User login
Should thiopurine methyltransferase (TPMT) activity be determined before prescribing azathioprine, mercaptopurine, or thioguanine?
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
Family history: Still relevant in the genomics era
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
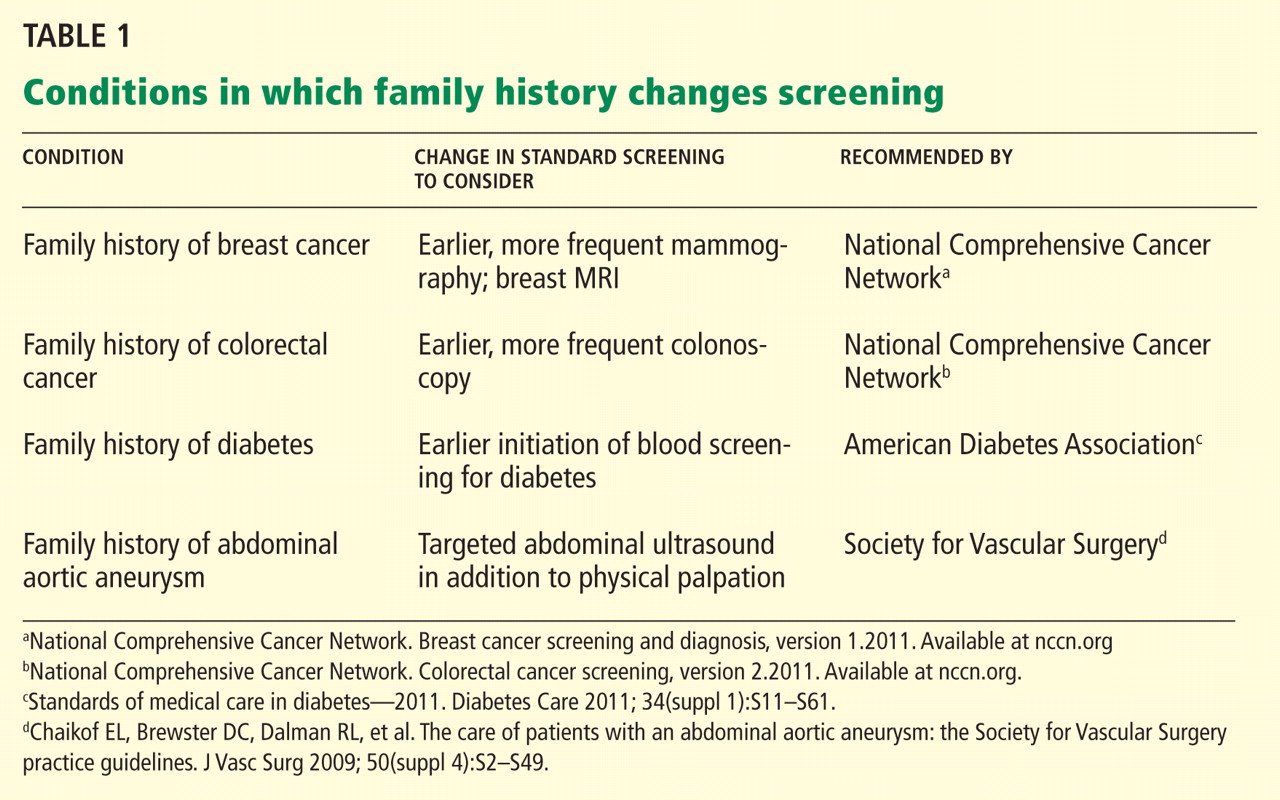
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
KEY POINTS
- The family history is an underused tool for predicting the risk of disease and for personalizing preventive care.
- Barriers to the appropriate collection and use of the family history include concerns over the reliability of patient reporting, a lack of time and reimbursement, and provider knowledge gaps.
- Use of family history to inform genetic testing for hereditary cancer syndromes has been shown to improve outcomes and may reduce overall health care costs.
- Future solutions need to focus on creating time-effective ways to collect and analyze the family history, and on developing innovative methods of educating medical providers at all levels of training as to how to apply the family history in clinical practice.
Building an innovative model for personalized healthcare
Personalized healthcare is the tailoring of medical management and patient care to the individual characteristics of each patient. This is achieved by incorporating the genetic and genomic makeup of an individual and his or her family medical history, environment, health-related behaviors, culture, and values into a complete health picture that can be used to customize care. Another level of personalization, often called personalized medicine, involves the selection of drug therapy through the use of tests to determine the genes and gene interactions that can reliably predict an individual’s response to a given therapy. This white paper focuses largely on the use of personalized healthcare as a risk prediction tool.
CURRENT STATUS OF PERSONALIZED HEALTHCARE

The case for personalized healthcare: Seeking value
To fully appreciate the need to advance the adoption of personalized healthcare into the delivery of medicine, one must consider the operation of our current healthcare system and its inefficiencies in terms of delivery and cost, its imprecision in the selection of therapies, and its inability to optimize outcomes. The framework of the US healthcare system as it is now constructed is expensive, disease-directed (instead of health- and wellness-directed), fragmented, and complex. While gross domestic product (GDP) in the United States has increased by approximately 3% per year,2 the compounded growth rate of healthcare expenditures is 6.1% per year. Healthcare in the aggregate now represents 17.6% of GDP and 27% of spending by the federal government and consumes 28% of the average household’s discretionary spending, surpassed only by housing.3
Personalized healthcare can potentially address the need for value consistent with the healthcare system’s prominent share of the US economy. The growth in healthcare spending is certain to be a target of the newly created Joint Select Committee on Deficit Reduction (created by the Budget Control Act of 2011), which is tasked with deficit reduction of at least $1.5 trillion over a 10-year period.
The need to address healthcare costs has been recognized in the Patient Protection and Affordable Care Act, a central feature of which is the creation of integrated health systems that pay for value based on quality, cost containment, and consumer experience. The legislation was enacted to transform healthcare in a variety of ways to make it more sustainable. The Patient Protection and Affordable Care Act seeks to end fragmentation by expanding the use of information technology to reorganize the delivery system and to prevent errors, shifting from volume-based incentives to incentives based on performance and outcomes, and rewarding effective healthcare delivery measures and good patient outcomes.
A shift from reactive to proactive
The premise behind personalized healthcare is the potential for more efficient healthcare, with the assumption that efficiency translates to lower cost and improved patient care.
Although healthcare reform is most often referred to in the context of improving access to care through insurance coverage mandates, true healthcare reform shifts current healthcare models from the practice of reactive medicine to the practice of proactive medicine, in which the tools of personalized healthcare (ie, genetics, genomics, and other molecular diagnostics) enable not only better quality of care but also less expensive care.
Several personalized tools have long been accepted into mainstream medicine. Two examples are the family history, which is the least expensive and most available genetic evaluation tool, and ABO blood typing for safe transfusions (as ABO blood types are alleles of a gene). In fact, much of what is now considered mainstream medical management was at one time considered new. To allow further evolution of medical practice, our challenge is to open our minds to the possibility that personalized proactive medicine can improve healthcare.
The new vision: More precise management
The trial-and-error approach to treating disease is inefficient and costly. Many drugs are effective for only about 50% of patients, often leading to switching or intensification of therapy that requires multiple patient visits.
Personalized medicine considers pharmacokinetic and other characteristics in selection of drug dosages. Genomic testing has the potential to provide clearer insight into the more successful use of currently available medicines. Treatment decisions (ie, drug and drug dosage choice) made on the basis of pharmacogenomic testing should increase adherence through greater effectiveness and fewer adverse drug reactions.
A massive amount of waste is related to pharmaceutical nonadherence and noncompliance. The New England Healthcare Institute has estimated that medication nonadherence costs the healthcare system $290 billion annually.4 Methodologies targeted at individual patients to improve adherence to drug regimens could save the healthcare system a tremendous amount of money.
Cancer management as a model for personalized healthcare. Personalization of therapy is especially suited to cancer management, given that the response to nonspecific cancer chemotherapy is suboptimal in most patients yet exposes them to adverse effects.5 Large-scale sequencing of human cancer genomes is rapidly changing the understanding of cancer biology and is identifying new targets in difficult-to-treat diseases and causes of drug resistance. Applying this information can achieve cost savings by avoiding the use of treatments that are ineffective in particular patients.
Overexpression of genetic mutations renders some cancers less susceptible to certain treatments, but has opened the door to individualized molecularly guided treatment strategies. For example, among patients with non–small cell lung cancer, mutations in the epidermal growth factor receptor (EGFR) tyrosine kinase domain predict response to EGFR tyrosine kinase inhibitors, and anaplastic lymphoma kinase (ALK) inhibitors induce response in patients harboring a mutation in EML4-ALK genes. The recognition that human epidermal growth factor receptor (HER)-2 overexpression as a result of ERBB2 gene amplification occurs in as many as 20% of human breast cancers paved the way for the development of HER-2–targeted therapies. Patients with advanced colorectal cancer whose tumors express the KRAS gene mutation do not benefit from an EGFR inhibitor, whereas those with wild-type KRAS have improved survival with EGFR inhibitor treatment.6
BARRIERS TO THE APPLICATION OF PERSONALIZED HEALTHCARE
The availability and potential of personalized healthcare services and technology is not universally recognized or appreciated by consumers and clinicians. This lack of awareness contributes to a shortage of public support and limited demand for such services. Other barriers include misperceptions regarding the impact of personalized healthcare on disease management, limited incentives to use the available technology, and a knowledge gap among healthcare providers.
Lack of awareness and support
As applications of personalized healthcare advance to the point of clinical relevance, it is important to consider strategies for effective implementation into healthcare practice. Personalized healthcare, when more fully implemented, promises to accelerate the progress that healthcare reform hopes to achieve.
A major challenge to widespread adoption of personalized healthcare is limited recognition by the public and some healthcare providers that personalized healthcare can help to achieve better value. For personalized medicine to be embraced, the concept of “helix to health,” or translation of knowledge to the clinical setting, must resonate with the general public. Despite lack of public and provider awareness, the Personalized Medicine Coalition (PMC) has documented the existence of 56 personalized treatment and diagnostic products. Further, more than 200 product labels now recommend genetic testing prior to use to identify likely responders or inform of the influence of genetic variation on safety and effectiveness.
Consumers’ confidence in the efficacy and safety of medicines they take might contribute to the absence of public support for personalized healthcare. Similarly, despite the availability of genomic tests and tools, many physicians who might be advocates for personalized healthcare do not see the relevance of genomic medicine to their practices in terms of direct benefit to patient care.7
Apart from clinicians and consumers, support is also weak among health insurers and employers, even though the return on investment for personalized healthcare may be profound. Payers await the economic outcomes data that are crucial for their commitment to personalized healthcare. In addition, some have concerns about the ethical implications of personalized healthcare (see “Managing Genomic Information Responsibly”).
Perception of impact on treatment and prevention
A frequent criticism of genomics in medicine is that a genetic diagnosis does not help with patient management. In fact, surveillance and management of patients and family members often changes in response to a genetic diagnosis; knowing which gene is involved personalizes medical management. An example is the management of hereditary nonpolyposis colorectal cancer (HNPCC), or Lynch syndrome, which is the most common form of hereditary colon cancer. For a person with HNPCC, the lifetime risk of developing colorectal cancer is approximately 80%. Lynch syndrome is caused by germline mutations in one of three major mismatch repair (MMR) genes (MLH1, MSH2, and MSH6), and it predisposes to other cancers—uterine, stomach, and ovarian—as well. In women with Lynch syndrome, the lifetime risk for uterine cancer is 40%, compared with 4% in the general population.
At least 90% of patients with Lynch syndrome can be detected through MMR testing via microsatellite instability (MSI) or immunohistochemistry (IHC).8 MSI is a cellular phenotype that indicates a deficiency in at least one DNA MMR protein.
Although 5-fluorouracil–based chemo therapy is the standard of care for treatment of colorectal cancer, it confers no survival advantage in patients with MMR-IHC null (lack of expression of the gene) or MSI-high sporadic colorectal cancer.9,10 Knowing the status of MMR proteins, therefore, would alter the decision regarding neoadjuvant and adjuvant chemotherapy.
Perception of value
Implementation of pharmacogenomics into clinical practice has lagged. One major reason is the lack of an obvious business model for a product that may only be required once in an individual patient’s lifetime.11
A second barrier to integration lies in the limited demand for pharmacogenomics from physicians. This may be related partly to limited expertise in genetics among many physicians and to significant pushback from payers against today’s costs. Without reimbursement, little incentive exists for pharmacogenomics diagnostics. The incentive for physicians is further depressed, perhaps appropriately, when randomized controlled studies fail to demonstrate improved clinical outcomes with the use of pharmacogenomicbased treatment strategies. Two such examples are genotype-guided warfarin dosing, which failed in a randomized controlled trial to improve the proportion of international normalized ratios in the therapeutic range,12 and dosing of clopidogrel based on platelet reactivity, which did not improve outcomes after percutaneous coronary intervention compared with standard dosing in a randomized double-blind clinical trial.13
A significant delay in obtaining the results of pharmacogenomics testing, which also postpones the prescribing encounter, is another major drawback.

A knowledge gap persists
At present, delivery of personalized healthcare is not part of the usual training of physicians and other healthcare providers who are the gatekeepers of medicine. Few medical schools incorporate human and medical genetics, genomics, and pharmacogenomics into their curricula. Genetics is inadequately emphasized in residency curricula outside of pediatrics, family medicine, and obstetrics/gynecology.
The resulting knowledge gap is a fundamental factor in the lack of interest in using genomics in clinical medicine. Educating consumers and physicians at all levels, including specialty societies as well as insurers, will be key to expanding utilization of personalized healthcare. Educating payers and providing them with more data on economic outcomes associated with personalized healthcare will be necessary for adoption into clinical practice; implementation will lag as long as reimbursement decisions do not support personalized approaches to medicine.
As DNA sequencing technology has become less expensive and more powerful, companies have begun to market personal genomic testing. As a result, patients who use these services will increasingly want to discuss the results with their physicians. A significant number of clinicians are unfamiliar with personal genomic testing and emerging genetic testing options. In one survey of physicians who attended educational sessions that discussed recent developments in clinical genetics, only 37% indicated that they were familiar with recent genetic research that affected their patients.14
Targeted education will enhance physicians’ understanding of probabilities and risk estimates from the use of genomic testing; it will also improve recognition of potential causes of patient anxiety, gene variants of unknown significance, and follow-up tests and procedures that can add to expense. Nonphysician healthcare providers (ie, nurses and physician assistants) of direct care also will benefit from education.
INTEGRATING PERSONALIZED HEALTHCARE INTO CLINICAL PRACTICE
Practice standardization and an overhaul of the health information technology (HIT) infrastructure are needed if we are to reap the potential benefits of personalized healthcare. Creative approaches to practitioner education, which are being used in some institutions, must become more widespread. Similarly, the models for successful integration of personalized healthcare that have been achieved in some settings also can be implemented in other institutions.
Data collection and integration must be prioritized
Personalized healthcare can be both predictive and preventive, but moving past the disruptive phase of personalized healthcare will require a radical transformation of the healthcare “ecosystem” and HIT infrastructure.
Although data collection in the current system is extensive, data sharing and data management are inadequate. The pace at which HIT links clinical and genetic information must be accelerated. HIT will expedite innovation and implementation of personalized healthcare, allowing greater integration of data to permit improved data analysis capability. The ultimate goal is to create an interoperable system that connects these data across hospitals and clinicians to help clinicians interpret genomic and other risk information to better inform patient care.
Fully integrated health systems support better coordination of care and optimize the treatment of individual patients: linking research findings, treatment guidelines, treatment outcomes based on genetic profiles, and the individual patient’s own genetic profile will help to personalize treatments. Genomic information added to an individual’s electronic medical record along with improved data-sharing will facilitate clinicians’ ability to retrieve outcomes data based on patient characteristics.
Care models must be standardized, evidence-based practices must be executed, and care must be coordinated yet decentralized. In this way, clinicians can use the electronic medical record as an interoperable patient record to determine a personalized pathway to patient management. Standardization reduces variability in practice and permits seamless execution of care. Automation is imperative to achieving standardization, irrespective of the care supervisor. Investments must therefore be made to stimulate electronic medical record decision support.
In addition, larger data sets will be needed to identify the types of patients likely to respond to a treatment. Ideal data sets would be large enough to have adequate statistical power, be publicly available, standardize the collection of data with respect to response to therapy and toxicity, and contain data on concomitant collections of biologic samples.
Reimbursement must keep pace with medical advances
Payer willingness to reimburse for genomic tests and treatments will determine the pace of integration of personalized healthcare into clinical practice. Evidence that enhanced value can be derived from personalized approaches to medicine must be generated before personalized healthcare gains widespread acceptance by payers.
In addition, care-coordinated models must be developed to promote a value-based agenda that facilitates physician accountability and encourages clinical integration.
Innovative approaches are needed to educate providers
Development of point-of-care tools. Because information overload and lack of time are obstacles to clinicians’ efforts to incorporate genomic information into clinical practice, emphasis must be placed on genomic applications that have demonstrated utility. Engaging busy clinicians with point-of-care tools will maximize the relevance of the genomic information they receive and encourage effective use of their time. Decision-making should be supported through automatic risk assessment and management recommendations.
Educational tools. The National Coalition for Health Professional Education in Genetics (NCHPEG) was borne out of the recognition that the pace of genomic discovery far exceeds the pace at which healthcare providers can be educated. Its vision is to improve healthcare through informed use of genomic resources. NCHPEG is a member-based organization whose stakeholders include professional societies, hospitals, advocacy groups, and industry; it attempts to identify the specific educational needs for particular target audiences and then address these needs. It achieves its goals through the use of point-of-care tools and educational programs for continuing medical education credit.
One NCHPEG tool is the Pregnancy and Health Profile, which is a risk assessment and screening tool that attempts to improve the identification of women and babies at risk of developing genetic disease. It collects personal and family history information, performs a risk assessment for the clinician, and provides clinical decision support and education.
Another example of an educational tool is the “Genes to Society” curriculum initiated by The Johns Hopkins University School of Medicine in August 2009. The curriculum is being used as “the foundation for the scientific and clinical career development of future physicians.”15
Using personal genomic testing for education. The number of direct-to-consumer genomic tests is growing, and their market penetration will only increase as the cost of supplying a personal genome continues to decline. Whole genome scanning is being offered with the promise of identifying genetic predisposition to multiple diseases.
Participation in personal genomic testing may be a useful educational tool. Medical students, residents, and practicing physicians who participate in testing may be better equipped to advise patients about the processes involved and the potential utility and limitations of direct-to-consumer genotyping.14
Some companies that offer direct-to-consumer genomic testing provide telephone support from genetic counselors to help clients and their healthcare providers manage genetic information. Counselor services include identifying hereditary risks and reviewing diagnostic, preventive, and early-detection options.
Implementing pharmacogenomics into practice: Decision support systems are needed
A genomic decision support system that guides medication prescribing is needed to implement pharmacogenomic diagnostics. For such a system to achieve the goal of selecting the best medication for each individual, it must do the following:
- Test all polymorphisms relevant to the prescribing of any medication
- Be completed with no out-of-pocket cost to the patient
- Be performed before the patient requires the medication
- Provide results that will be interpreted as part of an individualized pharmacogenomics consult.11

The 1200 Patients Project, a pilot research study under way at the Center for Personalized Therapeutics at the University of Chicago, is attempting to demonstrate the feasibility of incorporating pharmacogenomic testing into routine clinical practice for medication treatment decisions. DNA samples from patients who are taking at least one prescription medication are being tested for differences in genes that may suggest greater effectiveness or an increased risk of side effects from certain medications.
Solutions in practice
Cleveland Clinic’s genetics-based management of Lynch syndrome, the integration of genetics services during patient appointments at Cleveland Clinic, and a coordinated approach at The Ohio State University Medical Center are examples of practical applications of personalized healthcare.
Colorectal cancer management. One example of a personalized approach to medicine that improves health outcome while achieving cost savings is the genetics-based approach to HNPCC (Lynch syndrome) at Cleveland Clinic.
Early identification of Lynch syndrome by screening all colorectal cancer patients has been shown to save $250,000 per life-year gained in the United States.16 All colorectal cancers resected at the Cleveland Clinic main campus are routinely screened for MSI and IHC, and the process is embedded into the routine pathology workflow. With the patients’ foreknowledge, a gastrointestinal cancer genetics counselor scans the list of MSI and IHC results each week. Patients who are MSI-high or IHC-null are invited to receive genetic counseling and consider germline single-gene testing guided by the IHC results. With this active approach, patient uptake is 80%; in comparison, with a passive approach (MSI/IHC results are placed in the pathology report), the uptake is 14%17 (B. Leach and C. Eng, unpublished data, 2011).The successful application of the active approach requires the close cooperation of multiple disciplines, including members of the Cleveland Clinic Genomic Medicine, Pathology & Laboratory Medicine, and Digestive Disease Institutes.18
Integrating genetics-based care at Cleveland Clinic. Time delays for genetics services and limited collaboration with managing physicians who are not genetics specialists reduces genetics-based access and availability. Broad access to genetics clinical services is a means of clinical integration of genetics-enabled care. Providing patients and healthcare providers with easy access and short wait times is vital for clinical integration of genetics-enabled personalized healthcare.
