User login
When should an indwelling pleural catheter be considered for malignant pleural effusion?
An indwelling pleural catheter should be considered when a malignant pleural effusion causes symptoms and recurs after thoracentesis, especially in patients with short to intermediate life expectancy or trapped lung, or who underwent unsuccessful pleurodesis.1
MALIGNANT PLEURAL EFFUSION
Malignant pleural effusion affects about 150,000 people in the United States each year. It occurs in 15% of patients with advanced malignancies, most often lung cancer, breast cancer, lymphoma, and ovarian cancer, which account for more than 50% of cases.2
In most patients with malignant pleural effusion, disabling dyspnea causes poor quality of life. The prognosis is unfavorable, with life expectancy of 3 to 12 months. Patients with poor performance status and lower glucose concentrations in the pleural fluid face a worse prognosis and a shorter life expectancy.2

In general, management focuses on relieving symptoms rather than on cure. Symptoms can be controlled by thoracentesis, but if the effusion recurs, the patient needs repeated visits to the emergency room or clinic or a hospital admission to drain the fluid. Frequent hospital visits can be grueling for a patient with a poor functional status, and so can the adverse effects of repeated thoracentesis. For that reason, an early palliative approach to malignant pleural effusion in patients with cancer and a poor prognosis leads to better symptom control and a better quality of life.3 Multiple treatments can be offered to control the symptoms in patients with recurrent malignant pleural effusion (Table 1).
PLEURODESIS HAS BEEN THE TREATMENT OF CHOICE
Pleurodesis has been the treatment of choice for malignant pleural effusion for decades. In this procedure, adhesion of the visceral and parietal pleura is achxieved by inducing inflammation either mechanically or chemically between the pleural surfaces. Injection of a sclerosant into the pleural space generates the inflammation. The sclerosant can be introduced through a chest tube or thoracoscope such as in video-assisted thoracic surgery or medical pleuroscopy. The use of talc is associated with a higher success rate than other sclerosing agents such as bleomycin and doxycycline.4
The downside of this procedure is that pleural effusion recurs in 10% to 40% of cases, and patients require 2 to 4 days in the hospital. Also, the use of talc can lead to acute lung injury–acute respiratory distress syndrome, a rare but potentially life-threatening complication. The incidence of this complication may be related to particle size, with small particles posing a higher risk than large ones.5,6
PLACEMENT OF AN INDWELLING PLEURAL CATHETER
Indwelling pleural catheters are currently used as palliative therapy for patients with recurrent malignant pleural effusion who suffer from respiratory distress due to rapid reaccumulation of pleural fluids that require multiple thoracentesis procedures.
An indwelling pleural catheter is contraindicated in patients with uncontrolled coagulopathy, multiloculated pleural effusions, or extensive malignancy in the skin.3 Other factors that need to be considered are the patient’s social circumstances: ie, the patient must be in a clean and safe environment and must have insurance coverage for the supplies.
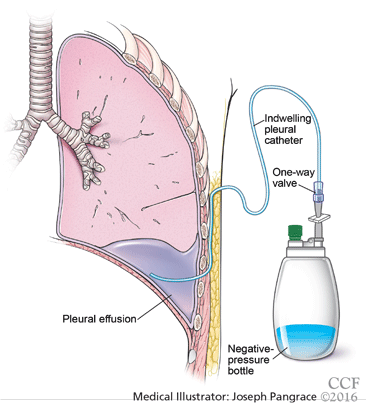
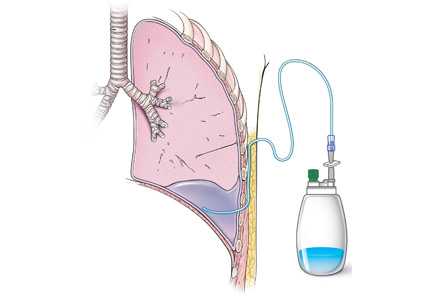
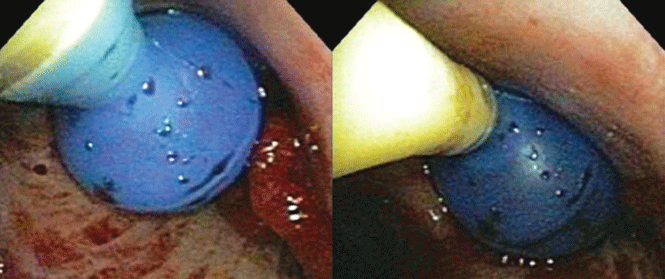
Catheters are 66 cm long and 15.5F and are made of silicone rubber with fenestrations along the distal 24 cm. They have a one-way valve at the proximal end that allows fluids and air to go out but not in (Figure 1).1 Several systems are commercially available in the United States.
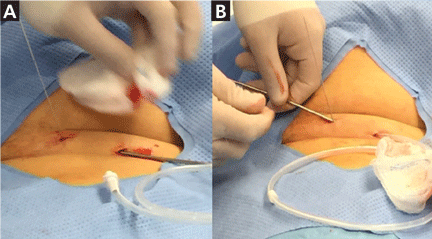
The catheter is inserted and tunneled percutaneously with the patient under local anesthesia and conscious sedation (Figure 2). Insertion is a same-day outpatient procedure, and intermittent pleural fluid drainage can be done at home by a home heathcare provider or a trained family member.7
In a meta-analysis, insertion difficulties were reported in only 4% of cases, particularly in patients who underwent prior pleural interventions. Spontaneous pleurodesis occurred in 45% of patients at a mean of 52 days after insertion.8
After catheter insertion, the pleural space should be drained three times a week. No more than 1,000 mL of fluid should be removed at a time—or less if drainage causes chest pain or cough secondary to trapped lung (see below). When the drainage declines to 150 mL per session, the sessions can be reduced to twice a week. If the volume drops to less than 50 mL per session, imaging (computed tomography or bedside thoracic ultrasonography) is recommended to ensure the achievement of pleurodesis and to rule out catheter blockage.
A large multicenter randomized controlled trial9 compared indwelling pleural catheter therapy and chest tube insertion with talc pleurodesis. Both procedures relieved symptoms for the first 42 days, and there was no significant difference in quality of life. However, the median length of hospital stay was 4 days for the talc pleurodesis group compared with 0 days for the indwelling pleural catheter group. Twenty-two percent of the talc group required a further pleural procedure such as a video-assisted thoracic surgery or thoracoscopy, compared with 6% of the indwelling catheter group. On the other hand, 36% of those in the indwelling catheter group experienced nonserious adverse events such as pleural infections that mandated outpatient oral antibiotic therapy, cellulitis, and catheter blockage, compared with 7% of the talc group.9
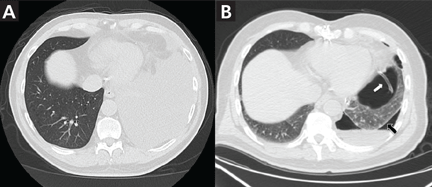
Symptomatic, inoperable trapped lung is another condition for which an indwelling pleural catheter is a reasonable strategy compared with pleurodesis. Trapped lung is a condition in which the lung fails to fully expand despite proper pleural fluid removal, creating a vacuum space between the parietal and visceral pleura (Figure 3).
Patients with trapped lung complain of severe dull or sharp pain during drainage of pleural fluids due to stretching of the visceral pleura against the intrathoracic vacuum space. Trapped lung can be detected objectively by using intrathoracic manometry while draining fluids, looking for more than a 20-cm H2O drop in the intrathoracic pressure. Radiographically, this may be identified as a pneumothorax ex vacuo10 (ie, caused by inability of the lung to expand to fill the thoracic cavity after pleural fluid has been drained) and is not a procedure complication.
Placement of an indwelling pleural catheter is the treatment of choice for trapped lung, since chemical pleurodesis is not feasible without the potential of parietal and visceral pleural apposition. In a retrospective study of indwelling catheter placement for palliative symptom control, a catheter relieved symptoms, improved quality of life, and afforded a substantial increase in mobility.1,11
In another multicenter pilot study,12 rapid pleurodesis was achieved in 30 patients with recurrent malignant pleural effusion by combining chemical pleurodesis and indwelling catheter placement. Both were done under direct vision with medical thoracoscopy. Pleurodesis succeeded in 92% of patients by day 8 after the procedure. The hospital stay was reduced to a mean of 2 days after the procedure. In the catheter group, fluids were drained three times in the first day after the procedure and twice a day on the second and third days. Of the 30 patients in this study, 2 had fever, 1 needed to have the catheter replaced, and 1 contracted empyema.
AN EFFECTIVE INITIAL TREATMENT
Placement of an indwelling pleural catheter is an effective initial treatment for recurrent malignant pleural effusion. Compared with chemical pleurodesis, it has a comparable success rate and complication rate. It offers the advantages of being a same-day surgical procedure entailing a shorter hospital stay and less need for further pleural intervention. This treatment should be considered for patients with symptomatic malignant pleural effusion, especially those in whom symptomatic malignant pleural effusion recurred after thoracentesis.8
- Roberts ME, Neville E, Berrisford RG, Antunes G, Ali NJ; BTS Pleural Disease Guideline Group. Management of a malignant pleural effusion: British Thoracic Society Pleural Disease Guideline 2010. Thorax 2010; 65(suppl 2):ii32–ii40.
- Thomas JM, Musani AI. Malignant pleural effusions: a review. Clin Chest Med 2013; 34:459–471.
- Thomas R, Francis R, Davies HE, Lee YC. Interventional therapies for malignant pleural effusions: the present and the future. Respirology 2014; 19:809–822.
- Rodriguez-Panadero F, Montes-Worboys A. Mechanisms of pleurodesis. Respiration 2012; 83:91–98.
- Gonzalez AV, Bezwada V, Beamis JF Jr, Villanueva AG. Lung injury following thoracoscopic talc insufflation: experience of a single North American center. Chest 2010; 137:1375–1381.
- Rossi VF, Vargas FS, Marchi E, et al. Acute inflammatory response secondary to intrapleural administration of two types of talc. Eur Respir J 2010; 35:396–401.
- Fysh ET, Waterer GW, Kendall PA, et al. Indwelling pleural catheters reduce inpatient days over pleurodesis for malignant pleural effusion. Chest 2012; 142:394–400.
- Kheir F, Shawwa K, Alokla K, Omballi M, Alraiyes AH. Tunneled pleural catheter for the treatment of malignant pleural effusion: a systematic review and meta-analysis. Am J Ther 2015 Feb 2. [Epub ahead of print]
- Davies HE, Mishra EK, Kahan BC, et al. Effect of an indwelling pleural catheter vs chest tube and talc pleurodesis for relieving dyspnea in patients with malignant pleural effusion: the TIME2 randomized controlled trial. JAMA 2012; 307:2383–2389.
- Ponrartana S, Laberge JM, Kerlan RK, Wilson MW, Gordon RL. Management of patients with “ex vacuo” pneumothorax after thoracentesis. Acad Radiol 2005; 12:980–986.
- Efthymiou CA, Masudi T, Thorpe JA, Papagiannopoulos K. Malignant pleural effusion in the presence of trapped lung. Five-year experience of PleurX tunnelled catheters. Interact Cardiovasc Thorac Surg 2009; 9:961–964.
- Reddy C, Ernst A, Lamb C, Feller-Kopman D. Rapid pleurodesis for malignant pleural effusions: a pilot study. Chest 2011; 139:1419–1423.
An indwelling pleural catheter should be considered when a malignant pleural effusion causes symptoms and recurs after thoracentesis, especially in patients with short to intermediate life expectancy or trapped lung, or who underwent unsuccessful pleurodesis.1
MALIGNANT PLEURAL EFFUSION
Malignant pleural effusion affects about 150,000 people in the United States each year. It occurs in 15% of patients with advanced malignancies, most often lung cancer, breast cancer, lymphoma, and ovarian cancer, which account for more than 50% of cases.2
In most patients with malignant pleural effusion, disabling dyspnea causes poor quality of life. The prognosis is unfavorable, with life expectancy of 3 to 12 months. Patients with poor performance status and lower glucose concentrations in the pleural fluid face a worse prognosis and a shorter life expectancy.2
In general, management focuses on relieving symptoms rather than on cure. Symptoms can be controlled by thoracentesis, but if the effusion recurs, the patient needs repeated visits to the emergency room or clinic or a hospital admission to drain the fluid. Frequent hospital visits can be grueling for a patient with a poor functional status, and so can the adverse effects of repeated thoracentesis. For that reason, an early palliative approach to malignant pleural effusion in patients with cancer and a poor prognosis leads to better symptom control and a better quality of life.3 Multiple treatments can be offered to control the symptoms in patients with recurrent malignant pleural effusion (Table 1).
