User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Generalized Fixed Drug Eruptions Require Urgent Care: A Case Series
Recognizing cutaneous drug eruptions is important for treatment and prevention of recurrence. Fixed drug eruptions (FDEs) typically are harmless but can have major negative cosmetic consequences for patients. In its more severe forms, patients are at risk for widespread epithelial necrosis with accompanying complications. We report 1 patient with generalized FDE and 2 with generalized bullous FDE. We also discuss the recognition and treatment of the condition. Two patients previously had been diagnosed with systemic lupus erythematosus (SLE).
Case Series
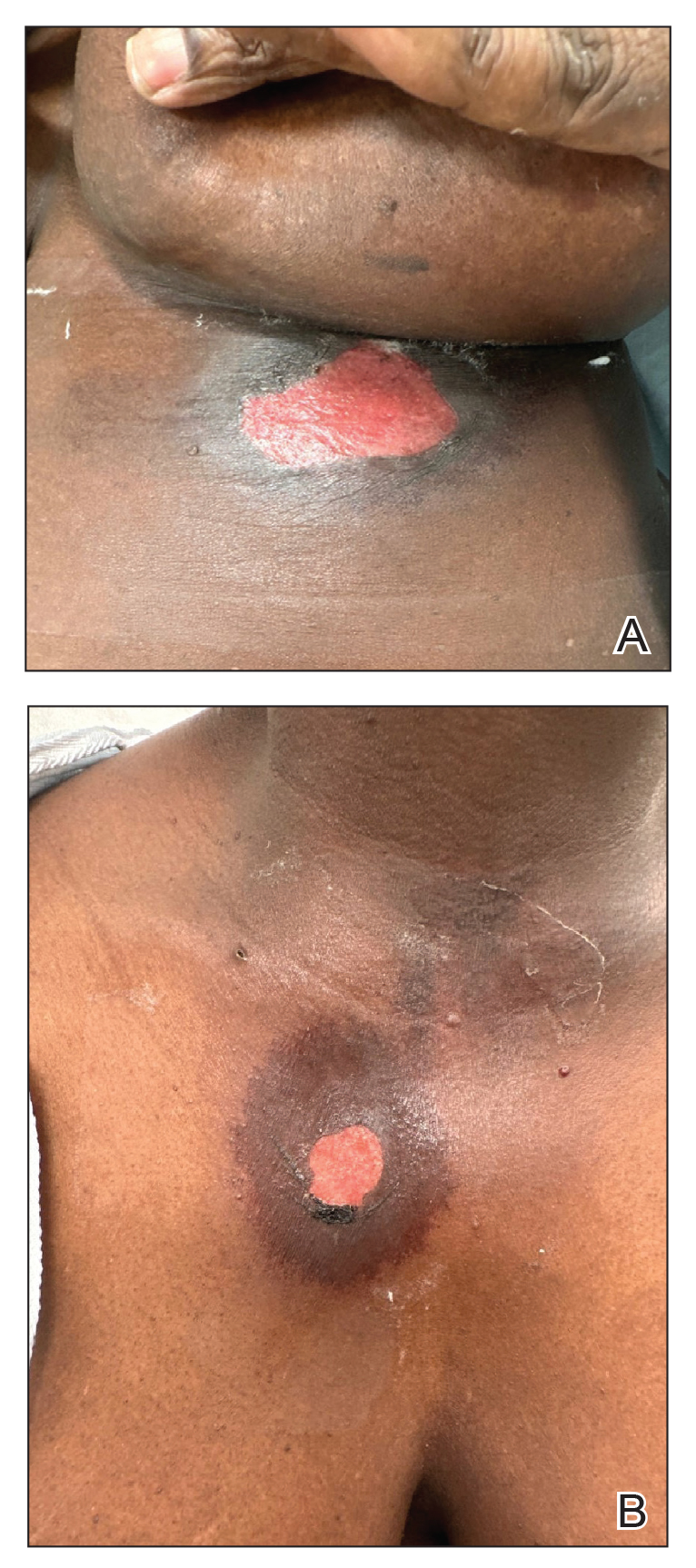
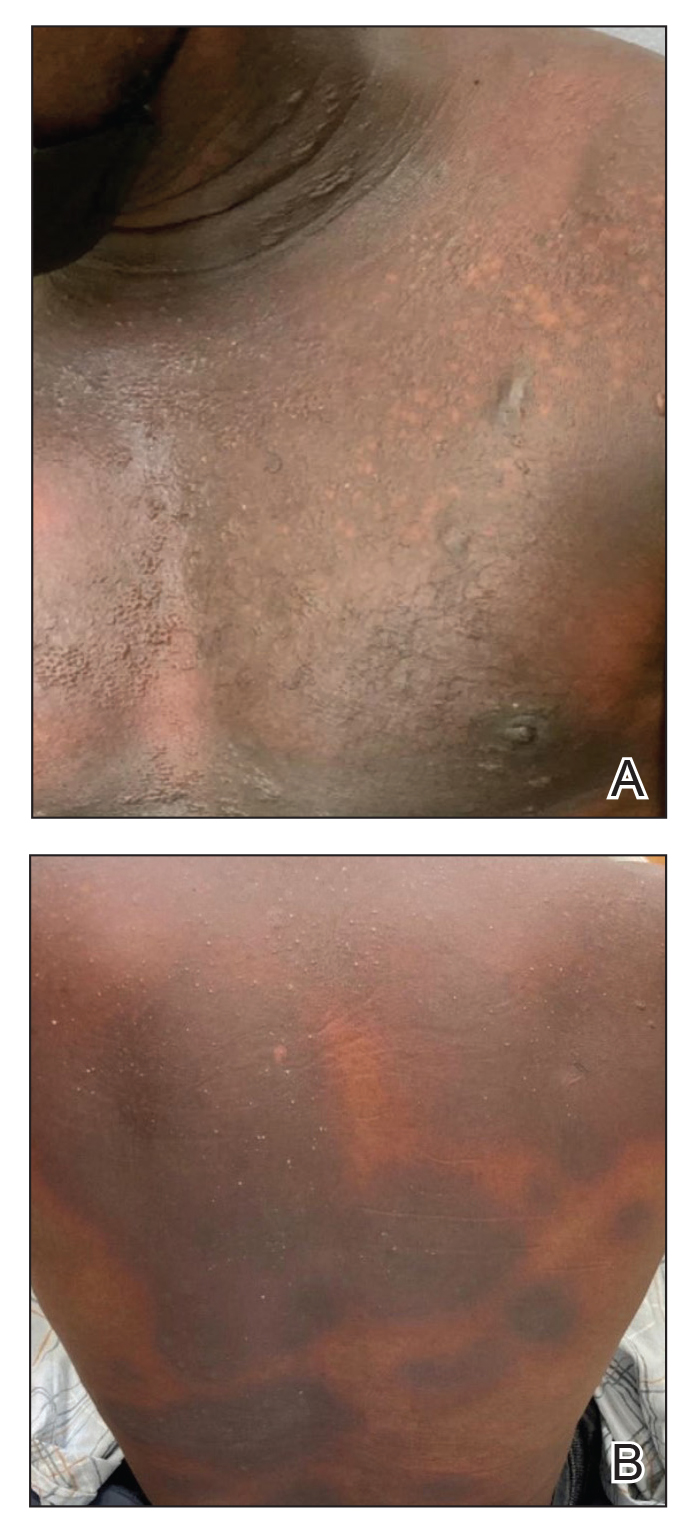
Patient 1—A 60-year-old woman presented to dermatology with a rash on the trunk and groin folds of 4 days’ duration. She had a history of SLE and cutaneous lupus treated with hydroxychloroquine 200 mg twice daily and topical corticosteroids. She had started sulfamethoxazole-trimethoprim for a urinary tract infection with a rash appearing 1 day later. She reported burning skin pain with progression to blisters that “sloughed” off. She denied any known history of allergy to sulfa drugs. Prior to evaluation by dermatology, she visited an urgent care facility and was prescribed hydroxyzine and intramuscular corticosteroids. At presentation to dermatology 3 days after taking sulfamethoxazole-trimethoprim, she had annular flaccid bullae and superficial erosions with dusky borders on the right posterior thigh, right side of the chest, left inframammary fold, and right inguinal fold (Figure 1). She had no ocular, oral, or vaginal erosions. A diagnosis of generalized bullous FDE was favored over erythema multiforme or Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). Shave biopsies from lesions on the right posterior thigh and right inguinal fold demonstrated interface dermatitis with epidermal necrosis, pigment incontinence, and numerous eosinophils. Direct immunofluorescence of the perilesional skin was negative for immunoprotein deposition. These findings were consistent with the clinical impression of generalized bullous FDE. Prior to receiving the histopathology report, the patient was initiated on a regimen of cyclosporine 5 mg/kg/d in the setting of normal renal function and followed until the eruption resolved completely. Cyclosporine was tapered at 2 weeks and discontinued at 3 weeks.
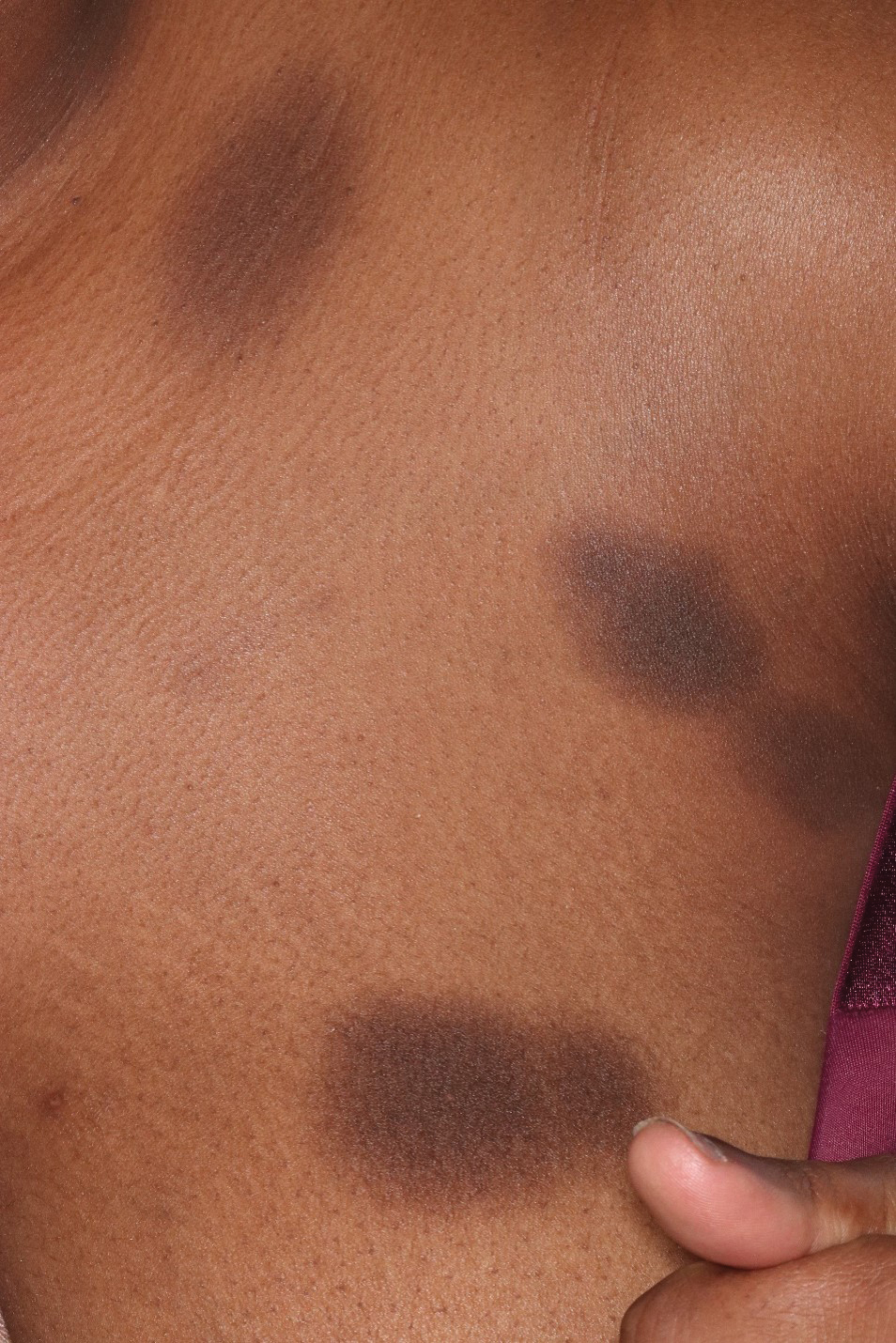
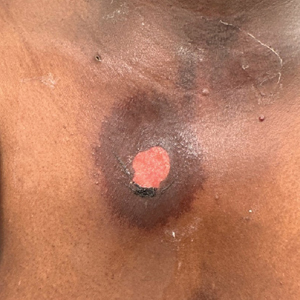
Patient 2—A 32-year-old woman presented for follow-up management of discoid lupus erythematosus. She had a history of systemic and cutaneous lupus, juvenile rheumatoid arthritis, and mixed connective tissue disease managed with prednisone, hydroxychloroquine, azathioprine, and belimumab. Physical examination revealed scarring alopecia with dyspigmentation and active inflammation consistent with uncontrolled cutaneous lupus. However, she also had oval-shaped hyperpigmented patches over the left breast, clavicle, and anterior chest consistent with a generalized FDE (Figure 2). The patient did not recall a history of similar lesions and could not identify a possible trigger. She was counseled on possible culprits and advised to avoid unnecessary medications. She had an unremarkable clinical course; therefore, no further intervention was necessary.