As part of a patient-centered focus on medicine, clinical genetics services have been integrated throughout Cleveland Clinic. The system has two genetics clinics at its main campus and has embedded multiple genetics satellites within its nongenetics clinics, easing access. Genetics counselors are stationed in the same areas of practice as referring providers. Although patient encounters have increased at the medical genetics clinic in the Genomic Medicine Institute, genetics consultations no longer require an extra trip to the clinic since they are integrated into existing appointments. With this approach, large numbers of patients can be seen with no wait times.
Coordinated care at The Ohio State University Medical Center. The Center for Personalized Health Care at The Ohio State University Medical Center (OSUMC) embraces a systems-based care-coordinated model that improves care by executing standardized processes and automating routine tasks. The Institute for Systems Biology, which was established to develop genomics, wellness, and chronic disease biomarkers, collaborates with OSUMC on pilot projects in chronic disease, including cancer.
The OSUMC has a closed system in which it is the payer, employer, and provider of healthcare. This closed system serves as an ideal testing ground for reform. Goals include intervention in disease before symptoms appear and maintenance of wellness. The data from these demonstration projects should facilitate adoption of personalized healthcare by improving physician acceptance of personalized approaches and satisfying payers that personalized healthcare is cost-effective.

- Personalized medicine. Coriell Institute for Medical Research Web site. http://www.coriell.org/personalized-medicine. Updated 2011. Accessed December 27, 2011.
- The 2012 Statistical Abstract. U.S. Census Bureau Web site. http://www.census.gov/compendia/statab/cats/income_expenditures_poverty_wealth/gross_domestic_product_gdp.html. Updated September 27, 2011. Accessed December 22, 2011.
- National health expenditure fact sheet. Center for Medicare & Medicaid Services (CMS) Web site. https://www.cms.gov/NationalHealthExpendData/25_NHE_Fact_Sheet.asp. Updated November 4, 2011. Accessed December 22, 2011.
- New England Healthcare Institute (NEHI). Thinking outside the pillbox: A system-wide approach to improving patient medication adherence for chronic disease. NEHI Web site. http://www.nehi.net/publications/44/thinking_outside_the_pillbox_a_systemwide_approach_to_improving_patient_medication_adherence_for_chronic_disease. Published August 12, 2009. Accessed December 22, 2011.
- Spear BB, Heath-Chiozzi M, Huff J. Clinical application of pharmacogenetics. Trends Mol Med 2001; 7:201–204.
- Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757–1765.
- Feero WG, Green ED. Genomics education for healthcare professionals in the 21st century. JAMA 2011; 306:989–990.
- Meyers M, Wagner MW, Hwang HS, Kinsella TJ, Boothman DA. Role of the hMLH1 DNA mismatch repair protein in flouropyrimidine-mediated cell death and cell cycle responses. Cancer Res 2001; 61:5193–5201.
- Carethers JM, Chauhan DP, Fink D, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology 1999; 117:123–131.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003; 349:247–257.
- Ratain MJ. Personalized medicine: building the GPS to take us there. Clin Pharmacol Ther 2007; 81:321–322.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation [published online ahead of print November 7, 2007]. Circulation 2007; 116:2563–2570. doi: 10.1161/CIRCULATIONAHA.107.737312
- Price MJ, Angiolillo DJ, Teirstein PS, et al .Platelet reactivity and cardiovascular outcomes after percutaneous coronary intervention: a time-dependent analysis of the Gauging Responsiveness with a VerifyNow P2Y12 assay: Impact on Thrombosis and Safety (GRAVITAS) trial [published online ahead of print August 29, 2011]. Circulation 2011; 124:1132–1137. doi: 10.1161/CIRCULATIONAHA.111.029165
- Sharp RR, Goldlust ME, Eng C. Addressing gaps in physician education using personal genomic testing. Genet Med 2011; 13:750–751.
- Wiener CM, Thomas PA, Goodspeed E, Valle D, Nichols DG. “Genes to society”—the logic and process of the new curriculum for the Johns Hopkins University School of Medicine. Acad Med 2010; 85:498–506.
- Ladabaum U, Wang G, Terdiman J, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer: a costeffectiveness analysis. Ann Intern Med 2011; 155:69–79.
- Leach B, Eng C, Kalady M, et al. Sharing the responsibility: multidisciplinary model improves colorectal cancer microsatellite testing. Paper presented at: InSight 2009 Annual Conference: September 2009; Orlando, FL.
- Manolio TA, Chisolm R, Ozenberger B, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med Forthcoming.
Personalized healthcare is the tailoring of medical management and patient care to the individual characteristics of each patient. This is achieved by incorporating the genetic and genomic makeup of an individual and his or her family medical history, environment, health-related behaviors, culture, and values into a complete health picture that can be used to customize care. Another level of personalization, often called personalized medicine, involves the selection of drug therapy through the use of tests to determine the genes and gene interactions that can reliably predict an individual’s response to a given therapy. This white paper focuses largely on the use of personalized healthcare as a risk prediction tool.
CURRENT STATUS OF PERSONALIZED HEALTHCARE
The case for personalized healthcare: Seeking value
To fully appreciate the need to advance the adoption of personalized healthcare into the delivery of medicine, one must consider the operation of our current healthcare system and its inefficiencies in terms of delivery and cost, its imprecision in the selection of therapies, and its inability to optimize outcomes. The framework of the US healthcare system as it is now constructed is expensive, disease-directed (instead of health- and wellness-directed), fragmented, and complex. While gross domestic product (GDP) in the United States has increased by approximately 3% per year,2 the compounded growth rate of healthcare expenditures is 6.1% per year. Healthcare in the aggregate now represents 17.6% of GDP and 27% of spending by the federal government and consumes 28% of the average household’s discretionary spending, surpassed only by housing.3
Personalized healthcare can potentially address the need for value consistent with the healthcare system’s prominent share of the US economy. The growth in healthcare spending is certain to be a target of the newly created Joint Select Committee on Deficit Reduction (created by the Budget Control Act of 2011), which is tasked with deficit reduction of at least $1.5 trillion over a 10-year period.
The need to address healthcare costs has been recognized in the Patient Protection and Affordable Care Act, a central feature of which is the creation of integrated health systems that pay for value based on quality, cost containment, and consumer experience. The legislation was enacted to transform healthcare in a variety of ways to make it more sustainable. The Patient Protection and Affordable Care Act seeks to end fragmentation by expanding the use of information technology to reorganize the delivery system and to prevent errors, shifting from volume-based incentives to incentives based on performance and outcomes, and rewarding effective healthcare delivery measures and good patient outcomes.
A shift from reactive to proactive
The premise behind personalized healthcare is the potential for more efficient healthcare, with the assumption that efficiency translates to lower cost and improved patient care.
Although healthcare reform is most often referred to in the context of improving access to care through insurance coverage mandates, true healthcare reform shifts current healthcare models from the practice of reactive medicine to the practice of proactive medicine, in which the tools of personalized healthcare (ie, genetics, genomics, and other molecular diagnostics) enable not only better quality of care but also less expensive care.
Several personalized tools have long been accepted into mainstream medicine. Two examples are the family history, which is the least expensive and most available genetic evaluation tool, and ABO blood typing for safe transfusions (as ABO blood types are alleles of a gene). In fact, much of what is now considered mainstream medical management was at one time considered new. To allow further evolution of medical practice, our challenge is to open our minds to the possibility that personalized proactive medicine can improve healthcare.
The new vision: More precise management
The trial-and-error approach to treating disease is inefficient and costly. Many drugs are effective for only about 50% of patients, often leading to switching or intensification of therapy that requires multiple patient visits.
Personalized medicine considers pharmacokinetic and other characteristics in selection of drug dosages. Genomic testing has the potential to provide clearer insight into the more successful use of currently available medicines. Treatment decisions (ie, drug and drug dosage choice) made on the basis of pharmacogenomic testing should increase adherence through greater effectiveness and fewer adverse drug reactions.
A massive amount of waste is related to pharmaceutical nonadherence and noncompliance. The New England Healthcare Institute has estimated that medication nonadherence costs the healthcare system $290 billion annually.4 Methodologies targeted at individual patients to improve adherence to drug regimens could save the healthcare system a tremendous amount of money.
Cancer management as a model for personalized healthcare. Personalization of therapy is especially suited to cancer management, given that the response to nonspecific cancer chemotherapy is suboptimal in most patients yet exposes them to adverse effects.5 Large-scale sequencing of human cancer genomes is rapidly changing the understanding of cancer biology and is identifying new targets in difficult-to-treat diseases and causes of drug resistance. Applying this information can achieve cost savings by avoiding the use of treatments that are ineffective in particular patients.
Overexpression of genetic mutations renders some cancers less susceptible to certain treatments, but has opened the door to individualized molecularly guided treatment strategies. For example, among patients with non–small cell lung cancer, mutations in the epidermal growth factor receptor (EGFR) tyrosine kinase domain predict response to EGFR tyrosine kinase inhibitors, and anaplastic lymphoma kinase (ALK) inhibitors induce response in patients harboring a mutation in EML4-ALK genes. The recognition that human epidermal growth factor receptor (HER)-2 overexpression as a result of ERBB2 gene amplification occurs in as many as 20% of human breast cancers paved the way for the development of HER-2–targeted therapies. Patients with advanced colorectal cancer whose tumors express the KRAS gene mutation do not benefit from an EGFR inhibitor, whereas those with wild-type KRAS have improved survival with EGFR inhibitor treatment.6
BARRIERS TO THE APPLICATION OF PERSONALIZED HEALTHCARE
The availability and potential of personalized healthcare services and technology is not universally recognized or appreciated by consumers and clinicians. This lack of awareness contributes to a shortage of public support and limited demand for such services. Other barriers include misperceptions regarding the impact of personalized healthcare on disease management, limited incentives to use the available technology, and a knowledge gap among healthcare providers.
Lack of awareness and support
As applications of personalized healthcare advance to the point of clinical relevance, it is important to consider strategies for effective implementation into healthcare practice. Personalized healthcare, when more fully implemented, promises to accelerate the progress that healthcare reform hopes to achieve.
A major challenge to widespread adoption of personalized healthcare is limited recognition by the public and some healthcare providers that personalized healthcare can help to achieve better value. For personalized medicine to be embraced, the concept of “helix to health,” or translation of knowledge to the clinical setting, must resonate with the general public. Despite lack of public and provider awareness, the Personalized Medicine Coalition (PMC) has documented the existence of 56 personalized treatment and diagnostic products. Further, more than 200 product labels now recommend genetic testing prior to use to identify likely responders or inform of the influence of genetic variation on safety and effectiveness.
Consumers’ confidence in the efficacy and safety of medicines they take might contribute to the absence of public support for personalized healthcare. Similarly, despite the availability of genomic tests and tools, many physicians who might be advocates for personalized healthcare do not see the relevance of genomic medicine to their practices in terms of direct benefit to patient care.7
Apart from clinicians and consumers, support is also weak among health insurers and employers, even though the return on investment for personalized healthcare may be profound. Payers await the economic outcomes data that are crucial for their commitment to personalized healthcare. In addition, some have concerns about the ethical implications of personalized healthcare (see “Managing Genomic Information Responsibly”).
Perception of impact on treatment and prevention
A frequent criticism of genomics in medicine is that a genetic diagnosis does not help with patient management. In fact, surveillance and management of patients and family members often changes in response to a genetic diagnosis; knowing which gene is involved personalizes medical management. An example is the management of hereditary nonpolyposis colorectal cancer (HNPCC), or Lynch syndrome, which is the most common form of hereditary colon cancer. For a person with HNPCC, the lifetime risk of developing colorectal cancer is approximately 80%. Lynch syndrome is caused by germline mutations in one of three major mismatch repair (MMR) genes (MLH1, MSH2, and MSH6), and it predisposes to other cancers—uterine, stomach, and ovarian—as well. In women with Lynch syndrome, the lifetime risk for uterine cancer is 40%, compared with 4% in the general population.
At least 90% of patients with Lynch syndrome can be detected through MMR testing via microsatellite instability (MSI) or immunohistochemistry (IHC).8 MSI is a cellular phenotype that indicates a deficiency in at least one DNA MMR protein.
Although 5-fluorouracil–based chemo therapy is the standard of care for treatment of colorectal cancer, it confers no survival advantage in patients with MMR-IHC null (lack of expression of the gene) or MSI-high sporadic colorectal cancer.9,10 Knowing the status of MMR proteins, therefore, would alter the decision regarding neoadjuvant and adjuvant chemotherapy.
Perception of value
Implementation of pharmacogenomics into clinical practice has lagged. One major reason is the lack of an obvious business model for a product that may only be required once in an individual patient’s lifetime.11
A second barrier to integration lies in the limited demand for pharmacogenomics from physicians. This may be related partly to limited expertise in genetics among many physicians and to significant pushback from payers against today’s costs. Without reimbursement, little incentive exists for pharmacogenomics diagnostics. The incentive for physicians is further depressed, perhaps appropriately, when randomized controlled studies fail to demonstrate improved clinical outcomes with the use of pharmacogenomicbased treatment strategies. Two such examples are genotype-guided warfarin dosing, which failed in a randomized controlled trial to improve the proportion of international normalized ratios in the therapeutic range,12 and dosing of clopidogrel based on platelet reactivity, which did not improve outcomes after percutaneous coronary intervention compared with standard dosing in a randomized double-blind clinical trial.13
A significant delay in obtaining the results of pharmacogenomics testing, which also postpones the prescribing encounter, is another major drawback.
A knowledge gap persists
At present, delivery of personalized healthcare is not part of the usual training of physicians and other healthcare providers who are the gatekeepers of medicine. Few medical schools incorporate human and medical genetics, genomics, and pharmacogenomics into their curricula. Genetics is inadequately emphasized in residency curricula outside of pediatrics, family medicine, and obstetrics/gynecology.
The resulting knowledge gap is a fundamental factor in the lack of interest in using genomics in clinical medicine. Educating consumers and physicians at all levels, including specialty societies as well as insurers, will be key to expanding utilization of personalized healthcare. Educating payers and providing them with more data on economic outcomes associated with personalized healthcare will be necessary for adoption into clinical practice; implementation will lag as long as reimbursement decisions do not support personalized approaches to medicine.
As DNA sequencing technology has become less expensive and more powerful, companies have begun to market personal genomic testing. As a result, patients who use these services will increasingly want to discuss the results with their physicians. A significant number of clinicians are unfamiliar with personal genomic testing and emerging genetic testing options. In one survey of physicians who attended educational sessions that discussed recent developments in clinical genetics, only 37% indicated that they were familiar with recent genetic research that affected their patients.14
Targeted education will enhance physicians’ understanding of probabilities and risk estimates from the use of genomic testing; it will also improve recognition of potential causes of patient anxiety, gene variants of unknown significance, and follow-up tests and procedures that can add to expense. Nonphysician healthcare providers (ie, nurses and physician assistants) of direct care also will benefit from education.
INTEGRATING PERSONALIZED HEALTHCARE INTO CLINICAL PRACTICE
Practice standardization and an overhaul of the health information technology (HIT) infrastructure are needed if we are to reap the potential benefits of personalized healthcare. Creative approaches to practitioner education, which are being used in some institutions, must become more widespread. Similarly, the models for successful integration of personalized healthcare that have been achieved in some settings also can be implemented in other institutions.
Data collection and integration must be prioritized
Personalized healthcare can be both predictive and preventive, but moving past the disruptive phase of personalized healthcare will require a radical transformation of the healthcare “ecosystem” and HIT infrastructure.
Although data collection in the current system is extensive, data sharing and data management are inadequate. The pace at which HIT links clinical and genetic information must be accelerated. HIT will expedite innovation and implementation of personalized healthcare, allowing greater integration of data to permit improved data analysis capability. The ultimate goal is to create an interoperable system that connects these data across hospitals and clinicians to help clinicians interpret genomic and other risk information to better inform patient care.
Fully integrated health systems support better coordination of care and optimize the treatment of individual patients: linking research findings, treatment guidelines, treatment outcomes based on genetic profiles, and the individual patient’s own genetic profile will help to personalize treatments. Genomic information added to an individual’s electronic medical record along with improved data-sharing will facilitate clinicians’ ability to retrieve outcomes data based on patient characteristics.
Care models must be standardized, evidence-based practices must be executed, and care must be coordinated yet decentralized. In this way, clinicians can use the electronic medical record as an interoperable patient record to determine a personalized pathway to patient management. Standardization reduces variability in practice and permits seamless execution of care. Automation is imperative to achieving standardization, irrespective of the care supervisor. Investments must therefore be made to stimulate electronic medical record decision support.
In addition, larger data sets will be needed to identify the types of patients likely to respond to a treatment. Ideal data sets would be large enough to have adequate statistical power, be publicly available, standardize the collection of data with respect to response to therapy and toxicity, and contain data on concomitant collections of biologic samples.
Reimbursement must keep pace with medical advances
Payer willingness to reimburse for genomic tests and treatments will determine the pace of integration of personalized healthcare into clinical practice. Evidence that enhanced value can be derived from personalized approaches to medicine must be generated before personalized healthcare gains widespread acceptance by payers.
In addition, care-coordinated models must be developed to promote a value-based agenda that facilitates physician accountability and encourages clinical integration.
Innovative approaches are needed to educate providers
Development of point-of-care tools. Because information overload and lack of time are obstacles to clinicians’ efforts to incorporate genomic information into clinical practice, emphasis must be placed on genomic applications that have demonstrated utility. Engaging busy clinicians with point-of-care tools will maximize the relevance of the genomic information they receive and encourage effective use of their time. Decision-making should be supported through automatic risk assessment and management recommendations.
Educational tools. The National Coalition for Health Professional Education in Genetics (NCHPEG) was borne out of the recognition that the pace of genomic discovery far exceeds the pace at which healthcare providers can be educated. Its vision is to improve healthcare through informed use of genomic resources. NCHPEG is a member-based organization whose stakeholders include professional societies, hospitals, advocacy groups, and industry; it attempts to identify the specific educational needs for particular target audiences and then address these needs. It achieves its goals through the use of point-of-care tools and educational programs for continuing medical education credit.
One NCHPEG tool is the Pregnancy and Health Profile, which is a risk assessment and screening tool that attempts to improve the identification of women and babies at risk of developing genetic disease. It collects personal and family history information, performs a risk assessment for the clinician, and provides clinical decision support and education.
Another example of an educational tool is the “Genes to Society” curriculum initiated by The Johns Hopkins University School of Medicine in August 2009. The curriculum is being used as “the foundation for the scientific and clinical career development of future physicians.”15
Using personal genomic testing for education. The number of direct-to-consumer genomic tests is growing, and their market penetration will only increase as the cost of supplying a personal genome continues to decline. Whole genome scanning is being offered with the promise of identifying genetic predisposition to multiple diseases.
Participation in personal genomic testing may be a useful educational tool. Medical students, residents, and practicing physicians who participate in testing may be better equipped to advise patients about the processes involved and the potential utility and limitations of direct-to-consumer genotyping.14
Some companies that offer direct-to-consumer genomic testing provide telephone support from genetic counselors to help clients and their healthcare providers manage genetic information. Counselor services include identifying hereditary risks and reviewing diagnostic, preventive, and early-detection options.
Implementing pharmacogenomics into practice: Decision support systems are needed
A genomic decision support system that guides medication prescribing is needed to implement pharmacogenomic diagnostics. For such a system to achieve the goal of selecting the best medication for each individual, it must do the following:
- Test all polymorphisms relevant to the prescribing of any medication
- Be completed with no out-of-pocket cost to the patient
- Be performed before the patient requires the medication
- Provide results that will be interpreted as part of an individualized pharmacogenomics consult.11
The 1200 Patients Project, a pilot research study under way at the Center for Personalized Therapeutics at the University of Chicago, is attempting to demonstrate the feasibility of incorporating pharmacogenomic testing into routine clinical practice for medication treatment decisions. DNA samples from patients who are taking at least one prescription medication are being tested for differences in genes that may suggest greater effectiveness or an increased risk of side effects from certain medications.
