User login
Multiple Nodules on the Scrotum
The Diagnosis: Scrotal Calcinosis
Scrotal calcinosis is a rare benign disease that results from the deposition of calcium, magnesium, phosphate, and carbonate within the dermis and subcutaneous layer of the skin in the absence of underlying systemic disease or serum calcium and phosphorus abnormalities.1,2 Lesions usually are asymptomatic but can be mildly painful or pruritic. They usually present in childhood or early adulthood as yellow-white firm nodules ranging in size from a few millimeters to a few centimeters that increase in size and number over time. Additionally, lesions can ulcerate and discharge a chalklike exudative material. Although benign in nature, the quality-of-life impact in patients with this condition can be substantial, specifically regarding cosmesis, which may cause patients to feel embarrassed and even avoid sexual activity. This condition rarely has been associated with infection.1
Our patient elected to undergo surgical excision under local anesthesia, and the lesions were sent for histopathologic examination. His postoperative course was unremarkable, and he was pleased with the cosmetic result of the surgery (Figure 1). Histopathology revealed calcified deposits that appeared as intradermal basophilic nodules lacking an epithelial lining (Figure 2), consistent with the diagnosis of scrotal calcinosis.2 No recurrence of the lesions was documented over the course of 18 months.
 following the removal of scrotal calcinosis nodules.")
The pathogenesis of this condition is not clear. Most research supports scrotal calcinosis resulting from dystrophic calcification of epidermal inclusion cysts.3 There have been cases of scrotal calcinosis coinciding with epidermal inclusion cysts of the scrotum in varying stages of inflammation (some intact and some ruptured).2 Some research also suggests dystrophic calcification of eccrine epithelial cysts and degenerated dartos muscle as the origin of scrotal calcinosis.3
.")
The differential diagnosis for this case included calcified steatocystoma multiplex, eruptive xanthomas, nodular scabies, and epidermal inclusion cysts. Steatocystoma multiplex can be inherited in an autosomal-dominant fashion or can develop sporadically with mutations in the KRT17 gene.4 It is characterized by multiple sebum-filled, cystic lesions of the pilosebaceous unit that may become calcified. Calcified lesions appear as yellow, firm, irregularly shaped papules or nodules ranging from a few millimeters to centimeters in size. Cysts can develop anywhere on the body with a predilection for the chest, upper extremities, axillae, trunk, groin, and scrotum.4 Histologically, our patient’s lesions were not associated with the pilosebaceous unit. Additionally, our patient denied a family history of similar skin lesions, which made calcified steatocystoma multiplex an unlikely diagnosis.
Eruptive xanthomas result from localized deposition of lipids within the dermis, typically in the setting of dyslipidemia or poorly controlled diabetes mellitus. They commonly appear on the extremities or buttocks as pruritic crops of yellow-red papules or nodules that are a few millimeters in size. Although our patient has a history of hyperlipidemia, his lesions differed substantially from eruptive xanthomas in clinical presentation.
Nodular scabies is a manifestation of classic scabies that presents with intensely pruritic erythematous papules and nodules that are a few millimeters in size and commonly occur on the axillae, groin, and genitalia. Our patient’s skin lesions were not pruritic and differed in appearance from nodular scabies.
Although research indicates scrotal calcinosis may result from dystrophic calcification of epidermal inclusion cysts,2 the latter present as dome-shaped, flesh-colored nodules with central pores representing the opening of hair follicles. Our patient lacked characteristic findings of epidermal inclusion cysts on histology.
The preferred treatment for scrotal calcinosis is surgical excision, which improves the aesthetic appearance, relieves itch, and removes ulcerative lesions.5 Additionally, surgical excision provides histological diagnostic confirmation. Recurrence with incomplete excision is possible; therefore, all lesions should be completely excised to reduce the risk for recurrence.3
- Pompeo A, Molina WR, Pohlman GD, et al. Idiopathic scrotal calcinosis: a rare entity and a review of the literature. Can Urol Assoc J. 2013;7:E439-E441. doi:10.5489/cuaj.1387
- Swinehart JM, Golitz LE. Scrotal calcinosis: dystrophic calcification of epidermoid cysts. Arch Dermatol. 1982;118:985-988. doi:10.1001 /archderm.1982.01650240029016
- Khallouk A, Yazami OE, Mellas S, et al. Idiopathic scrotal calcinosis: a nonelucidated pathogenesis and its surgical treatment. Rev Urol. 2011;13:95-97.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480. doi:10.1046/j.1365-2133.1998.02413.x
- Solanki A, Narang S, Kathpalia R, et al. Scrotal calcinosis: pathogenetic link with epidermal cyst. BMJ Case Rep. 2015;2015:bcr2015211163. doi:10.1136/bcr-2015-211163
The Diagnosis: Scrotal Calcinosis
Scrotal calcinosis is a rare benign disease that results from the deposition of calcium, magnesium, phosphate, and carbonate within the dermis and subcutaneous layer of the skin in the absence of underlying systemic disease or serum calcium and phosphorus abnormalities.1,2 Lesions usually are asymptomatic but can be mildly painful or pruritic. They usually present in childhood or early adulthood as yellow-white firm nodules ranging in size from a few millimeters to a few centimeters that increase in size and number over time. Additionally, lesions can ulcerate and discharge a chalklike exudative material. Although benign in nature, the quality-of-life impact in patients with this condition can be substantial, specifically regarding cosmesis, which may cause patients to feel embarrassed and even avoid sexual activity. This condition rarely has been associated with infection.1
Our patient elected to undergo surgical excision under local anesthesia, and the lesions were sent for histopathologic examination. His postoperative course was unremarkable, and he was pleased with the cosmetic result of the surgery (Figure 1). Histopathology revealed calcified deposits that appeared as intradermal basophilic nodules lacking an epithelial lining (Figure 2), consistent with the diagnosis of scrotal calcinosis.2 No recurrence of the lesions was documented over the course of 18 months.
The pathogenesis of this condition is not clear. Most research supports scrotal calcinosis resulting from dystrophic calcification of epidermal inclusion cysts.3 There have been cases of scrotal calcinosis coinciding with epidermal inclusion cysts of the scrotum in varying stages of inflammation (some intact and some ruptured).2 Some research also suggests dystrophic calcification of eccrine epithelial cysts and degenerated dartos muscle as the origin of scrotal calcinosis.3
The differential diagnosis for this case included calcified steatocystoma multiplex, eruptive xanthomas, nodular scabies, and epidermal inclusion cysts. Steatocystoma multiplex can be inherited in an autosomal-dominant fashion or can develop sporadically with mutations in the KRT17 gene.4 It is characterized by multiple sebum-filled, cystic lesions of the pilosebaceous unit that may become calcified. Calcified lesions appear as yellow, firm, irregularly shaped papules or nodules ranging from a few millimeters to centimeters in size. Cysts can develop anywhere on the body with a predilection for the chest, upper extremities, axillae, trunk, groin, and scrotum.4 Histologically, our patient’s lesions were not associated with the pilosebaceous unit. Additionally, our patient denied a family history of similar skin lesions, which made calcified steatocystoma multiplex an unlikely diagnosis.
Eruptive xanthomas result from localized deposition of lipids within the dermis, typically in the setting of dyslipidemia or poorly controlled diabetes mellitus. They commonly appear on the extremities or buttocks as pruritic crops of yellow-red papules or nodules that are a few millimeters in size. Although our patient has a history of hyperlipidemia, his lesions differed substantially from eruptive xanthomas in clinical presentation.
Nodular scabies is a manifestation of classic scabies that presents with intensely pruritic erythematous papules and nodules that are a few millimeters in size and commonly occur on the axillae, groin, and genitalia. Our patient’s skin lesions were not pruritic and differed in appearance from nodular scabies.
Although research indicates scrotal calcinosis may result from dystrophic calcification of epidermal inclusion cysts,2 the latter present as dome-shaped, flesh-colored nodules with central pores representing the opening of hair follicles. Our patient lacked characteristic findings of epidermal inclusion cysts on histology.
The preferred treatment for scrotal calcinosis is surgical excision, which improves the aesthetic appearance, relieves itch, and removes ulcerative lesions.5 Additionally, surgical excision provides histological diagnostic confirmation. Recurrence with incomplete excision is possible; therefore, all lesions should be completely excised to reduce the risk for recurrence.3
The Diagnosis: Scrotal Calcinosis
Scrotal calcinosis is a rare benign disease that results from the deposition of calcium, magnesium, phosphate, and carbonate within the dermis and subcutaneous layer of the skin in the absence of underlying systemic disease or serum calcium and phosphorus abnormalities.1,2 Lesions usually are asymptomatic but can be mildly painful or pruritic. They usually present in childhood or early adulthood as yellow-white firm nodules ranging in size from a few millimeters to a few centimeters that increase in size and number over time. Additionally, lesions can ulcerate and discharge a chalklike exudative material. Although benign in nature, the quality-of-life impact in patients with this condition can be substantial, specifically regarding cosmesis, which may cause patients to feel embarrassed and even avoid sexual activity. This condition rarely has been associated with infection.1
Our patient elected to undergo surgical excision under local anesthesia, and the lesions were sent for histopathologic examination. His postoperative course was unremarkable, and he was pleased with the cosmetic result of the surgery (Figure 1). Histopathology revealed calcified deposits that appeared as intradermal basophilic nodules lacking an epithelial lining (Figure 2), consistent with the diagnosis of scrotal calcinosis.2 No recurrence of the lesions was documented over the course of 18 months.
The pathogenesis of this condition is not clear. Most research supports scrotal calcinosis resulting from dystrophic calcification of epidermal inclusion cysts.3 There have been cases of scrotal calcinosis coinciding with epidermal inclusion cysts of the scrotum in varying stages of inflammation (some intact and some ruptured).2 Some research also suggests dystrophic calcification of eccrine epithelial cysts and degenerated dartos muscle as the origin of scrotal calcinosis.3
The differential diagnosis for this case included calcified steatocystoma multiplex, eruptive xanthomas, nodular scabies, and epidermal inclusion cysts. Steatocystoma multiplex can be inherited in an autosomal-dominant fashion or can develop sporadically with mutations in the KRT17 gene.4 It is characterized by multiple sebum-filled, cystic lesions of the pilosebaceous unit that may become calcified. Calcified lesions appear as yellow, firm, irregularly shaped papules or nodules ranging from a few millimeters to centimeters in size. Cysts can develop anywhere on the body with a predilection for the chest, upper extremities, axillae, trunk, groin, and scrotum.4 Histologically, our patient’s lesions were not associated with the pilosebaceous unit. Additionally, our patient denied a family history of similar skin lesions, which made calcified steatocystoma multiplex an unlikely diagnosis.
Eruptive xanthomas result from localized deposition of lipids within the dermis, typically in the setting of dyslipidemia or poorly controlled diabetes mellitus. They commonly appear on the extremities or buttocks as pruritic crops of yellow-red papules or nodules that are a few millimeters in size. Although our patient has a history of hyperlipidemia, his lesions differed substantially from eruptive xanthomas in clinical presentation.
Nodular scabies is a manifestation of classic scabies that presents with intensely pruritic erythematous papules and nodules that are a few millimeters in size and commonly occur on the axillae, groin, and genitalia. Our patient’s skin lesions were not pruritic and differed in appearance from nodular scabies.
Although research indicates scrotal calcinosis may result from dystrophic calcification of epidermal inclusion cysts,2 the latter present as dome-shaped, flesh-colored nodules with central pores representing the opening of hair follicles. Our patient lacked characteristic findings of epidermal inclusion cysts on histology.
The preferred treatment for scrotal calcinosis is surgical excision, which improves the aesthetic appearance, relieves itch, and removes ulcerative lesions.5 Additionally, surgical excision provides histological diagnostic confirmation. Recurrence with incomplete excision is possible; therefore, all lesions should be completely excised to reduce the risk for recurrence.3
- Pompeo A, Molina WR, Pohlman GD, et al. Idiopathic scrotal calcinosis: a rare entity and a review of the literature. Can Urol Assoc J. 2013;7:E439-E441. doi:10.5489/cuaj.1387
- Swinehart JM, Golitz LE. Scrotal calcinosis: dystrophic calcification of epidermoid cysts. Arch Dermatol. 1982;118:985-988. doi:10.1001 /archderm.1982.01650240029016
- Khallouk A, Yazami OE, Mellas S, et al. Idiopathic scrotal calcinosis: a nonelucidated pathogenesis and its surgical treatment. Rev Urol. 2011;13:95-97.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480. doi:10.1046/j.1365-2133.1998.02413.x
- Solanki A, Narang S, Kathpalia R, et al. Scrotal calcinosis: pathogenetic link with epidermal cyst. BMJ Case Rep. 2015;2015:bcr2015211163. doi:10.1136/bcr-2015-211163
- Pompeo A, Molina WR, Pohlman GD, et al. Idiopathic scrotal calcinosis: a rare entity and a review of the literature. Can Urol Assoc J. 2013;7:E439-E441. doi:10.5489/cuaj.1387
- Swinehart JM, Golitz LE. Scrotal calcinosis: dystrophic calcification of epidermoid cysts. Arch Dermatol. 1982;118:985-988. doi:10.1001 /archderm.1982.01650240029016
- Khallouk A, Yazami OE, Mellas S, et al. Idiopathic scrotal calcinosis: a nonelucidated pathogenesis and its surgical treatment. Rev Urol. 2011;13:95-97.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480. doi:10.1046/j.1365-2133.1998.02413.x
- Solanki A, Narang S, Kathpalia R, et al. Scrotal calcinosis: pathogenetic link with epidermal cyst. BMJ Case Rep. 2015;2015:bcr2015211163. doi:10.1136/bcr-2015-211163
A 33-year-old man presented with progressively enlarging bumps on the scrotum that were present since adolescence. He had a history of hyperlipidemia but no history of systemic or autoimmune disease. The lesions were asymptomatic without associated pruritus, pain, or discharge. No treatments had been administered, and he had no known personal or family history of similar skin conditions or skin cancer. He endorsed a monogamous relationship with his wife. Physical examination revealed 15 firm, yellow-white, subcutaneous nodules on the scrotum that varied in size.


Porcelain White, Crinkled, Violaceous Patches on the Inner Thighs
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).

Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.

Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).
Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.
Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).
Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.
Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
A 50-year-old woman presented with multiple pruritic lesions on the right inner thigh of 2 years’ duration. Physical examination revealed porcelain white, crinkled, violaceous patches extending from the right inner thigh to the inguinal fold (top). Dermoscopic examination revealed follicular plugs, white structureless areas, white lines, and a rainbow pattern arranged over white polygonal clods on polarized mode (bottom).


Genital Ulcerations With Swelling
The Diagnosis: Mpox (Monkeypox)
Tests for active herpes simplex virus (HHV), gonorrhea, chlamydia, HIV, and syphilis were negative. Swabs from the penile lesion demonstrated positivity for the West African clade of mpox (monkeypox) virus (MPXV) by polymerase chain reaction. The patient was treated supportively without the addition of antiviral therapy, and he experienced a complete recovery.
Mpox virus was first isolated in 1958 in a research facility and was named after the laboratory animals that were housed there. The first human documentation of the disease occurred in 1970, and it was first documented in the United States in 2003 in an infection that was traced to a shipment of small mammals from Ghana to Texas.1 The disease has always been endemic to Africa; however, the incidence has been increasing.2 A new MPXV outbreak was reported in many countries in early 2022, including the United States.1
The MPXV is a double-stranded DNA virus of the genus Orthopoxvirus, and 2 genetic clades have been identified: clade I (formerly the Central African clade) and clade II (formerly the West African clade). The virus has the capability to infect many mammals; however, its host remains unidentified.1 The exact mechanism of transmission from infected animals to humans largely is unknown; however, direct or indirect contact with infected animals likely is responsible. Human-to-human transmission can occur by many mechanisms including contact with large respiratory droplets, bodily fluids, and contaminated surfaces. The incubation period is 5 to 21 days, and the symptoms last 2 to 5 weeks.1
.")
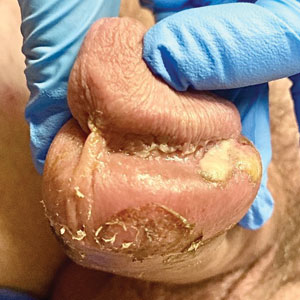
The clinical manifestations of MPXV during the most recent outbreak differ from prior outbreaks. Patients are more likely to experience minimal to no systemic symptoms, and cutaneous lesions can be few and localized to a focal area, especially on the face and in the anogenital region,3 similar to the presentation in our patient (Figure 1). Cutaneous lesions of the most recent MPXV outbreak also include painless ulcerations similar to syphilitic chancres and lesions that are in various stages of healing.3 Lesions often begin as pseudopustules, which are firm white papules with or without a necrotic center that resemble pustules; unlike true pustules, there is no identifiable purulent material within it. Bacterial superinfection of the lesions is not uncommon.4 Over time, a secondary pustular eruption resembling folliculitis also may occur,4 as noted in our patient (Figure 2).
 on the arms.")
