User login
Measuring Restrictive Lung Disease Severity Using FEV1 vs TLC
Respiratory diseases have varied clinical presentations and are classified as restrictive, obstructive, mixed, or normal. Restrictive lung diseases have reduced lung volumes, either due to an alteration in lung parenchyma or a disease of the pleura, chest wall, or neuromuscular apparatus. If caused by parenchymal lung disease, restrictive lung disorders are accompanied by reduced gas transfer, which may be portrayed clinically by desaturation after exercise. Based on anatomical structures, the causes of lung volume reduction may be intrinsic or extrinsic. Intrinsic causes correspond to diseases of the lung parenchyma, such as idiopathic fibrotic diseases, connective-tissue diseases, drug-induced lung diseases, and other primary diseases of the lungs. Extrinsic causes refer to disorders outside the lungs or extra-pulmonary diseases such as neuromuscular and nonmuscular diseases of the chest wall.1 For example, obesity and myasthenia gravis can cause restrictive lung diseases, one through mechanical interference of lung expansion and the other through neuromuscular impedance of thoracic cage expansion. All these diseases eventually result in lung restriction, impaired lung function, and respiratory failure. This heterogenicity of disease makes establishing a single severity criterion difficult.
Laboratory testing, imaging studies, and examinations are important for determining the pulmonary disease and its course and progression. The pulmonary function test (PFT), which consists of multiple procedures that are performed depending on the information needed, has been an essential tool in practice for the pulmonologist. The PFT includes spirometry, lung volume measurement, respiratory muscle strength, diffusion capacity, and a broncho-provocation test. Each test has a particular role in assisting the diagnosis and/or follow-up of the patient. Spirometry is frequently used due to its range of dynamic physiological parameters, ease of use, and accessibility. It is used for the diagnosis of pulmonary symptoms, in the assessment of disability, and preoperatory evaluation, including lung resection surgery, assisting in the diagnosis, monitoring, and therapy response of pulmonary diseases.
A systematic approach to PFT interpretation is recommended by several societies, such as the American Thoracic Society (ATS) and the European Respiratory Society (ERS).2 The pulmonary function test results must be reproducible and meet established standards to ensure reliable and consistent clinical outcomes. A restrictive respiratory disease is defined by a decrease in total lung capacity (TLC) (< 5% of predicted value) and a normal forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio.2 Although other findings—such as a decrease in vital capacity—should prompt an investigation into whether the patient has a possible restrictive respiratory disease, the sole presence of this parameter is not definitive or diagnostic of a restrictive impairment.2-4 The assessment of severity is typically determined by TLC. Unfortunately, the severity of a restrictive respiratory disease and the degree of patient discomfort do not always correlate when utilizing just TLC. Pulmonary sarcoidosis, for example, is a granulomatous lung disease with a restrictive PFT pattern and a disease burden that may vary over time. Having a more consistent method of grading the severity of the restrictive lung disease may help guide treatment. The modified Medical Research Council (mMRC) scale, a 5-point dyspnea scale, is widely used in assessing the severity of dyspnea in various respiratory conditions, including chronic obstructive pulmonary disease (COPD), where its scores have been associated with patient mortality.1,5 The goal of this study was to document the associations between objective parameters obtained through PFT and other variables, with an established measurement of dyspnea to assess the severity grade of restrictive lung diseases.
Methods
This retrospective record review at the Veterans Affairs Caribbean Healthcare System (VACHS) in San Juan, Puerto Rico, wasconducted using the Veterans Health Information Systems and Technology Architecture to identify patients with a PFT, including spirometry, that indicated a restrictive ventilator pattern based on the current ATS/ERS Task Force on Lung Function Testing.2 Patients were included if they were aged ≥ 21 years, PFT with TLC ≤ 80% predicted, mMRC score documented on PFT, and documented diffusing capacity of the lung for carbon monoxide (DLCO). Patients were excluded if their FEV1/vital capacity (VC) was < 70% predicted using the largest VC, or no mMRC score was available. All patients meeting the inclusion criteria were considered regardless of comorbidities.
The PFT results of all adult patients, including those performed between June 1, 2013, and January 6, 2016, were submitted to spirometry, and lung volume measurements were analyzed. Sociodemographic information was collected, including sex, ethnicity, age, height, weight, and basal metabolic index. Other data found in PFTs, such as smoking status, smoking in packs/year, mMRC score, predicted TLC value, imaging present (chest X-ray, computed tomography), and hospitalizations and exacerbations within 1 year were collected. In addition, we examined the predicted values for FEV1, DLCO, and DLCO/VA (calculated using the Ayer equation), FVC (calculated using the Knudson equation), expiratory reserve volume, inspiratory VC, and slow VC. PaO2, PaCO2, and Alveolar-arterial gradients also were collected.6-9 Information about heart failure status was gathered through medical evaluation of notes and cardiac studies. All categorical variables were correlated with Spearman analysis and quantitative variables with average percentages. P values were calculated with analysis of variance.
Results
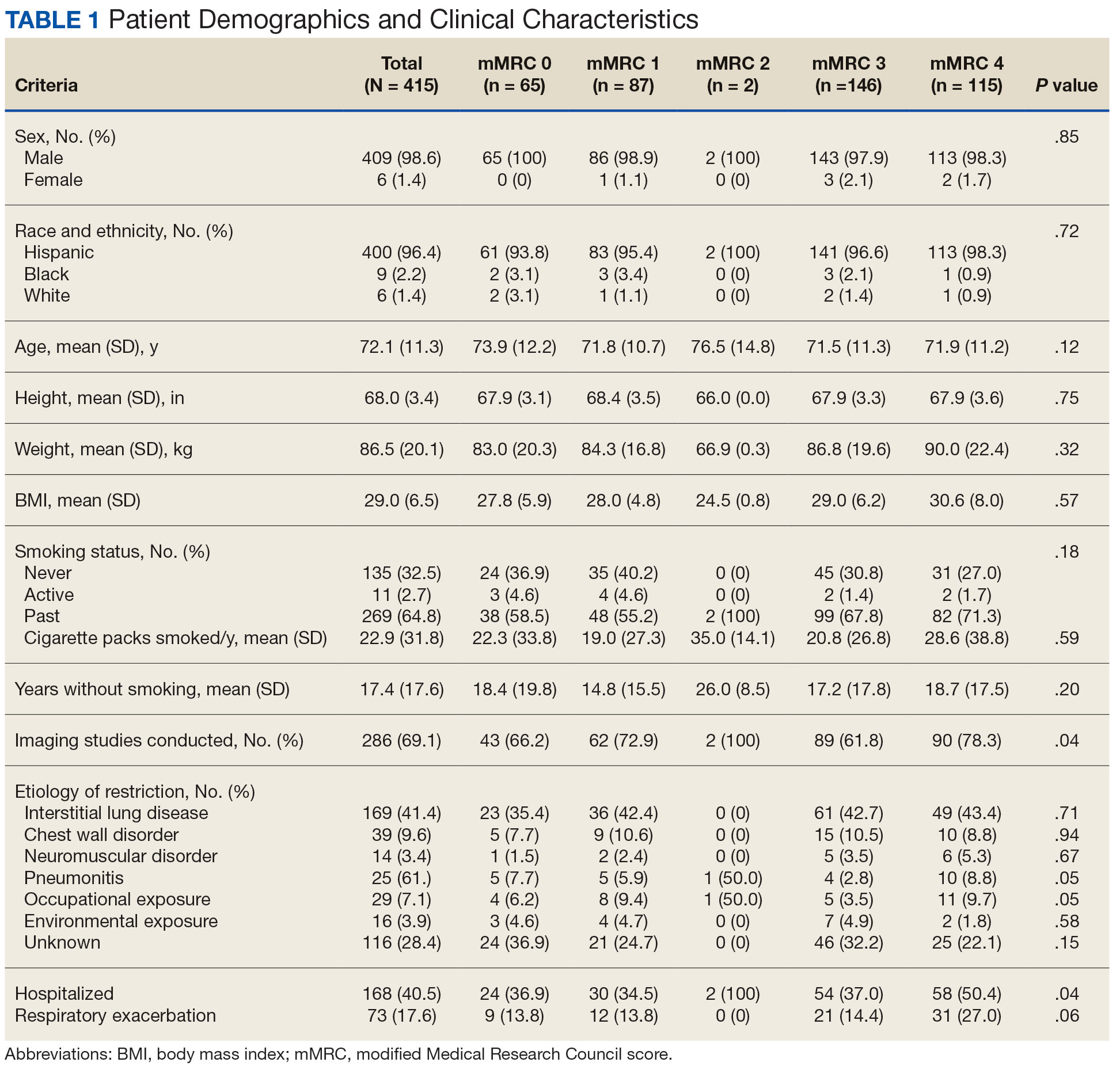
Of 6461 VACHS patient records reviewed, 415 met the inclusion criteria. Patients were divided according to their mMRC score: 65 had mMRC score of 0, 87 had an mMRC score of 1, 2 had an mMRC score of 2, 146 had an mMRC of 3, and 115 had an mMRC score of 4. The population was primarily male (98.6%) and of Hispanic ethnicity (96.4%), with a mean age of 72 years (Table 1). Most patients (n = 269, 64.0%) were prior smokers, while 135 patients (32.5%) had never smoked, and 11 (2.7%) were current smokers. At baseline, 169 patients (41.4%) had interstitial lung disease, 39 (9.6%) had chest wall disorders, 29 (7.1%) had occupational exposure, 25 (6.1%) had pneumonitis, and 14 (3.4%) had neuromuscular disorders.
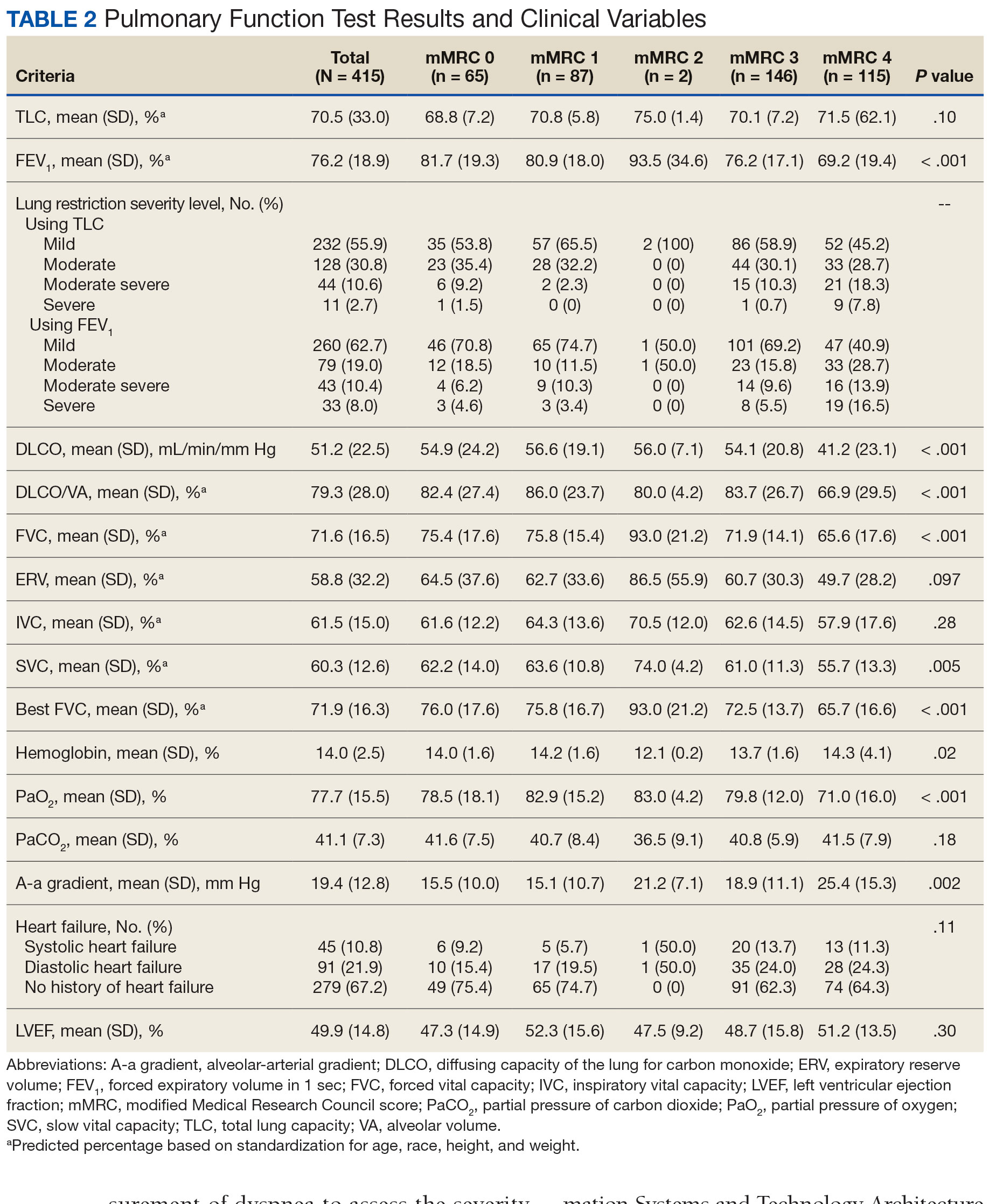
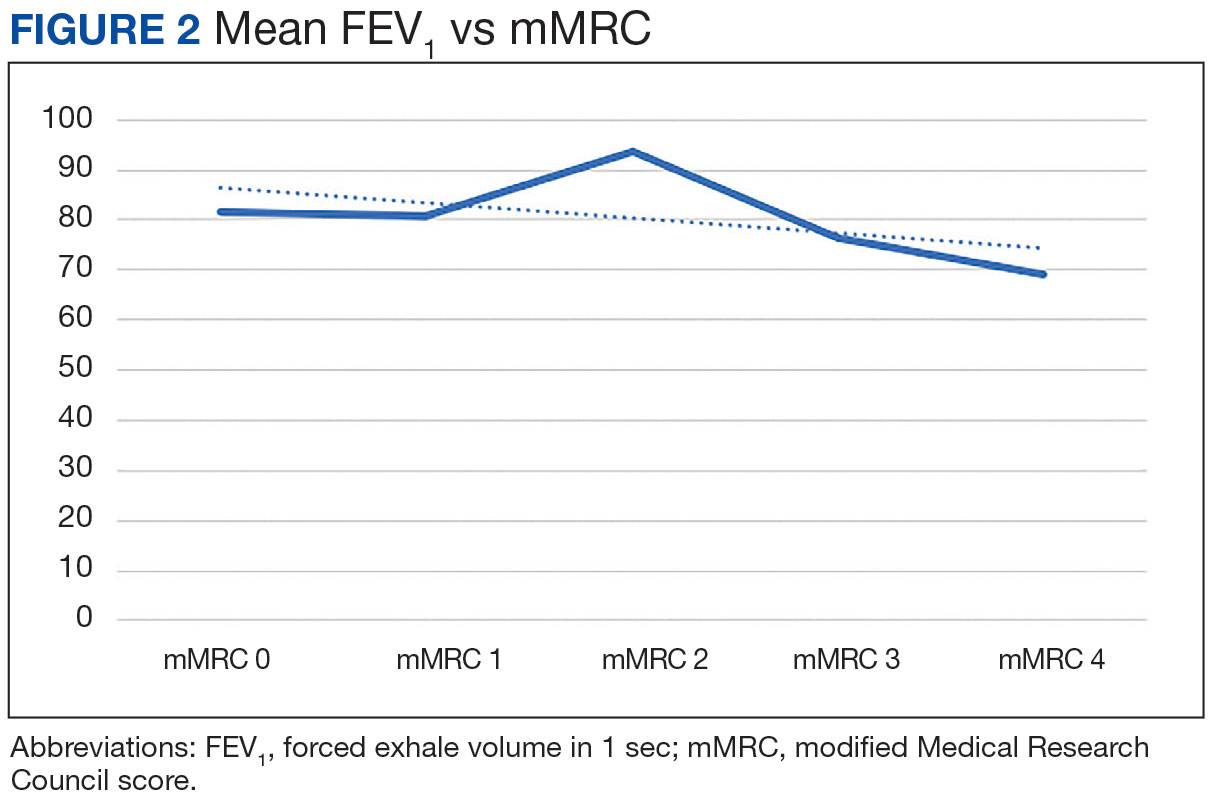
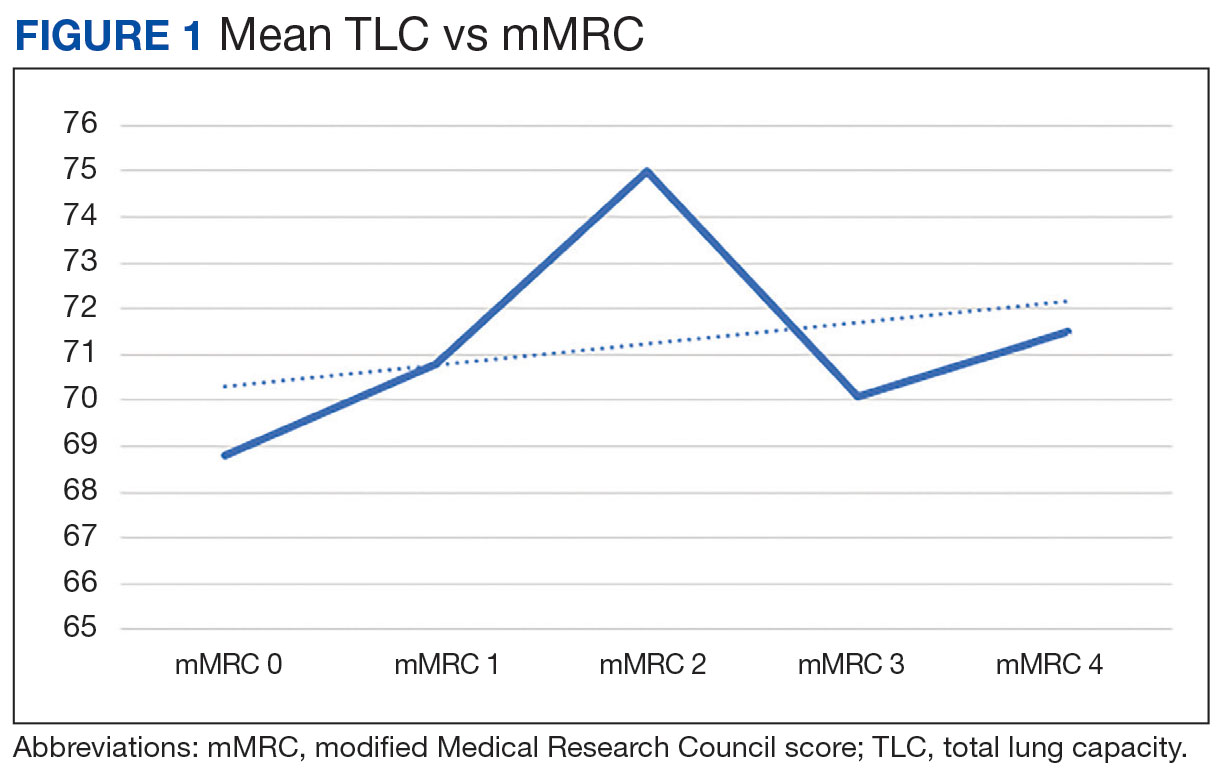
There was a statistically significant relationship between mMRC score and hospitalization and FEV1 but not TLC (Table 2). As mMRC increased, so did hospitalizations: a total of 168 patients (40.5%) were hospitalized; 24 patients (36.9%) had an mMRC score of 0, 30 patients (34.0%) had an mMRC score of 1, 2 patients (100%) had an mMRC score of 2, 54 patients (37.0%) had an mMRC score of 3, and 58 patients (50.0%) had an mMRC score of 4 (P = .04). Mean (SD) TLC values increased as mMRC scores increased. Mean (SD) TLC was 70.5% (33.0) for the entire population; 68.8% (7.2) for patients with an mMRC score of 0, 70.8% (5.8) for patients with an mMRC score of 1, 75.0% (1.4) for patients with an mMRC score of 2, 70.1% (7.2) for patients with an mMRC score of 3, and 71.5% (62.1) for patients with an mMRC score of 4 (P = .10) (Figure 1). There was an associated decrease in mean (SD) FEV1 with mMRC. Mean (SD) FEV1 was 76.2% (18.9) for the entire population; 81.7% (19.3) for patients with an mMRC score of 0, 80.9% (18) for patients with an mMRC score of 1, 93.5% (34.6) for patients with an mMRC score of 2, 76.2% (17.1) for patients with an mMRC score of 3, and 69.2% (19.4) for patients with an mMRC score of 4; (P < .001) (Figure 2).
The correlation between mMRC and FEV1 (r = 0.25, P < .001) was stronger than the correlation between mMRC and TLC (r = 0.15, P < .001). The correlations for DLCO (P < .001), DLCO/VA (P < .001), hemoglobin (P < .02), and PaO2 (P < .001) were all statistically significant (P < .005), but with no strong identifiable trend.