Solutions in practice
Cleveland Clinic’s genetics-based management of Lynch syndrome, the integration of genetics services during patient appointments at Cleveland Clinic, and a coordinated approach at The Ohio State University Medical Center are examples of practical applications of personalized healthcare.
Colorectal cancer management. One example of a personalized approach to medicine that improves health outcome while achieving cost savings is the genetics-based approach to HNPCC (Lynch syndrome) at Cleveland Clinic.
Early identification of Lynch syndrome by screening all colorectal cancer patients has been shown to save $250,000 per life-year gained in the United States.16 All colorectal cancers resected at the Cleveland Clinic main campus are routinely screened for MSI and IHC, and the process is embedded into the routine pathology workflow. With the patients’ foreknowledge, a gastrointestinal cancer genetics counselor scans the list of MSI and IHC results each week. Patients who are MSI-high or IHC-null are invited to receive genetic counseling and consider germline single-gene testing guided by the IHC results. With this active approach, patient uptake is 80%; in comparison, with a passive approach (MSI/IHC results are placed in the pathology report), the uptake is 14%17 (B. Leach and C. Eng, unpublished data, 2011).The successful application of the active approach requires the close cooperation of multiple disciplines, including members of the Cleveland Clinic Genomic Medicine, Pathology & Laboratory Medicine, and Digestive Disease Institutes.18
Integrating genetics-based care at Cleveland Clinic. Time delays for genetics services and limited collaboration with managing physicians who are not genetics specialists reduces genetics-based access and availability. Broad access to genetics clinical services is a means of clinical integration of genetics-enabled care. Providing patients and healthcare providers with easy access and short wait times is vital for clinical integration of genetics-enabled personalized healthcare.
As part of a patient-centered focus on medicine, clinical genetics services have been integrated throughout Cleveland Clinic. The system has two genetics clinics at its main campus and has embedded multiple genetics satellites within its nongenetics clinics, easing access. Genetics counselors are stationed in the same areas of practice as referring providers. Although patient encounters have increased at the medical genetics clinic in the Genomic Medicine Institute, genetics consultations no longer require an extra trip to the clinic since they are integrated into existing appointments. With this approach, large numbers of patients can be seen with no wait times.
Coordinated care at The Ohio State University Medical Center. The Center for Personalized Health Care at The Ohio State University Medical Center (OSUMC) embraces a systems-based care-coordinated model that improves care by executing standardized processes and automating routine tasks. The Institute for Systems Biology, which was established to develop genomics, wellness, and chronic disease biomarkers, collaborates with OSUMC on pilot projects in chronic disease, including cancer.
The OSUMC has a closed system in which it is the payer, employer, and provider of healthcare. This closed system serves as an ideal testing ground for reform. Goals include intervention in disease before symptoms appear and maintenance of wellness. The data from these demonstration projects should facilitate adoption of personalized healthcare by improving physician acceptance of personalized approaches and satisfying payers that personalized healthcare is cost-effective.
Personalized healthcare is the tailoring of medical management and patient care to the individual characteristics of each patient. This is achieved by incorporating the genetic and genomic makeup of an individual and his or her family medical history, environment, health-related behaviors, culture, and values into a complete health picture that can be used to customize care. Another level of personalization, often called personalized medicine, involves the selection of drug therapy through the use of tests to determine the genes and gene interactions that can reliably predict an individual’s response to a given therapy. This white paper focuses largely on the use of personalized healthcare as a risk prediction tool.
CURRENT STATUS OF PERSONALIZED HEALTHCARE
The case for personalized healthcare: Seeking value
To fully appreciate the need to advance the adoption of personalized healthcare into the delivery of medicine, one must consider the operation of our current healthcare system and its inefficiencies in terms of delivery and cost, its imprecision in the selection of therapies, and its inability to optimize outcomes. The framework of the US healthcare system as it is now constructed is expensive, disease-directed (instead of health- and wellness-directed), fragmented, and complex. While gross domestic product (GDP) in the United States has increased by approximately 3% per year,2 the compounded growth rate of healthcare expenditures is 6.1% per year. Healthcare in the aggregate now represents 17.6% of GDP and 27% of spending by the federal government and consumes 28% of the average household’s discretionary spending, surpassed only by housing.3
Personalized healthcare can potentially address the need for value consistent with the healthcare system’s prominent share of the US economy. The growth in healthcare spending is certain to be a target of the newly created Joint Select Committee on Deficit Reduction (created by the Budget Control Act of 2011), which is tasked with deficit reduction of at least $1.5 trillion over a 10-year period.
The need to address healthcare costs has been recognized in the Patient Protection and Affordable Care Act, a central feature of which is the creation of integrated health systems that pay for value based on quality, cost containment, and consumer experience. The legislation was enacted to transform healthcare in a variety of ways to make it more sustainable. The Patient Protection and Affordable Care Act seeks to end fragmentation by expanding the use of information technology to reorganize the delivery system and to prevent errors, shifting from volume-based incentives to incentives based on performance and outcomes, and rewarding effective healthcare delivery measures and good patient outcomes.
A shift from reactive to proactive
The premise behind personalized healthcare is the potential for more efficient healthcare, with the assumption that efficiency translates to lower cost and improved patient care.
Although healthcare reform is most often referred to in the context of improving access to care through insurance coverage mandates, true healthcare reform shifts current healthcare models from the practice of reactive medicine to the practice of proactive medicine, in which the tools of personalized healthcare (ie, genetics, genomics, and other molecular diagnostics) enable not only better quality of care but also less expensive care.
Several personalized tools have long been accepted into mainstream medicine. Two examples are the family history, which is the least expensive and most available genetic evaluation tool, and ABO blood typing for safe transfusions (as ABO blood types are alleles of a gene). In fact, much of what is now considered mainstream medical management was at one time considered new. To allow further evolution of medical practice, our challenge is to open our minds to the possibility that personalized proactive medicine can improve healthcare.
The new vision: More precise management
The trial-and-error approach to treating disease is inefficient and costly. Many drugs are effective for only about 50% of patients, often leading to switching or intensification of therapy that requires multiple patient visits.
Personalized medicine considers pharmacokinetic and other characteristics in selection of drug dosages. Genomic testing has the potential to provide clearer insight into the more successful use of currently available medicines. Treatment decisions (ie, drug and drug dosage choice) made on the basis of pharmacogenomic testing should increase adherence through greater effectiveness and fewer adverse drug reactions.
A massive amount of waste is related to pharmaceutical nonadherence and noncompliance. The New England Healthcare Institute has estimated that medication nonadherence costs the healthcare system $290 billion annually.4 Methodologies targeted at individual patients to improve adherence to drug regimens could save the healthcare system a tremendous amount of money.
Cancer management as a model for personalized healthcare. Personalization of therapy is especially suited to cancer management, given that the response to nonspecific cancer chemotherapy is suboptimal in most patients yet exposes them to adverse effects.5 Large-scale sequencing of human cancer genomes is rapidly changing the understanding of cancer biology and is identifying new targets in difficult-to-treat diseases and causes of drug resistance. Applying this information can achieve cost savings by avoiding the use of treatments that are ineffective in particular patients.
Overexpression of genetic mutations renders some cancers less susceptible to certain treatments, but has opened the door to individualized molecularly guided treatment strategies. For example, among patients with non–small cell lung cancer, mutations in the epidermal growth factor receptor (EGFR) tyrosine kinase domain predict response to EGFR tyrosine kinase inhibitors, and anaplastic lymphoma kinase (ALK) inhibitors induce response in patients harboring a mutation in EML4-ALK genes. The recognition that human epidermal growth factor receptor (HER)-2 overexpression as a result of ERBB2 gene amplification occurs in as many as 20% of human breast cancers paved the way for the development of HER-2–targeted therapies. Patients with advanced colorectal cancer whose tumors express the KRAS gene mutation do not benefit from an EGFR inhibitor, whereas those with wild-type KRAS have improved survival with EGFR inhibitor treatment.6
BARRIERS TO THE APPLICATION OF PERSONALIZED HEALTHCARE
The availability and potential of personalized healthcare services and technology is not universally recognized or appreciated by consumers and clinicians. This lack of awareness contributes to a shortage of public support and limited demand for such services. Other barriers include misperceptions regarding the impact of personalized healthcare on disease management, limited incentives to use the available technology, and a knowledge gap among healthcare providers.
Lack of awareness and support
As applications of personalized healthcare advance to the point of clinical relevance, it is important to consider strategies for effective implementation into healthcare practice. Personalized healthcare, when more fully implemented, promises to accelerate the progress that healthcare reform hopes to achieve.
A major challenge to widespread adoption of personalized healthcare is limited recognition by the public and some healthcare providers that personalized healthcare can help to achieve better value. For personalized medicine to be embraced, the concept of “helix to health,” or translation of knowledge to the clinical setting, must resonate with the general public. Despite lack of public and provider awareness, the Personalized Medicine Coalition (PMC) has documented the existence of 56 personalized treatment and diagnostic products. Further, more than 200 product labels now recommend genetic testing prior to use to identify likely responders or inform of the influence of genetic variation on safety and effectiveness.
Consumers’ confidence in the efficacy and safety of medicines they take might contribute to the absence of public support for personalized healthcare. Similarly, despite the availability of genomic tests and tools, many physicians who might be advocates for personalized healthcare do not see the relevance of genomic medicine to their practices in terms of direct benefit to patient care.7
Apart from clinicians and consumers, support is also weak among health insurers and employers, even though the return on investment for personalized healthcare may be profound. Payers await the economic outcomes data that are crucial for their commitment to personalized healthcare. In addition, some have concerns about the ethical implications of personalized healthcare (see “Managing Genomic Information Responsibly”).
Perception of impact on treatment and prevention
A frequent criticism of genomics in medicine is that a genetic diagnosis does not help with patient management. In fact, surveillance and management of patients and family members often changes in response to a genetic diagnosis; knowing which gene is involved personalizes medical management. An example is the management of hereditary nonpolyposis colorectal cancer (HNPCC), or Lynch syndrome, which is the most common form of hereditary colon cancer. For a person with HNPCC, the lifetime risk of developing colorectal cancer is approximately 80%. Lynch syndrome is caused by germline mutations in one of three major mismatch repair (MMR) genes (MLH1, MSH2, and MSH6), and it predisposes to other cancers—uterine, stomach, and ovarian—as well. In women with Lynch syndrome, the lifetime risk for uterine cancer is 40%, compared with 4% in the general population.
At least 90% of patients with Lynch syndrome can be detected through MMR testing via microsatellite instability (MSI) or immunohistochemistry (IHC).8 MSI is a cellular phenotype that indicates a deficiency in at least one DNA MMR protein.
Although 5-fluorouracil–based chemo therapy is the standard of care for treatment of colorectal cancer, it confers no survival advantage in patients with MMR-IHC null (lack of expression of the gene) or MSI-high sporadic colorectal cancer.9,10 Knowing the status of MMR proteins, therefore, would alter the decision regarding neoadjuvant and adjuvant chemotherapy.
Perception of value
Implementation of pharmacogenomics into clinical practice has lagged. One major reason is the lack of an obvious business model for a product that may only be required once in an individual patient’s lifetime.11
A second barrier to integration lies in the limited demand for pharmacogenomics from physicians. This may be related partly to limited expertise in genetics among many physicians and to significant pushback from payers against today’s costs. Without reimbursement, little incentive exists for pharmacogenomics diagnostics. The incentive for physicians is further depressed, perhaps appropriately, when randomized controlled studies fail to demonstrate improved clinical outcomes with the use of pharmacogenomicbased treatment strategies. Two such examples are genotype-guided warfarin dosing, which failed in a randomized controlled trial to improve the proportion of international normalized ratios in the therapeutic range,12 and dosing of clopidogrel based on platelet reactivity, which did not improve outcomes after percutaneous coronary intervention compared with standard dosing in a randomized double-blind clinical trial.13
A significant delay in obtaining the results of pharmacogenomics testing, which also postpones the prescribing encounter, is another major drawback.
A knowledge gap persists
At present, delivery of personalized healthcare is not part of the usual training of physicians and other healthcare providers who are the gatekeepers of medicine. Few medical schools incorporate human and medical genetics, genomics, and pharmacogenomics into their curricula. Genetics is inadequately emphasized in residency curricula outside of pediatrics, family medicine, and obstetrics/gynecology.
The resulting knowledge gap is a fundamental factor in the lack of interest in using genomics in clinical medicine. Educating consumers and physicians at all levels, including specialty societies as well as insurers, will be key to expanding utilization of personalized healthcare. Educating payers and providing them with more data on economic outcomes associated with personalized healthcare will be necessary for adoption into clinical practice; implementation will lag as long as reimbursement decisions do not support personalized approaches to medicine.
As DNA sequencing technology has become less expensive and more powerful, companies have begun to market personal genomic testing. As a result, patients who use these services will increasingly want to discuss the results with their physicians. A significant number of clinicians are unfamiliar with personal genomic testing and emerging genetic testing options. In one survey of physicians who attended educational sessions that discussed recent developments in clinical genetics, only 37% indicated that they were familiar with recent genetic research that affected their patients.14
Targeted education will enhance physicians’ understanding of probabilities and risk estimates from the use of genomic testing; it will also improve recognition of potential causes of patient anxiety, gene variants of unknown significance, and follow-up tests and procedures that can add to expense. Nonphysician healthcare providers (ie, nurses and physician assistants) of direct care also will benefit from education.
INTEGRATING PERSONALIZED HEALTHCARE INTO CLINICAL PRACTICE
Practice standardization and an overhaul of the health information technology (HIT) infrastructure are needed if we are to reap the potential benefits of personalized healthcare. Creative approaches to practitioner education, which are being used in some institutions, must become more widespread. Similarly, the models for successful integration of personalized healthcare that have been achieved in some settings also can be implemented in other institutions.
Data collection and integration must be prioritized
Personalized healthcare can be both predictive and preventive, but moving past the disruptive phase of personalized healthcare will require a radical transformation of the healthcare “ecosystem” and HIT infrastructure.
Although data collection in the current system is extensive, data sharing and data management are inadequate. The pace at which HIT links clinical and genetic information must be accelerated. HIT will expedite innovation and implementation of personalized healthcare, allowing greater integration of data to permit improved data analysis capability. The ultimate goal is to create an interoperable system that connects these data across hospitals and clinicians to help clinicians interpret genomic and other risk information to better inform patient care.
Fully integrated health systems support better coordination of care and optimize the treatment of individual patients: linking research findings, treatment guidelines, treatment outcomes based on genetic profiles, and the individual patient’s own genetic profile will help to personalize treatments. Genomic information added to an individual’s electronic medical record along with improved data-sharing will facilitate clinicians’ ability to retrieve outcomes data based on patient characteristics.
Care models must be standardized, evidence-based practices must be executed, and care must be coordinated yet decentralized. In this way, clinicians can use the electronic medical record as an interoperable patient record to determine a personalized pathway to patient management. Standardization reduces variability in practice and permits seamless execution of care. Automation is imperative to achieving standardization, irrespective of the care supervisor. Investments must therefore be made to stimulate electronic medical record decision support.
In addition, larger data sets will be needed to identify the types of patients likely to respond to a treatment. Ideal data sets would be large enough to have adequate statistical power, be publicly available, standardize the collection of data with respect to response to therapy and toxicity, and contain data on concomitant collections of biologic samples.
Reimbursement must keep pace with medical advances
Payer willingness to reimburse for genomic tests and treatments will determine the pace of integration of personalized healthcare into clinical practice. Evidence that enhanced value can be derived from personalized approaches to medicine must be generated before personalized healthcare gains widespread acceptance by payers.
In addition, care-coordinated models must be developed to promote a value-based agenda that facilitates physician accountability and encourages clinical integration.
Innovative approaches are needed to educate providers
Development of point-of-care tools. Because information overload and lack of time are obstacles to clinicians’ efforts to incorporate genomic information into clinical practice, emphasis must be placed on genomic applications that have demonstrated utility. Engaging busy clinicians with point-of-care tools will maximize the relevance of the genomic information they receive and encourage effective use of their time. Decision-making should be supported through automatic risk assessment and management recommendations.
Educational tools. The National Coalition for Health Professional Education in Genetics (NCHPEG) was borne out of the recognition that the pace of genomic discovery far exceeds the pace at which healthcare providers can be educated. Its vision is to improve healthcare through informed use of genomic resources. NCHPEG is a member-based organization whose stakeholders include professional societies, hospitals, advocacy groups, and industry; it attempts to identify the specific educational needs for particular target audiences and then address these needs. It achieves its goals through the use of point-of-care tools and educational programs for continuing medical education credit.
One NCHPEG tool is the Pregnancy and Health Profile, which is a risk assessment and screening tool that attempts to improve the identification of women and babies at risk of developing genetic disease. It collects personal and family history information, performs a risk assessment for the clinician, and provides clinical decision support and education.
Another example of an educational tool is the “Genes to Society” curriculum initiated by The Johns Hopkins University School of Medicine in August 2009. The curriculum is being used as “the foundation for the scientific and clinical career development of future physicians.”15
Using personal genomic testing for education. The number of direct-to-consumer genomic tests is growing, and their market penetration will only increase as the cost of supplying a personal genome continues to decline. Whole genome scanning is being offered with the promise of identifying genetic predisposition to multiple diseases.
Participation in personal genomic testing may be a useful educational tool. Medical students, residents, and practicing physicians who participate in testing may be better equipped to advise patients about the processes involved and the potential utility and limitations of direct-to-consumer genotyping.14
Some companies that offer direct-to-consumer genomic testing provide telephone support from genetic counselors to help clients and their healthcare providers manage genetic information. Counselor services include identifying hereditary risks and reviewing diagnostic, preventive, and early-detection options.
Implementing pharmacogenomics into practice: Decision support systems are needed
A genomic decision support system that guides medication prescribing is needed to implement pharmacogenomic diagnostics. For such a system to achieve the goal of selecting the best medication for each individual, it must do the following:
- Test all polymorphisms relevant to the prescribing of any medication
- Be completed with no out-of-pocket cost to the patient
- Be performed before the patient requires the medication
- Provide results that will be interpreted as part of an individualized pharmacogenomics consult.11
The 1200 Patients Project, a pilot research study under way at the Center for Personalized Therapeutics at the University of Chicago, is attempting to demonstrate the feasibility of incorporating pharmacogenomic testing into routine clinical practice for medication treatment decisions. DNA samples from patients who are taking at least one prescription medication are being tested for differences in genes that may suggest greater effectiveness or an increased risk of side effects from certain medications.
Solutions in practice
Cleveland Clinic’s genetics-based management of Lynch syndrome, the integration of genetics services during patient appointments at Cleveland Clinic, and a coordinated approach at The Ohio State University Medical Center are examples of practical applications of personalized healthcare.
Colorectal cancer management. One example of a personalized approach to medicine that improves health outcome while achieving cost savings is the genetics-based approach to HNPCC (Lynch syndrome) at Cleveland Clinic.
Early identification of Lynch syndrome by screening all colorectal cancer patients has been shown to save $250,000 per life-year gained in the United States.16 All colorectal cancers resected at the Cleveland Clinic main campus are routinely screened for MSI and IHC, and the process is embedded into the routine pathology workflow. With the patients’ foreknowledge, a gastrointestinal cancer genetics counselor scans the list of MSI and IHC results each week. Patients who are MSI-high or IHC-null are invited to receive genetic counseling and consider germline single-gene testing guided by the IHC results. With this active approach, patient uptake is 80%; in comparison, with a passive approach (MSI/IHC results are placed in the pathology report), the uptake is 14%17 (B. Leach and C. Eng, unpublished data, 2011).The successful application of the active approach requires the close cooperation of multiple disciplines, including members of the Cleveland Clinic Genomic Medicine, Pathology & Laboratory Medicine, and Digestive Disease Institutes.18
Integrating genetics-based care at Cleveland Clinic. Time delays for genetics services and limited collaboration with managing physicians who are not genetics specialists reduces genetics-based access and availability. Broad access to genetics clinical services is a means of clinical integration of genetics-enabled care. Providing patients and healthcare providers with easy access and short wait times is vital for clinical integration of genetics-enabled personalized healthcare.
As part of a patient-centered focus on medicine, clinical genetics services have been integrated throughout Cleveland Clinic. The system has two genetics clinics at its main campus and has embedded multiple genetics satellites within its nongenetics clinics, easing access. Genetics counselors are stationed in the same areas of practice as referring providers. Although patient encounters have increased at the medical genetics clinic in the Genomic Medicine Institute, genetics consultations no longer require an extra trip to the clinic since they are integrated into existing appointments. With this approach, large numbers of patients can be seen with no wait times.