PLEURODESIS HAS BEEN THE TREATMENT OF CHOICE
Pleurodesis has been the treatment of choice for malignant pleural effusion for decades. In this procedure, adhesion of the visceral and parietal pleura is achxieved by inducing inflammation either mechanically or chemically between the pleural surfaces. Injection of a sclerosant into the pleural space generates the inflammation. The sclerosant can be introduced through a chest tube or thoracoscope such as in video-assisted thoracic surgery or medical pleuroscopy. The use of talc is associated with a higher success rate than other sclerosing agents such as bleomycin and doxycycline.4
The downside of this procedure is that pleural effusion recurs in 10% to 40% of cases, and patients require 2 to 4 days in the hospital. Also, the use of talc can lead to acute lung injury–acute respiratory distress syndrome, a rare but potentially life-threatening complication. The incidence of this complication may be related to particle size, with small particles posing a higher risk than large ones.5,6
PLACEMENT OF AN INDWELLING PLEURAL CATHETER
Indwelling pleural catheters are currently used as palliative therapy for patients with recurrent malignant pleural effusion who suffer from respiratory distress due to rapid reaccumulation of pleural fluids that require multiple thoracentesis procedures.
An indwelling pleural catheter is contraindicated in patients with uncontrolled coagulopathy, multiloculated pleural effusions, or extensive malignancy in the skin.3 Other factors that need to be considered are the patient’s social circumstances: ie, the patient must be in a clean and safe environment and must have insurance coverage for the supplies.
Catheters are 66 cm long and 15.5F and are made of silicone rubber with fenestrations along the distal 24 cm. They have a one-way valve at the proximal end that allows fluids and air to go out but not in (Figure 1).1 Several systems are commercially available in the United States.
The catheter is inserted and tunneled percutaneously with the patient under local anesthesia and conscious sedation (Figure 2). Insertion is a same-day outpatient procedure, and intermittent pleural fluid drainage can be done at home by a home heathcare provider or a trained family member.7
In a meta-analysis, insertion difficulties were reported in only 4% of cases, particularly in patients who underwent prior pleural interventions. Spontaneous pleurodesis occurred in 45% of patients at a mean of 52 days after insertion.8
After catheter insertion, the pleural space should be drained three times a week. No more than 1,000 mL of fluid should be removed at a time—or less if drainage causes chest pain or cough secondary to trapped lung (see below). When the drainage declines to 150 mL per session, the sessions can be reduced to twice a week. If the volume drops to less than 50 mL per session, imaging (computed tomography or bedside thoracic ultrasonography) is recommended to ensure the achievement of pleurodesis and to rule out catheter blockage.
A large multicenter randomized controlled trial9 compared indwelling pleural catheter therapy and chest tube insertion with talc pleurodesis. Both procedures relieved symptoms for the first 42 days, and there was no significant difference in quality of life. However, the median length of hospital stay was 4 days for the talc pleurodesis group compared with 0 days for the indwelling pleural catheter group. Twenty-two percent of the talc group required a further pleural procedure such as a video-assisted thoracic surgery or thoracoscopy, compared with 6% of the indwelling catheter group. On the other hand, 36% of those in the indwelling catheter group experienced nonserious adverse events such as pleural infections that mandated outpatient oral antibiotic therapy, cellulitis, and catheter blockage, compared with 7% of the talc group.9
Symptomatic, inoperable trapped lung is another condition for which an indwelling pleural catheter is a reasonable strategy compared with pleurodesis. Trapped lung is a condition in which the lung fails to fully expand despite proper pleural fluid removal, creating a vacuum space between the parietal and visceral pleura (Figure 3).
Patients with trapped lung complain of severe dull or sharp pain during drainage of pleural fluids due to stretching of the visceral pleura against the intrathoracic vacuum space. Trapped lung can be detected objectively by using intrathoracic manometry while draining fluids, looking for more than a 20-cm H2O drop in the intrathoracic pressure. Radiographically, this may be identified as a pneumothorax ex vacuo10 (ie, caused by inability of the lung to expand to fill the thoracic cavity after pleural fluid has been drained) and is not a procedure complication.
Placement of an indwelling pleural catheter is the treatment of choice for trapped lung, since chemical pleurodesis is not feasible without the potential of parietal and visceral pleural apposition. In a retrospective study of indwelling catheter placement for palliative symptom control, a catheter relieved symptoms, improved quality of life, and afforded a substantial increase in mobility.1,11
In another multicenter pilot study,12 rapid pleurodesis was achieved in 30 patients with recurrent malignant pleural effusion by combining chemical pleurodesis and indwelling catheter placement. Both were done under direct vision with medical thoracoscopy. Pleurodesis succeeded in 92% of patients by day 8 after the procedure. The hospital stay was reduced to a mean of 2 days after the procedure. In the catheter group, fluids were drained three times in the first day after the procedure and twice a day on the second and third days. Of the 30 patients in this study, 2 had fever, 1 needed to have the catheter replaced, and 1 contracted empyema.
AN EFFECTIVE INITIAL TREATMENT
Placement of an indwelling pleural catheter is an effective initial treatment for recurrent malignant pleural effusion. Compared with chemical pleurodesis, it has a comparable success rate and complication rate. It offers the advantages of being a same-day surgical procedure entailing a shorter hospital stay and less need for further pleural intervention. This treatment should be considered for patients with symptomatic malignant pleural effusion, especially those in whom symptomatic malignant pleural effusion recurred after thoracentesis.8
An indwelling pleural catheter should be considered when a malignant pleural effusion causes symptoms and recurs after thoracentesis, especially in patients with short to intermediate life expectancy or trapped lung, or who underwent unsuccessful pleurodesis.1
MALIGNANT PLEURAL EFFUSION
Malignant pleural effusion affects about 150,000 people in the United States each year. It occurs in 15% of patients with advanced malignancies, most often lung cancer, breast cancer, lymphoma, and ovarian cancer, which account for more than 50% of cases.2
In most patients with malignant pleural effusion, disabling dyspnea causes poor quality of life. The prognosis is unfavorable, with life expectancy of 3 to 12 months. Patients with poor performance status and lower glucose concentrations in the pleural fluid face a worse prognosis and a shorter life expectancy.2
In general, management focuses on relieving symptoms rather than on cure. Symptoms can be controlled by thoracentesis, but if the effusion recurs, the patient needs repeated visits to the emergency room or clinic or a hospital admission to drain the fluid. Frequent hospital visits can be grueling for a patient with a poor functional status, and so can the adverse effects of repeated thoracentesis. For that reason, an early palliative approach to malignant pleural effusion in patients with cancer and a poor prognosis leads to better symptom control and a better quality of life.3 Multiple treatments can be offered to control the symptoms in patients with recurrent malignant pleural effusion (Table 1).
PLEURODESIS HAS BEEN THE TREATMENT OF CHOICE
Pleurodesis has been the treatment of choice for malignant pleural effusion for decades. In this procedure, adhesion of the visceral and parietal pleura is achxieved by inducing inflammation either mechanically or chemically between the pleural surfaces. Injection of a sclerosant into the pleural space generates the inflammation. The sclerosant can be introduced through a chest tube or thoracoscope such as in video-assisted thoracic surgery or medical pleuroscopy. The use of talc is associated with a higher success rate than other sclerosing agents such as bleomycin and doxycycline.4
The downside of this procedure is that pleural effusion recurs in 10% to 40% of cases, and patients require 2 to 4 days in the hospital. Also, the use of talc can lead to acute lung injury–acute respiratory distress syndrome, a rare but potentially life-threatening complication. The incidence of this complication may be related to particle size, with small particles posing a higher risk than large ones.5,6
PLACEMENT OF AN INDWELLING PLEURAL CATHETER
Indwelling pleural catheters are currently used as palliative therapy for patients with recurrent malignant pleural effusion who suffer from respiratory distress due to rapid reaccumulation of pleural fluids that require multiple thoracentesis procedures.
An indwelling pleural catheter is contraindicated in patients with uncontrolled coagulopathy, multiloculated pleural effusions, or extensive malignancy in the skin.3 Other factors that need to be considered are the patient’s social circumstances: ie, the patient must be in a clean and safe environment and must have insurance coverage for the supplies.
Catheters are 66 cm long and 15.5F and are made of silicone rubber with fenestrations along the distal 24 cm. They have a one-way valve at the proximal end that allows fluids and air to go out but not in (Figure 1).1 Several systems are commercially available in the United States.
The catheter is inserted and tunneled percutaneously with the patient under local anesthesia and conscious sedation (Figure 2). Insertion is a same-day outpatient procedure, and intermittent pleural fluid drainage can be done at home by a home heathcare provider or a trained family member.7
In a meta-analysis, insertion difficulties were reported in only 4% of cases, particularly in patients who underwent prior pleural interventions. Spontaneous pleurodesis occurred in 45% of patients at a mean of 52 days after insertion.8
After catheter insertion, the pleural space should be drained three times a week. No more than 1,000 mL of fluid should be removed at a time—or less if drainage causes chest pain or cough secondary to trapped lung (see below). When the drainage declines to 150 mL per session, the sessions can be reduced to twice a week. If the volume drops to less than 50 mL per session, imaging (computed tomography or bedside thoracic ultrasonography) is recommended to ensure the achievement of pleurodesis and to rule out catheter blockage.
A large multicenter randomized controlled trial9 compared indwelling pleural catheter therapy and chest tube insertion with talc pleurodesis. Both procedures relieved symptoms for the first 42 days, and there was no significant difference in quality of life. However, the median length of hospital stay was 4 days for the talc pleurodesis group compared with 0 days for the indwelling pleural catheter group. Twenty-two percent of the talc group required a further pleural procedure such as a video-assisted thoracic surgery or thoracoscopy, compared with 6% of the indwelling catheter group. On the other hand, 36% of those in the indwelling catheter group experienced nonserious adverse events such as pleural infections that mandated outpatient oral antibiotic therapy, cellulitis, and catheter blockage, compared with 7% of the talc group.9
Symptomatic, inoperable trapped lung is another condition for which an indwelling pleural catheter is a reasonable strategy compared with pleurodesis. Trapped lung is a condition in which the lung fails to fully expand despite proper pleural fluid removal, creating a vacuum space between the parietal and visceral pleura (Figure 3).
Patients with trapped lung complain of severe dull or sharp pain during drainage of pleural fluids due to stretching of the visceral pleura against the intrathoracic vacuum space. Trapped lung can be detected objectively by using intrathoracic manometry while draining fluids, looking for more than a 20-cm H2O drop in the intrathoracic pressure. Radiographically, this may be identified as a pneumothorax ex vacuo10 (ie, caused by inability of the lung to expand to fill the thoracic cavity after pleural fluid has been drained) and is not a procedure complication.
Placement of an indwelling pleural catheter is the treatment of choice for trapped lung, since chemical pleurodesis is not feasible without the potential of parietal and visceral pleural apposition. In a retrospective study of indwelling catheter placement for palliative symptom control, a catheter relieved symptoms, improved quality of life, and afforded a substantial increase in mobility.1,11
In another multicenter pilot study,12 rapid pleurodesis was achieved in 30 patients with recurrent malignant pleural effusion by combining chemical pleurodesis and indwelling catheter placement. Both were done under direct vision with medical thoracoscopy. Pleurodesis succeeded in 92% of patients by day 8 after the procedure. The hospital stay was reduced to a mean of 2 days after the procedure. In the catheter group, fluids were drained three times in the first day after the procedure and twice a day on the second and third days. Of the 30 patients in this study, 2 had fever, 1 needed to have the catheter replaced, and 1 contracted empyema.
