User login
Sustained Control with Investigational Monoclonal Antibody for Myasthenia Gravis
SAVANNAH, GEORGIA – , according to topline results from the phase 3 VIVACITY-MG3 study.
The VIVACITY-MG3 trial is the first registrational study of a neonatal fragment crystallizable receptor (FcRn) blocker to show sustained efficacy through 6 months of fixed schedule dosing.
Lead investigator Tuan Vu, MD, professor of neurology at the University of South Florida in Tampa, presented the findings at the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) 2024.
Autoantibody Depletion
FcRN plays a crucial role in the transport of immunoglobulin G. Blocking it can reduce circulating immunoglobulin G antibodies, including pathogenic gMG autoantibodies.
The double-blind, placebo-controlled trial included 196 adults with a broad range of seropositive gMG – who account for approximately 95% of the gMG patient population – and 42 seronegative patients.
The mean age was 52 years, 92% were female, and 63% were White. The mean disease duration was about 8 years. Among seropositive patients, 87.6% were acetylcholine receptor autoantibody-positive (AChR+), 10.5% were muscle-specific kinase autoantibody-positive (MuSK+), and 2% were low-density lipoprotein receptor-related protein 4 antibody positive.
They were randomly assigned 1:1 to receive either nipocalimab IV plus standard of care, or placebo plus standard of care for 24 weeks. A total of 87 patients in the nipocalimab arm and 82 in the placebo arm completed the study.
The primary efficacy endpoint was the Myasthenia Gravis Activities of Daily Living (MG-ADL) score. Participants treated with nipocalimab demonstrated a statistically significant improvement of 4.70 points from baseline, compared to the 3.25-point improvement in those treated with placebo (P =.002).
Clinically Meaningful Changes?
“For someone living with gMG, a 1 to 2-point improvement on MG-ADL may be the difference between normal eating and frequent choking on food, or shortness of breath at rest and being on a ventilator,” the drug’s manufacturer noted in a release.
Secondary endpoints were also better in the nipocalimab group, compared with participants on placebo. Specifically, on the 13-item clinician assessed Quantitative Myasthenia Gravis disease severity score, patients who received nipocalimab had an average reduction of 4.86 points from baseline compared to a reduction of 2.05 points in the placebo arm (P <.001).
Similarly, MG-ADL response (defined as ≥ 2-point improvement from baseline) was significantly greater in the nipocalimab versus placebo arms (68.8% vs 52.6%; P =.021).
Subgroup analysis revealed similar results for the different types of seropositive patients, but there was no statistically significant difference in results for seronegative patients treated with nipocalimab versus placebo.
“The drug was pretty well tolerated and there was little difference, other than more patients with muscle spasm in the nipocalimab group (12.2% vs 3.1%),” said Vu.
In addition, peripheral edema occurred in 11.2% of the nipocalimab group and none of the placebo-treated patients. Cholesterol levels were also higher in the nipocalimab arm, but there were no cardiac side effects, he added.
Encouraging Findings
Commenting on the findings, Neelam Goyal, MD, clinical professor of neurology and neurological sciences at Stanford University School of Medicine in Palo Alto, California, was encouraged.
“It’s a phase 3 trial, it’s positive, which is great, so it’ll be another drug on the market, another option for our patients,” she said. However, she cautioned, “their placebo arm did better than most placebos, so I think the delta is not as robust, but it was still statistically significant.”
Goyal noted that, if approved, nipocalimab will be the third FcRn inhibitor in the MG field, preceded by efgartigimod (Vyvgart), which is approved for AChR antibody-positive disease, and rozanolixizumab-noli (Rystiggo) which is approved for both for AChR and MUSK antibody positive disease.
“Its target of action is similar to the two drugs that are already on the market, but one thing that is unique about nipocalimab is that it is continuous dosing versus the other two medications that are given cyclically,” she said.
“The reason that’s an upside, is that with cyclical dosing, patients have a return of symptoms. We treat, they get better, and then they get worse. That’s very disconcerting to patients. So, they want to be treated continuously.”
Additionally, she said there are some early data suggesting its safety in pregnancy.
Vu disclosed he is the USF Site Principal Investigator for MG clinical trials sponsored by Alexion/ AstraZeneca Rare Disease, Amgen, argenx, Cartesian Therapeutics, COUR Pharmaceuticals, Dianthus Therapeutics, Immunovant, Johnson & Johnson, NMD Pharmaceuticals, Regeneron Pharmaceuticals, and UCB, and has served as a speaker for Alexion/AstraZeneca Rare Disease, argenx, and CSL Behring. He performs consulting work for Alexion/AstraZeneca Rare Disease, argenx, Dianthus Therapeutics, ImmunAbs, and UCB. Goyal disclosed consultant, advisory or grant support from argenx, UCB, Alexion, and Janssen. The study was funded by Janssen.
A version of this article appeared on Medscape.com.
SAVANNAH, GEORGIA – , according to topline results from the phase 3 VIVACITY-MG3 study.
The VIVACITY-MG3 trial is the first registrational study of a neonatal fragment crystallizable receptor (FcRn) blocker to show sustained efficacy through 6 months of fixed schedule dosing.
Lead investigator Tuan Vu, MD, professor of neurology at the University of South Florida in Tampa, presented the findings at the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) 2024.
Autoantibody Depletion
FcRN plays a crucial role in the transport of immunoglobulin G. Blocking it can reduce circulating immunoglobulin G antibodies, including pathogenic gMG autoantibodies.
The double-blind, placebo-controlled trial included 196 adults with a broad range of seropositive gMG – who account for approximately 95% of the gMG patient population – and 42 seronegative patients.
The mean age was 52 years, 92% were female, and 63% were White. The mean disease duration was about 8 years. Among seropositive patients, 87.6% were acetylcholine receptor autoantibody-positive (AChR+), 10.5% were muscle-specific kinase autoantibody-positive (MuSK+), and 2% were low-density lipoprotein receptor-related protein 4 antibody positive.
They were randomly assigned 1:1 to receive either nipocalimab IV plus standard of care, or placebo plus standard of care for 24 weeks. A total of 87 patients in the nipocalimab arm and 82 in the placebo arm completed the study.
The primary efficacy endpoint was the Myasthenia Gravis Activities of Daily Living (MG-ADL) score. Participants treated with nipocalimab demonstrated a statistically significant improvement of 4.70 points from baseline, compared to the 3.25-point improvement in those treated with placebo (P =.002).
Clinically Meaningful Changes?
“For someone living with gMG, a 1 to 2-point improvement on MG-ADL may be the difference between normal eating and frequent choking on food, or shortness of breath at rest and being on a ventilator,” the drug’s manufacturer noted in a release.
Secondary endpoints were also better in the nipocalimab group, compared with participants on placebo. Specifically, on the 13-item clinician assessed Quantitative Myasthenia Gravis disease severity score, patients who received nipocalimab had an average reduction of 4.86 points from baseline compared to a reduction of 2.05 points in the placebo arm (P <.001).
Similarly, MG-ADL response (defined as ≥ 2-point improvement from baseline) was significantly greater in the nipocalimab versus placebo arms (68.8% vs 52.6%; P =.021).
Subgroup analysis revealed similar results for the different types of seropositive patients, but there was no statistically significant difference in results for seronegative patients treated with nipocalimab versus placebo.
“The drug was pretty well tolerated and there was little difference, other than more patients with muscle spasm in the nipocalimab group (12.2% vs 3.1%),” said Vu.
In addition, peripheral edema occurred in 11.2% of the nipocalimab group and none of the placebo-treated patients. Cholesterol levels were also higher in the nipocalimab arm, but there were no cardiac side effects, he added.
Encouraging Findings
Commenting on the findings, Neelam Goyal, MD, clinical professor of neurology and neurological sciences at Stanford University School of Medicine in Palo Alto, California, was encouraged.
“It’s a phase 3 trial, it’s positive, which is great, so it’ll be another drug on the market, another option for our patients,” she said. However, she cautioned, “their placebo arm did better than most placebos, so I think the delta is not as robust, but it was still statistically significant.”
Goyal noted that, if approved, nipocalimab will be the third FcRn inhibitor in the MG field, preceded by efgartigimod (Vyvgart), which is approved for AChR antibody-positive disease, and rozanolixizumab-noli (Rystiggo) which is approved for both for AChR and MUSK antibody positive disease.
“Its target of action is similar to the two drugs that are already on the market, but one thing that is unique about nipocalimab is that it is continuous dosing versus the other two medications that are given cyclically,” she said.
“The reason that’s an upside, is that with cyclical dosing, patients have a return of symptoms. We treat, they get better, and then they get worse. That’s very disconcerting to patients. So, they want to be treated continuously.”
Additionally, she said there are some early data suggesting its safety in pregnancy.
Vu disclosed he is the USF Site Principal Investigator for MG clinical trials sponsored by Alexion/ AstraZeneca Rare Disease, Amgen, argenx, Cartesian Therapeutics, COUR Pharmaceuticals, Dianthus Therapeutics, Immunovant, Johnson & Johnson, NMD Pharmaceuticals, Regeneron Pharmaceuticals, and UCB, and has served as a speaker for Alexion/AstraZeneca Rare Disease, argenx, and CSL Behring. He performs consulting work for Alexion/AstraZeneca Rare Disease, argenx, Dianthus Therapeutics, ImmunAbs, and UCB. Goyal disclosed consultant, advisory or grant support from argenx, UCB, Alexion, and Janssen. The study was funded by Janssen.
A version of this article appeared on Medscape.com.
SAVANNAH, GEORGIA – , according to topline results from the phase 3 VIVACITY-MG3 study.
The VIVACITY-MG3 trial is the first registrational study of a neonatal fragment crystallizable receptor (FcRn) blocker to show sustained efficacy through 6 months of fixed schedule dosing.
Lead investigator Tuan Vu, MD, professor of neurology at the University of South Florida in Tampa, presented the findings at the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) 2024.
Autoantibody Depletion
FcRN plays a crucial role in the transport of immunoglobulin G. Blocking it can reduce circulating immunoglobulin G antibodies, including pathogenic gMG autoantibodies.
The double-blind, placebo-controlled trial included 196 adults with a broad range of seropositive gMG – who account for approximately 95% of the gMG patient population – and 42 seronegative patients.
The mean age was 52 years, 92% were female, and 63% were White. The mean disease duration was about 8 years. Among seropositive patients, 87.6% were acetylcholine receptor autoantibody-positive (AChR+), 10.5% were muscle-specific kinase autoantibody-positive (MuSK+), and 2% were low-density lipoprotein receptor-related protein 4 antibody positive.
They were randomly assigned 1:1 to receive either nipocalimab IV plus standard of care, or placebo plus standard of care for 24 weeks. A total of 87 patients in the nipocalimab arm and 82 in the placebo arm completed the study.
The primary efficacy endpoint was the Myasthenia Gravis Activities of Daily Living (MG-ADL) score. Participants treated with nipocalimab demonstrated a statistically significant improvement of 4.70 points from baseline, compared to the 3.25-point improvement in those treated with placebo (P =.002).
Clinically Meaningful Changes?
“For someone living with gMG, a 1 to 2-point improvement on MG-ADL may be the difference between normal eating and frequent choking on food, or shortness of breath at rest and being on a ventilator,” the drug’s manufacturer noted in a release.
Secondary endpoints were also better in the nipocalimab group, compared with participants on placebo. Specifically, on the 13-item clinician assessed Quantitative Myasthenia Gravis disease severity score, patients who received nipocalimab had an average reduction of 4.86 points from baseline compared to a reduction of 2.05 points in the placebo arm (P <.001).
Similarly, MG-ADL response (defined as ≥ 2-point improvement from baseline) was significantly greater in the nipocalimab versus placebo arms (68.8% vs 52.6%; P =.021).
Subgroup analysis revealed similar results for the different types of seropositive patients, but there was no statistically significant difference in results for seronegative patients treated with nipocalimab versus placebo.
“The drug was pretty well tolerated and there was little difference, other than more patients with muscle spasm in the nipocalimab group (12.2% vs 3.1%),” said Vu.
In addition, peripheral edema occurred in 11.2% of the nipocalimab group and none of the placebo-treated patients. Cholesterol levels were also higher in the nipocalimab arm, but there were no cardiac side effects, he added.
Encouraging Findings
Commenting on the findings, Neelam Goyal, MD, clinical professor of neurology and neurological sciences at Stanford University School of Medicine in Palo Alto, California, was encouraged.
“It’s a phase 3 trial, it’s positive, which is great, so it’ll be another drug on the market, another option for our patients,” she said. However, she cautioned, “their placebo arm did better than most placebos, so I think the delta is not as robust, but it was still statistically significant.”
Goyal noted that, if approved, nipocalimab will be the third FcRn inhibitor in the MG field, preceded by efgartigimod (Vyvgart), which is approved for AChR antibody-positive disease, and rozanolixizumab-noli (Rystiggo) which is approved for both for AChR and MUSK antibody positive disease.
“Its target of action is similar to the two drugs that are already on the market, but one thing that is unique about nipocalimab is that it is continuous dosing versus the other two medications that are given cyclically,” she said.
“The reason that’s an upside, is that with cyclical dosing, patients have a return of symptoms. We treat, they get better, and then they get worse. That’s very disconcerting to patients. So, they want to be treated continuously.”
Additionally, she said there are some early data suggesting its safety in pregnancy.
Vu disclosed he is the USF Site Principal Investigator for MG clinical trials sponsored by Alexion/ AstraZeneca Rare Disease, Amgen, argenx, Cartesian Therapeutics, COUR Pharmaceuticals, Dianthus Therapeutics, Immunovant, Johnson & Johnson, NMD Pharmaceuticals, Regeneron Pharmaceuticals, and UCB, and has served as a speaker for Alexion/AstraZeneca Rare Disease, argenx, and CSL Behring. He performs consulting work for Alexion/AstraZeneca Rare Disease, argenx, Dianthus Therapeutics, ImmunAbs, and UCB. Goyal disclosed consultant, advisory or grant support from argenx, UCB, Alexion, and Janssen. The study was funded by Janssen.
A version of this article appeared on Medscape.com.
FROM AANEM 2024
First-in-Class B-Cell Depleting Agent Promising for Myasthenia Gravis
SAVANNAH, GEORGIA — , new phase 3 data showed.
“Based on these results, we have demonstrated that targeting B cells, including the antibody-secreting cells, is beneficial, and there is likely a role for this kind of therapeutic strategy for patients with myasthenia gravis,” said senior investigator Richard Nowak, MD.
The findings were published and presented at the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) 2024.
Largest Cohort of Muscle-Specific Kinase (MuSK) Antibody–Positive Disease
The Myasthenia Gravis INebilizumab Trial study enrolled 238 participants, 60.8% women, mean age 47.5 years, from 79 sites in 18 countries. The participants were divided into two cohorts: 190 acetylcholine receptor (AChR) autoantibody–positive patients and 48 MuSK autoantibody–positive patients.
“This is the largest enrolled cohort of MuSK antibody–positive disease in a placebo-controlled trial to date,” said Nowak, director of the Yale Myasthenia Gravis Clinic and associate professor of neurology at Yale School of Medicine, in New Haven, Connecticut.
Both groups had similar gMG duration (mean 6.7 and 5.2 years for AChR+ and MuSK+ patients, respectively) and disease severity based on Myasthenia Gravis Activities of Daily Living (MG-ADL) and Quantitative Myasthenia Gravis (QMG) baseline score. In addition, more than 80% of participants were on a prednisone equivalent dose greater than 5 mg daily at study entry.
Participants were randomly assigned to receive intravenous (IV) inebilizumab or IV placebo for 52 weeks (AChR+ group) or 26 weeks (MuSK+ group). In addition, study participants who were taking corticosteroids were tapered down starting at week 4 to prednisone 5 mg per day by week 24.
The trial met its primary endpoint, with a statistically significant change from baseline in MG-ADL and with a reduction of 4.2 points for inebilizumab versus 2.2 for placebo (P < .0001) at week 26 for the combined study population.
“You can see that the trend is actually going toward separation of the two groups after week 8 in the combined population,” said Nowak. Key secondary endpoints also showed statistically significant and clinically meaningful change from baseline compared with placebo.
This included a statistically significant change in QMG score inebilizumab compared with placebo for the combined population, a reduction of 4.8 versus 2.3 points, respectively, at week 26 (P = .0002).
In addition, both MG-ADL and QMG scores in the AChR+ subgroup were superior for inebilizumab versus placebo at week 26, with reductions of 4.2 versus 2.4, and 4.4 versus 2.0; P = .0015 and P = .0011, respectively.
In the MuSK+ subgroup, inebilizumab-treated patients had better MG-ADL scores than placebo-treated patients, with reductions of 3.9 versus 1.7 points, respectively, at week 26, although this difference did not meet statistical significance.
“There were no increased safety incidents in the inebilizumab-treated patients versus placebo, and a similar percentage of safety incidents in the AChR–positive and MuSK–positive groups. There were three deaths reported, all likely related to myasthenic crisis,” he said.
Nowak said that inebilizumab is “unique from the other currently FDA-approved medications for myasthenia gravis in that it’s targeting the upstream immunopathogenic mechanism of disease, specifically B cells — and B cells that are actually antibody-secreting cells.”
“It is targeting the factories of autoantibody production, whereas an FcRn antagonist, for example, is not targeting those factories but rather targeting what’s being produced — the immunoglobulins, IgGs in general,” he added.
Nowak said that what is particularly exciting about the drug is that the schedule is not very frequent. It begins with an initial IV infusion, followed by a second infusion 2 weeks later and a third infusion 6 months after that, so that patients are treated approximately every 6 months. This is in contrast to some other targeted therapies, where failing to address the underlying factors driving immunopathogenesis necessitates more regular and frequent medication administration.
New, Novel, Exciting
Commenting on the research, Neelam Goyal, MD, who chaired the session, said, “It’s definitely new, novel, interesting, exciting.”
Goyal, clinical professor of neurology and neurological sciences at Stanford University School of Medicine in Palo Alto, California, also noted that while B-cell depletion has shown some previous success in MG, it was with rituximab, a CD20 B-cell depleting agent.
She noted that unlike rituximab, which targets CD20, inebilizumab targets CD19, although both medications lead to B-cell depletion. Rituximab has proven effective for MUSK–positive MG, which accounts for approximately 5% of cases.
However, Goyal noted that the results for AChR–positive MG have been mixed — “the BeatMG trial was negative and the RINOMAX trial was positive. So, I think this is really interesting. It is exciting, and this drug is already on the market.”
She added that although inebilizumab is already US Food and Drug Administration–approved for the treatment of neuromyelitis optica, it still faces approval and indication hurdles for MG.
The future of this drug in the management algorithm for MG remains uncertain. Goyal noted that it’s “quite costly,” and although its benefits are evident — particularly for FcRn and complement inhibitors — some early data from chimeric antigen receptor T-cell therapy studies appear significantly more impressive.
Nowak disclosed research support from the National Institutes of Health, Genentech, Alexion Pharmaceuticals, argenx, Annexon Biosciences, Ra Pharmaceuticals (now UCB S.A.), the Myasthenia Gravis Foundation of America, Momenta Pharmaceuticals (now Janssen), Immunovant, Grifols, S.A., and Viela Bio, Horizon Therapeutics (now Amgen). Served as a consultant and advisor for Alexion Pharmaceuticals, argenx, Cabaletta Bio, Cour Pharmaceuticals, Ra Pharmaceuticals (now UCB S.A.), Immunovant, Momenta Pharmaceuticals (now Janssen), and Viela Bio (Horizon Therapeutics, now Amgen).
Goyal disclosed consultant, advisory, or grant support from argenx, UCB, Alexion, and Janssen. The study was funded by Amgen.
A version of this article appeared on Medscape.com.
SAVANNAH, GEORGIA — , new phase 3 data showed.
“Based on these results, we have demonstrated that targeting B cells, including the antibody-secreting cells, is beneficial, and there is likely a role for this kind of therapeutic strategy for patients with myasthenia gravis,” said senior investigator Richard Nowak, MD.
The findings were published and presented at the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) 2024.
Largest Cohort of Muscle-Specific Kinase (MuSK) Antibody–Positive Disease
The Myasthenia Gravis INebilizumab Trial study enrolled 238 participants, 60.8% women, mean age 47.5 years, from 79 sites in 18 countries. The participants were divided into two cohorts: 190 acetylcholine receptor (AChR) autoantibody–positive patients and 48 MuSK autoantibody–positive patients.
“This is the largest enrolled cohort of MuSK antibody–positive disease in a placebo-controlled trial to date,” said Nowak, director of the Yale Myasthenia Gravis Clinic and associate professor of neurology at Yale School of Medicine, in New Haven, Connecticut.
Both groups had similar gMG duration (mean 6.7 and 5.2 years for AChR+ and MuSK+ patients, respectively) and disease severity based on Myasthenia Gravis Activities of Daily Living (MG-ADL) and Quantitative Myasthenia Gravis (QMG) baseline score. In addition, more than 80% of participants were on a prednisone equivalent dose greater than 5 mg daily at study entry.
Participants were randomly assigned to receive intravenous (IV) inebilizumab or IV placebo for 52 weeks (AChR+ group) or 26 weeks (MuSK+ group). In addition, study participants who were taking corticosteroids were tapered down starting at week 4 to prednisone 5 mg per day by week 24.
The trial met its primary endpoint, with a statistically significant change from baseline in MG-ADL and with a reduction of 4.2 points for inebilizumab versus 2.2 for placebo (P < .0001) at week 26 for the combined study population.
“You can see that the trend is actually going toward separation of the two groups after week 8 in the combined population,” said Nowak. Key secondary endpoints also showed statistically significant and clinically meaningful change from baseline compared with placebo.
This included a statistically significant change in QMG score inebilizumab compared with placebo for the combined population, a reduction of 4.8 versus 2.3 points, respectively, at week 26 (P = .0002).
In addition, both MG-ADL and QMG scores in the AChR+ subgroup were superior for inebilizumab versus placebo at week 26, with reductions of 4.2 versus 2.4, and 4.4 versus 2.0; P = .0015 and P = .0011, respectively.
In the MuSK+ subgroup, inebilizumab-treated patients had better MG-ADL scores than placebo-treated patients, with reductions of 3.9 versus 1.7 points, respectively, at week 26, although this difference did not meet statistical significance.
“There were no increased safety incidents in the inebilizumab-treated patients versus placebo, and a similar percentage of safety incidents in the AChR–positive and MuSK–positive groups. There were three deaths reported, all likely related to myasthenic crisis,” he said.
Nowak said that inebilizumab is “unique from the other currently FDA-approved medications for myasthenia gravis in that it’s targeting the upstream immunopathogenic mechanism of disease, specifically B cells — and B cells that are actually antibody-secreting cells.”
“It is targeting the factories of autoantibody production, whereas an FcRn antagonist, for example, is not targeting those factories but rather targeting what’s being produced — the immunoglobulins, IgGs in general,” he added.
Nowak said that what is particularly exciting about the drug is that the schedule is not very frequent. It begins with an initial IV infusion, followed by a second infusion 2 weeks later and a third infusion 6 months after that, so that patients are treated approximately every 6 months. This is in contrast to some other targeted therapies, where failing to address the underlying factors driving immunopathogenesis necessitates more regular and frequent medication administration.
New, Novel, Exciting
Commenting on the research, Neelam Goyal, MD, who chaired the session, said, “It’s definitely new, novel, interesting, exciting.”
Goyal, clinical professor of neurology and neurological sciences at Stanford University School of Medicine in Palo Alto, California, also noted that while B-cell depletion has shown some previous success in MG, it was with rituximab, a CD20 B-cell depleting agent.
She noted that unlike rituximab, which targets CD20, inebilizumab targets CD19, although both medications lead to B-cell depletion. Rituximab has proven effective for MUSK–positive MG, which accounts for approximately 5% of cases.
However, Goyal noted that the results for AChR–positive MG have been mixed — “the BeatMG trial was negative and the RINOMAX trial was positive. So, I think this is really interesting. It is exciting, and this drug is already on the market.”
She added that although inebilizumab is already US Food and Drug Administration–approved for the treatment of neuromyelitis optica, it still faces approval and indication hurdles for MG.
The future of this drug in the management algorithm for MG remains uncertain. Goyal noted that it’s “quite costly,” and although its benefits are evident — particularly for FcRn and complement inhibitors — some early data from chimeric antigen receptor T-cell therapy studies appear significantly more impressive.
Nowak disclosed research support from the National Institutes of Health, Genentech, Alexion Pharmaceuticals, argenx, Annexon Biosciences, Ra Pharmaceuticals (now UCB S.A.), the Myasthenia Gravis Foundation of America, Momenta Pharmaceuticals (now Janssen), Immunovant, Grifols, S.A., and Viela Bio, Horizon Therapeutics (now Amgen). Served as a consultant and advisor for Alexion Pharmaceuticals, argenx, Cabaletta Bio, Cour Pharmaceuticals, Ra Pharmaceuticals (now UCB S.A.), Immunovant, Momenta Pharmaceuticals (now Janssen), and Viela Bio (Horizon Therapeutics, now Amgen).
Goyal disclosed consultant, advisory, or grant support from argenx, UCB, Alexion, and Janssen. The study was funded by Amgen.
A version of this article appeared on Medscape.com.
SAVANNAH, GEORGIA — , new phase 3 data showed.
“Based on these results, we have demonstrated that targeting B cells, including the antibody-secreting cells, is beneficial, and there is likely a role for this kind of therapeutic strategy for patients with myasthenia gravis,” said senior investigator Richard Nowak, MD.
The findings were published and presented at the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM) 2024.
Largest Cohort of Muscle-Specific Kinase (MuSK) Antibody–Positive Disease
The Myasthenia Gravis INebilizumab Trial study enrolled 238 participants, 60.8% women, mean age 47.5 years, from 79 sites in 18 countries. The participants were divided into two cohorts: 190 acetylcholine receptor (AChR) autoantibody–positive patients and 48 MuSK autoantibody–positive patients.
“This is the largest enrolled cohort of MuSK antibody–positive disease in a placebo-controlled trial to date,” said Nowak, director of the Yale Myasthenia Gravis Clinic and associate professor of neurology at Yale School of Medicine, in New Haven, Connecticut.
Both groups had similar gMG duration (mean 6.7 and 5.2 years for AChR+ and MuSK+ patients, respectively) and disease severity based on Myasthenia Gravis Activities of Daily Living (MG-ADL) and Quantitative Myasthenia Gravis (QMG) baseline score. In addition, more than 80% of participants were on a prednisone equivalent dose greater than 5 mg daily at study entry.
Participants were randomly assigned to receive intravenous (IV) inebilizumab or IV placebo for 52 weeks (AChR+ group) or 26 weeks (MuSK+ group). In addition, study participants who were taking corticosteroids were tapered down starting at week 4 to prednisone 5 mg per day by week 24.
The trial met its primary endpoint, with a statistically significant change from baseline in MG-ADL and with a reduction of 4.2 points for inebilizumab versus 2.2 for placebo (P < .0001) at week 26 for the combined study population.
“You can see that the trend is actually going toward separation of the two groups after week 8 in the combined population,” said Nowak. Key secondary endpoints also showed statistically significant and clinically meaningful change from baseline compared with placebo.
This included a statistically significant change in QMG score inebilizumab compared with placebo for the combined population, a reduction of 4.8 versus 2.3 points, respectively, at week 26 (P = .0002).
In addition, both MG-ADL and QMG scores in the AChR+ subgroup were superior for inebilizumab versus placebo at week 26, with reductions of 4.2 versus 2.4, and 4.4 versus 2.0; P = .0015 and P = .0011, respectively.
In the MuSK+ subgroup, inebilizumab-treated patients had better MG-ADL scores than placebo-treated patients, with reductions of 3.9 versus 1.7 points, respectively, at week 26, although this difference did not meet statistical significance.
“There were no increased safety incidents in the inebilizumab-treated patients versus placebo, and a similar percentage of safety incidents in the AChR–positive and MuSK–positive groups. There were three deaths reported, all likely related to myasthenic crisis,” he said.
Nowak said that inebilizumab is “unique from the other currently FDA-approved medications for myasthenia gravis in that it’s targeting the upstream immunopathogenic mechanism of disease, specifically B cells — and B cells that are actually antibody-secreting cells.”
“It is targeting the factories of autoantibody production, whereas an FcRn antagonist, for example, is not targeting those factories but rather targeting what’s being produced — the immunoglobulins, IgGs in general,” he added.
Nowak said that what is particularly exciting about the drug is that the schedule is not very frequent. It begins with an initial IV infusion, followed by a second infusion 2 weeks later and a third infusion 6 months after that, so that patients are treated approximately every 6 months. This is in contrast to some other targeted therapies, where failing to address the underlying factors driving immunopathogenesis necessitates more regular and frequent medication administration.
New, Novel, Exciting
Commenting on the research, Neelam Goyal, MD, who chaired the session, said, “It’s definitely new, novel, interesting, exciting.”
Goyal, clinical professor of neurology and neurological sciences at Stanford University School of Medicine in Palo Alto, California, also noted that while B-cell depletion has shown some previous success in MG, it was with rituximab, a CD20 B-cell depleting agent.
She noted that unlike rituximab, which targets CD20, inebilizumab targets CD19, although both medications lead to B-cell depletion. Rituximab has proven effective for MUSK–positive MG, which accounts for approximately 5% of cases.
However, Goyal noted that the results for AChR–positive MG have been mixed — “the BeatMG trial was negative and the RINOMAX trial was positive. So, I think this is really interesting. It is exciting, and this drug is already on the market.”
She added that although inebilizumab is already US Food and Drug Administration–approved for the treatment of neuromyelitis optica, it still faces approval and indication hurdles for MG.
The future of this drug in the management algorithm for MG remains uncertain. Goyal noted that it’s “quite costly,” and although its benefits are evident — particularly for FcRn and complement inhibitors — some early data from chimeric antigen receptor T-cell therapy studies appear significantly more impressive.
Nowak disclosed research support from the National Institutes of Health, Genentech, Alexion Pharmaceuticals, argenx, Annexon Biosciences, Ra Pharmaceuticals (now UCB S.A.), the Myasthenia Gravis Foundation of America, Momenta Pharmaceuticals (now Janssen), Immunovant, Grifols, S.A., and Viela Bio, Horizon Therapeutics (now Amgen). Served as a consultant and advisor for Alexion Pharmaceuticals, argenx, Cabaletta Bio, Cour Pharmaceuticals, Ra Pharmaceuticals (now UCB S.A.), Immunovant, Momenta Pharmaceuticals (now Janssen), and Viela Bio (Horizon Therapeutics, now Amgen).
Goyal disclosed consultant, advisory, or grant support from argenx, UCB, Alexion, and Janssen. The study was funded by Amgen.
A version of this article appeared on Medscape.com.
FROM AANEM 2024
Myasthenia Gravis: Patient Choice, Cultural Change
Used appropriately, newer treatments can provide dramatic results faster and more safely than broad immunosuppressants. However, according to experts, payers’ willingness to cover costly new therapies remains a work in progress.
The availability of more effective treatments with fewer side effects has brought about a cultural shift, said James F. Howard, Jr, MD. “The physician’s goal now is for the patient to be symptom free with grade 1 or less adverse events. And patients are demanding freedom from all the side effects that our usual course of immune therapy produces.” Dr. Howard is professor of neurology, medicine and allied health and director of the Myasthenia Gravis Clinical Trials and Translational Research Program at the University of North Carolina at Chapel Hill.
The shift has been long in coming. Although myasthenia gravis was identified in the mid-1600s, it took more than 340 years to get the first drug approved specifically for the disorder.
Worldwide prevalence estimates vary widely, from less than 200,000 to 700,000 cases.1,2 Pathophysiologically, myasthenia gravis stems from autoimmune destruction of neuromuscular junctions (NMJs), which transmit motor neuron impulses to muscle fibers.1 Symptoms include variable skeletal muscle weakness that can range from mild and transient to life-threatening.
