User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
nav[contains(@class, 'nav-ce-stack nav-ce-stack__large-screen')]
header[@id='header']
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
COVID vaccines safe for young children, study finds
TOPLINE:
COVID-19 vaccines from Moderna and Pfizer-BioNTech are safe for children under age 5 years, according to findings from a study funded by the Centers for Disease Control and Prevention.
METHODOLOGY:
- Data came from the Vaccine Safety Datalink, which gathers information from eight health systems in the United States.
- Analyzed data from 135,005 doses given to children age 4 and younger who received the Pfizer-BioNTech , and 112,006 doses given to children aged 5 and younger who received the Moderna version.
- Assessed for 23 safety outcomes, including myocarditis, pericarditis, and seizures.
TAKEAWAY:
- One case of hemorrhagic stroke and one case of pulmonary embolism occurred after vaccination but these were linked to preexisting congenital abnormalities.
IN PRACTICE:
“These results can provide reassurance to clinicians, parents, and policymakers alike.”
STUDY DETAILS:
The study was led by Kristin Goddard, MPH, a researcher at the Kaiser Permanente Vaccine Study Center in Oakland, Calif., and was funded by the Centers for Disease Control and Prevention.
LIMITATIONS:
The researchers reported low statistical power for early analysis, especially for rare outcomes. In addition, fewer than 25% of children in the database had received a vaccine at the time of analysis.
DISCLOSURES:
A coauthor reported receiving funding from Janssen Vaccines and Prevention for a study unrelated to COVID-19 vaccines. Another coauthor reported receiving grants from Pfizer in 2019 for clinical trials for coronavirus vaccines, and from Merck, GSK, and Sanofi Pasteur for unrelated research.
A version of this article first appeared on Medscape.com.
TOPLINE:
COVID-19 vaccines from Moderna and Pfizer-BioNTech are safe for children under age 5 years, according to findings from a study funded by the Centers for Disease Control and Prevention.
METHODOLOGY:
- Data came from the Vaccine Safety Datalink, which gathers information from eight health systems in the United States.
- Analyzed data from 135,005 doses given to children age 4 and younger who received the Pfizer-BioNTech , and 112,006 doses given to children aged 5 and younger who received the Moderna version.
- Assessed for 23 safety outcomes, including myocarditis, pericarditis, and seizures.
TAKEAWAY:
- One case of hemorrhagic stroke and one case of pulmonary embolism occurred after vaccination but these were linked to preexisting congenital abnormalities.
IN PRACTICE:
“These results can provide reassurance to clinicians, parents, and policymakers alike.”
STUDY DETAILS:
The study was led by Kristin Goddard, MPH, a researcher at the Kaiser Permanente Vaccine Study Center in Oakland, Calif., and was funded by the Centers for Disease Control and Prevention.
LIMITATIONS:
The researchers reported low statistical power for early analysis, especially for rare outcomes. In addition, fewer than 25% of children in the database had received a vaccine at the time of analysis.
DISCLOSURES:
A coauthor reported receiving funding from Janssen Vaccines and Prevention for a study unrelated to COVID-19 vaccines. Another coauthor reported receiving grants from Pfizer in 2019 for clinical trials for coronavirus vaccines, and from Merck, GSK, and Sanofi Pasteur for unrelated research.
A version of this article first appeared on Medscape.com.
TOPLINE:
COVID-19 vaccines from Moderna and Pfizer-BioNTech are safe for children under age 5 years, according to findings from a study funded by the Centers for Disease Control and Prevention.
METHODOLOGY:
- Data came from the Vaccine Safety Datalink, which gathers information from eight health systems in the United States.
- Analyzed data from 135,005 doses given to children age 4 and younger who received the Pfizer-BioNTech , and 112,006 doses given to children aged 5 and younger who received the Moderna version.
- Assessed for 23 safety outcomes, including myocarditis, pericarditis, and seizures.
TAKEAWAY:
- One case of hemorrhagic stroke and one case of pulmonary embolism occurred after vaccination but these were linked to preexisting congenital abnormalities.
IN PRACTICE:
“These results can provide reassurance to clinicians, parents, and policymakers alike.”
STUDY DETAILS:
The study was led by Kristin Goddard, MPH, a researcher at the Kaiser Permanente Vaccine Study Center in Oakland, Calif., and was funded by the Centers for Disease Control and Prevention.
LIMITATIONS:
The researchers reported low statistical power for early analysis, especially for rare outcomes. In addition, fewer than 25% of children in the database had received a vaccine at the time of analysis.
DISCLOSURES:
A coauthor reported receiving funding from Janssen Vaccines and Prevention for a study unrelated to COVID-19 vaccines. Another coauthor reported receiving grants from Pfizer in 2019 for clinical trials for coronavirus vaccines, and from Merck, GSK, and Sanofi Pasteur for unrelated research.
A version of this article first appeared on Medscape.com.
FROM PEDIATRICS
Medicaid patients with heart failure get poor follow-up after hospital discharge
Nearly 60% of Medicaid-covered adults with concurrent diabetes and heart failure did not receive guideline-concordant postdischarge care within 7-10 days of leaving the hospital, according to a large Alabama study. Moreover, affected Black and Hispanic/other Alabamians were less likely than were their White counterparts to receive recommended postdischarge care.
In comparison with White participants, Black and Hispanic adults were less likely to have any postdischarge ambulatory care visits after HF hospitalization or had a delayed visit, according to researchers led by Yulia Khodneva, MD, PhD, an internist at the University of Alabama at Birmingham. “This is likely a reflection of a structural racism and implicit bias against racial and ethnic minorities that persists in the U.S. health care system,” she and her colleagues wrote.
The findings point to the need for strategies to improve access to postdischarge care for lower-income HF patients.
Among U.S. states, Alabama is the sixth-poorest, the third in diabetes prevalence (14%), and has the highest rates of heart failure hospitalizations and cardiovascular mortality, the authors noted.
Study details
The cohort included 9,857 adults with diabetes and first hospitalizations for heart failure who were covered by Alabama Medicaid during 2010-2019. The investigators analyzed patients’ claims for ambulatory care (any, primary, cardiology, or endocrinology) within 60 days of discharge.
The mean age of participants was 53.7 years; 47.3% were Black; 41.8% non-Hispanic White; and 10.9% Hispanic/other, with other including those identifying as non-White Hispanic, American Indian, Pacific Islander, and Asian. About two-thirds (65.4%) of participants were women.
Analysis revealed low rates of follow-up care after hospital discharge; 26.7% had an ambulatory visit within 0-7 days, 15.2% within 8-14 days, 31.3% within 15-60 days, and 26.8% had no follow-up visit at all. Of those having a follow-up visit, 71% saw a primary care physician and 12% saw a cardiologist.
In contrast, a much higher proportion of heart failure patients in a Swedish registry – 63% – received ambulatory follow-up in cardiology.
Ethnic/gender/age disparities
Black and Hispanic/other adults were less likely to have any postdischarge ambulatory visit (P <.0001) or had the visit delayed by 1.8 days (P = .0006) and 2.8 days (P = .0016), respectively. They were less likely to see a primary care physician than were non-Hispanic White adults: adjusted incidence rate ratio, 0.96 (95% confidence interval [CI], 0.91-1.00) and 0.91 (95% CI, 0.89-0.98), respectively.
Men and those with longer-standing heart failure were less likely to be seen in primary care, while the presence of multiple comorbidities was associated with a higher likelihood of a postdischarge primary care visit. Men were more likely to be seen by a cardiologist, while older discharged patients were less likely to be seen by an endocrinologist within 60 days. There was a U-shaped relationship between the timing of the first postdischarge ambulatory visit and all-cause mortality among adults with diabetes and heart failure. Higher rates of 60-day all-cause mortality were observed both in those who had seen a provider within 0-7 days after discharge and in those who had not seen any provider during the 60-day study period compared with those having an ambulatory care visit within 7-14 or 15-60 days. “The group with early follow-up (0-7 days) likely represents a sicker population of patients with heart failure with more comorbidity burden and higher overall health care use, including readmissions, as was demonstrated in our analysis,” Dr. Khodneva and associates wrote. “Interventions that improve access to postdischarge ambulatory care for low-income patients with diabetes and heart failure and eliminate racial and ethnic disparities may be warranted,” they added.
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases and the University of Alabama at Birmingham Diabetes Research Center. Dr. Khodneva reported funding from the University of Alabama at Birmingham and the Forge Ahead Center as well as from the NIDDK, the National Institutes of Health, the Agency for Healthcare Research and Quality, and the Alabama Medicaid Agency. Coauthor Emily Levitan, ScD, reported research funding from Amgen and has served on Amgen advisory boards. She has also served as a scientific consultant for a research project funded by Novartis.
Nearly 60% of Medicaid-covered adults with concurrent diabetes and heart failure did not receive guideline-concordant postdischarge care within 7-10 days of leaving the hospital, according to a large Alabama study. Moreover, affected Black and Hispanic/other Alabamians were less likely than were their White counterparts to receive recommended postdischarge care.
In comparison with White participants, Black and Hispanic adults were less likely to have any postdischarge ambulatory care visits after HF hospitalization or had a delayed visit, according to researchers led by Yulia Khodneva, MD, PhD, an internist at the University of Alabama at Birmingham. “This is likely a reflection of a structural racism and implicit bias against racial and ethnic minorities that persists in the U.S. health care system,” she and her colleagues wrote.
The findings point to the need for strategies to improve access to postdischarge care for lower-income HF patients.
Among U.S. states, Alabama is the sixth-poorest, the third in diabetes prevalence (14%), and has the highest rates of heart failure hospitalizations and cardiovascular mortality, the authors noted.
Study details
The cohort included 9,857 adults with diabetes and first hospitalizations for heart failure who were covered by Alabama Medicaid during 2010-2019. The investigators analyzed patients’ claims for ambulatory care (any, primary, cardiology, or endocrinology) within 60 days of discharge.
The mean age of participants was 53.7 years; 47.3% were Black; 41.8% non-Hispanic White; and 10.9% Hispanic/other, with other including those identifying as non-White Hispanic, American Indian, Pacific Islander, and Asian. About two-thirds (65.4%) of participants were women.
Analysis revealed low rates of follow-up care after hospital discharge; 26.7% had an ambulatory visit within 0-7 days, 15.2% within 8-14 days, 31.3% within 15-60 days, and 26.8% had no follow-up visit at all. Of those having a follow-up visit, 71% saw a primary care physician and 12% saw a cardiologist.
In contrast, a much higher proportion of heart failure patients in a Swedish registry – 63% – received ambulatory follow-up in cardiology.
Ethnic/gender/age disparities
Black and Hispanic/other adults were less likely to have any postdischarge ambulatory visit (P <.0001) or had the visit delayed by 1.8 days (P = .0006) and 2.8 days (P = .0016), respectively. They were less likely to see a primary care physician than were non-Hispanic White adults: adjusted incidence rate ratio, 0.96 (95% confidence interval [CI], 0.91-1.00) and 0.91 (95% CI, 0.89-0.98), respectively.
Men and those with longer-standing heart failure were less likely to be seen in primary care, while the presence of multiple comorbidities was associated with a higher likelihood of a postdischarge primary care visit. Men were more likely to be seen by a cardiologist, while older discharged patients were less likely to be seen by an endocrinologist within 60 days. There was a U-shaped relationship between the timing of the first postdischarge ambulatory visit and all-cause mortality among adults with diabetes and heart failure. Higher rates of 60-day all-cause mortality were observed both in those who had seen a provider within 0-7 days after discharge and in those who had not seen any provider during the 60-day study period compared with those having an ambulatory care visit within 7-14 or 15-60 days. “The group with early follow-up (0-7 days) likely represents a sicker population of patients with heart failure with more comorbidity burden and higher overall health care use, including readmissions, as was demonstrated in our analysis,” Dr. Khodneva and associates wrote. “Interventions that improve access to postdischarge ambulatory care for low-income patients with diabetes and heart failure and eliminate racial and ethnic disparities may be warranted,” they added.
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases and the University of Alabama at Birmingham Diabetes Research Center. Dr. Khodneva reported funding from the University of Alabama at Birmingham and the Forge Ahead Center as well as from the NIDDK, the National Institutes of Health, the Agency for Healthcare Research and Quality, and the Alabama Medicaid Agency. Coauthor Emily Levitan, ScD, reported research funding from Amgen and has served on Amgen advisory boards. She has also served as a scientific consultant for a research project funded by Novartis.
Nearly 60% of Medicaid-covered adults with concurrent diabetes and heart failure did not receive guideline-concordant postdischarge care within 7-10 days of leaving the hospital, according to a large Alabama study. Moreover, affected Black and Hispanic/other Alabamians were less likely than were their White counterparts to receive recommended postdischarge care.
In comparison with White participants, Black and Hispanic adults were less likely to have any postdischarge ambulatory care visits after HF hospitalization or had a delayed visit, according to researchers led by Yulia Khodneva, MD, PhD, an internist at the University of Alabama at Birmingham. “This is likely a reflection of a structural racism and implicit bias against racial and ethnic minorities that persists in the U.S. health care system,” she and her colleagues wrote.
The findings point to the need for strategies to improve access to postdischarge care for lower-income HF patients.
Among U.S. states, Alabama is the sixth-poorest, the third in diabetes prevalence (14%), and has the highest rates of heart failure hospitalizations and cardiovascular mortality, the authors noted.
Study details
The cohort included 9,857 adults with diabetes and first hospitalizations for heart failure who were covered by Alabama Medicaid during 2010-2019. The investigators analyzed patients’ claims for ambulatory care (any, primary, cardiology, or endocrinology) within 60 days of discharge.
The mean age of participants was 53.7 years; 47.3% were Black; 41.8% non-Hispanic White; and 10.9% Hispanic/other, with other including those identifying as non-White Hispanic, American Indian, Pacific Islander, and Asian. About two-thirds (65.4%) of participants were women.
Analysis revealed low rates of follow-up care after hospital discharge; 26.7% had an ambulatory visit within 0-7 days, 15.2% within 8-14 days, 31.3% within 15-60 days, and 26.8% had no follow-up visit at all. Of those having a follow-up visit, 71% saw a primary care physician and 12% saw a cardiologist.
In contrast, a much higher proportion of heart failure patients in a Swedish registry – 63% – received ambulatory follow-up in cardiology.
Ethnic/gender/age disparities
Black and Hispanic/other adults were less likely to have any postdischarge ambulatory visit (P <.0001) or had the visit delayed by 1.8 days (P = .0006) and 2.8 days (P = .0016), respectively. They were less likely to see a primary care physician than were non-Hispanic White adults: adjusted incidence rate ratio, 0.96 (95% confidence interval [CI], 0.91-1.00) and 0.91 (95% CI, 0.89-0.98), respectively.
Men and those with longer-standing heart failure were less likely to be seen in primary care, while the presence of multiple comorbidities was associated with a higher likelihood of a postdischarge primary care visit. Men were more likely to be seen by a cardiologist, while older discharged patients were less likely to be seen by an endocrinologist within 60 days. There was a U-shaped relationship between the timing of the first postdischarge ambulatory visit and all-cause mortality among adults with diabetes and heart failure. Higher rates of 60-day all-cause mortality were observed both in those who had seen a provider within 0-7 days after discharge and in those who had not seen any provider during the 60-day study period compared with those having an ambulatory care visit within 7-14 or 15-60 days. “The group with early follow-up (0-7 days) likely represents a sicker population of patients with heart failure with more comorbidity burden and higher overall health care use, including readmissions, as was demonstrated in our analysis,” Dr. Khodneva and associates wrote. “Interventions that improve access to postdischarge ambulatory care for low-income patients with diabetes and heart failure and eliminate racial and ethnic disparities may be warranted,” they added.
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases and the University of Alabama at Birmingham Diabetes Research Center. Dr. Khodneva reported funding from the University of Alabama at Birmingham and the Forge Ahead Center as well as from the NIDDK, the National Institutes of Health, the Agency for Healthcare Research and Quality, and the Alabama Medicaid Agency. Coauthor Emily Levitan, ScD, reported research funding from Amgen and has served on Amgen advisory boards. She has also served as a scientific consultant for a research project funded by Novartis.
