User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Slowly Enlarging Nodule on the Neck
The Diagnosis: Microsecretory Adenocarcinoma
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
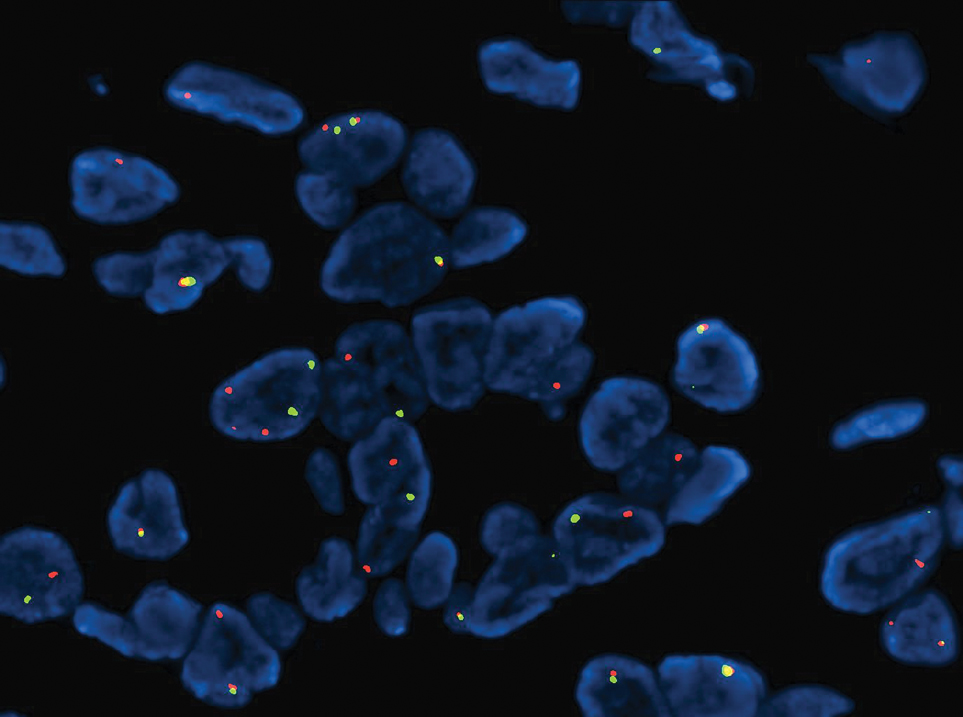
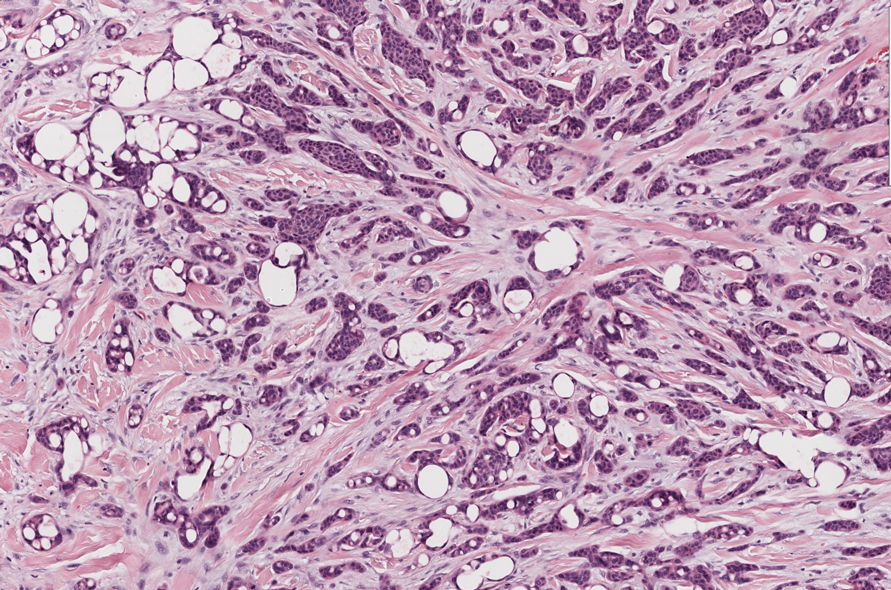
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
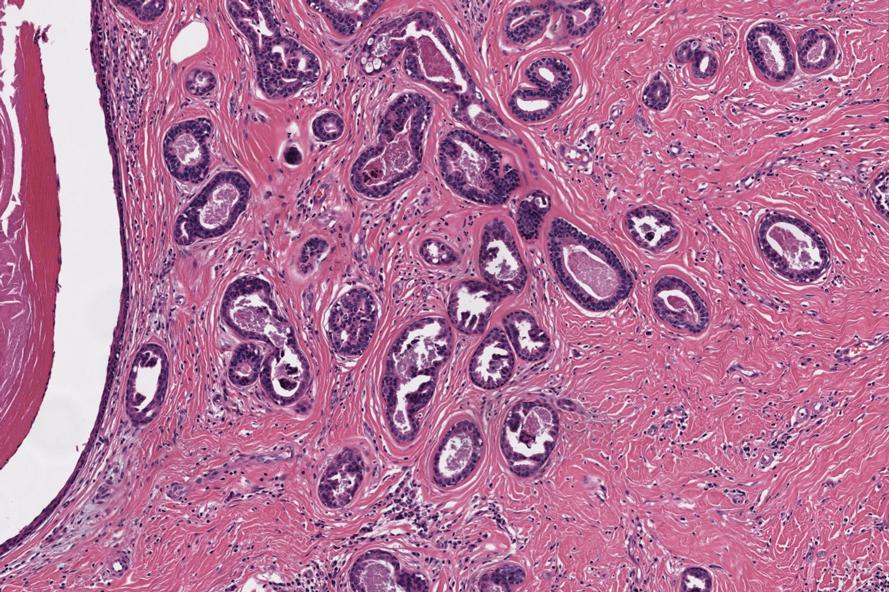
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
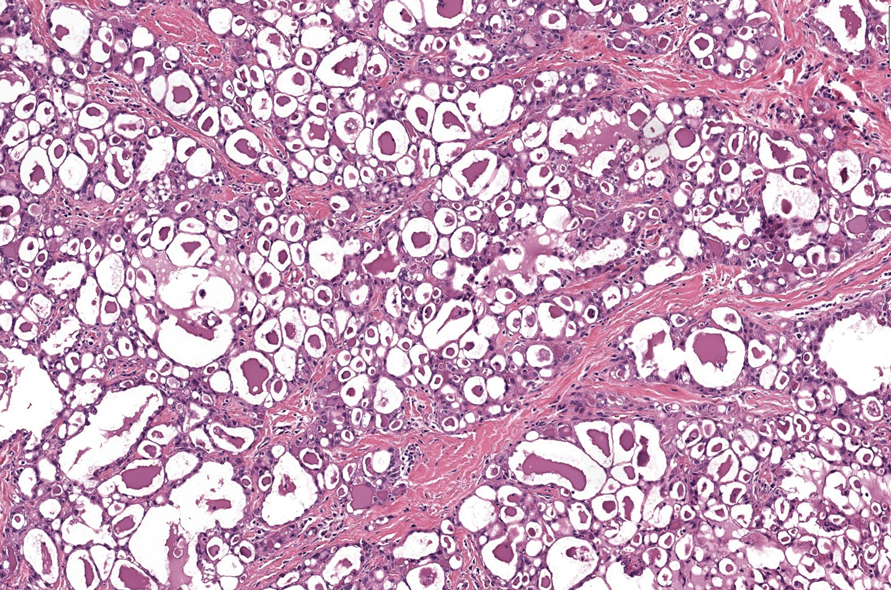
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
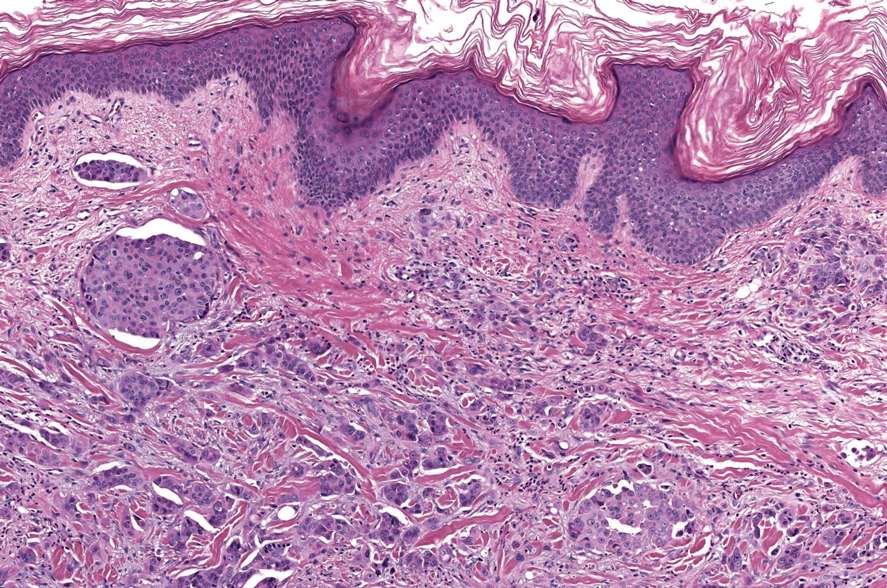
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
The Diagnosis: Microsecretory Adenocarcinoma
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
The Diagnosis: Microsecretory Adenocarcinoma
Microscopically, the tumor was relatively well circumscribed but had irregular borders. It consisted of microcysts and tubules lined by flattened to plump eosinophilic cells with mildly enlarged nuclei and intraluminal basophilic secretions. Peripheral lymphocytic aggregates also were seen in the mid and deep reticular dermis. Tumor necrosis, lymphovascular invasion, and notable mitotic activity were absent. Immunohistochemistry was diffusely positive for cytokeratin (CK) 7 and CK5/6. Occasional tumor cells showed variable expression of alpha smooth muscle actin, S-100 protein, and p40 and p63 antibodies. Immunohistochemistry was negative for CK20; GATA binding protein 3; MYB proto-oncogene, transcription factor; and insulinoma-associated protein 1. A dual-color, break-apart fluorescence in situ hybridization probe identified a rearrangement of the SS18 (SYT) gene locus on chromosome 18. The nodule was excised with clear surgical margins, and the patient had no evidence of recurrent disease or metastasis at 2-year follow-up.
In recent years, there has been a growing recognition of the pivotal role played by gene fusions in driving oncogenesis, encompassing a diverse range of benign and malignant cutaneous neoplasms. These investigations have shed light on previously unknown mechanisms and pathways contributing to the pathogenesis of these neoplastic conditions, offering invaluable insights into their underlying biology. As a result, our ability to classify and diagnose these cutaneous tumors has improved. A notable example of how our current understanding has evolved is the discovery of the new cutaneous adnexal tumor microsecretory adenocarcinoma (MSA). Initially described by Bishop et al1 in 2019 as predominantly occurring in the intraoral minor salivary glands, rare instances of primary cutaneous MSA involving the head and neck regions also have been reported.2 Microsecretory adenocarcinoma represents an important addition to the group of fusion-driven tumors with both salivary gland and cutaneous adnexal analogues, characterized by a MEF2C::SS18 gene fusion. This entity is now recognized as a group of cutaneous adnexal tumors with distinct gene fusions, including both relatively recently discovered entities (eg, secretory carcinoma with NTRK fusions) and previously known entities with newly identified gene fusions (eg, poroid neoplasms with NUTM1, YAP1, or WWTR1 fusions; hidradenomatous neoplasms with CRTC1::MAML2 fusions; and adenoid cystic carcinoma with MYB, MYBL1, and/or NFIB rearrangements).3
Microsecretory adenocarcinoma exhibits a high degree of morphologic consistency, characterized by a microcystic-predominant growth pattern, uniform intercalated ductlike tumor cells with attenuated eosinophilic to clear cytoplasm, monotonous oval hyperchromatic nuclei with indistinct nucleoli, abundant basophilic luminal secretions, and a variably cellular fibromyxoid stroma. It also shows rounded borders with subtle infiltrative growth. Occasionally, pseudoepitheliomatous hyperplasia, tumor-associated lymphoid proliferation, or metaplastic bone formation may accompany MSA. Perineural invasion is rare, necrosis is absent, and mitotic rates generally are low, contributing to its distinctive histopathologic features that aid in accurate diagnosis and differentiation from other entities. Immunohistochemistry reveals diffuse positivity for CK7 and patchy to diffuse expression of S-100 in tumor cells as well as variable expression of p40 and p63. Highly specific SS18 gene translocations at chromosome 18q are useful for diagnosing MSA when found alongside its characteristic appearance, and SS18 break-apart fluorescence in situ hybridization can serve reliably as an accurate diagnostic method (Figure 1).4 Our case illustrates how molecular analysis assists in distinguishing MSA from other cutaneous adnexal tumors, exemplifying the power of our evolving understanding in refining diagnostic accuracy and guiding targeted therapies in clinical practice.
The differential diagnosis of MSA includes tubular adenoma, secretory carcinoma, cribriform tumor (previously carcinoma), and metastatic adenocarcinoma. Tubular adenoma is a rare benign neoplasm that predominantly affects females and can manifest at any age in adulthood. It typically manifests as a slow-growing, occasionally pedunculated nodule, often measuring less than 2 cm. Although it most commonly manifests on the scalp, tubular adenoma also may arise in diverse sites such as the face, axillae, lower extremities, or genitalia.
Notably, scalp lesions often are associated with nevus sebaceus of Jadassohn or syringocystadenoma papilliferum. Microscopically, tubular adenoma is well circumscribed within the dermis and may extend into the subcutis in some cases. Its distinctive appearance consists of variably sized tubules lined by a double or multilayered cuboidal to columnar epithelium, frequently displaying apocrine decapitation secretion (Figure 2). Cystic changes and intraluminal papillae devoid of true fibrovascular cores frequently are observed. Immunohistochemically, luminal epithelial cells express epithelial membrane antigen and carcinoembryonic antigen, while the myoepithelial layer expresses smooth muscle markers, p40, and S-100 protein. BRAF V600E mutation can be detected using immunohistochemistry, with excellent sensitivity and specificity using the anti-BRAF V600E antibody (clone VE1).5 Distinguishing tubular adenoma from MSA is achievable by observing its larger, more variable tubules, along with the consistent presence of a peripheral myoepithelial layer.
Secretory carcinoma is recognized as a low-grade gene fusion–driven carcinoma that primarily arises in salivary glands (both major and minor), with occasional occurrences in the breast and extremely rare instances in other locations such as the skin, thyroid gland, and lung.6 Although the axilla is the most common cutaneous site, diverse locations such as the neck, eyelids, extremities, and nipples also have been documented. Secretory carcinoma affects individuals across a wide age range (13–71 years).6 The hallmark tumors exhibit densely packed, sievelike microcystic glands and tubular spaces filled with abundant eosinophilic intraluminal secretions (Figure 3). Additionally, morphologic variants, such as predominantly papillary, papillary-cystic, macrocystic, solid, partially mucinous, and mixed-pattern neoplasms, have been described. Secretory carcinoma shares certain features with MSA; however, it is distinguished by the presence of pronounced eosinophilic secretions, plump and vacuolated cytoplasm, and a less conspicuous fibromyxoid stroma. Immunohistochemistry reveals tumor cells that are positive for CK7, SOX-10, S-100, mammaglobin, MUC4, and variably GATA-3. Genetically, secretory carcinoma exhibits distinct characteristics, commonly showing the ETV6::NTRK3 fusion, detectable through molecular techniques or pan-TRK immunohistochemistry, while RET fusions and other rare variants are less frequent.7
In 1998, Requena et al8 introduced the concept of primary cutaneous cribriform carcinoma. Despite initially being classified as a carcinoma, the malignant potential of this tumor remains uncertain. Consequently, the term cribriform tumor now has become the preferred terminology for denoting this rare entity.9 Primary cutaneous cribriform tumors are observed more commonly in women and typically affect individuals aged 20 to 55 years (mean, 44 years). Predominant locations include the upper and lower extremities, especially the thighs, knees, and legs, with additional cases occurring on the head and trunk. Microscopically, cribriform tumor is characterized by a partially circumscribed, unencapsulated dermal nodule composed of round or oval nuclei displaying hyperchromatism and mild pleomorphism. The defining aspect of its morphology revolves around interspersed small round cavities that give rise to the hallmark cribriform pattern (Figure 4). Although MSA occasionally may exhibit a cribriform architectural pattern, it typically lacks the distinctive feature of thin, threadlike, intraluminal bridging strands observed in cribriform tumors. Similarly, luminal cells within the cribriform tumor express CK7 and exhibit variable S-100 expression. It is recognized as an indolent neoplasm with uncertain malignant potential.
The histopathologic features of metastatic carcinomas can overlap with those of primary cutaneous tumors, particularly adnexal neoplasms.10 However, several key features can aid in the differentiation of cutaneous metastases, including a dermal-based growth pattern with or without subcutaneous involvement, the presence of multiple lesions, and the occurrence of lymphovascular invasion (Figure 5). Conversely, features that suggest a primary cutaneous adnexal neoplasm include the presence of superimposed in situ disease, carcinoma developing within a benign adnexal neoplasm, and notable stromal and/or vascular hyalinization within benign-appearing areas. In some cases, it can be difficult to determine the primary site of origin of a metastatic carcinoma to the skin based on morphologic features alone. In these cases, immunohistochemistry can be helpful. The most cost-effective and time-efficient approach to accurate diagnosis is to obtain a comprehensive clinical history. If there is a known history of cancer, a small panel of organ-specific immunohistochemical studies can be performed to confirm the diagnosis. If there is no known history, an algorithmic approach can be used to identify the primary site of origin. In all circumstances, it cannot be stressed enough that acquiring a thorough clinical history before conducting any diagnostic examinations is paramount.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
- Bishop JA, Weinreb I, Swanson D, et al. Microsecretory adenocarcinoma: a novel salivary gland tumor characterized by a recurrent MEF2C-SS18 fusion. Am J Surg Pathol. 2019;43:1023-1032.
- Bishop JA, Williams EA, McLean AC, et al. Microsecretory adenocarcinoma of the skin harboring recurrent SS18 fusions: a cutaneous analog to a newly described salivary gland tumor. J Cutan Pathol. 2023;50:134-139.
- Macagno N, Sohier Pierre, Kervarrec T, et al. Recent advances on immunohistochemistry and molecular biology for the diagnosis of adnexal sweat gland tumors. Cancers (Basel). 2022;14:476.
- Bishop JA, Koduru P, Veremis BM, et al. SS18 break-apart fluorescence in situ hybridization is a practical and effective method for diagnosing microsecretory adenocarcinoma of salivary glands. Head Neck Pathol. 2021;15:723-726.
- Liau JY, Tsai JH, Huang WC, et al. BRAF and KRAS mutations in tubular apocrine adenoma and papillary eccrine adenoma of the skin. Hum Pathol. 2018;73:59-65.
- Chang MD, Arthur AK, Garcia JJ, et al. ETV6 rearrangement in a case of mammary analogue secretory carcinoma of the skin. J Cutan Pathol. 2016;43:1045-1049.
- Skalova A, Baneckova M, Thompson LDR, et al. Expanding the molecular spectrum of secretory carcinoma of salivary glands with a novel VIM-RET fusion. Am J Surg Pathol. 2020;44:1295-1307.
- Requena L, Kiryu H, Ackerman AB. Neoplasms With Apocrine Differentiation. Lippencott-Raven; 1998.
- Kazakov DV, Llamas-Velasco M, Fernandez-Flores A, et al. Cribriform tumour (previously carcinoma). In: WHO Classification of Tumours: Skin Tumours. 5th ed. International Agency for Research on Cancer; 2024.
- Habaermehl G, Ko J. Cutaneous metastases: a review and diagnostic approach to tumors of unknown origin. Arch Pathol Lab Med. 2019;143:943-957.
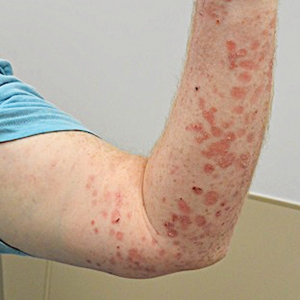
A 74-year-old man presented with an asymptomatic nodule on the left neck measuring approximately 2 cm. An excisional biopsy was obtained for histopathologic evaluation.
Epidermal Tumors Arising on Donor Sites From Autologous Skin Grafts: A Systematic Review
Skin grafting is a surgical technique used to cover skin defects resulting from the removal of skin tumors, ulcers, or burn injuries.1-3 Complications can occur at both donor and recipient sites and may include bleeding, hematoma/seroma formation, postoperative pain, infection, scarring, paresthesia, skin pigmentation, graft contracture, and graft failure.1,2,4,5 The development of epidermal tumors is not commonly reported among the complications of skin grafting; however, cases of epidermal tumor development on skin graft donor sites during the postoperative period have been reported.6-12
We performed a systematic review of the literature for cases of epidermal tumor development on skin graft donor sites in patients undergoing autologous skin graft surgery. We present the clinical characteristics of these cases and discuss the nature of these tumors.
Methods
Search Strategy and Study Selection—A literature search was conducted by 2 independent researchers (Z.P. and V.P.) for articles published before December 2022 in the following databases: MEDLINE/PubMed, Web of Science, Scopus, Cochrane Library, OpenGrey, Google Scholar, and WorldCat. Search terms included all possible combinations of the following: keratoacanthoma, molluscum sebaceum, basal cell carcinoma, squamous cell carcinoma, acanthoma, wart, Merkel cell carcinoma, verruca, Bowen disease, keratosis, skin cancer, cutaneous cancer, skin neoplasia, cutaneous neoplasia, and skin tumor. The literature search terms were selected based on the World Health Organization classification of skin tumors.13 Manual bibliography checks were performed on all eligible search results for possible relevant studies. Discrepancies were resolved through discussion and, if needed, mediation by a third researcher (N.C.). To be included, a study had to report a case(s) of epidermal tumor(s) that was confirmed by histopathology and arose on a graft donor site in a patient receiving autologous skin grafts for any reason. No language, geographic, or report date restrictions were set.
Data Extraction, Quality Assessment, and Statistical Analysis—We adhered to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.14 Two independent researchers (Z.P. and V.P.) retrieved the data from the included studies. We have used the terms case and patient interchangeably, and 1 month was measured as 4 weeks for simplicity. Disagreements were resolved by discussion and mediation by a third researcher (N.C.). The quality of the included studies was assessed by 2 researchers (M.P. and V.P.) using the tool proposed by Murad et al.15
We used descriptive statistical analysis to analyze clinical characteristics of the included cases. We performed separate descriptive analyses based on the most frequently reported types of epidermal tumors and compared the differences between different groups using the Mann-Whitney U test, χ2 test, and Fisher exact test. The level of significance was set at P<.05. All statistical analyses were conducted using SPSS (version 29).
Results
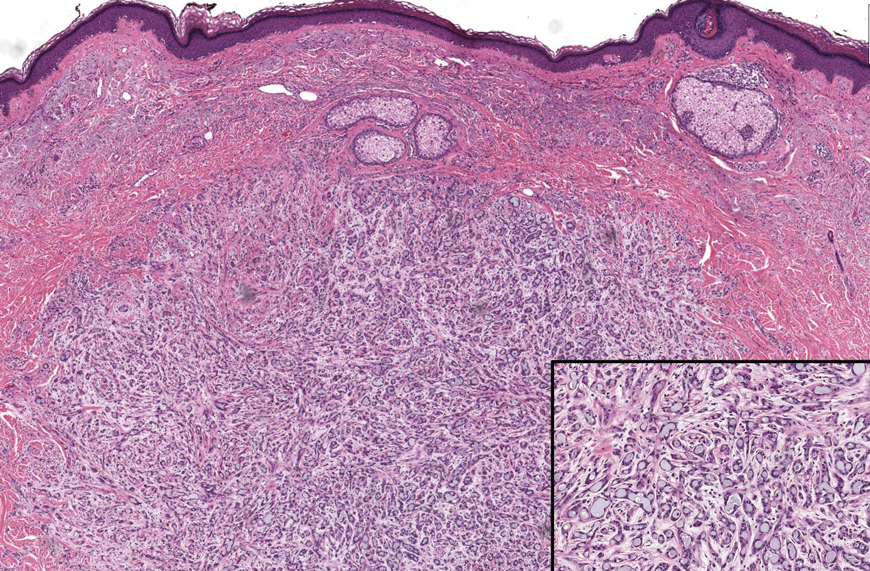
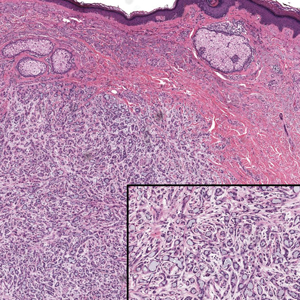
Literature Search and Characteristics of Included Studies—The initial literature search identified 1378 studies, which were screened based on title and abstract. After removing duplicate and irrelevant studies and evaluating the full text of eligible studies, 31 studies (4 case series and 27 case reports) were included in the systematic review (Figure).6-12,16-39 Quality assessment of the included studies is presented in Table 1.