Patient 3—A 33-year-old man presented to the emergency department with a painful rash on the chest and back of 2 days’ duration that began 1 hour after taking naproxen (dosage unknown) for back pain. He had no notable medical history. The patient stated that the rash had slowly worsened and started to develop blisters. He visited an urgent care facility 1 day prior to the current presentation and was started on a 5-day course of prednisone 40 mg daily; the first 2 doses did not help. He denied any mucosal involvement apart from a tender lesion on the penis. He reported a history of an allergic reaction to penicillin. Physical examination revealed extensive dusky violaceous annular plaques with erythematous borders across the anterior and posterior trunk (Figure 3). Multiple flaccid bullae developed within these plaques, involving 15% of the body surface area. He was diagnosed with generalized bullous FDE based on the clinical history and histopathology. He was admitted to the burn intensive care unit and treated with cyclosporine 3 mg/kg/d with subsequent resolution of the eruption.
Comment
Presentation of FDEs—A fixed drug eruption manifests with 1 or more well-demarcated, red or violaceous, annular patches that resolve with postinflammatory hyperpigmentation; it occasionally may manifest with bullae. Initial eruptions may occur up to 2 weeks following medication exposure, but recurrent eruptions usually happen within minutes to hours later. They often are in the same location as prior lesions. A fixed drug eruption can be solitary, scattered, or generalized; a generalized FDE typically demonstrates multiple bilateral lesions that may itch, burn, or cause no symptoms. Patients can experience an FDE at any age, though the median age is reported as 35 to 60 years of age.1 A fixed drug eruption usually occurs after ingestion of oral medications, though there have been a few reports with iodinated contrast.2 Well-known culprits include antibiotics (eg, sulfamethoxazole-trimethoprim, tetracyclines, penicillins/cephalosporins, quinolones, dapsone), nonsteroidal anti-inflammatory drugs, acetaminophen (eg, paracetamol), barbiturates, antimalarials, and anticonvulsants. It also can occur with vaccines or with certain foods (fixed food eruption).3,4 Clinicians may try an oral drug challenge to identify the cause of an FDE, but in patients with a history of a generalized FDE, the risk for developing an increasingly severe reaction with repeated exposure to the medication is too high.5
Histopathology—Patch testing at the site of prior eruption with suspected drug culprits may be useful.6 Histopathology of FDE typically demonstrates vacuolar changes at the dermoepidermal junction with a lichenoid lymphocytic infiltrate. Early lesions often show a predominance of eosinophils. Subepidermal clefting is a feature of the bullous variant. In an active lesion, there are large numbers of CD8+ T lymphocytes expressing natural killer cell–associated molecules.7 The pathologic mechanism is not well understood, though it has been hypothesized that memory CD8+ cells are maintained in specific regions of the epidermis by IL-15 produced in the microenvironment and are activated upon rechallenge.7Considerations in Generalized Bullous FDE—Generalized FDE is defined in the literature as an FDE with involvement of 3 of 6 body areas: head, neck, trunk, upper limbs, lower limbs, and genital area. It may cover more or less than 10% of the body surface area.8-10 Although an isolated FDE frequently is asymptomatic and may not be cause for alarm, recurring drug eruptions increase the risk for development of generalized bullous FDE. Generalized bullous FDE is a rare subset. It is frequently misdiagnosed, and data on its incidence are uncertain.11 Of note, several pathologies causing bullous lesions may be in the differential diagnosis, including bullous pemphigoid; pemphigus vulgaris; bullous SLE; or bullae from cutaneous lupus, staphylococcal scalded skin syndrome, erythema multiforme, or SJS/TEN.12 When matched for body surface area involvement with SJS/TEN, generalized bullous FDE shares nearly identical mortality rates10; therefore, these patients should be treated with the same level of urgency and admitted to a critical care or burn unit, as they are at serious risk for infection and other complications.13
Clinical history and presentation along with histopathologic findings help to narrow down the differential diagnosis. Clinically, generalized bullous FDE does not affect the surrounding skin and manifests sooner after drug exposure (1–24 hours) with less mucosal involvement than SJS/TEN.9 Additionally, SJS/TEN patients frequently have generalized malaise and/or fever, while generalized bullous FDE patients do not. Finally, patients with generalized bullous FDE may report a history of a cutaneous eruption similar in morphology or in the same location.
Histopathologically, generalized bullous FDE may be similar to FDE with the addition of a subepidermal blister. Generalized bullous FDE patients have greater eosinophil infiltration and dermal melanophages than patients with SJS/TEN.9 Cellular infiltrates in generalized bullous FDE include more dermal CD41 cells, such as Foxp31 regulatory T cells; fewer intraepidermal CD561 cells; and fewer intraepidermal cells with granulysin.9 Occasionally, generalized bullous FDE causes full-thickness necrosis. In those cases, generalized bullous FDE cannot reliably be distinguished from other conditions with epidermal necrolysis on histopathology.13
FDE Diagnostics—A cytotoxin produced by
Management—Avoidance of the inciting drug often is sufficient for patients with an FDE, as demonstrated in patient 2 in our case series. Clinicians also should counsel patients on avoidance of potential cross-reacting drugs. Symptomatic treatment for itch or pain is appropriate and may include antihistamines or topical steroids. Nonsteroidal anti-inflammatory drugs may exacerbate or be causative of FDE. For generalized bullous FDE, cyclosporine is favored in the literature15,16 and was used to successfully treat both patients 1 and 3 in our case series. A short course of systemic corticosteroids or intravenous immunoglobulin also may be considered. Mild cases of generalized bullous FDE may be treated with close outpatient follow-up (patient 1), while severe cases require inpatient or even critical care monitoring with aggressive medical management to prevent the progression of skin desquamation (patient 3). Patients with severe oral lesions may require inpatient support for fluid maintenance.
Lupus History—Two patients in our case series had a history of lupus. Lupus itself can cause primary bullous lesions. Similar to FDE, bullous SLE can involve sun-exposed and nonexposed areas of the skin as well as the mucous membranes with a predilection for the lower vermilion lip.17 In bullous SLE, tense subepidermal blisters with a neutrophil-rich infiltrate form due to circulating antibodies to type VII collagen. These blisters have an erythematous or urticated base, most commonly on the face, upper trunk, and proximal extremities.18 In both SLE with skin manifestations and lupus limited to the skin, bullae may form due to extensive vacuolar degeneration. Similar to TEN, they can form rapidly in a widespread distribution.17 However, there is limited mucosal involvement, no clear drug association, and a better prognosis. Bullae caused by lupus will frequently demonstrate deposition of immunoproteins IgG, IgM, IgA, and complement component 3 at the basement membrane zone in perilesional skin on direct immunofluorescence. However, negative direct immunofluorescence does not rule out lupus.12 At the same time, patients with lupus frequently have comorbidities requiring multiple medications; the need for these medications may predispose patients to higher rates of cutaneous drug eruptions.19 To our knowledge, there is no known association between FDE and lupus.
Conclusion
Patients with acute eruptions following the initiation of a new prescription or over-the-counter medication require urgent evaluation. Generalized bullous FDE requires timely diagnosis and intervention. Patients with lupus have an increased risk for cutaneous drug eruptions due to polypharmacy. Further investigation is necessary to determine if there is a pathophysiologic mechanism responsible for the development of FDE in lupus patients.
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925.
- Gavin M, Sharp L, Walker K, et al. Contrast-induced generalized bullous fixed drug eruption resembling Stevens-Johnson syndrome. Proc (Bayl Univ Med Cent). 2019;32:601-602.
- Kabir S, Feit EJ, Heilman ER. Generalized fixed drug eruption following Pfizer-BioNtech COVID-19 vaccination. Clin Case Rep. 2022;10:E6684.
- Choi S, Kim SH, Hwang JH, et al. Rapidly progressing generalized bullous fixed drug eruption after the first dose of COVID-19 messenger RNA vaccination. J Dermatol. 2023;50:1190-1193.
- Mahboob A, Haroon TS. Drugs causing fixed eruptions: a study of 450 cases. Int J Dermatol. 1998;37:833-838.
- Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
- Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br J Dermatol. 2008;158:1230-1238.
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15.
- Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ranario JS, Smith JL. Bullous lesions in a patient with systemic lupus erythematosus. J Clin Aesthet Dermatol. 2014;7:44-49.
- Perron E, Viarnaud A, Marciano L, et al. Clinical and histological features of fixed drug eruption: a single-centre series of 73 cases with comparison between bullous and non-bullous forms. Eur J Dermatol. 2021;31:372-380.
- Chen CB, Kuo KL, Wang CW, et al. Detecting lesional granulysin levels for rapid diagnosis of cytotoxic T lymphocyte-mediated bullous skin disorders. J Allergy Clin Immunol Pract. 2021;9:1327-1337.e3.
- Beniwal R, Gupta LK, Khare AK, et al. Cyclosporine in generalized bullous-fixed drug eruption. Indian J Dermatol. 2018;63:432-433.
- Vargas Mora P, García S, Valenzuela F, et al. Generalized bullous fixed drug eruption successfully treated with cyclosporine. Dermatol Ther. 2020;33:E13492.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Zonzits E, Aberer W, Tappeiner G. Drug eruptions from mesna. After cyclophosphamide treatment of patients with systemic lupus erythematosus and dermatomyositis. Arch Dermatol. 1992;128:80-82.
Recognizing cutaneous drug eruptions is important for treatment and prevention of recurrence. Fixed drug eruptions (FDEs) typically are harmless but can have major negative cosmetic consequences for patients. In its more severe forms, patients are at risk for widespread epithelial necrosis with accompanying complications. We report 1 patient with generalized FDE and 2 with generalized bullous FDE. We also discuss the recognition and treatment of the condition. Two patients previously had been diagnosed with systemic lupus erythematosus (SLE).
Case Series
Patient 1—A 60-year-old woman presented to dermatology with a rash on the trunk and groin folds of 4 days’ duration. She had a history of SLE and cutaneous lupus treated with hydroxychloroquine 200 mg twice daily and topical corticosteroids. She had started sulfamethoxazole-trimethoprim for a urinary tract infection with a rash appearing 1 day later. She reported burning skin pain with progression to blisters that “sloughed” off. She denied any known history of allergy to sulfa drugs. Prior to evaluation by dermatology, she visited an urgent care facility and was prescribed hydroxyzine and intramuscular corticosteroids. At presentation to dermatology 3 days after taking sulfamethoxazole-trimethoprim, she had annular flaccid bullae and superficial erosions with dusky borders on the right posterior thigh, right side of the chest, left inframammary fold, and right inguinal fold (Figure 1). She had no ocular, oral, or vaginal erosions. A diagnosis of generalized bullous FDE was favored over erythema multiforme or Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). Shave biopsies from lesions on the right posterior thigh and right inguinal fold demonstrated interface dermatitis with epidermal necrosis, pigment incontinence, and numerous eosinophils. Direct immunofluorescence of the perilesional skin was negative for immunoprotein deposition. These findings were consistent with the clinical impression of generalized bullous FDE. Prior to receiving the histopathology report, the patient was initiated on a regimen of cyclosporine 5 mg/kg/d in the setting of normal renal function and followed until the eruption resolved completely. Cyclosporine was tapered at 2 weeks and discontinued at 3 weeks.
