User login
What’s Eating You? Blister Beetles Revisited
Classification
Blister beetles are both a scourge and the source of medical cantharidin (Figure 1). The term blister beetle refers to 3 families of the order Coleoptera: Meloidae, Oedemeridae, and Staphylinidae (Figure 2).

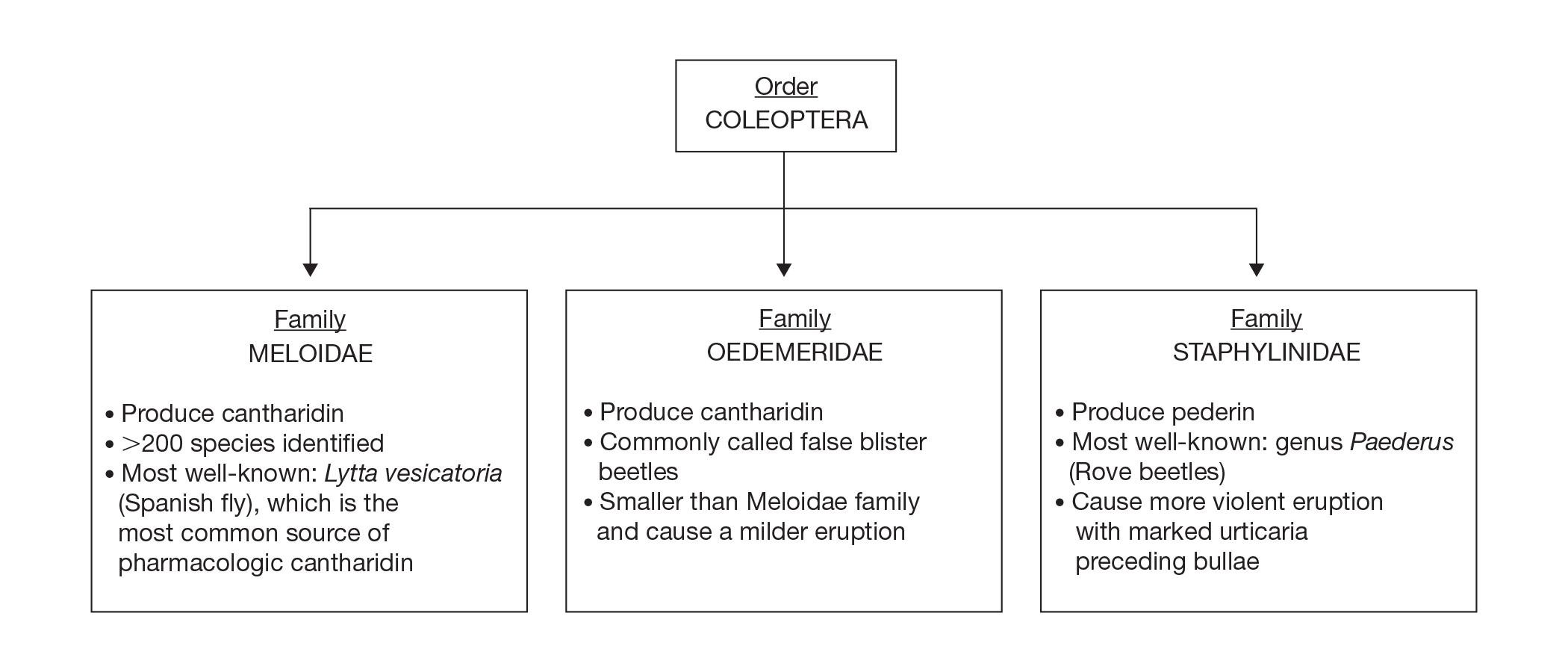
Meloidae is the most well-known family of blister beetles, with more than 200 species worldwide identified as a cause of blistering dermatitis.1 The most notorious is Lytta vesicatoria, also known as the Spanish fly. Although some blister beetles are inconspicuous in appearance, most are brightly colored and easily spotted, making them attractive to small children.2 They may be attracted to fluorescent lighting and commonly enter through open windows.3 Blister beetles do not bite or sting; rather, they release cantharidin via hemolymph, an oily yellow substance that is copiously expressed from the leg joints when disturbed by rubbing or pressing.1,3-5 Blistering also is associated with exposure to the contents of crushed beetles, which is the source of pharmacologic cantharidin.2,4,6
Oedemeridae are the smallest and least known beetles within the Coleoptera family. They often are called false blister beetles, which is a misnomer. Oedemeridae beetles also produce cantharidin, similar to Meloidae beetles; however, because Oedemeridae beetles are smaller, the vesiculobullous eruptions also are less pronounced.1
A third well-known family of blister beetles is the Staphylinidae family. Rove beetles (genus Paederus) differ from the Meloidae and Oedemeridae families in that they produce the vesicant pederin rather than cantharidin. Pederin causes a more violent eruption called dermatitis linearis, which often is associated with intense urticaria prior to blistering.1 Rove beetles are found in moist environments and tend to favor tropical and subtropical climates. They may emerge in large numbers after heavy rainfall.
Clinical Presentation
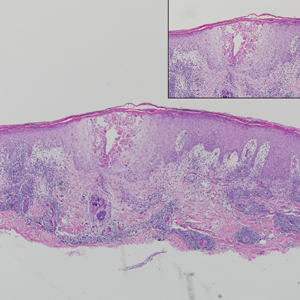
Blistering dermatitis—caused by exposure to cantharidin (families Meloidae and Oedemeridae) or pederin (genus Paederus)—begins as burning and tingling within minutes of exposure. Bullae later develop, often in a linear fashion, with subsequent bursting and crust formation.1,3 Secondary infection can occur.5 Exposure occurring on the elbows, knees, or mirroring skin folds results in lesions on opposing surfaces that come into contact, otherwise known as kissing lesions.1,3 Acantholysis of suprabasal keratinocytes can be seen on histologic sections of blisters (Figure 3)1,7 due to cantharidin activation of the serine/threonine protein phosphatases that cause detachment of tonofilaments from desmosomes.1,8 Washing of exposed sites with soap or alcohol can potentially prevent development of blistering dermatitis; however, lesions usually heal without complication when treated with topical antibiotics.3
If blister beetles are ingested, they can cause poisoning characterized by abdominal pain and hematuria.1,3,9,10 In fact, blister beetle ingestion by animals consuming baled hay is an important agricultural and economical implication. Several cases of fatal farm animal ingestion resulting in gastrointestinal erosion have been reported.4,6
Medicinal Properties
Medicinal use of cantharidin has been recorded as early as the 19th century and is believed to have been part of ancient medicinal practices in China, Spain, South Africa, and pre-Columbian America for its vesicant, abortifacient, and supposed aphrodisiac properties.2,4,8
Toxicity from internal ingestion has been well documented.1,3,9,10 The Mylabris and Epicauta genera of the family Meloidae, most commonly the Spanish fly, are used for modern extraction of cantharidin. Up to 5% of the dry weight of a blister beetle can be constituted by cantharidin.4
Although its use has diminished due to limited availability, cantharidin is still used for treatment of common warts, periungual warts, and molluscum contagiosum. It is a favorable choice for treating pediatric patients because of the tolerability and painlessness of application. Due to its acantholytic properties, warts generally slough with the cantharidin-induced blister.11
In recent years, cantharidin has been studied for its anticancer properties. It has been shown to weaken cancer cell antioxidant properties by interfering with glutathione-related enzymes, thus inducing oxidative damage. Additionally, cantharidin is associated with decreased mitochondrial cytochrome C and increased cytosolic cytochrome C, an important signal for apoptosis.12 Furthermore, cantharidin recently was shown to increase expression of proapoptotic proteins and decrease expression of antiapoptotic proteins causing cell death in nasopharyngeal carcinoma, making it a potential anticancer treatment.13
Conclusion
Blister beetles, long known for their production of vesicant agents, are both a cause of as well as a potential treatment of dermatologic disease. The blistering associated with exposure to a disturbed beetle generally is mild and heals without scarring if vital areas such as the eyelids are not affected.
- Nicholls DS, Christmas TI, Greig DE. Oedemerid blister beetle dermatosis: a review. J Am Acad Dermatol. 1990;22:815-819.
- Percino-Daniel N, Buckley D, García-París M. Pharmacological properties of blister beetles (Coleoptera: Meloidae) promoted their integration into the cultural heritage of native rural Spain as inferred by vernacular names diversity, traditions, and mitochondrial DNA. J Ethnopharmacol. 2013;147:570-583.
- James WD, Berger TG, Elston DM. Parasitic infestations, stings, and bites. In: James WD, Berger TG, Elston DM, eds. Andrew’s Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016:418-450.
- Selander RB, Fasulo TR. Featured creatures: blister beetles. University of Florida/IFAS Entomology and Nematology website. http://entnemdept.ufl.edu/creatures/urban/medical/blister_beetles.htm. Published October 2000. Revised September 2010. Accessed November 12, 2019.
- Wijerathne BTB. Blister mystery. Wilderness Environ Med. 2017;28:271-272.
- Penrith ML, Naude TW. Mortality in chickens associated with blister beetle consumption. J S Afr Vet Assoc. 1996;67:97-99.
- Yell JA, Burge SM, Dean D. Cantharidin-induced acantholysis: adhesion molecules, proteases, and related proteins. Br J Dermatol. 1994;130:148-157.
- Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine-threonine protein phosphatases types 1 and 2a. FEBS Letters. 1993;330:283-286.
- Al-Binali AM, Shabana M, Al-Fifi S, et al. Cantharidin poisoning due to blister beetle ingestion in children: two case reports and a review of clinical presentations. Sultan Qaboos Univ Med J. 2010;10:258-261.
- Tagwireyi D, Ball DE, Loga PJ, et al. Cantharidin poisoning due to “blister beetle” ingestion. Toxicon. 2000;38:1865-1869.
- Al-Dawsari NA, Masterpol KS. Cantharidin in dermatology. Skinmed. 2016;14:111-114.
- Verma AK, Prasad SB. Changes in glutathione, oxidative stress and mitochondrial membrane potential in apoptosis involving the anticancer activity of cantharidin isolated from redheaded blister beetles, epicauta hirticornis. Anticancer Agents Med Chem. 2013;13:1096-1114.
- Chen AW, Tseng YS, Lin CC, et al. Norcantharidin induce apoptosis in human nasopharyngeal carcinoma through caspase and mitochondrial pathway. Environ Toxicol. 2018;33:343-350.
Classification
Blister beetles are both a scourge and the source of medical cantharidin (Figure 1). The term blister beetle refers to 3 families of the order Coleoptera: Meloidae, Oedemeridae, and Staphylinidae (Figure 2).
Meloidae is the most well-known family of blister beetles, with more than 200 species worldwide identified as a cause of blistering dermatitis.1 The most notorious is Lytta vesicatoria, also known as the Spanish fly. Although some blister beetles are inconspicuous in appearance, most are brightly colored and easily spotted, making them attractive to small children.2 They may be attracted to fluorescent lighting and commonly enter through open windows.3 Blister beetles do not bite or sting; rather, they release cantharidin via hemolymph, an oily yellow substance that is copiously expressed from the leg joints when disturbed by rubbing or pressing.1,3-5 Blistering also is associated with exposure to the contents of crushed beetles, which is the source of pharmacologic cantharidin.2,4,6
Oedemeridae are the smallest and least known beetles within the Coleoptera family. They often are called false blister beetles, which is a misnomer. Oedemeridae beetles also produce cantharidin, similar to Meloidae beetles; however, because Oedemeridae beetles are smaller, the vesiculobullous eruptions also are less pronounced.1
A third well-known family of blister beetles is the Staphylinidae family. Rove beetles (genus Paederus) differ from the Meloidae and Oedemeridae families in that they produce the vesicant pederin rather than cantharidin. Pederin causes a more violent eruption called dermatitis linearis, which often is associated with intense urticaria prior to blistering.1 Rove beetles are found in moist environments and tend to favor tropical and subtropical climates. They may emerge in large numbers after heavy rainfall.
Clinical Presentation
Blistering dermatitis—caused by exposure to cantharidin (families Meloidae and Oedemeridae) or pederin (genus Paederus)—begins as burning and tingling within minutes of exposure. Bullae later develop, often in a linear fashion, with subsequent bursting and crust formation.1,3 Secondary infection can occur.5 Exposure occurring on the elbows, knees, or mirroring skin folds results in lesions on opposing surfaces that come into contact, otherwise known as kissing lesions.1,3 Acantholysis of suprabasal keratinocytes can be seen on histologic sections of blisters (Figure 3)1,7 due to cantharidin activation of the serine/threonine protein phosphatases that cause detachment of tonofilaments from desmosomes.1,8 Washing of exposed sites with soap or alcohol can potentially prevent development of blistering dermatitis; however, lesions usually heal without complication when treated with topical antibiotics.3
If blister beetles are ingested, they can cause poisoning characterized by abdominal pain and hematuria.1,3,9,10 In fact, blister beetle ingestion by animals consuming baled hay is an important agricultural and economical implication. Several cases of fatal farm animal ingestion resulting in gastrointestinal erosion have been reported.4,6
Medicinal Properties
Medicinal use of cantharidin has been recorded as early as the 19th century and is believed to have been part of ancient medicinal practices in China, Spain, South Africa, and pre-Columbian America for its vesicant, abortifacient, and supposed aphrodisiac properties.2,4,8
Toxicity from internal ingestion has been well documented.1,3,9,10 The Mylabris and Epicauta genera of the family Meloidae, most commonly the Spanish fly, are used for modern extraction of cantharidin. Up to 5% of the dry weight of a blister beetle can be constituted by cantharidin.4
Although its use has diminished due to limited availability, cantharidin is still used for treatment of common warts, periungual warts, and molluscum contagiosum. It is a favorable choice for treating pediatric patients because of the tolerability and painlessness of application. Due to its acantholytic properties, warts generally slough with the cantharidin-induced blister.11
In recent years, cantharidin has been studied for its anticancer properties. It has been shown to weaken cancer cell antioxidant properties by interfering with glutathione-related enzymes, thus inducing oxidative damage. Additionally, cantharidin is associated with decreased mitochondrial cytochrome C and increased cytosolic cytochrome C, an important signal for apoptosis.12 Furthermore, cantharidin recently was shown to increase expression of proapoptotic proteins and decrease expression of antiapoptotic proteins causing cell death in nasopharyngeal carcinoma, making it a potential anticancer treatment.13
Conclusion
Blister beetles, long known for their production of vesicant agents, are both a cause of as well as a potential treatment of dermatologic disease. The blistering associated with exposure to a disturbed beetle generally is mild and heals without scarring if vital areas such as the eyelids are not affected.
Classification
Blister beetles are both a scourge and the source of medical cantharidin (Figure 1). The term blister beetle refers to 3 families of the order Coleoptera: Meloidae, Oedemeridae, and Staphylinidae (Figure 2).
Meloidae is the most well-known family of blister beetles, with more than 200 species worldwide identified as a cause of blistering dermatitis.1 The most notorious is Lytta vesicatoria, also known as the Spanish fly. Although some blister beetles are inconspicuous in appearance, most are brightly colored and easily spotted, making them attractive to small children.2 They may be attracted to fluorescent lighting and commonly enter through open windows.3 Blister beetles do not bite or sting; rather, they release cantharidin via hemolymph, an oily yellow substance that is copiously expressed from the leg joints when disturbed by rubbing or pressing.1,3-5 Blistering also is associated with exposure to the contents of crushed beetles, which is the source of pharmacologic cantharidin.2,4,6
Oedemeridae are the smallest and least known beetles within the Coleoptera family. They often are called false blister beetles, which is a misnomer. Oedemeridae beetles also produce cantharidin, similar to Meloidae beetles; however, because Oedemeridae beetles are smaller, the vesiculobullous eruptions also are less pronounced.1
A third well-known family of blister beetles is the Staphylinidae family. Rove beetles (genus Paederus) differ from the Meloidae and Oedemeridae families in that they produce the vesicant pederin rather than cantharidin. Pederin causes a more violent eruption called dermatitis linearis, which often is associated with intense urticaria prior to blistering.1 Rove beetles are found in moist environments and tend to favor tropical and subtropical climates. They may emerge in large numbers after heavy rainfall.
Clinical Presentation
Blistering dermatitis—caused by exposure to cantharidin (families Meloidae and Oedemeridae) or pederin (genus Paederus)—begins as burning and tingling within minutes of exposure. Bullae later develop, often in a linear fashion, with subsequent bursting and crust formation.1,3 Secondary infection can occur.5 Exposure occurring on the elbows, knees, or mirroring skin folds results in lesions on opposing surfaces that come into contact, otherwise known as kissing lesions.1,3 Acantholysis of suprabasal keratinocytes can be seen on histologic sections of blisters (Figure 3)1,7 due to cantharidin activation of the serine/threonine protein phosphatases that cause detachment of tonofilaments from desmosomes.1,8 Washing of exposed sites with soap or alcohol can potentially prevent development of blistering dermatitis; however, lesions usually heal without complication when treated with topical antibiotics.3
If blister beetles are ingested, they can cause poisoning characterized by abdominal pain and hematuria.1,3,9,10 In fact, blister beetle ingestion by animals consuming baled hay is an important agricultural and economical implication. Several cases of fatal farm animal ingestion resulting in gastrointestinal erosion have been reported.4,6
Medicinal Properties
Medicinal use of cantharidin has been recorded as early as the 19th century and is believed to have been part of ancient medicinal practices in China, Spain, South Africa, and pre-Columbian America for its vesicant, abortifacient, and supposed aphrodisiac properties.2,4,8
Toxicity from internal ingestion has been well documented.1,3,9,10 The Mylabris and Epicauta genera of the family Meloidae, most commonly the Spanish fly, are used for modern extraction of cantharidin. Up to 5% of the dry weight of a blister beetle can be constituted by cantharidin.4
Although its use has diminished due to limited availability, cantharidin is still used for treatment of common warts, periungual warts, and molluscum contagiosum. It is a favorable choice for treating pediatric patients because of the tolerability and painlessness of application. Due to its acantholytic properties, warts generally slough with the cantharidin-induced blister.11
In recent years, cantharidin has been studied for its anticancer properties. It has been shown to weaken cancer cell antioxidant properties by interfering with glutathione-related enzymes, thus inducing oxidative damage. Additionally, cantharidin is associated with decreased mitochondrial cytochrome C and increased cytosolic cytochrome C, an important signal for apoptosis.12 Furthermore, cantharidin recently was shown to increase expression of proapoptotic proteins and decrease expression of antiapoptotic proteins causing cell death in nasopharyngeal carcinoma, making it a potential anticancer treatment.13
Conclusion
Blister beetles, long known for their production of vesicant agents, are both a cause of as well as a potential treatment of dermatologic disease. The blistering associated with exposure to a disturbed beetle generally is mild and heals without scarring if vital areas such as the eyelids are not affected.
- Nicholls DS, Christmas TI, Greig DE. Oedemerid blister beetle dermatosis: a review. J Am Acad Dermatol. 1990;22:815-819.
- Percino-Daniel N, Buckley D, García-París M. Pharmacological properties of blister beetles (Coleoptera: Meloidae) promoted their integration into the cultural heritage of native rural Spain as inferred by vernacular names diversity, traditions, and mitochondrial DNA. J Ethnopharmacol. 2013;147:570-583.
- James WD, Berger TG, Elston DM. Parasitic infestations, stings, and bites. In: James WD, Berger TG, Elston DM, eds. Andrew’s Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016:418-450.
- Selander RB, Fasulo TR. Featured creatures: blister beetles. University of Florida/IFAS Entomology and Nematology website. http://entnemdept.ufl.edu/creatures/urban/medical/blister_beetles.htm. Published October 2000. Revised September 2010. Accessed November 12, 2019.
- Wijerathne BTB. Blister mystery. Wilderness Environ Med. 2017;28:271-272.
- Penrith ML, Naude TW. Mortality in chickens associated with blister beetle consumption. J S Afr Vet Assoc. 1996;67:97-99.
- Yell JA, Burge SM, Dean D. Cantharidin-induced acantholysis: adhesion molecules, proteases, and related proteins. Br J Dermatol. 1994;130:148-157.
- Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine-threonine protein phosphatases types 1 and 2a. FEBS Letters. 1993;330:283-286.
- Al-Binali AM, Shabana M, Al-Fifi S, et al. Cantharidin poisoning due to blister beetle ingestion in children: two case reports and a review of clinical presentations. Sultan Qaboos Univ Med J. 2010;10:258-261.
- Tagwireyi D, Ball DE, Loga PJ, et al. Cantharidin poisoning due to “blister beetle” ingestion. Toxicon. 2000;38:1865-1869.
- Al-Dawsari NA, Masterpol KS. Cantharidin in dermatology. Skinmed. 2016;14:111-114.