Although we did not have a biopsy to support the diagnosis of associated erythema multiforme (EM) in our patient, features supportive of this diagnosis included the classic clinical appearance of typical, well-defined, targetoid plaques with 3 distinct zones (Figure 3); the association with a known infection; the distribution on the arms with truncal sparing; and self-limited lesions. More than 90% of EM cases are associated with infection, with HHV representing the most common culprit5; therefore, the relationship with a different virus is not an unreasonable suggestion. Additionally, there have been rare reports of EM in association with MPXV.4
.")
Histopathology of MPXV may have distinctive features. Lesions often demonstrate keratinocytic necrosis and basal layer vacuolization with an associated superficial and deep perivascular lymphohistiocytic infiltrate. When the morphology of the lesion is vesicular, histopathology reveals spongiosis and ballooning degeneration with epidermal necrosis. Viral inclusion bodies within keratinocytes may be identified.1 Death rates from MPXV has been reported from 1% to 11%, with increased mortality among high-risk populations including children and immunocompromised individuals. Treatment of the disease largely consists of supportive care and management of any associated complications including bacterial infection, pneumonia, and encephalitis.1
The differential diagnosis of MPXV includes other ulcerative lesions that can occur on the genital skin. Fixed drug eruptions often present on the penis,6 but there was no identifiable inciting drug in our patient. Herpes simplex virus infection was very high on the differential given our patient’s history of recurrent infections and association with a targetoid rash, but HHV type 1 and HHV type 2 testing of the lesion was negative. A syphilitic chancre also may present with the nontender genital ulceration7 that was seen in our patient, but serology did not support this diagnosis. Cutaneous Crohn disease also may manifest with genital ulceration even before a diagnosis of Crohn disease is made, but these lesions often present as linear knife-cut ulcerations of the anogenital region.8
Our case further supports a clinical presentation that diverges from the more traditional cases of MPXV. Additionally, associated EM may be a clue to infection, especially in cases of negative HHV and other sexually transmitted infection testing.
- Bunge EM, Hoet B, Chen L, et al. The changing epidemiology of human monkeypox—a potential threat? a systematic review. PLoS Negl Trop Dis. 2022;16:E0010141.
- Kumar N, Acharya A, Gendelman HE, et al. The 2022 outbreak and the pathobiology of the monkeypox virus. J Autoimmun. 2022;131:102855.
- Eisenstadt R, Liszewski WJ, Nguyen CV. Recognizing minimal cutaneous involvement or systemic symptoms in monkeypox. JAMA Dermatol. 2022;158:1457-1458.
- Català A, Clavo-Escribano P, Riera-Monroig J, et al. Monkeypox outbreak in Spain: clinical and epidemiological findings in a prospective cross-sectional study of 185 cases [published online August 2, 2022]. Br J Dermatol. 2022;187:765-772.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Waleryie-Allanore L, Obeid G, Revuz J. Drug reactions. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:348-375.
- Stary G, Stary A. Sexually transmitted infections. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1447-1469.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1644-1663.
The Diagnosis: Mpox (Monkeypox)
Tests for active herpes simplex virus (HHV), gonorrhea, chlamydia, HIV, and syphilis were negative. Swabs from the penile lesion demonstrated positivity for the West African clade of mpox (monkeypox) virus (MPXV) by polymerase chain reaction. The patient was treated supportively without the addition of antiviral therapy, and he experienced a complete recovery.
Mpox virus was first isolated in 1958 in a research facility and was named after the laboratory animals that were housed there. The first human documentation of the disease occurred in 1970, and it was first documented in the United States in 2003 in an infection that was traced to a shipment of small mammals from Ghana to Texas.1 The disease has always been endemic to Africa; however, the incidence has been increasing.2 A new MPXV outbreak was reported in many countries in early 2022, including the United States.1
The MPXV is a double-stranded DNA virus of the genus Orthopoxvirus, and 2 genetic clades have been identified: clade I (formerly the Central African clade) and clade II (formerly the West African clade). The virus has the capability to infect many mammals; however, its host remains unidentified.1 The exact mechanism of transmission from infected animals to humans largely is unknown; however, direct or indirect contact with infected animals likely is responsible. Human-to-human transmission can occur by many mechanisms including contact with large respiratory droplets, bodily fluids, and contaminated surfaces. The incubation period is 5 to 21 days, and the symptoms last 2 to 5 weeks.1
The clinical manifestations of MPXV during the most recent outbreak differ from prior outbreaks. Patients are more likely to experience minimal to no systemic symptoms, and cutaneous lesions can be few and localized to a focal area, especially on the face and in the anogenital region,3 similar to the presentation in our patient (Figure 1). Cutaneous lesions of the most recent MPXV outbreak also include painless ulcerations similar to syphilitic chancres and lesions that are in various stages of healing.3 Lesions often begin as pseudopustules, which are firm white papules with or without a necrotic center that resemble pustules; unlike true pustules, there is no identifiable purulent material within it. Bacterial superinfection of the lesions is not uncommon.4 Over time, a secondary pustular eruption resembling folliculitis also may occur,4 as noted in our patient (Figure 2).
Although we did not have a biopsy to support the diagnosis of associated erythema multiforme (EM) in our patient, features supportive of this diagnosis included the classic clinical appearance of typical, well-defined, targetoid plaques with 3 distinct zones (Figure 3); the association with a known infection; the distribution on the arms with truncal sparing; and self-limited lesions. More than 90% of EM cases are associated with infection, with HHV representing the most common culprit5; therefore, the relationship with a different virus is not an unreasonable suggestion. Additionally, there have been rare reports of EM in association with MPXV.4
Histopathology of MPXV may have distinctive features. Lesions often demonstrate keratinocytic necrosis and basal layer vacuolization with an associated superficial and deep perivascular lymphohistiocytic infiltrate. When the morphology of the lesion is vesicular, histopathology reveals spongiosis and ballooning degeneration with epidermal necrosis. Viral inclusion bodies within keratinocytes may be identified.1 Death rates from MPXV has been reported from 1% to 11%, with increased mortality among high-risk populations including children and immunocompromised individuals. Treatment of the disease largely consists of supportive care and management of any associated complications including bacterial infection, pneumonia, and encephalitis.1
The differential diagnosis of MPXV includes other ulcerative lesions that can occur on the genital skin. Fixed drug eruptions often present on the penis,6 but there was no identifiable inciting drug in our patient. Herpes simplex virus infection was very high on the differential given our patient’s history of recurrent infections and association with a targetoid rash, but HHV type 1 and HHV type 2 testing of the lesion was negative. A syphilitic chancre also may present with the nontender genital ulceration7 that was seen in our patient, but serology did not support this diagnosis. Cutaneous Crohn disease also may manifest with genital ulceration even before a diagnosis of Crohn disease is made, but these lesions often present as linear knife-cut ulcerations of the anogenital region.8
Our case further supports a clinical presentation that diverges from the more traditional cases of MPXV. Additionally, associated EM may be a clue to infection, especially in cases of negative HHV and other sexually transmitted infection testing.
The Diagnosis: Mpox (Monkeypox)
Tests for active herpes simplex virus (HHV), gonorrhea, chlamydia, HIV, and syphilis were negative. Swabs from the penile lesion demonstrated positivity for the West African clade of mpox (monkeypox) virus (MPXV) by polymerase chain reaction. The patient was treated supportively without the addition of antiviral therapy, and he experienced a complete recovery.
Mpox virus was first isolated in 1958 in a research facility and was named after the laboratory animals that were housed there. The first human documentation of the disease occurred in 1970, and it was first documented in the United States in 2003 in an infection that was traced to a shipment of small mammals from Ghana to Texas.1 The disease has always been endemic to Africa; however, the incidence has been increasing.2 A new MPXV outbreak was reported in many countries in early 2022, including the United States.1
The MPXV is a double-stranded DNA virus of the genus Orthopoxvirus, and 2 genetic clades have been identified: clade I (formerly the Central African clade) and clade II (formerly the West African clade). The virus has the capability to infect many mammals; however, its host remains unidentified.1 The exact mechanism of transmission from infected animals to humans largely is unknown; however, direct or indirect contact with infected animals likely is responsible. Human-to-human transmission can occur by many mechanisms including contact with large respiratory droplets, bodily fluids, and contaminated surfaces. The incubation period is 5 to 21 days, and the symptoms last 2 to 5 weeks.1
The clinical manifestations of MPXV during the most recent outbreak differ from prior outbreaks. Patients are more likely to experience minimal to no systemic symptoms, and cutaneous lesions can be few and localized to a focal area, especially on the face and in the anogenital region,3 similar to the presentation in our patient (Figure 1). Cutaneous lesions of the most recent MPXV outbreak also include painless ulcerations similar to syphilitic chancres and lesions that are in various stages of healing.3 Lesions often begin as pseudopustules, which are firm white papules with or without a necrotic center that resemble pustules; unlike true pustules, there is no identifiable purulent material within it. Bacterial superinfection of the lesions is not uncommon.4 Over time, a secondary pustular eruption resembling folliculitis also may occur,4 as noted in our patient (Figure 2).
Although we did not have a biopsy to support the diagnosis of associated erythema multiforme (EM) in our patient, features supportive of this diagnosis included the classic clinical appearance of typical, well-defined, targetoid plaques with 3 distinct zones (Figure 3); the association with a known infection; the distribution on the arms with truncal sparing; and self-limited lesions. More than 90% of EM cases are associated with infection, with HHV representing the most common culprit5; therefore, the relationship with a different virus is not an unreasonable suggestion. Additionally, there have been rare reports of EM in association with MPXV.4
Histopathology of MPXV may have distinctive features. Lesions often demonstrate keratinocytic necrosis and basal layer vacuolization with an associated superficial and deep perivascular lymphohistiocytic infiltrate. When the morphology of the lesion is vesicular, histopathology reveals spongiosis and ballooning degeneration with epidermal necrosis. Viral inclusion bodies within keratinocytes may be identified.1 Death rates from MPXV has been reported from 1% to 11%, with increased mortality among high-risk populations including children and immunocompromised individuals. Treatment of the disease largely consists of supportive care and management of any associated complications including bacterial infection, pneumonia, and encephalitis.1
The differential diagnosis of MPXV includes other ulcerative lesions that can occur on the genital skin. Fixed drug eruptions often present on the penis,6 but there was no identifiable inciting drug in our patient. Herpes simplex virus infection was very high on the differential given our patient’s history of recurrent infections and association with a targetoid rash, but HHV type 1 and HHV type 2 testing of the lesion was negative. A syphilitic chancre also may present with the nontender genital ulceration7 that was seen in our patient, but serology did not support this diagnosis. Cutaneous Crohn disease also may manifest with genital ulceration even before a diagnosis of Crohn disease is made, but these lesions often present as linear knife-cut ulcerations of the anogenital region.8
Our case further supports a clinical presentation that diverges from the more traditional cases of MPXV. Additionally, associated EM may be a clue to infection, especially in cases of negative HHV and other sexually transmitted infection testing.
- Bunge EM, Hoet B, Chen L, et al. The changing epidemiology of human monkeypox—a potential threat? a systematic review. PLoS Negl Trop Dis. 2022;16:E0010141.
- Kumar N, Acharya A, Gendelman HE, et al. The 2022 outbreak and the pathobiology of the monkeypox virus. J Autoimmun. 2022;131:102855.
- Eisenstadt R, Liszewski WJ, Nguyen CV. Recognizing minimal cutaneous involvement or systemic symptoms in monkeypox. JAMA Dermatol. 2022;158:1457-1458.
- Català A, Clavo-Escribano P, Riera-Monroig J, et al. Monkeypox outbreak in Spain: clinical and epidemiological findings in a prospective cross-sectional study of 185 cases [published online August 2, 2022]. Br J Dermatol. 2022;187:765-772.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Waleryie-Allanore L, Obeid G, Revuz J. Drug reactions. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:348-375.
- Stary G, Stary A. Sexually transmitted infections. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1447-1469.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1644-1663.
- Bunge EM, Hoet B, Chen L, et al. The changing epidemiology of human monkeypox—a potential threat? a systematic review. PLoS Negl Trop Dis. 2022;16:E0010141.
- Kumar N, Acharya A, Gendelman HE, et al. The 2022 outbreak and the pathobiology of the monkeypox virus. J Autoimmun. 2022;131:102855.
- Eisenstadt R, Liszewski WJ, Nguyen CV. Recognizing minimal cutaneous involvement or systemic symptoms in monkeypox. JAMA Dermatol. 2022;158:1457-1458.
- Català A, Clavo-Escribano P, Riera-Monroig J, et al. Monkeypox outbreak in Spain: clinical and epidemiological findings in a prospective cross-sectional study of 185 cases [published online August 2, 2022]. Br J Dermatol. 2022;187:765-772.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Waleryie-Allanore L, Obeid G, Revuz J. Drug reactions. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:348-375.
- Stary G, Stary A. Sexually transmitted infections. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1447-1469.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1644-1663.
A 50-year-old man with a history of recurrent genital herpes simplex virus infections presented to the hospital with genital lesions and swelling of 5 days’ duration. Prior to admission, the patient was treated with a course of valacyclovir by an urgent care physician without improvement. Physical examination revealed a 3-cm, nontender, shallow, ulcerative plaque with irregular borders and a purulent yellow base distributed on the distal shaft of the penis with extension into the coronal sulcus. A few other scattered erosions were noted on the distal penile shaft. He had associated diffuse nonpitting edema of the penis and scrotum as well as tender bilateral inguinal lymphadenopathy. Three days after the genital ulcerations began, the patient developed a nontender erythematous papule with a necrotic center on the right jaw followed by an eruption of erythematous papulopustules on the arms and trunk. The patient denied dysuria, purulent penile discharge, fevers, chills, headaches, myalgia, arthralgia, nausea, vomiting, or diarrhea. The patient was sexually active exclusively with females and had more than 10 partners in the prior year. Shortly after hospital admission, the patient developed red targetoid plaques on the groin, trunk, and arms. No oral mucosal lesions were identified.

Ulcerated Nodule on the Lip
The Diagnosis: Cutaneous Metastasis
A shave biopsy of the lip revealed a diffuse cellular infiltrate filling the superficial and deep dermis (Figure 1A). Morphologically, the cells had abundant clear cytoplasm with eccentrically located, pleomorphic, hyperchromatic nuclei with occasional prominent nucleoli (Figure 1B). The cells stained positive for AE1/ AE3 on immunohistochemistry (Figure 2). A punch biopsy of the nodule in the right axillary vault revealed a morphologically similar proliferation of cells. A colonoscopy revealed a completely obstructing circumferential mass in the distal ascending colon. A biopsy of the mass confirmed invasive adenocarcinoma, supporting a diagnosis of cutaneous metastases from adenocarcinoma of the colon. The patient underwent resection of the lip tumor and started multiagent chemotherapy for his newly diagnosed stage IV adenocarcinoma of the colon. The patient died, despite therapy.

Cutaneous metastasis from solid malignancies is uncommon, as only 1.3% of them exhibit cutaneous manifestations at presentation.1 Cutaneous metastasis from signet ring cell adenocarcinoma (SRCA) of the colon is uncommon, and cutaneous metastasis of colorectal SRCA rarely precedes the diagnosis of the primary lesion.2 Among the colorectal cancers that metastasize to the skin, metastasis to the face occurs in only 0.5% of patients.3
.")