In approximately 80% of cases, autoimmune antibodies target the postsynaptic acetylcholine receptor (AChR). Additional autoimmune targets mainly include muscle-specific kinase (MuSK) and lipoprotein receptor-related protein 4 (LRP4). However, around 10% of patients are seronegative, lacking autoantibodies detectable through conventional radioimmunoassays. Clinical disease does not always correspond with circulating antibody levels, and pathogenesis may require cooperation between multiple autoantibodies attacking the same target.3 Around 10% of MG cases are associated with thymomas.
Among myasthenia gravis treatments, immunosuppressants typically take 4-10 months to begin working and 18-36 months for maximum benefit. “Our new targeted therapies work within 1-2 weeks, with maximum improvement occurring somewhere between 8 and 12 weeks,” Dr. Howard said. Quick onset makes these drugs well suited for primary therapy in recalcitrant myasthenia gravis or as bridges to standard immunotherapy. Targeted drugs also appear to provide effective rescue therapy, although head-to-head studies are needed.
Complement Inhibition
In AChR antibody–positive myasthenia gravis, autoantibody binding with the postsynaptic AChR receptor activates complement to attack postsynaptic neuronal membrane. Complement inhibitors approved to date block activation of the terminal complement protein C5.

For many patients, complement inhibitors deliver dramatic results. Henry J. Kaminski, MD, said that the first patient for whom he prescribed a complement inhibitor outside a clinical trial went from being miserable to traveling internationally within a month. Dr. Kaminski is Meta A. Neumann Professor of Neurology at George Washington University, Washington, DC.
Eculizumab (Soliris, Alexion), earned Food and Drug Administration (FDA) approval for myasthenia gravis in 2017. Week 26 results in the phase 3 REGAIN trial showed no significant difference in Myasthenia Gravis–Activities of Daily Living (MG-ADL) scores between treatment and placebo. However, said Dr. Howard, primary investigator on the study, the negative result was a statistical aberration stemming from the FDA’s requirement to use worst-rank analysis rather than absolute change scores. What got eculizumab approved were highly positive results in the overwhelming majority of secondary endpoints.4 Subsequently, the FDA had the manufacturer rewrite the package insert using common statistical methods, which yielded positive primary results.
Ravulizumab (Ultomiris, Alexion), approved for myasthenia gravis in 2022, reduces eculizumab’s twice-monthly intravenous dosing to every 2 months (after loading doses), with very similar efficacy. The newest complement inhibitor, zilucoplan (Zilbrysq, UCB), administered once daily subcutaneously, earned FDA approval in 2023. Daily subcutaneous dosing provides patient convenience, said Dr. Howard. Because the body does not clear this small molecule as it would a full-size antibody, it is the only complement inhibitor that can be combined with a fragment crystallizable neonatal receptor (FcRn) inhibitor.
FcRn Inhibition
The FcRn exists on the surface and intracellular vesicles of many cells, including B cells, but not T cells.5FcRn inhibitors block binding of circulating IgG antibodies to the FcRn, preventing their normal recycling, significantly reducing circulating antibodies within days of treatment.
Efgartigimod (Vyvgart, Argenx), earned FDA approval in intravenous form in 2021, followed by a subcutaneous formulation that includes hyaluronidase (Vyvgart Hytrulo) in 2023. Rozanolixizumab (Rystiggo, UCB) earned FDA approval for both AChR antibody–positive and MuSK antibody–positive myasthenia gravis in 2023.
Along with rapid response, said Dr. Howard, complement inhibitors and FcRn inhibitors offer a “hugely improved” side-effect profile. In phase 3 research, the most common side effects for both classes included headache, nausea, and diarrhea.4,6,7 Because complement inhibitors increase the risk of Neisseria infection, users require immunization against meningococcal infection (or concurrent antibiotic prophylaxis) while on complement inhibitors.
Insurance Issues
With many clinicians wondering which targeted therapy to choose for a particular patient, said Dr. Howard and Dr. Kaminski, the main obstacle to wider use of these treatments is payer attitudes and practices. “While many of us would like to see these drugs used earlier in the course of disease,” Dr. Howard explained, “there are numerous restrictions placed on the physician and the patient by whatever insurance the individual has.”
Dr. Kaminski said: “There’s a lot of variability among insurance companies regarding what is expected in terms of getting approval for a certain medication. It frustrates me, thinking this patient may do well with a complement inhibitor or an FcRn inhibitor, but it takes weeks to get them approved.”
Some of his patients have been approved for, and flourished on, complement inhibitors and FcRn inhibitors, he added, and then denied a second round of treatment. Dr. Kaminski said he does not know why these patients were denied, and every time he requests reevaluation, the decision is reversed. “That’s a significant time frame for me and my staff to manage.”
When asked what can be done to address high drug prices, Dr. Howard replied, “I have no idea. I’m not an advocate of high drug prices. But I don’t think people realize the cost of doing clinical trials, which is hundreds of millions of dollars, particularly in rare diseases.”
Presently, Dr. Howard said, FcRn inhibitors are used more frequently than complement inhibitors solely because of cost. Zilucoplan will be priced below existing complement inhibitors, although it is too soon to compare its price with those of FcRn inhibitors.
When eculizumab debuted, said Dr. Howard, it cost nearly $750,000 annually. “But if you look at the number of patients treated, the cost of the drug over this population is probably less than the cost for using a cholesterol-lowering agent to treat hyperlipidemia.”
An Institute for Clinical and Economic Review (ICER) report stated that eculizumab and efgartigimod should both cost less than $20,000 annually to meet commonly used cost-effectiveness thresholds.8 However, Dr. Howard said ICER used models based on common diseases and ignored the economic impact of patients’ losing fewer workdays and avoiding long-term immunosuppressant side effects such as diabetes and osteoporosis and related treatment costs. “We’ve got to start looking at total societal cost,” he said.
Leapfrogging Ahead
Not all the new drugs work in every indicated patient, Dr. Howard said. For example, up to 30% of patients do not respond to complement inhibitors. “We don’t understand why. It’s as if we have leapfrogged way ahead in terms of therapeutics, and now we have to go back and answer all the questions – the who, what, where, and why of an individual drug and its response in folks.”
In this climate, said Dr. Kaminski, heavy direct-to-consumer advertising of newer myasthenia gravis therapies creates complications. “My patients are highly excited to see, ‘that’s my disease being advertised on Jeopardy.’ ” Many patients are frustrated with the general lack of awareness regarding myasthenia gravis, he added. “But then I’ve had patients who clearly would never qualify for a certain medication getting mailings to their homes.”
Dr. Howard countered that broader awareness of myasthenia gravis can only help. “There’s increasing recognition of the disease, not only by patients, but to some extent, by the treating clinician. Patients are coming to our offices and saying, ‘am I a candidate for this new drug?’ It’s the responsibility of the clinician to decide.”
Individual physicians’ practice patterns vary greatly, said Dr. Kaminski, and very little quantitative data exist here. But based on personal communications, academic-center neurologists tend to use targeted treatments on patients who have failed conventional treatments.
Conversely, Dr. Howard said that, because community physicians rarely see myasthenia gravis, and targeted treatments remain relatively new, many of these providers rely on prednisone, azathioprine, and mycophenolate mofetil.
B-Cell Blockers in Development
Overall, said Dr. Howard, the field of myasthenia gravis treatment development is “very rich. And pharma’s interest in myasthenia has taken off like a rocket. It’s exceptionally gratifying to those of us who take care of these patients whose life is miserable” because of adverse effects and/or nonresponse to current drugs.
“In myasthenia,” added Dr. Kaminski, “we know that T cells are promoting the activity of these auto-reactive B cells.” Many drugs currently in phase 2 or 3 development aim to eliminate B cells or signaling between T and B cells, he said. “That’s where most of the drug development is.”
Leading candidates include telitacicept (Tai’ai, RemeGen), which is both a B-lymphocyte stimulator and a proliferation-inducing ligand. A phase 3 trial (NCT05737160) is ongoing, with primary completion expected in late 2026. A second phase 3 trial (NCT06456580) recently began enrolling. Dr. Howard said that, although early results warranted phase 3 analysis, telitacicept’s phase 2 trial was open label and lacked a placebo group.9 The latter is a critical concern because placebo response rates in myasthenia gravis trials average 35%-40%.
Combined with standard care, the FcRn inhibitor nipocalimab (Johnson & Johnson) enabled patients with AChR, MuSK, and/or LRP4 autoantibodies to improve by 4.70 points on the MG-ADL vs 3.25 points for placebo (P = .002) over 24 weeks in phase 3.10All FcRn inhibitors in development can broadly reduce autoantibody levels, said Dr. Howard. “But what role they will play in myasthenia gravis when they’re several years behind leaders in the field in terms of capturing market remains to be seen.”
Additionally, batoclimab (Immunovant/Harbour BioMed) showed positive topline results in phase 3, and an elevated rate of hypercholesterolemia in treated patients that was transient and consistent with previous research.11 Subsequent to efgartigimod, Dr. Howard said, FcRn inhibitors are full-size antibodies. “I believe that contributes to the adverse events that we see. Efgartigimod is a small FcRn fragment. That’s why it’s a cleaner drug, if you will.”
FcRn inhibitors require periodic retreatment. For example, said Dr. Howard, the ADAPT phase 3 trial of efgartigimod, on which he was lead investigator, employed a cyclic dosing schedule – 4 weeks’ treatment, then observation until patients needed retreatment — because patients demanded it.12 In clinical practice, some patients have gone more than 25 weeks before needing retreatment. One of his patients went beyond 40 weeks. “Others only get around 6-9 weeks. So patient choice again enters the decision-making process.”
Rituximab targets the CD20 protein on B cells nonspecifically, producing general immunosuppression. “That’s problematic in producing significant immunosuppression,” said Dr. Kaminski. Nevertheless, he said, rituximab is very effective for most patients with MuSK-specific MG, and its application to this indication has revealed differences between the MuSK subtype and AChR antibody–positive myasthenia. Specifically, MuSK antibody–positive patients have short-lived plasmablasts, which rituximab eliminates.13
Conversely, said Dr. Kaminski, patients with AChR antibody-positive myasthenia, especially long-term, likely have long-lived plasmablasts producing antibodies. This fact, and these patients’ lack of CD20, likely explain their poor response to rituximab.
A phase 3 trial (NCT04524273) of the CD19 blocker inebilizumab (Uplinza, Amgen) reached primary completion in May. Dr. Howard said that if topline results (unreleased at press time) prove positive, inebilizumab could replace rituximab in MG — provided payers do not reject inebilizumab because of cost.
Packed Early-Development Pipeline
Regarding early-stage projects, said Dr. Howard, the pipeline is packed with compounds that target various aspects of the immune system. “The real question with those is, what’s going to be the side effect profile? All of the trials are very early. We need bigger trials with much longer observation for safety, durability, and degree of efficacy.”
The next potential B cell–targeting game changer, he said, is chimeric antigen receptor (CAR) T cell–based therapy. In a phase 2b trial of Descartes-08 (Cartesian Therapeutics), 71% of treated patients experienced clinically meaningful improvement in MG Composite score at 3 months vs 25% for placebo.14
In early clinical trials, said Dr. Howard, patients treated with Descartes-08 — which uses autologous mRNA to target B-cell maturation antigen — have shown “exceptional improvement” lasting 20 or more months. Because the drug is not ingrained permanently into the genome, Descartes-08 avoids potentially severe side effects of DNA-targeting CAR T candidates. Dr. Howard hopes a phase 3 trial will commence around January 2025.
The tolerance approach exemplified by CNP-106 (COUR Pharmaceuticals) and a myasthenia gravis tolerogen (Toleranzia) seeks to prevent the immune system from recognizing and reacting to the NMJ abnormalities that produce myasthenia gravis, potentially providing a cure. “We look forward to those trials as they come online in the next 1-2 years,” said Dr. Howard.
Unmet Needs
Historically, neurologists believed that all myasthenia gravis symptoms stemmed from muscle fatigue — the more active the muscle, the weaker it gets. However, said Dr. Kaminski, some patients might lack measurable weakness but still complain of fatigue.
Elevated levels of cytokines such as interleukin (IL)–6 or IL-17 also can produce fatigue, he noted. “With the drugs we’re using, certainly the new ones, we’re not specifically targeting this fatigue phenomenon, which has been studied in a very limited fashion.”
In the RAISE-XT zilucoplan trial, participants experienced significant improvement in fatigue scores for up to 60 weeks.15 Although zilucoplan does not address fatigue directly, said Dr. Howard, improving myasthenia gravis overall helps reduce fatigue.
The Myasthenia Gravis Symptoms Patient Reported Outcome (MG Symptoms PRO), which Dr. Kaminski helped develop, includes questions designed to distinguish muscular fatigue from overall physical fatigue.16 “I’m very interested in some of the information that’s coming out on long COVID and its effect on muscle,” Dr. Kaminski added. “We might be able to learn from there that there’s still some pathology going on beyond the neuromuscular junction.”
What the field desperately needs, said Dr. Howard, are biomarkers to identify which patients will and will not respond to certain therapeutics. “We’re not there yet.” Such biomarkers are at least 3-7 years from becoming clinical reality.
Promising antibody-independent serum markers include circulating microRNAs. For example, miRNA-150-5p and miRNA-21-5p are elevated in generalized AChR-positive myasthenia gravis and early-onset myasthenia gravis (occurring before age 50) and decline after immunosuppression and thymectomy.17
Among diagnostic modalities for patients with seronegative myasthenia gravis, said Dr. Kaminski, single-fiber EMG is the most sensitive, at approximately 95%. “It’s not perfect.” Moreover, he said, performing this test accurately requires a highly experienced expert, which many treatment centers lack.
Presently, added Dr. Kaminski, orbital MRI is neither specific nor sensitive enough to be clinically useful. “One needs to be careful with these specialized tests that are published from the best laboratory in the world that does the test, and does it repetitively.” As the search for effective myasthenia gravis biomarkers continues, avoiding false-positive results is as important as avoiding false negatives.
References
1. Bubuioc AM et al. J Med Life. 2021 Jan-Mar;14(1):7-16. doi: 10.25122/jml-2020-0145.
2. Deenen JC et al. J Neuromuscul Dis. 2015;2(1):73-85. doi: 10.3233/JND-140045.
3. Kaminski HJ et al. J Clin Invest. 2024 Jun 17;134(12):e179742. doi: 10.1172/JCI179742.
4. Howard JF Jr et al. Lancet Neurol. 2017 Dec;16(12):976-986. doi: 10.1016/S1474-4422(17)30369-1.
5. Huda R. Front Immunol. 2020 Feb 21:11:240. doi: 10.3389/fimmu.2020.00240.
6. Howard JF Jr et al. Lancet Neurol. 2023 May;22(5):395-406. doi: 10.1016/S1474-4422(23)00080-7.
7. Vu T et al. NEJM Evid. 2022 May;1(5):EVIDoa2100066. doi: 10.1056/EVIDoa2100066.
8. Tice JA et al. October 20, 2021. https://icer.org/assessment/myasthenia-gravis/.
9. Yin J et al. Eur J Neurol. 2024 Aug;31(8):e16322. doi: 10.1111/ene.16322.
10. Antozzi C et al. EAN 2024, Abstract EPR-116. https://www.neurology.org/doi/10.1212/WNL.0000000000203660.
11. Yan C et al. JAMA Neurol. 2024 Mar 4;81(4):336-345. doi: 10.1001/jamaneurol.2024.0044.
12. Howard JF Jr et al. Lancet Neurol. 2021 Jul;20(7):526-536. doi: 10.1016/S1474-4422(21)00159-9.
13. Stathopoulos P et al. JCI Insight. 2017 Sep 7;2(17):e94263. doi: 10.1172/jci.insight.94263.
14. Cartesian Therapeutics. Cartesian Therapeutics announces positive topline results from phase 2b trial of Descartes-08 in patients with myasthenia gravis. 2024 Jul 2. https://ir.cartesiantherapeutics.com/news-releases/news-release-details/cartesian-therapeutics-announces-positive-topline-results-phase.
15. Howard JF Jr et al. Ther Adv Neurol Disord. 2024 Apr 17:17:17562864241243186. doi: 10.1177/17562864241243186.
16. Cleanthous S et al. Orphanet J Rare Dis. 2021 Oct 30;16(1):457. doi: 10.1186/s13023-021-02064-0.
17. Sabre L et al. Front Immunol. 2020 Mar 4:11:213. doi: 10.3389/fimmu.2020.00213.
Used appropriately, newer treatments can provide dramatic results faster and more safely than broad immunosuppressants. However, according to experts, payers’ willingness to cover costly new therapies remains a work in progress.
The availability of more effective treatments with fewer side effects has brought about a cultural shift, said James F. Howard, Jr, MD. “The physician’s goal now is for the patient to be symptom free with grade 1 or less adverse events. And patients are demanding freedom from all the side effects that our usual course of immune therapy produces.” Dr. Howard is professor of neurology, medicine and allied health and director of the Myasthenia Gravis Clinical Trials and Translational Research Program at the University of North Carolina at Chapel Hill.
The shift has been long in coming. Although myasthenia gravis was identified in the mid-1600s, it took more than 340 years to get the first drug approved specifically for the disorder.
Worldwide prevalence estimates vary widely, from less than 200,000 to 700,000 cases.1,2 Pathophysiologically, myasthenia gravis stems from autoimmune destruction of neuromuscular junctions (NMJs), which transmit motor neuron impulses to muscle fibers.1 Symptoms include variable skeletal muscle weakness that can range from mild and transient to life-threatening.
In approximately 80% of cases, autoimmune antibodies target the postsynaptic acetylcholine receptor (AChR). Additional autoimmune targets mainly include muscle-specific kinase (MuSK) and lipoprotein receptor-related protein 4 (LRP4). However, around 10% of patients are seronegative, lacking autoantibodies detectable through conventional radioimmunoassays. Clinical disease does not always correspond with circulating antibody levels, and pathogenesis may require cooperation between multiple autoantibodies attacking the same target.3 Around 10% of MG cases are associated with thymomas.
Among myasthenia gravis treatments, immunosuppressants typically take 4-10 months to begin working and 18-36 months for maximum benefit. “Our new targeted therapies work within 1-2 weeks, with maximum improvement occurring somewhere between 8 and 12 weeks,” Dr. Howard said. Quick onset makes these drugs well suited for primary therapy in recalcitrant myasthenia gravis or as bridges to standard immunotherapy. Targeted drugs also appear to provide effective rescue therapy, although head-to-head studies are needed.
Complement Inhibition
In AChR antibody–positive myasthenia gravis, autoantibody binding with the postsynaptic AChR receptor activates complement to attack postsynaptic neuronal membrane. Complement inhibitors approved to date block activation of the terminal complement protein C5.
For many patients, complement inhibitors deliver dramatic results. Henry J. Kaminski, MD, said that the first patient for whom he prescribed a complement inhibitor outside a clinical trial went from being miserable to traveling internationally within a month. Dr. Kaminski is Meta A. Neumann Professor of Neurology at George Washington University, Washington, DC.
Eculizumab (Soliris, Alexion), earned Food and Drug Administration (FDA) approval for myasthenia gravis in 2017. Week 26 results in the phase 3 REGAIN trial showed no significant difference in Myasthenia Gravis–Activities of Daily Living (MG-ADL) scores between treatment and placebo. However, said Dr. Howard, primary investigator on the study, the negative result was a statistical aberration stemming from the FDA’s requirement to use worst-rank analysis rather than absolute change scores. What got eculizumab approved were highly positive results in the overwhelming majority of secondary endpoints.4 Subsequently, the FDA had the manufacturer rewrite the package insert using common statistical methods, which yielded positive primary results.
Ravulizumab (Ultomiris, Alexion), approved for myasthenia gravis in 2022, reduces eculizumab’s twice-monthly intravenous dosing to every 2 months (after loading doses), with very similar efficacy. The newest complement inhibitor, zilucoplan (Zilbrysq, UCB), administered once daily subcutaneously, earned FDA approval in 2023. Daily subcutaneous dosing provides patient convenience, said Dr. Howard. Because the body does not clear this small molecule as it would a full-size antibody, it is the only complement inhibitor that can be combined with a fragment crystallizable neonatal receptor (FcRn) inhibitor.
FcRn Inhibition
The FcRn exists on the surface and intracellular vesicles of many cells, including B cells, but not T cells.5FcRn inhibitors block binding of circulating IgG antibodies to the FcRn, preventing their normal recycling, significantly reducing circulating antibodies within days of treatment.
Efgartigimod (Vyvgart, Argenx), earned FDA approval in intravenous form in 2021, followed by a subcutaneous formulation that includes hyaluronidase (Vyvgart Hytrulo) in 2023. Rozanolixizumab (Rystiggo, UCB) earned FDA approval for both AChR antibody–positive and MuSK antibody–positive myasthenia gravis in 2023.
Along with rapid response, said Dr. Howard, complement inhibitors and FcRn inhibitors offer a “hugely improved” side-effect profile. In phase 3 research, the most common side effects for both classes included headache, nausea, and diarrhea.4,6,7 Because complement inhibitors increase the risk of Neisseria infection, users require immunization against meningococcal infection (or concurrent antibiotic prophylaxis) while on complement inhibitors.
Insurance Issues
With many clinicians wondering which targeted therapy to choose for a particular patient, said Dr. Howard and Dr. Kaminski, the main obstacle to wider use of these treatments is payer attitudes and practices. “While many of us would like to see these drugs used earlier in the course of disease,” Dr. Howard explained, “there are numerous restrictions placed on the physician and the patient by whatever insurance the individual has.”
Dr. Kaminski said: “There’s a lot of variability among insurance companies regarding what is expected in terms of getting approval for a certain medication. It frustrates me, thinking this patient may do well with a complement inhibitor or an FcRn inhibitor, but it takes weeks to get them approved.”
Some of his patients have been approved for, and flourished on, complement inhibitors and FcRn inhibitors, he added, and then denied a second round of treatment. Dr. Kaminski said he does not know why these patients were denied, and every time he requests reevaluation, the decision is reversed. “That’s a significant time frame for me and my staff to manage.”
When asked what can be done to address high drug prices, Dr. Howard replied, “I have no idea. I’m not an advocate of high drug prices. But I don’t think people realize the cost of doing clinical trials, which is hundreds of millions of dollars, particularly in rare diseases.”
Presently, Dr. Howard said, FcRn inhibitors are used more frequently than complement inhibitors solely because of cost. Zilucoplan will be priced below existing complement inhibitors, although it is too soon to compare its price with those of FcRn inhibitors.
When eculizumab debuted, said Dr. Howard, it cost nearly $750,000 annually. “But if you look at the number of patients treated, the cost of the drug over this population is probably less than the cost for using a cholesterol-lowering agent to treat hyperlipidemia.”
An Institute for Clinical and Economic Review (ICER) report stated that eculizumab and efgartigimod should both cost less than $20,000 annually to meet commonly used cost-effectiveness thresholds.8 However, Dr. Howard said ICER used models based on common diseases and ignored the economic impact of patients’ losing fewer workdays and avoiding long-term immunosuppressant side effects such as diabetes and osteoporosis and related treatment costs. “We’ve got to start looking at total societal cost,” he said.
Leapfrogging Ahead
Not all the new drugs work in every indicated patient, Dr. Howard said. For example, up to 30% of patients do not respond to complement inhibitors. “We don’t understand why. It’s as if we have leapfrogged way ahead in terms of therapeutics, and now we have to go back and answer all the questions – the who, what, where, and why of an individual drug and its response in folks.”
In this climate, said Dr. Kaminski, heavy direct-to-consumer advertising of newer myasthenia gravis therapies creates complications. “My patients are highly excited to see, ‘that’s my disease being advertised on Jeopardy.’ ” Many patients are frustrated with the general lack of awareness regarding myasthenia gravis, he added. “But then I’ve had patients who clearly would never qualify for a certain medication getting mailings to their homes.”
Dr. Howard countered that broader awareness of myasthenia gravis can only help. “There’s increasing recognition of the disease, not only by patients, but to some extent, by the treating clinician. Patients are coming to our offices and saying, ‘am I a candidate for this new drug?’ It’s the responsibility of the clinician to decide.”
Individual physicians’ practice patterns vary greatly, said Dr. Kaminski, and very little quantitative data exist here. But based on personal communications, academic-center neurologists tend to use targeted treatments on patients who have failed conventional treatments.
Conversely, Dr. Howard said that, because community physicians rarely see myasthenia gravis, and targeted treatments remain relatively new, many of these providers rely on prednisone, azathioprine, and mycophenolate mofetil.
B-Cell Blockers in Development
Overall, said Dr. Howard, the field of myasthenia gravis treatment development is “very rich. And pharma’s interest in myasthenia has taken off like a rocket. It’s exceptionally gratifying to those of us who take care of these patients whose life is miserable” because of adverse effects and/or nonresponse to current drugs.
“In myasthenia,” added Dr. Kaminski, “we know that T cells are promoting the activity of these auto-reactive B cells.” Many drugs currently in phase 2 or 3 development aim to eliminate B cells or signaling between T and B cells, he said. “That’s where most of the drug development is.”
Leading candidates include telitacicept (Tai’ai, RemeGen), which is both a B-lymphocyte stimulator and a proliferation-inducing ligand. A phase 3 trial (NCT05737160) is ongoing, with primary completion expected in late 2026. A second phase 3 trial (NCT06456580) recently began enrolling. Dr. Howard said that, although early results warranted phase 3 analysis, telitacicept’s phase 2 trial was open label and lacked a placebo group.9 The latter is a critical concern because placebo response rates in myasthenia gravis trials average 35%-40%.
Combined with standard care, the FcRn inhibitor nipocalimab (Johnson & Johnson) enabled patients with AChR, MuSK, and/or LRP4 autoantibodies to improve by 4.70 points on the MG-ADL vs 3.25 points for placebo (P = .002) over 24 weeks in phase 3.10All FcRn inhibitors in development can broadly reduce autoantibody levels, said Dr. Howard. “But what role they will play in myasthenia gravis when they’re several years behind leaders in the field in terms of capturing market remains to be seen.”
Additionally, batoclimab (Immunovant/Harbour BioMed) showed positive topline results in phase 3, and an elevated rate of hypercholesterolemia in treated patients that was transient and consistent with previous research.11 Subsequent to efgartigimod, Dr. Howard said, FcRn inhibitors are full-size antibodies. “I believe that contributes to the adverse events that we see. Efgartigimod is a small FcRn fragment. That’s why it’s a cleaner drug, if you will.”
FcRn inhibitors require periodic retreatment. For example, said Dr. Howard, the ADAPT phase 3 trial of efgartigimod, on which he was lead investigator, employed a cyclic dosing schedule – 4 weeks’ treatment, then observation until patients needed retreatment — because patients demanded it.12 In clinical practice, some patients have gone more than 25 weeks before needing retreatment. One of his patients went beyond 40 weeks. “Others only get around 6-9 weeks. So patient choice again enters the decision-making process.”
Rituximab targets the CD20 protein on B cells nonspecifically, producing general immunosuppression. “That’s problematic in producing significant immunosuppression,” said Dr. Kaminski. Nevertheless, he said, rituximab is very effective for most patients with MuSK-specific MG, and its application to this indication has revealed differences between the MuSK subtype and AChR antibody–positive myasthenia. Specifically, MuSK antibody–positive patients have short-lived plasmablasts, which rituximab eliminates.13
Conversely, said Dr. Kaminski, patients with AChR antibody-positive myasthenia, especially long-term, likely have long-lived plasmablasts producing antibodies. This fact, and these patients’ lack of CD20, likely explain their poor response to rituximab.
A phase 3 trial (NCT04524273) of the CD19 blocker inebilizumab (Uplinza, Amgen) reached primary completion in May. Dr. Howard said that if topline results (unreleased at press time) prove positive, inebilizumab could replace rituximab in MG — provided payers do not reject inebilizumab because of cost.
Packed Early-Development Pipeline
Regarding early-stage projects, said Dr. Howard, the pipeline is packed with compounds that target various aspects of the immune system. “The real question with those is, what’s going to be the side effect profile? All of the trials are very early. We need bigger trials with much longer observation for safety, durability, and degree of efficacy.”
The next potential B cell–targeting game changer, he said, is chimeric antigen receptor (CAR) T cell–based therapy. In a phase 2b trial of Descartes-08 (Cartesian Therapeutics), 71% of treated patients experienced clinically meaningful improvement in MG Composite score at 3 months vs 25% for placebo.14
In early clinical trials, said Dr. Howard, patients treated with Descartes-08 — which uses autologous mRNA to target B-cell maturation antigen — have shown “exceptional improvement” lasting 20 or more months. Because the drug is not ingrained permanently into the genome, Descartes-08 avoids potentially severe side effects of DNA-targeting CAR T candidates. Dr. Howard hopes a phase 3 trial will commence around January 2025.
The tolerance approach exemplified by CNP-106 (COUR Pharmaceuticals) and a myasthenia gravis tolerogen (Toleranzia) seeks to prevent the immune system from recognizing and reacting to the NMJ abnormalities that produce myasthenia gravis, potentially providing a cure. “We look forward to those trials as they come online in the next 1-2 years,” said Dr. Howard.
Unmet Needs
Historically, neurologists believed that all myasthenia gravis symptoms stemmed from muscle fatigue — the more active the muscle, the weaker it gets. However, said Dr. Kaminski, some patients might lack measurable weakness but still complain of fatigue.
Elevated levels of cytokines such as interleukin (IL)–6 or IL-17 also can produce fatigue, he noted. “With the drugs we’re using, certainly the new ones, we’re not specifically targeting this fatigue phenomenon, which has been studied in a very limited fashion.”
In the RAISE-XT zilucoplan trial, participants experienced significant improvement in fatigue scores for up to 60 weeks.15 Although zilucoplan does not address fatigue directly, said Dr. Howard, improving myasthenia gravis overall helps reduce fatigue.
The Myasthenia Gravis Symptoms Patient Reported Outcome (MG Symptoms PRO), which Dr. Kaminski helped develop, includes questions designed to distinguish muscular fatigue from overall physical fatigue.16 “I’m very interested in some of the information that’s coming out on long COVID and its effect on muscle,” Dr. Kaminski added. “We might be able to learn from there that there’s still some pathology going on beyond the neuromuscular junction.”
What the field desperately needs, said Dr. Howard, are biomarkers to identify which patients will and will not respond to certain therapeutics. “We’re not there yet.” Such biomarkers are at least 3-7 years from becoming clinical reality.
Promising antibody-independent serum markers include circulating microRNAs. For example, miRNA-150-5p and miRNA-21-5p are elevated in generalized AChR-positive myasthenia gravis and early-onset myasthenia gravis (occurring before age 50) and decline after immunosuppression and thymectomy.17
Among diagnostic modalities for patients with seronegative myasthenia gravis, said Dr. Kaminski, single-fiber EMG is the most sensitive, at approximately 95%. “It’s not perfect.” Moreover, he said, performing this test accurately requires a highly experienced expert, which many treatment centers lack.
Presently, added Dr. Kaminski, orbital MRI is neither specific nor sensitive enough to be clinically useful. “One needs to be careful with these specialized tests that are published from the best laboratory in the world that does the test, and does it repetitively.” As the search for effective myasthenia gravis biomarkers continues, avoiding false-positive results is as important as avoiding false negatives.