AN EFFECTIVE INITIAL TREATMENT
Placement of an indwelling pleural catheter is an effective initial treatment for recurrent malignant pleural effusion. Compared with chemical pleurodesis, it has a comparable success rate and complication rate. It offers the advantages of being a same-day surgical procedure entailing a shorter hospital stay and less need for further pleural intervention. This treatment should be considered for patients with symptomatic malignant pleural effusion, especially those in whom symptomatic malignant pleural effusion recurred after thoracentesis.8
- Roberts ME, Neville E, Berrisford RG, Antunes G, Ali NJ; BTS Pleural Disease Guideline Group. Management of a malignant pleural effusion: British Thoracic Society Pleural Disease Guideline 2010. Thorax 2010; 65(suppl 2):ii32–ii40.
- Thomas JM, Musani AI. Malignant pleural effusions: a review. Clin Chest Med 2013; 34:459–471.
- Thomas R, Francis R, Davies HE, Lee YC. Interventional therapies for malignant pleural effusions: the present and the future. Respirology 2014; 19:809–822.
- Rodriguez-Panadero F, Montes-Worboys A. Mechanisms of pleurodesis. Respiration 2012; 83:91–98.
- Gonzalez AV, Bezwada V, Beamis JF Jr, Villanueva AG. Lung injury following thoracoscopic talc insufflation: experience of a single North American center. Chest 2010; 137:1375–1381.
- Rossi VF, Vargas FS, Marchi E, et al. Acute inflammatory response secondary to intrapleural administration of two types of talc. Eur Respir J 2010; 35:396–401.
- Fysh ET, Waterer GW, Kendall PA, et al. Indwelling pleural catheters reduce inpatient days over pleurodesis for malignant pleural effusion. Chest 2012; 142:394–400.
- Kheir F, Shawwa K, Alokla K, Omballi M, Alraiyes AH. Tunneled pleural catheter for the treatment of malignant pleural effusion: a systematic review and meta-analysis. Am J Ther 2015 Feb 2. [Epub ahead of print]
- Davies HE, Mishra EK, Kahan BC, et al. Effect of an indwelling pleural catheter vs chest tube and talc pleurodesis for relieving dyspnea in patients with malignant pleural effusion: the TIME2 randomized controlled trial. JAMA 2012; 307:2383–2389.
- Ponrartana S, Laberge JM, Kerlan RK, Wilson MW, Gordon RL. Management of patients with “ex vacuo” pneumothorax after thoracentesis. Acad Radiol 2005; 12:980–986.
- Efthymiou CA, Masudi T, Thorpe JA, Papagiannopoulos K. Malignant pleural effusion in the presence of trapped lung. Five-year experience of PleurX tunnelled catheters. Interact Cardiovasc Thorac Surg 2009; 9:961–964.
- Reddy C, Ernst A, Lamb C, Feller-Kopman D. Rapid pleurodesis for malignant pleural effusions: a pilot study. Chest 2011; 139:1419–1423.
- Roberts ME, Neville E, Berrisford RG, Antunes G, Ali NJ; BTS Pleural Disease Guideline Group. Management of a malignant pleural effusion: British Thoracic Society Pleural Disease Guideline 2010. Thorax 2010; 65(suppl 2):ii32–ii40.
- Thomas JM, Musani AI. Malignant pleural effusions: a review. Clin Chest Med 2013; 34:459–471.
- Thomas R, Francis R, Davies HE, Lee YC. Interventional therapies for malignant pleural effusions: the present and the future. Respirology 2014; 19:809–822.
- Rodriguez-Panadero F, Montes-Worboys A. Mechanisms of pleurodesis. Respiration 2012; 83:91–98.
- Gonzalez AV, Bezwada V, Beamis JF Jr, Villanueva AG. Lung injury following thoracoscopic talc insufflation: experience of a single North American center. Chest 2010; 137:1375–1381.
- Rossi VF, Vargas FS, Marchi E, et al. Acute inflammatory response secondary to intrapleural administration of two types of talc. Eur Respir J 2010; 35:396–401.
- Fysh ET, Waterer GW, Kendall PA, et al. Indwelling pleural catheters reduce inpatient days over pleurodesis for malignant pleural effusion. Chest 2012; 142:394–400.
- Kheir F, Shawwa K, Alokla K, Omballi M, Alraiyes AH. Tunneled pleural catheter for the treatment of malignant pleural effusion: a systematic review and meta-analysis. Am J Ther 2015 Feb 2. [Epub ahead of print]
- Davies HE, Mishra EK, Kahan BC, et al. Effect of an indwelling pleural catheter vs chest tube and talc pleurodesis for relieving dyspnea in patients with malignant pleural effusion: the TIME2 randomized controlled trial. JAMA 2012; 307:2383–2389.
- Ponrartana S, Laberge JM, Kerlan RK, Wilson MW, Gordon RL. Management of patients with “ex vacuo” pneumothorax after thoracentesis. Acad Radiol 2005; 12:980–986.
- Efthymiou CA, Masudi T, Thorpe JA, Papagiannopoulos K. Malignant pleural effusion in the presence of trapped lung. Five-year experience of PleurX tunnelled catheters. Interact Cardiovasc Thorac Surg 2009; 9:961–964.
- Reddy C, Ernst A, Lamb C, Feller-Kopman D. Rapid pleurodesis for malignant pleural effusions: a pilot study. Chest 2011; 139:1419–1423.
Does stenting of severe renal artery stenosis improve outomes compared with medical therapy alone?
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
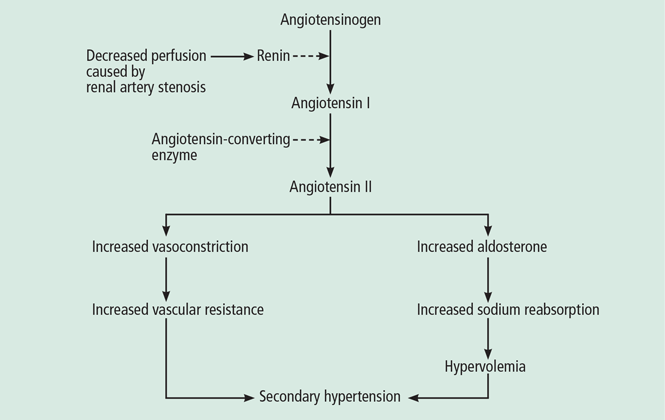
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
Does massive hemoptysis always merit diagnostic bronchoscopy?
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.

Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
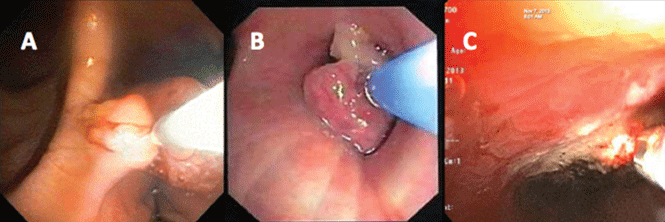
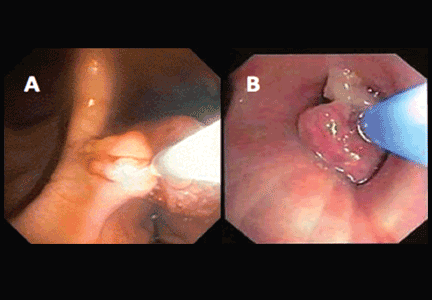
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.
Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.
Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.
In reply: Stress ulcer prophylaxis
In Reply: We welcome the comments from Dr. Chongnarungsin on our article and the opportunity to further discuss our opinions.
In our paper, we discussed current recommendations for prophylaxis of stress ulcer-related bleeding in hospitalized patients and advocated against the blind administration of drugs without risk stratification.
The landmark trial that provides the most-cited definitions and the risk factors for clinically significant stress ulcer-related bleeding in critically ill patients was published in 1994 by Cook et al.1 In their multicenter prospective cohort study of 2,252 patients, the authors reported that prolonged mechanical ventilation is an important risk factor for clinically significant stress ulcer-related bleeding.
Another major prospective cohort study observed an incidence rate of clinically significant stress ulcer-related bleeding of 3.5%.2
Dr. Chongnarungsin cites another prospective cohort study of 183 patients from the same era,3 wherein the authors defined stress ulcer-related bleeding as bleeding requiring transfusion of packed red blood cells, found on endoscopy or on postmortem evaluation. This was in contrast to the 1994 study of Cook et al,1 who had a more rigorous and comprehensive definition for overt and clinically significant stress ulcer-related bleeding, applied by up to three independent adjudicators not involved in the patients’ care. Their definition not only entailed a more accurate transfusion-dependent bleeding criterion, but also included hemodynamic and laboratory criteria. As such, the “very low rate” of stress ulcer-related bleeding reported by Zandstra et al3 should be critically appraised. Of note, the authors in that study did not report the rates of patients who received early enteral feeding, and their patients received cefotaxime for digestive tract decontamination, an important confounder to the interpretation of the study results.
Indeed, the remarkable variation in estimates of the incidence of stress ulcer-related bleeding is probably related to the lack of a uniform definition. Even when rates of endoscopic and occult bleeding are set aside, agreement is lacking as to which category of bleeding is clinically significant.
Dr. Chongnarungsin also cites the study by Ellison et al4 of a cohort of 874 patients who had no previous gastrointestinal bleeding or peptic ulcer disease and who were enrolled in a multicenter randomized controlled trial of prophylactic intravenous immune globulin to prevent infections associated with an intensive care unit. In a secondary objective, the authors did not identify coagulopathy or prolonged mechanical ventilation as a principal risk factor for bleeding. The authors ascribed this discrepancy with previously published literature to their unique study population, which consisted predominantly of elderly men and rarely included trauma patients. In light of these unique peculiarities of their population, the lack of an association between prolonged mechanical ventilation and stress ulcer-related bleeding cannot be determined. Moreover, that study showed that prolonged nasogastric tube insertion was one of the risk factors for increased risk of gastrointestinal bleeding, and not the risk factor for development of stress ulcer as stated by Dr. Chongnarungsin.
The decrease in the incidence of stress ulcer-related bleeding in critically ill patients over the years could be attributed to an era effect, from advances in critical care medicine and prophylactic methods.5 We agree with Dr. Chongnarungsin that the increased introduction of early enteral feeding may have also contributed to the reduced incidence of stress ulcer-related bleeding.6 However, we think the conclusion that “mechanical ventilation for more than 48 hours does not seem to increase the risk of stress ulcer” is overelaborated, and we believe that strong evidence demonstrates this association.1,2
Alternatively, we recognize the lack of mortality-benefit evidence for stress ulcer prophylaxis. This notwithstanding, according to recent Surviving Sepsis Campaign guidelines, the use of stress ulcer prophylaxis is listed as a 1B recommendation (strong recommendation) for severely septic patients who require prolonged mechanical ventilation. In addition, the updated 2014 guidelines of the American Society of Health-System Pharmacists7 continue to recommend stress ulcer prophylaxis in the context of mechanical ventilation, with H2 receptor antagonists being the preferred first-line agents.8
It is important to acknowledge that these recommendations were endorsed despite the lack of obvious mortality benefit, and it is our opinion that large randomized controlled studies are needed to evaluate the risks and mortality benefit of these prophylaxis methods.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Cook DJ, Griffith LE, Walter SD, et al. The attributable mortality and length of intensive care unit stay of clinically important gastrointestinal bleeding in critically ill patients. Crit Care 2001; 5:368–375.
- Zandstra DF, Stoutenbeek CP. The virtual absence of stress-ulceration related bleeding in ICU patients receiving prolonged mechanical ventilation without any prophylaxis. A prospective cohort study. Intensive Care Med 1994; 20:335–340.
- Ellison RT, Perez-Perez G, Welsh CH, et al. Risk factors for upper gastrointestinal bleeding in intensive care unit patients: role of Helicobacter pylori. Federal Hyperimmune Immunoglobulin Therapy Study Group. Crit Care Med 1996; 24:1974–1981.
- Duerksen DR. Stress-related mucosal disease in critically ill patients. Best Pract Res Clin Gastroenterol 2003; 17:327–344.
- Marik PE, Vasu T, Hirani A, Pachinburavan M. Stress ulcer prophylaxis in the new millennium: a systematic review and meta-analysis. Crit Care Med 2010; 38:2222–2228.
- Cohen H, editor. Stop stressing out: the new stress ulcer prophylaxis (SUP) guidelines are finally here! ASHP Midyear Clinical Meeting; 2013 11 Dec 2013; Orlando, FL.
In Reply: We welcome the comments from Dr. Chongnarungsin on our article and the opportunity to further discuss our opinions.