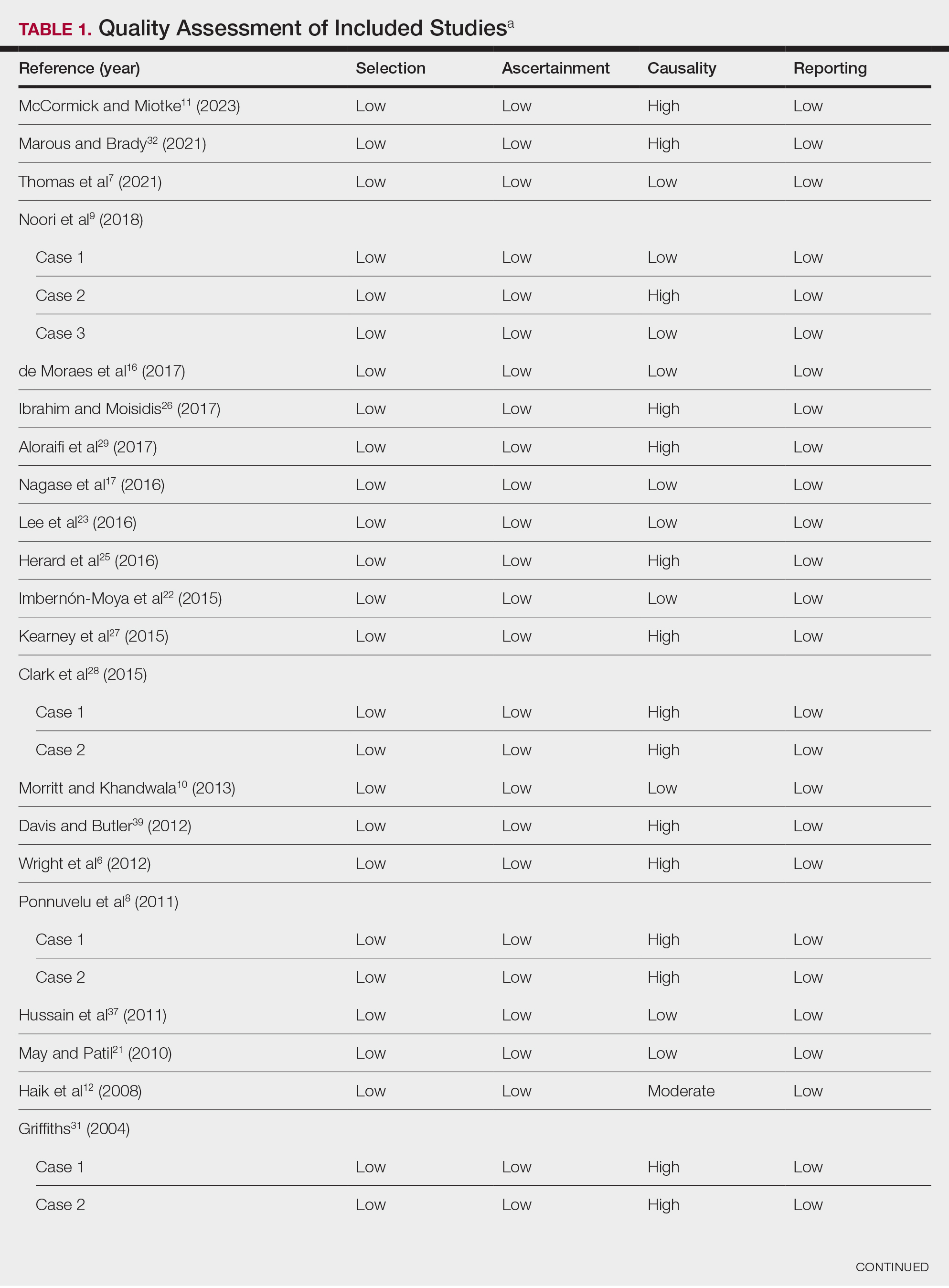
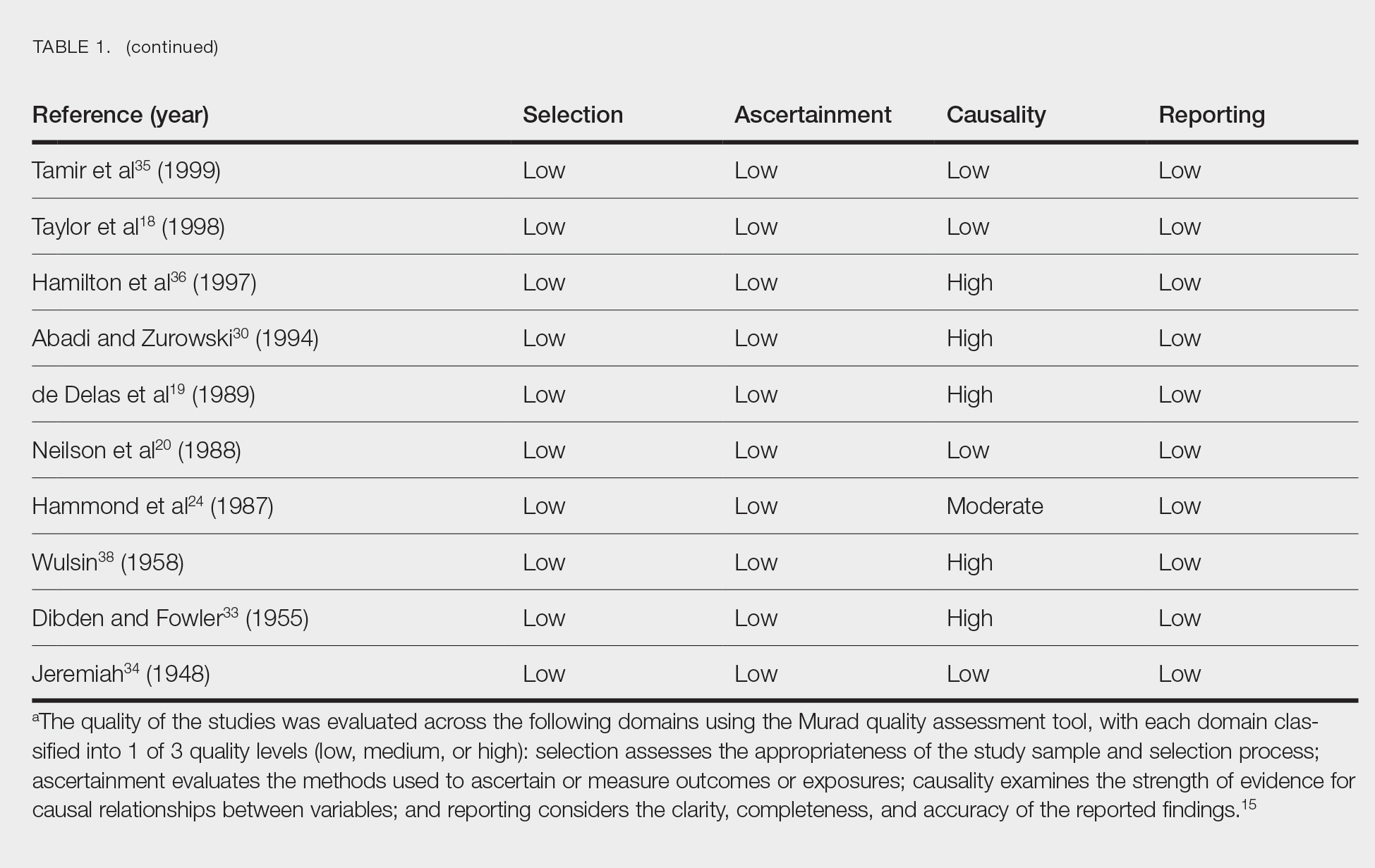
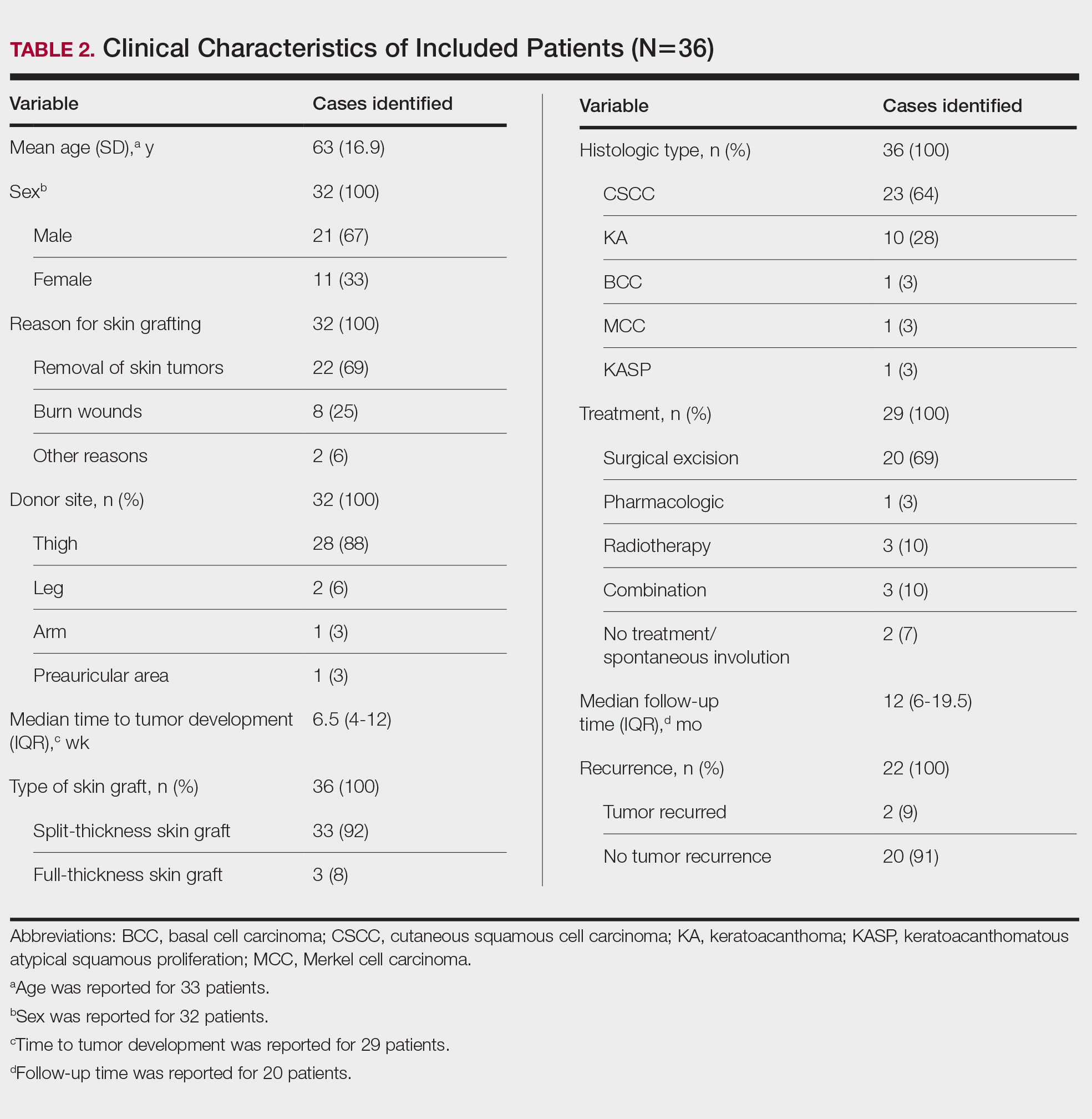
Clinical Characteristics of Included Patients—Our systematic review included 36 patients with a mean age of 63 years and a male to female ratio of 2:1. The 2 most common causes for skin grafting were burn wounds and surgical excision of skin tumors. Most grafts were harvested from the thighs. The development of a solitary lesion on the donor area was reported in two-thirds of the patients, while more than 1 lesion developed in the remaining one-third of patients. The median time to tumor development was 6.5 weeks. In most cases, a split-thickness skin graft was used.
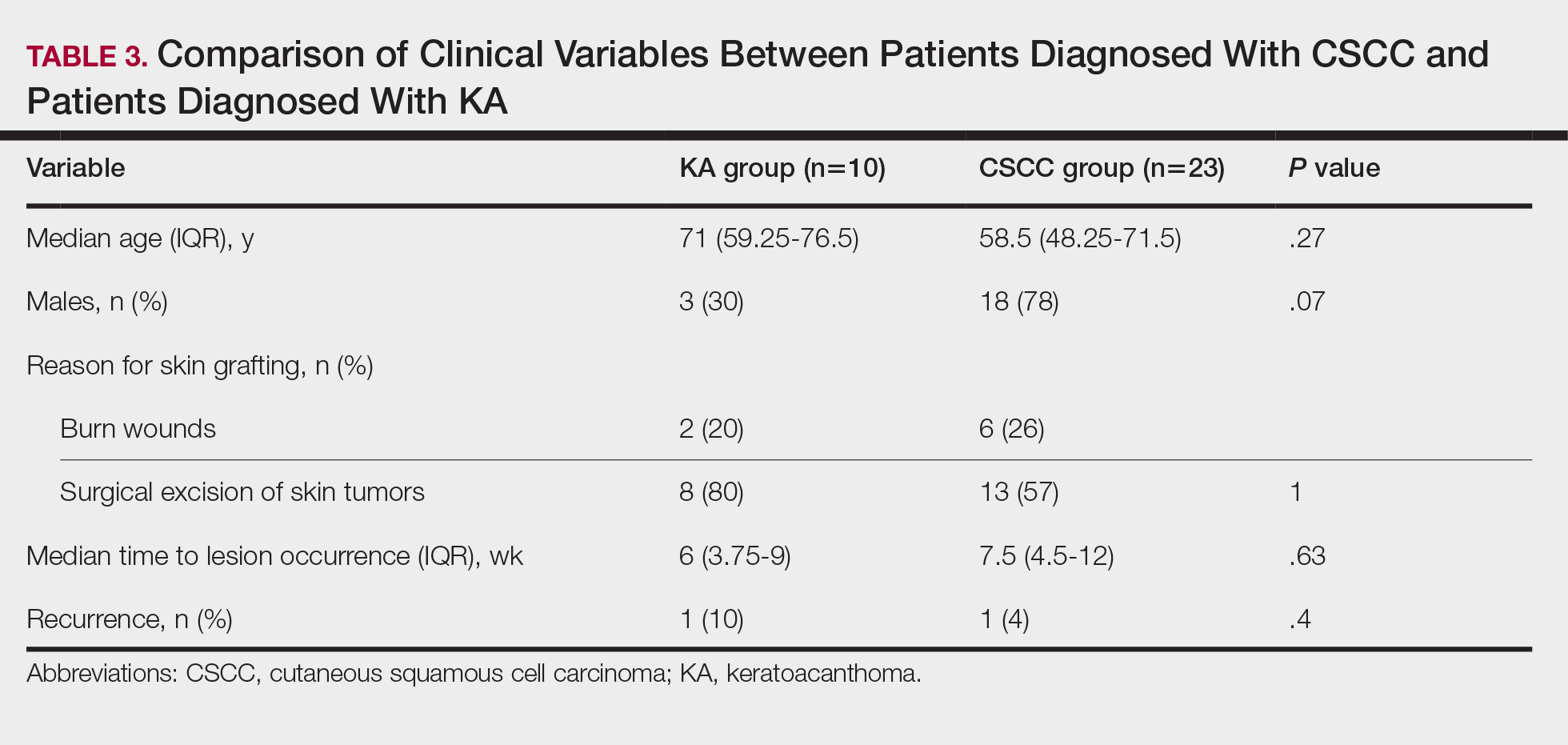
Cutaneous squamous cell carcinomas (CSCCs) were found in 23 patients, with well-differentiated CSCCs in 19 of these cases. Additionally, keratoacanthomas (KAs) were found in 10 patients. The majority of patients underwent surgical excision of the tumor. The median follow-up time was 12 months, during which recurrences were noted in a small percentage of cases. Clinical characteristics of included patients are presented in Table 2.
Comment
Reasons for Tumor Development on Skin Graft Donor Sites—The etiology behind epidermal tumor development on graft donor sites is unclear. According to one theory, iatrogenic contamination of the donor site during the removal of a primary epidermal tumor could be responsible. However, contemporary surgical procedures dictate the use of different sets of instruments for separate surgical sites. Moreover, this theory cannot explain the occurrence of epidermal tumors on donor sites in patients who have undergone skin grafting for the repair of burn wounds.37
Another theory suggests that hematogenous and/or lymphatic spread can occur from the site of the primary epidermal tumor to the donor site, which has increased vascularization.16,37 However, this theory also fails to provide an explanation for the development of epidermal tumors in patients who receive skin grafts for burn wounds.
A third theory states that the microenvironment of the donor site is key to tumor development. The donor site undergoes acute inflammation due to the trauma from harvesting the skin graft. According to this theory, acute inflammation could promote neoplastic growth and thus explain the development of epidermal tumors on the donor site.8,26 However, the relationship between acute inflammation and carcinogenesis remains unclear. What is known to date is that the development of CSCC has been documented primarily in chronically inflamed tissues, whereas the development of KA—a variant of CSCC with distinctive and more benign clinical characteristics—can be expected in the setting of acute trauma-related inflammation.13,40,41
Based on our systematic review, we propose that well-differentiated CSCC on graft donor sites might actually be misdiagnosed KA, given that the histopathologic differential diagnosis between CSCC and KA is extremely challenging.42 This hypothesis could explain the development of well-differentiated CSCC and KA on graft donor sites.
Conclusion
Development of CSCC and KA on graft donor sites can be listed among the postoperative complications of autologous skin grafting. Patients and physicians should be aware of this potential complication, and donor sites should be monitored for the occurrence of epidermal tumors.
- Adams DC, Ramsey ML. Grafts in dermatologic surgery: review and update on full- and split-thickness skin grafts, free cartilage grafts, and composite grafts. Dermatologic Surg. 2005;31(8, pt 2):1055-1067. doi:10.1111/j.1524-4725.2005.31831
- Shimizu R, Kishi K. Skin graft. Plast Surg Int. 2012;2012:563493. doi:10.1155/2012/563493
- Reddy S, El-Haddawi F, Fancourt M, et al. The incidence and risk factors for lower limb skin graft failure. Dermatol Res Pract. 2014;2014:582080. doi:10.1155/2014/582080
- Coughlin MJ, Dockery GD, Crawford ME, et al. Lower Extremity Soft Tissue & Cutaneous Plastic Surgery. 2nd ed. Saunders Ltd; 2012.
- Herskovitz I, Hughes OB, Macquhae F, et al. Epidermal skin grafting. Int Wound J. 2016;13(suppl 3):52-56. doi:10.1111/iwj.12631
- Wright H, McKinnell TH, Dunkin C. Recurrence of cutaneous squamous cell carcinoma at remote limb donor site. J Plast Reconstr Aesthet Surg. 2012;65:1265-1266. doi:10.1016/j.bjps.2012.01.022
- Thomas W, Rezzadeh K, Rossi K, et al. Squamous cell carcinoma arising at a skin graft donor site: case report and review of the literature. Plast Surg Case Stud. 2021;7:2513826X211008425. doi:10.1177/2513826X211008425
- Ponnuvelu G, Ng MFY, Connolly CM, et al. Inflammation to skin malignancy, time to rethink the link: SCC in skin graft donor sites. Surgeon. 2011;9:168-169. doi:10.1016/j.surge.2010.08.006
- Noori VJ, Trehan K, Savetamal A, et al. New onset squamous cell carcinoma in previous split-thickness skin graft donor site. Int J Surg. 2018;52:16-19. doi:10.1016/j.ijsu.2018.01.047
- Morritt DG, Khandwala AR. The development of squamous cell carcinomas in split-thickness skin graft donor sites. Eur J Plast Surg. 2013;36:377-380.
- McCormick M, Miotke S. Squamous cell carcinoma at split thickness skin graft donor site: a case report and review of the literature. J Burn Care Res. 2023;44:210-213. doi:10.1093/jbcr/irac137
- Haik J, Georgiou I, Farber N, et al. Squamous cell carcinoma arising in a split-thickness skin graft donor site. Burns. 2008;34:891-893. doi:10.1016/j.burns.2007.06.006
- Elder DE, Massi D, Scolyer RA WR. WHO Classification of Skin Tumours. 4th ed. IARC Press; 2018.
- Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151:264-269, W64. doi:10.7326/0003-4819-151-4-200908180-00135
- Murad MH, Sultan S, Haffar S, et al. Methodological quality and synthesis of case series and case reports. BMJ. 2018;23:60-63. doi:10.1136/bmjebm-2017-110853
- de Moraes LPB, Burchett I, Nicholls S, et al. Large solitary distant metastasis of cutaneous squamous cell carcinoma to skin graft site with complete response following definitive radiotherapy. Int J Bioautomation. 2017;21:103-108.
- Nagase K, Suzuki Y, Misago N, et al. Acute development of keratoacanthoma at a full-thickness skin graft donor site shortly after surgery. J Dermatol. 2016;43:1232-1233. doi:10.1111/1346-8138.13368
- Taylor CD, Snelling CF, Nickerson D, et al. Acute development of invasive squamous cell carcinoma in a split-thickness skin graft donor site. J Burn Care Rehabil. 1998;19:382-385. doi:10.1097/00004630-199809000-00004
- de Delas J, Leache A, Vazquez Doval J, et al. Keratoacanthoma over the donor site of a laminar skin graft. Med Cutan Ibero Lat Am. 1989;17:225-228.
- Neilson D, Emerson DJ, Dunn L. Squamous cell carcinoma of skin developing in a skin graft donor site. Br J Plast Surg. 1988;41:417-419. doi:10.1016/0007-1226(88)90086-0
- May JT, Patil YJ. Keratoacanthoma-type squamous cell carcinoma developing in a skin graft donor site after tumor extirpation at a distant site. Ear Nose Throat J. 2010;89:E11-E13.
- Imbernón-Moya A, Vargas-Laguna E, Lobato-Berezo A, et al. Simultaneous onset of basal cell carcinoma over skin graft and donor site. JAAD Case Rep. 2015;1:244-246. doi:10.1016/j.jdcr.2015.05.004
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:e117-e119. doi:10.1111/ajd.12501
- Hammond JS, Thomsen S, Ward CG. Scar carcinoma arising acutelyin a skin graft donor site. J Trauma. 1987;27:681-683. doi:10.1097/00005373-198706000-00017
- Herard C, Arnaud D, Goga D, et al. Rapid onset of squamous cell carcinoma in a thin skin graft donor site. Ann Dermatol Venereol. 2016;143:457-461. doi:10.1016/j.annder.2015.03.027
- Ibrahim A, Moisidis E. Case series: rapidly growing squamous cell carcinoma after cutaneous surgical intervention. JPRAS Open. 2017;14:27-32. doi:10.1016/j.jpra.2017.08.004
- Kearney L, Dolan RT, Parfrey NA, et al. Squamous cell carcinoma arising in a skin graft donor site following melanoma extirpation at a distant site: a case report and review of the literature. JPRAS Open. 2015;3:35-38. doi:10.1016/j.jpra.2015.02.002
- Clark MA, Guitart J, Gerami P, et al. Eruptive keratoacanthomatous atypical squamous proliferations (KASPs) arising in skin graft sites. JAAD Case Rep. 2015;1:274-276. doi:10.1016/j.jdcr.2015.06.009
- Aloraifi F, Mulgrew S, James NK. Secondary Merkel cell carcinoma arising from a graft donor site. J Cutan Med Surg. 2017;21:167-169. doi:10.1177/1203475416676805
- Abadir R, Zurowski S. Case report: squamous cell carcinoma of the skin in both palms, axillary node, donor skin graft site and both soles—associated hyperkeratosis and porokeratosis. Br J Radiol. 1994;67:507-510. doi:10.1259/0007-1285-67-797-507
- Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501. doi:10.1016/j.bjps.2004.05.007
- Marous M, Brady K. Cutaneous squamous cell carcinoma arising in a split thickness skin graft donor site in a patient with systemic lupus erythematosus. Dermatologic Surg. 2021;47:1106-1107. doi:10.1097/DSS.0000000000002955
- Dibden FA, Fowler M. The multiple growth of molluscum sebaceum in donor and recipient sites of skin graft. Aust N Z J Surg. 1955;25:157-159. doi:10.1111/j.1445-2197.1955.tb05122.x
- Jeremiah BS. Squamous cell carcinoma development on donor area following removal of a split thickness skin graft. Plast Reconstr Surg. 1948;3:718-721.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;40(5, pt 2):870-871. doi:10.1053/jd.1999.v40.a94419
- Hamilton SA, Dickson WA, O’Brien CJ. Keratoacanthoma developing in a split skin graft donor site. Br J Plast Surg. 1997;50:560-561. doi:10.1016/s0007-1226(97)91308-4
- Hussain A, Ekwobi C, Watson S. Metastatic implantation squamous cell carcinoma in a split-thickness skin graft donor site. J Plast Reconstr Aesthet Surg. 2011;64:690-692. doi:10.1016/j.bjps.2010.06.004
- Wulsin JH. Keratoacanthoma: a benign cutaneous tumors arising in a skin graft donor site. Am Surg. 1958;24:689-692.
- Davis L, Butler D. Acute development of squamous cell carcinoma in a split-thickness skin graft donor site [abstract]. J Am Acad Dermatol. 2012;66:AB208. doi:10.1016/j.jaad.2011.11.874
- Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park). 2002;16:217-226, 229; discussion 230-232.
- Piotrowski I, Kulcenty K, Suchorska W. Interplay between inflammation and cancer. Reports Pract Oncol Radiother. 2020;25:422-427. doi:10.1016/j.rpor.2020.04.004
- Carr RA, Houghton JP. Histopathologists’ approach to keratoacanthoma: a multisite survey of regional variation in Great Britain and Ireland. J Clin Pathol. 2014;67:637-638. doi:10.1136/jclinpath-2014-202255
Skin grafting is a surgical technique used to cover skin defects resulting from the removal of skin tumors, ulcers, or burn injuries.1-3 Complications can occur at both donor and recipient sites and may include bleeding, hematoma/seroma formation, postoperative pain, infection, scarring, paresthesia, skin pigmentation, graft contracture, and graft failure.1,2,4,5 The development of epidermal tumors is not commonly reported among the complications of skin grafting; however, cases of epidermal tumor development on skin graft donor sites during the postoperative period have been reported.6-12
We performed a systematic review of the literature for cases of epidermal tumor development on skin graft donor sites in patients undergoing autologous skin graft surgery. We present the clinical characteristics of these cases and discuss the nature of these tumors.
Methods
Search Strategy and Study Selection—A literature search was conducted by 2 independent researchers (Z.P. and V.P.) for articles published before December 2022 in the following databases: MEDLINE/PubMed, Web of Science, Scopus, Cochrane Library, OpenGrey, Google Scholar, and WorldCat. Search terms included all possible combinations of the following: keratoacanthoma, molluscum sebaceum, basal cell carcinoma, squamous cell carcinoma, acanthoma, wart, Merkel cell carcinoma, verruca, Bowen disease, keratosis, skin cancer, cutaneous cancer, skin neoplasia, cutaneous neoplasia, and skin tumor. The literature search terms were selected based on the World Health Organization classification of skin tumors.13 Manual bibliography checks were performed on all eligible search results for possible relevant studies. Discrepancies were resolved through discussion and, if needed, mediation by a third researcher (N.C.). To be included, a study had to report a case(s) of epidermal tumor(s) that was confirmed by histopathology and arose on a graft donor site in a patient receiving autologous skin grafts for any reason. No language, geographic, or report date restrictions were set.
Data Extraction, Quality Assessment, and Statistical Analysis—We adhered to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.14 Two independent researchers (Z.P. and V.P.) retrieved the data from the included studies. We have used the terms case and patient interchangeably, and 1 month was measured as 4 weeks for simplicity. Disagreements were resolved by discussion and mediation by a third researcher (N.C.). The quality of the included studies was assessed by 2 researchers (M.P. and V.P.) using the tool proposed by Murad et al.15
We used descriptive statistical analysis to analyze clinical characteristics of the included cases. We performed separate descriptive analyses based on the most frequently reported types of epidermal tumors and compared the differences between different groups using the Mann-Whitney U test, χ2 test, and Fisher exact test. The level of significance was set at P<.05. All statistical analyses were conducted using SPSS (version 29).