References
1. Bubuioc AM et al. J Med Life. 2021 Jan-Mar;14(1):7-16. doi: 10.25122/jml-2020-0145.
2. Deenen JC et al. J Neuromuscul Dis. 2015;2(1):73-85. doi: 10.3233/JND-140045.
3. Kaminski HJ et al. J Clin Invest. 2024 Jun 17;134(12):e179742. doi: 10.1172/JCI179742.
4. Howard JF Jr et al. Lancet Neurol. 2017 Dec;16(12):976-986. doi: 10.1016/S1474-4422(17)30369-1.
5. Huda R. Front Immunol. 2020 Feb 21:11:240. doi: 10.3389/fimmu.2020.00240.
6. Howard JF Jr et al. Lancet Neurol. 2023 May;22(5):395-406. doi: 10.1016/S1474-4422(23)00080-7.
7. Vu T et al. NEJM Evid. 2022 May;1(5):EVIDoa2100066. doi: 10.1056/EVIDoa2100066.
8. Tice JA et al. October 20, 2021. https://icer.org/assessment/myasthenia-gravis/.
9. Yin J et al. Eur J Neurol. 2024 Aug;31(8):e16322. doi: 10.1111/ene.16322.
10. Antozzi C et al. EAN 2024, Abstract EPR-116. https://www.neurology.org/doi/10.1212/WNL.0000000000203660.
11. Yan C et al. JAMA Neurol. 2024 Mar 4;81(4):336-345. doi: 10.1001/jamaneurol.2024.0044.
12. Howard JF Jr et al. Lancet Neurol. 2021 Jul;20(7):526-536. doi: 10.1016/S1474-4422(21)00159-9.
13. Stathopoulos P et al. JCI Insight. 2017 Sep 7;2(17):e94263. doi: 10.1172/jci.insight.94263.
14. Cartesian Therapeutics. Cartesian Therapeutics announces positive topline results from phase 2b trial of Descartes-08 in patients with myasthenia gravis. 2024 Jul 2. https://ir.cartesiantherapeutics.com/news-releases/news-release-details/cartesian-therapeutics-announces-positive-topline-results-phase.
15. Howard JF Jr et al. Ther Adv Neurol Disord. 2024 Apr 17:17:17562864241243186. doi: 10.1177/17562864241243186.
16. Cleanthous S et al. Orphanet J Rare Dis. 2021 Oct 30;16(1):457. doi: 10.1186/s13023-021-02064-0.
17. Sabre L et al. Front Immunol. 2020 Mar 4:11:213. doi: 10.3389/fimmu.2020.00213.
Used appropriately, newer treatments can provide dramatic results faster and more safely than broad immunosuppressants. However, according to experts, payers’ willingness to cover costly new therapies remains a work in progress.
The availability of more effective treatments with fewer side effects has brought about a cultural shift, said James F. Howard, Jr, MD. “The physician’s goal now is for the patient to be symptom free with grade 1 or less adverse events. And patients are demanding freedom from all the side effects that our usual course of immune therapy produces.” Dr. Howard is professor of neurology, medicine and allied health and director of the Myasthenia Gravis Clinical Trials and Translational Research Program at the University of North Carolina at Chapel Hill.
The shift has been long in coming. Although myasthenia gravis was identified in the mid-1600s, it took more than 340 years to get the first drug approved specifically for the disorder.
Worldwide prevalence estimates vary widely, from less than 200,000 to 700,000 cases.1,2 Pathophysiologically, myasthenia gravis stems from autoimmune destruction of neuromuscular junctions (NMJs), which transmit motor neuron impulses to muscle fibers.1 Symptoms include variable skeletal muscle weakness that can range from mild and transient to life-threatening.
In approximately 80% of cases, autoimmune antibodies target the postsynaptic acetylcholine receptor (AChR). Additional autoimmune targets mainly include muscle-specific kinase (MuSK) and lipoprotein receptor-related protein 4 (LRP4). However, around 10% of patients are seronegative, lacking autoantibodies detectable through conventional radioimmunoassays. Clinical disease does not always correspond with circulating antibody levels, and pathogenesis may require cooperation between multiple autoantibodies attacking the same target.3 Around 10% of MG cases are associated with thymomas.
Among myasthenia gravis treatments, immunosuppressants typically take 4-10 months to begin working and 18-36 months for maximum benefit. “Our new targeted therapies work within 1-2 weeks, with maximum improvement occurring somewhere between 8 and 12 weeks,” Dr. Howard said. Quick onset makes these drugs well suited for primary therapy in recalcitrant myasthenia gravis or as bridges to standard immunotherapy. Targeted drugs also appear to provide effective rescue therapy, although head-to-head studies are needed.
Complement Inhibition
In AChR antibody–positive myasthenia gravis, autoantibody binding with the postsynaptic AChR receptor activates complement to attack postsynaptic neuronal membrane. Complement inhibitors approved to date block activation of the terminal complement protein C5.
For many patients, complement inhibitors deliver dramatic results. Henry J. Kaminski, MD, said that the first patient for whom he prescribed a complement inhibitor outside a clinical trial went from being miserable to traveling internationally within a month. Dr. Kaminski is Meta A. Neumann Professor of Neurology at George Washington University, Washington, DC.
Eculizumab (Soliris, Alexion), earned Food and Drug Administration (FDA) approval for myasthenia gravis in 2017. Week 26 results in the phase 3 REGAIN trial showed no significant difference in Myasthenia Gravis–Activities of Daily Living (MG-ADL) scores between treatment and placebo. However, said Dr. Howard, primary investigator on the study, the negative result was a statistical aberration stemming from the FDA’s requirement to use worst-rank analysis rather than absolute change scores. What got eculizumab approved were highly positive results in the overwhelming majority of secondary endpoints.4 Subsequently, the FDA had the manufacturer rewrite the package insert using common statistical methods, which yielded positive primary results.
Ravulizumab (Ultomiris, Alexion), approved for myasthenia gravis in 2022, reduces eculizumab’s twice-monthly intravenous dosing to every 2 months (after loading doses), with very similar efficacy. The newest complement inhibitor, zilucoplan (Zilbrysq, UCB), administered once daily subcutaneously, earned FDA approval in 2023. Daily subcutaneous dosing provides patient convenience, said Dr. Howard. Because the body does not clear this small molecule as it would a full-size antibody, it is the only complement inhibitor that can be combined with a fragment crystallizable neonatal receptor (FcRn) inhibitor.
FcRn Inhibition
The FcRn exists on the surface and intracellular vesicles of many cells, including B cells, but not T cells.5FcRn inhibitors block binding of circulating IgG antibodies to the FcRn, preventing their normal recycling, significantly reducing circulating antibodies within days of treatment.
Efgartigimod (Vyvgart, Argenx), earned FDA approval in intravenous form in 2021, followed by a subcutaneous formulation that includes hyaluronidase (Vyvgart Hytrulo) in 2023. Rozanolixizumab (Rystiggo, UCB) earned FDA approval for both AChR antibody–positive and MuSK antibody–positive myasthenia gravis in 2023.
Along with rapid response, said Dr. Howard, complement inhibitors and FcRn inhibitors offer a “hugely improved” side-effect profile. In phase 3 research, the most common side effects for both classes included headache, nausea, and diarrhea.4,6,7 Because complement inhibitors increase the risk of Neisseria infection, users require immunization against meningococcal infection (or concurrent antibiotic prophylaxis) while on complement inhibitors.
Insurance Issues
With many clinicians wondering which targeted therapy to choose for a particular patient, said Dr. Howard and Dr. Kaminski, the main obstacle to wider use of these treatments is payer attitudes and practices. “While many of us would like to see these drugs used earlier in the course of disease,” Dr. Howard explained, “there are numerous restrictions placed on the physician and the patient by whatever insurance the individual has.”
Dr. Kaminski said: “There’s a lot of variability among insurance companies regarding what is expected in terms of getting approval for a certain medication. It frustrates me, thinking this patient may do well with a complement inhibitor or an FcRn inhibitor, but it takes weeks to get them approved.”
Some of his patients have been approved for, and flourished on, complement inhibitors and FcRn inhibitors, he added, and then denied a second round of treatment. Dr. Kaminski said he does not know why these patients were denied, and every time he requests reevaluation, the decision is reversed. “That’s a significant time frame for me and my staff to manage.”
When asked what can be done to address high drug prices, Dr. Howard replied, “I have no idea. I’m not an advocate of high drug prices. But I don’t think people realize the cost of doing clinical trials, which is hundreds of millions of dollars, particularly in rare diseases.”
Presently, Dr. Howard said, FcRn inhibitors are used more frequently than complement inhibitors solely because of cost. Zilucoplan will be priced below existing complement inhibitors, although it is too soon to compare its price with those of FcRn inhibitors.
When eculizumab debuted, said Dr. Howard, it cost nearly $750,000 annually. “But if you look at the number of patients treated, the cost of the drug over this population is probably less than the cost for using a cholesterol-lowering agent to treat hyperlipidemia.”
An Institute for Clinical and Economic Review (ICER) report stated that eculizumab and efgartigimod should both cost less than $20,000 annually to meet commonly used cost-effectiveness thresholds.8 However, Dr. Howard said ICER used models based on common diseases and ignored the economic impact of patients’ losing fewer workdays and avoiding long-term immunosuppressant side effects such as diabetes and osteoporosis and related treatment costs. “We’ve got to start looking at total societal cost,” he said.
Leapfrogging Ahead
Not all the new drugs work in every indicated patient, Dr. Howard said. For example, up to 30% of patients do not respond to complement inhibitors. “We don’t understand why. It’s as if we have leapfrogged way ahead in terms of therapeutics, and now we have to go back and answer all the questions – the who, what, where, and why of an individual drug and its response in folks.”
In this climate, said Dr. Kaminski, heavy direct-to-consumer advertising of newer myasthenia gravis therapies creates complications. “My patients are highly excited to see, ‘that’s my disease being advertised on Jeopardy.’ ” Many patients are frustrated with the general lack of awareness regarding myasthenia gravis, he added. “But then I’ve had patients who clearly would never qualify for a certain medication getting mailings to their homes.”
Dr. Howard countered that broader awareness of myasthenia gravis can only help. “There’s increasing recognition of the disease, not only by patients, but to some extent, by the treating clinician. Patients are coming to our offices and saying, ‘am I a candidate for this new drug?’ It’s the responsibility of the clinician to decide.”
Individual physicians’ practice patterns vary greatly, said Dr. Kaminski, and very little quantitative data exist here. But based on personal communications, academic-center neurologists tend to use targeted treatments on patients who have failed conventional treatments.
Conversely, Dr. Howard said that, because community physicians rarely see myasthenia gravis, and targeted treatments remain relatively new, many of these providers rely on prednisone, azathioprine, and mycophenolate mofetil.
B-Cell Blockers in Development
Overall, said Dr. Howard, the field of myasthenia gravis treatment development is “very rich. And pharma’s interest in myasthenia has taken off like a rocket. It’s exceptionally gratifying to those of us who take care of these patients whose life is miserable” because of adverse effects and/or nonresponse to current drugs.
“In myasthenia,” added Dr. Kaminski, “we know that T cells are promoting the activity of these auto-reactive B cells.” Many drugs currently in phase 2 or 3 development aim to eliminate B cells or signaling between T and B cells, he said. “That’s where most of the drug development is.”
Leading candidates include telitacicept (Tai’ai, RemeGen), which is both a B-lymphocyte stimulator and a proliferation-inducing ligand. A phase 3 trial (NCT05737160) is ongoing, with primary completion expected in late 2026. A second phase 3 trial (NCT06456580) recently began enrolling. Dr. Howard said that, although early results warranted phase 3 analysis, telitacicept’s phase 2 trial was open label and lacked a placebo group.9 The latter is a critical concern because placebo response rates in myasthenia gravis trials average 35%-40%.
Combined with standard care, the FcRn inhibitor nipocalimab (Johnson & Johnson) enabled patients with AChR, MuSK, and/or LRP4 autoantibodies to improve by 4.70 points on the MG-ADL vs 3.25 points for placebo (P = .002) over 24 weeks in phase 3.10All FcRn inhibitors in development can broadly reduce autoantibody levels, said Dr. Howard. “But what role they will play in myasthenia gravis when they’re several years behind leaders in the field in terms of capturing market remains to be seen.”
Additionally, batoclimab (Immunovant/Harbour BioMed) showed positive topline results in phase 3, and an elevated rate of hypercholesterolemia in treated patients that was transient and consistent with previous research.11 Subsequent to efgartigimod, Dr. Howard said, FcRn inhibitors are full-size antibodies. “I believe that contributes to the adverse events that we see. Efgartigimod is a small FcRn fragment. That’s why it’s a cleaner drug, if you will.”
FcRn inhibitors require periodic retreatment. For example, said Dr. Howard, the ADAPT phase 3 trial of efgartigimod, on which he was lead investigator, employed a cyclic dosing schedule – 4 weeks’ treatment, then observation until patients needed retreatment — because patients demanded it.12 In clinical practice, some patients have gone more than 25 weeks before needing retreatment. One of his patients went beyond 40 weeks. “Others only get around 6-9 weeks. So patient choice again enters the decision-making process.”
Rituximab targets the CD20 protein on B cells nonspecifically, producing general immunosuppression. “That’s problematic in producing significant immunosuppression,” said Dr. Kaminski. Nevertheless, he said, rituximab is very effective for most patients with MuSK-specific MG, and its application to this indication has revealed differences between the MuSK subtype and AChR antibody–positive myasthenia. Specifically, MuSK antibody–positive patients have short-lived plasmablasts, which rituximab eliminates.13
Conversely, said Dr. Kaminski, patients with AChR antibody-positive myasthenia, especially long-term, likely have long-lived plasmablasts producing antibodies. This fact, and these patients’ lack of CD20, likely explain their poor response to rituximab.
A phase 3 trial (NCT04524273) of the CD19 blocker inebilizumab (Uplinza, Amgen) reached primary completion in May. Dr. Howard said that if topline results (unreleased at press time) prove positive, inebilizumab could replace rituximab in MG — provided payers do not reject inebilizumab because of cost.
Packed Early-Development Pipeline
Regarding early-stage projects, said Dr. Howard, the pipeline is packed with compounds that target various aspects of the immune system. “The real question with those is, what’s going to be the side effect profile? All of the trials are very early. We need bigger trials with much longer observation for safety, durability, and degree of efficacy.”
The next potential B cell–targeting game changer, he said, is chimeric antigen receptor (CAR) T cell–based therapy. In a phase 2b trial of Descartes-08 (Cartesian Therapeutics), 71% of treated patients experienced clinically meaningful improvement in MG Composite score at 3 months vs 25% for placebo.14
In early clinical trials, said Dr. Howard, patients treated with Descartes-08 — which uses autologous mRNA to target B-cell maturation antigen — have shown “exceptional improvement” lasting 20 or more months. Because the drug is not ingrained permanently into the genome, Descartes-08 avoids potentially severe side effects of DNA-targeting CAR T candidates. Dr. Howard hopes a phase 3 trial will commence around January 2025.
The tolerance approach exemplified by CNP-106 (COUR Pharmaceuticals) and a myasthenia gravis tolerogen (Toleranzia) seeks to prevent the immune system from recognizing and reacting to the NMJ abnormalities that produce myasthenia gravis, potentially providing a cure. “We look forward to those trials as they come online in the next 1-2 years,” said Dr. Howard.
Unmet Needs
Historically, neurologists believed that all myasthenia gravis symptoms stemmed from muscle fatigue — the more active the muscle, the weaker it gets. However, said Dr. Kaminski, some patients might lack measurable weakness but still complain of fatigue.
Elevated levels of cytokines such as interleukin (IL)–6 or IL-17 also can produce fatigue, he noted. “With the drugs we’re using, certainly the new ones, we’re not specifically targeting this fatigue phenomenon, which has been studied in a very limited fashion.”
In the RAISE-XT zilucoplan trial, participants experienced significant improvement in fatigue scores for up to 60 weeks.15 Although zilucoplan does not address fatigue directly, said Dr. Howard, improving myasthenia gravis overall helps reduce fatigue.
The Myasthenia Gravis Symptoms Patient Reported Outcome (MG Symptoms PRO), which Dr. Kaminski helped develop, includes questions designed to distinguish muscular fatigue from overall physical fatigue.16 “I’m very interested in some of the information that’s coming out on long COVID and its effect on muscle,” Dr. Kaminski added. “We might be able to learn from there that there’s still some pathology going on beyond the neuromuscular junction.”
What the field desperately needs, said Dr. Howard, are biomarkers to identify which patients will and will not respond to certain therapeutics. “We’re not there yet.” Such biomarkers are at least 3-7 years from becoming clinical reality.
Promising antibody-independent serum markers include circulating microRNAs. For example, miRNA-150-5p and miRNA-21-5p are elevated in generalized AChR-positive myasthenia gravis and early-onset myasthenia gravis (occurring before age 50) and decline after immunosuppression and thymectomy.17
Among diagnostic modalities for patients with seronegative myasthenia gravis, said Dr. Kaminski, single-fiber EMG is the most sensitive, at approximately 95%. “It’s not perfect.” Moreover, he said, performing this test accurately requires a highly experienced expert, which many treatment centers lack.
Presently, added Dr. Kaminski, orbital MRI is neither specific nor sensitive enough to be clinically useful. “One needs to be careful with these specialized tests that are published from the best laboratory in the world that does the test, and does it repetitively.” As the search for effective myasthenia gravis biomarkers continues, avoiding false-positive results is as important as avoiding false negatives.
References
1. Bubuioc AM et al. J Med Life. 2021 Jan-Mar;14(1):7-16. doi: 10.25122/jml-2020-0145.
2. Deenen JC et al. J Neuromuscul Dis. 2015;2(1):73-85. doi: 10.3233/JND-140045.
3. Kaminski HJ et al. J Clin Invest. 2024 Jun 17;134(12):e179742. doi: 10.1172/JCI179742.
4. Howard JF Jr et al. Lancet Neurol. 2017 Dec;16(12):976-986. doi: 10.1016/S1474-4422(17)30369-1.
5. Huda R. Front Immunol. 2020 Feb 21:11:240. doi: 10.3389/fimmu.2020.00240.
6. Howard JF Jr et al. Lancet Neurol. 2023 May;22(5):395-406. doi: 10.1016/S1474-4422(23)00080-7.
7. Vu T et al. NEJM Evid. 2022 May;1(5):EVIDoa2100066. doi: 10.1056/EVIDoa2100066.
8. Tice JA et al. October 20, 2021. https://icer.org/assessment/myasthenia-gravis/.
9. Yin J et al. Eur J Neurol. 2024 Aug;31(8):e16322. doi: 10.1111/ene.16322.
10. Antozzi C et al. EAN 2024, Abstract EPR-116. https://www.neurology.org/doi/10.1212/WNL.0000000000203660.
11. Yan C et al. JAMA Neurol. 2024 Mar 4;81(4):336-345. doi: 10.1001/jamaneurol.2024.0044.
12. Howard JF Jr et al. Lancet Neurol. 2021 Jul;20(7):526-536. doi: 10.1016/S1474-4422(21)00159-9.
13. Stathopoulos P et al. JCI Insight. 2017 Sep 7;2(17):e94263. doi: 10.1172/jci.insight.94263.
14. Cartesian Therapeutics. Cartesian Therapeutics announces positive topline results from phase 2b trial of Descartes-08 in patients with myasthenia gravis. 2024 Jul 2. https://ir.cartesiantherapeutics.com/news-releases/news-release-details/cartesian-therapeutics-announces-positive-topline-results-phase.
15. Howard JF Jr et al. Ther Adv Neurol Disord. 2024 Apr 17:17:17562864241243186. doi: 10.1177/17562864241243186.
16. Cleanthous S et al. Orphanet J Rare Dis. 2021 Oct 30;16(1):457. doi: 10.1186/s13023-021-02064-0.
17. Sabre L et al. Front Immunol. 2020 Mar 4:11:213. doi: 10.3389/fimmu.2020.00213.
Untangling CIDP
Chronic inflammatory demyelinating polyradiculoneuropathy, or CIDP, is a rare immune-mediated nerve disorder characterized by progressive weakness and sensory impairment in the arms and legs, the result of an autoimmune attack on myelin.
Though some clustering of cases may occur in families, and susceptibility genes have been found, it is not considered a genetic disease. It can strike patients of either sex at any age, though most cases will occur in or after midlife.
Complicating matters further, CIDP has several variants whose symptoms differ from classical presentations.
Many patients who do not have CIDP end up being treated for it, and many CIDP patients experience delays to diagnosis and treatment that can potentially result in greater nerve damage and worse outcomes.
The good news, CIDP experts say, is that the last few years have seen important advances in diagnosis and treatment – including comprehensive new clinical guidelines and the June 2024 approval by the Food and Drug Administration of a new treatment, efgartigimod alfa and hyaluronidase-qvfc (Vyvgart, argenx). This antibody fragment represents the first non-steroid, non-immunoglobulin option for CIDP.
Despite the difficulties of recruiting patients with a tough-to-confirm disease that affects between 2 and 9 of every 100,000 people, according to the GPS-CIDP Foundation clinical trials have been successfully carried out in CIDP, and new ones continue to recruit. The experimental therapies being explored are based on a wide range of proposed disease pathways.
“It’s a very exciting time,” said Jeffrey Allen, MD, a neurologist at the University of Minnesota, Minneapolis, one of three CIDP experts who spoke about this challenging but treatable syndrome, its diagnosis and management, and the research questions that they hope to see answered.
Refining Diagnosis
In classical or typical CIDP, which accounts for most cases, patients present with progressive weakness and numbness that affects the arms and legs symmetrically, with the weakness being both proximal and distal. The disease usually evolves over a period of months, which helps distinguish it from Gullain-Barré syndrome, whose onset is more sudden and progression is less than 4 weeks.
CIDP was first described in the 1970s, and since that time more than a dozen sets of diagnostic criteria have been published. Starting about a decade ago, Dr. Allen and neurologist Richard Lewis, MD, of Cedars-Sinai Medical Center in Los Angeles, California, helped launch an effort to improve them.
“Experts in the field who were seeing patients with CIDP recognized that a lot of referrals coming to them were of people who actually didn’t have it, or they had the disease and were treated for it but didn’t need to be on treatment, or their treatment was very unconventional,” Dr. Allen said. “We wanted to try to put some data behind that.” In 2015 Dr. Allen and Dr. Lewis published a paper that found that nearly half of patients referred with a diagnosis of CIDP failed to meet basic diagnostic requirements.
Erroneous interpretation of nerve conduction studies “was a significant factor” contributing to the misdiagnoses, Dr. Lewis said. And another major problem was that patients’ response to standard treatment with intravenous immunoglobulins (current treatments have also come to include subcutaneous immunoglobulins) was not being measured objectively. Instead of evaluating patients using grip strength, walking tests, or other objective instruments, clinicians asked patients whether they felt better. “The problem is that IVIg makes people feel good,” Dr. Lewis said, “possibly by reducing normal inflammatory agents in the body.”
The 2015 paper caught the attention of neurologists and neuromuscular specialists worldwide, who reported similar problems with misdiagnosis. “And from there we did other work to try to dissect out what the more specific issues are,” Dr. Allen said. “The electrophysiology was a big one.”
Neurologist Nicholas Silvestri, MD, of the University at Buffalo in New York, one of the centers of excellence recognized by the CIDP-GBS Foundation, affirmed that nerve conduction studies, which essential to diagnosing CIDP, “are not as objective as we think they are. They’re very prone to user error and overinterpretation error. If they’re not performed appropriately, things can look like CIDP when they’re not. Very common forms of neuropathy, like diabetic neuropathy, can be misinterpreted as CIDP.”
The Challenge of Variants
After their 2015 paper on diagnostic pitfalls, Dr. Lewis and Dr. Allen, along with colleagues in the United States and Europe, started looking deeper into outcome measures and how to better follow and track patients with CIDP. In 2021 they helped create the first comprehensive clinical guidelines for CIDP in over a decade.
Much of their effort focused on atypical presentations, or what are now called variants, of CIDP — people with predominantly distal disease, asymmetrical symptomology, focal symptoms, or exclusively motor or sensory symptoms. With classical CIDP, “we don’t really have a problem with misdiagnosis,” Dr. Lewis said. With variants, however, misdiagnoses are extremely common. The 2021 guidelines try to address this, proposing differential diagnoses for each of the variants and ways to investigate them.
The guidelines also removed a subgroup of patients previously included as having CIDP. These patients, who comprise about 10% of cases, have antibodies to components of the Node of Ranvier, part of the axonal membrane, and the paranodal myelin. The autoimmune nodopathies do not respond to treatment with immunoglobulins or steroids in the way classical CIDP and its variants do. However, many patients have seen success with the immunotherapy rituximab.
“CIDP is a syndrome, not one disease,” Dr. Lewis said. “So it has been difficult to get guidelines or criteria that are sensitive to all the different forms of the disease, and yet specific for the disease and not overlapping. The nodopathies were pulled out because they don’t respond to usual treatments for CIDP. Hopefully over the years we’ll have even more specific diagnoses and can split out more patients.”
A Need for Better Biomarkers
With the neuromuscular autoimmune disease myasthenia gravis, 85% of patients have antibodies against the muscle acetylcholine receptor (AChR). Another 6% will have antibodies against muscle-specific kinase (MuSK).
Antibody profiles have long guided treatment decisions in myasthenia gravis, with AChR-positive patients responding to corticosteroids, IVIg, complement inhibitors, and other agents. MuSK-positive myasthenia gravis patients, similar to people with autoimmune nodopathies, respond poorly to IVIg but can have dramatic responses when treated with B cell–depleting therapies like rituximab.
Antibodies to nodal proteins neurofascin-155 and contactin-1 have been shown to be involved with the nodopathies. Assays for these are now commercially available, and Dr. Allen recommended that clinicians seek them for patients with a more rapid course, with tremor and ataxia, or who do not respond to standard CIDP treatments.
Still, no dominant autoantibody has been identified for the majority of presentations, including classical presentations. “I suspect it’s a heterogeneous group of multiple antibodies causing the disease,” Dr. Silvestri said. “That may explain to an extent the different manifestations and the different responses to treatment.”
Dr. Lewis said he thinks that, while more antibodies are likely to be discovered in the coming years, “we’re still identifying fewer than 20% of CIDP patients by specific antibodies, so we have a long way to go.”
Promising Trial Landscape
“CIDP is a challenging disease to study because of the diagnostic issues,” Dr. Allen said. “We know that a [nontrivial] percentage of patients ... can go into a drug-free remission. They actually don’t need treatment during that time. We don’t have any way to measure that. And if you put them in a clinical trial, it’s difficult to measure changes in the trial if they didn’t need the drug in the first place.”
In the global ADHERE trail, which looked at efgartigimod alfa and hyaluronidase-qvfc in CIDP patients, the investigators, led by Dr. Allen and Dr. Lewis, challenged patients to be off therapy for 12 weeks and allowed only those with active disease to enroll. They also used an adjudication panel of CIDP experts to review the records of each patient to assure patients had CIDP.
If two experts on the panel independently agreed that it was CIDP, Dr. Lewis said, then patients were eligible for enrollment. “If they both said they weren’t CIDP, they were not eligible. And if there was an argument between the two of them, then a third adjudicator would come in.”
About half of patients screened (n = 221) ended up included, and adjudication panels are now used in most CIDP trials.
The trial saw a positive outcome for efgartigimod alfa and hyaluronidase-qvfc, an antibody fragment that targets neonatal Fc receptor (FcRn), as a way to reduce to levels of pathogenic IgG autoantibodies. (The treatment was previously approved for myasthenia gravis.) The fact that two thirds of participants in the trial responded pointed to the likelihood that most CIDP patients have an IgG-related disease, Dr. Lewis said.
Different types of therapies are now being investigated in CIDP, among them other FcRn-inhibiting drugs and drugs inhibiting complement. Results from these trials may shed more light on the pathophysiology of the disease, which Dr. Silvestri said would be welcome.
“If I can test for antibodies, I can make a more timely diagnosis,” he said. “I’m assuming that some people with CIDP have non–antibody-driven disease. And in those cases, I want to avoid using drugs like Vyvgart, which are targeting antibodies. I want to give them a different therapy.”
Management: A Delicate Dance
Since the 1990s, the standard of care for CIDP has been IVIg and steroids. Newer subcutaneous immunoglobulin products, which take less time to administer, may be more convenient for patients than traditional IVIg and mitigate some concerning side effects.
Efgartigimod alfa and hyaluronidase-qvfc now offers an entirely different option that, while too new for clinicians to have much experience with in CIDP, represents further convenience for patients, with dosing in one 90-second subcutaneous injection per week.
In general, the sooner people are diagnosed and on therapy, the better they are likely to do, with fewer risks of irreversible axonal loss and disability. Referring to CIDP centers of excellence can help speed a definitive diagnosis.
Some patients will see a complete or near-complete recovery, while others will not. “It’s important to be up front with patients about what the benefit of treatments are, what are the expectations of treatment, what we can potentially get back, and what’s unlikely to come back,” Dr. Allen said. “We know that irreversible deficits are not uncommon in folks with CIDP. Part of that is driven by how severe their disease is or how long they’ve had it.”
Good CIDP management, according to the 2021 guidelines, involves making periodic dose reductions or withdrawing therapies on a trial basis, because people can and do experience remission. “We don’t have any test that tells us if somebody needs treatment or not. So this is the best we can do right now,” Dr. Allen said.
This process can be anxiety provoking for patients. “In my practice, there are no surprises,” he said. “We don’t typically say, ‘we’re going to stop your treatment today.’ It’s a discussion with a lead up that’s usually many months long.”
Management of CIDP also requires discussions to elicit when and whether worsening is occurring, along with a clear sense, by both patient and clinician, of what constitutes worsening.
Serial nerve conduction studies are not very useful, Dr. Lewis said, but objective disability measures are and should be more broadly adopted. These include the Medical Research Council sumscore, a test of 12 muscles that can determine weakness; a hand grip test; and functional disability scales such as Inflammatory Rasch Overall Disability Scale and Inflammatory Neuropathy Cause and Treatment scale. All are quick to administer in the office, and some can be done by the patient at home, providing the clinician useful information between visits.
“We could do a better job with educating [clinicians] on the value of different outcome measures that can really quantify disease activity,” Dr. Allen said, and pointed to the GBS-CIDP Foundation centers of excellence, which exist in most regions of the United States, as an outstanding resource for anyone wanting to know more.
“The centers are really, really helpful when you’re trying to work through some of these issues,” he said.
Suggested Reading
Allen JA and Lewis RA. Neurology. 2015 Aug 11;85(6):498-504. doi: 10.1212/WNL.0000000000001833.
Allen J et al. Neurology. 2024;102(17_supplement_1). doi: 10.1212/WNL-.0000000000206324.
Van den Bergh PYK et al. J Peripher Nerv Syst. 2021 Sep;26(3):242-268. doi: 10.1111/jns.12455.
Chronic inflammatory demyelinating polyradiculoneuropathy, or CIDP, is a rare immune-mediated nerve disorder characterized by progressive weakness and sensory impairment in the arms and legs, the result of an autoimmune attack on myelin.
Though some clustering of cases may occur in families, and susceptibility genes have been found, it is not considered a genetic disease. It can strike patients of either sex at any age, though most cases will occur in or after midlife.
Complicating matters further, CIDP has several variants whose symptoms differ from classical presentations.
Many patients who do not have CIDP end up being treated for it, and many CIDP patients experience delays to diagnosis and treatment that can potentially result in greater nerve damage and worse outcomes.
The good news, CIDP experts say, is that the last few years have seen important advances in diagnosis and treatment – including comprehensive new clinical guidelines and the June 2024 approval by the Food and Drug Administration of a new treatment, efgartigimod alfa and hyaluronidase-qvfc (Vyvgart, argenx). This antibody fragment represents the first non-steroid, non-immunoglobulin option for CIDP.
Despite the difficulties of recruiting patients with a tough-to-confirm disease that affects between 2 and 9 of every 100,000 people, according to the GPS-CIDP Foundation clinical trials have been successfully carried out in CIDP, and new ones continue to recruit. The experimental therapies being explored are based on a wide range of proposed disease pathways.