Coordinated care at The Ohio State University Medical Center. The Center for Personalized Health Care at The Ohio State University Medical Center (OSUMC) embraces a systems-based care-coordinated model that improves care by executing standardized processes and automating routine tasks. The Institute for Systems Biology, which was established to develop genomics, wellness, and chronic disease biomarkers, collaborates with OSUMC on pilot projects in chronic disease, including cancer.
The OSUMC has a closed system in which it is the payer, employer, and provider of healthcare. This closed system serves as an ideal testing ground for reform. Goals include intervention in disease before symptoms appear and maintenance of wellness. The data from these demonstration projects should facilitate adoption of personalized healthcare by improving physician acceptance of personalized approaches and satisfying payers that personalized healthcare is cost-effective.
- Personalized medicine. Coriell Institute for Medical Research Web site. http://www.coriell.org/personalized-medicine. Updated 2011. Accessed December 27, 2011.
- The 2012 Statistical Abstract. U.S. Census Bureau Web site. http://www.census.gov/compendia/statab/cats/income_expenditures_poverty_wealth/gross_domestic_product_gdp.html. Updated September 27, 2011. Accessed December 22, 2011.
- National health expenditure fact sheet. Center for Medicare & Medicaid Services (CMS) Web site. https://www.cms.gov/NationalHealthExpendData/25_NHE_Fact_Sheet.asp. Updated November 4, 2011. Accessed December 22, 2011.
- New England Healthcare Institute (NEHI). Thinking outside the pillbox: A system-wide approach to improving patient medication adherence for chronic disease. NEHI Web site. http://www.nehi.net/publications/44/thinking_outside_the_pillbox_a_systemwide_approach_to_improving_patient_medication_adherence_for_chronic_disease. Published August 12, 2009. Accessed December 22, 2011.
- Spear BB, Heath-Chiozzi M, Huff J. Clinical application of pharmacogenetics. Trends Mol Med 2001; 7:201–204.
- Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757–1765.
- Feero WG, Green ED. Genomics education for healthcare professionals in the 21st century. JAMA 2011; 306:989–990.
- Meyers M, Wagner MW, Hwang HS, Kinsella TJ, Boothman DA. Role of the hMLH1 DNA mismatch repair protein in flouropyrimidine-mediated cell death and cell cycle responses. Cancer Res 2001; 61:5193–5201.
- Carethers JM, Chauhan DP, Fink D, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology 1999; 117:123–131.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003; 349:247–257.
- Ratain MJ. Personalized medicine: building the GPS to take us there. Clin Pharmacol Ther 2007; 81:321–322.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation [published online ahead of print November 7, 2007]. Circulation 2007; 116:2563–2570. doi: 10.1161/CIRCULATIONAHA.107.737312
- Price MJ, Angiolillo DJ, Teirstein PS, et al .Platelet reactivity and cardiovascular outcomes after percutaneous coronary intervention: a time-dependent analysis of the Gauging Responsiveness with a VerifyNow P2Y12 assay: Impact on Thrombosis and Safety (GRAVITAS) trial [published online ahead of print August 29, 2011]. Circulation 2011; 124:1132–1137. doi: 10.1161/CIRCULATIONAHA.111.029165
- Sharp RR, Goldlust ME, Eng C. Addressing gaps in physician education using personal genomic testing. Genet Med 2011; 13:750–751.
- Wiener CM, Thomas PA, Goodspeed E, Valle D, Nichols DG. “Genes to society”—the logic and process of the new curriculum for the Johns Hopkins University School of Medicine. Acad Med 2010; 85:498–506.
- Ladabaum U, Wang G, Terdiman J, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer: a costeffectiveness analysis. Ann Intern Med 2011; 155:69–79.
- Leach B, Eng C, Kalady M, et al. Sharing the responsibility: multidisciplinary model improves colorectal cancer microsatellite testing. Paper presented at: InSight 2009 Annual Conference: September 2009; Orlando, FL.
- Manolio TA, Chisolm R, Ozenberger B, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med Forthcoming.
- Personalized medicine. Coriell Institute for Medical Research Web site. http://www.coriell.org/personalized-medicine. Updated 2011. Accessed December 27, 2011.
- The 2012 Statistical Abstract. U.S. Census Bureau Web site. http://www.census.gov/compendia/statab/cats/income_expenditures_poverty_wealth/gross_domestic_product_gdp.html. Updated September 27, 2011. Accessed December 22, 2011.
- National health expenditure fact sheet. Center for Medicare & Medicaid Services (CMS) Web site. https://www.cms.gov/NationalHealthExpendData/25_NHE_Fact_Sheet.asp. Updated November 4, 2011. Accessed December 22, 2011.
- New England Healthcare Institute (NEHI). Thinking outside the pillbox: A system-wide approach to improving patient medication adherence for chronic disease. NEHI Web site. http://www.nehi.net/publications/44/thinking_outside_the_pillbox_a_systemwide_approach_to_improving_patient_medication_adherence_for_chronic_disease. Published August 12, 2009. Accessed December 22, 2011.
- Spear BB, Heath-Chiozzi M, Huff J. Clinical application of pharmacogenetics. Trends Mol Med 2001; 7:201–204.
- Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757–1765.
- Feero WG, Green ED. Genomics education for healthcare professionals in the 21st century. JAMA 2011; 306:989–990.
- Meyers M, Wagner MW, Hwang HS, Kinsella TJ, Boothman DA. Role of the hMLH1 DNA mismatch repair protein in flouropyrimidine-mediated cell death and cell cycle responses. Cancer Res 2001; 61:5193–5201.
- Carethers JM, Chauhan DP, Fink D, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology 1999; 117:123–131.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003; 349:247–257.
- Ratain MJ. Personalized medicine: building the GPS to take us there. Clin Pharmacol Ther 2007; 81:321–322.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation [published online ahead of print November 7, 2007]. Circulation 2007; 116:2563–2570. doi: 10.1161/CIRCULATIONAHA.107.737312
- Price MJ, Angiolillo DJ, Teirstein PS, et al .Platelet reactivity and cardiovascular outcomes after percutaneous coronary intervention: a time-dependent analysis of the Gauging Responsiveness with a VerifyNow P2Y12 assay: Impact on Thrombosis and Safety (GRAVITAS) trial [published online ahead of print August 29, 2011]. Circulation 2011; 124:1132–1137. doi: 10.1161/CIRCULATIONAHA.111.029165
- Sharp RR, Goldlust ME, Eng C. Addressing gaps in physician education using personal genomic testing. Genet Med 2011; 13:750–751.
- Wiener CM, Thomas PA, Goodspeed E, Valle D, Nichols DG. “Genes to society”—the logic and process of the new curriculum for the Johns Hopkins University School of Medicine. Acad Med 2010; 85:498–506.
- Ladabaum U, Wang G, Terdiman J, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer: a costeffectiveness analysis. Ann Intern Med 2011; 155:69–79.
- Leach B, Eng C, Kalady M, et al. Sharing the responsibility: multidisciplinary model improves colorectal cancer microsatellite testing. Paper presented at: InSight 2009 Annual Conference: September 2009; Orlando, FL.
- Manolio TA, Chisolm R, Ozenberger B, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med Forthcoming.
Pharmacogenomics for the primary care provider: Why should we care?
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
Since the human genome was sequenced in 2000, the American public has continued to hold hope that our growing understanding of genetics will revolutionize the practice of medicine.
One way genetics promises to improve the quality and value of health care is in personalized medicine, by helping us tailor treatment to a person’s individual genetic makeup. One such approach is called pharmacogenomics.
Pharmacogenomics uses knowledge of a person’s genetics to understand how a particular drug will work, or not work, in his or her body. For instance, some people might carry genes that make them more sensitive than average to a drug, and therefore they would require a lower dose. Others might have genes that make them resistant to the drug, meaning the drug is ineffective unless they receive a higher dose.
Adverse drug reactions are a leading cause of death in hospitalized patients in the United States and are responsible for billions of dollars in health care costs.1,2 Our current practice of prescribing for adult patients is largely trial-and-error, with the same dose given to all patients, in many cases with little regard even to sex, height, or weight.
Pharmacogenomics promises to change this way of prescribing to a customized approach that uses genetic information to predict an individual’s response to medications. It is one piece of an overall initiative to personalize patient care based on the patient’s individual characteristics and preferences.
OVERCOMING BARRIERS TO USING PHARMACOGENOMICS IN PRACTICE
If personalized medicine has promised to improve the quality and value of health care for our patients, why have we been so slow to adopt this information in clinical practice?
The usual barriers to clinical adoption certainly exist. We need further studies to determine whether genetic-based prescribing is truly valid, and for which patient populations. We need to determine whether this approach is cost-effective and better than the current standard of care. We need to work on payment options.
However, one of the largest barriers for busy primary care physicians is the lack of time to keep up with new information. Many practicing physicians were taught little about formal genetics in medical school. The body of scientific literature on pharmacogenomics is fragmented, and it crosses disease states and specialties, making it difficult to unite. Given the breadth of diseases treated and drugs prescribed by primary care physicians, it is unrealistic for most to keep track of the vast body of literature of pharmacogenomic testing and to decipher how to apply this to clinical practice.
In this issue of the Journal, Kitzmiller et al3 provide one solution to this problem, giving an overview of pharmacogenomic applications that might be pertinent to practicing physicians. However, as we try to make pharmacogenomics accessible to busy physicians, we need other solutions to integrate pharmacogenomic information efficiently into the clinical work flow. One approach might be to build pharmacogenomics into the electronic medical record. We can also store the integrated information in research databases and provide clinical recommendations on Internet sites such as www.pharmgkb.org, and we can develop applications to run on cell phones and iPads.
QUESTIONS REMAIN
Kitzmiller et al discuss an important step in this process, highlighting several key questions:
Should we seek genetics-based information to personalize drug selection? Based on the information presented in the literature and in the Kitzmiller paper, there may be circumstances when it is appropriate to consider doing so. While the evidence is not yet compelling to order these tests on a regular basis in clinical practice, this information might be helpful in some situations, such as for patients who have had adverse effects from minimal doses of antidepressants.
For now, clinicians should not abandon their current practice of personalizing patient care on the basis of personal, cultural, and economic preferences. Rather, they should consider pharmacogenomic information an additional piece of information when selecting drug therapy. We should also encourage health care systems and interested providers to be early adopters and to study how their outcomes compare with the standard of care.
Once we have this information, what is our obligation to use it? An increasing number of patients already have genetic information in their health record, either ordered by or provided to their physicians. However, there is little in the scientific literature to guide us in this arena.
Yet most of us would agree that if we have information (genetic or otherwise) that can help to select a drug type or dose or reduce adverse events or costs, we should consider this information in our decision-making. Several circumstances are documented in this paper and in the literature in which prior knowledge about drug metabolism can help in selecting a dose of medication. One example would be the 50% recommended reduction in tricyclic antidepressant dose if the patient is a CYP2D6 poor metabolizer.4
MOVING FORWARD AS A TEAM
In summary, Kitzmiller et al bring to light the promise and the uncertainties that currently exist in the field of pharmacogenomics. While it is unclear if we should incorporate pharmacogenomic tests into standard medical practice at this time, it is clear that this information is becoming more readily available, whether or not we have requested it. Some would argue that, once we have the information, we have an obligation to use it, just as we use other information in our clinical decision-making. This means we need to develop tools and resources to help practitioners evaluate pharmacogenomic data and incorporate it into clinical care in an efficient manner.
The authors also highlight the need for more education about drug metabolism in general, and they cite several instances in which knowledge of drug interactions and metabolism can clearly influence decision-making. An example is paroxetine (Paxil) inhibition of tamoxifen (Nolvadex).5
Lastly, regardless of our personal feelings about the clinical usefulness of genetic testing in large populations, we need to work together to determine clinical utility and validity and to develop efficient ways to put into practice findings that could affect patient care. As we move forward, we need to work as a team, utilizing our clinical partners—pharmacists, pharmacologists, metabolism and health information technology experts, and medical geneticists. Working as a team, pooling our resources and tools, we move closer to providing world-class personalized health care.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998; 279:1200–1205.
- Field TS, Gilman BH, Subramanian S, Fuller JC, Bates DW, Gurwitz JH. The costs associated with adverse drug events among older adults in the ambulatory setting. Med Care 2005; 43:1171–1176.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice. Why drugs work in some patients but not others. Cleve Clin J Med 2011; 78:243–257.
- Thuerauf N, Lunkenheimer J. The impact of the CYP2D6-polymorphism on dose recommendations for current antidepressants. Eur Arch Psychiatry Clin Neurosci 2006; 256:287–293.
- Schwarz EB, McNamara M, Miller RG, Walsh JM. Update in women’s health for the general internist. J Gen Intern Med201; 26:207–213.
Premenopausal osteoporosis, an overlooked consequence of anorexia nervosa
Among the devastating effects of anorexia nervosa, and one that is easily overlooked, is its impact on bone.
Probably more than half of young women with anorexia nervosa develop osteoporosis, and relatively quickly. Baker et al1 obtained bone scans in a series of 56 young women, mean age 27 years, who had had an eating disorder for a mean of about 10 years, and found that the bone mineral density in the femur was below the critical fracture threshold in 42 (75%).
Osteoporosis is particularly common and worrisome in female athletes (and is becoming increasingly common in male athletes as well). Female athletes have a much higher incidence of disordered eating than their peers2 and therefore are at a much higher risk of fractures.
This review summarizes the factors affecting the development of osteoporosis in these patients and discusses potential targets for intervention.
ANOREXIA AND BONE HEALTH: A COMPLEX RELATIONSHIP
Anorexia nervosa is characterized by an intense fear of gaining weight, a body weight less than 85% of expected, a distorted self-image, and, in women, missing three consecutive menstrual periods.3 The lifetime prevalence in women is about 0.5%; it is much lower in men.3 The prevalence of eating disorders in female athletes is much higher, estimated at 15% to 62%.2
The etiology of osteoporosis in patients with anorexia nervosa is complex and multifaceted. In these patients, bone resorption is increased without a concomitant increase in bone formation, resulting in a net loss of bone.4 Thus, markers of bone resorption such as N-teleopeptide and deoxypyridoline are elevated, but markers of bone formation such as osteocalcin are not.4
The loss of bone may be rapid and can occur relatively early in the disease. Some studies suggest that an illness duration longer than 12 months predicts significant loss of bone density.5 Thus, early diagnosis and intervention are important to minimize bone loss.
Women gain from 40% to 60% of their bone mass during the pubertal growth spurt in ages 11 to 14, the time when anorexia nervosa is most prevalent.6 Peak bone mass is attained by the third decade of life, but the rate of growth of bone mass is highest during adolescence and early adulthood.7 Hence, it is important to optimize bone mass during this time, as small differences in bone density can have significant clinical implications later in life: a 5% increase in bone density significantly decreases fracture risk, whereas a 10% decrease in adult bone mineral density is associated with a two to three times higher risk of fracture (reviewed by Rome and Ammerman6).
What is the role of amenorrhea in the development of osteoporosis in premenopausal patients?
Given that two of the most characteristic manifestations of anorexia nervosa are low body weight and the absence of menses, these factors have been hypothesized to be potential causes of osteoporosis.
In general, young women who present with amenorrhea should be evaluated to determine if the amenorrhea is primary or secondary. Primary amenorrhea is the absence of menarche by age 16; secondary amenorrhea is the absence of menses for more than three cycles or more than 6 months in someone who previously had had menses. The most common causes of secondary amenorrhea are ovarian disease, hypothalamic or pituitary disease, and uterine disease. Anorexia nervosa causes hypothalamic dysfunction and is a cause of secondary amenorrhea. In clinical practice, it is also important to remember that pregnancy can occur even in the setting of amenorrhea.
Amenorrhea in patients with anorexia nervosa is related to hypothalamic suppression of the release of gonadotropin-releasing hormone, resulting in lower levels of follicle-stimulating hormone and luteinizing hormone and a resultant prepubertal low-estrogen state.
In a study of 73 women with anorexia nervosa and a mean age of 17.2 years,8 20 months of amenorrhea was the threshold above which the most severe osteopenia was seen, implying that the duration of amenorrhea affects bone health.
Which factors besides amenorrhea influence bone density in premenopausal women?
Undernutrition. Body weight has been suggested to have an independent effect on bone mineral density, and density has been found to increase following weight gain, even before the return of menses.1 Once a regular menstrual cycle has been restored, significant increases in trabecular and cortical bone have been detected.1
Deficiency of insulin-like growth factor 1 (IGF-1). Anorexia nervosa is associated with decreased hepatic synthesis of IGF-I.9 Low levels of IGF-I reduce the levels of osteocalcin, a marker of bone formation, and cause abnormalities in osteoblast function.10 This deficiency is associated with the development of osteopenia in patients with anorexia nervosa.11
Low androgen levels are present in patients with anorexia nervosa, and levels appear to be further reduced by oral contraceptives.12 It remains to be determined whether the further reduction in androgens in women with anorexia nervosa using oral contraceptives is harmful to skeletal health. Low testosterone levels in boys with anorexia nervosa have been associated with lower libido, fewer erections, and potentially lower bone density.13
Hypercortisolemia. Elevated levels of total and free serum cortisol and high 24-hour urinary free cortisol excretion have been noted in anorectic patients. Levels of cortisol are inversely related to levels of osteocalcin, and hypercortisolism has been shown to be associated with osteoporosis.14,15 However, no study has yet shown causality in this population.
Osteoprotegerin has been recognized as an important regulator of bone resorption. Osteoprotegerin inhibits osteoclast differentiation and activation and stimulates osteoclast apoptosis, helping to preserve bone density.
Misra et al16 showed that adolescent girls with anorexia nervosa have higher serum osteoprotegerin levels than controls and that osteoprotegerin levels correlate inversely with markers of nutritional status and lumbar bone density Z scores.16 They and other investigators17 postulate that osteoprotegerin may be released as a compensatory response to the bone loss seen in these patients in an attempt to preserve bone health.
Leptin is an adipocyte-derived hormone that acts on receptors in the hypothalamus, decreasing food intake and increasing energy expenditure. Low leptin levels are a key endocrinologic feature of anorexia nervosa.18 Leptin helps to induce weight loss by stimulating neurons in the hypothalamus that express “weight-loss-inducing” neuropeptides such as pro-opiomelanocortin and inhibiting “weight-gain-inducing” peptides such as neuropeptide Y.19
Although leptin was first believed to be a hormone released to counteract obesity, recent studies19,20 suggest that it is part of a major signaling system that controls adaptation to starvation. These studies have shown that the body senses its corporeal fat through leptin and inhibits ovulation when fat reserves are low.19 In addition, luteinizing hormone and leptin levels have been shown to increase in parallel in patients with anorexia nervosa when weight is restored.20 Thus, rising leptin levels correlate with the resumption of menses in women with anorexia nervosa and in turn have potential consequences for bone health.
Not enough ghrelin, too much obestatin? Ghrelin, a gastric hormone, acts as a natural antagonist to leptin, resulting in an increase in food intake and body weight.19 Circulating ghrelin levels are higher in illness-induced anorexia as well as in anorexia nervosa, and they normalize with weight gain, perhaps as an adaptive mechanism to compensate for a negative energy balance.21
Several in vitro studies suggest that ghrelin directly promotes osteoblast proliferation and differentiation.22 However, human studies of ghrelin’s effects on bone are limited. In a study of healthy younger women, healthy boys, and anorexia nervosa patients, plasma ghrelin levels were only weakly associated with bone mineral density.23
The effects of obestatin, another gastric hormone, are still being investigated. Obestatin was initially shown to oppose the effects of ghrelin by decreasing appetite and weight gain. When given with ghrelin, obestatin appears to work with ghrelin at the hypothalamic level to modulate food intake and growth hormone secretion.24
Interestingly, obestatin and the ratio of ghrelin to obestatin are decreased in patients with anorexia nervosa, but the ratio is unchanged in thin women who have an equivalent body mass index but no eating disorder.25 It has been hypothesized that the ghrelin-obestatin ratio may be the key to explaining the eating restriction and reduced motivation to eat despite high ghrelin levels seen in anorexia nervosa.26 Further studies are needed to determine the role of obestatin and the ghrelin-obestatin ratio in the bone health of women with anorexia nervosa.

HOW SHOULD WE DIAGNOSE OSTEOPOROSIS IN PREMENOPAUSAL PATIENTS?