- Verma AK, Prasad SB. Changes in glutathione, oxidative stress and mitochondrial membrane potential in apoptosis involving the anticancer activity of cantharidin isolated from redheaded blister beetles, epicauta hirticornis. Anticancer Agents Med Chem. 2013;13:1096-1114.
- Chen AW, Tseng YS, Lin CC, et al. Norcantharidin induce apoptosis in human nasopharyngeal carcinoma through caspase and mitochondrial pathway. Environ Toxicol. 2018;33:343-350.
- Nicholls DS, Christmas TI, Greig DE. Oedemerid blister beetle dermatosis: a review. J Am Acad Dermatol. 1990;22:815-819.
- Percino-Daniel N, Buckley D, García-París M. Pharmacological properties of blister beetles (Coleoptera: Meloidae) promoted their integration into the cultural heritage of native rural Spain as inferred by vernacular names diversity, traditions, and mitochondrial DNA. J Ethnopharmacol. 2013;147:570-583.
- James WD, Berger TG, Elston DM. Parasitic infestations, stings, and bites. In: James WD, Berger TG, Elston DM, eds. Andrew’s Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2016:418-450.
- Selander RB, Fasulo TR. Featured creatures: blister beetles. University of Florida/IFAS Entomology and Nematology website. http://entnemdept.ufl.edu/creatures/urban/medical/blister_beetles.htm. Published October 2000. Revised September 2010. Accessed November 12, 2019.
- Wijerathne BTB. Blister mystery. Wilderness Environ Med. 2017;28:271-272.
- Penrith ML, Naude TW. Mortality in chickens associated with blister beetle consumption. J S Afr Vet Assoc. 1996;67:97-99.
- Yell JA, Burge SM, Dean D. Cantharidin-induced acantholysis: adhesion molecules, proteases, and related proteins. Br J Dermatol. 1994;130:148-157.
- Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine-threonine protein phosphatases types 1 and 2a. FEBS Letters. 1993;330:283-286.
- Al-Binali AM, Shabana M, Al-Fifi S, et al. Cantharidin poisoning due to blister beetle ingestion in children: two case reports and a review of clinical presentations. Sultan Qaboos Univ Med J. 2010;10:258-261.
- Tagwireyi D, Ball DE, Loga PJ, et al. Cantharidin poisoning due to “blister beetle” ingestion. Toxicon. 2000;38:1865-1869.
- Al-Dawsari NA, Masterpol KS. Cantharidin in dermatology. Skinmed. 2016;14:111-114.
- Verma AK, Prasad SB. Changes in glutathione, oxidative stress and mitochondrial membrane potential in apoptosis involving the anticancer activity of cantharidin isolated from redheaded blister beetles, epicauta hirticornis. Anticancer Agents Med Chem. 2013;13:1096-1114.
- Chen AW, Tseng YS, Lin CC, et al. Norcantharidin induce apoptosis in human nasopharyngeal carcinoma through caspase and mitochondrial pathway. Environ Toxicol. 2018;33:343-350.
Practice Points
- Exposure to cantharidin presents initially with burning and tingling before progressing to bullae formation, sometimes in a linear fashion. Rupture of bullae and crust formation can occur.
- Washing of the exposed skin with soap and water may prevent development of blistering dermatitis.
- Clinical use of cantharidin is favorable in treating pediatric patients with common warts and molluscum contagiosum due to tolerability and painlessness of application.

Solitary Papule on the Nose
The Diagnosis: Sclerosing Perineurioma
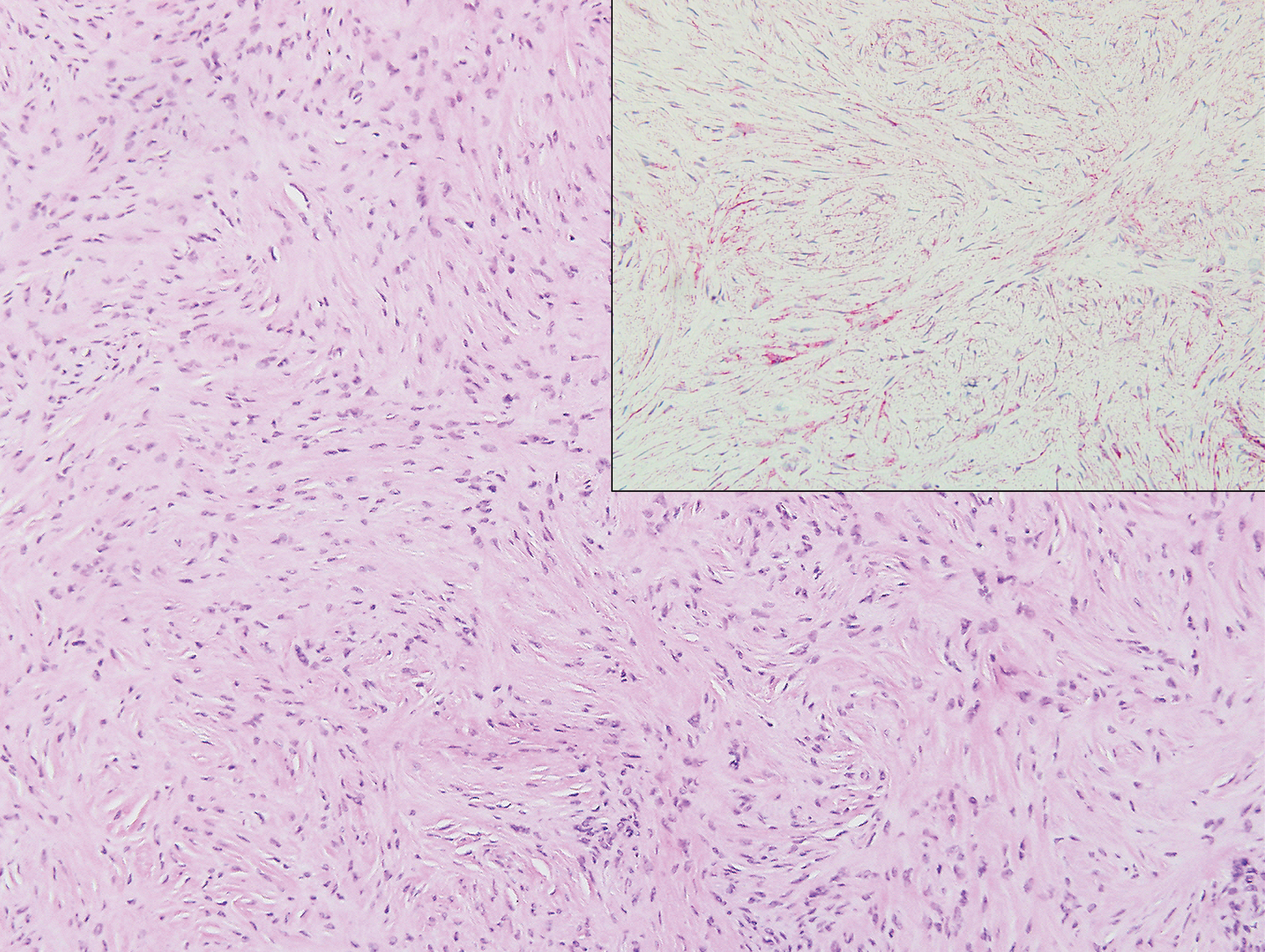
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
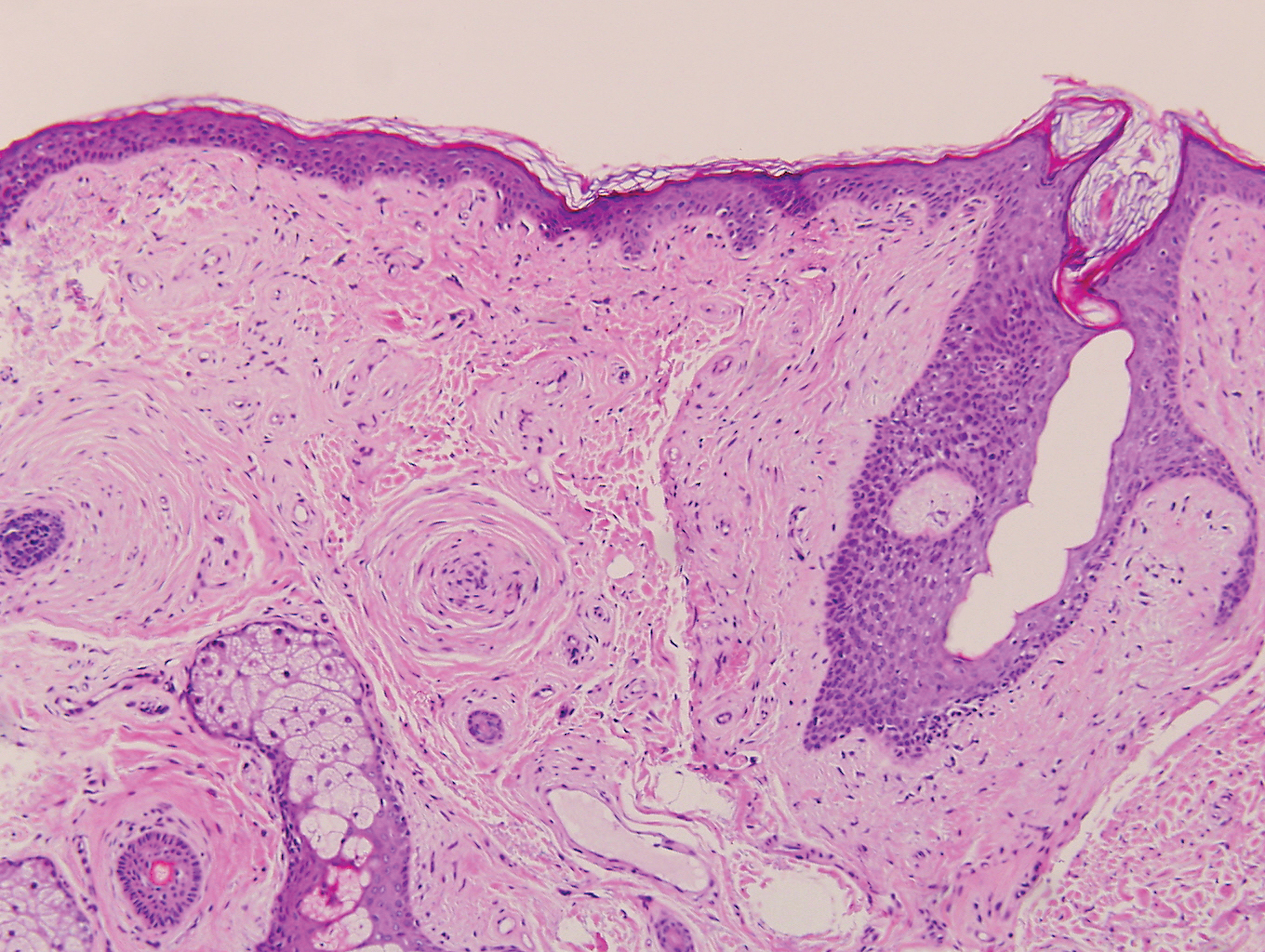
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
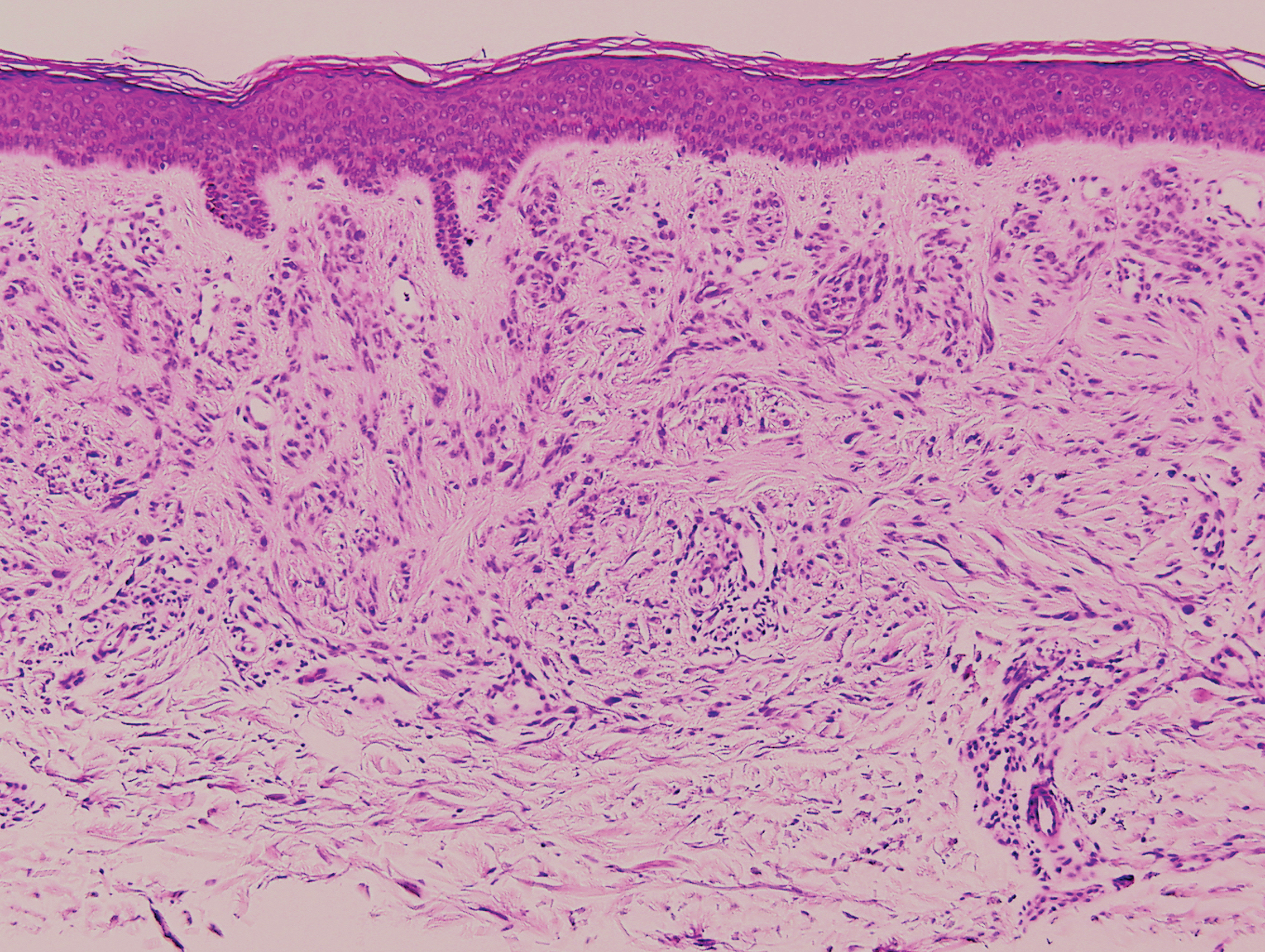
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
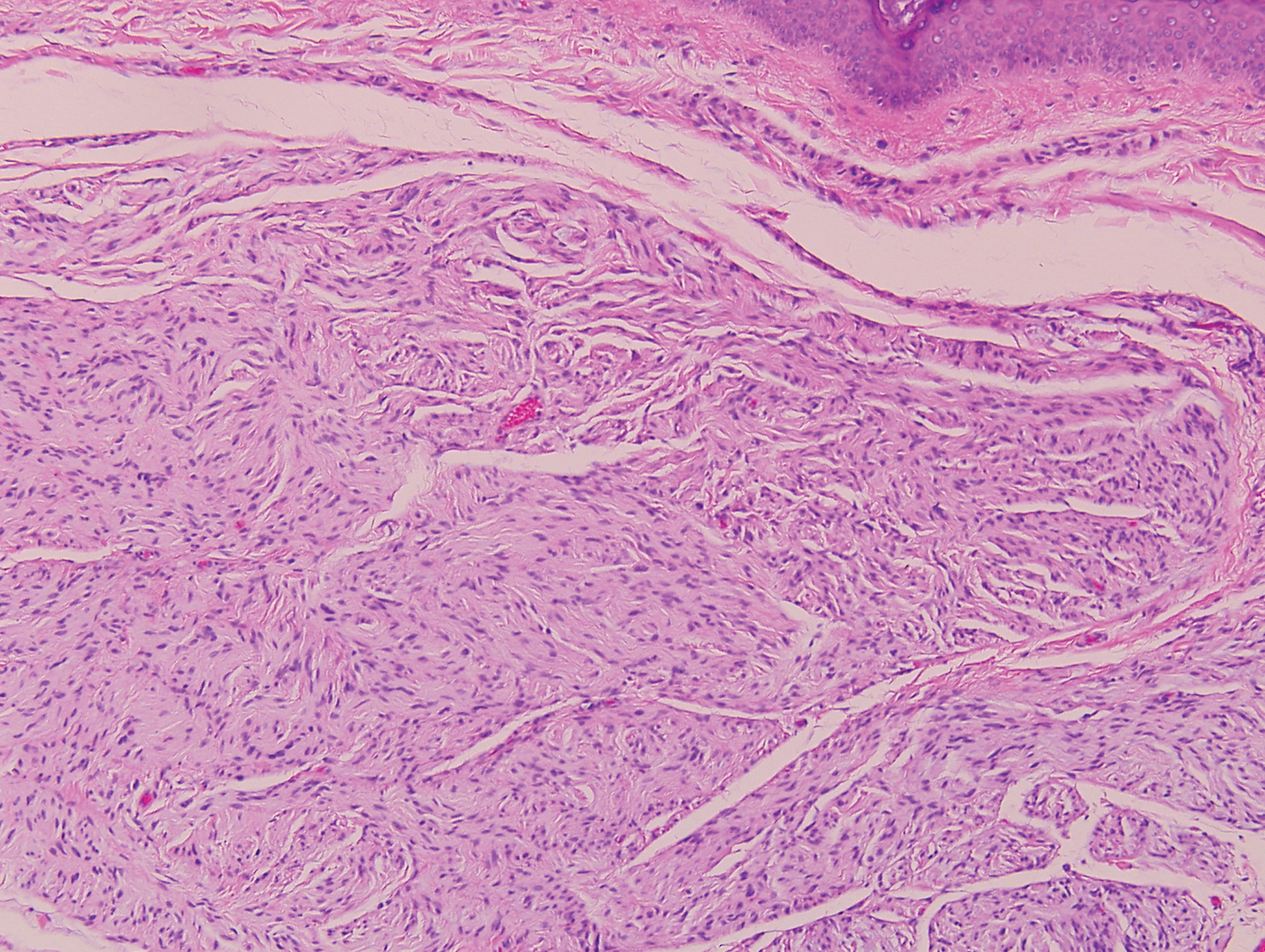
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
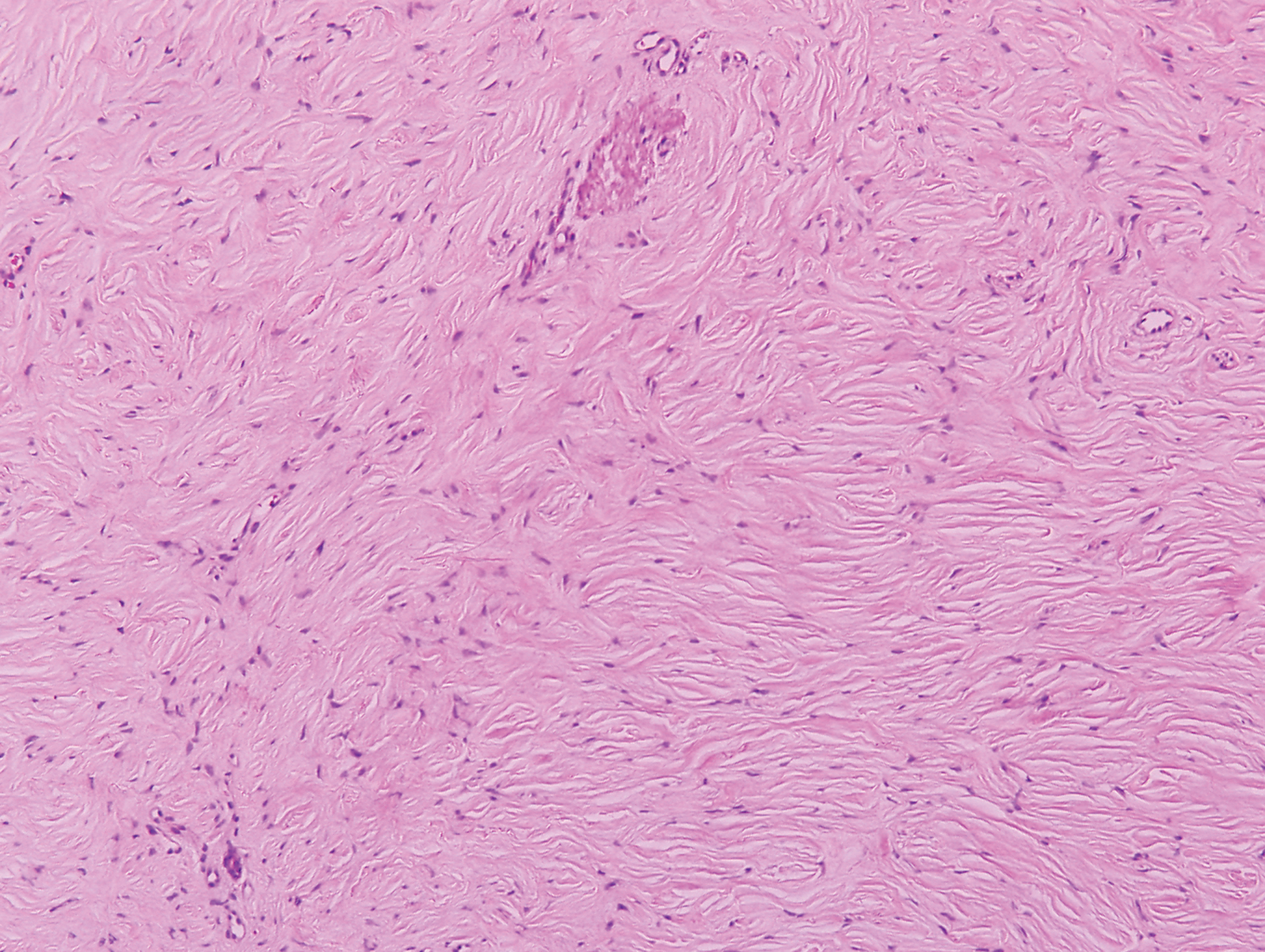
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
A 25-year-old man presented with a flesh-colored papule on the left side of the nose of 2 years' duration.