In our paper, we discussed current recommendations for prophylaxis of stress ulcer-related bleeding in hospitalized patients and advocated against the blind administration of drugs without risk stratification.
The landmark trial that provides the most-cited definitions and the risk factors for clinically significant stress ulcer-related bleeding in critically ill patients was published in 1994 by Cook et al.1 In their multicenter prospective cohort study of 2,252 patients, the authors reported that prolonged mechanical ventilation is an important risk factor for clinically significant stress ulcer-related bleeding.
Another major prospective cohort study observed an incidence rate of clinically significant stress ulcer-related bleeding of 3.5%.2
Dr. Chongnarungsin cites another prospective cohort study of 183 patients from the same era,3 wherein the authors defined stress ulcer-related bleeding as bleeding requiring transfusion of packed red blood cells, found on endoscopy or on postmortem evaluation. This was in contrast to the 1994 study of Cook et al,1 who had a more rigorous and comprehensive definition for overt and clinically significant stress ulcer-related bleeding, applied by up to three independent adjudicators not involved in the patients’ care. Their definition not only entailed a more accurate transfusion-dependent bleeding criterion, but also included hemodynamic and laboratory criteria. As such, the “very low rate” of stress ulcer-related bleeding reported by Zandstra et al3 should be critically appraised. Of note, the authors in that study did not report the rates of patients who received early enteral feeding, and their patients received cefotaxime for digestive tract decontamination, an important confounder to the interpretation of the study results.
Indeed, the remarkable variation in estimates of the incidence of stress ulcer-related bleeding is probably related to the lack of a uniform definition. Even when rates of endoscopic and occult bleeding are set aside, agreement is lacking as to which category of bleeding is clinically significant.
Dr. Chongnarungsin also cites the study by Ellison et al4 of a cohort of 874 patients who had no previous gastrointestinal bleeding or peptic ulcer disease and who were enrolled in a multicenter randomized controlled trial of prophylactic intravenous immune globulin to prevent infections associated with an intensive care unit. In a secondary objective, the authors did not identify coagulopathy or prolonged mechanical ventilation as a principal risk factor for bleeding. The authors ascribed this discrepancy with previously published literature to their unique study population, which consisted predominantly of elderly men and rarely included trauma patients. In light of these unique peculiarities of their population, the lack of an association between prolonged mechanical ventilation and stress ulcer-related bleeding cannot be determined. Moreover, that study showed that prolonged nasogastric tube insertion was one of the risk factors for increased risk of gastrointestinal bleeding, and not the risk factor for development of stress ulcer as stated by Dr. Chongnarungsin.
The decrease in the incidence of stress ulcer-related bleeding in critically ill patients over the years could be attributed to an era effect, from advances in critical care medicine and prophylactic methods.5 We agree with Dr. Chongnarungsin that the increased introduction of early enteral feeding may have also contributed to the reduced incidence of stress ulcer-related bleeding.6 However, we think the conclusion that “mechanical ventilation for more than 48 hours does not seem to increase the risk of stress ulcer” is overelaborated, and we believe that strong evidence demonstrates this association.1,2
Alternatively, we recognize the lack of mortality-benefit evidence for stress ulcer prophylaxis. This notwithstanding, according to recent Surviving Sepsis Campaign guidelines, the use of stress ulcer prophylaxis is listed as a 1B recommendation (strong recommendation) for severely septic patients who require prolonged mechanical ventilation. In addition, the updated 2014 guidelines of the American Society of Health-System Pharmacists7 continue to recommend stress ulcer prophylaxis in the context of mechanical ventilation, with H2 receptor antagonists being the preferred first-line agents.8
It is important to acknowledge that these recommendations were endorsed despite the lack of obvious mortality benefit, and it is our opinion that large randomized controlled studies are needed to evaluate the risks and mortality benefit of these prophylaxis methods.
In Reply: We welcome the comments from Dr. Chongnarungsin on our article and the opportunity to further discuss our opinions.
In our paper, we discussed current recommendations for prophylaxis of stress ulcer-related bleeding in hospitalized patients and advocated against the blind administration of drugs without risk stratification.
The landmark trial that provides the most-cited definitions and the risk factors for clinically significant stress ulcer-related bleeding in critically ill patients was published in 1994 by Cook et al.1 In their multicenter prospective cohort study of 2,252 patients, the authors reported that prolonged mechanical ventilation is an important risk factor for clinically significant stress ulcer-related bleeding.
Another major prospective cohort study observed an incidence rate of clinically significant stress ulcer-related bleeding of 3.5%.2
Dr. Chongnarungsin cites another prospective cohort study of 183 patients from the same era,3 wherein the authors defined stress ulcer-related bleeding as bleeding requiring transfusion of packed red blood cells, found on endoscopy or on postmortem evaluation. This was in contrast to the 1994 study of Cook et al,1 who had a more rigorous and comprehensive definition for overt and clinically significant stress ulcer-related bleeding, applied by up to three independent adjudicators not involved in the patients’ care. Their definition not only entailed a more accurate transfusion-dependent bleeding criterion, but also included hemodynamic and laboratory criteria. As such, the “very low rate” of stress ulcer-related bleeding reported by Zandstra et al3 should be critically appraised. Of note, the authors in that study did not report the rates of patients who received early enteral feeding, and their patients received cefotaxime for digestive tract decontamination, an important confounder to the interpretation of the study results.
Indeed, the remarkable variation in estimates of the incidence of stress ulcer-related bleeding is probably related to the lack of a uniform definition. Even when rates of endoscopic and occult bleeding are set aside, agreement is lacking as to which category of bleeding is clinically significant.
Dr. Chongnarungsin also cites the study by Ellison et al4 of a cohort of 874 patients who had no previous gastrointestinal bleeding or peptic ulcer disease and who were enrolled in a multicenter randomized controlled trial of prophylactic intravenous immune globulin to prevent infections associated with an intensive care unit. In a secondary objective, the authors did not identify coagulopathy or prolonged mechanical ventilation as a principal risk factor for bleeding. The authors ascribed this discrepancy with previously published literature to their unique study population, which consisted predominantly of elderly men and rarely included trauma patients. In light of these unique peculiarities of their population, the lack of an association between prolonged mechanical ventilation and stress ulcer-related bleeding cannot be determined. Moreover, that study showed that prolonged nasogastric tube insertion was one of the risk factors for increased risk of gastrointestinal bleeding, and not the risk factor for development of stress ulcer as stated by Dr. Chongnarungsin.
The decrease in the incidence of stress ulcer-related bleeding in critically ill patients over the years could be attributed to an era effect, from advances in critical care medicine and prophylactic methods.5 We agree with Dr. Chongnarungsin that the increased introduction of early enteral feeding may have also contributed to the reduced incidence of stress ulcer-related bleeding.6 However, we think the conclusion that “mechanical ventilation for more than 48 hours does not seem to increase the risk of stress ulcer” is overelaborated, and we believe that strong evidence demonstrates this association.1,2
Alternatively, we recognize the lack of mortality-benefit evidence for stress ulcer prophylaxis. This notwithstanding, according to recent Surviving Sepsis Campaign guidelines, the use of stress ulcer prophylaxis is listed as a 1B recommendation (strong recommendation) for severely septic patients who require prolonged mechanical ventilation. In addition, the updated 2014 guidelines of the American Society of Health-System Pharmacists7 continue to recommend stress ulcer prophylaxis in the context of mechanical ventilation, with H2 receptor antagonists being the preferred first-line agents.8
It is important to acknowledge that these recommendations were endorsed despite the lack of obvious mortality benefit, and it is our opinion that large randomized controlled studies are needed to evaluate the risks and mortality benefit of these prophylaxis methods.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Cook DJ, Griffith LE, Walter SD, et al. The attributable mortality and length of intensive care unit stay of clinically important gastrointestinal bleeding in critically ill patients. Crit Care 2001; 5:368–375.
- Zandstra DF, Stoutenbeek CP. The virtual absence of stress-ulceration related bleeding in ICU patients receiving prolonged mechanical ventilation without any prophylaxis. A prospective cohort study. Intensive Care Med 1994; 20:335–340.
- Ellison RT, Perez-Perez G, Welsh CH, et al. Risk factors for upper gastrointestinal bleeding in intensive care unit patients: role of Helicobacter pylori. Federal Hyperimmune Immunoglobulin Therapy Study Group. Crit Care Med 1996; 24:1974–1981.
- Duerksen DR. Stress-related mucosal disease in critically ill patients. Best Pract Res Clin Gastroenterol 2003; 17:327–344.
- Marik PE, Vasu T, Hirani A, Pachinburavan M. Stress ulcer prophylaxis in the new millennium: a systematic review and meta-analysis. Crit Care Med 2010; 38:2222–2228.
- Cohen H, editor. Stop stressing out: the new stress ulcer prophylaxis (SUP) guidelines are finally here! ASHP Midyear Clinical Meeting; 2013 11 Dec 2013; Orlando, FL.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Cook DJ, Griffith LE, Walter SD, et al. The attributable mortality and length of intensive care unit stay of clinically important gastrointestinal bleeding in critically ill patients. Crit Care 2001; 5:368–375.
- Zandstra DF, Stoutenbeek CP. The virtual absence of stress-ulceration related bleeding in ICU patients receiving prolonged mechanical ventilation without any prophylaxis. A prospective cohort study. Intensive Care Med 1994; 20:335–340.
- Ellison RT, Perez-Perez G, Welsh CH, et al. Risk factors for upper gastrointestinal bleeding in intensive care unit patients: role of Helicobacter pylori. Federal Hyperimmune Immunoglobulin Therapy Study Group. Crit Care Med 1996; 24:1974–1981.
- Duerksen DR. Stress-related mucosal disease in critically ill patients. Best Pract Res Clin Gastroenterol 2003; 17:327–344.
- Marik PE, Vasu T, Hirani A, Pachinburavan M. Stress ulcer prophylaxis in the new millennium: a systematic review and meta-analysis. Crit Care Med 2010; 38:2222–2228.
- Cohen H, editor. Stop stressing out: the new stress ulcer prophylaxis (SUP) guidelines are finally here! ASHP Midyear Clinical Meeting; 2013 11 Dec 2013; Orlando, FL.
Is hemoglobin A1c an accurate measure of glycemic control in all diabetic patients?
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
Do all hospitalized patients need stress ulcer prophylaxis?
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
Deep T waves and chest pain
A 67-year-old man with a history of hypertension and hyperlipidemia presented to the emergency department after 3 hours of what he described as a burning sensation in his chest that woke him from sleep. He attributed it at first to a late-night meal and treated himself with some milk and yogurt, which seemed to relieve the symptoms. However, the pain recurred and was associated with difficulty breathing. At that point, he drove himself to the emergency department.
On arrival, his temperature was 36.5°C (97.7°F), blood pressure 134/67 mm Hg, heart rate 89 bpm, respirations 18/min, and oxygen saturation 98% on room air. His cardiovascular, lung, and neurologic examinations were normal. His cardiac enzyme levels (creatine kinase, creatine kinase MB fraction, and troponin T) were within normal limits.
Figure 1 depicts his initial electrocardiogram. It showed deep, symmetric T-wave inversions in the precordial leads especially in V2 and V3, changes known as Wellens syndrome. The ST-T changes in lead V1 suggested a very proximal lesion in the left anterior descending artery (LAD), before the first septal perforator. Also, lateral and high lateral (V5 and V6) findings indicated stenoses of the branching diagonals and left circumflex myocardial territory. Furthermore, the inferior ST-T changes indicated that his LAD may have wrapped around the cardiac apex. All of these findings were prognostically significant.
The patient was given aspirin and was started on intravenous unfractionated heparin and nitroglycerin. He was sent for urgent left-heart catheterization, which showed a 50% to 60% stenosis in the left main coronary artery, with involvement of the left circumflex artery proximally, in addition to a “tight” first-diagonal stenosis, a 90% stenosis in a large (> 3.0-mm) proximal segment of the LAD, an 80% stenosis in a large (> 3.0-mm) mid-LAD segment, and a 40% stenosis in a large (> 3.0-mm) second diagonal artery (Figure 2).
He was referred for cardiac surgery and underwent triple coronary artery bypass grafting: the left internal thoracic artery was grafted to the LAD, a reverse saphenous vein graft was performed to the diagonal artery, and a reverse saphenous vein graft was performed to the obtuse marginal artery.