“It’s a very exciting time,” said Jeffrey Allen, MD, a neurologist at the University of Minnesota, Minneapolis, one of three CIDP experts who spoke about this challenging but treatable syndrome, its diagnosis and management, and the research questions that they hope to see answered.
Refining Diagnosis
In classical or typical CIDP, which accounts for most cases, patients present with progressive weakness and numbness that affects the arms and legs symmetrically, with the weakness being both proximal and distal. The disease usually evolves over a period of months, which helps distinguish it from Gullain-Barré syndrome, whose onset is more sudden and progression is less than 4 weeks.
CIDP was first described in the 1970s, and since that time more than a dozen sets of diagnostic criteria have been published. Starting about a decade ago, Dr. Allen and neurologist Richard Lewis, MD, of Cedars-Sinai Medical Center in Los Angeles, California, helped launch an effort to improve them.
“Experts in the field who were seeing patients with CIDP recognized that a lot of referrals coming to them were of people who actually didn’t have it, or they had the disease and were treated for it but didn’t need to be on treatment, or their treatment was very unconventional,” Dr. Allen said. “We wanted to try to put some data behind that.” In 2015 Dr. Allen and Dr. Lewis published a paper that found that nearly half of patients referred with a diagnosis of CIDP failed to meet basic diagnostic requirements.
Erroneous interpretation of nerve conduction studies “was a significant factor” contributing to the misdiagnoses, Dr. Lewis said. And another major problem was that patients’ response to standard treatment with intravenous immunoglobulins (current treatments have also come to include subcutaneous immunoglobulins) was not being measured objectively. Instead of evaluating patients using grip strength, walking tests, or other objective instruments, clinicians asked patients whether they felt better. “The problem is that IVIg makes people feel good,” Dr. Lewis said, “possibly by reducing normal inflammatory agents in the body.”
The 2015 paper caught the attention of neurologists and neuromuscular specialists worldwide, who reported similar problems with misdiagnosis. “And from there we did other work to try to dissect out what the more specific issues are,” Dr. Allen said. “The electrophysiology was a big one.”
Neurologist Nicholas Silvestri, MD, of the University at Buffalo in New York, one of the centers of excellence recognized by the CIDP-GBS Foundation, affirmed that nerve conduction studies, which essential to diagnosing CIDP, “are not as objective as we think they are. They’re very prone to user error and overinterpretation error. If they’re not performed appropriately, things can look like CIDP when they’re not. Very common forms of neuropathy, like diabetic neuropathy, can be misinterpreted as CIDP.”
The Challenge of Variants
After their 2015 paper on diagnostic pitfalls, Dr. Lewis and Dr. Allen, along with colleagues in the United States and Europe, started looking deeper into outcome measures and how to better follow and track patients with CIDP. In 2021 they helped create the first comprehensive clinical guidelines for CIDP in over a decade.
Much of their effort focused on atypical presentations, or what are now called variants, of CIDP — people with predominantly distal disease, asymmetrical symptomology, focal symptoms, or exclusively motor or sensory symptoms. With classical CIDP, “we don’t really have a problem with misdiagnosis,” Dr. Lewis said. With variants, however, misdiagnoses are extremely common. The 2021 guidelines try to address this, proposing differential diagnoses for each of the variants and ways to investigate them.
The guidelines also removed a subgroup of patients previously included as having CIDP. These patients, who comprise about 10% of cases, have antibodies to components of the Node of Ranvier, part of the axonal membrane, and the paranodal myelin. The autoimmune nodopathies do not respond to treatment with immunoglobulins or steroids in the way classical CIDP and its variants do. However, many patients have seen success with the immunotherapy rituximab.
“CIDP is a syndrome, not one disease,” Dr. Lewis said. “So it has been difficult to get guidelines or criteria that are sensitive to all the different forms of the disease, and yet specific for the disease and not overlapping. The nodopathies were pulled out because they don’t respond to usual treatments for CIDP. Hopefully over the years we’ll have even more specific diagnoses and can split out more patients.”
A Need for Better Biomarkers
With the neuromuscular autoimmune disease myasthenia gravis, 85% of patients have antibodies against the muscle acetylcholine receptor (AChR). Another 6% will have antibodies against muscle-specific kinase (MuSK).
Antibody profiles have long guided treatment decisions in myasthenia gravis, with AChR-positive patients responding to corticosteroids, IVIg, complement inhibitors, and other agents. MuSK-positive myasthenia gravis patients, similar to people with autoimmune nodopathies, respond poorly to IVIg but can have dramatic responses when treated with B cell–depleting therapies like rituximab.
Antibodies to nodal proteins neurofascin-155 and contactin-1 have been shown to be involved with the nodopathies. Assays for these are now commercially available, and Dr. Allen recommended that clinicians seek them for patients with a more rapid course, with tremor and ataxia, or who do not respond to standard CIDP treatments.
Still, no dominant autoantibody has been identified for the majority of presentations, including classical presentations. “I suspect it’s a heterogeneous group of multiple antibodies causing the disease,” Dr. Silvestri said. “That may explain to an extent the different manifestations and the different responses to treatment.”
Dr. Lewis said he thinks that, while more antibodies are likely to be discovered in the coming years, “we’re still identifying fewer than 20% of CIDP patients by specific antibodies, so we have a long way to go.”
Promising Trial Landscape
“CIDP is a challenging disease to study because of the diagnostic issues,” Dr. Allen said. “We know that a [nontrivial] percentage of patients ... can go into a drug-free remission. They actually don’t need treatment during that time. We don’t have any way to measure that. And if you put them in a clinical trial, it’s difficult to measure changes in the trial if they didn’t need the drug in the first place.”
In the global ADHERE trail, which looked at efgartigimod alfa and hyaluronidase-qvfc in CIDP patients, the investigators, led by Dr. Allen and Dr. Lewis, challenged patients to be off therapy for 12 weeks and allowed only those with active disease to enroll. They also used an adjudication panel of CIDP experts to review the records of each patient to assure patients had CIDP.
If two experts on the panel independently agreed that it was CIDP, Dr. Lewis said, then patients were eligible for enrollment. “If they both said they weren’t CIDP, they were not eligible. And if there was an argument between the two of them, then a third adjudicator would come in.”
About half of patients screened (n = 221) ended up included, and adjudication panels are now used in most CIDP trials.
The trial saw a positive outcome for efgartigimod alfa and hyaluronidase-qvfc, an antibody fragment that targets neonatal Fc receptor (FcRn), as a way to reduce to levels of pathogenic IgG autoantibodies. (The treatment was previously approved for myasthenia gravis.) The fact that two thirds of participants in the trial responded pointed to the likelihood that most CIDP patients have an IgG-related disease, Dr. Lewis said.
Different types of therapies are now being investigated in CIDP, among them other FcRn-inhibiting drugs and drugs inhibiting complement. Results from these trials may shed more light on the pathophysiology of the disease, which Dr. Silvestri said would be welcome.
“If I can test for antibodies, I can make a more timely diagnosis,” he said. “I’m assuming that some people with CIDP have non–antibody-driven disease. And in those cases, I want to avoid using drugs like Vyvgart, which are targeting antibodies. I want to give them a different therapy.”
Management: A Delicate Dance
Since the 1990s, the standard of care for CIDP has been IVIg and steroids. Newer subcutaneous immunoglobulin products, which take less time to administer, may be more convenient for patients than traditional IVIg and mitigate some concerning side effects.
Efgartigimod alfa and hyaluronidase-qvfc now offers an entirely different option that, while too new for clinicians to have much experience with in CIDP, represents further convenience for patients, with dosing in one 90-second subcutaneous injection per week.
In general, the sooner people are diagnosed and on therapy, the better they are likely to do, with fewer risks of irreversible axonal loss and disability. Referring to CIDP centers of excellence can help speed a definitive diagnosis.
Some patients will see a complete or near-complete recovery, while others will not. “It’s important to be up front with patients about what the benefit of treatments are, what are the expectations of treatment, what we can potentially get back, and what’s unlikely to come back,” Dr. Allen said. “We know that irreversible deficits are not uncommon in folks with CIDP. Part of that is driven by how severe their disease is or how long they’ve had it.”
Good CIDP management, according to the 2021 guidelines, involves making periodic dose reductions or withdrawing therapies on a trial basis, because people can and do experience remission. “We don’t have any test that tells us if somebody needs treatment or not. So this is the best we can do right now,” Dr. Allen said.
This process can be anxiety provoking for patients. “In my practice, there are no surprises,” he said. “We don’t typically say, ‘we’re going to stop your treatment today.’ It’s a discussion with a lead up that’s usually many months long.”
Management of CIDP also requires discussions to elicit when and whether worsening is occurring, along with a clear sense, by both patient and clinician, of what constitutes worsening.
Serial nerve conduction studies are not very useful, Dr. Lewis said, but objective disability measures are and should be more broadly adopted. These include the Medical Research Council sumscore, a test of 12 muscles that can determine weakness; a hand grip test; and functional disability scales such as Inflammatory Rasch Overall Disability Scale and Inflammatory Neuropathy Cause and Treatment scale. All are quick to administer in the office, and some can be done by the patient at home, providing the clinician useful information between visits.
“We could do a better job with educating [clinicians] on the value of different outcome measures that can really quantify disease activity,” Dr. Allen said, and pointed to the GBS-CIDP Foundation centers of excellence, which exist in most regions of the United States, as an outstanding resource for anyone wanting to know more.
“The centers are really, really helpful when you’re trying to work through some of these issues,” he said.
Suggested Reading
Allen JA and Lewis RA. Neurology. 2015 Aug 11;85(6):498-504. doi: 10.1212/WNL.0000000000001833.
Allen J et al. Neurology. 2024;102(17_supplement_1). doi: 10.1212/WNL-.0000000000206324.
Van den Bergh PYK et al. J Peripher Nerv Syst. 2021 Sep;26(3):242-268. doi: 10.1111/jns.12455.
Chronic inflammatory demyelinating polyradiculoneuropathy, or CIDP, is a rare immune-mediated nerve disorder characterized by progressive weakness and sensory impairment in the arms and legs, the result of an autoimmune attack on myelin.
Though some clustering of cases may occur in families, and susceptibility genes have been found, it is not considered a genetic disease. It can strike patients of either sex at any age, though most cases will occur in or after midlife.
Complicating matters further, CIDP has several variants whose symptoms differ from classical presentations.
Many patients who do not have CIDP end up being treated for it, and many CIDP patients experience delays to diagnosis and treatment that can potentially result in greater nerve damage and worse outcomes.
The good news, CIDP experts say, is that the last few years have seen important advances in diagnosis and treatment – including comprehensive new clinical guidelines and the June 2024 approval by the Food and Drug Administration of a new treatment, efgartigimod alfa and hyaluronidase-qvfc (Vyvgart, argenx). This antibody fragment represents the first non-steroid, non-immunoglobulin option for CIDP.
Despite the difficulties of recruiting patients with a tough-to-confirm disease that affects between 2 and 9 of every 100,000 people, according to the GPS-CIDP Foundation clinical trials have been successfully carried out in CIDP, and new ones continue to recruit. The experimental therapies being explored are based on a wide range of proposed disease pathways.
“It’s a very exciting time,” said Jeffrey Allen, MD, a neurologist at the University of Minnesota, Minneapolis, one of three CIDP experts who spoke about this challenging but treatable syndrome, its diagnosis and management, and the research questions that they hope to see answered.
Refining Diagnosis
In classical or typical CIDP, which accounts for most cases, patients present with progressive weakness and numbness that affects the arms and legs symmetrically, with the weakness being both proximal and distal. The disease usually evolves over a period of months, which helps distinguish it from Gullain-Barré syndrome, whose onset is more sudden and progression is less than 4 weeks.
CIDP was first described in the 1970s, and since that time more than a dozen sets of diagnostic criteria have been published. Starting about a decade ago, Dr. Allen and neurologist Richard Lewis, MD, of Cedars-Sinai Medical Center in Los Angeles, California, helped launch an effort to improve them.
“Experts in the field who were seeing patients with CIDP recognized that a lot of referrals coming to them were of people who actually didn’t have it, or they had the disease and were treated for it but didn’t need to be on treatment, or their treatment was very unconventional,” Dr. Allen said. “We wanted to try to put some data behind that.” In 2015 Dr. Allen and Dr. Lewis published a paper that found that nearly half of patients referred with a diagnosis of CIDP failed to meet basic diagnostic requirements.
Erroneous interpretation of nerve conduction studies “was a significant factor” contributing to the misdiagnoses, Dr. Lewis said. And another major problem was that patients’ response to standard treatment with intravenous immunoglobulins (current treatments have also come to include subcutaneous immunoglobulins) was not being measured objectively. Instead of evaluating patients using grip strength, walking tests, or other objective instruments, clinicians asked patients whether they felt better. “The problem is that IVIg makes people feel good,” Dr. Lewis said, “possibly by reducing normal inflammatory agents in the body.”
The 2015 paper caught the attention of neurologists and neuromuscular specialists worldwide, who reported similar problems with misdiagnosis. “And from there we did other work to try to dissect out what the more specific issues are,” Dr. Allen said. “The electrophysiology was a big one.”
Neurologist Nicholas Silvestri, MD, of the University at Buffalo in New York, one of the centers of excellence recognized by the CIDP-GBS Foundation, affirmed that nerve conduction studies, which essential to diagnosing CIDP, “are not as objective as we think they are. They’re very prone to user error and overinterpretation error. If they’re not performed appropriately, things can look like CIDP when they’re not. Very common forms of neuropathy, like diabetic neuropathy, can be misinterpreted as CIDP.”
The Challenge of Variants
After their 2015 paper on diagnostic pitfalls, Dr. Lewis and Dr. Allen, along with colleagues in the United States and Europe, started looking deeper into outcome measures and how to better follow and track patients with CIDP. In 2021 they helped create the first comprehensive clinical guidelines for CIDP in over a decade.
Much of their effort focused on atypical presentations, or what are now called variants, of CIDP — people with predominantly distal disease, asymmetrical symptomology, focal symptoms, or exclusively motor or sensory symptoms. With classical CIDP, “we don’t really have a problem with misdiagnosis,” Dr. Lewis said. With variants, however, misdiagnoses are extremely common. The 2021 guidelines try to address this, proposing differential diagnoses for each of the variants and ways to investigate them.
The guidelines also removed a subgroup of patients previously included as having CIDP. These patients, who comprise about 10% of cases, have antibodies to components of the Node of Ranvier, part of the axonal membrane, and the paranodal myelin. The autoimmune nodopathies do not respond to treatment with immunoglobulins or steroids in the way classical CIDP and its variants do. However, many patients have seen success with the immunotherapy rituximab.
“CIDP is a syndrome, not one disease,” Dr. Lewis said. “So it has been difficult to get guidelines or criteria that are sensitive to all the different forms of the disease, and yet specific for the disease and not overlapping. The nodopathies were pulled out because they don’t respond to usual treatments for CIDP. Hopefully over the years we’ll have even more specific diagnoses and can split out more patients.”
A Need for Better Biomarkers
With the neuromuscular autoimmune disease myasthenia gravis, 85% of patients have antibodies against the muscle acetylcholine receptor (AChR). Another 6% will have antibodies against muscle-specific kinase (MuSK).
Antibody profiles have long guided treatment decisions in myasthenia gravis, with AChR-positive patients responding to corticosteroids, IVIg, complement inhibitors, and other agents. MuSK-positive myasthenia gravis patients, similar to people with autoimmune nodopathies, respond poorly to IVIg but can have dramatic responses when treated with B cell–depleting therapies like rituximab.
Antibodies to nodal proteins neurofascin-155 and contactin-1 have been shown to be involved with the nodopathies. Assays for these are now commercially available, and Dr. Allen recommended that clinicians seek them for patients with a more rapid course, with tremor and ataxia, or who do not respond to standard CIDP treatments.
Still, no dominant autoantibody has been identified for the majority of presentations, including classical presentations. “I suspect it’s a heterogeneous group of multiple antibodies causing the disease,” Dr. Silvestri said. “That may explain to an extent the different manifestations and the different responses to treatment.”
Dr. Lewis said he thinks that, while more antibodies are likely to be discovered in the coming years, “we’re still identifying fewer than 20% of CIDP patients by specific antibodies, so we have a long way to go.”
Promising Trial Landscape
“CIDP is a challenging disease to study because of the diagnostic issues,” Dr. Allen said. “We know that a [nontrivial] percentage of patients ... can go into a drug-free remission. They actually don’t need treatment during that time. We don’t have any way to measure that. And if you put them in a clinical trial, it’s difficult to measure changes in the trial if they didn’t need the drug in the first place.”
In the global ADHERE trail, which looked at efgartigimod alfa and hyaluronidase-qvfc in CIDP patients, the investigators, led by Dr. Allen and Dr. Lewis, challenged patients to be off therapy for 12 weeks and allowed only those with active disease to enroll. They also used an adjudication panel of CIDP experts to review the records of each patient to assure patients had CIDP.
If two experts on the panel independently agreed that it was CIDP, Dr. Lewis said, then patients were eligible for enrollment. “If they both said they weren’t CIDP, they were not eligible. And if there was an argument between the two of them, then a third adjudicator would come in.”
About half of patients screened (n = 221) ended up included, and adjudication panels are now used in most CIDP trials.
The trial saw a positive outcome for efgartigimod alfa and hyaluronidase-qvfc, an antibody fragment that targets neonatal Fc receptor (FcRn), as a way to reduce to levels of pathogenic IgG autoantibodies. (The treatment was previously approved for myasthenia gravis.) The fact that two thirds of participants in the trial responded pointed to the likelihood that most CIDP patients have an IgG-related disease, Dr. Lewis said.
Different types of therapies are now being investigated in CIDP, among them other FcRn-inhibiting drugs and drugs inhibiting complement. Results from these trials may shed more light on the pathophysiology of the disease, which Dr. Silvestri said would be welcome.
“If I can test for antibodies, I can make a more timely diagnosis,” he said. “I’m assuming that some people with CIDP have non–antibody-driven disease. And in those cases, I want to avoid using drugs like Vyvgart, which are targeting antibodies. I want to give them a different therapy.”
Management: A Delicate Dance
Since the 1990s, the standard of care for CIDP has been IVIg and steroids. Newer subcutaneous immunoglobulin products, which take less time to administer, may be more convenient for patients than traditional IVIg and mitigate some concerning side effects.
Efgartigimod alfa and hyaluronidase-qvfc now offers an entirely different option that, while too new for clinicians to have much experience with in CIDP, represents further convenience for patients, with dosing in one 90-second subcutaneous injection per week.
In general, the sooner people are diagnosed and on therapy, the better they are likely to do, with fewer risks of irreversible axonal loss and disability. Referring to CIDP centers of excellence can help speed a definitive diagnosis.
Some patients will see a complete or near-complete recovery, while others will not. “It’s important to be up front with patients about what the benefit of treatments are, what are the expectations of treatment, what we can potentially get back, and what’s unlikely to come back,” Dr. Allen said. “We know that irreversible deficits are not uncommon in folks with CIDP. Part of that is driven by how severe their disease is or how long they’ve had it.”
Good CIDP management, according to the 2021 guidelines, involves making periodic dose reductions or withdrawing therapies on a trial basis, because people can and do experience remission. “We don’t have any test that tells us if somebody needs treatment or not. So this is the best we can do right now,” Dr. Allen said.
This process can be anxiety provoking for patients. “In my practice, there are no surprises,” he said. “We don’t typically say, ‘we’re going to stop your treatment today.’ It’s a discussion with a lead up that’s usually many months long.”
Management of CIDP also requires discussions to elicit when and whether worsening is occurring, along with a clear sense, by both patient and clinician, of what constitutes worsening.
Serial nerve conduction studies are not very useful, Dr. Lewis said, but objective disability measures are and should be more broadly adopted. These include the Medical Research Council sumscore, a test of 12 muscles that can determine weakness; a hand grip test; and functional disability scales such as Inflammatory Rasch Overall Disability Scale and Inflammatory Neuropathy Cause and Treatment scale. All are quick to administer in the office, and some can be done by the patient at home, providing the clinician useful information between visits.
“We could do a better job with educating [clinicians] on the value of different outcome measures that can really quantify disease activity,” Dr. Allen said, and pointed to the GBS-CIDP Foundation centers of excellence, which exist in most regions of the United States, as an outstanding resource for anyone wanting to know more.
“The centers are really, really helpful when you’re trying to work through some of these issues,” he said.
Suggested Reading
Allen JA and Lewis RA. Neurology. 2015 Aug 11;85(6):498-504. doi: 10.1212/WNL.0000000000001833.
Allen J et al. Neurology. 2024;102(17_supplement_1). doi: 10.1212/WNL-.0000000000206324.
Van den Bergh PYK et al. J Peripher Nerv Syst. 2021 Sep;26(3):242-268. doi: 10.1111/jns.12455.
Diagnosing and Managing Duchenne Muscular Dystrophy: Tips for Practicing Clinicians
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)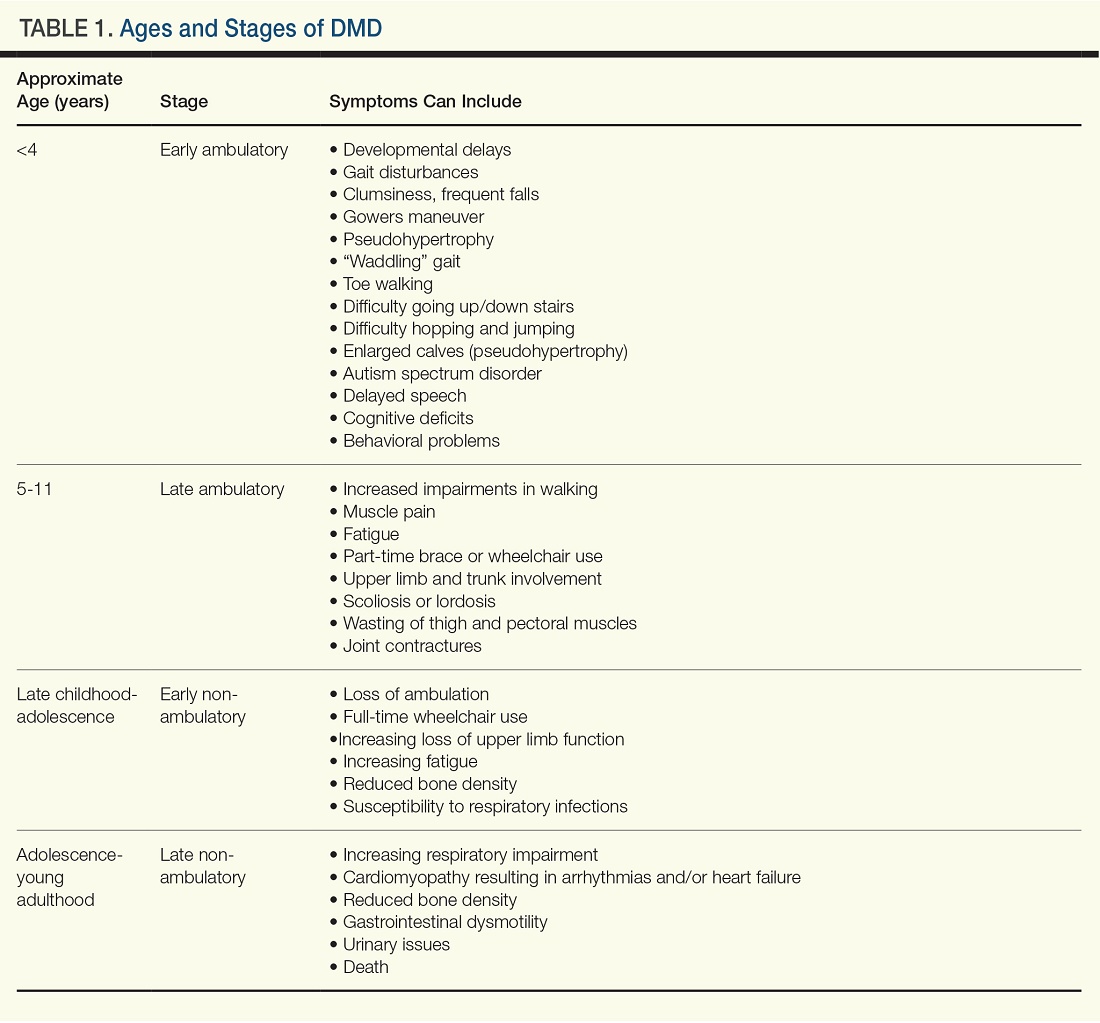
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.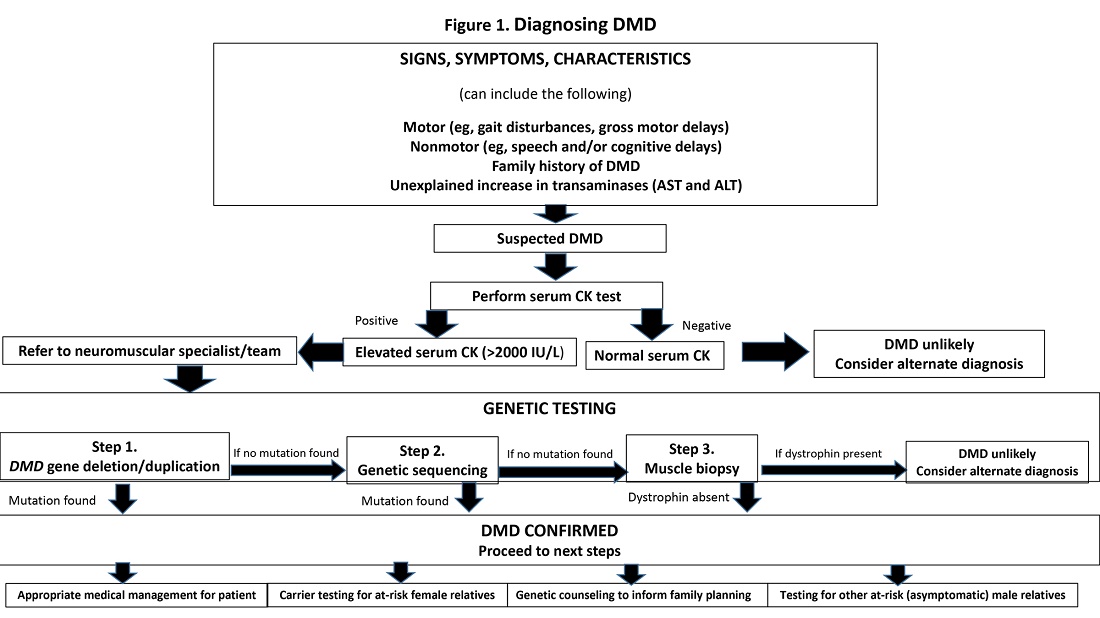
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.

“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”
Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.
“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”
Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.
Duchenne muscular dystrophy (DMD) is a severe progressive inherited disease characterized by muscle wasting and ultimately culminating in death. It’s a common enough neuromuscular disorder that pediatricians and family practice physicians are likely to see at least a couple of patients with DMD over the course of their career,” John Brandsema, MD, Neuromuscular Section Head, Division of Neurology, Children’s Hospital of Philadelphia in Pennsylvania, said in an interview. Healthcare providers should therefore be familiar with the disorder so as to provide timely diagnosis and early intervention as well as practical and emotional support to the patient and family/caregivers as they traverse the challenging and often heartbreaking journey with this condition.
Pathophysiology and Disease Trajectory
DMD is caused by pathogenic variants in the X-linked DMD gene, leading to reduction in dystrophin, a protein that serves as a cytoskeletal integrator, stabilizing the plasma membrane of striated muscle cells. Dystrophin is critical for muscle membrane stability.2 In particular, mutations in the gene that encodes for dystrophin lead to dysfunction in Dp427m, which is the muscle isoform of dystrophin.3,4
DMD is one of several types of muscular dystrophies. All are progressive disorders. Over time, healthy muscle fibers disappear and are replaced by fibrotic tissue and fat, making the muscles “less able to generate force for everyday activity.”2 Ultimately, the skeletal muscle dysfunction affects not only the patient’s day-to-day mobility but other systems as well. Most patients with DMD eventually die of cardiac and/or respiratory failure between the ages of 20 and 40 years, with a median life expectancy of 22 years — although children born after 1990 have a somewhat higher median life expectancy (28 years), because of the improving standard of care.3,5
Typically, DMD first presents with developmental delays and weakness in skeletal leg muscles. As the disease goes through stages of progression, it starts involving upper extremities and other systems. (Table 1)
Genetic Causes of DMD
The DMD gene, located on the X chromosome, encodes for the production of dystrophin. Variants of this gene result in the lack of dystrophin protein, leading in turn to muscle fiber degeneration and the progressive symptoms of DMD. Because of the gene’s location on the X chromosome, males (who don’t have a second copy of the X chromosome) cannot compensate for the mutated gene, which is why the disease affects male children. Females with this mutation are carriers and typically do not develop the same severity of symptoms, although they might have milder muscle cramps, weakness, and cardiac issues.3
A female carrier with DMD (or any other X-linked disorder) has a 25% chance to have a carrier daughter, a 25% change of having a noncarrier daughter, a 25% chance of having an affected son, and a 25% chance of having a nonaffected son. A male with the disorder will pass the mutated gene on to his daughters who then become carriers. He cannot pass the disorder on to his sons because males inherit only the Y chromosome from their fathers.3
Diagnosing DMD
“It can take as long as 1-3 years for a child to be diagnosed with DMD,” Dr. Brandsema said. “Parents typically have concerns and know that something is ‘off’ about their child and they’re sent to various specialists, but it usually takes time for an accurate diagnosis to be made.” The mean age at diagnosis of DMD is between ages 4 and 5 years.6
Early identification of infants at risk for developing DMD can help move the needle toward earlier diagnosis. Newborn screening for DMD has been researched and piloted in several programs.6 In 2023, DMD was nominated for inclusion in the Recommended Universal Screening Panel (RUSP) for universal newborn screening. But in May 2024, the advisory committee on Heritable Disorders in Newborns and Children decided to postpone the vote to include DMD in the RUSP, requesting additional information to ensure an evidence-based decision.
In the absence of universal newborn screening for DMD, alternative approaches have been proposed to reduce the delay in clinical diagnosis and specialist referral, including increasing awareness among healthcare providers (eg, pediatricians, pediatric neurologists, and primary care physicians).6
The National Task Force for Early Identification of Childhood Neuromuscular Disorders delineates the steps necessary to identify pediatric muscle weakness and signs of neuromuscular disease. Primary care providers are encouraged to engage in regular developmental surveillance. A surveillance aid lays out the timetable for recommended visits, typical developmental milestones, and components of surveillance. Clinical evaluation includes a detailed patient history, family history, and physical examination.
If a neuromuscular condition is suspected, laboratory work should include creatinine phosphokinase (CK).6 Elevated serum CK points to leakage of CK through the muscle membrane, suggesting muscle damage. If CK is elevated, genetic testing should be performed; and, if negative, it should be followed by genetic sequencing that tests for small-scale mutations in the DMD gene. If that test is negative, a muscle biopsy should be performed to test for deep intronic mutations in the DMD gene.4
The diagnostic process and immediate steps after a confirmed DMD diagnosis is found in Figure 1.