Patient 2—A 32-year-old woman presented for follow-up management of discoid lupus erythematosus. She had a history of systemic and cutaneous lupus, juvenile rheumatoid arthritis, and mixed connective tissue disease managed with prednisone, hydroxychloroquine, azathioprine, and belimumab. Physical examination revealed scarring alopecia with dyspigmentation and active inflammation consistent with uncontrolled cutaneous lupus. However, she also had oval-shaped hyperpigmented patches over the left breast, clavicle, and anterior chest consistent with a generalized FDE (Figure 2). The patient did not recall a history of similar lesions and could not identify a possible trigger. She was counseled on possible culprits and advised to avoid unnecessary medications. She had an unremarkable clinical course; therefore, no further intervention was necessary.
Patient 3—A 33-year-old man presented to the emergency department with a painful rash on the chest and back of 2 days’ duration that began 1 hour after taking naproxen (dosage unknown) for back pain. He had no notable medical history. The patient stated that the rash had slowly worsened and started to develop blisters. He visited an urgent care facility 1 day prior to the current presentation and was started on a 5-day course of prednisone 40 mg daily; the first 2 doses did not help. He denied any mucosal involvement apart from a tender lesion on the penis. He reported a history of an allergic reaction to penicillin. Physical examination revealed extensive dusky violaceous annular plaques with erythematous borders across the anterior and posterior trunk (Figure 3). Multiple flaccid bullae developed within these plaques, involving 15% of the body surface area. He was diagnosed with generalized bullous FDE based on the clinical history and histopathology. He was admitted to the burn intensive care unit and treated with cyclosporine 3 mg/kg/d with subsequent resolution of the eruption.
Comment
Presentation of FDEs—A fixed drug eruption manifests with 1 or more well-demarcated, red or violaceous, annular patches that resolve with postinflammatory hyperpigmentation; it occasionally may manifest with bullae. Initial eruptions may occur up to 2 weeks following medication exposure, but recurrent eruptions usually happen within minutes to hours later. They often are in the same location as prior lesions. A fixed drug eruption can be solitary, scattered, or generalized; a generalized FDE typically demonstrates multiple bilateral lesions that may itch, burn, or cause no symptoms. Patients can experience an FDE at any age, though the median age is reported as 35 to 60 years of age.1 A fixed drug eruption usually occurs after ingestion of oral medications, though there have been a few reports with iodinated contrast.2 Well-known culprits include antibiotics (eg, sulfamethoxazole-trimethoprim, tetracyclines, penicillins/cephalosporins, quinolones, dapsone), nonsteroidal anti-inflammatory drugs, acetaminophen (eg, paracetamol), barbiturates, antimalarials, and anticonvulsants. It also can occur with vaccines or with certain foods (fixed food eruption).3,4 Clinicians may try an oral drug challenge to identify the cause of an FDE, but in patients with a history of a generalized FDE, the risk for developing an increasingly severe reaction with repeated exposure to the medication is too high.5
Histopathology—Patch testing at the site of prior eruption with suspected drug culprits may be useful.6 Histopathology of FDE typically demonstrates vacuolar changes at the dermoepidermal junction with a lichenoid lymphocytic infiltrate. Early lesions often show a predominance of eosinophils. Subepidermal clefting is a feature of the bullous variant. In an active lesion, there are large numbers of CD8+ T lymphocytes expressing natural killer cell–associated molecules.7 The pathologic mechanism is not well understood, though it has been hypothesized that memory CD8+ cells are maintained in specific regions of the epidermis by IL-15 produced in the microenvironment and are activated upon rechallenge.7Considerations in Generalized Bullous FDE—Generalized FDE is defined in the literature as an FDE with involvement of 3 of 6 body areas: head, neck, trunk, upper limbs, lower limbs, and genital area. It may cover more or less than 10% of the body surface area.8-10 Although an isolated FDE frequently is asymptomatic and may not be cause for alarm, recurring drug eruptions increase the risk for development of generalized bullous FDE. Generalized bullous FDE is a rare subset. It is frequently misdiagnosed, and data on its incidence are uncertain.11 Of note, several pathologies causing bullous lesions may be in the differential diagnosis, including bullous pemphigoid; pemphigus vulgaris; bullous SLE; or bullae from cutaneous lupus, staphylococcal scalded skin syndrome, erythema multiforme, or SJS/TEN.12 When matched for body surface area involvement with SJS/TEN, generalized bullous FDE shares nearly identical mortality rates10; therefore, these patients should be treated with the same level of urgency and admitted to a critical care or burn unit, as they are at serious risk for infection and other complications.13
Clinical history and presentation along with histopathologic findings help to narrow down the differential diagnosis. Clinically, generalized bullous FDE does not affect the surrounding skin and manifests sooner after drug exposure (1–24 hours) with less mucosal involvement than SJS/TEN.9 Additionally, SJS/TEN patients frequently have generalized malaise and/or fever, while generalized bullous FDE patients do not. Finally, patients with generalized bullous FDE may report a history of a cutaneous eruption similar in morphology or in the same location.
Histopathologically, generalized bullous FDE may be similar to FDE with the addition of a subepidermal blister. Generalized bullous FDE patients have greater eosinophil infiltration and dermal melanophages than patients with SJS/TEN.9 Cellular infiltrates in generalized bullous FDE include more dermal CD41 cells, such as Foxp31 regulatory T cells; fewer intraepidermal CD561 cells; and fewer intraepidermal cells with granulysin.9 Occasionally, generalized bullous FDE causes full-thickness necrosis. In those cases, generalized bullous FDE cannot reliably be distinguished from other conditions with epidermal necrolysis on histopathology.13
FDE Diagnostics—A cytotoxin produced by
Management—Avoidance of the inciting drug often is sufficient for patients with an FDE, as demonstrated in patient 2 in our case series. Clinicians also should counsel patients on avoidance of potential cross-reacting drugs. Symptomatic treatment for itch or pain is appropriate and may include antihistamines or topical steroids. Nonsteroidal anti-inflammatory drugs may exacerbate or be causative of FDE. For generalized bullous FDE, cyclosporine is favored in the literature15,16 and was used to successfully treat both patients 1 and 3 in our case series. A short course of systemic corticosteroids or intravenous immunoglobulin also may be considered. Mild cases of generalized bullous FDE may be treated with close outpatient follow-up (patient 1), while severe cases require inpatient or even critical care monitoring with aggressive medical management to prevent the progression of skin desquamation (patient 3). Patients with severe oral lesions may require inpatient support for fluid maintenance.
Lupus History—Two patients in our case series had a history of lupus. Lupus itself can cause primary bullous lesions. Similar to FDE, bullous SLE can involve sun-exposed and nonexposed areas of the skin as well as the mucous membranes with a predilection for the lower vermilion lip.17 In bullous SLE, tense subepidermal blisters with a neutrophil-rich infiltrate form due to circulating antibodies to type VII collagen. These blisters have an erythematous or urticated base, most commonly on the face, upper trunk, and proximal extremities.18 In both SLE with skin manifestations and lupus limited to the skin, bullae may form due to extensive vacuolar degeneration. Similar to TEN, they can form rapidly in a widespread distribution.17 However, there is limited mucosal involvement, no clear drug association, and a better prognosis. Bullae caused by lupus will frequently demonstrate deposition of immunoproteins IgG, IgM, IgA, and complement component 3 at the basement membrane zone in perilesional skin on direct immunofluorescence. However, negative direct immunofluorescence does not rule out lupus.12 At the same time, patients with lupus frequently have comorbidities requiring multiple medications; the need for these medications may predispose patients to higher rates of cutaneous drug eruptions.19 To our knowledge, there is no known association between FDE and lupus.
Conclusion
Patients with acute eruptions following the initiation of a new prescription or over-the-counter medication require urgent evaluation. Generalized bullous FDE requires timely diagnosis and intervention. Patients with lupus have an increased risk for cutaneous drug eruptions due to polypharmacy. Further investigation is necessary to determine if there is a pathophysiologic mechanism responsible for the development of FDE in lupus patients.
Recognizing cutaneous drug eruptions is important for treatment and prevention of recurrence. Fixed drug eruptions (FDEs) typically are harmless but can have major negative cosmetic consequences for patients. In its more severe forms, patients are at risk for widespread epithelial necrosis with accompanying complications. We report 1 patient with generalized FDE and 2 with generalized bullous FDE. We also discuss the recognition and treatment of the condition. Two patients previously had been diagnosed with systemic lupus erythematosus (SLE).
Case Series
Patient 1—A 60-year-old woman presented to dermatology with a rash on the trunk and groin folds of 4 days’ duration. She had a history of SLE and cutaneous lupus treated with hydroxychloroquine 200 mg twice daily and topical corticosteroids. She had started sulfamethoxazole-trimethoprim for a urinary tract infection with a rash appearing 1 day later. She reported burning skin pain with progression to blisters that “sloughed” off. She denied any known history of allergy to sulfa drugs. Prior to evaluation by dermatology, she visited an urgent care facility and was prescribed hydroxyzine and intramuscular corticosteroids. At presentation to dermatology 3 days after taking sulfamethoxazole-trimethoprim, she had annular flaccid bullae and superficial erosions with dusky borders on the right posterior thigh, right side of the chest, left inframammary fold, and right inguinal fold (Figure 1). She had no ocular, oral, or vaginal erosions. A diagnosis of generalized bullous FDE was favored over erythema multiforme or Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). Shave biopsies from lesions on the right posterior thigh and right inguinal fold demonstrated interface dermatitis with epidermal necrosis, pigment incontinence, and numerous eosinophils. Direct immunofluorescence of the perilesional skin was negative for immunoprotein deposition. These findings were consistent with the clinical impression of generalized bullous FDE. Prior to receiving the histopathology report, the patient was initiated on a regimen of cyclosporine 5 mg/kg/d in the setting of normal renal function and followed until the eruption resolved completely. Cyclosporine was tapered at 2 weeks and discontinued at 3 weeks.
Patient 2—A 32-year-old woman presented for follow-up management of discoid lupus erythematosus. She had a history of systemic and cutaneous lupus, juvenile rheumatoid arthritis, and mixed connective tissue disease managed with prednisone, hydroxychloroquine, azathioprine, and belimumab. Physical examination revealed scarring alopecia with dyspigmentation and active inflammation consistent with uncontrolled cutaneous lupus. However, she also had oval-shaped hyperpigmented patches over the left breast, clavicle, and anterior chest consistent with a generalized FDE (Figure 2). The patient did not recall a history of similar lesions and could not identify a possible trigger. She was counseled on possible culprits and advised to avoid unnecessary medications. She had an unremarkable clinical course; therefore, no further intervention was necessary.
Patient 3—A 33-year-old man presented to the emergency department with a painful rash on the chest and back of 2 days’ duration that began 1 hour after taking naproxen (dosage unknown) for back pain. He had no notable medical history. The patient stated that the rash had slowly worsened and started to develop blisters. He visited an urgent care facility 1 day prior to the current presentation and was started on a 5-day course of prednisone 40 mg daily; the first 2 doses did not help. He denied any mucosal involvement apart from a tender lesion on the penis. He reported a history of an allergic reaction to penicillin. Physical examination revealed extensive dusky violaceous annular plaques with erythematous borders across the anterior and posterior trunk (Figure 3). Multiple flaccid bullae developed within these plaques, involving 15% of the body surface area. He was diagnosed with generalized bullous FDE based on the clinical history and histopathology. He was admitted to the burn intensive care unit and treated with cyclosporine 3 mg/kg/d with subsequent resolution of the eruption.