Discussion
The patient population of this study was primarily older males of Hispanic ethnicity with a history of smoking. There was no association between body mass index or smoking status with worsening dyspnea as measured with mMRC scores. We observed no significant correlation between mMRC scores and various factors such as comorbidities including heart conditions, and epidemiological factors like the etiology of lung disease, including both intrinsic and extrinsic causes. This lack of association was anticipated, as restrictive lung diseases in our study predominantly arose from intrinsic pulmonary etiologies, such as interstitial lung disease. A difference between more hospitalizations and worsening dyspnea was identified. There was a slightly higher correlation between FEV1 and mMRC scores when compared with TLC and mMRC scores concerning worsening dyspnea, which could indicate that the use of FEV1 should be preferred over previous recommendations to use TLC.10 Other guidelines have utilized exercise capacity via the 6-minute walk test as a marker of severity with spirometry values and found that DLCO was correlated with severity.11
The latest ERS/ATS guidelines recommend z scores for grading the severity of obstructive lung diseases but do not recommend them for the diagnosis of restrictive lung diseases.12 A z score encompasses diverse variables (eg, age, sex, and ethnicity) to provide more uniform and consistent results. Other studies have been done to relate z scores to other spirometry variables with restrictive lung disease. One such study indicates the potential benefit of using FVC alone to grade restrictive lung diseases.13 There continues to be great diversity in the interpretation of pulmonary function tests, and we believe the information gathered can provide valuable insight for managing patients with restrictive lung diseases.
Limitations
Only 2 patients reported an mMRC score of 2 in our study. This may have affected statistical outcomes. It also may reveal possible deficits in the efficacy of patient education on the mMRC scale. This study was also limited by its small sample size, single center location, and the distribution of patients that reported an mMRC favored either low or high values. The patients in this study, who were all veterans, may not be representative of other patient populations.
Conclusions
There continue to be few factors associated with the physiological severity of the defective oxygen delivery and reported dyspnea of a patient with restrictive lung disease that allows for an accurate, repeatable grading of severity. Using FEV1 instead of TLC to determine the severity of a restrictive lung disease should be reconsidered. We could not find any other strong correlation among other factors studied. Further research should be conducted to continue looking for variables that more accurately depict patient dyspnea in restrictive lung disease.
Acknowledgments
This study is based upon work supported by the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, and is the result of work supported by Pulmonary & Critical Care Medicine service, with resources and the use of its facilities.
1. Hegewald MJ, Crapo RO. Pulmonary function testing. In: Broaddus VC, Ernst JD, King Jr TE, eds. Murray and Nadel’s Textbook of Respiratory Medicine. 5th ed. Saunders; 2010:522-553.
2. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26(5):948-968. doi:10.1183/09031936.05.00035205
3. Rabe KF, Beghé B, Luppi F, Fabbri LM. Update in chronic obstructive pulmonary disease 2006. Am J Respir Crit Care Med. 2007;175(12):1222-1232. doi:10.1164/rccm.200704-586UP
4. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Spirometry for health care providers Accessed April 30, 2024. https://goldcopd.org/wp-content/uploads/2016/04/GOLD_Spirometry_2010.pdf
5. Mannino DM, Holguin F, Pavlin BI, Ferdinands JM. Risk factors for prevalence of and mortality related to restriction on spirometry: findings from the First National Health and Nutrition Examination Survey and follow-up. Int J Tuberc Lung Dis. 2005;9(6):613-621.
6. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734. doi:10.1164/arrd.1983.127.6.725
7. Knudson RJ, Burrows B, Lebowitz MD. The maximal expiratory flow-volume curve: its use in the detection of ventilatory abnormalities in a population study. Am Rev Respir Dis. 1976;114(5):871-879. doi:10.1164/arrd.1976.114.5.871
8. Knudson RJ, Lebowitz MD, Burton AP, Knudson DE. The closing volume test: evaluation of nitrogen and bolus methods in a random population. Am Rev Respir Dis. 1977;115(3):423-434. doi:10.1164/arrd.1977.115.3.423
9. Ayers LN, Ginsberg ML, Fein J, Wasserman K. Diffusing capacity, specific diffusing capacity and interpretation of diffusion defects. West J Med. 1975;123(4):255-264.
10. Lung function testing: selection of reference values and interpretative strategies. American Thoracic Society. Am Rev Respir Dis. 1991;144(5):1202-1218. doi:10.1164/ajrccm/144.5.1202
11. Larson J, Wrzos K, Corazalla E, Wang Q, Kim HJ, Cho RJ. Should FEV1 be used to grade restrictive impairment? A single-center comparison of lung function parameters to 6-minute walk test in patients with restrictive lung disease. HSOA J Pulm Med Respir Res. 2023;9:082. doi:10.24966/PMRR-0177/100082
12. Stanojevic S, Kaminsky DA, Miller MR, et al. ERS/ATS technical standard on interpretive strategies for routine lung function tests. Eur Respir J. 2022;60(1):2101499. Published 2022 Jul 13. doi:10.1183/13993003.01499-2021
13. Myrberg T, Lindberg A, Eriksson B, et al. Restrictive spirometry versus restrictive lung function using the GLI reference values. Clin Physiol Funct Imaging. 2022;42(3):181-189. doi:10.1111/cpf.12745
Rebeca Vazquez-Nieves, MDa; Vanessa Fonseca-Ferrer, MDa; Juan Irizarry-Nieves, MDa; Edgardo Adorno-Fontanez, MDa; William Rodriguez-Cintron, MDa,b,c
Correspondence: Juan Irizarry-Nieves (juan.irizarry-nieves@va.gov)
aVeterans Affairs Caribbean Healthcare System, San Juan, Puerto Rico
bUniversity of Puerto Rico School of Medicine, San Juan
cUniversidad Central del Caribe School of Medicine, San Juan, Puerto Rico
 Author disclosures
Author disclosures
The authors report no actual or potential conflicts of interest or outside sources of funding with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Ethics and consent
All documentation was approved by the Veterans Affairs Caribbean Healthcare System institutional review board.Appropriate waivers were obtained and there are no findings of incompliance present.
Rebeca Vazquez-Nieves, MDa; Vanessa Fonseca-Ferrer, MDa; Juan Irizarry-Nieves, MDa; Edgardo Adorno-Fontanez, MDa; William Rodriguez-Cintron, MDa,b,c
Correspondence: Juan Irizarry-Nieves (juan.irizarry-nieves@va.gov)
aVeterans Affairs Caribbean Healthcare System, San Juan, Puerto Rico
bUniversity of Puerto Rico School of Medicine, San Juan
cUniversidad Central del Caribe School of Medicine, San Juan, Puerto Rico
Author disclosures
The authors report no actual or potential conflicts of interest or outside sources of funding with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Ethics and consent
All documentation was approved by the Veterans Affairs Caribbean Healthcare System institutional review board.Appropriate waivers were obtained and there are no findings of incompliance present.
Rebeca Vazquez-Nieves, MDa; Vanessa Fonseca-Ferrer, MDa; Juan Irizarry-Nieves, MDa; Edgardo Adorno-Fontanez, MDa; William Rodriguez-Cintron, MDa,b,c
Correspondence: Juan Irizarry-Nieves (juan.irizarry-nieves@va.gov)
aVeterans Affairs Caribbean Healthcare System, San Juan, Puerto Rico
bUniversity of Puerto Rico School of Medicine, San Juan
cUniversidad Central del Caribe School of Medicine, San Juan, Puerto Rico
Author disclosures
The authors report no actual or potential conflicts of interest or outside sources of funding with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Ethics and consent
All documentation was approved by the Veterans Affairs Caribbean Healthcare System institutional review board.Appropriate waivers were obtained and there are no findings of incompliance present.
Respiratory diseases have varied clinical presentations and are classified as restrictive, obstructive, mixed, or normal. Restrictive lung diseases have reduced lung volumes, either due to an alteration in lung parenchyma or a disease of the pleura, chest wall, or neuromuscular apparatus. If caused by parenchymal lung disease, restrictive lung disorders are accompanied by reduced gas transfer, which may be portrayed clinically by desaturation after exercise. Based on anatomical structures, the causes of lung volume reduction may be intrinsic or extrinsic. Intrinsic causes correspond to diseases of the lung parenchyma, such as idiopathic fibrotic diseases, connective-tissue diseases, drug-induced lung diseases, and other primary diseases of the lungs. Extrinsic causes refer to disorders outside the lungs or extra-pulmonary diseases such as neuromuscular and nonmuscular diseases of the chest wall.1 For example, obesity and myasthenia gravis can cause restrictive lung diseases, one through mechanical interference of lung expansion and the other through neuromuscular impedance of thoracic cage expansion. All these diseases eventually result in lung restriction, impaired lung function, and respiratory failure. This heterogenicity of disease makes establishing a single severity criterion difficult.
Laboratory testing, imaging studies, and examinations are important for determining the pulmonary disease and its course and progression. The pulmonary function test (PFT), which consists of multiple procedures that are performed depending on the information needed, has been an essential tool in practice for the pulmonologist. The PFT includes spirometry, lung volume measurement, respiratory muscle strength, diffusion capacity, and a broncho-provocation test. Each test has a particular role in assisting the diagnosis and/or follow-up of the patient. Spirometry is frequently used due to its range of dynamic physiological parameters, ease of use, and accessibility. It is used for the diagnosis of pulmonary symptoms, in the assessment of disability, and preoperatory evaluation, including lung resection surgery, assisting in the diagnosis, monitoring, and therapy response of pulmonary diseases.
A systematic approach to PFT interpretation is recommended by several societies, such as the American Thoracic Society (ATS) and the European Respiratory Society (ERS).2 The pulmonary function test results must be reproducible and meet established standards to ensure reliable and consistent clinical outcomes. A restrictive respiratory disease is defined by a decrease in total lung capacity (TLC) (< 5% of predicted value) and a normal forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio.2 Although other findings—such as a decrease in vital capacity—should prompt an investigation into whether the patient has a possible restrictive respiratory disease, the sole presence of this parameter is not definitive or diagnostic of a restrictive impairment.2-4 The assessment of severity is typically determined by TLC. Unfortunately, the severity of a restrictive respiratory disease and the degree of patient discomfort do not always correlate when utilizing just TLC. Pulmonary sarcoidosis, for example, is a granulomatous lung disease with a restrictive PFT pattern and a disease burden that may vary over time. Having a more consistent method of grading the severity of the restrictive lung disease may help guide treatment. The modified Medical Research Council (mMRC) scale, a 5-point dyspnea scale, is widely used in assessing the severity of dyspnea in various respiratory conditions, including chronic obstructive pulmonary disease (COPD), where its scores have been associated with patient mortality.1,5 The goal of this study was to document the associations between objective parameters obtained through PFT and other variables, with an established measurement of dyspnea to assess the severity grade of restrictive lung diseases.
Methods
This retrospective record review at the Veterans Affairs Caribbean Healthcare System (VACHS) in San Juan, Puerto Rico, wasconducted using the Veterans Health Information Systems and Technology Architecture to identify patients with a PFT, including spirometry, that indicated a restrictive ventilator pattern based on the current ATS/ERS Task Force on Lung Function Testing.2 Patients were included if they were aged ≥ 21 years, PFT with TLC ≤ 80% predicted, mMRC score documented on PFT, and documented diffusing capacity of the lung for carbon monoxide (DLCO). Patients were excluded if their FEV1/vital capacity (VC) was < 70% predicted using the largest VC, or no mMRC score was available. All patients meeting the inclusion criteria were considered regardless of comorbidities.
The PFT results of all adult patients, including those performed between June 1, 2013, and January 6, 2016, were submitted to spirometry, and lung volume measurements were analyzed. Sociodemographic information was collected, including sex, ethnicity, age, height, weight, and basal metabolic index. Other data found in PFTs, such as smoking status, smoking in packs/year, mMRC score, predicted TLC value, imaging present (chest X-ray, computed tomography), and hospitalizations and exacerbations within 1 year were collected. In addition, we examined the predicted values for FEV1, DLCO, and DLCO/VA (calculated using the Ayer equation), FVC (calculated using the Knudson equation), expiratory reserve volume, inspiratory VC, and slow VC. PaO2, PaCO2, and Alveolar-arterial gradients also were collected.6-9 Information about heart failure status was gathered through medical evaluation of notes and cardiac studies. All categorical variables were correlated with Spearman analysis and quantitative variables with average percentages. P values were calculated with analysis of variance.
Results
Of 6461 VACHS patient records reviewed, 415 met the inclusion criteria. Patients were divided according to their mMRC score: 65 had mMRC score of 0, 87 had an mMRC score of 1, 2 had an mMRC score of 2, 146 had an mMRC of 3, and 115 had an mMRC score of 4. The population was primarily male (98.6%) and of Hispanic ethnicity (96.4%), with a mean age of 72 years (Table 1). Most patients (n = 269, 64.0%) were prior smokers, while 135 patients (32.5%) had never smoked, and 11 (2.7%) were current smokers. At baseline, 169 patients (41.4%) had interstitial lung disease, 39 (9.6%) had chest wall disorders, 29 (7.1%) had occupational exposure, 25 (6.1%) had pneumonitis, and 14 (3.4%) had neuromuscular disorders.
There was a statistically significant relationship between mMRC score and hospitalization and FEV1 but not TLC (Table 2). As mMRC increased, so did hospitalizations: a total of 168 patients (40.5%) were hospitalized; 24 patients (36.9%) had an mMRC score of 0, 30 patients (34.0%) had an mMRC score of 1, 2 patients (100%) had an mMRC score of 2, 54 patients (37.0%) had an mMRC score of 3, and 58 patients (50.0%) had an mMRC score of 4 (P = .04). Mean (SD) TLC values increased as mMRC scores increased. Mean (SD) TLC was 70.5% (33.0) for the entire population; 68.8% (7.2) for patients with an mMRC score of 0, 70.8% (5.8) for patients with an mMRC score of 1, 75.0% (1.4) for patients with an mMRC score of 2, 70.1% (7.2) for patients with an mMRC score of 3, and 71.5% (62.1) for patients with an mMRC score of 4 (P = .10) (Figure 1). There was an associated decrease in mean (SD) FEV1 with mMRC. Mean (SD) FEV1 was 76.2% (18.9) for the entire population; 81.7% (19.3) for patients with an mMRC score of 0, 80.9% (18) for patients with an mMRC score of 1, 93.5% (34.6) for patients with an mMRC score of 2, 76.2% (17.1) for patients with an mMRC score of 3, and 69.2% (19.4) for patients with an mMRC score of 4; (P < .001) (Figure 2).
The correlation between mMRC and FEV1 (r = 0.25, P < .001) was stronger than the correlation between mMRC and TLC (r = 0.15, P < .001). The correlations for DLCO (P < .001), DLCO/VA (P < .001), hemoglobin (P < .02), and PaO2 (P < .001) were all statistically significant (P < .005), but with no strong identifiable trend.
Discussion
The patient population of this study was primarily older males of Hispanic ethnicity with a history of smoking. There was no association between body mass index or smoking status with worsening dyspnea as measured with mMRC scores. We observed no significant correlation between mMRC scores and various factors such as comorbidities including heart conditions, and epidemiological factors like the etiology of lung disease, including both intrinsic and extrinsic causes. This lack of association was anticipated, as restrictive lung diseases in our study predominantly arose from intrinsic pulmonary etiologies, such as interstitial lung disease. A difference between more hospitalizations and worsening dyspnea was identified. There was a slightly higher correlation between FEV1 and mMRC scores when compared with TLC and mMRC scores concerning worsening dyspnea, which could indicate that the use of FEV1 should be preferred over previous recommendations to use TLC.10 Other guidelines have utilized exercise capacity via the 6-minute walk test as a marker of severity with spirometry values and found that DLCO was correlated with severity.11
The latest ERS/ATS guidelines recommend z scores for grading the severity of obstructive lung diseases but do not recommend them for the diagnosis of restrictive lung diseases.12 A z score encompasses diverse variables (eg, age, sex, and ethnicity) to provide more uniform and consistent results. Other studies have been done to relate z scores to other spirometry variables with restrictive lung disease. One such study indicates the potential benefit of using FVC alone to grade restrictive lung diseases.13 There continues to be great diversity in the interpretation of pulmonary function tests, and we believe the information gathered can provide valuable insight for managing patients with restrictive lung diseases.
Limitations
Only 2 patients reported an mMRC score of 2 in our study. This may have affected statistical outcomes. It also may reveal possible deficits in the efficacy of patient education on the mMRC scale. This study was also limited by its small sample size, single center location, and the distribution of patients that reported an mMRC favored either low or high values. The patients in this study, who were all veterans, may not be representative of other patient populations.
Conclusions
There continue to be few factors associated with the physiological severity of the defective oxygen delivery and reported dyspnea of a patient with restrictive lung disease that allows for an accurate, repeatable grading of severity. Using FEV1 instead of TLC to determine the severity of a restrictive lung disease should be reconsidered. We could not find any other strong correlation among other factors studied. Further research should be conducted to continue looking for variables that more accurately depict patient dyspnea in restrictive lung disease.
Acknowledgments
This study is based upon work supported by the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, and is the result of work supported by Pulmonary & Critical Care Medicine service, with resources and the use of its facilities.
Respiratory diseases have varied clinical presentations and are classified as restrictive, obstructive, mixed, or normal. Restrictive lung diseases have reduced lung volumes, either due to an alteration in lung parenchyma or a disease of the pleura, chest wall, or neuromuscular apparatus. If caused by parenchymal lung disease, restrictive lung disorders are accompanied by reduced gas transfer, which may be portrayed clinically by desaturation after exercise. Based on anatomical structures, the causes of lung volume reduction may be intrinsic or extrinsic. Intrinsic causes correspond to diseases of the lung parenchyma, such as idiopathic fibrotic diseases, connective-tissue diseases, drug-induced lung diseases, and other primary diseases of the lungs. Extrinsic causes refer to disorders outside the lungs or extra-pulmonary diseases such as neuromuscular and nonmuscular diseases of the chest wall.1 For example, obesity and myasthenia gravis can cause restrictive lung diseases, one through mechanical interference of lung expansion and the other through neuromuscular impedance of thoracic cage expansion. All these diseases eventually result in lung restriction, impaired lung function, and respiratory failure. This heterogenicity of disease makes establishing a single severity criterion difficult.
Laboratory testing, imaging studies, and examinations are important for determining the pulmonary disease and its course and progression. The pulmonary function test (PFT), which consists of multiple procedures that are performed depending on the information needed, has been an essential tool in practice for the pulmonologist. The PFT includes spirometry, lung volume measurement, respiratory muscle strength, diffusion capacity, and a broncho-provocation test. Each test has a particular role in assisting the diagnosis and/or follow-up of the patient. Spirometry is frequently used due to its range of dynamic physiological parameters, ease of use, and accessibility. It is used for the diagnosis of pulmonary symptoms, in the assessment of disability, and preoperatory evaluation, including lung resection surgery, assisting in the diagnosis, monitoring, and therapy response of pulmonary diseases.
A systematic approach to PFT interpretation is recommended by several societies, such as the American Thoracic Society (ATS) and the European Respiratory Society (ERS).2 The pulmonary function test results must be reproducible and meet established standards to ensure reliable and consistent clinical outcomes. A restrictive respiratory disease is defined by a decrease in total lung capacity (TLC) (< 5% of predicted value) and a normal forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio.2 Although other findings—such as a decrease in vital capacity—should prompt an investigation into whether the patient has a possible restrictive respiratory disease, the sole presence of this parameter is not definitive or diagnostic of a restrictive impairment.2-4 The assessment of severity is typically determined by TLC. Unfortunately, the severity of a restrictive respiratory disease and the degree of patient discomfort do not always correlate when utilizing just TLC. Pulmonary sarcoidosis, for example, is a granulomatous lung disease with a restrictive PFT pattern and a disease burden that may vary over time. Having a more consistent method of grading the severity of the restrictive lung disease may help guide treatment. The modified Medical Research Council (mMRC) scale, a 5-point dyspnea scale, is widely used in assessing the severity of dyspnea in various respiratory conditions, including chronic obstructive pulmonary disease (COPD), where its scores have been associated with patient mortality.1,5 The goal of this study was to document the associations between objective parameters obtained through PFT and other variables, with an established measurement of dyspnea to assess the severity grade of restrictive lung diseases.
Methods
This retrospective record review at the Veterans Affairs Caribbean Healthcare System (VACHS) in San Juan, Puerto Rico, wasconducted using the Veterans Health Information Systems and Technology Architecture to identify patients with a PFT, including spirometry, that indicated a restrictive ventilator pattern based on the current ATS/ERS Task Force on Lung Function Testing.2 Patients were included if they were aged ≥ 21 years, PFT with TLC ≤ 80% predicted, mMRC score documented on PFT, and documented diffusing capacity of the lung for carbon monoxide (DLCO). Patients were excluded if their FEV1/vital capacity (VC) was < 70% predicted using the largest VC, or no mMRC score was available. All patients meeting the inclusion criteria were considered regardless of comorbidities.
The PFT results of all adult patients, including those performed between June 1, 2013, and January 6, 2016, were submitted to spirometry, and lung volume measurements were analyzed. Sociodemographic information was collected, including sex, ethnicity, age, height, weight, and basal metabolic index. Other data found in PFTs, such as smoking status, smoking in packs/year, mMRC score, predicted TLC value, imaging present (chest X-ray, computed tomography), and hospitalizations and exacerbations within 1 year were collected. In addition, we examined the predicted values for FEV1, DLCO, and DLCO/VA (calculated using the Ayer equation), FVC (calculated using the Knudson equation), expiratory reserve volume, inspiratory VC, and slow VC. PaO2, PaCO2, and Alveolar-arterial gradients also were collected.6-9 Information about heart failure status was gathered through medical evaluation of notes and cardiac studies. All categorical variables were correlated with Spearman analysis and quantitative variables with average percentages. P values were calculated with analysis of variance.