A PRECURSOR TO INFARCTION
Wellens et al described specific precordial T-wave changes in patients with unstable angina who subsequently developed anterior wall myocardial infarction.1
The importance of Wellens syndrome is that it occurs in the pain-free interval when no other evidence of ischemia or angina may be present.1 Cardiac enzyme levels are typically normal or only minimally elevated; only 12% of patients with this syndrome have elevated cardiac biomarker levels.2
Given the extent of myocardial injury, urgent echocardiography can show a wall-motion abnormality even if cardiac enzyme levels are normal. This gives important insight into electrocardiographic changes and should prompt consideration of revascularization.
Even with extensive medical management, Wellens syndrome progresses to acute anterior wall ischemia. About 75% of patients with Wellens syndrome who receive medical management but do not undergo revascularization (eg, coronary artery bypass grafting, percutaneous coronary intervention) develop extensive anterior wall infarction within days.1,3 Despite negative cardiac biomarkers, Wellens syndrome is considered an acute coronary syndrome requiring urgent cardiac intervention.
- Movahed MR. Wellens’ syndrome or inverted U-waves? Clin Cardiol 2008; 31:133–134.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
A 67-year-old man with a history of hypertension and hyperlipidemia presented to the emergency department after 3 hours of what he described as a burning sensation in his chest that woke him from sleep. He attributed it at first to a late-night meal and treated himself with some milk and yogurt, which seemed to relieve the symptoms. However, the pain recurred and was associated with difficulty breathing. At that point, he drove himself to the emergency department.
On arrival, his temperature was 36.5°C (97.7°F), blood pressure 134/67 mm Hg, heart rate 89 bpm, respirations 18/min, and oxygen saturation 98% on room air. His cardiovascular, lung, and neurologic examinations were normal. His cardiac enzyme levels (creatine kinase, creatine kinase MB fraction, and troponin T) were within normal limits.
Figure 1 depicts his initial electrocardiogram. It showed deep, symmetric T-wave inversions in the precordial leads especially in V2 and V3, changes known as Wellens syndrome. The ST-T changes in lead V1 suggested a very proximal lesion in the left anterior descending artery (LAD), before the first septal perforator. Also, lateral and high lateral (V5 and V6) findings indicated stenoses of the branching diagonals and left circumflex myocardial territory. Furthermore, the inferior ST-T changes indicated that his LAD may have wrapped around the cardiac apex. All of these findings were prognostically significant.
The patient was given aspirin and was started on intravenous unfractionated heparin and nitroglycerin. He was sent for urgent left-heart catheterization, which showed a 50% to 60% stenosis in the left main coronary artery, with involvement of the left circumflex artery proximally, in addition to a “tight” first-diagonal stenosis, a 90% stenosis in a large (> 3.0-mm) proximal segment of the LAD, an 80% stenosis in a large (> 3.0-mm) mid-LAD segment, and a 40% stenosis in a large (> 3.0-mm) second diagonal artery (Figure 2).
He was referred for cardiac surgery and underwent triple coronary artery bypass grafting: the left internal thoracic artery was grafted to the LAD, a reverse saphenous vein graft was performed to the diagonal artery, and a reverse saphenous vein graft was performed to the obtuse marginal artery.
A PRECURSOR TO INFARCTION
Wellens et al described specific precordial T-wave changes in patients with unstable angina who subsequently developed anterior wall myocardial infarction.1
The importance of Wellens syndrome is that it occurs in the pain-free interval when no other evidence of ischemia or angina may be present.1 Cardiac enzyme levels are typically normal or only minimally elevated; only 12% of patients with this syndrome have elevated cardiac biomarker levels.2
Given the extent of myocardial injury, urgent echocardiography can show a wall-motion abnormality even if cardiac enzyme levels are normal. This gives important insight into electrocardiographic changes and should prompt consideration of revascularization.
Even with extensive medical management, Wellens syndrome progresses to acute anterior wall ischemia. About 75% of patients with Wellens syndrome who receive medical management but do not undergo revascularization (eg, coronary artery bypass grafting, percutaneous coronary intervention) develop extensive anterior wall infarction within days.1,3 Despite negative cardiac biomarkers, Wellens syndrome is considered an acute coronary syndrome requiring urgent cardiac intervention.
A 67-year-old man with a history of hypertension and hyperlipidemia presented to the emergency department after 3 hours of what he described as a burning sensation in his chest that woke him from sleep. He attributed it at first to a late-night meal and treated himself with some milk and yogurt, which seemed to relieve the symptoms. However, the pain recurred and was associated with difficulty breathing. At that point, he drove himself to the emergency department.
On arrival, his temperature was 36.5°C (97.7°F), blood pressure 134/67 mm Hg, heart rate 89 bpm, respirations 18/min, and oxygen saturation 98% on room air. His cardiovascular, lung, and neurologic examinations were normal. His cardiac enzyme levels (creatine kinase, creatine kinase MB fraction, and troponin T) were within normal limits.
Figure 1 depicts his initial electrocardiogram. It showed deep, symmetric T-wave inversions in the precordial leads especially in V2 and V3, changes known as Wellens syndrome. The ST-T changes in lead V1 suggested a very proximal lesion in the left anterior descending artery (LAD), before the first septal perforator. Also, lateral and high lateral (V5 and V6) findings indicated stenoses of the branching diagonals and left circumflex myocardial territory. Furthermore, the inferior ST-T changes indicated that his LAD may have wrapped around the cardiac apex. All of these findings were prognostically significant.
The patient was given aspirin and was started on intravenous unfractionated heparin and nitroglycerin. He was sent for urgent left-heart catheterization, which showed a 50% to 60% stenosis in the left main coronary artery, with involvement of the left circumflex artery proximally, in addition to a “tight” first-diagonal stenosis, a 90% stenosis in a large (> 3.0-mm) proximal segment of the LAD, an 80% stenosis in a large (> 3.0-mm) mid-LAD segment, and a 40% stenosis in a large (> 3.0-mm) second diagonal artery (Figure 2).
He was referred for cardiac surgery and underwent triple coronary artery bypass grafting: the left internal thoracic artery was grafted to the LAD, a reverse saphenous vein graft was performed to the diagonal artery, and a reverse saphenous vein graft was performed to the obtuse marginal artery.
A PRECURSOR TO INFARCTION
Wellens et al described specific precordial T-wave changes in patients with unstable angina who subsequently developed anterior wall myocardial infarction.1
The importance of Wellens syndrome is that it occurs in the pain-free interval when no other evidence of ischemia or angina may be present.1 Cardiac enzyme levels are typically normal or only minimally elevated; only 12% of patients with this syndrome have elevated cardiac biomarker levels.2
Given the extent of myocardial injury, urgent echocardiography can show a wall-motion abnormality even if cardiac enzyme levels are normal. This gives important insight into electrocardiographic changes and should prompt consideration of revascularization.
Even with extensive medical management, Wellens syndrome progresses to acute anterior wall ischemia. About 75% of patients with Wellens syndrome who receive medical management but do not undergo revascularization (eg, coronary artery bypass grafting, percutaneous coronary intervention) develop extensive anterior wall infarction within days.1,3 Despite negative cardiac biomarkers, Wellens syndrome is considered an acute coronary syndrome requiring urgent cardiac intervention.
- Movahed MR. Wellens’ syndrome or inverted U-waves? Clin Cardiol 2008; 31:133–134.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
- Movahed MR. Wellens’ syndrome or inverted U-waves? Clin Cardiol 2008; 31:133–134.
- de Zwaan C, Bär FW, Janssen JH, et al. Angiographic and clinical characteristics of patients with unstable angina showing an ECG pattern indicating critical narrowing of the proximal LAD coronary artery. Am Heart J 1989; 117:657–665.
- de Zwaan C, Bär FW, Wellens HJ. Characteristic electrocardiographic pattern indicating a critical stenosis high in left anterior descending coronary artery in patients admitted because of impending myocardial infarction. Am Heart J 1982; 103:730–736.
Peripheral opacity on plain chest radiography
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
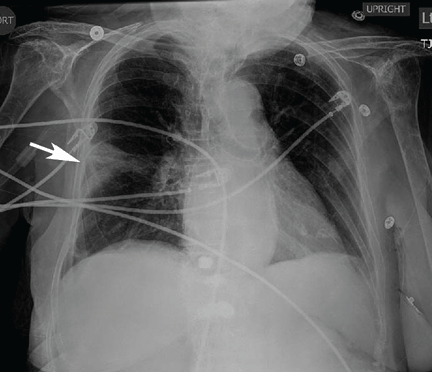
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
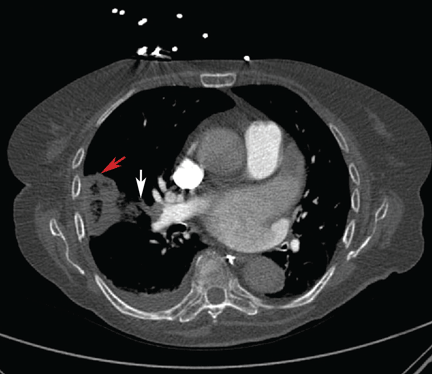
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
An 82-year-old woman was admitted to the hospital with dyspnea and chest discomfort over the past 24 hours. She was known to have paroxysmal atrial fibrillation and was taking warfarin, but that had been stopped 2 weeks earlier because of an acute ischemic stroke.
At the time of admission, she had no fever, cough, orthopnea, or leg swelling. Her physical activity was restricted, with residual right-sided weakness after her stroke. Her heart rate was 125 bpm; her oxygen saturation level was 98% on 2 L of oxygen per minute via nasal cannula. She had an irregularly irregular rhythm, a jugular venous pressure of 7 cm H2O, and no cardiac murmurs. Lung sounds were reduced at the bases, with faint crepitations.
Her hemoglobin concentration and white blood cell count were normal. Her brain-natriuretic peptide level was elevated at 2,648 pg/mL (reference range < 167), but cardiac enzyme levels were normal.
Electrocardiography showed atrial fibrillation with rapid ventricular response.
Plain chest radiography showed a 3-cm wedge-shaped opacity in the right mid-thorax (Figure 1), a finding known as the Hampton hump—a sign of pulmonary infarction caused by embolism.
Contrast-enhanced computed tomography (CT) of the chest showed acute thromboembolism in the right interlobar artery and wedge-shaped consolidation in the right-middle lobe (Figure 2), indicating pulmonary infarction.
Brain CT showed a stable infarction. Anticoagulation was restarted, and the patient was discharged in stable condition.
THE HAMPTON HUMP IN PULMONARY EMBOLISM
Because the lungs have a dual blood supply, pulmonary infarction is seen in only a minority of cases of pulmonary embolism. Infarction is more common in patients with peripheral pulmonary embolism, owing to the rapid inflow of bronchial blood, and in patients with medical comorbidities such as heart failure and chronic lung disease.2
The Hampton hump, first described by Aubrey Otis Hampton in 1940, is a peripheral (pleural-based) opacity that represents alveolar hemorrhage from underlying pulmonary infarction. It is one of several radiographic features that have been associated with pulmonary embolism; another is the Westermark sign, indicating oligemia.3
Worsley et al4 examined the diagnostic value of these radiographic features and found that the Hampton hump had a sensitivity of 22% and a specificity of 82% for detecting pulmonary embolism in the right hemithorax, and 24% and 82%, respectively, in the left hemithorax. The prevalence of pleural-based opacities was not significantly different in patients with or without pulmonary embolism. The authors concluded that chest radiography has limited diagnostic value in excluding or diagnosing pulmonary embolism.
In contrast, computed tomographic pulmonary angiography is the first-line imaging test in patients with suspected pulmonary embolism, because of its high sensitivity and specificity.1
We were not specifically looking for a pulmonary embolism when we found this new opacity on our patient’s radiograph, but this prompted further imaging, which led to the diagnosis. Although a near-normal chest radiograph is the most common radiologic finding in pulmonary embolism, this case shows how careful observation can detect unusual signs.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
- Mos IC, Klok FA, Kroft LJ, de Roos A, Huisman MV. Imaging tests in the diagnosis of pulmonary embolism. Semin Respir Crit Care Med 2012; 33:138–143.