Comment
Presentation of FDEs—A fixed drug eruption manifests with 1 or more well-demarcated, red or violaceous, annular patches that resolve with postinflammatory hyperpigmentation; it occasionally may manifest with bullae. Initial eruptions may occur up to 2 weeks following medication exposure, but recurrent eruptions usually happen within minutes to hours later. They often are in the same location as prior lesions. A fixed drug eruption can be solitary, scattered, or generalized; a generalized FDE typically demonstrates multiple bilateral lesions that may itch, burn, or cause no symptoms. Patients can experience an FDE at any age, though the median age is reported as 35 to 60 years of age.1 A fixed drug eruption usually occurs after ingestion of oral medications, though there have been a few reports with iodinated contrast.2 Well-known culprits include antibiotics (eg, sulfamethoxazole-trimethoprim, tetracyclines, penicillins/cephalosporins, quinolones, dapsone), nonsteroidal anti-inflammatory drugs, acetaminophen (eg, paracetamol), barbiturates, antimalarials, and anticonvulsants. It also can occur with vaccines or with certain foods (fixed food eruption).3,4 Clinicians may try an oral drug challenge to identify the cause of an FDE, but in patients with a history of a generalized FDE, the risk for developing an increasingly severe reaction with repeated exposure to the medication is too high.5
Histopathology—Patch testing at the site of prior eruption with suspected drug culprits may be useful.6 Histopathology of FDE typically demonstrates vacuolar changes at the dermoepidermal junction with a lichenoid lymphocytic infiltrate. Early lesions often show a predominance of eosinophils. Subepidermal clefting is a feature of the bullous variant. In an active lesion, there are large numbers of CD8+ T lymphocytes expressing natural killer cell–associated molecules.7 The pathologic mechanism is not well understood, though it has been hypothesized that memory CD8+ cells are maintained in specific regions of the epidermis by IL-15 produced in the microenvironment and are activated upon rechallenge.7Considerations in Generalized Bullous FDE—Generalized FDE is defined in the literature as an FDE with involvement of 3 of 6 body areas: head, neck, trunk, upper limbs, lower limbs, and genital area. It may cover more or less than 10% of the body surface area.8-10 Although an isolated FDE frequently is asymptomatic and may not be cause for alarm, recurring drug eruptions increase the risk for development of generalized bullous FDE. Generalized bullous FDE is a rare subset. It is frequently misdiagnosed, and data on its incidence are uncertain.11 Of note, several pathologies causing bullous lesions may be in the differential diagnosis, including bullous pemphigoid; pemphigus vulgaris; bullous SLE; or bullae from cutaneous lupus, staphylococcal scalded skin syndrome, erythema multiforme, or SJS/TEN.12 When matched for body surface area involvement with SJS/TEN, generalized bullous FDE shares nearly identical mortality rates10; therefore, these patients should be treated with the same level of urgency and admitted to a critical care or burn unit, as they are at serious risk for infection and other complications.13
Clinical history and presentation along with histopathologic findings help to narrow down the differential diagnosis. Clinically, generalized bullous FDE does not affect the surrounding skin and manifests sooner after drug exposure (1–24 hours) with less mucosal involvement than SJS/TEN.9 Additionally, SJS/TEN patients frequently have generalized malaise and/or fever, while generalized bullous FDE patients do not. Finally, patients with generalized bullous FDE may report a history of a cutaneous eruption similar in morphology or in the same location.
Histopathologically, generalized bullous FDE may be similar to FDE with the addition of a subepidermal blister. Generalized bullous FDE patients have greater eosinophil infiltration and dermal melanophages than patients with SJS/TEN.9 Cellular infiltrates in generalized bullous FDE include more dermal CD41 cells, such as Foxp31 regulatory T cells; fewer intraepidermal CD561 cells; and fewer intraepidermal cells with granulysin.9 Occasionally, generalized bullous FDE causes full-thickness necrosis. In those cases, generalized bullous FDE cannot reliably be distinguished from other conditions with epidermal necrolysis on histopathology.13
FDE Diagnostics—A cytotoxin produced by
Management—Avoidance of the inciting drug often is sufficient for patients with an FDE, as demonstrated in patient 2 in our case series. Clinicians also should counsel patients on avoidance of potential cross-reacting drugs. Symptomatic treatment for itch or pain is appropriate and may include antihistamines or topical steroids. Nonsteroidal anti-inflammatory drugs may exacerbate or be causative of FDE. For generalized bullous FDE, cyclosporine is favored in the literature15,16 and was used to successfully treat both patients 1 and 3 in our case series. A short course of systemic corticosteroids or intravenous immunoglobulin also may be considered. Mild cases of generalized bullous FDE may be treated with close outpatient follow-up (patient 1), while severe cases require inpatient or even critical care monitoring with aggressive medical management to prevent the progression of skin desquamation (patient 3). Patients with severe oral lesions may require inpatient support for fluid maintenance.
Lupus History—Two patients in our case series had a history of lupus. Lupus itself can cause primary bullous lesions. Similar to FDE, bullous SLE can involve sun-exposed and nonexposed areas of the skin as well as the mucous membranes with a predilection for the lower vermilion lip.17 In bullous SLE, tense subepidermal blisters with a neutrophil-rich infiltrate form due to circulating antibodies to type VII collagen. These blisters have an erythematous or urticated base, most commonly on the face, upper trunk, and proximal extremities.18 In both SLE with skin manifestations and lupus limited to the skin, bullae may form due to extensive vacuolar degeneration. Similar to TEN, they can form rapidly in a widespread distribution.17 However, there is limited mucosal involvement, no clear drug association, and a better prognosis. Bullae caused by lupus will frequently demonstrate deposition of immunoproteins IgG, IgM, IgA, and complement component 3 at the basement membrane zone in perilesional skin on direct immunofluorescence. However, negative direct immunofluorescence does not rule out lupus.12 At the same time, patients with lupus frequently have comorbidities requiring multiple medications; the need for these medications may predispose patients to higher rates of cutaneous drug eruptions.19 To our knowledge, there is no known association between FDE and lupus.
Conclusion
Patients with acute eruptions following the initiation of a new prescription or over-the-counter medication require urgent evaluation. Generalized bullous FDE requires timely diagnosis and intervention. Patients with lupus have an increased risk for cutaneous drug eruptions due to polypharmacy. Further investigation is necessary to determine if there is a pathophysiologic mechanism responsible for the development of FDE in lupus patients.
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925.
- Gavin M, Sharp L, Walker K, et al. Contrast-induced generalized bullous fixed drug eruption resembling Stevens-Johnson syndrome. Proc (Bayl Univ Med Cent). 2019;32:601-602.
- Kabir S, Feit EJ, Heilman ER. Generalized fixed drug eruption following Pfizer-BioNtech COVID-19 vaccination. Clin Case Rep. 2022;10:E6684.
- Choi S, Kim SH, Hwang JH, et al. Rapidly progressing generalized bullous fixed drug eruption after the first dose of COVID-19 messenger RNA vaccination. J Dermatol. 2023;50:1190-1193.
- Mahboob A, Haroon TS. Drugs causing fixed eruptions: a study of 450 cases. Int J Dermatol. 1998;37:833-838.
- Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
- Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br J Dermatol. 2008;158:1230-1238.
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15.
- Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ranario JS, Smith JL. Bullous lesions in a patient with systemic lupus erythematosus. J Clin Aesthet Dermatol. 2014;7:44-49.
- Perron E, Viarnaud A, Marciano L, et al. Clinical and histological features of fixed drug eruption: a single-centre series of 73 cases with comparison between bullous and non-bullous forms. Eur J Dermatol. 2021;31:372-380.
- Chen CB, Kuo KL, Wang CW, et al. Detecting lesional granulysin levels for rapid diagnosis of cytotoxic T lymphocyte-mediated bullous skin disorders. J Allergy Clin Immunol Pract. 2021;9:1327-1337.e3.
- Beniwal R, Gupta LK, Khare AK, et al. Cyclosporine in generalized bullous-fixed drug eruption. Indian J Dermatol. 2018;63:432-433.
- Vargas Mora P, García S, Valenzuela F, et al. Generalized bullous fixed drug eruption successfully treated with cyclosporine. Dermatol Ther. 2020;33:E13492.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Zonzits E, Aberer W, Tappeiner G. Drug eruptions from mesna. After cyclophosphamide treatment of patients with systemic lupus erythematosus and dermatomyositis. Arch Dermatol. 1992;128:80-82.
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925.
- Gavin M, Sharp L, Walker K, et al. Contrast-induced generalized bullous fixed drug eruption resembling Stevens-Johnson syndrome. Proc (Bayl Univ Med Cent). 2019;32:601-602.
- Kabir S, Feit EJ, Heilman ER. Generalized fixed drug eruption following Pfizer-BioNtech COVID-19 vaccination. Clin Case Rep. 2022;10:E6684.
- Choi S, Kim SH, Hwang JH, et al. Rapidly progressing generalized bullous fixed drug eruption after the first dose of COVID-19 messenger RNA vaccination. J Dermatol. 2023;50:1190-1193.
- Mahboob A, Haroon TS. Drugs causing fixed eruptions: a study of 450 cases. Int J Dermatol. 1998;37:833-838.
- Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
- Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br J Dermatol. 2008;158:1230-1238.
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15.
- Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ranario JS, Smith JL. Bullous lesions in a patient with systemic lupus erythematosus. J Clin Aesthet Dermatol. 2014;7:44-49.
- Perron E, Viarnaud A, Marciano L, et al. Clinical and histological features of fixed drug eruption: a single-centre series of 73 cases with comparison between bullous and non-bullous forms. Eur J Dermatol. 2021;31:372-380.
- Chen CB, Kuo KL, Wang CW, et al. Detecting lesional granulysin levels for rapid diagnosis of cytotoxic T lymphocyte-mediated bullous skin disorders. J Allergy Clin Immunol Pract. 2021;9:1327-1337.e3.
- Beniwal R, Gupta LK, Khare AK, et al. Cyclosporine in generalized bullous-fixed drug eruption. Indian J Dermatol. 2018;63:432-433.
- Vargas Mora P, García S, Valenzuela F, et al. Generalized bullous fixed drug eruption successfully treated with cyclosporine. Dermatol Ther. 2020;33:E13492.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Zonzits E, Aberer W, Tappeiner G. Drug eruptions from mesna. After cyclophosphamide treatment of patients with systemic lupus erythematosus and dermatomyositis. Arch Dermatol. 1992;128:80-82.
Practice Points
- Although localized fixed drug eruption (FDE) is a relatively benign diagnosis, generalized bullous FDE requires urgent management and may necessitate intensive burn care.
- Patients with lupus are at increased risk for drug eruptions due to polypharmacy, and there is a wide differential for bullous eruptions in these patients.
Mycobacterium interjectum Infection in an Immunocompetent Host Following Contact With Aquarium Fish
To the Editor:
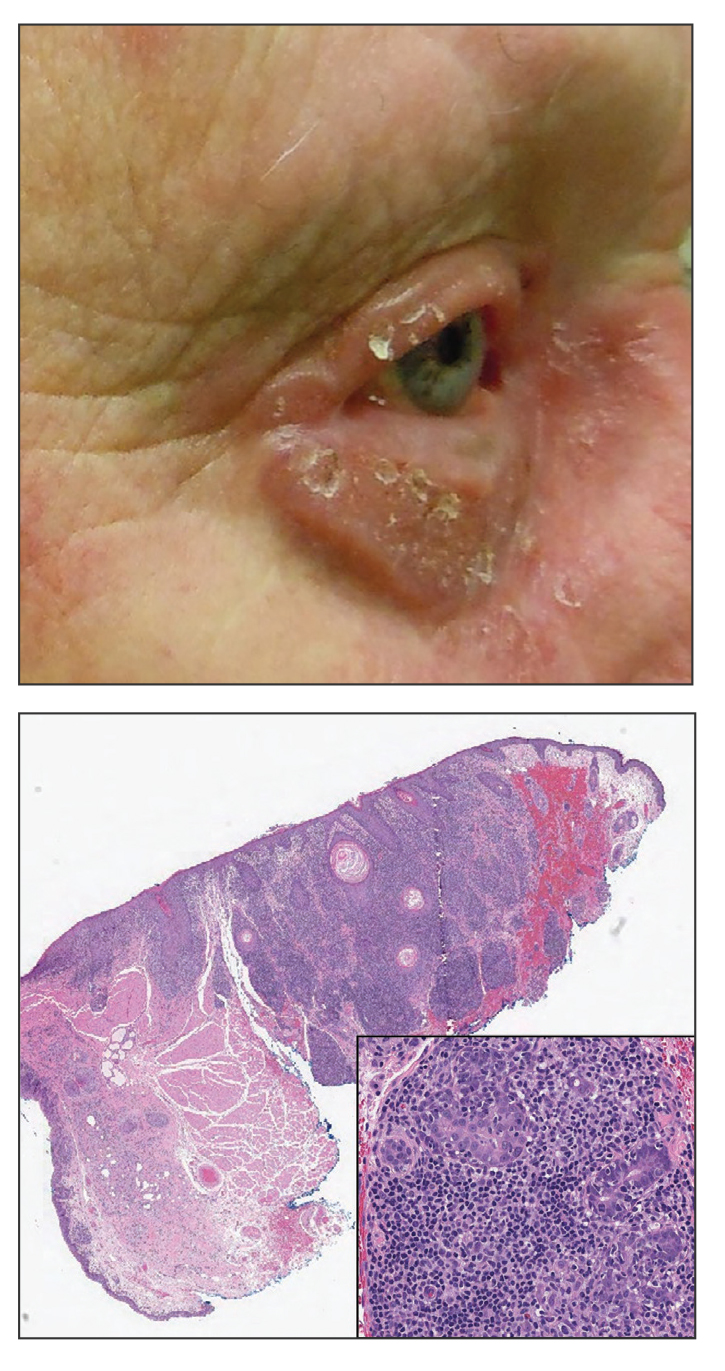
A 48-year-old man presented with nodular lesions in a sporotrichoid pattern on the right hand and forearm of 3 months’ duration (Figure). There were no lymphadeno-pathies, and he had no notable medical history. He denied fever and other systemic symptoms. The patient recently had manipulated a warm water fish aquarium. Although he did not recall a clear injury, inadvertent mild trauma was a possibility. He denied other contact or trauma in relation to animals or vegetables.