Results
Of 6461 VACHS patient records reviewed, 415 met the inclusion criteria. Patients were divided according to their mMRC score: 65 had mMRC score of 0, 87 had an mMRC score of 1, 2 had an mMRC score of 2, 146 had an mMRC of 3, and 115 had an mMRC score of 4. The population was primarily male (98.6%) and of Hispanic ethnicity (96.4%), with a mean age of 72 years (Table 1). Most patients (n = 269, 64.0%) were prior smokers, while 135 patients (32.5%) had never smoked, and 11 (2.7%) were current smokers. At baseline, 169 patients (41.4%) had interstitial lung disease, 39 (9.6%) had chest wall disorders, 29 (7.1%) had occupational exposure, 25 (6.1%) had pneumonitis, and 14 (3.4%) had neuromuscular disorders.
There was a statistically significant relationship between mMRC score and hospitalization and FEV1 but not TLC (Table 2). As mMRC increased, so did hospitalizations: a total of 168 patients (40.5%) were hospitalized; 24 patients (36.9%) had an mMRC score of 0, 30 patients (34.0%) had an mMRC score of 1, 2 patients (100%) had an mMRC score of 2, 54 patients (37.0%) had an mMRC score of 3, and 58 patients (50.0%) had an mMRC score of 4 (P = .04). Mean (SD) TLC values increased as mMRC scores increased. Mean (SD) TLC was 70.5% (33.0) for the entire population; 68.8% (7.2) for patients with an mMRC score of 0, 70.8% (5.8) for patients with an mMRC score of 1, 75.0% (1.4) for patients with an mMRC score of 2, 70.1% (7.2) for patients with an mMRC score of 3, and 71.5% (62.1) for patients with an mMRC score of 4 (P = .10) (Figure 1). There was an associated decrease in mean (SD) FEV1 with mMRC. Mean (SD) FEV1 was 76.2% (18.9) for the entire population; 81.7% (19.3) for patients with an mMRC score of 0, 80.9% (18) for patients with an mMRC score of 1, 93.5% (34.6) for patients with an mMRC score of 2, 76.2% (17.1) for patients with an mMRC score of 3, and 69.2% (19.4) for patients with an mMRC score of 4; (P < .001) (Figure 2).
The correlation between mMRC and FEV1 (r = 0.25, P < .001) was stronger than the correlation between mMRC and TLC (r = 0.15, P < .001). The correlations for DLCO (P < .001), DLCO/VA (P < .001), hemoglobin (P < .02), and PaO2 (P < .001) were all statistically significant (P < .005), but with no strong identifiable trend.
Discussion
The patient population of this study was primarily older males of Hispanic ethnicity with a history of smoking. There was no association between body mass index or smoking status with worsening dyspnea as measured with mMRC scores. We observed no significant correlation between mMRC scores and various factors such as comorbidities including heart conditions, and epidemiological factors like the etiology of lung disease, including both intrinsic and extrinsic causes. This lack of association was anticipated, as restrictive lung diseases in our study predominantly arose from intrinsic pulmonary etiologies, such as interstitial lung disease. A difference between more hospitalizations and worsening dyspnea was identified. There was a slightly higher correlation between FEV1 and mMRC scores when compared with TLC and mMRC scores concerning worsening dyspnea, which could indicate that the use of FEV1 should be preferred over previous recommendations to use TLC.10 Other guidelines have utilized exercise capacity via the 6-minute walk test as a marker of severity with spirometry values and found that DLCO was correlated with severity.11
The latest ERS/ATS guidelines recommend z scores for grading the severity of obstructive lung diseases but do not recommend them for the diagnosis of restrictive lung diseases.12 A z score encompasses diverse variables (eg, age, sex, and ethnicity) to provide more uniform and consistent results. Other studies have been done to relate z scores to other spirometry variables with restrictive lung disease. One such study indicates the potential benefit of using FVC alone to grade restrictive lung diseases.13 There continues to be great diversity in the interpretation of pulmonary function tests, and we believe the information gathered can provide valuable insight for managing patients with restrictive lung diseases.
Limitations
Only 2 patients reported an mMRC score of 2 in our study. This may have affected statistical outcomes. It also may reveal possible deficits in the efficacy of patient education on the mMRC scale. This study was also limited by its small sample size, single center location, and the distribution of patients that reported an mMRC favored either low or high values. The patients in this study, who were all veterans, may not be representative of other patient populations.
Conclusions
There continue to be few factors associated with the physiological severity of the defective oxygen delivery and reported dyspnea of a patient with restrictive lung disease that allows for an accurate, repeatable grading of severity. Using FEV1 instead of TLC to determine the severity of a restrictive lung disease should be reconsidered. We could not find any other strong correlation among other factors studied. Further research should be conducted to continue looking for variables that more accurately depict patient dyspnea in restrictive lung disease.
Acknowledgments
This study is based upon work supported by the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico, and is the result of work supported by Pulmonary & Critical Care Medicine service, with resources and the use of its facilities.
1. Hegewald MJ, Crapo RO. Pulmonary function testing. In: Broaddus VC, Ernst JD, King Jr TE, eds. Murray and Nadel’s Textbook of Respiratory Medicine. 5th ed. Saunders; 2010:522-553.
2. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26(5):948-968. doi:10.1183/09031936.05.00035205
3. Rabe KF, Beghé B, Luppi F, Fabbri LM. Update in chronic obstructive pulmonary disease 2006. Am J Respir Crit Care Med. 2007;175(12):1222-1232. doi:10.1164/rccm.200704-586UP
4. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Spirometry for health care providers Accessed April 30, 2024. https://goldcopd.org/wp-content/uploads/2016/04/GOLD_Spirometry_2010.pdf
5. Mannino DM, Holguin F, Pavlin BI, Ferdinands JM. Risk factors for prevalence of and mortality related to restriction on spirometry: findings from the First National Health and Nutrition Examination Survey and follow-up. Int J Tuberc Lung Dis. 2005;9(6):613-621.
6. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734. doi:10.1164/arrd.1983.127.6.725
7. Knudson RJ, Burrows B, Lebowitz MD. The maximal expiratory flow-volume curve: its use in the detection of ventilatory abnormalities in a population study. Am Rev Respir Dis. 1976;114(5):871-879. doi:10.1164/arrd.1976.114.5.871
8. Knudson RJ, Lebowitz MD, Burton AP, Knudson DE. The closing volume test: evaluation of nitrogen and bolus methods in a random population. Am Rev Respir Dis. 1977;115(3):423-434. doi:10.1164/arrd.1977.115.3.423
9. Ayers LN, Ginsberg ML, Fein J, Wasserman K. Diffusing capacity, specific diffusing capacity and interpretation of diffusion defects. West J Med. 1975;123(4):255-264.
10. Lung function testing: selection of reference values and interpretative strategies. American Thoracic Society. Am Rev Respir Dis. 1991;144(5):1202-1218. doi:10.1164/ajrccm/144.5.1202
11. Larson J, Wrzos K, Corazalla E, Wang Q, Kim HJ, Cho RJ. Should FEV1 be used to grade restrictive impairment? A single-center comparison of lung function parameters to 6-minute walk test in patients with restrictive lung disease. HSOA J Pulm Med Respir Res. 2023;9:082. doi:10.24966/PMRR-0177/100082
12. Stanojevic S, Kaminsky DA, Miller MR, et al. ERS/ATS technical standard on interpretive strategies for routine lung function tests. Eur Respir J. 2022;60(1):2101499. Published 2022 Jul 13. doi:10.1183/13993003.01499-2021
13. Myrberg T, Lindberg A, Eriksson B, et al. Restrictive spirometry versus restrictive lung function using the GLI reference values. Clin Physiol Funct Imaging. 2022;42(3):181-189. doi:10.1111/cpf.12745
1. Hegewald MJ, Crapo RO. Pulmonary function testing. In: Broaddus VC, Ernst JD, King Jr TE, eds. Murray and Nadel’s Textbook of Respiratory Medicine. 5th ed. Saunders; 2010:522-553.
2. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26(5):948-968. doi:10.1183/09031936.05.00035205
3. Rabe KF, Beghé B, Luppi F, Fabbri LM. Update in chronic obstructive pulmonary disease 2006. Am J Respir Crit Care Med. 2007;175(12):1222-1232. doi:10.1164/rccm.200704-586UP
4. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Spirometry for health care providers Accessed April 30, 2024. https://goldcopd.org/wp-content/uploads/2016/04/GOLD_Spirometry_2010.pdf
5. Mannino DM, Holguin F, Pavlin BI, Ferdinands JM. Risk factors for prevalence of and mortality related to restriction on spirometry: findings from the First National Health and Nutrition Examination Survey and follow-up. Int J Tuberc Lung Dis. 2005;9(6):613-621.
6. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734. doi:10.1164/arrd.1983.127.6.725
7. Knudson RJ, Burrows B, Lebowitz MD. The maximal expiratory flow-volume curve: its use in the detection of ventilatory abnormalities in a population study. Am Rev Respir Dis. 1976;114(5):871-879. doi:10.1164/arrd.1976.114.5.871
8. Knudson RJ, Lebowitz MD, Burton AP, Knudson DE. The closing volume test: evaluation of nitrogen and bolus methods in a random population. Am Rev Respir Dis. 1977;115(3):423-434. doi:10.1164/arrd.1977.115.3.423
9. Ayers LN, Ginsberg ML, Fein J, Wasserman K. Diffusing capacity, specific diffusing capacity and interpretation of diffusion defects. West J Med. 1975;123(4):255-264.
10. Lung function testing: selection of reference values and interpretative strategies. American Thoracic Society. Am Rev Respir Dis. 1991;144(5):1202-1218. doi:10.1164/ajrccm/144.5.1202
11. Larson J, Wrzos K, Corazalla E, Wang Q, Kim HJ, Cho RJ. Should FEV1 be used to grade restrictive impairment? A single-center comparison of lung function parameters to 6-minute walk test in patients with restrictive lung disease. HSOA J Pulm Med Respir Res. 2023;9:082. doi:10.24966/PMRR-0177/100082
12. Stanojevic S, Kaminsky DA, Miller MR, et al. ERS/ATS technical standard on interpretive strategies for routine lung function tests. Eur Respir J. 2022;60(1):2101499. Published 2022 Jul 13. doi:10.1183/13993003.01499-2021
13. Myrberg T, Lindberg A, Eriksson B, et al. Restrictive spirometry versus restrictive lung function using the GLI reference values. Clin Physiol Funct Imaging. 2022;42(3):181-189. doi:10.1111/cpf.12745
Hemophagocytic Lymphohistiocytosis: Early Treatment Leading to an Excellent Outcome
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
HLH is a rare and deadly disease increasingly more present in adults, but following treatment protocol may yield favorable results.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and deadly disease in which unregulated proliferation of histiocytes and T-cell infiltration takes place. It is known as a pediatric disease in which gene defects result in impaired cytotoxic NK- and T-cell function. It has been associated with autosomal recessive inheritance pattern. Without therapy, survival for these patients with active familial HLH is approximately 2 months.
Recognition of the disease has increased over the years, and as a result the diagnosis of HLH in adults also has increased. An acquired form can be triggered by viruses like Epstein-Barr virus, influenza, HIV, lymphoid malignancies, rheumatologic disorders, or immunodeficiency disorders. Survival rates for untreated HLH have been reported at < 5%.1 Despite early recognition and adequate treatment, HLH carries an overall mortality of 50% in the initial presentation, 90% die in the first 8 weeks of treatment due to uncontrolled disease.2
Case Presentation
A 56-year-old man with no active medical issues except for a remote history of non-Hodgkin lymphoma treated with chemotherapy and splenectomy in 1990 presented to the Veterans Affairs Caribbean Healthcare System in San Juan, Puerto Rico. He was admitted to the medicine ward due to community acquired pneumonia. Three days into admission his clinical status deteriorated, and the patient was transferred to the intensive care unit (ICU) due to acute respiratory failure and sepsis secondary to worsening pneumonia. Chest imaging demonstrated rapidly progressing diffuse bilateral infiltrates. Due to the severity of the chest imaging, a diagnostic bronchoscopy was performed.
The patient’s antibiotics regimen was empirically escalated to vancomycin 1500 mg IV every 12 hours and meropenem 2 g IV every 8 hours. Despite optimization of therapy, the patient did not show clinical signs of improvement. Febrile episodes persisted, pulmonary infiltrates and hypoxemia worsened, and the patient required a neuromuscular blockade. Since the bronchoscopy was nondiagnostic and deterioration persistent, the differential diagnosis was broadened. This led to the ordering of inflammatory markers. Laboratory testing showed ferritin levels > 16,000 ng/mL, pointing to HLH as a possible diagnosis. Further workup was remarkable for triglycerides of 1234 mg/dL and a fibrinogen of 0.77 g/L. In the setting of bicytopenia and persistent fever, HLH-94 regimen was started with dexamethasone 40 mg daily and etoposide 100 mg/m2. CD25 levels of 154,701 pg/mL were demonstrated as well as a decreased immunoglobulin (Ig) G levels with absent IgM and IgA. Bone marrow biopsy was consistent with hemophagocytosis. The patient eventually was extubated and sent to the oncology ward to continue chemotherapy.
Discussion
A high clinical suspicion is warranted for rapid diagnosis and treatment as HLH evolves in most cases to multiorgan failure and death. The diagnostic criteria for HLH was developed by the Histiocyte Society in 1991 and then restructured in 2004.3,4 In the first diagnostic tool developed in 1991, diagnosis was based on 5 criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). Three additional laboratory findings were also described as part of HLH diagnosis since 2004: low or absent NK-cell-activity, hyperferritinemia of > 500 ng/dL, and high-soluble interleukin-2-receptor levels (CD25) > 2400 U/mL. Overall, 5 of 8 criteria are needed for the HLH diagnosis.
Despite the common use of these diagnostic criteria, they were developed for the pediatric population but have not been validated for adult patients.5 For adult patients, the HScore was developed in 2014. It has 9 variables: 3 are based on clinical findings (known underlying immunosuppression, high temperature, and organomegaly; 5 are based on laboratory values (ferritin, serum glutamic oxaloacetic transaminase, cytopenia, triglycerides, and fibrinogen levels); the last variable uses cytologic findings in the bone marrow. In the initial study, probability of having HLH ranged from < 1% with an HScore of ≤ 90% to > 99% with an HScore of ≥ 250 in noncritically ill adults.5 A recently published retrospective study demonstrated the diagnostic reliability of both the HLH-2004 criteria and HScore in critically ill adult patients. This study concluded that the best prediction accuracy of HLH diagnosis for a cutoff of 4 fulfilled HLH-2004 criteria had a 95.0% sensitivity and 93.6% specificity and HScore cutoff of 168 reached a 100% sensitivity and 94.1% specificity.6
The early negative bronchoscopy lowered the possibility of an infection as the etiology of the clinical presentation and narrowed the hyperferritinemia differential diagnosis. Hyperferritinemia has a sensitivity and specificity of > 90% for diagnosis when above 10,000 ng/dL in the pediatric population.7 This is not the case in adults. Hyperferritinemia is a marker of different inflammatory responses, such as histoplasmosis infection, malignancy, or iron overload rather than an isolated diagnostic tool for HLH.8 It has been reported that CD25 levels less than the diagnostic threshold of 2400 U/mL have a 100% sensitivity for the diagnosis and therefore can rule out the diagnosis. When this is taken into consideration, it can be concluded that CD25 level is a better diagnostic tool when compared with ferritin, but its main limitation is its lack of widespread availability.9 Still, there is a limited number of pathologies that are associated with marked hyperferritinemia, specifically using thresholds of more than 6000 ng/dL.10 Taking into consideration the high mortality of untreated HLH, isolated hyperferritinemia still warrants HLH workup to aggressively pursue the diagnosis and improve outcomes.
The goal of therapy in HLH is prompt inactivation of the dysregulated inflammation with aggressive immunosuppression. In our deteriorating patient, the treatment was started with only 4 of the 8 HLH-2004 diagnostic criteria being met. As per the 2018 Histiocyte Society consensus statement, the decision to start the HLH-94 treatment relies on not only the HLH-2004 diagnostic criteria, but also the patient’s clinical evolution.11 In 1994 the Histiocyte Society also published a treatment protocol termed HLH-94. A Korean retrospective study demonstrated that this protocol led to a 5-year survival rate of 60 to 80% depending on the HLH trigger and response to initial treatment.12 The protocol consists of etoposide at 150 mg/m2, 2 weekly doses in the first 2 weeks and then 1 dose weekly for the next 6 weeks. Dexamethasone is the steroid of choice as it readily crosses the blood-brain barrier. Its dosage consists of 10 mg/m2 for the first 2 weeks and then it is halved every 2 weeks until the eighth week of treatment. A slow taper follows to avoid adrenal insufficiency. Once 8 weeks of treatment have been completed, cyclosporine is added to a goal trough of 200 mcg/dL. If there is central nervous system (CNS) involvement, early aggressive treatment with intrathecal methotrexate is indicated if no improvement is noted during initial therapy.11
In 2004 the Histiocyte Society restructured the HLH-94 treatment protocol with the aim of presenting a more aggressive treatment strategy. The protocol added cyclosporine to the initial induction therapy, rather than later in the ninth week as HLH-94. Neither the use of cyclosporine nor the HLH-2004 have been demonstrated to be superior to the use of etoposide and dexamethasone alone or in the HLH-94 protocol, respectively.13 Cyclosporine is associated with adverse effects (AEs) and may have many contraindications in the acute phase of the disease. Therefore, the HLH-94 protocol is still the recommended regimen.11
To assess adequate clinical response, several clinical and laboratory parameters are followed. Clinically, resolution of fever, improvement in hepatosplenomegaly, lymphadenopathy, and mental status can be useful. Laboratories can be used to assess improvement from organ specific damage such as hepatic involvement or cytopenia. The limitation of these diagnostic studies is that they could falsely suggest an inadequate response to treatment due to concomitant infection or medication AEs. Other markers such as ferritin levels, CD25, and NK cell activity levels are more specific to HLH. Out of them, a decreasing ferritin level has the needed specificity and widespread availability for repeated assessment. On the other hand, both CD25 and NK cell activity are readily available only in specialized centers. An initial high ferritin level is a marker for a poor prognosis, and the rate of decline correlates with mortality. Studies have demonstrated that persistently elevated ferritin levels after treatment initiation are associated with worse outcomes.14,15
Several salvage treatments have been identified in recalcitrant or relapsing disease. In general, chemotherapy needs to be intensified, either by returning to the initial high dosage if recurrence occurs in the weaning phase of treatment or adding other agents if no response was initially achieved. Emapalumab, an interferon γ antibody, was approved by the US Food and Drug Administration for the treatment of intractable HLH after it demonstrated that when added to dexamethasone, it lead to treatment response in 17 out of 27 pediatric patients, with a relatively safe AE profile.16 The goal of intensifying chemotherapy is to have the patient tolerate allogenic stem cell transplant, which is clinically indicated in familial HLH, malignancy induced HLH, and recalcitrant cases. In patients who undergo hematopoietic cell transplantation (HCT) there is a tendency to increase survival to 66% at 5 years.12
Conclusions
HLH is a rare and deadly disease increasingly more present in adults. Our patient who initially presented with a sepsis diagnosis was suspected of having a hematologic etiology for his clinical findings due to markedly elevated ferritin levels. In our patient, the HLH-94 treatment protocol was used, yielding favorable results. Given the lack of specific scientific data backing updated protocols such as HLH-2004 and a comparatively favorable safety profile, current guidelines still recommend using the HLH-94 treatment protocol. Decreasing ferritin levels may be used in conjunction with clinical improvement to demonstrate therapeutic response. Persistence of disease despite standard treatment may warrant novel therapies, such as emapalumab or HCT. Physicians need to be wary of an HLH diagnosis as early identification and treatment may improve its otherwise grim prognosis.
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
1. Chen TY, Hsu MH, Kuo HC, Sheen JM, Cheng MC, Lin YJ. Outcome analysis of pediatric hemophagocytic lymphohistiocytosis. J Formos Med Assoc. 2021;120(1, pt 1):172-179. doi:10.1016/j.jfma.2020.03.025
2. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. doi:10.1182/blood-2002-01-0172
3. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
4. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039
5. Knaak C, Nyvlt P, Schuster FS, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. 2020;24(1):244. Published 2020 May 24. doi:10.1186/s13054-020-02941-3
6. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620. doi:10.1002/art.38690
7. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477. doi:10.1182/blood.2018894618
8. Schaffner M, Rosenstein L, Ballas Z, Suneja M. Significance of Hyperferritinemia in Hospitalized Adults. Am J Med Sci. 2017;354(2):152-158. doi:10.1016/j.amjms.2017.04.016
9. Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1(26):2529-2534. Published 2017 Dec 6. doi:10.1182/bloodadvances.2017012310
10. Belfeki N, Strazzulla A, Picque M, Diamantis S. Extreme hyperferritinemia: etiological spectrum and impact on prognosis. Reumatismo. 2020;71(4):199-202. Published 2020 Jan 28. doi:10.4081/reumatismo.2019.1221
11. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508-1517. doi:10.1016/j.jaip.2018.05.031
12. Yoon JH, Park SS, Jeon YW, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269-276. doi:10.3324/haematol.2018.198655
13. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728-2738. doi:10.1182/blood-2017-06-788349
14. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154-155. doi:10.1002/pbc.22774
15. Zhou J, Zhou J, Shen DT, Goyal H, Wu ZQ, Xu HG. Development and validation of the prognostic value of ferritin in adult patients with Hemophagocytic Lymphohistiocytosis. Orphanet J Rare Dis. 2020;15(1):71. Published 2020 Mar 12. doi:10.1186/s13023-020-1336-616. Locatelli F, Jordan MB, Allen CE, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Presented at: American Society of Hematology Annual Meeting, November 29, 2018. Blood. 2018;132(suppl 1):LBA-6. doi:10.1182/blood-2018-120810
Flexible Bronchoscopic Removal of 3 Foreign Objects
Consider flexible bronchoscopy as an option to retrieve aspirated foreign bodies in the airway.