Targeting Inflammation in DMD
Traditionally, corticosteroids have been the only available medical treatment for DMD and they remain a cornerstone of DMD management. A meta-analysis found “moderate evidence” that corticosteroid therapy improves muscle strength and function in the short term (12 months), and strength up to 2 years.10
The two most common corticosteroids for DMD are prednisone and deflazacort. Deflazacort (Emflaza, PTC Therapeutics) was approved in 2017 to treat patients ages 5 years and older with DMD, subsequently expanded to 2 years and older. Deflazacort has been found to be more effective than prednisone in improving functional outcomes, delaying the onset of cardiomyopathy, and improving overall survival, with fewer adverse effects.11
In 2023, vamorolone (Agamree, Catalyst Pharmaceuticals) was approved by the Food and Drug Administration (FDA) to treat DMD patients (ages 2 years and older). Vamorolone is a dissociative steroidal anti-inflammatory that reduces bone morbidities and is regarded as a safer alternative than prednisone. A clinical trial comparing two doses of vamorolone with prednisone for 24 weeks found that vamorolone 6 mg/kg per day met the primary endpoint (time to stand velocity) and four sequential secondary motor function endpoints, with less bone morbidity, compared to prednisone.12 A more recent trial found improvements in motor outcomes at 48 weeks with a dose of 6 mg/kg per day of vamorolone. Bone morbidities of prednisone were reversed when the patient transitioned to vamorolone.13
“Steroid treatment has been proven to help, usually taken daily, although other schedules have been tried,” Dr. Brandsema said. However, all steroids are fraught with adverse effects and are suboptimal in the long term in reducing the disease burden.
The anti-inflammatory agent givinostat (Duvyzat, ITF Therapeutics), an oral histone deacetylase (HDAC) inhibitor, was approved in March 2024 for the treatment of DMD in patients 6 years of age and older. It is the first nonsteroidal drug to treat patients with all genetic variants of the disease, and it has a unique mechanism of action. Deficits in dystrophin can lead to increased HDAC activity in DMD, reducing the expression of genes involved in muscle regeneration. Givinostat therefore can help to counteract the pathogenic events downstream of dystrophin deficiency by inhibiting HDAC.14
Approval for givinostat was based on the phase 3 EPIDYS trial, which randomized 179 boys with DMD to receive either givinostat or placebo. Although results of a functional task worsened in both groups over the 12-month study period, the decline was significantly smaller with givinostat versus placebo. The most common adverse events were diarrhea and vomiting.14 Dr. Brandsema noted that monitoring of triglycerides and platelet count is required, as hypertriglyceridemia and thrombocytopenia can occur. This treatment was studied in tandem with corticosteroids as a combination approach to muscle stabilization.
New Pharmacotherapeutic Options: Exon-Skipping Agents
Today’s treatments have expanded beyond corticosteroids, with newer therapeutic options that include targeted exon-skipping therapies and, more recently, gene therapies. “These new treatment paradigms have changed the face of DMD treatment,” Dr. Brandsema said.
“Exon-skipping drugs in their current form have only a modest effect, but at least they’re a step in the right direction and a breakthrough, in terms of slowing disease progression,” Dr. Brandsema said.
Current exon-skipping agents use antisense phosphorodiamidate morpholino oligomers (PMOs) to restore a DMD open reading frame. Next-generation drugs called cell-penetrating peptide-conjugated PMOs (PPMOs) are being actively researched, Dr. Brandsema said. These agents have shown enhanced cellular uptake and more efficient dystrophin restoration, compared with unconjugated PMOs.15
There are currently four FDA-approved exon-skipping agents for DMD, all of which are administered via a weekly intravenous infusion: Casimersen (Amondys-45, SRP-4045), approved by the FDA in 2021; Eteplirsen (Exondys 51), approved in 2016; Golodirsen (Vyondys 53,SRP-4053), approved in 2019; and Vitolarsen (Viltepso), approved in 2020. They can be associated with multiple side effects, depending on the drug, including upper respiratory infection, fever, cough, rash, and gastrointestinal issues.16 These agents have the potential to help 30% of DMD patients, restoring low levels of dystrophin.16
Gene Transfer Therapies
Gene transfer therapies, a new class of agents, utilize a nonpathogenic viral vector (adeno-associated virus) to transfer specific genes to patients with DMD. Gene therapy involves overexpressing the micro-dystrophin gene to restore functional dystrophin expression.16
Multiple clinical trials of gene therapy are currently in progress. In 2023, delandistrogene moxeparvovec-rokl (Elevidys, Serepta) was granted accelerated FDA approval for ambulatory individuals with DMD between the ages of 4 and 5 years of age and a confirmed mutation in the DMD gene. It received expanded approval in June 2024 to include ambulatory and nonambulatory individuals aged 4 years and older with DMD and a confirmed mutation in the DMD gene (with the exception of exon 8 or 9 mutations).
The approval was based on preliminary data from two double-blind, placebo-controlled studies and two open-label studies, which enrolled a total of 218 male patients (including those who received placebo) with a confirmed disease-causing mutation in the DMD gene.
Delandistrogene moxeparvovec-rokl is delivered as a one-time infusion and has been associated with side effects and “a lot of potential issues,” Dr. Brandsema said. “We’ve seen cardiac effects, immune system effects, increased muscle inflammation and hepatic complications, and some people who became quite unwell were hospitalized for a long time.”
Fortunately, he added, “these seem to be rare but they do happen. Once the medication has been delivered, it’s permanently in the body, so you’re managing the side effects potentially on a long-term basis.”
It is critical to discuss the risks and benefits of this treatment with the family and caregivers and with the patient as well, if he old enough and able to participate in the decision-making progress. “We don’t want to give unrealistic expectations and we want people to be aware of the potential downside of this treatment,” he said. “This is a very complex discussion because the trajectory of the disease is so devastating and this treatment does hold out hope that other therapies don’t necessarily have.”
Nonpharmacologic Interventions
Physical therapy is a mainstay in DMD treatment, addressing protection of fragile muscles, preservation of strength, and prevention of muscle contractures.16 Given the respiratory impairments that occur with DMD progression, respiratory monitoring and therapy are essential; however, the number and type of evaluations and interventions vary with the stage of the disease, intensifying as the disease progresses.16 Similarly, cardiac monitoring should begin early, with patients screened for cardiac complications, and should intensify through the stages of disease progression.16
Bone health is compromised in patients with DMD, both as a result of corticosteroid treatment and as part of the disease itself. Fractures may be asymptomatic and may go unnoticed. Thus, bone health surveillance and maintenance are critical components of DMD management.16
Patients with DMD often experience gastrointestinal issues. They may experience weight gain because of lack of mobility and corticosteroid use in early stages, or weight loss as a result of diet or fluid imbalance, low bone density, or dysphagia in later stages. Patients should be closely followed by a nutritionist, a gastroenterologist as needed, and a physical therapist.16
Psychosocial support “should be developed and implemented across the lifespan in a manner that promotes thinking about the future and sets expectations that individuals will actively participate in their care and daily activities.”9 This includes psychological care, neuropsychological evaluations, and educational support.
Assisting Patients and Families Through the DMD Journey
DMD care is best delivered in a multidisciplinary setting, where physicians of relevant specialties, physical and occupational therapists, nutritionists, social workers, and genetic counselors collaborate. At Children’s Hospital of Philadelphia, DMD care is delivered through this collaborative model.
Unfortunately, Dr. Brandsema said, many patients don’t have this type of multidisciplinary resource available. “One specialist, such as a pulmonologist or neurologist, might have to be the sole source of care.” Or parents may have to ferry their child to multiple specialists in disparate locations, placing extra stress on an already-stressed family system.
“It’s helpful to connect the family with a comprehensive care center, if possible,” Dr. Brandsema advised. If that’s not available, then he suggests recommending educational opportunities and resources through national organizations such as the Muscular Dystrophy Association; Parent Project MD; NORD; Friends, Family and Duchenne; and Cure Duchenne. Families and caregivers, along with affected individuals, can get education and support from people who understand the day-to-day reality of living with this disease.
One of the major challenges that families face is navigating the high cost of treating DMD, especially the new medications, Dr. Brandsema said. “The authorization process can be intensive and long, and the family may need to take an active role, together with the provider team, in advocating for the patient to get access.”
Among her many responsibilities, Ms. Kaschak engages in care coordination tasks and management, helps patients and caregivers understand care plans, and provides psychosocial support and education about the disease process. She assists families in completing paperwork and navigating specialty authorizations, helping families understand and navigate the complex insurance process. “My role is to bridge gaps in care,” she said.
Dr. Brandsema noted that it’s important for couples to receive genetic counseling if they’re planning to have multiple children because there is a 50% chance that their next boy will be affected. About two thirds of mothers with children who have DMD are carriers, but many are not aware of it. Receiving counseling will enable them to understand their own risks of health complications, as well as the risk to future children.
Managing DMD Across the Lifespan
Another dimension of DMD care is providing resources and help to young people with DMD as they transition into adulthood. “In the past, we had limited treatment and mortality typically took place in the early 20s, so there weren’t a lot of patients who were adults. But as medication options have expanded and management of cardiac and respiratory failure has improved, we see a more significant proportion of adults who require adult-appropriate clinics — or, at the very least, specialists who are conversant in care or can provide care across the lifespan,” Dr. Brandsema said.
The DMD Care Considerations Working Group provides recommendations regarding care across the lifespan,9 as does the Adult North Star Network, of Muscular Dystrophy UK.17,18
Dr. Brandsema emphasized that, despite their disability, many adults with DMD “still engage with the community, and live life to its fullest.” It is to be hoped that, with ongoing research, earlier diagnosis, and improved treatment options, the future will look bright for people with DMD.
Dr. Brandsema has served as a consultant for Audentes, AveXis/Novartis, Biogen, Cytokinetics, Dyne, Edgewise, Fibrogen, Genentech, Marathon, Momenta/Janssen, NS Pharma, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Takeda, and WaVe. He is on the medical advisory council member for Cure SMA and is a site investigator for clinical trials with Alexion, Astellas, AveXis/Novartis, Biogen, Biohaven, Catabasis, CSL Behring, Cytokinetics, Dyne, Fibrogen, Genentech, Ionis, Lilly, ML Bio, Pfizer, PTC Therapeutics, Sarepta, Scholar Rock, Summit, and WaVe. Ms. Kaschak has nothing to disclose.
References
1. Venugopal V and Pavlakis S. Duchenne Muscular Dystrophy. 2023 Jul 10. In: StatPearls [Internet]. Treasure Island, Florida: StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK482346/.
2. Gao QQ and McNally EM. Compr Physiol. 2015 Jul 1;5(3):1223-39. doi: 10.1002/cphy.c140048.
3. Duan D et al. Nat Rev Dis Primers. 2021 Feb 18;7(1):13. doi: 10.1038/s41572-021-00248-3.
4. Aartsma-Rus A et al. J Pediatr. 2019 Jan:204:305-313.e14. doi: 10.1016/j.jpeds.2018.10.043.
5. Broomfield J et al. Neurology. 2021 Dec 7;97(23):e2304-e2314. doi: 10.1212/WNL.0000000000012910.
6. Mercuri E et al. Front Pediatr. 2023 Nov 10:11:1276144. doi: 10.1212/WNL.0000000000012910.
7. Birnkrant DJ et al. Lancet Neurol. 2018 Mar;17(3):251-267. doi: 10.1016/S1474-4422(18)30024-3.
8. Birnkrant DJ et al. Lancet Neurol. 2018 Apr;17(4):347-361. doi: 10.1016/S1474-4422(18)30025-5.
9. Birnkrant DJ et al. Lancet Neurol. 2018 May;17(5):445-455. doi: 10.1016/S1474-4422(18)30026-7.
10. Matthews E et al. Cochrane Database Syst Rev. 2016 May 5;2016(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
11. Bylo M et al. Ann Pharmacother. 2020 Aug;54(8):788-794. doi: 10.1177/1060028019900500.
12. Guglieri M et al. JAMA Neurol. 2022 Oct 1;79(10):1005-1014. doi: 10.1001/jamaneurol.2022.2480.
13. Dang UJ et al. Neurology. 2024 Mar 12;102(5):e208112. doi: 10.1212/WNL.0000000000208112.
14. Mercuri E et al. Lancet Neurol. 2024 Apr;23(4):393-403. doi: 10.1016/S1474-4422(24)00036-X.
15. Gushchina LV et al. Mol Ther Nucleic Acids. 2022 Nov 9:30:479-492. doi: 10.1016/j.omtn.2022.10.025.
16. Patterson G et al. Eur J Pharmacol. 2023 May 15:947:175675. doi: 10.1016/j.ejphar.2023.175675.
17. Quinlivan R et al. J Neuromuscul Dis. 2021;8(6):899-926. doi: 10.3233/JND-200609.
18. Narayan S et al. J Neuromuscul Dis. 2022;9(3):365-381. doi: 10.3233/JND-210707.
Promise for Disease-Modifying Therapies to Tame Huntington’s Disease
Much progress has been made in managing the symptoms of Huntington’s disease, but the real excitement lies in the development of disease-modifying drugs and genetic therapy.
In April 1872, The Medical and Surgical Reporter of Philadelphia published a roughly 3,000-word paper, titled “On Chorea,” by George Huntington, a 22-year-old family practice physician recently graduated from Columbia University, New York City.
“Chorea is essentially a disease of the nervous system. The name ‘chorea’ is given to the disease on account of the dancing propensities of those who are affected by it, and it is a very appropriate designation,” he wrote in the introduction.
Toward the end of the paper Dr. Huntington described a “hereditary chorea” that he had observed while on professional rounds with his father, also a physician, in towns on the eastern end of Long Island in New York.
“It is spoken of by those in whose veins the seeds of the disease are known to exist, with a kind of horror, and not at all alluded to except through dire necessity, when it is mentioned as ‘that disorder,’ ” he wrote, noting later that “I have never known a recovery or even an amelioration of symptoms in this form of chorea; when once it begins it clings to the bitter end.”1
It wasn’t until 1993 that a team of investigators identified the gene responsible for the neurodegenerative disorder we now know as Huntington’s disease.
That discovery sparked hope for better treatments and a cure, but progress over the last 31 years has been incremental. Nonetheless, recent intensive research into novel approaches for treating Huntington’s disease have considerably brightened prospects for patients and caregivers, experts say.
“This is a devastating neurodegenerative disease and in talking to families that I have had the privilege and honor of following, I hear from them that ‘in 1993 when the gene was identified and my family member — parent, grandparent, aunt, uncle — had Huntington’s, we thought that there would be a curative or disease-modifying therapy in no time,’ ” said Erin Furr Stimming, MD, FAAN, FANA professor of neurology and Memorial Hermann Endowed Chair at the McGovern Medical School at University of Texas Health Science Center in Houston.
“Here we are in 2024. I think our families are still so incredibly resilient and courageous, and they are willing and able to participate in clinical trials. Families in the Huntington’s disease community at large are really ready to have trials that do, in fact, demonstrate some evidence of slowing of disease progression. It’s an exciting time,” she said in an interview.
Repeating Nucleotides
Huntington’s disease is an autosomal dominant neurodegenerative disorder caused by an expansion of a CAG trinucleotide repeat in exon 1 of the huntingtin gene (HTT), which encodes for the huntingtin protein (HTT). The multiple repeats result in expanded expression of mutant HTT. The mutated protein disrupts normal cellular processes, alters intracellular calcium homeostasis, and interferes with gene transcription, leading to progressive degeneration of neurons, and to a hallmark Huntington’s disease triad consisting of movement disorders, cognitive decline, and mood/behavioral issues.
The number of repeats determines the severity of disease and the age of onset. In nonaffected persons the gene contains about 20 CAG repeats, but as genetics research dating from the 1990s has shown, a single HTT allele containing more than 40 CAG repeats will inevitably result in disease, whereas carriers with fewer than 36 repeats on both alleles will remain unaffected.2
The prevalence of the mutation in Western populations is estimated to be from 4 to 10 per 100,000.3
The disease usually manifests first in adults aged 30-50 years but may also occur in children or adolescents and young adults. Early symptoms often include a decline in executive function that may be noticeable to the patient’s family and friends, mood changes, and chorea.
“Because of the uncontrolled movements (chorea), a person with Huntington’s disease may lose a lot of weight without intending to, and may have trouble walking, balancing, and moving around safely. They will eventually lose the ability to work, drive, and manage tasks at home, and may qualify for disability benefits. Over time, the individual will develop difficulty with speaking and swallowing, and their movements will become slow and stiff. People with advanced Huntington’s disease need full-time care to help with their day-to-day activities, and they ultimately succumb to pneumonia, heart failure, or other complications,” according to the Huntington’s Disease Society of America.4
Managing Symptoms
say neurologists.
There are currently three medications approved for the treatment of chorea, all in the class of agents known as vesicular monoamine transporter 2 inhibitors. These agents are tetrabenazine (Xenazine, Lundbeck), deutetrabenazine (Austedo XR, Teva), and valbenazine (Ingrezza, Neurocrine Biosciences).
“These drugs can treat the chorea, but they also can help with some of the other motor features. For example, if a patient has chorea in the legs and you treat it, then maybe they’re walking will get better, or if they have chorea in the mouth and you treat the chorea, then maybe their speech and swallowing may improve,” Victor Sung, MD, professor of neurology and director of the Huntington’s Disease Clinic at the University of Alabama at Birmingham, said in an interview.
Mood and behavioral symptoms associated with Huntington’s disease – depression, anxiety, irritability, impulsivity, etc – can be managed with off-label use of antidepressants, mood stabilizers, and antipsychotic agents.
“When it comes to the cognitive symptoms, that’s a big gap where we don’t have anything that’s [Food and Drug Administration] approved for Huntington’s disease,” Dr. Sung said.
Agents used to treat Alzheimer’s disease, such as cholinesterase inhibitors and the glutamate receptor antagonist memantine, have been studied extensively for preservation of cognition in Huntington’s disease, but have not shown significant benefit.
Dr. Sung said that one promising approach to the problem of cognitive protection in Huntington’s disease is the investigational agent dalzanemdor (SAGE-718), an N-methyl-D-aspartic (NMDA) acid receptor positive allosteric modulator. The drug is in development for cognitive disorders associated with NMDA receptor dysfunction, including Huntington’s disease and Alzheimer’s disease.
In the phase 2 SURVEYOR trial, which was not powered to show efficacy of dalzanemdor over placebo, patients with Huntington’s disease reportedly tolerated the drug well, with treatment-related adverse events primarily mild or moderate in severity.
As of this writing dalzanemdor is being evaluated for efficacy, compared with placebo, in the phase 2 DIMENSION trial. The primary endpoint of this trial is a change from baseline in composite score of the Huntington’s Disease Cognitive Assessment Battery.
Disease-Modifying Therapies
As previously noted, although there is no cure for Huntington’s disease, neurology investigators are developing new strategies for delaying, stalling, or even preventing disease progression.

“There are so many different approaches, it’s hard to know where to start,” Christopher A. Ross, MD, director of the Huntington’s Disease Center at Johns Hopkins University in Baltimore, Maryland, said in an interview.
The most actively investigated approach may be huntingtin-lowering therapies, based on the supposition that mutant huntingtin protein (mHTT) is the primary toxin in Huntington’s disease.
“What you need to do is in some way lower it, and there are a number of different ways to do that: antinsense olignoclueotides, small interfering RNA, CRISPR Cas, or other gene-editing techniques, as well as gene therapy with an adenoviral vector” he said.
Gene Therapy
Dr. Ross cited as promising a phase 1/2 clinical trial of an investigational gene therapy, AMT-130 (uniQure), which consists of an adeno-associated virus vector and a gene encoding a microRNA that is designed to recognize, bind, and nonselectively lower both mHTT and wild-type HTT.
The compound is injected directly into the corpus striatum. It was demonstrated to decrease signs of Huntington’s disease in animal models, and interim data on 29 patients with Huntington’s disease followed for up to 24 months showed a statically significant, dose-dependent slowing of Huntington’s disease progression and lowering of neurofilament light protein, a marker for neuronal degeneration in Huntington’s disease, in cerebrospinal fluid (CSF).
AMT-130 targets exon 1 of the HTT gene, which appears to be an important target, Dr. Ross commented.
“The huntingtin protein is really big. The polyglutamine expansion, which is what causes the toxicity, is right at the N-terminus of exon 1, and there is increasingly good evidence that you can have an exon 1 misspliced protein product which causes the toxicity, so it may be especially important to lower Huntington by targeting exon 1,” he said.
Oral Splicing Modifier
PTC Therapeutics, based in New Jersey, is developing PTC518, an oral small molecule drug that can cross the blood-brain barrier and is reported to target mutant huntingtin protein.
The drug is a splicing modifier that promotes insertion of a premature stop codon to HTT mRNA, thereby degrading and lowering the HTT levels.5
In June 2024, the company reported that, in the phase 2a PIVOT-HD study of PTC518 in patients with Huntington’s disease, 12 months of treatment was associated with a dose-dependent lowering of mHTT in blood and CSF in an interim cohort.
“In addition, favorable trends were demonstrated on several relevant Huntington’s disease clinical assessments including Total Motor Score and Composite Unified Huntington’s Disease Rating Scale. Furthermore, following 12 months of treatment, PTC518 continues to be safe and well tolerated,” the company stated in a press release.
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are short strands of DNA or RNA that bind to RNA sequences in faulty genes to modify production of target proteins.
One such drug, tominersen, developed jointly by Ionis and Roche, initially showed promise in a phase 1/2 trial for lowering mHTT in CSF without serious adverse events. But in a phase 3 trial, the intrathecally delivered agent was halted after an independent data monitoring committee recommended halting the trial, which Roche ended in 2021. The company reported in a letter to The New England Journal of Medicine that people in the high-dosage treatment group did measurably worse – although it remains unclear whether this was caused by excess protein lowering or an off-target effect.6 The tominersen program was the first to demonstrate that it was possible to lower HTT with an intervention, and the companies reported that they are continuing the agent’s development program.
Wave Life Sciences is developing an ASO, labeled WVE-003, designed to target a single-nucleotide polymorphism associated with the mHTT mRNA transcript within HTT. The company says that targeting the single-nucleotide polymorphism should allow lowering of expression of the mHTT will preserving wild-type HTT. This approach has the potential for therapies to prevent disease progression during the prodromal period, the company states.
Somatic Expansion
Dr. Furr Stimming and Dr. Ross both noted that there is considerable research interest into recently identified genetic modifiers that are believed to influence somatic instability, which in turn leads to somatic expansion.
“That seems to happen selectively in the neurons that are affected in Huntington’s disease. So a big puzzle for all of the neurodegenerative diseases is why are certain regions of the brain affected and other regions not? And it looks like, for the repeat expansion, this idea of somatic expansion seems to be increasingly central,” Dr. Ross said.
“The really exciting idea here is that, if somatic expansion is critical to the disease process and you could slow it down or stop it, you could go very early and potentially not just slow the progression of the disease once it starts, but conceivably even delay or possibly prevent the onset of Huntington’s disease,” he said.
Does HTT Lowering Mediate Progression?
“The next question is what does this mean clinically? Does lowering mutant huntingtin protein levels, and wild type for that matter, actually slow disease progression, which can be challenging to measure in a disease that is relatively slowly progressive?,” Dr. Furr Stimming said.
One important tool to help answer this question, she noted, is the Huntington’s Disease Integrated Staging System (HDISS), first described in 2022.7 The consensus-based system incorporates biological, clinical, and functional assessments, and characterizes patients from birth by stages, from stage 0 (persons who carry the mutation but have no detectable pathology), to stage 1 (measurable indicators of underlying pathophysiology), stage 2 (a detectable clinical phenotype), and finally to stage 3 (decline in function).
“The goal of the HDISS, which is designed for clinical research purposes, is to try to enroll individuals into clinical trials earlier rather than later, trying to get pharmaceutical companies and others to take advantage of the period prior to significant neurodegeneration occurring. Like Alzheimer’s disease and Parkinson’s disease, by the time we make a clinical diagnose significant neurodegeneration has occurred, so we really want to take advantage of the prodromal period to intervene with these potential disease-modifying therapies,” Dr. Furr Stimming said.
“At the end of the day, I suspect that we will not have just one effective disease-modifying therapy. I suspect that it will be a multifacted approach. We envision that we would hit the huntingtin protein from a few different angles. I envision that we would not only modify in some form or fashion production of the mutant huntingtin protein, but also try to influence the genetic modifiers that we think are important in somatic instability and expansion, which likely contributes to the rate of progression and symptom severity,” Dr. Furr Stimming said.
References
1. Huntington G. On Chorea. Reprinted in The Journal of Neuropsychiatry and Clinical Neurosciences. 2003;15(1):109-112. doi: 10.1176/jnp.15.1.109.
2. Kaemmerer WF and Grondin RC. Degener Neurol Neuromuscul Dis. 2019 Mar 8;9:3-17. doi: 10.2147/DNND.S163808.
3. Jimenez-Sanchez M et al. Cold Spring Harb Perspect Med. 2017 Jul 5;7(7):a024240. doi: 10.1101/cshperspect.a024240.
4. Huntington’s Disease Society of America. Overview of Huntington’s Disease. https://hdsa.org/what-is-hd/overview-of-huntingtons-disease/.
5. Beers B et al. J Neurol Neurosurg Psychiatry. 2022;93:A96. https://jnnp.bmj.com/content/93/Suppl_1/A96.1.
6. McColgan P et al. N Engl J Med. 2023 Dec 7;389(23):2203-2205. doi: 10.1056/NEJMc2300400.
7. Tabrizi SJ et al. Lancet Neurol. 2022 Jul;21(7):632-644. doi: 10.1016/S1474-4422(22)00120-X.
Much progress has been made in managing the symptoms of Huntington’s disease, but the real excitement lies in the development of disease-modifying drugs and genetic therapy.
In April 1872, The Medical and Surgical Reporter of Philadelphia published a roughly 3,000-word paper, titled “On Chorea,” by George Huntington, a 22-year-old family practice physician recently graduated from Columbia University, New York City.
“Chorea is essentially a disease of the nervous system. The name ‘chorea’ is given to the disease on account of the dancing propensities of those who are affected by it, and it is a very appropriate designation,” he wrote in the introduction.
Toward the end of the paper Dr. Huntington described a “hereditary chorea” that he had observed while on professional rounds with his father, also a physician, in towns on the eastern end of Long Island in New York.
“It is spoken of by those in whose veins the seeds of the disease are known to exist, with a kind of horror, and not at all alluded to except through dire necessity, when it is mentioned as ‘that disorder,’ ” he wrote, noting later that “I have never known a recovery or even an amelioration of symptoms in this form of chorea; when once it begins it clings to the bitter end.”1
It wasn’t until 1993 that a team of investigators identified the gene responsible for the neurodegenerative disorder we now know as Huntington’s disease.
That discovery sparked hope for better treatments and a cure, but progress over the last 31 years has been incremental. Nonetheless, recent intensive research into novel approaches for treating Huntington’s disease have considerably brightened prospects for patients and caregivers, experts say.
“This is a devastating neurodegenerative disease and in talking to families that I have had the privilege and honor of following, I hear from them that ‘in 1993 when the gene was identified and my family member — parent, grandparent, aunt, uncle — had Huntington’s, we thought that there would be a curative or disease-modifying therapy in no time,’ ” said Erin Furr Stimming, MD, FAAN, FANA professor of neurology and Memorial Hermann Endowed Chair at the McGovern Medical School at University of Texas Health Science Center in Houston.
“Here we are in 2024. I think our families are still so incredibly resilient and courageous, and they are willing and able to participate in clinical trials. Families in the Huntington’s disease community at large are really ready to have trials that do, in fact, demonstrate some evidence of slowing of disease progression. It’s an exciting time,” she said in an interview.
Repeating Nucleotides
Huntington’s disease is an autosomal dominant neurodegenerative disorder caused by an expansion of a CAG trinucleotide repeat in exon 1 of the huntingtin gene (HTT), which encodes for the huntingtin protein (HTT). The multiple repeats result in expanded expression of mutant HTT. The mutated protein disrupts normal cellular processes, alters intracellular calcium homeostasis, and interferes with gene transcription, leading to progressive degeneration of neurons, and to a hallmark Huntington’s disease triad consisting of movement disorders, cognitive decline, and mood/behavioral issues.
The number of repeats determines the severity of disease and the age of onset. In nonaffected persons the gene contains about 20 CAG repeats, but as genetics research dating from the 1990s has shown, a single HTT allele containing more than 40 CAG repeats will inevitably result in disease, whereas carriers with fewer than 36 repeats on both alleles will remain unaffected.2
The prevalence of the mutation in Western populations is estimated to be from 4 to 10 per 100,000.3
The disease usually manifests first in adults aged 30-50 years but may also occur in children or adolescents and young adults. Early symptoms often include a decline in executive function that may be noticeable to the patient’s family and friends, mood changes, and chorea.
“Because of the uncontrolled movements (chorea), a person with Huntington’s disease may lose a lot of weight without intending to, and may have trouble walking, balancing, and moving around safely. They will eventually lose the ability to work, drive, and manage tasks at home, and may qualify for disability benefits. Over time, the individual will develop difficulty with speaking and swallowing, and their movements will become slow and stiff. People with advanced Huntington’s disease need full-time care to help with their day-to-day activities, and they ultimately succumb to pneumonia, heart failure, or other complications,” according to the Huntington’s Disease Society of America.4
Managing Symptoms
say neurologists.
There are currently three medications approved for the treatment of chorea, all in the class of agents known as vesicular monoamine transporter 2 inhibitors. These agents are tetrabenazine (Xenazine, Lundbeck), deutetrabenazine (Austedo XR, Teva), and valbenazine (Ingrezza, Neurocrine Biosciences).
“These drugs can treat the chorea, but they also can help with some of the other motor features. For example, if a patient has chorea in the legs and you treat it, then maybe they’re walking will get better, or if they have chorea in the mouth and you treat the chorea, then maybe their speech and swallowing may improve,” Victor Sung, MD, professor of neurology and director of the Huntington’s Disease Clinic at the University of Alabama at Birmingham, said in an interview.
Mood and behavioral symptoms associated with Huntington’s disease – depression, anxiety, irritability, impulsivity, etc – can be managed with off-label use of antidepressants, mood stabilizers, and antipsychotic agents.
“When it comes to the cognitive symptoms, that’s a big gap where we don’t have anything that’s [Food and Drug Administration] approved for Huntington’s disease,” Dr. Sung said.
Agents used to treat Alzheimer’s disease, such as cholinesterase inhibitors and the glutamate receptor antagonist memantine, have been studied extensively for preservation of cognition in Huntington’s disease, but have not shown significant benefit.
Dr. Sung said that one promising approach to the problem of cognitive protection in Huntington’s disease is the investigational agent dalzanemdor (SAGE-718), an N-methyl-D-aspartic (NMDA) acid receptor positive allosteric modulator. The drug is in development for cognitive disorders associated with NMDA receptor dysfunction, including Huntington’s disease and Alzheimer’s disease.
In the phase 2 SURVEYOR trial, which was not powered to show efficacy of dalzanemdor over placebo, patients with Huntington’s disease reportedly tolerated the drug well, with treatment-related adverse events primarily mild or moderate in severity.
As of this writing dalzanemdor is being evaluated for efficacy, compared with placebo, in the phase 2 DIMENSION trial. The primary endpoint of this trial is a change from baseline in composite score of the Huntington’s Disease Cognitive Assessment Battery.
Disease-Modifying Therapies
As previously noted, although there is no cure for Huntington’s disease, neurology investigators are developing new strategies for delaying, stalling, or even preventing disease progression.
“There are so many different approaches, it’s hard to know where to start,” Christopher A. Ross, MD, director of the Huntington’s Disease Center at Johns Hopkins University in Baltimore, Maryland, said in an interview.
The most actively investigated approach may be huntingtin-lowering therapies, based on the supposition that mutant huntingtin protein (mHTT) is the primary toxin in Huntington’s disease.
“What you need to do is in some way lower it, and there are a number of different ways to do that: antinsense olignoclueotides, small interfering RNA, CRISPR Cas, or other gene-editing techniques, as well as gene therapy with an adenoviral vector” he said.
Gene Therapy
Dr. Ross cited as promising a phase 1/2 clinical trial of an investigational gene therapy, AMT-130 (uniQure), which consists of an adeno-associated virus vector and a gene encoding a microRNA that is designed to recognize, bind, and nonselectively lower both mHTT and wild-type HTT.