FROM JOURNAL OF THE AMERICAN HEART ASSOCIATION
Investigational uricase-based gout drug meets primary endpoints in phase 3 trials
MILAN – Serum uric acid of less than 6 mg/dL was achieved and maintained for a substantial period of time with a once-monthly infusion of SEL-212 in patients with refractory gout, according to results of the two phase 3 DISSOLVE I and II trials.
Both trials met their primary endpoints. In DISSOLVE I – the U.S. study – 56% of patients on SEL-212 at 0.15 mg/kg (high dose) achieved a response, defined as achievement and maintenance of a reduction in serum urate to less than 6 mg/dL for at least 80% of the time during month 6 of treatment. In DISSOLVE II – the global study – 46% of patients on SEL-212 on the 0.15-mg/kg dose achieved response.
In participants aged 50 years or older, there was a statistically significant higher response rate at the high dose of SEL-212 in both DISSOLVE I and II of 65% and 47%, respectively, compared with placebo.
Herbert S.B. Baraf, MD, clinical professor of medicine at George Washington University, Washington, and principal investigator of the DISSOLVE program, presented results of the two phase 3 trials during a late-breaking session at the annual European Congress of Rheumatology.
“The top-line data from the two SEL-212 phase 3 studies are encouraging. They show that induction of immunotolerance with an infusion of a rapamycin-containing nanoparticle (SEL-110), followed immediately by an infusion of pegadricase, a potent but immunogenic uricase, allows for a strong and sustained uric acid–lowering effect without the development of anti-drug antibodies,” Dr. Baraf said in an interview.
SEL-212 is a monthly two-part infusion therapy – a combination of Selecta Biosciences’s ImmTOR immune tolerance platform, and a therapeutic uricase enzyme (pegadricase), designed to treat refractory gout. SEL-110 (ImmTOR) is an immune-tolerizing, nanoencapsulated rapamycin administered 30 minutes before pegadricase and inhibits anti-pegadricase antibodies. SEL-37 is a pegylated uricase (pegadricase) that converts uric acid to excretable allantoin.
SEL-212 was originally developed by Selecta. Swedish Orphan Biovitrum (Sobi) licensed SEL-212 from Selecta in June 2020 and is responsible for development, regulatory, and commercial activities in all markets outside of China. Selecta is responsible for ImmTOR manufacturing. The phase 3 program for SEL-212 was run by Selecta and funded by Sobi.
It is understood that a biologic license application will be submitted to the Food and Drug Administration, most likely next year, and if approved, “the SEL-212 two-component infusion treatment would provide a monthly alternative to twice-monthly pegloticase, for patients with refractory gout,” Dr. Baraf added.
Details of the trials
The two DISSOLVE studies replicate double-blind, placebo-controlled trials in patients with chronic refractory gout. DISSOLVE I was carried out in 112 patients across 29 sites in the United States, and DISSOLVE II tested the two-part treatment in 153 patients across 37 sites in the United States, Russia, Ukraine, Georgia, and Serbia.
Both studies randomized patients 1:1:1 to a high dose (SEL-110 of 0.15 mg/kg plus SEL-037 of 0.2 mg/kg), low dose (SEL-110 of 0.1 mg/kg plus SEL-037 of 0.2 mg/kg), or placebo (saline) infused every 28 days for 6 months. Prophylaxis against infusion reactions and gout flares were given to all participants.
Adult patients had a 10- to 14-year history of symptomatic gout, with three or more flares over the 18 months prior to screening, or one or more tophus, or a diagnosis of gouty arthritis. They were also required to have chronic refractory gout with a failure to normalize serum uric acid with any xanthine oxidase inhibitor (for example, allopurinol) and to have not been previously exposed to uricase-based therapy. Serum uric acid had to be at least 7 mg/dL. Participants were balanced for age, body mass index, and sex across treatment groups. Gout severity was greater in DISSOLVE II, Dr. Baraf reported.
Both studies treated patients for 6 months, but DISSOLVE 1 continued with a 6-month, blinded safety extension. The primary endpoint in both studies was serum urate control during month 6, and secondary endpoints included tender and swollen joint counts, tophus burden, patient-reported outcomes of activity limitation, quality of life, and gout flare incidence.
In DISSOLVE I, patients on SEL-212 had a statistically significant higher response rate during month 6 of 56% with the high dose (P < .0001) and 48% with the low dose (P < .0001), compared with 4% of patients randomized to receive placebo. In DISSOLVE II, participants on SEL-212 had a statistically significant higher response rate during month 6 of 46% with the high dose (P = .0002) and 40% with the low dose (P = .0008), compared with 11% of patients randomized to receive placebo.
“We also saw significant reductions in serum uric acid for all treatment groups, compared with placebo,” Dr. Baraf reported. Mean percentage change was –62.3% and –58.3% in the high- and low-dose groups, respectively, in DISSOLVE I, and –58.1% and –52.2% in DISSOLVE II, respectively.
SEL-212 had a favorable safety profile with adverse events as expected across both doses, including mild to moderate stomatitis (3.4% in the low-dose group and 9.2% in the high-dose group versus 0% in the placebo group), and a greater number of infusion reactions at 24 hours and 1 hour after drug administration in both treatment groups versus placebo. Six patients had treatment-related serious adverse events, including two cases of anaphylaxis and one gout flare in both the high- and low-dose treatment groups. The 6-month extension period in the DISSOLVE I trial showed that the majority (75%) of patients who completed 6 months of SEL-212 treatment as a responder continued to be successfully treated through 12 months with no infusion reactions or safety signals.
“I expect more data will be forthcoming on the important clinical secondary endpoints targeted by SEL-212 therapy,” Dr. Baraf noted.
Need control arm taking allopurinol?
Roy Fleischmann, MD, clinical professor of medicine at the University of Texas Southwestern Medical Center and codirector of the Metroplex Clinical Research Center, both in Dallas, commented on the study methods after the presentation. “The major problem with this study is that they say the patients had had insufficient response to allopurinol, and my guess is most had received 100-200 mg of allopurinol but were not titrated up to the maximum tolerated dose,” he said, adding: “they should have had a control arm of patients on allopurinol and titrated to the maximum tolerated dose. So, I don’t know what this is really telling us with respect to allopurinol, which is a relatively cheap drug.”
Dr. Baraf reported consulting with Horizon, Sobi, and Selecta; serving on Horizon’s speakers bureau, and receiving grant/research support from Horizon and Sobi. Dr. Fleischmann reported no financial relationship of relevance to this study.
MILAN – Serum uric acid of less than 6 mg/dL was achieved and maintained for a substantial period of time with a once-monthly infusion of SEL-212 in patients with refractory gout, according to results of the two phase 3 DISSOLVE I and II trials.
Both trials met their primary endpoints. In DISSOLVE I – the U.S. study – 56% of patients on SEL-212 at 0.15 mg/kg (high dose) achieved a response, defined as achievement and maintenance of a reduction in serum urate to less than 6 mg/dL for at least 80% of the time during month 6 of treatment. In DISSOLVE II – the global study – 46% of patients on SEL-212 on the 0.15-mg/kg dose achieved response.
In participants aged 50 years or older, there was a statistically significant higher response rate at the high dose of SEL-212 in both DISSOLVE I and II of 65% and 47%, respectively, compared with placebo.
Herbert S.B. Baraf, MD, clinical professor of medicine at George Washington University, Washington, and principal investigator of the DISSOLVE program, presented results of the two phase 3 trials during a late-breaking session at the annual European Congress of Rheumatology.
“The top-line data from the two SEL-212 phase 3 studies are encouraging. They show that induction of immunotolerance with an infusion of a rapamycin-containing nanoparticle (SEL-110), followed immediately by an infusion of pegadricase, a potent but immunogenic uricase, allows for a strong and sustained uric acid–lowering effect without the development of anti-drug antibodies,” Dr. Baraf said in an interview.
SEL-212 is a monthly two-part infusion therapy – a combination of Selecta Biosciences’s ImmTOR immune tolerance platform, and a therapeutic uricase enzyme (pegadricase), designed to treat refractory gout. SEL-110 (ImmTOR) is an immune-tolerizing, nanoencapsulated rapamycin administered 30 minutes before pegadricase and inhibits anti-pegadricase antibodies. SEL-37 is a pegylated uricase (pegadricase) that converts uric acid to excretable allantoin.
SEL-212 was originally developed by Selecta. Swedish Orphan Biovitrum (Sobi) licensed SEL-212 from Selecta in June 2020 and is responsible for development, regulatory, and commercial activities in all markets outside of China. Selecta is responsible for ImmTOR manufacturing. The phase 3 program for SEL-212 was run by Selecta and funded by Sobi.
It is understood that a biologic license application will be submitted to the Food and Drug Administration, most likely next year, and if approved, “the SEL-212 two-component infusion treatment would provide a monthly alternative to twice-monthly pegloticase, for patients with refractory gout,” Dr. Baraf added.
Details of the trials
The two DISSOLVE studies replicate double-blind, placebo-controlled trials in patients with chronic refractory gout. DISSOLVE I was carried out in 112 patients across 29 sites in the United States, and DISSOLVE II tested the two-part treatment in 153 patients across 37 sites in the United States, Russia, Ukraine, Georgia, and Serbia.
Both studies randomized patients 1:1:1 to a high dose (SEL-110 of 0.15 mg/kg plus SEL-037 of 0.2 mg/kg), low dose (SEL-110 of 0.1 mg/kg plus SEL-037 of 0.2 mg/kg), or placebo (saline) infused every 28 days for 6 months. Prophylaxis against infusion reactions and gout flares were given to all participants.
Adult patients had a 10- to 14-year history of symptomatic gout, with three or more flares over the 18 months prior to screening, or one or more tophus, or a diagnosis of gouty arthritis. They were also required to have chronic refractory gout with a failure to normalize serum uric acid with any xanthine oxidase inhibitor (for example, allopurinol) and to have not been previously exposed to uricase-based therapy. Serum uric acid had to be at least 7 mg/dL. Participants were balanced for age, body mass index, and sex across treatment groups. Gout severity was greater in DISSOLVE II, Dr. Baraf reported.
Both studies treated patients for 6 months, but DISSOLVE 1 continued with a 6-month, blinded safety extension. The primary endpoint in both studies was serum urate control during month 6, and secondary endpoints included tender and swollen joint counts, tophus burden, patient-reported outcomes of activity limitation, quality of life, and gout flare incidence.
In DISSOLVE I, patients on SEL-212 had a statistically significant higher response rate during month 6 of 56% with the high dose (P < .0001) and 48% with the low dose (P < .0001), compared with 4% of patients randomized to receive placebo. In DISSOLVE II, participants on SEL-212 had a statistically significant higher response rate during month 6 of 46% with the high dose (P = .0002) and 40% with the low dose (P = .0008), compared with 11% of patients randomized to receive placebo.
“We also saw significant reductions in serum uric acid for all treatment groups, compared with placebo,” Dr. Baraf reported. Mean percentage change was –62.3% and –58.3% in the high- and low-dose groups, respectively, in DISSOLVE I, and –58.1% and –52.2% in DISSOLVE II, respectively.
SEL-212 had a favorable safety profile with adverse events as expected across both doses, including mild to moderate stomatitis (3.4% in the low-dose group and 9.2% in the high-dose group versus 0% in the placebo group), and a greater number of infusion reactions at 24 hours and 1 hour after drug administration in both treatment groups versus placebo. Six patients had treatment-related serious adverse events, including two cases of anaphylaxis and one gout flare in both the high- and low-dose treatment groups. The 6-month extension period in the DISSOLVE I trial showed that the majority (75%) of patients who completed 6 months of SEL-212 treatment as a responder continued to be successfully treated through 12 months with no infusion reactions or safety signals.
“I expect more data will be forthcoming on the important clinical secondary endpoints targeted by SEL-212 therapy,” Dr. Baraf noted.
Need control arm taking allopurinol?
Roy Fleischmann, MD, clinical professor of medicine at the University of Texas Southwestern Medical Center and codirector of the Metroplex Clinical Research Center, both in Dallas, commented on the study methods after the presentation. “The major problem with this study is that they say the patients had had insufficient response to allopurinol, and my guess is most had received 100-200 mg of allopurinol but were not titrated up to the maximum tolerated dose,” he said, adding: “they should have had a control arm of patients on allopurinol and titrated to the maximum tolerated dose. So, I don’t know what this is really telling us with respect to allopurinol, which is a relatively cheap drug.”
Dr. Baraf reported consulting with Horizon, Sobi, and Selecta; serving on Horizon’s speakers bureau, and receiving grant/research support from Horizon and Sobi. Dr. Fleischmann reported no financial relationship of relevance to this study.
MILAN – Serum uric acid of less than 6 mg/dL was achieved and maintained for a substantial period of time with a once-monthly infusion of SEL-212 in patients with refractory gout, according to results of the two phase 3 DISSOLVE I and II trials.
Both trials met their primary endpoints. In DISSOLVE I – the U.S. study – 56% of patients on SEL-212 at 0.15 mg/kg (high dose) achieved a response, defined as achievement and maintenance of a reduction in serum urate to less than 6 mg/dL for at least 80% of the time during month 6 of treatment. In DISSOLVE II – the global study – 46% of patients on SEL-212 on the 0.15-mg/kg dose achieved response.
In participants aged 50 years or older, there was a statistically significant higher response rate at the high dose of SEL-212 in both DISSOLVE I and II of 65% and 47%, respectively, compared with placebo.
Herbert S.B. Baraf, MD, clinical professor of medicine at George Washington University, Washington, and principal investigator of the DISSOLVE program, presented results of the two phase 3 trials during a late-breaking session at the annual European Congress of Rheumatology.
“The top-line data from the two SEL-212 phase 3 studies are encouraging. They show that induction of immunotolerance with an infusion of a rapamycin-containing nanoparticle (SEL-110), followed immediately by an infusion of pegadricase, a potent but immunogenic uricase, allows for a strong and sustained uric acid–lowering effect without the development of anti-drug antibodies,” Dr. Baraf said in an interview.
SEL-212 is a monthly two-part infusion therapy – a combination of Selecta Biosciences’s ImmTOR immune tolerance platform, and a therapeutic uricase enzyme (pegadricase), designed to treat refractory gout. SEL-110 (ImmTOR) is an immune-tolerizing, nanoencapsulated rapamycin administered 30 minutes before pegadricase and inhibits anti-pegadricase antibodies. SEL-37 is a pegylated uricase (pegadricase) that converts uric acid to excretable allantoin.
SEL-212 was originally developed by Selecta. Swedish Orphan Biovitrum (Sobi) licensed SEL-212 from Selecta in June 2020 and is responsible for development, regulatory, and commercial activities in all markets outside of China. Selecta is responsible for ImmTOR manufacturing. The phase 3 program for SEL-212 was run by Selecta and funded by Sobi.
It is understood that a biologic license application will be submitted to the Food and Drug Administration, most likely next year, and if approved, “the SEL-212 two-component infusion treatment would provide a monthly alternative to twice-monthly pegloticase, for patients with refractory gout,” Dr. Baraf added.
Details of the trials
The two DISSOLVE studies replicate double-blind, placebo-controlled trials in patients with chronic refractory gout. DISSOLVE I was carried out in 112 patients across 29 sites in the United States, and DISSOLVE II tested the two-part treatment in 153 patients across 37 sites in the United States, Russia, Ukraine, Georgia, and Serbia.
Both studies randomized patients 1:1:1 to a high dose (SEL-110 of 0.15 mg/kg plus SEL-037 of 0.2 mg/kg), low dose (SEL-110 of 0.1 mg/kg plus SEL-037 of 0.2 mg/kg), or placebo (saline) infused every 28 days for 6 months. Prophylaxis against infusion reactions and gout flares were given to all participants.