Systemic Epstein-Barr Virus–Positive T-cell Lymphoma of Childhood
Case Report
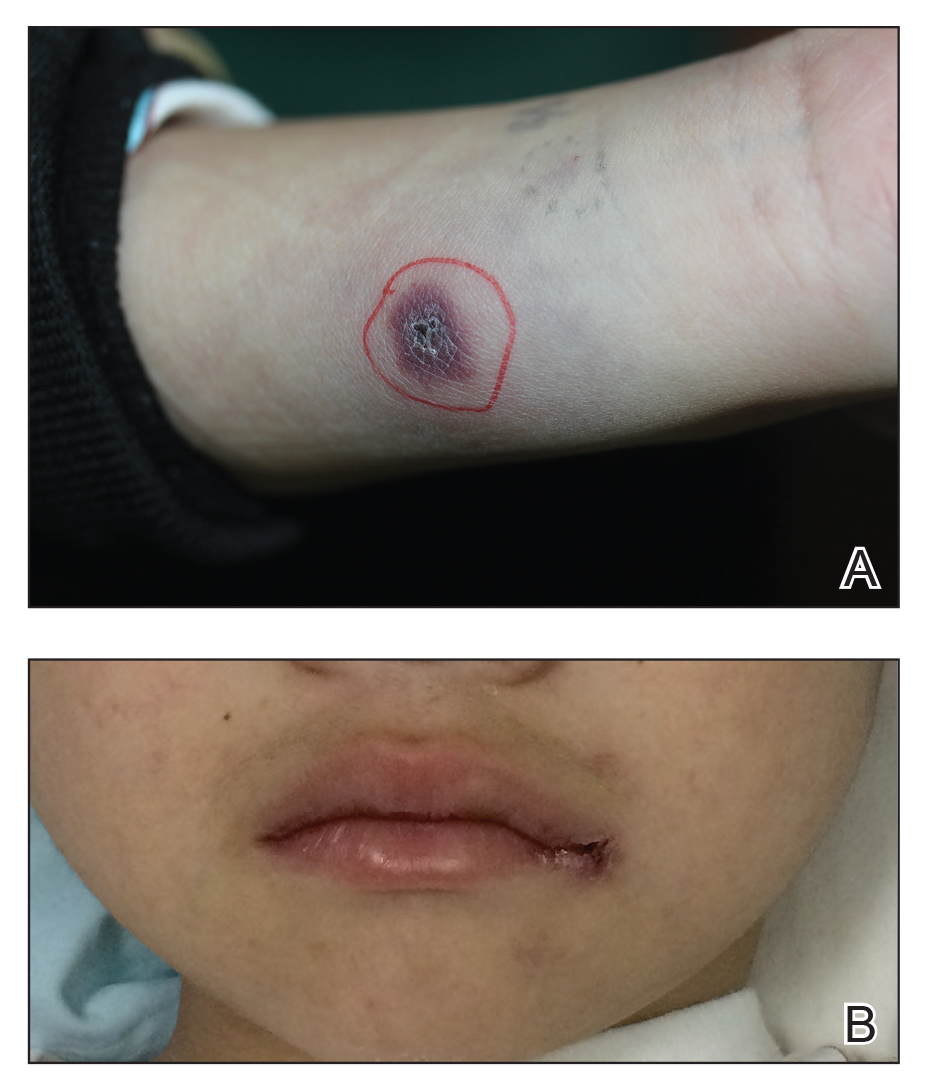
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
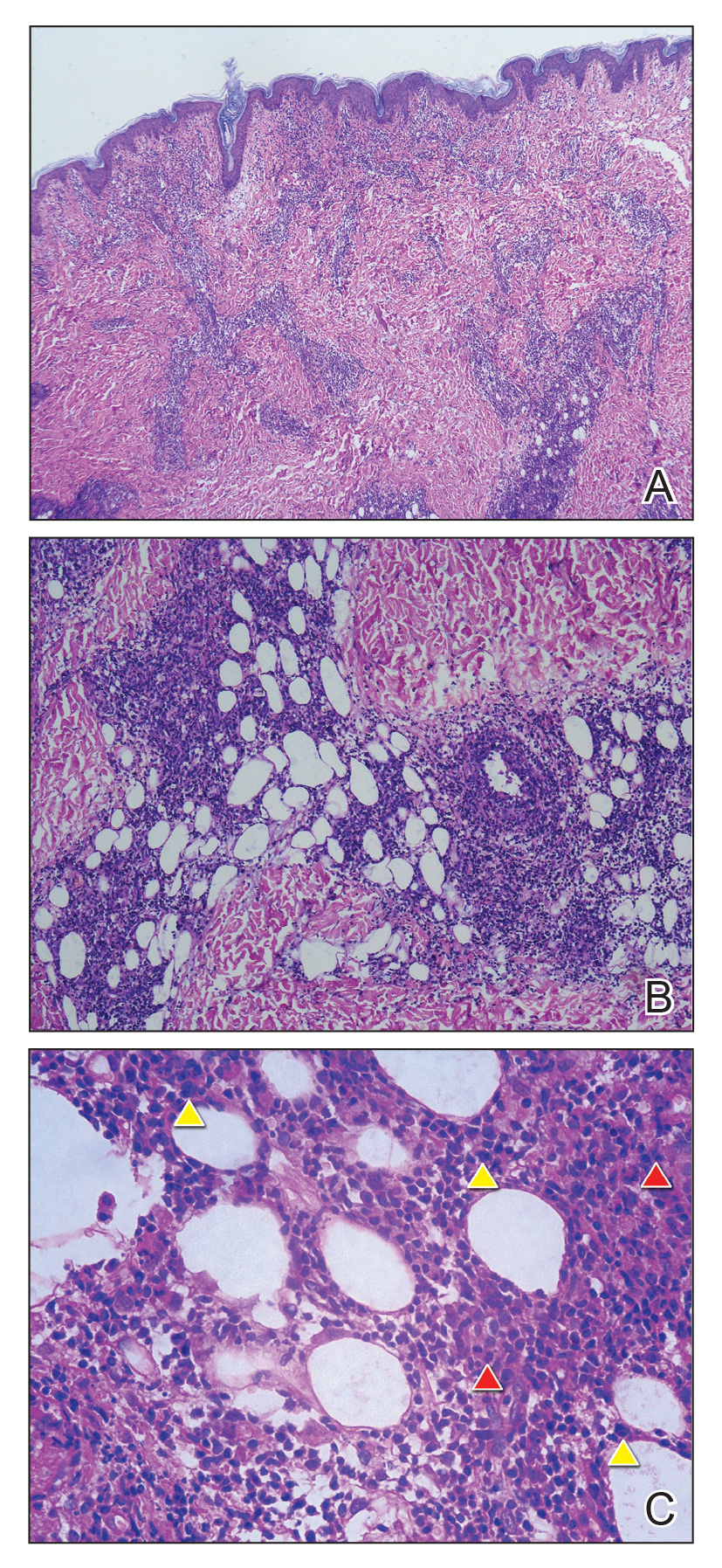
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
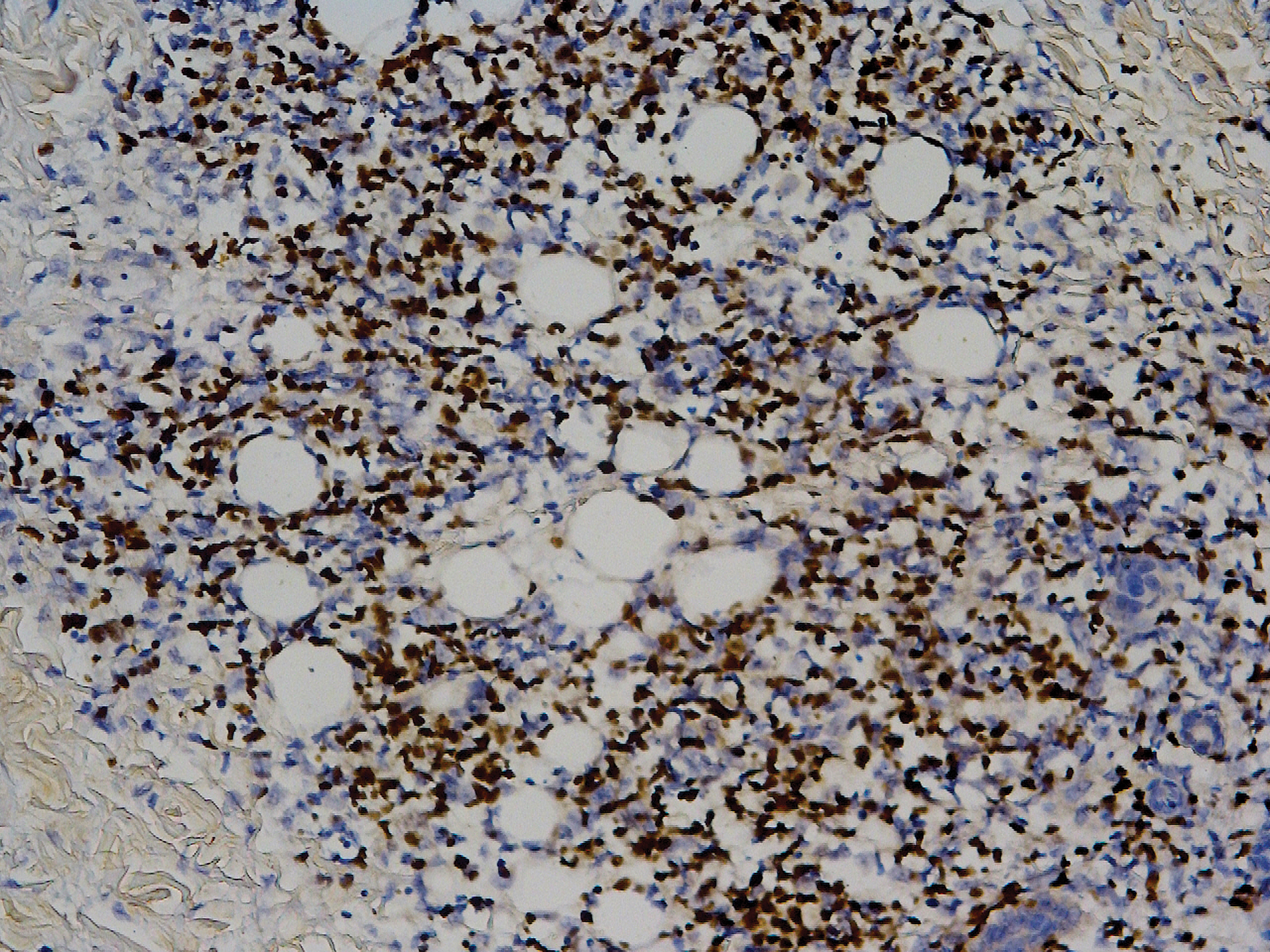
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Practice Points
- Systemic Epstein-Barr virus (EBV)–positive T-cell lymphoma of childhood is a fulminant illness with a predilection for Asians and indigenous populations from Mexico and Central and South America. In most patients, the disease has an aggressive clinical course with high mortality.
- The disease often is complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. When these severe complications are absent, the prognosis might be better.
- In situ hybridization for EBV-encoded RNA and for T-cell receptor gene rearrangements is an important tool to establish the diagnosis as well as for treatment options and predicting the prognosis.
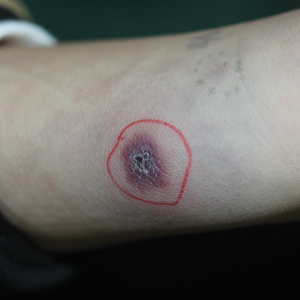
What’s Eating You? The South African Fattail Scorpion Revisited
Identification
The South African fattail scorpion (Parabuthus transvaalicus)(Figure) is one of the most poisonous scorpions in southern Africa.1 A member of the Buthidae scorpion family, it can grow as long as 15 cm and is dark brown-black with lighter red-brown pincers. Similar to other fattail scorpions, it has slender pincers (pedipalps) and a thick square tail (the telson). Parabuthus transvaalicus inhabits hot dry deserts, scrublands, and semiarid regions.1,2 It also is popular in exotic pet collections, the most common source of stings in the United States.

Stings and Envenomation
Scorpions with thicker tails generally have more potent venom than those with slender tails and thick pincers. Venom is injected by a stinger at the tip of the telson1; P transvaalicus also can spray venom as far as 3 m.1,2 Venom is not known to cause toxicity through skin contact but could represent a hazard if sprayed in the eye.
Scorpion toxins are a group of complex neurotoxins that act on sodium channels, either retarding inactivation (α toxin) or enhancing activation (β toxin), causing massive depolarization of excitable cells.1,3 The toxin causes neurons to fire repetitively.4 Neurotransmitters—noradrenaline, adrenaline, and acetylcholine—cause the observed sympathetic, parasympathetic, and skeletal muscle effects.1
Incidence
Worldwide, more than 1.2 million individuals are stung by a scorpion annually, causing more than 3250 deaths a year.5 Adults are stung more often, but children experience more severe envenomation, are more likely to develop severe illness requiring intensive supportive care, and have a higher mortality.4
As many as one-third of patients stung by a Parabuthus scorpion develop neuromuscular toxicity, which can be life-threatening.6 In a study of 277 envenomations by P transvaalicus, 10% of patients developed severe symptoms and 5 died. Children younger than 10 years and adults older than 50 years are at greatest risk for
Clinical Presentation
The clinical presentation of scorpion envenomation varies with the species involved, the amount of venom injected, and the victim’s weight and baseline health.1 Scorpion envenomation is divided into 4 grades based on the severity of a sting:
• Grade I: pain and paresthesia at the envenomation site; usually, no local inflammation
• Grade II: local symptoms as well as more remote pain and paresthesia; pain can radiate up the affected limb
• Grade III: cranial nerve or somatic skeletal neuromuscular dysfunction; either presentation can have associated autonomic dysfunction
• Grade IV: both cranial nerve and somatic skeletal neuromuscular dysfunction, with associated auto-nomic dysfunction
The initial symptom of a scorpion sting is intense burning pain. The sting site might be unimpressive, with only a mild local reaction. Symptoms usually progress to maximum severity within 5 hours.1 Muscle pain, cramps, and weakness are prominent. The patient might have difficulty walking and swallowing, with increased salivation and drooling, and visual disturbance with abnormal eye movements. Pulse, blood pressure, and temperature often are elevated. The patient might be hyperreflexic with clonus.1,6
Symptoms of increased sympathetic activity are hypertension, tachycardia, cardiac dysrhythmia, perspiration, hyperglycemia, and restlessness.1,2 Parasympathetic effects are increased salivation, hypotension, bradycardia, and gastric distension. Skeletal muscle effects include tremors and involuntary muscle movement, which can be severe. Cranial nerve dysfunction may manifest as dysphagia, drooling, abnormal eye movements, blurred vision, slurred speech, and tongue fasciculations. Subsequent development of muscle weakness, bulbar paralysis, and difficulty breathing may be caused by depletion of neurotransmitters after prolonged excessive neuronal activity.1
Distinctive Signs in Younger Patients
A child who is stung by a scorpion might have symptoms similar to those seen in an adult victim but can also experience an extreme form of restlessness that indicates severe envenomation characterized by inability to lay still, violent muscle twitching, and uncontrollable flailing of extremities. The child might have facial grimacing, with lip-smacking and chewing motions. In addition, bulbar paralysis and respiratory distress are more likely in children who have been stung than in adults.1,2
Management
Treatment of a P transvaalicus sting is directed at “scorpionism,” envenomation that is associated with systemic symptoms that can be life-threatening. Treatment comprises support of vital functions, symptomatic measures, and injection of antivenin.8
Support of Vital Functions
In adults, systemic symptoms can be delayed as long as 8 hours after the sting. However, most severe cases usually are evident within 60 minutes; infants can reach grade IV as quickly as 15 to 30 minutes.9,10 Loss of pharyngeal reflexes and development of respiratory distress are ominous warning signs requiring immediate respiratory support. Respiratory failure is the most common cause of death.1 An asymptomatic child should be admitted to a hospital for observation for a minimum of 12 hours if the species of scorpion was not identified.2
Pain Relief
Most patients cannot tolerate an ice pack because of severe hyperesthesia. Infiltration of the local sting site with an anesthetic generally is safe and can provide some local pain relief. Intravenous fentanyl has been used in closely monitored patients because the drug is not associated with histamine release. Medications that cause release of histamine, such as morphine, can exacerbate or confuse the clinical picture.
Antivenin
Scorpion antivenin contains purified IgG fragments; allergic reactions are now rare. The sooner antivenin is administered, the greater the benefit. When administered early, it can prevent many of the most serious complications.7 In a randomized, double-blind study of critically ill children with clinically significant signs of scorpion envenomation, intravenous administration of scorpion-specific fragment antigen-binding 2 (F[(ab’]2) antivenin resulted in resolution of clinical symptoms within 4 hours.11
When managing grade III or IV scorpion envenomation, all patients should be admitted to a medical facility equipped to provide intensive supportive care; consider consultation with a regional poison control center. The World Health Organization maintains an international poison control center (at https://www.who.int/ipcs/poisons/centre/en/) with regional telephone numbers; alternatively, in the United States, call the nationwide telephone number of the Poison Control Center (800-222-1222).
The World Health Organization has identified declining production of antivenin as a crisis.12
Resolution
Symptoms of envenomation typically resolve 9 to 30 hours after a sting in a patient with grade III or IV envenomation not treated with antivenin.4 However, pain and paresthesia occasionally last as long as 2 weeks. In rare cases, more long-term sequelae of burning paresthesia persist for months.4
Conclusion
It is important for dermatologists to be aware of the potential for life-threatening envenomation by certain scorpion species native to southern Africa. In the United States, stings of these species most often are seen in patients with a pet collection, but late sequelae also can be seen in travelers returning from an endemic region. The site of a sting often appears unimpressive initially, but severe hyperesthesia is common. Patients with cardiac, neurologic, or respiratory symptoms require intensive supportive care. Proper care can be lifesaving.
- Müller GJ, Modler H, Wium CA, et al. Scorpion sting in southern Africa: diagnosis and management. Continuing Medical Education. 2012;30:356-361.
- Müller GJ. Scorpionism in South Africa. a report of 42 serious scorpion envenomations. S Afr Med J. 1993;83:405-411.
- Quintero-Hernández V, Jiménez-Vargas JM, Gurrola GB, et al. Scorpion venom components that affect ion-channels function. Toxicon. 2013;76:328-342.
- LoVecchio F, McBride C. Scorpion envenomations in young children in central Arizona. J Toxicol Clin Toxicol. 2003;41:937-940.
- Chippaux JP, Goyffon M. Epidemiology of scorpionism: a global appraisal. Acta Trop. 2008;107:71-79.
- Bergman NJ. Clinical description of Parabuthus transvaalicus scorpionism in Zimbabwe. Toxicon. 1997;35:759-771.