Histopathology from a punch biopsy of the forearm revealed a granulomatous infiltrate with necrosis at the deep dermis level at the interface with the subcutaneous cellular tissue that was composed of mainly epithelioid cells with a few multinucleated giant cells. No acid-fast bacilli or fungi were observed with special stains.
A polymerase chain reaction assay for atypical mycobacteria was positive for Mycobacterium interjectum. The culture of the skin biopsy was negative for fungi and mycobacteria after long incubation (6 weeks) on 2 occasions, and an antibiogram was not available. Complementary tests including hemogram, HIV serology, and chest and upper extremity radiographs did not reveal any abnormalities.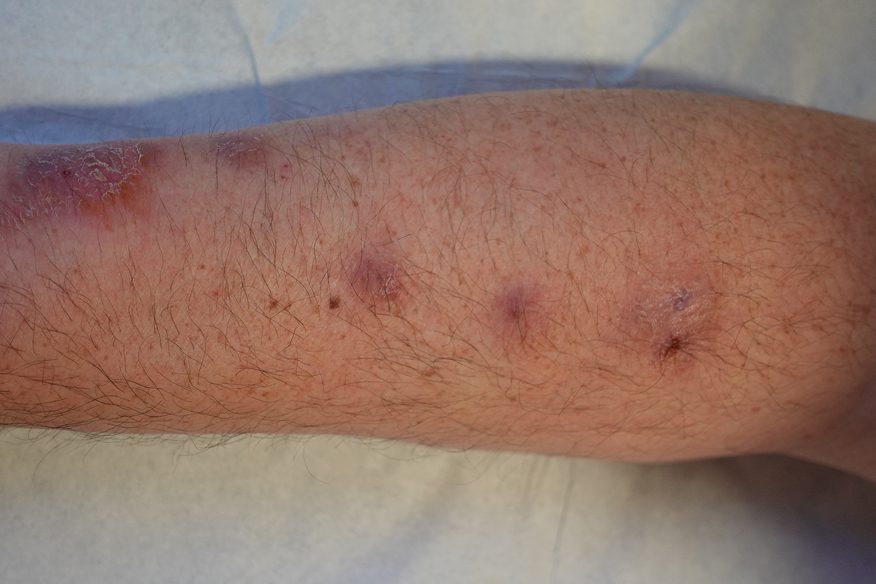
The patient was treated with rifampicin 600 mg/d, clarithromycin 500 mg every 12 hours, and co-trimoxazole 160/800 mg every 12 hours for 9 months with some resolution but persistence of some residual scarring lesions. There was no recurrence at 6-month follow-up.
Mycobacterium interjectum is a rare, slow-growing, scotochromogenic mycobacteria. Case reports usually refer to lymphadenitis in healthy children and pulmonary infections in immunocompromised or immunocompetent adults.1,2 A case of M interjectum with cutaneous involvement was reported by Fukuoka et al,3 with ulcerated nodules and abscesses on the leg identified in an immunocompromised patient. Our patient did not present with any cause of immunosuppression or clear injury predisposing him to infection. This microorganism has been detected in water, soil,3 and aquarium fish,4 the latter being the most likely source of infection in our patient. Given its slow growth rate and the need for a specific polymerase chain reaction assay, which is not widely available, M interjectum infection may be underdiagnosed.
No standard antibiotic regimen has been established, but M interjectum has proven to be a multidrug-resistant bacterium with frequent therapy failures. Treatment options have ranged from standard tuberculostatic therapy to combination therapy with medications such as amikacin, levofloxacin, rifampicin, and co-trimoxazole.1 Because an antibiogram was not available for our patient, empiric treatment with rifampicin, clarithromycin, and co-trimoxazole was prescribed for 9 months, with satisfactory response and tolerance. These drugs were selected because of their susceptibility profile in the literature.1,5
- Sotello D, Hata DJ, Reza M, et al. Disseminated Mycobacterium interjectum infection with bacteremia, hepatic and pulmonary involvement associated with a long-term catheter infection. Case Rep Infect Dis. 2017;2017:1-5.
- Dholakia YN. Mycobacterium interjectum isolated from an immunocompetent host with lung infection. Int J Mycobacteriol. 2017;6:401-403.
- Fukuoka M, Matsumura Y, Kore-eda S, et al. Cutaneous infection due to Mycobacterium interjectum in an immunosuppressed patient with microscopic polyangiitis. Br J Dermatol. 2008;159:1382-1384.
- Zanoni RG, Florio D, Fioravanti ML, et al. Occurrence of Mycobacterium spp. in ornamental fish in Italy. J Fish Dis. 2008;31:433-441.
- Emler S, Rochat T, Rohner P, et al. Chronic destructive lung disease associated with a novel mycobacterium. Am J Respir Crit Care Med. 1994;150:261-265.
To the Editor:
A 48-year-old man presented with nodular lesions in a sporotrichoid pattern on the right hand and forearm of 3 months’ duration (Figure). There were no lymphadeno-pathies, and he had no notable medical history. He denied fever and other systemic symptoms. The patient recently had manipulated a warm water fish aquarium. Although he did not recall a clear injury, inadvertent mild trauma was a possibility. He denied other contact or trauma in relation to animals or vegetables.
Histopathology from a punch biopsy of the forearm revealed a granulomatous infiltrate with necrosis at the deep dermis level at the interface with the subcutaneous cellular tissue that was composed of mainly epithelioid cells with a few multinucleated giant cells. No acid-fast bacilli or fungi were observed with special stains.
A polymerase chain reaction assay for atypical mycobacteria was positive for Mycobacterium interjectum. The culture of the skin biopsy was negative for fungi and mycobacteria after long incubation (6 weeks) on 2 occasions, and an antibiogram was not available. Complementary tests including hemogram, HIV serology, and chest and upper extremity radiographs did not reveal any abnormalities.
The patient was treated with rifampicin 600 mg/d, clarithromycin 500 mg every 12 hours, and co-trimoxazole 160/800 mg every 12 hours for 9 months with some resolution but persistence of some residual scarring lesions. There was no recurrence at 6-month follow-up.
Mycobacterium interjectum is a rare, slow-growing, scotochromogenic mycobacteria. Case reports usually refer to lymphadenitis in healthy children and pulmonary infections in immunocompromised or immunocompetent adults.1,2 A case of M interjectum with cutaneous involvement was reported by Fukuoka et al,3 with ulcerated nodules and abscesses on the leg identified in an immunocompromised patient. Our patient did not present with any cause of immunosuppression or clear injury predisposing him to infection. This microorganism has been detected in water, soil,3 and aquarium fish,4 the latter being the most likely source of infection in our patient. Given its slow growth rate and the need for a specific polymerase chain reaction assay, which is not widely available, M interjectum infection may be underdiagnosed.
No standard antibiotic regimen has been established, but M interjectum has proven to be a multidrug-resistant bacterium with frequent therapy failures. Treatment options have ranged from standard tuberculostatic therapy to combination therapy with medications such as amikacin, levofloxacin, rifampicin, and co-trimoxazole.1 Because an antibiogram was not available for our patient, empiric treatment with rifampicin, clarithromycin, and co-trimoxazole was prescribed for 9 months, with satisfactory response and tolerance. These drugs were selected because of their susceptibility profile in the literature.1,5
To the Editor:
A 48-year-old man presented with nodular lesions in a sporotrichoid pattern on the right hand and forearm of 3 months’ duration (Figure). There were no lymphadeno-pathies, and he had no notable medical history. He denied fever and other systemic symptoms. The patient recently had manipulated a warm water fish aquarium. Although he did not recall a clear injury, inadvertent mild trauma was a possibility. He denied other contact or trauma in relation to animals or vegetables.
Histopathology from a punch biopsy of the forearm revealed a granulomatous infiltrate with necrosis at the deep dermis level at the interface with the subcutaneous cellular tissue that was composed of mainly epithelioid cells with a few multinucleated giant cells. No acid-fast bacilli or fungi were observed with special stains.
A polymerase chain reaction assay for atypical mycobacteria was positive for Mycobacterium interjectum. The culture of the skin biopsy was negative for fungi and mycobacteria after long incubation (6 weeks) on 2 occasions, and an antibiogram was not available. Complementary tests including hemogram, HIV serology, and chest and upper extremity radiographs did not reveal any abnormalities.
The patient was treated with rifampicin 600 mg/d, clarithromycin 500 mg every 12 hours, and co-trimoxazole 160/800 mg every 12 hours for 9 months with some resolution but persistence of some residual scarring lesions. There was no recurrence at 6-month follow-up.
Mycobacterium interjectum is a rare, slow-growing, scotochromogenic mycobacteria. Case reports usually refer to lymphadenitis in healthy children and pulmonary infections in immunocompromised or immunocompetent adults.1,2 A case of M interjectum with cutaneous involvement was reported by Fukuoka et al,3 with ulcerated nodules and abscesses on the leg identified in an immunocompromised patient. Our patient did not present with any cause of immunosuppression or clear injury predisposing him to infection. This microorganism has been detected in water, soil,3 and aquarium fish,4 the latter being the most likely source of infection in our patient. Given its slow growth rate and the need for a specific polymerase chain reaction assay, which is not widely available, M interjectum infection may be underdiagnosed.
No standard antibiotic regimen has been established, but M interjectum has proven to be a multidrug-resistant bacterium with frequent therapy failures. Treatment options have ranged from standard tuberculostatic therapy to combination therapy with medications such as amikacin, levofloxacin, rifampicin, and co-trimoxazole.1 Because an antibiogram was not available for our patient, empiric treatment with rifampicin, clarithromycin, and co-trimoxazole was prescribed for 9 months, with satisfactory response and tolerance. These drugs were selected because of their susceptibility profile in the literature.1,5
- Sotello D, Hata DJ, Reza M, et al. Disseminated Mycobacterium interjectum infection with bacteremia, hepatic and pulmonary involvement associated with a long-term catheter infection. Case Rep Infect Dis. 2017;2017:1-5.
- Dholakia YN. Mycobacterium interjectum isolated from an immunocompetent host with lung infection. Int J Mycobacteriol. 2017;6:401-403.
- Fukuoka M, Matsumura Y, Kore-eda S, et al. Cutaneous infection due to Mycobacterium interjectum in an immunosuppressed patient with microscopic polyangiitis. Br J Dermatol. 2008;159:1382-1384.
- Zanoni RG, Florio D, Fioravanti ML, et al. Occurrence of Mycobacterium spp. in ornamental fish in Italy. J Fish Dis. 2008;31:433-441.
- Emler S, Rochat T, Rohner P, et al. Chronic destructive lung disease associated with a novel mycobacterium. Am J Respir Crit Care Med. 1994;150:261-265.
- Sotello D, Hata DJ, Reza M, et al. Disseminated Mycobacterium interjectum infection with bacteremia, hepatic and pulmonary involvement associated with a long-term catheter infection. Case Rep Infect Dis. 2017;2017:1-5.
- Dholakia YN. Mycobacterium interjectum isolated from an immunocompetent host with lung infection. Int J Mycobacteriol. 2017;6:401-403.
- Fukuoka M, Matsumura Y, Kore-eda S, et al. Cutaneous infection due to Mycobacterium interjectum in an immunosuppressed patient with microscopic polyangiitis. Br J Dermatol. 2008;159:1382-1384.
- Zanoni RG, Florio D, Fioravanti ML, et al. Occurrence of Mycobacterium spp. in ornamental fish in Italy. J Fish Dis. 2008;31:433-441.
- Emler S, Rochat T, Rohner P, et al. Chronic destructive lung disease associated with a novel mycobacterium. Am J Respir Crit Care Med. 1994;150:261-265.
Practice Points
- Mycobacterium interjectum can cause cutaneous nodules in a sporotrichoid or lymphocutaneous pattern and may affect immunocompromised and immunocompetent patients.