- Cha SI, Shin KM, Lee J, et al. Clinical relevance of pulmonary infarction in patients with pulmonary embolism. Thromb Res 2012; 130:e1–e5.
- Algın O, GÖkalp G, Topal U. Signs in chest imaging. Diagn Interv Radiol 2011; 17:18–29.
- Worsley DF, Alavi A, Aronchick JM, Chen JT, Greenspan RH, Ravin CE. Chest radiographic findings in patients with acute pulmonary embolism: observations from the PIOPED study. Radiology 1993; 189:133–136.
Lung air-fluid level in a smoker
A 49-year-old man was referred for evaluation of an abnormal chest radiograph. A 25-pack-year smoker, he had a history of chronic shortness of breath on exertion with occasional coughing and whitish sputum production. He also had a history of hypertension. He had not had hemoptysis, fever, chills, weight loss, or other symptoms, and he had not traveled recently.
On examination, he appeared comfortable. His breath sounds were decreased bilaterally; the rest of his physical examination was normal. His medical history, social history, and review of systems were otherwise unremarkable.
His white blood cell count was 9.4 × 109/L (reference range 4.5–11.0), with a normal differential. His hemoglobin concentration was 166 g/L (140–175).
Pulmonary function testing demonstrated moderate obstruction, with the following values:
- Forced expiratory volume in the first second of expiration/ forced vital capacity 0.65
- Forced expiratory volume in the first second of expiration 2.40 L (72% of predicted)
- Total lung capacity 7.11 L (92% of predicted)
- Diffusing capacity of lung for carbon monoxide 58% of predicted.
He underwent radiography (Figure 1) and computed tomography of the chest (Figure 2).
DIAGNOSIS: INFECTED EMPHYSEMATOUS BULLAE
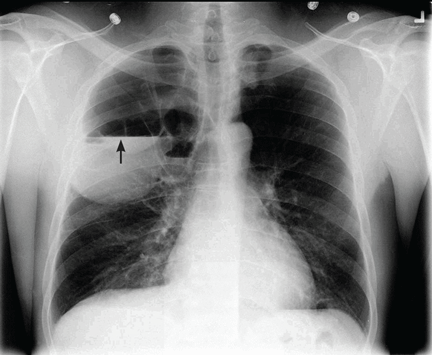
The patient had infected emphysematous bullae.
The diagnosis can typically be made by the new development of an air-fluid level in a patient known to have preexisting emphysematous bullae.1 If previous images are not available, the presence of other bullae in a patient with established chronic obstructive pulmonary disease, a thin-walled cavity, and a disproportionate presentation with impressive radiographic findings along with a subtle clinical picture can support the diagnosis.2 In most reported cases, patients are not significantly symptomatic or ill.3 The differential diagnosis includes loculated parapneumonic pleural effusion,4 lung abscess,5 tuberculosis,6 and infected pneumatocele.
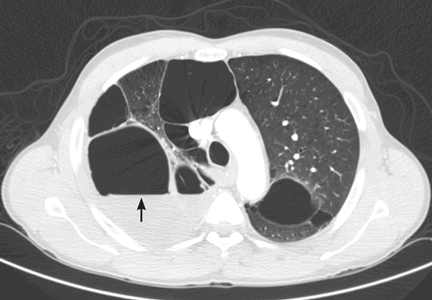
Since percutaneous aspiration of the bullae has been discouraged,2 the causative organism is often not identified. Also, the role of bronchoscopy in the diagnostic evaluation and treatment of infected emphysematous bullae appears to be limited.7
Our patient had minimal symptoms and did not appear ill; he had a relatively unremarkable physical examination, no leukocytosis, and negative blood and sputum cultures, suggesting a benign presentation. In addition, chest radiography a few months before this presentation showed multiple large emphysematous bullae (Figure 3). The current chest radiograph suggested multiple thin-walled cavitary lesions with an air-fluid level, which was confirmed on computed tomography.
TREATMENT OF INFECTED EMPHYSEMATOUS BULLAE
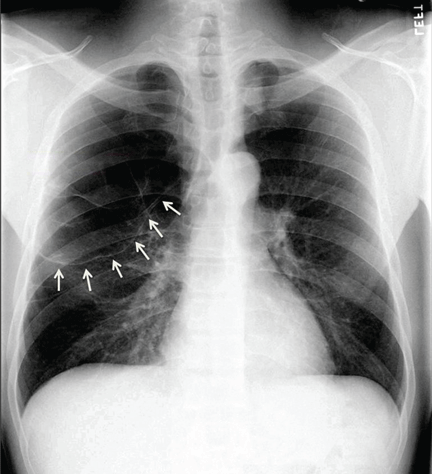
Currently, there is no established therapy for infected emphysematous bullae. Because the presentation is usually relatively benign in most case series, conservative treatment with a prolonged course of antibiotics alone seems to be the most appropriate initial course of action. A follow-up evaluation with chest imaging is recommended. On the other hand, in patients with worse symptoms, percutaneous aspiration of the bullae should be considered, as it may guide antibiotic therapy.8
We started our patient on clindamycin and scheduled him for follow-up chest imaging in 6 weeks.
- Burgener FA. Pulmonary cavitary and cystic lesions. In:Burgener FA, Kormano M, Pudas T, editors. Differential Diagnosis in Conventional Radiology. 3rd ed. New York, Thieme; 2008: chap.24.
- Chandra D, Soubra SH, Musher DM. A 57-year-old man with a fluid-containing lung cavity: infection of an emphysematous bulla with methicillin-resistant Staphylococcus aureus. Chest 2006; 130:1942–1946.
- Leatherman JW, McDonald FM, Niewohner DE. Fluid-containing bullae in the lung. South Med J 1985; 78:708–710.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Hammond JM, Potgieter PD, Hanslo D, Scott H, Roditi D. The etiology and antimicrobial susceptibility patterns of microorganisms in acute community-acquired lung abscess. Chest 1995; 108:937–941.
- Woodring JH, Vandiviere HM, Fried AM, Dillon ML, Williams TD, Melvin IG. Update: the radiographic features of pulmonary tuberculosis. AJR Am J Roentgenol 1986; 146:497–506.
- Chandra D, Rose SR, Carter RB, Musher DM, Hamill RJ. Fluid-containing emphysematous bullae: a spectrum of illness. Eur Respir J 2008; 32:303–306.
- Henao-Martinez AF, Fernandez JF, Adams SG, Restrepo C. Lung bullae with air-fluid levels: what is the appropriate therapeutic approach? Respir Care 2012; 57:642–645.
A 49-year-old man was referred for evaluation of an abnormal chest radiograph. A 25-pack-year smoker, he had a history of chronic shortness of breath on exertion with occasional coughing and whitish sputum production. He also had a history of hypertension. He had not had hemoptysis, fever, chills, weight loss, or other symptoms, and he had not traveled recently.
On examination, he appeared comfortable. His breath sounds were decreased bilaterally; the rest of his physical examination was normal. His medical history, social history, and review of systems were otherwise unremarkable.
His white blood cell count was 9.4 × 109/L (reference range 4.5–11.0), with a normal differential. His hemoglobin concentration was 166 g/L (140–175).
Pulmonary function testing demonstrated moderate obstruction, with the following values:
- Forced expiratory volume in the first second of expiration/ forced vital capacity 0.65
- Forced expiratory volume in the first second of expiration 2.40 L (72% of predicted)
- Total lung capacity 7.11 L (92% of predicted)
- Diffusing capacity of lung for carbon monoxide 58% of predicted.
He underwent radiography (Figure 1) and computed tomography of the chest (Figure 2).
DIAGNOSIS: INFECTED EMPHYSEMATOUS BULLAE
The patient had infected emphysematous bullae.
The diagnosis can typically be made by the new development of an air-fluid level in a patient known to have preexisting emphysematous bullae.1 If previous images are not available, the presence of other bullae in a patient with established chronic obstructive pulmonary disease, a thin-walled cavity, and a disproportionate presentation with impressive radiographic findings along with a subtle clinical picture can support the diagnosis.2 In most reported cases, patients are not significantly symptomatic or ill.3 The differential diagnosis includes loculated parapneumonic pleural effusion,4 lung abscess,5 tuberculosis,6 and infected pneumatocele.
Since percutaneous aspiration of the bullae has been discouraged,2 the causative organism is often not identified. Also, the role of bronchoscopy in the diagnostic evaluation and treatment of infected emphysematous bullae appears to be limited.7
Our patient had minimal symptoms and did not appear ill; he had a relatively unremarkable physical examination, no leukocytosis, and negative blood and sputum cultures, suggesting a benign presentation. In addition, chest radiography a few months before this presentation showed multiple large emphysematous bullae (Figure 3). The current chest radiograph suggested multiple thin-walled cavitary lesions with an air-fluid level, which was confirmed on computed tomography.
TREATMENT OF INFECTED EMPHYSEMATOUS BULLAE
Currently, there is no established therapy for infected emphysematous bullae. Because the presentation is usually relatively benign in most case series, conservative treatment with a prolonged course of antibiotics alone seems to be the most appropriate initial course of action. A follow-up evaluation with chest imaging is recommended. On the other hand, in patients with worse symptoms, percutaneous aspiration of the bullae should be considered, as it may guide antibiotic therapy.8
We started our patient on clindamycin and scheduled him for follow-up chest imaging in 6 weeks.
A 49-year-old man was referred for evaluation of an abnormal chest radiograph. A 25-pack-year smoker, he had a history of chronic shortness of breath on exertion with occasional coughing and whitish sputum production. He also had a history of hypertension. He had not had hemoptysis, fever, chills, weight loss, or other symptoms, and he had not traveled recently.
On examination, he appeared comfortable. His breath sounds were decreased bilaterally; the rest of his physical examination was normal. His medical history, social history, and review of systems were otherwise unremarkable.
His white blood cell count was 9.4 × 109/L (reference range 4.5–11.0), with a normal differential. His hemoglobin concentration was 166 g/L (140–175).
Pulmonary function testing demonstrated moderate obstruction, with the following values:
- Forced expiratory volume in the first second of expiration/ forced vital capacity 0.65
- Forced expiratory volume in the first second of expiration 2.40 L (72% of predicted)
- Total lung capacity 7.11 L (92% of predicted)
- Diffusing capacity of lung for carbon monoxide 58% of predicted.
He underwent radiography (Figure 1) and computed tomography of the chest (Figure 2).
DIAGNOSIS: INFECTED EMPHYSEMATOUS BULLAE
The patient had infected emphysematous bullae.
The diagnosis can typically be made by the new development of an air-fluid level in a patient known to have preexisting emphysematous bullae.1 If previous images are not available, the presence of other bullae in a patient with established chronic obstructive pulmonary disease, a thin-walled cavity, and a disproportionate presentation with impressive radiographic findings along with a subtle clinical picture can support the diagnosis.2 In most reported cases, patients are not significantly symptomatic or ill.3 The differential diagnosis includes loculated parapneumonic pleural effusion,4 lung abscess,5 tuberculosis,6 and infected pneumatocele.
Since percutaneous aspiration of the bullae has been discouraged,2 the causative organism is often not identified. Also, the role of bronchoscopy in the diagnostic evaluation and treatment of infected emphysematous bullae appears to be limited.7
Our patient had minimal symptoms and did not appear ill; he had a relatively unremarkable physical examination, no leukocytosis, and negative blood and sputum cultures, suggesting a benign presentation. In addition, chest radiography a few months before this presentation showed multiple large emphysematous bullae (Figure 3). The current chest radiograph suggested multiple thin-walled cavitary lesions with an air-fluid level, which was confirmed on computed tomography.
TREATMENT OF INFECTED EMPHYSEMATOUS BULLAE
Currently, there is no established therapy for infected emphysematous bullae. Because the presentation is usually relatively benign in most case series, conservative treatment with a prolonged course of antibiotics alone seems to be the most appropriate initial course of action. A follow-up evaluation with chest imaging is recommended. On the other hand, in patients with worse symptoms, percutaneous aspiration of the bullae should be considered, as it may guide antibiotic therapy.8
We started our patient on clindamycin and scheduled him for follow-up chest imaging in 6 weeks.
- Burgener FA. Pulmonary cavitary and cystic lesions. In:Burgener FA, Kormano M, Pudas T, editors. Differential Diagnosis in Conventional Radiology. 3rd ed. New York, Thieme; 2008: chap.24.