Airway foreign-body aspiration may cause no symptoms, although it can produce acute and life-threatening central airway obstruction.1 In the US, at least 2,700 people, including more than 300 children, die of foreign-body aspiration each year.2 Most foreign-body aspirations occur in children and elderly patients.3 In adults, dementia, drug intoxication, strokes, seizures, and neurologic disorders may predispose patients to aspiration.3 Some of the consequences of an aspirated object are complete or partial airway obstruction, respiratory distress and failure, pneumothorax, and hemorrhage.2 In addition, inadvertent aspiration of foreign objects in asymptomatic patients may not be evident for months, resulting in late complications as postobstructive pneumonia, bronchiectasis, or lung abscess.2
We present a case of a patient with documented schizophrenia with nonadherence to his antipsychotic medications who aspirated different objects. Flexible bronchoscopy was performed since rigid bronchoscopy is not available at our institution. Several bronchoscopy tools were required to successfully remove the objects and avoid further invasive interventions, such as cardiothoracic surgery.
Case Presentation
A 55-year-old man with schizophrenia on antipsychotics developed cough, shortness of breath, and dysphagia of 1-month of evolution. Because his symptoms worsened, his mother brought him to the emergency department. Peripheral oxygen saturation was 97% at room air. Lung auscultation was remarkable for bilateral scattered rhonchi and wheezes.
Laboratory results showed leukocytosis with neutrophilia and hypotonic hypovolemic hyponatremia. 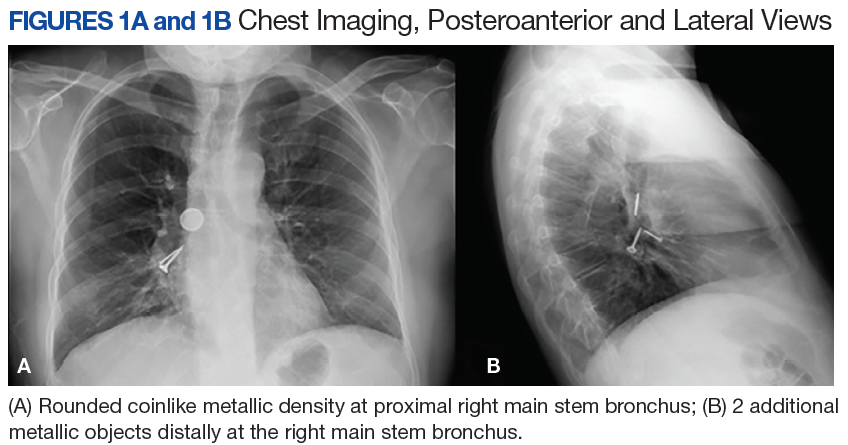
The patient stated that he did not remember swallowing any objects, although his mother confirmed that he was not adherent with his antipsychotic medications, which could have predisposed him to aspiration secondary to possible psychotic episodes. 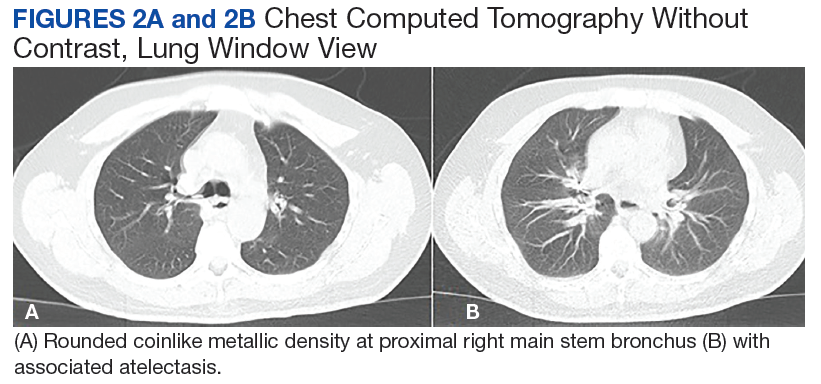
Piperacillin/tazobactam 4.5 g every 8 hours was started to cover anaerobic bacterial organisms causing abscess, and IV fluids were given for hypovolemia. Flexible bronchoscopy (rigid bronchoscopy is superior although not available at our institution) was planned to be performed in the operating room (OR) because we predicted a difficult and prolonged retrieval in view of multiple and different-sized objects.
A bronchoscopy was performed, showing a disk-shaped metallic foreign body at the right main stem bronchus. After multiple attempts using the tripod retrieval tool, a coin was removed (Figures 3A and 3B). 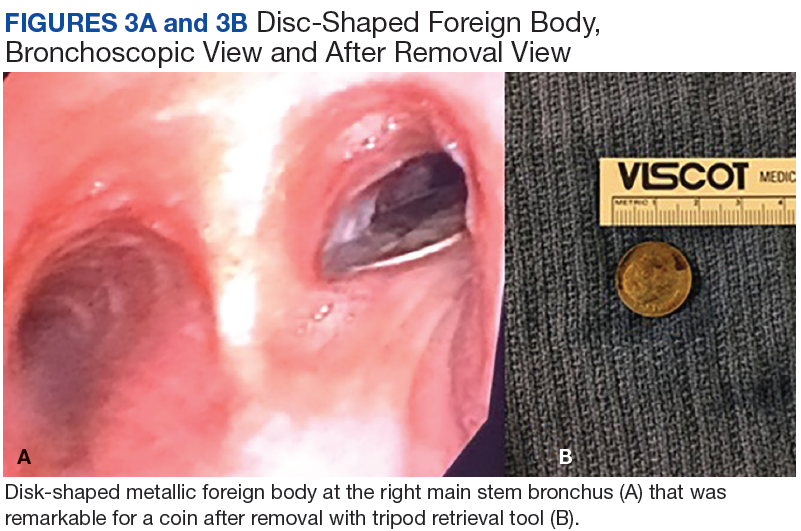
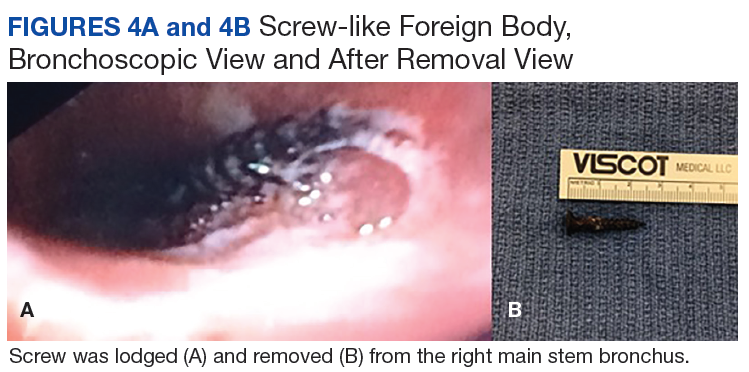

The patient was reintubated without any complications. A postprocedure chest radiograph showed the absence of foreign bodies and no pneumothorax. The patient completed IV antibiotic with piperacillin/tazobactam and supportive therapy with clinical improvement and successful extubation within 2 days. Cardiothoracic surgery was not required. Psychiatry service recommended to continue the same antipsychotic medications, administered only by his mother to assure adherence and to avoid similar future events. The patient was discharged home without any immediate complications despite having had a coin, nail, and screw aspiration (Figure 6).
Discussion
More than 50% of foreign bodies lodge at the right main stem bronchus due to the trachea’s anatomical position.2,4 In adults, foreign-body aspiration may present with nonspecific symptoms, such as cough and dyspnea.4 Other symptoms might include wheezes, chest discomfort, and sputum production. A chest radiograph is helpful as part of the initial diagnostic workup. A chest CT scan without contrast should be performed to confirm the diagnosis and to plan possible foreign-body retrieval.
Bronchoscopy is the gold standard for diagnosis and management of foreign-body aspiration.1 Rigid bronchoscopy is superior to flexible bronchoscopy in removal of large airway foreign bodies.1 The rigid bronchoscopy provides the ability to function as an endotracheal tube, thus allowing control of the airway and a conduit through which foreign bodies can be removed.1 Nonetheless, sometimes retrieval of foreign bodies deeper into the subsegmental bronchi cannot be achieved.1 Moreover, the required equipment or knowledgeable staff is not always available.1 Therefore, flexible bronchoscopy is an option to retrieve airway foreign bodies especially those located distal in the airway and for those medical centers without rigid bronchoscopy as is the case in our institution.
In our case, flexible bronchoscopy was performed in the OR because we predicted a difficult and prolonged retrieval in view of multiple and different-sized objects. Anesthesia Service assistance was requested anticipating need for patient sedation and intubation. We used the tripod and snare retrieval tools to remove 3 foreign objects located at the right main stem bronchus. Even though multiple attempts were made and endotracheal intubation was required, a successful retrieval with flexible bronchoscopy was performed. Moreover, cardiothoracic surgery was not required avoiding more invasive interventions with subsequent morbidity and mortality.
Conclusion
Flexible bronchoscopy is an important tool within the arsenal of the Pulmonology Service. The management of the underlying etiology also should be performed. In our case, the Psychiatry Service recommended that the patient’s medications should be administered by his mother to avoid similar events in the future. Flexible bronchoscopy can be a valuable option for foreign objects removal, especially those distally located in the lung segments as well as in those medical centers where rigid bronchoscopy is not available.
1. Mehta D, Mehta C, Bansal S, Singla S, Tangri N. Flexible bronchoscopic removal of a three piece foreign body from a child’s bronchus. Australas Med J. 2012;5(4):227-230.
2. Mercado JA, Rodríguez W. Occult aspiration of a chicken wishbone as a cause of hemoptysis. P R Health Sci J. 1999;18(1):71-73.
3. Robles-Arias CM, Campos-Santiago Z, Vega MT, Rosa-Cruz F, Rodríguez-Cintrón W. Aspiration of a dental tool during a crown placement procedure. Fed Pract. 2014;31(6):12-14.
4. Blanco-Ramos M, Botana-Rial M, García-Fontán E, Fernández-Villar A, Gallas-Torreira M. Update in the extraction of airway foreign bodies in adults. J Thorac Dis. 2016;8(11):3452-3456.
Consider flexible bronchoscopy as an option to retrieve aspirated foreign bodies in the airway.
Consider flexible bronchoscopy as an option to retrieve aspirated foreign bodies in the airway.
Airway foreign-body aspiration may cause no symptoms, although it can produce acute and life-threatening central airway obstruction.1 In the US, at least 2,700 people, including more than 300 children, die of foreign-body aspiration each year.2 Most foreign-body aspirations occur in children and elderly patients.3 In adults, dementia, drug intoxication, strokes, seizures, and neurologic disorders may predispose patients to aspiration.3 Some of the consequences of an aspirated object are complete or partial airway obstruction, respiratory distress and failure, pneumothorax, and hemorrhage.2 In addition, inadvertent aspiration of foreign objects in asymptomatic patients may not be evident for months, resulting in late complications as postobstructive pneumonia, bronchiectasis, or lung abscess.2
We present a case of a patient with documented schizophrenia with nonadherence to his antipsychotic medications who aspirated different objects. Flexible bronchoscopy was performed since rigid bronchoscopy is not available at our institution. Several bronchoscopy tools were required to successfully remove the objects and avoid further invasive interventions, such as cardiothoracic surgery.
Case Presentation
A 55-year-old man with schizophrenia on antipsychotics developed cough, shortness of breath, and dysphagia of 1-month of evolution. Because his symptoms worsened, his mother brought him to the emergency department. Peripheral oxygen saturation was 97% at room air. Lung auscultation was remarkable for bilateral scattered rhonchi and wheezes.
Laboratory results showed leukocytosis with neutrophilia and hypotonic hypovolemic hyponatremia.
The patient stated that he did not remember swallowing any objects, although his mother confirmed that he was not adherent with his antipsychotic medications, which could have predisposed him to aspiration secondary to possible psychotic episodes.
Piperacillin/tazobactam 4.5 g every 8 hours was started to cover anaerobic bacterial organisms causing abscess, and IV fluids were given for hypovolemia. Flexible bronchoscopy (rigid bronchoscopy is superior although not available at our institution) was planned to be performed in the operating room (OR) because we predicted a difficult and prolonged retrieval in view of multiple and different-sized objects.
A bronchoscopy was performed, showing a disk-shaped metallic foreign body at the right main stem bronchus. After multiple attempts using the tripod retrieval tool, a coin was removed (Figures 3A and 3B).
The patient was reintubated without any complications. A postprocedure chest radiograph showed the absence of foreign bodies and no pneumothorax. The patient completed IV antibiotic with piperacillin/tazobactam and supportive therapy with clinical improvement and successful extubation within 2 days. Cardiothoracic surgery was not required. Psychiatry service recommended to continue the same antipsychotic medications, administered only by his mother to assure adherence and to avoid similar future events. The patient was discharged home without any immediate complications despite having had a coin, nail, and screw aspiration (Figure 6).
Discussion
More than 50% of foreign bodies lodge at the right main stem bronchus due to the trachea’s anatomical position.2,4 In adults, foreign-body aspiration may present with nonspecific symptoms, such as cough and dyspnea.4 Other symptoms might include wheezes, chest discomfort, and sputum production. A chest radiograph is helpful as part of the initial diagnostic workup. A chest CT scan without contrast should be performed to confirm the diagnosis and to plan possible foreign-body retrieval.
Bronchoscopy is the gold standard for diagnosis and management of foreign-body aspiration.1 Rigid bronchoscopy is superior to flexible bronchoscopy in removal of large airway foreign bodies.1 The rigid bronchoscopy provides the ability to function as an endotracheal tube, thus allowing control of the airway and a conduit through which foreign bodies can be removed.1 Nonetheless, sometimes retrieval of foreign bodies deeper into the subsegmental bronchi cannot be achieved.1 Moreover, the required equipment or knowledgeable staff is not always available.1 Therefore, flexible bronchoscopy is an option to retrieve airway foreign bodies especially those located distal in the airway and for those medical centers without rigid bronchoscopy as is the case in our institution.
In our case, flexible bronchoscopy was performed in the OR because we predicted a difficult and prolonged retrieval in view of multiple and different-sized objects. Anesthesia Service assistance was requested anticipating need for patient sedation and intubation. We used the tripod and snare retrieval tools to remove 3 foreign objects located at the right main stem bronchus. Even though multiple attempts were made and endotracheal intubation was required, a successful retrieval with flexible bronchoscopy was performed. Moreover, cardiothoracic surgery was not required avoiding more invasive interventions with subsequent morbidity and mortality.
Conclusion
Flexible bronchoscopy is an important tool within the arsenal of the Pulmonology Service. The management of the underlying etiology also should be performed. In our case, the Psychiatry Service recommended that the patient’s medications should be administered by his mother to avoid similar events in the future. Flexible bronchoscopy can be a valuable option for foreign objects removal, especially those distally located in the lung segments as well as in those medical centers where rigid bronchoscopy is not available.
Airway foreign-body aspiration may cause no symptoms, although it can produce acute and life-threatening central airway obstruction.1 In the US, at least 2,700 people, including more than 300 children, die of foreign-body aspiration each year.2 Most foreign-body aspirations occur in children and elderly patients.3 In adults, dementia, drug intoxication, strokes, seizures, and neurologic disorders may predispose patients to aspiration.3 Some of the consequences of an aspirated object are complete or partial airway obstruction, respiratory distress and failure, pneumothorax, and hemorrhage.2 In addition, inadvertent aspiration of foreign objects in asymptomatic patients may not be evident for months, resulting in late complications as postobstructive pneumonia, bronchiectasis, or lung abscess.2
We present a case of a patient with documented schizophrenia with nonadherence to his antipsychotic medications who aspirated different objects. Flexible bronchoscopy was performed since rigid bronchoscopy is not available at our institution. Several bronchoscopy tools were required to successfully remove the objects and avoid further invasive interventions, such as cardiothoracic surgery.
Case Presentation
A 55-year-old man with schizophrenia on antipsychotics developed cough, shortness of breath, and dysphagia of 1-month of evolution. Because his symptoms worsened, his mother brought him to the emergency department. Peripheral oxygen saturation was 97% at room air. Lung auscultation was remarkable for bilateral scattered rhonchi and wheezes.
Laboratory results showed leukocytosis with neutrophilia and hypotonic hypovolemic hyponatremia.
The patient stated that he did not remember swallowing any objects, although his mother confirmed that he was not adherent with his antipsychotic medications, which could have predisposed him to aspiration secondary to possible psychotic episodes.
Piperacillin/tazobactam 4.5 g every 8 hours was started to cover anaerobic bacterial organisms causing abscess, and IV fluids were given for hypovolemia. Flexible bronchoscopy (rigid bronchoscopy is superior although not available at our institution) was planned to be performed in the operating room (OR) because we predicted a difficult and prolonged retrieval in view of multiple and different-sized objects.
A bronchoscopy was performed, showing a disk-shaped metallic foreign body at the right main stem bronchus. After multiple attempts using the tripod retrieval tool, a coin was removed (Figures 3A and 3B).
The patient was reintubated without any complications. A postprocedure chest radiograph showed the absence of foreign bodies and no pneumothorax. The patient completed IV antibiotic with piperacillin/tazobactam and supportive therapy with clinical improvement and successful extubation within 2 days. Cardiothoracic surgery was not required. Psychiatry service recommended to continue the same antipsychotic medications, administered only by his mother to assure adherence and to avoid similar future events. The patient was discharged home without any immediate complications despite having had a coin, nail, and screw aspiration (Figure 6).
Discussion
More than 50% of foreign bodies lodge at the right main stem bronchus due to the trachea’s anatomical position.2,4 In adults, foreign-body aspiration may present with nonspecific symptoms, such as cough and dyspnea.4 Other symptoms might include wheezes, chest discomfort, and sputum production. A chest radiograph is helpful as part of the initial diagnostic workup. A chest CT scan without contrast should be performed to confirm the diagnosis and to plan possible foreign-body retrieval.
Bronchoscopy is the gold standard for diagnosis and management of foreign-body aspiration.1 Rigid bronchoscopy is superior to flexible bronchoscopy in removal of large airway foreign bodies.1 The rigid bronchoscopy provides the ability to function as an endotracheal tube, thus allowing control of the airway and a conduit through which foreign bodies can be removed.1 Nonetheless, sometimes retrieval of foreign bodies deeper into the subsegmental bronchi cannot be achieved.1 Moreover, the required equipment or knowledgeable staff is not always available.1 Therefore, flexible bronchoscopy is an option to retrieve airway foreign bodies especially those located distal in the airway and for those medical centers without rigid bronchoscopy as is the case in our institution.
In our case, flexible bronchoscopy was performed in the OR because we predicted a difficult and prolonged retrieval in view of multiple and different-sized objects. Anesthesia Service assistance was requested anticipating need for patient sedation and intubation. We used the tripod and snare retrieval tools to remove 3 foreign objects located at the right main stem bronchus. Even though multiple attempts were made and endotracheal intubation was required, a successful retrieval with flexible bronchoscopy was performed. Moreover, cardiothoracic surgery was not required avoiding more invasive interventions with subsequent morbidity and mortality.
Conclusion
Flexible bronchoscopy is an important tool within the arsenal of the Pulmonology Service. The management of the underlying etiology also should be performed. In our case, the Psychiatry Service recommended that the patient’s medications should be administered by his mother to avoid similar events in the future. Flexible bronchoscopy can be a valuable option for foreign objects removal, especially those distally located in the lung segments as well as in those medical centers where rigid bronchoscopy is not available.
1. Mehta D, Mehta C, Bansal S, Singla S, Tangri N. Flexible bronchoscopic removal of a three piece foreign body from a child’s bronchus. Australas Med J. 2012;5(4):227-230.
2. Mercado JA, Rodríguez W. Occult aspiration of a chicken wishbone as a cause of hemoptysis. P R Health Sci J. 1999;18(1):71-73.
3. Robles-Arias CM, Campos-Santiago Z, Vega MT, Rosa-Cruz F, Rodríguez-Cintrón W. Aspiration of a dental tool during a crown placement procedure. Fed Pract. 2014;31(6):12-14.
4. Blanco-Ramos M, Botana-Rial M, García-Fontán E, Fernández-Villar A, Gallas-Torreira M. Update in the extraction of airway foreign bodies in adults. J Thorac Dis. 2016;8(11):3452-3456.
1. Mehta D, Mehta C, Bansal S, Singla S, Tangri N. Flexible bronchoscopic removal of a three piece foreign body from a child’s bronchus. Australas Med J. 2012;5(4):227-230.
2. Mercado JA, Rodríguez W. Occult aspiration of a chicken wishbone as a cause of hemoptysis. P R Health Sci J. 1999;18(1):71-73.
3. Robles-Arias CM, Campos-Santiago Z, Vega MT, Rosa-Cruz F, Rodríguez-Cintrón W. Aspiration of a dental tool during a crown placement procedure. Fed Pract. 2014;31(6):12-14.
4. Blanco-Ramos M, Botana-Rial M, García-Fontán E, Fernández-Villar A, Gallas-Torreira M. Update in the extraction of airway foreign bodies in adults. J Thorac Dis. 2016;8(11):3452-3456.
A Forgotten Cause of Cardiac Tamponade
Purulent pericarditis is an infection within the pericardial space rarely seen in the modern antibiotic era. Most cases are secondary to another infectious process of bacterial, viral, fungal, or parasitic origin.1,2 Predisposing factors include malignancy, chronic kidney disease, immunosuppression, diabetes mellitus, and alcohol misuse disorder.1 Although purulent pericarditis has been described extensively in the literature, it is a challenging diagnosis if it is not initially considered within the differential diagnosis repertoire.1-4 Most authors agree that this may be because it has become an infrequent diagnosis.1,2 In addition, purulent pericarditis may have an atypical presentation when compared with a classic case of pericarditis.2,3 The authors believe that this forgotten entity will be revisited through this case.
