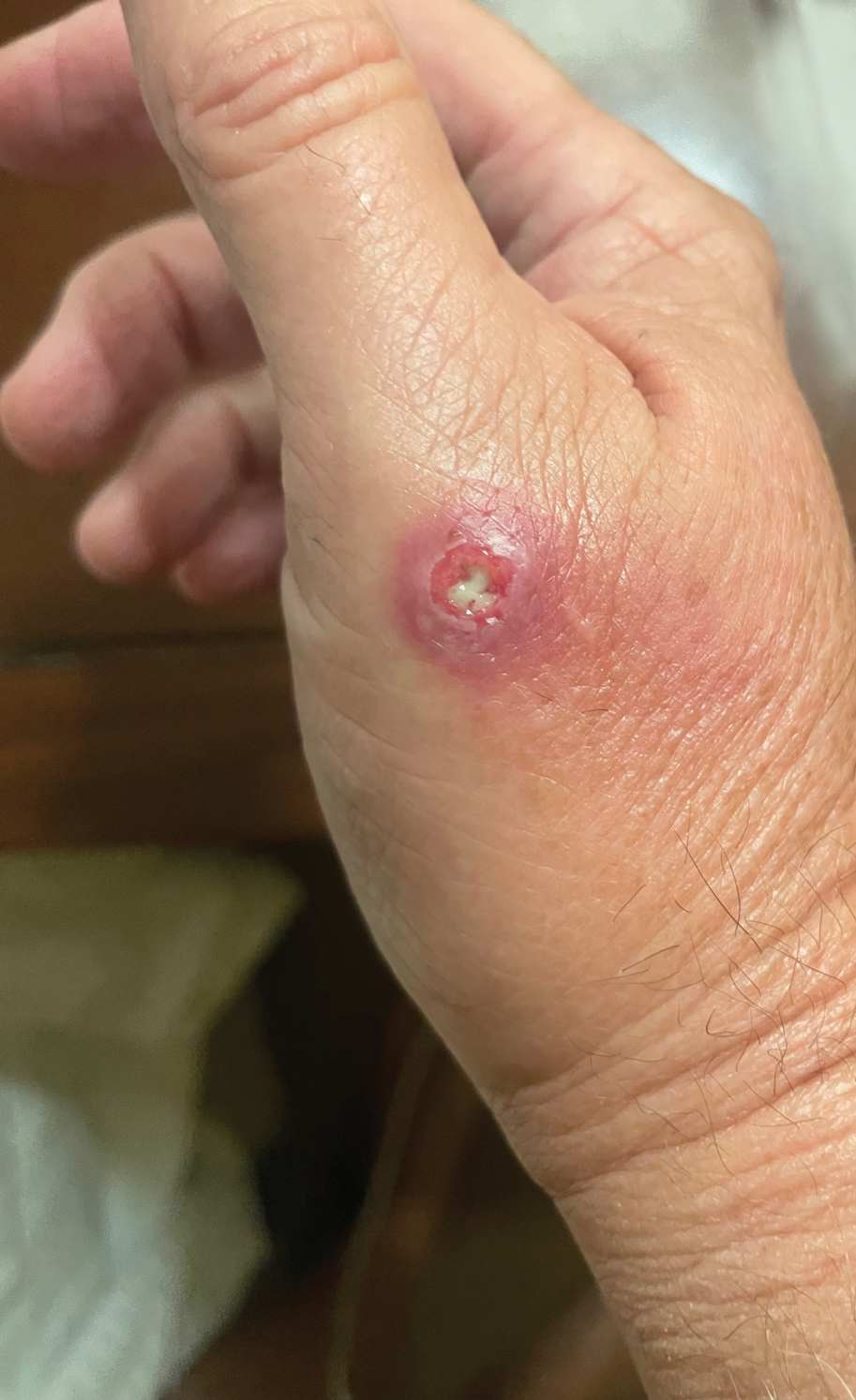
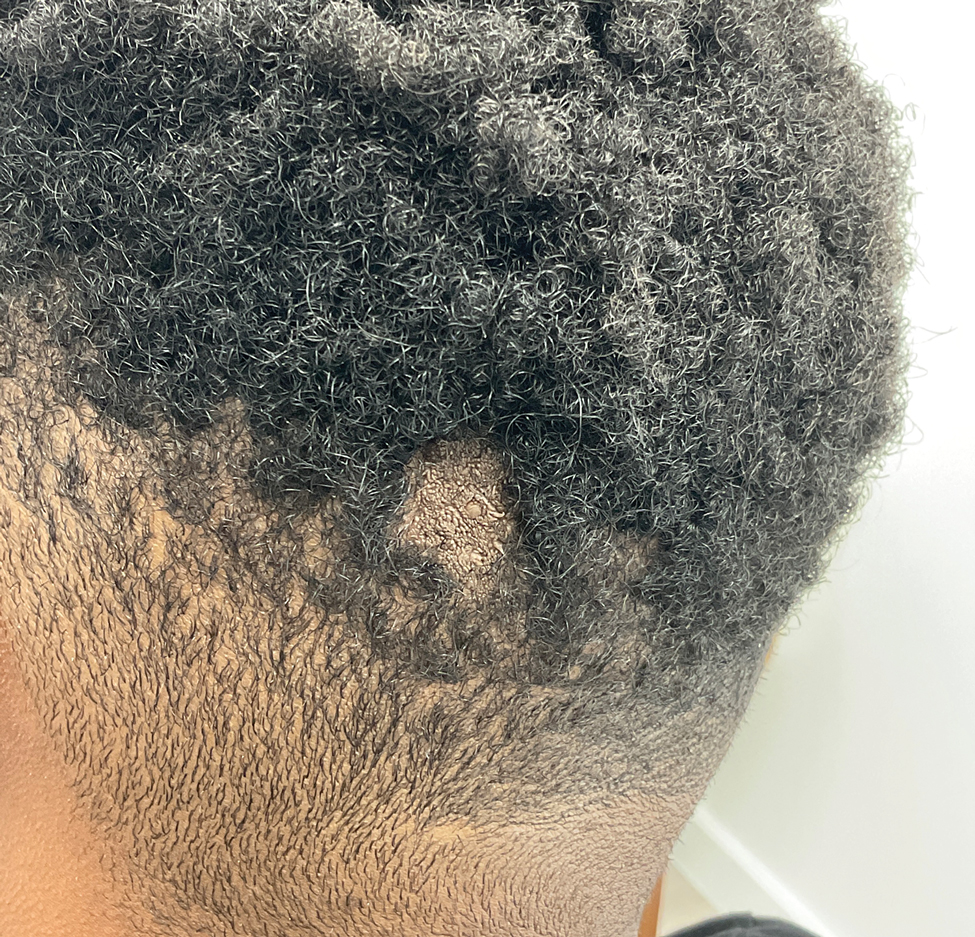
Hairless Scalp Lesion
The Diagnosis: Nevus Sebaceus of Jadassohn
The diagnosis of nevus sebaceus of Jadassohn was made clinically based on the lesion’s appearance and presence since birth as well as the absence of systemic symptoms. Clinically, nevus sebaceus of Jadassohn typically manifests as a well-demarcated, yellow- brown plaque often located on the scalp, as was seen in our patient. The lack of pruritus and pain further supported the diagnosis in our patient. No biopsy was performed, as the presentation was considered classic for this condition. Our patient opted to forgo surgery and will be routinely monitored for any changes, as nevus sebaceus has a potential risk, albeit low, for malignant transformation later in life. No changes have been observed since the initial presentation, and regular follow-ups are planned to monitor for future developments.
Nevus sebaceus of Jadassohn is a hamartomatous lesion involving the pilosebaceous follicle and adjacent adnexal structures.1-3 It most commonly forms on the scalp (59.3%) and is accompanied by partial or total alopecia. 3,4 It is seen less often on the face, periauricular area, or neck1,4; thorax or limbs5; and oral or genital mucosae.6 Nevus sebaceus of Jadassohn affects approximately 0.3% of newborns,1 usually as a solitary lesion that can form an extensive plaque. The male-to-female occurrence ratio has been reported as equal to slightly more predominant in females; all races and ethnicities are affected.1,5
Nevus sebaceus of Jadassohn follows 3 stages of clinical development: infantile, adolescent, and adulthood. It manifests at birth or shortly afterward as a smooth hairless patch or plaque that is yellowish and can be hyperpigmented in Black patients.5 It may have an oval or linear configuration, typically is asymptomatic, and often arises along the Blaschko lines when it occurs as multiple lesions (a rare manifestation).1 During puberty, hormonal changes cause accelerated growth, sebaceous gland maturation, and epidermal hyperplasia. 7 Nevus sebaceus of Jadassohn often is not identified until this stage, when its classic wartlike appearance has fully developed.1
Patients with nevus sebaceus of Jadassohn have a 10% to 20% risk for tumor development in adulthood.2,7 Trichoblastoma and syringocystadenoma papilliferum are the most frequently described neoplasms.8 Basal cell carcinoma is the most common malignant secondary neoplasm with an occurrence rate of 0.8%.6,9 However, basal cell carcinoma and trichoblastoma may share histopathologic features, which may lead to misdiagnosis and a higher reported incidence of basal cell carcinoma in adults than is accurate.2
Early prophylactic surgical removal of nevus sebaceus of Jadassohn has been recommended; however, surgical management is controversial because the risk for a benign secondary neoplasm remains relatively high while the risk for malignancy is much lower.2,7 Surgical excision remains an acceptable option once the patient is mature enough to tolerate the procedure.1 However, patient education regarding watchful waiting vs a surgical approach— and the risks of each—is critical to ensure shared decision-making and a management plan tailored to the individual.
The differential diagnosis includes hypertrophic lichen planus, Langerhans cell histiocytosis (Letterer-Siwe disease type), epidermal nevus, and seborrheic keratosis. Hypertrophic lichen planus often occurs symmetrically on the dorsal feet and shins with thick, scaly, and extremely pruritic plaques. The lesions often persist for an average of 6 years and may lead to multiple keratoacanthomas or follicular base squamous cell carcinomas. Langerhans cell histiocytosis (Letterer-Siwe disease type) manifests with acute, disseminated, visceral, and cutaneous lesions before 2 years of age. These lesions appear as 1- to 2-mm, pink, seborrheic papules, pustules, or vesicles on the scalp, flexural neck, axilla, perineum, and trunk; they often are associated with petechiae, purpura, scale, crust, erosion, impetiginization, and tender fissures. Epidermal nevus occurs within the first year of life and is a hamartoma of the epidermis and papillary dermis. It manifests as papillomatous pigmented linear lines along the Blaschko lines. Seborrheic keratosis manifests as well-demarcated, waxy/verrucous, brown papules with a “stuck on” appearance on hair-bearing skin sparing the mucosae. They are common benign lesions associated with sun exposure and often manifest in the fourth decade of life.10
- Baigrie D, Troxell T, Cook C. Nevus sebaceus. StatPearls [Internet]. Updated August 16, 2023. Accessed September 12, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482493/
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239. doi:10.1097/01 .scs.0000221531.56529.cc
- Kelati A, Baybay H, Gallouj S, et al. Dermoscopic analysis of nevus sebaceus of Jadassohn: a study of 13 cases. Skin Appendage Disord. 2017;3:83-91. doi:10.1159/000460258
- Ugras N, Ozgun G, Adim SB, et al. Nevus sebaceous at unusual location: a rare presentation. Indian J Pathol Microbiol. 2012;55:419-420. doi:10.4103/0377-4929.101768
- Serpas de Lopez RM, Hernandez-Perez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72. doi:10.1111/j.1524-4725 .1985.tb02893.x
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268. doi:10.1016/S0190-9622(00)90136-1
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660. doi:10.1097/00001665-200309000-00010
- Chahboun F, Eljazouly M, Elomari M, et al. Trichoblastoma arising from the nevus sebaceus of Jadassohn. Cureus. 2021;13:E15325. doi:10.7759/cureus.15325
- Cazzato G, Cimmino A, Colagrande A, et al. The multiple faces of nodular trichoblastoma: review of the literature with case presentation. Dermatopathology (Basel). 2021;8:265-270. doi:10.3390 /dermatopathology8030032
- Dandekar MN, Gandhi RK. Neoplastic dermatology. In: Alikhan A, Hocker TLH (eds). Review of Dermatology. Elsevier; 2016: 321-366.
The Diagnosis: Nevus Sebaceus of Jadassohn
The diagnosis of nevus sebaceus of Jadassohn was made clinically based on the lesion’s appearance and presence since birth as well as the absence of systemic symptoms. Clinically, nevus sebaceus of Jadassohn typically manifests as a well-demarcated, yellow- brown plaque often located on the scalp, as was seen in our patient. The lack of pruritus and pain further supported the diagnosis in our patient. No biopsy was performed, as the presentation was considered classic for this condition. Our patient opted to forgo surgery and will be routinely monitored for any changes, as nevus sebaceus has a potential risk, albeit low, for malignant transformation later in life. No changes have been observed since the initial presentation, and regular follow-ups are planned to monitor for future developments.
Nevus sebaceus of Jadassohn is a hamartomatous lesion involving the pilosebaceous follicle and adjacent adnexal structures.1-3 It most commonly forms on the scalp (59.3%) and is accompanied by partial or total alopecia. 3,4 It is seen less often on the face, periauricular area, or neck1,4; thorax or limbs5; and oral or genital mucosae.6 Nevus sebaceus of Jadassohn affects approximately 0.3% of newborns,1 usually as a solitary lesion that can form an extensive plaque. The male-to-female occurrence ratio has been reported as equal to slightly more predominant in females; all races and ethnicities are affected.1,5
Nevus sebaceus of Jadassohn follows 3 stages of clinical development: infantile, adolescent, and adulthood. It manifests at birth or shortly afterward as a smooth hairless patch or plaque that is yellowish and can be hyperpigmented in Black patients.5 It may have an oval or linear configuration, typically is asymptomatic, and often arises along the Blaschko lines when it occurs as multiple lesions (a rare manifestation).1 During puberty, hormonal changes cause accelerated growth, sebaceous gland maturation, and epidermal hyperplasia. 7 Nevus sebaceus of Jadassohn often is not identified until this stage, when its classic wartlike appearance has fully developed.1
Patients with nevus sebaceus of Jadassohn have a 10% to 20% risk for tumor development in adulthood.2,7 Trichoblastoma and syringocystadenoma papilliferum are the most frequently described neoplasms.8 Basal cell carcinoma is the most common malignant secondary neoplasm with an occurrence rate of 0.8%.6,9 However, basal cell carcinoma and trichoblastoma may share histopathologic features, which may lead to misdiagnosis and a higher reported incidence of basal cell carcinoma in adults than is accurate.2
Early prophylactic surgical removal of nevus sebaceus of Jadassohn has been recommended; however, surgical management is controversial because the risk for a benign secondary neoplasm remains relatively high while the risk for malignancy is much lower.2,7 Surgical excision remains an acceptable option once the patient is mature enough to tolerate the procedure.1 However, patient education regarding watchful waiting vs a surgical approach— and the risks of each—is critical to ensure shared decision-making and a management plan tailored to the individual.
The differential diagnosis includes hypertrophic lichen planus, Langerhans cell histiocytosis (Letterer-Siwe disease type), epidermal nevus, and seborrheic keratosis. Hypertrophic lichen planus often occurs symmetrically on the dorsal feet and shins with thick, scaly, and extremely pruritic plaques. The lesions often persist for an average of 6 years and may lead to multiple keratoacanthomas or follicular base squamous cell carcinomas. Langerhans cell histiocytosis (Letterer-Siwe disease type) manifests with acute, disseminated, visceral, and cutaneous lesions before 2 years of age. These lesions appear as 1- to 2-mm, pink, seborrheic papules, pustules, or vesicles on the scalp, flexural neck, axilla, perineum, and trunk; they often are associated with petechiae, purpura, scale, crust, erosion, impetiginization, and tender fissures. Epidermal nevus occurs within the first year of life and is a hamartoma of the epidermis and papillary dermis. It manifests as papillomatous pigmented linear lines along the Blaschko lines. Seborrheic keratosis manifests as well-demarcated, waxy/verrucous, brown papules with a “stuck on” appearance on hair-bearing skin sparing the mucosae. They are common benign lesions associated with sun exposure and often manifest in the fourth decade of life.10
The Diagnosis: Nevus Sebaceus of Jadassohn
The diagnosis of nevus sebaceus of Jadassohn was made clinically based on the lesion’s appearance and presence since birth as well as the absence of systemic symptoms. Clinically, nevus sebaceus of Jadassohn typically manifests as a well-demarcated, yellow- brown plaque often located on the scalp, as was seen in our patient. The lack of pruritus and pain further supported the diagnosis in our patient. No biopsy was performed, as the presentation was considered classic for this condition. Our patient opted to forgo surgery and will be routinely monitored for any changes, as nevus sebaceus has a potential risk, albeit low, for malignant transformation later in life. No changes have been observed since the initial presentation, and regular follow-ups are planned to monitor for future developments.
Nevus sebaceus of Jadassohn is a hamartomatous lesion involving the pilosebaceous follicle and adjacent adnexal structures.1-3 It most commonly forms on the scalp (59.3%) and is accompanied by partial or total alopecia. 3,4 It is seen less often on the face, periauricular area, or neck1,4; thorax or limbs5; and oral or genital mucosae.6 Nevus sebaceus of Jadassohn affects approximately 0.3% of newborns,1 usually as a solitary lesion that can form an extensive plaque. The male-to-female occurrence ratio has been reported as equal to slightly more predominant in females; all races and ethnicities are affected.1,5
Nevus sebaceus of Jadassohn follows 3 stages of clinical development: infantile, adolescent, and adulthood. It manifests at birth or shortly afterward as a smooth hairless patch or plaque that is yellowish and can be hyperpigmented in Black patients.5 It may have an oval or linear configuration, typically is asymptomatic, and often arises along the Blaschko lines when it occurs as multiple lesions (a rare manifestation).1 During puberty, hormonal changes cause accelerated growth, sebaceous gland maturation, and epidermal hyperplasia. 7 Nevus sebaceus of Jadassohn often is not identified until this stage, when its classic wartlike appearance has fully developed.1
Patients with nevus sebaceus of Jadassohn have a 10% to 20% risk for tumor development in adulthood.2,7 Trichoblastoma and syringocystadenoma papilliferum are the most frequently described neoplasms.8 Basal cell carcinoma is the most common malignant secondary neoplasm with an occurrence rate of 0.8%.6,9 However, basal cell carcinoma and trichoblastoma may share histopathologic features, which may lead to misdiagnosis and a higher reported incidence of basal cell carcinoma in adults than is accurate.2
Early prophylactic surgical removal of nevus sebaceus of Jadassohn has been recommended; however, surgical management is controversial because the risk for a benign secondary neoplasm remains relatively high while the risk for malignancy is much lower.2,7 Surgical excision remains an acceptable option once the patient is mature enough to tolerate the procedure.1 However, patient education regarding watchful waiting vs a surgical approach— and the risks of each—is critical to ensure shared decision-making and a management plan tailored to the individual.
The differential diagnosis includes hypertrophic lichen planus, Langerhans cell histiocytosis (Letterer-Siwe disease type), epidermal nevus, and seborrheic keratosis. Hypertrophic lichen planus often occurs symmetrically on the dorsal feet and shins with thick, scaly, and extremely pruritic plaques. The lesions often persist for an average of 6 years and may lead to multiple keratoacanthomas or follicular base squamous cell carcinomas. Langerhans cell histiocytosis (Letterer-Siwe disease type) manifests with acute, disseminated, visceral, and cutaneous lesions before 2 years of age. These lesions appear as 1- to 2-mm, pink, seborrheic papules, pustules, or vesicles on the scalp, flexural neck, axilla, perineum, and trunk; they often are associated with petechiae, purpura, scale, crust, erosion, impetiginization, and tender fissures. Epidermal nevus occurs within the first year of life and is a hamartoma of the epidermis and papillary dermis. It manifests as papillomatous pigmented linear lines along the Blaschko lines. Seborrheic keratosis manifests as well-demarcated, waxy/verrucous, brown papules with a “stuck on” appearance on hair-bearing skin sparing the mucosae. They are common benign lesions associated with sun exposure and often manifest in the fourth decade of life.10
- Baigrie D, Troxell T, Cook C. Nevus sebaceus. StatPearls [Internet]. Updated August 16, 2023. Accessed September 12, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482493/
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239. doi:10.1097/01 .scs.0000221531.56529.cc
- Kelati A, Baybay H, Gallouj S, et al. Dermoscopic analysis of nevus sebaceus of Jadassohn: a study of 13 cases. Skin Appendage Disord. 2017;3:83-91. doi:10.1159/000460258
- Ugras N, Ozgun G, Adim SB, et al. Nevus sebaceous at unusual location: a rare presentation. Indian J Pathol Microbiol. 2012;55:419-420. doi:10.4103/0377-4929.101768
- Serpas de Lopez RM, Hernandez-Perez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72. doi:10.1111/j.1524-4725 .1985.tb02893.x
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268. doi:10.1016/S0190-9622(00)90136-1
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660. doi:10.1097/00001665-200309000-00010
- Chahboun F, Eljazouly M, Elomari M, et al. Trichoblastoma arising from the nevus sebaceus of Jadassohn. Cureus. 2021;13:E15325. doi:10.7759/cureus.15325
- Cazzato G, Cimmino A, Colagrande A, et al. The multiple faces of nodular trichoblastoma: review of the literature with case presentation. Dermatopathology (Basel). 2021;8:265-270. doi:10.3390 /dermatopathology8030032
- Dandekar MN, Gandhi RK. Neoplastic dermatology. In: Alikhan A, Hocker TLH (eds). Review of Dermatology. Elsevier; 2016: 321-366.
- Baigrie D, Troxell T, Cook C. Nevus sebaceus. StatPearls [Internet]. Updated August 16, 2023. Accessed September 12, 2024. https://www.ncbi.nlm.nih.gov/books/NBK482493/
- Terenzi V, Indrizzi E, Buonaccorsi S, et al. Nevus sebaceus of Jadassohn. J Craniofac Surg. 2006;17:1234-1239. doi:10.1097/01 .scs.0000221531.56529.cc
- Kelati A, Baybay H, Gallouj S, et al. Dermoscopic analysis of nevus sebaceus of Jadassohn: a study of 13 cases. Skin Appendage Disord. 2017;3:83-91. doi:10.1159/000460258
- Ugras N, Ozgun G, Adim SB, et al. Nevus sebaceous at unusual location: a rare presentation. Indian J Pathol Microbiol. 2012;55:419-420. doi:10.4103/0377-4929.101768
- Serpas de Lopez RM, Hernandez-Perez E. Jadassohn’s sebaceous nevus. J Dermatol Surg Oncol. 1985;11:68-72. doi:10.1111/j.1524-4725 .1985.tb02893.x
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268. doi:10.1016/S0190-9622(00)90136-1
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660. doi:10.1097/00001665-200309000-00010
- Chahboun F, Eljazouly M, Elomari M, et al. Trichoblastoma arising from the nevus sebaceus of Jadassohn. Cureus. 2021;13:E15325. doi:10.7759/cureus.15325
- Cazzato G, Cimmino A, Colagrande A, et al. The multiple faces of nodular trichoblastoma: review of the literature with case presentation. Dermatopathology (Basel). 2021;8:265-270. doi:10.3390 /dermatopathology8030032
- Dandekar MN, Gandhi RK. Neoplastic dermatology. In: Alikhan A, Hocker TLH (eds). Review of Dermatology. Elsevier; 2016: 321-366.
A 23-year-old man presented to the dermatology clinic with hair loss on the scalp of several years’ duration. The patient reported persistent pigmented bumps on the back of the scalp. He denied any pruritus or pain and had no systemic symptoms or comorbidities. Physical examination revealed a 1×1.5-cm, yellow-brown, hairless plaque on the left parietal scalp.