Our approach to screening for and diagnosing osteoporosis is still largely based on measuring bone mineral density, although density by itself is not a perfect tool for predicting who will or will not experience a fracture, particularly in premenopausal women.26,27 Most premenopausal women with low bone mineral density but no other risk factors for fracture such as previous fractures or glucocorticoid therapy are at very low short-term risk of fracture.26
For these reasons, in premenopausal women and adolescents, the International Society for Clinical Densitometry28 advises against diagnosing osteoporosis on the basis of bone mineral density alone. Instead, it should be diagnosed in this population only if the bone mineral density is low (defined as a Z score below −2.0) and the patient has risk factors that suggest a higher short-term risk of bone mineral loss and fracture. Risk factors include chronic malnutrition, eating disorders, hypogonadism, glucocorticoid exposure, and previous fracture.29
A pitfall in interpreting low bone mineral density in premenopausal women younger than age 30 is the possibility that they may not yet have reached their peak bone mass. In addition, small stature and body size (and therefore bone size) also influence the results of dual-energy x-ray absorptiometry. This test may not be able to distinguish bone that is small but of normal density from bone that is of low density.26
Despite its limitations, until newer risk assessment tools are available for this patient population, measuring bone mineral density is still recommended in addition to assessing clinical risk factors to diagnose osteoporosis. Also, changes in bone mineral density over time can help to assess risk and guide treatment.
When should a patient with anorexia be screened for osteoporosis?
Because bone loss may begin early in the course of anorexia and progress rapidly (potentially inexorably), baseline screening is recommended for all patients who have had anorexia nervosa or amenorrhea for more than 6 to 12 months.30 The National Osteoporosis Foundation recommends screening in women under age 65 who have a low body weight.31 The American College of Sports Medicine recommends screening for osteoporosis in athletes with a history of hypoestrogenism or disordered eating for a cumulative total of 6 months or more, or with a history of stress fracture or fracture from minimal trauma.32
Knowledge of low bone mineral density and fracture risk can often guide treatment and prompt behavioral change. Given that most osteoporosis treatments do not lead to detectable changes in bone density until 18 months to 2 years, it is reasonable to repeat testing at this interval.33
NEW AND OLD TREATMENTS FOR LOW BONE DENSITY IN ANOREXIA NERVOSA
Weight restoration is the cornerstone
Restoration of body weight and nutritional rehabilitation remain the cornerstones of treatment. All patients with anorexia nervosa should be referred to a nutritionist to develop a meal plan that is adjusted for the amount of energy expended. The challenges lie in managing the complications of refeeding and the high relapse rate. The treatment goals in disordered eating are to optimize the overall nutritional status, normalize eating behavior, modify unhealthy thought processes that maintain the disorder, and treat possible emotional issues that help create or maintain the disorder.
The younger the patient, the more the family’s involvement is recommended. In addition to nutritional counseling, the care team should include a psychotherapist, a psychiatrist, and a primary care physician to assist with management and screening of medical complications.
Vitamin D for all
Low vitamin D levels have long been associated with low bone mineral density and risk of hip fracture.34
Vitamin D insufficiency is very common. More than 90% of blacks, Hispanics, and Asians and nearly 75% of whites have insufficient levels of vitamin D (25-hydroxyvitamin D3 level < 30 ng/mL).35 In a study of 307 healthy adolescents, vitamin D insufficiency (a 25-hydroxyvitamin D3 level < 20 ng/mL) was found in 42% and vitamin D deficiency (a level < 15 ng/mL) in 24.1%.36 In addition, this study confirmed an inverse correlation between body mass index and serum 25-hydroxyvitamin D3 concentration.
Therefore, while vitamin D supplementation has not been consistently shown to improve bone loss in anorectic patients,9 given the prevalence of vitamin D deficiency and insufficiency, supplementation is almost universally recommended.
There is no consensus as to the amount of supplementation to recommend for women with anorexia nervosa. The American College of Sports Medicine recommends a total daily intake of 800 IU of vitamin D (ie, from diet and supplements). Therapy should be titrated to doses that result in normocalcemia and a serum 25-hydroxyvitamin D3 concentration of at least 30 ng/mL.37,38
Does hormone treatment improve bone density?
In postmenopausal osteoporosis, estrogen therapy maintains or improves bone density and appears to reduce the rate of vertebral fractures.39,40 Perhaps not so with premenopausal osteoporosis due to anorexia nervosa.
Why should this be? In postmenopausal women, estrogen therapy appears to work by impairing osteoclast-mediated bone resorption, but it has only limited effects on bone formation. In premenopausal anorexia, however, bone loss appears to be due to a unique uncoupling of osteoblastic and osteoclastic functions, resulting in both reduced bone formation and increased bone resorption, which estrogen therapy may not improve.5
Despite the documented association between anorexia nervosa and estrogen deficiency and the strong correlation between osteoporosis and the duration of amenorrhea, most studies have found no improvement in bone mass with hormonal therapy.9 In particular, three randomized, placebo-controlled trials have been published to date, and not one showed a significant improvement in bone mineral density with estrogen therapy compared with placebo in patients with anorexia nervosa.41–43
Klibanski et al,41 in the first of these trials, found no significant difference in spinal bone mineral density between treated patients and controls. However, estrogen-treated patients whose initial body weight was very low (< 70% of expected) had a significant increase in their bone mineral density, whereas those in the control group lost bone density.
Baker et al44 suggest that hormone therapy might protect bone mass in athletes with amenorrhea, citing a study that found that women with a history of stress fractures were less likely to have used oral contraceptives previously than athletes without fractures.45 However, no prospective randomized study to date has established that hormone therapy effectively preserves bone mass in athletes with amenorrhea.
Based on the data presented above, we have little evidence for using estrogen to treat or prevent premenopausal osteoporosis.
The American College of Sports Medicine32 recommends consideration of estrogen therapy if there is evidence of a decline in bone mineral density in an athlete over the age of 16 with persistent functional hypothalamic amenorrhea despite adequate nutritional intake and weight. However, it acknowledges that restoring regular menstrual cycles with oral contraceptive pills will not normalize the metabolic factors that impair bone formation, health, and performance and is not likely to fully reverse low bone mineral density in this population.
What is the effect of exercise on bone health in these patients?
Several studies have examined the effect of weight-bearing exercise on bone density.
Young et al46 compared normal teenagers, ballet dancers, and young women with anorexia nervosa and found that weight-bearing exercise protected against osteoporosis, but only at weight-bearing sites. Athletes in weight-bearing sports had a 5% to 15% higher bone mineral density in weight-bearing sites (ie, the femur) compared with nonathletes, but had lower bone mineral density in the spine.
Therefore, a Z score below −1.0 in an athlete, especially in a distal site, warrants further investigation and treatment.32 In general, exercise does not necessarily protect against osteoporosis in this patient population, and it can sometimes mask underlying bone loss. In addition, keep in mind that many of these patients exercise compulsively, using it as a form of purging.
Insulin-like growth factor-1: More study needed
IGF-1 contributes to bone growth by stimulating osteoblasts, and patients with anorexia nervosa have been shown to have low levels of IGF-1.9
Grinspoon et al10 randomized 60 patients with anorexia nervosa to receive IGF-1 alone, IGF-1 plus an oral contraceptive, an oral contraceptive alone, or placebo. All patients were given calcium and vitamin D and were followed for 9 months. Total bone mass increased significantly in those taking IGF-1 compared with those taking placebo. Those taking an IGF-1 and an oral contraceptive had a significant increase in spinal bone mineral density compared with those on placebo group. At other skeletal sites, however, IGF-1 plus an oral contraceptive and IGF-1 alone failed to produce significant increases in bone mineral density compared with placebo.
Further study is needed to determine the role of IGF-1 in treating low bone mineral density in anorexia nervosa.
Bisphosphonates: Not approved for this indication
In premenopausal women, bisphosphonates are approved by the US Food and Drug Administration (FDA) for use only in those taking glucocorticoids. Although bisphosphonates have been shown to significantly increase bone mineral density in young women with anorexia nervosa,26 they should be used with caution in patients of childbearing age because they are teratogenic. Bisphosphonates have a long half-life and may continue to affect bone turnover for up to 2 years after they are discontinued.47 In addition, they are not recommended in patients with a history of purging via vomiting, due to a risk of esophageal ulceration.
Parathyroid hormone therapy: Studies ongoing
The parathyroid hormone fragment teriparatide (Forteo) is widely used for treating postmenopausal osteoporosis.
Before teriparatide was approved, there was concern that it might increase the risk of osteosarcoma, as almost 45% of rats treated with this drug at the highest-tested dose level developed this aggressive form of bone cancer.48 Balancing the proven benefits of teriparatide shown by clinical trials with the theoretic risk of teriparatide-induced osteosarcoma, the FDA mandated a “black-box” warning about this potential effect.
Studies of parathyroid hormone treatment in anorexia nervosa and other premenopausal patients are ongoing.26
Leptin: More study needed
Leptin is a potent stimulator of bone growth and has been shown to increase bone mineral density in vitro and in vivo.19 However, concerns have been raised about giving supra-physiologic doses of leptin to patients with anorexia nervosa, as this may increase the risk of further weight loss and relapse.
More work is needed to determine the role of leptin for the treatment of osteoporosis in anorexia nervosa.
Ghrelin: Probably not effective as a single agent
Pharmacologic use of ghrelin increases food intake in healthy humans,49 and it has been proposed as a treatment for weight restoration and bone health in anorexia nervosa. Preliminary studies have not shown it to increase appetite or weight gain,50 but it did increase slow-wave sleep.
Based on these studies, it is unlikely that ghrelin will be effective as a single agent to stimulate appetite, but it may be helpful in conjunction with other therapies.
Cannabinoids: Little ongoing research
Cannabinoids have been proposed as a treatment for anorexia nervosa in the hope that they would induce weight gain and in turn prevent osteoporosis.
Interest in their use in anorexia nervosa stems from the discovery of two cannabinoid receptors (CB1 and CB2) located in the brain and peripheral organ systems. Anorexia nervosa has been associated with different alleles of the CB1 gene,51 but the therapeutic implications of this are far from clear.
Cannabinoids appear to regulate eating behavior at several levels within the brain and periphery: the hypothalamus and hindbrain (integrative functions), the limbic system (for hedonic evaluation of foods), the intestinal system, and adipose tissue.52 At each of these levels, the endocannabinoid system interacts with a number of better known peptides involved in appetite regulation, including leptin, ghrelin, and the melanocortins. In mouse studies, genetic leptin deficiency is associated with elevated hypothalamic endocannabinoid levels.
Appetite stimulation by cannabinoids has been studied for several decades, particularly in relation to cachexia and malnutrition associated with cancer. Very few trials have studied cannabinoids for anorexia nervosa.
In a 4-week crossover trial in 11 patients with anorexia nervosa,53 tetrahydrocannabinol (THC) treatment resulted in an increase in sleep disturbances and interpersonal sensitivity, but it had no significant effect on weight gain compared with diazepam treatment.53
Another pilot study of nine outpatients with anorexia nervosa treated with THC showed a significant improvement in depression and perfectionism scores without any significant weight gain.19
Although this research was once promising, the risk was felt to outweigh the benefit, as cannabinoids may induce dependency in this patient group, who may already be at high risk of drug addiction, and very few have continued this line of investigation.
WHAT CAN WE DO FOR NOW?
- Weight restoration and nutritional rehabilitation remain the keys to treatment of low bone density to reduce the risk of osteoporosis in patients with anorexia nervosa. However, as many as one-third of patients with anorexia nervosa relapse during their lifetime, and other treatments are needed to stabilize and prevent bone loss.
- Vitamin D deficiency is clearly associated with a risk of osteoporosis and fracture, and patients with vitamin D deficiency should be treated with supplementation.
- Standard therapies in postmenopausal patients (such as bisphosphonates and teriparatide) should be used with caution in premenopausal anorexia nervosa patients because of potential long-term health risks.

- As we learn more about hormonal factors in anorexia nervosa, we hope to identify interventions that will help restore weight and decrease the risk of osteoporosis. A summary of potential treatment strategies and targets for prevention of osteoporosis in anorexia nervosa is presented in Table 2.

Acknowledgment: The author thanks the General Internal Medicine Works in Progress Group for its editorial comments, and Dr. Ellen Rome for her mentorship and support.
- Baker D, Roberts R, Towell T. Factors predictive of bone mineral density in eating-disordered women: a longitudinal study. Int J Eat Disord 2000; 27:29–35.
- Rome ES. Eating disorders. Obstet Gynecol Clin North Am 2003; 30:353–377.
- First MB, editor. Diagnostic and Statistical Manual of Mental Disorders—4th edition. Washington, DC: American Psychiatric Association, 2000.
- Mehler PS, MacKenzie TD. Treatment of osteopenia and osteoporosis in anorexia nervosa: a systematic review of the literature. Int J Eat Disord 2009; 42:195–201.
- Wong JC, Lewindon P, Mortimer R, Shepherd R. Bone mineral density in adolescent females with recently diagnosed anorexia nervosa. Int J Eat Disord 2001; 29:11–16.
- Rome ES, Ammerman S. Medical complications of eating disorders: an update. J Adolesc Health 2003; 33:418–426.
- Recker RR, Davies KM, Hinders SM, Heaney RP, Stegman MR, Kimmel DB. Bone gain in young adult women. JAMA 1992; 268:2403–2408.
- Sterling WM, Golden NH, Jacobson MS, Ornstein RM, Hertz SM. Metabolic assessment of menstruating and nonmenstruating normal weight adolescents. Int J Eat Disord 2009; 42:658–663.
- Legroux-Gerot I, Vignau J, Collier F, Cortet B. Bone loss associated with anorexia nervosa. Joint Bone Spine 2005; 72:489–495.
- Grinspoon S, Thomas L, Miller K, Herzog D, Klibanski A. Effects of recombinant human IGF-I and oral contraceptive administration on bone density in anorexia nervosa. J Clin Endocrinol Metab 2002; 87:2883–2891.
- Soyka LA, Grinspoon S, Levitsky LL, Herzog DB, Klibanski A. The effects of anorexia nervosa on bone metabolism in female adolescents. J Clin Endocrinol Metab 1999; 84:4489–4496.
- Miller KK, Lawson EA, Mathur V, et al. Androgens in women with anorexia nervosa and normal-weight women with hypothalamic amenorrhea. J Clin Endocrinol Metab 2007; 92:1334–1339.
- Misra M, Katzman DK, Cord J, et al. Bone metabolism in adolescent boys with anorexia nervosa. J Clin Endocrinol Metab 2008; 93:3029–3036.
- Misra M, Miller KK, Almazan C, et al. Alterations in cortisol secretory dynamics in adolescent girls with anorexia nervosa and effects on bone metabolism. J Clin Endocrinol Metab 2004; 89:4972–4980.
- Chiodini I, Mascia ML, Muscarella S, et al. Subclinical hypercortisolism among outpatients referred for osteoporosis. Ann Intern Med 2007; 147:541–548.
- Misra M, Soyka LA, Miller KK, et al. Serum osteoprotegerin in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab 2003; 88:3816–3822.
- Ohwada R, Hotta M, Sato K, Shibasaki T, Takano K. The relationship between serum levels of estradiol and osteoprotegerin in patients with anorexia nervosa. Endocr J 2007; 54:953–959.
- Müller TD, Föcker M, Holtkamp K, Herpertz-Dahlmann B, Hebebrand J. Leptin-mediated neuroendocrine alterations in anorexia nervosa: somatic and behavioral implications. Child Adolesc Psychiatr Clin N Am 2009; 18:117–129.
- Støving RK, Andries A, Brixen K, Flyvbjerg A, Hørder K, Frystyk J. Leptin, ghrelin, and endocannabinoids: potential therapeutic targets in anorexia nervosa. J Psychiatr Res 2009; 43:671–679.
- Audi L, Mantzoros CS, Vidal-Puig A, Vargas D, Gussinye M, Carrascosa A. Leptin in relation to resumption of menses in women with anorexia nervosa. Mol Psychiatry 1998; 3:544–547.
- Wong IP, Baldock PA, Herzog H. Gastrointestinal peptides and bone health. Curr Opin Endocrinol Diabetes Obes 2010; 17:44–50.
- Fukushima N, Hanada R, Teranishi H, et al. Ghrelin directly regulates bone formation. J Bone Miner Res 2005; 20:790–798.
- Makovey J, Naganathan V, Seibel M, Sambrook P. Gender differences in plasma ghrelin and its relations to body composition and bone—an opposite-sex twin study. Clin Endocrinol (Oxf) 2007; 66:530–537.
- Hassouna R, Zizzari P, Tolle V. The ghrelin/obestatin balance in the physiological and pathological control of growth hormone secretion, body composition and food intake. J Neuroendocrinol 2010; 22:793–804.
- Germain N, Galusca B, Grouselle D, et al. Ghrelin/obestatin ratio in two populations with low body weight: constitutional thinness and anorexia nervosa. Psychoneuroendocrinology 2009; 34:413–419.
- Cohen A, Shane E. Treatment of premenopausal women with low bone mineral density. Curr Osteoporos Rep 2008; 6:39–46.
- Licata A. Bone density vs bone quality: what’s a clinician to do? Cleve Clin J Med 2009; 76:331–336.
- Bianchi ML, Baim S, Bishop NJ, et al. Official positions of the International Society for Clinical Densitometry (ISCD) on DXA evaluation in children and adolescents. Pediatr Nephrol 2010; 25:37–47.
- Mendelsohn FA, Warren MP. Anorexia, bulimia, and the female athlete triad: evaluation and management. Endocrinol Metab Clin North Am 2010; 39:155–167.
- Mehler PS, Krantz M. Anorexia nervosa medical issues. J Womens Health (Larchmt) 2003; 12:331–340.
- Watts NB, Lewiecki EM, Miller PD, Baim S. National Osteoporosis Foundation 2008 Clinician’s Guide to Prevention and Treatment of Osteoporosis and the World Health Organization Fracture Risk Assessment Tool (FRAX): what they mean to the bone densitometrist and bone technologist. J Clin Densitom 2008; 11:473–477.
- Nattiv A, Loucks AB, Manore MM, Sanborn CF, Sundgot-Borgen J, Warren MP; American College of Sports Medicine. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 2007; 39:1867–1882.
- Cummings SR, Palermo L, Browner W, et al. Monitoring osteoporosis therapy with bone densitometry: misleading changes and regression to the mean. Fracture Intervention Trial Research Group. JAMA 2000; 283:1318–1321.
- LeBoff MS, Kohlmeier L, Hurwitz S, Franklin J, Wright J, Glowacki J. Occult vitamin D deficiency in postmenopausal US women with acute hip fracture. JAMA 1999; 281:1505–1511.
- Adams JS, Hewison M. Update in vitamin D. J Clin Endocrinol Metab 2010; 95:471–478.
- Gordon CM, DePeter KC, Feldman HA, Grace E, Emans SJ. Prevalence of vitamin D deficiency among healthy adolescents. Arch Pediatr Adolesc Med 2004; 158:531–537.
- Hofbauer LC, Hamann C, Ebeling PR. Approach to the patient with secondary osteoporosis. Eur J Endocrinol 2010; 162:1009–1020.
- Stoffman N, Gordon CM. Vitamin D and adolescents: what do we know? Curr Opin Pediatr 2009; 21:465–471.
- Bone HG, Greenspan SL, McKeever C, et al. Alendronate and estrogen effects in postmenopausal women with low bone mineral density. Alendronate/Estrogen Study Group. J Clin Endocrinol Metab 2000; 85:720–726.
- Barrett-Connor E, Wenger NK, Grady D, et al. Hormone and nonhormone therapy for the maintenance of postmenopausal health: the need for randomized controlled trials of estrogen and raloxifene. J Womens Health 1998; 7:839–847.
- Klibanski A, Biller BM, Schoenfeld DA, Herzog DB, Saxe VC. The effects of estrogen administration on trabecular bone loss in young women with anorexia nervosa. J Clin Endocrinol Metab 1995; 80:898–904.
- Stokosch GR, Friedman AJ, Wu SC, Kamin M. Effects of an oral contraceptive (norgestimate/ethinyl estradiol) on bone mineral density in adolescent females with anorexia nervosa: double-blind, placebo-controlled study. J Adolesc Health 2006; 39:819–827.
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15:135–143.
- Robinson E, Bachrach LK, Katzman DK. Use of hormone replacement therapy to reduce the risk of osteopenia in adolescent girls with anorexia nervosa. J Adolesc Health 2000; 26:343–348.
- Myburgh KH, Hutchins J, Fataar AB, et al. Low bone density is an etiologic factor for stress fractures in athletes. Ann Intern Med 1990; 113:754–759.
- Young N, Formica C, Szmukler G, Seeman E. Bone density at weight-bearing and nonweight-bearing sites in ballet dancers: the effects of exercise, hypogonadism, and body weight. J Clin Endocrinol Metab 1994; 78:449–454.
- Stock JL, Bell NH, Chesnut CH, et al. Increments in bone mineral density of the lumbar spine and hip and suppression of bone turnover are maintained after discontinuation of alendronate in postmenopausal women. Am J Med 1997; 103:291–297.