Adult patients had a 10- to 14-year history of symptomatic gout, with three or more flares over the 18 months prior to screening, or one or more tophus, or a diagnosis of gouty arthritis. They were also required to have chronic refractory gout with a failure to normalize serum uric acid with any xanthine oxidase inhibitor (for example, allopurinol) and to have not been previously exposed to uricase-based therapy. Serum uric acid had to be at least 7 mg/dL. Participants were balanced for age, body mass index, and sex across treatment groups. Gout severity was greater in DISSOLVE II, Dr. Baraf reported.
Both studies treated patients for 6 months, but DISSOLVE 1 continued with a 6-month, blinded safety extension. The primary endpoint in both studies was serum urate control during month 6, and secondary endpoints included tender and swollen joint counts, tophus burden, patient-reported outcomes of activity limitation, quality of life, and gout flare incidence.
In DISSOLVE I, patients on SEL-212 had a statistically significant higher response rate during month 6 of 56% with the high dose (P < .0001) and 48% with the low dose (P < .0001), compared with 4% of patients randomized to receive placebo. In DISSOLVE II, participants on SEL-212 had a statistically significant higher response rate during month 6 of 46% with the high dose (P = .0002) and 40% with the low dose (P = .0008), compared with 11% of patients randomized to receive placebo.
“We also saw significant reductions in serum uric acid for all treatment groups, compared with placebo,” Dr. Baraf reported. Mean percentage change was –62.3% and –58.3% in the high- and low-dose groups, respectively, in DISSOLVE I, and –58.1% and –52.2% in DISSOLVE II, respectively.
SEL-212 had a favorable safety profile with adverse events as expected across both doses, including mild to moderate stomatitis (3.4% in the low-dose group and 9.2% in the high-dose group versus 0% in the placebo group), and a greater number of infusion reactions at 24 hours and 1 hour after drug administration in both treatment groups versus placebo. Six patients had treatment-related serious adverse events, including two cases of anaphylaxis and one gout flare in both the high- and low-dose treatment groups. The 6-month extension period in the DISSOLVE I trial showed that the majority (75%) of patients who completed 6 months of SEL-212 treatment as a responder continued to be successfully treated through 12 months with no infusion reactions or safety signals.
“I expect more data will be forthcoming on the important clinical secondary endpoints targeted by SEL-212 therapy,” Dr. Baraf noted.
Need control arm taking allopurinol?
Roy Fleischmann, MD, clinical professor of medicine at the University of Texas Southwestern Medical Center and codirector of the Metroplex Clinical Research Center, both in Dallas, commented on the study methods after the presentation. “The major problem with this study is that they say the patients had had insufficient response to allopurinol, and my guess is most had received 100-200 mg of allopurinol but were not titrated up to the maximum tolerated dose,” he said, adding: “they should have had a control arm of patients on allopurinol and titrated to the maximum tolerated dose. So, I don’t know what this is really telling us with respect to allopurinol, which is a relatively cheap drug.”
Dr. Baraf reported consulting with Horizon, Sobi, and Selecta; serving on Horizon’s speakers bureau, and receiving grant/research support from Horizon and Sobi. Dr. Fleischmann reported no financial relationship of relevance to this study.
AT EULAR 2023
Lower racial disparity in melanoma diagnoses in vets than U.S. men overall, study finds
a new analysis shows.
“The trend of a lower racial disparity in the VA in the proportion of melanomas with local disease and in the proportion of distant metastasis at presentation was observed across age groups,” wrote Martin A. Weinstock MD, PhD, and Rachel K. Lim, of the department of dermatology at Brown University, Providence, R.I., and the Center for Dermatoepidemiology at the VA Providence Healthcare System. The study was published online in the Journal of the American Academy of Dermatology.
“Melanoma was the fourth-most common cancer [diagnosed] in male VA patients in 2010,” wrote the authors, who also pointed out that “prior surveys found that 11%-13% of U.S. active-duty personnel routinely use sunscreen despite significant occupational sun exposure. Racial disparities are important concerns in the VA and elsewhere.”
To compare the stage of melanoma at presentation among White and non-Whites patients in the VA and in the general U.S. population, the researchers identified invasive cutaneous melanoma cases from 2000 to 2019 in the VA Corporate Data Warehouse and the Surveillance, Epidemiology and End Results Program (SEER).
They restricted the analysis to men because of the small proportion of women in the at-risk veteran population and excluded cases with an age younger than 20, those with unknown histology, and melanoma in situ. The researchers performed two-tailed z-tests to evaluate the difference in proportions of melanoma stages between the veteran population and the general population.
The analysis included 44,077 cases of invasive melanoma in the VA and 217,030 in SEER. Racial disparities in melanoma staging were substantially less pronounced in the VA than in SEER.
In the VA, localized disease represented 77.9% of melanomas among Whites versus 71.0% among non-Whites. But in SEER, localized disease represented 80.7% of melanomas among Whites versus 61.5% in non-Whites – over double the VA disparity (P < .0001).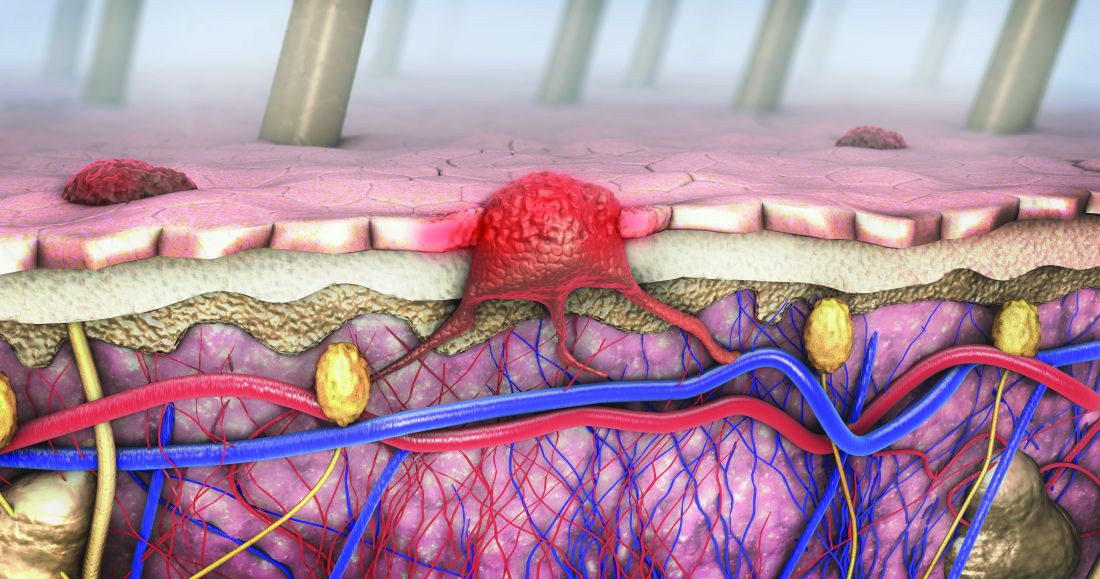
Likewise, the disparity between Whites and nonwhites observed for regional or distant metastatic disease at presentation in the VA was lower than the disparity observed in SEER. For example, in the VA, distant metastatic disease at presentation represented 6.1% of melanomas among Whites versus 8.6% among non-Whites, while in SEER it represented 4.8% of melanomas among Whites versus 11.3% in non-Whites – again, more than double the VA disparity (P < .0001).
“These differences between the VA and SEER were less marked” among those older than 65 years, the researchers wrote. “Notably, the differences between VA and SEER in racial disparities among those greater than 65 in age were still significant for localized disease and for distant metastasis.”
The findings suggest that the VA “may be more effective in reducing racial disparities in melanoma stage at diagnosis, potentially due to all patients in the VA dataset having insured access to health care, regardless of socioeconomic status,” the researchers concluded. Similarly, the decreased difference in racial disparities observed in patients older than 65 across systems “may be related to the availability of Medicare to the older general populations. The authors acknowledged several study limitations, such as the predominantly elderly and male VA population, potentially underreported utilization of non-VA dermatologic care, and variation in geographic regions covered by each database.
Travis W. Blalock, MD, director of dermatologic surgery, Mohs micrographic surgery, and cutaneous oncology at Emory University, Atlanta, who was asked to comment on the work, said in an interview he would have liked to see a more detailed breakdown of the younger patients, “for those in their 30s and 40s, to see if this trend held up.”
He would have also liked to see how the data trended over time, adding, “while this, broadly, may be good news for our veterans, attributing this finding to a reduction in access disparity or some other organizational intervention seems a little premature. Regardless, Dr. Weinstock has given us, once again, information from our veterans to probe for the betterment of all patients.”
The researchers reported having no relevant disclosures and the study had no funding. Dr. Blalock disclosed that he has served as a principal investigator for Castle Biosciences.
a new analysis shows.
“The trend of a lower racial disparity in the VA in the proportion of melanomas with local disease and in the proportion of distant metastasis at presentation was observed across age groups,” wrote Martin A. Weinstock MD, PhD, and Rachel K. Lim, of the department of dermatology at Brown University, Providence, R.I., and the Center for Dermatoepidemiology at the VA Providence Healthcare System. The study was published online in the Journal of the American Academy of Dermatology.
“Melanoma was the fourth-most common cancer [diagnosed] in male VA patients in 2010,” wrote the authors, who also pointed out that “prior surveys found that 11%-13% of U.S. active-duty personnel routinely use sunscreen despite significant occupational sun exposure. Racial disparities are important concerns in the VA and elsewhere.”
To compare the stage of melanoma at presentation among White and non-Whites patients in the VA and in the general U.S. population, the researchers identified invasive cutaneous melanoma cases from 2000 to 2019 in the VA Corporate Data Warehouse and the Surveillance, Epidemiology and End Results Program (SEER).
They restricted the analysis to men because of the small proportion of women in the at-risk veteran population and excluded cases with an age younger than 20, those with unknown histology, and melanoma in situ. The researchers performed two-tailed z-tests to evaluate the difference in proportions of melanoma stages between the veteran population and the general population.
The analysis included 44,077 cases of invasive melanoma in the VA and 217,030 in SEER. Racial disparities in melanoma staging were substantially less pronounced in the VA than in SEER.
In the VA, localized disease represented 77.9% of melanomas among Whites versus 71.0% among non-Whites. But in SEER, localized disease represented 80.7% of melanomas among Whites versus 61.5% in non-Whites – over double the VA disparity (P < .0001).
Likewise, the disparity between Whites and nonwhites observed for regional or distant metastatic disease at presentation in the VA was lower than the disparity observed in SEER. For example, in the VA, distant metastatic disease at presentation represented 6.1% of melanomas among Whites versus 8.6% among non-Whites, while in SEER it represented 4.8% of melanomas among Whites versus 11.3% in non-Whites – again, more than double the VA disparity (P < .0001).
“These differences between the VA and SEER were less marked” among those older than 65 years, the researchers wrote. “Notably, the differences between VA and SEER in racial disparities among those greater than 65 in age were still significant for localized disease and for distant metastasis.”
The findings suggest that the VA “may be more effective in reducing racial disparities in melanoma stage at diagnosis, potentially due to all patients in the VA dataset having insured access to health care, regardless of socioeconomic status,” the researchers concluded. Similarly, the decreased difference in racial disparities observed in patients older than 65 across systems “may be related to the availability of Medicare to the older general populations. The authors acknowledged several study limitations, such as the predominantly elderly and male VA population, potentially underreported utilization of non-VA dermatologic care, and variation in geographic regions covered by each database.
Travis W. Blalock, MD, director of dermatologic surgery, Mohs micrographic surgery, and cutaneous oncology at Emory University, Atlanta, who was asked to comment on the work, said in an interview he would have liked to see a more detailed breakdown of the younger patients, “for those in their 30s and 40s, to see if this trend held up.”
He would have also liked to see how the data trended over time, adding, “while this, broadly, may be good news for our veterans, attributing this finding to a reduction in access disparity or some other organizational intervention seems a little premature. Regardless, Dr. Weinstock has given us, once again, information from our veterans to probe for the betterment of all patients.”
The researchers reported having no relevant disclosures and the study had no funding. Dr. Blalock disclosed that he has served as a principal investigator for Castle Biosciences.
a new analysis shows.
“The trend of a lower racial disparity in the VA in the proportion of melanomas with local disease and in the proportion of distant metastasis at presentation was observed across age groups,” wrote Martin A. Weinstock MD, PhD, and Rachel K. Lim, of the department of dermatology at Brown University, Providence, R.I., and the Center for Dermatoepidemiology at the VA Providence Healthcare System. The study was published online in the Journal of the American Academy of Dermatology.
“Melanoma was the fourth-most common cancer [diagnosed] in male VA patients in 2010,” wrote the authors, who also pointed out that “prior surveys found that 11%-13% of U.S. active-duty personnel routinely use sunscreen despite significant occupational sun exposure. Racial disparities are important concerns in the VA and elsewhere.”
To compare the stage of melanoma at presentation among White and non-Whites patients in the VA and in the general U.S. population, the researchers identified invasive cutaneous melanoma cases from 2000 to 2019 in the VA Corporate Data Warehouse and the Surveillance, Epidemiology and End Results Program (SEER).
They restricted the analysis to men because of the small proportion of women in the at-risk veteran population and excluded cases with an age younger than 20, those with unknown histology, and melanoma in situ. The researchers performed two-tailed z-tests to evaluate the difference in proportions of melanoma stages between the veteran population and the general population.
The analysis included 44,077 cases of invasive melanoma in the VA and 217,030 in SEER. Racial disparities in melanoma staging were substantially less pronounced in the VA than in SEER.
In the VA, localized disease represented 77.9% of melanomas among Whites versus 71.0% among non-Whites. But in SEER, localized disease represented 80.7% of melanomas among Whites versus 61.5% in non-Whites – over double the VA disparity (P < .0001).
Likewise, the disparity between Whites and nonwhites observed for regional or distant metastatic disease at presentation in the VA was lower than the disparity observed in SEER. For example, in the VA, distant metastatic disease at presentation represented 6.1% of melanomas among Whites versus 8.6% among non-Whites, while in SEER it represented 4.8% of melanomas among Whites versus 11.3% in non-Whites – again, more than double the VA disparity (P < .0001).
“These differences between the VA and SEER were less marked” among those older than 65 years, the researchers wrote. “Notably, the differences between VA and SEER in racial disparities among those greater than 65 in age were still significant for localized disease and for distant metastasis.”
The findings suggest that the VA “may be more effective in reducing racial disparities in melanoma stage at diagnosis, potentially due to all patients in the VA dataset having insured access to health care, regardless of socioeconomic status,” the researchers concluded. Similarly, the decreased difference in racial disparities observed in patients older than 65 across systems “may be related to the availability of Medicare to the older general populations. The authors acknowledged several study limitations, such as the predominantly elderly and male VA population, potentially underreported utilization of non-VA dermatologic care, and variation in geographic regions covered by each database.
Travis W. Blalock, MD, director of dermatologic surgery, Mohs micrographic surgery, and cutaneous oncology at Emory University, Atlanta, who was asked to comment on the work, said in an interview he would have liked to see a more detailed breakdown of the younger patients, “for those in their 30s and 40s, to see if this trend held up.”
He would have also liked to see how the data trended over time, adding, “while this, broadly, may be good news for our veterans, attributing this finding to a reduction in access disparity or some other organizational intervention seems a little premature. Regardless, Dr. Weinstock has given us, once again, information from our veterans to probe for the betterment of all patients.”
The researchers reported having no relevant disclosures and the study had no funding. Dr. Blalock disclosed that he has served as a principal investigator for Castle Biosciences.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Abrocitinib remains effective at 96 weeks, in older as well as younger adults
WASHINGTON – A substantial proportion of , Andrew F. Alexis, MD, MPH, reported in a late-breaker abstract session at the annual Revolutionizing Atopic Dermatitis conference.
The analysis stratified patients by age – 18-50 and over 50 years – and found that the sustained improvement with the JAK-1 selective inhibitor as monotherapy was seen regardless of age. “In practice, patients who are older tend to have had AD for a longer period of time and tend to be more difficult to treat so it’s reassuring to see that even in the over-50 age group, they show substantial responses, even with more stringent endpoints,” said Dr. Alexis, professor of clinical dermatology at Weill Cornell Medical College, New York.