Signet ring cell adenocarcinomas are poorly differentiated adenocarcinomas histologically characterized by the neoplastic cells’ circular to ovoid appearance with a flattened top.4,5 This distinctive shape is from the displacement of the nucleus to the periphery of the cell due to the accumulation of intracytoplasmic mucin.4 Classically, malignancies are characterized as an SRCA if more than 50% of the cells have a signet ring cell morphology; if the signet ring cells comprise less than 50% of the neoplasm, the tumor is designated as an adenocarcinoma with signet ring morphology.4 The most common cause of cutaneous metastasis with signet ring morphology is gastric cancer, while colorectal carcinoma is less common.1 Colorectal SRCAs usually are found in the right colon or the rectum in comparison to other colonic sites.6
Clinically, cutaneous metastasis can present in a variety of ways. The most common presentation is nodular lesions that may coalesce to become zosteriform in configuration or lesions that mimic inflammatory dermatoses.7 Cutaneous metastasis is more common in breast and lung cancer, and when it occurs secondary to colorectal cancer, cutaneous metastasis rarely predates the detection of the primary neoplasm.2
The clinical appearance of metastasis is not specific and can mimic many entities8; therefore, a high index of suspicion must be employed when managing patients, even those without a history of internal malignancy. In our patient, the smooth nodular lesion appeared similar to a basal cell carcinoma; however, basal cell carcinomas appear more pearly, and arborizing telangiectasia often is seen.9 Merkel cell carcinoma is common on sundamaged skin of the head and neck but clinically appears more violaceous than the lesion seen in our patient.10 Paracoccidioidomycosis may form ulcerated papulonodules or plaques, especially around the nose and mouth. In many of these cases, lesions develop from contiguous lesions of the oral mucosa; therefore, the presence of oral lesions will help distinguish this infectious entity from cutaneous metastasis. Multiple lesions usually are identified when there is hematogenous dissemination.11 Mycosis fungoides is a subtype of cutaneous T-cell lymphoma and is characterized by multiple patches, plaques, and nodules on sun-protected areas. Involvement of the head and neck is not common, except in the folliculotropic subtype, which has a separate and distinct clinical morphology.12
The development of signet ring morphology from an adenocarcinoma can be attributed to the activation of phosphatidylinositol 3-kinase (PI3K), which leads to downstream activation of mitogen-activated protein kinase (MAPK) and the subsequent loss of intercellular tight junctions. The mucin 4 gene, MUC4, also is upregulated by PI3K activation and possesses antiapoptotic and mitogenic effects in addition to its mucin secretory function.13
The neoplastic cells in SRCAs stain positive for mucicarmine, Alcian blue, and periodic acid–Schiff, which highlights the mucinous component of the cells.7 Immunohistochemical stains with CK7, CK20, AE1/AE3, and epithelial membrane antigen can be implemented to confirm an epithelial origin of the primary cancer.7,13 CK20 is a low-molecular-weight cytokeratin normally expressed by Merkel cells and by the epithelium of the gastrointestinal tract and urothelium, whereas CK7 expression typically is expressed in the lungs, ovaries, endometrium, and breasts, but not in the lower gastrointestinal tract.14 Differentiating primary cutaneous adenocarcinoma from cutaneous metastasis can be accomplished with a thorough clinical history; however, p63 positivity supports a primary cutaneous lesion and may be useful in certain situations.7 CDX2 stains can be utilized to aid in localizing the primary neoplasm when it is unknown, and when positive, it suggests a lower gastrointestinal tract origin. However, special AT-rich sequence-binding protein 2 (SATB2) recently has been proposed as a replacement immunohistochemical marker for CDX2, as it has greater specificity for SRCA of the lower gastrointestinal tract.15 Benign entities with signet ring cell morphology are difficult to distinguish from SRCA; however, malignant lesions are more likely to demonstrate an infiltrative growth pattern, frequent mitotic figures, and apoptosis. Immunohistochemistry also can be utilized to support the diagnosis of benign proliferation with signet ring morphology, as benign lesions often will demonstrate E-cadherin positivity and negativity for p53 and Ki-67.13
Cutaneous metastasis usually correlates to advanced disease and generally indicates a worse prognosis.13 Signet ring cell morphology in both gastric and colorectal cancer portends a poor prognosis, and there is a lower overall survival in patients with these malignancies compared to cancers of the same organ with non–signet ring cell morphology.4,8
- Mandzhieva B, Jalil A, Nadeem M, et al. Most common pathway of metastasis of rectal signet ring cell carcinoma to the skin: hematogenous. Cureus. 2020;12:E6890.
- Parente P, Ciardiello D, Reggiani Bonetti L, et al. Cutaneous metastasis from colorectal cancer: making light on an unusual and misdiagnosed event. Life. 2021;11:954.
- Picciariello A, Tomasicchio G, Lantone G, et al. Synchronous “skip” facial metastases from colorectal adenocarcinoma: a case report and review of literature. BMC Gastroenterol. 2022;22:68.
- Benesch MGK, Mathieson A. Epidemiology of signet ring cell adenocarcinomas. Cancers. 2020;12:1544.
- Xu Q, Karouji Y, Kobayashi M, et al. The PI 3-kinase-Rac-p38 MAP kinase pathway is involved in the formation of signet-ring cell carcinoma. Oncogene. 2003;22:5537-5544.
- Morales-Cruz M, Salgado-Nesme N, Trolle-Silva AM, et al. Signet ring cell carcinoma of the rectum: atypical metastatic presentation. BMJ Case Rep CP. 2019;12:E229135.
- Demirciog˘lu D, Öztürk Durmaz E, Demirkesen C, et al. Livedoid cutaneous metastasis of signet‐ring cell gastric carcinoma. J Cutan Pathol. 2021;48:785-788.
- Dong X, Sun G, Qu H, et al. Prognostic significance of signet-ring cell components in patients with gastric carcinoma of different stages. Front Surg. 2021;8:642468.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Nguyen AH, Tahseen AI, Vaudreuil AM, et al. Clinical features and treatment of vulvar Merkel cell carcinoma: a systematic review. Gynecol Oncol Res Pract. 2017;4:2.
- Marques, SA. Paracoccidioidomycosis. Clin Dermatol. 2012;30:610-615.
- Larocca C, Kupper T. Mycosis fungoides and Sézary syndrome. Hematol Oncol Clin. 2019;33:103-120.
- Gündüz Ö, Emeksiz MC, Atasoy P, et al. Signet-ring cells in the skin: a case of late-onset cutaneous metastasis of gastric carcinoma and a brief review of histological approach. Dermatol Rep. 2017;8:6819.
- Al-Taee A, Almukhtar R, Lai J, et al. Metastatic signet ring cell carcinoma of unknown primary origin: a case report and review of the literature. Ann Transl Med. 2016;4:283.
- Ma C, Lowenthal BM, Pai RK. SATB2 is superior to CDX2 in distinguishing signet ring cell carcinoma of the upper gastrointestinal tract and lower gastrointestinal tract. Am J Surg Pathol. 2018; 42:1715-1722.
The Diagnosis: Cutaneous Metastasis
A shave biopsy of the lip revealed a diffuse cellular infiltrate filling the superficial and deep dermis (Figure 1A). Morphologically, the cells had abundant clear cytoplasm with eccentrically located, pleomorphic, hyperchromatic nuclei with occasional prominent nucleoli (Figure 1B). The cells stained positive for AE1/ AE3 on immunohistochemistry (Figure 2). A punch biopsy of the nodule in the right axillary vault revealed a morphologically similar proliferation of cells. A colonoscopy revealed a completely obstructing circumferential mass in the distal ascending colon. A biopsy of the mass confirmed invasive adenocarcinoma, supporting a diagnosis of cutaneous metastases from adenocarcinoma of the colon. The patient underwent resection of the lip tumor and started multiagent chemotherapy for his newly diagnosed stage IV adenocarcinoma of the colon. The patient died, despite therapy.
Cutaneous metastasis from solid malignancies is uncommon, as only 1.3% of them exhibit cutaneous manifestations at presentation.1 Cutaneous metastasis from signet ring cell adenocarcinoma (SRCA) of the colon is uncommon, and cutaneous metastasis of colorectal SRCA rarely precedes the diagnosis of the primary lesion.2 Among the colorectal cancers that metastasize to the skin, metastasis to the face occurs in only 0.5% of patients.3
Signet ring cell adenocarcinomas are poorly differentiated adenocarcinomas histologically characterized by the neoplastic cells’ circular to ovoid appearance with a flattened top.4,5 This distinctive shape is from the displacement of the nucleus to the periphery of the cell due to the accumulation of intracytoplasmic mucin.4 Classically, malignancies are characterized as an SRCA if more than 50% of the cells have a signet ring cell morphology; if the signet ring cells comprise less than 50% of the neoplasm, the tumor is designated as an adenocarcinoma with signet ring morphology.4 The most common cause of cutaneous metastasis with signet ring morphology is gastric cancer, while colorectal carcinoma is less common.1 Colorectal SRCAs usually are found in the right colon or the rectum in comparison to other colonic sites.6
Clinically, cutaneous metastasis can present in a variety of ways. The most common presentation is nodular lesions that may coalesce to become zosteriform in configuration or lesions that mimic inflammatory dermatoses.7 Cutaneous metastasis is more common in breast and lung cancer, and when it occurs secondary to colorectal cancer, cutaneous metastasis rarely predates the detection of the primary neoplasm.2
The clinical appearance of metastasis is not specific and can mimic many entities8; therefore, a high index of suspicion must be employed when managing patients, even those without a history of internal malignancy. In our patient, the smooth nodular lesion appeared similar to a basal cell carcinoma; however, basal cell carcinomas appear more pearly, and arborizing telangiectasia often is seen.9 Merkel cell carcinoma is common on sundamaged skin of the head and neck but clinically appears more violaceous than the lesion seen in our patient.10 Paracoccidioidomycosis may form ulcerated papulonodules or plaques, especially around the nose and mouth. In many of these cases, lesions develop from contiguous lesions of the oral mucosa; therefore, the presence of oral lesions will help distinguish this infectious entity from cutaneous metastasis. Multiple lesions usually are identified when there is hematogenous dissemination.11 Mycosis fungoides is a subtype of cutaneous T-cell lymphoma and is characterized by multiple patches, plaques, and nodules on sun-protected areas. Involvement of the head and neck is not common, except in the folliculotropic subtype, which has a separate and distinct clinical morphology.12
The development of signet ring morphology from an adenocarcinoma can be attributed to the activation of phosphatidylinositol 3-kinase (PI3K), which leads to downstream activation of mitogen-activated protein kinase (MAPK) and the subsequent loss of intercellular tight junctions. The mucin 4 gene, MUC4, also is upregulated by PI3K activation and possesses antiapoptotic and mitogenic effects in addition to its mucin secretory function.13
The neoplastic cells in SRCAs stain positive for mucicarmine, Alcian blue, and periodic acid–Schiff, which highlights the mucinous component of the cells.7 Immunohistochemical stains with CK7, CK20, AE1/AE3, and epithelial membrane antigen can be implemented to confirm an epithelial origin of the primary cancer.7,13 CK20 is a low-molecular-weight cytokeratin normally expressed by Merkel cells and by the epithelium of the gastrointestinal tract and urothelium, whereas CK7 expression typically is expressed in the lungs, ovaries, endometrium, and breasts, but not in the lower gastrointestinal tract.14 Differentiating primary cutaneous adenocarcinoma from cutaneous metastasis can be accomplished with a thorough clinical history; however, p63 positivity supports a primary cutaneous lesion and may be useful in certain situations.7 CDX2 stains can be utilized to aid in localizing the primary neoplasm when it is unknown, and when positive, it suggests a lower gastrointestinal tract origin. However, special AT-rich sequence-binding protein 2 (SATB2) recently has been proposed as a replacement immunohistochemical marker for CDX2, as it has greater specificity for SRCA of the lower gastrointestinal tract.15 Benign entities with signet ring cell morphology are difficult to distinguish from SRCA; however, malignant lesions are more likely to demonstrate an infiltrative growth pattern, frequent mitotic figures, and apoptosis. Immunohistochemistry also can be utilized to support the diagnosis of benign proliferation with signet ring morphology, as benign lesions often will demonstrate E-cadherin positivity and negativity for p53 and Ki-67.13
Cutaneous metastasis usually correlates to advanced disease and generally indicates a worse prognosis.13 Signet ring cell morphology in both gastric and colorectal cancer portends a poor prognosis, and there is a lower overall survival in patients with these malignancies compared to cancers of the same organ with non–signet ring cell morphology.4,8
The Diagnosis: Cutaneous Metastasis
A shave biopsy of the lip revealed a diffuse cellular infiltrate filling the superficial and deep dermis (Figure 1A). Morphologically, the cells had abundant clear cytoplasm with eccentrically located, pleomorphic, hyperchromatic nuclei with occasional prominent nucleoli (Figure 1B). The cells stained positive for AE1/ AE3 on immunohistochemistry (Figure 2). A punch biopsy of the nodule in the right axillary vault revealed a morphologically similar proliferation of cells. A colonoscopy revealed a completely obstructing circumferential mass in the distal ascending colon. A biopsy of the mass confirmed invasive adenocarcinoma, supporting a diagnosis of cutaneous metastases from adenocarcinoma of the colon. The patient underwent resection of the lip tumor and started multiagent chemotherapy for his newly diagnosed stage IV adenocarcinoma of the colon. The patient died, despite therapy.
Cutaneous metastasis from solid malignancies is uncommon, as only 1.3% of them exhibit cutaneous manifestations at presentation.1 Cutaneous metastasis from signet ring cell adenocarcinoma (SRCA) of the colon is uncommon, and cutaneous metastasis of colorectal SRCA rarely precedes the diagnosis of the primary lesion.2 Among the colorectal cancers that metastasize to the skin, metastasis to the face occurs in only 0.5% of patients.3
Signet ring cell adenocarcinomas are poorly differentiated adenocarcinomas histologically characterized by the neoplastic cells’ circular to ovoid appearance with a flattened top.4,5 This distinctive shape is from the displacement of the nucleus to the periphery of the cell due to the accumulation of intracytoplasmic mucin.4 Classically, malignancies are characterized as an SRCA if more than 50% of the cells have a signet ring cell morphology; if the signet ring cells comprise less than 50% of the neoplasm, the tumor is designated as an adenocarcinoma with signet ring morphology.4 The most common cause of cutaneous metastasis with signet ring morphology is gastric cancer, while colorectal carcinoma is less common.1 Colorectal SRCAs usually are found in the right colon or the rectum in comparison to other colonic sites.6
Clinically, cutaneous metastasis can present in a variety of ways. The most common presentation is nodular lesions that may coalesce to become zosteriform in configuration or lesions that mimic inflammatory dermatoses.7 Cutaneous metastasis is more common in breast and lung cancer, and when it occurs secondary to colorectal cancer, cutaneous metastasis rarely predates the detection of the primary neoplasm.2
The clinical appearance of metastasis is not specific and can mimic many entities8; therefore, a high index of suspicion must be employed when managing patients, even those without a history of internal malignancy. In our patient, the smooth nodular lesion appeared similar to a basal cell carcinoma; however, basal cell carcinomas appear more pearly, and arborizing telangiectasia often is seen.9 Merkel cell carcinoma is common on sundamaged skin of the head and neck but clinically appears more violaceous than the lesion seen in our patient.10 Paracoccidioidomycosis may form ulcerated papulonodules or plaques, especially around the nose and mouth. In many of these cases, lesions develop from contiguous lesions of the oral mucosa; therefore, the presence of oral lesions will help distinguish this infectious entity from cutaneous metastasis. Multiple lesions usually are identified when there is hematogenous dissemination.11 Mycosis fungoides is a subtype of cutaneous T-cell lymphoma and is characterized by multiple patches, plaques, and nodules on sun-protected areas. Involvement of the head and neck is not common, except in the folliculotropic subtype, which has a separate and distinct clinical morphology.12
The development of signet ring morphology from an adenocarcinoma can be attributed to the activation of phosphatidylinositol 3-kinase (PI3K), which leads to downstream activation of mitogen-activated protein kinase (MAPK) and the subsequent loss of intercellular tight junctions. The mucin 4 gene, MUC4, also is upregulated by PI3K activation and possesses antiapoptotic and mitogenic effects in addition to its mucin secretory function.13
The neoplastic cells in SRCAs stain positive for mucicarmine, Alcian blue, and periodic acid–Schiff, which highlights the mucinous component of the cells.7 Immunohistochemical stains with CK7, CK20, AE1/AE3, and epithelial membrane antigen can be implemented to confirm an epithelial origin of the primary cancer.7,13 CK20 is a low-molecular-weight cytokeratin normally expressed by Merkel cells and by the epithelium of the gastrointestinal tract and urothelium, whereas CK7 expression typically is expressed in the lungs, ovaries, endometrium, and breasts, but not in the lower gastrointestinal tract.14 Differentiating primary cutaneous adenocarcinoma from cutaneous metastasis can be accomplished with a thorough clinical history; however, p63 positivity supports a primary cutaneous lesion and may be useful in certain situations.7 CDX2 stains can be utilized to aid in localizing the primary neoplasm when it is unknown, and when positive, it suggests a lower gastrointestinal tract origin. However, special AT-rich sequence-binding protein 2 (SATB2) recently has been proposed as a replacement immunohistochemical marker for CDX2, as it has greater specificity for SRCA of the lower gastrointestinal tract.15 Benign entities with signet ring cell morphology are difficult to distinguish from SRCA; however, malignant lesions are more likely to demonstrate an infiltrative growth pattern, frequent mitotic figures, and apoptosis. Immunohistochemistry also can be utilized to support the diagnosis of benign proliferation with signet ring morphology, as benign lesions often will demonstrate E-cadherin positivity and negativity for p53 and Ki-67.13
Cutaneous metastasis usually correlates to advanced disease and generally indicates a worse prognosis.13 Signet ring cell morphology in both gastric and colorectal cancer portends a poor prognosis, and there is a lower overall survival in patients with these malignancies compared to cancers of the same organ with non–signet ring cell morphology.4,8
- Mandzhieva B, Jalil A, Nadeem M, et al. Most common pathway of metastasis of rectal signet ring cell carcinoma to the skin: hematogenous. Cureus. 2020;12:E6890.
- Parente P, Ciardiello D, Reggiani Bonetti L, et al. Cutaneous metastasis from colorectal cancer: making light on an unusual and misdiagnosed event. Life. 2021;11:954.
- Picciariello A, Tomasicchio G, Lantone G, et al. Synchronous “skip” facial metastases from colorectal adenocarcinoma: a case report and review of literature. BMC Gastroenterol. 2022;22:68.
- Benesch MGK, Mathieson A. Epidemiology of signet ring cell adenocarcinomas. Cancers. 2020;12:1544.