- Chippaux JP. Emerging options for the management of scorpion stings. Drug Des Devel Ther. 2012;6:165-173.
- Santos MS, Silva CG, Neto BS, et al. Clinical and epidemiological aspects of scorpionism in the world: a systematic review. Wilderness Environ Med. 2016;27:504-518.
- Amaral CF, Rezende NA. Both cardiogenic and non-cardiogenic factors are involved in the pathogenesis of pulmonary oedema after scorpion envenoming. Toxicon. 1997;35:997-998.
- Bergman NJ. Scorpion sting in Zimbabwe. S Afr Med J. 1997;87:163-167.
- Boyer LV, Theodorou AA, Berg RA, et al; Arizona Envenomation Investigators. antivenom for critically ill children with neurotoxicity from scorpion stings. N Engl J Med. 2009;360:2090-2098.
- Theakston RD, Warrell DA, Griffiths E. Report of a WHO workshop on the standardization and control of antivenoms. Toxicon. 2003;41:541-557.
Identification
The South African fattail scorpion (Parabuthus transvaalicus)(Figure) is one of the most poisonous scorpions in southern Africa.1 A member of the Buthidae scorpion family, it can grow as long as 15 cm and is dark brown-black with lighter red-brown pincers. Similar to other fattail scorpions, it has slender pincers (pedipalps) and a thick square tail (the telson). Parabuthus transvaalicus inhabits hot dry deserts, scrublands, and semiarid regions.1,2 It also is popular in exotic pet collections, the most common source of stings in the United States.
Stings and Envenomation
Scorpions with thicker tails generally have more potent venom than those with slender tails and thick pincers. Venom is injected by a stinger at the tip of the telson1; P transvaalicus also can spray venom as far as 3 m.1,2 Venom is not known to cause toxicity through skin contact but could represent a hazard if sprayed in the eye.
Scorpion toxins are a group of complex neurotoxins that act on sodium channels, either retarding inactivation (α toxin) or enhancing activation (β toxin), causing massive depolarization of excitable cells.1,3 The toxin causes neurons to fire repetitively.4 Neurotransmitters—noradrenaline, adrenaline, and acetylcholine—cause the observed sympathetic, parasympathetic, and skeletal muscle effects.1
Incidence
Worldwide, more than 1.2 million individuals are stung by a scorpion annually, causing more than 3250 deaths a year.5 Adults are stung more often, but children experience more severe envenomation, are more likely to develop severe illness requiring intensive supportive care, and have a higher mortality.4
As many as one-third of patients stung by a Parabuthus scorpion develop neuromuscular toxicity, which can be life-threatening.6 In a study of 277 envenomations by P transvaalicus, 10% of patients developed severe symptoms and 5 died. Children younger than 10 years and adults older than 50 years are at greatest risk for
Clinical Presentation
The clinical presentation of scorpion envenomation varies with the species involved, the amount of venom injected, and the victim’s weight and baseline health.1 Scorpion envenomation is divided into 4 grades based on the severity of a sting:
• Grade I: pain and paresthesia at the envenomation site; usually, no local inflammation
• Grade II: local symptoms as well as more remote pain and paresthesia; pain can radiate up the affected limb
• Grade III: cranial nerve or somatic skeletal neuromuscular dysfunction; either presentation can have associated autonomic dysfunction
• Grade IV: both cranial nerve and somatic skeletal neuromuscular dysfunction, with associated auto-nomic dysfunction
The initial symptom of a scorpion sting is intense burning pain. The sting site might be unimpressive, with only a mild local reaction. Symptoms usually progress to maximum severity within 5 hours.1 Muscle pain, cramps, and weakness are prominent. The patient might have difficulty walking and swallowing, with increased salivation and drooling, and visual disturbance with abnormal eye movements. Pulse, blood pressure, and temperature often are elevated. The patient might be hyperreflexic with clonus.1,6
Symptoms of increased sympathetic activity are hypertension, tachycardia, cardiac dysrhythmia, perspiration, hyperglycemia, and restlessness.1,2 Parasympathetic effects are increased salivation, hypotension, bradycardia, and gastric distension. Skeletal muscle effects include tremors and involuntary muscle movement, which can be severe. Cranial nerve dysfunction may manifest as dysphagia, drooling, abnormal eye movements, blurred vision, slurred speech, and tongue fasciculations. Subsequent development of muscle weakness, bulbar paralysis, and difficulty breathing may be caused by depletion of neurotransmitters after prolonged excessive neuronal activity.1
Distinctive Signs in Younger Patients
A child who is stung by a scorpion might have symptoms similar to those seen in an adult victim but can also experience an extreme form of restlessness that indicates severe envenomation characterized by inability to lay still, violent muscle twitching, and uncontrollable flailing of extremities. The child might have facial grimacing, with lip-smacking and chewing motions. In addition, bulbar paralysis and respiratory distress are more likely in children who have been stung than in adults.1,2
Management
Treatment of a P transvaalicus sting is directed at “scorpionism,” envenomation that is associated with systemic symptoms that can be life-threatening. Treatment comprises support of vital functions, symptomatic measures, and injection of antivenin.8
Support of Vital Functions
In adults, systemic symptoms can be delayed as long as 8 hours after the sting. However, most severe cases usually are evident within 60 minutes; infants can reach grade IV as quickly as 15 to 30 minutes.9,10 Loss of pharyngeal reflexes and development of respiratory distress are ominous warning signs requiring immediate respiratory support. Respiratory failure is the most common cause of death.1 An asymptomatic child should be admitted to a hospital for observation for a minimum of 12 hours if the species of scorpion was not identified.2
Pain Relief
Most patients cannot tolerate an ice pack because of severe hyperesthesia. Infiltration of the local sting site with an anesthetic generally is safe and can provide some local pain relief. Intravenous fentanyl has been used in closely monitored patients because the drug is not associated with histamine release. Medications that cause release of histamine, such as morphine, can exacerbate or confuse the clinical picture.
Antivenin
Scorpion antivenin contains purified IgG fragments; allergic reactions are now rare. The sooner antivenin is administered, the greater the benefit. When administered early, it can prevent many of the most serious complications.7 In a randomized, double-blind study of critically ill children with clinically significant signs of scorpion envenomation, intravenous administration of scorpion-specific fragment antigen-binding 2 (F[(ab’]2) antivenin resulted in resolution of clinical symptoms within 4 hours.11
When managing grade III or IV scorpion envenomation, all patients should be admitted to a medical facility equipped to provide intensive supportive care; consider consultation with a regional poison control center. The World Health Organization maintains an international poison control center (at https://www.who.int/ipcs/poisons/centre/en/) with regional telephone numbers; alternatively, in the United States, call the nationwide telephone number of the Poison Control Center (800-222-1222).
The World Health Organization has identified declining production of antivenin as a crisis.12
Resolution
Symptoms of envenomation typically resolve 9 to 30 hours after a sting in a patient with grade III or IV envenomation not treated with antivenin.4 However, pain and paresthesia occasionally last as long as 2 weeks. In rare cases, more long-term sequelae of burning paresthesia persist for months.4
Conclusion
It is important for dermatologists to be aware of the potential for life-threatening envenomation by certain scorpion species native to southern Africa. In the United States, stings of these species most often are seen in patients with a pet collection, but late sequelae also can be seen in travelers returning from an endemic region. The site of a sting often appears unimpressive initially, but severe hyperesthesia is common. Patients with cardiac, neurologic, or respiratory symptoms require intensive supportive care. Proper care can be lifesaving.
Identification
The South African fattail scorpion (Parabuthus transvaalicus)(Figure) is one of the most poisonous scorpions in southern Africa.1 A member of the Buthidae scorpion family, it can grow as long as 15 cm and is dark brown-black with lighter red-brown pincers. Similar to other fattail scorpions, it has slender pincers (pedipalps) and a thick square tail (the telson). Parabuthus transvaalicus inhabits hot dry deserts, scrublands, and semiarid regions.1,2 It also is popular in exotic pet collections, the most common source of stings in the United States.
Stings and Envenomation
Scorpions with thicker tails generally have more potent venom than those with slender tails and thick pincers. Venom is injected by a stinger at the tip of the telson1; P transvaalicus also can spray venom as far as 3 m.1,2 Venom is not known to cause toxicity through skin contact but could represent a hazard if sprayed in the eye.
Scorpion toxins are a group of complex neurotoxins that act on sodium channels, either retarding inactivation (α toxin) or enhancing activation (β toxin), causing massive depolarization of excitable cells.1,3 The toxin causes neurons to fire repetitively.4 Neurotransmitters—noradrenaline, adrenaline, and acetylcholine—cause the observed sympathetic, parasympathetic, and skeletal muscle effects.1
Incidence
Worldwide, more than 1.2 million individuals are stung by a scorpion annually, causing more than 3250 deaths a year.5 Adults are stung more often, but children experience more severe envenomation, are more likely to develop severe illness requiring intensive supportive care, and have a higher mortality.4
As many as one-third of patients stung by a Parabuthus scorpion develop neuromuscular toxicity, which can be life-threatening.6 In a study of 277 envenomations by P transvaalicus, 10% of patients developed severe symptoms and 5 died. Children younger than 10 years and adults older than 50 years are at greatest risk for
Clinical Presentation
The clinical presentation of scorpion envenomation varies with the species involved, the amount of venom injected, and the victim’s weight and baseline health.1 Scorpion envenomation is divided into 4 grades based on the severity of a sting:
• Grade I: pain and paresthesia at the envenomation site; usually, no local inflammation
• Grade II: local symptoms as well as more remote pain and paresthesia; pain can radiate up the affected limb
• Grade III: cranial nerve or somatic skeletal neuromuscular dysfunction; either presentation can have associated autonomic dysfunction
• Grade IV: both cranial nerve and somatic skeletal neuromuscular dysfunction, with associated auto-nomic dysfunction
The initial symptom of a scorpion sting is intense burning pain. The sting site might be unimpressive, with only a mild local reaction. Symptoms usually progress to maximum severity within 5 hours.1 Muscle pain, cramps, and weakness are prominent. The patient might have difficulty walking and swallowing, with increased salivation and drooling, and visual disturbance with abnormal eye movements. Pulse, blood pressure, and temperature often are elevated. The patient might be hyperreflexic with clonus.1,6
Symptoms of increased sympathetic activity are hypertension, tachycardia, cardiac dysrhythmia, perspiration, hyperglycemia, and restlessness.1,2 Parasympathetic effects are increased salivation, hypotension, bradycardia, and gastric distension. Skeletal muscle effects include tremors and involuntary muscle movement, which can be severe. Cranial nerve dysfunction may manifest as dysphagia, drooling, abnormal eye movements, blurred vision, slurred speech, and tongue fasciculations. Subsequent development of muscle weakness, bulbar paralysis, and difficulty breathing may be caused by depletion of neurotransmitters after prolonged excessive neuronal activity.1
Distinctive Signs in Younger Patients
A child who is stung by a scorpion might have symptoms similar to those seen in an adult victim but can also experience an extreme form of restlessness that indicates severe envenomation characterized by inability to lay still, violent muscle twitching, and uncontrollable flailing of extremities. The child might have facial grimacing, with lip-smacking and chewing motions. In addition, bulbar paralysis and respiratory distress are more likely in children who have been stung than in adults.1,2
Management
Treatment of a P transvaalicus sting is directed at “scorpionism,” envenomation that is associated with systemic symptoms that can be life-threatening. Treatment comprises support of vital functions, symptomatic measures, and injection of antivenin.8
Support of Vital Functions
In adults, systemic symptoms can be delayed as long as 8 hours after the sting. However, most severe cases usually are evident within 60 minutes; infants can reach grade IV as quickly as 15 to 30 minutes.9,10 Loss of pharyngeal reflexes and development of respiratory distress are ominous warning signs requiring immediate respiratory support. Respiratory failure is the most common cause of death.1 An asymptomatic child should be admitted to a hospital for observation for a minimum of 12 hours if the species of scorpion was not identified.2
Pain Relief
Most patients cannot tolerate an ice pack because of severe hyperesthesia. Infiltration of the local sting site with an anesthetic generally is safe and can provide some local pain relief. Intravenous fentanyl has been used in closely monitored patients because the drug is not associated with histamine release. Medications that cause release of histamine, such as morphine, can exacerbate or confuse the clinical picture.
Antivenin
Scorpion antivenin contains purified IgG fragments; allergic reactions are now rare. The sooner antivenin is administered, the greater the benefit. When administered early, it can prevent many of the most serious complications.7 In a randomized, double-blind study of critically ill children with clinically significant signs of scorpion envenomation, intravenous administration of scorpion-specific fragment antigen-binding 2 (F[(ab’]2) antivenin resulted in resolution of clinical symptoms within 4 hours.11
When managing grade III or IV scorpion envenomation, all patients should be admitted to a medical facility equipped to provide intensive supportive care; consider consultation with a regional poison control center. The World Health Organization maintains an international poison control center (at https://www.who.int/ipcs/poisons/centre/en/) with regional telephone numbers; alternatively, in the United States, call the nationwide telephone number of the Poison Control Center (800-222-1222).
The World Health Organization has identified declining production of antivenin as a crisis.12
Resolution
Symptoms of envenomation typically resolve 9 to 30 hours after a sting in a patient with grade III or IV envenomation not treated with antivenin.4 However, pain and paresthesia occasionally last as long as 2 weeks. In rare cases, more long-term sequelae of burning paresthesia persist for months.4
Conclusion
It is important for dermatologists to be aware of the potential for life-threatening envenomation by certain scorpion species native to southern Africa. In the United States, stings of these species most often are seen in patients with a pet collection, but late sequelae also can be seen in travelers returning from an endemic region. The site of a sting often appears unimpressive initially, but severe hyperesthesia is common. Patients with cardiac, neurologic, or respiratory symptoms require intensive supportive care. Proper care can be lifesaving.
- Müller GJ, Modler H, Wium CA, et al. Scorpion sting in southern Africa: diagnosis and management. Continuing Medical Education. 2012;30:356-361.
- Müller GJ. Scorpionism in South Africa. a report of 42 serious scorpion envenomations. S Afr Med J. 1993;83:405-411.
- Quintero-Hernández V, Jiménez-Vargas JM, Gurrola GB, et al. Scorpion venom components that affect ion-channels function. Toxicon. 2013;76:328-342.
- LoVecchio F, McBride C. Scorpion envenomations in young children in central Arizona. J Toxicol Clin Toxicol. 2003;41:937-940.
- Chippaux JP, Goyffon M. Epidemiology of scorpionism: a global appraisal. Acta Trop. 2008;107:71-79.
- Bergman NJ. Clinical description of Parabuthus transvaalicus scorpionism in Zimbabwe. Toxicon. 1997;35:759-771.
- Chippaux JP. Emerging options for the management of scorpion stings. Drug Des Devel Ther. 2012;6:165-173.
- Santos MS, Silva CG, Neto BS, et al. Clinical and epidemiological aspects of scorpionism in the world: a systematic review. Wilderness Environ Med. 2016;27:504-518.
- Amaral CF, Rezende NA. Both cardiogenic and non-cardiogenic factors are involved in the pathogenesis of pulmonary oedema after scorpion envenoming. Toxicon. 1997;35:997-998.
- Bergman NJ. Scorpion sting in Zimbabwe. S Afr Med J. 1997;87:163-167.
- Boyer LV, Theodorou AA, Berg RA, et al; Arizona Envenomation Investigators. antivenom for critically ill children with neurotoxicity from scorpion stings. N Engl J Med. 2009;360:2090-2098.
- Theakston RD, Warrell DA, Griffiths E. Report of a WHO workshop on the standardization and control of antivenoms. Toxicon. 2003;41:541-557.
- Müller GJ, Modler H, Wium CA, et al. Scorpion sting in southern Africa: diagnosis and management. Continuing Medical Education. 2012;30:356-361.
- Müller GJ. Scorpionism in South Africa. a report of 42 serious scorpion envenomations. S Afr Med J. 1993;83:405-411.
- Quintero-Hernández V, Jiménez-Vargas JM, Gurrola GB, et al. Scorpion venom components that affect ion-channels function. Toxicon. 2013;76:328-342.
- LoVecchio F, McBride C. Scorpion envenomations in young children in central Arizona. J Toxicol Clin Toxicol. 2003;41:937-940.
- Chippaux JP, Goyffon M. Epidemiology of scorpionism: a global appraisal. Acta Trop. 2008;107:71-79.
- Bergman NJ. Clinical description of Parabuthus transvaalicus scorpionism in Zimbabwe. Toxicon. 1997;35:759-771.
- Chippaux JP. Emerging options for the management of scorpion stings. Drug Des Devel Ther. 2012;6:165-173.
- Santos MS, Silva CG, Neto BS, et al. Clinical and epidemiological aspects of scorpionism in the world: a systematic review. Wilderness Environ Med. 2016;27:504-518.
- Amaral CF, Rezende NA. Both cardiogenic and non-cardiogenic factors are involved in the pathogenesis of pulmonary oedema after scorpion envenoming. Toxicon. 1997;35:997-998.
- Bergman NJ. Scorpion sting in Zimbabwe. S Afr Med J. 1997;87:163-167.
- Boyer LV, Theodorou AA, Berg RA, et al; Arizona Envenomation Investigators. antivenom for critically ill children with neurotoxicity from scorpion stings. N Engl J Med. 2009;360:2090-2098.
- Theakston RD, Warrell DA, Griffiths E. Report of a WHO workshop on the standardization and control of antivenoms. Toxicon. 2003;41:541-557.
Practice Points
- Exotic and dangerous pets are becoming more popular. Scorpion stings cause potentially life-threatening neurotoxicity, with children particularly susceptible.
- Fattail scorpions are particularly dangerous and physicians should be aware that their stings may be encountered worldwide.
- Symptoms present 1 to 8 hours after envenomation, with severe cases showing hyperreflexia, clonus, difficulty swallowing, and respiratory distress. The sting site may be unimpressive.