At week 96, for instance, the proportion of patients who achieved at least a 75% improvement from baseline on the Eczema Area and Severity Index (EASI-75) was 73% with the 100-mg dose and 85% with the 200-mg dose in the younger age group, and 86% and 89%, respectively, in the older age group.
An EASI-90 response – one of the more stringent outcomes – was achieved by 45% and 58% in the 18-50 group and 58% and 73% in the over 50 group (for 100-mg and 200-mg doses, respectively), Dr. Alexis reported.
The interim analysis also showed dose-dependent efficacy overall up to 96 weeks in the younger age group but only up to 48 weeks in the older age group. Response to some outcome measures in patients over age 50 years was “less clearly dose dependent after week 48” than earlier, Dr. Alexis said.
The ongoing JADE EXTEND trial enrolled patients who had participated in the phase 3 JADE clinical trials. This analysis covered 1,309 patients who were enrolled by a September 2021 cutoff. The patient population leaned young: Eighty percent (1,046) were aged 18-50, and 20% (263) were over 50.
Patients who were randomly assigned to abrocitinib 200 mg or 100 mg in the parent trials continued to receive the same dose in JADE EXTEND with blinding maintained. Those who received placebo in the qualifying trial were randomly assigned to abrocitinib 200 mg or 100 mg. And patients from JADE DARE continued with their dosing of 200 mg. Grouping by age for the analysis was made based on the age recorded at the screening visit of the qualifying trial.
IGA, PP-NRS, and DLQI results
At week 96, the proportion of patients 18-50 years of age who achieved the Investigator’s Global Assessment (IGA) score of 0 or 1 (clear or almost clear) with at least a 2-grade improvement from baseline was 44% in the 100-mg group and 55% in the 200-mg group. Among patients over 50, these proportions were 51% and 58%, respectively.
The proportion of patients who achieved at least a 4-point improvement from baseline in the Peak Pruritus Numerical Rating Scale (PP-NRS) score was 54% and 66% (on 100 mg and 200 mg, respectively) among those aged 18-50, and 79% and 80%, respectively, among those over 50.
Looking at more stringent outcomes, 26% and 38% in the 18-50 group on 100 mg and 200 mg, respectively, achieved a PP-NRS of 0/1, as did 54% and 44% in the over-50 group.
Lastly, a score of less than 2 on the Dermatology Life Quality Index (DLQI 0/1) was achieved by 32% and 41% of patients aged 18-50 and by 51% and 48% of patients over 50, for the 100-mg and 200-mg doses, respectively.
The decline in dose-dependent efficacy in the older age group after 48 weeks may be due to the smaller sample of older patients and/or the fact that a higher proportion of older patients had moderate baseline disease per their IGA score, versus severe disease, compared with the younger patients, Dr. Alexis said. “We see a skewing toward a bit more severe [disease] in the younger age group compared to the older,” he noted.
Abrocitinib (Cibinqo) is approved for the treatment of moderate to severe AD in adolescents aged 12 and up and adults whose disease is not adequately controlled with other systemic treatments or those for whom the use of these drugs is not advised. It is available in a 50-mg dose for dose adjustments in special populations, but this dose was not studied in the clinical trials, Dr. Alexis noted. The interim analysis did not include safety data.
In a separate presentation in which he reviewed long-term data on AD medications, Raj Chovatiya, MD, PhD, assistant professor of dermatology at Northwestern University, Chicago, said that most patients who meet defined endpoints at week 12 of treatment with abrocitinib maintain that response over time. “By and large, there’s a steep initial rise that flattens over the long run, which is what you want to see. People getting that response are generally staying there over the course of treatment,” he said, referring to the JADE EXTEND data up to week 48.
It’s important to also appreciate, however, that the proportion of patients meeting efficacy outcomes in the trials of abrocitinib has grown well beyond 12 weeks, Dr. Chovatiya said.
Pointing to data presented at a 2021 RAD meeting depicting the proportion of 12-week nonresponders achieving a response at weeks 24 and 48 on IGA 0/1, EASI-75, and PP-NRS, Dr. Chovatiya said the level of response grew at both time points. “You’re capturing a chunk of people well beyond the primary endpoint if you keep them on therapy continuously, suggesting that ... we may need to reframe how we’re thinking about oral JAK inhibitors,” he said. “Not only are they rapidly acting, but they are medications that can provide good control and changes in the long run.”
Dr. Alexis and Dr. Chovatiya disclosed ties with Pfizer, which funded the study.
WASHINGTON – A substantial proportion of , Andrew F. Alexis, MD, MPH, reported in a late-breaker abstract session at the annual Revolutionizing Atopic Dermatitis conference.
The analysis stratified patients by age – 18-50 and over 50 years – and found that the sustained improvement with the JAK-1 selective inhibitor as monotherapy was seen regardless of age. “In practice, patients who are older tend to have had AD for a longer period of time and tend to be more difficult to treat so it’s reassuring to see that even in the over-50 age group, they show substantial responses, even with more stringent endpoints,” said Dr. Alexis, professor of clinical dermatology at Weill Cornell Medical College, New York.
At week 96, for instance, the proportion of patients who achieved at least a 75% improvement from baseline on the Eczema Area and Severity Index (EASI-75) was 73% with the 100-mg dose and 85% with the 200-mg dose in the younger age group, and 86% and 89%, respectively, in the older age group.
An EASI-90 response – one of the more stringent outcomes – was achieved by 45% and 58% in the 18-50 group and 58% and 73% in the over 50 group (for 100-mg and 200-mg doses, respectively), Dr. Alexis reported.
The interim analysis also showed dose-dependent efficacy overall up to 96 weeks in the younger age group but only up to 48 weeks in the older age group. Response to some outcome measures in patients over age 50 years was “less clearly dose dependent after week 48” than earlier, Dr. Alexis said.
The ongoing JADE EXTEND trial enrolled patients who had participated in the phase 3 JADE clinical trials. This analysis covered 1,309 patients who were enrolled by a September 2021 cutoff. The patient population leaned young: Eighty percent (1,046) were aged 18-50, and 20% (263) were over 50.
Patients who were randomly assigned to abrocitinib 200 mg or 100 mg in the parent trials continued to receive the same dose in JADE EXTEND with blinding maintained. Those who received placebo in the qualifying trial were randomly assigned to abrocitinib 200 mg or 100 mg. And patients from JADE DARE continued with their dosing of 200 mg. Grouping by age for the analysis was made based on the age recorded at the screening visit of the qualifying trial.
IGA, PP-NRS, and DLQI results
At week 96, the proportion of patients 18-50 years of age who achieved the Investigator’s Global Assessment (IGA) score of 0 or 1 (clear or almost clear) with at least a 2-grade improvement from baseline was 44% in the 100-mg group and 55% in the 200-mg group. Among patients over 50, these proportions were 51% and 58%, respectively.
The proportion of patients who achieved at least a 4-point improvement from baseline in the Peak Pruritus Numerical Rating Scale (PP-NRS) score was 54% and 66% (on 100 mg and 200 mg, respectively) among those aged 18-50, and 79% and 80%, respectively, among those over 50.
Looking at more stringent outcomes, 26% and 38% in the 18-50 group on 100 mg and 200 mg, respectively, achieved a PP-NRS of 0/1, as did 54% and 44% in the over-50 group.
Lastly, a score of less than 2 on the Dermatology Life Quality Index (DLQI 0/1) was achieved by 32% and 41% of patients aged 18-50 and by 51% and 48% of patients over 50, for the 100-mg and 200-mg doses, respectively.
The decline in dose-dependent efficacy in the older age group after 48 weeks may be due to the smaller sample of older patients and/or the fact that a higher proportion of older patients had moderate baseline disease per their IGA score, versus severe disease, compared with the younger patients, Dr. Alexis said. “We see a skewing toward a bit more severe [disease] in the younger age group compared to the older,” he noted.
Abrocitinib (Cibinqo) is approved for the treatment of moderate to severe AD in adolescents aged 12 and up and adults whose disease is not adequately controlled with other systemic treatments or those for whom the use of these drugs is not advised. It is available in a 50-mg dose for dose adjustments in special populations, but this dose was not studied in the clinical trials, Dr. Alexis noted. The interim analysis did not include safety data.
In a separate presentation in which he reviewed long-term data on AD medications, Raj Chovatiya, MD, PhD, assistant professor of dermatology at Northwestern University, Chicago, said that most patients who meet defined endpoints at week 12 of treatment with abrocitinib maintain that response over time. “By and large, there’s a steep initial rise that flattens over the long run, which is what you want to see. People getting that response are generally staying there over the course of treatment,” he said, referring to the JADE EXTEND data up to week 48.
It’s important to also appreciate, however, that the proportion of patients meeting efficacy outcomes in the trials of abrocitinib has grown well beyond 12 weeks, Dr. Chovatiya said.
Pointing to data presented at a 2021 RAD meeting depicting the proportion of 12-week nonresponders achieving a response at weeks 24 and 48 on IGA 0/1, EASI-75, and PP-NRS, Dr. Chovatiya said the level of response grew at both time points. “You’re capturing a chunk of people well beyond the primary endpoint if you keep them on therapy continuously, suggesting that ... we may need to reframe how we’re thinking about oral JAK inhibitors,” he said. “Not only are they rapidly acting, but they are medications that can provide good control and changes in the long run.”
Dr. Alexis and Dr. Chovatiya disclosed ties with Pfizer, which funded the study.
WASHINGTON – A substantial proportion of , Andrew F. Alexis, MD, MPH, reported in a late-breaker abstract session at the annual Revolutionizing Atopic Dermatitis conference.
The analysis stratified patients by age – 18-50 and over 50 years – and found that the sustained improvement with the JAK-1 selective inhibitor as monotherapy was seen regardless of age. “In practice, patients who are older tend to have had AD for a longer period of time and tend to be more difficult to treat so it’s reassuring to see that even in the over-50 age group, they show substantial responses, even with more stringent endpoints,” said Dr. Alexis, professor of clinical dermatology at Weill Cornell Medical College, New York.
At week 96, for instance, the proportion of patients who achieved at least a 75% improvement from baseline on the Eczema Area and Severity Index (EASI-75) was 73% with the 100-mg dose and 85% with the 200-mg dose in the younger age group, and 86% and 89%, respectively, in the older age group.
An EASI-90 response – one of the more stringent outcomes – was achieved by 45% and 58% in the 18-50 group and 58% and 73% in the over 50 group (for 100-mg and 200-mg doses, respectively), Dr. Alexis reported.
The interim analysis also showed dose-dependent efficacy overall up to 96 weeks in the younger age group but only up to 48 weeks in the older age group. Response to some outcome measures in patients over age 50 years was “less clearly dose dependent after week 48” than earlier, Dr. Alexis said.
The ongoing JADE EXTEND trial enrolled patients who had participated in the phase 3 JADE clinical trials. This analysis covered 1,309 patients who were enrolled by a September 2021 cutoff. The patient population leaned young: Eighty percent (1,046) were aged 18-50, and 20% (263) were over 50.
Patients who were randomly assigned to abrocitinib 200 mg or 100 mg in the parent trials continued to receive the same dose in JADE EXTEND with blinding maintained. Those who received placebo in the qualifying trial were randomly assigned to abrocitinib 200 mg or 100 mg. And patients from JADE DARE continued with their dosing of 200 mg. Grouping by age for the analysis was made based on the age recorded at the screening visit of the qualifying trial.
IGA, PP-NRS, and DLQI results
At week 96, the proportion of patients 18-50 years of age who achieved the Investigator’s Global Assessment (IGA) score of 0 or 1 (clear or almost clear) with at least a 2-grade improvement from baseline was 44% in the 100-mg group and 55% in the 200-mg group. Among patients over 50, these proportions were 51% and 58%, respectively.
The proportion of patients who achieved at least a 4-point improvement from baseline in the Peak Pruritus Numerical Rating Scale (PP-NRS) score was 54% and 66% (on 100 mg and 200 mg, respectively) among those aged 18-50, and 79% and 80%, respectively, among those over 50.
Looking at more stringent outcomes, 26% and 38% in the 18-50 group on 100 mg and 200 mg, respectively, achieved a PP-NRS of 0/1, as did 54% and 44% in the over-50 group.
Lastly, a score of less than 2 on the Dermatology Life Quality Index (DLQI 0/1) was achieved by 32% and 41% of patients aged 18-50 and by 51% and 48% of patients over 50, for the 100-mg and 200-mg doses, respectively.
The decline in dose-dependent efficacy in the older age group after 48 weeks may be due to the smaller sample of older patients and/or the fact that a higher proportion of older patients had moderate baseline disease per their IGA score, versus severe disease, compared with the younger patients, Dr. Alexis said. “We see a skewing toward a bit more severe [disease] in the younger age group compared to the older,” he noted.
Abrocitinib (Cibinqo) is approved for the treatment of moderate to severe AD in adolescents aged 12 and up and adults whose disease is not adequately controlled with other systemic treatments or those for whom the use of these drugs is not advised. It is available in a 50-mg dose for dose adjustments in special populations, but this dose was not studied in the clinical trials, Dr. Alexis noted. The interim analysis did not include safety data.
In a separate presentation in which he reviewed long-term data on AD medications, Raj Chovatiya, MD, PhD, assistant professor of dermatology at Northwestern University, Chicago, said that most patients who meet defined endpoints at week 12 of treatment with abrocitinib maintain that response over time. “By and large, there’s a steep initial rise that flattens over the long run, which is what you want to see. People getting that response are generally staying there over the course of treatment,” he said, referring to the JADE EXTEND data up to week 48.
It’s important to also appreciate, however, that the proportion of patients meeting efficacy outcomes in the trials of abrocitinib has grown well beyond 12 weeks, Dr. Chovatiya said.
Pointing to data presented at a 2021 RAD meeting depicting the proportion of 12-week nonresponders achieving a response at weeks 24 and 48 on IGA 0/1, EASI-75, and PP-NRS, Dr. Chovatiya said the level of response grew at both time points. “You’re capturing a chunk of people well beyond the primary endpoint if you keep them on therapy continuously, suggesting that ... we may need to reframe how we’re thinking about oral JAK inhibitors,” he said. “Not only are they rapidly acting, but they are medications that can provide good control and changes in the long run.”
Dr. Alexis and Dr. Chovatiya disclosed ties with Pfizer, which funded the study.
AT RAD 2023
We may need a new defense against new COVID variants
At the end of 2022, the European Medicines Agency’s Emergency Task Force warned European regulatory bodies, governments, and doctors that Antiviral drugs remain available but have many limitations. And, of course, there are still vaccines, which can significantly reduce (but not remove) the risk of severe cases and decrease the number of deaths, although they have lost the efficacy that they once had in countering the original virus.
Research therefore continues. Immunologists continue to search for new targets to synthesize broadly neutralizing monoclonal antibodies for treating or preventing the infection. These results could also lead to new vaccines that induce longer-lasting immunity not only against the thousands of subvariants and recombinant versions of SARS-CoV-2 being identified around the world, but also possibly against other coronaviruses that could emerge in the coming years. A study conducted at Stanford (Calif.) University and published in the journal Science Translational Medicine has afforded a glimmer of hope by discovering the broadly neutralizing efficacy of some antibodies produced by macaque monkeys in response to vaccination with AS03 (squalene) adjuvanted monovalent subunit vaccines.
The speed with which the virus continues to evolve has rendered the plan for annual vaccine updates, which initially was envisioned early in the pandemic, unfeasible for the time being. In 2020, scientists were considering updating vaccines annually based on the prevalent variants of the disease, similar to the approach to the flu. Perhaps that day will come, but in the meantime, laboratories are pursuing other routes: finding spike epitopes that are preserved more than others each time the virus evolves or focusing on other virus proteins that still manage to induce a neutralizing antibody response.
Eventually, artificial intelligence might be able to custom design monoclonal antibodies that are even more effective than natural ones. Or researchers could completely change tack and shift their attention to the host, rather than the virus itself.