Results
Literature Search and Characteristics of Included Studies—The initial literature search identified 1378 studies, which were screened based on title and abstract. After removing duplicate and irrelevant studies and evaluating the full text of eligible studies, 31 studies (4 case series and 27 case reports) were included in the systematic review (Figure).6-12,16-39 Quality assessment of the included studies is presented in Table 1.
Clinical Characteristics of Included Patients—Our systematic review included 36 patients with a mean age of 63 years and a male to female ratio of 2:1. The 2 most common causes for skin grafting were burn wounds and surgical excision of skin tumors. Most grafts were harvested from the thighs. The development of a solitary lesion on the donor area was reported in two-thirds of the patients, while more than 1 lesion developed in the remaining one-third of patients. The median time to tumor development was 6.5 weeks. In most cases, a split-thickness skin graft was used.
Cutaneous squamous cell carcinomas (CSCCs) were found in 23 patients, with well-differentiated CSCCs in 19 of these cases. Additionally, keratoacanthomas (KAs) were found in 10 patients. The majority of patients underwent surgical excision of the tumor. The median follow-up time was 12 months, during which recurrences were noted in a small percentage of cases. Clinical characteristics of included patients are presented in Table 2.
Comment
Reasons for Tumor Development on Skin Graft Donor Sites—The etiology behind epidermal tumor development on graft donor sites is unclear. According to one theory, iatrogenic contamination of the donor site during the removal of a primary epidermal tumor could be responsible. However, contemporary surgical procedures dictate the use of different sets of instruments for separate surgical sites. Moreover, this theory cannot explain the occurrence of epidermal tumors on donor sites in patients who have undergone skin grafting for the repair of burn wounds.37
Another theory suggests that hematogenous and/or lymphatic spread can occur from the site of the primary epidermal tumor to the donor site, which has increased vascularization.16,37 However, this theory also fails to provide an explanation for the development of epidermal tumors in patients who receive skin grafts for burn wounds.
A third theory states that the microenvironment of the donor site is key to tumor development. The donor site undergoes acute inflammation due to the trauma from harvesting the skin graft. According to this theory, acute inflammation could promote neoplastic growth and thus explain the development of epidermal tumors on the donor site.8,26 However, the relationship between acute inflammation and carcinogenesis remains unclear. What is known to date is that the development of CSCC has been documented primarily in chronically inflamed tissues, whereas the development of KA—a variant of CSCC with distinctive and more benign clinical characteristics—can be expected in the setting of acute trauma-related inflammation.13,40,41
Based on our systematic review, we propose that well-differentiated CSCC on graft donor sites might actually be misdiagnosed KA, given that the histopathologic differential diagnosis between CSCC and KA is extremely challenging.42 This hypothesis could explain the development of well-differentiated CSCC and KA on graft donor sites.
Conclusion
Development of CSCC and KA on graft donor sites can be listed among the postoperative complications of autologous skin grafting. Patients and physicians should be aware of this potential complication, and donor sites should be monitored for the occurrence of epidermal tumors.
Skin grafting is a surgical technique used to cover skin defects resulting from the removal of skin tumors, ulcers, or burn injuries.1-3 Complications can occur at both donor and recipient sites and may include bleeding, hematoma/seroma formation, postoperative pain, infection, scarring, paresthesia, skin pigmentation, graft contracture, and graft failure.1,2,4,5 The development of epidermal tumors is not commonly reported among the complications of skin grafting; however, cases of epidermal tumor development on skin graft donor sites during the postoperative period have been reported.6-12
We performed a systematic review of the literature for cases of epidermal tumor development on skin graft donor sites in patients undergoing autologous skin graft surgery. We present the clinical characteristics of these cases and discuss the nature of these tumors.
Methods
Search Strategy and Study Selection—A literature search was conducted by 2 independent researchers (Z.P. and V.P.) for articles published before December 2022 in the following databases: MEDLINE/PubMed, Web of Science, Scopus, Cochrane Library, OpenGrey, Google Scholar, and WorldCat. Search terms included all possible combinations of the following: keratoacanthoma, molluscum sebaceum, basal cell carcinoma, squamous cell carcinoma, acanthoma, wart, Merkel cell carcinoma, verruca, Bowen disease, keratosis, skin cancer, cutaneous cancer, skin neoplasia, cutaneous neoplasia, and skin tumor. The literature search terms were selected based on the World Health Organization classification of skin tumors.13 Manual bibliography checks were performed on all eligible search results for possible relevant studies. Discrepancies were resolved through discussion and, if needed, mediation by a third researcher (N.C.). To be included, a study had to report a case(s) of epidermal tumor(s) that was confirmed by histopathology and arose on a graft donor site in a patient receiving autologous skin grafts for any reason. No language, geographic, or report date restrictions were set.
Data Extraction, Quality Assessment, and Statistical Analysis—We adhered to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.14 Two independent researchers (Z.P. and V.P.) retrieved the data from the included studies. We have used the terms case and patient interchangeably, and 1 month was measured as 4 weeks for simplicity. Disagreements were resolved by discussion and mediation by a third researcher (N.C.). The quality of the included studies was assessed by 2 researchers (M.P. and V.P.) using the tool proposed by Murad et al.15
We used descriptive statistical analysis to analyze clinical characteristics of the included cases. We performed separate descriptive analyses based on the most frequently reported types of epidermal tumors and compared the differences between different groups using the Mann-Whitney U test, χ2 test, and Fisher exact test. The level of significance was set at P<.05. All statistical analyses were conducted using SPSS (version 29).
Results
Literature Search and Characteristics of Included Studies—The initial literature search identified 1378 studies, which were screened based on title and abstract. After removing duplicate and irrelevant studies and evaluating the full text of eligible studies, 31 studies (4 case series and 27 case reports) were included in the systematic review (Figure).6-12,16-39 Quality assessment of the included studies is presented in Table 1.
Clinical Characteristics of Included Patients—Our systematic review included 36 patients with a mean age of 63 years and a male to female ratio of 2:1. The 2 most common causes for skin grafting were burn wounds and surgical excision of skin tumors. Most grafts were harvested from the thighs. The development of a solitary lesion on the donor area was reported in two-thirds of the patients, while more than 1 lesion developed in the remaining one-third of patients. The median time to tumor development was 6.5 weeks. In most cases, a split-thickness skin graft was used.
Cutaneous squamous cell carcinomas (CSCCs) were found in 23 patients, with well-differentiated CSCCs in 19 of these cases. Additionally, keratoacanthomas (KAs) were found in 10 patients. The majority of patients underwent surgical excision of the tumor. The median follow-up time was 12 months, during which recurrences were noted in a small percentage of cases. Clinical characteristics of included patients are presented in Table 2.
Comment
Reasons for Tumor Development on Skin Graft Donor Sites—The etiology behind epidermal tumor development on graft donor sites is unclear. According to one theory, iatrogenic contamination of the donor site during the removal of a primary epidermal tumor could be responsible. However, contemporary surgical procedures dictate the use of different sets of instruments for separate surgical sites. Moreover, this theory cannot explain the occurrence of epidermal tumors on donor sites in patients who have undergone skin grafting for the repair of burn wounds.37
Another theory suggests that hematogenous and/or lymphatic spread can occur from the site of the primary epidermal tumor to the donor site, which has increased vascularization.16,37 However, this theory also fails to provide an explanation for the development of epidermal tumors in patients who receive skin grafts for burn wounds.
A third theory states that the microenvironment of the donor site is key to tumor development. The donor site undergoes acute inflammation due to the trauma from harvesting the skin graft. According to this theory, acute inflammation could promote neoplastic growth and thus explain the development of epidermal tumors on the donor site.8,26 However, the relationship between acute inflammation and carcinogenesis remains unclear. What is known to date is that the development of CSCC has been documented primarily in chronically inflamed tissues, whereas the development of KA—a variant of CSCC with distinctive and more benign clinical characteristics—can be expected in the setting of acute trauma-related inflammation.13,40,41
Based on our systematic review, we propose that well-differentiated CSCC on graft donor sites might actually be misdiagnosed KA, given that the histopathologic differential diagnosis between CSCC and KA is extremely challenging.42 This hypothesis could explain the development of well-differentiated CSCC and KA on graft donor sites.
Conclusion
Development of CSCC and KA on graft donor sites can be listed among the postoperative complications of autologous skin grafting. Patients and physicians should be aware of this potential complication, and donor sites should be monitored for the occurrence of epidermal tumors.
- Adams DC, Ramsey ML. Grafts in dermatologic surgery: review and update on full- and split-thickness skin grafts, free cartilage grafts, and composite grafts. Dermatologic Surg. 2005;31(8, pt 2):1055-1067. doi:10.1111/j.1524-4725.2005.31831
- Shimizu R, Kishi K. Skin graft. Plast Surg Int. 2012;2012:563493. doi:10.1155/2012/563493
- Reddy S, El-Haddawi F, Fancourt M, et al. The incidence and risk factors for lower limb skin graft failure. Dermatol Res Pract. 2014;2014:582080. doi:10.1155/2014/582080
- Coughlin MJ, Dockery GD, Crawford ME, et al. Lower Extremity Soft Tissue & Cutaneous Plastic Surgery. 2nd ed. Saunders Ltd; 2012.
- Herskovitz I, Hughes OB, Macquhae F, et al. Epidermal skin grafting. Int Wound J. 2016;13(suppl 3):52-56. doi:10.1111/iwj.12631
- Wright H, McKinnell TH, Dunkin C. Recurrence of cutaneous squamous cell carcinoma at remote limb donor site. J Plast Reconstr Aesthet Surg. 2012;65:1265-1266. doi:10.1016/j.bjps.2012.01.022
- Thomas W, Rezzadeh K, Rossi K, et al. Squamous cell carcinoma arising at a skin graft donor site: case report and review of the literature. Plast Surg Case Stud. 2021;7:2513826X211008425. doi:10.1177/2513826X211008425
- Ponnuvelu G, Ng MFY, Connolly CM, et al. Inflammation to skin malignancy, time to rethink the link: SCC in skin graft donor sites. Surgeon. 2011;9:168-169. doi:10.1016/j.surge.2010.08.006
- Noori VJ, Trehan K, Savetamal A, et al. New onset squamous cell carcinoma in previous split-thickness skin graft donor site. Int J Surg. 2018;52:16-19. doi:10.1016/j.ijsu.2018.01.047
- Morritt DG, Khandwala AR. The development of squamous cell carcinomas in split-thickness skin graft donor sites. Eur J Plast Surg. 2013;36:377-380.
- McCormick M, Miotke S. Squamous cell carcinoma at split thickness skin graft donor site: a case report and review of the literature. J Burn Care Res. 2023;44:210-213. doi:10.1093/jbcr/irac137
- Haik J, Georgiou I, Farber N, et al. Squamous cell carcinoma arising in a split-thickness skin graft donor site. Burns. 2008;34:891-893. doi:10.1016/j.burns.2007.06.006
- Elder DE, Massi D, Scolyer RA WR. WHO Classification of Skin Tumours. 4th ed. IARC Press; 2018.
- Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151:264-269, W64. doi:10.7326/0003-4819-151-4-200908180-00135
- Murad MH, Sultan S, Haffar S, et al. Methodological quality and synthesis of case series and case reports. BMJ. 2018;23:60-63. doi:10.1136/bmjebm-2017-110853
- de Moraes LPB, Burchett I, Nicholls S, et al. Large solitary distant metastasis of cutaneous squamous cell carcinoma to skin graft site with complete response following definitive radiotherapy. Int J Bioautomation. 2017;21:103-108.
- Nagase K, Suzuki Y, Misago N, et al. Acute development of keratoacanthoma at a full-thickness skin graft donor site shortly after surgery. J Dermatol. 2016;43:1232-1233. doi:10.1111/1346-8138.13368
- Taylor CD, Snelling CF, Nickerson D, et al. Acute development of invasive squamous cell carcinoma in a split-thickness skin graft donor site. J Burn Care Rehabil. 1998;19:382-385. doi:10.1097/00004630-199809000-00004
- de Delas J, Leache A, Vazquez Doval J, et al. Keratoacanthoma over the donor site of a laminar skin graft. Med Cutan Ibero Lat Am. 1989;17:225-228.
- Neilson D, Emerson DJ, Dunn L. Squamous cell carcinoma of skin developing in a skin graft donor site. Br J Plast Surg. 1988;41:417-419. doi:10.1016/0007-1226(88)90086-0
- May JT, Patil YJ. Keratoacanthoma-type squamous cell carcinoma developing in a skin graft donor site after tumor extirpation at a distant site. Ear Nose Throat J. 2010;89:E11-E13.
- Imbernón-Moya A, Vargas-Laguna E, Lobato-Berezo A, et al. Simultaneous onset of basal cell carcinoma over skin graft and donor site. JAAD Case Rep. 2015;1:244-246. doi:10.1016/j.jdcr.2015.05.004
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:e117-e119. doi:10.1111/ajd.12501
- Hammond JS, Thomsen S, Ward CG. Scar carcinoma arising acutelyin a skin graft donor site. J Trauma. 1987;27:681-683. doi:10.1097/00005373-198706000-00017
- Herard C, Arnaud D, Goga D, et al. Rapid onset of squamous cell carcinoma in a thin skin graft donor site. Ann Dermatol Venereol. 2016;143:457-461. doi:10.1016/j.annder.2015.03.027
- Ibrahim A, Moisidis E. Case series: rapidly growing squamous cell carcinoma after cutaneous surgical intervention. JPRAS Open. 2017;14:27-32. doi:10.1016/j.jpra.2017.08.004
- Kearney L, Dolan RT, Parfrey NA, et al. Squamous cell carcinoma arising in a skin graft donor site following melanoma extirpation at a distant site: a case report and review of the literature. JPRAS Open. 2015;3:35-38. doi:10.1016/j.jpra.2015.02.002
- Clark MA, Guitart J, Gerami P, et al. Eruptive keratoacanthomatous atypical squamous proliferations (KASPs) arising in skin graft sites. JAAD Case Rep. 2015;1:274-276. doi:10.1016/j.jdcr.2015.06.009
- Aloraifi F, Mulgrew S, James NK. Secondary Merkel cell carcinoma arising from a graft donor site. J Cutan Med Surg. 2017;21:167-169. doi:10.1177/1203475416676805
- Abadir R, Zurowski S. Case report: squamous cell carcinoma of the skin in both palms, axillary node, donor skin graft site and both soles—associated hyperkeratosis and porokeratosis. Br J Radiol. 1994;67:507-510. doi:10.1259/0007-1285-67-797-507
- Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501. doi:10.1016/j.bjps.2004.05.007
- Marous M, Brady K. Cutaneous squamous cell carcinoma arising in a split thickness skin graft donor site in a patient with systemic lupus erythematosus. Dermatologic Surg. 2021;47:1106-1107. doi:10.1097/DSS.0000000000002955
- Dibden FA, Fowler M. The multiple growth of molluscum sebaceum in donor and recipient sites of skin graft. Aust N Z J Surg. 1955;25:157-159. doi:10.1111/j.1445-2197.1955.tb05122.x
- Jeremiah BS. Squamous cell carcinoma development on donor area following removal of a split thickness skin graft. Plast Reconstr Surg. 1948;3:718-721.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;40(5, pt 2):870-871. doi:10.1053/jd.1999.v40.a94419
- Hamilton SA, Dickson WA, O’Brien CJ. Keratoacanthoma developing in a split skin graft donor site. Br J Plast Surg. 1997;50:560-561. doi:10.1016/s0007-1226(97)91308-4
- Hussain A, Ekwobi C, Watson S. Metastatic implantation squamous cell carcinoma in a split-thickness skin graft donor site. J Plast Reconstr Aesthet Surg. 2011;64:690-692. doi:10.1016/j.bjps.2010.06.004
- Wulsin JH. Keratoacanthoma: a benign cutaneous tumors arising in a skin graft donor site. Am Surg. 1958;24:689-692.
- Davis L, Butler D. Acute development of squamous cell carcinoma in a split-thickness skin graft donor site [abstract]. J Am Acad Dermatol. 2012;66:AB208. doi:10.1016/j.jaad.2011.11.874
- Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park). 2002;16:217-226, 229; discussion 230-232.
- Piotrowski I, Kulcenty K, Suchorska W. Interplay between inflammation and cancer. Reports Pract Oncol Radiother. 2020;25:422-427. doi:10.1016/j.rpor.2020.04.004
- Carr RA, Houghton JP. Histopathologists’ approach to keratoacanthoma: a multisite survey of regional variation in Great Britain and Ireland. J Clin Pathol. 2014;67:637-638. doi:10.1136/jclinpath-2014-202255
- Adams DC, Ramsey ML. Grafts in dermatologic surgery: review and update on full- and split-thickness skin grafts, free cartilage grafts, and composite grafts. Dermatologic Surg. 2005;31(8, pt 2):1055-1067. doi:10.1111/j.1524-4725.2005.31831
- Shimizu R, Kishi K. Skin graft. Plast Surg Int. 2012;2012:563493. doi:10.1155/2012/563493
- Reddy S, El-Haddawi F, Fancourt M, et al. The incidence and risk factors for lower limb skin graft failure. Dermatol Res Pract. 2014;2014:582080. doi:10.1155/2014/582080
- Coughlin MJ, Dockery GD, Crawford ME, et al. Lower Extremity Soft Tissue & Cutaneous Plastic Surgery. 2nd ed. Saunders Ltd; 2012.
- Herskovitz I, Hughes OB, Macquhae F, et al. Epidermal skin grafting. Int Wound J. 2016;13(suppl 3):52-56. doi:10.1111/iwj.12631
- Wright H, McKinnell TH, Dunkin C. Recurrence of cutaneous squamous cell carcinoma at remote limb donor site. J Plast Reconstr Aesthet Surg. 2012;65:1265-1266. doi:10.1016/j.bjps.2012.01.022
- Thomas W, Rezzadeh K, Rossi K, et al. Squamous cell carcinoma arising at a skin graft donor site: case report and review of the literature. Plast Surg Case Stud. 2021;7:2513826X211008425. doi:10.1177/2513826X211008425
- Ponnuvelu G, Ng MFY, Connolly CM, et al. Inflammation to skin malignancy, time to rethink the link: SCC in skin graft donor sites. Surgeon. 2011;9:168-169. doi:10.1016/j.surge.2010.08.006
- Noori VJ, Trehan K, Savetamal A, et al. New onset squamous cell carcinoma in previous split-thickness skin graft donor site. Int J Surg. 2018;52:16-19. doi:10.1016/j.ijsu.2018.01.047
- Morritt DG, Khandwala AR. The development of squamous cell carcinomas in split-thickness skin graft donor sites. Eur J Plast Surg. 2013;36:377-380.
- McCormick M, Miotke S. Squamous cell carcinoma at split thickness skin graft donor site: a case report and review of the literature. J Burn Care Res. 2023;44:210-213. doi:10.1093/jbcr/irac137
- Haik J, Georgiou I, Farber N, et al. Squamous cell carcinoma arising in a split-thickness skin graft donor site. Burns. 2008;34:891-893. doi:10.1016/j.burns.2007.06.006
- Elder DE, Massi D, Scolyer RA WR. WHO Classification of Skin Tumours. 4th ed. IARC Press; 2018.
- Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151:264-269, W64. doi:10.7326/0003-4819-151-4-200908180-00135
- Murad MH, Sultan S, Haffar S, et al. Methodological quality and synthesis of case series and case reports. BMJ. 2018;23:60-63. doi:10.1136/bmjebm-2017-110853
- de Moraes LPB, Burchett I, Nicholls S, et al. Large solitary distant metastasis of cutaneous squamous cell carcinoma to skin graft site with complete response following definitive radiotherapy. Int J Bioautomation. 2017;21:103-108.
- Nagase K, Suzuki Y, Misago N, et al. Acute development of keratoacanthoma at a full-thickness skin graft donor site shortly after surgery. J Dermatol. 2016;43:1232-1233. doi:10.1111/1346-8138.13368
- Taylor CD, Snelling CF, Nickerson D, et al. Acute development of invasive squamous cell carcinoma in a split-thickness skin graft donor site. J Burn Care Rehabil. 1998;19:382-385. doi:10.1097/00004630-199809000-00004
- de Delas J, Leache A, Vazquez Doval J, et al. Keratoacanthoma over the donor site of a laminar skin graft. Med Cutan Ibero Lat Am. 1989;17:225-228.
- Neilson D, Emerson DJ, Dunn L. Squamous cell carcinoma of skin developing in a skin graft donor site. Br J Plast Surg. 1988;41:417-419. doi:10.1016/0007-1226(88)90086-0
- May JT, Patil YJ. Keratoacanthoma-type squamous cell carcinoma developing in a skin graft donor site after tumor extirpation at a distant site. Ear Nose Throat J. 2010;89:E11-E13.
- Imbernón-Moya A, Vargas-Laguna E, Lobato-Berezo A, et al. Simultaneous onset of basal cell carcinoma over skin graft and donor site. JAAD Case Rep. 2015;1:244-246. doi:10.1016/j.jdcr.2015.05.004
- Lee S, Coutts I, Ryan A, et al. Keratoacanthoma formation after skin grafting: a brief report and pathophysiological hypothesis. Australas J Dermatol. 2017;58:e117-e119. doi:10.1111/ajd.12501
- Hammond JS, Thomsen S, Ward CG. Scar carcinoma arising acutelyin a skin graft donor site. J Trauma. 1987;27:681-683. doi:10.1097/00005373-198706000-00017
- Herard C, Arnaud D, Goga D, et al. Rapid onset of squamous cell carcinoma in a thin skin graft donor site. Ann Dermatol Venereol. 2016;143:457-461. doi:10.1016/j.annder.2015.03.027
- Ibrahim A, Moisidis E. Case series: rapidly growing squamous cell carcinoma after cutaneous surgical intervention. JPRAS Open. 2017;14:27-32. doi:10.1016/j.jpra.2017.08.004
- Kearney L, Dolan RT, Parfrey NA, et al. Squamous cell carcinoma arising in a skin graft donor site following melanoma extirpation at a distant site: a case report and review of the literature. JPRAS Open. 2015;3:35-38. doi:10.1016/j.jpra.2015.02.002
- Clark MA, Guitart J, Gerami P, et al. Eruptive keratoacanthomatous atypical squamous proliferations (KASPs) arising in skin graft sites. JAAD Case Rep. 2015;1:274-276. doi:10.1016/j.jdcr.2015.06.009
- Aloraifi F, Mulgrew S, James NK. Secondary Merkel cell carcinoma arising from a graft donor site. J Cutan Med Surg. 2017;21:167-169. doi:10.1177/1203475416676805
- Abadir R, Zurowski S. Case report: squamous cell carcinoma of the skin in both palms, axillary node, donor skin graft site and both soles—associated hyperkeratosis and porokeratosis. Br J Radiol. 1994;67:507-510. doi:10.1259/0007-1285-67-797-507
- Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501. doi:10.1016/j.bjps.2004.05.007
- Marous M, Brady K. Cutaneous squamous cell carcinoma arising in a split thickness skin graft donor site in a patient with systemic lupus erythematosus. Dermatologic Surg. 2021;47:1106-1107. doi:10.1097/DSS.0000000000002955
- Dibden FA, Fowler M. The multiple growth of molluscum sebaceum in donor and recipient sites of skin graft. Aust N Z J Surg. 1955;25:157-159. doi:10.1111/j.1445-2197.1955.tb05122.x
- Jeremiah BS. Squamous cell carcinoma development on donor area following removal of a split thickness skin graft. Plast Reconstr Surg. 1948;3:718-721.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;40(5, pt 2):870-871. doi:10.1053/jd.1999.v40.a94419
- Hamilton SA, Dickson WA, O’Brien CJ. Keratoacanthoma developing in a split skin graft donor site. Br J Plast Surg. 1997;50:560-561. doi:10.1016/s0007-1226(97)91308-4
- Hussain A, Ekwobi C, Watson S. Metastatic implantation squamous cell carcinoma in a split-thickness skin graft donor site. J Plast Reconstr Aesthet Surg. 2011;64:690-692. doi:10.1016/j.bjps.2010.06.004
- Wulsin JH. Keratoacanthoma: a benign cutaneous tumors arising in a skin graft donor site. Am Surg. 1958;24:689-692.
- Davis L, Butler D. Acute development of squamous cell carcinoma in a split-thickness skin graft donor site [abstract]. J Am Acad Dermatol. 2012;66:AB208. doi:10.1016/j.jaad.2011.11.874
- Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park). 2002;16:217-226, 229; discussion 230-232.
- Piotrowski I, Kulcenty K, Suchorska W. Interplay between inflammation and cancer. Reports Pract Oncol Radiother. 2020;25:422-427. doi:10.1016/j.rpor.2020.04.004
- Carr RA, Houghton JP. Histopathologists’ approach to keratoacanthoma: a multisite survey of regional variation in Great Britain and Ireland. J Clin Pathol. 2014;67:637-638. doi:10.1136/jclinpath-2014-202255
Practice Points
- Donor site cutaneous squamous cell carcinoma (CSCC) and keratoacanthoma (KA) can be postoperative complications of autologous skin grafting.
- Surgical excision of donor site CSCC and KA typically is curative.