Solitary Papule on the Leg
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
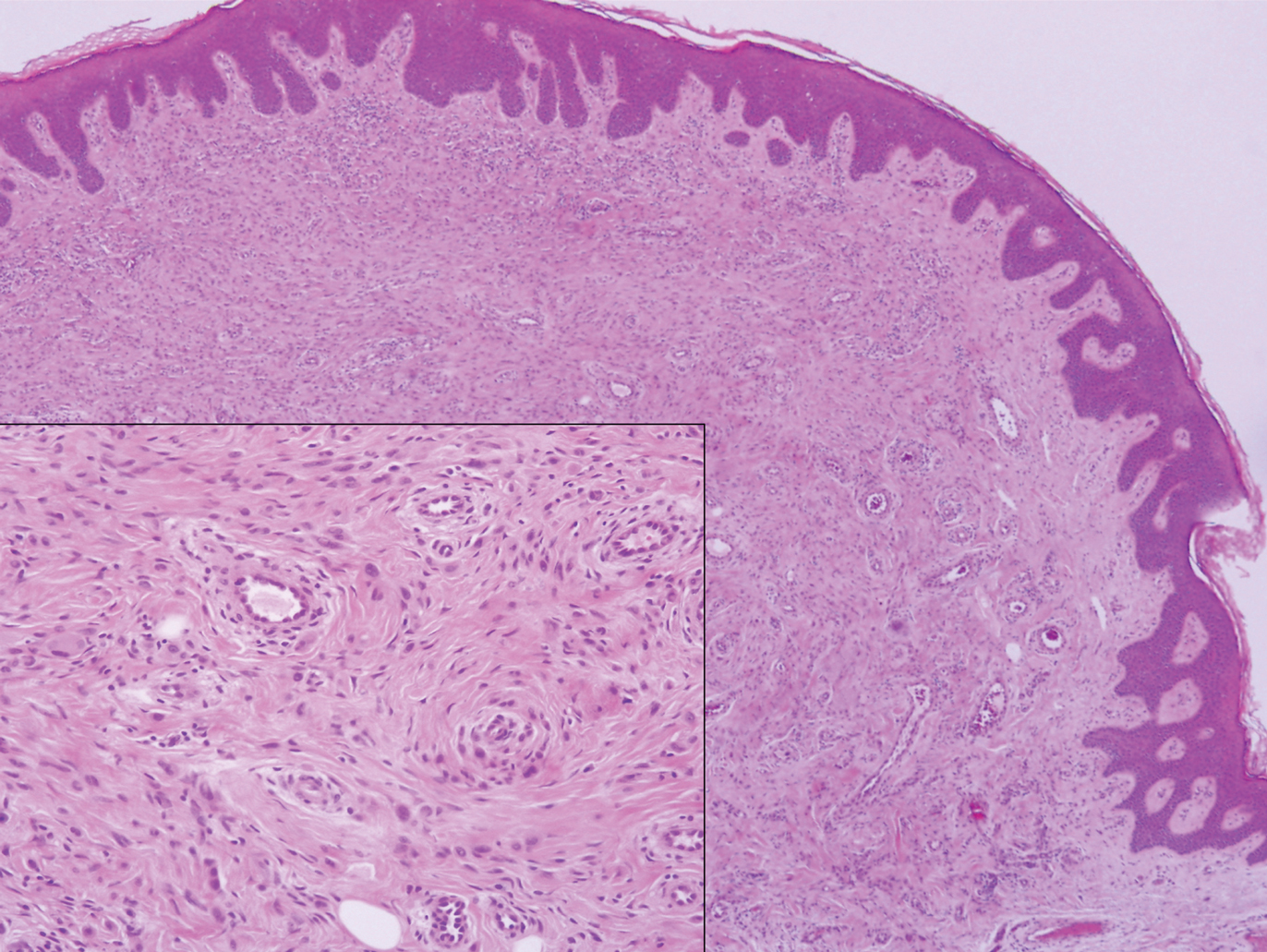
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18

Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22

Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
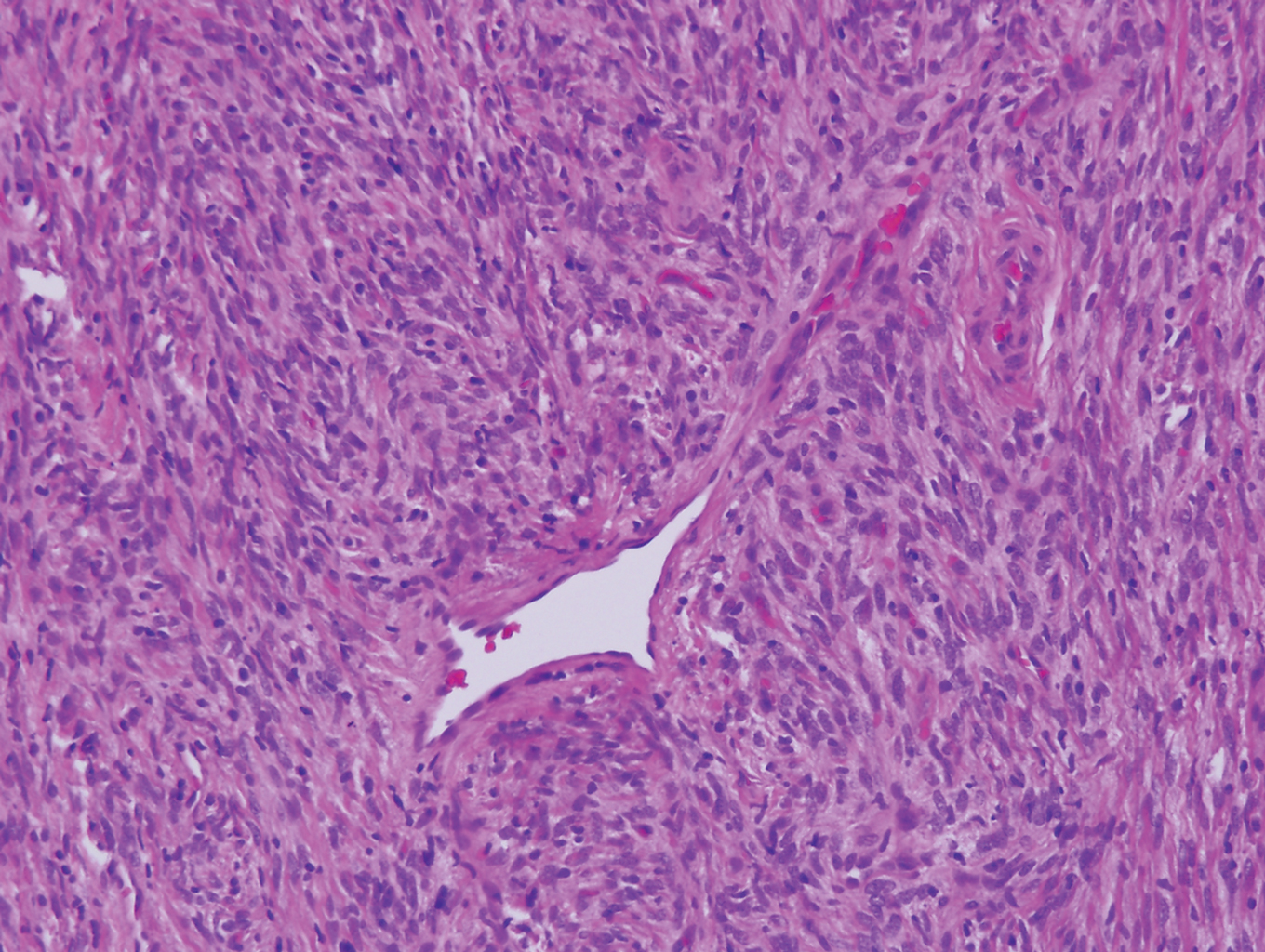
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
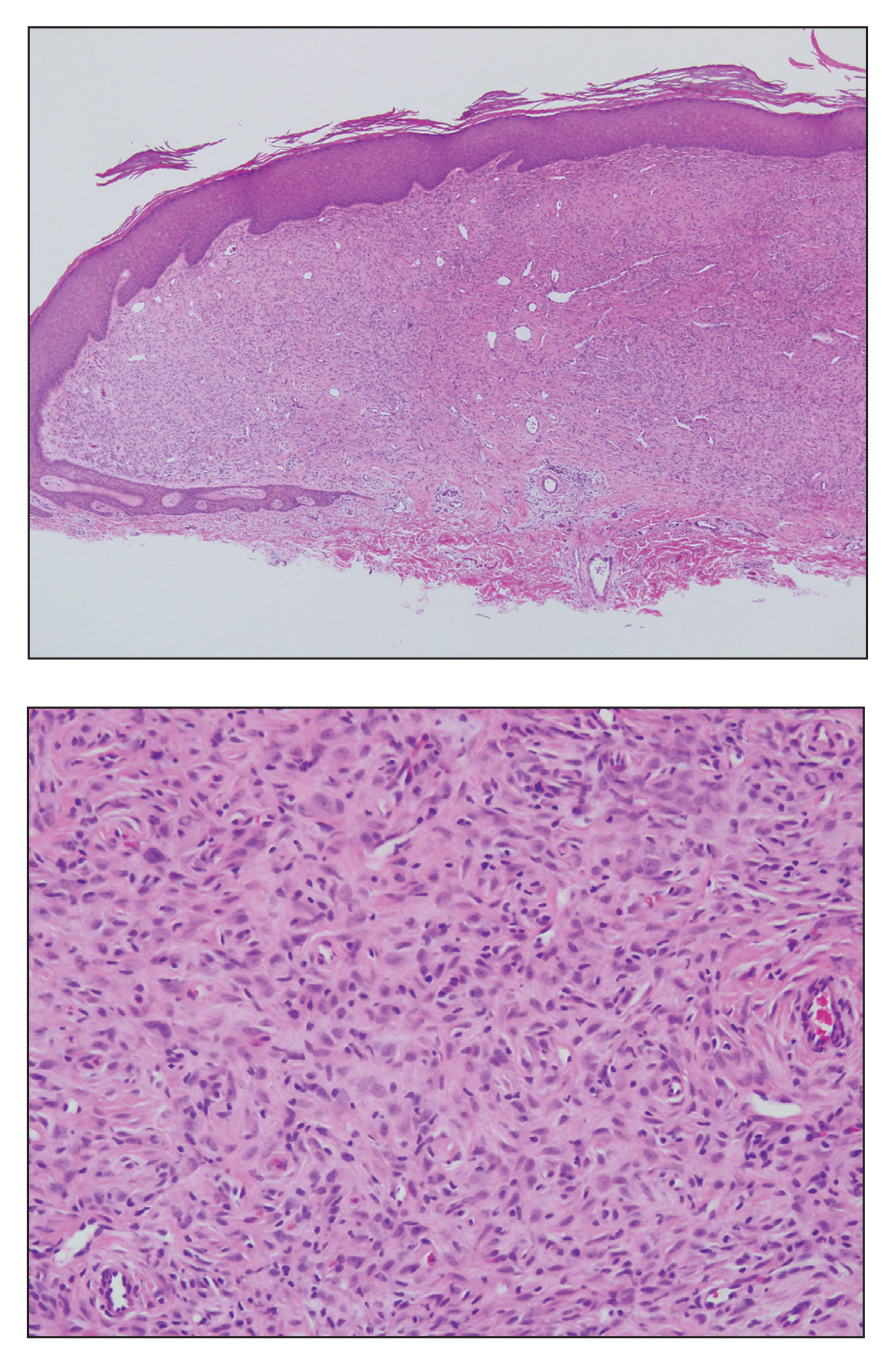
A 28-year-old man presented with a growing asymptomatic papule on the right leg.
What’s Eating You? Cat Flea (Ctenocephalides felis) Revisited
Fleas of the order Siphonaptera are insects that feed on the blood of a mammalian host. They have no wings but jump to near 150 times their body lengths to reach potential hosts.1 An epidemiologic survey performed in 2016 demonstrated that 96% of fleas in the United States are cat fleas (Ctenocephalides felis).2 The bites often present as pruritic, nonfollicular-based, excoriated papules; papular urticaria; or vesiculobullous lesions distributed across the lower legs. Antihistamines and topical steroids may be helpful for symptomatic relief, but flea eradication is key.


Identification
Ctenocephalides fleas, including the common cat flea and the dog flea, have a characteristic pronotal comb that resembles a mane of hair (Figure 1) and genal comb that resembles a mustache. Compared to the dog flea (Ctenocephalides canis), cat fleas have a flatter head and fewer hair-bearing notches on the dorsal hind tibia (the dog flea has 8 notches and the cat flea has 6 notches)(Figure 2).
Flea Prevention and Eradication
Effective management of flea bites requires avoidance of infested areas and eradication of fleas from the home and pets. Home treatment should be performed by a qualified specialist and a veterinarian should treat the pet, but the dermatologist must be knowledgeable about treatment options. Flea pupae can lie dormant between floorboards for extended periods of time and hatch rapidly when new tenants enter a house or apartment. Insecticidal dusts and spray formulations frequently are used to treat infested homes. It also is important to reduce flea egg numbers by vacuuming carpets and areas where pets sleep.3 Rodents often introduce fleas to households and pets, so eliminating them from the area may play an important role in flea control. Consulting with a veterinarian is important, as treatment directed at pets is critical to control flea populations. Oral agents, including fluralaner, afoxolaner, sarolaner, and spinosad, can reduce flea populations on animals by as much as 99.3% after 7 days.4,5 Fast-acting pulicidal agents, such as the combination of dinotefuran and fipronil, demonstrate curative activity as soon as 3 hours after treatment, which also may prevent reinfestation for as long as 6 weeks after treatment.6
Vector-Borne Disease
Fleas living on animals in close contact with humans, such as cats and dogs, can transmit zoonotic pathogens. Around 12,000 outpatients and 500 inpatients are diagnosed with cat scratch disease, a form of bartonellosis, annually. Ctenocephalides felis transmits Bartonella henselae from cat-to-cat and often cat-to-human through infected flea feces, causing a primary inoculation lesion and lymphadenitis. Of 3011 primary care providers surveyed from 2014 to 2015, 37.2% had treated at least 1 patient with cat scratch disease, yet knowledge gaps remain regarding the proper treatment and preventative measures for the disease.7 Current recommendations for the treatment of lymphadenitis caused by B henselae include a 5-day course of oral azithromycin.8 The preferred dosing regimen in adults is 500 mg on day 1 and 250 mg on days 2 through 5. Pediatric patients weighing less than 45.5 kg should receive 10 mg/kg on day 1 and 5 mg/kg on days 2 through 5.8 Additionally, less than one-third of the primary care providers surveyed from 2014 to 2015 said they would discuss the importance of pet flea control with immunocompromised patients who own cats, despite evidence implicating fleas in disease transmission.7 Pet-directed topical therapy with agents such as selamectin prescribed by a qualified veterinarian can prevent transmission of B henselae in cats exposed to fleas infected with the bacteria,9 which supports the importance of patient education and flea control, especially in pets owned by immunocompromised patients. Patients who are immunocompromised are at increased risk for persistent or disseminated bartonellosis, including endocarditis, in addition to cat scratch disease. Although arriving at a diagnosis may be difficult, one study found that bartonellosis in 13 renal transplant recipients was best diagnosed using both serology and polymerase chain reaction via DNA extraction of tissue specimens.10 These findings may enhance diagnostic yield for similar patients when bartonellosis is suspected.
Flea-borne typhus is endemic to Texas and Southern California.11,12 Evidence suggests that the pathogenic bacteria, Rickettsia typhi and Rickettsia felis, also commonly infect fleas in the Great Plains area.13 Opossums carry R felis, and the fleas transmit murine or endemic typhus. A retrospective case series in Texas identified 11 cases of fatal flea-borne typhus from 1985 to 2015.11 More than half of the patients reported contact with animals or fleas prior to the illness. Patients with typhus may present with fever, nausea, vomiting, rash (macular, maculopapular, papular, petechial, or morbilliform), respiratory or neurologic symptoms, thrombocytopenia, and elevated hepatic liver enzymes. Unfortunately, there often is a notable delay in initiation of treatment with the appropriate class of antibiotics—tetracyclines—and such delays can prove fatal.11 The current recommendation for nonpregnant adults is oral doxycycline 100 mg twice daily continued 48 hours after the patient becomes afebrile or for 7 days, whichever therapy duration is longer.14 Because of the consequences of delayed treatment, it is important for clinicians to consider a diagnosis of vector-borne illness in a febrile patient with other associated gastrointestinal, cutaneous, respiratory, or neurologic symptoms, especially if they have animal or flea exposures. Flea control and exposure awareness remains paramount in preventing and treating this illness.
Yersinia pestis causes the plague, an important re-emerging disease that causes infection through flea bites, inhalation, or ingestion.15 From 2000 to 2009, 56 cases and 7 deaths in the United States—New Mexico, Arizona, Colorado, California, and Texas—and 21,725 cases and 1612 deaths worldwide were attributed to Y pestis. Most patients present with the bubonic form of the disease, with fever and an enlarging painful femoral or inguinal lymph node due to leg flea bites.16 Other forms of disease, including septicemic and pneumonic plague, are less common but relevant, as one-third of cases in the United States present with septicemia.15,17,18 Although molecular diagnosis and immunohistochemistry play important roles, the diagnosis of Y pestis infection often is still accomplished with culture. A 2012 survey of 392 strains from 17 countries demonstrated that Y pestis remained susceptible to the antibiotics currently used to treat the disease, including doxycycline, streptomycin, gentamicin, tetracycline, trimethoprim-sulfamethoxazole, and ciprofloxacin.19
Human infection with Dipylidium caninum, a dog tapeworm, has been reported after suspected accidental ingestion of cat fleas carrying the parasite.20 Children, who may present with diarrhea or white worms in their feces, are more susceptible to the infection, perhaps due to accidental flea consumption while being licked by the pet.20,21
Conclusion
Cat fleas may act as a pruritic nuisance for pet owners and even deliver deadly pathogens to immunocompromised patients. Providers can minimize their impact by educating patients on flea prevention and eradication as well as astutely recognizing and treating flea-borne diseases.
- Cadiergues MC. A comparison of jump performances of the dog flea, Ctenocephalides canis (Curtis, 1826) and the cat flea, Ctenocephalides felis (Bouché, 1835). Vet Parasitol. 2000;92:239-241.
- Blagburn B, Butler J, Land T, et al. Who’s who and where: prevalence of Ctenocephalides felis and Ctenocephalides canis in shelter dogs and cats in the United States. Presented at: American Association of Veterinary Parasitologists 61st Annual Meeting; August 6-9, 2016; San Antonio, TX. P9.
- Bitam I, Dittmar K, Parola P, et al. Fleas and flea-borne diseases. Int J Infect Dis. 2010;14:E667-E676.
- Dryden MW, Canfield MS, Niedfeldt E, et al. Evaluation of sarolaner and spinosad oral treatments to eliminate fleas, reduce dermatologic lesions and minimize pruritus in naturally infested dogs in west Central Florida, USA. Parasit Vectors. 2017;10:389.
- Dryden MW, Canfield MS, Kalosy K, et al. Evaluation of fluralaner and afoxolaner treatments to control flea populations, reduce pruritus and minimize dermatologic lesions in naturally infested dogs in private residences in west Central Florida, USA. Parasit Vectors. 2016;9:365.
- Delcombel R, Karembe H, Nare B, et al. Synergy between dinotefuran and fipronil against the cat flea (Ctenocephalides felis): improved onset of action and residual speed of kill in adult cats. Parasit Vectors. 2017;10:341.
- Nelson CA, Moore AR, Perea AE, et al. Cat scratch disease: U.S. clinicians’ experience and knowledge. Zoonoses Public Health. 2018;65:67-73.
- Spach DH, Kaplan SL. Treatment of cat scratch disease. UpToDate. https://www.uptodate.com/contents/treatment-of-cat-scratch-disease?search=treatment%20of%20cat%20scratch&source=search_result&selectedTitle=1~59&usage_type=default&display_rank=1.Updated June 12, 2019. Accessed August 15, 2019.
- Bouhsira E, Franc M, Lienard E, et al. The efficacy of a selamectin (Stronghold®) spot on treatment in the prevention of Bartonella henselae transmission by Ctenocephalides felis in cats, using a new high-challenge model. Parasitol Res. 2015;114:1045-1050.
- Shamekhi Amiri F. Bartonellosis in chronic kidney disease: an unrecognized and unsuspected diagnosis. Ther Apher Dial. 2017;21:430-440.
- Pieracci EG, Evert N, Drexler NA, et al. Fatal flea-borne typhus in Texas: a retrospective case series, 1985-2015. American J Trop Med Hyg. 2017;96:1088-1093.
- Maina AN, Fogarty C, Krueger L, et al. Rickettsial infections among Ctenocephalides felis and host animals during a flea-borne rickettsioses outbreak in Orange County, California. PLoS One. 2016;11:e0160604.
- Noden BH, Davidson S, Smith JL, et al. First detection of Rickettsia typhi and Rickettsia felis in fleas collected from client-owned companion animals in the Southern Great Plains. J Med Entomol. 2017;54:1093-1097.
- Sexton DJ. Murine typhus. UpToDate. https://www.uptodate.com/contents/murine-typhus?search=diagnosis-and-treatment-of-murine-typhus&source=search_result&selectedTitle=1~21&usage_type=default&display_rank=1. Updated January 17, 2019. Accessed August 15, 2019.
- Riehm JM, Löscher T. Human plague and pneumonic plague: pathogenicity, epidemiology, clinical presentations and therapy [in German]. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2015;58:721-729.
- Butler T. Plague gives surprises in the first decade of the 21st century in the United States and worldwide. Am J Trop Med Hyg. 2013;89:788-793.
- Gould LH, Pape J, Ettestad P, Griffith KS, et al. Dog-associated risk factors for human plague. Zoonoses Public Health. 2008;55:448-454.
- Margolis DA, Burns J, Reed SL, et al. Septicemic plague in a community hospital in California. Am J Trop Med Hyg. 2008;78:868-871.
- Urich SK, Chalcraft L, Schriefer ME, et al. Lack of antimicrobial resistance in Yersinia pestis isolates from 17 countries in the Americas, Africa, and Asia. Antimicrob Agents Chemother. 2012;56:555-558.
- Jiang P, Zhang X, Liu RD, et al. A human case of zoonotic dog tapeworm, Dipylidium caninum (Eucestoda: Dilepidiidae), in China. Korean J Parasitol. 2017;55:61-64.
- Roberts LS, Janovy J Jr, eds. Foundations of Parasitology. 8th ed. New York, NY: McGraw-Hill; 2009.
Fleas of the order Siphonaptera are insects that feed on the blood of a mammalian host. They have no wings but jump to near 150 times their body lengths to reach potential hosts.1 An epidemiologic survey performed in 2016 demonstrated that 96% of fleas in the United States are cat fleas (Ctenocephalides felis).2 The bites often present as pruritic, nonfollicular-based, excoriated papules; papular urticaria; or vesiculobullous lesions distributed across the lower legs. Antihistamines and topical steroids may be helpful for symptomatic relief, but flea eradication is key.
Identification
Ctenocephalides fleas, including the common cat flea and the dog flea, have a characteristic pronotal comb that resembles a mane of hair (Figure 1) and genal comb that resembles a mustache. Compared to the dog flea (Ctenocephalides canis), cat fleas have a flatter head and fewer hair-bearing notches on the dorsal hind tibia (the dog flea has 8 notches and the cat flea has 6 notches)(Figure 2).