The compound is injected directly into the corpus striatum. It was demonstrated to decrease signs of Huntington’s disease in animal models, and interim data on 29 patients with Huntington’s disease followed for up to 24 months showed a statically significant, dose-dependent slowing of Huntington’s disease progression and lowering of neurofilament light protein, a marker for neuronal degeneration in Huntington’s disease, in cerebrospinal fluid (CSF).
AMT-130 targets exon 1 of the HTT gene, which appears to be an important target, Dr. Ross commented.
“The huntingtin protein is really big. The polyglutamine expansion, which is what causes the toxicity, is right at the N-terminus of exon 1, and there is increasingly good evidence that you can have an exon 1 misspliced protein product which causes the toxicity, so it may be especially important to lower Huntington by targeting exon 1,” he said.
Oral Splicing Modifier
PTC Therapeutics, based in New Jersey, is developing PTC518, an oral small molecule drug that can cross the blood-brain barrier and is reported to target mutant huntingtin protein.
The drug is a splicing modifier that promotes insertion of a premature stop codon to HTT mRNA, thereby degrading and lowering the HTT levels.5
In June 2024, the company reported that, in the phase 2a PIVOT-HD study of PTC518 in patients with Huntington’s disease, 12 months of treatment was associated with a dose-dependent lowering of mHTT in blood and CSF in an interim cohort.
“In addition, favorable trends were demonstrated on several relevant Huntington’s disease clinical assessments including Total Motor Score and Composite Unified Huntington’s Disease Rating Scale. Furthermore, following 12 months of treatment, PTC518 continues to be safe and well tolerated,” the company stated in a press release.
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are short strands of DNA or RNA that bind to RNA sequences in faulty genes to modify production of target proteins.
One such drug, tominersen, developed jointly by Ionis and Roche, initially showed promise in a phase 1/2 trial for lowering mHTT in CSF without serious adverse events. But in a phase 3 trial, the intrathecally delivered agent was halted after an independent data monitoring committee recommended halting the trial, which Roche ended in 2021. The company reported in a letter to The New England Journal of Medicine that people in the high-dosage treatment group did measurably worse – although it remains unclear whether this was caused by excess protein lowering or an off-target effect.6 The tominersen program was the first to demonstrate that it was possible to lower HTT with an intervention, and the companies reported that they are continuing the agent’s development program.
Wave Life Sciences is developing an ASO, labeled WVE-003, designed to target a single-nucleotide polymorphism associated with the mHTT mRNA transcript within HTT. The company says that targeting the single-nucleotide polymorphism should allow lowering of expression of the mHTT will preserving wild-type HTT. This approach has the potential for therapies to prevent disease progression during the prodromal period, the company states.
Somatic Expansion
Dr. Furr Stimming and Dr. Ross both noted that there is considerable research interest into recently identified genetic modifiers that are believed to influence somatic instability, which in turn leads to somatic expansion.
“That seems to happen selectively in the neurons that are affected in Huntington’s disease. So a big puzzle for all of the neurodegenerative diseases is why are certain regions of the brain affected and other regions not? And it looks like, for the repeat expansion, this idea of somatic expansion seems to be increasingly central,” Dr. Ross said.
“The really exciting idea here is that, if somatic expansion is critical to the disease process and you could slow it down or stop it, you could go very early and potentially not just slow the progression of the disease once it starts, but conceivably even delay or possibly prevent the onset of Huntington’s disease,” he said.
Does HTT Lowering Mediate Progression?
“The next question is what does this mean clinically? Does lowering mutant huntingtin protein levels, and wild type for that matter, actually slow disease progression, which can be challenging to measure in a disease that is relatively slowly progressive?,” Dr. Furr Stimming said.
One important tool to help answer this question, she noted, is the Huntington’s Disease Integrated Staging System (HDISS), first described in 2022.7 The consensus-based system incorporates biological, clinical, and functional assessments, and characterizes patients from birth by stages, from stage 0 (persons who carry the mutation but have no detectable pathology), to stage 1 (measurable indicators of underlying pathophysiology), stage 2 (a detectable clinical phenotype), and finally to stage 3 (decline in function).
“The goal of the HDISS, which is designed for clinical research purposes, is to try to enroll individuals into clinical trials earlier rather than later, trying to get pharmaceutical companies and others to take advantage of the period prior to significant neurodegeneration occurring. Like Alzheimer’s disease and Parkinson’s disease, by the time we make a clinical diagnose significant neurodegeneration has occurred, so we really want to take advantage of the prodromal period to intervene with these potential disease-modifying therapies,” Dr. Furr Stimming said.
“At the end of the day, I suspect that we will not have just one effective disease-modifying therapy. I suspect that it will be a multifacted approach. We envision that we would hit the huntingtin protein from a few different angles. I envision that we would not only modify in some form or fashion production of the mutant huntingtin protein, but also try to influence the genetic modifiers that we think are important in somatic instability and expansion, which likely contributes to the rate of progression and symptom severity,” Dr. Furr Stimming said.
References
1. Huntington G. On Chorea. Reprinted in The Journal of Neuropsychiatry and Clinical Neurosciences. 2003;15(1):109-112. doi: 10.1176/jnp.15.1.109.
2. Kaemmerer WF and Grondin RC. Degener Neurol Neuromuscul Dis. 2019 Mar 8;9:3-17. doi: 10.2147/DNND.S163808.
3. Jimenez-Sanchez M et al. Cold Spring Harb Perspect Med. 2017 Jul 5;7(7):a024240. doi: 10.1101/cshperspect.a024240.
4. Huntington’s Disease Society of America. Overview of Huntington’s Disease. https://hdsa.org/what-is-hd/overview-of-huntingtons-disease/.
5. Beers B et al. J Neurol Neurosurg Psychiatry. 2022;93:A96. https://jnnp.bmj.com/content/93/Suppl_1/A96.1.
6. McColgan P et al. N Engl J Med. 2023 Dec 7;389(23):2203-2205. doi: 10.1056/NEJMc2300400.
7. Tabrizi SJ et al. Lancet Neurol. 2022 Jul;21(7):632-644. doi: 10.1016/S1474-4422(22)00120-X.
Much progress has been made in managing the symptoms of Huntington’s disease, but the real excitement lies in the development of disease-modifying drugs and genetic therapy.
In April 1872, The Medical and Surgical Reporter of Philadelphia published a roughly 3,000-word paper, titled “On Chorea,” by George Huntington, a 22-year-old family practice physician recently graduated from Columbia University, New York City.
“Chorea is essentially a disease of the nervous system. The name ‘chorea’ is given to the disease on account of the dancing propensities of those who are affected by it, and it is a very appropriate designation,” he wrote in the introduction.
Toward the end of the paper Dr. Huntington described a “hereditary chorea” that he had observed while on professional rounds with his father, also a physician, in towns on the eastern end of Long Island in New York.
“It is spoken of by those in whose veins the seeds of the disease are known to exist, with a kind of horror, and not at all alluded to except through dire necessity, when it is mentioned as ‘that disorder,’ ” he wrote, noting later that “I have never known a recovery or even an amelioration of symptoms in this form of chorea; when once it begins it clings to the bitter end.”1
It wasn’t until 1993 that a team of investigators identified the gene responsible for the neurodegenerative disorder we now know as Huntington’s disease.
That discovery sparked hope for better treatments and a cure, but progress over the last 31 years has been incremental. Nonetheless, recent intensive research into novel approaches for treating Huntington’s disease have considerably brightened prospects for patients and caregivers, experts say.
“This is a devastating neurodegenerative disease and in talking to families that I have had the privilege and honor of following, I hear from them that ‘in 1993 when the gene was identified and my family member — parent, grandparent, aunt, uncle — had Huntington’s, we thought that there would be a curative or disease-modifying therapy in no time,’ ” said Erin Furr Stimming, MD, FAAN, FANA professor of neurology and Memorial Hermann Endowed Chair at the McGovern Medical School at University of Texas Health Science Center in Houston.
“Here we are in 2024. I think our families are still so incredibly resilient and courageous, and they are willing and able to participate in clinical trials. Families in the Huntington’s disease community at large are really ready to have trials that do, in fact, demonstrate some evidence of slowing of disease progression. It’s an exciting time,” she said in an interview.
Repeating Nucleotides
Huntington’s disease is an autosomal dominant neurodegenerative disorder caused by an expansion of a CAG trinucleotide repeat in exon 1 of the huntingtin gene (HTT), which encodes for the huntingtin protein (HTT). The multiple repeats result in expanded expression of mutant HTT. The mutated protein disrupts normal cellular processes, alters intracellular calcium homeostasis, and interferes with gene transcription, leading to progressive degeneration of neurons, and to a hallmark Huntington’s disease triad consisting of movement disorders, cognitive decline, and mood/behavioral issues.
The number of repeats determines the severity of disease and the age of onset. In nonaffected persons the gene contains about 20 CAG repeats, but as genetics research dating from the 1990s has shown, a single HTT allele containing more than 40 CAG repeats will inevitably result in disease, whereas carriers with fewer than 36 repeats on both alleles will remain unaffected.2
The prevalence of the mutation in Western populations is estimated to be from 4 to 10 per 100,000.3
The disease usually manifests first in adults aged 30-50 years but may also occur in children or adolescents and young adults. Early symptoms often include a decline in executive function that may be noticeable to the patient’s family and friends, mood changes, and chorea.
“Because of the uncontrolled movements (chorea), a person with Huntington’s disease may lose a lot of weight without intending to, and may have trouble walking, balancing, and moving around safely. They will eventually lose the ability to work, drive, and manage tasks at home, and may qualify for disability benefits. Over time, the individual will develop difficulty with speaking and swallowing, and their movements will become slow and stiff. People with advanced Huntington’s disease need full-time care to help with their day-to-day activities, and they ultimately succumb to pneumonia, heart failure, or other complications,” according to the Huntington’s Disease Society of America.4
Managing Symptoms
say neurologists.
There are currently three medications approved for the treatment of chorea, all in the class of agents known as vesicular monoamine transporter 2 inhibitors. These agents are tetrabenazine (Xenazine, Lundbeck), deutetrabenazine (Austedo XR, Teva), and valbenazine (Ingrezza, Neurocrine Biosciences).
“These drugs can treat the chorea, but they also can help with some of the other motor features. For example, if a patient has chorea in the legs and you treat it, then maybe they’re walking will get better, or if they have chorea in the mouth and you treat the chorea, then maybe their speech and swallowing may improve,” Victor Sung, MD, professor of neurology and director of the Huntington’s Disease Clinic at the University of Alabama at Birmingham, said in an interview.
Mood and behavioral symptoms associated with Huntington’s disease – depression, anxiety, irritability, impulsivity, etc – can be managed with off-label use of antidepressants, mood stabilizers, and antipsychotic agents.
“When it comes to the cognitive symptoms, that’s a big gap where we don’t have anything that’s [Food and Drug Administration] approved for Huntington’s disease,” Dr. Sung said.
Agents used to treat Alzheimer’s disease, such as cholinesterase inhibitors and the glutamate receptor antagonist memantine, have been studied extensively for preservation of cognition in Huntington’s disease, but have not shown significant benefit.
Dr. Sung said that one promising approach to the problem of cognitive protection in Huntington’s disease is the investigational agent dalzanemdor (SAGE-718), an N-methyl-D-aspartic (NMDA) acid receptor positive allosteric modulator. The drug is in development for cognitive disorders associated with NMDA receptor dysfunction, including Huntington’s disease and Alzheimer’s disease.
In the phase 2 SURVEYOR trial, which was not powered to show efficacy of dalzanemdor over placebo, patients with Huntington’s disease reportedly tolerated the drug well, with treatment-related adverse events primarily mild or moderate in severity.
As of this writing dalzanemdor is being evaluated for efficacy, compared with placebo, in the phase 2 DIMENSION trial. The primary endpoint of this trial is a change from baseline in composite score of the Huntington’s Disease Cognitive Assessment Battery.
Disease-Modifying Therapies
As previously noted, although there is no cure for Huntington’s disease, neurology investigators are developing new strategies for delaying, stalling, or even preventing disease progression.
“There are so many different approaches, it’s hard to know where to start,” Christopher A. Ross, MD, director of the Huntington’s Disease Center at Johns Hopkins University in Baltimore, Maryland, said in an interview.
The most actively investigated approach may be huntingtin-lowering therapies, based on the supposition that mutant huntingtin protein (mHTT) is the primary toxin in Huntington’s disease.
“What you need to do is in some way lower it, and there are a number of different ways to do that: antinsense olignoclueotides, small interfering RNA, CRISPR Cas, or other gene-editing techniques, as well as gene therapy with an adenoviral vector” he said.
Gene Therapy
Dr. Ross cited as promising a phase 1/2 clinical trial of an investigational gene therapy, AMT-130 (uniQure), which consists of an adeno-associated virus vector and a gene encoding a microRNA that is designed to recognize, bind, and nonselectively lower both mHTT and wild-type HTT.
The compound is injected directly into the corpus striatum. It was demonstrated to decrease signs of Huntington’s disease in animal models, and interim data on 29 patients with Huntington’s disease followed for up to 24 months showed a statically significant, dose-dependent slowing of Huntington’s disease progression and lowering of neurofilament light protein, a marker for neuronal degeneration in Huntington’s disease, in cerebrospinal fluid (CSF).
AMT-130 targets exon 1 of the HTT gene, which appears to be an important target, Dr. Ross commented.
“The huntingtin protein is really big. The polyglutamine expansion, which is what causes the toxicity, is right at the N-terminus of exon 1, and there is increasingly good evidence that you can have an exon 1 misspliced protein product which causes the toxicity, so it may be especially important to lower Huntington by targeting exon 1,” he said.
Oral Splicing Modifier
PTC Therapeutics, based in New Jersey, is developing PTC518, an oral small molecule drug that can cross the blood-brain barrier and is reported to target mutant huntingtin protein.
The drug is a splicing modifier that promotes insertion of a premature stop codon to HTT mRNA, thereby degrading and lowering the HTT levels.5
In June 2024, the company reported that, in the phase 2a PIVOT-HD study of PTC518 in patients with Huntington’s disease, 12 months of treatment was associated with a dose-dependent lowering of mHTT in blood and CSF in an interim cohort.
“In addition, favorable trends were demonstrated on several relevant Huntington’s disease clinical assessments including Total Motor Score and Composite Unified Huntington’s Disease Rating Scale. Furthermore, following 12 months of treatment, PTC518 continues to be safe and well tolerated,” the company stated in a press release.
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are short strands of DNA or RNA that bind to RNA sequences in faulty genes to modify production of target proteins.
One such drug, tominersen, developed jointly by Ionis and Roche, initially showed promise in a phase 1/2 trial for lowering mHTT in CSF without serious adverse events. But in a phase 3 trial, the intrathecally delivered agent was halted after an independent data monitoring committee recommended halting the trial, which Roche ended in 2021. The company reported in a letter to The New England Journal of Medicine that people in the high-dosage treatment group did measurably worse – although it remains unclear whether this was caused by excess protein lowering or an off-target effect.6 The tominersen program was the first to demonstrate that it was possible to lower HTT with an intervention, and the companies reported that they are continuing the agent’s development program.
Wave Life Sciences is developing an ASO, labeled WVE-003, designed to target a single-nucleotide polymorphism associated with the mHTT mRNA transcript within HTT. The company says that targeting the single-nucleotide polymorphism should allow lowering of expression of the mHTT will preserving wild-type HTT. This approach has the potential for therapies to prevent disease progression during the prodromal period, the company states.
Somatic Expansion
Dr. Furr Stimming and Dr. Ross both noted that there is considerable research interest into recently identified genetic modifiers that are believed to influence somatic instability, which in turn leads to somatic expansion.
“That seems to happen selectively in the neurons that are affected in Huntington’s disease. So a big puzzle for all of the neurodegenerative diseases is why are certain regions of the brain affected and other regions not? And it looks like, for the repeat expansion, this idea of somatic expansion seems to be increasingly central,” Dr. Ross said.
“The really exciting idea here is that, if somatic expansion is critical to the disease process and you could slow it down or stop it, you could go very early and potentially not just slow the progression of the disease once it starts, but conceivably even delay or possibly prevent the onset of Huntington’s disease,” he said.
Does HTT Lowering Mediate Progression?
“The next question is what does this mean clinically? Does lowering mutant huntingtin protein levels, and wild type for that matter, actually slow disease progression, which can be challenging to measure in a disease that is relatively slowly progressive?,” Dr. Furr Stimming said.
One important tool to help answer this question, she noted, is the Huntington’s Disease Integrated Staging System (HDISS), first described in 2022.7 The consensus-based system incorporates biological, clinical, and functional assessments, and characterizes patients from birth by stages, from stage 0 (persons who carry the mutation but have no detectable pathology), to stage 1 (measurable indicators of underlying pathophysiology), stage 2 (a detectable clinical phenotype), and finally to stage 3 (decline in function).
“The goal of the HDISS, which is designed for clinical research purposes, is to try to enroll individuals into clinical trials earlier rather than later, trying to get pharmaceutical companies and others to take advantage of the period prior to significant neurodegeneration occurring. Like Alzheimer’s disease and Parkinson’s disease, by the time we make a clinical diagnose significant neurodegeneration has occurred, so we really want to take advantage of the prodromal period to intervene with these potential disease-modifying therapies,” Dr. Furr Stimming said.
“At the end of the day, I suspect that we will not have just one effective disease-modifying therapy. I suspect that it will be a multifacted approach. We envision that we would hit the huntingtin protein from a few different angles. I envision that we would not only modify in some form or fashion production of the mutant huntingtin protein, but also try to influence the genetic modifiers that we think are important in somatic instability and expansion, which likely contributes to the rate of progression and symptom severity,” Dr. Furr Stimming said.
References
1. Huntington G. On Chorea. Reprinted in The Journal of Neuropsychiatry and Clinical Neurosciences. 2003;15(1):109-112. doi: 10.1176/jnp.15.1.109.
2. Kaemmerer WF and Grondin RC. Degener Neurol Neuromuscul Dis. 2019 Mar 8;9:3-17. doi: 10.2147/DNND.S163808.
3. Jimenez-Sanchez M et al. Cold Spring Harb Perspect Med. 2017 Jul 5;7(7):a024240. doi: 10.1101/cshperspect.a024240.
4. Huntington’s Disease Society of America. Overview of Huntington’s Disease. https://hdsa.org/what-is-hd/overview-of-huntingtons-disease/.
5. Beers B et al. J Neurol Neurosurg Psychiatry. 2022;93:A96. https://jnnp.bmj.com/content/93/Suppl_1/A96.1.
6. McColgan P et al. N Engl J Med. 2023 Dec 7;389(23):2203-2205. doi: 10.1056/NEJMc2300400.
7. Tabrizi SJ et al. Lancet Neurol. 2022 Jul;21(7):632-644. doi: 10.1016/S1474-4422(22)00120-X.
Genetic Testing for ALS, Now a Standard, Creates a Path Toward Individualized Care
The first therapy targeted at modifying a mutant gene associated with amyotrophic lateral sclerosis (ALS), approved in early 2023, has offered reassurance that the biology of ALS, when known, is targetable. Historically, the disease has been considered a clinical diagnosis, but the
Despite a narrow indication, the only therapy targeted at an ALS-associated gene so far, SOD1 ALS, supports the premise that the biology of ALS can be modified, according to Christina N. Fournier, MD, an associate professor in the Department of Neurology, Emory University, Atlanta, Georgia.

Rather than a single pathological entity, ALS is best understood as the end result of many different pathological processes. Each might require its own targeted therapy in order to interrupt the upstream biological pathways that drive disease.
About 15% of ALS Has An Identifiable Genetic Cause
A family history of ALS is present in about 10% of cases. A genetic cause can be identified in approximately 15%. Cases without an identifiable genetic etiology are considered sporadic. So far, the only approved therapy that modifies the function of a gene associated with ALS is tofersen (Qalsody, Biogen), an antisense oligonucleotide. Tofersen inhibits RNA transcription of the superoxide dismutase 1 (SOD1) gene to decrease production of the SOD1 protein.
This first gene therapy for ALS is a breakthrough, but it is indicated for only a small proportion of ALS patients. Even though SOD1 gene mutations represent the second most common genetic cause of ALS after the C90rf72 gene, the proportion of patients who are candidates for tofersen is low. Efficacy is expected only in about 1% of those with familial ALS and 1% of those with sporadic ALS, or about 2% of all patients with ALS.
The evidence of benefit from a treatment with a specific target has provided the basis for concluding that “we are onto something,” Dr. Fournier said. An expert in ALS, she sees reason for excitement about the prospects in treatment with the growing focus on the underlying pathways of disease rather than the downstream consequences.
“The hope is that new gene-targeted therapies will be developed in the future to treat the broader ALS population,” said Dr. Fournier, explaining that the move toward rationally targeted treatments, whether related to gene mutations or independent molecular pathways of ALS progression, has created excitement in the field.
Numerous Disease Processes Are Potentially Targetable
As treatments are developed to address nongenetic molecular processes that contribute to the risk or progression of ALS, such as neuroinflammation or abnormal protein misfolding and aggregation, individualized treatment is likely to become key. Just as not all genetic cases share mutations in the same gene, the key molecular drivers of disease are likely to differ between patients. If so, it is hoped that biomarkers reflective of this underlying biology can be identified to appropriately target treatments.
“The excitement behind the newer targets in clinical trials is based on both basic science and early clinical data that support treatment based on specific drivers of disease,” Dr. Fournier said.
In 2023 and just prior to the FDA approval of tofersen, a set of expert consensus guidelines were published calling for genetic testing to be offered to all patients with ALS. These recommendations suggested that SOD1, C9orf72, FUS, and TARDBP should be included routinely into the panel of genes evaluated, calling for additional genes to be added as they emerge as potential therapeutic targets.
Even before these guidelines were released, genetic testing was already being offered at many centers with expertise in ALS. The rationale was to differentiate ALS with a genetic etiology from that with a nongenetic etiology, as well as to counsel family members when genetic risk was identified, but genetic testing has now assumed new urgency. In addition to the potential for offering a specific treatment for SOD1-related ALS, patients with other genetic forms of disease might be candidates for genetically focused clinical trials.
Genetic testing should be performed as soon as a diagnosis of ALS is made, according to Dr. Fournier. Although not all patients have accepted genetic testing, particularly in the past when there was no immediate clinical gain from establishing the presence of a genetic mutation, she said there is no longer any controversy about clinical relevance.
Genetic Testing Is Key to Genetic Therapies
“We do not want to miss the opportunity to treat patients when we have the chance,” said Dr. Fournier, referring to both the likely advantage of an early start of the approved gene therapy as well as the opportunity to participate in a clinical trial with other gene therapies in development.
Prior to the approval of tofersen, riluzole and edaravone had been the only disease-modifying agents in widespread use, but these drugs are nonspecific. There are no established biomarkers for establishing which patients are most likely to benefit.
In the case of riluzole, a pivotal trial conducted 30 years ago showed a survival benefit relative to placebo at 12 months (74% vs. 58%; P = 0.014). In a retrospective study published in 2022 that evaluated survival in a database of 4778 ALS patients of whom 3446 received riluzole, early diagnosis of ALS and prompt treatment with riluzole was associated with longer survival than delayed treatment. The benefit of edaravone has been validated with clinical measures, such as the revised Amyotrophic Lateral Sclerosis Functional Scale (ALSFRS-R).
The retrospective study of riluzole provides the basis for predicting better benefits from disease-modifying therapies if started earlier in the course of ALS. The same premise will be explored with newer therapies that target ALS-associated genes.
Early Treatment Presumed More Effective
“We think that earlier treatment in the course of ALS is probably better for gene therapies as well,” Dr. Fournier said. She cautioned that follow-up is not yet long enough to confirm a survival benefit with tofersen, but she said it is reasonable to anticipate better and longer response when neurologic damage is limited. Citing the effect of gene therapy in spinal muscular atrophy (SMA), where progression is halted if gene therapy is initiated early in life, Dr. Fourier suggested that the emphasis on early treatment stems from the low likelihood for treatments to reverse functional impairments.
“It is conceivable that future treatments might be developed to reverse symptoms, but current drug development is largely aimed at slowing progression,” she explained. Under some circumstances, halting progression has the potential to allow some function to be regained, but as the etiologies of ALS and the pathways of progression are better understood, she believes that all targeted therapy will be started as early as possible to prevent rather than treat neurological damage.
Tofersen, the gene therapy for SOD1-ALS, has provided an opportunity to test the idea that it may be possible to prevent ALS. In a phase 3 trial called ATLAS, unaffected carriers of SOD1 variants that are associated with aggressive disease and high or complete penetrance are enrolled for a run-in phase (Part A) during which participants are followed for a rise in neurofilament light chain (NfL) levels. Based on a previous natural history study called the Pre-Symptomatic Familial ALS (Pre-fALS) study, NfL rises in the serum of unaffected SOD1 carriers prior to phenoconversion. A low NfL is an entry criterion for ATLAS.
ATLAS End Point Is Reduction in Phenoconversion to Clinically Manifest ALS
People in whom NfL rises above a predefined threshold during the run-in stage will be eligible for randomization (Part B) to receive either tofersen or placebo. Efficacy will be measured by comparing the rates of phenoconversion to clinically manifest ALS between those who receive placebo and those who receive tofersen.
Two other groups enrolled in ATLAS will be followed on open-label tofersen. One comprises people who phenoconvert during Part B and the other comprises those who develop ALS during the run-in and therefore are not enrolled in Part B. These patients, forming Parts C and D of the study, provide another set of data to evaluate whether earlier rather than later introduction of therapy provides better outcomes.
“There is a lot of interest and optimism about the trial,” said Dr. Fournier, who praised the trial design and thinks the hypothesis being explored “makes sense.”
Michael Benatar, MD, PhD, professor of neurology and public health, University of Miami School of Medicine, Miami, Florida, is the principal investigator of ATLAS and also leads the Pre-Symptomatic Familial ALS study together with a colleague, Joanne Wuu, Associate Director of Research at the University of Miami ALS Center. The hope from these initiatives, according to Dr. Fournier, is that ATLAS will offer broader learnings beyond just the SOD1 population, providing critical information about the optimal timing of treatment initiation.
The benefit from targeting genes considered causative for ALS is not yet a sure thing. A clinical trial targeting C9orf72, for example, failed to support an approvable therapy. There is a trial of a gene therapy for the FUS variant that is ongoing. Yet, the introduction of a gene therapy for SOD1 variant ALS has already established that highly targeted therapies can be effective, an important step forward after so many failed treatment trials with nonspecific drugs.
“We are seeing more and more therapies being developed to address specific ALS biology,” said Dr. Fournier, who predicts a pivot toward conceptualizing ALS as an array of pathologies rather than one disorder driven by a single mechanism. More effort is being directed to recognizing phenotypes as well as genotypes. Hopefully, more biomarkers that distinguish between ALS variants will emerge and help in individualizing treatment.
“We are not there yet, but I think many of us in the field see this as a way forward,” she said.
Multidisciplinary Care, Symptomatic Management, and Palliative Care Are Still Essential for ALS
Disease-modifying therapies are the ultimate goal in ALS, but Dr. Fournier said that the other side of the equation is multidisciplinary and palliative care. To the extent that almost all ALS therapies only modify the course of disease modestly, palliative care remains the cornerstone of day-to-day care.
“Multidisciplinary and palliative care are not necessarily novel, but they are still critically important. There are clear data to show that multidisciplinary care improves functional status and quality of life, and that this is meaningful to patients,” Dr. Fournier said.
There have been numerous improvements in the areas of multidisciplinary and palliative care, some of which can be credited to advancing technology. In centers of excellence, the multidisciplinary approach has been focused on helping patients sustain a sense of independence and self-worth.
Now robotics, devices, and software are being increasingly employed to extend patient capabilities even in relatively advanced stages of disease, according to Dr. Fournier. As one example, she cited current work in brain-computer interfaces to record electrical activity in the central nervous system to allow patients to communicate even when speech is impaired.
A focus on patient-centered clinical care is appropriate because it is the best current opportunity to improve the lives of patients with ALS. Clinically, this work is very rewarding, according to Dr. Fournier, who described ALS patients overall as generally ”very invested in advocacy and research initiatives and motivated to help others,” Dr. Fournier said.
“The diagnosis can be tough, but there is satisfaction in helping these patients navigate toward an acceptable and meaningful quality of life. They typically give a lot back,” she added.
Overall, there is a sense of progress in ALS, even though it remains a uniformly fatal disease. Dr. Fournier expressed hope that clinical research is reaching a tipping point and an emphasis on targeted treatments after a long list of failed trials over the past 30 years. However, with only one approved therapy modifying an ALS-associated gene, this approach is still in its early stages.
Dr. Fournier has financial relationships with Amylyx, Biogen, Corcept, Denali, Mitsubishi QurAlis, and Tanabe.
Suggested Reading
Benatar M et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: the ATLAS Study. Neurotherapeutics. 2022 Jul;19(4):1248-1258. doi: 10.1007/s13311-022-01237-4.
Geronimo A et al. Ten Years of Riluzole Use in a Tertiary ALS Clinic. Muscle Nerve. 2022 Jun;65(6):659-666. doi: 10.1002/mus.27541.
Roggenbuck J et al. Evidence-Based Consensus Guidelines for ALS Genetic Testing and Counseling. Ann Clin Transl Neurol. 2023 Nov;10(11):2074-2091. doi: 10.1002/acn3.51895.
The first therapy targeted at modifying a mutant gene associated with amyotrophic lateral sclerosis (ALS), approved in early 2023, has offered reassurance that the biology of ALS, when known, is targetable. Historically, the disease has been considered a clinical diagnosis, but the
Despite a narrow indication, the only therapy targeted at an ALS-associated gene so far, SOD1 ALS, supports the premise that the biology of ALS can be modified, according to Christina N. Fournier, MD, an associate professor in the Department of Neurology, Emory University, Atlanta, Georgia.
Rather than a single pathological entity, ALS is best understood as the end result of many different pathological processes. Each might require its own targeted therapy in order to interrupt the upstream biological pathways that drive disease.
About 15% of ALS Has An Identifiable Genetic Cause
A family history of ALS is present in about 10% of cases. A genetic cause can be identified in approximately 15%. Cases without an identifiable genetic etiology are considered sporadic. So far, the only approved therapy that modifies the function of a gene associated with ALS is tofersen (Qalsody, Biogen), an antisense oligonucleotide. Tofersen inhibits RNA transcription of the superoxide dismutase 1 (SOD1) gene to decrease production of the SOD1 protein.
This first gene therapy for ALS is a breakthrough, but it is indicated for only a small proportion of ALS patients. Even though SOD1 gene mutations represent the second most common genetic cause of ALS after the C90rf72 gene, the proportion of patients who are candidates for tofersen is low. Efficacy is expected only in about 1% of those with familial ALS and 1% of those with sporadic ALS, or about 2% of all patients with ALS.
The evidence of benefit from a treatment with a specific target has provided the basis for concluding that “we are onto something,” Dr. Fournier said. An expert in ALS, she sees reason for excitement about the prospects in treatment with the growing focus on the underlying pathways of disease rather than the downstream consequences.
“The hope is that new gene-targeted therapies will be developed in the future to treat the broader ALS population,” said Dr. Fournier, explaining that the move toward rationally targeted treatments, whether related to gene mutations or independent molecular pathways of ALS progression, has created excitement in the field.
Numerous Disease Processes Are Potentially Targetable
As treatments are developed to address nongenetic molecular processes that contribute to the risk or progression of ALS, such as neuroinflammation or abnormal protein misfolding and aggregation, individualized treatment is likely to become key. Just as not all genetic cases share mutations in the same gene, the key molecular drivers of disease are likely to differ between patients. If so, it is hoped that biomarkers reflective of this underlying biology can be identified to appropriately target treatments.