Case Presentation
A 66-year-old-man was transferred to Veterans Affairs Caribbean Healthcare System (VACHS) from a community hospital with a diagnosis of community-acquired pneumonia (CAP) and bilateral pleural effusions. Four days prior to arrival at the community hospital, the patient had developed diffuse, watery diarrhea, which resolved in 3 days. After resolution of diarrhea, he began experiencing shortness of breath on exertion that progressed to onset at rest. The patient reported no fever, chills, nausea, vomiting, cough, or contact with others who were not healthy. He had a history of alcohol misuse without liver cirrhosis and reported no chronic diseases or use of medications. The patient had no history of tuberculosis exposure or pneumococcal vaccination, and had a negative interferon gamma release assay.
On admission to the community hospital, the patient was treated for CAP with ceftriaxone and azithromycin. On hospital day 3, the patient developed hypoxemia and an altered mental status. He was started on supplemental oxygen and transferred to the intensive care unit (ICU). Antibiotic therapy consequently was changed to levofloxacin and meropenem. However, no clinical improvement was noted on the following days.
On hospital day 7, the patient developed acute respiratory failure that required mechanical ventilation while being transferred to VACHS via air ambulance. His vital signs on arrival were the following: temperature, 97° F; heart rate, 86 beats/min; blood pressure, 103/61 mm Hg; respiratory rate, 14 breaths/min and SaO2 of 97%, measured while he breathed supplemental oxygen at an FiO2 of 0.4. 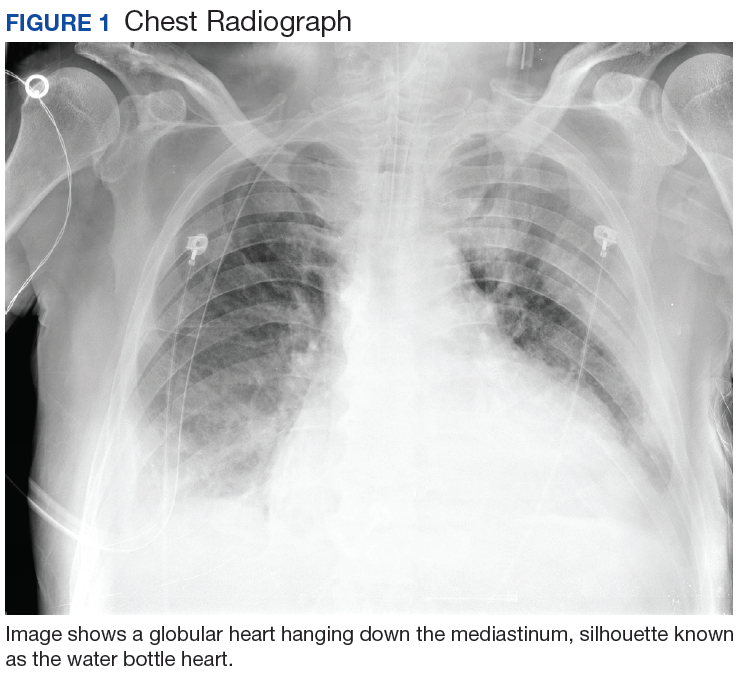
Hours after arrival, the patient developed sinus tachycardia and hypotension. A bedside 2D echocardiogram demonstrated a large pericardial effusion with diastolic collapse of the right atrium (Figure 2). 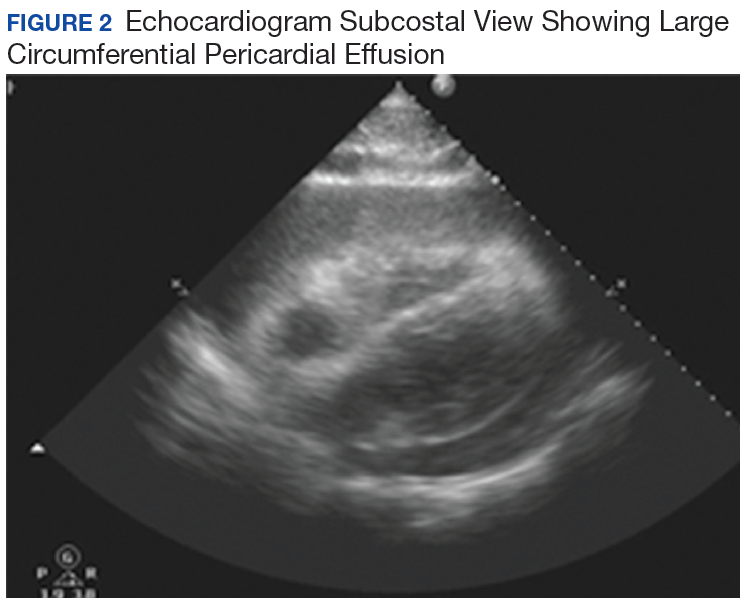
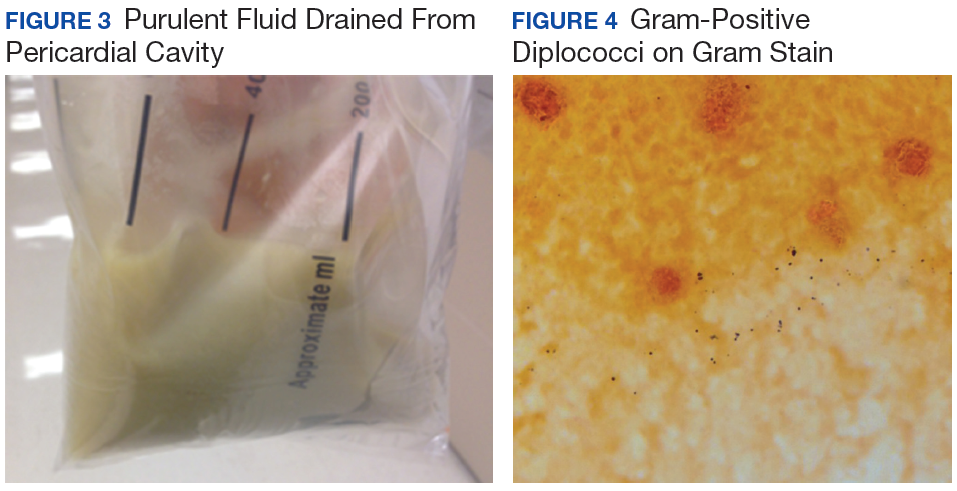
The patient’s clinical condition improved following drainage of pericardial fluid, with no further need for inotropic support. Antibiotic therapy was changed to vancomycin and meropenem. Initial microbiologic samples from pericardial fluid demonstrated Gram-positive diplococci, suggestive of Streptococcus pneumoniae (S pneumoniae) (Figure 4). Other diagnostic pericardial fluid test results included: WBC count 25,330 cmm, with 99% neutrophils and 1% lymphocytes; total protein, 3.8 mg/dL; glucose, < 2.0 mg/dL,LDH, > 2,500 U/L, potassium hydroxide preparation. The tests found no fungus, and the acid fast bacilli smear revealed no Bacillus. However, the pericardial fluid culture failed to demonstrate growth of any organism. Blood cultures also were negative.
The patient underwent anterior thoracotomy with partial pericardiectomy, and a pericardial tube was left in place connected to drainage. During the procedure, an abundant amount of fibrinous tissue was evacuated from the pericardial space (Figure 5). 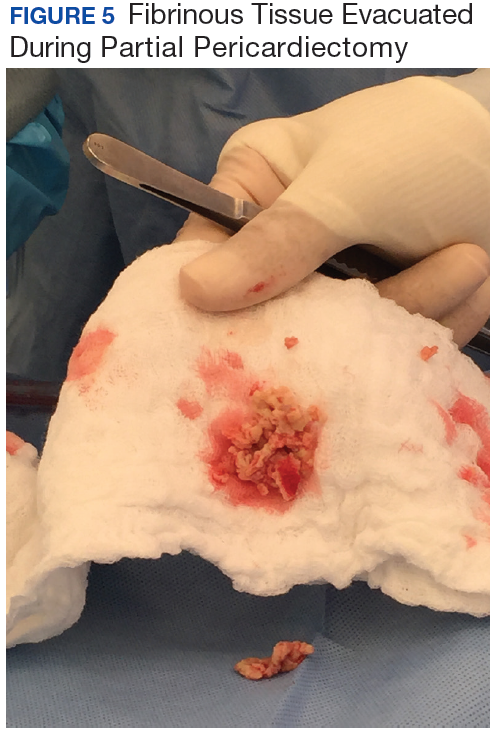
The patient was extubated, pericardial and pleural tubes were removed, and he was transferred to the internal medicine ward 24 days after admission to the ICU. He received in-patient physical rehabilitation while completing a 6-week course of IV antibiotics (vancomycin and meropenem). After completion of therapy, the patient received the pneumococcal polysaccharide vaccination, and an echocardiography was repeated. No significant re-accumulation of pericardial effusion or constrictive pattern was evidenced. The patient was discharged to his out-of-state home, and follow-up was consequently lost.
Discussion
Purulent pericarditis is an infection localized within the pericardial space. Most cases are secondary to an infectious process elsewhere, which could be of bacterial, viral, fungal, or parasitic etiology.1 Five mechanisms could lead the infecting organism to infect the pericardial space; contiguous spread from intrathoracic site, hematogenous spread, extension from myocardial site, perforating injury or surgery, and extension from a subdiaphragmatic site.1 Predisposing factors for the development of this condition include malignancy, chronic kidney disease, immunosuppression, diabetes mellitus, and alcohol misuse. Pericarditis is an infection localized within the pericardial space.
Purulent pericarditis has become a rare entity in the antibiotic era.2 Prior to the development of antibiotics, most cases were secondary to S pneumoniae.1,2,5,6 As per Cilloniz and colleagues, about 40% to 50% of all cases of purulent pericarditis are caused by Gram-positive bacteria, mostly S pneumoniae.5 In this case study, bacterial culture did not reveal growth of an organism—most likely because the patient had received antibiotics elsewhere. However, Gram-positive cocci were seen within the initial pericardial aspirate. This organism was suspected to have spread contiguously from a pulmonary focus, which also led to pleural effusions.
Since the patient in this case study had no history of thoracic surgery, malignancy, or other immunosuppression, the patient’s history of alcohol misuse was the only predisposing factor for development of purulent pericarditis. Contrary to the common presentation of pericarditis, purulent pericarditis may not have the common clinical findings, such as chest pain, pericardial friction rub, and distended neck veins.2,3 Furthermore, according to Parikh and colleagues, about 35% of affected patients may have a normal electrocardiogram.2 Hence, the diagnosis of purulent pericarditis often is missed because the classic signs of pericarditis are often absent, and other nonspecific symptoms are attributed to initial underlying infection.7
A high index of suspicion is needed to diagnose purulent pericarditis. Once a diagnosis is made, initial treatment should consist of prompt drainage of pericardial fluid combined with systemic antibiotic therapy. Vancomycin and a third-generation cephalosporin may be started empirically until results of pericardial fluid cultures become available.3 Drainage can be achieved by pericardiocentesis, pericardiotomy, or pericardiectomy (partial or total).1 In cases of hemodynamic instability due to cardiac tamponade, sonographically guided pericardiocentesis should be undertaken and an indwelling pericardial catheter left in place.1 Although this is the simplest and fastest method of evacuation, it may not be effective when dealing with thick, fibrinous fluid. In such cases, intrapericardial fibrinolysis may be considered. This approach may be undertaken early in the process, after drainage insertion, or as salvage therapy, when there has been incomplete evacuation of purulent material or open surgical drainage is not available.
Streptokinase, urokinase, and tissue plasminogen activator have been used for intrapericadial fibrinolysis.1 However, there is no definite data on dosage or frequency at which these medications should be administered. No matter the therapeutic approach, effective drainage of the pericardial fluid is crucial to avoid the development of pericardial constriction. Constrictive pericarditis occurs when fibrosis and adhesions create a dense pericardium that encases the heart. This causes impaired ventricular filling that can lead eventually to heart failure.4 Pericardiectomy is the definitive treatment for constrictive pericarditis.
Conclusion
Although purulent pericarditis has become a rare diagnosis since the development of antibiotics, knowledge of how to identify it is essential since mortality reaches 100% if the diagnosis is missed.4 Even when the condition is promptly diagnosed and treated, mortality is 40%, mainly due to cardiac tamponade, septic shock, or constriction.1 The case presented here illustrates the clinical features associated with this condition. Knowing these features can translate in a successful patient outcome.
1. Ferreira dos Santos L, Moreira D, Ribeiro P, et al. Purulent pericarditis: a rare diagnosis [in Portuguese]. Rev Port Cardiol. 2013;32(9):721-727.
2. Parikh SV, Memon N, Echols M, Shah J, McGuire DK, Keeley EC. Purulent pericarditis: report of 2 cases and review of the literature. Medicine (Baltimore). 2009;88(1):52–65.
3. Go C, Asnis DS, Saltzman H. Pneumococcal pericarditis since 1980. Clin Infect Dis. 1998;27(5):1338-1340.
4. Wada A, Craft J, Mazzaferri EL. Purulent pericarditis leading to constriction. Cardiol Res. 2014;5(6):188-190.
5. Cillóniz C, Rangel E, Barlascini C, Piroddi IMG, Torres A, Nicolini A. Streptococcus pneumoniae-associated pneumonia complicated by purulent pericarditis: case series [in English, Portuguese]. J Bras Pneumol. 2015;41(4):389-394.
6. Saenz RE, Sanders CV, Aldridge KE, Patel MM. Purulent pericarditis with associated cardiac tamponade caused by a Streptococcus pneumoniae strain highly resistant to penicillin, cefotaxime, and ceftriaxone. Clin Infect Dis. 1998;26(3):762–763.
7. Sagristà-Sauleda J, Barrabés JA, Permanyer-Miralda G, Soler-Soler J. Purulent pericarditis: review of a 20-year experience in a general hospital. J Am Coll Cardiol. 1993; 22(6):1661-1665.
Purulent pericarditis is an infection within the pericardial space rarely seen in the modern antibiotic era. Most cases are secondary to another infectious process of bacterial, viral, fungal, or parasitic origin.1,2 Predisposing factors include malignancy, chronic kidney disease, immunosuppression, diabetes mellitus, and alcohol misuse disorder.1 Although purulent pericarditis has been described extensively in the literature, it is a challenging diagnosis if it is not initially considered within the differential diagnosis repertoire.1-4 Most authors agree that this may be because it has become an infrequent diagnosis.1,2 In addition, purulent pericarditis may have an atypical presentation when compared with a classic case of pericarditis.2,3 The authors believe that this forgotten entity will be revisited through this case.
Case Presentation
A 66-year-old-man was transferred to Veterans Affairs Caribbean Healthcare System (VACHS) from a community hospital with a diagnosis of community-acquired pneumonia (CAP) and bilateral pleural effusions. Four days prior to arrival at the community hospital, the patient had developed diffuse, watery diarrhea, which resolved in 3 days. After resolution of diarrhea, he began experiencing shortness of breath on exertion that progressed to onset at rest. The patient reported no fever, chills, nausea, vomiting, cough, or contact with others who were not healthy. He had a history of alcohol misuse without liver cirrhosis and reported no chronic diseases or use of medications. The patient had no history of tuberculosis exposure or pneumococcal vaccination, and had a negative interferon gamma release assay.
On admission to the community hospital, the patient was treated for CAP with ceftriaxone and azithromycin. On hospital day 3, the patient developed hypoxemia and an altered mental status. He was started on supplemental oxygen and transferred to the intensive care unit (ICU). Antibiotic therapy consequently was changed to levofloxacin and meropenem. However, no clinical improvement was noted on the following days.
On hospital day 7, the patient developed acute respiratory failure that required mechanical ventilation while being transferred to VACHS via air ambulance. His vital signs on arrival were the following: temperature, 97° F; heart rate, 86 beats/min; blood pressure, 103/61 mm Hg; respiratory rate, 14 breaths/min and SaO2 of 97%, measured while he breathed supplemental oxygen at an FiO2 of 0.4.
Hours after arrival, the patient developed sinus tachycardia and hypotension. A bedside 2D echocardiogram demonstrated a large pericardial effusion with diastolic collapse of the right atrium (Figure 2).
The patient’s clinical condition improved following drainage of pericardial fluid, with no further need for inotropic support. Antibiotic therapy was changed to vancomycin and meropenem. Initial microbiologic samples from pericardial fluid demonstrated Gram-positive diplococci, suggestive of Streptococcus pneumoniae (S pneumoniae) (Figure 4). Other diagnostic pericardial fluid test results included: WBC count 25,330 cmm, with 99% neutrophils and 1% lymphocytes; total protein, 3.8 mg/dL; glucose, < 2.0 mg/dL,LDH, > 2,500 U/L, potassium hydroxide preparation. The tests found no fungus, and the acid fast bacilli smear revealed no Bacillus. However, the pericardial fluid culture failed to demonstrate growth of any organism. Blood cultures also were negative.
The patient underwent anterior thoracotomy with partial pericardiectomy, and a pericardial tube was left in place connected to drainage. During the procedure, an abundant amount of fibrinous tissue was evacuated from the pericardial space (Figure 5).
The patient was extubated, pericardial and pleural tubes were removed, and he was transferred to the internal medicine ward 24 days after admission to the ICU. He received in-patient physical rehabilitation while completing a 6-week course of IV antibiotics (vancomycin and meropenem). After completion of therapy, the patient received the pneumococcal polysaccharide vaccination, and an echocardiography was repeated. No significant re-accumulation of pericardial effusion or constrictive pattern was evidenced. The patient was discharged to his out-of-state home, and follow-up was consequently lost.
Discussion
Purulent pericarditis is an infection localized within the pericardial space. Most cases are secondary to an infectious process elsewhere, which could be of bacterial, viral, fungal, or parasitic etiology.1 Five mechanisms could lead the infecting organism to infect the pericardial space; contiguous spread from intrathoracic site, hematogenous spread, extension from myocardial site, perforating injury or surgery, and extension from a subdiaphragmatic site.1 Predisposing factors for the development of this condition include malignancy, chronic kidney disease, immunosuppression, diabetes mellitus, and alcohol misuse. Pericarditis is an infection localized within the pericardial space.
Purulent pericarditis has become a rare entity in the antibiotic era.2 Prior to the development of antibiotics, most cases were secondary to S pneumoniae.1,2,5,6 As per Cilloniz and colleagues, about 40% to 50% of all cases of purulent pericarditis are caused by Gram-positive bacteria, mostly S pneumoniae.5 In this case study, bacterial culture did not reveal growth of an organism—most likely because the patient had received antibiotics elsewhere. However, Gram-positive cocci were seen within the initial pericardial aspirate. This organism was suspected to have spread contiguously from a pulmonary focus, which also led to pleural effusions.
Since the patient in this case study had no history of thoracic surgery, malignancy, or other immunosuppression, the patient’s history of alcohol misuse was the only predisposing factor for development of purulent pericarditis. Contrary to the common presentation of pericarditis, purulent pericarditis may not have the common clinical findings, such as chest pain, pericardial friction rub, and distended neck veins.2,3 Furthermore, according to Parikh and colleagues, about 35% of affected patients may have a normal electrocardiogram.2 Hence, the diagnosis of purulent pericarditis often is missed because the classic signs of pericarditis are often absent, and other nonspecific symptoms are attributed to initial underlying infection.7
A high index of suspicion is needed to diagnose purulent pericarditis. Once a diagnosis is made, initial treatment should consist of prompt drainage of pericardial fluid combined with systemic antibiotic therapy. Vancomycin and a third-generation cephalosporin may be started empirically until results of pericardial fluid cultures become available.3 Drainage can be achieved by pericardiocentesis, pericardiotomy, or pericardiectomy (partial or total).1 In cases of hemodynamic instability due to cardiac tamponade, sonographically guided pericardiocentesis should be undertaken and an indwelling pericardial catheter left in place.1 Although this is the simplest and fastest method of evacuation, it may not be effective when dealing with thick, fibrinous fluid. In such cases, intrapericardial fibrinolysis may be considered. This approach may be undertaken early in the process, after drainage insertion, or as salvage therapy, when there has been incomplete evacuation of purulent material or open surgical drainage is not available.
Streptokinase, urokinase, and tissue plasminogen activator have been used for intrapericadial fibrinolysis.1 However, there is no definite data on dosage or frequency at which these medications should be administered. No matter the therapeutic approach, effective drainage of the pericardial fluid is crucial to avoid the development of pericardial constriction. Constrictive pericarditis occurs when fibrosis and adhesions create a dense pericardium that encases the heart. This causes impaired ventricular filling that can lead eventually to heart failure.4 Pericardiectomy is the definitive treatment for constrictive pericarditis.
Conclusion
Although purulent pericarditis has become a rare diagnosis since the development of antibiotics, knowledge of how to identify it is essential since mortality reaches 100% if the diagnosis is missed.4 Even when the condition is promptly diagnosed and treated, mortality is 40%, mainly due to cardiac tamponade, septic shock, or constriction.1 The case presented here illustrates the clinical features associated with this condition. Knowing these features can translate in a successful patient outcome.