Purpuric Lesions on the Leg
THE DIAGNOSIS: Dengue Hemorrhagic Fever
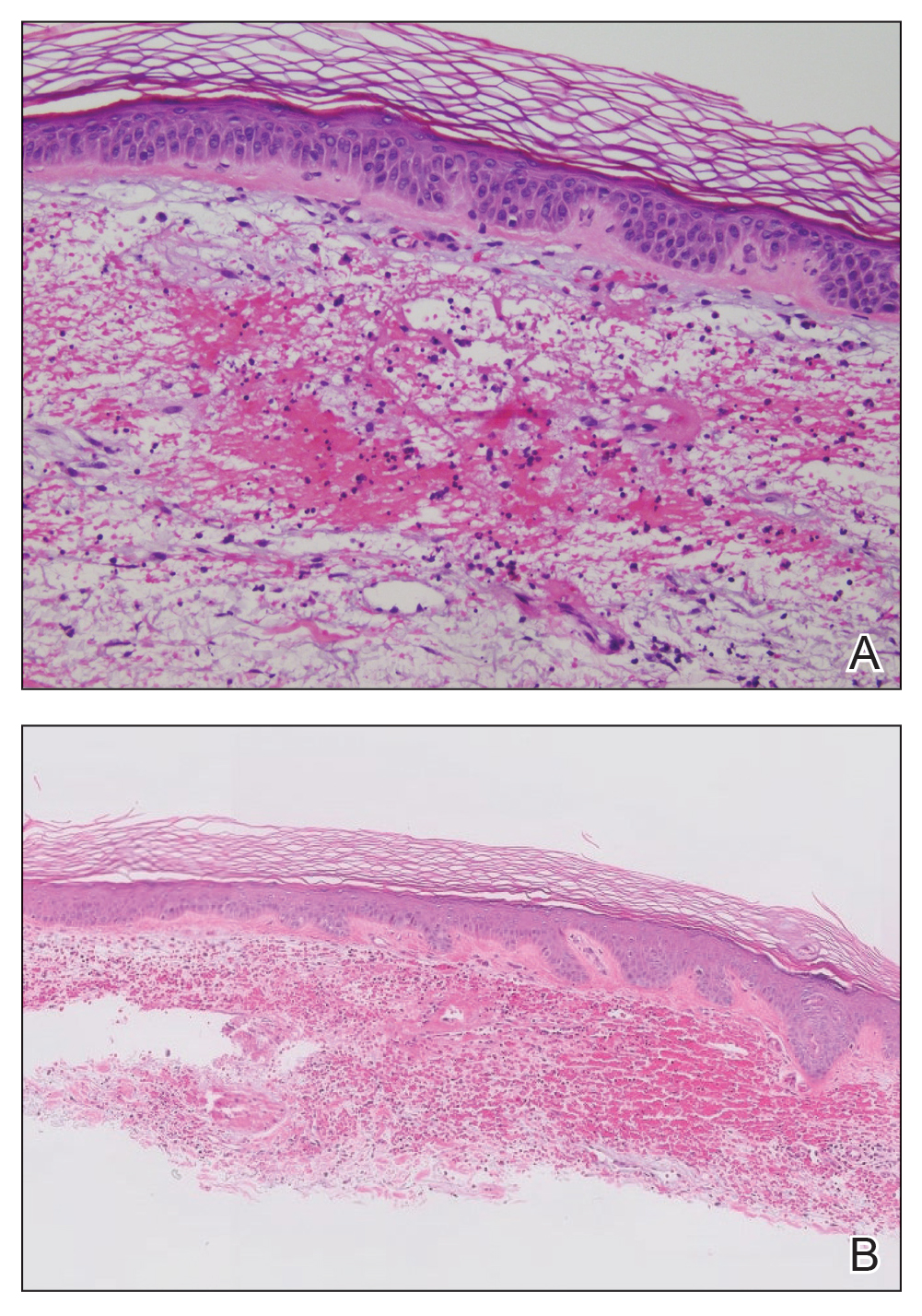
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
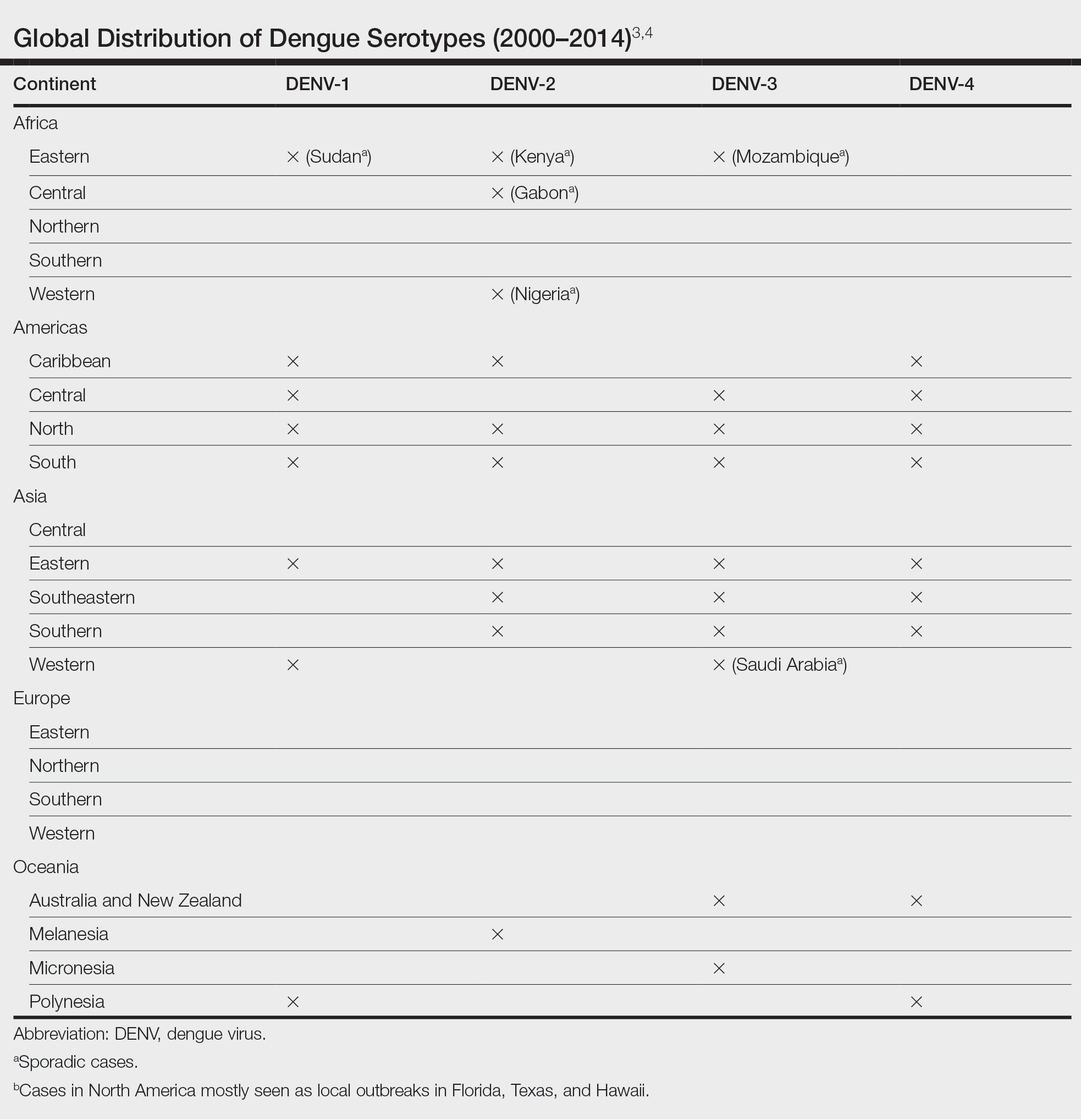
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
THE DIAGNOSIS: Dengue Hemorrhagic Fever
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
THE DIAGNOSIS: Dengue Hemorrhagic Fever
The retiform purpura observed in our patient was suggestive of a vasculitic, thrombotic, or embolic etiology. Dengue IgM serologic testing performed based on her extensive travel history and recent return from a dengue-endemic area was positive, indicating acute infection. A clinical diagnosis of dengue hemorrhagic fever (DHF) was made based on the hemorrhagic appearance of the lesion. Histopathology revealed leukocytoclastic vasculitis (Figure). Anti–double-stranded DNA, antideoxyribonuclease, C3 and C4, CH50 (total hemolytic complement), antineutrophil cytoplasmic antibodies, HIV, and hepatitis B virus tests were normal. Direct immunofluorescence was negative.
Dengue virus is a single-stranded RNA virus transmitted by Aedes aegypti and Aedes albopictus mosquitoes and is one of the most prevalent arthropod-borne viruses affecting humans today.1,2 Infection with the dengue virus generally is seen in travelers visiting tropical regions of Africa, Mexico, South America, South and Central Asia, Southeast Asia, and the Caribbean.1 The Table shows the global distribution of dengue serotypes from 2000 to 2014.3,4 There are 4 serotypes of the dengue virus: DENV-1 to DENV-4. Infection with 1 strain elicits longlasting immunity to that strain, but subsequent infection with another strain can result in severe DHF due to antibody cross-reaction.1
Dengue virus infection ranges from mildly symptomatic to a spectrum of increasingly severe conditions that comprise dengue fever (DF) and DHF, as well as dengue shock syndrome and brain stem hemorrhage, which may be fatal.2,5 Dengue fever manifests as severe myalgia, fever, headache (usually retro-orbital), arthralgia, erythema, and rubelliform exanthema.6 The frequency of skin eruptions in patients with DF varies with the virus strain and outbreaks.7 The lesions initially develop with the onset of fever and manifest as flushing or erythematous mottling of the face, neck, and chest areas.1,7 The morbilliform eruption develops 2 to 6 days after the onset of the fever, beginning on the trunk and spreading to the face and extremities.1,7 The rash may become confluent with characteristic sparing of small round areas of normal skin described as white islands in a sea of red.2 Verrucous papules on the ears also have been described and may resemble those seen in Cowden syndrome. In patients with prior infection with a different strain of the virus, hemorrhagic lesions may develop, including characteristic retiform purpura, a positive tourniquet test, and the appearance of petechiae on the lower legs. Pruritus and desquamation, especially on the palms and soles, may follow the termination of the eruption.7
The differential diagnosis of DF includes measles, rubella, enteroviruses, and influenza. Chikungunya and West Nile viruses in Asia and Africa and the O’nyong-nyong virus in Africa are also arboviruses that cause a clinical picture similar to DF but not DHF. Other diagnostic considerations include phases of scarlet fever, typhoid, malaria, leptospirosis, hepatitis A, and trypanosomal and rickettsial diseases.7 The differential diagnosis of DHF includes antineutrophil cytoplasmic antibody–associated vasculitis, rheumatoid vasculitis, and bacterial septic vasculitis.
Acute clinical diagnosis of DF can be challenging because of the nonspecific symptoms that can be seen in almost every infectious disease. Clinical presentation assessment should be confirmed with laboratory testing.6 Dengue virus infection usually is confirmed by the identification of viral genomic RNA, antigens, or the antibodies it elicits. Enzyme-linked immunosorbent assay–based serologic tests are cost-effective and easy to perform.5 IgM antibodies usually show cross-reactivity with platelets, but the antibody levels are not positively correlated with the severity of DF.8 Primary infection with the dengue virus is characterized by the elevation of specific IgM levels that usually occurs 3 to 5 days after symptom onset and persists during the postfebrile stage (up to 30 to 60 days). In secondary infections, the IgM levels usually rise more slowly and reach a lower level than in primary infections.9 For both primary and secondary infections, testing IgM levels after the febrile stage may be helpful with the laboratory diagnosis.
Currently, there is no antiviral drug available for dengue. Treatment of dengue infection is symptomatic and supportive.2
Dengue hemorrhagic fever is indicated by a rising hematocrit (≥20%) and a falling platelet count (>100,000/mm3) accompanying clinical signs of hemorrhage. Treatment includes intravenous fluid replacement and careful clinical monitoring of hematocrit levels, platelet count, vitals, urine output, and other signs of shock.5 For patients with a history of dengue infection, travel to areas with other serotypes is not recommended.
If any travel to a high-risk area is planned, countryspecific travel recommendations and warnings should be reviewed from the Centers for Disease Control and Prevention’s website (https://wwwnc.cdc.gov/travel/notices/level1/dengue-global). Use of an Environmental Protection Agency–registered insect repellent to avoid mosquito bites and acetaminophen for managing symptoms is advised. During travel, staying in places with window and door screens and using a bed net during sleep are suggested. Long-sleeved shirts and long pants also are preferred. Travelers should see a health care provider if they have symptoms of dengue.10
African tick bite fever (ATBF) is caused by Rickettsia africae transmitted by Amblyomma ticks. Skin findings in ATBF include erythematous, firm, tender papules with central eschars consistent with the feeding patterns of ticks.11 Histopathology of ATBF usually includes fibrinoid necrosis of vessels in the dermis with a perivascular inflammatory infiltrate and coagulation necrosis of the surrounding dermis consistent with eschar formation.12 The lack of an eschar weighs against this diagnosis.
African trypanosomiasis (also known as sleeping sickness) is caused by protozoa transmitted by the tsetse fly. A chancrelike, circumscribed, rubbery, indurated red or violaceous nodule measuring 2 to 5 cm in diameter often develops as the earliest cutaneous sign of the disease.13 Nonspecific histopathologic findings, such as infiltration of lymphocytes and macrophages and proliferation of endothelial cells and fibroblasts, may be observed.14 Extravascular parasites have been noted in skin biopsies.15 In later stages, skin lesions called trypanids may be observed as macular, papular, annular, targetoid, purpuric, and erythematous lesions, and histopathologic findings consistent with vasculitis also may be seen.13
Chikungunya virus infection is an acute-onset, mosquito-borne viral disease. Skin manifestations may start with nonspecific, generalized, morbilliform, maculopapular rashes coinciding with fever, which also may be seen initially with DHF. Skin hyperpigmentation, mostly centrofacial and involving the nose (chik sign); purpuric and ecchymotic lesions over the trunk and flexors of limbs in adults, often surmounted by subepidermal bullae and lesions resembling toxic epidermal necrolysis; and nonhealing ulcers in the genital and groin areas are common skin manifestations of chikungunya infection.16 Intraepithelial splitting with acantholysis and perivascular lymphohistiocytic infiltration may be observed in the histopathology of blistering lesions, which are not consistent with DHF.17
Zika virus infection is caused by an arbovirus within the Flaviviridae family, which also includes the dengue virus. Initial mucocutaneous findings of the Zika virus include nonspecific diffuse maculopapular eruptions. The eruption generally spares the palms and soles; however, various manifestations including involvement of the palms and soles have been reported.18 The morbilliform eruption begins on the face and extends to the trunk and extremities. Mild hemorrhagic manifestations, including petechiae and bleeding gums, may be observed. Distinguishing between dengue and Zika virus infection relies on the severity of symptoms and laboratory tests, including polymerase chain reaction or IgM antibody testing.19 The other conditions listed do not produce hemorrhagic fever.
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
- Pincus LB, Grossman ME, Fox LP. The exanthem of dengue fever: clinical features of two US tourists traveling abroad. J Am Acad Dermatol. 2008;58:308-316. doi:10.1016/j.jaad.2007.08.042
- Radakovic-Fijan S, Graninger W, Müller C, et al. Dengue hemorrhagic fever in a British travel guide. J Am Acad Dermatol. 2002;46:430-433. doi:10.1067/mjd.2002.111904
- Yamashita A, Sakamoto T, Sekizuka T, et al. DGV: dengue genographic viewer. Front Microbiol. 2016;7:875. doi:10.3389/fmicb.2016.00875
- Centers for Disease and Prevention. Dengue in the US states and territories. Updated October 7, 2020. Accessed September 30, 2024. https://www.cdc.gov/dengue/data-research/facts-stats/?CDC_AAref_Val=https://www.cdc.gov/dengue/areaswithrisk/in-the-us.html
- Khetarpal N, Khanna I. Dengue fever: causes, complications, and vaccine strategies. J Immunol Res. 2016;2016:6803098. doi:10.1155/2016/6803098
- Muller DA, Depelsenaire AC, Young PR. Clinical and laboratory diagnosis of dengue virus infection. J Infect Dis. 2017;215(suppl 2):S89-S95. doi:10.1093/infdis/jiw649
- Waterman SH, Gubler DJ. Dengue fever. Clin Dermatol. 1989;7:117-122. doi:10.1016/0738-081x(89)90034-5
- Lin CF, Lei HY, Liu CC, et al. Generation of IgM anti-platelet autoantibody in dengue patients. J Med Virol. 2001;63:143-149. doi:10.1002/1096- 9071(20000201)63:2<143::AID-JMV1009>3.0.CO;2-L
- Tripathi NK, Shrivastava A, Dash PK, et al. Detection of dengue virus. Methods Mol Biol. 2011;665:51-64. doi:10.1007/978-1-60761-817-1_4
- Centers for Disease Control and Prevention. Plan for travel. Accessed September 30, 2024. https://wwwnc.cdc.gov/travel
- Mack I, Ritz N. African tick-bite fever. N Engl J Med. 2019;380:960. doi:10.1056/NEJMicm1810093
- Lepidi H, Fournier PE, Raoult D. Histologic features and immunodetection of African tick-bite fever eschar. Emerg Infect Dis. 2006;12:1332- 1337. doi:10.3201/eid1209.051540
- McGovern TW, Williams W, Fitzpatrick JE, et al. Cutaneous manifestations of African trypanosomiasis. Arch Dermatol. 1995;131:1178-1182.
- Kristensson K, Bentivoglio M. Pathology of African trypanosomiasis. In: Dumas M, Bouteille B, Buguet A, eds. Progress in Human African Trypanosomiasis, Sleeping Sickness. Springer; 1999:157-181.
- Capewell P, Cren-Travaillé C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5:e17716. doi:10.7554/eLife.17716
- Singal A. Chikungunya and skin: current perspective. Indian Dermatol Online J. 2017;8:307-309. doi:10.4103/idoj.IDOJ_93_17
- Robin S, Ramful D, Zettor J, et al. Severe bullous skin lesions associated with chikungunya virus infection in small infants. Eur J Pediatr. 2009;169:67-72. doi:10.1007/s00431-009-0986-0
- Hussain A, Ali F, Latiwesh OB, et al. A comprehensive review of the manifestations and pathogenesis of Zika virus in neonates and adults. Cureus. 2018;10:E3290. doi:10.7759/cureus.3290
- Farahnik B, Beroukhim K, Blattner CM, et al. Cutaneous manifestations of the Zika virus. J Am Acad Dermatol. 2016;74:1286-1287. doi:10.1016/j.jaad.2016.02.1232
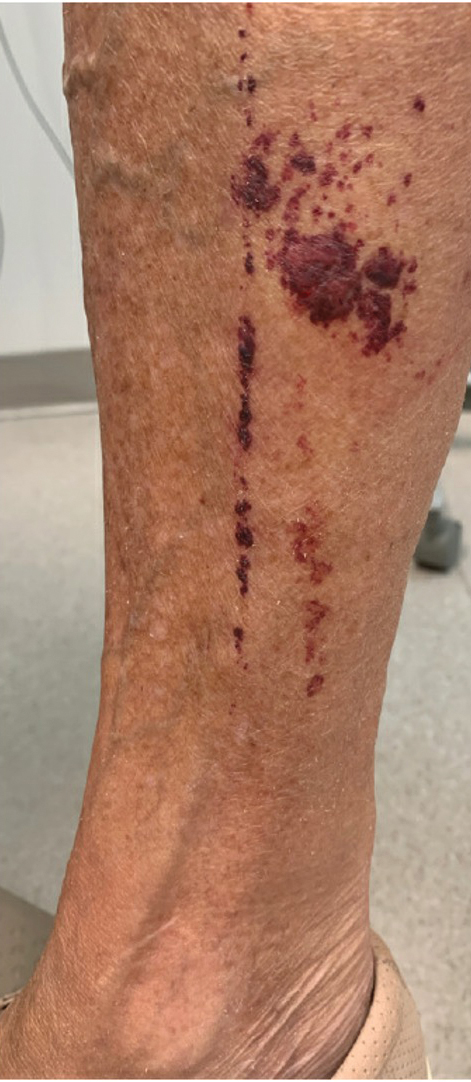
A 74-year-old woman who frequently traveled abroad presented to the dermatology department with retiform purpura of the lower leg along with gastrointestinal cramps, fatigue, and myalgia. The patient reported that the symptoms had started 10 days after returning from a recent trip to Africa.
Nonscaly Red-Brown Macules on the Feet and Ankles
THE DIAGNOSIS: Secondary Syphilis
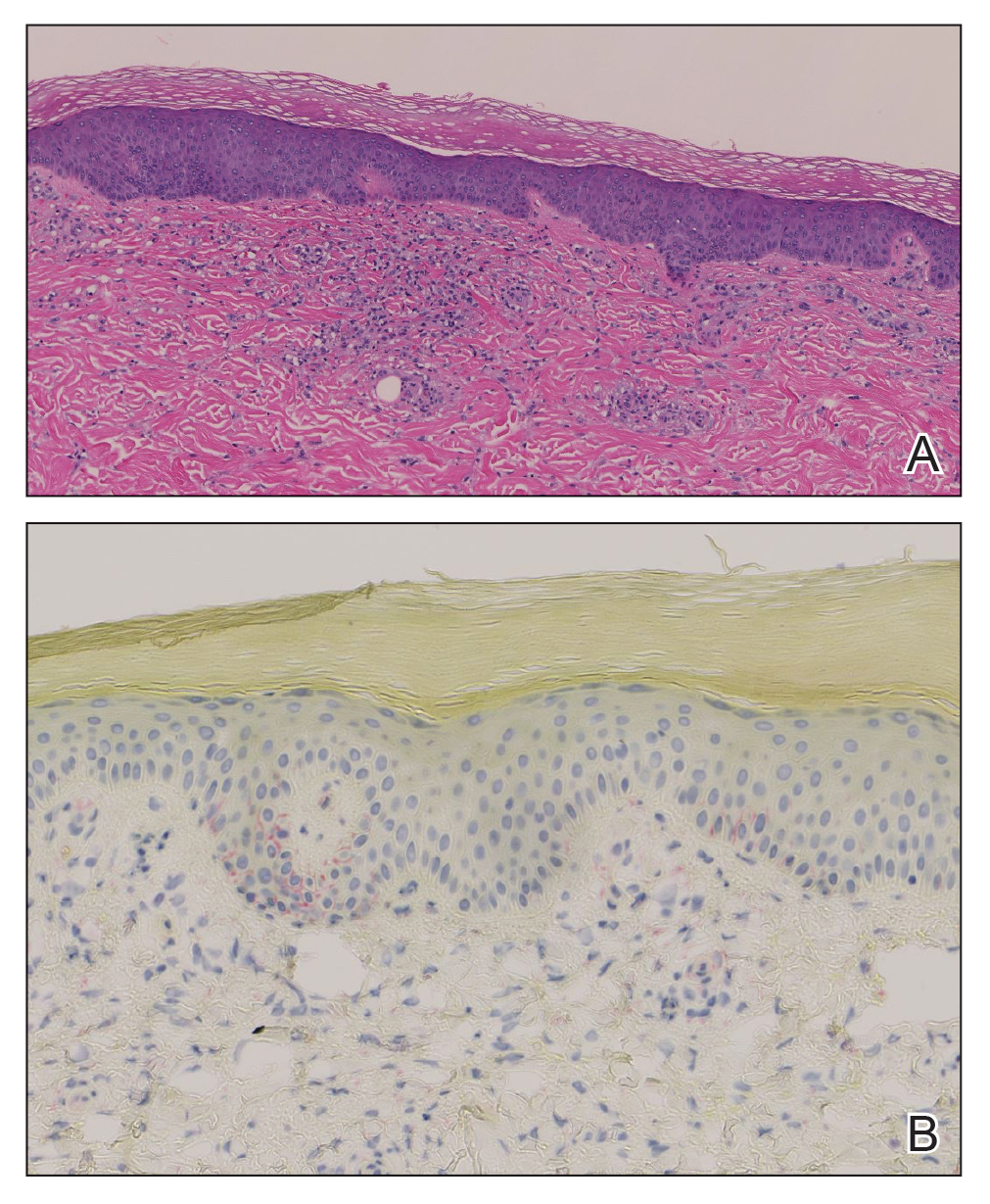
Histopathology demonstrated a mild superficial perivascular and interstitial infiltrate composed of lymphocytes, histiocytes, and rare plasma cells with a background of extravasated erythrocytes (Figure, A). Treponema pallidum staining highlighted multiple spirochetes along the dermoepidermal junction and in the superficial dermis (Figure, B). Direct immunofluorescence was negative. Laboratory workup revealed a reactive rapid plasma reagin screen with a titer of 1:16 and positive IgG and IgM treponemal antibodies. The patient was diagnosed with secondary syphilis and was treated with a single dose of 2.4 million U of intramuscular benzathine penicillin G, with notable improvement of the rash and arthritis symptoms at 2-week follow-up.
Syphilis is a sexually transmitted infection caused by the spirochete T pallidum that progresses through active and latent stages. The incidence of both the primary and secondary stages of syphilis was at a historic low in the year 2000 and has increased annually since then.1 Syphilis is more common in men, and men who have sex with men (MSM) are disproportionately affected. Although the incidence of syphilis in MSM has increased since 2000, rates have slowed, with slight decreases in this population between 2019 and 2020.1 Conversely, rates among women have increased substantially in recent years, suggesting a more recent epidemic affecting heterosexual men and women.2
Classically, the primary stage of syphilis manifests as an asymptomatic papule followed by a painless ulcer (chancre) that heals spontaneously. The secondary stage of syphilis results from dissemination of T pallidum and is characterized by a wide range of mucocutaneous manifestations and prodromal symptoms. The most common cutaneous manifestation is a diffuse, nonpruritic, papulosquamous rash with red-brown scaly macules or papules on the trunk and extremities.3 The palms and soles commonly are involved. Mucosal patches, “snail-track” ulcers in the mouth, and condylomata lata are the characteristic mucosal lesions of secondary syphilis. Mucocutaneous findings typically are preceded by systemic signs including fever, malaise, myalgia, and generalized lymphadenopathy. However, syphilis is considered “the great mimicker,” with new reports of unusual presentations of the disease. In addition to papulosquamous morphologies, pustular, targetoid, psoriasiform, and noduloulcerative (also known as lues maligna) forms of syphilis have been reported.3-5
The histopathologic features of secondary syphilis also are variable. Classically, secondary syphilis demonstrates vacuolar interface dermatitis and acanthosis with slender elongated rete ridges. Other well-known features include endothelial swelling and the presence of plasma cells in most cases.6 However, the histopathologic features of secondary syphilis may vary depending on the morphology of the skin eruption and when the biopsy is taken. Our patient lacked the classic histopathologic features of secondary syphilis. However, because syphilis was in the clinical differential diagnosis, a treponemal stain was ordered and confirmed the diagnosis. Immunohistochemical stains using antibodies to treponemal antigens have a reported sensitivity of 71% to 100% and are highly specific.7 Although the combination of endothelial swelling, interstitial inflammation, irregular acanthosis, and elongated rete ridges should raise the possibility of syphilis, a treponemal stain may be useful to identify spirochetes if clinical suspicion exists.8
Given our patient’s known history of GPA, leukocytoclastic vasculitis was high on the list of differential diagnoses. However, leukocytoclastic vasculitis most classically manifests as petechiae and palpable purpura, and unlike in secondary syphilis, the palms and soles are less commonly involved. Because our patient’s rash was mainly localized to the lower limbs, the differential also included 2 pigmented purpuric dermatoses (PPDs): progressive pigmentary purpura (Schamberg disease) and purpura annularis telangiectodes (Majocchi disease). Progressive pigmentary purpura is the most common manifestation of PPD and appears as cayenne pepper–colored macules that coalesce into golden brown–pigmented patches on the legs.9 Purpura annularis telangiectodes is another variant of PPD that manifests as pinpoint telangiectatic macules that progress to annular hyperpigmented patches with central clearing. Although PPDs frequently occur on the lower extremities, reports of plantar involvement are rare.10 Annular lichen planus manifests as violaceous papules with a clear center; however, it would be atypical for these lesions to be restricted to the feet and ankles. Palmoplantar lichen planus can mimic secondary syphilis clinically, but these cases manifest as hyperkeratotic pruritic papules on the palms and soles in contrast to the faint brown asymptomatic macules noted in our case.11
Our case highlights an unusual presentation of secondary syphilis and demonstrates the challenge of diagnosing this entity on clinical presentation alone. Because this patient lacked the classic clinical and histopathologic features of secondary syphilis, a skin biopsy with positive immunohistochemical staining for treponemal antigens was necessary to make the diagnosis. Given the variability in presentation of secondary syphilis, a biopsy or serologic testing may be necessary to make a proper diagnosis.
- Centers for Disease Control and Prevention. Sexually transmitted disease surveillance 2020. Accessed September 4, 2024. https://www.cdc.gov/std/statistics/2020/2020-SR-4-10-2023.pdf
- Ghanem KG, Ram S, Rice PA. The modern epidemic of syphilis. N Engl J Med. 2020;382:845-854. doi:10.1056/NEJMra1901593
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: historical aspects, microbiology, epidemiology, and clinical manifestations. J Am Acad Dermatol. 2020;82:1-14. doi:10.1016/j.jaad.2019.02.073
- Wu MC, Hsu CK, Lee JY, et al. Erythema multiforme-like secondary syphilis in a HIV-positive bisexual man. Acta Derm Venereol. 2010;90:647-648. doi:10.2340/00015555-0920
- Kopelman H, Lin A, Jorizzo JL. A pemphigus-like presentation of secondary syphilis. JAAD Case Rep. 2019;5:861-864. doi:10.1016/j.jdcr.2019.07.021
- Liu XK, Li J. Histologic features of secondary syphilis. Dermatology. 2020;236:145-150. doi:10.1159/000502641
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: laboratory diagnosis, management, and prevention. J Am Acad Dermatol. 2020;82:17-28. doi:10.1016/j.jaad.2019.02.074
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:1025-1030. doi:10.1016/j.jaad.2015.08.062
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410. doi:10.5021/ad.2015.27.4.404
- Sivendran M, Mowad C. Hyperpigmented patches on shins, palms, and soles. JAMA Dermatol. 2013;149:223. doi:10.1001/2013.jamadermatol.652a
- Kim YS, Kim MH, Kim CW, et al. A case of palmoplantar lichen planus mimicking secondary syphilis. Ann Dermatol. 2009;21:429-431.doi:10.5021/ad.2009.21.4.429
THE DIAGNOSIS: Secondary Syphilis
Histopathology demonstrated a mild superficial perivascular and interstitial infiltrate composed of lymphocytes, histiocytes, and rare plasma cells with a background of extravasated erythrocytes (Figure, A). Treponema pallidum staining highlighted multiple spirochetes along the dermoepidermal junction and in the superficial dermis (Figure, B). Direct immunofluorescence was negative. Laboratory workup revealed a reactive rapid plasma reagin screen with a titer of 1:16 and positive IgG and IgM treponemal antibodies. The patient was diagnosed with secondary syphilis and was treated with a single dose of 2.4 million U of intramuscular benzathine penicillin G, with notable improvement of the rash and arthritis symptoms at 2-week follow-up.