“The excitement behind the newer targets in clinical trials is based on both basic science and early clinical data that support treatment based on specific drivers of disease,” Dr. Fournier said.
In 2023 and just prior to the FDA approval of tofersen, a set of expert consensus guidelines were published calling for genetic testing to be offered to all patients with ALS. These recommendations suggested that SOD1, C9orf72, FUS, and TARDBP should be included routinely into the panel of genes evaluated, calling for additional genes to be added as they emerge as potential therapeutic targets.
Even before these guidelines were released, genetic testing was already being offered at many centers with expertise in ALS. The rationale was to differentiate ALS with a genetic etiology from that with a nongenetic etiology, as well as to counsel family members when genetic risk was identified, but genetic testing has now assumed new urgency. In addition to the potential for offering a specific treatment for SOD1-related ALS, patients with other genetic forms of disease might be candidates for genetically focused clinical trials.
Genetic testing should be performed as soon as a diagnosis of ALS is made, according to Dr. Fournier. Although not all patients have accepted genetic testing, particularly in the past when there was no immediate clinical gain from establishing the presence of a genetic mutation, she said there is no longer any controversy about clinical relevance.
Genetic Testing Is Key to Genetic Therapies
“We do not want to miss the opportunity to treat patients when we have the chance,” said Dr. Fournier, referring to both the likely advantage of an early start of the approved gene therapy as well as the opportunity to participate in a clinical trial with other gene therapies in development.
Prior to the approval of tofersen, riluzole and edaravone had been the only disease-modifying agents in widespread use, but these drugs are nonspecific. There are no established biomarkers for establishing which patients are most likely to benefit.
In the case of riluzole, a pivotal trial conducted 30 years ago showed a survival benefit relative to placebo at 12 months (74% vs. 58%; P = 0.014). In a retrospective study published in 2022 that evaluated survival in a database of 4778 ALS patients of whom 3446 received riluzole, early diagnosis of ALS and prompt treatment with riluzole was associated with longer survival than delayed treatment. The benefit of edaravone has been validated with clinical measures, such as the revised Amyotrophic Lateral Sclerosis Functional Scale (ALSFRS-R).
The retrospective study of riluzole provides the basis for predicting better benefits from disease-modifying therapies if started earlier in the course of ALS. The same premise will be explored with newer therapies that target ALS-associated genes.
Early Treatment Presumed More Effective
“We think that earlier treatment in the course of ALS is probably better for gene therapies as well,” Dr. Fournier said. She cautioned that follow-up is not yet long enough to confirm a survival benefit with tofersen, but she said it is reasonable to anticipate better and longer response when neurologic damage is limited. Citing the effect of gene therapy in spinal muscular atrophy (SMA), where progression is halted if gene therapy is initiated early in life, Dr. Fourier suggested that the emphasis on early treatment stems from the low likelihood for treatments to reverse functional impairments.
“It is conceivable that future treatments might be developed to reverse symptoms, but current drug development is largely aimed at slowing progression,” she explained. Under some circumstances, halting progression has the potential to allow some function to be regained, but as the etiologies of ALS and the pathways of progression are better understood, she believes that all targeted therapy will be started as early as possible to prevent rather than treat neurological damage.
Tofersen, the gene therapy for SOD1-ALS, has provided an opportunity to test the idea that it may be possible to prevent ALS. In a phase 3 trial called ATLAS, unaffected carriers of SOD1 variants that are associated with aggressive disease and high or complete penetrance are enrolled for a run-in phase (Part A) during which participants are followed for a rise in neurofilament light chain (NfL) levels. Based on a previous natural history study called the Pre-Symptomatic Familial ALS (Pre-fALS) study, NfL rises in the serum of unaffected SOD1 carriers prior to phenoconversion. A low NfL is an entry criterion for ATLAS.
ATLAS End Point Is Reduction in Phenoconversion to Clinically Manifest ALS
People in whom NfL rises above a predefined threshold during the run-in stage will be eligible for randomization (Part B) to receive either tofersen or placebo. Efficacy will be measured by comparing the rates of phenoconversion to clinically manifest ALS between those who receive placebo and those who receive tofersen.
Two other groups enrolled in ATLAS will be followed on open-label tofersen. One comprises people who phenoconvert during Part B and the other comprises those who develop ALS during the run-in and therefore are not enrolled in Part B. These patients, forming Parts C and D of the study, provide another set of data to evaluate whether earlier rather than later introduction of therapy provides better outcomes.
“There is a lot of interest and optimism about the trial,” said Dr. Fournier, who praised the trial design and thinks the hypothesis being explored “makes sense.”
Michael Benatar, MD, PhD, professor of neurology and public health, University of Miami School of Medicine, Miami, Florida, is the principal investigator of ATLAS and also leads the Pre-Symptomatic Familial ALS study together with a colleague, Joanne Wuu, Associate Director of Research at the University of Miami ALS Center. The hope from these initiatives, according to Dr. Fournier, is that ATLAS will offer broader learnings beyond just the SOD1 population, providing critical information about the optimal timing of treatment initiation.
The benefit from targeting genes considered causative for ALS is not yet a sure thing. A clinical trial targeting C9orf72, for example, failed to support an approvable therapy. There is a trial of a gene therapy for the FUS variant that is ongoing. Yet, the introduction of a gene therapy for SOD1 variant ALS has already established that highly targeted therapies can be effective, an important step forward after so many failed treatment trials with nonspecific drugs.
“We are seeing more and more therapies being developed to address specific ALS biology,” said Dr. Fournier, who predicts a pivot toward conceptualizing ALS as an array of pathologies rather than one disorder driven by a single mechanism. More effort is being directed to recognizing phenotypes as well as genotypes. Hopefully, more biomarkers that distinguish between ALS variants will emerge and help in individualizing treatment.
“We are not there yet, but I think many of us in the field see this as a way forward,” she said.
Multidisciplinary Care, Symptomatic Management, and Palliative Care Are Still Essential for ALS
Disease-modifying therapies are the ultimate goal in ALS, but Dr. Fournier said that the other side of the equation is multidisciplinary and palliative care. To the extent that almost all ALS therapies only modify the course of disease modestly, palliative care remains the cornerstone of day-to-day care.
“Multidisciplinary and palliative care are not necessarily novel, but they are still critically important. There are clear data to show that multidisciplinary care improves functional status and quality of life, and that this is meaningful to patients,” Dr. Fournier said.
There have been numerous improvements in the areas of multidisciplinary and palliative care, some of which can be credited to advancing technology. In centers of excellence, the multidisciplinary approach has been focused on helping patients sustain a sense of independence and self-worth.
Now robotics, devices, and software are being increasingly employed to extend patient capabilities even in relatively advanced stages of disease, according to Dr. Fournier. As one example, she cited current work in brain-computer interfaces to record electrical activity in the central nervous system to allow patients to communicate even when speech is impaired.
A focus on patient-centered clinical care is appropriate because it is the best current opportunity to improve the lives of patients with ALS. Clinically, this work is very rewarding, according to Dr. Fournier, who described ALS patients overall as generally ”very invested in advocacy and research initiatives and motivated to help others,” Dr. Fournier said.
“The diagnosis can be tough, but there is satisfaction in helping these patients navigate toward an acceptable and meaningful quality of life. They typically give a lot back,” she added.
Overall, there is a sense of progress in ALS, even though it remains a uniformly fatal disease. Dr. Fournier expressed hope that clinical research is reaching a tipping point and an emphasis on targeted treatments after a long list of failed trials over the past 30 years. However, with only one approved therapy modifying an ALS-associated gene, this approach is still in its early stages.
Dr. Fournier has financial relationships with Amylyx, Biogen, Corcept, Denali, Mitsubishi QurAlis, and Tanabe.
Suggested Reading
Benatar M et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: the ATLAS Study. Neurotherapeutics. 2022 Jul;19(4):1248-1258. doi: 10.1007/s13311-022-01237-4.
Geronimo A et al. Ten Years of Riluzole Use in a Tertiary ALS Clinic. Muscle Nerve. 2022 Jun;65(6):659-666. doi: 10.1002/mus.27541.
Roggenbuck J et al. Evidence-Based Consensus Guidelines for ALS Genetic Testing and Counseling. Ann Clin Transl Neurol. 2023 Nov;10(11):2074-2091. doi: 10.1002/acn3.51895.
The first therapy targeted at modifying a mutant gene associated with amyotrophic lateral sclerosis (ALS), approved in early 2023, has offered reassurance that the biology of ALS, when known, is targetable. Historically, the disease has been considered a clinical diagnosis, but the
Despite a narrow indication, the only therapy targeted at an ALS-associated gene so far, SOD1 ALS, supports the premise that the biology of ALS can be modified, according to Christina N. Fournier, MD, an associate professor in the Department of Neurology, Emory University, Atlanta, Georgia.
Rather than a single pathological entity, ALS is best understood as the end result of many different pathological processes. Each might require its own targeted therapy in order to interrupt the upstream biological pathways that drive disease.
About 15% of ALS Has An Identifiable Genetic Cause
A family history of ALS is present in about 10% of cases. A genetic cause can be identified in approximately 15%. Cases without an identifiable genetic etiology are considered sporadic. So far, the only approved therapy that modifies the function of a gene associated with ALS is tofersen (Qalsody, Biogen), an antisense oligonucleotide. Tofersen inhibits RNA transcription of the superoxide dismutase 1 (SOD1) gene to decrease production of the SOD1 protein.
This first gene therapy for ALS is a breakthrough, but it is indicated for only a small proportion of ALS patients. Even though SOD1 gene mutations represent the second most common genetic cause of ALS after the C90rf72 gene, the proportion of patients who are candidates for tofersen is low. Efficacy is expected only in about 1% of those with familial ALS and 1% of those with sporadic ALS, or about 2% of all patients with ALS.
The evidence of benefit from a treatment with a specific target has provided the basis for concluding that “we are onto something,” Dr. Fournier said. An expert in ALS, she sees reason for excitement about the prospects in treatment with the growing focus on the underlying pathways of disease rather than the downstream consequences.
“The hope is that new gene-targeted therapies will be developed in the future to treat the broader ALS population,” said Dr. Fournier, explaining that the move toward rationally targeted treatments, whether related to gene mutations or independent molecular pathways of ALS progression, has created excitement in the field.
Numerous Disease Processes Are Potentially Targetable
As treatments are developed to address nongenetic molecular processes that contribute to the risk or progression of ALS, such as neuroinflammation or abnormal protein misfolding and aggregation, individualized treatment is likely to become key. Just as not all genetic cases share mutations in the same gene, the key molecular drivers of disease are likely to differ between patients. If so, it is hoped that biomarkers reflective of this underlying biology can be identified to appropriately target treatments.
“The excitement behind the newer targets in clinical trials is based on both basic science and early clinical data that support treatment based on specific drivers of disease,” Dr. Fournier said.
In 2023 and just prior to the FDA approval of tofersen, a set of expert consensus guidelines were published calling for genetic testing to be offered to all patients with ALS. These recommendations suggested that SOD1, C9orf72, FUS, and TARDBP should be included routinely into the panel of genes evaluated, calling for additional genes to be added as they emerge as potential therapeutic targets.
Even before these guidelines were released, genetic testing was already being offered at many centers with expertise in ALS. The rationale was to differentiate ALS with a genetic etiology from that with a nongenetic etiology, as well as to counsel family members when genetic risk was identified, but genetic testing has now assumed new urgency. In addition to the potential for offering a specific treatment for SOD1-related ALS, patients with other genetic forms of disease might be candidates for genetically focused clinical trials.
Genetic testing should be performed as soon as a diagnosis of ALS is made, according to Dr. Fournier. Although not all patients have accepted genetic testing, particularly in the past when there was no immediate clinical gain from establishing the presence of a genetic mutation, she said there is no longer any controversy about clinical relevance.
Genetic Testing Is Key to Genetic Therapies
“We do not want to miss the opportunity to treat patients when we have the chance,” said Dr. Fournier, referring to both the likely advantage of an early start of the approved gene therapy as well as the opportunity to participate in a clinical trial with other gene therapies in development.
Prior to the approval of tofersen, riluzole and edaravone had been the only disease-modifying agents in widespread use, but these drugs are nonspecific. There are no established biomarkers for establishing which patients are most likely to benefit.
In the case of riluzole, a pivotal trial conducted 30 years ago showed a survival benefit relative to placebo at 12 months (74% vs. 58%; P = 0.014). In a retrospective study published in 2022 that evaluated survival in a database of 4778 ALS patients of whom 3446 received riluzole, early diagnosis of ALS and prompt treatment with riluzole was associated with longer survival than delayed treatment. The benefit of edaravone has been validated with clinical measures, such as the revised Amyotrophic Lateral Sclerosis Functional Scale (ALSFRS-R).
The retrospective study of riluzole provides the basis for predicting better benefits from disease-modifying therapies if started earlier in the course of ALS. The same premise will be explored with newer therapies that target ALS-associated genes.
Early Treatment Presumed More Effective
“We think that earlier treatment in the course of ALS is probably better for gene therapies as well,” Dr. Fournier said. She cautioned that follow-up is not yet long enough to confirm a survival benefit with tofersen, but she said it is reasonable to anticipate better and longer response when neurologic damage is limited. Citing the effect of gene therapy in spinal muscular atrophy (SMA), where progression is halted if gene therapy is initiated early in life, Dr. Fourier suggested that the emphasis on early treatment stems from the low likelihood for treatments to reverse functional impairments.
“It is conceivable that future treatments might be developed to reverse symptoms, but current drug development is largely aimed at slowing progression,” she explained. Under some circumstances, halting progression has the potential to allow some function to be regained, but as the etiologies of ALS and the pathways of progression are better understood, she believes that all targeted therapy will be started as early as possible to prevent rather than treat neurological damage.
Tofersen, the gene therapy for SOD1-ALS, has provided an opportunity to test the idea that it may be possible to prevent ALS. In a phase 3 trial called ATLAS, unaffected carriers of SOD1 variants that are associated with aggressive disease and high or complete penetrance are enrolled for a run-in phase (Part A) during which participants are followed for a rise in neurofilament light chain (NfL) levels. Based on a previous natural history study called the Pre-Symptomatic Familial ALS (Pre-fALS) study, NfL rises in the serum of unaffected SOD1 carriers prior to phenoconversion. A low NfL is an entry criterion for ATLAS.
ATLAS End Point Is Reduction in Phenoconversion to Clinically Manifest ALS
People in whom NfL rises above a predefined threshold during the run-in stage will be eligible for randomization (Part B) to receive either tofersen or placebo. Efficacy will be measured by comparing the rates of phenoconversion to clinically manifest ALS between those who receive placebo and those who receive tofersen.
Two other groups enrolled in ATLAS will be followed on open-label tofersen. One comprises people who phenoconvert during Part B and the other comprises those who develop ALS during the run-in and therefore are not enrolled in Part B. These patients, forming Parts C and D of the study, provide another set of data to evaluate whether earlier rather than later introduction of therapy provides better outcomes.
“There is a lot of interest and optimism about the trial,” said Dr. Fournier, who praised the trial design and thinks the hypothesis being explored “makes sense.”
Michael Benatar, MD, PhD, professor of neurology and public health, University of Miami School of Medicine, Miami, Florida, is the principal investigator of ATLAS and also leads the Pre-Symptomatic Familial ALS study together with a colleague, Joanne Wuu, Associate Director of Research at the University of Miami ALS Center. The hope from these initiatives, according to Dr. Fournier, is that ATLAS will offer broader learnings beyond just the SOD1 population, providing critical information about the optimal timing of treatment initiation.
The benefit from targeting genes considered causative for ALS is not yet a sure thing. A clinical trial targeting C9orf72, for example, failed to support an approvable therapy. There is a trial of a gene therapy for the FUS variant that is ongoing. Yet, the introduction of a gene therapy for SOD1 variant ALS has already established that highly targeted therapies can be effective, an important step forward after so many failed treatment trials with nonspecific drugs.
“We are seeing more and more therapies being developed to address specific ALS biology,” said Dr. Fournier, who predicts a pivot toward conceptualizing ALS as an array of pathologies rather than one disorder driven by a single mechanism. More effort is being directed to recognizing phenotypes as well as genotypes. Hopefully, more biomarkers that distinguish between ALS variants will emerge and help in individualizing treatment.
“We are not there yet, but I think many of us in the field see this as a way forward,” she said.
Multidisciplinary Care, Symptomatic Management, and Palliative Care Are Still Essential for ALS
Disease-modifying therapies are the ultimate goal in ALS, but Dr. Fournier said that the other side of the equation is multidisciplinary and palliative care. To the extent that almost all ALS therapies only modify the course of disease modestly, palliative care remains the cornerstone of day-to-day care.
“Multidisciplinary and palliative care are not necessarily novel, but they are still critically important. There are clear data to show that multidisciplinary care improves functional status and quality of life, and that this is meaningful to patients,” Dr. Fournier said.
There have been numerous improvements in the areas of multidisciplinary and palliative care, some of which can be credited to advancing technology. In centers of excellence, the multidisciplinary approach has been focused on helping patients sustain a sense of independence and self-worth.
Now robotics, devices, and software are being increasingly employed to extend patient capabilities even in relatively advanced stages of disease, according to Dr. Fournier. As one example, she cited current work in brain-computer interfaces to record electrical activity in the central nervous system to allow patients to communicate even when speech is impaired.
A focus on patient-centered clinical care is appropriate because it is the best current opportunity to improve the lives of patients with ALS. Clinically, this work is very rewarding, according to Dr. Fournier, who described ALS patients overall as generally ”very invested in advocacy and research initiatives and motivated to help others,” Dr. Fournier said.
“The diagnosis can be tough, but there is satisfaction in helping these patients navigate toward an acceptable and meaningful quality of life. They typically give a lot back,” she added.
Overall, there is a sense of progress in ALS, even though it remains a uniformly fatal disease. Dr. Fournier expressed hope that clinical research is reaching a tipping point and an emphasis on targeted treatments after a long list of failed trials over the past 30 years. However, with only one approved therapy modifying an ALS-associated gene, this approach is still in its early stages.
Dr. Fournier has financial relationships with Amylyx, Biogen, Corcept, Denali, Mitsubishi QurAlis, and Tanabe.
Suggested Reading
Benatar M et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: the ATLAS Study. Neurotherapeutics. 2022 Jul;19(4):1248-1258. doi: 10.1007/s13311-022-01237-4.
Geronimo A et al. Ten Years of Riluzole Use in a Tertiary ALS Clinic. Muscle Nerve. 2022 Jun;65(6):659-666. doi: 10.1002/mus.27541.
Roggenbuck J et al. Evidence-Based Consensus Guidelines for ALS Genetic Testing and Counseling. Ann Clin Transl Neurol. 2023 Nov;10(11):2074-2091. doi: 10.1002/acn3.51895.
Hearing Loss, Neuropathy Cut Survival in Older Adults
TOPLINE:
METHODOLOGY:
- Researchers analyzed 793 older adults recruited from primary care practices participating in the OKLAHOMA Studies in 1999.
- Participants completed a questionnaire and underwent a physical examination; timed gait assessments (50 ft); and tests for peripheral nerve function, balance, and hearing.
- Hearing thresholds were tested at 20, 25, and 40 dB, respectively, and at sound frequencies of 500, 1000, 2000, and 4000 Hz.
- Researchers tracked mortality data over 22 years.
TAKEAWAY:
- Overall, 83% participants experienced hearing loss. Regular use of hearing aids was low, reported in 19% and 55% of those with moderate and severe hearing loss, respectively.
- Hearing loss was linked to impaired balance (P = .0014), slower walking (P = .0024), and reduced survival time (P = .0001). Moderate to severe hearing loss was strongly associated with reduced survival time (odds ratio, 1.36; P = .001), independent of the use of hearing aids.
- Peripheral neuropathy was present in 32% participants. The condition also increased the risk for death over the study period (hazard ratio [HR], 1.32; P = .003). Participants with both hearing loss and peripheral neuropathy showed reduced balance and survival time compared with people with either condition alone (HR, 1.55; P < .0001).
IN PRACTICE:
“Like peripheral neuropathy, advanced-age hearing loss is associated with reduced life expectancy, probably mediated in part through an adverse impact on balance,” the authors wrote. “Greater appreciation for the serious impacts of hearing loss and peripheral neuropathy could lead to further efforts to understand their causes and improve prevention and treatment strategies.”
SOURCE:
The study was led by James W. Mold, MD, MPH, of the University of Oklahoma Health Sciences Center, Oklahoma City. It was published online in the Journal of the American Geriatrics Society.
LIMITATIONS:
The dataset was collected in 1999 and may not entirely represent the current cohorts of older primary care patients. The absence of soundproof rooms and the exclusion of some components of the standard audiometric evaluation may have affected low-frequency sound measurements. Furthermore, physical examination was a less accurate measure of peripheral neuropathy. Information on the duration or severity of predictors and causes of death was not available.
DISCLOSURES:
The study was funded by the Presbyterian Health Foundation. The authors did not disclose any competing interests.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Researchers analyzed 793 older adults recruited from primary care practices participating in the OKLAHOMA Studies in 1999.
- Participants completed a questionnaire and underwent a physical examination; timed gait assessments (50 ft); and tests for peripheral nerve function, balance, and hearing.
- Hearing thresholds were tested at 20, 25, and 40 dB, respectively, and at sound frequencies of 500, 1000, 2000, and 4000 Hz.
- Researchers tracked mortality data over 22 years.
TAKEAWAY:
- Overall, 83% participants experienced hearing loss. Regular use of hearing aids was low, reported in 19% and 55% of those with moderate and severe hearing loss, respectively.
- Hearing loss was linked to impaired balance (P = .0014), slower walking (P = .0024), and reduced survival time (P = .0001). Moderate to severe hearing loss was strongly associated with reduced survival time (odds ratio, 1.36; P = .001), independent of the use of hearing aids.
- Peripheral neuropathy was present in 32% participants. The condition also increased the risk for death over the study period (hazard ratio [HR], 1.32; P = .003). Participants with both hearing loss and peripheral neuropathy showed reduced balance and survival time compared with people with either condition alone (HR, 1.55; P < .0001).
IN PRACTICE:
“Like peripheral neuropathy, advanced-age hearing loss is associated with reduced life expectancy, probably mediated in part through an adverse impact on balance,” the authors wrote. “Greater appreciation for the serious impacts of hearing loss and peripheral neuropathy could lead to further efforts to understand their causes and improve prevention and treatment strategies.”
SOURCE:
The study was led by James W. Mold, MD, MPH, of the University of Oklahoma Health Sciences Center, Oklahoma City. It was published online in the Journal of the American Geriatrics Society.
LIMITATIONS:
The dataset was collected in 1999 and may not entirely represent the current cohorts of older primary care patients. The absence of soundproof rooms and the exclusion of some components of the standard audiometric evaluation may have affected low-frequency sound measurements. Furthermore, physical examination was a less accurate measure of peripheral neuropathy. Information on the duration or severity of predictors and causes of death was not available.
DISCLOSURES:
The study was funded by the Presbyterian Health Foundation. The authors did not disclose any competing interests.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Researchers analyzed 793 older adults recruited from primary care practices participating in the OKLAHOMA Studies in 1999.
- Participants completed a questionnaire and underwent a physical examination; timed gait assessments (50 ft); and tests for peripheral nerve function, balance, and hearing.
- Hearing thresholds were tested at 20, 25, and 40 dB, respectively, and at sound frequencies of 500, 1000, 2000, and 4000 Hz.
- Researchers tracked mortality data over 22 years.
TAKEAWAY:
- Overall, 83% participants experienced hearing loss. Regular use of hearing aids was low, reported in 19% and 55% of those with moderate and severe hearing loss, respectively.
- Hearing loss was linked to impaired balance (P = .0014), slower walking (P = .0024), and reduced survival time (P = .0001). Moderate to severe hearing loss was strongly associated with reduced survival time (odds ratio, 1.36; P = .001), independent of the use of hearing aids.
- Peripheral neuropathy was present in 32% participants. The condition also increased the risk for death over the study period (hazard ratio [HR], 1.32; P = .003). Participants with both hearing loss and peripheral neuropathy showed reduced balance and survival time compared with people with either condition alone (HR, 1.55; P < .0001).
IN PRACTICE:
“Like peripheral neuropathy, advanced-age hearing loss is associated with reduced life expectancy, probably mediated in part through an adverse impact on balance,” the authors wrote. “Greater appreciation for the serious impacts of hearing loss and peripheral neuropathy could lead to further efforts to understand their causes and improve prevention and treatment strategies.”
SOURCE:
The study was led by James W. Mold, MD, MPH, of the University of Oklahoma Health Sciences Center, Oklahoma City. It was published online in the Journal of the American Geriatrics Society.
LIMITATIONS:
The dataset was collected in 1999 and may not entirely represent the current cohorts of older primary care patients. The absence of soundproof rooms and the exclusion of some components of the standard audiometric evaluation may have affected low-frequency sound measurements. Furthermore, physical examination was a less accurate measure of peripheral neuropathy. Information on the duration or severity of predictors and causes of death was not available.
DISCLOSURES:
The study was funded by the Presbyterian Health Foundation. The authors did not disclose any competing interests.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Mobile App Shows Promise in Managing Fibromyalgia Symptoms
TOPLINE:
A smartphone app that delivers acceptance and commitment therapy (ACT), a type of cognitive behavioral therapy, improves overall well-being and reduces the severity of pain, fatigue, sleep issues, and depression to a greater extent than daily symptom tracking in patients with fibromyalgia.
METHODOLOGY:
- Researchers conducted the phase 3 PROSPER-FM trial at 25 community sites in the United States to assess the efficacy and safety of digital ACT for patients with fibromyalgia.
- A total of 275 adult patients aged 22-75 years with fibromyalgia were randomly assigned to either the digital ACT group (n = 140) or the active control group (n = 135) for 12 weeks.
- Patients in the digital ACT group received a self-guided, smartphone-delivered program in which they learned and practiced the core ACT skills of acceptance, values, mindfulness, defusion, self as context, and willingness and committed action to build psychological flexibility, while the control group underwent daily symptom tracking and received educational materials.
- The primary endpoint was the response rate on the Patient Global Impression of Change (PGIC) at week 12, which is an indicator of patient well-being.
- The secondary endpoints included changes in the Revised Fibromyalgia Impact Questionnaire (FIQ-R) total score and pain intensity, pain interference, and sleep interference scores.
TAKEAWAY:
- At week 12, 71% of the patients in the digital ACT group responded with a minimally improved or better change in the PGIC response, compared with only 22% of the patients in the control group (P < .0001).
- The digital ACT group showed a significant reduction in the impact of fibromyalgia, with a between-group effect size of d = 0.65 (P < .0001) at week 12. The FIQ-R total score significantly improved within 3 weeks of using the self-guided digital ACT app.
- The use of digital ACT also demonstrated positive effects on the levels of weekly pain intensity (P = .001) and depression (P < .0001), compared with the control group.
- No serious adverse effects related to the app were reported, and both groups demonstrated high rates of adherence, with most (72%) participants in the digital ACT group completing at least 42 sessions.
IN PRACTICE:
“The results found in the study are essential for professionals who care for patients with fibromyalgia as they present a new viable treatment alternative,” Guilherme Torres Vilarino, PhD, Santa Catarina State University, Florianópolis, Brazil, wrote in an accompanying editorial.
SOURCE:
This study was led by R. Michael Gendreau, MD, PhD, Gendreau Consulting, Poway, California. It was published online in The Lancet.
LIMITATIONS:
The study population predominantly consisted of women and White individuals, which may limit the generalizability of the findings to more diverse populations. Additionally, the study was conducted in the United States, and the results may thus not be applicable to other countries with different racial, ethnic, educational, and economic characteristics. The study duration was 12 weeks, and the long-term benefits of digital ACT have not yet been shown.
DISCLOSURES:
This study was funded by Swing Therapeutics. Seven authors declared having stock options and/or receiving salary from Swing Therapeutics. Other authors reported having many ties with several sources, including Swing Therapeutics.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
A smartphone app that delivers acceptance and commitment therapy (ACT), a type of cognitive behavioral therapy, improves overall well-being and reduces the severity of pain, fatigue, sleep issues, and depression to a greater extent than daily symptom tracking in patients with fibromyalgia.
METHODOLOGY:
- Researchers conducted the phase 3 PROSPER-FM trial at 25 community sites in the United States to assess the efficacy and safety of digital ACT for patients with fibromyalgia.
- A total of 275 adult patients aged 22-75 years with fibromyalgia were randomly assigned to either the digital ACT group (n = 140) or the active control group (n = 135) for 12 weeks.
- Patients in the digital ACT group received a self-guided, smartphone-delivered program in which they learned and practiced the core ACT skills of acceptance, values, mindfulness, defusion, self as context, and willingness and committed action to build psychological flexibility, while the control group underwent daily symptom tracking and received educational materials.
- The primary endpoint was the response rate on the Patient Global Impression of Change (PGIC) at week 12, which is an indicator of patient well-being.
- The secondary endpoints included changes in the Revised Fibromyalgia Impact Questionnaire (FIQ-R) total score and pain intensity, pain interference, and sleep interference scores.
TAKEAWAY:
- At week 12, 71% of the patients in the digital ACT group responded with a minimally improved or better change in the PGIC response, compared with only 22% of the patients in the control group (P < .0001).
- The digital ACT group showed a significant reduction in the impact of fibromyalgia, with a between-group effect size of d = 0.65 (P < .0001) at week 12. The FIQ-R total score significantly improved within 3 weeks of using the self-guided digital ACT app.
- The use of digital ACT also demonstrated positive effects on the levels of weekly pain intensity (P = .001) and depression (P < .0001), compared with the control group.
- No serious adverse effects related to the app were reported, and both groups demonstrated high rates of adherence, with most (72%) participants in the digital ACT group completing at least 42 sessions.
IN PRACTICE:
“The results found in the study are essential for professionals who care for patients with fibromyalgia as they present a new viable treatment alternative,” Guilherme Torres Vilarino, PhD, Santa Catarina State University, Florianópolis, Brazil, wrote in an accompanying editorial.
SOURCE:
This study was led by R. Michael Gendreau, MD, PhD, Gendreau Consulting, Poway, California. It was published online in The Lancet.
LIMITATIONS:
The study population predominantly consisted of women and White individuals, which may limit the generalizability of the findings to more diverse populations. Additionally, the study was conducted in the United States, and the results may thus not be applicable to other countries with different racial, ethnic, educational, and economic characteristics. The study duration was 12 weeks, and the long-term benefits of digital ACT have not yet been shown.
DISCLOSURES:
This study was funded by Swing Therapeutics. Seven authors declared having stock options and/or receiving salary from Swing Therapeutics. Other authors reported having many ties with several sources, including Swing Therapeutics.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
A smartphone app that delivers acceptance and commitment therapy (ACT), a type of cognitive behavioral therapy, improves overall well-being and reduces the severity of pain, fatigue, sleep issues, and depression to a greater extent than daily symptom tracking in patients with fibromyalgia.
METHODOLOGY:
- Researchers conducted the phase 3 PROSPER-FM trial at 25 community sites in the United States to assess the efficacy and safety of digital ACT for patients with fibromyalgia.
- A total of 275 adult patients aged 22-75 years with fibromyalgia were randomly assigned to either the digital ACT group (n = 140) or the active control group (n = 135) for 12 weeks.
- Patients in the digital ACT group received a self-guided, smartphone-delivered program in which they learned and practiced the core ACT skills of acceptance, values, mindfulness, defusion, self as context, and willingness and committed action to build psychological flexibility, while the control group underwent daily symptom tracking and received educational materials.
- The primary endpoint was the response rate on the Patient Global Impression of Change (PGIC) at week 12, which is an indicator of patient well-being.
- The secondary endpoints included changes in the Revised Fibromyalgia Impact Questionnaire (FIQ-R) total score and pain intensity, pain interference, and sleep interference scores.