- Xu Q, Karouji Y, Kobayashi M, et al. The PI 3-kinase-Rac-p38 MAP kinase pathway is involved in the formation of signet-ring cell carcinoma. Oncogene. 2003;22:5537-5544.
- Morales-Cruz M, Salgado-Nesme N, Trolle-Silva AM, et al. Signet ring cell carcinoma of the rectum: atypical metastatic presentation. BMJ Case Rep CP. 2019;12:E229135.
- Demirciog˘lu D, Öztürk Durmaz E, Demirkesen C, et al. Livedoid cutaneous metastasis of signet‐ring cell gastric carcinoma. J Cutan Pathol. 2021;48:785-788.
- Dong X, Sun G, Qu H, et al. Prognostic significance of signet-ring cell components in patients with gastric carcinoma of different stages. Front Surg. 2021;8:642468.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Nguyen AH, Tahseen AI, Vaudreuil AM, et al. Clinical features and treatment of vulvar Merkel cell carcinoma: a systematic review. Gynecol Oncol Res Pract. 2017;4:2.
- Marques, SA. Paracoccidioidomycosis. Clin Dermatol. 2012;30:610-615.
- Larocca C, Kupper T. Mycosis fungoides and Sézary syndrome. Hematol Oncol Clin. 2019;33:103-120.
- Gündüz Ö, Emeksiz MC, Atasoy P, et al. Signet-ring cells in the skin: a case of late-onset cutaneous metastasis of gastric carcinoma and a brief review of histological approach. Dermatol Rep. 2017;8:6819.
- Al-Taee A, Almukhtar R, Lai J, et al. Metastatic signet ring cell carcinoma of unknown primary origin: a case report and review of the literature. Ann Transl Med. 2016;4:283.
- Ma C, Lowenthal BM, Pai RK. SATB2 is superior to CDX2 in distinguishing signet ring cell carcinoma of the upper gastrointestinal tract and lower gastrointestinal tract. Am J Surg Pathol. 2018; 42:1715-1722.
- Mandzhieva B, Jalil A, Nadeem M, et al. Most common pathway of metastasis of rectal signet ring cell carcinoma to the skin: hematogenous. Cureus. 2020;12:E6890.
- Parente P, Ciardiello D, Reggiani Bonetti L, et al. Cutaneous metastasis from colorectal cancer: making light on an unusual and misdiagnosed event. Life. 2021;11:954.
- Picciariello A, Tomasicchio G, Lantone G, et al. Synchronous “skip” facial metastases from colorectal adenocarcinoma: a case report and review of literature. BMC Gastroenterol. 2022;22:68.
- Benesch MGK, Mathieson A. Epidemiology of signet ring cell adenocarcinomas. Cancers. 2020;12:1544.
- Xu Q, Karouji Y, Kobayashi M, et al. The PI 3-kinase-Rac-p38 MAP kinase pathway is involved in the formation of signet-ring cell carcinoma. Oncogene. 2003;22:5537-5544.
- Morales-Cruz M, Salgado-Nesme N, Trolle-Silva AM, et al. Signet ring cell carcinoma of the rectum: atypical metastatic presentation. BMJ Case Rep CP. 2019;12:E229135.
- Demirciog˘lu D, Öztürk Durmaz E, Demirkesen C, et al. Livedoid cutaneous metastasis of signet‐ring cell gastric carcinoma. J Cutan Pathol. 2021;48:785-788.
- Dong X, Sun G, Qu H, et al. Prognostic significance of signet-ring cell components in patients with gastric carcinoma of different stages. Front Surg. 2021;8:642468.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Nguyen AH, Tahseen AI, Vaudreuil AM, et al. Clinical features and treatment of vulvar Merkel cell carcinoma: a systematic review. Gynecol Oncol Res Pract. 2017;4:2.
- Marques, SA. Paracoccidioidomycosis. Clin Dermatol. 2012;30:610-615.
- Larocca C, Kupper T. Mycosis fungoides and Sézary syndrome. Hematol Oncol Clin. 2019;33:103-120.
- Gündüz Ö, Emeksiz MC, Atasoy P, et al. Signet-ring cells in the skin: a case of late-onset cutaneous metastasis of gastric carcinoma and a brief review of histological approach. Dermatol Rep. 2017;8:6819.
- Al-Taee A, Almukhtar R, Lai J, et al. Metastatic signet ring cell carcinoma of unknown primary origin: a case report and review of the literature. Ann Transl Med. 2016;4:283.
- Ma C, Lowenthal BM, Pai RK. SATB2 is superior to CDX2 in distinguishing signet ring cell carcinoma of the upper gastrointestinal tract and lower gastrointestinal tract. Am J Surg Pathol. 2018; 42:1715-1722.
A 79-year-old man with a medical history of type 2 diabetes mellitus, hypothyroidism, and atrial fibrillation presented with an enlarging lesion on the right side of the upper cutaneous lip of 5 weeks’ duration. He had no personal history of skin cancer or other malignancy and was up to date on all routine cancer screenings. He reported associated lip and oral cavity tenderness, weakness, and a 13.6-kg (30-lb) unintentional weight loss over the last 6 months. He had used over-the-counter bacitracin ointment on the lesion without relief. A full-body skin examination revealed a firm, mobile, flesh-colored, nondraining nodule in the right axillary vault.


White Spots on the Extremities
The Diagnosis: Hypopigmented Mycosis Fungoides
Histopathology showed an atypical lymphoid infiltrate with expanded cytoplasm and hyperchromatic nuclei of irregular contours in the dermoepidermal junction (Figure 1). Immunohistochemical stains of atypical lymphocytes demonstrated the presence of CD3, CD8, and CD5, as well as the absence of CD7 and CD4 lymphocytes (Figure 2). The T-cell γ rearrangement showed polyclonal lymphocytes with 5% tumor cells. The histologic and clinical findings along with our patient’s medical history led to a diagnosis of stage IA (<10% body surface area involvement) hypopigmented mycosis fungoides (hMF).1 Our patient was treated with triamcinolone cream 0.1%; she noted an improvement in her symptoms at 2-month follow-up.
.")
Hypopigmented MF is an uncommon manifestation of MF with unknown prevalence and incidence rates. Mycosis fungoides is considered the most common subtype of cutaneous T-cell lymphoma that classically presents as a chronic, indolent, hypopigmented or depigmented macule or patch, commonly with scaling, in sunprotected areas such as the trunk and proximal arms and legs. It predominantly affects younger adults with darker skin tones and may be present in the pediatric population within the first decade of life.1 Classically, MF affects White patients aged 55 to 60 years. Disease progression is slow, with an incidence rate of 10% of tumor or extracutaneous involvement in the early stages of disease. A lack of specificity on the clinical and histopathologic findings in the initial stage often contributes to the diagnostic delay of hMF. As seen in our patient, this disease can be misdiagnosed as tinea versicolor, postinflammatory hypopigmentation, vitiligo, pityriasis alba, subcutaneous lupus erythematosus, or Hansen disease due to prolonged hypopigmented lesions.2 The clinical findings and histopathologic results including immunohistochemistry confirmed the diagnosis of hMF and ruled out pityriasis alba, postinflammatory hypopigmentation, subcutaneous lupus erythematosus, and vitiligo.
.")
The etiology and pathophysiology of hMF are not fully understood; however, it is hypothesized that melanocyte degeneration, abnormal melanogenesis, and disturbance of melanosome transfer result from the clonal expansion of T helper memory cells. T-cell dyscrasia has been reported to evolve into hMF during etanercept therapy.3 Clinically, hMF presents as hypopigmented papulosquamous, eczematous, or erythrodermic patches, plaques, and tumors with poorly defined atrophied borders. Multiple biopsies of steroid-naive lesions are needed for the diagnosis, as the initial hMF histologic finding cannot be specific for diagnostic confirmation. Common histopathologic findings include a bandlike lymphocytic infiltrate with epidermotropism, intraepidermal nests of atypical cells, or cerebriform nuclei lymphocytes on hematoxylin and eosin staining. In comparison to classical MF epidermotropism, CD4− and CD8+ atypical cells aid in the diagnosis of hMF. Although hMF carries a good prognosis and a benign clinical course,4 full-body computed tomography or positron emission tomography/computed tomography as well as laboratory analysis for lactate dehydrogenase should be pursued if lymphadenopathy, systemic symptoms, or advancedstage hMF are present.
Treatment of hMF depends on the disease stage. Psoralen plus UVA and narrowband UVB can be utilized for the initial stages with a relatively fast response and remission of lesions as early as the first 2 months of treatment. In addition to phototherapy, stage IA to IIA mycosis fungoides with localized skin lesions can benefit from topical steroids, topical retinoids, imiquimod, nitrogen mustard, and carmustine. For advanced stages of mycosis fungoides, combination therapy consisting of psoralen plus UVA with an oral retinoid, interferon alfa, and systemic chemotherapy commonly are prescribed. Maintenance therapy is used for prolonging remission; however, long-term phototherapy is not recommended due to the risk for skin cancer. Unfortunately, hMF requires long-term treatment due to its waxing and waning course, and recurrence may occur after complete resolution.5
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Lambroza E, Cohen SR, Lebwohl M, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- Chuang GS, Wasserman DI, Byers HR, et al. Hypopigmented T-cell dyscrasia evolving to hypopigmented mycosis fungoides during etanercept therapy. J Am Acad Dermatol. 2008;59(5 suppl):S121-S122.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/ European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014; 70:223.e1-17; quiz 240-242.
The Diagnosis: Hypopigmented Mycosis Fungoides
Histopathology showed an atypical lymphoid infiltrate with expanded cytoplasm and hyperchromatic nuclei of irregular contours in the dermoepidermal junction (Figure 1). Immunohistochemical stains of atypical lymphocytes demonstrated the presence of CD3, CD8, and CD5, as well as the absence of CD7 and CD4 lymphocytes (Figure 2). The T-cell γ rearrangement showed polyclonal lymphocytes with 5% tumor cells. The histologic and clinical findings along with our patient’s medical history led to a diagnosis of stage IA (<10% body surface area involvement) hypopigmented mycosis fungoides (hMF).1 Our patient was treated with triamcinolone cream 0.1%; she noted an improvement in her symptoms at 2-month follow-up.
Hypopigmented MF is an uncommon manifestation of MF with unknown prevalence and incidence rates. Mycosis fungoides is considered the most common subtype of cutaneous T-cell lymphoma that classically presents as a chronic, indolent, hypopigmented or depigmented macule or patch, commonly with scaling, in sunprotected areas such as the trunk and proximal arms and legs. It predominantly affects younger adults with darker skin tones and may be present in the pediatric population within the first decade of life.1 Classically, MF affects White patients aged 55 to 60 years. Disease progression is slow, with an incidence rate of 10% of tumor or extracutaneous involvement in the early stages of disease. A lack of specificity on the clinical and histopathologic findings in the initial stage often contributes to the diagnostic delay of hMF. As seen in our patient, this disease can be misdiagnosed as tinea versicolor, postinflammatory hypopigmentation, vitiligo, pityriasis alba, subcutaneous lupus erythematosus, or Hansen disease due to prolonged hypopigmented lesions.2 The clinical findings and histopathologic results including immunohistochemistry confirmed the diagnosis of hMF and ruled out pityriasis alba, postinflammatory hypopigmentation, subcutaneous lupus erythematosus, and vitiligo.
The etiology and pathophysiology of hMF are not fully understood; however, it is hypothesized that melanocyte degeneration, abnormal melanogenesis, and disturbance of melanosome transfer result from the clonal expansion of T helper memory cells. T-cell dyscrasia has been reported to evolve into hMF during etanercept therapy.3 Clinically, hMF presents as hypopigmented papulosquamous, eczematous, or erythrodermic patches, plaques, and tumors with poorly defined atrophied borders. Multiple biopsies of steroid-naive lesions are needed for the diagnosis, as the initial hMF histologic finding cannot be specific for diagnostic confirmation. Common histopathologic findings include a bandlike lymphocytic infiltrate with epidermotropism, intraepidermal nests of atypical cells, or cerebriform nuclei lymphocytes on hematoxylin and eosin staining. In comparison to classical MF epidermotropism, CD4− and CD8+ atypical cells aid in the diagnosis of hMF. Although hMF carries a good prognosis and a benign clinical course,4 full-body computed tomography or positron emission tomography/computed tomography as well as laboratory analysis for lactate dehydrogenase should be pursued if lymphadenopathy, systemic symptoms, or advancedstage hMF are present.
Treatment of hMF depends on the disease stage. Psoralen plus UVA and narrowband UVB can be utilized for the initial stages with a relatively fast response and remission of lesions as early as the first 2 months of treatment. In addition to phototherapy, stage IA to IIA mycosis fungoides with localized skin lesions can benefit from topical steroids, topical retinoids, imiquimod, nitrogen mustard, and carmustine. For advanced stages of mycosis fungoides, combination therapy consisting of psoralen plus UVA with an oral retinoid, interferon alfa, and systemic chemotherapy commonly are prescribed. Maintenance therapy is used for prolonging remission; however, long-term phototherapy is not recommended due to the risk for skin cancer. Unfortunately, hMF requires long-term treatment due to its waxing and waning course, and recurrence may occur after complete resolution.5
The Diagnosis: Hypopigmented Mycosis Fungoides
Histopathology showed an atypical lymphoid infiltrate with expanded cytoplasm and hyperchromatic nuclei of irregular contours in the dermoepidermal junction (Figure 1). Immunohistochemical stains of atypical lymphocytes demonstrated the presence of CD3, CD8, and CD5, as well as the absence of CD7 and CD4 lymphocytes (Figure 2). The T-cell γ rearrangement showed polyclonal lymphocytes with 5% tumor cells. The histologic and clinical findings along with our patient’s medical history led to a diagnosis of stage IA (<10% body surface area involvement) hypopigmented mycosis fungoides (hMF).1 Our patient was treated with triamcinolone cream 0.1%; she noted an improvement in her symptoms at 2-month follow-up.
Hypopigmented MF is an uncommon manifestation of MF with unknown prevalence and incidence rates. Mycosis fungoides is considered the most common subtype of cutaneous T-cell lymphoma that classically presents as a chronic, indolent, hypopigmented or depigmented macule or patch, commonly with scaling, in sunprotected areas such as the trunk and proximal arms and legs. It predominantly affects younger adults with darker skin tones and may be present in the pediatric population within the first decade of life.1 Classically, MF affects White patients aged 55 to 60 years. Disease progression is slow, with an incidence rate of 10% of tumor or extracutaneous involvement in the early stages of disease. A lack of specificity on the clinical and histopathologic findings in the initial stage often contributes to the diagnostic delay of hMF. As seen in our patient, this disease can be misdiagnosed as tinea versicolor, postinflammatory hypopigmentation, vitiligo, pityriasis alba, subcutaneous lupus erythematosus, or Hansen disease due to prolonged hypopigmented lesions.2 The clinical findings and histopathologic results including immunohistochemistry confirmed the diagnosis of hMF and ruled out pityriasis alba, postinflammatory hypopigmentation, subcutaneous lupus erythematosus, and vitiligo.
The etiology and pathophysiology of hMF are not fully understood; however, it is hypothesized that melanocyte degeneration, abnormal melanogenesis, and disturbance of melanosome transfer result from the clonal expansion of T helper memory cells. T-cell dyscrasia has been reported to evolve into hMF during etanercept therapy.3 Clinically, hMF presents as hypopigmented papulosquamous, eczematous, or erythrodermic patches, plaques, and tumors with poorly defined atrophied borders. Multiple biopsies of steroid-naive lesions are needed for the diagnosis, as the initial hMF histologic finding cannot be specific for diagnostic confirmation. Common histopathologic findings include a bandlike lymphocytic infiltrate with epidermotropism, intraepidermal nests of atypical cells, or cerebriform nuclei lymphocytes on hematoxylin and eosin staining. In comparison to classical MF epidermotropism, CD4− and CD8+ atypical cells aid in the diagnosis of hMF. Although hMF carries a good prognosis and a benign clinical course,4 full-body computed tomography or positron emission tomography/computed tomography as well as laboratory analysis for lactate dehydrogenase should be pursued if lymphadenopathy, systemic symptoms, or advancedstage hMF are present.
Treatment of hMF depends on the disease stage. Psoralen plus UVA and narrowband UVB can be utilized for the initial stages with a relatively fast response and remission of lesions as early as the first 2 months of treatment. In addition to phototherapy, stage IA to IIA mycosis fungoides with localized skin lesions can benefit from topical steroids, topical retinoids, imiquimod, nitrogen mustard, and carmustine. For advanced stages of mycosis fungoides, combination therapy consisting of psoralen plus UVA with an oral retinoid, interferon alfa, and systemic chemotherapy commonly are prescribed. Maintenance therapy is used for prolonging remission; however, long-term phototherapy is not recommended due to the risk for skin cancer. Unfortunately, hMF requires long-term treatment due to its waxing and waning course, and recurrence may occur after complete resolution.5
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Lambroza E, Cohen SR, Lebwohl M, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- Chuang GS, Wasserman DI, Byers HR, et al. Hypopigmented T-cell dyscrasia evolving to hypopigmented mycosis fungoides during etanercept therapy. J Am Acad Dermatol. 2008;59(5 suppl):S121-S122.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/ European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014; 70:223.e1-17; quiz 240-242.