- Subbiah V, Madsen VS, Raymond AK, Benjamin RS, Ludwig JA. Of mice and men: divergent risks of teriparatide-induced osteosarcoma. Osteoporos Int 2010; 21:1041–1045.
- Wren AM, Seal LJ, Cohen MA, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 2001; 86:5992.
- Miljic D, Pekic S, Djurovic M, et al. Ghrelin has partial or no effect on appetite, growth hormone, prolactin, and cortisol release in patients with anorexia nervosa. J Clin Endocrinol Metab 2006; 914:1491–1495.
- Siegfried Z, Kanyas K, Latzer Y, et al. Association study of cannabinoid receptor gene (CNR1) alleles and anorexia nervosa: differences between restricting and binging/purging subtypes. Am J Med Genet B Neuropsychiatr Genet 2004; 125B:126–130.
- Cota D, Marsicano G, Lutz B, et al. Endogenous cannabinoid system as a modulator of food intake. Int J Obes Relat Metab Disord 2003; 27:289–301.
- Gross H, Ebert MH, Faden VB, et al. A double-blind trial of delta 9-tetrahydrocannabinol in primary anorexia nervosa. J Clin Psychopharmacol 1983; 3:165–171.
Among the devastating effects of anorexia nervosa, and one that is easily overlooked, is its impact on bone.
Probably more than half of young women with anorexia nervosa develop osteoporosis, and relatively quickly. Baker et al1 obtained bone scans in a series of 56 young women, mean age 27 years, who had had an eating disorder for a mean of about 10 years, and found that the bone mineral density in the femur was below the critical fracture threshold in 42 (75%).
Osteoporosis is particularly common and worrisome in female athletes (and is becoming increasingly common in male athletes as well). Female athletes have a much higher incidence of disordered eating than their peers2 and therefore are at a much higher risk of fractures.
This review summarizes the factors affecting the development of osteoporosis in these patients and discusses potential targets for intervention.
ANOREXIA AND BONE HEALTH: A COMPLEX RELATIONSHIP
Anorexia nervosa is characterized by an intense fear of gaining weight, a body weight less than 85% of expected, a distorted self-image, and, in women, missing three consecutive menstrual periods.3 The lifetime prevalence in women is about 0.5%; it is much lower in men.3 The prevalence of eating disorders in female athletes is much higher, estimated at 15% to 62%.2
The etiology of osteoporosis in patients with anorexia nervosa is complex and multifaceted. In these patients, bone resorption is increased without a concomitant increase in bone formation, resulting in a net loss of bone.4 Thus, markers of bone resorption such as N-teleopeptide and deoxypyridoline are elevated, but markers of bone formation such as osteocalcin are not.4
The loss of bone may be rapid and can occur relatively early in the disease. Some studies suggest that an illness duration longer than 12 months predicts significant loss of bone density.5 Thus, early diagnosis and intervention are important to minimize bone loss.
Women gain from 40% to 60% of their bone mass during the pubertal growth spurt in ages 11 to 14, the time when anorexia nervosa is most prevalent.6 Peak bone mass is attained by the third decade of life, but the rate of growth of bone mass is highest during adolescence and early adulthood.7 Hence, it is important to optimize bone mass during this time, as small differences in bone density can have significant clinical implications later in life: a 5% increase in bone density significantly decreases fracture risk, whereas a 10% decrease in adult bone mineral density is associated with a two to three times higher risk of fracture (reviewed by Rome and Ammerman6).
What is the role of amenorrhea in the development of osteoporosis in premenopausal patients?
Given that two of the most characteristic manifestations of anorexia nervosa are low body weight and the absence of menses, these factors have been hypothesized to be potential causes of osteoporosis.
In general, young women who present with amenorrhea should be evaluated to determine if the amenorrhea is primary or secondary. Primary amenorrhea is the absence of menarche by age 16; secondary amenorrhea is the absence of menses for more than three cycles or more than 6 months in someone who previously had had menses. The most common causes of secondary amenorrhea are ovarian disease, hypothalamic or pituitary disease, and uterine disease. Anorexia nervosa causes hypothalamic dysfunction and is a cause of secondary amenorrhea. In clinical practice, it is also important to remember that pregnancy can occur even in the setting of amenorrhea.
Amenorrhea in patients with anorexia nervosa is related to hypothalamic suppression of the release of gonadotropin-releasing hormone, resulting in lower levels of follicle-stimulating hormone and luteinizing hormone and a resultant prepubertal low-estrogen state.
In a study of 73 women with anorexia nervosa and a mean age of 17.2 years,8 20 months of amenorrhea was the threshold above which the most severe osteopenia was seen, implying that the duration of amenorrhea affects bone health.
Which factors besides amenorrhea influence bone density in premenopausal women?
Undernutrition. Body weight has been suggested to have an independent effect on bone mineral density, and density has been found to increase following weight gain, even before the return of menses.1 Once a regular menstrual cycle has been restored, significant increases in trabecular and cortical bone have been detected.1
Deficiency of insulin-like growth factor 1 (IGF-1). Anorexia nervosa is associated with decreased hepatic synthesis of IGF-I.9 Low levels of IGF-I reduce the levels of osteocalcin, a marker of bone formation, and cause abnormalities in osteoblast function.10 This deficiency is associated with the development of osteopenia in patients with anorexia nervosa.11
Low androgen levels are present in patients with anorexia nervosa, and levels appear to be further reduced by oral contraceptives.12 It remains to be determined whether the further reduction in androgens in women with anorexia nervosa using oral contraceptives is harmful to skeletal health. Low testosterone levels in boys with anorexia nervosa have been associated with lower libido, fewer erections, and potentially lower bone density.13
Hypercortisolemia. Elevated levels of total and free serum cortisol and high 24-hour urinary free cortisol excretion have been noted in anorectic patients. Levels of cortisol are inversely related to levels of osteocalcin, and hypercortisolism has been shown to be associated with osteoporosis.14,15 However, no study has yet shown causality in this population.
Osteoprotegerin has been recognized as an important regulator of bone resorption. Osteoprotegerin inhibits osteoclast differentiation and activation and stimulates osteoclast apoptosis, helping to preserve bone density.
Misra et al16 showed that adolescent girls with anorexia nervosa have higher serum osteoprotegerin levels than controls and that osteoprotegerin levels correlate inversely with markers of nutritional status and lumbar bone density Z scores.16 They and other investigators17 postulate that osteoprotegerin may be released as a compensatory response to the bone loss seen in these patients in an attempt to preserve bone health.
Leptin is an adipocyte-derived hormone that acts on receptors in the hypothalamus, decreasing food intake and increasing energy expenditure. Low leptin levels are a key endocrinologic feature of anorexia nervosa.18 Leptin helps to induce weight loss by stimulating neurons in the hypothalamus that express “weight-loss-inducing” neuropeptides such as pro-opiomelanocortin and inhibiting “weight-gain-inducing” peptides such as neuropeptide Y.19
Although leptin was first believed to be a hormone released to counteract obesity, recent studies19,20 suggest that it is part of a major signaling system that controls adaptation to starvation. These studies have shown that the body senses its corporeal fat through leptin and inhibits ovulation when fat reserves are low.19 In addition, luteinizing hormone and leptin levels have been shown to increase in parallel in patients with anorexia nervosa when weight is restored.20 Thus, rising leptin levels correlate with the resumption of menses in women with anorexia nervosa and in turn have potential consequences for bone health.
Not enough ghrelin, too much obestatin? Ghrelin, a gastric hormone, acts as a natural antagonist to leptin, resulting in an increase in food intake and body weight.19 Circulating ghrelin levels are higher in illness-induced anorexia as well as in anorexia nervosa, and they normalize with weight gain, perhaps as an adaptive mechanism to compensate for a negative energy balance.21
Several in vitro studies suggest that ghrelin directly promotes osteoblast proliferation and differentiation.22 However, human studies of ghrelin’s effects on bone are limited. In a study of healthy younger women, healthy boys, and anorexia nervosa patients, plasma ghrelin levels were only weakly associated with bone mineral density.23
The effects of obestatin, another gastric hormone, are still being investigated. Obestatin was initially shown to oppose the effects of ghrelin by decreasing appetite and weight gain. When given with ghrelin, obestatin appears to work with ghrelin at the hypothalamic level to modulate food intake and growth hormone secretion.24
Interestingly, obestatin and the ratio of ghrelin to obestatin are decreased in patients with anorexia nervosa, but the ratio is unchanged in thin women who have an equivalent body mass index but no eating disorder.25 It has been hypothesized that the ghrelin-obestatin ratio may be the key to explaining the eating restriction and reduced motivation to eat despite high ghrelin levels seen in anorexia nervosa.26 Further studies are needed to determine the role of obestatin and the ghrelin-obestatin ratio in the bone health of women with anorexia nervosa.
HOW SHOULD WE DIAGNOSE OSTEOPOROSIS IN PREMENOPAUSAL PATIENTS?
Our approach to screening for and diagnosing osteoporosis is still largely based on measuring bone mineral density, although density by itself is not a perfect tool for predicting who will or will not experience a fracture, particularly in premenopausal women.26,27 Most premenopausal women with low bone mineral density but no other risk factors for fracture such as previous fractures or glucocorticoid therapy are at very low short-term risk of fracture.26
For these reasons, in premenopausal women and adolescents, the International Society for Clinical Densitometry28 advises against diagnosing osteoporosis on the basis of bone mineral density alone. Instead, it should be diagnosed in this population only if the bone mineral density is low (defined as a Z score below −2.0) and the patient has risk factors that suggest a higher short-term risk of bone mineral loss and fracture. Risk factors include chronic malnutrition, eating disorders, hypogonadism, glucocorticoid exposure, and previous fracture.29
A pitfall in interpreting low bone mineral density in premenopausal women younger than age 30 is the possibility that they may not yet have reached their peak bone mass. In addition, small stature and body size (and therefore bone size) also influence the results of dual-energy x-ray absorptiometry. This test may not be able to distinguish bone that is small but of normal density from bone that is of low density.26
Despite its limitations, until newer risk assessment tools are available for this patient population, measuring bone mineral density is still recommended in addition to assessing clinical risk factors to diagnose osteoporosis. Also, changes in bone mineral density over time can help to assess risk and guide treatment.
When should a patient with anorexia be screened for osteoporosis?
Because bone loss may begin early in the course of anorexia and progress rapidly (potentially inexorably), baseline screening is recommended for all patients who have had anorexia nervosa or amenorrhea for more than 6 to 12 months.30 The National Osteoporosis Foundation recommends screening in women under age 65 who have a low body weight.31 The American College of Sports Medicine recommends screening for osteoporosis in athletes with a history of hypoestrogenism or disordered eating for a cumulative total of 6 months or more, or with a history of stress fracture or fracture from minimal trauma.32
Knowledge of low bone mineral density and fracture risk can often guide treatment and prompt behavioral change. Given that most osteoporosis treatments do not lead to detectable changes in bone density until 18 months to 2 years, it is reasonable to repeat testing at this interval.33
NEW AND OLD TREATMENTS FOR LOW BONE DENSITY IN ANOREXIA NERVOSA
Weight restoration is the cornerstone
Restoration of body weight and nutritional rehabilitation remain the cornerstones of treatment. All patients with anorexia nervosa should be referred to a nutritionist to develop a meal plan that is adjusted for the amount of energy expended. The challenges lie in managing the complications of refeeding and the high relapse rate. The treatment goals in disordered eating are to optimize the overall nutritional status, normalize eating behavior, modify unhealthy thought processes that maintain the disorder, and treat possible emotional issues that help create or maintain the disorder.
The younger the patient, the more the family’s involvement is recommended. In addition to nutritional counseling, the care team should include a psychotherapist, a psychiatrist, and a primary care physician to assist with management and screening of medical complications.
Vitamin D for all
Low vitamin D levels have long been associated with low bone mineral density and risk of hip fracture.34
Vitamin D insufficiency is very common. More than 90% of blacks, Hispanics, and Asians and nearly 75% of whites have insufficient levels of vitamin D (25-hydroxyvitamin D3 level < 30 ng/mL).35 In a study of 307 healthy adolescents, vitamin D insufficiency (a 25-hydroxyvitamin D3 level < 20 ng/mL) was found in 42% and vitamin D deficiency (a level < 15 ng/mL) in 24.1%.36 In addition, this study confirmed an inverse correlation between body mass index and serum 25-hydroxyvitamin D3 concentration.
Therefore, while vitamin D supplementation has not been consistently shown to improve bone loss in anorectic patients,9 given the prevalence of vitamin D deficiency and insufficiency, supplementation is almost universally recommended.
There is no consensus as to the amount of supplementation to recommend for women with anorexia nervosa. The American College of Sports Medicine recommends a total daily intake of 800 IU of vitamin D (ie, from diet and supplements). Therapy should be titrated to doses that result in normocalcemia and a serum 25-hydroxyvitamin D3 concentration of at least 30 ng/mL.37,38
Does hormone treatment improve bone density?
In postmenopausal osteoporosis, estrogen therapy maintains or improves bone density and appears to reduce the rate of vertebral fractures.39,40 Perhaps not so with premenopausal osteoporosis due to anorexia nervosa.
Why should this be? In postmenopausal women, estrogen therapy appears to work by impairing osteoclast-mediated bone resorption, but it has only limited effects on bone formation. In premenopausal anorexia, however, bone loss appears to be due to a unique uncoupling of osteoblastic and osteoclastic functions, resulting in both reduced bone formation and increased bone resorption, which estrogen therapy may not improve.5
Despite the documented association between anorexia nervosa and estrogen deficiency and the strong correlation between osteoporosis and the duration of amenorrhea, most studies have found no improvement in bone mass with hormonal therapy.9 In particular, three randomized, placebo-controlled trials have been published to date, and not one showed a significant improvement in bone mineral density with estrogen therapy compared with placebo in patients with anorexia nervosa.41–43
Klibanski et al,41 in the first of these trials, found no significant difference in spinal bone mineral density between treated patients and controls. However, estrogen-treated patients whose initial body weight was very low (< 70% of expected) had a significant increase in their bone mineral density, whereas those in the control group lost bone density.
Baker et al44 suggest that hormone therapy might protect bone mass in athletes with amenorrhea, citing a study that found that women with a history of stress fractures were less likely to have used oral contraceptives previously than athletes without fractures.45 However, no prospective randomized study to date has established that hormone therapy effectively preserves bone mass in athletes with amenorrhea.
Based on the data presented above, we have little evidence for using estrogen to treat or prevent premenopausal osteoporosis.
The American College of Sports Medicine32 recommends consideration of estrogen therapy if there is evidence of a decline in bone mineral density in an athlete over the age of 16 with persistent functional hypothalamic amenorrhea despite adequate nutritional intake and weight. However, it acknowledges that restoring regular menstrual cycles with oral contraceptive pills will not normalize the metabolic factors that impair bone formation, health, and performance and is not likely to fully reverse low bone mineral density in this population.
What is the effect of exercise on bone health in these patients?
Several studies have examined the effect of weight-bearing exercise on bone density.
Young et al46 compared normal teenagers, ballet dancers, and young women with anorexia nervosa and found that weight-bearing exercise protected against osteoporosis, but only at weight-bearing sites. Athletes in weight-bearing sports had a 5% to 15% higher bone mineral density in weight-bearing sites (ie, the femur) compared with nonathletes, but had lower bone mineral density in the spine.
Therefore, a Z score below −1.0 in an athlete, especially in a distal site, warrants further investigation and treatment.32 In general, exercise does not necessarily protect against osteoporosis in this patient population, and it can sometimes mask underlying bone loss. In addition, keep in mind that many of these patients exercise compulsively, using it as a form of purging.
Insulin-like growth factor-1: More study needed
IGF-1 contributes to bone growth by stimulating osteoblasts, and patients with anorexia nervosa have been shown to have low levels of IGF-1.9
Grinspoon et al10 randomized 60 patients with anorexia nervosa to receive IGF-1 alone, IGF-1 plus an oral contraceptive, an oral contraceptive alone, or placebo. All patients were given calcium and vitamin D and were followed for 9 months. Total bone mass increased significantly in those taking IGF-1 compared with those taking placebo. Those taking an IGF-1 and an oral contraceptive had a significant increase in spinal bone mineral density compared with those on placebo group. At other skeletal sites, however, IGF-1 plus an oral contraceptive and IGF-1 alone failed to produce significant increases in bone mineral density compared with placebo.
Further study is needed to determine the role of IGF-1 in treating low bone mineral density in anorexia nervosa.
Bisphosphonates: Not approved for this indication
In premenopausal women, bisphosphonates are approved by the US Food and Drug Administration (FDA) for use only in those taking glucocorticoids. Although bisphosphonates have been shown to significantly increase bone mineral density in young women with anorexia nervosa,26 they should be used with caution in patients of childbearing age because they are teratogenic. Bisphosphonates have a long half-life and may continue to affect bone turnover for up to 2 years after they are discontinued.47 In addition, they are not recommended in patients with a history of purging via vomiting, due to a risk of esophageal ulceration.
Parathyroid hormone therapy: Studies ongoing
The parathyroid hormone fragment teriparatide (Forteo) is widely used for treating postmenopausal osteoporosis.
Before teriparatide was approved, there was concern that it might increase the risk of osteosarcoma, as almost 45% of rats treated with this drug at the highest-tested dose level developed this aggressive form of bone cancer.48 Balancing the proven benefits of teriparatide shown by clinical trials with the theoretic risk of teriparatide-induced osteosarcoma, the FDA mandated a “black-box” warning about this potential effect.
Studies of parathyroid hormone treatment in anorexia nervosa and other premenopausal patients are ongoing.26
Leptin: More study needed
Leptin is a potent stimulator of bone growth and has been shown to increase bone mineral density in vitro and in vivo.19 However, concerns have been raised about giving supra-physiologic doses of leptin to patients with anorexia nervosa, as this may increase the risk of further weight loss and relapse.
More work is needed to determine the role of leptin for the treatment of osteoporosis in anorexia nervosa.
Ghrelin: Probably not effective as a single agent
Pharmacologic use of ghrelin increases food intake in healthy humans,49 and it has been proposed as a treatment for weight restoration and bone health in anorexia nervosa. Preliminary studies have not shown it to increase appetite or weight gain,50 but it did increase slow-wave sleep.
Based on these studies, it is unlikely that ghrelin will be effective as a single agent to stimulate appetite, but it may be helpful in conjunction with other therapies.
Cannabinoids: Little ongoing research
Cannabinoids have been proposed as a treatment for anorexia nervosa in the hope that they would induce weight gain and in turn prevent osteoporosis.
Interest in their use in anorexia nervosa stems from the discovery of two cannabinoid receptors (CB1 and CB2) located in the brain and peripheral organ systems. Anorexia nervosa has been associated with different alleles of the CB1 gene,51 but the therapeutic implications of this are far from clear.
Cannabinoids appear to regulate eating behavior at several levels within the brain and periphery: the hypothalamus and hindbrain (integrative functions), the limbic system (for hedonic evaluation of foods), the intestinal system, and adipose tissue.52 At each of these levels, the endocannabinoid system interacts with a number of better known peptides involved in appetite regulation, including leptin, ghrelin, and the melanocortins. In mouse studies, genetic leptin deficiency is associated with elevated hypothalamic endocannabinoid levels.
Appetite stimulation by cannabinoids has been studied for several decades, particularly in relation to cachexia and malnutrition associated with cancer. Very few trials have studied cannabinoids for anorexia nervosa.
In a 4-week crossover trial in 11 patients with anorexia nervosa,53 tetrahydrocannabinol (THC) treatment resulted in an increase in sleep disturbances and interpersonal sensitivity, but it had no significant effect on weight gain compared with diazepam treatment.53
Another pilot study of nine outpatients with anorexia nervosa treated with THC showed a significant improvement in depression and perfectionism scores without any significant weight gain.19
Although this research was once promising, the risk was felt to outweigh the benefit, as cannabinoids may induce dependency in this patient group, who may already be at high risk of drug addiction, and very few have continued this line of investigation.
WHAT CAN WE DO FOR NOW?
- Weight restoration and nutritional rehabilitation remain the keys to treatment of low bone density to reduce the risk of osteoporosis in patients with anorexia nervosa. However, as many as one-third of patients with anorexia nervosa relapse during their lifetime, and other treatments are needed to stabilize and prevent bone loss.
- Vitamin D deficiency is clearly associated with a risk of osteoporosis and fracture, and patients with vitamin D deficiency should be treated with supplementation.
- Standard therapies in postmenopausal patients (such as bisphosphonates and teriparatide) should be used with caution in premenopausal anorexia nervosa patients because of potential long-term health risks.
- As we learn more about hormonal factors in anorexia nervosa, we hope to identify interventions that will help restore weight and decrease the risk of osteoporosis. A summary of potential treatment strategies and targets for prevention of osteoporosis in anorexia nervosa is presented in Table 2.