TAKEAWAY:
- At week 12, 71% of the patients in the digital ACT group responded with a minimally improved or better change in the PGIC response, compared with only 22% of the patients in the control group (P < .0001).
- The digital ACT group showed a significant reduction in the impact of fibromyalgia, with a between-group effect size of d = 0.65 (P < .0001) at week 12. The FIQ-R total score significantly improved within 3 weeks of using the self-guided digital ACT app.
- The use of digital ACT also demonstrated positive effects on the levels of weekly pain intensity (P = .001) and depression (P < .0001), compared with the control group.
- No serious adverse effects related to the app were reported, and both groups demonstrated high rates of adherence, with most (72%) participants in the digital ACT group completing at least 42 sessions.
IN PRACTICE:
“The results found in the study are essential for professionals who care for patients with fibromyalgia as they present a new viable treatment alternative,” Guilherme Torres Vilarino, PhD, Santa Catarina State University, Florianópolis, Brazil, wrote in an accompanying editorial.
SOURCE:
This study was led by R. Michael Gendreau, MD, PhD, Gendreau Consulting, Poway, California. It was published online in The Lancet.
LIMITATIONS:
The study population predominantly consisted of women and White individuals, which may limit the generalizability of the findings to more diverse populations. Additionally, the study was conducted in the United States, and the results may thus not be applicable to other countries with different racial, ethnic, educational, and economic characteristics. The study duration was 12 weeks, and the long-term benefits of digital ACT have not yet been shown.
DISCLOSURES:
This study was funded by Swing Therapeutics. Seven authors declared having stock options and/or receiving salary from Swing Therapeutics. Other authors reported having many ties with several sources, including Swing Therapeutics.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
‘Doesn’t Fit Anything I Trained for’: Committee Examines Treatment for Chronic Illness After Lyme Disease
WASHINGTON — Advancing treatment for what has been variably called chronic Lyme and posttreatment Lyme disease (PTLD) is under the eyes of a National Academies of Science, Engineering, and Medicine (NASEM) committee of experts for the first time — a year after the NASEM shone a spotlight on the need to accelerate research on chronic illnesses that follow known or suspected infections.
The committee will not make recommendations on specific approaches to diagnosis and treatment when it issues a report in early 2025 but will instead present “consensus findings” on treatment for chronic illness associated with Lyme disease, including recommendations for advancing treatment.
It’s an area void of the US Food and Drug Administration–approved therapies, void of any consensus on the off-label use of medications, and without any current standard of care or proven mechanisms and pathophysiology, said John Aucott, MD, director of the Johns Hopkins Medicine Lyme Disease Clinical Research Center, Baltimore, one of the invited speakers at a public meeting held by the NASEM in Washington, DC.
“The best way to look at this illness is not from the silos of infectious disease or the silos of rheumatology; you have to look across disciplines,” Dr. Aucott, also associate professor of medicine in the Division of Rheumatology, told the committee. “The story doesn’t fit anything I trained for in my infectious disease fellowship. Even today, I’d posit that PTLD is like an island — it’s still not connected to a lot of the mainstream of medicine.”
Rhisa Parera, who wrote and directed a 2021 documentary, Your Labs Are Normal, was one of several invited speakers who amplified the patient voice. Starting around age 7, she had pain in her knees, spine, and hips and vivid nightmares. In high school, she developed gastrointestinal issues, and in college, she developed debilitating neurologic symptoms.
Depression was her eventual diagnosis after having seen “every specialist in the book,” she said. At age 29, she received a positive western blot test and a Lyme disease diagnosis, at which point “I was prescribed 4 weeks of doxycycline and left in the dark,” the 34-year-old Black patient told the committee. Her health improved only after she began working with an “LLMD,” or Lyme-literate medical doctor (a term used in the patient community), while she lived with her mother and did not work, she said.
“I don’t share my Lyme disease history with other doctors. It’s pointless when you have those who will laugh at you, say you’re fine if you were treated, or just deny the disease completely,” Ms. Parera said. “We need this to be taught in medical school. It’s a literal emergency.”
Incidence and Potential Mechanisms
Limited research has suggested that 10%-20% of patients with Lyme disease develop persistent symptoms after standard antibiotic treatment advised by the Infectious Diseases Society of America (IDSA), Dr. Aucott said. (On its web page on chronic symptoms, the Centers for Disease Control and Prevention presents a more conservative range of 5%-10%.)
His own prospective cohort study at Johns Hopkins, published in 2022, found that 13.7% of 234 patients with prior Lyme disease met symptom and functional impact criteria for PTLD, compared with 4.1% of 49 participants without a history of Lyme disease — a statistically significant difference that he said should “put to rest” the question of “is it real?”
PTLD is the research case definition proposed by the IDSA in 2006; it requires that patients have prior documented Lyme disease, no other specific comorbidities, and specific symptoms (fatigue, widespread musculoskeletal pain, and/or cognitive difficulties) causing significant functional impact at least 6 months from their initial diagnosis and treatment.
In the real world, however, where diagnostics for acute Lyme disease are often inaccurate, erythema migrans is often absent, and the symptomatology of Lyme IACI is variable (and where there is no approved laboratory test or objective biomarker for diagnosing Lyme IACI), PTLD represents only a subset of a broader, heterogeneous population with persistent symptoms.
The term “Lyme IACI,” pronounced “Lyme eye-ACK-ee” at the meeting, builds on conversations at the 2023 NASEM workshop on infection-associated chronic illnesses and “encompasses a variety of terms that are used,” including PTLD, PTLD syndrome, persistent Lyme disease, and chronic Lyme disease, according to committee documents. Symptoms are distinct from the known complications of Lyme disease, such as arthritis or carditis.
The findings from Dr. Aucott’s SLICE cohort likely represent “the best outcome,” he said. They’re “probably not generalizable to a community setting where we see lots of missed diagnoses and delayed diagnoses,” as well as other tick-borne coinfections.
One of the challenges in designing future trials, in fact, relates to enrollment criteria and whether to use strict inclusion and exclusion criteria associated with the IDSA definition or take a broader approach to trial enrollment, he and others said. “You want to enroll patients for whom there’s no controversy that they’ve had Lyme infection ... for a study people believe in,” Dr. Aucott said during a discussion period, noting that it’s typical to screen over 100 patients to find one enrollee. “But it’s a tension we’re having.”
Timothy Sellati, PhD, chief scientific officer of the Global Lyme Alliance, urged change. “It’s really important to try to figure out how to alter our thinking on identifying and diagnosing chronic Lyme patients because they need to be recruited into clinical trials,” he said during his presentation.
“We think the best way to do this is to [develop and] employ composite diagnostic testing” that looks at unique Borrelia signatures (eg, protein, DNA, RNA, or metabolites), genetic and/or epigenetic signatures, inflammation signatures, T-cell-independent antibody signatures, and other elements, Dr. Sellati said.
Researchers designing treatment trials also face unknowns, Dr. Aucott and others said, about the role of potential mechanisms of Lyme IACI, from persistent Borrelia burgdorferi (or Borrelia mayonii) infection or the persistence of bacterial remnants (eg, nucleic acids or peptidoglycans) to infection-triggered pathology such as persistent immune dysregulation, chronic inflammation, autoimmunity, microbiome alterations, and dysautonomia and other neural network alterations.
The NASEM’s spotlight on Lyme IACI follows its long COVID-driven push last year to advance a common research agenda in infection-associated chronic illnesses. Investigators see common symptoms and potential shared mechanisms between long COVID, Lyme IACI, myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), and other complex chronic illnesses following infections.
At the Lyme IACI meeting, invited speakers described parts of the research landscape. Avindra Nath, MD, of the National Institute of Neurological Disorders and Stroke, for instance, described a recently published deep phenotyping study of 17 patients with ME/CFS that found decreased central catecholamine synthesis, circuit dysfunction of integrative brain regions, and immune profiling differences (eg, defects in B-cell maturation or T-cell exhaustion), compared with matched controls, that suggest the persistence of microbial antigens.
And John Leong, MD, PhD, of Tufts University, Boston, described his lab’s focus on understanding the microbe-host interactions that enable bloodstream dissemination and tissue invasion of B burgdorferi to take hold, increasing the risk for persistent symptoms. Other research at Tufts, he noted during a discussion period, has demonstrated the persistence of B burgdorferi to antibiotics in microtiter dishes. “Those organisms that survive are really difficult to eradicate in vitro,” Dr. Leong said.
Other physician investigators described research on nociplastic pain — a category of pain that can be triggered by infections, causing both amplified sensory processing and augmented central nervous system pain — and on whether reactivation of the Epstein-Barr virus could potentiate autoimmunity in the context of Borrelia infection.
Researchers are ready to test therapies while pathophysiology is unraveled — provided there is funding, Dr. Aucott said. The Clinical Trials Network for Lyme and Other Tick-Borne Diseases, coordinated by Brian Fallon, MD, of Columbia University, New York City, and funded several years ago by the Steven & Alexandra Cohen Foundation, has a slate of small pilot studies underway or being planned that address potential mechanisms (eg, studies of pulse intravenous ceftriaxone, tetracycline, transauricular vagus nerve stimulation, and mast cell modulation). And should full multisite trials be designed and funded, the network is ready with an infrastructure.
Need for Patient-Centered Outcomes
Persistent symptomatology is on the NIH’s radar screen. Efforts to understand causes were part of a strategic tick-borne disease research plan developed by the NIH in 2019. And in 2023, the National Institute of Allergy and Infectious Diseases (NIAID) funded seven projects addressing persistent symptoms that will run through 2028, C. Benjamin Beard, PhD, deputy division director of the CDC’s Division of Vector-Borne Disease, said at the NASEM committee meeting.
Patient advocates maintained that too much emphasis is placed on tick biology and pathophysiology. When Wendy Adams, research grant director and advisory board member of the Bay Area Lyme Foundation, and a colleague analyzed NIAID tick-borne disease funding from 2013 to 2021, they found that 75% of the funding went toward basic research, 15% to translational research, and “only 3% went to clinical research,” Ms. Adams told the committee.
Only 3% of the basic research budget was spent on coinfections, she said, and only 1% was spent on neurologic disease associated with tick-borne infections, both of which are survey-defined patient priorities. Moreover, “12% of the overall NIAID [tick-borne diseases] budget was spent on tick biology,” she said.
Research needs to involve community physicians who are utilizing the guidelines and approaches of the International Lyme and Associated Diseases Society to treat most patients with Lyme IACI, Ms. Adams said. “They have data to be mined,” she said, as does LymeDisease.org, which maintains a patient registry, MyLymeData, with over 18,000 patients. The organization has published two treatment studies, including one on antibiotic treatment response.
Lorraine Johnson, JD, MBA, CEO of LymeDisease.org and principal investigator of MyLymeData, stressed the importance of using patient-centered outcomes that incorporate minimal clinically important differences (MCIDs). “A change in the SF-36 score [without consideration of MCIDs] is not inherently important or meaningful to patients,” she said, referring to the SF-36 survey of health-related quality of life.
“This may seem like an esoteric issue, but two of the four clinical trials done [on retreatment of] persistent Lyme disease used the SF-36 as their outcome measure, and those studies, led by [Mark] Klempner, concluded that retreatment was not effective,” Ms. Johnson said. “Patients have been and continue to be harmed by [this research] because they’re told by physicians that antibiotics don’t work.”
A 2012 biostatistical review of these four RCTs — trials that helped inform the 2006 IDSA treatment guidelines — concluded that the Klempner studies “set the bar for treatment success too high,” Ms. Johnson said. Three of the four trials were likely underpowered to detect clinically meaningful treatment effects, the review also found.
The NASEM committee will hold additional public meetings and review a wide range of literature through this year. The formation of the committee was recommended by the US Department of Health and Human Services Tick-Borne Disease Working Group that was established by Congress in 2016 and concluded its work in 2022. The committee’s work is funded by the Cohen Foundation.
A version of this article appeared on Medscape.com.
WASHINGTON — Advancing treatment for what has been variably called chronic Lyme and posttreatment Lyme disease (PTLD) is under the eyes of a National Academies of Science, Engineering, and Medicine (NASEM) committee of experts for the first time — a year after the NASEM shone a spotlight on the need to accelerate research on chronic illnesses that follow known or suspected infections.
The committee will not make recommendations on specific approaches to diagnosis and treatment when it issues a report in early 2025 but will instead present “consensus findings” on treatment for chronic illness associated with Lyme disease, including recommendations for advancing treatment.
It’s an area void of the US Food and Drug Administration–approved therapies, void of any consensus on the off-label use of medications, and without any current standard of care or proven mechanisms and pathophysiology, said John Aucott, MD, director of the Johns Hopkins Medicine Lyme Disease Clinical Research Center, Baltimore, one of the invited speakers at a public meeting held by the NASEM in Washington, DC.
“The best way to look at this illness is not from the silos of infectious disease or the silos of rheumatology; you have to look across disciplines,” Dr. Aucott, also associate professor of medicine in the Division of Rheumatology, told the committee. “The story doesn’t fit anything I trained for in my infectious disease fellowship. Even today, I’d posit that PTLD is like an island — it’s still not connected to a lot of the mainstream of medicine.”
Rhisa Parera, who wrote and directed a 2021 documentary, Your Labs Are Normal, was one of several invited speakers who amplified the patient voice. Starting around age 7, she had pain in her knees, spine, and hips and vivid nightmares. In high school, she developed gastrointestinal issues, and in college, she developed debilitating neurologic symptoms.
Depression was her eventual diagnosis after having seen “every specialist in the book,” she said. At age 29, she received a positive western blot test and a Lyme disease diagnosis, at which point “I was prescribed 4 weeks of doxycycline and left in the dark,” the 34-year-old Black patient told the committee. Her health improved only after she began working with an “LLMD,” or Lyme-literate medical doctor (a term used in the patient community), while she lived with her mother and did not work, she said.
“I don’t share my Lyme disease history with other doctors. It’s pointless when you have those who will laugh at you, say you’re fine if you were treated, or just deny the disease completely,” Ms. Parera said. “We need this to be taught in medical school. It’s a literal emergency.”
Incidence and Potential Mechanisms
Limited research has suggested that 10%-20% of patients with Lyme disease develop persistent symptoms after standard antibiotic treatment advised by the Infectious Diseases Society of America (IDSA), Dr. Aucott said. (On its web page on chronic symptoms, the Centers for Disease Control and Prevention presents a more conservative range of 5%-10%.)
His own prospective cohort study at Johns Hopkins, published in 2022, found that 13.7% of 234 patients with prior Lyme disease met symptom and functional impact criteria for PTLD, compared with 4.1% of 49 participants without a history of Lyme disease — a statistically significant difference that he said should “put to rest” the question of “is it real?”
PTLD is the research case definition proposed by the IDSA in 2006; it requires that patients have prior documented Lyme disease, no other specific comorbidities, and specific symptoms (fatigue, widespread musculoskeletal pain, and/or cognitive difficulties) causing significant functional impact at least 6 months from their initial diagnosis and treatment.
In the real world, however, where diagnostics for acute Lyme disease are often inaccurate, erythema migrans is often absent, and the symptomatology of Lyme IACI is variable (and where there is no approved laboratory test or objective biomarker for diagnosing Lyme IACI), PTLD represents only a subset of a broader, heterogeneous population with persistent symptoms.
The term “Lyme IACI,” pronounced “Lyme eye-ACK-ee” at the meeting, builds on conversations at the 2023 NASEM workshop on infection-associated chronic illnesses and “encompasses a variety of terms that are used,” including PTLD, PTLD syndrome, persistent Lyme disease, and chronic Lyme disease, according to committee documents. Symptoms are distinct from the known complications of Lyme disease, such as arthritis or carditis.
The findings from Dr. Aucott’s SLICE cohort likely represent “the best outcome,” he said. They’re “probably not generalizable to a community setting where we see lots of missed diagnoses and delayed diagnoses,” as well as other tick-borne coinfections.
One of the challenges in designing future trials, in fact, relates to enrollment criteria and whether to use strict inclusion and exclusion criteria associated with the IDSA definition or take a broader approach to trial enrollment, he and others said. “You want to enroll patients for whom there’s no controversy that they’ve had Lyme infection ... for a study people believe in,” Dr. Aucott said during a discussion period, noting that it’s typical to screen over 100 patients to find one enrollee. “But it’s a tension we’re having.”
Timothy Sellati, PhD, chief scientific officer of the Global Lyme Alliance, urged change. “It’s really important to try to figure out how to alter our thinking on identifying and diagnosing chronic Lyme patients because they need to be recruited into clinical trials,” he said during his presentation.
“We think the best way to do this is to [develop and] employ composite diagnostic testing” that looks at unique Borrelia signatures (eg, protein, DNA, RNA, or metabolites), genetic and/or epigenetic signatures, inflammation signatures, T-cell-independent antibody signatures, and other elements, Dr. Sellati said.
Researchers designing treatment trials also face unknowns, Dr. Aucott and others said, about the role of potential mechanisms of Lyme IACI, from persistent Borrelia burgdorferi (or Borrelia mayonii) infection or the persistence of bacterial remnants (eg, nucleic acids or peptidoglycans) to infection-triggered pathology such as persistent immune dysregulation, chronic inflammation, autoimmunity, microbiome alterations, and dysautonomia and other neural network alterations.
The NASEM’s spotlight on Lyme IACI follows its long COVID-driven push last year to advance a common research agenda in infection-associated chronic illnesses. Investigators see common symptoms and potential shared mechanisms between long COVID, Lyme IACI, myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), and other complex chronic illnesses following infections.
At the Lyme IACI meeting, invited speakers described parts of the research landscape. Avindra Nath, MD, of the National Institute of Neurological Disorders and Stroke, for instance, described a recently published deep phenotyping study of 17 patients with ME/CFS that found decreased central catecholamine synthesis, circuit dysfunction of integrative brain regions, and immune profiling differences (eg, defects in B-cell maturation or T-cell exhaustion), compared with matched controls, that suggest the persistence of microbial antigens.
And John Leong, MD, PhD, of Tufts University, Boston, described his lab’s focus on understanding the microbe-host interactions that enable bloodstream dissemination and tissue invasion of B burgdorferi to take hold, increasing the risk for persistent symptoms. Other research at Tufts, he noted during a discussion period, has demonstrated the persistence of B burgdorferi to antibiotics in microtiter dishes. “Those organisms that survive are really difficult to eradicate in vitro,” Dr. Leong said.
Other physician investigators described research on nociplastic pain — a category of pain that can be triggered by infections, causing both amplified sensory processing and augmented central nervous system pain — and on whether reactivation of the Epstein-Barr virus could potentiate autoimmunity in the context of Borrelia infection.
Researchers are ready to test therapies while pathophysiology is unraveled — provided there is funding, Dr. Aucott said. The Clinical Trials Network for Lyme and Other Tick-Borne Diseases, coordinated by Brian Fallon, MD, of Columbia University, New York City, and funded several years ago by the Steven & Alexandra Cohen Foundation, has a slate of small pilot studies underway or being planned that address potential mechanisms (eg, studies of pulse intravenous ceftriaxone, tetracycline, transauricular vagus nerve stimulation, and mast cell modulation). And should full multisite trials be designed and funded, the network is ready with an infrastructure.
Need for Patient-Centered Outcomes
Persistent symptomatology is on the NIH’s radar screen. Efforts to understand causes were part of a strategic tick-borne disease research plan developed by the NIH in 2019. And in 2023, the National Institute of Allergy and Infectious Diseases (NIAID) funded seven projects addressing persistent symptoms that will run through 2028, C. Benjamin Beard, PhD, deputy division director of the CDC’s Division of Vector-Borne Disease, said at the NASEM committee meeting.
Patient advocates maintained that too much emphasis is placed on tick biology and pathophysiology. When Wendy Adams, research grant director and advisory board member of the Bay Area Lyme Foundation, and a colleague analyzed NIAID tick-borne disease funding from 2013 to 2021, they found that 75% of the funding went toward basic research, 15% to translational research, and “only 3% went to clinical research,” Ms. Adams told the committee.
Only 3% of the basic research budget was spent on coinfections, she said, and only 1% was spent on neurologic disease associated with tick-borne infections, both of which are survey-defined patient priorities. Moreover, “12% of the overall NIAID [tick-borne diseases] budget was spent on tick biology,” she said.
Research needs to involve community physicians who are utilizing the guidelines and approaches of the International Lyme and Associated Diseases Society to treat most patients with Lyme IACI, Ms. Adams said. “They have data to be mined,” she said, as does LymeDisease.org, which maintains a patient registry, MyLymeData, with over 18,000 patients. The organization has published two treatment studies, including one on antibiotic treatment response.
Lorraine Johnson, JD, MBA, CEO of LymeDisease.org and principal investigator of MyLymeData, stressed the importance of using patient-centered outcomes that incorporate minimal clinically important differences (MCIDs). “A change in the SF-36 score [without consideration of MCIDs] is not inherently important or meaningful to patients,” she said, referring to the SF-36 survey of health-related quality of life.
“This may seem like an esoteric issue, but two of the four clinical trials done [on retreatment of] persistent Lyme disease used the SF-36 as their outcome measure, and those studies, led by [Mark] Klempner, concluded that retreatment was not effective,” Ms. Johnson said. “Patients have been and continue to be harmed by [this research] because they’re told by physicians that antibiotics don’t work.”
A 2012 biostatistical review of these four RCTs — trials that helped inform the 2006 IDSA treatment guidelines — concluded that the Klempner studies “set the bar for treatment success too high,” Ms. Johnson said. Three of the four trials were likely underpowered to detect clinically meaningful treatment effects, the review also found.
The NASEM committee will hold additional public meetings and review a wide range of literature through this year. The formation of the committee was recommended by the US Department of Health and Human Services Tick-Borne Disease Working Group that was established by Congress in 2016 and concluded its work in 2022. The committee’s work is funded by the Cohen Foundation.
A version of this article appeared on Medscape.com.
WASHINGTON — Advancing treatment for what has been variably called chronic Lyme and posttreatment Lyme disease (PTLD) is under the eyes of a National Academies of Science, Engineering, and Medicine (NASEM) committee of experts for the first time — a year after the NASEM shone a spotlight on the need to accelerate research on chronic illnesses that follow known or suspected infections.
The committee will not make recommendations on specific approaches to diagnosis and treatment when it issues a report in early 2025 but will instead present “consensus findings” on treatment for chronic illness associated with Lyme disease, including recommendations for advancing treatment.
It’s an area void of the US Food and Drug Administration–approved therapies, void of any consensus on the off-label use of medications, and without any current standard of care or proven mechanisms and pathophysiology, said John Aucott, MD, director of the Johns Hopkins Medicine Lyme Disease Clinical Research Center, Baltimore, one of the invited speakers at a public meeting held by the NASEM in Washington, DC.
“The best way to look at this illness is not from the silos of infectious disease or the silos of rheumatology; you have to look across disciplines,” Dr. Aucott, also associate professor of medicine in the Division of Rheumatology, told the committee. “The story doesn’t fit anything I trained for in my infectious disease fellowship. Even today, I’d posit that PTLD is like an island — it’s still not connected to a lot of the mainstream of medicine.”
Rhisa Parera, who wrote and directed a 2021 documentary, Your Labs Are Normal, was one of several invited speakers who amplified the patient voice. Starting around age 7, she had pain in her knees, spine, and hips and vivid nightmares. In high school, she developed gastrointestinal issues, and in college, she developed debilitating neurologic symptoms.
Depression was her eventual diagnosis after having seen “every specialist in the book,” she said. At age 29, she received a positive western blot test and a Lyme disease diagnosis, at which point “I was prescribed 4 weeks of doxycycline and left in the dark,” the 34-year-old Black patient told the committee. Her health improved only after she began working with an “LLMD,” or Lyme-literate medical doctor (a term used in the patient community), while she lived with her mother and did not work, she said.
“I don’t share my Lyme disease history with other doctors. It’s pointless when you have those who will laugh at you, say you’re fine if you were treated, or just deny the disease completely,” Ms. Parera said. “We need this to be taught in medical school. It’s a literal emergency.”
Incidence and Potential Mechanisms
Limited research has suggested that 10%-20% of patients with Lyme disease develop persistent symptoms after standard antibiotic treatment advised by the Infectious Diseases Society of America (IDSA), Dr. Aucott said. (On its web page on chronic symptoms, the Centers for Disease Control and Prevention presents a more conservative range of 5%-10%.)
His own prospective cohort study at Johns Hopkins, published in 2022, found that 13.7% of 234 patients with prior Lyme disease met symptom and functional impact criteria for PTLD, compared with 4.1% of 49 participants without a history of Lyme disease — a statistically significant difference that he said should “put to rest” the question of “is it real?”
PTLD is the research case definition proposed by the IDSA in 2006; it requires that patients have prior documented Lyme disease, no other specific comorbidities, and specific symptoms (fatigue, widespread musculoskeletal pain, and/or cognitive difficulties) causing significant functional impact at least 6 months from their initial diagnosis and treatment.
In the real world, however, where diagnostics for acute Lyme disease are often inaccurate, erythema migrans is often absent, and the symptomatology of Lyme IACI is variable (and where there is no approved laboratory test or objective biomarker for diagnosing Lyme IACI), PTLD represents only a subset of a broader, heterogeneous population with persistent symptoms.
The term “Lyme IACI,” pronounced “Lyme eye-ACK-ee” at the meeting, builds on conversations at the 2023 NASEM workshop on infection-associated chronic illnesses and “encompasses a variety of terms that are used,” including PTLD, PTLD syndrome, persistent Lyme disease, and chronic Lyme disease, according to committee documents. Symptoms are distinct from the known complications of Lyme disease, such as arthritis or carditis.
The findings from Dr. Aucott’s SLICE cohort likely represent “the best outcome,” he said. They’re “probably not generalizable to a community setting where we see lots of missed diagnoses and delayed diagnoses,” as well as other tick-borne coinfections.
One of the challenges in designing future trials, in fact, relates to enrollment criteria and whether to use strict inclusion and exclusion criteria associated with the IDSA definition or take a broader approach to trial enrollment, he and others said. “You want to enroll patients for whom there’s no controversy that they’ve had Lyme infection ... for a study people believe in,” Dr. Aucott said during a discussion period, noting that it’s typical to screen over 100 patients to find one enrollee. “But it’s a tension we’re having.”
Timothy Sellati, PhD, chief scientific officer of the Global Lyme Alliance, urged change. “It’s really important to try to figure out how to alter our thinking on identifying and diagnosing chronic Lyme patients because they need to be recruited into clinical trials,” he said during his presentation.
“We think the best way to do this is to [develop and] employ composite diagnostic testing” that looks at unique Borrelia signatures (eg, protein, DNA, RNA, or metabolites), genetic and/or epigenetic signatures, inflammation signatures, T-cell-independent antibody signatures, and other elements, Dr. Sellati said.
Researchers designing treatment trials also face unknowns, Dr. Aucott and others said, about the role of potential mechanisms of Lyme IACI, from persistent Borrelia burgdorferi (or Borrelia mayonii) infection or the persistence of bacterial remnants (eg, nucleic acids or peptidoglycans) to infection-triggered pathology such as persistent immune dysregulation, chronic inflammation, autoimmunity, microbiome alterations, and dysautonomia and other neural network alterations.
The NASEM’s spotlight on Lyme IACI follows its long COVID-driven push last year to advance a common research agenda in infection-associated chronic illnesses. Investigators see common symptoms and potential shared mechanisms between long COVID, Lyme IACI, myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), and other complex chronic illnesses following infections.
At the Lyme IACI meeting, invited speakers described parts of the research landscape. Avindra Nath, MD, of the National Institute of Neurological Disorders and Stroke, for instance, described a recently published deep phenotyping study of 17 patients with ME/CFS that found decreased central catecholamine synthesis, circuit dysfunction of integrative brain regions, and immune profiling differences (eg, defects in B-cell maturation or T-cell exhaustion), compared with matched controls, that suggest the persistence of microbial antigens.
And John Leong, MD, PhD, of Tufts University, Boston, described his lab’s focus on understanding the microbe-host interactions that enable bloodstream dissemination and tissue invasion of B burgdorferi to take hold, increasing the risk for persistent symptoms. Other research at Tufts, he noted during a discussion period, has demonstrated the persistence of B burgdorferi to antibiotics in microtiter dishes. “Those organisms that survive are really difficult to eradicate in vitro,” Dr. Leong said.
Other physician investigators described research on nociplastic pain — a category of pain that can be triggered by infections, causing both amplified sensory processing and augmented central nervous system pain — and on whether reactivation of the Epstein-Barr virus could potentiate autoimmunity in the context of Borrelia infection.
Researchers are ready to test therapies while pathophysiology is unraveled — provided there is funding, Dr. Aucott said. The Clinical Trials Network for Lyme and Other Tick-Borne Diseases, coordinated by Brian Fallon, MD, of Columbia University, New York City, and funded several years ago by the Steven & Alexandra Cohen Foundation, has a slate of small pilot studies underway or being planned that address potential mechanisms (eg, studies of pulse intravenous ceftriaxone, tetracycline, transauricular vagus nerve stimulation, and mast cell modulation). And should full multisite trials be designed and funded, the network is ready with an infrastructure.
Need for Patient-Centered Outcomes
Persistent symptomatology is on the NIH’s radar screen. Efforts to understand causes were part of a strategic tick-borne disease research plan developed by the NIH in 2019. And in 2023, the National Institute of Allergy and Infectious Diseases (NIAID) funded seven projects addressing persistent symptoms that will run through 2028, C. Benjamin Beard, PhD, deputy division director of the CDC’s Division of Vector-Borne Disease, said at the NASEM committee meeting.
Patient advocates maintained that too much emphasis is placed on tick biology and pathophysiology. When Wendy Adams, research grant director and advisory board member of the Bay Area Lyme Foundation, and a colleague analyzed NIAID tick-borne disease funding from 2013 to 2021, they found that 75% of the funding went toward basic research, 15% to translational research, and “only 3% went to clinical research,” Ms. Adams told the committee.
Only 3% of the basic research budget was spent on coinfections, she said, and only 1% was spent on neurologic disease associated with tick-borne infections, both of which are survey-defined patient priorities. Moreover, “12% of the overall NIAID [tick-borne diseases] budget was spent on tick biology,” she said.
Research needs to involve community physicians who are utilizing the guidelines and approaches of the International Lyme and Associated Diseases Society to treat most patients with Lyme IACI, Ms. Adams said. “They have data to be mined,” she said, as does LymeDisease.org, which maintains a patient registry, MyLymeData, with over 18,000 patients. The organization has published two treatment studies, including one on antibiotic treatment response.
Lorraine Johnson, JD, MBA, CEO of LymeDisease.org and principal investigator of MyLymeData, stressed the importance of using patient-centered outcomes that incorporate minimal clinically important differences (MCIDs). “A change in the SF-36 score [without consideration of MCIDs] is not inherently important or meaningful to patients,” she said, referring to the SF-36 survey of health-related quality of life.
“This may seem like an esoteric issue, but two of the four clinical trials done [on retreatment of] persistent Lyme disease used the SF-36 as their outcome measure, and those studies, led by [Mark] Klempner, concluded that retreatment was not effective,” Ms. Johnson said. “Patients have been and continue to be harmed by [this research] because they’re told by physicians that antibiotics don’t work.”
A 2012 biostatistical review of these four RCTs — trials that helped inform the 2006 IDSA treatment guidelines — concluded that the Klempner studies “set the bar for treatment success too high,” Ms. Johnson said. Three of the four trials were likely underpowered to detect clinically meaningful treatment effects, the review also found.
The NASEM committee will hold additional public meetings and review a wide range of literature through this year. The formation of the committee was recommended by the US Department of Health and Human Services Tick-Borne Disease Working Group that was established by Congress in 2016 and concluded its work in 2022. The committee’s work is funded by the Cohen Foundation.
A version of this article appeared on Medscape.com.