This is the approach taken by one study published in Nature Microbiology, which starts from a simple assumption: SARS-CoV-2 continues to modify its spike protein because of the evolutionary pressure of the antibodies produced by millions of infected people, but all these variants and subvariants, both present and future, enter cells by binding – not solely, but mostly – to the ACE2 receptor. Instead of neutralizing the virus, why not try to block its access to the cells occupying its route in? In this way, we could also be ready for future emerging sarbecoviruses that will have a spike sequence that cannot yet be predicted.
Researchers at Rockefeller University, New York, have generated six human monoclonal antibodies that bind to the ACE2 receptor, rather than to the spike, preventing infection by all sarbecoviruses tested, even at low concentrations, including the virus that originated in Wuhan, China; the aggressive Delta variant; and various forms of Omicron.
The monoclonal antibodies bind to the ACE2 receptor at a part of the protein that is distal to the active enzyme portion that converts angiotensin and does not modify its expression on the cell surface. Therefore, no adverse effects are expected at this level. In animal models, these monoclonal antibodies succeed in stopping the infection. Moving into the clinical phase will be needed to find out if it will be possible to create products adapted to preventing and treating all SARS-CoV-2 variants, and perhaps also the next coronavirus large enough to spill over into a new epidemic that threatens the human race.
This article was translated from Univadis Italy. A version appeared on Medscape.com.
At the end of 2022, the European Medicines Agency’s Emergency Task Force warned European regulatory bodies, governments, and doctors that Antiviral drugs remain available but have many limitations. And, of course, there are still vaccines, which can significantly reduce (but not remove) the risk of severe cases and decrease the number of deaths, although they have lost the efficacy that they once had in countering the original virus.
Research therefore continues. Immunologists continue to search for new targets to synthesize broadly neutralizing monoclonal antibodies for treating or preventing the infection. These results could also lead to new vaccines that induce longer-lasting immunity not only against the thousands of subvariants and recombinant versions of SARS-CoV-2 being identified around the world, but also possibly against other coronaviruses that could emerge in the coming years. A study conducted at Stanford (Calif.) University and published in the journal Science Translational Medicine has afforded a glimmer of hope by discovering the broadly neutralizing efficacy of some antibodies produced by macaque monkeys in response to vaccination with AS03 (squalene) adjuvanted monovalent subunit vaccines.
The speed with which the virus continues to evolve has rendered the plan for annual vaccine updates, which initially was envisioned early in the pandemic, unfeasible for the time being. In 2020, scientists were considering updating vaccines annually based on the prevalent variants of the disease, similar to the approach to the flu. Perhaps that day will come, but in the meantime, laboratories are pursuing other routes: finding spike epitopes that are preserved more than others each time the virus evolves or focusing on other virus proteins that still manage to induce a neutralizing antibody response.
Eventually, artificial intelligence might be able to custom design monoclonal antibodies that are even more effective than natural ones. Or researchers could completely change tack and shift their attention to the host, rather than the virus itself.
This is the approach taken by one study published in Nature Microbiology, which starts from a simple assumption: SARS-CoV-2 continues to modify its spike protein because of the evolutionary pressure of the antibodies produced by millions of infected people, but all these variants and subvariants, both present and future, enter cells by binding – not solely, but mostly – to the ACE2 receptor. Instead of neutralizing the virus, why not try to block its access to the cells occupying its route in? In this way, we could also be ready for future emerging sarbecoviruses that will have a spike sequence that cannot yet be predicted.
Researchers at Rockefeller University, New York, have generated six human monoclonal antibodies that bind to the ACE2 receptor, rather than to the spike, preventing infection by all sarbecoviruses tested, even at low concentrations, including the virus that originated in Wuhan, China; the aggressive Delta variant; and various forms of Omicron.
The monoclonal antibodies bind to the ACE2 receptor at a part of the protein that is distal to the active enzyme portion that converts angiotensin and does not modify its expression on the cell surface. Therefore, no adverse effects are expected at this level. In animal models, these monoclonal antibodies succeed in stopping the infection. Moving into the clinical phase will be needed to find out if it will be possible to create products adapted to preventing and treating all SARS-CoV-2 variants, and perhaps also the next coronavirus large enough to spill over into a new epidemic that threatens the human race.
This article was translated from Univadis Italy. A version appeared on Medscape.com.
At the end of 2022, the European Medicines Agency’s Emergency Task Force warned European regulatory bodies, governments, and doctors that Antiviral drugs remain available but have many limitations. And, of course, there are still vaccines, which can significantly reduce (but not remove) the risk of severe cases and decrease the number of deaths, although they have lost the efficacy that they once had in countering the original virus.
Research therefore continues. Immunologists continue to search for new targets to synthesize broadly neutralizing monoclonal antibodies for treating or preventing the infection. These results could also lead to new vaccines that induce longer-lasting immunity not only against the thousands of subvariants and recombinant versions of SARS-CoV-2 being identified around the world, but also possibly against other coronaviruses that could emerge in the coming years. A study conducted at Stanford (Calif.) University and published in the journal Science Translational Medicine has afforded a glimmer of hope by discovering the broadly neutralizing efficacy of some antibodies produced by macaque monkeys in response to vaccination with AS03 (squalene) adjuvanted monovalent subunit vaccines.
The speed with which the virus continues to evolve has rendered the plan for annual vaccine updates, which initially was envisioned early in the pandemic, unfeasible for the time being. In 2020, scientists were considering updating vaccines annually based on the prevalent variants of the disease, similar to the approach to the flu. Perhaps that day will come, but in the meantime, laboratories are pursuing other routes: finding spike epitopes that are preserved more than others each time the virus evolves or focusing on other virus proteins that still manage to induce a neutralizing antibody response.
Eventually, artificial intelligence might be able to custom design monoclonal antibodies that are even more effective than natural ones. Or researchers could completely change tack and shift their attention to the host, rather than the virus itself.
This is the approach taken by one study published in Nature Microbiology, which starts from a simple assumption: SARS-CoV-2 continues to modify its spike protein because of the evolutionary pressure of the antibodies produced by millions of infected people, but all these variants and subvariants, both present and future, enter cells by binding – not solely, but mostly – to the ACE2 receptor. Instead of neutralizing the virus, why not try to block its access to the cells occupying its route in? In this way, we could also be ready for future emerging sarbecoviruses that will have a spike sequence that cannot yet be predicted.
Researchers at Rockefeller University, New York, have generated six human monoclonal antibodies that bind to the ACE2 receptor, rather than to the spike, preventing infection by all sarbecoviruses tested, even at low concentrations, including the virus that originated in Wuhan, China; the aggressive Delta variant; and various forms of Omicron.
The monoclonal antibodies bind to the ACE2 receptor at a part of the protein that is distal to the active enzyme portion that converts angiotensin and does not modify its expression on the cell surface. Therefore, no adverse effects are expected at this level. In animal models, these monoclonal antibodies succeed in stopping the infection. Moving into the clinical phase will be needed to find out if it will be possible to create products adapted to preventing and treating all SARS-CoV-2 variants, and perhaps also the next coronavirus large enough to spill over into a new epidemic that threatens the human race.
This article was translated from Univadis Italy. A version appeared on Medscape.com.
COVID nonvaccination linked with avoidable hospitalizations
A retrospective, population-based cohort study in Alberta, Edmonton, found that between late September 2021 and late January 2022, eligible unvaccinated patients with COVID-19 had a nearly 10-fold higher risk for hospitalization than did patients who were fully vaccinated with two doses. Unvaccinated patients had a nearly 21-fold higher risk than did patients who were boosted with three doses.
“We have shown that eligible nonvaccinated persons, especially in the age strata 50-79 years, accounted for 3,000-4,000 potentially avoidable hospitalizations, 35,000-40,000 avoidable bed-days, and $100–$110 million [Canadian dollars] in avoidable health care costs during a 120-day period coinciding with the fourth (Delta) and fifth (Omicron) COVID-19 waves, respectively,” wrote Sean M. Bagshaw, MD, chair of critical care medicine at the University of Alberta, Edmonton, and colleagues.
The findings were published in the Canadian Journal of Public Health.
‘Unsatisfactory’ vaccine uptake
While a previous study by Dr. Bagshaw and colleagues recently showed that higher vaccine uptake could have avoided significant intensive care unit admissions and costs, the researchers sought to expand their analysis to include non-ICU use.
The current study examined data from the government of Alberta and the Discharge Abstract Database to assess vaccination status and hospitalization with confirmed SARS-CoV-2. Secondary outcomes included avoidable hospitalizations, avoidable hospital bed-days, and the potential cost avoidance related to COVID-19.
During the study period, “societal factors contributed to an unsatisfactory voluntary vaccine uptake, particularly in the province of Alberta,” wrote the authors, adding that “only 63.7% and 2.7% of the eligible population in Alberta [had] received two (full) and three (boosted) COVID-19 vaccine doses as of September 27, 2021.”
The analysis found the highest number of hospitalizations among unvaccinated patients (n = 3,835), compared with vaccinated (n = 1,907) and boosted patients (n = 481). This finding yielded a risk ratio (RR) of hospitalization of 9.7 for unvaccinated patients, compared with fully vaccinated patients, and an RR of 20.6, compared with patients who were boosted. Unvaccinated patients aged 60-69 years had the highest RR for hospitalization, compared with vaccinated (RR, 16.4) and boosted patients (RR, 151.9).
The estimated number of avoidable hospitalizations for unvaccinated patients was 3,439 (total of 36,331 bed-days), compared with vaccinated patients, and 3,764 (total of 40,185 bed-days), compared with boosted patients.
The avoidable hospitalization-related costs for unvaccinated patients totaled $101.4 million (Canadian dollars) if they had been vaccinated and $110.24 million if they had been boosted.
“Moreover, strained hospital systems and the widespread adoption of crisis standards of care in response to surges in COVID-19 hospitalizations have contributed to unnecessary excess deaths,” wrote the authors. “These are preventable and missed public health opportunities that provoked massive health system disruptions and resource diversions, including deferral of routine health services (e.g., cancer and chronic disease screening and monitoring and scheduled vaccinations), postponement of scheduled procedures and surgeries, and redeployment of health care professionals.”
Dr. Bagshaw said in an interview that he was not surprised by the findings. “However, I wonder whether the public and those who direct policy and make decisions about the health system would be interested in better understanding the scope and sheer disruption the health system suffered due to COVID-19,” he said.
The current study suggests that “at least some of this could have been avoided,” said Dr. Bagshaw. “I hope we – that is the public, users of the health system, decision-makers and health care professionals – can learn from our experiences.” Studies such as the current analysis “will reinforce the importance of timely and clearly articulated public health promotion, education, and policy,” he added.
Economic benefit underestimated
Commenting on the study, David Fisman, MD, MPH, an epidemiologist and professor at the University of Toronto, said: “The approach these investigators have taken is clear and straightforward. It is easy to reproduce. It is also entirely consistent with what other scientific groups have been demonstrating for a couple of years now.” Dr. Fisman was not involved with the study.
A group led by Dr. Fisman as senior author has just completed a study examining the effectiveness of the Canadian pandemic response, compared with responses in four peer countries. In the as-yet unpublished paper, the researchers concluded that “relative to the United States, United Kingdom, and France, the Canadian pandemic response was estimated to have averted 94,492, 64,306, and 13,641 deaths, respectively, with more than 480,000 hospitalizations averted and 1 million QALY [quality-adjusted life-years] saved, relative to the United States. A United States pandemic response applied to Canada would have resulted in more than $40 billion in economic losses due to healthcare expenditures and lost QALY; losses relative to the United Kingdom and France would have been $21 billion and $5 billion, respectively. By contrast, an Australian pandemic response would have averted over 28,000 additional deaths and averted nearly $9 billion in costs in Canada.”
Dr. Fisman added that while the current researchers focused their study on the direct protective effects of vaccines, “we know that, even with initial waves of Omicron, vaccinated individuals continued to be protected against infection as well as disease, and even if they were infected, we know from household contact studies that they were less infectious to others. That means that even though the implicit estimate of cost savings that could have been achieved through better coverage are pretty high in this paper, the economic benefit of vaccination is underestimated in this analysis, because we can’t quantify the infections that never happened because of vaccination.”
The study was supported by the Strategic Clinical Networks, Alberta Health Services. Dr. Bagshaw declared no relevant financial relationships. Dr. Fisman has taken part in advisory boards for Seqirus, Pfizer, AstraZeneca, Sanofi, and Merck vaccines during the past 3 years.
A version of this article first appeared on Medscape.com.
A retrospective, population-based cohort study in Alberta, Edmonton, found that between late September 2021 and late January 2022, eligible unvaccinated patients with COVID-19 had a nearly 10-fold higher risk for hospitalization than did patients who were fully vaccinated with two doses. Unvaccinated patients had a nearly 21-fold higher risk than did patients who were boosted with three doses.
“We have shown that eligible nonvaccinated persons, especially in the age strata 50-79 years, accounted for 3,000-4,000 potentially avoidable hospitalizations, 35,000-40,000 avoidable bed-days, and $100–$110 million [Canadian dollars] in avoidable health care costs during a 120-day period coinciding with the fourth (Delta) and fifth (Omicron) COVID-19 waves, respectively,” wrote Sean M. Bagshaw, MD, chair of critical care medicine at the University of Alberta, Edmonton, and colleagues.
The findings were published in the Canadian Journal of Public Health.
‘Unsatisfactory’ vaccine uptake
While a previous study by Dr. Bagshaw and colleagues recently showed that higher vaccine uptake could have avoided significant intensive care unit admissions and costs, the researchers sought to expand their analysis to include non-ICU use.
The current study examined data from the government of Alberta and the Discharge Abstract Database to assess vaccination status and hospitalization with confirmed SARS-CoV-2. Secondary outcomes included avoidable hospitalizations, avoidable hospital bed-days, and the potential cost avoidance related to COVID-19.
During the study period, “societal factors contributed to an unsatisfactory voluntary vaccine uptake, particularly in the province of Alberta,” wrote the authors, adding that “only 63.7% and 2.7% of the eligible population in Alberta [had] received two (full) and three (boosted) COVID-19 vaccine doses as of September 27, 2021.”
The analysis found the highest number of hospitalizations among unvaccinated patients (n = 3,835), compared with vaccinated (n = 1,907) and boosted patients (n = 481). This finding yielded a risk ratio (RR) of hospitalization of 9.7 for unvaccinated patients, compared with fully vaccinated patients, and an RR of 20.6, compared with patients who were boosted. Unvaccinated patients aged 60-69 years had the highest RR for hospitalization, compared with vaccinated (RR, 16.4) and boosted patients (RR, 151.9).
The estimated number of avoidable hospitalizations for unvaccinated patients was 3,439 (total of 36,331 bed-days), compared with vaccinated patients, and 3,764 (total of 40,185 bed-days), compared with boosted patients.
The avoidable hospitalization-related costs for unvaccinated patients totaled $101.4 million (Canadian dollars) if they had been vaccinated and $110.24 million if they had been boosted.
“Moreover, strained hospital systems and the widespread adoption of crisis standards of care in response to surges in COVID-19 hospitalizations have contributed to unnecessary excess deaths,” wrote the authors. “These are preventable and missed public health opportunities that provoked massive health system disruptions and resource diversions, including deferral of routine health services (e.g., cancer and chronic disease screening and monitoring and scheduled vaccinations), postponement of scheduled procedures and surgeries, and redeployment of health care professionals.”
Dr. Bagshaw said in an interview that he was not surprised by the findings. “However, I wonder whether the public and those who direct policy and make decisions about the health system would be interested in better understanding the scope and sheer disruption the health system suffered due to COVID-19,” he said.
The current study suggests that “at least some of this could have been avoided,” said Dr. Bagshaw. “I hope we – that is the public, users of the health system, decision-makers and health care professionals – can learn from our experiences.” Studies such as the current analysis “will reinforce the importance of timely and clearly articulated public health promotion, education, and policy,” he added.