Syphilis is a sexually transmitted infection caused by the spirochete T pallidum that progresses through active and latent stages. The incidence of both the primary and secondary stages of syphilis was at a historic low in the year 2000 and has increased annually since then.1 Syphilis is more common in men, and men who have sex with men (MSM) are disproportionately affected. Although the incidence of syphilis in MSM has increased since 2000, rates have slowed, with slight decreases in this population between 2019 and 2020.1 Conversely, rates among women have increased substantially in recent years, suggesting a more recent epidemic affecting heterosexual men and women.2
Classically, the primary stage of syphilis manifests as an asymptomatic papule followed by a painless ulcer (chancre) that heals spontaneously. The secondary stage of syphilis results from dissemination of T pallidum and is characterized by a wide range of mucocutaneous manifestations and prodromal symptoms. The most common cutaneous manifestation is a diffuse, nonpruritic, papulosquamous rash with red-brown scaly macules or papules on the trunk and extremities.3 The palms and soles commonly are involved. Mucosal patches, “snail-track” ulcers in the mouth, and condylomata lata are the characteristic mucosal lesions of secondary syphilis. Mucocutaneous findings typically are preceded by systemic signs including fever, malaise, myalgia, and generalized lymphadenopathy. However, syphilis is considered “the great mimicker,” with new reports of unusual presentations of the disease. In addition to papulosquamous morphologies, pustular, targetoid, psoriasiform, and noduloulcerative (also known as lues maligna) forms of syphilis have been reported.3-5
The histopathologic features of secondary syphilis also are variable. Classically, secondary syphilis demonstrates vacuolar interface dermatitis and acanthosis with slender elongated rete ridges. Other well-known features include endothelial swelling and the presence of plasma cells in most cases.6 However, the histopathologic features of secondary syphilis may vary depending on the morphology of the skin eruption and when the biopsy is taken. Our patient lacked the classic histopathologic features of secondary syphilis. However, because syphilis was in the clinical differential diagnosis, a treponemal stain was ordered and confirmed the diagnosis. Immunohistochemical stains using antibodies to treponemal antigens have a reported sensitivity of 71% to 100% and are highly specific.7 Although the combination of endothelial swelling, interstitial inflammation, irregular acanthosis, and elongated rete ridges should raise the possibility of syphilis, a treponemal stain may be useful to identify spirochetes if clinical suspicion exists.8
Given our patient’s known history of GPA, leukocytoclastic vasculitis was high on the list of differential diagnoses. However, leukocytoclastic vasculitis most classically manifests as petechiae and palpable purpura, and unlike in secondary syphilis, the palms and soles are less commonly involved. Because our patient’s rash was mainly localized to the lower limbs, the differential also included 2 pigmented purpuric dermatoses (PPDs): progressive pigmentary purpura (Schamberg disease) and purpura annularis telangiectodes (Majocchi disease). Progressive pigmentary purpura is the most common manifestation of PPD and appears as cayenne pepper–colored macules that coalesce into golden brown–pigmented patches on the legs.9 Purpura annularis telangiectodes is another variant of PPD that manifests as pinpoint telangiectatic macules that progress to annular hyperpigmented patches with central clearing. Although PPDs frequently occur on the lower extremities, reports of plantar involvement are rare.10 Annular lichen planus manifests as violaceous papules with a clear center; however, it would be atypical for these lesions to be restricted to the feet and ankles. Palmoplantar lichen planus can mimic secondary syphilis clinically, but these cases manifest as hyperkeratotic pruritic papules on the palms and soles in contrast to the faint brown asymptomatic macules noted in our case.11
Our case highlights an unusual presentation of secondary syphilis and demonstrates the challenge of diagnosing this entity on clinical presentation alone. Because this patient lacked the classic clinical and histopathologic features of secondary syphilis, a skin biopsy with positive immunohistochemical staining for treponemal antigens was necessary to make the diagnosis. Given the variability in presentation of secondary syphilis, a biopsy or serologic testing may be necessary to make a proper diagnosis.
THE DIAGNOSIS: Secondary Syphilis
Histopathology demonstrated a mild superficial perivascular and interstitial infiltrate composed of lymphocytes, histiocytes, and rare plasma cells with a background of extravasated erythrocytes (Figure, A). Treponema pallidum staining highlighted multiple spirochetes along the dermoepidermal junction and in the superficial dermis (Figure, B). Direct immunofluorescence was negative. Laboratory workup revealed a reactive rapid plasma reagin screen with a titer of 1:16 and positive IgG and IgM treponemal antibodies. The patient was diagnosed with secondary syphilis and was treated with a single dose of 2.4 million U of intramuscular benzathine penicillin G, with notable improvement of the rash and arthritis symptoms at 2-week follow-up.
Syphilis is a sexually transmitted infection caused by the spirochete T pallidum that progresses through active and latent stages. The incidence of both the primary and secondary stages of syphilis was at a historic low in the year 2000 and has increased annually since then.1 Syphilis is more common in men, and men who have sex with men (MSM) are disproportionately affected. Although the incidence of syphilis in MSM has increased since 2000, rates have slowed, with slight decreases in this population between 2019 and 2020.1 Conversely, rates among women have increased substantially in recent years, suggesting a more recent epidemic affecting heterosexual men and women.2
Classically, the primary stage of syphilis manifests as an asymptomatic papule followed by a painless ulcer (chancre) that heals spontaneously. The secondary stage of syphilis results from dissemination of T pallidum and is characterized by a wide range of mucocutaneous manifestations and prodromal symptoms. The most common cutaneous manifestation is a diffuse, nonpruritic, papulosquamous rash with red-brown scaly macules or papules on the trunk and extremities.3 The palms and soles commonly are involved. Mucosal patches, “snail-track” ulcers in the mouth, and condylomata lata are the characteristic mucosal lesions of secondary syphilis. Mucocutaneous findings typically are preceded by systemic signs including fever, malaise, myalgia, and generalized lymphadenopathy. However, syphilis is considered “the great mimicker,” with new reports of unusual presentations of the disease. In addition to papulosquamous morphologies, pustular, targetoid, psoriasiform, and noduloulcerative (also known as lues maligna) forms of syphilis have been reported.3-5
The histopathologic features of secondary syphilis also are variable. Classically, secondary syphilis demonstrates vacuolar interface dermatitis and acanthosis with slender elongated rete ridges. Other well-known features include endothelial swelling and the presence of plasma cells in most cases.6 However, the histopathologic features of secondary syphilis may vary depending on the morphology of the skin eruption and when the biopsy is taken. Our patient lacked the classic histopathologic features of secondary syphilis. However, because syphilis was in the clinical differential diagnosis, a treponemal stain was ordered and confirmed the diagnosis. Immunohistochemical stains using antibodies to treponemal antigens have a reported sensitivity of 71% to 100% and are highly specific.7 Although the combination of endothelial swelling, interstitial inflammation, irregular acanthosis, and elongated rete ridges should raise the possibility of syphilis, a treponemal stain may be useful to identify spirochetes if clinical suspicion exists.8
Given our patient’s known history of GPA, leukocytoclastic vasculitis was high on the list of differential diagnoses. However, leukocytoclastic vasculitis most classically manifests as petechiae and palpable purpura, and unlike in secondary syphilis, the palms and soles are less commonly involved. Because our patient’s rash was mainly localized to the lower limbs, the differential also included 2 pigmented purpuric dermatoses (PPDs): progressive pigmentary purpura (Schamberg disease) and purpura annularis telangiectodes (Majocchi disease). Progressive pigmentary purpura is the most common manifestation of PPD and appears as cayenne pepper–colored macules that coalesce into golden brown–pigmented patches on the legs.9 Purpura annularis telangiectodes is another variant of PPD that manifests as pinpoint telangiectatic macules that progress to annular hyperpigmented patches with central clearing. Although PPDs frequently occur on the lower extremities, reports of plantar involvement are rare.10 Annular lichen planus manifests as violaceous papules with a clear center; however, it would be atypical for these lesions to be restricted to the feet and ankles. Palmoplantar lichen planus can mimic secondary syphilis clinically, but these cases manifest as hyperkeratotic pruritic papules on the palms and soles in contrast to the faint brown asymptomatic macules noted in our case.11
Our case highlights an unusual presentation of secondary syphilis and demonstrates the challenge of diagnosing this entity on clinical presentation alone. Because this patient lacked the classic clinical and histopathologic features of secondary syphilis, a skin biopsy with positive immunohistochemical staining for treponemal antigens was necessary to make the diagnosis. Given the variability in presentation of secondary syphilis, a biopsy or serologic testing may be necessary to make a proper diagnosis.
- Centers for Disease Control and Prevention. Sexually transmitted disease surveillance 2020. Accessed September 4, 2024. https://www.cdc.gov/std/statistics/2020/2020-SR-4-10-2023.pdf
- Ghanem KG, Ram S, Rice PA. The modern epidemic of syphilis. N Engl J Med. 2020;382:845-854. doi:10.1056/NEJMra1901593
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: historical aspects, microbiology, epidemiology, and clinical manifestations. J Am Acad Dermatol. 2020;82:1-14. doi:10.1016/j.jaad.2019.02.073
- Wu MC, Hsu CK, Lee JY, et al. Erythema multiforme-like secondary syphilis in a HIV-positive bisexual man. Acta Derm Venereol. 2010;90:647-648. doi:10.2340/00015555-0920
- Kopelman H, Lin A, Jorizzo JL. A pemphigus-like presentation of secondary syphilis. JAAD Case Rep. 2019;5:861-864. doi:10.1016/j.jdcr.2019.07.021
- Liu XK, Li J. Histologic features of secondary syphilis. Dermatology. 2020;236:145-150. doi:10.1159/000502641
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: laboratory diagnosis, management, and prevention. J Am Acad Dermatol. 2020;82:17-28. doi:10.1016/j.jaad.2019.02.074
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:1025-1030. doi:10.1016/j.jaad.2015.08.062
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410. doi:10.5021/ad.2015.27.4.404
- Sivendran M, Mowad C. Hyperpigmented patches on shins, palms, and soles. JAMA Dermatol. 2013;149:223. doi:10.1001/2013.jamadermatol.652a
- Kim YS, Kim MH, Kim CW, et al. A case of palmoplantar lichen planus mimicking secondary syphilis. Ann Dermatol. 2009;21:429-431.doi:10.5021/ad.2009.21.4.429
- Centers for Disease Control and Prevention. Sexually transmitted disease surveillance 2020. Accessed September 4, 2024. https://www.cdc.gov/std/statistics/2020/2020-SR-4-10-2023.pdf
- Ghanem KG, Ram S, Rice PA. The modern epidemic of syphilis. N Engl J Med. 2020;382:845-854. doi:10.1056/NEJMra1901593
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: historical aspects, microbiology, epidemiology, and clinical manifestations. J Am Acad Dermatol. 2020;82:1-14. doi:10.1016/j.jaad.2019.02.073
- Wu MC, Hsu CK, Lee JY, et al. Erythema multiforme-like secondary syphilis in a HIV-positive bisexual man. Acta Derm Venereol. 2010;90:647-648. doi:10.2340/00015555-0920
- Kopelman H, Lin A, Jorizzo JL. A pemphigus-like presentation of secondary syphilis. JAAD Case Rep. 2019;5:861-864. doi:10.1016/j.jdcr.2019.07.021
- Liu XK, Li J. Histologic features of secondary syphilis. Dermatology. 2020;236:145-150. doi:10.1159/000502641
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: laboratory diagnosis, management, and prevention. J Am Acad Dermatol. 2020;82:17-28. doi:10.1016/j.jaad.2019.02.074
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:1025-1030. doi:10.1016/j.jaad.2015.08.062
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410. doi:10.5021/ad.2015.27.4.404
- Sivendran M, Mowad C. Hyperpigmented patches on shins, palms, and soles. JAMA Dermatol. 2013;149:223. doi:10.1001/2013.jamadermatol.652a
- Kim YS, Kim MH, Kim CW, et al. A case of palmoplantar lichen planus mimicking secondary syphilis. Ann Dermatol. 2009;21:429-431.doi:10.5021/ad.2009.21.4.429
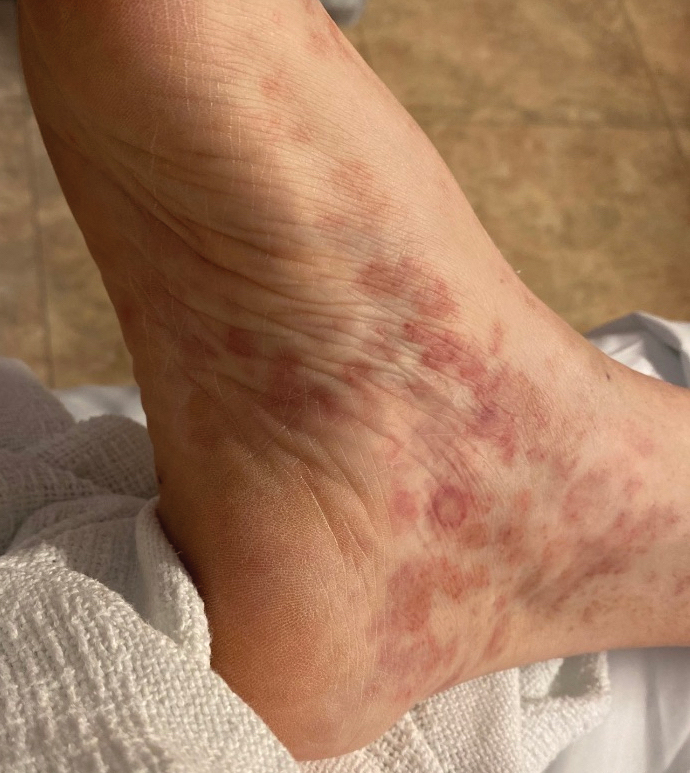
A 59-year-old man presented with a nontender nonpruritic rash on the feet of 2 days’ duration. The patient had a several-year history of granulomatosis with polyangiitis (GPA) and was taking methotrexate and prednisone. The rash appeared suddenly—first on the right foot and then on the left foot—and was preceded by 1 week of worsening polyarthralgia, most notably in the ankles. He denied any fever, chills, sore throat, or weight loss. His typical GPA symptoms included inflammatory arthritis, scleritis, leukocytoclastic vasculitis, and sinonasal and renal involvement. He recently experienced exacerbation of inflammatory arthritis that required an increase in the prednisone dosage (from 40 mg to 60 mg daily), but there were no other GPA symptoms. He had a history of multiple female sexual partners but no known history of HIV and no recent testing for sexually transmitted infections. Hepatitis C antibody testing performed 5 years earlier was nonreactive. He denied any illicit drug use, recent travel, sick contacts, or new medications.
Dermatologic examination revealed nonscaly, clustered, red-brown macules, some with central clearing, on the medial and lateral aspects of the feet and ankles with a few faint copper-colored macules on the palms and soles. The ankles had full range of motion with no edema or effusion. There were no oral or genital lesions. The remainder of the skin examination was normal. Punch biopsies of skin on the left foot were obtained for histopathology and direct immunofluorescence.
Multiple Draining Sinus Tracts on the Thigh
The Diagnosis: Mycobacterial Infection
An injury sustained in a wet environment that results in chronic indolent abscesses, nodules, or draining sinus tracts suggests a mycobacterial infection. In our patient, a culture revealed MycobacteriuM fortuitum, which is classified in the rapid grower nontuberculous mycobacteria (NTM) group, along with Mycobacterium chelonae and Mycobacterium abscessus.1 The patient’s history of skin injury while cutting wet grass and the common presence of M fortuitum in the environment suggested that the organism entered the wound. The patient healed completely following surgical excision and a 2-month course of clarithromycin 1 g daily and rifampin 600 mg daily.
MycobacteriuM fortuitum was first isolated from an amphibian source in 1905 and later identified in a human with cutaneous infection in 1938. It commonly is found in soil and water.2 Skin and soft-tissue infections with M fortuitum usually are acquired from direct entry of the organism through a damaged skin barrier from trauma, medical injection, surgery, or tattoo placement.2,3
Skin lesions caused by NTM often are nonspecific and can mimic a variety of other dermatologic conditions, making clinical diagnosis challenging. As such, cutaneous manifestations of M fortuitum infection can include recurrent cutaneous abscesses, nodular lesions, chronic discharging sinuses, cellulitis, and surgical site infections.4 Although cutaneous infection with M fortuitum classically manifests with a single subcutaneous nodule at the site of trauma or surgery,5 it also can manifest as multiple draining sinus tracts, as seen in our patient. Hence, the diagnosis and treatment of cutaneous NTM infection is challenging, especially when M fortuitum skin manifestations can take up to 4 to 6 weeks to develop after inoculation. Diagnosis often requires a detailed patient history, tissue cultures, and histopathology.5
In recent years, rapid detection with polymerase chain reaction (PCR) techniques has been employed more widely. Notably, a molecular system based on multiplex real-time PCR with high-resolution melting was shown to have a sensitivity of up to 54% for distinguishing M fortuitum from other NTM.6 More recently, a 2-step real-time PCR method has demonstrated diagnostic sensitivity and specificity for differentiating NTM from Mycobacterium tuberculosis infections and identifying the causative NTM agent.7
Compared to immunocompetent individuals, those who are immunocompromised are more susceptible to less pathogenic strains of NTM, which can cause dissemination and lead to tenosynovitis, myositis, osteomyelitis, and septic arthritis.8-12 Nonetheless, cases of infections with NTM—including M fortuitum—are becoming harder to treat. Several single nucleotide polymorphisms and point mutations have been demonstrated in the ribosomal RNA methylase gene erm(39) related to clarithromycin resistance and in the rrl gene related to linezolid resistance.13 Due to increasing inducible resistance to common classes of antibiotics, such as macrolides and linezolid, treatment of M fortuitum requires multidrug regimens.13,14 Drug susceptibility testing also may be required, as M fortuitum has shown low resistance to tigecycline, tetracycline, cefmetazole, imipenem, and aminoglycosides (eg, amikacin, tobramycin, neomycin, gentamycin). Surgery is an important adjunctive tool in treating M fortuitum infections; patients with a single lesion are more likely to undergo surgical treatment alone or in combination with antibiotic therapy.15 More recently, antimicrobial photodynamic therapy has been explored as an alternative to eliminate NTM, including M fortuitum.16
The differential diagnosis for skin lesions manifesting with draining fistulae and sinus tracts includes conditions with infectious (cellulitis and chromomycosis) and inflammatory (pyoderma gangrenosum [PG] and hidradenitis suppurativa [HS]) causes.
Cellulitis is a common infection of the skin and subcutaneous tissue that predominantly is caused by gram-positive organisms such as β-hemolytic streptococci.17 Clinical manifestations include acute skin erythema, swelling, tenderness, and warmth. The legs are the most common sites of infection, but any area of the skin can be involved.17 Cellulitis comprises 10% of all infectious disease hospitalizations and up to 11% of all dermatologic admissions.18,19 It frequently is misdiagnosed, perhaps due to the lack of a reliable confirmatory laboratory test or imaging study, in addition to the plethora of diseases that mimic cellulitis, such as stasis dermatitis, lipodermatosclerosis, contact dermatitis, lymphedema, eosinophilic cellulitis, and papular urticaria.20,21 The consequences of misdiagnosis include but are not limited to unnecessary hospitalizations, inappropriate antibiotic use, and delayed management of the disease; thus, there is an urgent need for a reliable standard test to confirm the diagnosis, especially among nonspecialist physicians. 20 Most patients with uncomplicated cellulitis can be treated with empiric oral antibiotics that target β-hemolytic streptococci (ie, penicillin V potassium, amoxicillin).17 Methicillin-resistant Staphylococcus aureus coverage generally is unnecessary for nonpurulent cellulitis, but clinicians can consider adding amoxicillin-clavulanate, dicloxacillin, and cephalexin to the regimen. For purulent cellulitis, incision and drainage should be performed. In severe cases that manifest with sepsis, altered mental status, or hemodynamic instability, inpatient management is required.17
Chromomycosis (also known as chromoblastomycosis) is a chronic, indolent, granulomatous, suppurative mycosis of the skin and subcutaneous tissue22 that is caused by traumatic inoculation of various fungi of the order Chaetothyriales and family Herpotrichiellaceae, which are present in soil, plants, and decomposing wood. Chromomycosis is prevalent in tropical and subtropical regions.23,24 Clinically, it manifests as oligosymptomatic or asymptomatic lesions around an infection site that can manifest as papules with centrifugal growth evolving into nodular, verrucous, plaque, tumoral, or atrophic forms.22 Diagnosis is made with direct microscopy using potassium hydroxide, which reveals muriform bodies. Fungal culture in Sabouraud agar also can be used to isolate the causative pathogen.22 Unfortunately, chromomycosis is difficult to treat, with low cure rates and high relapse rates. Antifungal agents combined with surgery, cryotherapy, or thermotherapy often are used, with cure rates ranging from 15% to 80%.22,25
Pyoderma gangrenosum is a reactive noninfectious inflammatory dermatosis associated with inflammatory bowel disease and rheumatoid arthritis. The exact etiology is not clearly understood, but it generally is considered an autoinflammatory disorder.26 The most common form—classical PG—occurs in approximately 85% of cases and manifests as a painful erythematous lesion that progresses to a blistered or necrotic ulcer. It primarily affects the lower legs but can occur in other body sites.27 The diagnosis is based on clinical symptoms after excluding other similar conditions; histopathology of biopsied wound tissues often are required for confirmation. Treatment of PG starts with fast-acting immunosuppressive drugs (corticosteroids and/or cyclosporine) followed by slowacting immunosuppressive drugs (biologics).26
Hidradenitis suppurativa is a chronic recurrent disease of the hair follicle unit that develops after puberty.28 Clinically, HS manifests with painful nodules, abscesses, chronically draining fistulas, and scarring in areas of the body rich in apocrine glands.29,30 Treatment of HS is challenging due to its diverse clinical manifestations and unclear etiology. Topical therapy, systemic treatments, biologic agents, surgery, and light therapy have shown variable results.28,31
- Franco-Paredes C, Marcos LA, Henao-Martínez AF, et al. Cutaneous mycobacterial infections. Clin Microbiol Rev. 2018;32: E00069-18. doi:10.1128/CMR.00069-18
- Brown TH. The rapidly growing mycobacteria—MycobacteriuM fortuitum and Mycobacterium chelonae. Infect Control. 1985;6:283-238. doi:10.1017/s0195941700061762
- Hooper J; Beltrami EJ; Santoro F; et al. Remember the fite: a case of cutaneous MycobacteriuM fortuitum infection. Am J Dermatopathol. 2023;45:214-215. doi:10.1097/DAD.0000000000002336
- Franco-Paredes C, Chastain DB, Allen L, et al. Overview of cutaneous mycobacterial infections. Curr Trop Med Rep. 2018;5:228-232. doi:10.1007/s40475-018-0161-7
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-77. doi:10.1016/j.det.2015.03.017
- Peixoto ADS, Montenegro LML, Lima AS, et al. Identification of nontuberculous mycobacteria species by multiplex real-time PCR with high-resolution melting. Rev Soc Bras Med Trop. 2020;53:E20200211. doi:10.1590/0037-8682-0211-2020
- Park J, Kwak N, Chae JC, et al. A two-step real-time PCR method to identify Mycobacterium tuberculosis infections and six dominant nontuberculous mycobacterial infections from clinical specimens. Microbiol Spectr. 2023:E0160623. doi:10.1128/spectrum.01606-23
- Fowler J, Mahlen SD. Localized cutaneous infections in immunocompetent individuals due to rapidly growing mycobacteria. Arch Pathol Lab Med. 2014;138:1106-1109. doi:10.5858/arpa.2012-0203-RS
- Gardini G, Gregori N, Matteelli A, et al. Mycobacterial skin infection. Curr Opin Infect Dis. 2022;35:79-87. doi:10.1097/QCO.0000000000000820
- Wang SH, Pancholi P. Mycobacterial skin and soft tissue infection. Curr Infect Dis Rep. 2014;16:438. doi:10.1007/s11908-014-0438-5
- Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416. doi:10.1164/rccm.200604-571ST
- Mougari F, Guglielmetti L, Raskine L, et al. Infections caused by Mycobacterium abscessus: epidemiology, diagnostic tools and treatment. Expert Rev Anti Infect Ther. 2016;14:1139-1154. doi:10.1080/14787210.201 6.1238304
- Tu HZ, Lee HS, Chen YS, et al. High rates of antimicrobial resistance in rapidly growing mycobacterial infections in Taiwan. Pathogens. 2022;11:969. doi:10.3390/pathogens11090969
- Hashemzadeh M, Zadegan Dezfuli AA, Khosravi AD, et al. F requency of mutations in erm(39) related to clarithromycin resistance and in rrl related to linezolid resistance in clinical isolates of MycobacteriuM fortuitum in Iran. Acta Microbiol Immunol Hung. 2023;70:167-176. doi:10.1556/030.2023.02020
- Uslan DZ, Kowalski TJ, Wengenack NL, et al. Skin and soft tissue infections due to rapidly growing mycobacteria: comparison of clinical features, treatment, and susceptibility. Arch Dermatol. 2006;142:1287-1292. doi:10.1001/archderm.142.10.1287
- Miretti M, Juri L, Peralta A, et al. Photoinactivation of non-tuberculous mycobacteria using Zn-phthalocyanine loaded into liposomes. Tuberculosis (Edinb). 2022;136:102247. doi:10.1016/j.tube.2022.102247
- Bystritsky RJ. Cellulitis. Infect Dis Clin North Am. 2021;35:49-60. doi:10.1016/j.idc.2020.10.002
- Christensen K, Holman R, Steiner C, et al. Infectious disease hospitalizations in the United States. Clin Infect Dis. 2009;49:1025-1035. doi:10.1086/605562
- Yang JJ, Maloney NJ, Bach DQ, et al. Dermatology in the emergency department: prescriptions, rates of inpatient admission, and predictors of high utilization in the United States from 1996 to 2012. J Am Acad Dermatol. 2021;84:1480-1483. doi:10.1016/J.JAAD.2020.07.055
- Cutler TS, Jannat-Khah DP, Kam B, et al. Prevalence of misdiagnosis of cellulitis: a systematic review and meta-analysis. J Hosp Med. 2023;18:254-261. doi:10.1002/jhm.12977
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-52. doi:10.3949/ccjm.79a.11121
- Brito AC, Bittencourt MJS. Chromoblastomycosis: an etiological, epidemiological, clinical, diagnostic, and treatment update. An Bras Dermatol. 2018;93:495-506. doi:10.1590/abd1806-4841.20187321
- McGinnis MR. Chromoblastomycosis and phaeohyphomycosis: new concepts, diagnosis, and mycology. J Am Acad Dermatol. 1983;8:1-16.