Flea Prevention and Eradication
Effective management of flea bites requires avoidance of infested areas and eradication of fleas from the home and pets. Home treatment should be performed by a qualified specialist and a veterinarian should treat the pet, but the dermatologist must be knowledgeable about treatment options. Flea pupae can lie dormant between floorboards for extended periods of time and hatch rapidly when new tenants enter a house or apartment. Insecticidal dusts and spray formulations frequently are used to treat infested homes. It also is important to reduce flea egg numbers by vacuuming carpets and areas where pets sleep.3 Rodents often introduce fleas to households and pets, so eliminating them from the area may play an important role in flea control. Consulting with a veterinarian is important, as treatment directed at pets is critical to control flea populations. Oral agents, including fluralaner, afoxolaner, sarolaner, and spinosad, can reduce flea populations on animals by as much as 99.3% after 7 days.4,5 Fast-acting pulicidal agents, such as the combination of dinotefuran and fipronil, demonstrate curative activity as soon as 3 hours after treatment, which also may prevent reinfestation for as long as 6 weeks after treatment.6
Vector-Borne Disease
Fleas living on animals in close contact with humans, such as cats and dogs, can transmit zoonotic pathogens. Around 12,000 outpatients and 500 inpatients are diagnosed with cat scratch disease, a form of bartonellosis, annually. Ctenocephalides felis transmits Bartonella henselae from cat-to-cat and often cat-to-human through infected flea feces, causing a primary inoculation lesion and lymphadenitis. Of 3011 primary care providers surveyed from 2014 to 2015, 37.2% had treated at least 1 patient with cat scratch disease, yet knowledge gaps remain regarding the proper treatment and preventative measures for the disease.7 Current recommendations for the treatment of lymphadenitis caused by B henselae include a 5-day course of oral azithromycin.8 The preferred dosing regimen in adults is 500 mg on day 1 and 250 mg on days 2 through 5. Pediatric patients weighing less than 45.5 kg should receive 10 mg/kg on day 1 and 5 mg/kg on days 2 through 5.8 Additionally, less than one-third of the primary care providers surveyed from 2014 to 2015 said they would discuss the importance of pet flea control with immunocompromised patients who own cats, despite evidence implicating fleas in disease transmission.7 Pet-directed topical therapy with agents such as selamectin prescribed by a qualified veterinarian can prevent transmission of B henselae in cats exposed to fleas infected with the bacteria,9 which supports the importance of patient education and flea control, especially in pets owned by immunocompromised patients. Patients who are immunocompromised are at increased risk for persistent or disseminated bartonellosis, including endocarditis, in addition to cat scratch disease. Although arriving at a diagnosis may be difficult, one study found that bartonellosis in 13 renal transplant recipients was best diagnosed using both serology and polymerase chain reaction via DNA extraction of tissue specimens.10 These findings may enhance diagnostic yield for similar patients when bartonellosis is suspected.
Flea-borne typhus is endemic to Texas and Southern California.11,12 Evidence suggests that the pathogenic bacteria, Rickettsia typhi and Rickettsia felis, also commonly infect fleas in the Great Plains area.13 Opossums carry R felis, and the fleas transmit murine or endemic typhus. A retrospective case series in Texas identified 11 cases of fatal flea-borne typhus from 1985 to 2015.11 More than half of the patients reported contact with animals or fleas prior to the illness. Patients with typhus may present with fever, nausea, vomiting, rash (macular, maculopapular, papular, petechial, or morbilliform), respiratory or neurologic symptoms, thrombocytopenia, and elevated hepatic liver enzymes. Unfortunately, there often is a notable delay in initiation of treatment with the appropriate class of antibiotics—tetracyclines—and such delays can prove fatal.11 The current recommendation for nonpregnant adults is oral doxycycline 100 mg twice daily continued 48 hours after the patient becomes afebrile or for 7 days, whichever therapy duration is longer.14 Because of the consequences of delayed treatment, it is important for clinicians to consider a diagnosis of vector-borne illness in a febrile patient with other associated gastrointestinal, cutaneous, respiratory, or neurologic symptoms, especially if they have animal or flea exposures. Flea control and exposure awareness remains paramount in preventing and treating this illness.
Yersinia pestis causes the plague, an important re-emerging disease that causes infection through flea bites, inhalation, or ingestion.15 From 2000 to 2009, 56 cases and 7 deaths in the United States—New Mexico, Arizona, Colorado, California, and Texas—and 21,725 cases and 1612 deaths worldwide were attributed to Y pestis. Most patients present with the bubonic form of the disease, with fever and an enlarging painful femoral or inguinal lymph node due to leg flea bites.16 Other forms of disease, including septicemic and pneumonic plague, are less common but relevant, as one-third of cases in the United States present with septicemia.15,17,18 Although molecular diagnosis and immunohistochemistry play important roles, the diagnosis of Y pestis infection often is still accomplished with culture. A 2012 survey of 392 strains from 17 countries demonstrated that Y pestis remained susceptible to the antibiotics currently used to treat the disease, including doxycycline, streptomycin, gentamicin, tetracycline, trimethoprim-sulfamethoxazole, and ciprofloxacin.19
Human infection with Dipylidium caninum, a dog tapeworm, has been reported after suspected accidental ingestion of cat fleas carrying the parasite.20 Children, who may present with diarrhea or white worms in their feces, are more susceptible to the infection, perhaps due to accidental flea consumption while being licked by the pet.20,21
Conclusion
Cat fleas may act as a pruritic nuisance for pet owners and even deliver deadly pathogens to immunocompromised patients. Providers can minimize their impact by educating patients on flea prevention and eradication as well as astutely recognizing and treating flea-borne diseases.
Fleas of the order Siphonaptera are insects that feed on the blood of a mammalian host. They have no wings but jump to near 150 times their body lengths to reach potential hosts.1 An epidemiologic survey performed in 2016 demonstrated that 96% of fleas in the United States are cat fleas (Ctenocephalides felis).2 The bites often present as pruritic, nonfollicular-based, excoriated papules; papular urticaria; or vesiculobullous lesions distributed across the lower legs. Antihistamines and topical steroids may be helpful for symptomatic relief, but flea eradication is key.
Identification
Ctenocephalides fleas, including the common cat flea and the dog flea, have a characteristic pronotal comb that resembles a mane of hair (Figure 1) and genal comb that resembles a mustache. Compared to the dog flea (Ctenocephalides canis), cat fleas have a flatter head and fewer hair-bearing notches on the dorsal hind tibia (the dog flea has 8 notches and the cat flea has 6 notches)(Figure 2).
Flea Prevention and Eradication
Effective management of flea bites requires avoidance of infested areas and eradication of fleas from the home and pets. Home treatment should be performed by a qualified specialist and a veterinarian should treat the pet, but the dermatologist must be knowledgeable about treatment options. Flea pupae can lie dormant between floorboards for extended periods of time and hatch rapidly when new tenants enter a house or apartment. Insecticidal dusts and spray formulations frequently are used to treat infested homes. It also is important to reduce flea egg numbers by vacuuming carpets and areas where pets sleep.3 Rodents often introduce fleas to households and pets, so eliminating them from the area may play an important role in flea control. Consulting with a veterinarian is important, as treatment directed at pets is critical to control flea populations. Oral agents, including fluralaner, afoxolaner, sarolaner, and spinosad, can reduce flea populations on animals by as much as 99.3% after 7 days.4,5 Fast-acting pulicidal agents, such as the combination of dinotefuran and fipronil, demonstrate curative activity as soon as 3 hours after treatment, which also may prevent reinfestation for as long as 6 weeks after treatment.6
Vector-Borne Disease
Fleas living on animals in close contact with humans, such as cats and dogs, can transmit zoonotic pathogens. Around 12,000 outpatients and 500 inpatients are diagnosed with cat scratch disease, a form of bartonellosis, annually. Ctenocephalides felis transmits Bartonella henselae from cat-to-cat and often cat-to-human through infected flea feces, causing a primary inoculation lesion and lymphadenitis. Of 3011 primary care providers surveyed from 2014 to 2015, 37.2% had treated at least 1 patient with cat scratch disease, yet knowledge gaps remain regarding the proper treatment and preventative measures for the disease.7 Current recommendations for the treatment of lymphadenitis caused by B henselae include a 5-day course of oral azithromycin.8 The preferred dosing regimen in adults is 500 mg on day 1 and 250 mg on days 2 through 5. Pediatric patients weighing less than 45.5 kg should receive 10 mg/kg on day 1 and 5 mg/kg on days 2 through 5.8 Additionally, less than one-third of the primary care providers surveyed from 2014 to 2015 said they would discuss the importance of pet flea control with immunocompromised patients who own cats, despite evidence implicating fleas in disease transmission.7 Pet-directed topical therapy with agents such as selamectin prescribed by a qualified veterinarian can prevent transmission of B henselae in cats exposed to fleas infected with the bacteria,9 which supports the importance of patient education and flea control, especially in pets owned by immunocompromised patients. Patients who are immunocompromised are at increased risk for persistent or disseminated bartonellosis, including endocarditis, in addition to cat scratch disease. Although arriving at a diagnosis may be difficult, one study found that bartonellosis in 13 renal transplant recipients was best diagnosed using both serology and polymerase chain reaction via DNA extraction of tissue specimens.10 These findings may enhance diagnostic yield for similar patients when bartonellosis is suspected.
Flea-borne typhus is endemic to Texas and Southern California.11,12 Evidence suggests that the pathogenic bacteria, Rickettsia typhi and Rickettsia felis, also commonly infect fleas in the Great Plains area.13 Opossums carry R felis, and the fleas transmit murine or endemic typhus. A retrospective case series in Texas identified 11 cases of fatal flea-borne typhus from 1985 to 2015.11 More than half of the patients reported contact with animals or fleas prior to the illness. Patients with typhus may present with fever, nausea, vomiting, rash (macular, maculopapular, papular, petechial, or morbilliform), respiratory or neurologic symptoms, thrombocytopenia, and elevated hepatic liver enzymes. Unfortunately, there often is a notable delay in initiation of treatment with the appropriate class of antibiotics—tetracyclines—and such delays can prove fatal.11 The current recommendation for nonpregnant adults is oral doxycycline 100 mg twice daily continued 48 hours after the patient becomes afebrile or for 7 days, whichever therapy duration is longer.14 Because of the consequences of delayed treatment, it is important for clinicians to consider a diagnosis of vector-borne illness in a febrile patient with other associated gastrointestinal, cutaneous, respiratory, or neurologic symptoms, especially if they have animal or flea exposures. Flea control and exposure awareness remains paramount in preventing and treating this illness.
Yersinia pestis causes the plague, an important re-emerging disease that causes infection through flea bites, inhalation, or ingestion.15 From 2000 to 2009, 56 cases and 7 deaths in the United States—New Mexico, Arizona, Colorado, California, and Texas—and 21,725 cases and 1612 deaths worldwide were attributed to Y pestis. Most patients present with the bubonic form of the disease, with fever and an enlarging painful femoral or inguinal lymph node due to leg flea bites.16 Other forms of disease, including septicemic and pneumonic plague, are less common but relevant, as one-third of cases in the United States present with septicemia.15,17,18 Although molecular diagnosis and immunohistochemistry play important roles, the diagnosis of Y pestis infection often is still accomplished with culture. A 2012 survey of 392 strains from 17 countries demonstrated that Y pestis remained susceptible to the antibiotics currently used to treat the disease, including doxycycline, streptomycin, gentamicin, tetracycline, trimethoprim-sulfamethoxazole, and ciprofloxacin.19
Human infection with Dipylidium caninum, a dog tapeworm, has been reported after suspected accidental ingestion of cat fleas carrying the parasite.20 Children, who may present with diarrhea or white worms in their feces, are more susceptible to the infection, perhaps due to accidental flea consumption while being licked by the pet.20,21
Conclusion
Cat fleas may act as a pruritic nuisance for pet owners and even deliver deadly pathogens to immunocompromised patients. Providers can minimize their impact by educating patients on flea prevention and eradication as well as astutely recognizing and treating flea-borne diseases.
- Cadiergues MC. A comparison of jump performances of the dog flea, Ctenocephalides canis (Curtis, 1826) and the cat flea, Ctenocephalides felis (Bouché, 1835). Vet Parasitol. 2000;92:239-241.
- Blagburn B, Butler J, Land T, et al. Who’s who and where: prevalence of Ctenocephalides felis and Ctenocephalides canis in shelter dogs and cats in the United States. Presented at: American Association of Veterinary Parasitologists 61st Annual Meeting; August 6-9, 2016; San Antonio, TX. P9.
- Bitam I, Dittmar K, Parola P, et al. Fleas and flea-borne diseases. Int J Infect Dis. 2010;14:E667-E676.
- Dryden MW, Canfield MS, Niedfeldt E, et al. Evaluation of sarolaner and spinosad oral treatments to eliminate fleas, reduce dermatologic lesions and minimize pruritus in naturally infested dogs in west Central Florida, USA. Parasit Vectors. 2017;10:389.
- Dryden MW, Canfield MS, Kalosy K, et al. Evaluation of fluralaner and afoxolaner treatments to control flea populations, reduce pruritus and minimize dermatologic lesions in naturally infested dogs in private residences in west Central Florida, USA. Parasit Vectors. 2016;9:365.
- Delcombel R, Karembe H, Nare B, et al. Synergy between dinotefuran and fipronil against the cat flea (Ctenocephalides felis): improved onset of action and residual speed of kill in adult cats. Parasit Vectors. 2017;10:341.
- Nelson CA, Moore AR, Perea AE, et al. Cat scratch disease: U.S. clinicians’ experience and knowledge. Zoonoses Public Health. 2018;65:67-73.
- Spach DH, Kaplan SL. Treatment of cat scratch disease. UpToDate. https://www.uptodate.com/contents/treatment-of-cat-scratch-disease?search=treatment%20of%20cat%20scratch&source=search_result&selectedTitle=1~59&usage_type=default&display_rank=1.Updated June 12, 2019. Accessed August 15, 2019.
- Bouhsira E, Franc M, Lienard E, et al. The efficacy of a selamectin (Stronghold®) spot on treatment in the prevention of Bartonella henselae transmission by Ctenocephalides felis in cats, using a new high-challenge model. Parasitol Res. 2015;114:1045-1050.
- Shamekhi Amiri F. Bartonellosis in chronic kidney disease: an unrecognized and unsuspected diagnosis. Ther Apher Dial. 2017;21:430-440.
- Pieracci EG, Evert N, Drexler NA, et al. Fatal flea-borne typhus in Texas: a retrospective case series, 1985-2015. American J Trop Med Hyg. 2017;96:1088-1093.
- Maina AN, Fogarty C, Krueger L, et al. Rickettsial infections among Ctenocephalides felis and host animals during a flea-borne rickettsioses outbreak in Orange County, California. PLoS One. 2016;11:e0160604.
- Noden BH, Davidson S, Smith JL, et al. First detection of Rickettsia typhi and Rickettsia felis in fleas collected from client-owned companion animals in the Southern Great Plains. J Med Entomol. 2017;54:1093-1097.
- Sexton DJ. Murine typhus. UpToDate. https://www.uptodate.com/contents/murine-typhus?search=diagnosis-and-treatment-of-murine-typhus&source=search_result&selectedTitle=1~21&usage_type=default&display_rank=1. Updated January 17, 2019. Accessed August 15, 2019.
- Riehm JM, Löscher T. Human plague and pneumonic plague: pathogenicity, epidemiology, clinical presentations and therapy [in German]. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2015;58:721-729.
- Butler T. Plague gives surprises in the first decade of the 21st century in the United States and worldwide. Am J Trop Med Hyg. 2013;89:788-793.
- Gould LH, Pape J, Ettestad P, Griffith KS, et al. Dog-associated risk factors for human plague. Zoonoses Public Health. 2008;55:448-454.
- Margolis DA, Burns J, Reed SL, et al. Septicemic plague in a community hospital in California. Am J Trop Med Hyg. 2008;78:868-871.
- Urich SK, Chalcraft L, Schriefer ME, et al. Lack of antimicrobial resistance in Yersinia pestis isolates from 17 countries in the Americas, Africa, and Asia. Antimicrob Agents Chemother. 2012;56:555-558.
- Jiang P, Zhang X, Liu RD, et al. A human case of zoonotic dog tapeworm, Dipylidium caninum (Eucestoda: Dilepidiidae), in China. Korean J Parasitol. 2017;55:61-64.
- Roberts LS, Janovy J Jr, eds. Foundations of Parasitology. 8th ed. New York, NY: McGraw-Hill; 2009.
- Cadiergues MC. A comparison of jump performances of the dog flea, Ctenocephalides canis (Curtis, 1826) and the cat flea, Ctenocephalides felis (Bouché, 1835). Vet Parasitol. 2000;92:239-241.
- Blagburn B, Butler J, Land T, et al. Who’s who and where: prevalence of Ctenocephalides felis and Ctenocephalides canis in shelter dogs and cats in the United States. Presented at: American Association of Veterinary Parasitologists 61st Annual Meeting; August 6-9, 2016; San Antonio, TX. P9.
- Bitam I, Dittmar K, Parola P, et al. Fleas and flea-borne diseases. Int J Infect Dis. 2010;14:E667-E676.
- Dryden MW, Canfield MS, Niedfeldt E, et al. Evaluation of sarolaner and spinosad oral treatments to eliminate fleas, reduce dermatologic lesions and minimize pruritus in naturally infested dogs in west Central Florida, USA. Parasit Vectors. 2017;10:389.
- Dryden MW, Canfield MS, Kalosy K, et al. Evaluation of fluralaner and afoxolaner treatments to control flea populations, reduce pruritus and minimize dermatologic lesions in naturally infested dogs in private residences in west Central Florida, USA. Parasit Vectors. 2016;9:365.
- Delcombel R, Karembe H, Nare B, et al. Synergy between dinotefuran and fipronil against the cat flea (Ctenocephalides felis): improved onset of action and residual speed of kill in adult cats. Parasit Vectors. 2017;10:341.
- Nelson CA, Moore AR, Perea AE, et al. Cat scratch disease: U.S. clinicians’ experience and knowledge. Zoonoses Public Health. 2018;65:67-73.
- Spach DH, Kaplan SL. Treatment of cat scratch disease. UpToDate. https://www.uptodate.com/contents/treatment-of-cat-scratch-disease?search=treatment%20of%20cat%20scratch&source=search_result&selectedTitle=1~59&usage_type=default&display_rank=1.Updated June 12, 2019. Accessed August 15, 2019.
- Bouhsira E, Franc M, Lienard E, et al. The efficacy of a selamectin (Stronghold®) spot on treatment in the prevention of Bartonella henselae transmission by Ctenocephalides felis in cats, using a new high-challenge model. Parasitol Res. 2015;114:1045-1050.
- Shamekhi Amiri F. Bartonellosis in chronic kidney disease: an unrecognized and unsuspected diagnosis. Ther Apher Dial. 2017;21:430-440.
- Pieracci EG, Evert N, Drexler NA, et al. Fatal flea-borne typhus in Texas: a retrospective case series, 1985-2015. American J Trop Med Hyg. 2017;96:1088-1093.
- Maina AN, Fogarty C, Krueger L, et al. Rickettsial infections among Ctenocephalides felis and host animals during a flea-borne rickettsioses outbreak in Orange County, California. PLoS One. 2016;11:e0160604.
- Noden BH, Davidson S, Smith JL, et al. First detection of Rickettsia typhi and Rickettsia felis in fleas collected from client-owned companion animals in the Southern Great Plains. J Med Entomol. 2017;54:1093-1097.
- Sexton DJ. Murine typhus. UpToDate. https://www.uptodate.com/contents/murine-typhus?search=diagnosis-and-treatment-of-murine-typhus&source=search_result&selectedTitle=1~21&usage_type=default&display_rank=1. Updated January 17, 2019. Accessed August 15, 2019.
- Riehm JM, Löscher T. Human plague and pneumonic plague: pathogenicity, epidemiology, clinical presentations and therapy [in German]. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2015;58:721-729.
- Butler T. Plague gives surprises in the first decade of the 21st century in the United States and worldwide. Am J Trop Med Hyg. 2013;89:788-793.
- Gould LH, Pape J, Ettestad P, Griffith KS, et al. Dog-associated risk factors for human plague. Zoonoses Public Health. 2008;55:448-454.
- Margolis DA, Burns J, Reed SL, et al. Septicemic plague in a community hospital in California. Am J Trop Med Hyg. 2008;78:868-871.
- Urich SK, Chalcraft L, Schriefer ME, et al. Lack of antimicrobial resistance in Yersinia pestis isolates from 17 countries in the Americas, Africa, and Asia. Antimicrob Agents Chemother. 2012;56:555-558.
- Jiang P, Zhang X, Liu RD, et al. A human case of zoonotic dog tapeworm, Dipylidium caninum (Eucestoda: Dilepidiidae), in China. Korean J Parasitol. 2017;55:61-64.