- This mycobacteria has been detected in water, soil, and aquarium fish. The latter could be a source of infection and should be taken into account in the anamnesis.
- There is no established therapeutic regimen for M interjectum infection. Combination therapy with rifampicin, clarithromycin, and co-trimoxazole could be an option, though it must always be adapted to an antibiogram if results are available.
Brazilian Peppertree: Watch Out for This Lesser-Known Relative of Poison Ivy
Brazilian peppertree (Schinus terebinthifolia), a member of the Anacardiaceae family, is an internationally invasive plant that causes allergic contact dermatitis (ACD) in susceptible individuals. This noxious weed has settled into the landscape of the southern United States and continues to expand. Its key identifying features include its year-round white flowers as well as a peppery and turpentinelike aroma created by cracking its bright red berries. The ACD associated with contact—primarily with the plant’s sap—stems from known alkenyl phenols, cardol and cardanol. Treatment of Brazilian peppertree–associated ACD parallels that for poison ivy. As this pest increases its range, dermatologists living in endemic areas should familiarize themselves with Brazilian peppertree and its potential for harm.
Brazilian Peppertree Morphology and Geography
Plants in the Anacardiaceae family contribute to more ACD than any other family, and its 80 genera include most of the urushiol-containing plants, such as Toxicodendron (poison ivy, poison oak, poison sumac, Japanese lacquer tree), Anacardium (cashew tree), Mangifera (mango fruit), Semecarpus (India marking nut tree), and Schinus (Brazilian peppertree). Deciduous and evergreen tree members of the Anacardiaceae family grow primarily in tropical and subtropical locations and produce thick resins, 5-petalled flowers, and small fruit known as drupes. The genus name for Brazilian peppertree, Schinus, derives from Latin and Greek words meaning “mastic tree,” a relative of the pistachio tree that the Brazilian peppertree resembles.1 Brazilian peppertree leaves look and smell similar to Pistacia terebinthus (turpentine tree or terebinth), from which the species name terebinthifolia derives.2
Brazilian peppertree originated in South America, particularly Brazil, Paraguay, and Argentina.3 Since the 1840s,4 it has been an invasive weed in the United States, notably in Florida, California, Hawaii, Alabama, Georgia,5 Arizona,6 Nevada,3 and Texas.5,7 The plant also grows throughout the world, including parts of Africa, Asia, Central America, Europe,6 New Zealand,8 Australia, and various islands.9 The plant expertly outcompetes neighboring plants and has prompted control and eradication efforts in many locations.3
Identifying Features and Allergenic Plant Parts
Brazilian peppertree can be either a shrub or tree up to 30 feet tall.4 As an evergreen, it retains its leaves year-round. During fruiting seasons (primarily December through March7), bright red or pink (depending on the variety3) berries appear (Figure 1A) and contribute to its nickname “Florida holly.” Although generally considered an unwelcome guest in Florida, it does display white flowers (Figure 1B) year-round, especially from September to November.9 It characteristically exhibits 3 to 13 leaflets per leaf.10 The leaflets’ ovoid and ridged edges, netlike vasculature, shiny hue, and aroma can help identify the plant (Figure 2A). For decades, the sap of the Brazilian peppertree has been associated with skin irritation (Figure 2B).6 Although the sap of the plant serves as the main culprit of Brazilian peppertree–associated ACD, it appears that other parts of the plant, including the fruit, can cause irritating effects to skin on contact.11,12 The leaves, trunk, and fruit can be harmful to both humans and animals.6 Chemicals from flowers and crushed fruit also can lead to irritating effects in the respiratory tract if aspirated.13


Urushiol, an oily resin present in most plants of the Anacardiaceae family,14 contains many chemicals, including allergenic phenols, catechols, and resorcinols.15 Urushiol-allergic individuals develop dermatitis upon exposure to Brazilian peppertree sap.6 Alkenyl phenols found in Brazilian peppertree lead to the cutaneous manifestations in sensitized patients.11,12 In 1983, Stahl et al11 identified a phenol, cardanol (chemical name 3-pentadecylphenol16) C15:1, in Brazilian peppertree fruit. The group further tested this compound’s effect on skin via patch testing, which showed an allergic response.11 Cashew nut shells (Anacardium occidentale) contain cardanol, anacardic acid (a phenolic acid), and cardol (a phenol with the chemical name 5-pentadecylresorcinol),15,16 though Stahl et al11 were unable to extract these 2 substances (if present) from Brazilian peppertree fruit. When exposed to cardol and anacardic acid, those allergic to poison ivy often develop ACD,15 and these 2 substances are more irritating than cardanol.11 A later study did identify cardol in addition to cardanol in Brazilian peppertree.12
Cutaneous Manifestations
Brazilian peppertree–induced ACD appears similar to other plant-induced ACD with linear streaks of erythema, juicy papules, vesicles, coalescing erythematous plaques, and/or occasional edema and bullae accompanied by intense pruritus.
Treatment
Avoiding contact with Brazilian peppertree is the first line of defense, and treatment for a reaction associated with exposure is similar to that of poison ivy.17 Application of cool compresses, calamine lotion, and topical astringents offer symptom alleviation, and topical steroids (eg, clobetasol propionate 0.05% twice daily) can improve mild localized ACD when given prior to formation of blisters. For more severe and diffuse ACD, oral steroids (eg, prednisone 1 mg/kg/d tapered over 2–3 weeks) likely are necessary, though intramuscular options greatly alleviate discomfort in more severe cases (eg, intramuscular triamcinolone acetonide 1 mg/kg combined with betamethasone 0.1 mg/kg). Physicians should monitor sites for any signs of superimposed bacterial infection and initiate antibiotics as necessary.17
- Zona S. The correct gender of Schinus (Anacardiaceae). Phytotaxa. 2015;222:075-077.
- Terebinth. Encyclopedia.com website. Updated May 17, 2018. Accessed July 9, 2024. https://www.encyclopedia.com/plants-and-animals/plants/plants/terebinth
- Brazilian pepper tree. iNaturalist website. Accessed July 1, 2024. https://www.inaturalist.org/guide_taxa/841531#:~:text=Throughout% 20South%20and%20Central%20America,and%20as%20a%20topical%20antiseptic
- Center for Aquatic and Invasive Plants. Schinus terebinthifolia. Brazilian peppertree. Accessed July 1, 2024. https://plants.ifas.ufl.edu/plant-directory/schinus-terebinthifolia/#:~:text=Species%20Overview&text=People%20sensitive%20to%20poison%20ivy,associated%20with%20its%20bloom%20period
- Brazilian peppertree (Schinus terebinthifolia). Early Detection & Distribution Mapping System. Accessed July 4, 2024. https://www.eddmaps.org/distribution/usstate.cfm?sub=78819
- Morton F. Brazilian pepper: its impact on people, animals, and the environment. Econ Bot. 1978;32:353-359.
- Fire Effects Information System. Schinus terebinthifolius. US Department of Agriculture website. Accessed July 4, 2024. https://www.fs.usda.gov/database/feis/plants/shrub/schter/all.html
- New Zealand Plant Conservation Network. Schinus terebinthifolius. Accessed July 1, 2024. https://www.nzpcn.org.nz/flora/species/schinus-terebinthifolius
- Rojas-Sandoval J, Acevedo-Rodriguez P. Schinus terebinthifolius (Brazilian pepper tree). CABI Compendium. July 23, 2014. Accessed July 1, 2024. https://www.cabidigitallibrary.org/doi/10.1079/cabicompendium.49031
- Patocka J, Diz de Almeida J. Brazilian peppertree: review of pharmacology. Mil Med Sci Lett. 2017;86:32-41.
- Stahl E, Keller K, Blinn C. Cardanol, a skin irritant in pink pepper. Plant Medica. 1983;48:5-9.
- Skopp G, Opferkuch H-J, Schqenker G. n-Alkylphenols from Schinus terebinthifolius Raddi (Anacardiaceae). In German. Zeitschrift für Naturforschung C. 1987;42:1-16. https://doi.org/10.1515/znc-1987-1-203.
- Lloyd HA, Jaouni TM, Evans SL, et al. Terpenes of Schinus terebinthifolius. Phytochemistry. 1977;16:1301-1302.
- Goon ATJ, Goh CL. Plant dermatitis: Asian perspective. Indian J Dermatol. 2011;56:707-710.
- Rozas-Muñoz E, Lepoittevin JP, Pujol RM, et al. Allergic contact dermatitis to plants: understanding the chemistry will help our diagnostic approach. Actas Dermosifiliogr. 2012;103:456-477.
- Caillol S. Cardanol: a promising building block for biobased polymers and additives. Curr Opin Green Sustain Chem. 2018;14: 26-32.
- Prok L, McGovern T. Poison ivy (Toxicodendron) dermatitis. UpToDate. Updated June 21, 2024. Accessed July 7, 2024. https://www.uptodate.com/contents/poison-ivy-toxicodendron-dermatitis#
Brazilian peppertree (Schinus terebinthifolia), a member of the Anacardiaceae family, is an internationally invasive plant that causes allergic contact dermatitis (ACD) in susceptible individuals. This noxious weed has settled into the landscape of the southern United States and continues to expand. Its key identifying features include its year-round white flowers as well as a peppery and turpentinelike aroma created by cracking its bright red berries. The ACD associated with contact—primarily with the plant’s sap—stems from known alkenyl phenols, cardol and cardanol. Treatment of Brazilian peppertree–associated ACD parallels that for poison ivy. As this pest increases its range, dermatologists living in endemic areas should familiarize themselves with Brazilian peppertree and its potential for harm.
Brazilian Peppertree Morphology and Geography
Plants in the Anacardiaceae family contribute to more ACD than any other family, and its 80 genera include most of the urushiol-containing plants, such as Toxicodendron (poison ivy, poison oak, poison sumac, Japanese lacquer tree), Anacardium (cashew tree), Mangifera (mango fruit), Semecarpus (India marking nut tree), and Schinus (Brazilian peppertree). Deciduous and evergreen tree members of the Anacardiaceae family grow primarily in tropical and subtropical locations and produce thick resins, 5-petalled flowers, and small fruit known as drupes. The genus name for Brazilian peppertree, Schinus, derives from Latin and Greek words meaning “mastic tree,” a relative of the pistachio tree that the Brazilian peppertree resembles.1 Brazilian peppertree leaves look and smell similar to Pistacia terebinthus (turpentine tree or terebinth), from which the species name terebinthifolia derives.2
Brazilian peppertree originated in South America, particularly Brazil, Paraguay, and Argentina.3 Since the 1840s,4 it has been an invasive weed in the United States, notably in Florida, California, Hawaii, Alabama, Georgia,5 Arizona,6 Nevada,3 and Texas.5,7 The plant also grows throughout the world, including parts of Africa, Asia, Central America, Europe,6 New Zealand,8 Australia, and various islands.9 The plant expertly outcompetes neighboring plants and has prompted control and eradication efforts in many locations.3
Identifying Features and Allergenic Plant Parts
Brazilian peppertree can be either a shrub or tree up to 30 feet tall.4 As an evergreen, it retains its leaves year-round. During fruiting seasons (primarily December through March7), bright red or pink (depending on the variety3) berries appear (Figure 1A) and contribute to its nickname “Florida holly.” Although generally considered an unwelcome guest in Florida, it does display white flowers (Figure 1B) year-round, especially from September to November.9 It characteristically exhibits 3 to 13 leaflets per leaf.10 The leaflets’ ovoid and ridged edges, netlike vasculature, shiny hue, and aroma can help identify the plant (Figure 2A). For decades, the sap of the Brazilian peppertree has been associated with skin irritation (Figure 2B).6 Although the sap of the plant serves as the main culprit of Brazilian peppertree–associated ACD, it appears that other parts of the plant, including the fruit, can cause irritating effects to skin on contact.11,12 The leaves, trunk, and fruit can be harmful to both humans and animals.6 Chemicals from flowers and crushed fruit also can lead to irritating effects in the respiratory tract if aspirated.13
Urushiol, an oily resin present in most plants of the Anacardiaceae family,14 contains many chemicals, including allergenic phenols, catechols, and resorcinols.15 Urushiol-allergic individuals develop dermatitis upon exposure to Brazilian peppertree sap.6 Alkenyl phenols found in Brazilian peppertree lead to the cutaneous manifestations in sensitized patients.11,12 In 1983, Stahl et al11 identified a phenol, cardanol (chemical name 3-pentadecylphenol16) C15:1, in Brazilian peppertree fruit. The group further tested this compound’s effect on skin via patch testing, which showed an allergic response.11 Cashew nut shells (Anacardium occidentale) contain cardanol, anacardic acid (a phenolic acid), and cardol (a phenol with the chemical name 5-pentadecylresorcinol),15,16 though Stahl et al11 were unable to extract these 2 substances (if present) from Brazilian peppertree fruit. When exposed to cardol and anacardic acid, those allergic to poison ivy often develop ACD,15 and these 2 substances are more irritating than cardanol.11 A later study did identify cardol in addition to cardanol in Brazilian peppertree.12
Cutaneous Manifestations
Brazilian peppertree–induced ACD appears similar to other plant-induced ACD with linear streaks of erythema, juicy papules, vesicles, coalescing erythematous plaques, and/or occasional edema and bullae accompanied by intense pruritus.