- Rubin HA, Bruce S, Rosen T, et al. Evidence for percutaneous inoculation as the mode of transmission for chromoblastomycosis. J Am Acad Dermatol. 1991;25:951-954.
- Bonifaz A, Paredes-Solís V, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Maverakis E, Marzano AV, Le ST, et al. Pyoderma gangrenosum. Nat Rev Dis Primers. 2020;6:81. doi:10.1038/s41572-020-0213-x
- George C, Deroide F, Rustin M. Pyoderma gangrenosum—a guide to diagnosis and management. Clin Med (Lond). 2019;19:224-228. doi:10.7861/clinmedicine.19-3-224
- Narla S, Lyons AB, Hamzavi IH. The most recent advances in understanding and managing hidradenitis suppurativa. F1000Res. 2020;9:F1000 Faculty Rev-1049. doi:10.12688/f1000research.26083.1
- Garg A, Lavian J, Lin G, et al. Incidence of hidradenitis suppurativa in the United States: a sex- and age-adjusted population analysis. J Am Acad Dermatol. 2017;77:118-122. doi:10.1016/j.jaad.2017.02.005
- Daxhelet M, Suppa M, White J, et al. Proposed definitions of typical lesions in hidradenitis suppurativa. Dermatology. 2020;236:431-438. doi:10.1159/000507348
- Amat-Samaranch V, Agut-Busquet E, Vilarrasa E, et al. New perspectives on the treatment of hidradenitis suppurativa. Ther Adv Chronic Dis. 2021;12:20406223211055920. doi:10.1177/20406223211055920
The Diagnosis: Mycobacterial Infection
An injury sustained in a wet environment that results in chronic indolent abscesses, nodules, or draining sinus tracts suggests a mycobacterial infection. In our patient, a culture revealed MycobacteriuM fortuitum, which is classified in the rapid grower nontuberculous mycobacteria (NTM) group, along with Mycobacterium chelonae and Mycobacterium abscessus.1 The patient’s history of skin injury while cutting wet grass and the common presence of M fortuitum in the environment suggested that the organism entered the wound. The patient healed completely following surgical excision and a 2-month course of clarithromycin 1 g daily and rifampin 600 mg daily.
MycobacteriuM fortuitum was first isolated from an amphibian source in 1905 and later identified in a human with cutaneous infection in 1938. It commonly is found in soil and water.2 Skin and soft-tissue infections with M fortuitum usually are acquired from direct entry of the organism through a damaged skin barrier from trauma, medical injection, surgery, or tattoo placement.2,3
Skin lesions caused by NTM often are nonspecific and can mimic a variety of other dermatologic conditions, making clinical diagnosis challenging. As such, cutaneous manifestations of M fortuitum infection can include recurrent cutaneous abscesses, nodular lesions, chronic discharging sinuses, cellulitis, and surgical site infections.4 Although cutaneous infection with M fortuitum classically manifests with a single subcutaneous nodule at the site of trauma or surgery,5 it also can manifest as multiple draining sinus tracts, as seen in our patient. Hence, the diagnosis and treatment of cutaneous NTM infection is challenging, especially when M fortuitum skin manifestations can take up to 4 to 6 weeks to develop after inoculation. Diagnosis often requires a detailed patient history, tissue cultures, and histopathology.5
In recent years, rapid detection with polymerase chain reaction (PCR) techniques has been employed more widely. Notably, a molecular system based on multiplex real-time PCR with high-resolution melting was shown to have a sensitivity of up to 54% for distinguishing M fortuitum from other NTM.6 More recently, a 2-step real-time PCR method has demonstrated diagnostic sensitivity and specificity for differentiating NTM from Mycobacterium tuberculosis infections and identifying the causative NTM agent.7
Compared to immunocompetent individuals, those who are immunocompromised are more susceptible to less pathogenic strains of NTM, which can cause dissemination and lead to tenosynovitis, myositis, osteomyelitis, and septic arthritis.8-12 Nonetheless, cases of infections with NTM—including M fortuitum—are becoming harder to treat. Several single nucleotide polymorphisms and point mutations have been demonstrated in the ribosomal RNA methylase gene erm(39) related to clarithromycin resistance and in the rrl gene related to linezolid resistance.13 Due to increasing inducible resistance to common classes of antibiotics, such as macrolides and linezolid, treatment of M fortuitum requires multidrug regimens.13,14 Drug susceptibility testing also may be required, as M fortuitum has shown low resistance to tigecycline, tetracycline, cefmetazole, imipenem, and aminoglycosides (eg, amikacin, tobramycin, neomycin, gentamycin). Surgery is an important adjunctive tool in treating M fortuitum infections; patients with a single lesion are more likely to undergo surgical treatment alone or in combination with antibiotic therapy.15 More recently, antimicrobial photodynamic therapy has been explored as an alternative to eliminate NTM, including M fortuitum.16
The differential diagnosis for skin lesions manifesting with draining fistulae and sinus tracts includes conditions with infectious (cellulitis and chromomycosis) and inflammatory (pyoderma gangrenosum [PG] and hidradenitis suppurativa [HS]) causes.
Cellulitis is a common infection of the skin and subcutaneous tissue that predominantly is caused by gram-positive organisms such as β-hemolytic streptococci.17 Clinical manifestations include acute skin erythema, swelling, tenderness, and warmth. The legs are the most common sites of infection, but any area of the skin can be involved.17 Cellulitis comprises 10% of all infectious disease hospitalizations and up to 11% of all dermatologic admissions.18,19 It frequently is misdiagnosed, perhaps due to the lack of a reliable confirmatory laboratory test or imaging study, in addition to the plethora of diseases that mimic cellulitis, such as stasis dermatitis, lipodermatosclerosis, contact dermatitis, lymphedema, eosinophilic cellulitis, and papular urticaria.20,21 The consequences of misdiagnosis include but are not limited to unnecessary hospitalizations, inappropriate antibiotic use, and delayed management of the disease; thus, there is an urgent need for a reliable standard test to confirm the diagnosis, especially among nonspecialist physicians. 20 Most patients with uncomplicated cellulitis can be treated with empiric oral antibiotics that target β-hemolytic streptococci (ie, penicillin V potassium, amoxicillin).17 Methicillin-resistant Staphylococcus aureus coverage generally is unnecessary for nonpurulent cellulitis, but clinicians can consider adding amoxicillin-clavulanate, dicloxacillin, and cephalexin to the regimen. For purulent cellulitis, incision and drainage should be performed. In severe cases that manifest with sepsis, altered mental status, or hemodynamic instability, inpatient management is required.17
Chromomycosis (also known as chromoblastomycosis) is a chronic, indolent, granulomatous, suppurative mycosis of the skin and subcutaneous tissue22 that is caused by traumatic inoculation of various fungi of the order Chaetothyriales and family Herpotrichiellaceae, which are present in soil, plants, and decomposing wood. Chromomycosis is prevalent in tropical and subtropical regions.23,24 Clinically, it manifests as oligosymptomatic or asymptomatic lesions around an infection site that can manifest as papules with centrifugal growth evolving into nodular, verrucous, plaque, tumoral, or atrophic forms.22 Diagnosis is made with direct microscopy using potassium hydroxide, which reveals muriform bodies. Fungal culture in Sabouraud agar also can be used to isolate the causative pathogen.22 Unfortunately, chromomycosis is difficult to treat, with low cure rates and high relapse rates. Antifungal agents combined with surgery, cryotherapy, or thermotherapy often are used, with cure rates ranging from 15% to 80%.22,25
Pyoderma gangrenosum is a reactive noninfectious inflammatory dermatosis associated with inflammatory bowel disease and rheumatoid arthritis. The exact etiology is not clearly understood, but it generally is considered an autoinflammatory disorder.26 The most common form—classical PG—occurs in approximately 85% of cases and manifests as a painful erythematous lesion that progresses to a blistered or necrotic ulcer. It primarily affects the lower legs but can occur in other body sites.27 The diagnosis is based on clinical symptoms after excluding other similar conditions; histopathology of biopsied wound tissues often are required for confirmation. Treatment of PG starts with fast-acting immunosuppressive drugs (corticosteroids and/or cyclosporine) followed by slowacting immunosuppressive drugs (biologics).26
Hidradenitis suppurativa is a chronic recurrent disease of the hair follicle unit that develops after puberty.28 Clinically, HS manifests with painful nodules, abscesses, chronically draining fistulas, and scarring in areas of the body rich in apocrine glands.29,30 Treatment of HS is challenging due to its diverse clinical manifestations and unclear etiology. Topical therapy, systemic treatments, biologic agents, surgery, and light therapy have shown variable results.28,31
The Diagnosis: Mycobacterial Infection
An injury sustained in a wet environment that results in chronic indolent abscesses, nodules, or draining sinus tracts suggests a mycobacterial infection. In our patient, a culture revealed MycobacteriuM fortuitum, which is classified in the rapid grower nontuberculous mycobacteria (NTM) group, along with Mycobacterium chelonae and Mycobacterium abscessus.1 The patient’s history of skin injury while cutting wet grass and the common presence of M fortuitum in the environment suggested that the organism entered the wound. The patient healed completely following surgical excision and a 2-month course of clarithromycin 1 g daily and rifampin 600 mg daily.
MycobacteriuM fortuitum was first isolated from an amphibian source in 1905 and later identified in a human with cutaneous infection in 1938. It commonly is found in soil and water.2 Skin and soft-tissue infections with M fortuitum usually are acquired from direct entry of the organism through a damaged skin barrier from trauma, medical injection, surgery, or tattoo placement.2,3
Skin lesions caused by NTM often are nonspecific and can mimic a variety of other dermatologic conditions, making clinical diagnosis challenging. As such, cutaneous manifestations of M fortuitum infection can include recurrent cutaneous abscesses, nodular lesions, chronic discharging sinuses, cellulitis, and surgical site infections.4 Although cutaneous infection with M fortuitum classically manifests with a single subcutaneous nodule at the site of trauma or surgery,5 it also can manifest as multiple draining sinus tracts, as seen in our patient. Hence, the diagnosis and treatment of cutaneous NTM infection is challenging, especially when M fortuitum skin manifestations can take up to 4 to 6 weeks to develop after inoculation. Diagnosis often requires a detailed patient history, tissue cultures, and histopathology.5
In recent years, rapid detection with polymerase chain reaction (PCR) techniques has been employed more widely. Notably, a molecular system based on multiplex real-time PCR with high-resolution melting was shown to have a sensitivity of up to 54% for distinguishing M fortuitum from other NTM.6 More recently, a 2-step real-time PCR method has demonstrated diagnostic sensitivity and specificity for differentiating NTM from Mycobacterium tuberculosis infections and identifying the causative NTM agent.7
Compared to immunocompetent individuals, those who are immunocompromised are more susceptible to less pathogenic strains of NTM, which can cause dissemination and lead to tenosynovitis, myositis, osteomyelitis, and septic arthritis.8-12 Nonetheless, cases of infections with NTM—including M fortuitum—are becoming harder to treat. Several single nucleotide polymorphisms and point mutations have been demonstrated in the ribosomal RNA methylase gene erm(39) related to clarithromycin resistance and in the rrl gene related to linezolid resistance.13 Due to increasing inducible resistance to common classes of antibiotics, such as macrolides and linezolid, treatment of M fortuitum requires multidrug regimens.13,14 Drug susceptibility testing also may be required, as M fortuitum has shown low resistance to tigecycline, tetracycline, cefmetazole, imipenem, and aminoglycosides (eg, amikacin, tobramycin, neomycin, gentamycin). Surgery is an important adjunctive tool in treating M fortuitum infections; patients with a single lesion are more likely to undergo surgical treatment alone or in combination with antibiotic therapy.15 More recently, antimicrobial photodynamic therapy has been explored as an alternative to eliminate NTM, including M fortuitum.16
The differential diagnosis for skin lesions manifesting with draining fistulae and sinus tracts includes conditions with infectious (cellulitis and chromomycosis) and inflammatory (pyoderma gangrenosum [PG] and hidradenitis suppurativa [HS]) causes.
Cellulitis is a common infection of the skin and subcutaneous tissue that predominantly is caused by gram-positive organisms such as β-hemolytic streptococci.17 Clinical manifestations include acute skin erythema, swelling, tenderness, and warmth. The legs are the most common sites of infection, but any area of the skin can be involved.17 Cellulitis comprises 10% of all infectious disease hospitalizations and up to 11% of all dermatologic admissions.18,19 It frequently is misdiagnosed, perhaps due to the lack of a reliable confirmatory laboratory test or imaging study, in addition to the plethora of diseases that mimic cellulitis, such as stasis dermatitis, lipodermatosclerosis, contact dermatitis, lymphedema, eosinophilic cellulitis, and papular urticaria.20,21 The consequences of misdiagnosis include but are not limited to unnecessary hospitalizations, inappropriate antibiotic use, and delayed management of the disease; thus, there is an urgent need for a reliable standard test to confirm the diagnosis, especially among nonspecialist physicians. 20 Most patients with uncomplicated cellulitis can be treated with empiric oral antibiotics that target β-hemolytic streptococci (ie, penicillin V potassium, amoxicillin).17 Methicillin-resistant Staphylococcus aureus coverage generally is unnecessary for nonpurulent cellulitis, but clinicians can consider adding amoxicillin-clavulanate, dicloxacillin, and cephalexin to the regimen. For purulent cellulitis, incision and drainage should be performed. In severe cases that manifest with sepsis, altered mental status, or hemodynamic instability, inpatient management is required.17
Chromomycosis (also known as chromoblastomycosis) is a chronic, indolent, granulomatous, suppurative mycosis of the skin and subcutaneous tissue22 that is caused by traumatic inoculation of various fungi of the order Chaetothyriales and family Herpotrichiellaceae, which are present in soil, plants, and decomposing wood. Chromomycosis is prevalent in tropical and subtropical regions.23,24 Clinically, it manifests as oligosymptomatic or asymptomatic lesions around an infection site that can manifest as papules with centrifugal growth evolving into nodular, verrucous, plaque, tumoral, or atrophic forms.22 Diagnosis is made with direct microscopy using potassium hydroxide, which reveals muriform bodies. Fungal culture in Sabouraud agar also can be used to isolate the causative pathogen.22 Unfortunately, chromomycosis is difficult to treat, with low cure rates and high relapse rates. Antifungal agents combined with surgery, cryotherapy, or thermotherapy often are used, with cure rates ranging from 15% to 80%.22,25
Pyoderma gangrenosum is a reactive noninfectious inflammatory dermatosis associated with inflammatory bowel disease and rheumatoid arthritis. The exact etiology is not clearly understood, but it generally is considered an autoinflammatory disorder.26 The most common form—classical PG—occurs in approximately 85% of cases and manifests as a painful erythematous lesion that progresses to a blistered or necrotic ulcer. It primarily affects the lower legs but can occur in other body sites.27 The diagnosis is based on clinical symptoms after excluding other similar conditions; histopathology of biopsied wound tissues often are required for confirmation. Treatment of PG starts with fast-acting immunosuppressive drugs (corticosteroids and/or cyclosporine) followed by slowacting immunosuppressive drugs (biologics).26
Hidradenitis suppurativa is a chronic recurrent disease of the hair follicle unit that develops after puberty.28 Clinically, HS manifests with painful nodules, abscesses, chronically draining fistulas, and scarring in areas of the body rich in apocrine glands.29,30 Treatment of HS is challenging due to its diverse clinical manifestations and unclear etiology. Topical therapy, systemic treatments, biologic agents, surgery, and light therapy have shown variable results.28,31
- Franco-Paredes C, Marcos LA, Henao-Martínez AF, et al. Cutaneous mycobacterial infections. Clin Microbiol Rev. 2018;32: E00069-18. doi:10.1128/CMR.00069-18
- Brown TH. The rapidly growing mycobacteria—MycobacteriuM fortuitum and Mycobacterium chelonae. Infect Control. 1985;6:283-238. doi:10.1017/s0195941700061762
- Hooper J; Beltrami EJ; Santoro F; et al. Remember the fite: a case of cutaneous MycobacteriuM fortuitum infection. Am J Dermatopathol. 2023;45:214-215. doi:10.1097/DAD.0000000000002336
- Franco-Paredes C, Chastain DB, Allen L, et al. Overview of cutaneous mycobacterial infections. Curr Trop Med Rep. 2018;5:228-232. doi:10.1007/s40475-018-0161-7
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-77. doi:10.1016/j.det.2015.03.017
- Peixoto ADS, Montenegro LML, Lima AS, et al. Identification of nontuberculous mycobacteria species by multiplex real-time PCR with high-resolution melting. Rev Soc Bras Med Trop. 2020;53:E20200211. doi:10.1590/0037-8682-0211-2020
- Park J, Kwak N, Chae JC, et al. A two-step real-time PCR method to identify Mycobacterium tuberculosis infections and six dominant nontuberculous mycobacterial infections from clinical specimens. Microbiol Spectr. 2023:E0160623. doi:10.1128/spectrum.01606-23
- Fowler J, Mahlen SD. Localized cutaneous infections in immunocompetent individuals due to rapidly growing mycobacteria. Arch Pathol Lab Med. 2014;138:1106-1109. doi:10.5858/arpa.2012-0203-RS
- Gardini G, Gregori N, Matteelli A, et al. Mycobacterial skin infection. Curr Opin Infect Dis. 2022;35:79-87. doi:10.1097/QCO.0000000000000820
- Wang SH, Pancholi P. Mycobacterial skin and soft tissue infection. Curr Infect Dis Rep. 2014;16:438. doi:10.1007/s11908-014-0438-5
- Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416. doi:10.1164/rccm.200604-571ST
- Mougari F, Guglielmetti L, Raskine L, et al. Infections caused by Mycobacterium abscessus: epidemiology, diagnostic tools and treatment. Expert Rev Anti Infect Ther. 2016;14:1139-1154. doi:10.1080/14787210.201 6.1238304
- Tu HZ, Lee HS, Chen YS, et al. High rates of antimicrobial resistance in rapidly growing mycobacterial infections in Taiwan. Pathogens. 2022;11:969. doi:10.3390/pathogens11090969
- Hashemzadeh M, Zadegan Dezfuli AA, Khosravi AD, et al. F requency of mutations in erm(39) related to clarithromycin resistance and in rrl related to linezolid resistance in clinical isolates of MycobacteriuM fortuitum in Iran. Acta Microbiol Immunol Hung. 2023;70:167-176. doi:10.1556/030.2023.02020
- Uslan DZ, Kowalski TJ, Wengenack NL, et al. Skin and soft tissue infections due to rapidly growing mycobacteria: comparison of clinical features, treatment, and susceptibility. Arch Dermatol. 2006;142:1287-1292. doi:10.1001/archderm.142.10.1287
- Miretti M, Juri L, Peralta A, et al. Photoinactivation of non-tuberculous mycobacteria using Zn-phthalocyanine loaded into liposomes. Tuberculosis (Edinb). 2022;136:102247. doi:10.1016/j.tube.2022.102247
- Bystritsky RJ. Cellulitis. Infect Dis Clin North Am. 2021;35:49-60. doi:10.1016/j.idc.2020.10.002
- Christensen K, Holman R, Steiner C, et al. Infectious disease hospitalizations in the United States. Clin Infect Dis. 2009;49:1025-1035. doi:10.1086/605562
- Yang JJ, Maloney NJ, Bach DQ, et al. Dermatology in the emergency department: prescriptions, rates of inpatient admission, and predictors of high utilization in the United States from 1996 to 2012. J Am Acad Dermatol. 2021;84:1480-1483. doi:10.1016/J.JAAD.2020.07.055
- Cutler TS, Jannat-Khah DP, Kam B, et al. Prevalence of misdiagnosis of cellulitis: a systematic review and meta-analysis. J Hosp Med. 2023;18:254-261. doi:10.1002/jhm.12977
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-52. doi:10.3949/ccjm.79a.11121
- Brito AC, Bittencourt MJS. Chromoblastomycosis: an etiological, epidemiological, clinical, diagnostic, and treatment update. An Bras Dermatol. 2018;93:495-506. doi:10.1590/abd1806-4841.20187321
- McGinnis MR. Chromoblastomycosis and phaeohyphomycosis: new concepts, diagnosis, and mycology. J Am Acad Dermatol. 1983;8:1-16.
- Rubin HA, Bruce S, Rosen T, et al. Evidence for percutaneous inoculation as the mode of transmission for chromoblastomycosis. J Am Acad Dermatol. 1991;25:951-954.
- Bonifaz A, Paredes-Solís V, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Maverakis E, Marzano AV, Le ST, et al. Pyoderma gangrenosum. Nat Rev Dis Primers. 2020;6:81. doi:10.1038/s41572-020-0213-x
- George C, Deroide F, Rustin M. Pyoderma gangrenosum—a guide to diagnosis and management. Clin Med (Lond). 2019;19:224-228. doi:10.7861/clinmedicine.19-3-224
- Narla S, Lyons AB, Hamzavi IH. The most recent advances in understanding and managing hidradenitis suppurativa. F1000Res. 2020;9:F1000 Faculty Rev-1049. doi:10.12688/f1000research.26083.1
- Garg A, Lavian J, Lin G, et al. Incidence of hidradenitis suppurativa in the United States: a sex- and age-adjusted population analysis. J Am Acad Dermatol. 2017;77:118-122. doi:10.1016/j.jaad.2017.02.005
- Daxhelet M, Suppa M, White J, et al. Proposed definitions of typical lesions in hidradenitis suppurativa. Dermatology. 2020;236:431-438. doi:10.1159/000507348
- Amat-Samaranch V, Agut-Busquet E, Vilarrasa E, et al. New perspectives on the treatment of hidradenitis suppurativa. Ther Adv Chronic Dis. 2021;12:20406223211055920. doi:10.1177/20406223211055920
- Franco-Paredes C, Marcos LA, Henao-Martínez AF, et al. Cutaneous mycobacterial infections. Clin Microbiol Rev. 2018;32: E00069-18. doi:10.1128/CMR.00069-18
- Brown TH. The rapidly growing mycobacteria—MycobacteriuM fortuitum and Mycobacterium chelonae. Infect Control. 1985;6:283-238. doi:10.1017/s0195941700061762
- Hooper J; Beltrami EJ; Santoro F; et al. Remember the fite: a case of cutaneous MycobacteriuM fortuitum infection. Am J Dermatopathol. 2023;45:214-215. doi:10.1097/DAD.0000000000002336
- Franco-Paredes C, Chastain DB, Allen L, et al. Overview of cutaneous mycobacterial infections. Curr Trop Med Rep. 2018;5:228-232. doi:10.1007/s40475-018-0161-7
- Gonzalez-Santiago TM, Drage LA. Nontuberculous mycobacteria: skin and soft tissue infections. Dermatol Clin. 2015;33:563-77. doi:10.1016/j.det.2015.03.017
- Peixoto ADS, Montenegro LML, Lima AS, et al. Identification of nontuberculous mycobacteria species by multiplex real-time PCR with high-resolution melting. Rev Soc Bras Med Trop. 2020;53:E20200211. doi:10.1590/0037-8682-0211-2020
- Park J, Kwak N, Chae JC, et al. A two-step real-time PCR method to identify Mycobacterium tuberculosis infections and six dominant nontuberculous mycobacterial infections from clinical specimens. Microbiol Spectr. 2023:E0160623. doi:10.1128/spectrum.01606-23
- Fowler J, Mahlen SD. Localized cutaneous infections in immunocompetent individuals due to rapidly growing mycobacteria. Arch Pathol Lab Med. 2014;138:1106-1109. doi:10.5858/arpa.2012-0203-RS
- Gardini G, Gregori N, Matteelli A, et al. Mycobacterial skin infection. Curr Opin Infect Dis. 2022;35:79-87. doi:10.1097/QCO.0000000000000820
- Wang SH, Pancholi P. Mycobacterial skin and soft tissue infection. Curr Infect Dis Rep. 2014;16:438. doi:10.1007/s11908-014-0438-5
- Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416. doi:10.1164/rccm.200604-571ST
- Mougari F, Guglielmetti L, Raskine L, et al. Infections caused by Mycobacterium abscessus: epidemiology, diagnostic tools and treatment. Expert Rev Anti Infect Ther. 2016;14:1139-1154. doi:10.1080/14787210.201 6.1238304
- Tu HZ, Lee HS, Chen YS, et al. High rates of antimicrobial resistance in rapidly growing mycobacterial infections in Taiwan. Pathogens. 2022;11:969. doi:10.3390/pathogens11090969
- Hashemzadeh M, Zadegan Dezfuli AA, Khosravi AD, et al. F requency of mutations in erm(39) related to clarithromycin resistance and in rrl related to linezolid resistance in clinical isolates of MycobacteriuM fortuitum in Iran. Acta Microbiol Immunol Hung. 2023;70:167-176. doi:10.1556/030.2023.02020
- Uslan DZ, Kowalski TJ, Wengenack NL, et al. Skin and soft tissue infections due to rapidly growing mycobacteria: comparison of clinical features, treatment, and susceptibility. Arch Dermatol. 2006;142:1287-1292. doi:10.1001/archderm.142.10.1287
- Miretti M, Juri L, Peralta A, et al. Photoinactivation of non-tuberculous mycobacteria using Zn-phthalocyanine loaded into liposomes. Tuberculosis (Edinb). 2022;136:102247. doi:10.1016/j.tube.2022.102247
- Bystritsky RJ. Cellulitis. Infect Dis Clin North Am. 2021;35:49-60. doi:10.1016/j.idc.2020.10.002
- Christensen K, Holman R, Steiner C, et al. Infectious disease hospitalizations in the United States. Clin Infect Dis. 2009;49:1025-1035. doi:10.1086/605562
- Yang JJ, Maloney NJ, Bach DQ, et al. Dermatology in the emergency department: prescriptions, rates of inpatient admission, and predictors of high utilization in the United States from 1996 to 2012. J Am Acad Dermatol. 2021;84:1480-1483. doi:10.1016/J.JAAD.2020.07.055
- Cutler TS, Jannat-Khah DP, Kam B, et al. Prevalence of misdiagnosis of cellulitis: a systematic review and meta-analysis. J Hosp Med. 2023;18:254-261. doi:10.1002/jhm.12977
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleve Clin J Med. 2012;79:547-52. doi:10.3949/ccjm.79a.11121
- Brito AC, Bittencourt MJS. Chromoblastomycosis: an etiological, epidemiological, clinical, diagnostic, and treatment update. An Bras Dermatol. 2018;93:495-506. doi:10.1590/abd1806-4841.20187321
- McGinnis MR. Chromoblastomycosis and phaeohyphomycosis: new concepts, diagnosis, and mycology. J Am Acad Dermatol. 1983;8:1-16.
- Rubin HA, Bruce S, Rosen T, et al. Evidence for percutaneous inoculation as the mode of transmission for chromoblastomycosis. J Am Acad Dermatol. 1991;25:951-954.