- Roberts LS, Janovy J Jr, eds. Foundations of Parasitology. 8th ed. New York, NY: McGraw-Hill; 2009.
Practice Points
- Cat fleas classically cause pruritic grouped papulovesicles on the lower legs of pet owners.
- Affected patients require thorough education on flea eradication.
- Cat fleas can transmit endemic typhus, cat scratch disease, and bubonic plague.

Acute-Onset Alopecia
The Diagnosis: Thallium-Induced Alopecia
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
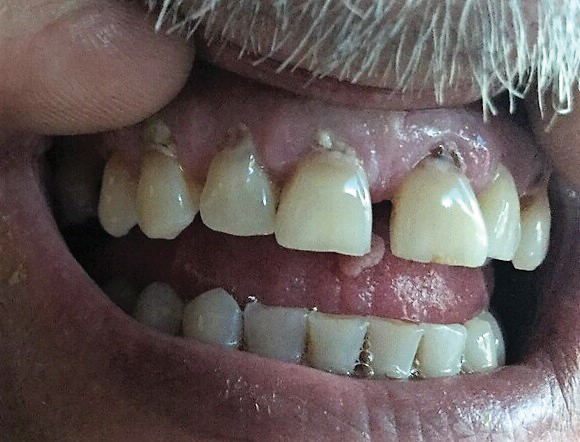
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.
The Diagnosis: Thallium-Induced Alopecia
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
The Diagnosis: Thallium-Induced Alopecia
At the time of presentation, a punch biopsy specimen of the scalp revealed nonscarring alopecia with increased catagen hairs; follicular miniaturization; peribulbar lymphoid infiltrates; and fibrous tract remnants containing melanin, lymphocytes, and occasional mast cells (Figure 1). The differential diagnosis included alopecia areata, syphilis, and toxin-mediated anagen effluvium (AE). Given the abrupt onset affecting multiple individuals in an industrial environment, heavy metal poisoning was suspected. Blood and urine testing was negative, but a few months had elapsed since exposure. Several months after his initial presentation, the patient reported problems with his teeth, thin brittle nails, and resolution of the visual changes. Photographs sent by the patient revealed darkening and degeneration of the gingival margin (Figure 2).
Environmental review revealed the patient was working on a demolition site of a 150-year-old electrical plant near a river. Inundation of rainfall caused a river swell and subsequent flooding of the work site. The patient reported working for more than 2 months in knee-deep muddy water, and he noted that water for consumption and showers was procured on-site from a well-based source that may have been contaminated by the floodwaters.
Acute nonscarring alopecia can be an AE or telogen effluvium (TE), also known as telogen defluvium. The key distinguishing factor is the mode of injury.1 In TE, medications, stress, hormonal shifts, or inflammation induce a synchronized and abrupt transition of hairs from anagen phase to catagen phase, a committed step that then must fully cycle through the telogen phase, culminating in the simultaneous shedding of numerous telogen hairs approximately 3 to 4 months later. Conversely, AE is caused by a sudden insult to the metabolic machinery of the hair matrix. Affected follicles rapidly produce thinner weaker shafts yielding Pohl-Pinkus constrictions or pencil point-shaped fractures that shed approximately 1 to 2 months after injury. The 10% of scalp hairs in the resting telogen phase have no matrix and thus are unaffected. Some etiologies can cause either AE or TE, depending on the dose and intensity of the insult. Common causes of AE include alopecia areata and syphilis, both consisting of abrupt severe bulbar inflammation.1 Other causes include chemotherapy, particularly antimetabolites, alkylating agents, and mitotic inhibitors; radiation; medications (eg, isoniazid); severe protein malnutrition; toxic chemicals (eg, boron/boric acid); and heavy metals (eg, thallium, mercury).
Thallium is one of the most common causes of heavy metal poisoning and is particularly dangerous due to its colorless, tasteless, and odorless characteristics. Although its common use as a rodenticide has dramatically decreased in the United States after it was banned in 1965, it is still used in this fashion in other countries and has a notable industrial presence, particularly in electronics, superconductors, and low-temperature thermometers. Accidental poisoning of a graduate chemistry student during copper research has been reported,2 highlighting that thallium can be inhaled, ingested, or absorbed through the skin. Thallium is even present in mycoplasma agar plates, the ingestion of which has resulted in poisoning.3
Systemic symptoms of thallium poisoning include somnolence, weakness, nausea, vomiting, stomatitis, abdominal pain, diarrhea, tachycardia, hypertension, and polyneuropathy.4-7 Neuropathy often manifests as painful acral dysesthesia and paresthesia, perioral numbness, optic neuropathy causing visual changes, and encephalopathy. Cutaneous findings include diffuse alopecia of the scalp and eyebrows, perioral dermatitis, glossitis, diffuse hyperpigmentation, oral hyperpigmentation (often as a stippled lead line along the gingival margin with subsequent alveolar damage and resorption), melanonychia, palmoplantar keratoderma, acneform or pustular eruption, and nail changes including Mees lines.2,4,5,7-9 Rarely, major organ failure and death may result.10
Toxin panels may not include thallium, and urine and serum tests may be negative if too much time has transpired since the acute exposure. Hair or nail analysis has proved useful in subacute cases11; however, most laboratories require a pencil-thick segment of hair cut at the roots and bundled, weighing at least 500 mg. Thallium poisoning is treated with activated charcoal, Prussian blue, and blood purification therapies (eg, hemodialysis, hemoperfusion, hemofiltration).4,7 Cutaneous findings typically resolve, but neuropathic changes may persist.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology With Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Campbell C, Bahrami S, Owen C. Anagen effluvium caused by thallium poisoning. JAMA Dermatol. 2016;152:724-726.
- Puschner B, Basso MM. Graham TW. Thallium toxicosis in a dog consequent to ingestion of Mycoplasma agar plates. J Vet Diagn Invest. 2012;24:227-230.
- Sojáková M, Zigrai M, Karaman A, et al. Thallium intoxication: case report. Neuro Endocrinol Lett. 2015;36:311-315.
- Lu Cl, Huang CC, Chang YC, et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Arch Dermatol. 2007;143:93-98.
- Liu EM, Rajagopal R, Grand MG. Optic nerve atrophy and hair loss in a young man. JAMA Ophthalmol. 2015;133:1469-1470.
- Zhang HT, Qiao BP, Liu BP, et al. Study on the treatment of acute thallium poisoning. Am J Med Sci. 2014;347:377-381.
- Misra UK, Kalita J, Yadav RK, et al. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. 2003;79:103-105.
- Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138:321-325.
- Li S, Huang W, Duan Y, et al. Human fatality due to thallium poisoning: autopsy, microscopy, and mass spectrometry assays. J Forensic Sci. 2015;60:247-251.
- Daniel CR 3rd, Piraccini BM, Tosti A. The nail and hair in forensic science. J Am Acad Dermatol. 2004;50:258-261.

A previously healthy 45-year-old man presented to the dermatology department with abrupt onset of patchy, progressively worsening alopecia of the scalp as well as nausea with emesis and blurry vision of a few weeks' duration. All symptoms were temporally associated with a new demolition job the patient had started at an industrial site. He reported 10 other contractors were similarly affected. The patient denied paresthesia or other skin changes. On physical examination, large patches of smooth alopecia without erythema, scale, scarring, tenderness, or edema that coalesced to involve the majority of the scalp, eyebrows, and eyelashes (inset) were noted.
Acquired Hypertrichosis of the Periorbital Area and Malar Cheek
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
The Diagnosis: Bimatoprost-Induced Hypertrichosis
Latanoprost, a prostaglandin analogue, typically is prescribed by ophthalmologists as eye drops to reduce intraocular pressure in open-angle glaucoma.1 Common adverse reactions of latanoprost drops include blurred vision, ocular irritation, darkening of the eyelid skin, and pigmentation of the iris.
In 1997, Johnstone2 reported hypertrichosis and increased pigmentation of the eyelashes of both eyes and adjacent skin after latanoprost drops were used in glaucoma patients. Subsequently, topical latanoprost and bimatoprost, a similar analogue, are now utilized for the cosmetic purpose of thickening and lengthening the eyelashes due to the hypertrichosis effect. Travoprost, another prostaglandin analogue used to treat glaucoma, also has been associated with periocular hypertrichosis.3 Concomitant poliosis of the eyelashes with hypertrichosis from latanoprost also has been reported.4 Our patient specifically purchased the eye drops (marketed as generic bimatoprost) to lengthen her eyelashes and had noticed an increase in length. She denied a family history of increased facial hair in females.
Along with gingival hyperplasia, systemic cyclosporine may cause generalized hypertrichosis consisting of terminal hair growth, particularly on the face and forearms. However, hypertrichosis from cyclosporine ophthalmic emulsion 0.05% rarely has been reported5 but would be more likely to occur in a patient reporting a history of chronic dry eye. Oral acetazolamide, not eye drops, is prescribed for glaucoma and typically is not associated with hypertrichosis. Betamethasone and timolol eye drops may cause burning, stinging, redness, or watering of the eyes, but they do not typically cause hypertrichosis.
Other systemic medications (eg, zidovudine, phenytoin, minoxidil, danazol, anabolic steroids) may cause hypertrichosis but not typically localized to the periocular area. Phenytoin usually causes hair growth on the limbs but not on the face and trunk. Oral minoxidil causes hypertrichosis, predominately on the face, lower legs, and forearms.
Systemic conditions such as endocrine abnormalities or porphyria cutanea tarda also may cause hypertrichosis; however, it typically does not present in small focal areas, and other stigmata often are present such as signs of virilization in hirsutism (ie, deepening of voice, pattern alopecia, acne) or liver disease with photosensitive erosions and bullae that leave scars and milia in porphyria cutanea tarda. Acquired hypertrichosis lanuginosa deserves consideration, in part due to its association with lung and colon cancers; however, it consists of softer, downy, nonterminal hairs (malignant down) and is more generalized on the face. Malnutrition from anorexia nervosa may similarly induce hypertrichosis lanuginose.
The molecular mechanism for latanoprost-induced hypertrichosis is unknown; however, it may promote anagen growth as well as hypertrophic changes in the affected follicles.6 Patients should use extreme caution when purchasing unregulated medications due to the risk for impurities, less stable formulation, or inaccurate concentrations. Comparison between brand name and approved generic latanoprost has found notable differences, including variations in active-ingredient concentration, poor stability in warmer temperatures, and higher levels of particulate matter.7 Some cosmetic eyelash enhancers sold over-the-counter or online may contain prostaglandin analogues, but they may not be listed as ingredients.8 One report noted a bimatoprost product with a concentration level double that of brand-name bimatoprost that was discovered using high-performance liquid chromatography-tandem mass spectrometry.9
Treatment options for eliminating the excess hairs include discontinuing the prostaglandin analogue or applying it only to the eyelid margin with an appropriate applicator. Waxing, manual extraction, laser hair removal, electrolysis, and depilatory creams are alternative treatments.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
- Alm A. Latanoprost in the treatment of glaucoma. Clin Ophthalmol. 2014;8:1967-1985.
- Johnstone MA. Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol. 1997;124:544-547.
- Ortiz-Perez S, Olver JM. Hypertrichosis of the upper cheek area associated with travoprost treatment of glaucoma. Ophthalmic Plast Reconstr Surg. 2010;26:376-377.
- Özyurt S, Çetinkaya GS. Hypertrichosis of the malar areas and poliosis of the eyelashes caused by latanoprost. Actas Dermosifiliogr. 2015;106:74-75.
- Lei HL, Ku WC, Sun MH, et al. Cyclosporine A eye drop-induced elongated eyelashes: a case report. Case Rep Ophthalmol. 2011;2:398-400.
- Johnstone MA, Albert DM. Prostaglandin-induced hair growth. Surv Ophthalmol. 2002;47(suppl 1):S185-S202.
- Kahook MY, Fechtner RD, Katz LJ, et al. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37:101-108.
- Swedish Medical Products Agency. Pharmaceutical ingredients in one out of three eyelash serums. https://www.dr-jetskeultee.nl/jetskeultee/download/common/artikel-wimpers-ingredients.pdf. Published April 15, 2013. Accessed April 11, 2019.
- Marchei E, De Orsi D, Guarino C, et al. High performance liquid chromatography tandem mass spectrometry measurement of bimatoprost, latanoprost and travoprost in eyelash enhancing cosmetic serums. Cosmetics. 2016;3:4.
An otherwise healthy woman in her late 50s with Fitzpatrick skin type II presented to the dermatology department for a scheduled cosmetic botulinum toxin injection. Her medical history was notable only for periodic nonsurgical cosmetic procedures including botulinum toxin and dermal fillers, and she was not taking any daily systemic medications. During the preoperative assessment, subtle bilateral and symmetric hypertrichosis with darker terminal hair formation was noted on the periorbital skin and zygomatic cheek. Upon inquiry, the patient admitted to purchasing a “special eye drop” from Mexico and using it regularly. After instillation of 2 to 3 drops per eye, she would laterally wipe the resulting excess drops away from the eyes with her hands and then wash her hands. She denied a change in eye color from their natural brown but did report using blue color contact lenses. She denied an increase in hair growth elsewhere including the upper lip, chin, upper chest, forearms, and hands. She denied deepening of her voice, acne, or hair thinning.
What’s Eating You? Millipede Burns
Clinical Presentation
Millipedes secrete a noxious toxin implicated in millipede burns. The toxic substance is benzoquinone, a strong irritant secreted from the repugnatorial glands contained in each segment of the arthropod (Figure 1). This compound serves as a natural insect repellant, acting as the millipede’s defense mechanism from potential predators.1 On human skin, benzoquinone causes localized pigmentary changes most commonly presenting on the feet and toes. Local lesions may be associated with pain or burning, but there are no known reports of adverse systemic effects.2 Affected patients experience cutaneous pigmentary changes, which may be dark red, blue, or black, and spontaneously resolve over time.2 The degree of pigment change may be associated with duration of skin contact with the toxin. The affected areas may resemble burns, dermatitis, or skin necrosis. More distal lesions may present similarly to blue toe syndrome or acute arterial occlusion but can be differentiated by the presence of intact peripheral pulses and lack of temperature discrepancy between the feet.3,4 Histologic evaluation of the lesions generally reveals nonspecific full-thickness epidermal necrosis, making clinical suspicion and physical examination paramount to the diagnosis of millipede burns.5

Diagnostic Difficulties
Accurate diagnosis of millipede burns is more difficult when the burn involves an unusual site. The most common site of involvement is the foot (Figure 2), followed by other commonly exposed areas such as the arms, face, and eyes.2,3,6,7 Covered parts of the body are much less commonly affected, requiring the arthropod to gain access via infiltration of clothing, often when hanging on a clothesline. In these cases, burns may be mistaken for child abuse, especially if certain areas of the body are involved, such as the groin and genitals.2 The well-defined arcuate lesions of the burns may resemble injuries from a wire or belt to the unsuspecting observer.
Conclusion
Although millipedes often are regarded as harmless, they are capable of causing adverse reactions through the secretion of toxic chemicals. Millipede burns cause localized pigmentary changes that may be associated with pain or burning in some patients. Because these burns may resemble child abuse in pediatric patients, physicians should be aware of this diagnosis when unusual parts of the body are involved.
- Kuwahara Y, Omura H, Tanabe T. 2-Nitroethenylbenzenes as naturalproducts in millipede defense secretions. Naturwissenschaften. 2002;89:308-310.
- De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190.
- Heeren Neto AS, Bernardes Filho F, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258.
- Lima CA, Cardoso JL, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda class (“millipedes”). An Bras Dermatol. 2010;85:391-392.
- Dar NR, Raza N, Rehman SB. Millipede burn at an unusual site mimicking child abuse in an 8-year-old girl. Clin Pediatr (Phila). 2008;47:490-492.
- Hendrickson RG. Millipede exposure. Clin Toxicol (Phila). 2005;43:211-212.
- Verma AK, Bourke B. Millipede burn masquerading as trash foot in a paediatric patient [published online October 29, 2013]. ANZ J Surg. 2014;84:388-390.
Clinical Presentation
Millipedes secrete a noxious toxin implicated in millipede burns. The toxic substance is benzoquinone, a strong irritant secreted from the repugnatorial glands contained in each segment of the arthropod (Figure 1). This compound serves as a natural insect repellant, acting as the millipede’s defense mechanism from potential predators.1 On human skin, benzoquinone causes localized pigmentary changes most commonly presenting on the feet and toes. Local lesions may be associated with pain or burning, but there are no known reports of adverse systemic effects.2 Affected patients experience cutaneous pigmentary changes, which may be dark red, blue, or black, and spontaneously resolve over time.2 The degree of pigment change may be associated with duration of skin contact with the toxin. The affected areas may resemble burns, dermatitis, or skin necrosis. More distal lesions may present similarly to blue toe syndrome or acute arterial occlusion but can be differentiated by the presence of intact peripheral pulses and lack of temperature discrepancy between the feet.3,4 Histologic evaluation of the lesions generally reveals nonspecific full-thickness epidermal necrosis, making clinical suspicion and physical examination paramount to the diagnosis of millipede burns.5
Diagnostic Difficulties
Accurate diagnosis of millipede burns is more difficult when the burn involves an unusual site. The most common site of involvement is the foot (Figure 2), followed by other commonly exposed areas such as the arms, face, and eyes.2,3,6,7 Covered parts of the body are much less commonly affected, requiring the arthropod to gain access via infiltration of clothing, often when hanging on a clothesline. In these cases, burns may be mistaken for child abuse, especially if certain areas of the body are involved, such as the groin and genitals.2 The well-defined arcuate lesions of the burns may resemble injuries from a wire or belt to the unsuspecting observer.
Conclusion
Although millipedes often are regarded as harmless, they are capable of causing adverse reactions through the secretion of toxic chemicals. Millipede burns cause localized pigmentary changes that may be associated with pain or burning in some patients. Because these burns may resemble child abuse in pediatric patients, physicians should be aware of this diagnosis when unusual parts of the body are involved.
Clinical Presentation
Millipedes secrete a noxious toxin implicated in millipede burns. The toxic substance is benzoquinone, a strong irritant secreted from the repugnatorial glands contained in each segment of the arthropod (Figure 1). This compound serves as a natural insect repellant, acting as the millipede’s defense mechanism from potential predators.1 On human skin, benzoquinone causes localized pigmentary changes most commonly presenting on the feet and toes. Local lesions may be associated with pain or burning, but there are no known reports of adverse systemic effects.2 Affected patients experience cutaneous pigmentary changes, which may be dark red, blue, or black, and spontaneously resolve over time.2 The degree of pigment change may be associated with duration of skin contact with the toxin. The affected areas may resemble burns, dermatitis, or skin necrosis. More distal lesions may present similarly to blue toe syndrome or acute arterial occlusion but can be differentiated by the presence of intact peripheral pulses and lack of temperature discrepancy between the feet.3,4 Histologic evaluation of the lesions generally reveals nonspecific full-thickness epidermal necrosis, making clinical suspicion and physical examination paramount to the diagnosis of millipede burns.5
Diagnostic Difficulties
Accurate diagnosis of millipede burns is more difficult when the burn involves an unusual site. The most common site of involvement is the foot (Figure 2), followed by other commonly exposed areas such as the arms, face, and eyes.2,3,6,7 Covered parts of the body are much less commonly affected, requiring the arthropod to gain access via infiltration of clothing, often when hanging on a clothesline. In these cases, burns may be mistaken for child abuse, especially if certain areas of the body are involved, such as the groin and genitals.2 The well-defined arcuate lesions of the burns may resemble injuries from a wire or belt to the unsuspecting observer.
Conclusion
Although millipedes often are regarded as harmless, they are capable of causing adverse reactions through the secretion of toxic chemicals. Millipede burns cause localized pigmentary changes that may be associated with pain or burning in some patients. Because these burns may resemble child abuse in pediatric patients, physicians should be aware of this diagnosis when unusual parts of the body are involved.
- Kuwahara Y, Omura H, Tanabe T. 2-Nitroethenylbenzenes as naturalproducts in millipede defense secretions. Naturwissenschaften. 2002;89:308-310.
- De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190.
- Heeren Neto AS, Bernardes Filho F, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258.
- Lima CA, Cardoso JL, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda class (“millipedes”). An Bras Dermatol. 2010;85:391-392.
- Dar NR, Raza N, Rehman SB. Millipede burn at an unusual site mimicking child abuse in an 8-year-old girl. Clin Pediatr (Phila). 2008;47:490-492.
- Hendrickson RG. Millipede exposure. Clin Toxicol (Phila). 2005;43:211-212.
- Verma AK, Bourke B. Millipede burn masquerading as trash foot in a paediatric patient [published online October 29, 2013]. ANZ J Surg. 2014;84:388-390.