Economic benefit underestimated
Commenting on the study, David Fisman, MD, MPH, an epidemiologist and professor at the University of Toronto, said: “The approach these investigators have taken is clear and straightforward. It is easy to reproduce. It is also entirely consistent with what other scientific groups have been demonstrating for a couple of years now.” Dr. Fisman was not involved with the study.
A group led by Dr. Fisman as senior author has just completed a study examining the effectiveness of the Canadian pandemic response, compared with responses in four peer countries. In the as-yet unpublished paper, the researchers concluded that “relative to the United States, United Kingdom, and France, the Canadian pandemic response was estimated to have averted 94,492, 64,306, and 13,641 deaths, respectively, with more than 480,000 hospitalizations averted and 1 million QALY [quality-adjusted life-years] saved, relative to the United States. A United States pandemic response applied to Canada would have resulted in more than $40 billion in economic losses due to healthcare expenditures and lost QALY; losses relative to the United Kingdom and France would have been $21 billion and $5 billion, respectively. By contrast, an Australian pandemic response would have averted over 28,000 additional deaths and averted nearly $9 billion in costs in Canada.”
Dr. Fisman added that while the current researchers focused their study on the direct protective effects of vaccines, “we know that, even with initial waves of Omicron, vaccinated individuals continued to be protected against infection as well as disease, and even if they were infected, we know from household contact studies that they were less infectious to others. That means that even though the implicit estimate of cost savings that could have been achieved through better coverage are pretty high in this paper, the economic benefit of vaccination is underestimated in this analysis, because we can’t quantify the infections that never happened because of vaccination.”
The study was supported by the Strategic Clinical Networks, Alberta Health Services. Dr. Bagshaw declared no relevant financial relationships. Dr. Fisman has taken part in advisory boards for Seqirus, Pfizer, AstraZeneca, Sanofi, and Merck vaccines during the past 3 years.
A version of this article first appeared on Medscape.com.
A retrospective, population-based cohort study in Alberta, Edmonton, found that between late September 2021 and late January 2022, eligible unvaccinated patients with COVID-19 had a nearly 10-fold higher risk for hospitalization than did patients who were fully vaccinated with two doses. Unvaccinated patients had a nearly 21-fold higher risk than did patients who were boosted with three doses.
“We have shown that eligible nonvaccinated persons, especially in the age strata 50-79 years, accounted for 3,000-4,000 potentially avoidable hospitalizations, 35,000-40,000 avoidable bed-days, and $100–$110 million [Canadian dollars] in avoidable health care costs during a 120-day period coinciding with the fourth (Delta) and fifth (Omicron) COVID-19 waves, respectively,” wrote Sean M. Bagshaw, MD, chair of critical care medicine at the University of Alberta, Edmonton, and colleagues.
The findings were published in the Canadian Journal of Public Health.
‘Unsatisfactory’ vaccine uptake
While a previous study by Dr. Bagshaw and colleagues recently showed that higher vaccine uptake could have avoided significant intensive care unit admissions and costs, the researchers sought to expand their analysis to include non-ICU use.
The current study examined data from the government of Alberta and the Discharge Abstract Database to assess vaccination status and hospitalization with confirmed SARS-CoV-2. Secondary outcomes included avoidable hospitalizations, avoidable hospital bed-days, and the potential cost avoidance related to COVID-19.
During the study period, “societal factors contributed to an unsatisfactory voluntary vaccine uptake, particularly in the province of Alberta,” wrote the authors, adding that “only 63.7% and 2.7% of the eligible population in Alberta [had] received two (full) and three (boosted) COVID-19 vaccine doses as of September 27, 2021.”
The analysis found the highest number of hospitalizations among unvaccinated patients (n = 3,835), compared with vaccinated (n = 1,907) and boosted patients (n = 481). This finding yielded a risk ratio (RR) of hospitalization of 9.7 for unvaccinated patients, compared with fully vaccinated patients, and an RR of 20.6, compared with patients who were boosted. Unvaccinated patients aged 60-69 years had the highest RR for hospitalization, compared with vaccinated (RR, 16.4) and boosted patients (RR, 151.9).
The estimated number of avoidable hospitalizations for unvaccinated patients was 3,439 (total of 36,331 bed-days), compared with vaccinated patients, and 3,764 (total of 40,185 bed-days), compared with boosted patients.
The avoidable hospitalization-related costs for unvaccinated patients totaled $101.4 million (Canadian dollars) if they had been vaccinated and $110.24 million if they had been boosted.
“Moreover, strained hospital systems and the widespread adoption of crisis standards of care in response to surges in COVID-19 hospitalizations have contributed to unnecessary excess deaths,” wrote the authors. “These are preventable and missed public health opportunities that provoked massive health system disruptions and resource diversions, including deferral of routine health services (e.g., cancer and chronic disease screening and monitoring and scheduled vaccinations), postponement of scheduled procedures and surgeries, and redeployment of health care professionals.”
Dr. Bagshaw said in an interview that he was not surprised by the findings. “However, I wonder whether the public and those who direct policy and make decisions about the health system would be interested in better understanding the scope and sheer disruption the health system suffered due to COVID-19,” he said.
The current study suggests that “at least some of this could have been avoided,” said Dr. Bagshaw. “I hope we – that is the public, users of the health system, decision-makers and health care professionals – can learn from our experiences.” Studies such as the current analysis “will reinforce the importance of timely and clearly articulated public health promotion, education, and policy,” he added.
Economic benefit underestimated
Commenting on the study, David Fisman, MD, MPH, an epidemiologist and professor at the University of Toronto, said: “The approach these investigators have taken is clear and straightforward. It is easy to reproduce. It is also entirely consistent with what other scientific groups have been demonstrating for a couple of years now.” Dr. Fisman was not involved with the study.
A group led by Dr. Fisman as senior author has just completed a study examining the effectiveness of the Canadian pandemic response, compared with responses in four peer countries. In the as-yet unpublished paper, the researchers concluded that “relative to the United States, United Kingdom, and France, the Canadian pandemic response was estimated to have averted 94,492, 64,306, and 13,641 deaths, respectively, with more than 480,000 hospitalizations averted and 1 million QALY [quality-adjusted life-years] saved, relative to the United States. A United States pandemic response applied to Canada would have resulted in more than $40 billion in economic losses due to healthcare expenditures and lost QALY; losses relative to the United Kingdom and France would have been $21 billion and $5 billion, respectively. By contrast, an Australian pandemic response would have averted over 28,000 additional deaths and averted nearly $9 billion in costs in Canada.”
Dr. Fisman added that while the current researchers focused their study on the direct protective effects of vaccines, “we know that, even with initial waves of Omicron, vaccinated individuals continued to be protected against infection as well as disease, and even if they were infected, we know from household contact studies that they were less infectious to others. That means that even though the implicit estimate of cost savings that could have been achieved through better coverage are pretty high in this paper, the economic benefit of vaccination is underestimated in this analysis, because we can’t quantify the infections that never happened because of vaccination.”
The study was supported by the Strategic Clinical Networks, Alberta Health Services. Dr. Bagshaw declared no relevant financial relationships. Dr. Fisman has taken part in advisory boards for Seqirus, Pfizer, AstraZeneca, Sanofi, and Merck vaccines during the past 3 years.
A version of this article first appeared on Medscape.com.
FROM CANADIAN JOURNAL OF PUBLIC HEALTH
The new vaccine your patients may not want
Compared with the complicated and ever-changing recommended vaccine schedule for infants and children, vaccines for adults have been straightforward. Adults without compromised immunity who received all their childhood vaccinations are eligible for a tetanus and diphtheria (Td) or tetanus, diphtheria, and pertussis (Tdap) booster every 10 years, recombinant herpes zoster vaccine at age 50, and pneumococcal vaccines at age 65, along with annual influenza and (likely) COVID-19 vaccines. Last year, due to rising rates of acute hepatitis B, the Centers for Disease Control and Prevention first recommended universal hepatitis B vaccination for adults aged 19-59 years without a record of previous hepatitis B infection or vaccination.
An additional routine vaccine for adults is now on the horizon. The U.S. Food and Drug Administration recently approved Arexvy, a vaccine against respiratory syncytial virus (RSV) for adults aged 60 years or older. Two more RSV vaccines are in the final stages of development. Why should family physicians prioritize vaccinating older adults against RSV, and how can we incorporate this new vaccine into our practices and overcome patient hesitancy to receive yet another vaccine?
Clinicians tend to think of RSV as a serious disease in young children – which it is – but data suggest that in 2019, RSV infection led to more than 100,000 hospitalizations and 7,700 deaths in older adults in the United States. In a randomized controlled trial of 25,000 adults aged 60 years or older with a median of 6.7 months of follow-up, Arexvy reduced severe RSV disease by 94% and RSV-related acute respiratory infections by 71%, with similar effectiveness in adults with underlying health conditions. That’s considerably better protection than current influenza vaccines and comparable to COVID-19 mRNA vaccines before variants became widespread. Pain and fatigue were the most common side effects and usually resolved within 1-2 days.
Although the seasonal pattern of RSV shifted during the COVID-19 pandemic, RSV season historically begins in October, peaks in December, and ends in April. If the vaccine is recommended by the CDC and is widely available by fall, as the manufacturer, GSK, expects, it could be administered around the same time as influenza and COVID-19 vaccines.
The challenges of incorporating this new vaccine into practice will feel familiar: Many of our patients won’t have heard about it, may feel that they don’t need it, or may decline it because of concerns about side effects, real or imagined. (Of note, the FDA is requiring GSK to perform a postmarketing study to rule out associations with rare cases of Guillain-Barré syndrome and acute disseminated encephalomyelitis, and the company also plans to monitor the incidence of atrial fibrillation, which was slightly more common in the vaccine group than the placebo group.)
While a strong recommendation from a family physician is often enough to convince patients to accept vaccination, rampant misinformation during the pandemic may have worsened vaccine hesitancy for some. It may feel like a fruitless exercise to try to convince adults who have refused COVID-19 and influenza vaccines to accept a newer vaccine against a respiratory virus that causes less serious illness overall. But with other RSV vaccines and monoclonal antibodies for older adults and infants likely to be approved soon, it’s important for us to start laying the groundwork now by educating colleagues, staff, and patients about preventing serious illness caused by RSV.
Dr. Lin is an associate professor in the Department of Family Medicine at Georgetown University and a staff physician atMedStar Health Center, both in Washington. He has received income from UpToDate, Wiley-Blackwell, and the American Academy of Family Physicians.
A version of this article first appeared on Medscape.com.
Compared with the complicated and ever-changing recommended vaccine schedule for infants and children, vaccines for adults have been straightforward. Adults without compromised immunity who received all their childhood vaccinations are eligible for a tetanus and diphtheria (Td) or tetanus, diphtheria, and pertussis (Tdap) booster every 10 years, recombinant herpes zoster vaccine at age 50, and pneumococcal vaccines at age 65, along with annual influenza and (likely) COVID-19 vaccines. Last year, due to rising rates of acute hepatitis B, the Centers for Disease Control and Prevention first recommended universal hepatitis B vaccination for adults aged 19-59 years without a record of previous hepatitis B infection or vaccination.
An additional routine vaccine for adults is now on the horizon. The U.S. Food and Drug Administration recently approved Arexvy, a vaccine against respiratory syncytial virus (RSV) for adults aged 60 years or older. Two more RSV vaccines are in the final stages of development. Why should family physicians prioritize vaccinating older adults against RSV, and how can we incorporate this new vaccine into our practices and overcome patient hesitancy to receive yet another vaccine?
Clinicians tend to think of RSV as a serious disease in young children – which it is – but data suggest that in 2019, RSV infection led to more than 100,000 hospitalizations and 7,700 deaths in older adults in the United States. In a randomized controlled trial of 25,000 adults aged 60 years or older with a median of 6.7 months of follow-up, Arexvy reduced severe RSV disease by 94% and RSV-related acute respiratory infections by 71%, with similar effectiveness in adults with underlying health conditions. That’s considerably better protection than current influenza vaccines and comparable to COVID-19 mRNA vaccines before variants became widespread. Pain and fatigue were the most common side effects and usually resolved within 1-2 days.
Although the seasonal pattern of RSV shifted during the COVID-19 pandemic, RSV season historically begins in October, peaks in December, and ends in April. If the vaccine is recommended by the CDC and is widely available by fall, as the manufacturer, GSK, expects, it could be administered around the same time as influenza and COVID-19 vaccines.
The challenges of incorporating this new vaccine into practice will feel familiar: Many of our patients won’t have heard about it, may feel that they don’t need it, or may decline it because of concerns about side effects, real or imagined. (Of note, the FDA is requiring GSK to perform a postmarketing study to rule out associations with rare cases of Guillain-Barré syndrome and acute disseminated encephalomyelitis, and the company also plans to monitor the incidence of atrial fibrillation, which was slightly more common in the vaccine group than the placebo group.)
While a strong recommendation from a family physician is often enough to convince patients to accept vaccination, rampant misinformation during the pandemic may have worsened vaccine hesitancy for some. It may feel like a fruitless exercise to try to convince adults who have refused COVID-19 and influenza vaccines to accept a newer vaccine against a respiratory virus that causes less serious illness overall. But with other RSV vaccines and monoclonal antibodies for older adults and infants likely to be approved soon, it’s important for us to start laying the groundwork now by educating colleagues, staff, and patients about preventing serious illness caused by RSV.
Dr. Lin is an associate professor in the Department of Family Medicine at Georgetown University and a staff physician atMedStar Health Center, both in Washington. He has received income from UpToDate, Wiley-Blackwell, and the American Academy of Family Physicians.
A version of this article first appeared on Medscape.com.
Compared with the complicated and ever-changing recommended vaccine schedule for infants and children, vaccines for adults have been straightforward. Adults without compromised immunity who received all their childhood vaccinations are eligible for a tetanus and diphtheria (Td) or tetanus, diphtheria, and pertussis (Tdap) booster every 10 years, recombinant herpes zoster vaccine at age 50, and pneumococcal vaccines at age 65, along with annual influenza and (likely) COVID-19 vaccines. Last year, due to rising rates of acute hepatitis B, the Centers for Disease Control and Prevention first recommended universal hepatitis B vaccination for adults aged 19-59 years without a record of previous hepatitis B infection or vaccination.
An additional routine vaccine for adults is now on the horizon. The U.S. Food and Drug Administration recently approved Arexvy, a vaccine against respiratory syncytial virus (RSV) for adults aged 60 years or older. Two more RSV vaccines are in the final stages of development. Why should family physicians prioritize vaccinating older adults against RSV, and how can we incorporate this new vaccine into our practices and overcome patient hesitancy to receive yet another vaccine?
Clinicians tend to think of RSV as a serious disease in young children – which it is – but data suggest that in 2019, RSV infection led to more than 100,000 hospitalizations and 7,700 deaths in older adults in the United States. In a randomized controlled trial of 25,000 adults aged 60 years or older with a median of 6.7 months of follow-up, Arexvy reduced severe RSV disease by 94% and RSV-related acute respiratory infections by 71%, with similar effectiveness in adults with underlying health conditions. That’s considerably better protection than current influenza vaccines and comparable to COVID-19 mRNA vaccines before variants became widespread. Pain and fatigue were the most common side effects and usually resolved within 1-2 days.
Although the seasonal pattern of RSV shifted during the COVID-19 pandemic, RSV season historically begins in October, peaks in December, and ends in April. If the vaccine is recommended by the CDC and is widely available by fall, as the manufacturer, GSK, expects, it could be administered around the same time as influenza and COVID-19 vaccines.
The challenges of incorporating this new vaccine into practice will feel familiar: Many of our patients won’t have heard about it, may feel that they don’t need it, or may decline it because of concerns about side effects, real or imagined. (Of note, the FDA is requiring GSK to perform a postmarketing study to rule out associations with rare cases of Guillain-Barré syndrome and acute disseminated encephalomyelitis, and the company also plans to monitor the incidence of atrial fibrillation, which was slightly more common in the vaccine group than the placebo group.)