Treatment
Avoiding contact with Brazilian peppertree is the first line of defense, and treatment for a reaction associated with exposure is similar to that of poison ivy.17 Application of cool compresses, calamine lotion, and topical astringents offer symptom alleviation, and topical steroids (eg, clobetasol propionate 0.05% twice daily) can improve mild localized ACD when given prior to formation of blisters. For more severe and diffuse ACD, oral steroids (eg, prednisone 1 mg/kg/d tapered over 2–3 weeks) likely are necessary, though intramuscular options greatly alleviate discomfort in more severe cases (eg, intramuscular triamcinolone acetonide 1 mg/kg combined with betamethasone 0.1 mg/kg). Physicians should monitor sites for any signs of superimposed bacterial infection and initiate antibiotics as necessary.17
Brazilian peppertree (Schinus terebinthifolia), a member of the Anacardiaceae family, is an internationally invasive plant that causes allergic contact dermatitis (ACD) in susceptible individuals. This noxious weed has settled into the landscape of the southern United States and continues to expand. Its key identifying features include its year-round white flowers as well as a peppery and turpentinelike aroma created by cracking its bright red berries. The ACD associated with contact—primarily with the plant’s sap—stems from known alkenyl phenols, cardol and cardanol. Treatment of Brazilian peppertree–associated ACD parallels that for poison ivy. As this pest increases its range, dermatologists living in endemic areas should familiarize themselves with Brazilian peppertree and its potential for harm.
Brazilian Peppertree Morphology and Geography
Plants in the Anacardiaceae family contribute to more ACD than any other family, and its 80 genera include most of the urushiol-containing plants, such as Toxicodendron (poison ivy, poison oak, poison sumac, Japanese lacquer tree), Anacardium (cashew tree), Mangifera (mango fruit), Semecarpus (India marking nut tree), and Schinus (Brazilian peppertree). Deciduous and evergreen tree members of the Anacardiaceae family grow primarily in tropical and subtropical locations and produce thick resins, 5-petalled flowers, and small fruit known as drupes. The genus name for Brazilian peppertree, Schinus, derives from Latin and Greek words meaning “mastic tree,” a relative of the pistachio tree that the Brazilian peppertree resembles.1 Brazilian peppertree leaves look and smell similar to Pistacia terebinthus (turpentine tree or terebinth), from which the species name terebinthifolia derives.2
Brazilian peppertree originated in South America, particularly Brazil, Paraguay, and Argentina.3 Since the 1840s,4 it has been an invasive weed in the United States, notably in Florida, California, Hawaii, Alabama, Georgia,5 Arizona,6 Nevada,3 and Texas.5,7 The plant also grows throughout the world, including parts of Africa, Asia, Central America, Europe,6 New Zealand,8 Australia, and various islands.9 The plant expertly outcompetes neighboring plants and has prompted control and eradication efforts in many locations.3
Identifying Features and Allergenic Plant Parts
Brazilian peppertree can be either a shrub or tree up to 30 feet tall.4 As an evergreen, it retains its leaves year-round. During fruiting seasons (primarily December through March7), bright red or pink (depending on the variety3) berries appear (Figure 1A) and contribute to its nickname “Florida holly.” Although generally considered an unwelcome guest in Florida, it does display white flowers (Figure 1B) year-round, especially from September to November.9 It characteristically exhibits 3 to 13 leaflets per leaf.10 The leaflets’ ovoid and ridged edges, netlike vasculature, shiny hue, and aroma can help identify the plant (Figure 2A). For decades, the sap of the Brazilian peppertree has been associated with skin irritation (Figure 2B).6 Although the sap of the plant serves as the main culprit of Brazilian peppertree–associated ACD, it appears that other parts of the plant, including the fruit, can cause irritating effects to skin on contact.11,12 The leaves, trunk, and fruit can be harmful to both humans and animals.6 Chemicals from flowers and crushed fruit also can lead to irritating effects in the respiratory tract if aspirated.13
Urushiol, an oily resin present in most plants of the Anacardiaceae family,14 contains many chemicals, including allergenic phenols, catechols, and resorcinols.15 Urushiol-allergic individuals develop dermatitis upon exposure to Brazilian peppertree sap.6 Alkenyl phenols found in Brazilian peppertree lead to the cutaneous manifestations in sensitized patients.11,12 In 1983, Stahl et al11 identified a phenol, cardanol (chemical name 3-pentadecylphenol16) C15:1, in Brazilian peppertree fruit. The group further tested this compound’s effect on skin via patch testing, which showed an allergic response.11 Cashew nut shells (Anacardium occidentale) contain cardanol, anacardic acid (a phenolic acid), and cardol (a phenol with the chemical name 5-pentadecylresorcinol),15,16 though Stahl et al11 were unable to extract these 2 substances (if present) from Brazilian peppertree fruit. When exposed to cardol and anacardic acid, those allergic to poison ivy often develop ACD,15 and these 2 substances are more irritating than cardanol.11 A later study did identify cardol in addition to cardanol in Brazilian peppertree.12
Cutaneous Manifestations
Brazilian peppertree–induced ACD appears similar to other plant-induced ACD with linear streaks of erythema, juicy papules, vesicles, coalescing erythematous plaques, and/or occasional edema and bullae accompanied by intense pruritus.
Treatment
Avoiding contact with Brazilian peppertree is the first line of defense, and treatment for a reaction associated with exposure is similar to that of poison ivy.17 Application of cool compresses, calamine lotion, and topical astringents offer symptom alleviation, and topical steroids (eg, clobetasol propionate 0.05% twice daily) can improve mild localized ACD when given prior to formation of blisters. For more severe and diffuse ACD, oral steroids (eg, prednisone 1 mg/kg/d tapered over 2–3 weeks) likely are necessary, though intramuscular options greatly alleviate discomfort in more severe cases (eg, intramuscular triamcinolone acetonide 1 mg/kg combined with betamethasone 0.1 mg/kg). Physicians should monitor sites for any signs of superimposed bacterial infection and initiate antibiotics as necessary.17
- Zona S. The correct gender of Schinus (Anacardiaceae). Phytotaxa. 2015;222:075-077.
- Terebinth. Encyclopedia.com website. Updated May 17, 2018. Accessed July 9, 2024. https://www.encyclopedia.com/plants-and-animals/plants/plants/terebinth
- Brazilian pepper tree. iNaturalist website. Accessed July 1, 2024. https://www.inaturalist.org/guide_taxa/841531#:~:text=Throughout% 20South%20and%20Central%20America,and%20as%20a%20topical%20antiseptic
- Center for Aquatic and Invasive Plants. Schinus terebinthifolia. Brazilian peppertree. Accessed July 1, 2024. https://plants.ifas.ufl.edu/plant-directory/schinus-terebinthifolia/#:~:text=Species%20Overview&text=People%20sensitive%20to%20poison%20ivy,associated%20with%20its%20bloom%20period
- Brazilian peppertree (Schinus terebinthifolia). Early Detection & Distribution Mapping System. Accessed July 4, 2024. https://www.eddmaps.org/distribution/usstate.cfm?sub=78819
- Morton F. Brazilian pepper: its impact on people, animals, and the environment. Econ Bot. 1978;32:353-359.
- Fire Effects Information System. Schinus terebinthifolius. US Department of Agriculture website. Accessed July 4, 2024. https://www.fs.usda.gov/database/feis/plants/shrub/schter/all.html
- New Zealand Plant Conservation Network. Schinus terebinthifolius. Accessed July 1, 2024. https://www.nzpcn.org.nz/flora/species/schinus-terebinthifolius
- Rojas-Sandoval J, Acevedo-Rodriguez P. Schinus terebinthifolius (Brazilian pepper tree). CABI Compendium. July 23, 2014. Accessed July 1, 2024. https://www.cabidigitallibrary.org/doi/10.1079/cabicompendium.49031
- Patocka J, Diz de Almeida J. Brazilian peppertree: review of pharmacology. Mil Med Sci Lett. 2017;86:32-41.
- Stahl E, Keller K, Blinn C. Cardanol, a skin irritant in pink pepper. Plant Medica. 1983;48:5-9.
- Skopp G, Opferkuch H-J, Schqenker G. n-Alkylphenols from Schinus terebinthifolius Raddi (Anacardiaceae). In German. Zeitschrift für Naturforschung C. 1987;42:1-16. https://doi.org/10.1515/znc-1987-1-203.
- Lloyd HA, Jaouni TM, Evans SL, et al. Terpenes of Schinus terebinthifolius. Phytochemistry. 1977;16:1301-1302.
- Goon ATJ, Goh CL. Plant dermatitis: Asian perspective. Indian J Dermatol. 2011;56:707-710.
- Rozas-Muñoz E, Lepoittevin JP, Pujol RM, et al. Allergic contact dermatitis to plants: understanding the chemistry will help our diagnostic approach. Actas Dermosifiliogr. 2012;103:456-477.
- Caillol S. Cardanol: a promising building block for biobased polymers and additives. Curr Opin Green Sustain Chem. 2018;14: 26-32.
- Prok L, McGovern T. Poison ivy (Toxicodendron) dermatitis. UpToDate. Updated June 21, 2024. Accessed July 7, 2024. https://www.uptodate.com/contents/poison-ivy-toxicodendron-dermatitis#
- Zona S. The correct gender of Schinus (Anacardiaceae). Phytotaxa. 2015;222:075-077.
- Terebinth. Encyclopedia.com website. Updated May 17, 2018. Accessed July 9, 2024. https://www.encyclopedia.com/plants-and-animals/plants/plants/terebinth
- Brazilian pepper tree. iNaturalist website. Accessed July 1, 2024. https://www.inaturalist.org/guide_taxa/841531#:~:text=Throughout% 20South%20and%20Central%20America,and%20as%20a%20topical%20antiseptic
- Center for Aquatic and Invasive Plants. Schinus terebinthifolia. Brazilian peppertree. Accessed July 1, 2024. https://plants.ifas.ufl.edu/plant-directory/schinus-terebinthifolia/#:~:text=Species%20Overview&text=People%20sensitive%20to%20poison%20ivy,associated%20with%20its%20bloom%20period
- Brazilian peppertree (Schinus terebinthifolia). Early Detection & Distribution Mapping System. Accessed July 4, 2024. https://www.eddmaps.org/distribution/usstate.cfm?sub=78819
- Morton F. Brazilian pepper: its impact on people, animals, and the environment. Econ Bot. 1978;32:353-359.
- Fire Effects Information System. Schinus terebinthifolius. US Department of Agriculture website. Accessed July 4, 2024. https://www.fs.usda.gov/database/feis/plants/shrub/schter/all.html
- New Zealand Plant Conservation Network. Schinus terebinthifolius. Accessed July 1, 2024. https://www.nzpcn.org.nz/flora/species/schinus-terebinthifolius
- Rojas-Sandoval J, Acevedo-Rodriguez P. Schinus terebinthifolius (Brazilian pepper tree). CABI Compendium. July 23, 2014. Accessed July 1, 2024. https://www.cabidigitallibrary.org/doi/10.1079/cabicompendium.49031
- Patocka J, Diz de Almeida J. Brazilian peppertree: review of pharmacology. Mil Med Sci Lett. 2017;86:32-41.
- Stahl E, Keller K, Blinn C. Cardanol, a skin irritant in pink pepper. Plant Medica. 1983;48:5-9.
- Skopp G, Opferkuch H-J, Schqenker G. n-Alkylphenols from Schinus terebinthifolius Raddi (Anacardiaceae). In German. Zeitschrift für Naturforschung C. 1987;42:1-16. https://doi.org/10.1515/znc-1987-1-203.
- Lloyd HA, Jaouni TM, Evans SL, et al. Terpenes of Schinus terebinthifolius. Phytochemistry. 1977;16:1301-1302.
- Goon ATJ, Goh CL. Plant dermatitis: Asian perspective. Indian J Dermatol. 2011;56:707-710.