Purulent pericarditis is an infection within the pericardial space rarely seen in the modern antibiotic era. Most cases are secondary to another infectious process of bacterial, viral, fungal, or parasitic origin.1,2 Predisposing factors include malignancy, chronic kidney disease, immunosuppression, diabetes mellitus, and alcohol misuse disorder.1 Although purulent pericarditis has been described extensively in the literature, it is a challenging diagnosis if it is not initially considered within the differential diagnosis repertoire.1-4 Most authors agree that this may be because it has become an infrequent diagnosis.1,2 In addition, purulent pericarditis may have an atypical presentation when compared with a classic case of pericarditis.2,3 The authors believe that this forgotten entity will be revisited through this case.
Case Presentation
A 66-year-old-man was transferred to Veterans Affairs Caribbean Healthcare System (VACHS) from a community hospital with a diagnosis of community-acquired pneumonia (CAP) and bilateral pleural effusions. Four days prior to arrival at the community hospital, the patient had developed diffuse, watery diarrhea, which resolved in 3 days. After resolution of diarrhea, he began experiencing shortness of breath on exertion that progressed to onset at rest. The patient reported no fever, chills, nausea, vomiting, cough, or contact with others who were not healthy. He had a history of alcohol misuse without liver cirrhosis and reported no chronic diseases or use of medications. The patient had no history of tuberculosis exposure or pneumococcal vaccination, and had a negative interferon gamma release assay.
On admission to the community hospital, the patient was treated for CAP with ceftriaxone and azithromycin. On hospital day 3, the patient developed hypoxemia and an altered mental status. He was started on supplemental oxygen and transferred to the intensive care unit (ICU). Antibiotic therapy consequently was changed to levofloxacin and meropenem. However, no clinical improvement was noted on the following days.
On hospital day 7, the patient developed acute respiratory failure that required mechanical ventilation while being transferred to VACHS via air ambulance. His vital signs on arrival were the following: temperature, 97° F; heart rate, 86 beats/min; blood pressure, 103/61 mm Hg; respiratory rate, 14 breaths/min and SaO2 of 97%, measured while he breathed supplemental oxygen at an FiO2 of 0.4.
Hours after arrival, the patient developed sinus tachycardia and hypotension. A bedside 2D echocardiogram demonstrated a large pericardial effusion with diastolic collapse of the right atrium (Figure 2).
The patient’s clinical condition improved following drainage of pericardial fluid, with no further need for inotropic support. Antibiotic therapy was changed to vancomycin and meropenem. Initial microbiologic samples from pericardial fluid demonstrated Gram-positive diplococci, suggestive of Streptococcus pneumoniae (S pneumoniae) (Figure 4). Other diagnostic pericardial fluid test results included: WBC count 25,330 cmm, with 99% neutrophils and 1% lymphocytes; total protein, 3.8 mg/dL; glucose, < 2.0 mg/dL,LDH, > 2,500 U/L, potassium hydroxide preparation. The tests found no fungus, and the acid fast bacilli smear revealed no Bacillus. However, the pericardial fluid culture failed to demonstrate growth of any organism. Blood cultures also were negative.
The patient underwent anterior thoracotomy with partial pericardiectomy, and a pericardial tube was left in place connected to drainage. During the procedure, an abundant amount of fibrinous tissue was evacuated from the pericardial space (Figure 5).
The patient was extubated, pericardial and pleural tubes were removed, and he was transferred to the internal medicine ward 24 days after admission to the ICU. He received in-patient physical rehabilitation while completing a 6-week course of IV antibiotics (vancomycin and meropenem). After completion of therapy, the patient received the pneumococcal polysaccharide vaccination, and an echocardiography was repeated. No significant re-accumulation of pericardial effusion or constrictive pattern was evidenced. The patient was discharged to his out-of-state home, and follow-up was consequently lost.
Discussion
Purulent pericarditis is an infection localized within the pericardial space. Most cases are secondary to an infectious process elsewhere, which could be of bacterial, viral, fungal, or parasitic etiology.1 Five mechanisms could lead the infecting organism to infect the pericardial space; contiguous spread from intrathoracic site, hematogenous spread, extension from myocardial site, perforating injury or surgery, and extension from a subdiaphragmatic site.1 Predisposing factors for the development of this condition include malignancy, chronic kidney disease, immunosuppression, diabetes mellitus, and alcohol misuse. Pericarditis is an infection localized within the pericardial space.
Purulent pericarditis has become a rare entity in the antibiotic era.2 Prior to the development of antibiotics, most cases were secondary to S pneumoniae.1,2,5,6 As per Cilloniz and colleagues, about 40% to 50% of all cases of purulent pericarditis are caused by Gram-positive bacteria, mostly S pneumoniae.5 In this case study, bacterial culture did not reveal growth of an organism—most likely because the patient had received antibiotics elsewhere. However, Gram-positive cocci were seen within the initial pericardial aspirate. This organism was suspected to have spread contiguously from a pulmonary focus, which also led to pleural effusions.
Since the patient in this case study had no history of thoracic surgery, malignancy, or other immunosuppression, the patient’s history of alcohol misuse was the only predisposing factor for development of purulent pericarditis. Contrary to the common presentation of pericarditis, purulent pericarditis may not have the common clinical findings, such as chest pain, pericardial friction rub, and distended neck veins.2,3 Furthermore, according to Parikh and colleagues, about 35% of affected patients may have a normal electrocardiogram.2 Hence, the diagnosis of purulent pericarditis often is missed because the classic signs of pericarditis are often absent, and other nonspecific symptoms are attributed to initial underlying infection.7
A high index of suspicion is needed to diagnose purulent pericarditis. Once a diagnosis is made, initial treatment should consist of prompt drainage of pericardial fluid combined with systemic antibiotic therapy. Vancomycin and a third-generation cephalosporin may be started empirically until results of pericardial fluid cultures become available.3 Drainage can be achieved by pericardiocentesis, pericardiotomy, or pericardiectomy (partial or total).1 In cases of hemodynamic instability due to cardiac tamponade, sonographically guided pericardiocentesis should be undertaken and an indwelling pericardial catheter left in place.1 Although this is the simplest and fastest method of evacuation, it may not be effective when dealing with thick, fibrinous fluid. In such cases, intrapericardial fibrinolysis may be considered. This approach may be undertaken early in the process, after drainage insertion, or as salvage therapy, when there has been incomplete evacuation of purulent material or open surgical drainage is not available.
Streptokinase, urokinase, and tissue plasminogen activator have been used for intrapericadial fibrinolysis.1 However, there is no definite data on dosage or frequency at which these medications should be administered. No matter the therapeutic approach, effective drainage of the pericardial fluid is crucial to avoid the development of pericardial constriction. Constrictive pericarditis occurs when fibrosis and adhesions create a dense pericardium that encases the heart. This causes impaired ventricular filling that can lead eventually to heart failure.4 Pericardiectomy is the definitive treatment for constrictive pericarditis.
Conclusion
Although purulent pericarditis has become a rare diagnosis since the development of antibiotics, knowledge of how to identify it is essential since mortality reaches 100% if the diagnosis is missed.4 Even when the condition is promptly diagnosed and treated, mortality is 40%, mainly due to cardiac tamponade, septic shock, or constriction.1 The case presented here illustrates the clinical features associated with this condition. Knowing these features can translate in a successful patient outcome.
1. Ferreira dos Santos L, Moreira D, Ribeiro P, et al. Purulent pericarditis: a rare diagnosis [in Portuguese]. Rev Port Cardiol. 2013;32(9):721-727.
2. Parikh SV, Memon N, Echols M, Shah J, McGuire DK, Keeley EC. Purulent pericarditis: report of 2 cases and review of the literature. Medicine (Baltimore). 2009;88(1):52–65.
3. Go C, Asnis DS, Saltzman H. Pneumococcal pericarditis since 1980. Clin Infect Dis. 1998;27(5):1338-1340.
4. Wada A, Craft J, Mazzaferri EL. Purulent pericarditis leading to constriction. Cardiol Res. 2014;5(6):188-190.
5. Cillóniz C, Rangel E, Barlascini C, Piroddi IMG, Torres A, Nicolini A. Streptococcus pneumoniae-associated pneumonia complicated by purulent pericarditis: case series [in English, Portuguese]. J Bras Pneumol. 2015;41(4):389-394.
6. Saenz RE, Sanders CV, Aldridge KE, Patel MM. Purulent pericarditis with associated cardiac tamponade caused by a Streptococcus pneumoniae strain highly resistant to penicillin, cefotaxime, and ceftriaxone. Clin Infect Dis. 1998;26(3):762–763.
7. Sagristà-Sauleda J, Barrabés JA, Permanyer-Miralda G, Soler-Soler J. Purulent pericarditis: review of a 20-year experience in a general hospital. J Am Coll Cardiol. 1993; 22(6):1661-1665.
1. Ferreira dos Santos L, Moreira D, Ribeiro P, et al. Purulent pericarditis: a rare diagnosis [in Portuguese]. Rev Port Cardiol. 2013;32(9):721-727.
2. Parikh SV, Memon N, Echols M, Shah J, McGuire DK, Keeley EC. Purulent pericarditis: report of 2 cases and review of the literature. Medicine (Baltimore). 2009;88(1):52–65.
3. Go C, Asnis DS, Saltzman H. Pneumococcal pericarditis since 1980. Clin Infect Dis. 1998;27(5):1338-1340.
4. Wada A, Craft J, Mazzaferri EL. Purulent pericarditis leading to constriction. Cardiol Res. 2014;5(6):188-190.
5. Cillóniz C, Rangel E, Barlascini C, Piroddi IMG, Torres A, Nicolini A. Streptococcus pneumoniae-associated pneumonia complicated by purulent pericarditis: case series [in English, Portuguese]. J Bras Pneumol. 2015;41(4):389-394.
6. Saenz RE, Sanders CV, Aldridge KE, Patel MM. Purulent pericarditis with associated cardiac tamponade caused by a Streptococcus pneumoniae strain highly resistant to penicillin, cefotaxime, and ceftriaxone. Clin Infect Dis. 1998;26(3):762–763.
7. Sagristà-Sauleda J, Barrabés JA, Permanyer-Miralda G, Soler-Soler J. Purulent pericarditis: review of a 20-year experience in a general hospital. J Am Coll Cardiol. 1993; 22(6):1661-1665.
Misleading Diagnosis of Idiopathic Pulmonary Fibrosis: A Clinical Concern
Sjogren syndrome (SS) is a chronic inflammatory autoimmune disorder characterized by lymphocytic infiltration of lacrimal and salivary glands causing sicca syndrome.¹ The disease can extend beyond the exocrine glands, and systemic manifestations, including vasculitis, lung, renal or neurologic involvement, can occur.² Lung disease associated with SS is more commonly seen in women aged ≥ 60 years. The most common symptoms include dry cough, chest pain, and dyspnea on exertion. Sjogren syndrome also may produce several respiratory complications, including bronchial hyperresponsiveness, bronchiolitis, bronchiectasis, pulmonary infections, pulmonary amyloidosis, pulmonary embolism, pulmonary hypertension, lymphomas, and interstitial lung diseases (ILD).² Although ILD typically occurs 5 to 10 years after the onset of SS, lung disease can precede SS.
Pulmonary involvement is associated with systemic manifestations, hypergammaglobulinemia, and anti-SSA and anti-SSB antibodies.² Laboratory tests that confirm a diagnosis of SS include antinuclear antibody (ANA), anti-Ro/SSA, and anti-La/SSB antibodies.² Pulmonary function test (PFT) results appear to reflect impairment of either the lung (restrictive syndrome) or airways (obstructive syndrome).² Imaging abnormalities may include ground-glass attenuation, subpleural small nodules, nonseptal linear opacities, interlobular septal thickening, bronchiectasis, and cysts.³ Therefore, many ILD cases show similar imaging and pathologic findings; nevertheless, they have identifiable etiology that are not idiopathic.
Case Presentation
A 67-year-old man with a medical history of hypertension, peripheral vascular disease, and keratoconjunctivitis sicca (treated with eye drops) developed progressive shortness of breath, dyspnea on exertion, and weight loss (40 pounds) over the course of 6 months. A pulmonary function test showed a restrictive abnormality with decreased diffusing capacity of the lungs for carbon monoxide (eFigure available online at www.fedprac.com). A chest computed tomography (CT) scan showed the presence of significant thickening of the interlobular septi that was more pronounced in the subpleural regions of the lungs and lower lobes, which was consistent with usual interstitial pneumonia. A chest X-ray conducted 4 months prior showed no significant acute cardiopulmonary abnormalities (Figure 1). An open lung wedge biopsy revealed chronic organizing pneumonia with mild interstitial chronic inflammation, smooth muscle hypertrophy, and honeycomb changes consistent with usual interstitial pneumonia. 
The patient had been diagnosed with idiopathic pulmonary fibrosis (IPF) by a private physician and started pirfenidone and oxygen therapy. Three months later the patient presented to the VA Caribbean Healthcare System in San Juan Puerto Rico when he developed an exacerbation of IPF. The patient reported having fever, chills, dry cough, night sweats, and marked shortness of breath. He was found hypoxemic (partial pressure of O2 was 50 mm Hg) and required a venturi mask set to 50% fractioned of inspired O2 to maintain a peripheral oxygen saturation around 90%. A chest X-ray showed decreased lung volume with bilateral interstitial and alveolar disease (Figure 2). Leukocytosis was present at 17×10-3/µl. The chest CT scan showed interval worsening of diffuse ground-glass airspace opacities and worsening of interstitial opacities; there 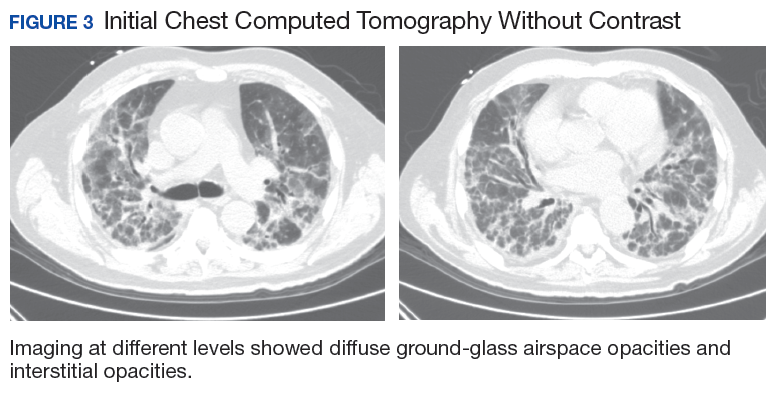
After careful clinical assessment (+ dry eyes) and radiographic pattern evaluation (diffuse bilateral interstitial and ground-glass opacities), the clinical diagnosis of IPF was queried after the patient’s rheumatologic workup came back positive for ANA and anti-Ro/SSA tests. Since the etiology of ILD was secondary to SS, pirfenidone was discontinued, and the patient was started on steroid therapy with subsequent marked clinical improvement. Parotid biopsy revealed the presence of inflammatory cells supporting the diagnosis of ILD associated to SS. The patient was discharged home on a tapering dose of steroids. Four months after therapy with steroids, a follow-up chest CT scan without contrast showed a chronic ILD with improved ground-glass opacities (Figure 4). The patient currently is in good health without oxygen supplementation.
Discussion
Diagnosis of SS is challenging, since it may mimic other conditions such as IPF. The most common type of SS-associated ILD is nonspecific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) can be visualized, as in this case study. Usual interstitial pneumonia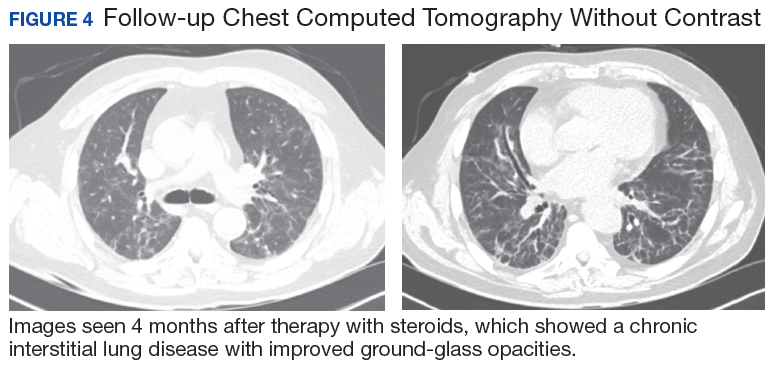
Diagnosing IPF cannot be solely based on a lung biopsy consistent with UIP. Appropriate diagnosis should consider the clinical presentation; PFT, laboratory findings (including rheumatologic workup), imaging (especially radiographic patterns), and biopsies. Moreover, the pathologic characteristic of IPF, which is UIP, can be found with other diseases, such as SS. Thus, it is important to make an accurate diagnosis to provide the appropriate treatment available. Patients with ILD associated with SS who have worsening symptoms, PFT, and radiographic abnormalities may be treated with oral prednisone (daily dose: 1 mg/kg).
Conclusion
This case highlights the importance of making an adequate diagnosis of ILD considering that available treatments differ for all possible etiologies other than IPF. This is a true clinical concern taking into account that many patients might be receiving inappropriate therapy for IPF diagnosis, as illustrated in the case study.
1. Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of primary Sjogren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171(6):632-638.
2. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren’s syndrome. Eur Respir Rev. 2016;25(140):110-123.
3. Koyama M, Johkoh T, Honda O, et al. Pulmonary involvement in primary Sjögren’s syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imaging. 2001;16(4):290-296.
4. Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014;23(133):308-319.
Sjogren syndrome (SS) is a chronic inflammatory autoimmune disorder characterized by lymphocytic infiltration of lacrimal and salivary glands causing sicca syndrome.¹ The disease can extend beyond the exocrine glands, and systemic manifestations, including vasculitis, lung, renal or neurologic involvement, can occur.² Lung disease associated with SS is more commonly seen in women aged ≥ 60 years. The most common symptoms include dry cough, chest pain, and dyspnea on exertion. Sjogren syndrome also may produce several respiratory complications, including bronchial hyperresponsiveness, bronchiolitis, bronchiectasis, pulmonary infections, pulmonary amyloidosis, pulmonary embolism, pulmonary hypertension, lymphomas, and interstitial lung diseases (ILD).² Although ILD typically occurs 5 to 10 years after the onset of SS, lung disease can precede SS.
Pulmonary involvement is associated with systemic manifestations, hypergammaglobulinemia, and anti-SSA and anti-SSB antibodies.² Laboratory tests that confirm a diagnosis of SS include antinuclear antibody (ANA), anti-Ro/SSA, and anti-La/SSB antibodies.² Pulmonary function test (PFT) results appear to reflect impairment of either the lung (restrictive syndrome) or airways (obstructive syndrome).² Imaging abnormalities may include ground-glass attenuation, subpleural small nodules, nonseptal linear opacities, interlobular septal thickening, bronchiectasis, and cysts.³ Therefore, many ILD cases show similar imaging and pathologic findings; nevertheless, they have identifiable etiology that are not idiopathic.
Case Presentation
A 67-year-old man with a medical history of hypertension, peripheral vascular disease, and keratoconjunctivitis sicca (treated with eye drops) developed progressive shortness of breath, dyspnea on exertion, and weight loss (40 pounds) over the course of 6 months. A pulmonary function test showed a restrictive abnormality with decreased diffusing capacity of the lungs for carbon monoxide (eFigure available online at www.fedprac.com). A chest computed tomography (CT) scan showed the presence of significant thickening of the interlobular septi that was more pronounced in the subpleural regions of the lungs and lower lobes, which was consistent with usual interstitial pneumonia. A chest X-ray conducted 4 months prior showed no significant acute cardiopulmonary abnormalities (Figure 1). An open lung wedge biopsy revealed chronic organizing pneumonia with mild interstitial chronic inflammation, smooth muscle hypertrophy, and honeycomb changes consistent with usual interstitial pneumonia.
The patient had been diagnosed with idiopathic pulmonary fibrosis (IPF) by a private physician and started pirfenidone and oxygen therapy. Three months later the patient presented to the VA Caribbean Healthcare System in San Juan Puerto Rico when he developed an exacerbation of IPF. The patient reported having fever, chills, dry cough, night sweats, and marked shortness of breath. He was found hypoxemic (partial pressure of O2 was 50 mm Hg) and required a venturi mask set to 50% fractioned of inspired O2 to maintain a peripheral oxygen saturation around 90%. A chest X-ray showed decreased lung volume with bilateral interstitial and alveolar disease (Figure 2). Leukocytosis was present at 17×10-3/µl. The chest CT scan showed interval worsening of diffuse ground-glass airspace opacities and worsening of interstitial opacities; there
After careful clinical assessment (+ dry eyes) and radiographic pattern evaluation (diffuse bilateral interstitial and ground-glass opacities), the clinical diagnosis of IPF was queried after the patient’s rheumatologic workup came back positive for ANA and anti-Ro/SSA tests. Since the etiology of ILD was secondary to SS, pirfenidone was discontinued, and the patient was started on steroid therapy with subsequent marked clinical improvement. Parotid biopsy revealed the presence of inflammatory cells supporting the diagnosis of ILD associated to SS. The patient was discharged home on a tapering dose of steroids. Four months after therapy with steroids, a follow-up chest CT scan without contrast showed a chronic ILD with improved ground-glass opacities (Figure 4). The patient currently is in good health without oxygen supplementation.
Discussion
Diagnosis of SS is challenging, since it may mimic other conditions such as IPF. The most common type of SS-associated ILD is nonspecific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) can be visualized, as in this case study. Usual interstitial pneumonia
Diagnosing IPF cannot be solely based on a lung biopsy consistent with UIP. Appropriate diagnosis should consider the clinical presentation; PFT, laboratory findings (including rheumatologic workup), imaging (especially radiographic patterns), and biopsies. Moreover, the pathologic characteristic of IPF, which is UIP, can be found with other diseases, such as SS. Thus, it is important to make an accurate diagnosis to provide the appropriate treatment available. Patients with ILD associated with SS who have worsening symptoms, PFT, and radiographic abnormalities may be treated with oral prednisone (daily dose: 1 mg/kg).