While a strong recommendation from a family physician is often enough to convince patients to accept vaccination, rampant misinformation during the pandemic may have worsened vaccine hesitancy for some. It may feel like a fruitless exercise to try to convince adults who have refused COVID-19 and influenza vaccines to accept a newer vaccine against a respiratory virus that causes less serious illness overall. But with other RSV vaccines and monoclonal antibodies for older adults and infants likely to be approved soon, it’s important for us to start laying the groundwork now by educating colleagues, staff, and patients about preventing serious illness caused by RSV.
Dr. Lin is an associate professor in the Department of Family Medicine at Georgetown University and a staff physician atMedStar Health Center, both in Washington. He has received income from UpToDate, Wiley-Blackwell, and the American Academy of Family Physicians.
A version of this article first appeared on Medscape.com.
URAT1 inhibitor shows ‘substantial’ uric acid reduction in phase 2 gout trial
MILAN – About 80% of patients with gout who took the investigational selective uric acid transporter 1 (URAT1) inhibitor AR882 over 3 months reduced their serum uric acid levels to below recommended thresholds (below 5 or 4 mg/dL) for better flare and tophi reduction in a phase 2b study.
The drug was well tolerated, and patients with comorbidities did not require any adjustments in disease management.
At 75 mg, the highest tested dose of AR882, 73% of patients had serum uric acid levels < 5 mg/dL and 55% had < 4 mg/dL by week 12 of therapy in the intent-to-treat population, whereas in the per-protocol analysis, 82% had serum uric acid levels < 5 mg/dL and 63% < 4 mg/dL.
“These efficacy results are not typically what you see with a once-daily oral medication, so it is really exciting,” Robert Keenan, MD, chief medical officer of Arthrosi Therapeutics, San Diego, said in presenting the results at the annual European Congress of Rheumatology.
“Regardless of whether you’re treating subclinical, hidden crystal deposition, versus clinically visible tophi, versus chronic, debilitating gout, we believe that AR882 has the potential to treat the entire gout spectrum with a once-daily monotherapy,” Dr. Keenan asserted.
“Currently, most gout patients around the world do not have a safe, effective, and easy to use alternative to allopurinol or febuxostat, which decrease the production of uric acid,” he said. “AR882 is a URAT1 inhibitor that goes to the root of the problem in over 90% of gout patients, helping the kidneys eliminate uric acid to levels similar to all those without hyperuricemia and gout.”
Abhishek Abhishek, MD, professor of rheumatology at the University of Nottingham (England), welcomed the study. “It’s a promising study and the reduction in uric acid was substantial. It was a small study and a larger phase 3 study is needed, but it does offer real hope for patients with gout as a third treatment option because we only have allopurinol and febuxostat, so if it is shown efficacious and safe and gets approved, then it’ll help more patients with gout.”
Anne-Kathrin Tausche, MD, a rheumatologist from University Clinic Dresden (Germany), said: “These results are really impressive. We’ve lost lesinurad now because Grünenthal no longer produces it, so this might be a good alternative for patients with severe gout.
“It is favorable with [few] side effects. With allopurinol, we have to titrate it in patients with poor renal function, but it doesn’t seem to be the case with this drug. I really hope they start phase 3 soon,” she added.
Phase 2 study, but promising results
Results of the global phase 2b, randomized, double-blind trial compared the safety, tolerability, and efficacy of AR882 against placebo in patients with gout.
A total of 140 patients with gout, aged 18-75 years with an estimated glomerular filtration rate (eGFR) > 30 mL/min, were recruited across the United States, Australia, and Taiwan. Patients received either once-daily AR882 50 mg (n = 46) for 12 weeks, AR882 50 mg for 2 weeks and then AR882 75 mg (n = 47) for 10 weeks, or matching placebo for 12 weeks (n = 47). Flare prophylaxis with daily colchicine started 10 days prior to the first dose and continued throughout the study.
“Patient characteristics were typical for gout trials except for having a very diverse population,” he said. The trial included 57.9% White, 27.9% Asian, and 15% Black patients. They had a mean age of 55 years, body mass index 31-32 kg/m2. There was a range of comorbidities including hypertension, hyperlipidemia, diabetes, and cardiovascular disease, evenly distributed across placebo and AR882 treatment groups.
The efficacy endpoints were the proportion of patients who reached serum uric acid below 6, 5, 4, and 3 mg/dL, at 6 weeks of therapy, while safety was also monitored throughout the study. Reductions in serum uric acid at weeks 2, 4, 6, 12 were exploratory endpoints.
The primary endpoint of the percentage of patients below < 6 mg/dL at 6 weeks in the intent-to-treat population was met by 66% with AR882 at the lower dose (50 mg) and 84% at the higher dose (75 mg).
With the 50-mg dose, serum uric acid was reduced at week 6 to < 5mg/dL by 41%, < 4mg/dL by 12%, and < 3mg/dL by 2%, whereas these percentages were 68%, 52%, and 23%, respectively, with the 75-mg dose.
Exploratory endpoints showed that by week 12, serum uric acid levels dropped from baseline 8.6 mg/dL to about 5.0 mg/dL with AR882 50 mg and from baseline 8.6 mg/dL to about 3.5 mg/dL with AR882 75 mg. Also at week 12, 55% and 23% reached serum uric acid levels of < 4mg/dL and < 3mg/dL in the intent-to-treat population. No change was observed in the placebo group.
All adverse events were mild to moderate, with the most prevalent being gout flares. There was little difference between doses. There were no clinically significant changes in a total of 778 post-dose measurements of alanine transaminase (ALT) and aspartate transaminase (AST) and 723 post-dose triplicated electrocardiogram (ECG) measurements.
Dr. Keenan is chief medical officer of Arthrosi Therapeutics. Dr. Tausche and Dr. Abhishek have no relevant financial relationships to disclose.
MILAN – About 80% of patients with gout who took the investigational selective uric acid transporter 1 (URAT1) inhibitor AR882 over 3 months reduced their serum uric acid levels to below recommended thresholds (below 5 or 4 mg/dL) for better flare and tophi reduction in a phase 2b study.
The drug was well tolerated, and patients with comorbidities did not require any adjustments in disease management.
At 75 mg, the highest tested dose of AR882, 73% of patients had serum uric acid levels < 5 mg/dL and 55% had < 4 mg/dL by week 12 of therapy in the intent-to-treat population, whereas in the per-protocol analysis, 82% had serum uric acid levels < 5 mg/dL and 63% < 4 mg/dL.
“These efficacy results are not typically what you see with a once-daily oral medication, so it is really exciting,” Robert Keenan, MD, chief medical officer of Arthrosi Therapeutics, San Diego, said in presenting the results at the annual European Congress of Rheumatology.
“Regardless of whether you’re treating subclinical, hidden crystal deposition, versus clinically visible tophi, versus chronic, debilitating gout, we believe that AR882 has the potential to treat the entire gout spectrum with a once-daily monotherapy,” Dr. Keenan asserted.
“Currently, most gout patients around the world do not have a safe, effective, and easy to use alternative to allopurinol or febuxostat, which decrease the production of uric acid,” he said. “AR882 is a URAT1 inhibitor that goes to the root of the problem in over 90% of gout patients, helping the kidneys eliminate uric acid to levels similar to all those without hyperuricemia and gout.”
Abhishek Abhishek, MD, professor of rheumatology at the University of Nottingham (England), welcomed the study. “It’s a promising study and the reduction in uric acid was substantial. It was a small study and a larger phase 3 study is needed, but it does offer real hope for patients with gout as a third treatment option because we only have allopurinol and febuxostat, so if it is shown efficacious and safe and gets approved, then it’ll help more patients with gout.”
Anne-Kathrin Tausche, MD, a rheumatologist from University Clinic Dresden (Germany), said: “These results are really impressive. We’ve lost lesinurad now because Grünenthal no longer produces it, so this might be a good alternative for patients with severe gout.
“It is favorable with [few] side effects. With allopurinol, we have to titrate it in patients with poor renal function, but it doesn’t seem to be the case with this drug. I really hope they start phase 3 soon,” she added.
Phase 2 study, but promising results
Results of the global phase 2b, randomized, double-blind trial compared the safety, tolerability, and efficacy of AR882 against placebo in patients with gout.
A total of 140 patients with gout, aged 18-75 years with an estimated glomerular filtration rate (eGFR) > 30 mL/min, were recruited across the United States, Australia, and Taiwan. Patients received either once-daily AR882 50 mg (n = 46) for 12 weeks, AR882 50 mg for 2 weeks and then AR882 75 mg (n = 47) for 10 weeks, or matching placebo for 12 weeks (n = 47). Flare prophylaxis with daily colchicine started 10 days prior to the first dose and continued throughout the study.
“Patient characteristics were typical for gout trials except for having a very diverse population,” he said. The trial included 57.9% White, 27.9% Asian, and 15% Black patients. They had a mean age of 55 years, body mass index 31-32 kg/m2. There was a range of comorbidities including hypertension, hyperlipidemia, diabetes, and cardiovascular disease, evenly distributed across placebo and AR882 treatment groups.
The efficacy endpoints were the proportion of patients who reached serum uric acid below 6, 5, 4, and 3 mg/dL, at 6 weeks of therapy, while safety was also monitored throughout the study. Reductions in serum uric acid at weeks 2, 4, 6, 12 were exploratory endpoints.
The primary endpoint of the percentage of patients below < 6 mg/dL at 6 weeks in the intent-to-treat population was met by 66% with AR882 at the lower dose (50 mg) and 84% at the higher dose (75 mg).
With the 50-mg dose, serum uric acid was reduced at week 6 to < 5mg/dL by 41%, < 4mg/dL by 12%, and < 3mg/dL by 2%, whereas these percentages were 68%, 52%, and 23%, respectively, with the 75-mg dose.
Exploratory endpoints showed that by week 12, serum uric acid levels dropped from baseline 8.6 mg/dL to about 5.0 mg/dL with AR882 50 mg and from baseline 8.6 mg/dL to about 3.5 mg/dL with AR882 75 mg. Also at week 12, 55% and 23% reached serum uric acid levels of < 4mg/dL and < 3mg/dL in the intent-to-treat population. No change was observed in the placebo group.
All adverse events were mild to moderate, with the most prevalent being gout flares. There was little difference between doses. There were no clinically significant changes in a total of 778 post-dose measurements of alanine transaminase (ALT) and aspartate transaminase (AST) and 723 post-dose triplicated electrocardiogram (ECG) measurements.
Dr. Keenan is chief medical officer of Arthrosi Therapeutics. Dr. Tausche and Dr. Abhishek have no relevant financial relationships to disclose.
MILAN – About 80% of patients with gout who took the investigational selective uric acid transporter 1 (URAT1) inhibitor AR882 over 3 months reduced their serum uric acid levels to below recommended thresholds (below 5 or 4 mg/dL) for better flare and tophi reduction in a phase 2b study.
The drug was well tolerated, and patients with comorbidities did not require any adjustments in disease management.
At 75 mg, the highest tested dose of AR882, 73% of patients had serum uric acid levels < 5 mg/dL and 55% had < 4 mg/dL by week 12 of therapy in the intent-to-treat population, whereas in the per-protocol analysis, 82% had serum uric acid levels < 5 mg/dL and 63% < 4 mg/dL.
“These efficacy results are not typically what you see with a once-daily oral medication, so it is really exciting,” Robert Keenan, MD, chief medical officer of Arthrosi Therapeutics, San Diego, said in presenting the results at the annual European Congress of Rheumatology.
“Regardless of whether you’re treating subclinical, hidden crystal deposition, versus clinically visible tophi, versus chronic, debilitating gout, we believe that AR882 has the potential to treat the entire gout spectrum with a once-daily monotherapy,” Dr. Keenan asserted.
“Currently, most gout patients around the world do not have a safe, effective, and easy to use alternative to allopurinol or febuxostat, which decrease the production of uric acid,” he said. “AR882 is a URAT1 inhibitor that goes to the root of the problem in over 90% of gout patients, helping the kidneys eliminate uric acid to levels similar to all those without hyperuricemia and gout.”
Abhishek Abhishek, MD, professor of rheumatology at the University of Nottingham (England), welcomed the study. “It’s a promising study and the reduction in uric acid was substantial. It was a small study and a larger phase 3 study is needed, but it does offer real hope for patients with gout as a third treatment option because we only have allopurinol and febuxostat, so if it is shown efficacious and safe and gets approved, then it’ll help more patients with gout.”
Anne-Kathrin Tausche, MD, a rheumatologist from University Clinic Dresden (Germany), said: “These results are really impressive. We’ve lost lesinurad now because Grünenthal no longer produces it, so this might be a good alternative for patients with severe gout.
“It is favorable with [few] side effects. With allopurinol, we have to titrate it in patients with poor renal function, but it doesn’t seem to be the case with this drug. I really hope they start phase 3 soon,” she added.
Phase 2 study, but promising results
Results of the global phase 2b, randomized, double-blind trial compared the safety, tolerability, and efficacy of AR882 against placebo in patients with gout.
A total of 140 patients with gout, aged 18-75 years with an estimated glomerular filtration rate (eGFR) > 30 mL/min, were recruited across the United States, Australia, and Taiwan. Patients received either once-daily AR882 50 mg (n = 46) for 12 weeks, AR882 50 mg for 2 weeks and then AR882 75 mg (n = 47) for 10 weeks, or matching placebo for 12 weeks (n = 47). Flare prophylaxis with daily colchicine started 10 days prior to the first dose and continued throughout the study.
“Patient characteristics were typical for gout trials except for having a very diverse population,” he said. The trial included 57.9% White, 27.9% Asian, and 15% Black patients. They had a mean age of 55 years, body mass index 31-32 kg/m2. There was a range of comorbidities including hypertension, hyperlipidemia, diabetes, and cardiovascular disease, evenly distributed across placebo and AR882 treatment groups.
The efficacy endpoints were the proportion of patients who reached serum uric acid below 6, 5, 4, and 3 mg/dL, at 6 weeks of therapy, while safety was also monitored throughout the study. Reductions in serum uric acid at weeks 2, 4, 6, 12 were exploratory endpoints.
The primary endpoint of the percentage of patients below < 6 mg/dL at 6 weeks in the intent-to-treat population was met by 66% with AR882 at the lower dose (50 mg) and 84% at the higher dose (75 mg).
With the 50-mg dose, serum uric acid was reduced at week 6 to < 5mg/dL by 41%, < 4mg/dL by 12%, and < 3mg/dL by 2%, whereas these percentages were 68%, 52%, and 23%, respectively, with the 75-mg dose.
Exploratory endpoints showed that by week 12, serum uric acid levels dropped from baseline 8.6 mg/dL to about 5.0 mg/dL with AR882 50 mg and from baseline 8.6 mg/dL to about 3.5 mg/dL with AR882 75 mg. Also at week 12, 55% and 23% reached serum uric acid levels of < 4mg/dL and < 3mg/dL in the intent-to-treat population. No change was observed in the placebo group.
All adverse events were mild to moderate, with the most prevalent being gout flares. There was little difference between doses. There were no clinically significant changes in a total of 778 post-dose measurements of alanine transaminase (ALT) and aspartate transaminase (AST) and 723 post-dose triplicated electrocardiogram (ECG) measurements.
Dr. Keenan is chief medical officer of Arthrosi Therapeutics. Dr. Tausche and Dr. Abhishek have no relevant financial relationships to disclose.
AT EULAR 2023
Flavanol supplement improves memory in adults with poor diets
Taking a daily flavanol supplement improves hippocampal-dependent memory in older adults who have a relatively poor diet, results of a large new study suggest.
There’s increasing evidence that certain nutrients are important for the aging body and brain, study investigator Scott Small, MD, the Boris and Rose Katz Professor of Neurology, Columbia University Vagelos College of Physicians and Surgeons, New York, told this news organization.
“With this new study, I think we can begin to say flavanols might be the first one that really is a nutrient for the aging brain.”
These findings, said Dr. Small, represent “the beginning of a new era” that will eventually lead to formal recommendations” related to ideal intake of flavanols to reduce cognitive aging.
The findings were published online in the Proceedings of the National Academy of Science.