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Lambroza E, Cohen SR, Lebwohl M, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- Chuang GS, Wasserman DI, Byers HR, et al. Hypopigmented T-cell dyscrasia evolving to hypopigmented mycosis fungoides during etanercept therapy. J Am Acad Dermatol. 2008;59(5 suppl):S121-S122.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/ European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014; 70:223.e1-17; quiz 240-242.
A 52-year-old Black woman presented with self-described whitened spots on the arms and legs of 2 years’ duration. She experienced no improvement with ketoconazole cream and topical calcineurin inhibitors prescribed during a prior dermatology visit at an outside institution. She denied pain or pruritus. A review of systems as well as the patient’s medical history were noncontributory. A prior biopsy at an outside institution revealed an interface dermatitis suggestive of cutaneous lupus erythematosus. The patient noted social drinking and denied tobacco use. She had no known allergies to medications and currently was on tamoxifen for breast cancer following a right mastectomy. Physical examination showed hypopigmented macules and patches on the left upper arm and right proximal leg. The center of the lesions was not erythematous or scaly. Palpation did not reveal enlarged lymph nodes, and laboratory analyses ruled out low levels of red blood cells, white blood cells, or platelets. Punch biopsies from the left arm and right thigh were performed.


Erythematous Dermal Facial Plaques in a Neutropenic Patient
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
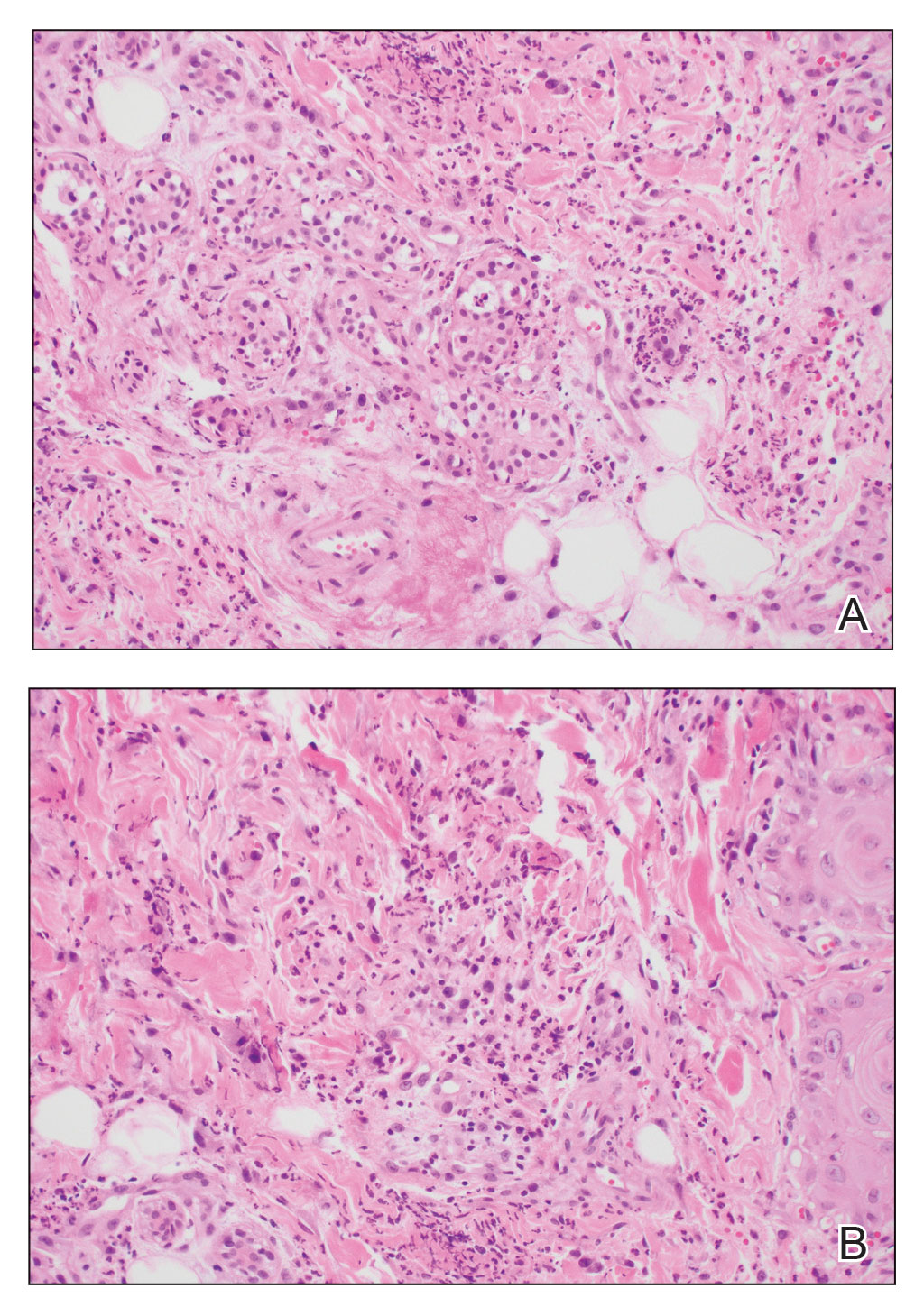
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
A 50-year-old woman undergoing cytarabine induction therapy for acute myeloid leukemia developed tender, erythematous, dermal plaques on the nasal dorsum, left medial eyebrow, left preauricular cheek, and right cheek. The rash erupted 7 days after receiving the cytarabine induction regimen. She had a fever (temperature, 39.9 °C [103.8 °F]) and also was neutropenic.
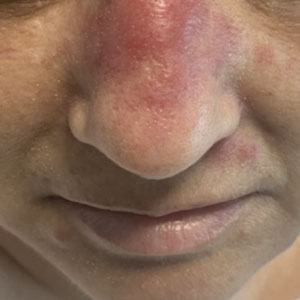
Oval Brown Plaque on the Palm
The Diagnosis: Poroma
Histopathology showed an endophytic expansion of the epidermis by bland, uniform, basaloid epithelial cells with focal ductal differentiation and an abrupt transition with surrounding epidermal keratinocytes (Figure), consistent with a diagnosis of poroma. The patient elected to monitor the lesion rather than to have it excised.

Eccrine poroma, used interchangeably with the term poroma, is a rare benign adnexal tumor of the eccrine sweat glands resulting from proliferation of the acrosyringium.1,2 It often occurs on the palms or soles, though it also can arise anywhere sweat glands are present.1 Eccrine poromas often appear in middle-aged individuals as singular, well-circumscribed, red-brown papules or nodules.3 A characteristic feature is a shallow, cup-shaped depression within the larger papule or nodule.1
Because the condition is benign and often asymptomatic, it can be safely monitored for progression.1 However, if the lesion is symptomatic or located in a sensitive area, complete excision is curative.4 Eccrine poromas can recur, making close monitoring following excision important.5 The development of bleeding, itching, or pain in a previously asymptomatic lesion may indicate possible malignant transformation, which occurs in only 18% of cases.6
The differential diagnosis includes basal cell carcinoma, circumscribed acral hypokeratosis, Kaposi sarcoma, and pyogenic granuloma. Basal cell carcinoma is the most common type of skin cancer.7 In rare cases it has been shown to present on the palms or soles as a slowgrowing, reddish-pink papule or plaque with central ulceration. It typically is asymptomatic. Histopathology shows dermal nests of basaloid cells with peripheral palisading, stromal mucin, and peritumoral clefts. Treatment is surgical excision.7
Circumscribed acral hypokeratosis presents on the palms or soles as a solitary, shallow, well-defined lesion with a flat base and raised border.8 It often is red-pink in color and most frequently occurs in middle-aged women. Although the cause of the condition is unknown, it is thought to be the result of trauma or human papillomavirus infection.8 Biopsy results characteristically show hypokeratosis demarcated by a sharp and frayed cutoff from uninvolved acral skin with discrete hypogranulosis, dilated blood vessels in the papillary dermis, and slightly thickened collagen fibers in the reticular dermis.9 Surgical excision is a potential treatment option, as topical corticosteroids, retinoids, and calcipotriene have not been shown to be effective; spontaneous resolution has been reported.8
Kaposi sarcoma is a vascular neoplasm that is associated with human herpesvirus 8 infection.10 It typically presents on mucocutaneous sites and the lower extremities. Palmar involvement has been reported in rare cases, occurring as a solitary, well-demarcated, violaceous macule or patch that may be painful.10-12 Characteristic histopathologic features include a proliferation in the dermis of slitlike vascular spaces and spindle cell proliferation.13 Treatment options include cryosurgery; pulsed dye laser; and topical, intralesional, or systemic chemotherapy agents, depending on the stage of the patient’s disease. Antiretroviral therapy is indicated for patients with Kaposi sarcoma secondary to AIDS.14
Pyogenic granuloma presents as a solitary red-brown or bluish-black papule or nodule that bleeds easily when manipulated.15 It commonly occurs following trauma, typically on the fingers, feet, and lips.6 Although benign, potential complications include ulceration and blood loss. Pyogenic granulomas can be treated via curettage and cautery, excision, cryosurgery, or pulsed dye laser.15
- Wankhade V, Singh R, Sadhwani V, et al. Eccrine poroma. Indian Dermatol Online J. 2015;6:304-305.
- Yorulmaz A, Aksoy GG, Ozhamam EU. A growing mass under the nail: subungual eccrine poroma. Skin Appendage Disord. 2020;6:254-257.
- Wang Y, Liu M, Zheng Y, et al. Eccrine poroma presented as spindleshaped plaque: a case report. Medicine (Baltimore). 2021;100:E25971. doi:10.1097/MD.0000000000025971
- Sharma M, Singh M, Gupta K, et al. Eccrine poroma of the eyelid. Indian J Ophthalmol. 2020;68:2522.
- Rasool MN, Hawary MB. Benign eccrine poroma in the palm of the hand. Ann Saudi Med. 2004;24:46-47.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms [published online April 2, 2014]. Int J Dermatol. 2014;53:1053-1061. doi:10.1111/ijd.12448
- López-Sánchez C, Ferguson P, Collgros H. Basal cell carcinoma of the palm: an unusual presentation of a common tumour [published online August 6, 2019]. Australas J Dermatol. 2020;61:69-70. doi:10.1111/ajd.13129
- Berk DR, Böer A, Bauschard FD, et al. Circumscribed acral hypokeratosis [published online April 6, 2007]. J Am Acad Dermatol. 2007;57:292-296. doi:10.1016/j.jaad.2007.02.022
- Majluf-Cáceres P, Vera-Kellet C, González-Bombardiere S. New dermoscopic keys for circumscribed acral hypokeratosis: report of four cases. Dermatol Pract Concept. 2021;11:E2021010. doi:10.5826/dpc.1102a10
- Simonart T, De Dobbeleer G, Stallenberg B. Classic Kaposi’s sarcoma of the palm in a metallurgist: role of iron filings in its development? Br J Dermatol. 2003;148:1061-1063. doi:10.1046/j.1365-2133.2003.05331.x
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Al Zolibani AA, Al Robaee AA. Primary palmoplantar Kaposi’s sarcoma: an unusual presentation. Skinmed. 2006;5:248-249. doi:10.1111/j.1540-9740.2006.04662.x
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Etemad SA, Dewan AK. Kaposi sarcoma updates [published online July 10, 2019]. Dermatol Clin. 2019;37:505-517. doi:10.1016/j. det.2019.05.008
- Murthy SC, Nagaraj A. Pyogenic granuloma. Indian Pediatr. 2012;49:855. doi:10.1007/s13312-012-0184-4
The Diagnosis: Poroma
Histopathology showed an endophytic expansion of the epidermis by bland, uniform, basaloid epithelial cells with focal ductal differentiation and an abrupt transition with surrounding epidermal keratinocytes (Figure), consistent with a diagnosis of poroma. The patient elected to monitor the lesion rather than to have it excised.
Eccrine poroma, used interchangeably with the term poroma, is a rare benign adnexal tumor of the eccrine sweat glands resulting from proliferation of the acrosyringium.1,2 It often occurs on the palms or soles, though it also can arise anywhere sweat glands are present.1 Eccrine poromas often appear in middle-aged individuals as singular, well-circumscribed, red-brown papules or nodules.3 A characteristic feature is a shallow, cup-shaped depression within the larger papule or nodule.1
Because the condition is benign and often asymptomatic, it can be safely monitored for progression.1 However, if the lesion is symptomatic or located in a sensitive area, complete excision is curative.4 Eccrine poromas can recur, making close monitoring following excision important.5 The development of bleeding, itching, or pain in a previously asymptomatic lesion may indicate possible malignant transformation, which occurs in only 18% of cases.6
The differential diagnosis includes basal cell carcinoma, circumscribed acral hypokeratosis, Kaposi sarcoma, and pyogenic granuloma. Basal cell carcinoma is the most common type of skin cancer.7 In rare cases it has been shown to present on the palms or soles as a slowgrowing, reddish-pink papule or plaque with central ulceration. It typically is asymptomatic. Histopathology shows dermal nests of basaloid cells with peripheral palisading, stromal mucin, and peritumoral clefts. Treatment is surgical excision.7
Circumscribed acral hypokeratosis presents on the palms or soles as a solitary, shallow, well-defined lesion with a flat base and raised border.8 It often is red-pink in color and most frequently occurs in middle-aged women. Although the cause of the condition is unknown, it is thought to be the result of trauma or human papillomavirus infection.8 Biopsy results characteristically show hypokeratosis demarcated by a sharp and frayed cutoff from uninvolved acral skin with discrete hypogranulosis, dilated blood vessels in the papillary dermis, and slightly thickened collagen fibers in the reticular dermis.9 Surgical excision is a potential treatment option, as topical corticosteroids, retinoids, and calcipotriene have not been shown to be effective; spontaneous resolution has been reported.8
Kaposi sarcoma is a vascular neoplasm that is associated with human herpesvirus 8 infection.10 It typically presents on mucocutaneous sites and the lower extremities. Palmar involvement has been reported in rare cases, occurring as a solitary, well-demarcated, violaceous macule or patch that may be painful.10-12 Characteristic histopathologic features include a proliferation in the dermis of slitlike vascular spaces and spindle cell proliferation.13 Treatment options include cryosurgery; pulsed dye laser; and topical, intralesional, or systemic chemotherapy agents, depending on the stage of the patient’s disease. Antiretroviral therapy is indicated for patients with Kaposi sarcoma secondary to AIDS.14
Pyogenic granuloma presents as a solitary red-brown or bluish-black papule or nodule that bleeds easily when manipulated.15 It commonly occurs following trauma, typically on the fingers, feet, and lips.6 Although benign, potential complications include ulceration and blood loss. Pyogenic granulomas can be treated via curettage and cautery, excision, cryosurgery, or pulsed dye laser.15
The Diagnosis: Poroma
Histopathology showed an endophytic expansion of the epidermis by bland, uniform, basaloid epithelial cells with focal ductal differentiation and an abrupt transition with surrounding epidermal keratinocytes (Figure), consistent with a diagnosis of poroma. The patient elected to monitor the lesion rather than to have it excised.
Eccrine poroma, used interchangeably with the term poroma, is a rare benign adnexal tumor of the eccrine sweat glands resulting from proliferation of the acrosyringium.1,2 It often occurs on the palms or soles, though it also can arise anywhere sweat glands are present.1 Eccrine poromas often appear in middle-aged individuals as singular, well-circumscribed, red-brown papules or nodules.3 A characteristic feature is a shallow, cup-shaped depression within the larger papule or nodule.1
Because the condition is benign and often asymptomatic, it can be safely monitored for progression.1 However, if the lesion is symptomatic or located in a sensitive area, complete excision is curative.4 Eccrine poromas can recur, making close monitoring following excision important.5 The development of bleeding, itching, or pain in a previously asymptomatic lesion may indicate possible malignant transformation, which occurs in only 18% of cases.6
The differential diagnosis includes basal cell carcinoma, circumscribed acral hypokeratosis, Kaposi sarcoma, and pyogenic granuloma. Basal cell carcinoma is the most common type of skin cancer.7 In rare cases it has been shown to present on the palms or soles as a slowgrowing, reddish-pink papule or plaque with central ulceration. It typically is asymptomatic. Histopathology shows dermal nests of basaloid cells with peripheral palisading, stromal mucin, and peritumoral clefts. Treatment is surgical excision.7
Circumscribed acral hypokeratosis presents on the palms or soles as a solitary, shallow, well-defined lesion with a flat base and raised border.8 It often is red-pink in color and most frequently occurs in middle-aged women. Although the cause of the condition is unknown, it is thought to be the result of trauma or human papillomavirus infection.8 Biopsy results characteristically show hypokeratosis demarcated by a sharp and frayed cutoff from uninvolved acral skin with discrete hypogranulosis, dilated blood vessels in the papillary dermis, and slightly thickened collagen fibers in the reticular dermis.9 Surgical excision is a potential treatment option, as topical corticosteroids, retinoids, and calcipotriene have not been shown to be effective; spontaneous resolution has been reported.8
Kaposi sarcoma is a vascular neoplasm that is associated with human herpesvirus 8 infection.10 It typically presents on mucocutaneous sites and the lower extremities. Palmar involvement has been reported in rare cases, occurring as a solitary, well-demarcated, violaceous macule or patch that may be painful.10-12 Characteristic histopathologic features include a proliferation in the dermis of slitlike vascular spaces and spindle cell proliferation.13 Treatment options include cryosurgery; pulsed dye laser; and topical, intralesional, or systemic chemotherapy agents, depending on the stage of the patient’s disease. Antiretroviral therapy is indicated for patients with Kaposi sarcoma secondary to AIDS.14
Pyogenic granuloma presents as a solitary red-brown or bluish-black papule or nodule that bleeds easily when manipulated.15 It commonly occurs following trauma, typically on the fingers, feet, and lips.6 Although benign, potential complications include ulceration and blood loss. Pyogenic granulomas can be treated via curettage and cautery, excision, cryosurgery, or pulsed dye laser.15
- Wankhade V, Singh R, Sadhwani V, et al. Eccrine poroma. Indian Dermatol Online J. 2015;6:304-305.