Acknowledgment: The author thanks the General Internal Medicine Works in Progress Group for its editorial comments, and Dr. Ellen Rome for her mentorship and support.
Among the devastating effects of anorexia nervosa, and one that is easily overlooked, is its impact on bone.
Probably more than half of young women with anorexia nervosa develop osteoporosis, and relatively quickly. Baker et al1 obtained bone scans in a series of 56 young women, mean age 27 years, who had had an eating disorder for a mean of about 10 years, and found that the bone mineral density in the femur was below the critical fracture threshold in 42 (75%).
Osteoporosis is particularly common and worrisome in female athletes (and is becoming increasingly common in male athletes as well). Female athletes have a much higher incidence of disordered eating than their peers2 and therefore are at a much higher risk of fractures.
This review summarizes the factors affecting the development of osteoporosis in these patients and discusses potential targets for intervention.
ANOREXIA AND BONE HEALTH: A COMPLEX RELATIONSHIP
Anorexia nervosa is characterized by an intense fear of gaining weight, a body weight less than 85% of expected, a distorted self-image, and, in women, missing three consecutive menstrual periods.3 The lifetime prevalence in women is about 0.5%; it is much lower in men.3 The prevalence of eating disorders in female athletes is much higher, estimated at 15% to 62%.2
The etiology of osteoporosis in patients with anorexia nervosa is complex and multifaceted. In these patients, bone resorption is increased without a concomitant increase in bone formation, resulting in a net loss of bone.4 Thus, markers of bone resorption such as N-teleopeptide and deoxypyridoline are elevated, but markers of bone formation such as osteocalcin are not.4
The loss of bone may be rapid and can occur relatively early in the disease. Some studies suggest that an illness duration longer than 12 months predicts significant loss of bone density.5 Thus, early diagnosis and intervention are important to minimize bone loss.
Women gain from 40% to 60% of their bone mass during the pubertal growth spurt in ages 11 to 14, the time when anorexia nervosa is most prevalent.6 Peak bone mass is attained by the third decade of life, but the rate of growth of bone mass is highest during adolescence and early adulthood.7 Hence, it is important to optimize bone mass during this time, as small differences in bone density can have significant clinical implications later in life: a 5% increase in bone density significantly decreases fracture risk, whereas a 10% decrease in adult bone mineral density is associated with a two to three times higher risk of fracture (reviewed by Rome and Ammerman6).
What is the role of amenorrhea in the development of osteoporosis in premenopausal patients?
Given that two of the most characteristic manifestations of anorexia nervosa are low body weight and the absence of menses, these factors have been hypothesized to be potential causes of osteoporosis.
In general, young women who present with amenorrhea should be evaluated to determine if the amenorrhea is primary or secondary. Primary amenorrhea is the absence of menarche by age 16; secondary amenorrhea is the absence of menses for more than three cycles or more than 6 months in someone who previously had had menses. The most common causes of secondary amenorrhea are ovarian disease, hypothalamic or pituitary disease, and uterine disease. Anorexia nervosa causes hypothalamic dysfunction and is a cause of secondary amenorrhea. In clinical practice, it is also important to remember that pregnancy can occur even in the setting of amenorrhea.
Amenorrhea in patients with anorexia nervosa is related to hypothalamic suppression of the release of gonadotropin-releasing hormone, resulting in lower levels of follicle-stimulating hormone and luteinizing hormone and a resultant prepubertal low-estrogen state.
In a study of 73 women with anorexia nervosa and a mean age of 17.2 years,8 20 months of amenorrhea was the threshold above which the most severe osteopenia was seen, implying that the duration of amenorrhea affects bone health.
Which factors besides amenorrhea influence bone density in premenopausal women?
Undernutrition. Body weight has been suggested to have an independent effect on bone mineral density, and density has been found to increase following weight gain, even before the return of menses.1 Once a regular menstrual cycle has been restored, significant increases in trabecular and cortical bone have been detected.1
Deficiency of insulin-like growth factor 1 (IGF-1). Anorexia nervosa is associated with decreased hepatic synthesis of IGF-I.9 Low levels of IGF-I reduce the levels of osteocalcin, a marker of bone formation, and cause abnormalities in osteoblast function.10 This deficiency is associated with the development of osteopenia in patients with anorexia nervosa.11
Low androgen levels are present in patients with anorexia nervosa, and levels appear to be further reduced by oral contraceptives.12 It remains to be determined whether the further reduction in androgens in women with anorexia nervosa using oral contraceptives is harmful to skeletal health. Low testosterone levels in boys with anorexia nervosa have been associated with lower libido, fewer erections, and potentially lower bone density.13
Hypercortisolemia. Elevated levels of total and free serum cortisol and high 24-hour urinary free cortisol excretion have been noted in anorectic patients. Levels of cortisol are inversely related to levels of osteocalcin, and hypercortisolism has been shown to be associated with osteoporosis.14,15 However, no study has yet shown causality in this population.
Osteoprotegerin has been recognized as an important regulator of bone resorption. Osteoprotegerin inhibits osteoclast differentiation and activation and stimulates osteoclast apoptosis, helping to preserve bone density.
Misra et al16 showed that adolescent girls with anorexia nervosa have higher serum osteoprotegerin levels than controls and that osteoprotegerin levels correlate inversely with markers of nutritional status and lumbar bone density Z scores.16 They and other investigators17 postulate that osteoprotegerin may be released as a compensatory response to the bone loss seen in these patients in an attempt to preserve bone health.
Leptin is an adipocyte-derived hormone that acts on receptors in the hypothalamus, decreasing food intake and increasing energy expenditure. Low leptin levels are a key endocrinologic feature of anorexia nervosa.18 Leptin helps to induce weight loss by stimulating neurons in the hypothalamus that express “weight-loss-inducing” neuropeptides such as pro-opiomelanocortin and inhibiting “weight-gain-inducing” peptides such as neuropeptide Y.19
Although leptin was first believed to be a hormone released to counteract obesity, recent studies19,20 suggest that it is part of a major signaling system that controls adaptation to starvation. These studies have shown that the body senses its corporeal fat through leptin and inhibits ovulation when fat reserves are low.19 In addition, luteinizing hormone and leptin levels have been shown to increase in parallel in patients with anorexia nervosa when weight is restored.20 Thus, rising leptin levels correlate with the resumption of menses in women with anorexia nervosa and in turn have potential consequences for bone health.
Not enough ghrelin, too much obestatin? Ghrelin, a gastric hormone, acts as a natural antagonist to leptin, resulting in an increase in food intake and body weight.19 Circulating ghrelin levels are higher in illness-induced anorexia as well as in anorexia nervosa, and they normalize with weight gain, perhaps as an adaptive mechanism to compensate for a negative energy balance.21
Several in vitro studies suggest that ghrelin directly promotes osteoblast proliferation and differentiation.22 However, human studies of ghrelin’s effects on bone are limited. In a study of healthy younger women, healthy boys, and anorexia nervosa patients, plasma ghrelin levels were only weakly associated with bone mineral density.23
The effects of obestatin, another gastric hormone, are still being investigated. Obestatin was initially shown to oppose the effects of ghrelin by decreasing appetite and weight gain. When given with ghrelin, obestatin appears to work with ghrelin at the hypothalamic level to modulate food intake and growth hormone secretion.24
Interestingly, obestatin and the ratio of ghrelin to obestatin are decreased in patients with anorexia nervosa, but the ratio is unchanged in thin women who have an equivalent body mass index but no eating disorder.25 It has been hypothesized that the ghrelin-obestatin ratio may be the key to explaining the eating restriction and reduced motivation to eat despite high ghrelin levels seen in anorexia nervosa.26 Further studies are needed to determine the role of obestatin and the ghrelin-obestatin ratio in the bone health of women with anorexia nervosa.
HOW SHOULD WE DIAGNOSE OSTEOPOROSIS IN PREMENOPAUSAL PATIENTS?
Our approach to screening for and diagnosing osteoporosis is still largely based on measuring bone mineral density, although density by itself is not a perfect tool for predicting who will or will not experience a fracture, particularly in premenopausal women.26,27 Most premenopausal women with low bone mineral density but no other risk factors for fracture such as previous fractures or glucocorticoid therapy are at very low short-term risk of fracture.26
For these reasons, in premenopausal women and adolescents, the International Society for Clinical Densitometry28 advises against diagnosing osteoporosis on the basis of bone mineral density alone. Instead, it should be diagnosed in this population only if the bone mineral density is low (defined as a Z score below −2.0) and the patient has risk factors that suggest a higher short-term risk of bone mineral loss and fracture. Risk factors include chronic malnutrition, eating disorders, hypogonadism, glucocorticoid exposure, and previous fracture.29
A pitfall in interpreting low bone mineral density in premenopausal women younger than age 30 is the possibility that they may not yet have reached their peak bone mass. In addition, small stature and body size (and therefore bone size) also influence the results of dual-energy x-ray absorptiometry. This test may not be able to distinguish bone that is small but of normal density from bone that is of low density.26
Despite its limitations, until newer risk assessment tools are available for this patient population, measuring bone mineral density is still recommended in addition to assessing clinical risk factors to diagnose osteoporosis. Also, changes in bone mineral density over time can help to assess risk and guide treatment.
When should a patient with anorexia be screened for osteoporosis?
Because bone loss may begin early in the course of anorexia and progress rapidly (potentially inexorably), baseline screening is recommended for all patients who have had anorexia nervosa or amenorrhea for more than 6 to 12 months.30 The National Osteoporosis Foundation recommends screening in women under age 65 who have a low body weight.31 The American College of Sports Medicine recommends screening for osteoporosis in athletes with a history of hypoestrogenism or disordered eating for a cumulative total of 6 months or more, or with a history of stress fracture or fracture from minimal trauma.32
Knowledge of low bone mineral density and fracture risk can often guide treatment and prompt behavioral change. Given that most osteoporosis treatments do not lead to detectable changes in bone density until 18 months to 2 years, it is reasonable to repeat testing at this interval.33
NEW AND OLD TREATMENTS FOR LOW BONE DENSITY IN ANOREXIA NERVOSA
Weight restoration is the cornerstone
Restoration of body weight and nutritional rehabilitation remain the cornerstones of treatment. All patients with anorexia nervosa should be referred to a nutritionist to develop a meal plan that is adjusted for the amount of energy expended. The challenges lie in managing the complications of refeeding and the high relapse rate. The treatment goals in disordered eating are to optimize the overall nutritional status, normalize eating behavior, modify unhealthy thought processes that maintain the disorder, and treat possible emotional issues that help create or maintain the disorder.
The younger the patient, the more the family’s involvement is recommended. In addition to nutritional counseling, the care team should include a psychotherapist, a psychiatrist, and a primary care physician to assist with management and screening of medical complications.
Vitamin D for all
Low vitamin D levels have long been associated with low bone mineral density and risk of hip fracture.34
Vitamin D insufficiency is very common. More than 90% of blacks, Hispanics, and Asians and nearly 75% of whites have insufficient levels of vitamin D (25-hydroxyvitamin D3 level < 30 ng/mL).35 In a study of 307 healthy adolescents, vitamin D insufficiency (a 25-hydroxyvitamin D3 level < 20 ng/mL) was found in 42% and vitamin D deficiency (a level < 15 ng/mL) in 24.1%.36 In addition, this study confirmed an inverse correlation between body mass index and serum 25-hydroxyvitamin D3 concentration.
Therefore, while vitamin D supplementation has not been consistently shown to improve bone loss in anorectic patients,9 given the prevalence of vitamin D deficiency and insufficiency, supplementation is almost universally recommended.
There is no consensus as to the amount of supplementation to recommend for women with anorexia nervosa. The American College of Sports Medicine recommends a total daily intake of 800 IU of vitamin D (ie, from diet and supplements). Therapy should be titrated to doses that result in normocalcemia and a serum 25-hydroxyvitamin D3 concentration of at least 30 ng/mL.37,38
Does hormone treatment improve bone density?
In postmenopausal osteoporosis, estrogen therapy maintains or improves bone density and appears to reduce the rate of vertebral fractures.39,40 Perhaps not so with premenopausal osteoporosis due to anorexia nervosa.
Why should this be? In postmenopausal women, estrogen therapy appears to work by impairing osteoclast-mediated bone resorption, but it has only limited effects on bone formation. In premenopausal anorexia, however, bone loss appears to be due to a unique uncoupling of osteoblastic and osteoclastic functions, resulting in both reduced bone formation and increased bone resorption, which estrogen therapy may not improve.5
Despite the documented association between anorexia nervosa and estrogen deficiency and the strong correlation between osteoporosis and the duration of amenorrhea, most studies have found no improvement in bone mass with hormonal therapy.9 In particular, three randomized, placebo-controlled trials have been published to date, and not one showed a significant improvement in bone mineral density with estrogen therapy compared with placebo in patients with anorexia nervosa.41–43
Klibanski et al,41 in the first of these trials, found no significant difference in spinal bone mineral density between treated patients and controls. However, estrogen-treated patients whose initial body weight was very low (< 70% of expected) had a significant increase in their bone mineral density, whereas those in the control group lost bone density.
Baker et al44 suggest that hormone therapy might protect bone mass in athletes with amenorrhea, citing a study that found that women with a history of stress fractures were less likely to have used oral contraceptives previously than athletes without fractures.45 However, no prospective randomized study to date has established that hormone therapy effectively preserves bone mass in athletes with amenorrhea.
Based on the data presented above, we have little evidence for using estrogen to treat or prevent premenopausal osteoporosis.
The American College of Sports Medicine32 recommends consideration of estrogen therapy if there is evidence of a decline in bone mineral density in an athlete over the age of 16 with persistent functional hypothalamic amenorrhea despite adequate nutritional intake and weight. However, it acknowledges that restoring regular menstrual cycles with oral contraceptive pills will not normalize the metabolic factors that impair bone formation, health, and performance and is not likely to fully reverse low bone mineral density in this population.
What is the effect of exercise on bone health in these patients?
Several studies have examined the effect of weight-bearing exercise on bone density.
Young et al46 compared normal teenagers, ballet dancers, and young women with anorexia nervosa and found that weight-bearing exercise protected against osteoporosis, but only at weight-bearing sites. Athletes in weight-bearing sports had a 5% to 15% higher bone mineral density in weight-bearing sites (ie, the femur) compared with nonathletes, but had lower bone mineral density in the spine.
Therefore, a Z score below −1.0 in an athlete, especially in a distal site, warrants further investigation and treatment.32 In general, exercise does not necessarily protect against osteoporosis in this patient population, and it can sometimes mask underlying bone loss. In addition, keep in mind that many of these patients exercise compulsively, using it as a form of purging.
Insulin-like growth factor-1: More study needed
IGF-1 contributes to bone growth by stimulating osteoblasts, and patients with anorexia nervosa have been shown to have low levels of IGF-1.9
Grinspoon et al10 randomized 60 patients with anorexia nervosa to receive IGF-1 alone, IGF-1 plus an oral contraceptive, an oral contraceptive alone, or placebo. All patients were given calcium and vitamin D and were followed for 9 months. Total bone mass increased significantly in those taking IGF-1 compared with those taking placebo. Those taking an IGF-1 and an oral contraceptive had a significant increase in spinal bone mineral density compared with those on placebo group. At other skeletal sites, however, IGF-1 plus an oral contraceptive and IGF-1 alone failed to produce significant increases in bone mineral density compared with placebo.
Further study is needed to determine the role of IGF-1 in treating low bone mineral density in anorexia nervosa.
Bisphosphonates: Not approved for this indication
In premenopausal women, bisphosphonates are approved by the US Food and Drug Administration (FDA) for use only in those taking glucocorticoids. Although bisphosphonates have been shown to significantly increase bone mineral density in young women with anorexia nervosa,26 they should be used with caution in patients of childbearing age because they are teratogenic. Bisphosphonates have a long half-life and may continue to affect bone turnover for up to 2 years after they are discontinued.47 In addition, they are not recommended in patients with a history of purging via vomiting, due to a risk of esophageal ulceration.
Parathyroid hormone therapy: Studies ongoing
The parathyroid hormone fragment teriparatide (Forteo) is widely used for treating postmenopausal osteoporosis.
Before teriparatide was approved, there was concern that it might increase the risk of osteosarcoma, as almost 45% of rats treated with this drug at the highest-tested dose level developed this aggressive form of bone cancer.48 Balancing the proven benefits of teriparatide shown by clinical trials with the theoretic risk of teriparatide-induced osteosarcoma, the FDA mandated a “black-box” warning about this potential effect.
Studies of parathyroid hormone treatment in anorexia nervosa and other premenopausal patients are ongoing.26
Leptin: More study needed
Leptin is a potent stimulator of bone growth and has been shown to increase bone mineral density in vitro and in vivo.19 However, concerns have been raised about giving supra-physiologic doses of leptin to patients with anorexia nervosa, as this may increase the risk of further weight loss and relapse.
More work is needed to determine the role of leptin for the treatment of osteoporosis in anorexia nervosa.
Ghrelin: Probably not effective as a single agent
Pharmacologic use of ghrelin increases food intake in healthy humans,49 and it has been proposed as a treatment for weight restoration and bone health in anorexia nervosa. Preliminary studies have not shown it to increase appetite or weight gain,50 but it did increase slow-wave sleep.
Based on these studies, it is unlikely that ghrelin will be effective as a single agent to stimulate appetite, but it may be helpful in conjunction with other therapies.
Cannabinoids: Little ongoing research
Cannabinoids have been proposed as a treatment for anorexia nervosa in the hope that they would induce weight gain and in turn prevent osteoporosis.
Interest in their use in anorexia nervosa stems from the discovery of two cannabinoid receptors (CB1 and CB2) located in the brain and peripheral organ systems. Anorexia nervosa has been associated with different alleles of the CB1 gene,51 but the therapeutic implications of this are far from clear.
Cannabinoids appear to regulate eating behavior at several levels within the brain and periphery: the hypothalamus and hindbrain (integrative functions), the limbic system (for hedonic evaluation of foods), the intestinal system, and adipose tissue.52 At each of these levels, the endocannabinoid system interacts with a number of better known peptides involved in appetite regulation, including leptin, ghrelin, and the melanocortins. In mouse studies, genetic leptin deficiency is associated with elevated hypothalamic endocannabinoid levels.
Appetite stimulation by cannabinoids has been studied for several decades, particularly in relation to cachexia and malnutrition associated with cancer. Very few trials have studied cannabinoids for anorexia nervosa.
In a 4-week crossover trial in 11 patients with anorexia nervosa,53 tetrahydrocannabinol (THC) treatment resulted in an increase in sleep disturbances and interpersonal sensitivity, but it had no significant effect on weight gain compared with diazepam treatment.53
Another pilot study of nine outpatients with anorexia nervosa treated with THC showed a significant improvement in depression and perfectionism scores without any significant weight gain.19
Although this research was once promising, the risk was felt to outweigh the benefit, as cannabinoids may induce dependency in this patient group, who may already be at high risk of drug addiction, and very few have continued this line of investigation.
WHAT CAN WE DO FOR NOW?
- Weight restoration and nutritional rehabilitation remain the keys to treatment of low bone density to reduce the risk of osteoporosis in patients with anorexia nervosa. However, as many as one-third of patients with anorexia nervosa relapse during their lifetime, and other treatments are needed to stabilize and prevent bone loss.
- Vitamin D deficiency is clearly associated with a risk of osteoporosis and fracture, and patients with vitamin D deficiency should be treated with supplementation.
- Standard therapies in postmenopausal patients (such as bisphosphonates and teriparatide) should be used with caution in premenopausal anorexia nervosa patients because of potential long-term health risks.
- As we learn more about hormonal factors in anorexia nervosa, we hope to identify interventions that will help restore weight and decrease the risk of osteoporosis. A summary of potential treatment strategies and targets for prevention of osteoporosis in anorexia nervosa is presented in Table 2.
Acknowledgment: The author thanks the General Internal Medicine Works in Progress Group for its editorial comments, and Dr. Ellen Rome for her mentorship and support.
- Baker D, Roberts R, Towell T. Factors predictive of bone mineral density in eating-disordered women: a longitudinal study. Int J Eat Disord 2000; 27:29–35.
- Rome ES. Eating disorders. Obstet Gynecol Clin North Am 2003; 30:353–377.
- First MB, editor. Diagnostic and Statistical Manual of Mental Disorders—4th edition. Washington, DC: American Psychiatric Association, 2000.
- Mehler PS, MacKenzie TD. Treatment of osteopenia and osteoporosis in anorexia nervosa: a systematic review of the literature. Int J Eat Disord 2009; 42:195–201.