Eruptive Syringoma Manifesting as a Widespread Rash in 3 Patients
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
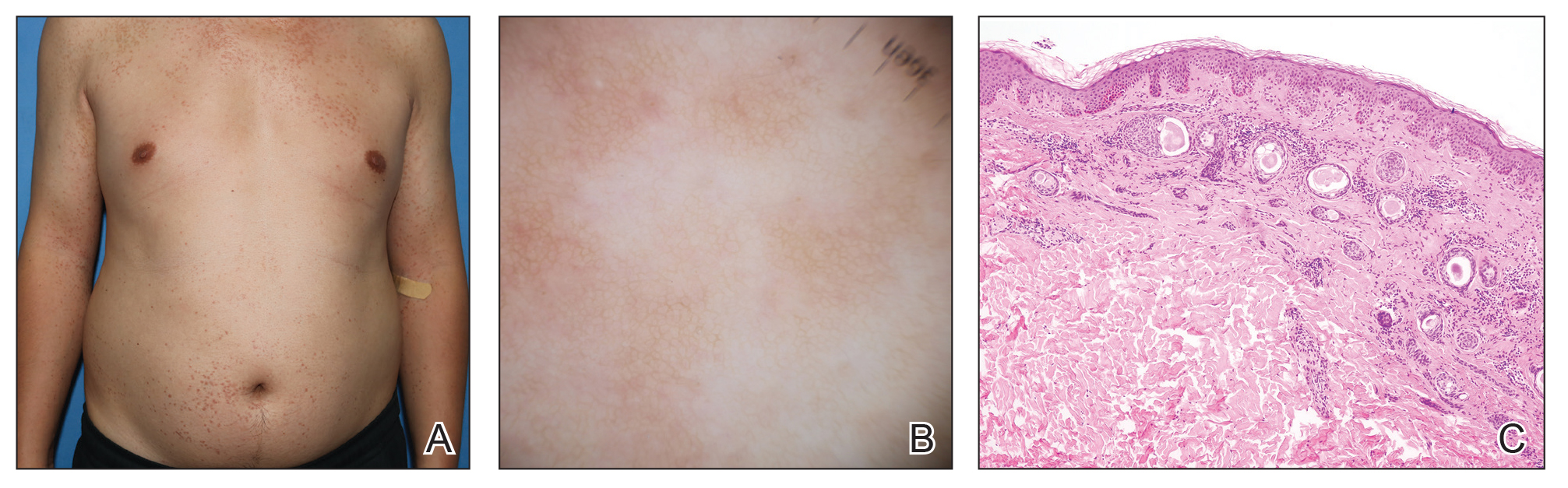
A 27-year-old woman presented with widespread asymptomatic papules

A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment. 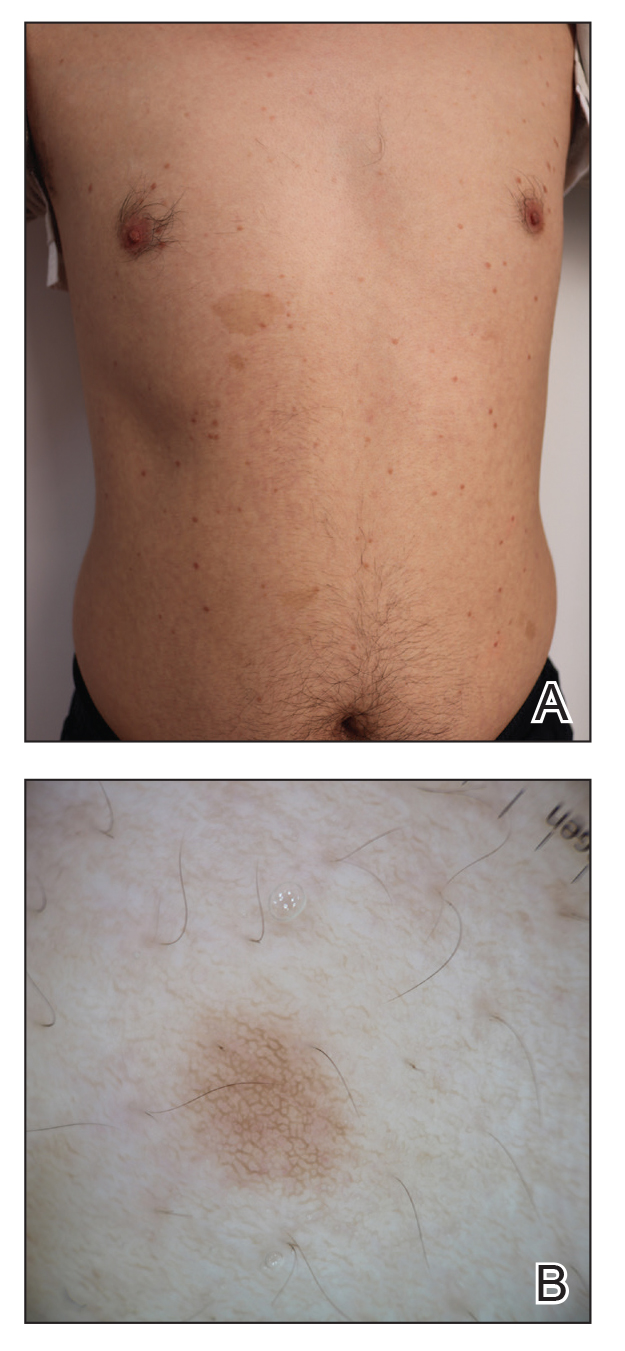
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
A 27-year-old woman presented with widespread asymptomatic papules
A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment.
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
A 27-year-old woman presented with widespread asymptomatic papules
A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment.
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
Practice Points
- Eruptive syringoma (ES) is a benign cutaneous adnexal neoplasm that typically does not require treatment.
- Dermoscopy and biopsy are helpful for the diagnosis of ES, which often is missed or misdiagnosed clinically.
Saxophone Penis: A Forgotten Manifestation of Hidradenitis Suppurativa
To the Editor:
Hidradenitis suppurativa (HS) is a multifactorial chronic inflammatory skin disease affecting 1% to 4% of Europeans. It is characterized by recurrent inflamed nodules, abscesses, and sinus tracts in intertriginous regions.1 The genital area is affected in 11% of cases2 and usually is connected to severe forms of HS in both men and women.3 The prevalence of HS-associated genital lymphedema remains unknown.
Saxophone penis is a specific penile malformation characterized by a saxophone shape due to inflammation of the major penile lymphatic vessels that cause fibrosis of the surrounding connective tissue. Poor blood flow further causes contracture and distortion of the penile axis.4 Saxophone penis also has been associated with primary lymphedema, lymphogranuloma venereum, filariasis,5 and administration of paraffin injections.6 We describe 3 men with HS who presented with saxophone penis.
A 33-year-old man with Hurley stage III HS presented with a medical history of groin lesions and progressive penoscrotal edema of 13 years’ duration. He had a body mass index (BMI) of 37, no family history of HS or comorbidities, and a 15-year history of smoking 20 cigarettes per day. After repeated surgical drainage of the HS lesions as well as antibiotic treatment with clindamycin 600 mg/d and rifampicin 600 mg/d, the patient was kept on a maintenance therapy with adalimumab 40 mg/wk. Due to lack of response, treatment was discontinued at week 16. Clindamycin and rifampicin 300 mg were immediately reintroduced with no benefit on the genital lesions. The patient underwent genital reconstruction, including penile degloving, scrotoplasty, infrapubic fat pad removal, and perineoplasty (Figure 1). The patient currently is not undergoing any therapies.
A 55-year-old man presented with Hurley stage II HS of 33 years’ duration. He had a BMI of 52; a history of hypertension, hyperuricemia, severe hip and knee osteoarthritis, and orchiopexy in childhood; a smoking history of 40 cigarettes per day; and an alcohol consumption history of 200 mL per day since 18 years of age. He had radical excision of axillary lesions 8 years prior. One year later, he was treated with concomitant clindamycin and rifampicin 300 mg twice daily for 3 months with no desirable effects. Adalimumab 40 mg/wk was initiated. After 12 weeks of treatment, he experienced 80% improvement in all areas except the genital region. He continued adalimumab for 3 years with good clinical response in all HS-affected sites except the genital region.
A 66-year-old man presented with Hurley stage III HS of 37 years’ duration. He had a smoking history of 10 cigarettes per day for 30 years, a BMI of 24.6, and a medical history of long-standing hypertension and hypothyroidism. A 3-month course of clindamycin and rifampicin 600 mg/d was ineffective; adalimumab 40 mg/wk was initiated. All affected areas improved, except for the saxophone penis. He continues his fifth year of therapy with adalimumab (Figure 2).

Hidradenitis suppurativa is associated with chronic pain, purulent malodor, and scarring with structural deformity. Repetitive inflammation causes fibrosis, scar formation, and soft-tissue destruction of lymphatic vessels, leading to lymphedema; primary lymphedema of the genitals in men has been reported to result in a saxophone penis.4
The only approved biologic treatments for moderate to severe HS are the tumor necrosis factor α inhibitor adalimumab and anti-IL-17 secukinumab.1 All 3 of our patients with HS were treated with adalimumab with reasonable success; however, the penile condition remained refractory, which we speculate may be due to adalimumab’s ability to control only active inflammatory lesions but not scars or fibrotic tissue.7 Higher adalimumab dosages were unlikely to be beneficial for their penile condition; some improvements have been reported following fluoroquinolone therapy. To our knowledge, there is no effective medical treatment for saxophone penis. However, surgery showed good results in one of our patients. Among our 3 adalimumab-treated patients, only 1 patient had corrective surgery that resulted in improvement in the penile deformity, further confirming adalimumab’s limited role in genital lymphedema.7 Extensive resection of the lymphedematous tissue, scrotoplasty, and Charles procedure are treatment options.8
Genital lymphedema has been associated with lymphangiectasia, lymphangioma circumscriptum, infections, and neoplasms such as lymphangiosarcoma and squamous cell carcinoma.9 Our patients reported discomfort, hygiene issues, and swelling. One patient reported micturition, and 2 patients reported sexual dysfunction.
Saxophone penis remains a disabling sequela of HS. Early diagnosis and treatment of HS may help prevent development of this condition.
- Lee EY, Alhusayen R, Lansang P, et al. What is hidradenitis suppurativa? Can Fam Physician. 2017;63:114-120.
- Fertitta L, Hotz C, Wolkenstein P, et al. Efficacy and satisfaction of surgical treatment for hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2020;34:839-845.
- Micieli R, Alavi A. Lymphedema in patients with hidradenitis suppurativa: a systematic review of published literature. Int J Dermatol. 2018;57:1471-1480.
- Maatouk I, Moutran R. Saxophone penis. JAMA Dermatol. 2013;149:802.
- Koley S, Mandal RK. Saxophone penis after unilateral inguinal bubo of lymphogranuloma venereum. Indian J Sex Transm Dis AIDS. 2013;34:149-151.
- D’Antuono A, Lambertini M, Gaspari V, et al. Visual dermatology: self-induced chronic saxophone penis due to paraffin injections. J Cutan Med Surg. 2019;23:330.
- Musumeci ML, Scilletta A, Sorci F, et al. Genital lymphedema associated with hidradenitis suppurativa unresponsive to adalimumab treatment. JAAD Case Rep. 2019;5:326-328.
- Jain V, Singh S, Garge S, et al. Saxophone penis due to primary lymphoedema. J Indian Assoc Pediatr Surg. 2009;14:230-231.
- Moosbrugger EA, Mutasim DF. Hidradenitis suppurativa complicated by severe lymphedema and lymphangiectasias. J Am Acad Dermatol. 2011;64:1223-1224.
To the Editor:
Hidradenitis suppurativa (HS) is a multifactorial chronic inflammatory skin disease affecting 1% to 4% of Europeans. It is characterized by recurrent inflamed nodules, abscesses, and sinus tracts in intertriginous regions.1 The genital area is affected in 11% of cases2 and usually is connected to severe forms of HS in both men and women.3 The prevalence of HS-associated genital lymphedema remains unknown.
Saxophone penis is a specific penile malformation characterized by a saxophone shape due to inflammation of the major penile lymphatic vessels that cause fibrosis of the surrounding connective tissue. Poor blood flow further causes contracture and distortion of the penile axis.4 Saxophone penis also has been associated with primary lymphedema, lymphogranuloma venereum, filariasis,5 and administration of paraffin injections.6 We describe 3 men with HS who presented with saxophone penis.
A 33-year-old man with Hurley stage III HS presented with a medical history of groin lesions and progressive penoscrotal edema of 13 years’ duration. He had a body mass index (BMI) of 37, no family history of HS or comorbidities, and a 15-year history of smoking 20 cigarettes per day. After repeated surgical drainage of the HS lesions as well as antibiotic treatment with clindamycin 600 mg/d and rifampicin 600 mg/d, the patient was kept on a maintenance therapy with adalimumab 40 mg/wk. Due to lack of response, treatment was discontinued at week 16. Clindamycin and rifampicin 300 mg were immediately reintroduced with no benefit on the genital lesions. The patient underwent genital reconstruction, including penile degloving, scrotoplasty, infrapubic fat pad removal, and perineoplasty (Figure 1). The patient currently is not undergoing any therapies.
A 55-year-old man presented with Hurley stage II HS of 33 years’ duration. He had a BMI of 52; a history of hypertension, hyperuricemia, severe hip and knee osteoarthritis, and orchiopexy in childhood; a smoking history of 40 cigarettes per day; and an alcohol consumption history of 200 mL per day since 18 years of age. He had radical excision of axillary lesions 8 years prior. One year later, he was treated with concomitant clindamycin and rifampicin 300 mg twice daily for 3 months with no desirable effects. Adalimumab 40 mg/wk was initiated. After 12 weeks of treatment, he experienced 80% improvement in all areas except the genital region. He continued adalimumab for 3 years with good clinical response in all HS-affected sites except the genital region.
A 66-year-old man presented with Hurley stage III HS of 37 years’ duration. He had a smoking history of 10 cigarettes per day for 30 years, a BMI of 24.6, and a medical history of long-standing hypertension and hypothyroidism. A 3-month course of clindamycin and rifampicin 600 mg/d was ineffective; adalimumab 40 mg/wk was initiated. All affected areas improved, except for the saxophone penis. He continues his fifth year of therapy with adalimumab (Figure 2).
Hidradenitis suppurativa is associated with chronic pain, purulent malodor, and scarring with structural deformity. Repetitive inflammation causes fibrosis, scar formation, and soft-tissue destruction of lymphatic vessels, leading to lymphedema; primary lymphedema of the genitals in men has been reported to result in a saxophone penis.4
The only approved biologic treatments for moderate to severe HS are the tumor necrosis factor α inhibitor adalimumab and anti-IL-17 secukinumab.1 All 3 of our patients with HS were treated with adalimumab with reasonable success; however, the penile condition remained refractory, which we speculate may be due to adalimumab’s ability to control only active inflammatory lesions but not scars or fibrotic tissue.7 Higher adalimumab dosages were unlikely to be beneficial for their penile condition; some improvements have been reported following fluoroquinolone therapy. To our knowledge, there is no effective medical treatment for saxophone penis. However, surgery showed good results in one of our patients. Among our 3 adalimumab-treated patients, only 1 patient had corrective surgery that resulted in improvement in the penile deformity, further confirming adalimumab’s limited role in genital lymphedema.7 Extensive resection of the lymphedematous tissue, scrotoplasty, and Charles procedure are treatment options.8
Genital lymphedema has been associated with lymphangiectasia, lymphangioma circumscriptum, infections, and neoplasms such as lymphangiosarcoma and squamous cell carcinoma.9 Our patients reported discomfort, hygiene issues, and swelling. One patient reported micturition, and 2 patients reported sexual dysfunction.
Saxophone penis remains a disabling sequela of HS. Early diagnosis and treatment of HS may help prevent development of this condition.
To the Editor:
Hidradenitis suppurativa (HS) is a multifactorial chronic inflammatory skin disease affecting 1% to 4% of Europeans. It is characterized by recurrent inflamed nodules, abscesses, and sinus tracts in intertriginous regions.1 The genital area is affected in 11% of cases2 and usually is connected to severe forms of HS in both men and women.3 The prevalence of HS-associated genital lymphedema remains unknown.
Saxophone penis is a specific penile malformation characterized by a saxophone shape due to inflammation of the major penile lymphatic vessels that cause fibrosis of the surrounding connective tissue. Poor blood flow further causes contracture and distortion of the penile axis.4 Saxophone penis also has been associated with primary lymphedema, lymphogranuloma venereum, filariasis,5 and administration of paraffin injections.6 We describe 3 men with HS who presented with saxophone penis.
A 33-year-old man with Hurley stage III HS presented with a medical history of groin lesions and progressive penoscrotal edema of 13 years’ duration. He had a body mass index (BMI) of 37, no family history of HS or comorbidities, and a 15-year history of smoking 20 cigarettes per day. After repeated surgical drainage of the HS lesions as well as antibiotic treatment with clindamycin 600 mg/d and rifampicin 600 mg/d, the patient was kept on a maintenance therapy with adalimumab 40 mg/wk. Due to lack of response, treatment was discontinued at week 16. Clindamycin and rifampicin 300 mg were immediately reintroduced with no benefit on the genital lesions. The patient underwent genital reconstruction, including penile degloving, scrotoplasty, infrapubic fat pad removal, and perineoplasty (Figure 1). The patient currently is not undergoing any therapies.
A 55-year-old man presented with Hurley stage II HS of 33 years’ duration. He had a BMI of 52; a history of hypertension, hyperuricemia, severe hip and knee osteoarthritis, and orchiopexy in childhood; a smoking history of 40 cigarettes per day; and an alcohol consumption history of 200 mL per day since 18 years of age. He had radical excision of axillary lesions 8 years prior. One year later, he was treated with concomitant clindamycin and rifampicin 300 mg twice daily for 3 months with no desirable effects. Adalimumab 40 mg/wk was initiated. After 12 weeks of treatment, he experienced 80% improvement in all areas except the genital region. He continued adalimumab for 3 years with good clinical response in all HS-affected sites except the genital region.
A 66-year-old man presented with Hurley stage III HS of 37 years’ duration. He had a smoking history of 10 cigarettes per day for 30 years, a BMI of 24.6, and a medical history of long-standing hypertension and hypothyroidism. A 3-month course of clindamycin and rifampicin 600 mg/d was ineffective; adalimumab 40 mg/wk was initiated. All affected areas improved, except for the saxophone penis. He continues his fifth year of therapy with adalimumab (Figure 2).
Hidradenitis suppurativa is associated with chronic pain, purulent malodor, and scarring with structural deformity. Repetitive inflammation causes fibrosis, scar formation, and soft-tissue destruction of lymphatic vessels, leading to lymphedema; primary lymphedema of the genitals in men has been reported to result in a saxophone penis.4
The only approved biologic treatments for moderate to severe HS are the tumor necrosis factor α inhibitor adalimumab and anti-IL-17 secukinumab.1 All 3 of our patients with HS were treated with adalimumab with reasonable success; however, the penile condition remained refractory, which we speculate may be due to adalimumab’s ability to control only active inflammatory lesions but not scars or fibrotic tissue.7 Higher adalimumab dosages were unlikely to be beneficial for their penile condition; some improvements have been reported following fluoroquinolone therapy. To our knowledge, there is no effective medical treatment for saxophone penis. However, surgery showed good results in one of our patients. Among our 3 adalimumab-treated patients, only 1 patient had corrective surgery that resulted in improvement in the penile deformity, further confirming adalimumab’s limited role in genital lymphedema.7 Extensive resection of the lymphedematous tissue, scrotoplasty, and Charles procedure are treatment options.8
Genital lymphedema has been associated with lymphangiectasia, lymphangioma circumscriptum, infections, and neoplasms such as lymphangiosarcoma and squamous cell carcinoma.9 Our patients reported discomfort, hygiene issues, and swelling. One patient reported micturition, and 2 patients reported sexual dysfunction.
Saxophone penis remains a disabling sequela of HS. Early diagnosis and treatment of HS may help prevent development of this condition.
- Lee EY, Alhusayen R, Lansang P, et al. What is hidradenitis suppurativa? Can Fam Physician. 2017;63:114-120.
- Fertitta L, Hotz C, Wolkenstein P, et al. Efficacy and satisfaction of surgical treatment for hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2020;34:839-845.
- Micieli R, Alavi A. Lymphedema in patients with hidradenitis suppurativa: a systematic review of published literature. Int J Dermatol. 2018;57:1471-1480.
- Maatouk I, Moutran R. Saxophone penis. JAMA Dermatol. 2013;149:802.
- Koley S, Mandal RK. Saxophone penis after unilateral inguinal bubo of lymphogranuloma venereum. Indian J Sex Transm Dis AIDS. 2013;34:149-151.
- D’Antuono A, Lambertini M, Gaspari V, et al. Visual dermatology: self-induced chronic saxophone penis due to paraffin injections. J Cutan Med Surg. 2019;23:330.
- Musumeci ML, Scilletta A, Sorci F, et al. Genital lymphedema associated with hidradenitis suppurativa unresponsive to adalimumab treatment. JAAD Case Rep. 2019;5:326-328.
- Jain V, Singh S, Garge S, et al. Saxophone penis due to primary lymphoedema. J Indian Assoc Pediatr Surg. 2009;14:230-231.
- Moosbrugger EA, Mutasim DF. Hidradenitis suppurativa complicated by severe lymphedema and lymphangiectasias. J Am Acad Dermatol. 2011;64:1223-1224.
- Lee EY, Alhusayen R, Lansang P, et al. What is hidradenitis suppurativa? Can Fam Physician. 2017;63:114-120.
- Fertitta L, Hotz C, Wolkenstein P, et al. Efficacy and satisfaction of surgical treatment for hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2020;34:839-845.
- Micieli R, Alavi A. Lymphedema in patients with hidradenitis suppurativa: a systematic review of published literature. Int J Dermatol. 2018;57:1471-1480.
- Maatouk I, Moutran R. Saxophone penis. JAMA Dermatol. 2013;149:802.
- Koley S, Mandal RK. Saxophone penis after unilateral inguinal bubo of lymphogranuloma venereum. Indian J Sex Transm Dis AIDS. 2013;34:149-151.
- D’Antuono A, Lambertini M, Gaspari V, et al. Visual dermatology: self-induced chronic saxophone penis due to paraffin injections. J Cutan Med Surg. 2019;23:330.
- Musumeci ML, Scilletta A, Sorci F, et al. Genital lymphedema associated with hidradenitis suppurativa unresponsive to adalimumab treatment. JAAD Case Rep. 2019;5:326-328.
- Jain V, Singh S, Garge S, et al. Saxophone penis due to primary lymphoedema. J Indian Assoc Pediatr Surg. 2009;14:230-231.
- Moosbrugger EA, Mutasim DF. Hidradenitis suppurativa complicated by severe lymphedema and lymphangiectasias. J Am Acad Dermatol. 2011;64:1223-1224.
Practice Points
- Hidradenitis suppurativa (HS) is a multifactorial chronic inflammatory skin disease.
- Saxophone penis is a specific penile malformation characterized by a saxophone shape due to inflammation.
- Repetitive inflammation within the context of HS may cause structural deformity of the penis, resulting in a saxophone penis.
- Early diagnosis and treatment of HS may help prevent development of this condition.
Anti-Smith and Anti–Double-Stranded DNA Antibodies in a Patient With Henoch-Schönlein Purpura Following COVID-19 Vaccination
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
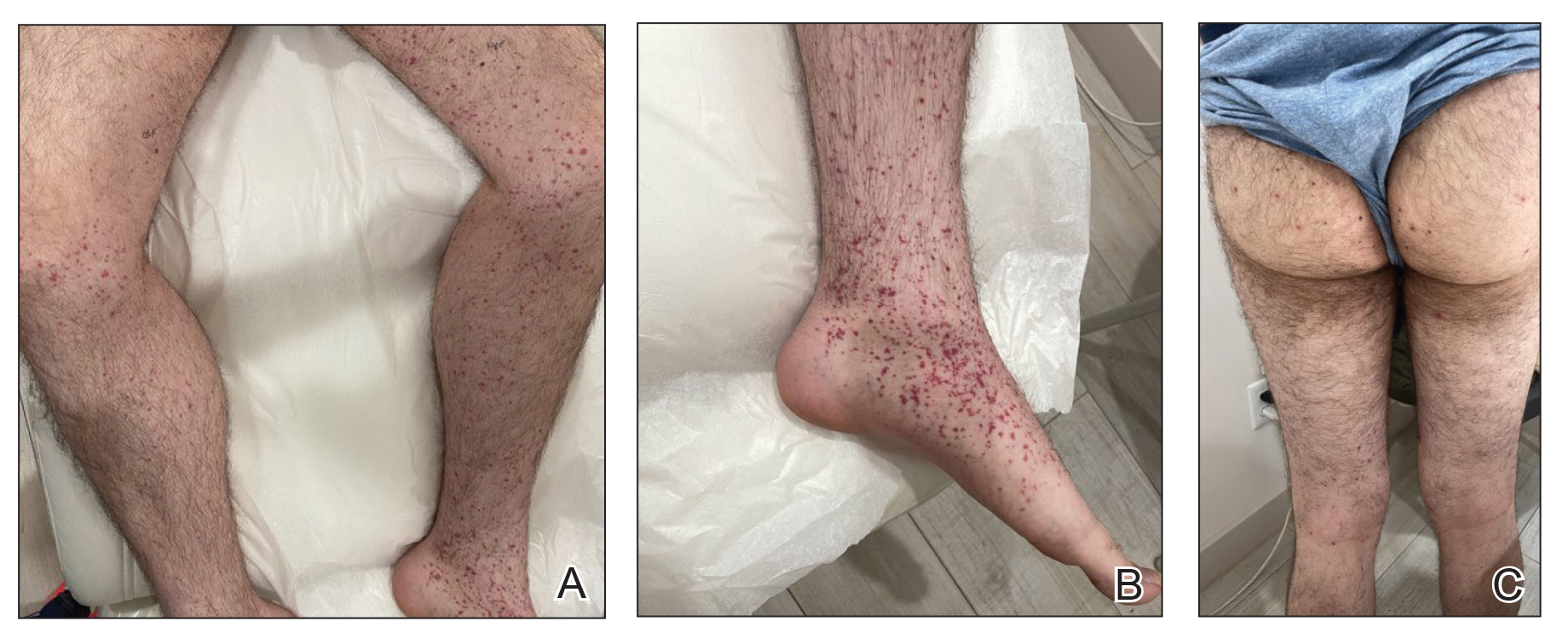
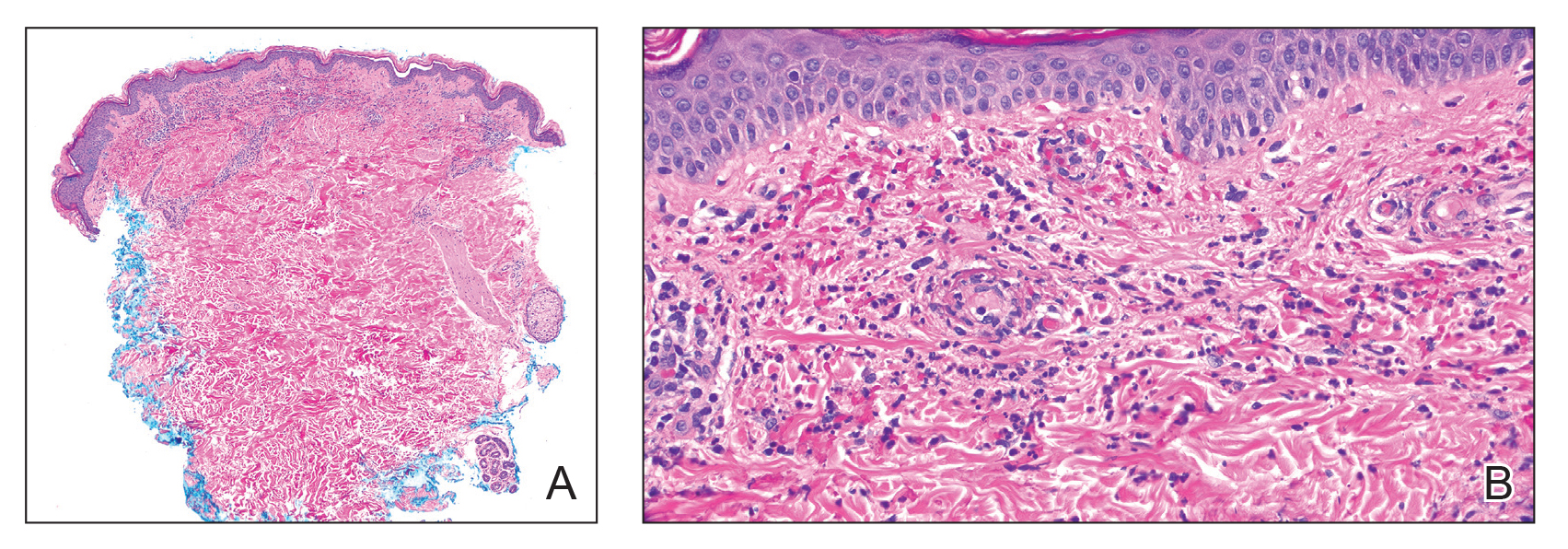
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
Practice Points
- Dermatologists should be vigilant for Henoch-Schönlein purpura (HSP) despite negative direct immunofluorescence of IgA deposition and unusual antibodies.