Better cognitive aging
Cognitive aging refers to the decline in cognitive abilities that are not thought to be caused by neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. Cognitive aging targets two areas of the brain: the hippocampus, which is related to memory function, and the prefrontal cortex, which is related to attention and executive function.
Previous research has linked flavanols, which are found in foods like apples, pears, berries, and cocoa beans, to improved cognitive aging. The evidence shows that consuming these nutrients might be associated with the hippocampal-dependent memory component of cognitive aging.
The new study, known as COcoa Supplement and Multivitamin Outcomes Study-Web (COSMOS-Web), included 3,562 generally healthy men and women, mean age 71 years, who were mostly well-educated and non-Hispanic/non-Latinx White individuals.
Participants were randomly assigned to receive oral flavanol-containing cocoa extract (500 mg of cocoa flavanols, including 80 mg of epicatechin) or a placebo daily.
The primary endpoint was hippocampal-dependent memory at year 1 as assessed with the ModRey, a neuropsychological test designed to measure hippocampal function.
Results showed participants in both groups had a typical learning (practice) effect, with similar improvements (d = 0.025; P = .42).
Researchers used other tests to measure cognition: the Color/Directional Flanker Task, a measure of prefrontal cortex function, and the ModBent, a measure that’s sensitive to dentate gyrus function. The flavanol intervention did not affect ModBent results or performance on the Flanker test after 1 year.
However, it was a different story for those with a poor diet at baseline. Researchers stratified participants into tertiles on the basis of diet quality as measured by the Healthy Eating Index (HEI) scores. Those in the lowest tertile had poorer baseline hippocampal-dependent memory performance but not memory related to the prefrontal cortex.
The flavanol intervention improved performance on the ModRey test, compared with placebo in participants in the low HEI tertile (overall effect: d = 0.086; P = .011) but not among those with a medium or high HEI at baseline.
“We confirmed that the flavanol intervention only benefits people who are relatively deficient at baseline,” said Dr. Small.
The correlation with hippocampal-dependent memory was confirmed in a subset of 1,361 study participants who provided a urine sample. Researchers measured urinary 5-(3′,4′-dihydroxyphenyl)-gamma-valerolactone metabolite (gVLM) concentrations, a validated biomarker of flavanol consumption.
After stratifying these results into tertiles, researchers found performance on the ModRey was significantly improved with the dietary flavanol intervention (overall effect: d = 0.141; P = .006) in the lowest gVLM tertile.
Memory restored
When participants in the lowest tertile consumed the supplement, “their flavanol levels went back to normal, and when that happened, their memory was restored,” said Dr. Small.
It appears that there is a sort of ceiling effect to the flavanol benefits. “It seems what you need to do is normalize your flavanol levels; if you go above normal, there was no evidence that your memory keeps on getting better,” said Dr. Small.
The study included only older adults, so it’s unclear what the impact of flavanol supplementation is in younger adults. But cognitive aging “begins its slippery side” in the 40s, said Dr. Small. “If this is truly a nutrient that is taken to prevent that slide from happening, it might be beneficial to start in our 40s.”
He recognized that the effect size is not large but said this is “very dependent” on baseline factors and most study participants had a rather healthy diet. “None of our participants were really highly deficient” in flavanols, he said.
“To see a stronger effect size, we need to do another study where we recruit people who are very low, truly deficient, in flavanols, and then see what happens.”
Showing that flavanols are linked to the hippocampal and not to the prefrontal component of cognitive aging “speaks to the mechanism,” said Dr. Small.
Though the exact mechanism linking flavanols with enhanced memory isn’t clear, there are some clues; for example, research suggests cognitive aging affects the dentate gyrus, a subregion of the hippocampus.
The flavanol supplements were well tolerated. “I can say with close to certainty that this is very safe,” said Dr. Small, adding the flavanols have now been used in numerous studies.
The findings suggest flavanol consumption might be part of future dietary guidelines. “I suspect that once there is sufficient evidence, flavanols will be part of the dietary recommendations for healthy aging,” said Dr. Small.
A word of caution
Heather M. Snyder, PhD, vice president of medical and scientific relations, Alzheimer’s Association, said that though science suggests a balanced diet is good for overall brain health, no single food, beverage, ingredient, vitamin, or supplement has yet been proven to prevent dementia, treat or cure Alzheimer’s, or benefit cognitive function or brain health.
Experts agree the best source of vitamins and other nutrients is from whole foods as part of a balanced diet. “We recognize that, for a variety of reasons, this may not always be possible,” said Dr. Snyder.
However, she noted, dietary supplements are not subject to the same rigorous review and regulation process as medications.
“The Alzheimer’s Association strongly encourages individuals to have conversations with their physicians about all medications and dietary supplements they are currently taking or interested in starting.”
COSMOS is supported by an investigator-initiated grant from Mars Edge, a segment of Mars, company engaged in flavanol research and flavanol-related commercial activities, which included infrastructure support and the donation of study pills and packaging. Small reports receiving an unrestricted research grant from Mars.
A version of this article first appeared on Medscape.com.
Taking a daily flavanol supplement improves hippocampal-dependent memory in older adults who have a relatively poor diet, results of a large new study suggest.
There’s increasing evidence that certain nutrients are important for the aging body and brain, study investigator Scott Small, MD, the Boris and Rose Katz Professor of Neurology, Columbia University Vagelos College of Physicians and Surgeons, New York, told this news organization.
“With this new study, I think we can begin to say flavanols might be the first one that really is a nutrient for the aging brain.”
These findings, said Dr. Small, represent “the beginning of a new era” that will eventually lead to formal recommendations” related to ideal intake of flavanols to reduce cognitive aging.
The findings were published online in the Proceedings of the National Academy of Science.
Better cognitive aging
Cognitive aging refers to the decline in cognitive abilities that are not thought to be caused by neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. Cognitive aging targets two areas of the brain: the hippocampus, which is related to memory function, and the prefrontal cortex, which is related to attention and executive function.
Previous research has linked flavanols, which are found in foods like apples, pears, berries, and cocoa beans, to improved cognitive aging. The evidence shows that consuming these nutrients might be associated with the hippocampal-dependent memory component of cognitive aging.
The new study, known as COcoa Supplement and Multivitamin Outcomes Study-Web (COSMOS-Web), included 3,562 generally healthy men and women, mean age 71 years, who were mostly well-educated and non-Hispanic/non-Latinx White individuals.
Participants were randomly assigned to receive oral flavanol-containing cocoa extract (500 mg of cocoa flavanols, including 80 mg of epicatechin) or a placebo daily.
The primary endpoint was hippocampal-dependent memory at year 1 as assessed with the ModRey, a neuropsychological test designed to measure hippocampal function.
Results showed participants in both groups had a typical learning (practice) effect, with similar improvements (d = 0.025; P = .42).
Researchers used other tests to measure cognition: the Color/Directional Flanker Task, a measure of prefrontal cortex function, and the ModBent, a measure that’s sensitive to dentate gyrus function. The flavanol intervention did not affect ModBent results or performance on the Flanker test after 1 year.
However, it was a different story for those with a poor diet at baseline. Researchers stratified participants into tertiles on the basis of diet quality as measured by the Healthy Eating Index (HEI) scores. Those in the lowest tertile had poorer baseline hippocampal-dependent memory performance but not memory related to the prefrontal cortex.
The flavanol intervention improved performance on the ModRey test, compared with placebo in participants in the low HEI tertile (overall effect: d = 0.086; P = .011) but not among those with a medium or high HEI at baseline.
“We confirmed that the flavanol intervention only benefits people who are relatively deficient at baseline,” said Dr. Small.
The correlation with hippocampal-dependent memory was confirmed in a subset of 1,361 study participants who provided a urine sample. Researchers measured urinary 5-(3′,4′-dihydroxyphenyl)-gamma-valerolactone metabolite (gVLM) concentrations, a validated biomarker of flavanol consumption.
After stratifying these results into tertiles, researchers found performance on the ModRey was significantly improved with the dietary flavanol intervention (overall effect: d = 0.141; P = .006) in the lowest gVLM tertile.
Memory restored
When participants in the lowest tertile consumed the supplement, “their flavanol levels went back to normal, and when that happened, their memory was restored,” said Dr. Small.
It appears that there is a sort of ceiling effect to the flavanol benefits. “It seems what you need to do is normalize your flavanol levels; if you go above normal, there was no evidence that your memory keeps on getting better,” said Dr. Small.
The study included only older adults, so it’s unclear what the impact of flavanol supplementation is in younger adults. But cognitive aging “begins its slippery side” in the 40s, said Dr. Small. “If this is truly a nutrient that is taken to prevent that slide from happening, it might be beneficial to start in our 40s.”
He recognized that the effect size is not large but said this is “very dependent” on baseline factors and most study participants had a rather healthy diet. “None of our participants were really highly deficient” in flavanols, he said.
“To see a stronger effect size, we need to do another study where we recruit people who are very low, truly deficient, in flavanols, and then see what happens.”
Showing that flavanols are linked to the hippocampal and not to the prefrontal component of cognitive aging “speaks to the mechanism,” said Dr. Small.
Though the exact mechanism linking flavanols with enhanced memory isn’t clear, there are some clues; for example, research suggests cognitive aging affects the dentate gyrus, a subregion of the hippocampus.
The flavanol supplements were well tolerated. “I can say with close to certainty that this is very safe,” said Dr. Small, adding the flavanols have now been used in numerous studies.
The findings suggest flavanol consumption might be part of future dietary guidelines. “I suspect that once there is sufficient evidence, flavanols will be part of the dietary recommendations for healthy aging,” said Dr. Small.
A word of caution
Heather M. Snyder, PhD, vice president of medical and scientific relations, Alzheimer’s Association, said that though science suggests a balanced diet is good for overall brain health, no single food, beverage, ingredient, vitamin, or supplement has yet been proven to prevent dementia, treat or cure Alzheimer’s, or benefit cognitive function or brain health.
Experts agree the best source of vitamins and other nutrients is from whole foods as part of a balanced diet. “We recognize that, for a variety of reasons, this may not always be possible,” said Dr. Snyder.
However, she noted, dietary supplements are not subject to the same rigorous review and regulation process as medications.
“The Alzheimer’s Association strongly encourages individuals to have conversations with their physicians about all medications and dietary supplements they are currently taking or interested in starting.”
COSMOS is supported by an investigator-initiated grant from Mars Edge, a segment of Mars, company engaged in flavanol research and flavanol-related commercial activities, which included infrastructure support and the donation of study pills and packaging. Small reports receiving an unrestricted research grant from Mars.
A version of this article first appeared on Medscape.com.
Taking a daily flavanol supplement improves hippocampal-dependent memory in older adults who have a relatively poor diet, results of a large new study suggest.
There’s increasing evidence that certain nutrients are important for the aging body and brain, study investigator Scott Small, MD, the Boris and Rose Katz Professor of Neurology, Columbia University Vagelos College of Physicians and Surgeons, New York, told this news organization.
“With this new study, I think we can begin to say flavanols might be the first one that really is a nutrient for the aging brain.”
These findings, said Dr. Small, represent “the beginning of a new era” that will eventually lead to formal recommendations” related to ideal intake of flavanols to reduce cognitive aging.
The findings were published online in the Proceedings of the National Academy of Science.
Better cognitive aging
Cognitive aging refers to the decline in cognitive abilities that are not thought to be caused by neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. Cognitive aging targets two areas of the brain: the hippocampus, which is related to memory function, and the prefrontal cortex, which is related to attention and executive function.
Previous research has linked flavanols, which are found in foods like apples, pears, berries, and cocoa beans, to improved cognitive aging. The evidence shows that consuming these nutrients might be associated with the hippocampal-dependent memory component of cognitive aging.
The new study, known as COcoa Supplement and Multivitamin Outcomes Study-Web (COSMOS-Web), included 3,562 generally healthy men and women, mean age 71 years, who were mostly well-educated and non-Hispanic/non-Latinx White individuals.
Participants were randomly assigned to receive oral flavanol-containing cocoa extract (500 mg of cocoa flavanols, including 80 mg of epicatechin) or a placebo daily.
The primary endpoint was hippocampal-dependent memory at year 1 as assessed with the ModRey, a neuropsychological test designed to measure hippocampal function.
Results showed participants in both groups had a typical learning (practice) effect, with similar improvements (d = 0.025; P = .42).
Researchers used other tests to measure cognition: the Color/Directional Flanker Task, a measure of prefrontal cortex function, and the ModBent, a measure that’s sensitive to dentate gyrus function. The flavanol intervention did not affect ModBent results or performance on the Flanker test after 1 year.
However, it was a different story for those with a poor diet at baseline. Researchers stratified participants into tertiles on the basis of diet quality as measured by the Healthy Eating Index (HEI) scores. Those in the lowest tertile had poorer baseline hippocampal-dependent memory performance but not memory related to the prefrontal cortex.
The flavanol intervention improved performance on the ModRey test, compared with placebo in participants in the low HEI tertile (overall effect: d = 0.086; P = .011) but not among those with a medium or high HEI at baseline.
“We confirmed that the flavanol intervention only benefits people who are relatively deficient at baseline,” said Dr. Small.
The correlation with hippocampal-dependent memory was confirmed in a subset of 1,361 study participants who provided a urine sample. Researchers measured urinary 5-(3′,4′-dihydroxyphenyl)-gamma-valerolactone metabolite (gVLM) concentrations, a validated biomarker of flavanol consumption.
After stratifying these results into tertiles, researchers found performance on the ModRey was significantly improved with the dietary flavanol intervention (overall effect: d = 0.141; P = .006) in the lowest gVLM tertile.
Memory restored
When participants in the lowest tertile consumed the supplement, “their flavanol levels went back to normal, and when that happened, their memory was restored,” said Dr. Small.
It appears that there is a sort of ceiling effect to the flavanol benefits. “It seems what you need to do is normalize your flavanol levels; if you go above normal, there was no evidence that your memory keeps on getting better,” said Dr. Small.
The study included only older adults, so it’s unclear what the impact of flavanol supplementation is in younger adults. But cognitive aging “begins its slippery side” in the 40s, said Dr. Small. “If this is truly a nutrient that is taken to prevent that slide from happening, it might be beneficial to start in our 40s.”
He recognized that the effect size is not large but said this is “very dependent” on baseline factors and most study participants had a rather healthy diet. “None of our participants were really highly deficient” in flavanols, he said.
“To see a stronger effect size, we need to do another study where we recruit people who are very low, truly deficient, in flavanols, and then see what happens.”
Showing that flavanols are linked to the hippocampal and not to the prefrontal component of cognitive aging “speaks to the mechanism,” said Dr. Small.
Though the exact mechanism linking flavanols with enhanced memory isn’t clear, there are some clues; for example, research suggests cognitive aging affects the dentate gyrus, a subregion of the hippocampus.
The flavanol supplements were well tolerated. “I can say with close to certainty that this is very safe,” said Dr. Small, adding the flavanols have now been used in numerous studies.
The findings suggest flavanol consumption might be part of future dietary guidelines. “I suspect that once there is sufficient evidence, flavanols will be part of the dietary recommendations for healthy aging,” said Dr. Small.
A word of caution
Heather M. Snyder, PhD, vice president of medical and scientific relations, Alzheimer’s Association, said that though science suggests a balanced diet is good for overall brain health, no single food, beverage, ingredient, vitamin, or supplement has yet been proven to prevent dementia, treat or cure Alzheimer’s, or benefit cognitive function or brain health.
Experts agree the best source of vitamins and other nutrients is from whole foods as part of a balanced diet. “We recognize that, for a variety of reasons, this may not always be possible,” said Dr. Snyder.
However, she noted, dietary supplements are not subject to the same rigorous review and regulation process as medications.
“The Alzheimer’s Association strongly encourages individuals to have conversations with their physicians about all medications and dietary supplements they are currently taking or interested in starting.”
COSMOS is supported by an investigator-initiated grant from Mars Edge, a segment of Mars, company engaged in flavanol research and flavanol-related commercial activities, which included infrastructure support and the donation of study pills and packaging. Small reports receiving an unrestricted research grant from Mars.
A version of this article first appeared on Medscape.com.