- Wong JC, Lewindon P, Mortimer R, Shepherd R. Bone mineral density in adolescent females with recently diagnosed anorexia nervosa. Int J Eat Disord 2001; 29:11–16.
- Rome ES, Ammerman S. Medical complications of eating disorders: an update. J Adolesc Health 2003; 33:418–426.
- Recker RR, Davies KM, Hinders SM, Heaney RP, Stegman MR, Kimmel DB. Bone gain in young adult women. JAMA 1992; 268:2403–2408.
- Sterling WM, Golden NH, Jacobson MS, Ornstein RM, Hertz SM. Metabolic assessment of menstruating and nonmenstruating normal weight adolescents. Int J Eat Disord 2009; 42:658–663.
- Legroux-Gerot I, Vignau J, Collier F, Cortet B. Bone loss associated with anorexia nervosa. Joint Bone Spine 2005; 72:489–495.
- Grinspoon S, Thomas L, Miller K, Herzog D, Klibanski A. Effects of recombinant human IGF-I and oral contraceptive administration on bone density in anorexia nervosa. J Clin Endocrinol Metab 2002; 87:2883–2891.
- Soyka LA, Grinspoon S, Levitsky LL, Herzog DB, Klibanski A. The effects of anorexia nervosa on bone metabolism in female adolescents. J Clin Endocrinol Metab 1999; 84:4489–4496.
- Miller KK, Lawson EA, Mathur V, et al. Androgens in women with anorexia nervosa and normal-weight women with hypothalamic amenorrhea. J Clin Endocrinol Metab 2007; 92:1334–1339.
- Misra M, Katzman DK, Cord J, et al. Bone metabolism in adolescent boys with anorexia nervosa. J Clin Endocrinol Metab 2008; 93:3029–3036.
- Misra M, Miller KK, Almazan C, et al. Alterations in cortisol secretory dynamics in adolescent girls with anorexia nervosa and effects on bone metabolism. J Clin Endocrinol Metab 2004; 89:4972–4980.
- Chiodini I, Mascia ML, Muscarella S, et al. Subclinical hypercortisolism among outpatients referred for osteoporosis. Ann Intern Med 2007; 147:541–548.
- Misra M, Soyka LA, Miller KK, et al. Serum osteoprotegerin in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab 2003; 88:3816–3822.
- Ohwada R, Hotta M, Sato K, Shibasaki T, Takano K. The relationship between serum levels of estradiol and osteoprotegerin in patients with anorexia nervosa. Endocr J 2007; 54:953–959.
- Müller TD, Föcker M, Holtkamp K, Herpertz-Dahlmann B, Hebebrand J. Leptin-mediated neuroendocrine alterations in anorexia nervosa: somatic and behavioral implications. Child Adolesc Psychiatr Clin N Am 2009; 18:117–129.
- Støving RK, Andries A, Brixen K, Flyvbjerg A, Hørder K, Frystyk J. Leptin, ghrelin, and endocannabinoids: potential therapeutic targets in anorexia nervosa. J Psychiatr Res 2009; 43:671–679.
- Audi L, Mantzoros CS, Vidal-Puig A, Vargas D, Gussinye M, Carrascosa A. Leptin in relation to resumption of menses in women with anorexia nervosa. Mol Psychiatry 1998; 3:544–547.
- Wong IP, Baldock PA, Herzog H. Gastrointestinal peptides and bone health. Curr Opin Endocrinol Diabetes Obes 2010; 17:44–50.
- Fukushima N, Hanada R, Teranishi H, et al. Ghrelin directly regulates bone formation. J Bone Miner Res 2005; 20:790–798.
- Makovey J, Naganathan V, Seibel M, Sambrook P. Gender differences in plasma ghrelin and its relations to body composition and bone—an opposite-sex twin study. Clin Endocrinol (Oxf) 2007; 66:530–537.
- Hassouna R, Zizzari P, Tolle V. The ghrelin/obestatin balance in the physiological and pathological control of growth hormone secretion, body composition and food intake. J Neuroendocrinol 2010; 22:793–804.
- Germain N, Galusca B, Grouselle D, et al. Ghrelin/obestatin ratio in two populations with low body weight: constitutional thinness and anorexia nervosa. Psychoneuroendocrinology 2009; 34:413–419.
- Cohen A, Shane E. Treatment of premenopausal women with low bone mineral density. Curr Osteoporos Rep 2008; 6:39–46.
- Licata A. Bone density vs bone quality: what’s a clinician to do? Cleve Clin J Med 2009; 76:331–336.
- Bianchi ML, Baim S, Bishop NJ, et al. Official positions of the International Society for Clinical Densitometry (ISCD) on DXA evaluation in children and adolescents. Pediatr Nephrol 2010; 25:37–47.
- Mendelsohn FA, Warren MP. Anorexia, bulimia, and the female athlete triad: evaluation and management. Endocrinol Metab Clin North Am 2010; 39:155–167.
- Mehler PS, Krantz M. Anorexia nervosa medical issues. J Womens Health (Larchmt) 2003; 12:331–340.
- Watts NB, Lewiecki EM, Miller PD, Baim S. National Osteoporosis Foundation 2008 Clinician’s Guide to Prevention and Treatment of Osteoporosis and the World Health Organization Fracture Risk Assessment Tool (FRAX): what they mean to the bone densitometrist and bone technologist. J Clin Densitom 2008; 11:473–477.
- Nattiv A, Loucks AB, Manore MM, Sanborn CF, Sundgot-Borgen J, Warren MP; American College of Sports Medicine. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 2007; 39:1867–1882.
- Cummings SR, Palermo L, Browner W, et al. Monitoring osteoporosis therapy with bone densitometry: misleading changes and regression to the mean. Fracture Intervention Trial Research Group. JAMA 2000; 283:1318–1321.
- LeBoff MS, Kohlmeier L, Hurwitz S, Franklin J, Wright J, Glowacki J. Occult vitamin D deficiency in postmenopausal US women with acute hip fracture. JAMA 1999; 281:1505–1511.
- Adams JS, Hewison M. Update in vitamin D. J Clin Endocrinol Metab 2010; 95:471–478.
- Gordon CM, DePeter KC, Feldman HA, Grace E, Emans SJ. Prevalence of vitamin D deficiency among healthy adolescents. Arch Pediatr Adolesc Med 2004; 158:531–537.
- Hofbauer LC, Hamann C, Ebeling PR. Approach to the patient with secondary osteoporosis. Eur J Endocrinol 2010; 162:1009–1020.
- Stoffman N, Gordon CM. Vitamin D and adolescents: what do we know? Curr Opin Pediatr 2009; 21:465–471.
- Bone HG, Greenspan SL, McKeever C, et al. Alendronate and estrogen effects in postmenopausal women with low bone mineral density. Alendronate/Estrogen Study Group. J Clin Endocrinol Metab 2000; 85:720–726.
- Barrett-Connor E, Wenger NK, Grady D, et al. Hormone and nonhormone therapy for the maintenance of postmenopausal health: the need for randomized controlled trials of estrogen and raloxifene. J Womens Health 1998; 7:839–847.
- Klibanski A, Biller BM, Schoenfeld DA, Herzog DB, Saxe VC. The effects of estrogen administration on trabecular bone loss in young women with anorexia nervosa. J Clin Endocrinol Metab 1995; 80:898–904.
- Stokosch GR, Friedman AJ, Wu SC, Kamin M. Effects of an oral contraceptive (norgestimate/ethinyl estradiol) on bone mineral density in adolescent females with anorexia nervosa: double-blind, placebo-controlled study. J Adolesc Health 2006; 39:819–827.
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15:135–143.
- Robinson E, Bachrach LK, Katzman DK. Use of hormone replacement therapy to reduce the risk of osteopenia in adolescent girls with anorexia nervosa. J Adolesc Health 2000; 26:343–348.
- Myburgh KH, Hutchins J, Fataar AB, et al. Low bone density is an etiologic factor for stress fractures in athletes. Ann Intern Med 1990; 113:754–759.
- Young N, Formica C, Szmukler G, Seeman E. Bone density at weight-bearing and nonweight-bearing sites in ballet dancers: the effects of exercise, hypogonadism, and body weight. J Clin Endocrinol Metab 1994; 78:449–454.
- Stock JL, Bell NH, Chesnut CH, et al. Increments in bone mineral density of the lumbar spine and hip and suppression of bone turnover are maintained after discontinuation of alendronate in postmenopausal women. Am J Med 1997; 103:291–297.
- Subbiah V, Madsen VS, Raymond AK, Benjamin RS, Ludwig JA. Of mice and men: divergent risks of teriparatide-induced osteosarcoma. Osteoporos Int 2010; 21:1041–1045.
- Wren AM, Seal LJ, Cohen MA, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 2001; 86:5992.
- Miljic D, Pekic S, Djurovic M, et al. Ghrelin has partial or no effect on appetite, growth hormone, prolactin, and cortisol release in patients with anorexia nervosa. J Clin Endocrinol Metab 2006; 914:1491–1495.
- Siegfried Z, Kanyas K, Latzer Y, et al. Association study of cannabinoid receptor gene (CNR1) alleles and anorexia nervosa: differences between restricting and binging/purging subtypes. Am J Med Genet B Neuropsychiatr Genet 2004; 125B:126–130.
- Cota D, Marsicano G, Lutz B, et al. Endogenous cannabinoid system as a modulator of food intake. Int J Obes Relat Metab Disord 2003; 27:289–301.
- Gross H, Ebert MH, Faden VB, et al. A double-blind trial of delta 9-tetrahydrocannabinol in primary anorexia nervosa. J Clin Psychopharmacol 1983; 3:165–171.
- Baker D, Roberts R, Towell T. Factors predictive of bone mineral density in eating-disordered women: a longitudinal study. Int J Eat Disord 2000; 27:29–35.
- Rome ES. Eating disorders. Obstet Gynecol Clin North Am 2003; 30:353–377.
- First MB, editor. Diagnostic and Statistical Manual of Mental Disorders—4th edition. Washington, DC: American Psychiatric Association, 2000.
- Mehler PS, MacKenzie TD. Treatment of osteopenia and osteoporosis in anorexia nervosa: a systematic review of the literature. Int J Eat Disord 2009; 42:195–201.
- Wong JC, Lewindon P, Mortimer R, Shepherd R. Bone mineral density in adolescent females with recently diagnosed anorexia nervosa. Int J Eat Disord 2001; 29:11–16.
- Rome ES, Ammerman S. Medical complications of eating disorders: an update. J Adolesc Health 2003; 33:418–426.
- Recker RR, Davies KM, Hinders SM, Heaney RP, Stegman MR, Kimmel DB. Bone gain in young adult women. JAMA 1992; 268:2403–2408.
- Sterling WM, Golden NH, Jacobson MS, Ornstein RM, Hertz SM. Metabolic assessment of menstruating and nonmenstruating normal weight adolescents. Int J Eat Disord 2009; 42:658–663.
- Legroux-Gerot I, Vignau J, Collier F, Cortet B. Bone loss associated with anorexia nervosa. Joint Bone Spine 2005; 72:489–495.
- Grinspoon S, Thomas L, Miller K, Herzog D, Klibanski A. Effects of recombinant human IGF-I and oral contraceptive administration on bone density in anorexia nervosa. J Clin Endocrinol Metab 2002; 87:2883–2891.
- Soyka LA, Grinspoon S, Levitsky LL, Herzog DB, Klibanski A. The effects of anorexia nervosa on bone metabolism in female adolescents. J Clin Endocrinol Metab 1999; 84:4489–4496.
- Miller KK, Lawson EA, Mathur V, et al. Androgens in women with anorexia nervosa and normal-weight women with hypothalamic amenorrhea. J Clin Endocrinol Metab 2007; 92:1334–1339.
- Misra M, Katzman DK, Cord J, et al. Bone metabolism in adolescent boys with anorexia nervosa. J Clin Endocrinol Metab 2008; 93:3029–3036.
- Misra M, Miller KK, Almazan C, et al. Alterations in cortisol secretory dynamics in adolescent girls with anorexia nervosa and effects on bone metabolism. J Clin Endocrinol Metab 2004; 89:4972–4980.
- Chiodini I, Mascia ML, Muscarella S, et al. Subclinical hypercortisolism among outpatients referred for osteoporosis. Ann Intern Med 2007; 147:541–548.
- Misra M, Soyka LA, Miller KK, et al. Serum osteoprotegerin in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab 2003; 88:3816–3822.
- Ohwada R, Hotta M, Sato K, Shibasaki T, Takano K. The relationship between serum levels of estradiol and osteoprotegerin in patients with anorexia nervosa. Endocr J 2007; 54:953–959.
- Müller TD, Föcker M, Holtkamp K, Herpertz-Dahlmann B, Hebebrand J. Leptin-mediated neuroendocrine alterations in anorexia nervosa: somatic and behavioral implications. Child Adolesc Psychiatr Clin N Am 2009; 18:117–129.
- Støving RK, Andries A, Brixen K, Flyvbjerg A, Hørder K, Frystyk J. Leptin, ghrelin, and endocannabinoids: potential therapeutic targets in anorexia nervosa. J Psychiatr Res 2009; 43:671–679.
- Audi L, Mantzoros CS, Vidal-Puig A, Vargas D, Gussinye M, Carrascosa A. Leptin in relation to resumption of menses in women with anorexia nervosa. Mol Psychiatry 1998; 3:544–547.
- Wong IP, Baldock PA, Herzog H. Gastrointestinal peptides and bone health. Curr Opin Endocrinol Diabetes Obes 2010; 17:44–50.
- Fukushima N, Hanada R, Teranishi H, et al. Ghrelin directly regulates bone formation. J Bone Miner Res 2005; 20:790–798.
- Makovey J, Naganathan V, Seibel M, Sambrook P. Gender differences in plasma ghrelin and its relations to body composition and bone—an opposite-sex twin study. Clin Endocrinol (Oxf) 2007; 66:530–537.
- Hassouna R, Zizzari P, Tolle V. The ghrelin/obestatin balance in the physiological and pathological control of growth hormone secretion, body composition and food intake. J Neuroendocrinol 2010; 22:793–804.
- Germain N, Galusca B, Grouselle D, et al. Ghrelin/obestatin ratio in two populations with low body weight: constitutional thinness and anorexia nervosa. Psychoneuroendocrinology 2009; 34:413–419.
- Cohen A, Shane E. Treatment of premenopausal women with low bone mineral density. Curr Osteoporos Rep 2008; 6:39–46.
- Licata A. Bone density vs bone quality: what’s a clinician to do? Cleve Clin J Med 2009; 76:331–336.
- Bianchi ML, Baim S, Bishop NJ, et al. Official positions of the International Society for Clinical Densitometry (ISCD) on DXA evaluation in children and adolescents. Pediatr Nephrol 2010; 25:37–47.
- Mendelsohn FA, Warren MP. Anorexia, bulimia, and the female athlete triad: evaluation and management. Endocrinol Metab Clin North Am 2010; 39:155–167.
- Mehler PS, Krantz M. Anorexia nervosa medical issues. J Womens Health (Larchmt) 2003; 12:331–340.
- Watts NB, Lewiecki EM, Miller PD, Baim S. National Osteoporosis Foundation 2008 Clinician’s Guide to Prevention and Treatment of Osteoporosis and the World Health Organization Fracture Risk Assessment Tool (FRAX): what they mean to the bone densitometrist and bone technologist. J Clin Densitom 2008; 11:473–477.
- Nattiv A, Loucks AB, Manore MM, Sanborn CF, Sundgot-Borgen J, Warren MP; American College of Sports Medicine. American College of Sports Medicine position stand. The female athlete triad. Med Sci Sports Exerc 2007; 39:1867–1882.
- Cummings SR, Palermo L, Browner W, et al. Monitoring osteoporosis therapy with bone densitometry: misleading changes and regression to the mean. Fracture Intervention Trial Research Group. JAMA 2000; 283:1318–1321.
- LeBoff MS, Kohlmeier L, Hurwitz S, Franklin J, Wright J, Glowacki J. Occult vitamin D deficiency in postmenopausal US women with acute hip fracture. JAMA 1999; 281:1505–1511.
- Adams JS, Hewison M. Update in vitamin D. J Clin Endocrinol Metab 2010; 95:471–478.
- Gordon CM, DePeter KC, Feldman HA, Grace E, Emans SJ. Prevalence of vitamin D deficiency among healthy adolescents. Arch Pediatr Adolesc Med 2004; 158:531–537.
- Hofbauer LC, Hamann C, Ebeling PR. Approach to the patient with secondary osteoporosis. Eur J Endocrinol 2010; 162:1009–1020.
- Stoffman N, Gordon CM. Vitamin D and adolescents: what do we know? Curr Opin Pediatr 2009; 21:465–471.
- Bone HG, Greenspan SL, McKeever C, et al. Alendronate and estrogen effects in postmenopausal women with low bone mineral density. Alendronate/Estrogen Study Group. J Clin Endocrinol Metab 2000; 85:720–726.
- Barrett-Connor E, Wenger NK, Grady D, et al. Hormone and nonhormone therapy for the maintenance of postmenopausal health: the need for randomized controlled trials of estrogen and raloxifene. J Womens Health 1998; 7:839–847.
- Klibanski A, Biller BM, Schoenfeld DA, Herzog DB, Saxe VC. The effects of estrogen administration on trabecular bone loss in young women with anorexia nervosa. J Clin Endocrinol Metab 1995; 80:898–904.
- Stokosch GR, Friedman AJ, Wu SC, Kamin M. Effects of an oral contraceptive (norgestimate/ethinyl estradiol) on bone mineral density in adolescent females with anorexia nervosa: double-blind, placebo-controlled study. J Adolesc Health 2006; 39:819–827.
- Golden NH, Lanzkowsky L, Schebendach J, Palestro CJ, Jacobson MS, Shenker IR. The effect of estrogen-progestin treatment on bone mineral density in anorexia nervosa. J Pediatr Adolesc Gynecol 2002; 15:135–143.
- Robinson E, Bachrach LK, Katzman DK. Use of hormone replacement therapy to reduce the risk of osteopenia in adolescent girls with anorexia nervosa. J Adolesc Health 2000; 26:343–348.
- Myburgh KH, Hutchins J, Fataar AB, et al. Low bone density is an etiologic factor for stress fractures in athletes. Ann Intern Med 1990; 113:754–759.
- Young N, Formica C, Szmukler G, Seeman E. Bone density at weight-bearing and nonweight-bearing sites in ballet dancers: the effects of exercise, hypogonadism, and body weight. J Clin Endocrinol Metab 1994; 78:449–454.
- Stock JL, Bell NH, Chesnut CH, et al. Increments in bone mineral density of the lumbar spine and hip and suppression of bone turnover are maintained after discontinuation of alendronate in postmenopausal women. Am J Med 1997; 103:291–297.
- Subbiah V, Madsen VS, Raymond AK, Benjamin RS, Ludwig JA. Of mice and men: divergent risks of teriparatide-induced osteosarcoma. Osteoporos Int 2010; 21:1041–1045.
- Wren AM, Seal LJ, Cohen MA, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 2001; 86:5992.
- Miljic D, Pekic S, Djurovic M, et al. Ghrelin has partial or no effect on appetite, growth hormone, prolactin, and cortisol release in patients with anorexia nervosa. J Clin Endocrinol Metab 2006; 914:1491–1495.
- Siegfried Z, Kanyas K, Latzer Y, et al. Association study of cannabinoid receptor gene (CNR1) alleles and anorexia nervosa: differences between restricting and binging/purging subtypes. Am J Med Genet B Neuropsychiatr Genet 2004; 125B:126–130.
- Cota D, Marsicano G, Lutz B, et al. Endogenous cannabinoid system as a modulator of food intake. Int J Obes Relat Metab Disord 2003; 27:289–301.
- Gross H, Ebert MH, Faden VB, et al. A double-blind trial of delta 9-tetrahydrocannabinol in primary anorexia nervosa. J Clin Psychopharmacol 1983; 3:165–171.
KEY POINTS
- Women gain 40% to 60% of their bone mass during adolescence, a time coinciding with the peak incidence of anorexia nervosa, and they attain their peak bone mass by the time they are in their 20s.
- The etiology of osteoporosis in anorexia nervosa is complex and multifaceted. Early detection and treatment are critical.
- Osteoporosis in premenopausal patients is defined as low bone mineral density (a Z score below −2.0) in combination with risk factors such as chronic malnutrition, eating disorders, hypogonadism, glucocorticoid exposure, and previous fractures.
- Restoring body weight is the key treatment. Vitamin D should be supplemented if low. Estrogen therapy has not been shown to be effective, and exercise may be counterproductive. Bisphosphonates and teriparatide should be used with caution, if at all.
A 43-year-old woman with chest pressure
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma

The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4

At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.

DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.