- Bonifaz A, Paredes-Solís V, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Maverakis E, Marzano AV, Le ST, et al. Pyoderma gangrenosum. Nat Rev Dis Primers. 2020;6:81. doi:10.1038/s41572-020-0213-x
- George C, Deroide F, Rustin M. Pyoderma gangrenosum—a guide to diagnosis and management. Clin Med (Lond). 2019;19:224-228. doi:10.7861/clinmedicine.19-3-224
- Narla S, Lyons AB, Hamzavi IH. The most recent advances in understanding and managing hidradenitis suppurativa. F1000Res. 2020;9:F1000 Faculty Rev-1049. doi:10.12688/f1000research.26083.1
- Garg A, Lavian J, Lin G, et al. Incidence of hidradenitis suppurativa in the United States: a sex- and age-adjusted population analysis. J Am Acad Dermatol. 2017;77:118-122. doi:10.1016/j.jaad.2017.02.005
- Daxhelet M, Suppa M, White J, et al. Proposed definitions of typical lesions in hidradenitis suppurativa. Dermatology. 2020;236:431-438. doi:10.1159/000507348
- Amat-Samaranch V, Agut-Busquet E, Vilarrasa E, et al. New perspectives on the treatment of hidradenitis suppurativa. Ther Adv Chronic Dis. 2021;12:20406223211055920. doi:10.1177/20406223211055920
A 40-year-old woman presented with multiple draining sinus tracts on the right thigh following an injury sustained weeks earlier while mowing wet grass.
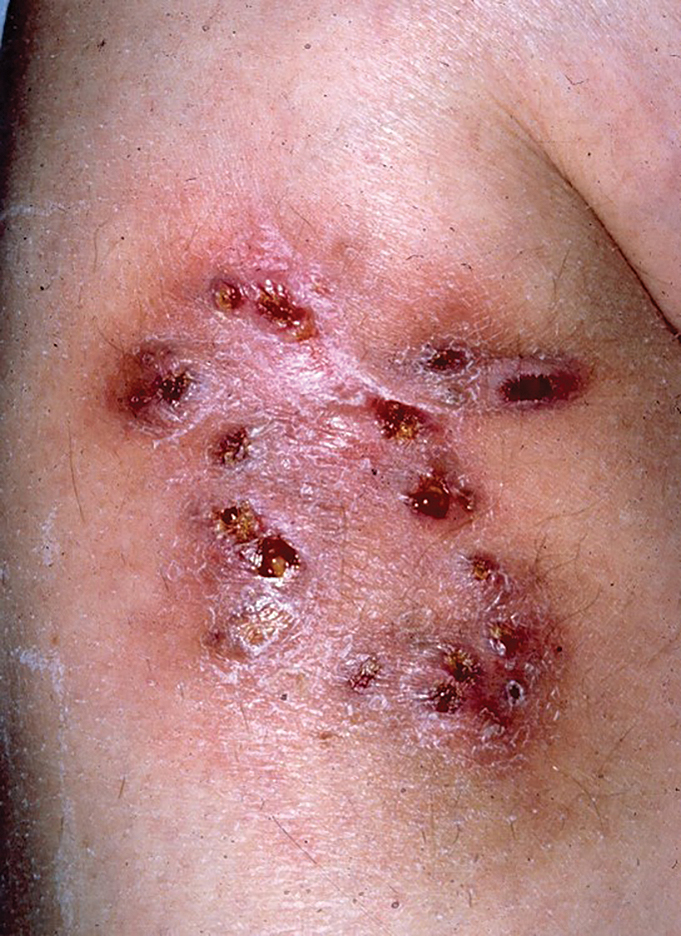
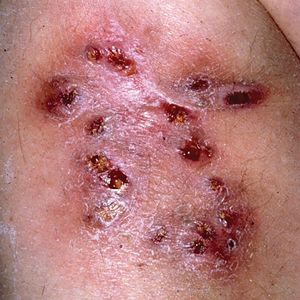
Necrotic Papules in a Pediatric Patient
The Diagnosis: Pityriasis Lichenoides et Varioliformis Acuta
Sectioned punch biopsies were performed on the patient’s right arm. Histopathology showed acanthosis and parakeratosis in the epidermis, with vacuolar degeneration and dyskeratosis in the basal layer. Dermal changes included extravasated red blood cells in the papillary dermis as well as perivascular lymphocytic infiltrates in both the papillary and reticular dermis (Figure). Direct immunofluorescence of a perilesional biopsy using anti–human IgG, IgM, IgA, C3, and fibrin conjugates showed no findings of immune deposition. Biopsy results were consistent with pityriasis lichenoides et varioliformis acuta (PLEVA), and the patient was treated with a 5-day course of oral azithromycin, triamcinolone ointment 0.1% twice daily, and phototherapy with narrowband UVB 3 times weekly. Rapid improvement was noted at 2-month follow-up.
Pityriasis lichenoides et varioliformis acuta is a form of pityriasis lichenoides, a group of inflammatory dermatoses that are characterized clinically by successive crops of morphologically diverse lesions. Epidemiologic studies have shown a slight male predominance. It primarily affects children and young adults, with peak ages of 8 and 32 years in pediatric and adult populations, respectively.1
The pathogenesis of PLEVA remains unclear. An abnormal immune response to Toxoplasma, Epstein-Barr virus, HIV, and other pathogens has been suggested based on serologic evidence of concurrent disease activity with the onset of lesions as well as cutaneous improvement in some patients after treatment of the infection.1 A T-cell lymphoproliferative etiology also has been considered based on histopathologic similarities between PLEVA and lymphomatoid papulosis (LyP) as well as findings of clonality in T-cell receptor gene rearrangement in many patients.1,2 Some clinicians consider LyP and PLEVA as separate entities on one disease spectrum.
Eruptions of PLEVA tend to favor the trunk and proximal extremities. Lesions may begin as macules measuring 2 to 3 mm in diameter that quickly evolve into papules with fine scale that remains attached centrally. Ulcerations with hemorrhagic crusts also may be noted as the lesions progress in stage. The rash may persist for weeks to years, and overlapping crops of macules and papules at varying stages of development may be seen in the same patient.1
Histopathologic findings of PLEVA include spongiosis, dyskeratosis, parakeratosis, and focal keratinocyte necrosis within the epidermis, as well as vacuolar degeneration of the basal layer. Lymphocyte and erythrocyte extravasation may extend into the epidermis. Dermal findings may include edema and wedge-shaped perivascular lymphocytic infiltrates extending into the reticular dermis.1

Important differential diagnoses to consider include LyP, mycosis fungoides (MF), pemphigus foliaceus, and varicella. Lymphomatoid papulosis is a benign CD30+ lymphoproliferative disorder that is characterized by an indolent course of recurrent, often self-resolving papules that occur most frequently on the trunk, arms, and legs of older patients. There are several histologic subtypes of LyP, but the most common (type A) may manifest with wedge-shaped perivascular lymphocytic infiltrates in the dermis, similar to PLEVA. T-cell receptor gene rearrangement studies characteristically reveal clonality in LyP, and clonality has been reported in PLEVA. However, LyP demonstrates a higher cytologic grade and lacks the characteristic parakeratotic scale and superficial dermal microhemorrhage of PLEVA.3
Mycosis fungoides is a malignant lymphoproliferative disorder that is characterized by an indolent clinical course of persistent patches, plaques, or tumors of various sizes that often manifest in non–sun-exposed areas of the skin. Early stages of MF are difficult to detect histologically, but biopsies may show atypical lymphocytes with hyperchromatic, irregularly contoured nuclei arranged along the basal layer of the epidermis. Epidermal aggregates of atypical lymphocytes (also known as Pautrier microabscesses) are considered highly specific for MF. T-cell receptor and immunopathologic studies also are important adjuncts in the diagnosis of MF.4
Pemphigus foliaceus is an autoimmune blistering disease caused by antibodies directed against desmoglein 1, which is found in the granular layer of the epidermis. It manifests with a subtle onset of scattered crusted lesions in the seborrheic areas, such as the scalp, face, chest, and upper back. Histopathologic findings of early blisters may include acantholysis and dyskeratosis in the stratum granulosum as well as vacuolization of the granular layer. The blisters may coalesce into superficial bullae containing fibrin and neutrophils. Immunofluorescence studies that demonstrate intraepidermal C3 and IgG deposition are key to the diagnosis of pemphigus.5
Varicella (also known as chickenpox) manifests with crops of vesicles on an erythematous base in a centripetal distribution favoring the trunk and proximal extremities. It often is preceded by prodromal fever, malaise, and myalgia. Histopathologic evaluation of varicella is uncommon but may reveal acantholysis, multinucleation, and nuclear margination of keratinocytes. Viral culture or nucleic acid amplification testing of lesions can be used to verify the diagnosis.6
Most cases of PLEVA resolve without intervention.7 Treatment is directed at speeding recovery, providing symptomatic relief, and limiting permanent sequelae. Topical steroids often are used to alleviate inflammation and pruritus. Systemic antibiotics such as doxycycline, minocycline, and erythromycin have been used for their anti-inflammatory properties. Phototherapy of various wavelengths, including broadband and narrowband UVB as well as psoralen plus UVA, have led to improvements in affected patients. Refractory disease may warrant consideration of therapy with methotrexate, acitretin, dapsone, or cyclosporine.7
There have been rare reports of PLEVA evolving into its potentially lethal variant, febrile ulceronecrotic Mucha-Habermann disease, which is differentiated by the presence of systemic manifestations, including high fever, sore throat, diarrhea, central nervous system symptoms, abdominal pain, interstitial pneumonitis, splenomegaly, arthritis, sepsis, megaloblastic anemia, or conjunctival ulcers. The orogenital mucosa may be affected. Cutaneous lesions may rapidly progress to large, generalized, coalescent ulcers with necrotic crusts and vasculitic features on biopsy.8 Malignant transformation of PLEVA into LyP or MF rarely may occur and warrants continued follow-up of unresolved lesions.9
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-572. doi:10.1016/j.jaad.2005.07.058
- Teklehaimanot F, Gade A, Rubenstein R. Pityriasis lichenoides et varioliformis acuta (PLEVA). In: StatPearls. StatPearls Publishing; 2023.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73. doi:10.1111/jdv.15931
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Lepe K, Yarrarapu SNS, Zito PM. Pemphigus foliaceus. In: StatPearls. StatPearls Publishing; 2023.
- Ayoade F, Kumar S. Varicella zoster (chickenpox). In: StatPearls. StatPearls Publishing; 2023.
- Bellinato F, Maurelli M, Gisondi P, et al. A systematic review of treatments for pityriasis lichenoides. J Eur Acad Dermatol Venereol. 2019;33:2039-2049. doi:10.1111/jdv.15813
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738. doi:10.1111/ijd.13195
- Thomson KF, Whittaker SJ, Russell-Jones R, et al. Childhood cutaneous T-cell lymphoma in association with pityriasis lichenoides chronica. Br J Dermatol. 1999;141:1136-1152. doi:10.1046/j.1365-2133.1999.03232.x
The Diagnosis: Pityriasis Lichenoides et Varioliformis Acuta
Sectioned punch biopsies were performed on the patient’s right arm. Histopathology showed acanthosis and parakeratosis in the epidermis, with vacuolar degeneration and dyskeratosis in the basal layer. Dermal changes included extravasated red blood cells in the papillary dermis as well as perivascular lymphocytic infiltrates in both the papillary and reticular dermis (Figure). Direct immunofluorescence of a perilesional biopsy using anti–human IgG, IgM, IgA, C3, and fibrin conjugates showed no findings of immune deposition. Biopsy results were consistent with pityriasis lichenoides et varioliformis acuta (PLEVA), and the patient was treated with a 5-day course of oral azithromycin, triamcinolone ointment 0.1% twice daily, and phototherapy with narrowband UVB 3 times weekly. Rapid improvement was noted at 2-month follow-up.
Pityriasis lichenoides et varioliformis acuta is a form of pityriasis lichenoides, a group of inflammatory dermatoses that are characterized clinically by successive crops of morphologically diverse lesions. Epidemiologic studies have shown a slight male predominance. It primarily affects children and young adults, with peak ages of 8 and 32 years in pediatric and adult populations, respectively.1
The pathogenesis of PLEVA remains unclear. An abnormal immune response to Toxoplasma, Epstein-Barr virus, HIV, and other pathogens has been suggested based on serologic evidence of concurrent disease activity with the onset of lesions as well as cutaneous improvement in some patients after treatment of the infection.1 A T-cell lymphoproliferative etiology also has been considered based on histopathologic similarities between PLEVA and lymphomatoid papulosis (LyP) as well as findings of clonality in T-cell receptor gene rearrangement in many patients.1,2 Some clinicians consider LyP and PLEVA as separate entities on one disease spectrum.
Eruptions of PLEVA tend to favor the trunk and proximal extremities. Lesions may begin as macules measuring 2 to 3 mm in diameter that quickly evolve into papules with fine scale that remains attached centrally. Ulcerations with hemorrhagic crusts also may be noted as the lesions progress in stage. The rash may persist for weeks to years, and overlapping crops of macules and papules at varying stages of development may be seen in the same patient.1
Histopathologic findings of PLEVA include spongiosis, dyskeratosis, parakeratosis, and focal keratinocyte necrosis within the epidermis, as well as vacuolar degeneration of the basal layer. Lymphocyte and erythrocyte extravasation may extend into the epidermis. Dermal findings may include edema and wedge-shaped perivascular lymphocytic infiltrates extending into the reticular dermis.1
Important differential diagnoses to consider include LyP, mycosis fungoides (MF), pemphigus foliaceus, and varicella. Lymphomatoid papulosis is a benign CD30+ lymphoproliferative disorder that is characterized by an indolent course of recurrent, often self-resolving papules that occur most frequently on the trunk, arms, and legs of older patients. There are several histologic subtypes of LyP, but the most common (type A) may manifest with wedge-shaped perivascular lymphocytic infiltrates in the dermis, similar to PLEVA. T-cell receptor gene rearrangement studies characteristically reveal clonality in LyP, and clonality has been reported in PLEVA. However, LyP demonstrates a higher cytologic grade and lacks the characteristic parakeratotic scale and superficial dermal microhemorrhage of PLEVA.3
Mycosis fungoides is a malignant lymphoproliferative disorder that is characterized by an indolent clinical course of persistent patches, plaques, or tumors of various sizes that often manifest in non–sun-exposed areas of the skin. Early stages of MF are difficult to detect histologically, but biopsies may show atypical lymphocytes with hyperchromatic, irregularly contoured nuclei arranged along the basal layer of the epidermis. Epidermal aggregates of atypical lymphocytes (also known as Pautrier microabscesses) are considered highly specific for MF. T-cell receptor and immunopathologic studies also are important adjuncts in the diagnosis of MF.4
Pemphigus foliaceus is an autoimmune blistering disease caused by antibodies directed against desmoglein 1, which is found in the granular layer of the epidermis. It manifests with a subtle onset of scattered crusted lesions in the seborrheic areas, such as the scalp, face, chest, and upper back. Histopathologic findings of early blisters may include acantholysis and dyskeratosis in the stratum granulosum as well as vacuolization of the granular layer. The blisters may coalesce into superficial bullae containing fibrin and neutrophils. Immunofluorescence studies that demonstrate intraepidermal C3 and IgG deposition are key to the diagnosis of pemphigus.5
Varicella (also known as chickenpox) manifests with crops of vesicles on an erythematous base in a centripetal distribution favoring the trunk and proximal extremities. It often is preceded by prodromal fever, malaise, and myalgia. Histopathologic evaluation of varicella is uncommon but may reveal acantholysis, multinucleation, and nuclear margination of keratinocytes. Viral culture or nucleic acid amplification testing of lesions can be used to verify the diagnosis.6
Most cases of PLEVA resolve without intervention.7 Treatment is directed at speeding recovery, providing symptomatic relief, and limiting permanent sequelae. Topical steroids often are used to alleviate inflammation and pruritus. Systemic antibiotics such as doxycycline, minocycline, and erythromycin have been used for their anti-inflammatory properties. Phototherapy of various wavelengths, including broadband and narrowband UVB as well as psoralen plus UVA, have led to improvements in affected patients. Refractory disease may warrant consideration of therapy with methotrexate, acitretin, dapsone, or cyclosporine.7
There have been rare reports of PLEVA evolving into its potentially lethal variant, febrile ulceronecrotic Mucha-Habermann disease, which is differentiated by the presence of systemic manifestations, including high fever, sore throat, diarrhea, central nervous system symptoms, abdominal pain, interstitial pneumonitis, splenomegaly, arthritis, sepsis, megaloblastic anemia, or conjunctival ulcers. The orogenital mucosa may be affected. Cutaneous lesions may rapidly progress to large, generalized, coalescent ulcers with necrotic crusts and vasculitic features on biopsy.8 Malignant transformation of PLEVA into LyP or MF rarely may occur and warrants continued follow-up of unresolved lesions.9
The Diagnosis: Pityriasis Lichenoides et Varioliformis Acuta
Sectioned punch biopsies were performed on the patient’s right arm. Histopathology showed acanthosis and parakeratosis in the epidermis, with vacuolar degeneration and dyskeratosis in the basal layer. Dermal changes included extravasated red blood cells in the papillary dermis as well as perivascular lymphocytic infiltrates in both the papillary and reticular dermis (Figure). Direct immunofluorescence of a perilesional biopsy using anti–human IgG, IgM, IgA, C3, and fibrin conjugates showed no findings of immune deposition. Biopsy results were consistent with pityriasis lichenoides et varioliformis acuta (PLEVA), and the patient was treated with a 5-day course of oral azithromycin, triamcinolone ointment 0.1% twice daily, and phototherapy with narrowband UVB 3 times weekly. Rapid improvement was noted at 2-month follow-up.
Pityriasis lichenoides et varioliformis acuta is a form of pityriasis lichenoides, a group of inflammatory dermatoses that are characterized clinically by successive crops of morphologically diverse lesions. Epidemiologic studies have shown a slight male predominance. It primarily affects children and young adults, with peak ages of 8 and 32 years in pediatric and adult populations, respectively.1
The pathogenesis of PLEVA remains unclear. An abnormal immune response to Toxoplasma, Epstein-Barr virus, HIV, and other pathogens has been suggested based on serologic evidence of concurrent disease activity with the onset of lesions as well as cutaneous improvement in some patients after treatment of the infection.1 A T-cell lymphoproliferative etiology also has been considered based on histopathologic similarities between PLEVA and lymphomatoid papulosis (LyP) as well as findings of clonality in T-cell receptor gene rearrangement in many patients.1,2 Some clinicians consider LyP and PLEVA as separate entities on one disease spectrum.
Eruptions of PLEVA tend to favor the trunk and proximal extremities. Lesions may begin as macules measuring 2 to 3 mm in diameter that quickly evolve into papules with fine scale that remains attached centrally. Ulcerations with hemorrhagic crusts also may be noted as the lesions progress in stage. The rash may persist for weeks to years, and overlapping crops of macules and papules at varying stages of development may be seen in the same patient.1
Histopathologic findings of PLEVA include spongiosis, dyskeratosis, parakeratosis, and focal keratinocyte necrosis within the epidermis, as well as vacuolar degeneration of the basal layer. Lymphocyte and erythrocyte extravasation may extend into the epidermis. Dermal findings may include edema and wedge-shaped perivascular lymphocytic infiltrates extending into the reticular dermis.1
Important differential diagnoses to consider include LyP, mycosis fungoides (MF), pemphigus foliaceus, and varicella. Lymphomatoid papulosis is a benign CD30+ lymphoproliferative disorder that is characterized by an indolent course of recurrent, often self-resolving papules that occur most frequently on the trunk, arms, and legs of older patients. There are several histologic subtypes of LyP, but the most common (type A) may manifest with wedge-shaped perivascular lymphocytic infiltrates in the dermis, similar to PLEVA. T-cell receptor gene rearrangement studies characteristically reveal clonality in LyP, and clonality has been reported in PLEVA. However, LyP demonstrates a higher cytologic grade and lacks the characteristic parakeratotic scale and superficial dermal microhemorrhage of PLEVA.3
Mycosis fungoides is a malignant lymphoproliferative disorder that is characterized by an indolent clinical course of persistent patches, plaques, or tumors of various sizes that often manifest in non–sun-exposed areas of the skin. Early stages of MF are difficult to detect histologically, but biopsies may show atypical lymphocytes with hyperchromatic, irregularly contoured nuclei arranged along the basal layer of the epidermis. Epidermal aggregates of atypical lymphocytes (also known as Pautrier microabscesses) are considered highly specific for MF. T-cell receptor and immunopathologic studies also are important adjuncts in the diagnosis of MF.4
Pemphigus foliaceus is an autoimmune blistering disease caused by antibodies directed against desmoglein 1, which is found in the granular layer of the epidermis. It manifests with a subtle onset of scattered crusted lesions in the seborrheic areas, such as the scalp, face, chest, and upper back. Histopathologic findings of early blisters may include acantholysis and dyskeratosis in the stratum granulosum as well as vacuolization of the granular layer. The blisters may coalesce into superficial bullae containing fibrin and neutrophils. Immunofluorescence studies that demonstrate intraepidermal C3 and IgG deposition are key to the diagnosis of pemphigus.5
Varicella (also known as chickenpox) manifests with crops of vesicles on an erythematous base in a centripetal distribution favoring the trunk and proximal extremities. It often is preceded by prodromal fever, malaise, and myalgia. Histopathologic evaluation of varicella is uncommon but may reveal acantholysis, multinucleation, and nuclear margination of keratinocytes. Viral culture or nucleic acid amplification testing of lesions can be used to verify the diagnosis.6
Most cases of PLEVA resolve without intervention.7 Treatment is directed at speeding recovery, providing symptomatic relief, and limiting permanent sequelae. Topical steroids often are used to alleviate inflammation and pruritus. Systemic antibiotics such as doxycycline, minocycline, and erythromycin have been used for their anti-inflammatory properties. Phototherapy of various wavelengths, including broadband and narrowband UVB as well as psoralen plus UVA, have led to improvements in affected patients. Refractory disease may warrant consideration of therapy with methotrexate, acitretin, dapsone, or cyclosporine.7
There have been rare reports of PLEVA evolving into its potentially lethal variant, febrile ulceronecrotic Mucha-Habermann disease, which is differentiated by the presence of systemic manifestations, including high fever, sore throat, diarrhea, central nervous system symptoms, abdominal pain, interstitial pneumonitis, splenomegaly, arthritis, sepsis, megaloblastic anemia, or conjunctival ulcers. The orogenital mucosa may be affected. Cutaneous lesions may rapidly progress to large, generalized, coalescent ulcers with necrotic crusts and vasculitic features on biopsy.8 Malignant transformation of PLEVA into LyP or MF rarely may occur and warrants continued follow-up of unresolved lesions.9
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-572. doi:10.1016/j.jaad.2005.07.058
- Teklehaimanot F, Gade A, Rubenstein R. Pityriasis lichenoides et varioliformis acuta (PLEVA). In: StatPearls. StatPearls Publishing; 2023.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73. doi:10.1111/jdv.15931
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Lepe K, Yarrarapu SNS, Zito PM. Pemphigus foliaceus. In: StatPearls. StatPearls Publishing; 2023.
- Ayoade F, Kumar S. Varicella zoster (chickenpox). In: StatPearls. StatPearls Publishing; 2023.
- Bellinato F, Maurelli M, Gisondi P, et al. A systematic review of treatments for pityriasis lichenoides. J Eur Acad Dermatol Venereol. 2019;33:2039-2049. doi:10.1111/jdv.15813
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738. doi:10.1111/ijd.13195
- Thomson KF, Whittaker SJ, Russell-Jones R, et al. Childhood cutaneous T-cell lymphoma in association with pityriasis lichenoides chronica. Br J Dermatol. 1999;141:1136-1152. doi:10.1046/j.1365-2133.1999.03232.x
- Bowers S, Warshaw EM. Pityriasis lichenoides and its subtypes. J Am Acad Dermatol. 2006;55:557-572. doi:10.1016/j.jaad.2005.07.058
- Teklehaimanot F, Gade A, Rubenstein R. Pityriasis lichenoides et varioliformis acuta (PLEVA). In: StatPearls. StatPearls Publishing; 2023.
- Martinez-Cabriales SA, Walsh S, Sade S, et al. Lymphomatoid papulosis: an update and review. J Eur Acad Dermatol Venereol. 2020;34:59-73. doi:10.1111/jdv.15931
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Lepe K, Yarrarapu SNS, Zito PM. Pemphigus foliaceus. In: StatPearls. StatPearls Publishing; 2023.
- Ayoade F, Kumar S. Varicella zoster (chickenpox). In: StatPearls. StatPearls Publishing; 2023.
- Bellinato F, Maurelli M, Gisondi P, et al. A systematic review of treatments for pityriasis lichenoides. J Eur Acad Dermatol Venereol. 2019;33:2039-2049. doi:10.1111/jdv.15813
- Nofal A, Assaf M, Alakad R, et al. Febrile ulceronecrotic Mucha-Habermann disease: proposed diagnostic criteria and therapeutic evaluation. Int J Dermatol. 2016;55:729-738. doi:10.1111/ijd.13195
- Thomson KF, Whittaker SJ, Russell-Jones R, et al. Childhood cutaneous T-cell lymphoma in association with pityriasis lichenoides chronica. Br J Dermatol. 1999;141:1136-1152. doi:10.1046/j.1365-2133.1999.03232.x
A 7-year-old boy was referred to the dermatology clinic for evaluation of a diffuse pruritic rash of 3 months’ duration. The rash began as scant erythematous papules on the face, and crops of similar lesions later erupted on the trunk, arms, and legs. He was treated previously by a pediatrician for scabies with topical permethrin followed by 2 doses of oral ivermectin 200 μg/kg without improvement. Physical examination revealed innumerable erythematous macules and papules with centrally adherent scaling distributed on the trunk, arms, and legs, as well as scant necrotic papules with a hemorrhagic crust and a peripheral rim of scale.


Scarring Head Wound
The Diagnosis: Brunsting-Perry Cicatricial Pemphigoid
Physical examination and histopathology are paramount in diagnosing Brunsting-Perry cicatricial pemphigoid (BPCP). In our patient, histopathology showed subepidermal blistering with a mixed superficial dermal inflammatory cell infiltrate. Direct immunofluorescence was positive for linear IgG and C3 antibodies along the basement membrane. The scarring erosions on the scalp combined with the autoantibody findings on direct immunofluorescence were consistent with BPCP. He was started on dapsone 100 mg daily and demonstrated complete resolution of symptoms after 10 months, with the exception of persistent scarring hair loss (Figure).

Brunsting-Perry cicatricial pemphigoid is a rare dermatologic condition. It was first defined in 1957 when Brunsting and Perry1 examined 7 patients with cicatricial pemphigoid that predominantly affected the head and neck region, with occasional mucous membrane involvement but no mucosal scarring. Characteristically, BPCP manifests as scarring herpetiform plaques with varied blisters, erosions, crusts, and scarring.1 It primarily affects middle-aged men.2
Historically, BPCP has been considered a variant of cicatricial pemphigoid (now known as mucous membrane pemphigoid), bullous pemphigoid, or epidermolysis bullosa acquisita.3 The antigen target has not been established clearly; however, autoantibodies against laminin 332, collagen VII, and BP180 and BP230 have been proposed.2,4,5 Jacoby et al6 described BPCP on a spectrum with bullous pemphigoid and cicatricial pemphigoid, with primarily circulating autoantibodies on one end and tissue-fixed autoantibodies on the other.