- Messenger RNA–based COVID-19 vaccines are associated with various cutaneous reactions, including HSP.
- Anti-Smith and anti–double-stranded DNA antibodies typically are not associated with HSP but may be seen in patients with coexisting systemic lupus erythematosus.
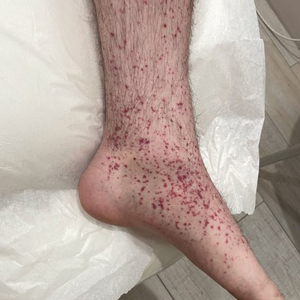
Painful Anal Lesions in a Patient With HIV
The Diagnosis: Condyloma Latum
Laboratory test results were notable for a rapid plasma reagin titer of 1:512, a positive Treponema pallidum particle agglutination test, negative rectal nucleic acid amplification tests for gonorrhea and chlamydia, and a negative herpes simplex virus polymerase chain reaction. A VDRL test of cerebrospinal fluid from a lumbar puncture was negative. Histopathology of the punch biopsy sample revealed marked verrucous epidermal hyperplasia without keratinocytic atypia and with mixed inflammation (Figure 1), while immunohistochemical staining showed numerus T pallidum organisms (Figure 2). A diagnosis of condyloma latum was made based on the laboratory, lumbar puncture, and punch biopsy results. Due to a penicillin allergy, the patient was treated with oral doxycycline for 14 days. On follow-up at day 12 of therapy, he reported cessation of rectal pain, and resolution of anal lesions was noted on physical examination.
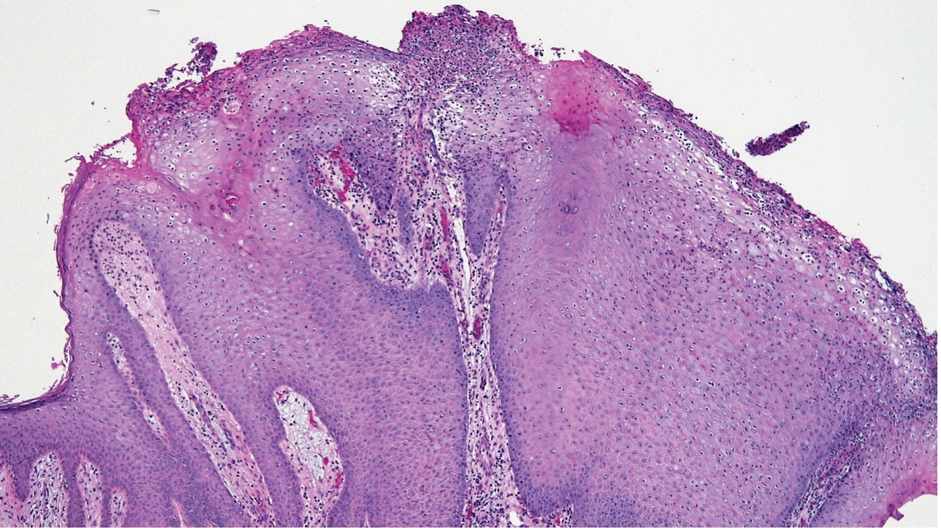
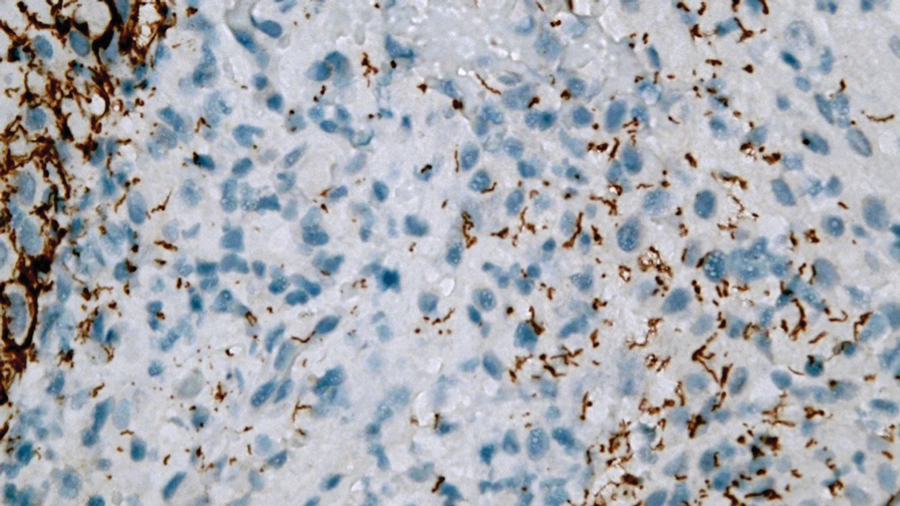
Condylomata lata are highly infectious cutaneous lesions that can manifest during secondary syphilis.1 They typically are described as white or gray, raised, flatappearing plaques and occur in moist areas or skin folds including the anus, scrotum, and vulva. However, these lesions also have been reported in the axillae, umbilicus, nasolabial folds, and other anatomic areas.1,2 The lesions can be painful and often manifest in multiples, especially in patients living with HIV.3
Condylomata lata can have a verrucous appearance and may mimic other anogenital lesions, such as condylomata acuminata, genital herpes, and malignant tumors, leading to an initial misdiagnosis.1,2 Condylomata lata should always be included in the differential when evaluating anogenital lesions. Other conditions in the differential diagnosis include psoriasis, typically manifesting as erythematous plaques with silver scale, and molluscum contagiosum, appearing as small umbilicated papules on physical examination.
Condylomata lata have been reported to occur in 6% to 23% of patients with secondary syphilis.1 Although secondary syphilis more typically manifests with a diffuse maculopapular rash, condylomata lata may be the sole dermatologic manifestation.4
Histopathology of condylomata lata consists of epithelial hyperplasia as well as lymphocytic and plasma cell infiltrates. It is diagnosed by serologic testing as well as immunohistochemical staining or dark-field microscopy.
First-line treatment of secondary syphilis is a single dose of benzathine penicillin G administered intramuscularly.5 However, a 14-day course of oral doxycycline can be used in patients with a penicillin allergy. When compliance and follow-up cannot be guaranteed, penicillin desensitization and treatment with benzathine penicillin G is recommended. Clinical evaluation and repeat serologic testing should be performed at 6 and 12 months follow-up, or more frequently if clinically indicated.5
- Pourang A, Fung MA, Tartar D, et al. Condyloma lata in secondary syphilis. JAAD Case Rep. 2021;10:18-21. doi:10.1016/j.jdcr.2021.01.025
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277. doi:10.4103/0378-6323.191129
- Rompalo AM, Joesoef MR, O’Donnell JA, et al; Syphilis and HIV Study Group. Clinical manifestations of early syphilis by HIV status and gender: results of the syphilis and HIV study. Sex Transm Dis.2001;28:158-165.
- Kumar P, Das A, Mondal A. Secondary syphilis: an unusual presentation. Indian J Sex Transm Dis AIDS. 2017;38:98-99. doi:10.4103/0253-7184.194318
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
The Diagnosis: Condyloma Latum
Laboratory test results were notable for a rapid plasma reagin titer of 1:512, a positive Treponema pallidum particle agglutination test, negative rectal nucleic acid amplification tests for gonorrhea and chlamydia, and a negative herpes simplex virus polymerase chain reaction. A VDRL test of cerebrospinal fluid from a lumbar puncture was negative. Histopathology of the punch biopsy sample revealed marked verrucous epidermal hyperplasia without keratinocytic atypia and with mixed inflammation (Figure 1), while immunohistochemical staining showed numerus T pallidum organisms (Figure 2). A diagnosis of condyloma latum was made based on the laboratory, lumbar puncture, and punch biopsy results. Due to a penicillin allergy, the patient was treated with oral doxycycline for 14 days. On follow-up at day 12 of therapy, he reported cessation of rectal pain, and resolution of anal lesions was noted on physical examination.
Condylomata lata are highly infectious cutaneous lesions that can manifest during secondary syphilis.1 They typically are described as white or gray, raised, flatappearing plaques and occur in moist areas or skin folds including the anus, scrotum, and vulva. However, these lesions also have been reported in the axillae, umbilicus, nasolabial folds, and other anatomic areas.1,2 The lesions can be painful and often manifest in multiples, especially in patients living with HIV.3
Condylomata lata can have a verrucous appearance and may mimic other anogenital lesions, such as condylomata acuminata, genital herpes, and malignant tumors, leading to an initial misdiagnosis.1,2 Condylomata lata should always be included in the differential when evaluating anogenital lesions. Other conditions in the differential diagnosis include psoriasis, typically manifesting as erythematous plaques with silver scale, and molluscum contagiosum, appearing as small umbilicated papules on physical examination.
Condylomata lata have been reported to occur in 6% to 23% of patients with secondary syphilis.1 Although secondary syphilis more typically manifests with a diffuse maculopapular rash, condylomata lata may be the sole dermatologic manifestation.4
Histopathology of condylomata lata consists of epithelial hyperplasia as well as lymphocytic and plasma cell infiltrates. It is diagnosed by serologic testing as well as immunohistochemical staining or dark-field microscopy.
First-line treatment of secondary syphilis is a single dose of benzathine penicillin G administered intramuscularly.5 However, a 14-day course of oral doxycycline can be used in patients with a penicillin allergy. When compliance and follow-up cannot be guaranteed, penicillin desensitization and treatment with benzathine penicillin G is recommended. Clinical evaluation and repeat serologic testing should be performed at 6 and 12 months follow-up, or more frequently if clinically indicated.5
The Diagnosis: Condyloma Latum
Laboratory test results were notable for a rapid plasma reagin titer of 1:512, a positive Treponema pallidum particle agglutination test, negative rectal nucleic acid amplification tests for gonorrhea and chlamydia, and a negative herpes simplex virus polymerase chain reaction. A VDRL test of cerebrospinal fluid from a lumbar puncture was negative. Histopathology of the punch biopsy sample revealed marked verrucous epidermal hyperplasia without keratinocytic atypia and with mixed inflammation (Figure 1), while immunohistochemical staining showed numerus T pallidum organisms (Figure 2). A diagnosis of condyloma latum was made based on the laboratory, lumbar puncture, and punch biopsy results. Due to a penicillin allergy, the patient was treated with oral doxycycline for 14 days. On follow-up at day 12 of therapy, he reported cessation of rectal pain, and resolution of anal lesions was noted on physical examination.
Condylomata lata are highly infectious cutaneous lesions that can manifest during secondary syphilis.1 They typically are described as white or gray, raised, flatappearing plaques and occur in moist areas or skin folds including the anus, scrotum, and vulva. However, these lesions also have been reported in the axillae, umbilicus, nasolabial folds, and other anatomic areas.1,2 The lesions can be painful and often manifest in multiples, especially in patients living with HIV.3
Condylomata lata can have a verrucous appearance and may mimic other anogenital lesions, such as condylomata acuminata, genital herpes, and malignant tumors, leading to an initial misdiagnosis.1,2 Condylomata lata should always be included in the differential when evaluating anogenital lesions. Other conditions in the differential diagnosis include psoriasis, typically manifesting as erythematous plaques with silver scale, and molluscum contagiosum, appearing as small umbilicated papules on physical examination.
Condylomata lata have been reported to occur in 6% to 23% of patients with secondary syphilis.1 Although secondary syphilis more typically manifests with a diffuse maculopapular rash, condylomata lata may be the sole dermatologic manifestation.4
Histopathology of condylomata lata consists of epithelial hyperplasia as well as lymphocytic and plasma cell infiltrates. It is diagnosed by serologic testing as well as immunohistochemical staining or dark-field microscopy.
First-line treatment of secondary syphilis is a single dose of benzathine penicillin G administered intramuscularly.5 However, a 14-day course of oral doxycycline can be used in patients with a penicillin allergy. When compliance and follow-up cannot be guaranteed, penicillin desensitization and treatment with benzathine penicillin G is recommended. Clinical evaluation and repeat serologic testing should be performed at 6 and 12 months follow-up, or more frequently if clinically indicated.5
- Pourang A, Fung MA, Tartar D, et al. Condyloma lata in secondary syphilis. JAAD Case Rep. 2021;10:18-21. doi:10.1016/j.jdcr.2021.01.025
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277. doi:10.4103/0378-6323.191129
- Rompalo AM, Joesoef MR, O’Donnell JA, et al; Syphilis and HIV Study Group. Clinical manifestations of early syphilis by HIV status and gender: results of the syphilis and HIV study. Sex Transm Dis.2001;28:158-165.
- Kumar P, Das A, Mondal A. Secondary syphilis: an unusual presentation. Indian J Sex Transm Dis AIDS. 2017;38:98-99. doi:10.4103/0253-7184.194318
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
- Pourang A, Fung MA, Tartar D, et al. Condyloma lata in secondary syphilis. JAAD Case Rep. 2021;10:18-21. doi:10.1016/j.jdcr.2021.01.025
- Liu Z, Wang L, Zhang G, et al. Warty mucosal lesions: oral condyloma lata of secondary syphilis. Indian J Dermatol Venereol Leprol. 2017;83:277. doi:10.4103/0378-6323.191129
- Rompalo AM, Joesoef MR, O’Donnell JA, et al; Syphilis and HIV Study Group. Clinical manifestations of early syphilis by HIV status and gender: results of the syphilis and HIV study. Sex Transm Dis.2001;28:158-165.
- Kumar P, Das A, Mondal A. Secondary syphilis: an unusual presentation. Indian J Sex Transm Dis AIDS. 2017;38:98-99. doi:10.4103/0253-7184.194318
- Workowski KA, Bachmann LH, Chan PA, et al. Sexually transmitted infections treatment guidelines, 2021. MMWR Recomm Rep. 2021;70:1-187. doi:10.15585/mmwr.rr7004a1
A 24-year-old man presented to the emergency department with rectal pain and lesions of 3 weeks’ duration that were progressively worsening. He had a medical history of poorly controlled HIV, cerebral toxoplasmosis, and genital herpes, as well as a social history of sexual activity with other men.
He had been diagnosed with HIV 7 years prior and had been off therapy until 1 year prior to the current presentation, when he was hospitalized with encephalopathy (CD4 count, <50 cells/mm3). A diagnosis of cerebral toxoplasmosis was made, and he began a treatment regimen of sulfadiazine, pyrimethamine, and leucovorin, as well as bictegravir, emtricitabine, and tenofovir alafenamide. Since then, the patient admitted to difficulty with medication adherence.
Rapid plasma reagin, gonorrhea, and chlamydia testing were negative during a routine workup 6 months prior to the current presentation. He initially presented to an urgent care clinic for evaluation of the rectal pain and lesions and was treated empirically with topical podofilox. He presented to the emergency department 1 week later (3 weeks after symptom onset) with anal warts and apparent vesicular lesions. Empiric treatment with oral valacyclovir was prescribed.
Despite these treatments, the rectal pain became severe—especially upon sitting, defecation, and physical exertion—prompting further evaluation. Physical examination revealed soft, flat-topped, moist-appearing, gray plaques with minimal surrounding erythema at the anus. Laboratory test results demonstrated a CD4 count of 161 cells/mm3 and an HIV viral load of 137 copies/mL.
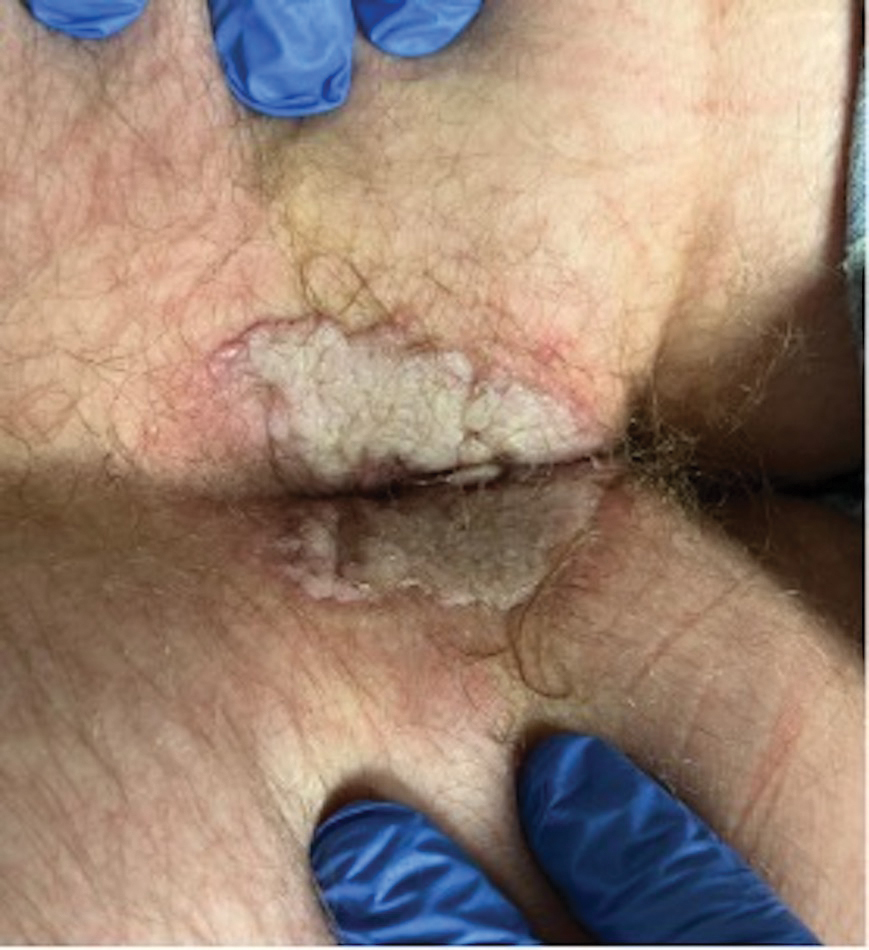
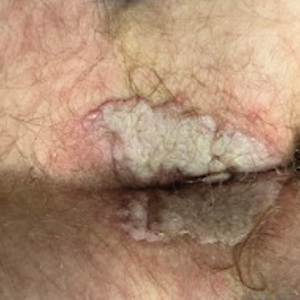
The Shield Sign of Cutaneous Metastases Is Associated With Carcinoma Hemorrhagiectoides
To the Editor:
We read with interest the Case Letter from Wang et al1 (Cutis. 2023;112:E13-E15) of a 60-year-old man whose metastatic salivary duct adenocarcinoma manifested with the shield sign as well as carcinoma hemorrhagiectoides. Cutaneous metastases have seldom been described in association with salivary duct carcinoma.2-7 In addition, carcinoma hemorrhagiectoides–associated shield sign has not been commonly reported.5,8-12
Salivary duct carcinoma—an uncommon head and neck malignancy characterized by androgen receptor expression—rarely is associated with cutaneous metastases. Based on a PubMed search of articles indexed for MEDLINE using the terms cutaneous, metastatic, salivary duct carcinoma, and/or skin, including the patient described by Wang et al,1 there have been 8 individuals with cutaneous metastases from this cancer. The morphology of the cutaneous metastases has varied from angiomatous to angiokeratomalike (black and keratotic) papules, bullae, macules (red), papules and nodules (erythematous and scaly), plaques (cellulitislike and confluent that were purpuric, hemorrhagic, and violaceous), pseudovesicles, purpuric papules, subcutaneous nodules, and an ulcer (superficial and mimicked a basal cell carcinoma).1-7 Remarkably, 4 of 8 patients (50%) with salivary duct carcinoma cutaneous metastases presented with a shield sign,5,7 including the case reported by Wang et al.1
The shield sign is a distinctive clinical manifestation of cutaneous metastasis.10 It was named to describe the skin metastases located predominantly on the chest area that would be covered by a medieval knight’s shield5,10,12; metastatic lesions also have been noted on the proximal arm and/or the upper back in a similar distribution.8,9 To date, based on a PubMed search of articles indexed for MEDLINE using the search terms breast cancer, carcinoma, hemorrhagiectoides, metastases, salivary duct carcinoma, shield, and/or sign, the shield sign has been described in 6 patients with cutaneous metastases either from salivary duct carcinoma (4 patients)1,5,7 or breast cancer (2 patients).8,9 The shield sign pathologically corresponds to carcinoma hemorrhagiectoides, an inflammatory pattern of cutaneous metastases.5,11
Inflammatory cutaneous metastatic carcinoma has 3 distinctive clinical and pathologic manifestations.11 Carcinoma erysipelatoides and carcinoma telangiectoides were the earlier described variants.11 In 2012, carcinoma hemorrhagiectoides was described as the third pattern of inflammatory cutaneous metastasis.5
Carcinoma erysipelatoides, which clinically mimics cutaneous streptococcal cellulitis, appears as a well-defined erythematous patch or plaque; the tumor cells can be found in the lymphatic vessels and either are absent or minimally present in the dermis. Carcinoma telangiectoides, which clinically mimics idiopathic telangiectases, appears as an erythematous patch with prominent telangiectases; the tumor cells can be found in the blood vessels and are either absent or minimally present in the dermis. Carcinoma hemorrhagiectoides appears as purpuric or violaceous indurated plaques; the tumor cells are not only found in the blood vessels, in the lymphatic vessels, or both, but also can be mildly to extensively present in the dermis.5,10,11
In conclusion, the shield sign is a unique presentation of inflammatory cutaneous metastatic carcinoma, which is associated with carcinoma hemorrhagiectoides. The clinical features of the infiltrated plaques correspond to the presence of tumor cells in the blood vessels, lymphatic vessels, and the dermis; in addition, the purpuric and violaceous appearance correlates with the presence of extravasated erythrocytes or hemorrhage in the dermis. To date, half of the patients with skin metastases from salivary duct carcinoma have presented with carcinoma hemorrhagiectoides–associated shield sign.
Authors’ Response
We appreciate and welcome the comments provided by the authors. Drawing attention to unusual pathologic manifestations of cutaneous metastatic salivary duct carcinoma manifesting with the shield sign, the authors present a comprehensive review of 3 distinctive presentations: carcinoma erysipelatoides, carcinoma telangiectoides, and carcinoma hemorrhagiectoides. The inclusion of these variants enriches the discussion and makes this letter a valuable addition to the literature on cutaneous metastatic carcinoma, particularly metastatic salivary duct carcinoma.
Xintong Wang, MD; William H. Westra, MD
From the Department of Pathology, Icahn School of Medicine at Mount Sinai, New York, New York.
The authors report no conflict of interest.
- Wang X, Vyas NS, Alghamdi AA, et al. Cutaneous presentation of metastatic salivary duct carcinoma. Cutis. 2023;112:E13-E15.
- Pollock JL, Catalano E. Metastatic ductal carcinoma of the parotid gland in a patient with sarcoidosis. Arch Dermatol. 1979;115:1098-1099.
- Pollock JL. Metastatic carcinoma of the parotid gland resembling carcinoma of the breast. J Am Acad Dermatol. 1996;34:1093.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Chakari W, Andersen L, Anderson JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Shin JY, Eun DH, Lee JY, et al. A case of cutaneous metastases of salivary duct carcinoma mimicking radiation recall dermatitis. Ann Dermatol. 2020;32:436-438.
- Aravena RC, Aravena DC, Velasco MJ, et al. Carcinoma hemorrhagiectoides: case report of an uncommon presentation of cutaneous metastatic breast carcinoma. Dermatol Online J. 2017;23:13030/qt3hn3z850.
- Smith KA, Basko-Plluska J, Kothari AD, et al. Cutaneous metastatic breast adenocarcinoma. Cutis. 2020;105:E20-E22.
- Cohen PR, Kurzrock R. Cutaneous metastatic cancer: carcinoma hemorrhagiectoides presenting as the shield sign. Cureus. 2021;13:e12627.
- Cohen PR. Pleomorphic appearance of breast cancer cutaneous metastases. Cureus. 2021;13:e20301.
- Cohen PR, Prieto VG, Kurzrock R. Tumor lysis syndrome: introduction of a cutaneous variant and a new classification system. Cureus. 2021;13:e13816.