- Chandra D, Soubra SH, Musher DM. A 57-year-old man with a fluid-containing lung cavity: infection of an emphysematous bulla with methicillin-resistant Staphylococcus aureus. Chest 2006; 130:1942–1946.
- Leatherman JW, McDonald FM, Niewohner DE. Fluid-containing bullae in the lung. South Med J 1985; 78:708–710.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Hammond JM, Potgieter PD, Hanslo D, Scott H, Roditi D. The etiology and antimicrobial susceptibility patterns of microorganisms in acute community-acquired lung abscess. Chest 1995; 108:937–941.
- Woodring JH, Vandiviere HM, Fried AM, Dillon ML, Williams TD, Melvin IG. Update: the radiographic features of pulmonary tuberculosis. AJR Am J Roentgenol 1986; 146:497–506.
- Chandra D, Rose SR, Carter RB, Musher DM, Hamill RJ. Fluid-containing emphysematous bullae: a spectrum of illness. Eur Respir J 2008; 32:303–306.
- Henao-Martinez AF, Fernandez JF, Adams SG, Restrepo C. Lung bullae with air-fluid levels: what is the appropriate therapeutic approach? Respir Care 2012; 57:642–645.
- Burgener FA. Pulmonary cavitary and cystic lesions. In:Burgener FA, Kormano M, Pudas T, editors. Differential Diagnosis in Conventional Radiology. 3rd ed. New York, Thieme; 2008: chap.24.
- Chandra D, Soubra SH, Musher DM. A 57-year-old man with a fluid-containing lung cavity: infection of an emphysematous bulla with methicillin-resistant Staphylococcus aureus. Chest 2006; 130:1942–1946.
- Leatherman JW, McDonald FM, Niewohner DE. Fluid-containing bullae in the lung. South Med J 1985; 78:708–710.
- Sahn SA. Diagnosis and management of parapneumonic effusions and empyema. Clin Infect Dis 2007; 45:1480–1486.
- Hammond JM, Potgieter PD, Hanslo D, Scott H, Roditi D. The etiology and antimicrobial susceptibility patterns of microorganisms in acute community-acquired lung abscess. Chest 1995; 108:937–941.
- Woodring JH, Vandiviere HM, Fried AM, Dillon ML, Williams TD, Melvin IG. Update: the radiographic features of pulmonary tuberculosis. AJR Am J Roentgenol 1986; 146:497–506.
- Chandra D, Rose SR, Carter RB, Musher DM, Hamill RJ. Fluid-containing emphysematous bullae: a spectrum of illness. Eur Respir J 2008; 32:303–306.
- Henao-Martinez AF, Fernandez JF, Adams SG, Restrepo C. Lung bullae with air-fluid levels: what is the appropriate therapeutic approach? Respir Care 2012; 57:642–645.
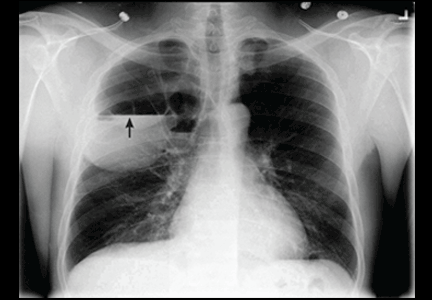
A 47-year-old man with chest and neck pain
A 47-year-old man presented with acute shortness of breath and chest and neck pain, which began after he heard popping sounds while boarding a bus. The pain was right-sided, sharp, worse with deep breathing, and associated with a sensation of fullness over the right chest.
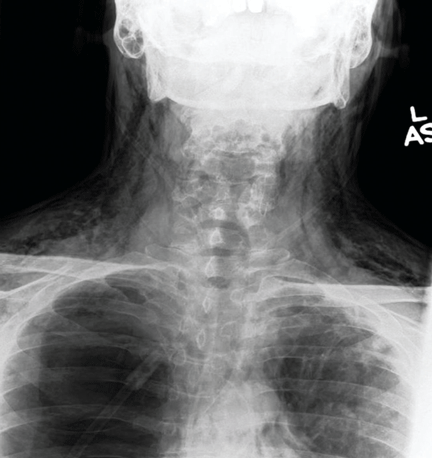
His medical conditions included controlled hypertension, gastroesophageal reflux disease, and chronic obstructive pulmonary disease (COPD). The COPD was managed with an albuterol inhaler only. He had a 50-pack-year history of smoking, and he drank alcohol occasionally.
On arrival, he was in mild respiratory distress, but his vital signs were stable. We could hear wheezing on both sides of his chest and feel subcutaneous crepitation on both sides of his chest and neck, the latter more on the right side. The rest of the examination was unremarkable.
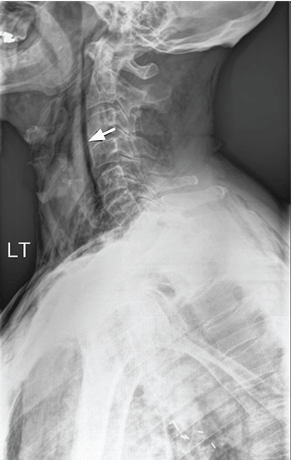
Results of a complete blood cell count and metabolic panel were within normal limits. Because of the above findings, nasopharyngeal radiogragraphy was ordered (Figures 1 and 2).
Q: What is the most likely cause of this presentation?
- Esophageal rupture
- Gas gangrene
- Asthma exacerbation
- Ruptured emphysematous bullae
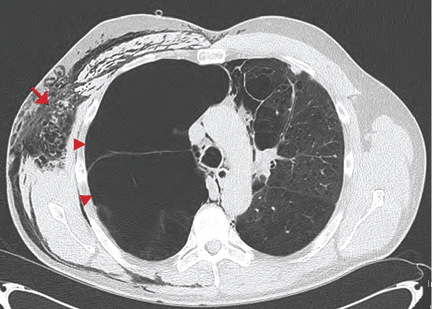
A: This patient had a history of COPD, which put him at risk of developing bullous emphysematous bullae that can rupture and cause subcutaneous emphysema. His nasopharyngeal radiograph (Figure1) showed bilateral extensive subcutaneous emphysema. His lateral nasopharyngeal radiograph (Figure 2) showed air-tracking within the mediastinum and into the retropharyngeal space (arrow). Computed tomography (Figure 3) showed extensive subcutaneous emphysema in the right lateral chest wall (arrow) and large bullae in the right upper lobe (arrow heads). As for the other possibilities:
Esophageal ruptures and tears are iatrogenic in most cases and usually occur after endoscopic procedures, but they can also occur in patients with intractable vomiting. Computed tomography often shows esophageal thickening, periesophageal fluid, mediastinal widening, and extraluminal air. However, in most cases, it is seen as pneumomediastinum and subcutaneous emphysema.1
Gas gangrene is a life-threatening soft-tissue and muscle infection caused by Clostridium perfringens in most cases.2 The pain is out of proportion to the findings on physical examination. Patients usually have toxic signs and symptoms such as fever and hypotension. Our patient was hemodynamically stable, with no changes in skin color.
Severe exacerbations of asthma can lead to alveolar rupture, pneumothorax, and subcutaneous emphysema, although this is a rare complication. Air can dissect along the bronchovascular sheaths into the neck and cause subcutaneous emphysema, or into the pleural space and cause pneumothorax. Our patient had no history of asthma and plainly had emphysematous bullae.3
SUBCUTANEOUS EMPHYSEMA
Subcutaneous emphysema is a collection of air within subcutaneous tissues. It usually presents as bloating of the skin around the neck and the chest wall. It is often seen in patients with pneumothorax.
The most common cause of subcutaneous emphysema is traumatic injury to the chest wall, such as from a motor vehicle accident or a stab wound,4 but it can also occur spontaneously in patients who have severe emphysema with large bullae. As the emphysema progresses, the bullae can easily rupture, and this can lead to pneumothorax, which can lead to subcutaneous emphysema. Primary spontaneous pneumothorax and subcutaneous emphysema can occur in people who have unrecognized lung disease and genetic disorders such as Marfan syndrome and Ehler-Danlos syndrome.5 Other causes include iatrogenic injury, Pneumocystis jirovecii pneumonia (common in patients with human immunodeficiency virus infection), and cystic fibrosis. Pneumothorax occurs in about 30% of cases of P jirovecii pneumonia,6 and in about 6% of patients with cystic fibrosis.7 Bronchocutaneous fistula is an extremely rare complication of lung cancer and can cause subcutaneous emphysema.8 Tuberculosis is another possible cause.9
Subcutaneous emphysema mainly presents with chest or neck pain and wheezing. In severe cases, air can track to the face, causing facial swelling and difficulty breathing due to compression of the larynx. Also, it can track down to the thighs, causing leg pain and swelling.10
On examination, subcutaneous emphysema can be detected by palpating the chest wall, which causes the air bubble to move and produce crackling sounds. Most cases of subcutaneous emphysema are diagnosed clinically. Chest radiography and computed tomography help identify the source of air leak. Ultrasonography is usually used in cases of blunt trauma to the chest as part of the Focal Assessment With Sonography for Trauma protocol.11
Subcutaneous emphysema can resolve spontaneously, requiring only pain management and supplemental oxygen.12 In severe cases, air collection can lead to what is called “massive subcutaneous emphysema,” which requires surgical drainage.
Our patient had large emphysematous bullae in the apical region of the right lung that ruptured and led to subcutaneous emphysema. After placement of a chest tube, he underwent right-sided thoracotomy with bullectomy. His postoperative course was uneventful, and he was discharged a few days later. Three weeks later, repeated chest radiography showed resolution of his subcutaneous emphysema (Figure 4).
- White CS, Templeton PA, Attar S. Esophageal perforation: CT findings. AJR Am J Roentgenol 1993; 160:767–770.
- Aggelidakis J, Lasithiotakis K, Topalidou A, Koutroumpas J, Kouvidis G, Katonis P. Limb salvage after gas gangrene: a case report and review of the literature. World J Emerg Surg 2011; 6:28.
- Romero KJ, Trujillo MH. Spontaneous pneumomediastinum and subcutaneous emphysema in asthma exacerbation: the Macklin effect. Heart Lung 2010; 39:444–447.
- Peart O. Subcutaneous emphysema. Radiol Technol 2006; 77:296.
- Chiu HT, Garcia CK. Familial spontaneous pneumothorax. Curr Opin Pulm Med 2006; 12:268–272.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Flume PA, Strange C, Ye X, Ebeling M, Hulsey T, Clark LL. Pneumothorax in cystic fibrosis. Chest 2005; 128:720–728.
- Yalçinkaya S, Vural AH, Göncü MT, Özyazicioglu AF. Cavitary lung cancer presenting as subcutaneous emphysema on the contralateral side. Interact Cardiovasc Thorac Surg 2012; 14:338–339.
- Shamaei M, Tabarsi P, Pojhan S, et al. Tuberculosis-associated secondary pneumothorax: a retrospective study of 53 patients. Respir Care 2011; 56:298–302.
- Sherif HM, Ott DA. The use of subcutaneous drains to manage subcutaneous emphysema. Tex Heart Inst J 1999; 26:129–131.
- Wilkerson RG, Stone MB. Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med 2010; 17:11–17.
- Mattox KL, Allen MK. Systematic approach to pneumothorax, haemothorax, pneumomediastinum and subcutaneous emphysema. Injury 1986; 17:309–312.
A 47-year-old man presented with acute shortness of breath and chest and neck pain, which began after he heard popping sounds while boarding a bus. The pain was right-sided, sharp, worse with deep breathing, and associated with a sensation of fullness over the right chest.
His medical conditions included controlled hypertension, gastroesophageal reflux disease, and chronic obstructive pulmonary disease (COPD). The COPD was managed with an albuterol inhaler only. He had a 50-pack-year history of smoking, and he drank alcohol occasionally.
On arrival, he was in mild respiratory distress, but his vital signs were stable. We could hear wheezing on both sides of his chest and feel subcutaneous crepitation on both sides of his chest and neck, the latter more on the right side. The rest of the examination was unremarkable.
Results of a complete blood cell count and metabolic panel were within normal limits. Because of the above findings, nasopharyngeal radiogragraphy was ordered (Figures 1 and 2).
Q: What is the most likely cause of this presentation?