- Kuwahara Y, Omura H, Tanabe T. 2-Nitroethenylbenzenes as naturalproducts in millipede defense secretions. Naturwissenschaften. 2002;89:308-310.
- De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190.
- Heeren Neto AS, Bernardes Filho F, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258.
- Lima CA, Cardoso JL, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda class (“millipedes”). An Bras Dermatol. 2010;85:391-392.
- Dar NR, Raza N, Rehman SB. Millipede burn at an unusual site mimicking child abuse in an 8-year-old girl. Clin Pediatr (Phila). 2008;47:490-492.
- Hendrickson RG. Millipede exposure. Clin Toxicol (Phila). 2005;43:211-212.
- Verma AK, Bourke B. Millipede burn masquerading as trash foot in a paediatric patient [published online October 29, 2013]. ANZ J Surg. 2014;84:388-390.
Practice Points
- The most common site of involvement of millipede burns is the foot, followed by other commonly exposed areas such as the arms, face, and eyes. Covered parts of the body are much less commonly affected.
- Millipede burns may resemble child abuse in pediatric patients; therefore, physicians should be aware of this diagnosis when unusual parts of the body are involved.
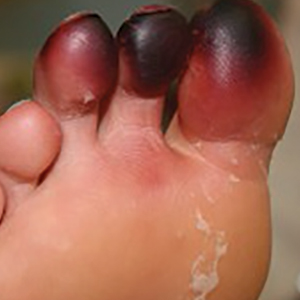
Indurated Plaque on the Shoulder
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
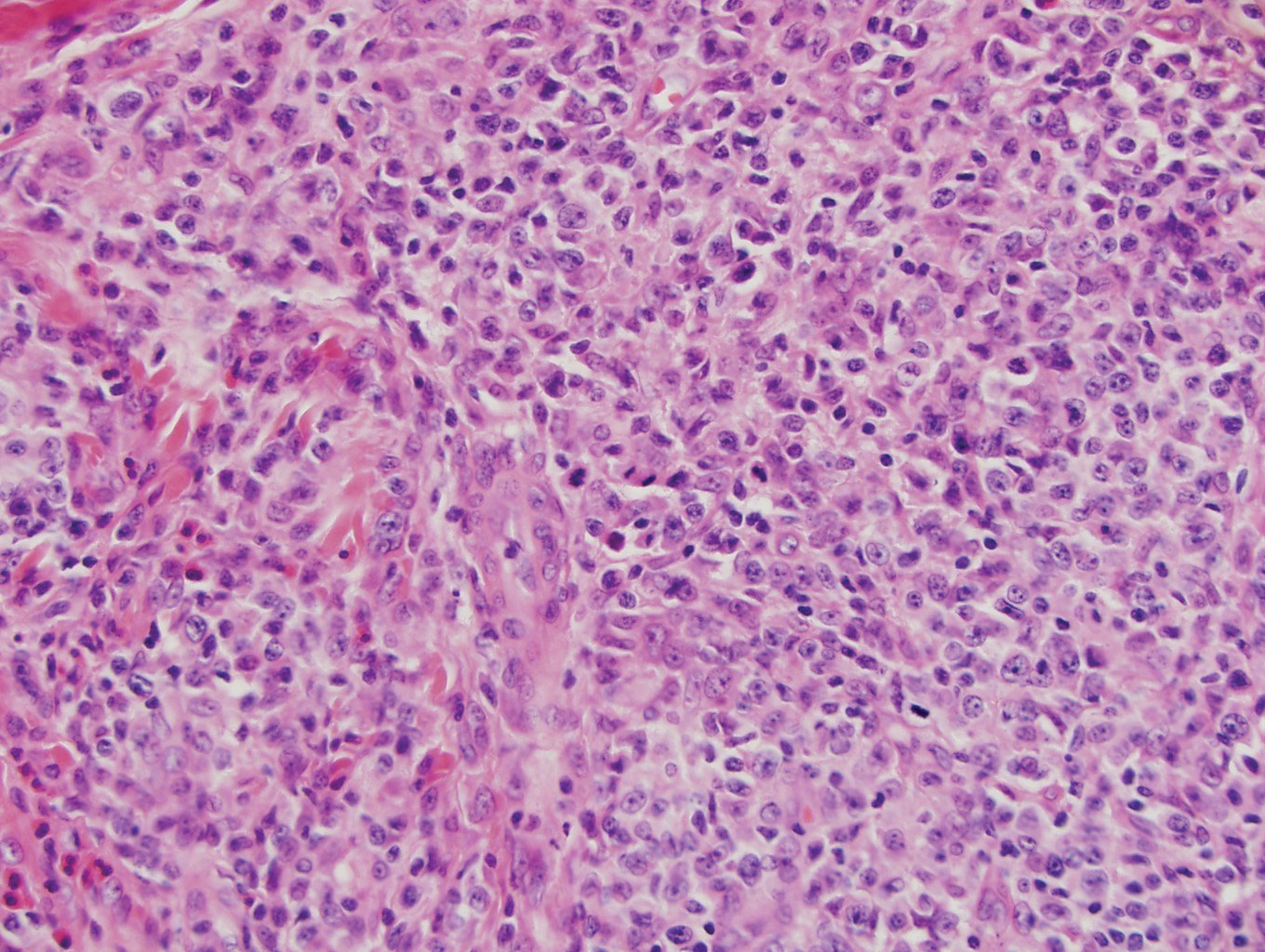
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
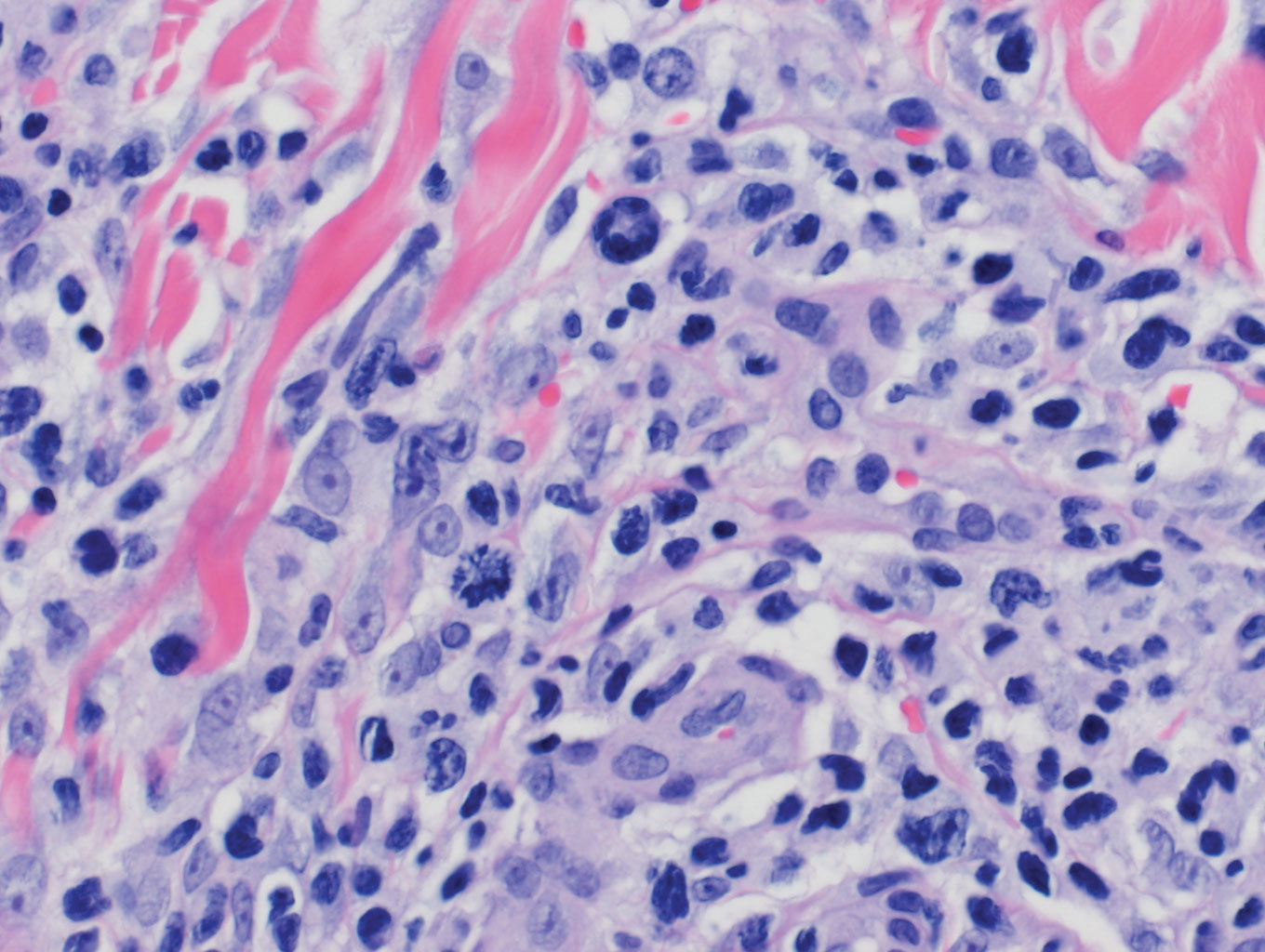
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
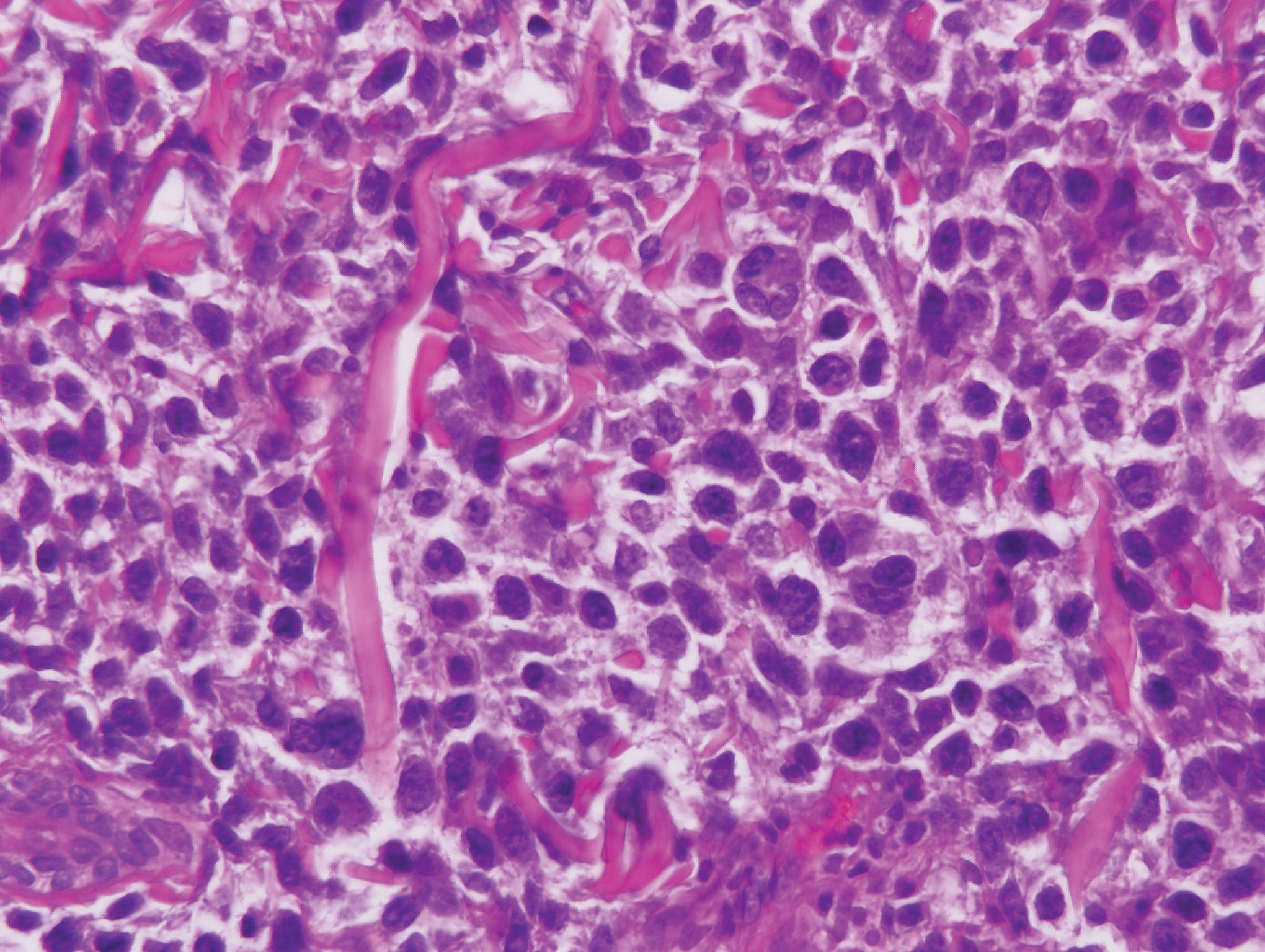
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
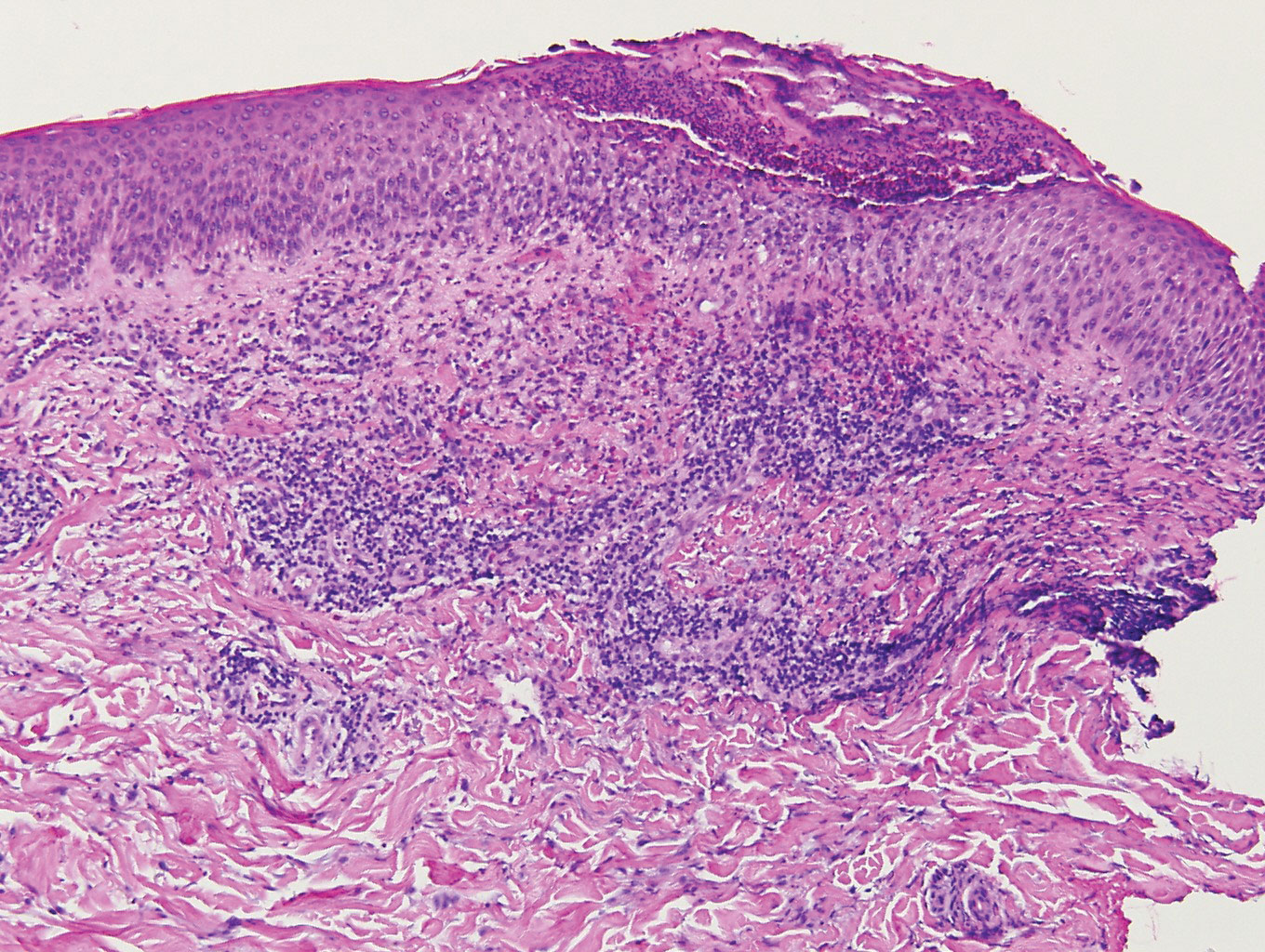
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
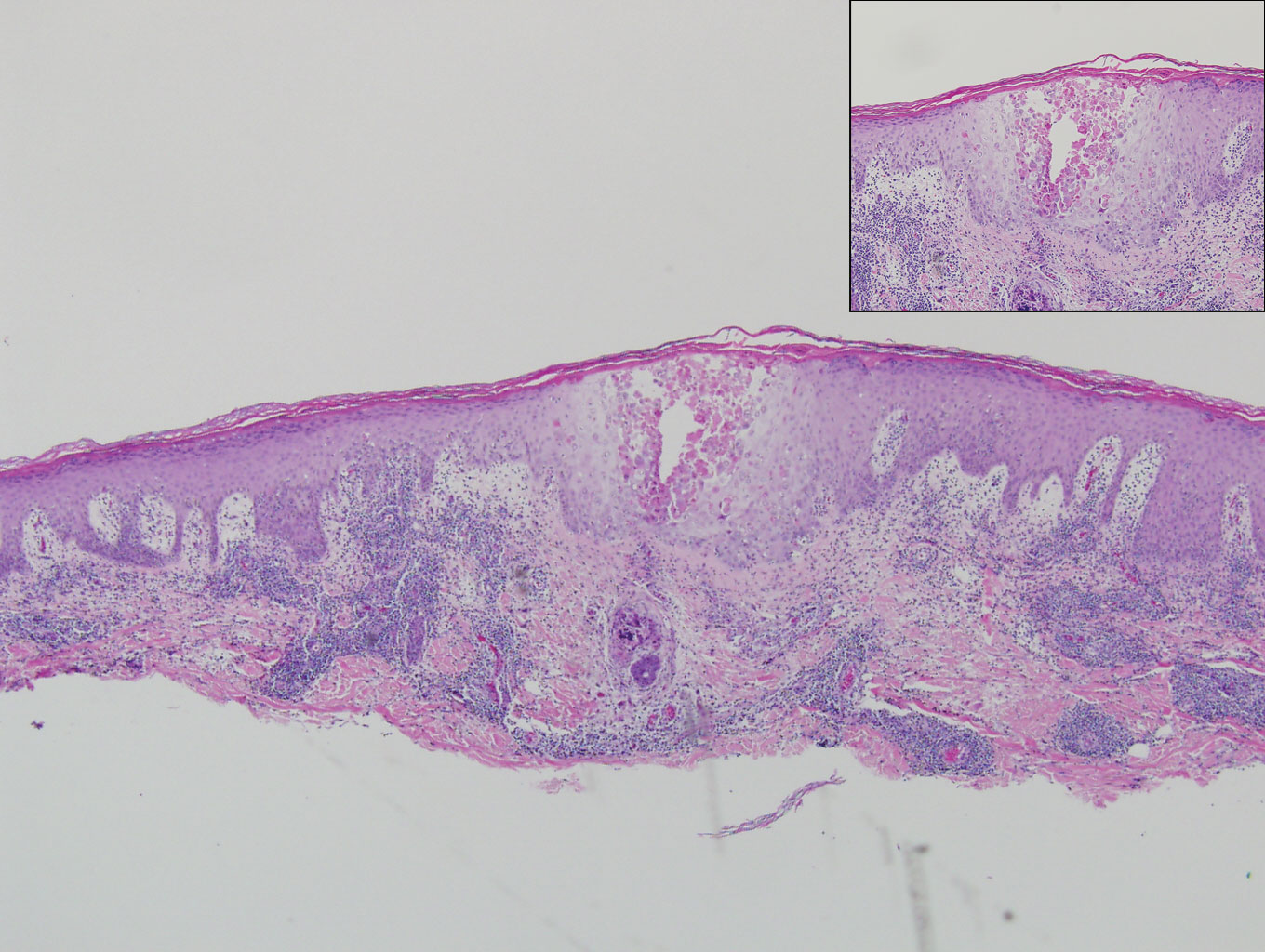
A 66-year-old man with mycosis fungoides presented with a new indurated plaque on the left shoulder. Biopsies of the left shoulder and back lesions were obtained.