To the Editor:
We read with interest the Case Letter from Wang et al1 (Cutis. 2023;112:E13-E15) of a 60-year-old man whose metastatic salivary duct adenocarcinoma manifested with the shield sign as well as carcinoma hemorrhagiectoides. Cutaneous metastases have seldom been described in association with salivary duct carcinoma.2-7 In addition, carcinoma hemorrhagiectoides–associated shield sign has not been commonly reported.5,8-12
Salivary duct carcinoma—an uncommon head and neck malignancy characterized by androgen receptor expression—rarely is associated with cutaneous metastases. Based on a PubMed search of articles indexed for MEDLINE using the terms cutaneous, metastatic, salivary duct carcinoma, and/or skin, including the patient described by Wang et al,1 there have been 8 individuals with cutaneous metastases from this cancer. The morphology of the cutaneous metastases has varied from angiomatous to angiokeratomalike (black and keratotic) papules, bullae, macules (red), papules and nodules (erythematous and scaly), plaques (cellulitislike and confluent that were purpuric, hemorrhagic, and violaceous), pseudovesicles, purpuric papules, subcutaneous nodules, and an ulcer (superficial and mimicked a basal cell carcinoma).1-7 Remarkably, 4 of 8 patients (50%) with salivary duct carcinoma cutaneous metastases presented with a shield sign,5,7 including the case reported by Wang et al.1
The shield sign is a distinctive clinical manifestation of cutaneous metastasis.10 It was named to describe the skin metastases located predominantly on the chest area that would be covered by a medieval knight’s shield5,10,12; metastatic lesions also have been noted on the proximal arm and/or the upper back in a similar distribution.8,9 To date, based on a PubMed search of articles indexed for MEDLINE using the search terms breast cancer, carcinoma, hemorrhagiectoides, metastases, salivary duct carcinoma, shield, and/or sign, the shield sign has been described in 6 patients with cutaneous metastases either from salivary duct carcinoma (4 patients)1,5,7 or breast cancer (2 patients).8,9 The shield sign pathologically corresponds to carcinoma hemorrhagiectoides, an inflammatory pattern of cutaneous metastases.5,11
Inflammatory cutaneous metastatic carcinoma has 3 distinctive clinical and pathologic manifestations.11 Carcinoma erysipelatoides and carcinoma telangiectoides were the earlier described variants.11 In 2012, carcinoma hemorrhagiectoides was described as the third pattern of inflammatory cutaneous metastasis.5
Carcinoma erysipelatoides, which clinically mimics cutaneous streptococcal cellulitis, appears as a well-defined erythematous patch or plaque; the tumor cells can be found in the lymphatic vessels and either are absent or minimally present in the dermis. Carcinoma telangiectoides, which clinically mimics idiopathic telangiectases, appears as an erythematous patch with prominent telangiectases; the tumor cells can be found in the blood vessels and are either absent or minimally present in the dermis. Carcinoma hemorrhagiectoides appears as purpuric or violaceous indurated plaques; the tumor cells are not only found in the blood vessels, in the lymphatic vessels, or both, but also can be mildly to extensively present in the dermis.5,10,11
In conclusion, the shield sign is a unique presentation of inflammatory cutaneous metastatic carcinoma, which is associated with carcinoma hemorrhagiectoides. The clinical features of the infiltrated plaques correspond to the presence of tumor cells in the blood vessels, lymphatic vessels, and the dermis; in addition, the purpuric and violaceous appearance correlates with the presence of extravasated erythrocytes or hemorrhage in the dermis. To date, half of the patients with skin metastases from salivary duct carcinoma have presented with carcinoma hemorrhagiectoides–associated shield sign.
Authors’ Response
We appreciate and welcome the comments provided by the authors. Drawing attention to unusual pathologic manifestations of cutaneous metastatic salivary duct carcinoma manifesting with the shield sign, the authors present a comprehensive review of 3 distinctive presentations: carcinoma erysipelatoides, carcinoma telangiectoides, and carcinoma hemorrhagiectoides. The inclusion of these variants enriches the discussion and makes this letter a valuable addition to the literature on cutaneous metastatic carcinoma, particularly metastatic salivary duct carcinoma.
Xintong Wang, MD; William H. Westra, MD
From the Department of Pathology, Icahn School of Medicine at Mount Sinai, New York, New York.
The authors report no conflict of interest.
To the Editor:
We read with interest the Case Letter from Wang et al1 (Cutis. 2023;112:E13-E15) of a 60-year-old man whose metastatic salivary duct adenocarcinoma manifested with the shield sign as well as carcinoma hemorrhagiectoides. Cutaneous metastases have seldom been described in association with salivary duct carcinoma.2-7 In addition, carcinoma hemorrhagiectoides–associated shield sign has not been commonly reported.5,8-12
Salivary duct carcinoma—an uncommon head and neck malignancy characterized by androgen receptor expression—rarely is associated with cutaneous metastases. Based on a PubMed search of articles indexed for MEDLINE using the terms cutaneous, metastatic, salivary duct carcinoma, and/or skin, including the patient described by Wang et al,1 there have been 8 individuals with cutaneous metastases from this cancer. The morphology of the cutaneous metastases has varied from angiomatous to angiokeratomalike (black and keratotic) papules, bullae, macules (red), papules and nodules (erythematous and scaly), plaques (cellulitislike and confluent that were purpuric, hemorrhagic, and violaceous), pseudovesicles, purpuric papules, subcutaneous nodules, and an ulcer (superficial and mimicked a basal cell carcinoma).1-7 Remarkably, 4 of 8 patients (50%) with salivary duct carcinoma cutaneous metastases presented with a shield sign,5,7 including the case reported by Wang et al.1
The shield sign is a distinctive clinical manifestation of cutaneous metastasis.10 It was named to describe the skin metastases located predominantly on the chest area that would be covered by a medieval knight’s shield5,10,12; metastatic lesions also have been noted on the proximal arm and/or the upper back in a similar distribution.8,9 To date, based on a PubMed search of articles indexed for MEDLINE using the search terms breast cancer, carcinoma, hemorrhagiectoides, metastases, salivary duct carcinoma, shield, and/or sign, the shield sign has been described in 6 patients with cutaneous metastases either from salivary duct carcinoma (4 patients)1,5,7 or breast cancer (2 patients).8,9 The shield sign pathologically corresponds to carcinoma hemorrhagiectoides, an inflammatory pattern of cutaneous metastases.5,11
Inflammatory cutaneous metastatic carcinoma has 3 distinctive clinical and pathologic manifestations.11 Carcinoma erysipelatoides and carcinoma telangiectoides were the earlier described variants.11 In 2012, carcinoma hemorrhagiectoides was described as the third pattern of inflammatory cutaneous metastasis.5
Carcinoma erysipelatoides, which clinically mimics cutaneous streptococcal cellulitis, appears as a well-defined erythematous patch or plaque; the tumor cells can be found in the lymphatic vessels and either are absent or minimally present in the dermis. Carcinoma telangiectoides, which clinically mimics idiopathic telangiectases, appears as an erythematous patch with prominent telangiectases; the tumor cells can be found in the blood vessels and are either absent or minimally present in the dermis. Carcinoma hemorrhagiectoides appears as purpuric or violaceous indurated plaques; the tumor cells are not only found in the blood vessels, in the lymphatic vessels, or both, but also can be mildly to extensively present in the dermis.5,10,11
In conclusion, the shield sign is a unique presentation of inflammatory cutaneous metastatic carcinoma, which is associated with carcinoma hemorrhagiectoides. The clinical features of the infiltrated plaques correspond to the presence of tumor cells in the blood vessels, lymphatic vessels, and the dermis; in addition, the purpuric and violaceous appearance correlates with the presence of extravasated erythrocytes or hemorrhage in the dermis. To date, half of the patients with skin metastases from salivary duct carcinoma have presented with carcinoma hemorrhagiectoides–associated shield sign.
Authors’ Response
We appreciate and welcome the comments provided by the authors. Drawing attention to unusual pathologic manifestations of cutaneous metastatic salivary duct carcinoma manifesting with the shield sign, the authors present a comprehensive review of 3 distinctive presentations: carcinoma erysipelatoides, carcinoma telangiectoides, and carcinoma hemorrhagiectoides. The inclusion of these variants enriches the discussion and makes this letter a valuable addition to the literature on cutaneous metastatic carcinoma, particularly metastatic salivary duct carcinoma.
Xintong Wang, MD; William H. Westra, MD
From the Department of Pathology, Icahn School of Medicine at Mount Sinai, New York, New York.
The authors report no conflict of interest.
- Wang X, Vyas NS, Alghamdi AA, et al. Cutaneous presentation of metastatic salivary duct carcinoma. Cutis. 2023;112:E13-E15.
- Pollock JL, Catalano E. Metastatic ductal carcinoma of the parotid gland in a patient with sarcoidosis. Arch Dermatol. 1979;115:1098-1099.
- Pollock JL. Metastatic carcinoma of the parotid gland resembling carcinoma of the breast. J Am Acad Dermatol. 1996;34:1093.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Chakari W, Andersen L, Anderson JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Shin JY, Eun DH, Lee JY, et al. A case of cutaneous metastases of salivary duct carcinoma mimicking radiation recall dermatitis. Ann Dermatol. 2020;32:436-438.
- Aravena RC, Aravena DC, Velasco MJ, et al. Carcinoma hemorrhagiectoides: case report of an uncommon presentation of cutaneous metastatic breast carcinoma. Dermatol Online J. 2017;23:13030/qt3hn3z850.
- Smith KA, Basko-Plluska J, Kothari AD, et al. Cutaneous metastatic breast adenocarcinoma. Cutis. 2020;105:E20-E22.
- Cohen PR, Kurzrock R. Cutaneous metastatic cancer: carcinoma hemorrhagiectoides presenting as the shield sign. Cureus. 2021;13:e12627.
- Cohen PR. Pleomorphic appearance of breast cancer cutaneous metastases. Cureus. 2021;13:e20301.
- Cohen PR, Prieto VG, Kurzrock R. Tumor lysis syndrome: introduction of a cutaneous variant and a new classification system. Cureus. 2021;13:e13816.
- Wang X, Vyas NS, Alghamdi AA, et al. Cutaneous presentation of metastatic salivary duct carcinoma. Cutis. 2023;112:E13-E15.
- Pollock JL, Catalano E. Metastatic ductal carcinoma of the parotid gland in a patient with sarcoidosis. Arch Dermatol. 1979;115:1098-1099.
- Pollock JL. Metastatic carcinoma of the parotid gland resembling carcinoma of the breast. J Am Acad Dermatol. 1996;34:1093.
- Aygit AC, Top H, Cakir B, et al. Salivary duct carcinoma of the parotid gland metastasizing to the skin: a case report and review of the literature. Am J Dermatopathol. 2005;27:48-50.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The “shield sign” in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Chakari W, Andersen L, Anderson JL. Cutaneous metastases from salivary duct carcinoma of the submandibular gland. Case Rep Dermatol. 2017;9:254-258.
- Shin JY, Eun DH, Lee JY, et al. A case of cutaneous metastases of salivary duct carcinoma mimicking radiation recall dermatitis. Ann Dermatol. 2020;32:436-438.
- Aravena RC, Aravena DC, Velasco MJ, et al. Carcinoma hemorrhagiectoides: case report of an uncommon presentation of cutaneous metastatic breast carcinoma. Dermatol Online J. 2017;23:13030/qt3hn3z850.
- Smith KA, Basko-Plluska J, Kothari AD, et al. Cutaneous metastatic breast adenocarcinoma. Cutis. 2020;105:E20-E22.
- Cohen PR, Kurzrock R. Cutaneous metastatic cancer: carcinoma hemorrhagiectoides presenting as the shield sign. Cureus. 2021;13:e12627.
- Cohen PR. Pleomorphic appearance of breast cancer cutaneous metastases. Cureus. 2021;13:e20301.
- Cohen PR, Prieto VG, Kurzrock R. Tumor lysis syndrome: introduction of a cutaneous variant and a new classification system. Cureus. 2021;13:e13816.
Pruritic Rash on the Neck and Back
The Diagnosis: Prurigo Pigmentosa
A comprehensive metabolic panel collected from our patient 1 month earlier did not reveal any abnormalities. Serum methylmalonic acid and homocysteine were both elevated at 417 nmol/L (reference range [for those aged 2–59 years], 55–335 nmol/L) and 23 μmol/L (reference range, 5–15 μmol/L), respectively. Serum folate and 25-hydroxyvitamin D were low at 3.1 ng/mL (reference range, >4.8 ng/mL) and 5 ng/mL (reference range, 30–80 ng/mL), respectively. Vitamin B12 was within reference range. Two 4-mm punch biopsies collected from the upper back showed spongiotic dermatitis.
Our patient’s histopathology results along with the rash distribution and medical history of anorexia increased suspicion for prurigo pigmentosa. A trial of oral doxycycline 100 mg twice daily for 2 weeks was prescribed. At 2-week follow-up, the patient’s mother revealed a history of ketosis in her daughter, solidifying the diagnosis. The patient was counseled on maintaining a healthy diet to prevent future breakouts. The patient’s rash resolved with diet modification and doxycycline; however, it recurred upon relapse of anorexia 4 months later.
Prurigo pigmentosa, originally identified in Japan by Nagashima et al,1 is an uncommon recurrent inflammatory disorder predominantly observed in young adults of Asian descent. Subsequently, it was reported to occur among individuals from different ethnic backgrounds, indicating potential underdiagnosis or misdiagnosis in Western countries.2 Although a direct pathogenic cause for prurigo pigmentosa has not been identified, a strong association has been linked to diet, specifically when ketosis is induced, such as in ketogenic diets and anorexia nervosa.3-5 Other possible causes include sunlight exposure, clothing friction, and sweating.1,5 The disease course is characterized by intermittent flares and spontaneous resolution, with recurrence in most cases. During the active phase, intensely pruritic, papulovesicular or urticarial papules are predominant and most often are localized to the upper body and torso, including the back, shoulders, neck, and chest.5 These flares can persist for several days but eventually subside, leaving behind a characteristic reticular pigmentation that can persist for months.5 First-line treatment often involves the use of tetracycline antibiotics, such as minocycline or doxycycline. 2,4,5 Dapsone often is used with successful resolution. 6 Dietary modifications also have been found to be effective in treating prurigo pigmentosa, particularly in patients presenting with dietary insufficiency.6,7 Increased carbohydrate intake has been shown to promote resolution. 6 Topical corticosteroids demonstrate limited efficacy in controlling flares.6,8
Histopathology has been variably described, with initial findings reported as nonspecific.1 However, it was later described as a distinct inflammatory disease of the skin with histologically distinct stages.2,9 Early stages reveal scattered dermal, dermal papillary, and perivascular neutrophilic infiltration.9 The lesions then progress and become fully developed, at which point neutrophilic infiltration becomes more prominent, accompanied by the presence of intraepidermal neutrophils and spongiosis. As the lesions resolve, the infiltration transitions to lymphocytic, and lichenoid changes can sometimes be appreciated along with epidermal hyperplasia, hyperpigmentation, and dermal melanophages.9 Although these findings aid in the diagnosis of prurigo pigmentosa, a clinicopathologic correlation is necessary to establish a definitive diagnosis.
Because prurigo pigmentosa is rare, it often is misdiagnosed as another condition with a similar presentation and nonspecific biopsy findings.6 Allergic contact dermatitis is a common type IV delayed hypersensitivity reaction that manifests similar to prurigo pigmentosa with pruritus and a well-demarcated distribution10 that is related to the pattern of allergen exposure; in the case of allergic contact dermatitis related to textiles, a well-demarcated rash will appear in the distribution area of the associated clothing (eg, shirt, pants, shorts).11 Development of allergy involves exposure and sensitization to an allergen, followed by subsequent re-exposure that results in cutaneous T-cell activation and inflammation. 10 Histopathology shows nonspecific spongiotic inflammation, and the gold standard for diagnosis is patch testing to identify the causative substance(s). Definitive treatment includes avoidance of identified allergies; however, if patients are unable to avoid the allergen or the cause is unknown, then corticosteroids, antihistamines, and/or calcineurin inhibitors are beneficial in controlling symptoms and flares.10
Pityrosporum folliculitis (also known as Malassezia folliculitis) is a fungal acneform condition that arises from overgrowth of normal skin flora Malassezia yeast,12 which may be due to occlusion of follicles or disruption of the normal flora composition. Clinically, the manifestation may resemble prurigo pigmentosa in distribution and presence of intense pruritus. However, pustular lesions and involvement of the face can aid in differentiating Pityrosporum from prurigo pigmentosa, which can be confirmed via periodic acid–Schiff staining with numerous round yeasts within affected follicles. Oral antifungal therapy typically yields rapid improvement and resolution of symptoms.12
Urticaria and prurigo pigmentosa share similar clinical characteristics, with symptoms of intense pruritus and urticarial lesions on the trunk.2,13 Urticaria is an IgEmediated type I hypersensitivity reaction characterized by wheals (ie, edematous red or pink lesions of variable size and shape that typically resolve spontaneously within 24–48 hours).13 Notably, urticaria will improve and in some cases completely resolve with antihistamines or anti-IgE antibody treatment, which may aid in distinguishing it from prurigo pigmentosa, as the latter typically exhibits limited response to such treatment.2 Histopathology also can assist in the diagnosis by ruling out other causes of similar rash; however, biopsies are not routinely done unless other inflammatory conditions are of high suspicion.13
Bullous pemphigoid is an autoimmune, subepidermal, blistering dermatosis that is most common among the elderly.14 It is characterized by the presence of IgG antibodies that target BP180 and BP230, which initiate inflammatory cascades that lead to tissue damage and blister formation. It typically manifests as pruritic blistering eruptions, primarily on the limbs and trunk, but may involve the head, neck, or palmoplantar regions.14 Although blistering eruptions are the prodrome of the disease, some cases may present with nonspecific urticarial or eczematous lesions14,15 that may resemble prurigo pigmentosa. The diagnosis is confirmed through direct immunofluorescence microscopy of biopsied lesions, which reveals IgG and/or C3 deposits along the dermoepidermal junction.14 Management of bullous pemphigoid involves timely initiation of dapsone or systemic corticosteroids, which have demonstrated high efficacy in controlling the disease and its associated symptoms.15
Our patient achieved a favorable response to diet modification and doxycycline therapy consistent with the diagnosis of prurigo pigmentosa. Unfortunately, the condition recurred following a relapse of anorexia. Management of prurigo pigmentosa necessitates not only accurate diagnosis but also addressing any underlying factors that may contribute to disease exacerbation. We anticipate the eating disorder will pose a major challenge in achieving long-term control of prurigo pigmentosa.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation. Jpn J Dermatol. 1971;81:38-39.
- Boer A, Asgari M. Prurigo pigmentosa: an underdiagnosed disease? Indian J Dermatol Venereol Leprol. 2006;72:405-409. doi:10.4103/0378-6323.29334
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet. Pediatr Dermatol. 2013;32:248-251. doi:10.1111/pde.12275
- Teraki Y, Teraki E, Kawashima M, et al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511. doi:10.1016/s0190-9622(96)90460-0
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79-87. doi:10.1016/j.jdin.2021.03.003
- Wong M, Lee E, Wu Y, et al. Treatment of prurigo pigmentosa with diet modification: a medical case study. Hawaii J Med Public Health. 2018;77:114-117.
- Almaani N, Al-Tarawneh AH, Msallam H. Prurigo pigmentosa: a clinicopathological report of three Middle Eastern patients. Case Rep Dermatol Med. 2018;2018:9406797. doi:10.1155/2018/9406797
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897. doi:10.1111/j.1346-8138.2012.01640.x
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74:1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Lazarov A, Cordoba M, Plosk N, et al. Atypical and unusual clinical manifestations of contact dermatitis to clothing (textile contact dermatitis)—case presentation and review of the literature. Dermatol Online J. 2003;9. doi:10.5070/d30kd1d259
- Rubenstein RM, Malerich SA. Malassezia (Pityrosporum) folliculitis. J Clin Aesthet Dermatol. 2014;7:37-41.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277. doi:10.1016/j.jaci.2014.02.036
- della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol. 2012;167:1111-1117. doi:10.1111/j.1365-2133.2012.11108.x
- Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, et al. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. Int J Dermatol. 1998;37:508-514.
The Diagnosis: Prurigo Pigmentosa
A comprehensive metabolic panel collected from our patient 1 month earlier did not reveal any abnormalities. Serum methylmalonic acid and homocysteine were both elevated at 417 nmol/L (reference range [for those aged 2–59 years], 55–335 nmol/L) and 23 μmol/L (reference range, 5–15 μmol/L), respectively. Serum folate and 25-hydroxyvitamin D were low at 3.1 ng/mL (reference range, >4.8 ng/mL) and 5 ng/mL (reference range, 30–80 ng/mL), respectively. Vitamin B12 was within reference range. Two 4-mm punch biopsies collected from the upper back showed spongiotic dermatitis.
Our patient’s histopathology results along with the rash distribution and medical history of anorexia increased suspicion for prurigo pigmentosa. A trial of oral doxycycline 100 mg twice daily for 2 weeks was prescribed. At 2-week follow-up, the patient’s mother revealed a history of ketosis in her daughter, solidifying the diagnosis. The patient was counseled on maintaining a healthy diet to prevent future breakouts. The patient’s rash resolved with diet modification and doxycycline; however, it recurred upon relapse of anorexia 4 months later.
Prurigo pigmentosa, originally identified in Japan by Nagashima et al,1 is an uncommon recurrent inflammatory disorder predominantly observed in young adults of Asian descent. Subsequently, it was reported to occur among individuals from different ethnic backgrounds, indicating potential underdiagnosis or misdiagnosis in Western countries.2 Although a direct pathogenic cause for prurigo pigmentosa has not been identified, a strong association has been linked to diet, specifically when ketosis is induced, such as in ketogenic diets and anorexia nervosa.3-5 Other possible causes include sunlight exposure, clothing friction, and sweating.1,5 The disease course is characterized by intermittent flares and spontaneous resolution, with recurrence in most cases. During the active phase, intensely pruritic, papulovesicular or urticarial papules are predominant and most often are localized to the upper body and torso, including the back, shoulders, neck, and chest.5 These flares can persist for several days but eventually subside, leaving behind a characteristic reticular pigmentation that can persist for months.5 First-line treatment often involves the use of tetracycline antibiotics, such as minocycline or doxycycline. 2,4,5 Dapsone often is used with successful resolution. 6 Dietary modifications also have been found to be effective in treating prurigo pigmentosa, particularly in patients presenting with dietary insufficiency.6,7 Increased carbohydrate intake has been shown to promote resolution. 6 Topical corticosteroids demonstrate limited efficacy in controlling flares.6,8
Histopathology has been variably described, with initial findings reported as nonspecific.1 However, it was later described as a distinct inflammatory disease of the skin with histologically distinct stages.2,9 Early stages reveal scattered dermal, dermal papillary, and perivascular neutrophilic infiltration.9 The lesions then progress and become fully developed, at which point neutrophilic infiltration becomes more prominent, accompanied by the presence of intraepidermal neutrophils and spongiosis. As the lesions resolve, the infiltration transitions to lymphocytic, and lichenoid changes can sometimes be appreciated along with epidermal hyperplasia, hyperpigmentation, and dermal melanophages.9 Although these findings aid in the diagnosis of prurigo pigmentosa, a clinicopathologic correlation is necessary to establish a definitive diagnosis.
Because prurigo pigmentosa is rare, it often is misdiagnosed as another condition with a similar presentation and nonspecific biopsy findings.6 Allergic contact dermatitis is a common type IV delayed hypersensitivity reaction that manifests similar to prurigo pigmentosa with pruritus and a well-demarcated distribution10 that is related to the pattern of allergen exposure; in the case of allergic contact dermatitis related to textiles, a well-demarcated rash will appear in the distribution area of the associated clothing (eg, shirt, pants, shorts).11 Development of allergy involves exposure and sensitization to an allergen, followed by subsequent re-exposure that results in cutaneous T-cell activation and inflammation. 10 Histopathology shows nonspecific spongiotic inflammation, and the gold standard for diagnosis is patch testing to identify the causative substance(s). Definitive treatment includes avoidance of identified allergies; however, if patients are unable to avoid the allergen or the cause is unknown, then corticosteroids, antihistamines, and/or calcineurin inhibitors are beneficial in controlling symptoms and flares.10
Pityrosporum folliculitis (also known as Malassezia folliculitis) is a fungal acneform condition that arises from overgrowth of normal skin flora Malassezia yeast,12 which may be due to occlusion of follicles or disruption of the normal flora composition. Clinically, the manifestation may resemble prurigo pigmentosa in distribution and presence of intense pruritus. However, pustular lesions and involvement of the face can aid in differentiating Pityrosporum from prurigo pigmentosa, which can be confirmed via periodic acid–Schiff staining with numerous round yeasts within affected follicles. Oral antifungal therapy typically yields rapid improvement and resolution of symptoms.12
Urticaria and prurigo pigmentosa share similar clinical characteristics, with symptoms of intense pruritus and urticarial lesions on the trunk.2,13 Urticaria is an IgEmediated type I hypersensitivity reaction characterized by wheals (ie, edematous red or pink lesions of variable size and shape that typically resolve spontaneously within 24–48 hours).13 Notably, urticaria will improve and in some cases completely resolve with antihistamines or anti-IgE antibody treatment, which may aid in distinguishing it from prurigo pigmentosa, as the latter typically exhibits limited response to such treatment.2 Histopathology also can assist in the diagnosis by ruling out other causes of similar rash; however, biopsies are not routinely done unless other inflammatory conditions are of high suspicion.13
Bullous pemphigoid is an autoimmune, subepidermal, blistering dermatosis that is most common among the elderly.14 It is characterized by the presence of IgG antibodies that target BP180 and BP230, which initiate inflammatory cascades that lead to tissue damage and blister formation. It typically manifests as pruritic blistering eruptions, primarily on the limbs and trunk, but may involve the head, neck, or palmoplantar regions.14 Although blistering eruptions are the prodrome of the disease, some cases may present with nonspecific urticarial or eczematous lesions14,15 that may resemble prurigo pigmentosa. The diagnosis is confirmed through direct immunofluorescence microscopy of biopsied lesions, which reveals IgG and/or C3 deposits along the dermoepidermal junction.14 Management of bullous pemphigoid involves timely initiation of dapsone or systemic corticosteroids, which have demonstrated high efficacy in controlling the disease and its associated symptoms.15
Our patient achieved a favorable response to diet modification and doxycycline therapy consistent with the diagnosis of prurigo pigmentosa. Unfortunately, the condition recurred following a relapse of anorexia. Management of prurigo pigmentosa necessitates not only accurate diagnosis but also addressing any underlying factors that may contribute to disease exacerbation. We anticipate the eating disorder will pose a major challenge in achieving long-term control of prurigo pigmentosa.
The Diagnosis: Prurigo Pigmentosa
A comprehensive metabolic panel collected from our patient 1 month earlier did not reveal any abnormalities. Serum methylmalonic acid and homocysteine were both elevated at 417 nmol/L (reference range [for those aged 2–59 years], 55–335 nmol/L) and 23 μmol/L (reference range, 5–15 μmol/L), respectively. Serum folate and 25-hydroxyvitamin D were low at 3.1 ng/mL (reference range, >4.8 ng/mL) and 5 ng/mL (reference range, 30–80 ng/mL), respectively. Vitamin B12 was within reference range. Two 4-mm punch biopsies collected from the upper back showed spongiotic dermatitis.
Our patient’s histopathology results along with the rash distribution and medical history of anorexia increased suspicion for prurigo pigmentosa. A trial of oral doxycycline 100 mg twice daily for 2 weeks was prescribed. At 2-week follow-up, the patient’s mother revealed a history of ketosis in her daughter, solidifying the diagnosis. The patient was counseled on maintaining a healthy diet to prevent future breakouts. The patient’s rash resolved with diet modification and doxycycline; however, it recurred upon relapse of anorexia 4 months later.