The differential for BPCP also includes anti-p200 pemphigoid and anti–laminin 332 pemphigoid. Anti-p200 pemphigoid also is known as bullous pemphigoid with antibodies against the 200-kDa protein.7 It may clinically manifest similar to bullous pemphigoid and other subepidermal autoimmune blistering diseases; thus, immunopathologic differentiation can be helpful. Anti–laminin 332 pemphigoid (also known as anti–laminin gamma-1 pemphigoid) is characterized by autoantibodies targeting the laminin 332 protein in the basement membrane zone, resulting in blistering and erosions.8 Similar to BPCP and epidermolysis bullosa aquisita, anti–laminin 332 pemphigoid may affect cephalic regions and mucous membrane surfaces, resulting in scarring and cicatricial changes. Anti–laminin 332 pemphigoid also has been associated with internal malignancy.8 The use of the salt-split skin technique can be utilized to differentiate these entities based on their autoantibody-binding patterns in relation to the lamina densa.
Treatment options for mild BPCP include potent topical or intralesional steroids and dapsone, while more severe cases may require systemic therapy with rituximab, azathioprine, mycophenolate mofetil, or cyclophosphamide.4
This case highlights the importance of histopathologic examination of skin lesions with an unusual history or clinical presentation. Dermatologists should consider BPCP when presented with erosions, ulcerations, or blisters of the head and neck in middle-aged male patients.
- Brunsting LA, Perry HO. Benign pemphigoid? a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm. 1957;75:489-501. doi:10.1001 /archderm.1957.01550160015002
- Jedlickova H, Neidermeier A, Zgažarová S, et al. Brunsting-Perry pemphigoid of the scalp with antibodies against laminin 332. Dermatology. 2011;222:193-195. doi:10.1159/000322842
- Eichhoff G. Brunsting-Perry pemphigoid as differential diagnosis of nonmelanoma skin cancer. Cureus. 2019;11:E5400. doi:10.7759/cureus.5400
- Asfour L, Chong H, Mee J, et al. Epidermolysis bullosa acquisita (Brunsting-Perry pemphigoid variant) localized to the face and diagnosed with antigen identification using skin deficient in type VII collagen. Am J Dermatopathol. 2017;39:e90-e96. doi:10.1097 /DAD.0000000000000829
- Zhou S, Zou Y, Pan M. Brunsting-Perry pemphigoid transitioning from previous bullous pemphigoid. JAAD Case Rep. 2020;6:192-194. doi:10.1016/j.jdcr.2019.12.018
- Jacoby WD Jr, Bartholome CW, Ramchand SC, et al. Cicatricial pemphigoid (Brunsting-Perry type). case report and immunofluorescence findings. Arch Dermatol. 1978;114:779-781. doi:10.1001/archderm.1978.01640170079018
- Kridin K, Ahmed AR. Anti-p200 pemphigoid: a systematic review. Front Immunol. 2019;10:2466. doi:10.3389/fimmu.2019.02466
- Shi L, Li X, Qian H. Anti-laminin 332-type mucous membrane pemphigoid. Biomolecules. 2022;12:1461. doi:10.3390/biom12101461
The Diagnosis: Brunsting-Perry Cicatricial Pemphigoid
Physical examination and histopathology are paramount in diagnosing Brunsting-Perry cicatricial pemphigoid (BPCP). In our patient, histopathology showed subepidermal blistering with a mixed superficial dermal inflammatory cell infiltrate. Direct immunofluorescence was positive for linear IgG and C3 antibodies along the basement membrane. The scarring erosions on the scalp combined with the autoantibody findings on direct immunofluorescence were consistent with BPCP. He was started on dapsone 100 mg daily and demonstrated complete resolution of symptoms after 10 months, with the exception of persistent scarring hair loss (Figure).
Brunsting-Perry cicatricial pemphigoid is a rare dermatologic condition. It was first defined in 1957 when Brunsting and Perry1 examined 7 patients with cicatricial pemphigoid that predominantly affected the head and neck region, with occasional mucous membrane involvement but no mucosal scarring. Characteristically, BPCP manifests as scarring herpetiform plaques with varied blisters, erosions, crusts, and scarring.1 It primarily affects middle-aged men.2
Historically, BPCP has been considered a variant of cicatricial pemphigoid (now known as mucous membrane pemphigoid), bullous pemphigoid, or epidermolysis bullosa acquisita.3 The antigen target has not been established clearly; however, autoantibodies against laminin 332, collagen VII, and BP180 and BP230 have been proposed.2,4,5 Jacoby et al6 described BPCP on a spectrum with bullous pemphigoid and cicatricial pemphigoid, with primarily circulating autoantibodies on one end and tissue-fixed autoantibodies on the other.
The differential for BPCP also includes anti-p200 pemphigoid and anti–laminin 332 pemphigoid. Anti-p200 pemphigoid also is known as bullous pemphigoid with antibodies against the 200-kDa protein.7 It may clinically manifest similar to bullous pemphigoid and other subepidermal autoimmune blistering diseases; thus, immunopathologic differentiation can be helpful. Anti–laminin 332 pemphigoid (also known as anti–laminin gamma-1 pemphigoid) is characterized by autoantibodies targeting the laminin 332 protein in the basement membrane zone, resulting in blistering and erosions.8 Similar to BPCP and epidermolysis bullosa aquisita, anti–laminin 332 pemphigoid may affect cephalic regions and mucous membrane surfaces, resulting in scarring and cicatricial changes. Anti–laminin 332 pemphigoid also has been associated with internal malignancy.8 The use of the salt-split skin technique can be utilized to differentiate these entities based on their autoantibody-binding patterns in relation to the lamina densa.
Treatment options for mild BPCP include potent topical or intralesional steroids and dapsone, while more severe cases may require systemic therapy with rituximab, azathioprine, mycophenolate mofetil, or cyclophosphamide.4
This case highlights the importance of histopathologic examination of skin lesions with an unusual history or clinical presentation. Dermatologists should consider BPCP when presented with erosions, ulcerations, or blisters of the head and neck in middle-aged male patients.
The Diagnosis: Brunsting-Perry Cicatricial Pemphigoid
Physical examination and histopathology are paramount in diagnosing Brunsting-Perry cicatricial pemphigoid (BPCP). In our patient, histopathology showed subepidermal blistering with a mixed superficial dermal inflammatory cell infiltrate. Direct immunofluorescence was positive for linear IgG and C3 antibodies along the basement membrane. The scarring erosions on the scalp combined with the autoantibody findings on direct immunofluorescence were consistent with BPCP. He was started on dapsone 100 mg daily and demonstrated complete resolution of symptoms after 10 months, with the exception of persistent scarring hair loss (Figure).
Brunsting-Perry cicatricial pemphigoid is a rare dermatologic condition. It was first defined in 1957 when Brunsting and Perry1 examined 7 patients with cicatricial pemphigoid that predominantly affected the head and neck region, with occasional mucous membrane involvement but no mucosal scarring. Characteristically, BPCP manifests as scarring herpetiform plaques with varied blisters, erosions, crusts, and scarring.1 It primarily affects middle-aged men.2
Historically, BPCP has been considered a variant of cicatricial pemphigoid (now known as mucous membrane pemphigoid), bullous pemphigoid, or epidermolysis bullosa acquisita.3 The antigen target has not been established clearly; however, autoantibodies against laminin 332, collagen VII, and BP180 and BP230 have been proposed.2,4,5 Jacoby et al6 described BPCP on a spectrum with bullous pemphigoid and cicatricial pemphigoid, with primarily circulating autoantibodies on one end and tissue-fixed autoantibodies on the other.
The differential for BPCP also includes anti-p200 pemphigoid and anti–laminin 332 pemphigoid. Anti-p200 pemphigoid also is known as bullous pemphigoid with antibodies against the 200-kDa protein.7 It may clinically manifest similar to bullous pemphigoid and other subepidermal autoimmune blistering diseases; thus, immunopathologic differentiation can be helpful. Anti–laminin 332 pemphigoid (also known as anti–laminin gamma-1 pemphigoid) is characterized by autoantibodies targeting the laminin 332 protein in the basement membrane zone, resulting in blistering and erosions.8 Similar to BPCP and epidermolysis bullosa aquisita, anti–laminin 332 pemphigoid may affect cephalic regions and mucous membrane surfaces, resulting in scarring and cicatricial changes. Anti–laminin 332 pemphigoid also has been associated with internal malignancy.8 The use of the salt-split skin technique can be utilized to differentiate these entities based on their autoantibody-binding patterns in relation to the lamina densa.
Treatment options for mild BPCP include potent topical or intralesional steroids and dapsone, while more severe cases may require systemic therapy with rituximab, azathioprine, mycophenolate mofetil, or cyclophosphamide.4
This case highlights the importance of histopathologic examination of skin lesions with an unusual history or clinical presentation. Dermatologists should consider BPCP when presented with erosions, ulcerations, or blisters of the head and neck in middle-aged male patients.
- Brunsting LA, Perry HO. Benign pemphigoid? a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm. 1957;75:489-501. doi:10.1001 /archderm.1957.01550160015002
- Jedlickova H, Neidermeier A, Zgažarová S, et al. Brunsting-Perry pemphigoid of the scalp with antibodies against laminin 332. Dermatology. 2011;222:193-195. doi:10.1159/000322842
- Eichhoff G. Brunsting-Perry pemphigoid as differential diagnosis of nonmelanoma skin cancer. Cureus. 2019;11:E5400. doi:10.7759/cureus.5400
- Asfour L, Chong H, Mee J, et al. Epidermolysis bullosa acquisita (Brunsting-Perry pemphigoid variant) localized to the face and diagnosed with antigen identification using skin deficient in type VII collagen. Am J Dermatopathol. 2017;39:e90-e96. doi:10.1097 /DAD.0000000000000829
- Zhou S, Zou Y, Pan M. Brunsting-Perry pemphigoid transitioning from previous bullous pemphigoid. JAAD Case Rep. 2020;6:192-194. doi:10.1016/j.jdcr.2019.12.018
- Jacoby WD Jr, Bartholome CW, Ramchand SC, et al. Cicatricial pemphigoid (Brunsting-Perry type). case report and immunofluorescence findings. Arch Dermatol. 1978;114:779-781. doi:10.1001/archderm.1978.01640170079018
- Kridin K, Ahmed AR. Anti-p200 pemphigoid: a systematic review. Front Immunol. 2019;10:2466. doi:10.3389/fimmu.2019.02466
- Shi L, Li X, Qian H. Anti-laminin 332-type mucous membrane pemphigoid. Biomolecules. 2022;12:1461. doi:10.3390/biom12101461
- Brunsting LA, Perry HO. Benign pemphigoid? a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm. 1957;75:489-501. doi:10.1001 /archderm.1957.01550160015002
- Jedlickova H, Neidermeier A, Zgažarová S, et al. Brunsting-Perry pemphigoid of the scalp with antibodies against laminin 332. Dermatology. 2011;222:193-195. doi:10.1159/000322842
- Eichhoff G. Brunsting-Perry pemphigoid as differential diagnosis of nonmelanoma skin cancer. Cureus. 2019;11:E5400. doi:10.7759/cureus.5400
- Asfour L, Chong H, Mee J, et al. Epidermolysis bullosa acquisita (Brunsting-Perry pemphigoid variant) localized to the face and diagnosed with antigen identification using skin deficient in type VII collagen. Am J Dermatopathol. 2017;39:e90-e96. doi:10.1097 /DAD.0000000000000829
- Zhou S, Zou Y, Pan M. Brunsting-Perry pemphigoid transitioning from previous bullous pemphigoid. JAAD Case Rep. 2020;6:192-194. doi:10.1016/j.jdcr.2019.12.018
- Jacoby WD Jr, Bartholome CW, Ramchand SC, et al. Cicatricial pemphigoid (Brunsting-Perry type). case report and immunofluorescence findings. Arch Dermatol. 1978;114:779-781. doi:10.1001/archderm.1978.01640170079018
- Kridin K, Ahmed AR. Anti-p200 pemphigoid: a systematic review. Front Immunol. 2019;10:2466. doi:10.3389/fimmu.2019.02466
- Shi L, Li X, Qian H. Anti-laminin 332-type mucous membrane pemphigoid. Biomolecules. 2022;12:1461. doi:10.3390/biom12101461
A 60-year-old man presented to a dermatology clinic with a wound on the scalp that had persisted for 11 months. The lesion started as a small erosion that eventually progressed to involve the entire parietal scalp. He had a history of type 2 diabetes mellitus, hypertension, and Graves disease. Physical examination demonstrated a large scar over the vertex scalp with central erosion, overlying crust, peripheral scalp atrophy, hypopigmentation at the periphery, and exaggerated superficial vasculature. Some oral erosions also were observed. A review of systems was negative for any constitutional symptoms. A month prior, the patient had been started on dapsone 50 mg with a prednisone taper by an outside dermatologist and noticed some improvement.


Painful Plaque on the Forearm
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
The Diagnosis: Mycobacterium marinum Infection
A repeat excisional biopsy showed suppurative granulomatous dermatitis with negative stains for infectious organisms; however, tissue culture grew Mycobacterium marinum. The patient had a history of exposure to fish tanks, which are a potential habitat for nontuberculous mycobacteria. These bacteria can enter the body through a minor laceration or cut in the skin, which was likely due to her occupation and pet care activities.1 Her fish tank exposure combined with the cutaneous findings of a long-standing indurated plaque with proximal nodular lymphangitis made M marinum infection the most likely diagnosis.2
Due to the limited specificity and sensitivity of patient symptoms, histologic staining, and direct microscopy, the gold standard for diagnosing acid-fast bacilli is tissue culture. 3 Tissue polymerase chain reaction testing is most useful in identifying the species of mycobacteria when histologic stains identify acid-fast bacilli but repeated tissue cultures are negative.4 With M marinum, a high clinical suspicion is needed to acquire a positive tissue culture because it needs to be grown for several weeks and at a temperature of 30 °C.5 Therefore, the physician should inform the laboratory if there is any suspicion for M marinum to increase the likelihood of obtaining a positive culture.
The differential diagnosis for M marinum infection includes other skin diseases that can cause nodular lymphangitis (also known as sporotrichoid spread) such as sporotrichosis, leishmaniasis, and certain bacterial and fungal infections. Although cat scratch disease, which is caused by Bartonella henselae, can appear similar to M marinum on histopathology, it clinically manifests with a single papulovesicular lesion at the site of inoculation that then forms a central eschar and resolves within a few weeks. Cat scratch disease typically causes painful lymphadenopathy, but it does not cause nodular lymphangitis or sporotrichoid spread.6 Sporotrichosis can have a similar clinical and histologic manifestation to M marinum infection, but the patient history typically includes exposure to Sporothrix schenckii through gardening or other contact with thorns, plants, or soil.2 Cutaneous sarcoidosis can have a similar clinical appearance to M marinum infection, but nodular lymphangitis does not occur and histopathology would demonstrate noncaseating epithelioid cell granulomas.7 Lastly, although vegetative pyoderma gangrenosum can have some of the same histologic findings as M marinum, it typically also demonstrates sinus tract formation, which was not present in our case. Additionally, vegetative pyoderma gangrenosum manifests with a verrucous and pustular plaque that would not have lymphocutaneous spread.8
Treatment of cutaneous M marinum infection is guided by antibiotic susceptibility testing. One regimen is clarithromycin (500 mg twice daily9) plus ethambutol. 10 Treatment often entails a multidrug combination due to the high rates of antibiotic resistance. Other antibiotics that potentially can be used include rifampin, trimethoprim-sulfamethoxazole, minocycline, and quinolones. The treatment duration typically is more than 3 months, and therapy is continued for 4 to 6 weeks after the skin lesions resolve.11 Excision of the lesion is reserved for patients with M marinum infection that fails to respond to antibiotic therapy.5
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
- Wayne LG, Sramek HA. Agents of newly recognized or infrequently encountered mycobacterial diseases. Clin Microbiol Rev. 1992;5:1-25. doi:10.1128/CMR.5.1.1
- Tobin EH, Jih WW. Sporotrichoid lymphocutaneous infections: etiology, diagnosis and therapy. Am Fam Physician. 2001;63:326-332.
- van Ingen J. Diagnosis of nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:103-109. doi:10.1055/s-0033-1333569
- Williamson H, Phillips R, Sarfo S, et al. Genetic diversity of PCR-positive, culture-negative and culture-positive Mycobacterium ulcerans isolated from Buruli ulcer patients in Ghana. PLoS One. 2014;9:E88007. doi:10.1371/journal.pone.0088007
- Aubry A, Mougari F, Reibel F, et al. Mycobacterium marinum. Microbiol Spectr. 2017;5. doi:10.1128/microbiolspec.TNMI7-0038-2016
- Baranowski K, Huang B. Cat scratch disease. StatPearls [Internet]. Updated June 12, 2023. Accessed July 15, 2024. https://www.ncbi.nlm .nih.gov/books/NBK482139/
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416. doi:10.1016/j.det.2015.03.006
- Borg Grech S, Vella Baldacchino A, Corso R, et al. Superficial granulomatous pyoderma successfully treated with intravenous immunoglobulin. Eur J Case Rep Intern Med. 2021;8:002656. doi:10.12890/2021_002656
- Krooks J, Weatherall A, Markowitz S. Complete resolution of Mycobacterium marinum infection with clarithromycin and ethambutol: a case report and a review of the literature. J Clin Aesthet Dermatol. 2018;11:48-51.
- Medel-Plaza M., Esteban J. Current treatment options for Mycobacterium marinum cutaneous infections. Expert Opin Pharmacother. 2023;24:1113-1123. doi:10.1080/14656566.2023.2211258
- Tirado-Sánchez A, Bonifaz A. Nodular lymphangitis (sporotrichoid lymphocutaneous infections): clues to differential diagnosis. J Fungi (Basel). 2018;4:56. doi:10.3390/jof4020056
A 30-year-old woman presented to the dermatology clinic with lesions on the right forearm of 2 years’ duration. Her medical history was unremarkable. She reported working as a chef and caring for multiple pets in her home, including 3 cats, 6 fish tanks, 3 dogs, and 3 lizards. Physical examination revealed a painful, indurated, red-violaceous plaque on the right forearm with satellite pink nodules that had been slowly migrating proximally up the forearm. An outside excisional biopsy performed 1 year prior had shown suppurative granulomatous dermatitis with negative stains for infectious organisms and negative tissue cultures. At that time, the patient was diagnosed with ruptured folliculitis; however, a subsequent lack of clinical improvement prompted her to seek a second opinion at our clinic.
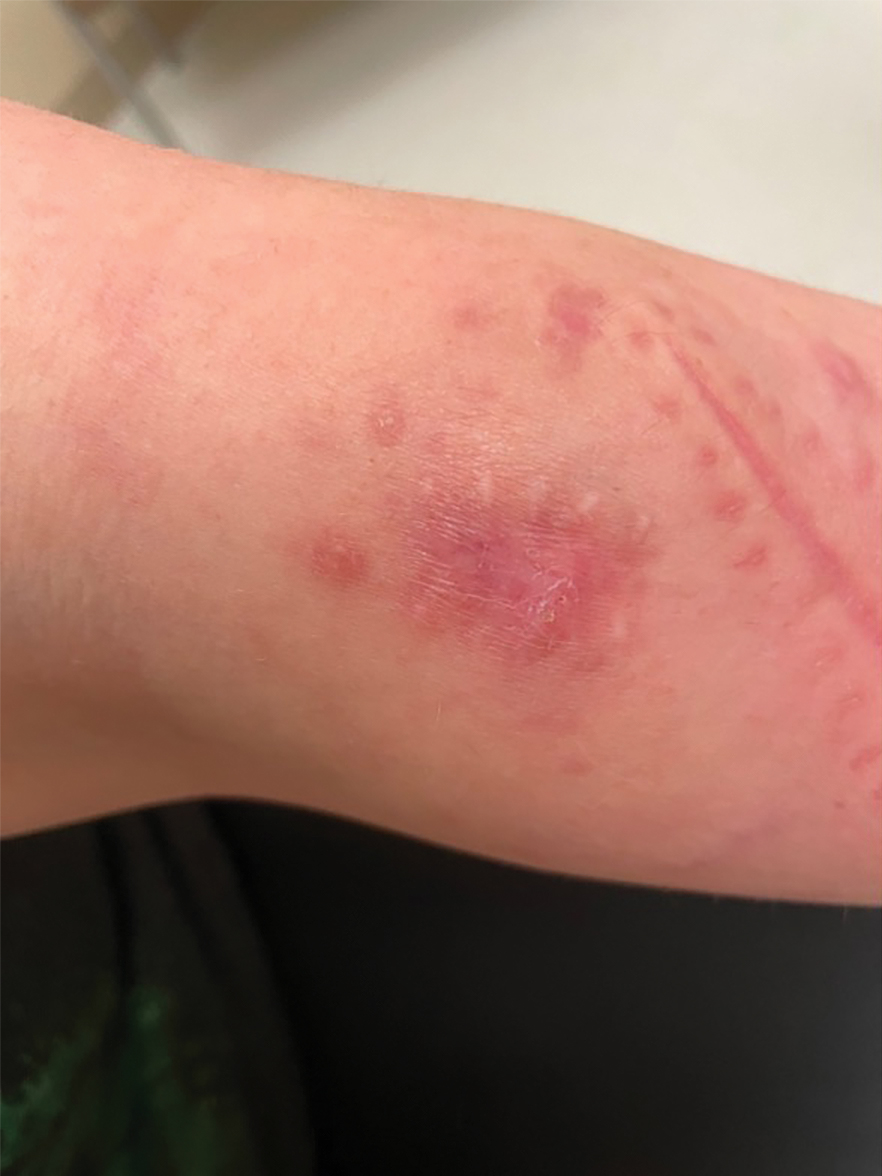
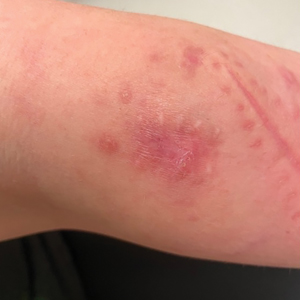
Painful Anal Lesions in a Patient With HIV
The Diagnosis: Condyloma Latum
Laboratory test results were notable for a rapid plasma reagin titer of 1:512, a positive Treponema pallidum particle agglutination test, negative rectal nucleic acid amplification tests for gonorrhea and chlamydia, and a negative herpes simplex virus polymerase chain reaction. A VDRL test of cerebrospinal fluid from a lumbar puncture was negative. Histopathology of the punch biopsy sample revealed marked verrucous epidermal hyperplasia without keratinocytic atypia and with mixed inflammation (Figure 1), while immunohistochemical staining showed numerus T pallidum organisms (Figure 2). A diagnosis of condyloma latum was made based on the laboratory, lumbar puncture, and punch biopsy results. Due to a penicillin allergy, the patient was treated with oral doxycycline for 14 days. On follow-up at day 12 of therapy, he reported cessation of rectal pain, and resolution of anal lesions was noted on physical examination.
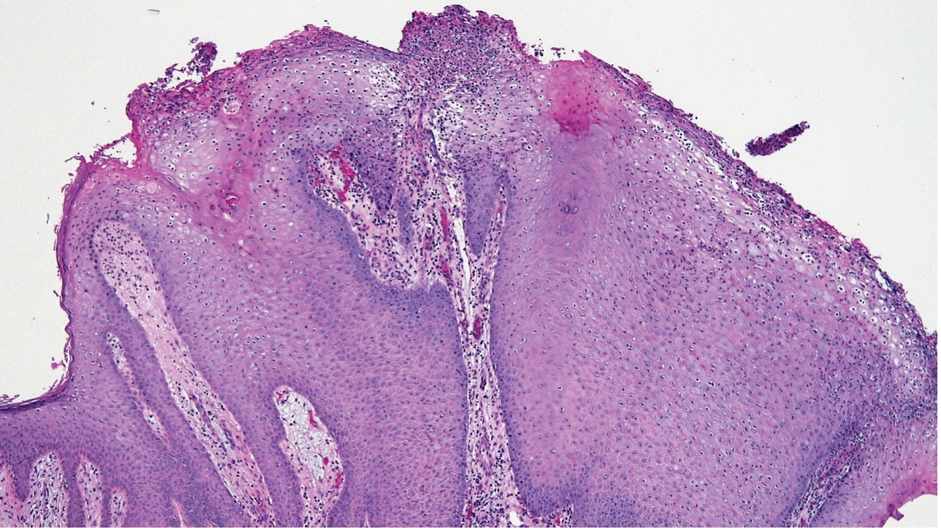
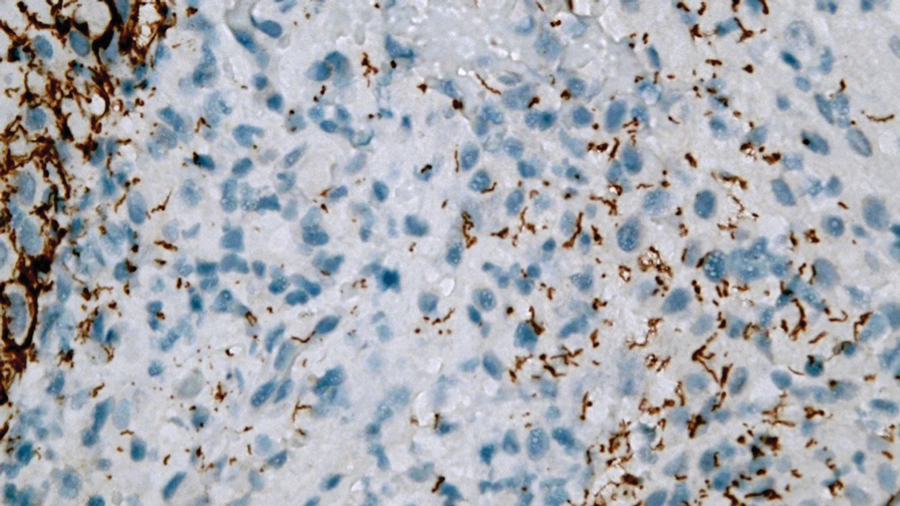
Condylomata lata are highly infectious cutaneous lesions that can manifest during secondary syphilis.1 They typically are described as white or gray, raised, flatappearing plaques and occur in moist areas or skin folds including the anus, scrotum, and vulva. However, these lesions also have been reported in the axillae, umbilicus, nasolabial folds, and other anatomic areas.1,2 The lesions can be painful and often manifest in multiples, especially in patients living with HIV.3
Condylomata lata can have a verrucous appearance and may mimic other anogenital lesions, such as condylomata acuminata, genital herpes, and malignant tumors, leading to an initial misdiagnosis.1,2 Condylomata lata should always be included in the differential when evaluating anogenital lesions. Other conditions in the differential diagnosis include psoriasis, typically manifesting as erythematous plaques with silver scale, and molluscum contagiosum, appearing as small umbilicated papules on physical examination.
Condylomata lata have been reported to occur in 6% to 23% of patients with secondary syphilis.1 Although secondary syphilis more typically manifests with a diffuse maculopapular rash, condylomata lata may be the sole dermatologic manifestation.4
Histopathology of condylomata lata consists of epithelial hyperplasia as well as lymphocytic and plasma cell infiltrates. It is diagnosed by serologic testing as well as immunohistochemical staining or dark-field microscopy.