- Yorulmaz A, Aksoy GG, Ozhamam EU. A growing mass under the nail: subungual eccrine poroma. Skin Appendage Disord. 2020;6:254-257.
- Wang Y, Liu M, Zheng Y, et al. Eccrine poroma presented as spindleshaped plaque: a case report. Medicine (Baltimore). 2021;100:E25971. doi:10.1097/MD.0000000000025971
- Sharma M, Singh M, Gupta K, et al. Eccrine poroma of the eyelid. Indian J Ophthalmol. 2020;68:2522.
- Rasool MN, Hawary MB. Benign eccrine poroma in the palm of the hand. Ann Saudi Med. 2004;24:46-47.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms [published online April 2, 2014]. Int J Dermatol. 2014;53:1053-1061. doi:10.1111/ijd.12448
- López-Sánchez C, Ferguson P, Collgros H. Basal cell carcinoma of the palm: an unusual presentation of a common tumour [published online August 6, 2019]. Australas J Dermatol. 2020;61:69-70. doi:10.1111/ajd.13129
- Berk DR, Böer A, Bauschard FD, et al. Circumscribed acral hypokeratosis [published online April 6, 2007]. J Am Acad Dermatol. 2007;57:292-296. doi:10.1016/j.jaad.2007.02.022
- Majluf-Cáceres P, Vera-Kellet C, González-Bombardiere S. New dermoscopic keys for circumscribed acral hypokeratosis: report of four cases. Dermatol Pract Concept. 2021;11:E2021010. doi:10.5826/dpc.1102a10
- Simonart T, De Dobbeleer G, Stallenberg B. Classic Kaposi’s sarcoma of the palm in a metallurgist: role of iron filings in its development? Br J Dermatol. 2003;148:1061-1063. doi:10.1046/j.1365-2133.2003.05331.x
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Al Zolibani AA, Al Robaee AA. Primary palmoplantar Kaposi’s sarcoma: an unusual presentation. Skinmed. 2006;5:248-249. doi:10.1111/j.1540-9740.2006.04662.x
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Etemad SA, Dewan AK. Kaposi sarcoma updates [published online July 10, 2019]. Dermatol Clin. 2019;37:505-517. doi:10.1016/j. det.2019.05.008
- Murthy SC, Nagaraj A. Pyogenic granuloma. Indian Pediatr. 2012;49:855. doi:10.1007/s13312-012-0184-4
- Wankhade V, Singh R, Sadhwani V, et al. Eccrine poroma. Indian Dermatol Online J. 2015;6:304-305.
- Yorulmaz A, Aksoy GG, Ozhamam EU. A growing mass under the nail: subungual eccrine poroma. Skin Appendage Disord. 2020;6:254-257.
- Wang Y, Liu M, Zheng Y, et al. Eccrine poroma presented as spindleshaped plaque: a case report. Medicine (Baltimore). 2021;100:E25971. doi:10.1097/MD.0000000000025971
- Sharma M, Singh M, Gupta K, et al. Eccrine poroma of the eyelid. Indian J Ophthalmol. 2020;68:2522.
- Rasool MN, Hawary MB. Benign eccrine poroma in the palm of the hand. Ann Saudi Med. 2004;24:46-47.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms [published online April 2, 2014]. Int J Dermatol. 2014;53:1053-1061. doi:10.1111/ijd.12448
- López-Sánchez C, Ferguson P, Collgros H. Basal cell carcinoma of the palm: an unusual presentation of a common tumour [published online August 6, 2019]. Australas J Dermatol. 2020;61:69-70. doi:10.1111/ajd.13129
- Berk DR, Böer A, Bauschard FD, et al. Circumscribed acral hypokeratosis [published online April 6, 2007]. J Am Acad Dermatol. 2007;57:292-296. doi:10.1016/j.jaad.2007.02.022
- Majluf-Cáceres P, Vera-Kellet C, González-Bombardiere S. New dermoscopic keys for circumscribed acral hypokeratosis: report of four cases. Dermatol Pract Concept. 2021;11:E2021010. doi:10.5826/dpc.1102a10
- Simonart T, De Dobbeleer G, Stallenberg B. Classic Kaposi’s sarcoma of the palm in a metallurgist: role of iron filings in its development? Br J Dermatol. 2003;148:1061-1063. doi:10.1046/j.1365-2133.2003.05331.x
- Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294. doi:10.5858/arpa.2012-0101-RS
- Al Zolibani AA, Al Robaee AA. Primary palmoplantar Kaposi’s sarcoma: an unusual presentation. Skinmed. 2006;5:248-249. doi:10.1111/j.1540-9740.2006.04662.x
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Etemad SA, Dewan AK. Kaposi sarcoma updates [published online July 10, 2019]. Dermatol Clin. 2019;37:505-517. doi:10.1016/j. det.2019.05.008
- Murthy SC, Nagaraj A. Pyogenic granuloma. Indian Pediatr. 2012;49:855. doi:10.1007/s13312-012-0184-4
A 43-year-old woman presented with a painful lesion on the palm of 30 years’ duration that had grown in size. Physical examination revealed an oval, brown, lobulated plaque with a hyperkeratotic rim on the left palm. She reported bleeding and pain. A shallow cup-shaped depression was noted within the plaque. A 4-mm punch biopsy was performed.


Pruritic Photosensitive Rash
The Diagnosis: Actinic Prurigo
Actinic prurigo is an idiopathic photodermatosis triggered by UV exposure that primarily affects sun-exposed areas of the skin.1,2 It typically presents as pruritic papules, plaques, and nodules, with most patients also experiencing oral tingling and pain.3 In more severe cases, it can progress to include conjunctival disease, scarring, and cheilitis.1 A study of ocular findings among children with actinic pruritus reported that photophobia was one of the most important features,4 which was present in our patient. The face, especially over the zygomatic arches, nasal bridge, and lower lip, commonly is affected.1 Secondary lichenification or eczematization may occur.5 In our patient, the combination of conjunctivitis, cheilitis, and an eruption on sun-exposed skin were crucial in making the diagnosis.
Most cases present in patients younger than 10 years. It most commonly is seen in American Indians in North America, Central America, and South America.2 After the diagnosis was considered in our patient, the family was asked about their ancestry and confirmed that both of the patient’s maternal and paternal grandparents were of American Indian descent. There also is a strong genetic component; the HLA-DR4 allele variant is present in 90% of cases, especially DRB1*0407, which is seen in 60% of cases.1,6 In our patient, testing revealed HLA-DR4, DRB1*04 positivity. We further hypothesized that his mother’s photosensitive rash may have been actinic prurigo as opposed to polymorphous light eruption, which could explain the lack of response to hydroxychloroquine.
The diagnosis of actinic prurigo usually is made clinically. A skin biopsy typically is not necessary but would show hyperkeratosis, spongiosis, and acanthosis with a lymphocytic perivascular infiltrate. Biopsies of the lip classically show lymphoid germinal centers in the lamina propria, which can help distinguish actinic prurigo from polymorphous light eruption.1
In our patient, the differential diagnosis included polymorphous light eruption, connective tissue disease such as lupus erythematosus or dermatomyositis, porphyria such as erythropoietic protoporphyria, and allergic contact dermatitis. Polymorphous light eruption was ruled out by the oral and ocular manifestations, which are not atypical for this diagnosis. The patient’s laboratory results displayed unremarkable antinuclear antibodies, creatine kinase, aldolase, and extractable nuclear antigens, which made connective tissue disease unlikely. Furthermore, a porphyria screen for total plasma porphyrins and whole blood protoporphyrin was negative, which helped rule out porphyria. Allergic contact dermatitis seemed less likely given the lack of improvement with topical steroids. Overall, the clinical presentation in a patient with relevant family ancestry and HLA-DR4 positivity supported a diagnosis of actinic prurigo.7
To manage the condition in our patient, strict photoprotection was recommended as well as the application of triamcinolone ointment 0.025% to the affected areas twice daily until the skin symptoms improved. For acute flares, other treatment considerations include topical tacrolimus, oral antihistamines, and oral corticosteroids. Some success has been reported with cyclosporine and azathioprine. For severe disease, thalidomide is the recommended treatment; it is effective in pediatric patients at dosages of 50 to 100 mg daily, but the dose has not yet been standardized for this age group.8,9 Many adult patients initially are controlled with 100 to 200 mg daily, which can be tapered down to a dosage of 25 to 50 mg weekly with few adverse effects; however, the overall substantial side effects of thalidomide limit its use in both pediatric and adult populations.1,2 Newer studies have suggested promising results with dupilumab, especially when actinic prurigo presents with high IgE levels or eosinophils on histology.7,10 In our patient, the IgE level was normal.
- Pile HD, Crane JS. Actinic prurigo. StatPearls. StatPearls Publishing; 2022.
- Valbuena MC, Muvdi S, Lim HW. Actinic prurigo. Dermatol Clin. 2014;32:335-344, viii.
- Vega Memije ME, Cuevas Gonzalez JC, Hojyo-Tomoka MT, et al. Actinic prurigo as a hypersensitivity reaction type 4. Int J Dermatol. 2017;56:E135-E136.
- Magaña M, Mendez Y, Rodriguez A, et al. The conjunctivitis of solar (actinic) prurigo. Pediatr Dermatol. 2000;17:432-435.
- Ross G, Foley P, Baker C. Actinic prurigo. Photodermatol Photoimmunol Photomed. 2008;24:272-275.
- Rodríguez-Carreón AA, Rodríguez-Lobato E, Rodríguez-Gutiérrez G, et al. Actinic prurigo. Skinmed. 2015;13:287-295.
- Balwani M, Bloomer J, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic protoporphyria, autosomal recessive. GeneReviews. University of Washington; 1993.
- Crouch RB, Foley PA, Ng JCH, et al. Thalidomide experience of a major Australian teaching hospital. Australas J Dermatol. 2002;43:278-284.
- Watts-Santos A, Martinez-Rico JC, Gomez-Flores M, et al. Thalidomide: an option for the pediatric patient with actinic prurigo. Pediatr Dermatol. 2020;37:362-365.
- Eickstaedt JB, Starke S, Krakora D, et al. Clearance of pediatric actinic prurigo with dupilumab. Pediatr Dermatol. 2020;37:1176-1178.
The Diagnosis: Actinic Prurigo
Actinic prurigo is an idiopathic photodermatosis triggered by UV exposure that primarily affects sun-exposed areas of the skin.1,2 It typically presents as pruritic papules, plaques, and nodules, with most patients also experiencing oral tingling and pain.3 In more severe cases, it can progress to include conjunctival disease, scarring, and cheilitis.1 A study of ocular findings among children with actinic pruritus reported that photophobia was one of the most important features,4 which was present in our patient. The face, especially over the zygomatic arches, nasal bridge, and lower lip, commonly is affected.1 Secondary lichenification or eczematization may occur.5 In our patient, the combination of conjunctivitis, cheilitis, and an eruption on sun-exposed skin were crucial in making the diagnosis.
Most cases present in patients younger than 10 years. It most commonly is seen in American Indians in North America, Central America, and South America.2 After the diagnosis was considered in our patient, the family was asked about their ancestry and confirmed that both of the patient’s maternal and paternal grandparents were of American Indian descent. There also is a strong genetic component; the HLA-DR4 allele variant is present in 90% of cases, especially DRB1*0407, which is seen in 60% of cases.1,6 In our patient, testing revealed HLA-DR4, DRB1*04 positivity. We further hypothesized that his mother’s photosensitive rash may have been actinic prurigo as opposed to polymorphous light eruption, which could explain the lack of response to hydroxychloroquine.
The diagnosis of actinic prurigo usually is made clinically. A skin biopsy typically is not necessary but would show hyperkeratosis, spongiosis, and acanthosis with a lymphocytic perivascular infiltrate. Biopsies of the lip classically show lymphoid germinal centers in the lamina propria, which can help distinguish actinic prurigo from polymorphous light eruption.1
In our patient, the differential diagnosis included polymorphous light eruption, connective tissue disease such as lupus erythematosus or dermatomyositis, porphyria such as erythropoietic protoporphyria, and allergic contact dermatitis. Polymorphous light eruption was ruled out by the oral and ocular manifestations, which are not atypical for this diagnosis. The patient’s laboratory results displayed unremarkable antinuclear antibodies, creatine kinase, aldolase, and extractable nuclear antigens, which made connective tissue disease unlikely. Furthermore, a porphyria screen for total plasma porphyrins and whole blood protoporphyrin was negative, which helped rule out porphyria. Allergic contact dermatitis seemed less likely given the lack of improvement with topical steroids. Overall, the clinical presentation in a patient with relevant family ancestry and HLA-DR4 positivity supported a diagnosis of actinic prurigo.7
To manage the condition in our patient, strict photoprotection was recommended as well as the application of triamcinolone ointment 0.025% to the affected areas twice daily until the skin symptoms improved. For acute flares, other treatment considerations include topical tacrolimus, oral antihistamines, and oral corticosteroids. Some success has been reported with cyclosporine and azathioprine. For severe disease, thalidomide is the recommended treatment; it is effective in pediatric patients at dosages of 50 to 100 mg daily, but the dose has not yet been standardized for this age group.8,9 Many adult patients initially are controlled with 100 to 200 mg daily, which can be tapered down to a dosage of 25 to 50 mg weekly with few adverse effects; however, the overall substantial side effects of thalidomide limit its use in both pediatric and adult populations.1,2 Newer studies have suggested promising results with dupilumab, especially when actinic prurigo presents with high IgE levels or eosinophils on histology.7,10 In our patient, the IgE level was normal.
The Diagnosis: Actinic Prurigo
Actinic prurigo is an idiopathic photodermatosis triggered by UV exposure that primarily affects sun-exposed areas of the skin.1,2 It typically presents as pruritic papules, plaques, and nodules, with most patients also experiencing oral tingling and pain.3 In more severe cases, it can progress to include conjunctival disease, scarring, and cheilitis.1 A study of ocular findings among children with actinic pruritus reported that photophobia was one of the most important features,4 which was present in our patient. The face, especially over the zygomatic arches, nasal bridge, and lower lip, commonly is affected.1 Secondary lichenification or eczematization may occur.5 In our patient, the combination of conjunctivitis, cheilitis, and an eruption on sun-exposed skin were crucial in making the diagnosis.
Most cases present in patients younger than 10 years. It most commonly is seen in American Indians in North America, Central America, and South America.2 After the diagnosis was considered in our patient, the family was asked about their ancestry and confirmed that both of the patient’s maternal and paternal grandparents were of American Indian descent. There also is a strong genetic component; the HLA-DR4 allele variant is present in 90% of cases, especially DRB1*0407, which is seen in 60% of cases.1,6 In our patient, testing revealed HLA-DR4, DRB1*04 positivity. We further hypothesized that his mother’s photosensitive rash may have been actinic prurigo as opposed to polymorphous light eruption, which could explain the lack of response to hydroxychloroquine.