- Esophageal rupture
- Gas gangrene
- Asthma exacerbation
- Ruptured emphysematous bullae
A: This patient had a history of COPD, which put him at risk of developing bullous emphysematous bullae that can rupture and cause subcutaneous emphysema. His nasopharyngeal radiograph (Figure1) showed bilateral extensive subcutaneous emphysema. His lateral nasopharyngeal radiograph (Figure 2) showed air-tracking within the mediastinum and into the retropharyngeal space (arrow). Computed tomography (Figure 3) showed extensive subcutaneous emphysema in the right lateral chest wall (arrow) and large bullae in the right upper lobe (arrow heads). As for the other possibilities:
Esophageal ruptures and tears are iatrogenic in most cases and usually occur after endoscopic procedures, but they can also occur in patients with intractable vomiting. Computed tomography often shows esophageal thickening, periesophageal fluid, mediastinal widening, and extraluminal air. However, in most cases, it is seen as pneumomediastinum and subcutaneous emphysema.1
Gas gangrene is a life-threatening soft-tissue and muscle infection caused by Clostridium perfringens in most cases.2 The pain is out of proportion to the findings on physical examination. Patients usually have toxic signs and symptoms such as fever and hypotension. Our patient was hemodynamically stable, with no changes in skin color.
Severe exacerbations of asthma can lead to alveolar rupture, pneumothorax, and subcutaneous emphysema, although this is a rare complication. Air can dissect along the bronchovascular sheaths into the neck and cause subcutaneous emphysema, or into the pleural space and cause pneumothorax. Our patient had no history of asthma and plainly had emphysematous bullae.3
SUBCUTANEOUS EMPHYSEMA
Subcutaneous emphysema is a collection of air within subcutaneous tissues. It usually presents as bloating of the skin around the neck and the chest wall. It is often seen in patients with pneumothorax.
The most common cause of subcutaneous emphysema is traumatic injury to the chest wall, such as from a motor vehicle accident or a stab wound,4 but it can also occur spontaneously in patients who have severe emphysema with large bullae. As the emphysema progresses, the bullae can easily rupture, and this can lead to pneumothorax, which can lead to subcutaneous emphysema. Primary spontaneous pneumothorax and subcutaneous emphysema can occur in people who have unrecognized lung disease and genetic disorders such as Marfan syndrome and Ehler-Danlos syndrome.5 Other causes include iatrogenic injury, Pneumocystis jirovecii pneumonia (common in patients with human immunodeficiency virus infection), and cystic fibrosis. Pneumothorax occurs in about 30% of cases of P jirovecii pneumonia,6 and in about 6% of patients with cystic fibrosis.7 Bronchocutaneous fistula is an extremely rare complication of lung cancer and can cause subcutaneous emphysema.8 Tuberculosis is another possible cause.9
Subcutaneous emphysema mainly presents with chest or neck pain and wheezing. In severe cases, air can track to the face, causing facial swelling and difficulty breathing due to compression of the larynx. Also, it can track down to the thighs, causing leg pain and swelling.10
On examination, subcutaneous emphysema can be detected by palpating the chest wall, which causes the air bubble to move and produce crackling sounds. Most cases of subcutaneous emphysema are diagnosed clinically. Chest radiography and computed tomography help identify the source of air leak. Ultrasonography is usually used in cases of blunt trauma to the chest as part of the Focal Assessment With Sonography for Trauma protocol.11
Subcutaneous emphysema can resolve spontaneously, requiring only pain management and supplemental oxygen.12 In severe cases, air collection can lead to what is called “massive subcutaneous emphysema,” which requires surgical drainage.
Our patient had large emphysematous bullae in the apical region of the right lung that ruptured and led to subcutaneous emphysema. After placement of a chest tube, he underwent right-sided thoracotomy with bullectomy. His postoperative course was uneventful, and he was discharged a few days later. Three weeks later, repeated chest radiography showed resolution of his subcutaneous emphysema (Figure 4).
A 47-year-old man presented with acute shortness of breath and chest and neck pain, which began after he heard popping sounds while boarding a bus. The pain was right-sided, sharp, worse with deep breathing, and associated with a sensation of fullness over the right chest.
His medical conditions included controlled hypertension, gastroesophageal reflux disease, and chronic obstructive pulmonary disease (COPD). The COPD was managed with an albuterol inhaler only. He had a 50-pack-year history of smoking, and he drank alcohol occasionally.
On arrival, he was in mild respiratory distress, but his vital signs were stable. We could hear wheezing on both sides of his chest and feel subcutaneous crepitation on both sides of his chest and neck, the latter more on the right side. The rest of the examination was unremarkable.
Results of a complete blood cell count and metabolic panel were within normal limits. Because of the above findings, nasopharyngeal radiogragraphy was ordered (Figures 1 and 2).
Q: What is the most likely cause of this presentation?
- Esophageal rupture
- Gas gangrene
- Asthma exacerbation
- Ruptured emphysematous bullae
A: This patient had a history of COPD, which put him at risk of developing bullous emphysematous bullae that can rupture and cause subcutaneous emphysema. His nasopharyngeal radiograph (Figure1) showed bilateral extensive subcutaneous emphysema. His lateral nasopharyngeal radiograph (Figure 2) showed air-tracking within the mediastinum and into the retropharyngeal space (arrow). Computed tomography (Figure 3) showed extensive subcutaneous emphysema in the right lateral chest wall (arrow) and large bullae in the right upper lobe (arrow heads). As for the other possibilities:
Esophageal ruptures and tears are iatrogenic in most cases and usually occur after endoscopic procedures, but they can also occur in patients with intractable vomiting. Computed tomography often shows esophageal thickening, periesophageal fluid, mediastinal widening, and extraluminal air. However, in most cases, it is seen as pneumomediastinum and subcutaneous emphysema.1
Gas gangrene is a life-threatening soft-tissue and muscle infection caused by Clostridium perfringens in most cases.2 The pain is out of proportion to the findings on physical examination. Patients usually have toxic signs and symptoms such as fever and hypotension. Our patient was hemodynamically stable, with no changes in skin color.
Severe exacerbations of asthma can lead to alveolar rupture, pneumothorax, and subcutaneous emphysema, although this is a rare complication. Air can dissect along the bronchovascular sheaths into the neck and cause subcutaneous emphysema, or into the pleural space and cause pneumothorax. Our patient had no history of asthma and plainly had emphysematous bullae.3
SUBCUTANEOUS EMPHYSEMA
Subcutaneous emphysema is a collection of air within subcutaneous tissues. It usually presents as bloating of the skin around the neck and the chest wall. It is often seen in patients with pneumothorax.
The most common cause of subcutaneous emphysema is traumatic injury to the chest wall, such as from a motor vehicle accident or a stab wound,4 but it can also occur spontaneously in patients who have severe emphysema with large bullae. As the emphysema progresses, the bullae can easily rupture, and this can lead to pneumothorax, which can lead to subcutaneous emphysema. Primary spontaneous pneumothorax and subcutaneous emphysema can occur in people who have unrecognized lung disease and genetic disorders such as Marfan syndrome and Ehler-Danlos syndrome.5 Other causes include iatrogenic injury, Pneumocystis jirovecii pneumonia (common in patients with human immunodeficiency virus infection), and cystic fibrosis. Pneumothorax occurs in about 30% of cases of P jirovecii pneumonia,6 and in about 6% of patients with cystic fibrosis.7 Bronchocutaneous fistula is an extremely rare complication of lung cancer and can cause subcutaneous emphysema.8 Tuberculosis is another possible cause.9
Subcutaneous emphysema mainly presents with chest or neck pain and wheezing. In severe cases, air can track to the face, causing facial swelling and difficulty breathing due to compression of the larynx. Also, it can track down to the thighs, causing leg pain and swelling.10
On examination, subcutaneous emphysema can be detected by palpating the chest wall, which causes the air bubble to move and produce crackling sounds. Most cases of subcutaneous emphysema are diagnosed clinically. Chest radiography and computed tomography help identify the source of air leak. Ultrasonography is usually used in cases of blunt trauma to the chest as part of the Focal Assessment With Sonography for Trauma protocol.11
Subcutaneous emphysema can resolve spontaneously, requiring only pain management and supplemental oxygen.12 In severe cases, air collection can lead to what is called “massive subcutaneous emphysema,” which requires surgical drainage.
Our patient had large emphysematous bullae in the apical region of the right lung that ruptured and led to subcutaneous emphysema. After placement of a chest tube, he underwent right-sided thoracotomy with bullectomy. His postoperative course was uneventful, and he was discharged a few days later. Three weeks later, repeated chest radiography showed resolution of his subcutaneous emphysema (Figure 4).
- White CS, Templeton PA, Attar S. Esophageal perforation: CT findings. AJR Am J Roentgenol 1993; 160:767–770.
- Aggelidakis J, Lasithiotakis K, Topalidou A, Koutroumpas J, Kouvidis G, Katonis P. Limb salvage after gas gangrene: a case report and review of the literature. World J Emerg Surg 2011; 6:28.
- Romero KJ, Trujillo MH. Spontaneous pneumomediastinum and subcutaneous emphysema in asthma exacerbation: the Macklin effect. Heart Lung 2010; 39:444–447.
- Peart O. Subcutaneous emphysema. Radiol Technol 2006; 77:296.
- Chiu HT, Garcia CK. Familial spontaneous pneumothorax. Curr Opin Pulm Med 2006; 12:268–272.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Flume PA, Strange C, Ye X, Ebeling M, Hulsey T, Clark LL. Pneumothorax in cystic fibrosis. Chest 2005; 128:720–728.
- Yalçinkaya S, Vural AH, Göncü MT, Özyazicioglu AF. Cavitary lung cancer presenting as subcutaneous emphysema on the contralateral side. Interact Cardiovasc Thorac Surg 2012; 14:338–339.
- Shamaei M, Tabarsi P, Pojhan S, et al. Tuberculosis-associated secondary pneumothorax: a retrospective study of 53 patients. Respir Care 2011; 56:298–302.
- Sherif HM, Ott DA. The use of subcutaneous drains to manage subcutaneous emphysema. Tex Heart Inst J 1999; 26:129–131.
- Wilkerson RG, Stone MB. Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med 2010; 17:11–17.
- Mattox KL, Allen MK. Systematic approach to pneumothorax, haemothorax, pneumomediastinum and subcutaneous emphysema. Injury 1986; 17:309–312.
- White CS, Templeton PA, Attar S. Esophageal perforation: CT findings. AJR Am J Roentgenol 1993; 160:767–770.
- Aggelidakis J, Lasithiotakis K, Topalidou A, Koutroumpas J, Kouvidis G, Katonis P. Limb salvage after gas gangrene: a case report and review of the literature. World J Emerg Surg 2011; 6:28.
- Romero KJ, Trujillo MH. Spontaneous pneumomediastinum and subcutaneous emphysema in asthma exacerbation: the Macklin effect. Heart Lung 2010; 39:444–447.
- Peart O. Subcutaneous emphysema. Radiol Technol 2006; 77:296.
- Chiu HT, Garcia CK. Familial spontaneous pneumothorax. Curr Opin Pulm Med 2006; 12:268–272.
- Sepkowitz KA, Telzak EE, Gold JW, et al. Pneumothorax in AIDS. Ann Intern Med 1991; 114:455–459.
- Flume PA, Strange C, Ye X, Ebeling M, Hulsey T, Clark LL. Pneumothorax in cystic fibrosis. Chest 2005; 128:720–728.
- Yalçinkaya S, Vural AH, Göncü MT, Özyazicioglu AF. Cavitary lung cancer presenting as subcutaneous emphysema on the contralateral side. Interact Cardiovasc Thorac Surg 2012; 14:338–339.
- Shamaei M, Tabarsi P, Pojhan S, et al. Tuberculosis-associated secondary pneumothorax: a retrospective study of 53 patients. Respir Care 2011; 56:298–302.
- Sherif HM, Ott DA. The use of subcutaneous drains to manage subcutaneous emphysema. Tex Heart Inst J 1999; 26:129–131.
- Wilkerson RG, Stone MB. Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med 2010; 17:11–17.
- Mattox KL, Allen MK. Systematic approach to pneumothorax, haemothorax, pneumomediastinum and subcutaneous emphysema. Injury 1986; 17:309–312.