First-line treatment of secondary syphilis is a single dose of benzathine penicillin G administered intramuscularly.5 However, a 14-day course of oral doxycycline can be used in patients with a penicillin allergy. When compliance and follow-up cannot be guaranteed, penicillin desensitization and treatment with benzathine penicillin G is recommended. Clinical evaluation and repeat serologic testing should be performed at 6 and 12 months follow-up, or more frequently if clinically indicated.5
- Pourang A, Fung MA, Tartar D, et al. Condyloma lata in secondary syphilis. JAAD Case Rep. 2021;10:18-21. doi:10.1016/j.jdcr.2021.01.025
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277. doi:10.4103/0378-6323.191129
- Rompalo AM, Joesoef MR, O’Donnell JA, et al; Syphilis and HIV Study Group. Clinical manifestations of early syphilis by HIV status and gender: results of the syphilis and HIV study. Sex Transm Dis.2001;28:158-165.
- Kumar P, Das A, Mondal A. Secondary syphilis: an unusual presentation. Indian J Sex Transm Dis AIDS. 2017;38:98-99. doi:10.4103/0253-7184.194318
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
The Diagnosis: Condyloma Latum
Laboratory test results were notable for a rapid plasma reagin titer of 1:512, a positive Treponema pallidum particle agglutination test, negative rectal nucleic acid amplification tests for gonorrhea and chlamydia, and a negative herpes simplex virus polymerase chain reaction. A VDRL test of cerebrospinal fluid from a lumbar puncture was negative. Histopathology of the punch biopsy sample revealed marked verrucous epidermal hyperplasia without keratinocytic atypia and with mixed inflammation (Figure 1), while immunohistochemical staining showed numerus T pallidum organisms (Figure 2). A diagnosis of condyloma latum was made based on the laboratory, lumbar puncture, and punch biopsy results. Due to a penicillin allergy, the patient was treated with oral doxycycline for 14 days. On follow-up at day 12 of therapy, he reported cessation of rectal pain, and resolution of anal lesions was noted on physical examination.
Condylomata lata are highly infectious cutaneous lesions that can manifest during secondary syphilis.1 They typically are described as white or gray, raised, flatappearing plaques and occur in moist areas or skin folds including the anus, scrotum, and vulva. However, these lesions also have been reported in the axillae, umbilicus, nasolabial folds, and other anatomic areas.1,2 The lesions can be painful and often manifest in multiples, especially in patients living with HIV.3
Condylomata lata can have a verrucous appearance and may mimic other anogenital lesions, such as condylomata acuminata, genital herpes, and malignant tumors, leading to an initial misdiagnosis.1,2 Condylomata lata should always be included in the differential when evaluating anogenital lesions. Other conditions in the differential diagnosis include psoriasis, typically manifesting as erythematous plaques with silver scale, and molluscum contagiosum, appearing as small umbilicated papules on physical examination.
Condylomata lata have been reported to occur in 6% to 23% of patients with secondary syphilis.1 Although secondary syphilis more typically manifests with a diffuse maculopapular rash, condylomata lata may be the sole dermatologic manifestation.4
Histopathology of condylomata lata consists of epithelial hyperplasia as well as lymphocytic and plasma cell infiltrates. It is diagnosed by serologic testing as well as immunohistochemical staining or dark-field microscopy.
First-line treatment of secondary syphilis is a single dose of benzathine penicillin G administered intramuscularly.5 However, a 14-day course of oral doxycycline can be used in patients with a penicillin allergy. When compliance and follow-up cannot be guaranteed, penicillin desensitization and treatment with benzathine penicillin G is recommended. Clinical evaluation and repeat serologic testing should be performed at 6 and 12 months follow-up, or more frequently if clinically indicated.5
The Diagnosis: Condyloma Latum
Laboratory test results were notable for a rapid plasma reagin titer of 1:512, a positive Treponema pallidum particle agglutination test, negative rectal nucleic acid amplification tests for gonorrhea and chlamydia, and a negative herpes simplex virus polymerase chain reaction. A VDRL test of cerebrospinal fluid from a lumbar puncture was negative. Histopathology of the punch biopsy sample revealed marked verrucous epidermal hyperplasia without keratinocytic atypia and with mixed inflammation (Figure 1), while immunohistochemical staining showed numerus T pallidum organisms (Figure 2). A diagnosis of condyloma latum was made based on the laboratory, lumbar puncture, and punch biopsy results. Due to a penicillin allergy, the patient was treated with oral doxycycline for 14 days. On follow-up at day 12 of therapy, he reported cessation of rectal pain, and resolution of anal lesions was noted on physical examination.
Condylomata lata are highly infectious cutaneous lesions that can manifest during secondary syphilis.1 They typically are described as white or gray, raised, flatappearing plaques and occur in moist areas or skin folds including the anus, scrotum, and vulva. However, these lesions also have been reported in the axillae, umbilicus, nasolabial folds, and other anatomic areas.1,2 The lesions can be painful and often manifest in multiples, especially in patients living with HIV.3
Condylomata lata can have a verrucous appearance and may mimic other anogenital lesions, such as condylomata acuminata, genital herpes, and malignant tumors, leading to an initial misdiagnosis.1,2 Condylomata lata should always be included in the differential when evaluating anogenital lesions. Other conditions in the differential diagnosis include psoriasis, typically manifesting as erythematous plaques with silver scale, and molluscum contagiosum, appearing as small umbilicated papules on physical examination.
Condylomata lata have been reported to occur in 6% to 23% of patients with secondary syphilis.1 Although secondary syphilis more typically manifests with a diffuse maculopapular rash, condylomata lata may be the sole dermatologic manifestation.4
Histopathology of condylomata lata consists of epithelial hyperplasia as well as lymphocytic and plasma cell infiltrates. It is diagnosed by serologic testing as well as immunohistochemical staining or dark-field microscopy.
First-line treatment of secondary syphilis is a single dose of benzathine penicillin G administered intramuscularly.5 However, a 14-day course of oral doxycycline can be used in patients with a penicillin allergy. When compliance and follow-up cannot be guaranteed, penicillin desensitization and treatment with benzathine penicillin G is recommended. Clinical evaluation and repeat serologic testing should be performed at 6 and 12 months follow-up, or more frequently if clinically indicated.5
- Pourang A, Fung MA, Tartar D, et al. Condyloma lata in secondary syphilis. JAAD Case Rep. 2021;10:18-21. doi:10.1016/j.jdcr.2021.01.025
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277. doi:10.4103/0378-6323.191129
- Rompalo AM, Joesoef MR, O’Donnell JA, et al; Syphilis and HIV Study Group. Clinical manifestations of early syphilis by HIV status and gender: results of the syphilis and HIV study. Sex Transm Dis.2001;28:158-165.
- Kumar P, Das A, Mondal A. Secondary syphilis: an unusual presentation. Indian J Sex Transm Dis AIDS. 2017;38:98-99. doi:10.4103/0253-7184.194318
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
- Pourang A, Fung MA, Tartar D, et al. Condyloma lata in secondary syphilis. JAAD Case Rep. 2021;10:18-21. doi:10.1016/j.jdcr.2021.01.025
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277. doi:10.4103/0378-6323.191129
- Rompalo AM, Joesoef MR, O’Donnell JA, et al; Syphilis and HIV Study Group. Clinical manifestations of early syphilis by HIV status and gender: results of the syphilis and HIV study. Sex Transm Dis.2001;28:158-165.
- Kumar P, Das A, Mondal A. Secondary syphilis: an unusual presentation. Indian J Sex Transm Dis AIDS. 2017;38:98-99. doi:10.4103/0253-7184.194318
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
A 24-year-old man presented to the emergency department with rectal pain and lesions of 3 weeks’ duration that were progressively worsening. He had a medical history of poorly controlled HIV, cerebral toxoplasmosis, and genital herpes, as well as a social history of sexual activity with other men.
He had been diagnosed with HIV 7 years prior and had been off therapy until 1 year prior to the current presentation, when he was hospitalized with encephalopathy (CD4 count, <50 cells/mm3). A diagnosis of cerebral toxoplasmosis was made, and he began a treatment regimen of sulfadiazine, pyrimethamine, and leucovorin, as well as bictegravir, emtricitabine, and tenofovir alafenamide. Since then, the patient admitted to difficulty with medication adherence.
Rapid plasma reagin, gonorrhea, and chlamydia testing were negative during a routine workup 6 months prior to the current presentation. He initially presented to an urgent care clinic for evaluation of the rectal pain and lesions and was treated empirically with topical podofilox. He presented to the emergency department 1 week later (3 weeks after symptom onset) with anal warts and apparent vesicular lesions. Empiric treatment with oral valacyclovir was prescribed.
Despite these treatments, the rectal pain became severe—especially upon sitting, defecation, and physical exertion—prompting further evaluation. Physical examination revealed soft, flat-topped, moist-appearing, gray plaques with minimal surrounding erythema at the anus. Laboratory test results demonstrated a CD4 count of 161 cells/mm3 and an HIV viral load of 137 copies/mL.
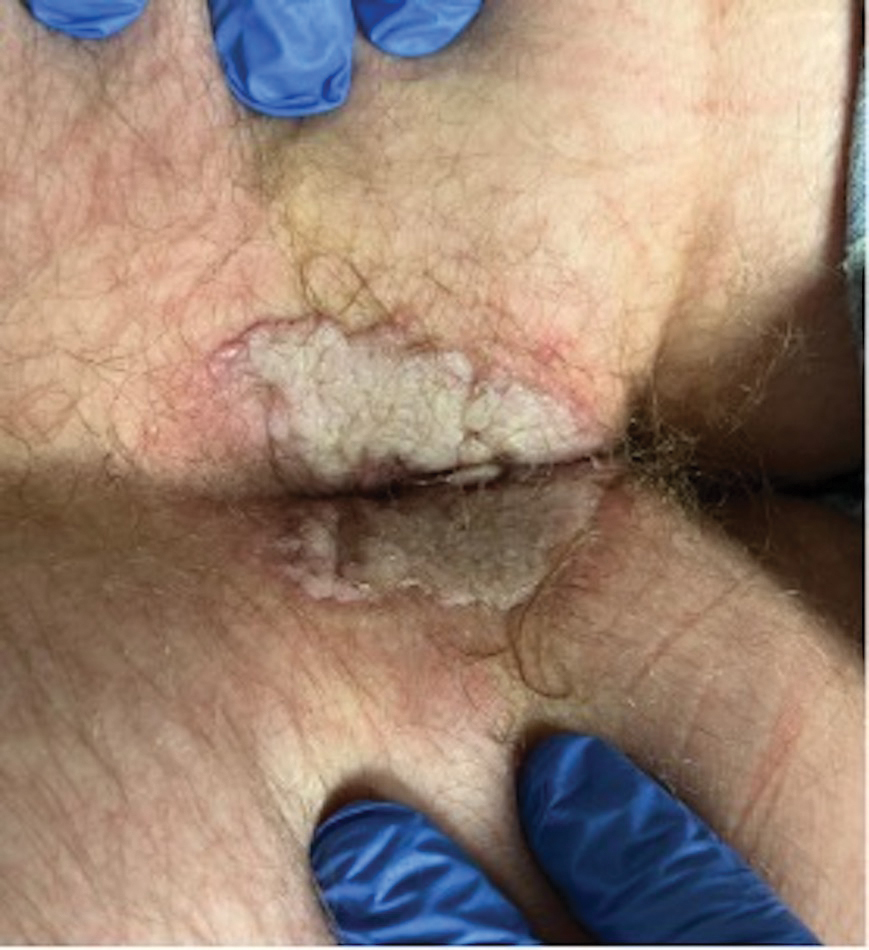
Pruritic Rash on the Neck and Back
The Diagnosis: Prurigo Pigmentosa
A comprehensive metabolic panel collected from our patient 1 month earlier did not reveal any abnormalities. Serum methylmalonic acid and homocysteine were both elevated at 417 nmol/L (reference range [for those aged 2–59 years], 55–335 nmol/L) and 23 μmol/L (reference range, 5–15 μmol/L), respectively. Serum folate and 25-hydroxyvitamin D were low at 3.1 ng/mL (reference range, >4.8 ng/mL) and 5 ng/mL (reference range, 30–80 ng/mL), respectively. Vitamin B12 was within reference range. Two 4-mm punch biopsies collected from the upper back showed spongiotic dermatitis.
Our patient’s histopathology results along with the rash distribution and medical history of anorexia increased suspicion for prurigo pigmentosa. A trial of oral doxycycline 100 mg twice daily for 2 weeks was prescribed. At 2-week follow-up, the patient’s mother revealed a history of ketosis in her daughter, solidifying the diagnosis. The patient was counseled on maintaining a healthy diet to prevent future breakouts. The patient’s rash resolved with diet modification and doxycycline; however, it recurred upon relapse of anorexia 4 months later.
Prurigo pigmentosa, originally identified in Japan by Nagashima et al,1 is an uncommon recurrent inflammatory disorder predominantly observed in young adults of Asian descent. Subsequently, it was reported to occur among individuals from different ethnic backgrounds, indicating potential underdiagnosis or misdiagnosis in Western countries.2 Although a direct pathogenic cause for prurigo pigmentosa has not been identified, a strong association has been linked to diet, specifically when ketosis is induced, such as in ketogenic diets and anorexia nervosa.3-5 Other possible causes include sunlight exposure, clothing friction, and sweating.1,5 The disease course is characterized by intermittent flares and spontaneous resolution, with recurrence in most cases. During the active phase, intensely pruritic, papulovesicular or urticarial papules are predominant and most often are localized to the upper body and torso, including the back, shoulders, neck, and chest.5 These flares can persist for several days but eventually subside, leaving behind a characteristic reticular pigmentation that can persist for months.5 First-line treatment often involves the use of tetracycline antibiotics, such as minocycline or doxycycline. 2,4,5 Dapsone often is used with successful resolution. 6 Dietary modifications also have been found to be effective in treating prurigo pigmentosa, particularly in patients presenting with dietary insufficiency.6,7 Increased carbohydrate intake has been shown to promote resolution. 6 Topical corticosteroids demonstrate limited efficacy in controlling flares.6,8
Histopathology has been variably described, with initial findings reported as nonspecific.1 However, it was later described as a distinct inflammatory disease of the skin with histologically distinct stages.2,9 Early stages reveal scattered dermal, dermal papillary, and perivascular neutrophilic infiltration.9 The lesions then progress and become fully developed, at which point neutrophilic infiltration becomes more prominent, accompanied by the presence of intraepidermal neutrophils and spongiosis. As the lesions resolve, the infiltration transitions to lymphocytic, and lichenoid changes can sometimes be appreciated along with epidermal hyperplasia, hyperpigmentation, and dermal melanophages.9 Although these findings aid in the diagnosis of prurigo pigmentosa, a clinicopathologic correlation is necessary to establish a definitive diagnosis.
Because prurigo pigmentosa is rare, it often is misdiagnosed as another condition with a similar presentation and nonspecific biopsy findings.6 Allergic contact dermatitis is a common type IV delayed hypersensitivity reaction that manifests similar to prurigo pigmentosa with pruritus and a well-demarcated distribution10 that is related to the pattern of allergen exposure; in the case of allergic contact dermatitis related to textiles, a well-demarcated rash will appear in the distribution area of the associated clothing (eg, shirt, pants, shorts).11 Development of allergy involves exposure and sensitization to an allergen, followed by subsequent re-exposure that results in cutaneous T-cell activation and inflammation. 10 Histopathology shows nonspecific spongiotic inflammation, and the gold standard for diagnosis is patch testing to identify the causative substance(s). Definitive treatment includes avoidance of identified allergies; however, if patients are unable to avoid the allergen or the cause is unknown, then corticosteroids, antihistamines, and/or calcineurin inhibitors are beneficial in controlling symptoms and flares.10
Pityrosporum folliculitis (also known as Malassezia folliculitis) is a fungal acneform condition that arises from overgrowth of normal skin flora Malassezia yeast,12 which may be due to occlusion of follicles or disruption of the normal flora composition. Clinically, the manifestation may resemble prurigo pigmentosa in distribution and presence of intense pruritus. However, pustular lesions and involvement of the face can aid in differentiating Pityrosporum from prurigo pigmentosa, which can be confirmed via periodic acid–Schiff staining with numerous round yeasts within affected follicles. Oral antifungal therapy typically yields rapid improvement and resolution of symptoms.12
Urticaria and prurigo pigmentosa share similar clinical characteristics, with symptoms of intense pruritus and urticarial lesions on the trunk.2,13 Urticaria is an IgEmediated type I hypersensitivity reaction characterized by wheals (ie, edematous red or pink lesions of variable size and shape that typically resolve spontaneously within 24–48 hours).13 Notably, urticaria will improve and in some cases completely resolve with antihistamines or anti-IgE antibody treatment, which may aid in distinguishing it from prurigo pigmentosa, as the latter typically exhibits limited response to such treatment.2 Histopathology also can assist in the diagnosis by ruling out other causes of similar rash; however, biopsies are not routinely done unless other inflammatory conditions are of high suspicion.13
Bullous pemphigoid is an autoimmune, subepidermal, blistering dermatosis that is most common among the elderly.14 It is characterized by the presence of IgG antibodies that target BP180 and BP230, which initiate inflammatory cascades that lead to tissue damage and blister formation. It typically manifests as pruritic blistering eruptions, primarily on the limbs and trunk, but may involve the head, neck, or palmoplantar regions.14 Although blistering eruptions are the prodrome of the disease, some cases may present with nonspecific urticarial or eczematous lesions14,15 that may resemble prurigo pigmentosa. The diagnosis is confirmed through direct immunofluorescence microscopy of biopsied lesions, which reveals IgG and/or C3 deposits along the dermoepidermal junction.14 Management of bullous pemphigoid involves timely initiation of dapsone or systemic corticosteroids, which have demonstrated high efficacy in controlling the disease and its associated symptoms.15
Our patient achieved a favorable response to diet modification and doxycycline therapy consistent with the diagnosis of prurigo pigmentosa. Unfortunately, the condition recurred following a relapse of anorexia. Management of prurigo pigmentosa necessitates not only accurate diagnosis but also addressing any underlying factors that may contribute to disease exacerbation. We anticipate the eating disorder will pose a major challenge in achieving long-term control of prurigo pigmentosa.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation. Jpn J Dermatol. 1971;81:38-39.
- Boer A, Asgari M. Prurigo pigmentosa: an underdiagnosed disease? Indian J Dermatol Venereol Leprol. 2006;72:405-409. doi:10.4103/0378-6323.29334
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet. Pediatr Dermatol. 2013;32:248-251. doi:10.1111/pde.12275
- Teraki Y, Teraki E, Kawashima M, et al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511. doi:10.1016/s0190-9622(96)90460-0
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79-87. doi:10.1016/j.jdin.2021.03.003
- Wong M, Lee E, Wu Y, et al. Treatment of prurigo pigmentosa with diet modification: a medical case study. Hawaii J Med Public Health. 2018;77:114-117.
- Almaani N, Al-Tarawneh AH, Msallam H. Prurigo pigmentosa: a clinicopathological report of three Middle Eastern patients. Case Rep Dermatol Med. 2018;2018:9406797. doi:10.1155/2018/9406797
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897. doi:10.1111/j.1346-8138.2012.01640.x
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74:1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Lazarov A, Cordoba M, Plosk N, et al. Atypical and unusual clinical manifestations of contact dermatitis to clothing (textile contact dermatitis)—case presentation and review of the literature. Dermatol Online J. 2003;9. doi:10.5070/d30kd1d259
- Rubenstein RM, Malerich SA. Malassezia (Pityrosporum) folliculitis. J Clin Aesthet Dermatol. 2014;7:37-41.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277. doi:10.1016/j.jaci.2014.02.036
- della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol. 2012;167:1111-1117. doi:10.1111/j.1365-2133.2012.11108.x
- Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, et al. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. Int J Dermatol. 1998;37:508-514.
The Diagnosis: Prurigo Pigmentosa
A comprehensive metabolic panel collected from our patient 1 month earlier did not reveal any abnormalities. Serum methylmalonic acid and homocysteine were both elevated at 417 nmol/L (reference range [for those aged 2–59 years], 55–335 nmol/L) and 23 μmol/L (reference range, 5–15 μmol/L), respectively. Serum folate and 25-hydroxyvitamin D were low at 3.1 ng/mL (reference range, >4.8 ng/mL) and 5 ng/mL (reference range, 30–80 ng/mL), respectively. Vitamin B12 was within reference range. Two 4-mm punch biopsies collected from the upper back showed spongiotic dermatitis.
Our patient’s histopathology results along with the rash distribution and medical history of anorexia increased suspicion for prurigo pigmentosa. A trial of oral doxycycline 100 mg twice daily for 2 weeks was prescribed. At 2-week follow-up, the patient’s mother revealed a history of ketosis in her daughter, solidifying the diagnosis. The patient was counseled on maintaining a healthy diet to prevent future breakouts. The patient’s rash resolved with diet modification and doxycycline; however, it recurred upon relapse of anorexia 4 months later.
Prurigo pigmentosa, originally identified in Japan by Nagashima et al,1 is an uncommon recurrent inflammatory disorder predominantly observed in young adults of Asian descent. Subsequently, it was reported to occur among individuals from different ethnic backgrounds, indicating potential underdiagnosis or misdiagnosis in Western countries.2 Although a direct pathogenic cause for prurigo pigmentosa has not been identified, a strong association has been linked to diet, specifically when ketosis is induced, such as in ketogenic diets and anorexia nervosa.3-5 Other possible causes include sunlight exposure, clothing friction, and sweating.1,5 The disease course is characterized by intermittent flares and spontaneous resolution, with recurrence in most cases. During the active phase, intensely pruritic, papulovesicular or urticarial papules are predominant and most often are localized to the upper body and torso, including the back, shoulders, neck, and chest.5 These flares can persist for several days but eventually subside, leaving behind a characteristic reticular pigmentation that can persist for months.5 First-line treatment often involves the use of tetracycline antibiotics, such as minocycline or doxycycline. 2,4,5 Dapsone often is used with successful resolution. 6 Dietary modifications also have been found to be effective in treating prurigo pigmentosa, particularly in patients presenting with dietary insufficiency.6,7 Increased carbohydrate intake has been shown to promote resolution. 6 Topical corticosteroids demonstrate limited efficacy in controlling flares.6,8
Histopathology has been variably described, with initial findings reported as nonspecific.1 However, it was later described as a distinct inflammatory disease of the skin with histologically distinct stages.2,9 Early stages reveal scattered dermal, dermal papillary, and perivascular neutrophilic infiltration.9 The lesions then progress and become fully developed, at which point neutrophilic infiltration becomes more prominent, accompanied by the presence of intraepidermal neutrophils and spongiosis. As the lesions resolve, the infiltration transitions to lymphocytic, and lichenoid changes can sometimes be appreciated along with epidermal hyperplasia, hyperpigmentation, and dermal melanophages.9 Although these findings aid in the diagnosis of prurigo pigmentosa, a clinicopathologic correlation is necessary to establish a definitive diagnosis.
Because prurigo pigmentosa is rare, it often is misdiagnosed as another condition with a similar presentation and nonspecific biopsy findings.6 Allergic contact dermatitis is a common type IV delayed hypersensitivity reaction that manifests similar to prurigo pigmentosa with pruritus and a well-demarcated distribution10 that is related to the pattern of allergen exposure; in the case of allergic contact dermatitis related to textiles, a well-demarcated rash will appear in the distribution area of the associated clothing (eg, shirt, pants, shorts).11 Development of allergy involves exposure and sensitization to an allergen, followed by subsequent re-exposure that results in cutaneous T-cell activation and inflammation. 10 Histopathology shows nonspecific spongiotic inflammation, and the gold standard for diagnosis is patch testing to identify the causative substance(s). Definitive treatment includes avoidance of identified allergies; however, if patients are unable to avoid the allergen or the cause is unknown, then corticosteroids, antihistamines, and/or calcineurin inhibitors are beneficial in controlling symptoms and flares.10
Pityrosporum folliculitis (also known as Malassezia folliculitis) is a fungal acneform condition that arises from overgrowth of normal skin flora Malassezia yeast,12 which may be due to occlusion of follicles or disruption of the normal flora composition. Clinically, the manifestation may resemble prurigo pigmentosa in distribution and presence of intense pruritus. However, pustular lesions and involvement of the face can aid in differentiating Pityrosporum from prurigo pigmentosa, which can be confirmed via periodic acid–Schiff staining with numerous round yeasts within affected follicles. Oral antifungal therapy typically yields rapid improvement and resolution of symptoms.12
Urticaria and prurigo pigmentosa share similar clinical characteristics, with symptoms of intense pruritus and urticarial lesions on the trunk.2,13 Urticaria is an IgEmediated type I hypersensitivity reaction characterized by wheals (ie, edematous red or pink lesions of variable size and shape that typically resolve spontaneously within 24–48 hours).13 Notably, urticaria will improve and in some cases completely resolve with antihistamines or anti-IgE antibody treatment, which may aid in distinguishing it from prurigo pigmentosa, as the latter typically exhibits limited response to such treatment.2 Histopathology also can assist in the diagnosis by ruling out other causes of similar rash; however, biopsies are not routinely done unless other inflammatory conditions are of high suspicion.13
Bullous pemphigoid is an autoimmune, subepidermal, blistering dermatosis that is most common among the elderly.14 It is characterized by the presence of IgG antibodies that target BP180 and BP230, which initiate inflammatory cascades that lead to tissue damage and blister formation. It typically manifests as pruritic blistering eruptions, primarily on the limbs and trunk, but may involve the head, neck, or palmoplantar regions.14 Although blistering eruptions are the prodrome of the disease, some cases may present with nonspecific urticarial or eczematous lesions14,15 that may resemble prurigo pigmentosa. The diagnosis is confirmed through direct immunofluorescence microscopy of biopsied lesions, which reveals IgG and/or C3 deposits along the dermoepidermal junction.14 Management of bullous pemphigoid involves timely initiation of dapsone or systemic corticosteroids, which have demonstrated high efficacy in controlling the disease and its associated symptoms.15
Our patient achieved a favorable response to diet modification and doxycycline therapy consistent with the diagnosis of prurigo pigmentosa. Unfortunately, the condition recurred following a relapse of anorexia. Management of prurigo pigmentosa necessitates not only accurate diagnosis but also addressing any underlying factors that may contribute to disease exacerbation. We anticipate the eating disorder will pose a major challenge in achieving long-term control of prurigo pigmentosa.
The Diagnosis: Prurigo Pigmentosa
A comprehensive metabolic panel collected from our patient 1 month earlier did not reveal any abnormalities. Serum methylmalonic acid and homocysteine were both elevated at 417 nmol/L (reference range [for those aged 2–59 years], 55–335 nmol/L) and 23 μmol/L (reference range, 5–15 μmol/L), respectively. Serum folate and 25-hydroxyvitamin D were low at 3.1 ng/mL (reference range, >4.8 ng/mL) and 5 ng/mL (reference range, 30–80 ng/mL), respectively. Vitamin B12 was within reference range. Two 4-mm punch biopsies collected from the upper back showed spongiotic dermatitis.
Our patient’s histopathology results along with the rash distribution and medical history of anorexia increased suspicion for prurigo pigmentosa. A trial of oral doxycycline 100 mg twice daily for 2 weeks was prescribed. At 2-week follow-up, the patient’s mother revealed a history of ketosis in her daughter, solidifying the diagnosis. The patient was counseled on maintaining a healthy diet to prevent future breakouts. The patient’s rash resolved with diet modification and doxycycline; however, it recurred upon relapse of anorexia 4 months later.