The diagnosis of actinic prurigo usually is made clinically. A skin biopsy typically is not necessary but would show hyperkeratosis, spongiosis, and acanthosis with a lymphocytic perivascular infiltrate. Biopsies of the lip classically show lymphoid germinal centers in the lamina propria, which can help distinguish actinic prurigo from polymorphous light eruption.1
In our patient, the differential diagnosis included polymorphous light eruption, connective tissue disease such as lupus erythematosus or dermatomyositis, porphyria such as erythropoietic protoporphyria, and allergic contact dermatitis. Polymorphous light eruption was ruled out by the oral and ocular manifestations, which are not atypical for this diagnosis. The patient’s laboratory results displayed unremarkable antinuclear antibodies, creatine kinase, aldolase, and extractable nuclear antigens, which made connective tissue disease unlikely. Furthermore, a porphyria screen for total plasma porphyrins and whole blood protoporphyrin was negative, which helped rule out porphyria. Allergic contact dermatitis seemed less likely given the lack of improvement with topical steroids. Overall, the clinical presentation in a patient with relevant family ancestry and HLA-DR4 positivity supported a diagnosis of actinic prurigo.7
To manage the condition in our patient, strict photoprotection was recommended as well as the application of triamcinolone ointment 0.025% to the affected areas twice daily until the skin symptoms improved. For acute flares, other treatment considerations include topical tacrolimus, oral antihistamines, and oral corticosteroids. Some success has been reported with cyclosporine and azathioprine. For severe disease, thalidomide is the recommended treatment; it is effective in pediatric patients at dosages of 50 to 100 mg daily, but the dose has not yet been standardized for this age group.8,9 Many adult patients initially are controlled with 100 to 200 mg daily, which can be tapered down to a dosage of 25 to 50 mg weekly with few adverse effects; however, the overall substantial side effects of thalidomide limit its use in both pediatric and adult populations.1,2 Newer studies have suggested promising results with dupilumab, especially when actinic prurigo presents with high IgE levels or eosinophils on histology.7,10 In our patient, the IgE level was normal.
- Pile HD, Crane JS. Actinic prurigo. StatPearls. StatPearls Publishing; 2022.
- Valbuena MC, Muvdi S, Lim HW. Actinic prurigo. Dermatol Clin. 2014;32:335-344, viii.
- Vega Memije ME, Cuevas Gonzalez JC, Hojyo-Tomoka MT, et al. Actinic prurigo as a hypersensitivity reaction type 4. Int J Dermatol. 2017;56:E135-E136.
- Magaña M, Mendez Y, Rodriguez A, et al. The conjunctivitis of solar (actinic) prurigo. Pediatr Dermatol. 2000;17:432-435.
- Ross G, Foley P, Baker C. Actinic prurigo. Photodermatol Photoimmunol Photomed. 2008;24:272-275.
- Rodríguez-Carreón AA, Rodríguez-Lobato E, Rodríguez-Gutiérrez G, et al. Actinic prurigo. Skinmed. 2015;13:287-295.
- Balwani M, Bloomer J, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic protoporphyria, autosomal recessive. GeneReviews. University of Washington; 1993.
- Crouch RB, Foley PA, Ng JCH, et al. Thalidomide experience of a major Australian teaching hospital. Australas J Dermatol. 2002;43:278-284.
- Watts-Santos A, Martinez-Rico JC, Gomez-Flores M, et al. Thalidomide: an option for the pediatric patient with actinic prurigo. Pediatr Dermatol. 2020;37:362-365.
- Eickstaedt JB, Starke S, Krakora D, et al. Clearance of pediatric actinic prurigo with dupilumab. Pediatr Dermatol. 2020;37:1176-1178.
- Pile HD, Crane JS. Actinic prurigo. StatPearls. StatPearls Publishing; 2022.
- Valbuena MC, Muvdi S, Lim HW. Actinic prurigo. Dermatol Clin. 2014;32:335-344, viii.
- Vega Memije ME, Cuevas Gonzalez JC, Hojyo-Tomoka MT, et al. Actinic prurigo as a hypersensitivity reaction type 4. Int J Dermatol. 2017;56:E135-E136.
- Magaña M, Mendez Y, Rodriguez A, et al. The conjunctivitis of solar (actinic) prurigo. Pediatr Dermatol. 2000;17:432-435.
- Ross G, Foley P, Baker C. Actinic prurigo. Photodermatol Photoimmunol Photomed. 2008;24:272-275.
- Rodríguez-Carreón AA, Rodríguez-Lobato E, Rodríguez-Gutiérrez G, et al. Actinic prurigo. Skinmed. 2015;13:287-295.
- Balwani M, Bloomer J, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic protoporphyria, autosomal recessive. GeneReviews. University of Washington; 1993.
- Crouch RB, Foley PA, Ng JCH, et al. Thalidomide experience of a major Australian teaching hospital. Australas J Dermatol. 2002;43:278-284.
- Watts-Santos A, Martinez-Rico JC, Gomez-Flores M, et al. Thalidomide: an option for the pediatric patient with actinic prurigo. Pediatr Dermatol. 2020;37:362-365.
- Eickstaedt JB, Starke S, Krakora D, et al. Clearance of pediatric actinic prurigo with dupilumab. Pediatr Dermatol. 2020;37:1176-1178.
A 6-year-old boy presented via telemedicine for evaluation of a recurring rash that first presented on the face 9 months prior to presentation and waxed and waned throughout the fall and winter seasons for about 5 months. His mother noted that on a warm and sunny day 5 months after its first appearance, the patient was at a dog park and developed the rash on the face and hands—the only areas that had been exposed to the sun—later that evening. The patient reported pruritus but no associated burning or stinging. He was evaluated by an allergist 1 month later and was treated with oral cefazolin and hydrocortisone ointment 2.5% for suspected impetiginized dermatitis without improvement. The rash persisted until evaluation by our clinic 2 months later. Photographs showed erythematous scaly plaques and papules scattered on the cheeks, nose, upper and lower lips, and vermilion borders, as well as the dorsal aspect of the hands. He also had conjunctival erythema, which his mother reported was particularly worse in the summer months and associated with photophobia. His mother also noted increased tear production when in the sun. There was no mucosal involvement. The patient had no notable medical history and was not taking any medications. His mother had a history of polymorphous light eruption that recently was treated with hydroxychloroquine but without benefit.


Papular Acneform Eruption With Mucositis
The Diagnosis: Syphilis
Histopathology revealed psoriasiform hyperplasia, endothelial cell swelling, and a brisk lichenoid inflammation with plasma cells (Figure, A). There also was pustular folliculitis in association with well-formed granulomatous inflammation and a prominent number of plasma cells (Figure, B). Treponema pallidum immunostaining showed numerous organisms in the epidermal and follicular epithelium. Rapid plasma reagin was found to be positive with a titer of 1:128. Evaluation for neurosyphilis through lumbar puncture was negative; the patient also was HIV negative. All of our patient’s skin lesions cleared after a 3-week course of weekly intramuscular benzathine G injections. Due to his substantial clinical improvement, the patient was subsequently lost to follow-up.

Syphilis, an infectious disease caused by the spirochete bacterium T pallidum, has a well-known natural history defined by various stages classically categorized as primary, secondary, latent, or late (tertiary).1 The classic lesion in primary syphilis is the chancre, a painless ulcer with raised borders that develops within approximately 3 weeks following the initial inoculation.2 Secondary syphilis manifests with mucocutaneous findings in up to 97% of patients, and untreated patients develop secondary syphilis at a rate of approximately 25%.3 Although mucocutaneous findings in secondary syphilis can vary widely, patients most commonly develop a diffuse maculopapular exanthem, and 40% develop mucosal findings including genital ulcers, mucous patches, and condylomata lata.1 In latent syphilis, there is seroreactivity, but otherwise there are no clinical symptoms. A clear symptomatic history of prior primary or secondary syphilis may be known or unknown. Latent syphilis is divided into early and late phases, and the World Health Organization designates 2 years after the first suspected exposure as the cutoff point for early and late latency.4 During the first 4 years of latent syphilis, patients may exhibit mucocutaneous relapses. Our patient denied any sexual activity for more than 3 years prior to presentation. Because of the start of iatrogenic immunosuppression during this period, this case was classified as late latent syphilis with mucocutaneous reactivation.
Behçet disease was included within the differential diagnosis but is characterized by multiorgan systemic vasculitis that causes various mucocutaneous findings including aphthous ulcers, papulopustular lesions, and genital ulcers.5 Histopathologic features are nonspecific, and the clinical finding of recurrent genital and oral ulceration should be present for diagnosis. This disease predominantly occurs in East Asian or Mediterranean populations and is otherwise rare in White individuals.
SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis) syndrome is a rare disorder consisting of skin, joint, and bone manifestations.6 Severe acne generally is accompanied by palmoplantar pustulosis along with pain and joint tenderness involving the anterior chest and axial skeleton, both of which were absent in our patient.
Pustular psoriasis can be localized or generalized. Localized presentations frequently are acral and may be associated with a variable degree of nail dystrophy and arthritis. Generalized presentations are characterized by hyperemic, well-defined patches with variable numbers of pustules.7 The pustules are the consequence of exuberate neutrophilic exocytosis into the epidermis and are nonfollicular.
Steroid-induced acne may be considered in the proper clinical setting of an acneform eruption with a prior history of systemic steroid treatment. However, additional findings of mucositis would not be expected, and although our patient was prescribed prednisone from his primary care physician prior to presentation to our clinic, this medication was given after the onset of the cutaneous eruption.
Syphilis commonly is referred to as the great mimicker due to its potential diverse morphologic presentations, which can involve acneform eruptions, though rare.8 In the setting of mucositis, generalized acneform eruptions should raise suspicion for the possibility of syphilis, even in the absence of other more classic cutaneous features.
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: historical aspects, microbiology, epidemiology, and clinical manifestations. J Am Acad Dermatol. 2020;82:1-14.
- Sparling PF. Natural history of syphilis. In: Holmes KK, Mardh PA, Sparling PF, et al, eds. Sexually Transmitted Diseases. McGraw Hill; 1990:213.
- Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: an epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
- Sule RR, Deshpande SG, Dharmadhikari NJ, et al. Late cutaneous syphilis. Cutis. 1997;59:135-137.
- Wilder EG, Frieder J, Sulhan S, et al. Spectrum of orocutaneous disease associations: genodermatoses and inflammatory conditions. J Am Acad Dermatol. 2017;77:809-830.
- Carneiro S, Sampaio-Barros PD. SAPHO syndrome. Rheum Dis Clin North Am. 2013;39:401-418.
- Bachelez H. Pustular psoriasis and related pustular skin diseases. Br J Dermatol. 2018;178:614-618.
- Domantay-Apostol GP, Handog EB, Gabriel MT. Syphilis: the international challenge of the great imitator. Dermatol Clin. 2008; 26:191-202, v. doi:10.1016/j.det.2007.12.001
The Diagnosis: Syphilis
Histopathology revealed psoriasiform hyperplasia, endothelial cell swelling, and a brisk lichenoid inflammation with plasma cells (Figure, A). There also was pustular folliculitis in association with well-formed granulomatous inflammation and a prominent number of plasma cells (Figure, B). Treponema pallidum immunostaining showed numerous organisms in the epidermal and follicular epithelium. Rapid plasma reagin was found to be positive with a titer of 1:128. Evaluation for neurosyphilis through lumbar puncture was negative; the patient also was HIV negative. All of our patient’s skin lesions cleared after a 3-week course of weekly intramuscular benzathine G injections. Due to his substantial clinical improvement, the patient was subsequently lost to follow-up.
Syphilis, an infectious disease caused by the spirochete bacterium T pallidum, has a well-known natural history defined by various stages classically categorized as primary, secondary, latent, or late (tertiary).1 The classic lesion in primary syphilis is the chancre, a painless ulcer with raised borders that develops within approximately 3 weeks following the initial inoculation.2 Secondary syphilis manifests with mucocutaneous findings in up to 97% of patients, and untreated patients develop secondary syphilis at a rate of approximately 25%.3 Although mucocutaneous findings in secondary syphilis can vary widely, patients most commonly develop a diffuse maculopapular exanthem, and 40% develop mucosal findings including genital ulcers, mucous patches, and condylomata lata.1 In latent syphilis, there is seroreactivity, but otherwise there are no clinical symptoms. A clear symptomatic history of prior primary or secondary syphilis may be known or unknown. Latent syphilis is divided into early and late phases, and the World Health Organization designates 2 years after the first suspected exposure as the cutoff point for early and late latency.4 During the first 4 years of latent syphilis, patients may exhibit mucocutaneous relapses. Our patient denied any sexual activity for more than 3 years prior to presentation. Because of the start of iatrogenic immunosuppression during this period, this case was classified as late latent syphilis with mucocutaneous reactivation.
Behçet disease was included within the differential diagnosis but is characterized by multiorgan systemic vasculitis that causes various mucocutaneous findings including aphthous ulcers, papulopustular lesions, and genital ulcers.5 Histopathologic features are nonspecific, and the clinical finding of recurrent genital and oral ulceration should be present for diagnosis. This disease predominantly occurs in East Asian or Mediterranean populations and is otherwise rare in White individuals.
SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis) syndrome is a rare disorder consisting of skin, joint, and bone manifestations.6 Severe acne generally is accompanied by palmoplantar pustulosis along with pain and joint tenderness involving the anterior chest and axial skeleton, both of which were absent in our patient.
Pustular psoriasis can be localized or generalized. Localized presentations frequently are acral and may be associated with a variable degree of nail dystrophy and arthritis. Generalized presentations are characterized by hyperemic, well-defined patches with variable numbers of pustules.7 The pustules are the consequence of exuberate neutrophilic exocytosis into the epidermis and are nonfollicular.
Steroid-induced acne may be considered in the proper clinical setting of an acneform eruption with a prior history of systemic steroid treatment. However, additional findings of mucositis would not be expected, and although our patient was prescribed prednisone from his primary care physician prior to presentation to our clinic, this medication was given after the onset of the cutaneous eruption.
Syphilis commonly is referred to as the great mimicker due to its potential diverse morphologic presentations, which can involve acneform eruptions, though rare.8 In the setting of mucositis, generalized acneform eruptions should raise suspicion for the possibility of syphilis, even in the absence of other more classic cutaneous features.
The Diagnosis: Syphilis
Histopathology revealed psoriasiform hyperplasia, endothelial cell swelling, and a brisk lichenoid inflammation with plasma cells (Figure, A). There also was pustular folliculitis in association with well-formed granulomatous inflammation and a prominent number of plasma cells (Figure, B). Treponema pallidum immunostaining showed numerous organisms in the epidermal and follicular epithelium. Rapid plasma reagin was found to be positive with a titer of 1:128. Evaluation for neurosyphilis through lumbar puncture was negative; the patient also was HIV negative. All of our patient’s skin lesions cleared after a 3-week course of weekly intramuscular benzathine G injections. Due to his substantial clinical improvement, the patient was subsequently lost to follow-up.
Syphilis, an infectious disease caused by the spirochete bacterium T pallidum, has a well-known natural history defined by various stages classically categorized as primary, secondary, latent, or late (tertiary).1 The classic lesion in primary syphilis is the chancre, a painless ulcer with raised borders that develops within approximately 3 weeks following the initial inoculation.2 Secondary syphilis manifests with mucocutaneous findings in up to 97% of patients, and untreated patients develop secondary syphilis at a rate of approximately 25%.3 Although mucocutaneous findings in secondary syphilis can vary widely, patients most commonly develop a diffuse maculopapular exanthem, and 40% develop mucosal findings including genital ulcers, mucous patches, and condylomata lata.1 In latent syphilis, there is seroreactivity, but otherwise there are no clinical symptoms. A clear symptomatic history of prior primary or secondary syphilis may be known or unknown. Latent syphilis is divided into early and late phases, and the World Health Organization designates 2 years after the first suspected exposure as the cutoff point for early and late latency.4 During the first 4 years of latent syphilis, patients may exhibit mucocutaneous relapses. Our patient denied any sexual activity for more than 3 years prior to presentation. Because of the start of iatrogenic immunosuppression during this period, this case was classified as late latent syphilis with mucocutaneous reactivation.
Behçet disease was included within the differential diagnosis but is characterized by multiorgan systemic vasculitis that causes various mucocutaneous findings including aphthous ulcers, papulopustular lesions, and genital ulcers.5 Histopathologic features are nonspecific, and the clinical finding of recurrent genital and oral ulceration should be present for diagnosis. This disease predominantly occurs in East Asian or Mediterranean populations and is otherwise rare in White individuals.
SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis) syndrome is a rare disorder consisting of skin, joint, and bone manifestations.6 Severe acne generally is accompanied by palmoplantar pustulosis along with pain and joint tenderness involving the anterior chest and axial skeleton, both of which were absent in our patient.
Pustular psoriasis can be localized or generalized. Localized presentations frequently are acral and may be associated with a variable degree of nail dystrophy and arthritis. Generalized presentations are characterized by hyperemic, well-defined patches with variable numbers of pustules.7 The pustules are the consequence of exuberate neutrophilic exocytosis into the epidermis and are nonfollicular.
Steroid-induced acne may be considered in the proper clinical setting of an acneform eruption with a prior history of systemic steroid treatment. However, additional findings of mucositis would not be expected, and although our patient was prescribed prednisone from his primary care physician prior to presentation to our clinic, this medication was given after the onset of the cutaneous eruption.
Syphilis commonly is referred to as the great mimicker due to its potential diverse morphologic presentations, which can involve acneform eruptions, though rare.8 In the setting of mucositis, generalized acneform eruptions should raise suspicion for the possibility of syphilis, even in the absence of other more classic cutaneous features.
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: historical aspects, microbiology, epidemiology, and clinical manifestations. J Am Acad Dermatol. 2020;82:1-14.
- Sparling PF. Natural history of syphilis. In: Holmes KK, Mardh PA, Sparling PF, et al, eds. Sexually Transmitted Diseases. McGraw Hill; 1990:213.
- Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: an epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
- Sule RR, Deshpande SG, Dharmadhikari NJ, et al. Late cutaneous syphilis. Cutis. 1997;59:135-137.
- Wilder EG, Frieder J, Sulhan S, et al. Spectrum of orocutaneous disease associations: genodermatoses and inflammatory conditions. J Am Acad Dermatol. 2017;77:809-830.
- Carneiro S, Sampaio-Barros PD. SAPHO syndrome. Rheum Dis Clin North Am. 2013;39:401-418.
- Bachelez H. Pustular psoriasis and related pustular skin diseases. Br J Dermatol. 2018;178:614-618.
- Domantay-Apostol GP, Handog EB, Gabriel MT. Syphilis: the international challenge of the great imitator. Dermatol Clin. 2008; 26:191-202, v. doi:10.1016/j.det.2007.12.001
- Forrestel AK, Kovarik CL, Katz KA. Sexually acquired syphilis: historical aspects, microbiology, epidemiology, and clinical manifestations. J Am Acad Dermatol. 2020;82:1-14.
- Sparling PF. Natural history of syphilis. In: Holmes KK, Mardh PA, Sparling PF, et al, eds. Sexually Transmitted Diseases. McGraw Hill; 1990:213.
- Clark EG, Danbolt N. The Oslo study of the natural course of untreated syphilis: an epidemiologic investigation based on a re-study of the Boeck-Bruusgaard material. Med Clin North Am. 1964;48:613.
- Sule RR, Deshpande SG, Dharmadhikari NJ, et al. Late cutaneous syphilis. Cutis. 1997;59:135-137.
- Wilder EG, Frieder J, Sulhan S, et al. Spectrum of orocutaneous disease associations: genodermatoses and inflammatory conditions. J Am Acad Dermatol. 2017;77:809-830.
- Carneiro S, Sampaio-Barros PD. SAPHO syndrome. Rheum Dis Clin North Am. 2013;39:401-418.
- Bachelez H. Pustular psoriasis and related pustular skin diseases. Br J Dermatol. 2018;178:614-618.
- Domantay-Apostol GP, Handog EB, Gabriel MT. Syphilis: the international challenge of the great imitator. Dermatol Clin. 2008; 26:191-202, v. doi:10.1016/j.det.2007.12.001
A 48-year-old man with a history of ulcerative colitis that was well-controlled with adalimumab presented with a generalized acneform eruption involving the face, chest (top) and back, as well as a well-defined ovoid ulcer on the anterior aspect of the tongue (bottom) of 2 months’ duration. Prior treatment with prednisone 60 mg daily for 14 days resulted in no improvement. He denied unintentional weight loss, cyclic fever, or arthritis. A complete blood cell count with differential showed mild anemia (hemoglobin, 11.6 g/dL [reference range, 13.2–16.6 g/dL]) with a differential cell count that was within reference range for each cell type. The erythrocyte sedimentation rate was elevated at 44 mm/h (reference range, 0–22 mm/h). A 4-mm punch biopsy specimen of an indurated cystic papule on the torso was obtained.


Persistent Wounds Refractory to Broad-Spectrum Antibiotics
The Diagnosis: PASH (Pyoderma Gangrenosum, Acne, Hidradenitis Suppurativa) Syndrome
Obtaining our patient’s history of hidradenitis suppurativa (HS), a hallmark sterile neutrophilic dermatosis, was key to making the correct diagnosis of PASH (pyoderma gangrenosum, acne, HS) syndrome. In our patient, the history of HS increased the consideration of pyoderma gangrenosum (PG) due to the persistent breast and leg wounds. Additionally, it was important to consider a diagnosis of PG in lesions that were not responding to broad-spectrum antimicrobial treatment. In our patient, the concurrent presentation of draining abscesses in the axillae (Figure, A) and inflammatory nodulocystic facial acne (Figure, B) were additional diagnostic clues that suggested the triad of PASH syndrome.

Although SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis) syndrome also can present with cutaneous features of acne and HS, the lack of bone and joint involvement in our patient made this diagnosis less likely. Calciphylaxis can present as ulcerations on the lower extremities, but it usually presents with a livedolike pattern with overlying black eschar and is unlikely in the absence of underlying metabolic or renal disease. PAPA (pyogenic arthritis, PG, acne) syndrome is characterized by recurrent joint involvement and lacks features of HS. Lastly, our patient was immunocompetent with no risk factors for mycobacterial infection.
PASH syndrome is a rare inherited syndrome, but its constituent inflammatory conditions are ubiquitous. They share a common underlying mechanism consisting of overactivation of the innate immune systems driven by increased production of the inflammatory cytokines IL-1, IL-17, and tumor necrosis factor α, resulting in sterile neutrophilic dermatoses.1 The diagnosis is based on the clinical presentation, as laboratory investigations are nondiagnostic. Biopsies and cultures can be performed to rule out infectious etiologies. Additionally, PASH syndrome is considered part of a larger spectrum of syndromes including PAPA and PAPASH (pyogenic arthritis, acne, PG, HS) syndromes. The absence of pyogenic arthritis distinguishes PASH syndrome from PAPA and PAPASH syndromes.2 Clinically, PASH syndrome and the related sterile neutrophilic dermatoses share the characteristic of pronounced cutaneous involvement that substantially alters the patient’s quality of life. Cigarette smoking is an exacerbating factor and has a well-established association with HS.3 Therefore, smoking cessation should be encouraged in these patients to avoid exacerbation of the disease process.
Maintaining adequate immunosuppression is key to managing the underlying disease processes. Classic immunosuppressive agents such as systemic glucocorticoids and methotrexate may fail to satisfactorily control the disease.4 Treatment options currently are somewhat limited and are aimed at targeting the inflammatory cytokines that propagate the disease. The most consistent responses have been observed with anti–tumor necrosis factor α antagonists such as adalimumab, infliximab, and etanercept.5 Additionally, there is varied response to anakinra, suggesting the importance of selectively targeting IL-1β.6 Unfortunately, misdiagnosis for an infectious etiology is common, and antibiotics and debridement are of limited use for the underlying pathophysiology of PASH syndrome. Importantly, biopsy and debridement often are discouraged due to the risk of pathergy.7
Our case demonstrates the importance of maintaining a high clinical suspicion for immune-mediated lesions that are refractory to antimicrobial agents. Additionally, prior history of multiple neutrophilic dermatoses should prompt consideration for the PASH/PAPA/PAPASH disease spectrum. Early and accurate identification of neutrophilic dermatoses such as PG and HS are crucial to initiating proper cytokine-targeting treatment and achieving disease remission.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Genovese G, Moltrasio C, Garcovich S, et al. PAPA spectrum disorders. G Ital Dermatol Venereol. 2020;155:542-550.
- König A, Lehmann C, Rompel R, et al. Cigarette smoking as a triggering factor of hidradenitis suppurativa. Dermatology. 1999;198:261-264.
- Ahn C, Negus D, Huang W. Pyoderma gangrenosum: a review of pathogenesis and treatment. Expert Rev Clin Immunol. 2018;14:225-233.
- Saint-Georges V, Peternel S, Kaštelan M, et al. Tumor necrosis factor antagonists in the treatment of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) syndrome. Acta Dermatovenerol Croat. 2018;26:173-178.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Patel DK, Locke M, Jarrett P. Pyoderma gangrenosum with pathergy: a potentially significant complication following breast reconstruction. J Plast Reconstr Aesthet Surg. 2017;70:884-892.
The Diagnosis: PASH (Pyoderma Gangrenosum, Acne, Hidradenitis Suppurativa) Syndrome
Obtaining our patient’s history of hidradenitis suppurativa (HS), a hallmark sterile neutrophilic dermatosis, was key to making the correct diagnosis of PASH (pyoderma gangrenosum, acne, HS) syndrome. In our patient, the history of HS increased the consideration of pyoderma gangrenosum (PG) due to the persistent breast and leg wounds. Additionally, it was important to consider a diagnosis of PG in lesions that were not responding to broad-spectrum antimicrobial treatment. In our patient, the concurrent presentation of draining abscesses in the axillae (Figure, A) and inflammatory nodulocystic facial acne (Figure, B) were additional diagnostic clues that suggested the triad of PASH syndrome.
Although SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis) syndrome also can present with cutaneous features of acne and HS, the lack of bone and joint involvement in our patient made this diagnosis less likely. Calciphylaxis can present as ulcerations on the lower extremities, but it usually presents with a livedolike pattern with overlying black eschar and is unlikely in the absence of underlying metabolic or renal disease. PAPA (pyogenic arthritis, PG, acne) syndrome is characterized by recurrent joint involvement and lacks features of HS. Lastly, our patient was immunocompetent with no risk factors for mycobacterial infection.
PASH syndrome is a rare inherited syndrome, but its constituent inflammatory conditions are ubiquitous. They share a common underlying mechanism consisting of overactivation of the innate immune systems driven by increased production of the inflammatory cytokines IL-1, IL-17, and tumor necrosis factor α, resulting in sterile neutrophilic dermatoses.1 The diagnosis is based on the clinical presentation, as laboratory investigations are nondiagnostic. Biopsies and cultures can be performed to rule out infectious etiologies. Additionally, PASH syndrome is considered part of a larger spectrum of syndromes including PAPA and PAPASH (pyogenic arthritis, acne, PG, HS) syndromes. The absence of pyogenic arthritis distinguishes PASH syndrome from PAPA and PAPASH syndromes.2 Clinically, PASH syndrome and the related sterile neutrophilic dermatoses share the characteristic of pronounced cutaneous involvement that substantially alters the patient’s quality of life. Cigarette smoking is an exacerbating factor and has a well-established association with HS.3 Therefore, smoking cessation should be encouraged in these patients to avoid exacerbation of the disease process.
Maintaining adequate immunosuppression is key to managing the underlying disease processes. Classic immunosuppressive agents such as systemic glucocorticoids and methotrexate may fail to satisfactorily control the disease.4 Treatment options currently are somewhat limited and are aimed at targeting the inflammatory cytokines that propagate the disease. The most consistent responses have been observed with anti–tumor necrosis factor α antagonists such as adalimumab, infliximab, and etanercept.5 Additionally, there is varied response to anakinra, suggesting the importance of selectively targeting IL-1β.6 Unfortunately, misdiagnosis for an infectious etiology is common, and antibiotics and debridement are of limited use for the underlying pathophysiology of PASH syndrome. Importantly, biopsy and debridement often are discouraged due to the risk of pathergy.7
Our case demonstrates the importance of maintaining a high clinical suspicion for immune-mediated lesions that are refractory to antimicrobial agents. Additionally, prior history of multiple neutrophilic dermatoses should prompt consideration for the PASH/PAPA/PAPASH disease spectrum. Early and accurate identification of neutrophilic dermatoses such as PG and HS are crucial to initiating proper cytokine-targeting treatment and achieving disease remission.
The Diagnosis: PASH (Pyoderma Gangrenosum, Acne, Hidradenitis Suppurativa) Syndrome
Obtaining our patient’s history of hidradenitis suppurativa (HS), a hallmark sterile neutrophilic dermatosis, was key to making the correct diagnosis of PASH (pyoderma gangrenosum, acne, HS) syndrome. In our patient, the history of HS increased the consideration of pyoderma gangrenosum (PG) due to the persistent breast and leg wounds. Additionally, it was important to consider a diagnosis of PG in lesions that were not responding to broad-spectrum antimicrobial treatment. In our patient, the concurrent presentation of draining abscesses in the axillae (Figure, A) and inflammatory nodulocystic facial acne (Figure, B) were additional diagnostic clues that suggested the triad of PASH syndrome.
Although SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis) syndrome also can present with cutaneous features of acne and HS, the lack of bone and joint involvement in our patient made this diagnosis less likely. Calciphylaxis can present as ulcerations on the lower extremities, but it usually presents with a livedolike pattern with overlying black eschar and is unlikely in the absence of underlying metabolic or renal disease. PAPA (pyogenic arthritis, PG, acne) syndrome is characterized by recurrent joint involvement and lacks features of HS. Lastly, our patient was immunocompetent with no risk factors for mycobacterial infection.
PASH syndrome is a rare inherited syndrome, but its constituent inflammatory conditions are ubiquitous. They share a common underlying mechanism consisting of overactivation of the innate immune systems driven by increased production of the inflammatory cytokines IL-1, IL-17, and tumor necrosis factor α, resulting in sterile neutrophilic dermatoses.1 The diagnosis is based on the clinical presentation, as laboratory investigations are nondiagnostic. Biopsies and cultures can be performed to rule out infectious etiologies. Additionally, PASH syndrome is considered part of a larger spectrum of syndromes including PAPA and PAPASH (pyogenic arthritis, acne, PG, HS) syndromes. The absence of pyogenic arthritis distinguishes PASH syndrome from PAPA and PAPASH syndromes.2 Clinically, PASH syndrome and the related sterile neutrophilic dermatoses share the characteristic of pronounced cutaneous involvement that substantially alters the patient’s quality of life. Cigarette smoking is an exacerbating factor and has a well-established association with HS.3 Therefore, smoking cessation should be encouraged in these patients to avoid exacerbation of the disease process.
Maintaining adequate immunosuppression is key to managing the underlying disease processes. Classic immunosuppressive agents such as systemic glucocorticoids and methotrexate may fail to satisfactorily control the disease.4 Treatment options currently are somewhat limited and are aimed at targeting the inflammatory cytokines that propagate the disease. The most consistent responses have been observed with anti–tumor necrosis factor α antagonists such as adalimumab, infliximab, and etanercept.5 Additionally, there is varied response to anakinra, suggesting the importance of selectively targeting IL-1β.6 Unfortunately, misdiagnosis for an infectious etiology is common, and antibiotics and debridement are of limited use for the underlying pathophysiology of PASH syndrome. Importantly, biopsy and debridement often are discouraged due to the risk of pathergy.7
Our case demonstrates the importance of maintaining a high clinical suspicion for immune-mediated lesions that are refractory to antimicrobial agents. Additionally, prior history of multiple neutrophilic dermatoses should prompt consideration for the PASH/PAPA/PAPASH disease spectrum. Early and accurate identification of neutrophilic dermatoses such as PG and HS are crucial to initiating proper cytokine-targeting treatment and achieving disease remission.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Genovese G, Moltrasio C, Garcovich S, et al. PAPA spectrum disorders. G Ital Dermatol Venereol. 2020;155:542-550.
- König A, Lehmann C, Rompel R, et al. Cigarette smoking as a triggering factor of hidradenitis suppurativa. Dermatology. 1999;198:261-264.
- Ahn C, Negus D, Huang W. Pyoderma gangrenosum: a review of pathogenesis and treatment. Expert Rev Clin Immunol. 2018;14:225-233.
- Saint-Georges V, Peternel S, Kaštelan M, et al. Tumor necrosis factor antagonists in the treatment of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) syndrome. Acta Dermatovenerol Croat. 2018;26:173-178.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Patel DK, Locke M, Jarrett P. Pyoderma gangrenosum with pathergy: a potentially significant complication following breast reconstruction. J Plast Reconstr Aesthet Surg. 2017;70:884-892.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Genovese G, Moltrasio C, Garcovich S, et al. PAPA spectrum disorders. G Ital Dermatol Venereol. 2020;155:542-550.
- König A, Lehmann C, Rompel R, et al. Cigarette smoking as a triggering factor of hidradenitis suppurativa. Dermatology. 1999;198:261-264.
- Ahn C, Negus D, Huang W. Pyoderma gangrenosum: a review of pathogenesis and treatment. Expert Rev Clin Immunol. 2018;14:225-233.
- Saint-Georges V, Peternel S, Kaštelan M, et al. Tumor necrosis factor antagonists in the treatment of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) syndrome. Acta Dermatovenerol Croat. 2018;26:173-178.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Patel DK, Locke M, Jarrett P. Pyoderma gangrenosum with pathergy: a potentially significant complication following breast reconstruction. J Plast Reconstr Aesthet Surg. 2017;70:884-892.
A 28-year-old Black woman presented to the hospital for evaluation of worsening leg wounds as well as a similar eroding plaque on the left breast of 1 month’s duration. Broad-spectrum antibiotics prescribed during a prior emergency department visit resulted in no improvement. Her medical history was notable for hidradenitis suppurativa that previously was well controlled on adalimumab prior to discontinuation 1 year prior. A review of systems was negative for fever, chills, shortness of breath, chest pain, night sweats, and arthralgia. The patient had discontinued the antibiotics and was not taking any other medications at the time of presentation. She reported a history of smoking cigarettes (5 pack years). Physical examination revealed hyperkeratotic eroded plaques with violaceous borders circumferentially around the left breast (top) and legs with notable undermining (bottom). Inflammatory nodulocystic acne of the face as well as sinus tract formation with purulent drainage in the axillae also were present. Laboratory workup revealed an elevated erythrocyte sedimentation rate (116 mm/h [reference range, <20 mm/h]). Computed tomography of the leg wound was negative for soft-tissue infection. Aerobic and anaerobic tissue cultures demonstrated no growth.