Prurigo pigmentosa, originally identified in Japan by Nagashima et al,1 is an uncommon recurrent inflammatory disorder predominantly observed in young adults of Asian descent. Subsequently, it was reported to occur among individuals from different ethnic backgrounds, indicating potential underdiagnosis or misdiagnosis in Western countries.2 Although a direct pathogenic cause for prurigo pigmentosa has not been identified, a strong association has been linked to diet, specifically when ketosis is induced, such as in ketogenic diets and anorexia nervosa.3-5 Other possible causes include sunlight exposure, clothing friction, and sweating.1,5 The disease course is characterized by intermittent flares and spontaneous resolution, with recurrence in most cases. During the active phase, intensely pruritic, papulovesicular or urticarial papules are predominant and most often are localized to the upper body and torso, including the back, shoulders, neck, and chest.5 These flares can persist for several days but eventually subside, leaving behind a characteristic reticular pigmentation that can persist for months.5 First-line treatment often involves the use of tetracycline antibiotics, such as minocycline or doxycycline. 2,4,5 Dapsone often is used with successful resolution. 6 Dietary modifications also have been found to be effective in treating prurigo pigmentosa, particularly in patients presenting with dietary insufficiency.6,7 Increased carbohydrate intake has been shown to promote resolution. 6 Topical corticosteroids demonstrate limited efficacy in controlling flares.6,8
Histopathology has been variably described, with initial findings reported as nonspecific.1 However, it was later described as a distinct inflammatory disease of the skin with histologically distinct stages.2,9 Early stages reveal scattered dermal, dermal papillary, and perivascular neutrophilic infiltration.9 The lesions then progress and become fully developed, at which point neutrophilic infiltration becomes more prominent, accompanied by the presence of intraepidermal neutrophils and spongiosis. As the lesions resolve, the infiltration transitions to lymphocytic, and lichenoid changes can sometimes be appreciated along with epidermal hyperplasia, hyperpigmentation, and dermal melanophages.9 Although these findings aid in the diagnosis of prurigo pigmentosa, a clinicopathologic correlation is necessary to establish a definitive diagnosis.
Because prurigo pigmentosa is rare, it often is misdiagnosed as another condition with a similar presentation and nonspecific biopsy findings.6 Allergic contact dermatitis is a common type IV delayed hypersensitivity reaction that manifests similar to prurigo pigmentosa with pruritus and a well-demarcated distribution10 that is related to the pattern of allergen exposure; in the case of allergic contact dermatitis related to textiles, a well-demarcated rash will appear in the distribution area of the associated clothing (eg, shirt, pants, shorts).11 Development of allergy involves exposure and sensitization to an allergen, followed by subsequent re-exposure that results in cutaneous T-cell activation and inflammation. 10 Histopathology shows nonspecific spongiotic inflammation, and the gold standard for diagnosis is patch testing to identify the causative substance(s). Definitive treatment includes avoidance of identified allergies; however, if patients are unable to avoid the allergen or the cause is unknown, then corticosteroids, antihistamines, and/or calcineurin inhibitors are beneficial in controlling symptoms and flares.10
Pityrosporum folliculitis (also known as Malassezia folliculitis) is a fungal acneform condition that arises from overgrowth of normal skin flora Malassezia yeast,12 which may be due to occlusion of follicles or disruption of the normal flora composition. Clinically, the manifestation may resemble prurigo pigmentosa in distribution and presence of intense pruritus. However, pustular lesions and involvement of the face can aid in differentiating Pityrosporum from prurigo pigmentosa, which can be confirmed via periodic acid–Schiff staining with numerous round yeasts within affected follicles. Oral antifungal therapy typically yields rapid improvement and resolution of symptoms.12
Urticaria and prurigo pigmentosa share similar clinical characteristics, with symptoms of intense pruritus and urticarial lesions on the trunk.2,13 Urticaria is an IgEmediated type I hypersensitivity reaction characterized by wheals (ie, edematous red or pink lesions of variable size and shape that typically resolve spontaneously within 24–48 hours).13 Notably, urticaria will improve and in some cases completely resolve with antihistamines or anti-IgE antibody treatment, which may aid in distinguishing it from prurigo pigmentosa, as the latter typically exhibits limited response to such treatment.2 Histopathology also can assist in the diagnosis by ruling out other causes of similar rash; however, biopsies are not routinely done unless other inflammatory conditions are of high suspicion.13
Bullous pemphigoid is an autoimmune, subepidermal, blistering dermatosis that is most common among the elderly.14 It is characterized by the presence of IgG antibodies that target BP180 and BP230, which initiate inflammatory cascades that lead to tissue damage and blister formation. It typically manifests as pruritic blistering eruptions, primarily on the limbs and trunk, but may involve the head, neck, or palmoplantar regions.14 Although blistering eruptions are the prodrome of the disease, some cases may present with nonspecific urticarial or eczematous lesions14,15 that may resemble prurigo pigmentosa. The diagnosis is confirmed through direct immunofluorescence microscopy of biopsied lesions, which reveals IgG and/or C3 deposits along the dermoepidermal junction.14 Management of bullous pemphigoid involves timely initiation of dapsone or systemic corticosteroids, which have demonstrated high efficacy in controlling the disease and its associated symptoms.15
Our patient achieved a favorable response to diet modification and doxycycline therapy consistent with the diagnosis of prurigo pigmentosa. Unfortunately, the condition recurred following a relapse of anorexia. Management of prurigo pigmentosa necessitates not only accurate diagnosis but also addressing any underlying factors that may contribute to disease exacerbation. We anticipate the eating disorder will pose a major challenge in achieving long-term control of prurigo pigmentosa.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation. Jpn J Dermatol. 1971;81:38-39.
- Boer A, Asgari M. Prurigo pigmentosa: an underdiagnosed disease? Indian J Dermatol Venereol Leprol. 2006;72:405-409. doi:10.4103/0378-6323.29334
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet. Pediatr Dermatol. 2013;32:248-251. doi:10.1111/pde.12275
- Teraki Y, Teraki E, Kawashima M, et al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511. doi:10.1016/s0190-9622(96)90460-0
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79-87. doi:10.1016/j.jdin.2021.03.003
- Wong M, Lee E, Wu Y, et al. Treatment of prurigo pigmentosa with diet modification: a medical case study. Hawaii J Med Public Health. 2018;77:114-117.
- Almaani N, Al-Tarawneh AH, Msallam H. Prurigo pigmentosa: a clinicopathological report of three Middle Eastern patients. Case Rep Dermatol Med. 2018;2018:9406797. doi:10.1155/2018/9406797
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897. doi:10.1111/j.1346-8138.2012.01640.x
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74:1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Lazarov A, Cordoba M, Plosk N, et al. Atypical and unusual clinical manifestations of contact dermatitis to clothing (textile contact dermatitis)—case presentation and review of the literature. Dermatol Online J. 2003;9. doi:10.5070/d30kd1d259
- Rubenstein RM, Malerich SA. Malassezia (Pityrosporum) folliculitis. J Clin Aesthet Dermatol. 2014;7:37-41.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277. doi:10.1016/j.jaci.2014.02.036
- della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol. 2012;167:1111-1117. doi:10.1111/j.1365-2133.2012.11108.x
- Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, et al. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. Int J Dermatol. 1998;37:508-514.
- Nagashima M, Ohshiro A, Shimizu N. A peculiar pruriginous dermatosis with gross reticular pigmentation. Jpn J Dermatol. 1971;81:38-39.
- Boer A, Asgari M. Prurigo pigmentosa: an underdiagnosed disease? Indian J Dermatol Venereol Leprol. 2006;72:405-409. doi:10.4103/0378-6323.29334
- Michaels JD, Hoss E, DiCaudo DJ, et al. Prurigo pigmentosa after a strict ketogenic diet. Pediatr Dermatol. 2013;32:248-251. doi:10.1111/pde.12275
- Teraki Y, Teraki E, Kawashima M, et al. Ketosis is involved in the origin of prurigo pigmentosa. J Am Acad Dermatol. 1996;34:509-511. doi:10.1016/s0190-9622(96)90460-0
- Böer A, Misago N, Wolter M, et al. Prurigo pigmentosa: a distinctive inflammatory disease of the skin. Am J Dermatopathol. 2003;25:117-129. doi:10.1097/00000372-200304000-00005
- Mufti A, Mirali S, Abduelmula A, et al. Clinical manifestations and treatment outcomes in prurigo pigmentosa (Nagashima disease): a systematic review of the literature. JAAD Int. 2021;3:79-87. doi:10.1016/j.jdin.2021.03.003
- Wong M, Lee E, Wu Y, et al. Treatment of prurigo pigmentosa with diet modification: a medical case study. Hawaii J Med Public Health. 2018;77:114-117.
- Almaani N, Al-Tarawneh AH, Msallam H. Prurigo pigmentosa: a clinicopathological report of three Middle Eastern patients. Case Rep Dermatol Med. 2018;2018:9406797. doi:10.1155/2018/9406797
- Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897. doi:10.1111/j.1346-8138.2012.01640.x
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient diagnosis and evaluation. J Am Acad Dermatol. 2016;74:1029-1040. doi:10.1016/j.jaad.2015.02.1139
- Lazarov A, Cordoba M, Plosk N, et al. Atypical and unusual clinical manifestations of contact dermatitis to clothing (textile contact dermatitis)—case presentation and review of the literature. Dermatol Online J. 2003;9. doi:10.5070/d30kd1d259
- Rubenstein RM, Malerich SA. Malassezia (Pityrosporum) folliculitis. J Clin Aesthet Dermatol. 2014;7:37-41.
- Bernstein JA, Lang DM, Khan DA, et al. The diagnosis and management of acute and chronic urticaria: 2014 update. J Allergy Clin Immunol. 2014;133:1270-1277. doi:10.1016/j.jaci.2014.02.036
- della Torre R, Combescure C, Cortés B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol. 2012;167:1111-1117. doi:10.1111/j.1365-2133.2012.11108.x
- Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, et al. Bullous pemphigoid presenting as generalized pruritus: observations in six patients. Int J Dermatol. 1998;37:508-514.
A 43-year-old woman presented with a pruritic rash across the neck and back of 6 months’ duration that progressively worsened. She had a medical history of anorexia nervosa, herpes zoster with a recent flare, and peripheral neuropathy. Physical examination showed numerous red scaly papules across the upper back and shoulders that coalesced in a reticular pattern. No similar papules were seen elsewhere on the body.
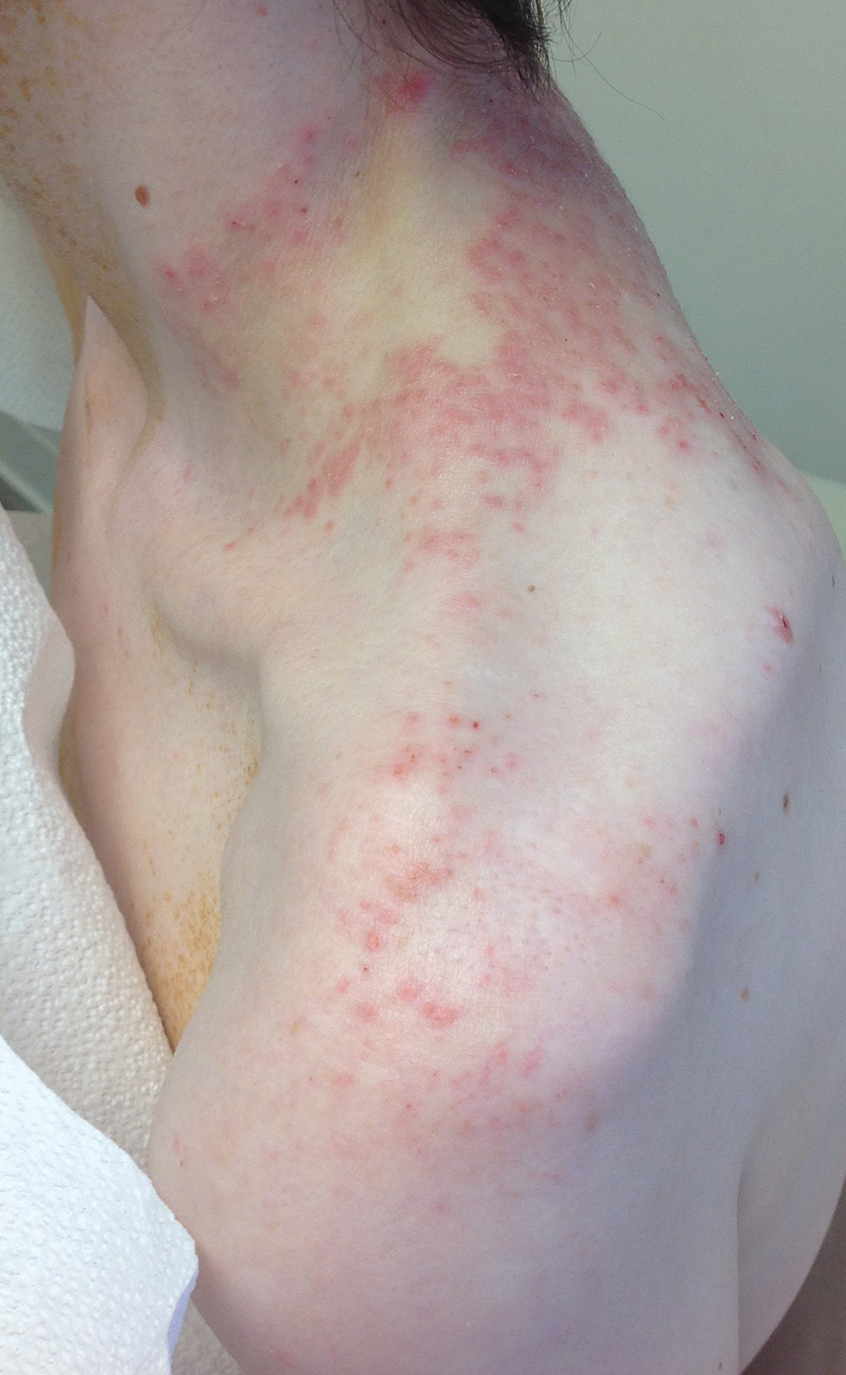
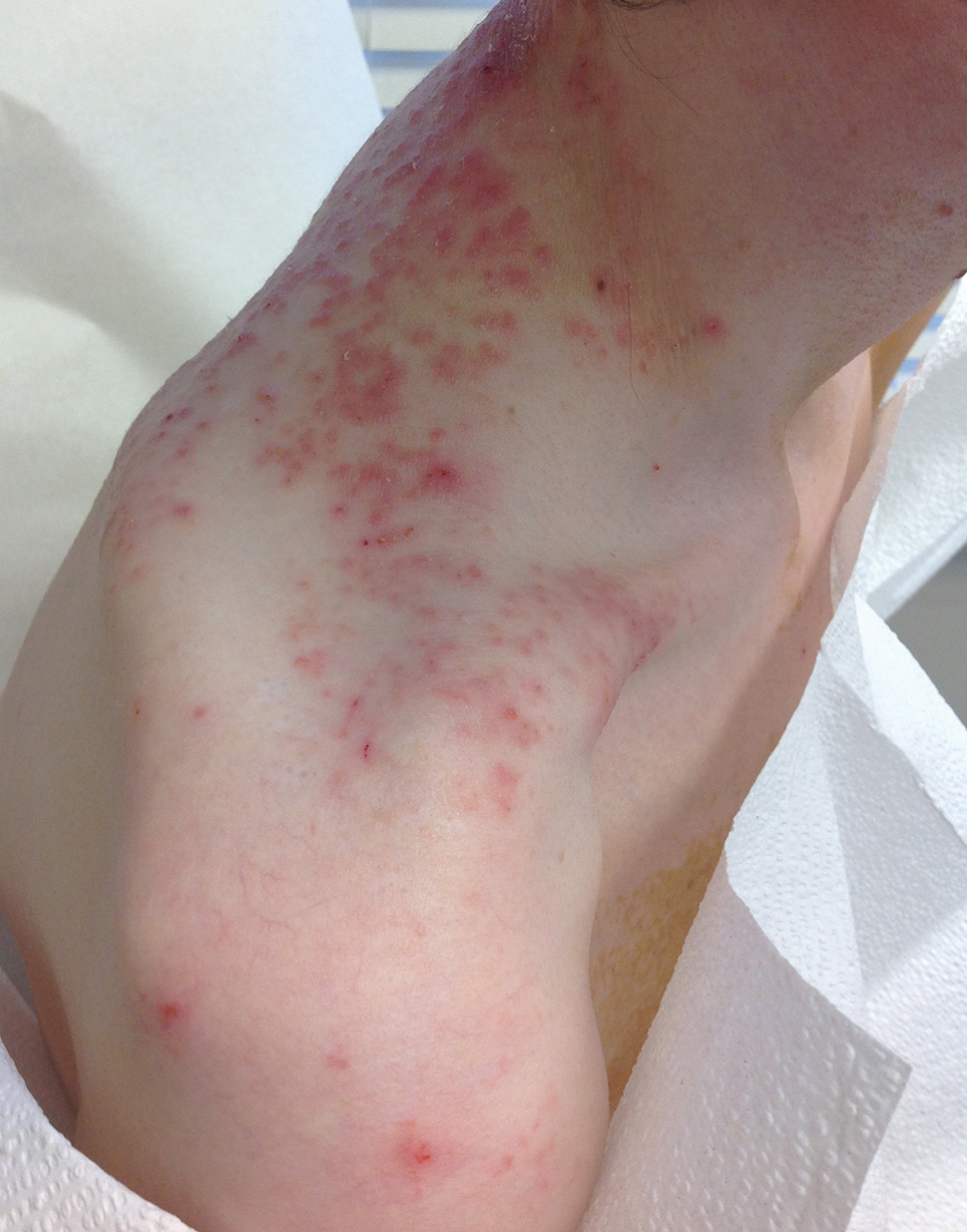
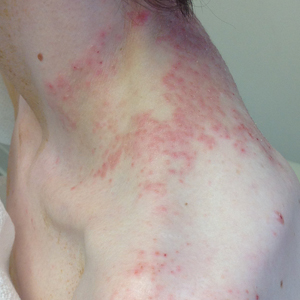
Cyclosporine for Recalcitrant Bullous Pemphigoid Induced by Nivolumab Therapy for Malignant Melanoma
To the Editor:
Immune checkpoint inhibitors have revolutionized the treatment of advanced-stage melanoma, with remarkably improved progression-free survival.1 Anti–programmed death receptor 1 (anti–PD-1) therapies, such as nivolumab and pembrolizumab, are a class of checkpoint inhibitors that have been approved by the US Food and Drug Administration for unresectable metastatic melanoma. Anti–PD-1 agents block the interaction of programmed death-ligand 1 (PD-L1) found on tumor cells with the PD-1 receptor on T cells, facilitating a positive immune response.2
Although these therapies have demonstrated notable antitumor efficacy, they also give rise to numerous immune-related adverse events (irAEs). As many as 70% of patients treated with PD-1/PD-L1 inhibitors experience some type of organ system irAE, of which 30% to 40% are cutaneous.3-6 Dermatologic adverse events are the most common irAEs, specifically spongiotic dermatitis, lichenoid dermatitis, pruritus, and vitiligo.7 Bullous pemphigoid (BP), an autoimmune bullous skin disorder caused by autoantibodies to basement membrane zone antigens, is a rare but potentially serious cutaneous irAE.8 Systemic corticosteroids commonly are used to treat immune checkpoint inhibitor–induced BP; other options include tetracyclines for maintenance therapy and rituximab for corticosteroid-refractory BP associated with anti-PD-1.9 We present a case of recalcitrant BP secondary to nivolumab therapy in a patient with metastatic melanoma who had near-complete resolution of BP following 2 months of cyclosporine.
A 41-year-old man presented with a generalized papular skin eruption of 1 month’s duration. He had a history of stage IIIC malignant melanoma of the lower right leg with positive sentinel lymph node biopsy. The largest lymph node deposit was 0.03 mm without extracapsular extension. Whole-body positron emission tomography–computed tomography showed no evidence of distant disease. The patient was treated with wide local excision with clear surgical margins plus 12 cycles of nivolumab, which was discontinued due to colitis. Four months after the final cycle of nivolumab, the patient developed widespread erythematous papules with hemorrhagic yellow crusting and no mucosal involvement. He was referred to dermatology by his primary oncologist for further evaluation.
A punch biopsy from the abdomen showed parakeratosis with leukocytoclasis and a superficial dermal infiltrate of neutrophils and eosinophils (Figure 1). Direct immunofluorescence revealed linear basement membrane deposits of IgG and C3, consistent with subepidermal blistering disease. Indirect immunofluorescence demonstrated trace IgG and IgG4 antibodies localized to the epidermal roof of salt-split skin and was negative for IgA antibodies. An enzyme-linked immunoassay was positive for BP antigen 2 (BP180) antibodies (98.4 U/mL [positive, ≥9 U/mL]) and negative for BP antigen 1 (BP230) antibodies (4.3 U/mL [positive, ≥9 U/mL]). Overall, these findings were consistent with a diagnosis of BP.
The patient was treated with prednisone 60 mg daily with initial response; however, there was disease recurrence with tapering. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily were added as steroid-sparing agents, as prednisone was discontinued due to mood changes. Three months after the prednisone taper, the patient continued to develop new blisters. He completed treatment with doxycycline and nicotinamide. Rituximab 375 mg weekly was then initiated for 4 weeks.
At 2-week follow-up after completing the rituximab course, the patient reported worsening symptoms and presented with new bullae on the abdomen and upper extremities (Figure 2). Because of the recent history of mood changes while taking prednisone, a trial of cyclosporine 100 mg twice daily (1.37 mg/kg/d) was initiated, with notable improvement within 2 weeks of treatment. After 2 months of cyclosporine, approximately 90% of the rash had resolved with a few tense bullae remaining on the left frontal scalp but no new flares (Figure 3). One month after treatment ended, the patient remained clear of lesions without relapse.
Programmed death receptor 1 inhibitors have shown dramatic efficacy for a growing number of solid and hematologic malignancies, especially malignant melanoma. However, their use is accompanied by nonspecific activation of the immune system, resulting in a variety of adverse events, many manifesting on the skin. Several cases of BP in patients treated with PD-1/PD-L1 inhibitors have been reported.9 Cutaneous irAEs usually manifest within 3 weeks of initiation of PD-1 inhibitor therapy; however, the onset of BP typically occurs later at approximately 21 weeks.4,9 Our patient developed cutaneous manifestations 4 months after cessation of nivolumab.
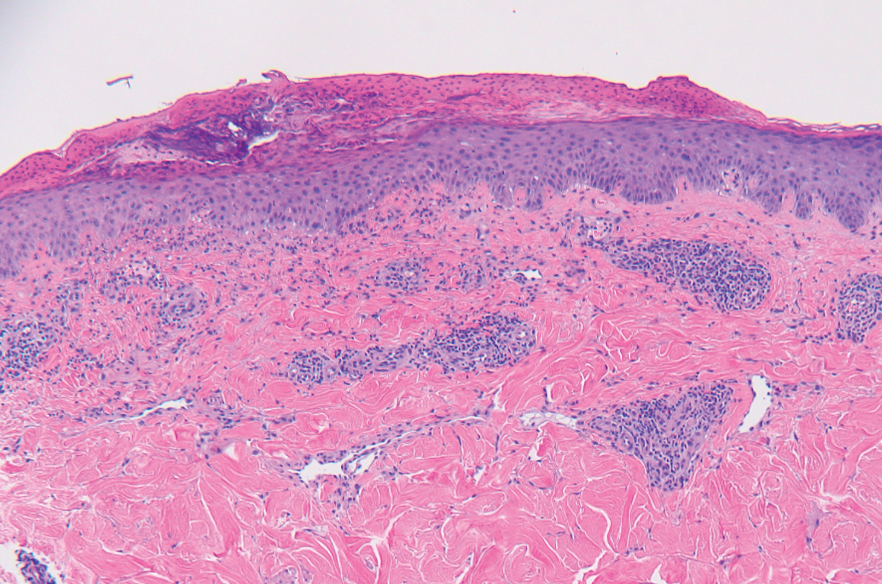
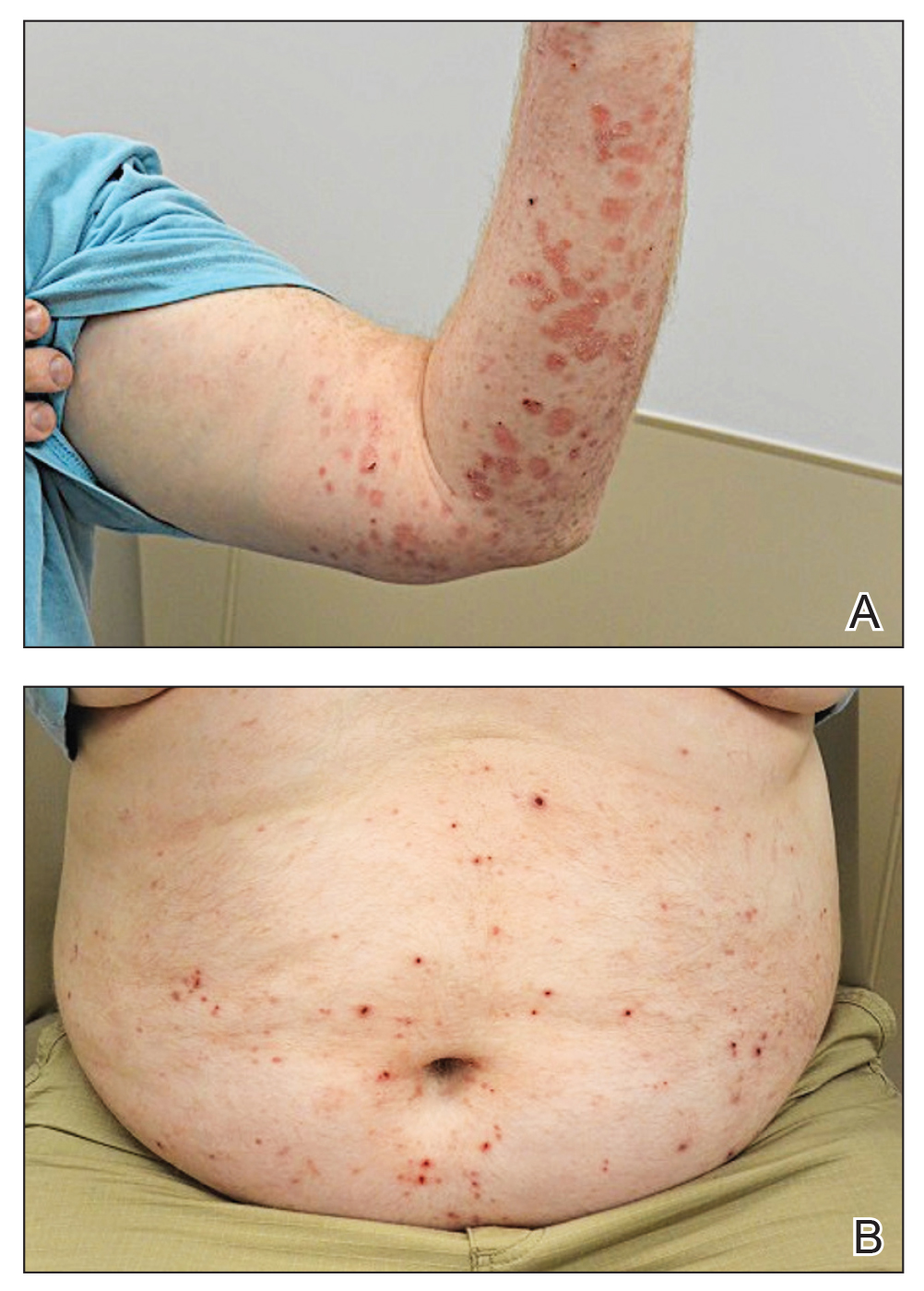
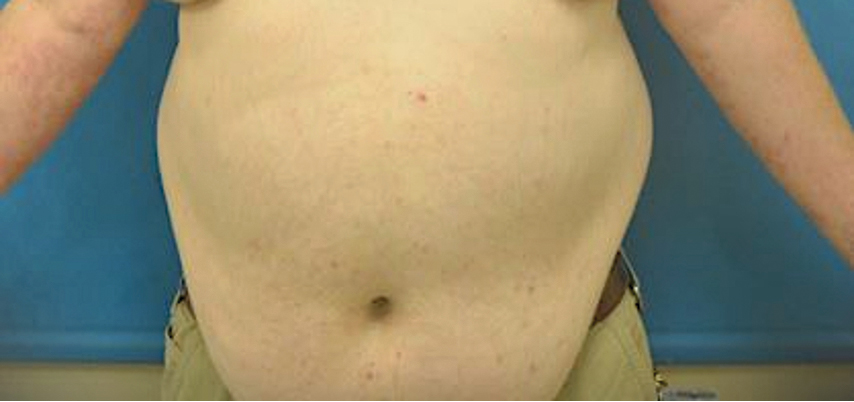
Bullous pemphigoid classically manifests with pruritus and tense bullae. Notably, our patient’s clinical presentation included a widespread eruption of papules without bullae, which was similar to a review by Tsiogka et al,9 which reported that one-third of patients first present with a nonspecific cutaneous eruption. Bullous pemphigoid induced by anti–PD-1 may manifest differently than traditional BP, illuminating the importance of a thorough diagnostic workup.