Conclusion
This case highlights the importance of making an adequate diagnosis of ILD considering that available treatments differ for all possible etiologies other than IPF. This is a true clinical concern taking into account that many patients might be receiving inappropriate therapy for IPF diagnosis, as illustrated in the case study.
Sjogren syndrome (SS) is a chronic inflammatory autoimmune disorder characterized by lymphocytic infiltration of lacrimal and salivary glands causing sicca syndrome.¹ The disease can extend beyond the exocrine glands, and systemic manifestations, including vasculitis, lung, renal or neurologic involvement, can occur.² Lung disease associated with SS is more commonly seen in women aged ≥ 60 years. The most common symptoms include dry cough, chest pain, and dyspnea on exertion. Sjogren syndrome also may produce several respiratory complications, including bronchial hyperresponsiveness, bronchiolitis, bronchiectasis, pulmonary infections, pulmonary amyloidosis, pulmonary embolism, pulmonary hypertension, lymphomas, and interstitial lung diseases (ILD).² Although ILD typically occurs 5 to 10 years after the onset of SS, lung disease can precede SS.
Pulmonary involvement is associated with systemic manifestations, hypergammaglobulinemia, and anti-SSA and anti-SSB antibodies.² Laboratory tests that confirm a diagnosis of SS include antinuclear antibody (ANA), anti-Ro/SSA, and anti-La/SSB antibodies.² Pulmonary function test (PFT) results appear to reflect impairment of either the lung (restrictive syndrome) or airways (obstructive syndrome).² Imaging abnormalities may include ground-glass attenuation, subpleural small nodules, nonseptal linear opacities, interlobular septal thickening, bronchiectasis, and cysts.³ Therefore, many ILD cases show similar imaging and pathologic findings; nevertheless, they have identifiable etiology that are not idiopathic.
Case Presentation
A 67-year-old man with a medical history of hypertension, peripheral vascular disease, and keratoconjunctivitis sicca (treated with eye drops) developed progressive shortness of breath, dyspnea on exertion, and weight loss (40 pounds) over the course of 6 months. A pulmonary function test showed a restrictive abnormality with decreased diffusing capacity of the lungs for carbon monoxide (eFigure available online at www.fedprac.com). A chest computed tomography (CT) scan showed the presence of significant thickening of the interlobular septi that was more pronounced in the subpleural regions of the lungs and lower lobes, which was consistent with usual interstitial pneumonia. A chest X-ray conducted 4 months prior showed no significant acute cardiopulmonary abnormalities (Figure 1). An open lung wedge biopsy revealed chronic organizing pneumonia with mild interstitial chronic inflammation, smooth muscle hypertrophy, and honeycomb changes consistent with usual interstitial pneumonia.
The patient had been diagnosed with idiopathic pulmonary fibrosis (IPF) by a private physician and started pirfenidone and oxygen therapy. Three months later the patient presented to the VA Caribbean Healthcare System in San Juan Puerto Rico when he developed an exacerbation of IPF. The patient reported having fever, chills, dry cough, night sweats, and marked shortness of breath. He was found hypoxemic (partial pressure of O2 was 50 mm Hg) and required a venturi mask set to 50% fractioned of inspired O2 to maintain a peripheral oxygen saturation around 90%. A chest X-ray showed decreased lung volume with bilateral interstitial and alveolar disease (Figure 2). Leukocytosis was present at 17×10-3/µl. The chest CT scan showed interval worsening of diffuse ground-glass airspace opacities and worsening of interstitial opacities; there
After careful clinical assessment (+ dry eyes) and radiographic pattern evaluation (diffuse bilateral interstitial and ground-glass opacities), the clinical diagnosis of IPF was queried after the patient’s rheumatologic workup came back positive for ANA and anti-Ro/SSA tests. Since the etiology of ILD was secondary to SS, pirfenidone was discontinued, and the patient was started on steroid therapy with subsequent marked clinical improvement. Parotid biopsy revealed the presence of inflammatory cells supporting the diagnosis of ILD associated to SS. The patient was discharged home on a tapering dose of steroids. Four months after therapy with steroids, a follow-up chest CT scan without contrast showed a chronic ILD with improved ground-glass opacities (Figure 4). The patient currently is in good health without oxygen supplementation.
Discussion
Diagnosis of SS is challenging, since it may mimic other conditions such as IPF. The most common type of SS-associated ILD is nonspecific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) can be visualized, as in this case study. Usual interstitial pneumonia
Diagnosing IPF cannot be solely based on a lung biopsy consistent with UIP. Appropriate diagnosis should consider the clinical presentation; PFT, laboratory findings (including rheumatologic workup), imaging (especially radiographic patterns), and biopsies. Moreover, the pathologic characteristic of IPF, which is UIP, can be found with other diseases, such as SS. Thus, it is important to make an accurate diagnosis to provide the appropriate treatment available. Patients with ILD associated with SS who have worsening symptoms, PFT, and radiographic abnormalities may be treated with oral prednisone (daily dose: 1 mg/kg).
Conclusion
This case highlights the importance of making an adequate diagnosis of ILD considering that available treatments differ for all possible etiologies other than IPF. This is a true clinical concern taking into account that many patients might be receiving inappropriate therapy for IPF diagnosis, as illustrated in the case study.
1. Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of primary Sjogren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171(6):632-638.
2. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren’s syndrome. Eur Respir Rev. 2016;25(140):110-123.
3. Koyama M, Johkoh T, Honda O, et al. Pulmonary involvement in primary Sjögren’s syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imaging. 2001;16(4):290-296.
4. Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014;23(133):308-319.
1. Ito I, Nagai S, Kitaichi M, et al. Pulmonary manifestations of primary Sjogren’s syndrome: a clinical, radiologic, and pathologic study. Am J Respir Crit Care Med. 2005;171(6):632-638.
2. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren’s syndrome. Eur Respir Rev. 2016;25(140):110-123.
3. Koyama M, Johkoh T, Honda O, et al. Pulmonary involvement in primary Sjögren’s syndrome: spectrum of pulmonary abnormalities and computed tomography findings in 60 patients. J Thorac Imaging. 2001;16(4):290-296.
4. Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014;23(133):308-319.
Unusual Congenital Pulmonary Anomaly in an Adult Patient With Dyspnea
Anatomic variations may result in abnormal return from the pulmonary veins to the right side of the heart. This group of congenital anomalies, also known as partial anomalous pulmonary venous return (PAPVR), may connect oxygenated blood from the pulmonary vein to a systemic vein before reaching the right atrium. The most common PAPVR is derived from the left upper pulmonary vein, which then connects to the left innominate vein and drains into the superior vena cava (SVC).
Scimitar syndrome is a rare PAPVR variant in which part of or the entire right lung is drained by the pulmonary vein into the inferior vena cava (IVC), giving the curvilinear dimension the appearance of a Middle Eastern sword (scimitar). The syndrome is frequently associated with other abnormalities, such as right lung hypoplasia and abnormal right lung lobation, dextroposition of the heart, right pulmonary artery hypoplasia, systemic arterial blood supply to the right lower lung from the infradiaphragmatic aorta, atrial septal defects of the secundum type, right-sided diaphragmatic hernia, and horseshoe lung.1,2 The syndrome was first described in 1836 by Cooper during an autopsy of an infant, and Dotter diagnosed the first symptomatic patient in 1949.3,4
Case Report
A 62-year-old man, former smoker (40 pack-year), with a past medical history of arterial hypertension and asthma visited the clinic, reporting exertional dyspnea. He also reported oppressive, retrosternally located exertional chest pain, 6/10 in intensity, of 3 minutes’ duration that radiated to the right chest and ameliorated with rest. Symptoms had occurred every other day for the past year. His physical exam was remarkable for central obesity. Lung auscultation was essentially clear. There was no jugular vein distention. The patient’s heart showed a regular rate and rhythm without evidence of murmurs or gallops. There was no evidence of leg edema or cyanosis. The patient’s resting oxygen saturation of 98% remained unchanged after exercise.
Related: Venous Thromboembolism Prophylaxis in Acutely Ill Veterans With Respiratory Disease
An electrocardiogram showed normal sinus rhythm with no ischemic changes. A pulmonary function test showed a forced expiratory volume (FEV1) of 1.44 L (61% of predicted), forced vital capacity (FVC) of 1.99 L (68% of predicted), and slow vital capacity (SVC) of 2.09 L (60% of predicted), with an FEV1/SVC ratio of 68% of predicted. These results suggested moderate-to-severe obstructive ventilatory impairment.
There was no response to bronchodilator therapy. Lung volumes were measured by plethysmography. The residual volume (RV), total lung capacity (TLC), and RV/TLC ratio were 2.57 L (147% of predicted), 4.66 L (88% of predicted), and 55%, respectively, suggesting severe air trapping. Diffusion lung capacity (DLCO) testing revealed 16.95 mL/min/mm Hg (73% of predicted) when corrected by hemoglobin and DLCO/alveolar volume of 4.97 mL/min/mm Hg/L (114% of predicted). This result was consistent with a mild reduction of gas transfer, which normalized when corrected by alveolar volume.
A posteroanterior chest radiograph image was remarkable for mediastinal shifting toward the right side, volume loss of the right lung, and evidence of a previous gunshot on the right chest wall (Figure 1). Previous chest imaging done in October 2009 showed an opacification of the right lower lung with indistinctness of the right cardiac border and partial obliteration of the right hemidiaphragm. The patient was treated with inhaled steroids and long- acting bronchodilators with partial improvement in dyspnea symptoms.
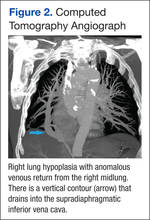
Myocardial perfusion imaging revealed scintigraphic evidence of heart rate-induced ischemia on the inferior and apical wall segments of the left ventricular myocardium. A transthoracic echocardiogram showed a very poor echocardiographic window. Left ventricular function seemed preserved. Transesophageal echocardiography was scheduled, but the patient missed the appointment.
Cardiac catheterization was only remarkable for 40% to 50% obstruction of the mid-left anterior descending artery, which did not explain the patient’s dyspnea or chest pain. Right side pressures were described as follows: right atrial mean, 10 mm Hg; right ventricle, 36/8 mm Hg; pulmonary artery, 33/16 mm Hg; pulmonary artery mean, 23 mm Hg; pulmonary capillary wedge pressure, 12 mm Hg; and a mean arterial pressure of 100 mm Hg. He had a left ventricle ejection fraction of 60%.

Because images suggested dextroposition of the heart and right lung hypoplasia, a chest computed tomography (CT) and angiography were done (Figure 2). The images showed hypoplasia of the right lung field with an anomalous venous return from the right midlung, having a vertical contour that drained into the supradiaphragmatic IVC. In addition, CT reconstruction demarcated the last mentioned contour draining into the IVC, consistent with scimitar syndrome (Figure 3). The patient was treated conservatively due to age, optimizing therapy for obstructive lung and cardiovascular disease.
Discussion
Partial anomalous pulmonary venous return is a relatively uncommon congenital anomaly, accounting for 0.5% to 1% of congenital heart disease.4,5 The characteristic abnormality is PAPVR of part of or the entire right lung to the IVC, either below the diaphragm or at the junction of the IVC and the right atrium. The rare combination (3%-5%) of an association of PAPVR, right lung hypoplasia, and dextroposition of the heart is designated scimitar syndrome. The scimitar vein sign is a characteristic chest roentgenographic finding of a crescentlike shadow in the right lower lung field where the curvilinear dimension gives the appearance of a scimitar sword.
Related: Another Reason Not to Smoke: Acute Eosinophilic Pneunomia
Normally, the pulmonary veins from the right and left lung carry oxygenated blood into the left atrium, then to the left ventricle, and then flowing out systemically. The SCV and IVC return the deoxygenated blood from the body system to the right atrium. From the right atrium, blood flows into the right ventricle, and then through pulmonary arteries, reaching the lungs where oxygenation occurs. In this syndrome, a left-to-right shunt is established when the anomalous pulmonary vein drains blood from the right lung into the IVC, resulting in an increased risk of developing right ventricular failure due to long-standing right ventricular volume overload.
Presentation and Diagnosis
There are two clinical presentations of scimitar syndrome: infantile and pediatric/adult. Infantile scimitar syndrome has a clinical presentation of tachypnea and heart failure within the first 2 months of life, with a high mortality rate. The pediatric/adult type is milder and frequently asymptomatic, and the diagnosis is usually incidental after performing an imaging study. Scimitar vein sign appears in 70% of the noninfantile cases, and lung hypoplasia is less severe. A spirometry may reveal mild deficits in vital capacity and FEV1. An electrocardiogram may show right ventricular hypertrophy.
Cardiac catheterization is required to confirm the diagnosis. Additionally, this procedure can help in the assessment of the pulmonary venous drainage course, pulmonary artery anatomy and pressure, scimitar vein stenosis, and presence of left-to-right shunt or other cardiac anomalies, if present. Other modalities have been suggested as alternative methods for diagnosing this condition, including the use of coronary CT and 3D echocardiography.6,7 However, these diagnostic tests are not available in all facilities and are very costly.
Treatment and Prognosis
Vida and colleagues conducted a multicentric study for the European Congenital Heart Surgeons Association on scimitar syndrome.8 Data were collected from 1997 to 2007 for 68 patients who underwent a surgical procedure. A total of 11 patients were categorized as late onset, and when compared with the infantile category, they had fewer postoperatory complications, hospital mortality, late mortality, and were less likely to develop pulmonary hypertension. Both pulmonary stenosis and pulmonary hypertension were linked with poor outcomes. It seems the younger the patient (infantile), the higher the possibility of complications and mortality. Adults who are incidentally diagnosed have a better outcome if asymptomatic. Findings such as hypoplastic lungs may predispose these patients to developing recurrent pneumonias.8,9
Related: Prevention of Venous Thromboembolism After Total Joint Replacement: A Rivaroxaban Update
Dusenbery and colleagues documented in a cohort study the relationship between poor survival and other variables. Significant variables included age at presentation, nonatrial septal defect (non-ASD) congenital heart disease, left pulmonary vein stenosis, and pulmonary artery pressure (PAP) at the time of presentation. Predictors of survival for nonsurgical patients were directly related to PAP at presentation and absence of non-ASD congenital heart disease. If the patient’s PAP is less than half of the systemic pressure, the survival is near 100% at 5 years from initial presentation.9
Surgery is the definitive treatment for PAPVR. However, asymptomatic patients with PAPVR with small left-to-right shunt do not require intervention, as the defect has no significant clinical impact, and patients have a normal life expectancy without correction.10
Surgical treatment may be considered in the following circumstances:
- A hemodynamically significant left-to-right shunt (a ratio of pulmonary to systemic blood flow is greater than 2:1), often manifested as right ventricular volume overload
- Recurrent pulmonary infections
- Compression or obstruction of surrounding structures caused by the anomalous vein
- During surgical repair of other major cardiac lesions, depending on the surgical risk of a repair and level and degree of shunting
Surgical options include redirecting the venous drainage to the left atria, ligation/embolization of vascular supply to the sequestered lobe, and pneumonectomy. The procedure complications may include thrombosis of the scimitar vein, lung infarct, hemoptysis, and pulmonary hypertension, which may lead to resection of the lung.11,12 Surgical procedures are recommended in cases where the patient has had recurrent lung infections or a significant degree of shunting. Studies have compared both approaches, demonstrating a better outcome after 10 years for those patients who were medically treated considering the aforementioned surgical indications.
Conclusion
Scimitar syndrome is a rare but welldescribed constellation of cardiopulmonary anomalies, accounting for 0.5% to 1% of congenital heart disease. It is a variant of PAPVR, in which part of or even the entire right lung is drained by right pulmonary veins that connect anomalously to the IVC. Although a diagnosis can be made by chest radiograph, further imaging is needed to corroborate the diagnosis and demonstrate other associated abnormalities.
Additional tests have been described in the literature, but these procedures are not available in all facilities and may incur a higher cost. Therefore, CT angiographic reconstruction is an alternative, noninvasive procedure. Surgery is the definitive treatment; however, asymptomatic patients with PAPVR and small left-to-right shunt do not require intervention.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Cooper G. Case of malformation of the thoracic viscera: consisting of imperfect development of the right lung, and transposition of the heart. London Med Gaz. 1836;18:600-602.
2. Spentzouris G, Zandian A, Cesmebasi A, et al. The clinical anatomy of the inferior vena cava: a review of common congenital anomalies and the considerations for clinicians. Clin Anat. 2014;27(8):1234-1243.
3. Neill CA, Ferencz C, Sabiston DC, Sheldon H. The familial occurrence of hypoplastic right lung with systemic arterial supply and venous drainage “scimitar syndrome.” Bull Johns Hopkins Hosp. 1960;107:1-21.
4. Ward KE, Mullins CE. Anomalous pulmonary venous connections, pulmonary vein stenosis, and atresia of the common pulmonary vein. In: Garson A, Bricker JT, Fisher DJ, Neish SR, eds. The Science and Practice of Pediatric Cardiology. 2nd ed. Baltimore, MD: Williams and Wilkins; 1998:1431-1461.
5. Garcia-Barreto L, Vega W, Deliz R, Rodriguez W. Right hilar abnormality in a young man. Respiration. 1996;63(4):246-250.
6. Simmons DB, Menon RS, Pomeroy WL, Batts TC, Slim AM. An unusual presentation of scimitar syndrome in a military service member. Case Rep Vasc Med. 2013;2013:632402.
7. Palios J, Pernetz MA, Clements S Jr, Lerakis S. Three-dimensional echocardiography images showing anomalous pulmonary venous return in an adult with scimitar syndrome. Echocardiography. 2014;31(3):E103.
8. Vida VL, Padalino MA, Boccuzzo G, et al. Scimitar syndrome: a European Congenital Heart Surgeons Association (ECHSA) multicentric study. Circulation. 2010;122(12):1159-1166.
9. Dusenbery SM, Geva T, Seale A, et al. Outcome predictors and implications for management of scimitar syndrome. Am Heart J. 2013;165(5):770-777.
10. Sehgal A, Loughran-Fowlds A. Scimitar syndrome. Indian J Pediatr. 2005;72(3):249-251.
11. Najm HK, Williams WG, Coles JG, Rebeyka IM, Freedom RM. Scimitar syndrome: twenty years’ experience and results of repair. J Thorac Cardiovasc Surg. 1996;112(5):1161-1169.
12. Dupuis C, Charaf LA, Brevière GM, Abou P, Rémy-Jardin M, Helmius G. The “adult” form of the scimitar syndrome. Am J Cardiol. 1992;70(4):502-507.
Anatomic variations may result in abnormal return from the pulmonary veins to the right side of the heart. This group of congenital anomalies, also known as partial anomalous pulmonary venous return (PAPVR), may connect oxygenated blood from the pulmonary vein to a systemic vein before reaching the right atrium. The most common PAPVR is derived from the left upper pulmonary vein, which then connects to the left innominate vein and drains into the superior vena cava (SVC).
Scimitar syndrome is a rare PAPVR variant in which part of or the entire right lung is drained by the pulmonary vein into the inferior vena cava (IVC), giving the curvilinear dimension the appearance of a Middle Eastern sword (scimitar). The syndrome is frequently associated with other abnormalities, such as right lung hypoplasia and abnormal right lung lobation, dextroposition of the heart, right pulmonary artery hypoplasia, systemic arterial blood supply to the right lower lung from the infradiaphragmatic aorta, atrial septal defects of the secundum type, right-sided diaphragmatic hernia, and horseshoe lung.1,2 The syndrome was first described in 1836 by Cooper during an autopsy of an infant, and Dotter diagnosed the first symptomatic patient in 1949.3,4
Case Report
A 62-year-old man, former smoker (40 pack-year), with a past medical history of arterial hypertension and asthma visited the clinic, reporting exertional dyspnea. He also reported oppressive, retrosternally located exertional chest pain, 6/10 in intensity, of 3 minutes’ duration that radiated to the right chest and ameliorated with rest. Symptoms had occurred every other day for the past year. His physical exam was remarkable for central obesity. Lung auscultation was essentially clear. There was no jugular vein distention. The patient’s heart showed a regular rate and rhythm without evidence of murmurs or gallops. There was no evidence of leg edema or cyanosis. The patient’s resting oxygen saturation of 98% remained unchanged after exercise.
Related: Venous Thromboembolism Prophylaxis in Acutely Ill Veterans With Respiratory Disease
An electrocardiogram showed normal sinus rhythm with no ischemic changes. A pulmonary function test showed a forced expiratory volume (FEV1) of 1.44 L (61% of predicted), forced vital capacity (FVC) of 1.99 L (68% of predicted), and slow vital capacity (SVC) of 2.09 L (60% of predicted), with an FEV1/SVC ratio of 68% of predicted. These results suggested moderate-to-severe obstructive ventilatory impairment.
There was no response to bronchodilator therapy. Lung volumes were measured by plethysmography. The residual volume (RV), total lung capacity (TLC), and RV/TLC ratio were 2.57 L (147% of predicted), 4.66 L (88% of predicted), and 55%, respectively, suggesting severe air trapping. Diffusion lung capacity (DLCO) testing revealed 16.95 mL/min/mm Hg (73% of predicted) when corrected by hemoglobin and DLCO/alveolar volume of 4.97 mL/min/mm Hg/L (114% of predicted). This result was consistent with a mild reduction of gas transfer, which normalized when corrected by alveolar volume.