Prurigo pigmentosa, originally identified in Japan by Nagashima et al,1 is an uncommon recurrent inflammatory disorder predominantly observed in young adults of Asian descent. Subsequently, it was reported to occur among individuals from different ethnic backgrounds, indicating potential underdiagnosis or misdiagnosis in Western countries.2 Although a direct pathogenic cause for prurigo pigmentosa has not been identified, a strong association has been linked to diet, specifically when ketosis is induced, such as in ketogenic diets and anorexia nervosa.3-5 Other possible causes include sunlight exposure, clothing friction, and sweating.1,5 The disease course is characterized by intermittent flares and spontaneous resolution, with recurrence in most cases. During the active phase, intensely pruritic, papulovesicular or urticarial papules are predominant and most often are localized to the upper body and torso, including the back, shoulders, neck, and chest.5 These flares can persist for several days but eventually subside, leaving behind a characteristic reticular pigmentation that can persist for months.5 First-line treatment often involves the use of tetracycline antibiotics, such as minocycline or doxycycline. 2,4,5 Dapsone often is used with successful resolution. 6 Dietary modifications also have been found to be effective in treating prurigo pigmentosa, particularly in patients presenting with dietary insufficiency.6,7 Increased carbohydrate intake has been shown to promote resolution. 6 Topical corticosteroids demonstrate limited efficacy in controlling flares.6,8
Histopathology has been variably described, with initial findings reported as nonspecific.1 However, it was later described as a distinct inflammatory disease of the skin with histologically distinct stages.2,9 Early stages reveal scattered dermal, dermal papillary, and perivascular neutrophilic infiltration.9 The lesions then progress and become fully developed, at which point neutrophilic infiltration becomes more prominent, accompanied by the presence of intraepidermal neutrophils and spongiosis. As the lesions resolve, the infiltration transitions to lymphocytic, and lichenoid changes can sometimes be appreciated along with epidermal hyperplasia, hyperpigmentation, and dermal melanophages.9 Although these findings aid in the diagnosis of prurigo pigmentosa, a clinicopathologic correlation is necessary to establish a definitive diagnosis.
Because prurigo pigmentosa is rare, it often is misdiagnosed as another condition with a similar presentation and nonspecific biopsy findings.6 Allergic contact dermatitis is a common type IV delayed hypersensitivity reaction that manifests similar to prurigo pigmentosa with pruritus and a well-demarcated distribution10 that is related to the pattern of allergen exposure; in the case of allergic contact dermatitis related to textiles, a well-demarcated rash will appear in the distribution area of the associated clothing (eg, shirt, pants, shorts).11 Development of allergy involves exposure and sensitization to an allergen, followed by subsequent re-exposure that results in cutaneous T-cell activation and inflammation. 10 Histopathology shows nonspecific spongiotic inflammation, and the gold standard for diagnosis is patch testing to identify the causative substance(s). Definitive treatment includes avoidance of identified allergies; however, if patients are unable to avoid the allergen or the cause is unknown, then corticosteroids, antihistamines, and/or calcineurin inhibitors are beneficial in controlling symptoms and flares.10
Pityrosporum folliculitis (also known as Malassezia folliculitis) is a fungal acneform condition that arises from overgrowth of normal skin flora Malassezia yeast,12 which may be due to occlusion of follicles or disruption of the normal flora composition. Clinically, the manifestation may resemble prurigo pigmentosa in distribution and presence of intense pruritus. However, pustular lesions and involvement of the face can aid in differentiating Pityrosporum from prurigo pigmentosa, which can be confirmed via periodic acid–Schiff staining with numerous round yeasts within affected follicles. Oral antifungal therapy typically yields rapid improvement and resolution of symptoms.12
Urticaria and prurigo pigmentosa share similar clinical characteristics, with symptoms of intense pruritus and urticarial lesions on the trunk.2,13 Urticaria is an IgEmediated type I hypersensitivity reaction characterized by wheals (ie, edematous red or pink lesions of variable size and shape that typically resolve spontaneously within 24–48 hours).13 Notably, urticaria will improve and in some cases completely resolve with antihistamines or anti-IgE antibody treatment, which may aid in distinguishing it from prurigo pigmentosa, as the latter typically exhibits limited response to such treatment.2 Histopathology also can assist in the diagnosis by ruling out other causes of similar rash; however, biopsies are not routinely done unless other inflammatory conditions are of high suspicion.13
Bullous pemphigoid is an autoimmune, subepidermal, blistering dermatosis that is most common among the elderly.14 It is characterized by the presence of IgG antibodies that target BP180 and BP230, which initiate inflammatory cascades that lead to tissue damage and blister formation. It typically manifests as pruritic blistering eruptions, primarily on the limbs and trunk, but may involve the head, neck, or palmoplantar regions.14 Although blistering eruptions are the prodrome of the disease, some cases may present with nonspecific urticarial or eczematous lesions14,15 that may resemble prurigo pigmentosa. The diagnosis is confirmed through direct immunofluorescence microscopy of biopsied lesions, which reveals IgG and/or C3 deposits along the dermoepidermal junction.14 Management of bullous pemphigoid involves timely initiation of dapsone or systemic corticosteroids, which have demonstrated high efficacy in controlling the disease and its associated symptoms.15
Our patient achieved a favorable response to diet modification and doxycycline therapy consistent with the diagnosis of prurigo pigmentosa. Unfortunately, the condition recurred following a relapse of anorexia. Management of prurigo pigmentosa necessitates not only accurate diagnosis but also addressing any underlying factors that may contribute to disease exacerbation. We anticipate the eating disorder will pose a major challenge in achieving long-term control of prurigo pigmentosa.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation. Jpn J Dermatol. 1971;81:38-39.
- Boer A, Asgari M. Prurigo pigmentosa: an underdiagnosed disease? Indian J Dermatol Venereol Leprol. 2006;72:405-409. doi:10.4103/0378-6323.29334
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet. Pediatr Dermatol. 2013;32:248-251. doi:10.1111/pde.12275
- Teraki Y, Teraki E, Kawashima M, et al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511. doi:10.1016/s0190-9622(96)90460-0
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79-87. doi:10.1016/j.jdin.2021.03.003
- Wong M, Lee E, Wu Y, et al. Treatment of prurigo pigmentosa with diet modification: a medical case study. Hawaii J Med Public Health. 2018;77:114-117.
- Almaani N, Al-Tarawneh AH, Msallam H. Prurigo pigmentosa: a clinicopathological report of three Middle Eastern patients. Case Rep Dermatol Med. 2018;2018:9406797. doi:10.1155/2018/9406797
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897. doi:10.1111/j.1346-8138.2012.01640.x
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74:1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Lazarov A, Cordoba M, Plosk N, et al. Atypical and unusual clinical manifestations of contact dermatitis to clothing (textile contact dermatitis)—case presentation and review of the literature. Dermatol Online J. 2003;9. doi:10.5070/d30kd1d259
- Rubenstein RM, Malerich SA. Malassezia (Pityrosporum) folliculitis. J Clin Aesthet Dermatol. 2014;7:37-41.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277. doi:10.1016/j.jaci.2014.02.036
- della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol. 2012;167:1111-1117. doi:10.1111/j.1365-2133.2012.11108.x
- Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, et al. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. Int J Dermatol. 1998;37:508-514.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation. Jpn J Dermatol. 1971;81:38-39.
- Boer A, Asgari M. Prurigo pigmentosa: an underdiagnosed disease? Indian J Dermatol Venereol Leprol. 2006;72:405-409. doi:10.4103/0378-6323.29334
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet. Pediatr Dermatol. 2013;32:248-251. doi:10.1111/pde.12275
- Teraki Y, Teraki E, Kawashima M, et al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511. doi:10.1016/s0190-9622(96)90460-0
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79-87. doi:10.1016/j.jdin.2021.03.003
- Wong M, Lee E, Wu Y, et al. Treatment of prurigo pigmentosa with diet modification: a medical case study. Hawaii J Med Public Health. 2018;77:114-117.
- Almaani N, Al-Tarawneh AH, Msallam H. Prurigo pigmentosa: a clinicopathological report of three Middle Eastern patients. Case Rep Dermatol Med. 2018;2018:9406797. doi:10.1155/2018/9406797
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897. doi:10.1111/j.1346-8138.2012.01640.x
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74:1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Lazarov A, Cordoba M, Plosk N, et al. Atypical and unusual clinical manifestations of contact dermatitis to clothing (textile contact dermatitis)—case presentation and review of the literature. Dermatol Online J. 2003;9. doi:10.5070/d30kd1d259
- Rubenstein RM, Malerich SA. Malassezia (Pityrosporum) folliculitis. J Clin Aesthet Dermatol. 2014;7:37-41.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277. doi:10.1016/j.jaci.2014.02.036
- della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol. 2012;167:1111-1117. doi:10.1111/j.1365-2133.2012.11108.x
- Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, et al. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. Int J Dermatol. 1998;37:508-514.
A 43-year-old woman presented with a pruritic rash across the neck and back of 6 months’ duration that progressively worsened. She had a medical history of anorexia nervosa, herpes zoster with a recent flare, and peripheral neuropathy. Physical examination showed numerous red scaly papules across the upper back and shoulders that coalesced in a reticular pattern. No similar papules were seen elsewhere on the body.
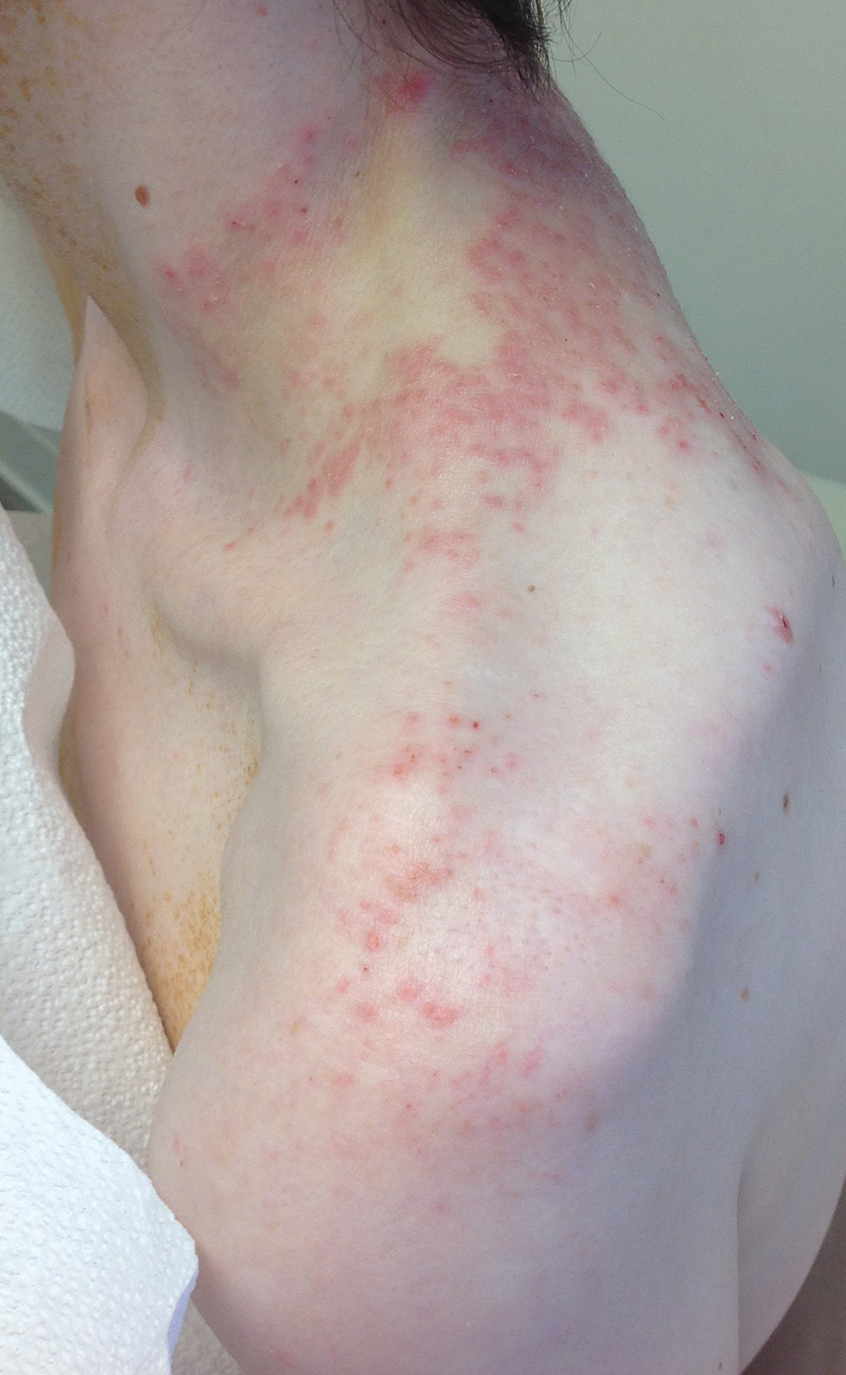
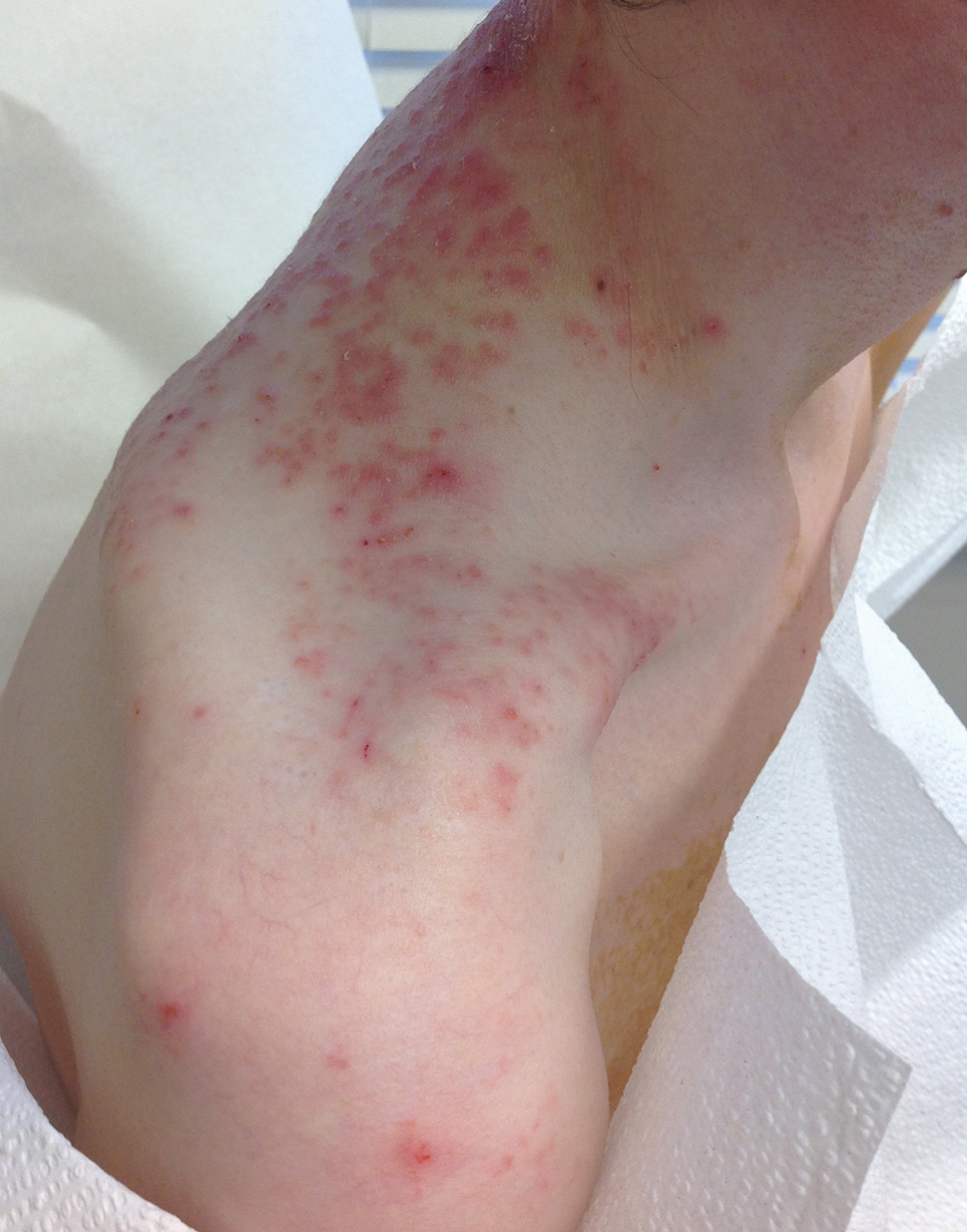
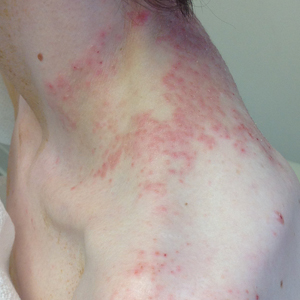
Draining Nodule of the Hand
The Diagnosis: Cutaneous Nocardiosis
The wound culture was positive for Nocardia farcinica. The patient received a 5-day course of intravenous sulfamethoxazole-trimethoprim in the hospital and was transitioned to oral sulfamethoxazoletrimethoprim (800 mg/160 mg taken as 1 tablet twice daily) for 6 months. Complete resolution of the infection was noted at 6-month follow-up (Figure).
Nocardia is a gram-positive, aerobic bacterium that typically is found in soil, water, and decaying organic matter.1 There are more than 50 species; N farcinica, Nocardia nova, and Nocardia asteroides are the leading causes of infection in humans and animals. Nocardia asteroides is the most common cause of infection in humans.1,2 Nocardiosis is an uncommon opportunistic infection that usually targets the skin, lungs, and central nervous system.3 Although it mainly affects individuals who are immunocompromised, up to 30% of infections can be seen in immunocompetent hosts who can contract cutaneous nocardiosis after experiencing traumatic injury to the skin.1
Nocardiosis is difficult to diagnose due to its diverse clinical presentations. For example, cutaneous nocardiosis can manifest similar to mycetoma, sporotrichosis, spider bites, nontuberculous mycobacteria such as Mycobacterium marinum, or methicillin-resistant Staphylococcus aureus infections, thus making cutaneous nocardiosis one of the great imitators.1 A culture is required for definitive diagnosis, as Nocardia grows well on nonselective media such as blood or Löwenstein-Jensen agar. It grows as waxy, pigmented, cerebriform colonies 3 to 5 days following incubation.3 The bacterium can be difficult to culture, and it is important to notify the microbiology laboratory if there is a high index of clinical suspicion for infection.
A history of exposure to gardening or handling animals can increase the risk for an individual contracting Nocardia.3 Although nocardiosis can be found across the world, it is native to tropical and subtropical climates such as those found in India, Africa, Latin America, and Southeast Asia.1 Infections mostly are observed in individuals aged 20 to 40 years and tend to affect men more than women. Lesions typically are seen on the lower extremities, but localized infections also can be found on the torso, neck, and upper extremities.1
Cutaneous nocardiosis is a granulomatous infection encompassing both cutaneous and subcutaneous tissue, which ultimately can lead to injury of bone and viscera.1 Primary cutaneous nocardiosis can manifest as tumors or nodules that have a sporotrichoid pattern, in which they ascend along the lymphatics. Histopathology of infected tissue frequently shows a subcutaneous dermal infiltrate of neutrophils accompanied with abscess formation, and everlasting lesions may show signs of chronic inflammation and nonspecific granulomas.3
Treatment of nocardiosis should be guided by in vitro susceptibility tests. Sulfamethoxazole-trimethoprim 800 mg/160 mg taken as 1 tablet twice daily is the first-line option. The treatment duration is contingent on the extent, severity, and complications of infection but typically is 3 to 6 months.1
- Yu Q, Song J, Liu Y, et al. Progressive primary cutaneous nocardiosis in an immunocompetent patient. Cutis. 2023;111:E22-E25.
- Gaines RJ, Randall CJ, Ruland RT. Lymphocutaneous nocardiosis from commercially treated lumber: a case report. Cutis. 2006;78:249-251.
- Riswold KJ, Tjarks BJ, Kerkvliet AM. Cutaneous nocardiosis in an immunocompromised patient. Cutis. 2019;104:226-229.
The Diagnosis: Cutaneous Nocardiosis
The wound culture was positive for Nocardia farcinica. The patient received a 5-day course of intravenous sulfamethoxazole-trimethoprim in the hospital and was transitioned to oral sulfamethoxazoletrimethoprim (800 mg/160 mg taken as 1 tablet twice daily) for 6 months. Complete resolution of the infection was noted at 6-month follow-up (Figure).
Nocardia is a gram-positive, aerobic bacterium that typically is found in soil, water, and decaying organic matter.1 There are more than 50 species; N farcinica, Nocardia nova, and Nocardia asteroides are the leading causes of infection in humans and animals. Nocardia asteroides is the most common cause of infection in humans.1,2 Nocardiosis is an uncommon opportunistic infection that usually targets the skin, lungs, and central nervous system.3 Although it mainly affects individuals who are immunocompromised, up to 30% of infections can be seen in immunocompetent hosts who can contract cutaneous nocardiosis after experiencing traumatic injury to the skin.1
Nocardiosis is difficult to diagnose due to its diverse clinical presentations. For example, cutaneous nocardiosis can manifest similar to mycetoma, sporotrichosis, spider bites, nontuberculous mycobacteria such as Mycobacterium marinum, or methicillin-resistant Staphylococcus aureus infections, thus making cutaneous nocardiosis one of the great imitators.1 A culture is required for definitive diagnosis, as Nocardia grows well on nonselective media such as blood or Löwenstein-Jensen agar. It grows as waxy, pigmented, cerebriform colonies 3 to 5 days following incubation.3 The bacterium can be difficult to culture, and it is important to notify the microbiology laboratory if there is a high index of clinical suspicion for infection.
A history of exposure to gardening or handling animals can increase the risk for an individual contracting Nocardia.3 Although nocardiosis can be found across the world, it is native to tropical and subtropical climates such as those found in India, Africa, Latin America, and Southeast Asia.1 Infections mostly are observed in individuals aged 20 to 40 years and tend to affect men more than women. Lesions typically are seen on the lower extremities, but localized infections also can be found on the torso, neck, and upper extremities.1
Cutaneous nocardiosis is a granulomatous infection encompassing both cutaneous and subcutaneous tissue, which ultimately can lead to injury of bone and viscera.1 Primary cutaneous nocardiosis can manifest as tumors or nodules that have a sporotrichoid pattern, in which they ascend along the lymphatics. Histopathology of infected tissue frequently shows a subcutaneous dermal infiltrate of neutrophils accompanied with abscess formation, and everlasting lesions may show signs of chronic inflammation and nonspecific granulomas.3
Treatment of nocardiosis should be guided by in vitro susceptibility tests. Sulfamethoxazole-trimethoprim 800 mg/160 mg taken as 1 tablet twice daily is the first-line option. The treatment duration is contingent on the extent, severity, and complications of infection but typically is 3 to 6 months.1
The Diagnosis: Cutaneous Nocardiosis
The wound culture was positive for Nocardia farcinica. The patient received a 5-day course of intravenous sulfamethoxazole-trimethoprim in the hospital and was transitioned to oral sulfamethoxazoletrimethoprim (800 mg/160 mg taken as 1 tablet twice daily) for 6 months. Complete resolution of the infection was noted at 6-month follow-up (Figure).
Nocardia is a gram-positive, aerobic bacterium that typically is found in soil, water, and decaying organic matter.1 There are more than 50 species; N farcinica, Nocardia nova, and Nocardia asteroides are the leading causes of infection in humans and animals. Nocardia asteroides is the most common cause of infection in humans.1,2 Nocardiosis is an uncommon opportunistic infection that usually targets the skin, lungs, and central nervous system.3 Although it mainly affects individuals who are immunocompromised, up to 30% of infections can be seen in immunocompetent hosts who can contract cutaneous nocardiosis after experiencing traumatic injury to the skin.1
Nocardiosis is difficult to diagnose due to its diverse clinical presentations. For example, cutaneous nocardiosis can manifest similar to mycetoma, sporotrichosis, spider bites, nontuberculous mycobacteria such as Mycobacterium marinum, or methicillin-resistant Staphylococcus aureus infections, thus making cutaneous nocardiosis one of the great imitators.1 A culture is required for definitive diagnosis, as Nocardia grows well on nonselective media such as blood or Löwenstein-Jensen agar. It grows as waxy, pigmented, cerebriform colonies 3 to 5 days following incubation.3 The bacterium can be difficult to culture, and it is important to notify the microbiology laboratory if there is a high index of clinical suspicion for infection.
A history of exposure to gardening or handling animals can increase the risk for an individual contracting Nocardia.3 Although nocardiosis can be found across the world, it is native to tropical and subtropical climates such as those found in India, Africa, Latin America, and Southeast Asia.1 Infections mostly are observed in individuals aged 20 to 40 years and tend to affect men more than women. Lesions typically are seen on the lower extremities, but localized infections also can be found on the torso, neck, and upper extremities.1
Cutaneous nocardiosis is a granulomatous infection encompassing both cutaneous and subcutaneous tissue, which ultimately can lead to injury of bone and viscera.1 Primary cutaneous nocardiosis can manifest as tumors or nodules that have a sporotrichoid pattern, in which they ascend along the lymphatics. Histopathology of infected tissue frequently shows a subcutaneous dermal infiltrate of neutrophils accompanied with abscess formation, and everlasting lesions may show signs of chronic inflammation and nonspecific granulomas.3
Treatment of nocardiosis should be guided by in vitro susceptibility tests. Sulfamethoxazole-trimethoprim 800 mg/160 mg taken as 1 tablet twice daily is the first-line option. The treatment duration is contingent on the extent, severity, and complications of infection but typically is 3 to 6 months.1
- Yu Q, Song J, Liu Y, et al. Progressive primary cutaneous nocardiosis in an immunocompetent patient. Cutis. 2023;111:E22-E25.
- Gaines RJ, Randall CJ, Ruland RT. Lymphocutaneous nocardiosis from commercially treated lumber: a case report. Cutis. 2006;78:249-251.
- Riswold KJ, Tjarks BJ, Kerkvliet AM. Cutaneous nocardiosis in an immunocompromised patient. Cutis. 2019;104:226-229.
- Yu Q, Song J, Liu Y, et al. Progressive primary cutaneous nocardiosis in an immunocompetent patient. Cutis. 2023;111:E22-E25.
- Gaines RJ, Randall CJ, Ruland RT. Lymphocutaneous nocardiosis from commercially treated lumber: a case report. Cutis. 2006;78:249-251.
- Riswold KJ, Tjarks BJ, Kerkvliet AM. Cutaneous nocardiosis in an immunocompromised patient. Cutis. 2019;104:226-229.
A 67-year-old man presented to the emergency department with a draining nodule on the right hand of 4 days’ duration. He reported that the swelling and redness started 1 hour after handling a succulent plant. The following day, the nodule increased in size and exudated yellow pus. He presented with swelling of the thumb and hand, which resulted in a decreased range of motion. He had a history of prediabetes and denied any recent travel, allergies, or animal exposures. Physical examination revealed a draining nodule on the dorsal aspect of the right hand that measured approximately 15×15 mm with surrounding erythema and tenderness. There also was progression of ascending erythema up to the axilla. The patient was admitted to the hospital.