Although the pathogenesis of immune checkpoint inhibitor–induced BP has not been fully elucidated, it is hypothesized to be caused by increased T cell cytotoxic activity leading to tumor lysis and release of numerous autoantigens. These autoantigens cause priming of abnormal T cells that can lead to further tissue damage in peripheral tissue and to generation of aberrant B cells and subsequent autoantibodies such as BP180 in germinal centers.4,10,11
Cyclosporine is a calcineurin inhibitor that reduces synthesis of IL-2, resulting in reduced cell activation.12 Therefore, cyclosporine may alleviate BP in patients who are being treated, or were previously treated, with an immune checkpoint inhibitor by suppressing T cell–mediated immune reaction and may be a rapid alternative for patients who cannot tolerate systemic steroids.
Treatment options for mild to moderate cases of BP include topical corticosteroids and antihistamines, while severe cases may require high-dose systemic corticosteroids. In recalcitrant cases, rituximab infusion with or without intravenous immunoglobulin often is utilized.8,13 The use of cyclosporine for various bullous disorders, including pemphigus vulgaris and epidermolysis bullosa acquisita, has been described.14 In recent years there has been a shift away from the use of cyclosporine for these conditions following the introduction of rituximab, a monoclonal antibody directed against the CD20 antigen on B lymphocytes. We utilized cyclosporine in our patient after he experienced worsening symptoms 1 month after completing treatment with rituximab.
Improvement from rituximab therapy may be delayed because it can take months to deplete CD20+ B lymphocytes from circulation, which may necessitate additional immunosuppressants or re-treatment with rituximab.15,16 In these instances, cyclosporine may be beneficial as a low-cost alternative in patients who are unable to tolerate systemic steroids, with a relatively good safety profile. The dosage of cyclosporine prescribed to the patient was chosen based on Joint American Academy of Dermatology–National Psoriasis Foundation management guidelines for psoriasis with systemic nonbiologic therapies, which recommends an initial dosage of 1 to 3 mg/kg/d in 2 divided doses.17
As immunotherapy for treating various cancers gains popularity, the frequency of dermatologic irAEs will increase. Therefore, dermatologists must be aware of the array of cutaneous manifestations, such as BP, and potential treatment options. When first-line and second-line therapies are contraindicated or do not provide notable improvement, cyclosporine may be an effective alternative for immune checkpoint inhibitor–induced BP.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34. doi:10.1056/NEJMoa1504030
- Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi:10.3389/fphar.2017.00561
- Puzanov I, Diab A, Abdallah K, et al; . Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95. doi:10.1186/s40425-017-0300-z
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4:560-575. doi:10.3978/j.issn.2218-6751.2015.06.06
- Kumar V, Chaudhary N, Garg M, et al. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. 2017;8:49. doi:10.3389/fphar.2017.00049
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Schauer F, Rafei-Shamsabadi D, Mai S, et al. Hemidesmosomal reactivity and treatment recommendations in immune checkpoint inhibitor-induced bullous pemphigoid—a retrospective, monocentric study. Front Immunol. 2022;13:953546. doi:10.3389/fimmu.2022.953546
- Tsiogka A, Bauer JW, Patsatsi A. Bullous pemphigoid associated with anti-programmed cell death protein 1 and anti-programmed cell death ligand 1 therapy: a review of the literature. Acta Derm Venereol. 2021;101:adv00377. doi:10.2340/00015555-3740
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669. doi:10.1111/ijd.13984
- Yang H, Yao Z, Zhou X, et al. Immune-related adverse events of checkpoint inhibitors: insights into immunological dysregulation. Clin Immunol. 2020;213:108377. doi:10.1016/j.clim.2020.108377
- Russell G, Graveley R, Seid J, et al. Mechanisms of action of cyclosporine and effects on connective tissues. Semin Arthritis Rheum. 1992;21(6 suppl 3):16-22. doi:10.1016/0049-0172(92)90009-3
- Ahmed AR, Shetty S, Kaveri S, et al. Treatment of recalcitrant bullous pemphigoid (BP) with a novel protocol: a retrospective study with a 6-year follow-up. J Am Acad Dermatol. 2016;74:700-708.e3. doi:10.1016/j.jaad.2015.11.030
- Amor KT, Ryan C, Menter A. The use of cyclosporine in dermatology: part I. J Am Acad Dermatol. 2010;63:925-946. doi:10.1016/j.jaad.2010.02.063
- Schmidt E, Hunzelmann N, Zillikens D, et al. Rituximab in refractory autoimmune bullous diseases. Clin Exp Dermatol. 2006;31:503-508. doi:10.1111/j.1365-2230.2006.02151.x
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, et al. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol. 2011;65:552-558.
- Menter A, Gelfand JM, Connor C, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82:1445-1486. doi:10.1016/j.jaad.2020.02.044
To the Editor:
Immune checkpoint inhibitors have revolutionized the treatment of advanced-stage melanoma, with remarkably improved progression-free survival.1 Anti–programmed death receptor 1 (anti–PD-1) therapies, such as nivolumab and pembrolizumab, are a class of checkpoint inhibitors that have been approved by the US Food and Drug Administration for unresectable metastatic melanoma. Anti–PD-1 agents block the interaction of programmed death-ligand 1 (PD-L1) found on tumor cells with the PD-1 receptor on T cells, facilitating a positive immune response.2
Although these therapies have demonstrated notable antitumor efficacy, they also give rise to numerous immune-related adverse events (irAEs). As many as 70% of patients treated with PD-1/PD-L1 inhibitors experience some type of organ system irAE, of which 30% to 40% are cutaneous.3-6 Dermatologic adverse events are the most common irAEs, specifically spongiotic dermatitis, lichenoid dermatitis, pruritus, and vitiligo.7 Bullous pemphigoid (BP), an autoimmune bullous skin disorder caused by autoantibodies to basement membrane zone antigens, is a rare but potentially serious cutaneous irAE.8 Systemic corticosteroids commonly are used to treat immune checkpoint inhibitor–induced BP; other options include tetracyclines for maintenance therapy and rituximab for corticosteroid-refractory BP associated with anti-PD-1.9 We present a case of recalcitrant BP secondary to nivolumab therapy in a patient with metastatic melanoma who had near-complete resolution of BP following 2 months of cyclosporine.
A 41-year-old man presented with a generalized papular skin eruption of 1 month’s duration. He had a history of stage IIIC malignant melanoma of the lower right leg with positive sentinel lymph node biopsy. The largest lymph node deposit was 0.03 mm without extracapsular extension. Whole-body positron emission tomography–computed tomography showed no evidence of distant disease. The patient was treated with wide local excision with clear surgical margins plus 12 cycles of nivolumab, which was discontinued due to colitis. Four months after the final cycle of nivolumab, the patient developed widespread erythematous papules with hemorrhagic yellow crusting and no mucosal involvement. He was referred to dermatology by his primary oncologist for further evaluation.
A punch biopsy from the abdomen showed parakeratosis with leukocytoclasis and a superficial dermal infiltrate of neutrophils and eosinophils (Figure 1). Direct immunofluorescence revealed linear basement membrane deposits of IgG and C3, consistent with subepidermal blistering disease. Indirect immunofluorescence demonstrated trace IgG and IgG4 antibodies localized to the epidermal roof of salt-split skin and was negative for IgA antibodies. An enzyme-linked immunoassay was positive for BP antigen 2 (BP180) antibodies (98.4 U/mL [positive, ≥9 U/mL]) and negative for BP antigen 1 (BP230) antibodies (4.3 U/mL [positive, ≥9 U/mL]). Overall, these findings were consistent with a diagnosis of BP.
The patient was treated with prednisone 60 mg daily with initial response; however, there was disease recurrence with tapering. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily were added as steroid-sparing agents, as prednisone was discontinued due to mood changes. Three months after the prednisone taper, the patient continued to develop new blisters. He completed treatment with doxycycline and nicotinamide. Rituximab 375 mg weekly was then initiated for 4 weeks.
At 2-week follow-up after completing the rituximab course, the patient reported worsening symptoms and presented with new bullae on the abdomen and upper extremities (Figure 2). Because of the recent history of mood changes while taking prednisone, a trial of cyclosporine 100 mg twice daily (1.37 mg/kg/d) was initiated, with notable improvement within 2 weeks of treatment. After 2 months of cyclosporine, approximately 90% of the rash had resolved with a few tense bullae remaining on the left frontal scalp but no new flares (Figure 3). One month after treatment ended, the patient remained clear of lesions without relapse.
Programmed death receptor 1 inhibitors have shown dramatic efficacy for a growing number of solid and hematologic malignancies, especially malignant melanoma. However, their use is accompanied by nonspecific activation of the immune system, resulting in a variety of adverse events, many manifesting on the skin. Several cases of BP in patients treated with PD-1/PD-L1 inhibitors have been reported.9 Cutaneous irAEs usually manifest within 3 weeks of initiation of PD-1 inhibitor therapy; however, the onset of BP typically occurs later at approximately 21 weeks.4,9 Our patient developed cutaneous manifestations 4 months after cessation of nivolumab.
Bullous pemphigoid classically manifests with pruritus and tense bullae. Notably, our patient’s clinical presentation included a widespread eruption of papules without bullae, which was similar to a review by Tsiogka et al,9 which reported that one-third of patients first present with a nonspecific cutaneous eruption. Bullous pemphigoid induced by anti–PD-1 may manifest differently than traditional BP, illuminating the importance of a thorough diagnostic workup.
Although the pathogenesis of immune checkpoint inhibitor–induced BP has not been fully elucidated, it is hypothesized to be caused by increased T cell cytotoxic activity leading to tumor lysis and release of numerous autoantigens. These autoantigens cause priming of abnormal T cells that can lead to further tissue damage in peripheral tissue and to generation of aberrant B cells and subsequent autoantibodies such as BP180 in germinal centers.4,10,11
Cyclosporine is a calcineurin inhibitor that reduces synthesis of IL-2, resulting in reduced cell activation.12 Therefore, cyclosporine may alleviate BP in patients who are being treated, or were previously treated, with an immune checkpoint inhibitor by suppressing T cell–mediated immune reaction and may be a rapid alternative for patients who cannot tolerate systemic steroids.
Treatment options for mild to moderate cases of BP include topical corticosteroids and antihistamines, while severe cases may require high-dose systemic corticosteroids. In recalcitrant cases, rituximab infusion with or without intravenous immunoglobulin often is utilized.8,13 The use of cyclosporine for various bullous disorders, including pemphigus vulgaris and epidermolysis bullosa acquisita, has been described.14 In recent years there has been a shift away from the use of cyclosporine for these conditions following the introduction of rituximab, a monoclonal antibody directed against the CD20 antigen on B lymphocytes. We utilized cyclosporine in our patient after he experienced worsening symptoms 1 month after completing treatment with rituximab.
Improvement from rituximab therapy may be delayed because it can take months to deplete CD20+ B lymphocytes from circulation, which may necessitate additional immunosuppressants or re-treatment with rituximab.15,16 In these instances, cyclosporine may be beneficial as a low-cost alternative in patients who are unable to tolerate systemic steroids, with a relatively good safety profile. The dosage of cyclosporine prescribed to the patient was chosen based on Joint American Academy of Dermatology–National Psoriasis Foundation management guidelines for psoriasis with systemic nonbiologic therapies, which recommends an initial dosage of 1 to 3 mg/kg/d in 2 divided doses.17
As immunotherapy for treating various cancers gains popularity, the frequency of dermatologic irAEs will increase. Therefore, dermatologists must be aware of the array of cutaneous manifestations, such as BP, and potential treatment options. When first-line and second-line therapies are contraindicated or do not provide notable improvement, cyclosporine may be an effective alternative for immune checkpoint inhibitor–induced BP.
To the Editor:
Immune checkpoint inhibitors have revolutionized the treatment of advanced-stage melanoma, with remarkably improved progression-free survival.1 Anti–programmed death receptor 1 (anti–PD-1) therapies, such as nivolumab and pembrolizumab, are a class of checkpoint inhibitors that have been approved by the US Food and Drug Administration for unresectable metastatic melanoma. Anti–PD-1 agents block the interaction of programmed death-ligand 1 (PD-L1) found on tumor cells with the PD-1 receptor on T cells, facilitating a positive immune response.2
Although these therapies have demonstrated notable antitumor efficacy, they also give rise to numerous immune-related adverse events (irAEs). As many as 70% of patients treated with PD-1/PD-L1 inhibitors experience some type of organ system irAE, of which 30% to 40% are cutaneous.3-6 Dermatologic adverse events are the most common irAEs, specifically spongiotic dermatitis, lichenoid dermatitis, pruritus, and vitiligo.7 Bullous pemphigoid (BP), an autoimmune bullous skin disorder caused by autoantibodies to basement membrane zone antigens, is a rare but potentially serious cutaneous irAE.8 Systemic corticosteroids commonly are used to treat immune checkpoint inhibitor–induced BP; other options include tetracyclines for maintenance therapy and rituximab for corticosteroid-refractory BP associated with anti-PD-1.9 We present a case of recalcitrant BP secondary to nivolumab therapy in a patient with metastatic melanoma who had near-complete resolution of BP following 2 months of cyclosporine.
A 41-year-old man presented with a generalized papular skin eruption of 1 month’s duration. He had a history of stage IIIC malignant melanoma of the lower right leg with positive sentinel lymph node biopsy. The largest lymph node deposit was 0.03 mm without extracapsular extension. Whole-body positron emission tomography–computed tomography showed no evidence of distant disease. The patient was treated with wide local excision with clear surgical margins plus 12 cycles of nivolumab, which was discontinued due to colitis. Four months after the final cycle of nivolumab, the patient developed widespread erythematous papules with hemorrhagic yellow crusting and no mucosal involvement. He was referred to dermatology by his primary oncologist for further evaluation.
A punch biopsy from the abdomen showed parakeratosis with leukocytoclasis and a superficial dermal infiltrate of neutrophils and eosinophils (Figure 1). Direct immunofluorescence revealed linear basement membrane deposits of IgG and C3, consistent with subepidermal blistering disease. Indirect immunofluorescence demonstrated trace IgG and IgG4 antibodies localized to the epidermal roof of salt-split skin and was negative for IgA antibodies. An enzyme-linked immunoassay was positive for BP antigen 2 (BP180) antibodies (98.4 U/mL [positive, ≥9 U/mL]) and negative for BP antigen 1 (BP230) antibodies (4.3 U/mL [positive, ≥9 U/mL]). Overall, these findings were consistent with a diagnosis of BP.
The patient was treated with prednisone 60 mg daily with initial response; however, there was disease recurrence with tapering. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily were added as steroid-sparing agents, as prednisone was discontinued due to mood changes. Three months after the prednisone taper, the patient continued to develop new blisters. He completed treatment with doxycycline and nicotinamide. Rituximab 375 mg weekly was then initiated for 4 weeks.
At 2-week follow-up after completing the rituximab course, the patient reported worsening symptoms and presented with new bullae on the abdomen and upper extremities (Figure 2). Because of the recent history of mood changes while taking prednisone, a trial of cyclosporine 100 mg twice daily (1.37 mg/kg/d) was initiated, with notable improvement within 2 weeks of treatment. After 2 months of cyclosporine, approximately 90% of the rash had resolved with a few tense bullae remaining on the left frontal scalp but no new flares (Figure 3). One month after treatment ended, the patient remained clear of lesions without relapse.
Programmed death receptor 1 inhibitors have shown dramatic efficacy for a growing number of solid and hematologic malignancies, especially malignant melanoma. However, their use is accompanied by nonspecific activation of the immune system, resulting in a variety of adverse events, many manifesting on the skin. Several cases of BP in patients treated with PD-1/PD-L1 inhibitors have been reported.9 Cutaneous irAEs usually manifest within 3 weeks of initiation of PD-1 inhibitor therapy; however, the onset of BP typically occurs later at approximately 21 weeks.4,9 Our patient developed cutaneous manifestations 4 months after cessation of nivolumab.
Bullous pemphigoid classically manifests with pruritus and tense bullae. Notably, our patient’s clinical presentation included a widespread eruption of papules without bullae, which was similar to a review by Tsiogka et al,9 which reported that one-third of patients first present with a nonspecific cutaneous eruption. Bullous pemphigoid induced by anti–PD-1 may manifest differently than traditional BP, illuminating the importance of a thorough diagnostic workup.
Although the pathogenesis of immune checkpoint inhibitor–induced BP has not been fully elucidated, it is hypothesized to be caused by increased T cell cytotoxic activity leading to tumor lysis and release of numerous autoantigens. These autoantigens cause priming of abnormal T cells that can lead to further tissue damage in peripheral tissue and to generation of aberrant B cells and subsequent autoantibodies such as BP180 in germinal centers.4,10,11
Cyclosporine is a calcineurin inhibitor that reduces synthesis of IL-2, resulting in reduced cell activation.12 Therefore, cyclosporine may alleviate BP in patients who are being treated, or were previously treated, with an immune checkpoint inhibitor by suppressing T cell–mediated immune reaction and may be a rapid alternative for patients who cannot tolerate systemic steroids.
Treatment options for mild to moderate cases of BP include topical corticosteroids and antihistamines, while severe cases may require high-dose systemic corticosteroids. In recalcitrant cases, rituximab infusion with or without intravenous immunoglobulin often is utilized.8,13 The use of cyclosporine for various bullous disorders, including pemphigus vulgaris and epidermolysis bullosa acquisita, has been described.14 In recent years there has been a shift away from the use of cyclosporine for these conditions following the introduction of rituximab, a monoclonal antibody directed against the CD20 antigen on B lymphocytes. We utilized cyclosporine in our patient after he experienced worsening symptoms 1 month after completing treatment with rituximab.
Improvement from rituximab therapy may be delayed because it can take months to deplete CD20+ B lymphocytes from circulation, which may necessitate additional immunosuppressants or re-treatment with rituximab.15,16 In these instances, cyclosporine may be beneficial as a low-cost alternative in patients who are unable to tolerate systemic steroids, with a relatively good safety profile. The dosage of cyclosporine prescribed to the patient was chosen based on Joint American Academy of Dermatology–National Psoriasis Foundation management guidelines for psoriasis with systemic nonbiologic therapies, which recommends an initial dosage of 1 to 3 mg/kg/d in 2 divided doses.17
As immunotherapy for treating various cancers gains popularity, the frequency of dermatologic irAEs will increase. Therefore, dermatologists must be aware of the array of cutaneous manifestations, such as BP, and potential treatment options. When first-line and second-line therapies are contraindicated or do not provide notable improvement, cyclosporine may be an effective alternative for immune checkpoint inhibitor–induced BP.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34. doi:10.1056/NEJMoa1504030
- Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi:10.3389/fphar.2017.00561
- Puzanov I, Diab A, Abdallah K, et al; . Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95. doi:10.1186/s40425-017-0300-z
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4:560-575. doi:10.3978/j.issn.2218-6751.2015.06.06
- Kumar V, Chaudhary N, Garg M, et al. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. 2017;8:49. doi:10.3389/fphar.2017.00049
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Schauer F, Rafei-Shamsabadi D, Mai S, et al. Hemidesmosomal reactivity and treatment recommendations in immune checkpoint inhibitor-induced bullous pemphigoid—a retrospective, monocentric study. Front Immunol. 2022;13:953546. doi:10.3389/fimmu.2022.953546
- Tsiogka A, Bauer JW, Patsatsi A. Bullous pemphigoid associated with anti-programmed cell death protein 1 and anti-programmed cell death ligand 1 therapy: a review of the literature. Acta Derm Venereol. 2021;101:adv00377. doi:10.2340/00015555-3740
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669. doi:10.1111/ijd.13984
- Yang H, Yao Z, Zhou X, et al. Immune-related adverse events of checkpoint inhibitors: insights into immunological dysregulation. Clin Immunol. 2020;213:108377. doi:10.1016/j.clim.2020.108377
- Russell G, Graveley R, Seid J, et al. Mechanisms of action of cyclosporine and effects on connective tissues. Semin Arthritis Rheum. 1992;21(6 suppl 3):16-22. doi:10.1016/0049-0172(92)90009-3
- Ahmed AR, Shetty S, Kaveri S, et al. Treatment of recalcitrant bullous pemphigoid (BP) with a novel protocol: a retrospective study with a 6-year follow-up. J Am Acad Dermatol. 2016;74:700-708.e3. doi:10.1016/j.jaad.2015.11.030
- Amor KT, Ryan C, Menter A. The use of cyclosporine in dermatology: part I. J Am Acad Dermatol. 2010;63:925-946. doi:10.1016/j.jaad.2010.02.063
- Schmidt E, Hunzelmann N, Zillikens D, et al. Rituximab in refractory autoimmune bullous diseases. Clin Exp Dermatol. 2006;31:503-508. doi:10.1111/j.1365-2230.2006.02151.x
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, et al. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol. 2011;65:552-558.
- Menter A, Gelfand JM, Connor C, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82:1445-1486. doi:10.1016/j.jaad.2020.02.044
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34. doi:10.1056/NEJMoa1504030
- Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi:10.3389/fphar.2017.00561
- Puzanov I, Diab A, Abdallah K, et al; . Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95. doi:10.1186/s40425-017-0300-z
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4:560-575. doi:10.3978/j.issn.2218-6751.2015.06.06
- Kumar V, Chaudhary N, Garg M, et al. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. 2017;8:49. doi:10.3389/fphar.2017.00049
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Schauer F, Rafei-Shamsabadi D, Mai S, et al. Hemidesmosomal reactivity and treatment recommendations in immune checkpoint inhibitor-induced bullous pemphigoid—a retrospective, monocentric study. Front Immunol. 2022;13:953546. doi:10.3389/fimmu.2022.953546
- Tsiogka A, Bauer JW, Patsatsi A. Bullous pemphigoid associated with anti-programmed cell death protein 1 and anti-programmed cell death ligand 1 therapy: a review of the literature. Acta Derm Venereol. 2021;101:adv00377. doi:10.2340/00015555-3740
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669. doi:10.1111/ijd.13984
- Yang H, Yao Z, Zhou X, et al. Immune-related adverse events of checkpoint inhibitors: insights into immunological dysregulation. Clin Immunol. 2020;213:108377. doi:10.1016/j.clim.2020.108377
- Russell G, Graveley R, Seid J, et al. Mechanisms of action of cyclosporine and effects on connective tissues. Semin Arthritis Rheum. 1992;21(6 suppl 3):16-22. doi:10.1016/0049-0172(92)90009-3
- Ahmed AR, Shetty S, Kaveri S, et al. Treatment of recalcitrant bullous pemphigoid (BP) with a novel protocol: a retrospective study with a 6-year follow-up. J Am Acad Dermatol. 2016;74:700-708.e3. doi:10.1016/j.jaad.2015.11.030
- Amor KT, Ryan C, Menter A. The use of cyclosporine in dermatology: part I. J Am Acad Dermatol. 2010;63:925-946. doi:10.1016/j.jaad.2010.02.063
- Schmidt E, Hunzelmann N, Zillikens D, et al. Rituximab in refractory autoimmune bullous diseases. Clin Exp Dermatol. 2006;31:503-508. doi:10.1111/j.1365-2230.2006.02151.x
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, et al. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol. 2011;65:552-558.
- Menter A, Gelfand JM, Connor C, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82:1445-1486. doi:10.1016/j.jaad.2020.02.044
Practice Points
- Bullous pemphigoid is a rare dermatologic immune-related adverse event that can occur secondary to anti–programmed death receptor 1 therapy.
- For cases of immune checkpoint inhibitor–induced bullous pemphigoid that are recalcitrant to corticosteroids and rituximab, cyclosporine might be an effective alternative.