A posteroanterior chest radiograph image was remarkable for mediastinal shifting toward the right side, volume loss of the right lung, and evidence of a previous gunshot on the right chest wall (Figure 1). Previous chest imaging done in October 2009 showed an opacification of the right lower lung with indistinctness of the right cardiac border and partial obliteration of the right hemidiaphragm. The patient was treated with inhaled steroids and long- acting bronchodilators with partial improvement in dyspnea symptoms.
Myocardial perfusion imaging revealed scintigraphic evidence of heart rate-induced ischemia on the inferior and apical wall segments of the left ventricular myocardium. A transthoracic echocardiogram showed a very poor echocardiographic window. Left ventricular function seemed preserved. Transesophageal echocardiography was scheduled, but the patient missed the appointment.
Cardiac catheterization was only remarkable for 40% to 50% obstruction of the mid-left anterior descending artery, which did not explain the patient’s dyspnea or chest pain. Right side pressures were described as follows: right atrial mean, 10 mm Hg; right ventricle, 36/8 mm Hg; pulmonary artery, 33/16 mm Hg; pulmonary artery mean, 23 mm Hg; pulmonary capillary wedge pressure, 12 mm Hg; and a mean arterial pressure of 100 mm Hg. He had a left ventricle ejection fraction of 60%.
Because images suggested dextroposition of the heart and right lung hypoplasia, a chest computed tomography (CT) and angiography were done (Figure 2). The images showed hypoplasia of the right lung field with an anomalous venous return from the right midlung, having a vertical contour that drained into the supradiaphragmatic IVC. In addition, CT reconstruction demarcated the last mentioned contour draining into the IVC, consistent with scimitar syndrome (Figure 3). The patient was treated conservatively due to age, optimizing therapy for obstructive lung and cardiovascular disease.
Discussion
Partial anomalous pulmonary venous return is a relatively uncommon congenital anomaly, accounting for 0.5% to 1% of congenital heart disease.4,5 The characteristic abnormality is PAPVR of part of or the entire right lung to the IVC, either below the diaphragm or at the junction of the IVC and the right atrium. The rare combination (3%-5%) of an association of PAPVR, right lung hypoplasia, and dextroposition of the heart is designated scimitar syndrome. The scimitar vein sign is a characteristic chest roentgenographic finding of a crescentlike shadow in the right lower lung field where the curvilinear dimension gives the appearance of a scimitar sword.
Related: Another Reason Not to Smoke: Acute Eosinophilic Pneunomia
Normally, the pulmonary veins from the right and left lung carry oxygenated blood into the left atrium, then to the left ventricle, and then flowing out systemically. The SCV and IVC return the deoxygenated blood from the body system to the right atrium. From the right atrium, blood flows into the right ventricle, and then through pulmonary arteries, reaching the lungs where oxygenation occurs. In this syndrome, a left-to-right shunt is established when the anomalous pulmonary vein drains blood from the right lung into the IVC, resulting in an increased risk of developing right ventricular failure due to long-standing right ventricular volume overload.
Presentation and Diagnosis
There are two clinical presentations of scimitar syndrome: infantile and pediatric/adult. Infantile scimitar syndrome has a clinical presentation of tachypnea and heart failure within the first 2 months of life, with a high mortality rate. The pediatric/adult type is milder and frequently asymptomatic, and the diagnosis is usually incidental after performing an imaging study. Scimitar vein sign appears in 70% of the noninfantile cases, and lung hypoplasia is less severe. A spirometry may reveal mild deficits in vital capacity and FEV1. An electrocardiogram may show right ventricular hypertrophy.
Cardiac catheterization is required to confirm the diagnosis. Additionally, this procedure can help in the assessment of the pulmonary venous drainage course, pulmonary artery anatomy and pressure, scimitar vein stenosis, and presence of left-to-right shunt or other cardiac anomalies, if present. Other modalities have been suggested as alternative methods for diagnosing this condition, including the use of coronary CT and 3D echocardiography.6,7 However, these diagnostic tests are not available in all facilities and are very costly.
Treatment and Prognosis
Vida and colleagues conducted a multicentric study for the European Congenital Heart Surgeons Association on scimitar syndrome.8 Data were collected from 1997 to 2007 for 68 patients who underwent a surgical procedure. A total of 11 patients were categorized as late onset, and when compared with the infantile category, they had fewer postoperatory complications, hospital mortality, late mortality, and were less likely to develop pulmonary hypertension. Both pulmonary stenosis and pulmonary hypertension were linked with poor outcomes. It seems the younger the patient (infantile), the higher the possibility of complications and mortality. Adults who are incidentally diagnosed have a better outcome if asymptomatic. Findings such as hypoplastic lungs may predispose these patients to developing recurrent pneumonias.8,9
Related: Prevention of Venous Thromboembolism After Total Joint Replacement: A Rivaroxaban Update
Dusenbery and colleagues documented in a cohort study the relationship between poor survival and other variables. Significant variables included age at presentation, nonatrial septal defect (non-ASD) congenital heart disease, left pulmonary vein stenosis, and pulmonary artery pressure (PAP) at the time of presentation. Predictors of survival for nonsurgical patients were directly related to PAP at presentation and absence of non-ASD congenital heart disease. If the patient’s PAP is less than half of the systemic pressure, the survival is near 100% at 5 years from initial presentation.9
Surgery is the definitive treatment for PAPVR. However, asymptomatic patients with PAPVR with small left-to-right shunt do not require intervention, as the defect has no significant clinical impact, and patients have a normal life expectancy without correction.10
Surgical treatment may be considered in the following circumstances:
- A hemodynamically significant left-to-right shunt (a ratio of pulmonary to systemic blood flow is greater than 2:1), often manifested as right ventricular volume overload
- Recurrent pulmonary infections
- Compression or obstruction of surrounding structures caused by the anomalous vein
- During surgical repair of other major cardiac lesions, depending on the surgical risk of a repair and level and degree of shunting
Surgical options include redirecting the venous drainage to the left atria, ligation/embolization of vascular supply to the sequestered lobe, and pneumonectomy. The procedure complications may include thrombosis of the scimitar vein, lung infarct, hemoptysis, and pulmonary hypertension, which may lead to resection of the lung.11,12 Surgical procedures are recommended in cases where the patient has had recurrent lung infections or a significant degree of shunting. Studies have compared both approaches, demonstrating a better outcome after 10 years for those patients who were medically treated considering the aforementioned surgical indications.
Conclusion
Scimitar syndrome is a rare but welldescribed constellation of cardiopulmonary anomalies, accounting for 0.5% to 1% of congenital heart disease. It is a variant of PAPVR, in which part of or even the entire right lung is drained by right pulmonary veins that connect anomalously to the IVC. Although a diagnosis can be made by chest radiograph, further imaging is needed to corroborate the diagnosis and demonstrate other associated abnormalities.
Additional tests have been described in the literature, but these procedures are not available in all facilities and may incur a higher cost. Therefore, CT angiographic reconstruction is an alternative, noninvasive procedure. Surgery is the definitive treatment; however, asymptomatic patients with PAPVR and small left-to-right shunt do not require intervention.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Anatomic variations may result in abnormal return from the pulmonary veins to the right side of the heart. This group of congenital anomalies, also known as partial anomalous pulmonary venous return (PAPVR), may connect oxygenated blood from the pulmonary vein to a systemic vein before reaching the right atrium. The most common PAPVR is derived from the left upper pulmonary vein, which then connects to the left innominate vein and drains into the superior vena cava (SVC).
Scimitar syndrome is a rare PAPVR variant in which part of or the entire right lung is drained by the pulmonary vein into the inferior vena cava (IVC), giving the curvilinear dimension the appearance of a Middle Eastern sword (scimitar). The syndrome is frequently associated with other abnormalities, such as right lung hypoplasia and abnormal right lung lobation, dextroposition of the heart, right pulmonary artery hypoplasia, systemic arterial blood supply to the right lower lung from the infradiaphragmatic aorta, atrial septal defects of the secundum type, right-sided diaphragmatic hernia, and horseshoe lung.1,2 The syndrome was first described in 1836 by Cooper during an autopsy of an infant, and Dotter diagnosed the first symptomatic patient in 1949.3,4
Case Report
A 62-year-old man, former smoker (40 pack-year), with a past medical history of arterial hypertension and asthma visited the clinic, reporting exertional dyspnea. He also reported oppressive, retrosternally located exertional chest pain, 6/10 in intensity, of 3 minutes’ duration that radiated to the right chest and ameliorated with rest. Symptoms had occurred every other day for the past year. His physical exam was remarkable for central obesity. Lung auscultation was essentially clear. There was no jugular vein distention. The patient’s heart showed a regular rate and rhythm without evidence of murmurs or gallops. There was no evidence of leg edema or cyanosis. The patient’s resting oxygen saturation of 98% remained unchanged after exercise.
Related: Venous Thromboembolism Prophylaxis in Acutely Ill Veterans With Respiratory Disease
An electrocardiogram showed normal sinus rhythm with no ischemic changes. A pulmonary function test showed a forced expiratory volume (FEV1) of 1.44 L (61% of predicted), forced vital capacity (FVC) of 1.99 L (68% of predicted), and slow vital capacity (SVC) of 2.09 L (60% of predicted), with an FEV1/SVC ratio of 68% of predicted. These results suggested moderate-to-severe obstructive ventilatory impairment.
There was no response to bronchodilator therapy. Lung volumes were measured by plethysmography. The residual volume (RV), total lung capacity (TLC), and RV/TLC ratio were 2.57 L (147% of predicted), 4.66 L (88% of predicted), and 55%, respectively, suggesting severe air trapping. Diffusion lung capacity (DLCO) testing revealed 16.95 mL/min/mm Hg (73% of predicted) when corrected by hemoglobin and DLCO/alveolar volume of 4.97 mL/min/mm Hg/L (114% of predicted). This result was consistent with a mild reduction of gas transfer, which normalized when corrected by alveolar volume.
A posteroanterior chest radiograph image was remarkable for mediastinal shifting toward the right side, volume loss of the right lung, and evidence of a previous gunshot on the right chest wall (Figure 1). Previous chest imaging done in October 2009 showed an opacification of the right lower lung with indistinctness of the right cardiac border and partial obliteration of the right hemidiaphragm. The patient was treated with inhaled steroids and long- acting bronchodilators with partial improvement in dyspnea symptoms.
Myocardial perfusion imaging revealed scintigraphic evidence of heart rate-induced ischemia on the inferior and apical wall segments of the left ventricular myocardium. A transthoracic echocardiogram showed a very poor echocardiographic window. Left ventricular function seemed preserved. Transesophageal echocardiography was scheduled, but the patient missed the appointment.
Cardiac catheterization was only remarkable for 40% to 50% obstruction of the mid-left anterior descending artery, which did not explain the patient’s dyspnea or chest pain. Right side pressures were described as follows: right atrial mean, 10 mm Hg; right ventricle, 36/8 mm Hg; pulmonary artery, 33/16 mm Hg; pulmonary artery mean, 23 mm Hg; pulmonary capillary wedge pressure, 12 mm Hg; and a mean arterial pressure of 100 mm Hg. He had a left ventricle ejection fraction of 60%.
Because images suggested dextroposition of the heart and right lung hypoplasia, a chest computed tomography (CT) and angiography were done (Figure 2). The images showed hypoplasia of the right lung field with an anomalous venous return from the right midlung, having a vertical contour that drained into the supradiaphragmatic IVC. In addition, CT reconstruction demarcated the last mentioned contour draining into the IVC, consistent with scimitar syndrome (Figure 3). The patient was treated conservatively due to age, optimizing therapy for obstructive lung and cardiovascular disease.
Discussion
Partial anomalous pulmonary venous return is a relatively uncommon congenital anomaly, accounting for 0.5% to 1% of congenital heart disease.4,5 The characteristic abnormality is PAPVR of part of or the entire right lung to the IVC, either below the diaphragm or at the junction of the IVC and the right atrium. The rare combination (3%-5%) of an association of PAPVR, right lung hypoplasia, and dextroposition of the heart is designated scimitar syndrome. The scimitar vein sign is a characteristic chest roentgenographic finding of a crescentlike shadow in the right lower lung field where the curvilinear dimension gives the appearance of a scimitar sword.
Related: Another Reason Not to Smoke: Acute Eosinophilic Pneunomia
Normally, the pulmonary veins from the right and left lung carry oxygenated blood into the left atrium, then to the left ventricle, and then flowing out systemically. The SCV and IVC return the deoxygenated blood from the body system to the right atrium. From the right atrium, blood flows into the right ventricle, and then through pulmonary arteries, reaching the lungs where oxygenation occurs. In this syndrome, a left-to-right shunt is established when the anomalous pulmonary vein drains blood from the right lung into the IVC, resulting in an increased risk of developing right ventricular failure due to long-standing right ventricular volume overload.
Presentation and Diagnosis
There are two clinical presentations of scimitar syndrome: infantile and pediatric/adult. Infantile scimitar syndrome has a clinical presentation of tachypnea and heart failure within the first 2 months of life, with a high mortality rate. The pediatric/adult type is milder and frequently asymptomatic, and the diagnosis is usually incidental after performing an imaging study. Scimitar vein sign appears in 70% of the noninfantile cases, and lung hypoplasia is less severe. A spirometry may reveal mild deficits in vital capacity and FEV1. An electrocardiogram may show right ventricular hypertrophy.
Cardiac catheterization is required to confirm the diagnosis. Additionally, this procedure can help in the assessment of the pulmonary venous drainage course, pulmonary artery anatomy and pressure, scimitar vein stenosis, and presence of left-to-right shunt or other cardiac anomalies, if present. Other modalities have been suggested as alternative methods for diagnosing this condition, including the use of coronary CT and 3D echocardiography.6,7 However, these diagnostic tests are not available in all facilities and are very costly.
Treatment and Prognosis
Vida and colleagues conducted a multicentric study for the European Congenital Heart Surgeons Association on scimitar syndrome.8 Data were collected from 1997 to 2007 for 68 patients who underwent a surgical procedure. A total of 11 patients were categorized as late onset, and when compared with the infantile category, they had fewer postoperatory complications, hospital mortality, late mortality, and were less likely to develop pulmonary hypertension. Both pulmonary stenosis and pulmonary hypertension were linked with poor outcomes. It seems the younger the patient (infantile), the higher the possibility of complications and mortality. Adults who are incidentally diagnosed have a better outcome if asymptomatic. Findings such as hypoplastic lungs may predispose these patients to developing recurrent pneumonias.8,9
Related: Prevention of Venous Thromboembolism After Total Joint Replacement: A Rivaroxaban Update
Dusenbery and colleagues documented in a cohort study the relationship between poor survival and other variables. Significant variables included age at presentation, nonatrial septal defect (non-ASD) congenital heart disease, left pulmonary vein stenosis, and pulmonary artery pressure (PAP) at the time of presentation. Predictors of survival for nonsurgical patients were directly related to PAP at presentation and absence of non-ASD congenital heart disease. If the patient’s PAP is less than half of the systemic pressure, the survival is near 100% at 5 years from initial presentation.9
Surgery is the definitive treatment for PAPVR. However, asymptomatic patients with PAPVR with small left-to-right shunt do not require intervention, as the defect has no significant clinical impact, and patients have a normal life expectancy without correction.10
Surgical treatment may be considered in the following circumstances:
- A hemodynamically significant left-to-right shunt (a ratio of pulmonary to systemic blood flow is greater than 2:1), often manifested as right ventricular volume overload
- Recurrent pulmonary infections
- Compression or obstruction of surrounding structures caused by the anomalous vein
- During surgical repair of other major cardiac lesions, depending on the surgical risk of a repair and level and degree of shunting
Surgical options include redirecting the venous drainage to the left atria, ligation/embolization of vascular supply to the sequestered lobe, and pneumonectomy. The procedure complications may include thrombosis of the scimitar vein, lung infarct, hemoptysis, and pulmonary hypertension, which may lead to resection of the lung.11,12 Surgical procedures are recommended in cases where the patient has had recurrent lung infections or a significant degree of shunting. Studies have compared both approaches, demonstrating a better outcome after 10 years for those patients who were medically treated considering the aforementioned surgical indications.
Conclusion
Scimitar syndrome is a rare but welldescribed constellation of cardiopulmonary anomalies, accounting for 0.5% to 1% of congenital heart disease. It is a variant of PAPVR, in which part of or even the entire right lung is drained by right pulmonary veins that connect anomalously to the IVC. Although a diagnosis can be made by chest radiograph, further imaging is needed to corroborate the diagnosis and demonstrate other associated abnormalities.
Additional tests have been described in the literature, but these procedures are not available in all facilities and may incur a higher cost. Therefore, CT angiographic reconstruction is an alternative, noninvasive procedure. Surgery is the definitive treatment; however, asymptomatic patients with PAPVR and small left-to-right shunt do not require intervention.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Cooper G. Case of malformation of the thoracic viscera: consisting of imperfect development of the right lung, and transposition of the heart. London Med Gaz. 1836;18:600-602.
2. Spentzouris G, Zandian A, Cesmebasi A, et al. The clinical anatomy of the inferior vena cava: a review of common congenital anomalies and the considerations for clinicians. Clin Anat. 2014;27(8):1234-1243.
3. Neill CA, Ferencz C, Sabiston DC, Sheldon H. The familial occurrence of hypoplastic right lung with systemic arterial supply and venous drainage “scimitar syndrome.” Bull Johns Hopkins Hosp. 1960;107:1-21.
4. Ward KE, Mullins CE. Anomalous pulmonary venous connections, pulmonary vein stenosis, and atresia of the common pulmonary vein. In: Garson A, Bricker JT, Fisher DJ, Neish SR, eds. The Science and Practice of Pediatric Cardiology. 2nd ed. Baltimore, MD: Williams and Wilkins; 1998:1431-1461.
5. Garcia-Barreto L, Vega W, Deliz R, Rodriguez W. Right hilar abnormality in a young man. Respiration. 1996;63(4):246-250.
6. Simmons DB, Menon RS, Pomeroy WL, Batts TC, Slim AM. An unusual presentation of scimitar syndrome in a military service member. Case Rep Vasc Med. 2013;2013:632402.
7. Palios J, Pernetz MA, Clements S Jr, Lerakis S. Three-dimensional echocardiography images showing anomalous pulmonary venous return in an adult with scimitar syndrome. Echocardiography. 2014;31(3):E103.
8. Vida VL, Padalino MA, Boccuzzo G, et al. Scimitar syndrome: a European Congenital Heart Surgeons Association (ECHSA) multicentric study. Circulation. 2010;122(12):1159-1166.
9. Dusenbery SM, Geva T, Seale A, et al. Outcome predictors and implications for management of scimitar syndrome. Am Heart J. 2013;165(5):770-777.
10. Sehgal A, Loughran-Fowlds A. Scimitar syndrome. Indian J Pediatr. 2005;72(3):249-251.
11. Najm HK, Williams WG, Coles JG, Rebeyka IM, Freedom RM. Scimitar syndrome: twenty years’ experience and results of repair. J Thorac Cardiovasc Surg. 1996;112(5):1161-1169.
12. Dupuis C, Charaf LA, Brevière GM, Abou P, Rémy-Jardin M, Helmius G. The “adult” form of the scimitar syndrome. Am J Cardiol. 1992;70(4):502-507.
1. Cooper G. Case of malformation of the thoracic viscera: consisting of imperfect development of the right lung, and transposition of the heart. London Med Gaz. 1836;18:600-602.
2. Spentzouris G, Zandian A, Cesmebasi A, et al. The clinical anatomy of the inferior vena cava: a review of common congenital anomalies and the considerations for clinicians. Clin Anat. 2014;27(8):1234-1243.
3. Neill CA, Ferencz C, Sabiston DC, Sheldon H. The familial occurrence of hypoplastic right lung with systemic arterial supply and venous drainage “scimitar syndrome.” Bull Johns Hopkins Hosp. 1960;107:1-21.
4. Ward KE, Mullins CE. Anomalous pulmonary venous connections, pulmonary vein stenosis, and atresia of the common pulmonary vein. In: Garson A, Bricker JT, Fisher DJ, Neish SR, eds. The Science and Practice of Pediatric Cardiology. 2nd ed. Baltimore, MD: Williams and Wilkins; 1998:1431-1461.
5. Garcia-Barreto L, Vega W, Deliz R, Rodriguez W. Right hilar abnormality in a young man. Respiration. 1996;63(4):246-250.
6. Simmons DB, Menon RS, Pomeroy WL, Batts TC, Slim AM. An unusual presentation of scimitar syndrome in a military service member. Case Rep Vasc Med. 2013;2013:632402.
7. Palios J, Pernetz MA, Clements S Jr, Lerakis S. Three-dimensional echocardiography images showing anomalous pulmonary venous return in an adult with scimitar syndrome. Echocardiography. 2014;31(3):E103.
8. Vida VL, Padalino MA, Boccuzzo G, et al. Scimitar syndrome: a European Congenital Heart Surgeons Association (ECHSA) multicentric study. Circulation. 2010;122(12):1159-1166.
9. Dusenbery SM, Geva T, Seale A, et al. Outcome predictors and implications for management of scimitar syndrome. Am Heart J. 2013;165(5):770-777.
10. Sehgal A, Loughran-Fowlds A. Scimitar syndrome. Indian J Pediatr. 2005;72(3):249-251.
11. Najm HK, Williams WG, Coles JG, Rebeyka IM, Freedom RM. Scimitar syndrome: twenty years’ experience and results of repair. J Thorac Cardiovasc Surg. 1996;112(5):1161-1169.
12. Dupuis C, Charaf LA, Brevière GM, Abou P, Rémy-Jardin M, Helmius G. The “adult” form of the scimitar syndrome. Am J Cardiol. 1992;70(4):502-507.