- Rozas-Muñoz E, Lepoittevin JP, Pujol RM, et al. Allergic contact dermatitis to plants: understanding the chemistry will help our diagnostic approach. Actas Dermosifiliogr. 2012;103:456-477.
- Caillol S. Cardanol: a promising building block for biobased polymers and additives. Curr Opin Green Sustain Chem. 2018;14: 26-32.
- Prok L, McGovern T. Poison ivy (Toxicodendron) dermatitis. UpToDate. Updated June 21, 2024. Accessed July 7, 2024. https://www.uptodate.com/contents/poison-ivy-toxicodendron-dermatitis#
Practice Points
- The Anacardiaceae family contains several plants, including Brazilian peppertree and poison ivy, that have the potential to cause allergic contact dermatitis (ACD).
- Hot spots for Brazilian peppertree include Florida and California, though it also has been reported in Texas, Hawaii, Georgia, Alabama, Arkansas, Nevada, and Arizona.
- Alkenyl phenols (eg, cardol, cardanol) are the key sensitizers found in Brazilian peppertree.
- Treatment consists of supportive care and either topical, oral, or intramuscular steroids depending on the extent and severity of the ACD.

Barriers to Mohs Micrographic Surgery in Japanese Patients With Basal Cell Carcinoma
Margin-controlled surgery for squamous cell carcinoma (SCC) on the lower lip was first performed by Dr. Frederic Mohs on June 30, 1936. Since then, thousands of skin cancer surgeons have refined and adopted the technique. Due to the high cure rate and sparing of normal tissue, Mohs micrographic surgery (MMS) has become the gold standard treatment for facial and special-site nonmelanoma skin cancer worldwide. Mohs micrographic surgery is performed on more than 876,000 tumors annually in the United States.1 Among 3.5 million Americans diagnosed with nonmelanoma skin cancer in 2006, one-quarter were treated with MMS.2 In Japan, basal cell carcinoma (BCC) is the most common skin malignancy, with an incidence of 3.34 cases per 100,000 individuals; SCC is the second most common, with an incidence of 2.5 cases per 100,000 individuals.3
The essential element that makes MMS unique is the careful microscopic examination of the entire margin of the removed specimen. Tissue processing is done with careful en face orientation to ensure that circumferential and deep margins are entirely visible. The surgeon interprets the slides and proceeds to remove the additional tumor as necessary. Because the same physician performs both the surgery and the pathologic assessment throughout the procedure, a precise correlation between the microscopic and surgical findings can be made. The surgeon can begin with smaller margins, removing minimal healthy tissue while removing all the cancer cells, which results in the smallest-possible skin defect and the best prognosis for the malignancy (Figure 1).
At the only facility in Japan offering MMS, the lead author (S.S.) has treated 52 lesions with MMS in 46 patients (2020-2022). Of these patients, 40 were White, 5 were Japanese, and 1 was of African descent. In this case series, we present 5 Japanese patients who had BCC treated with MMS.
Case Series
Patient 1—A 50-year-old Japanese woman presented to dermatology with a brown papule on the nasal tip of 1.25 year’s duration (Figure 2). A biopsy revealed infiltrative BCC (Figure 3), and the patient was referred to the dermatology department at a nearby university hospital. Because the BCC was an aggressive variant, wide local excision (WLE) with subsequent flap reconstruction was recommended as well as radiation therapy. The patient learned about MMS through an internet search and refused both options, seeking MMS treatment at our clinic. Although Japanese health insurance does not cover MMS, the patient had supplemental private insurance that did cover the cost. She provided consent to undergo the procedure. Physical examination revealed a 7.5×6-mm, brown-red macule with ill-defined borders on the tip of the nose. We used a 1.5-mm margin for the first stage of MMS (Figure 4A). The frozen section revealed that the tumor had been entirely excised in the first stage, leaving only a 10.5×9-mm skin defect that was reconstructed with a Dufourmentel flap (Figure 4B). No signs of recurrence were noted at 3.5-year follow-up, and the cosmetic outcome was favorable (Figure 4C). National Comprehensive Cancer Network guidelines recommend a margin greater than 4 mm for infiltrative BCCs4; therefore, our technique reduced the total defect by at least 4 mm in a cosmetically sensitive area. The patient also did not need radiation therapy, which reduced morbidity. She continues to be recurrence free at 3.5-year follow-up.
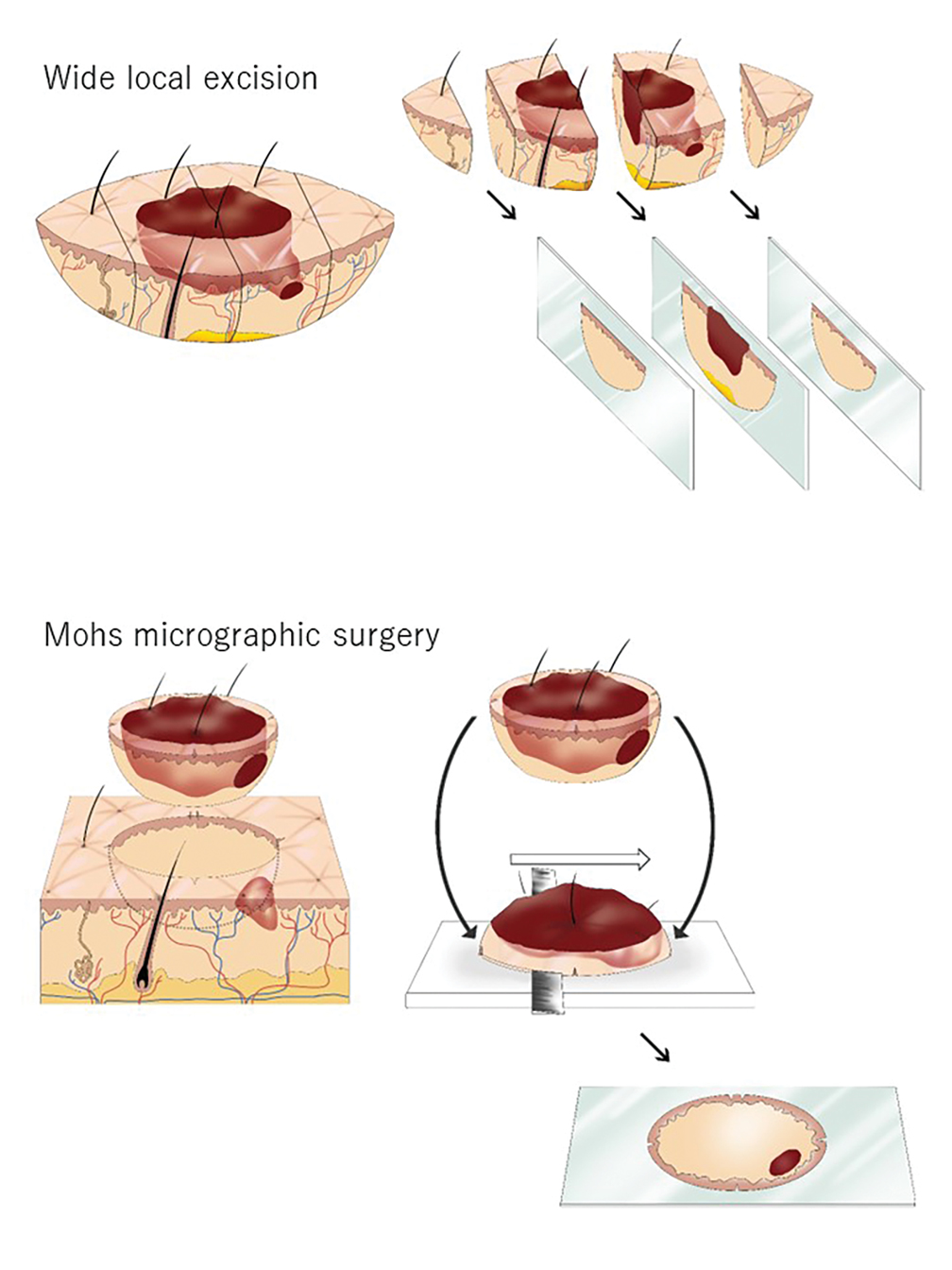
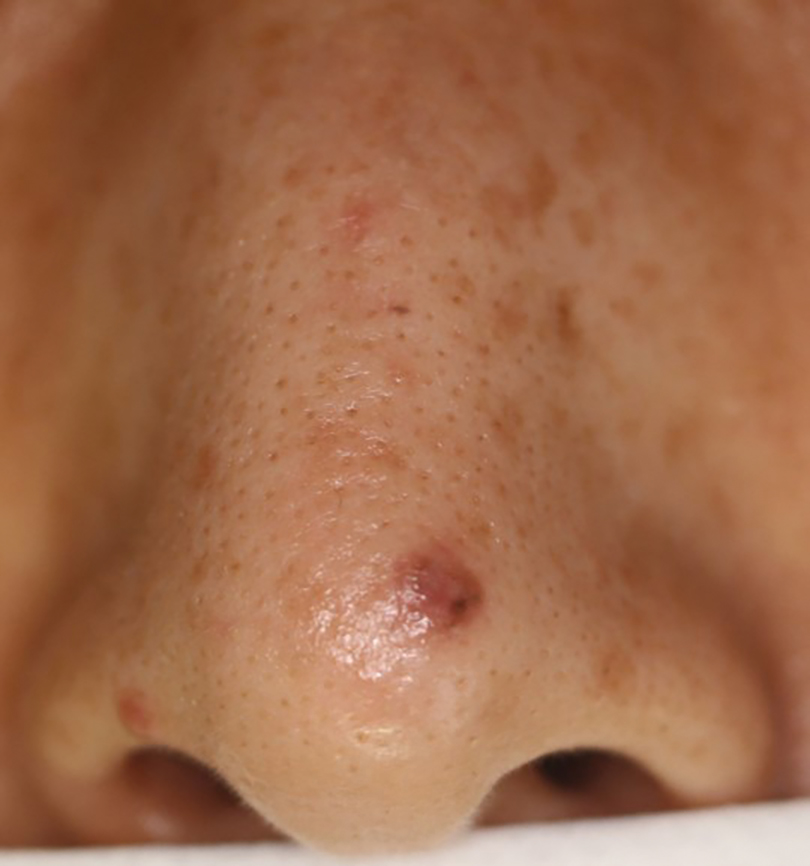
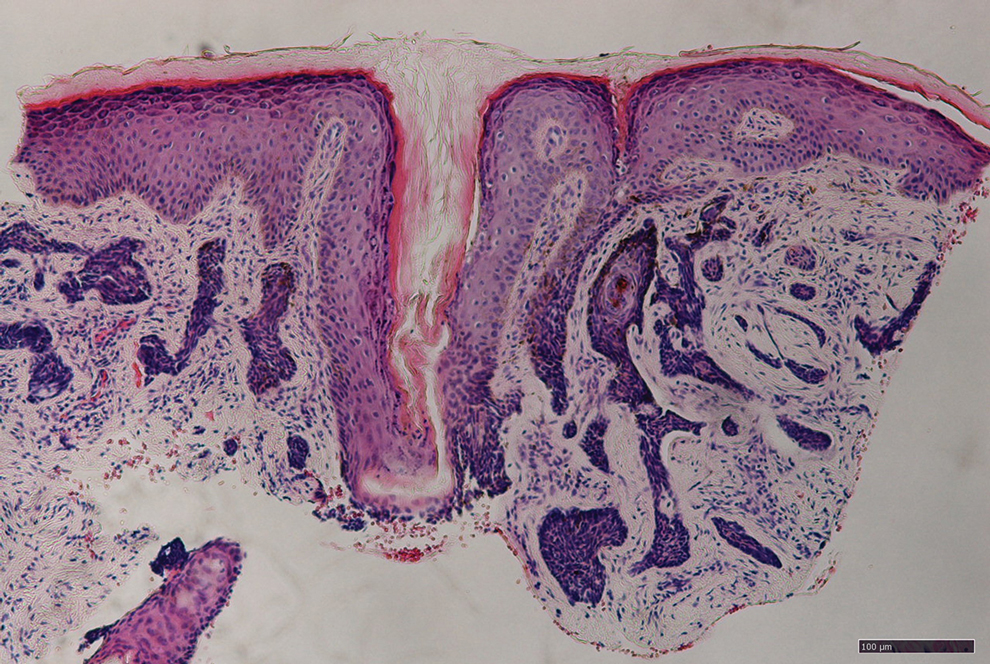
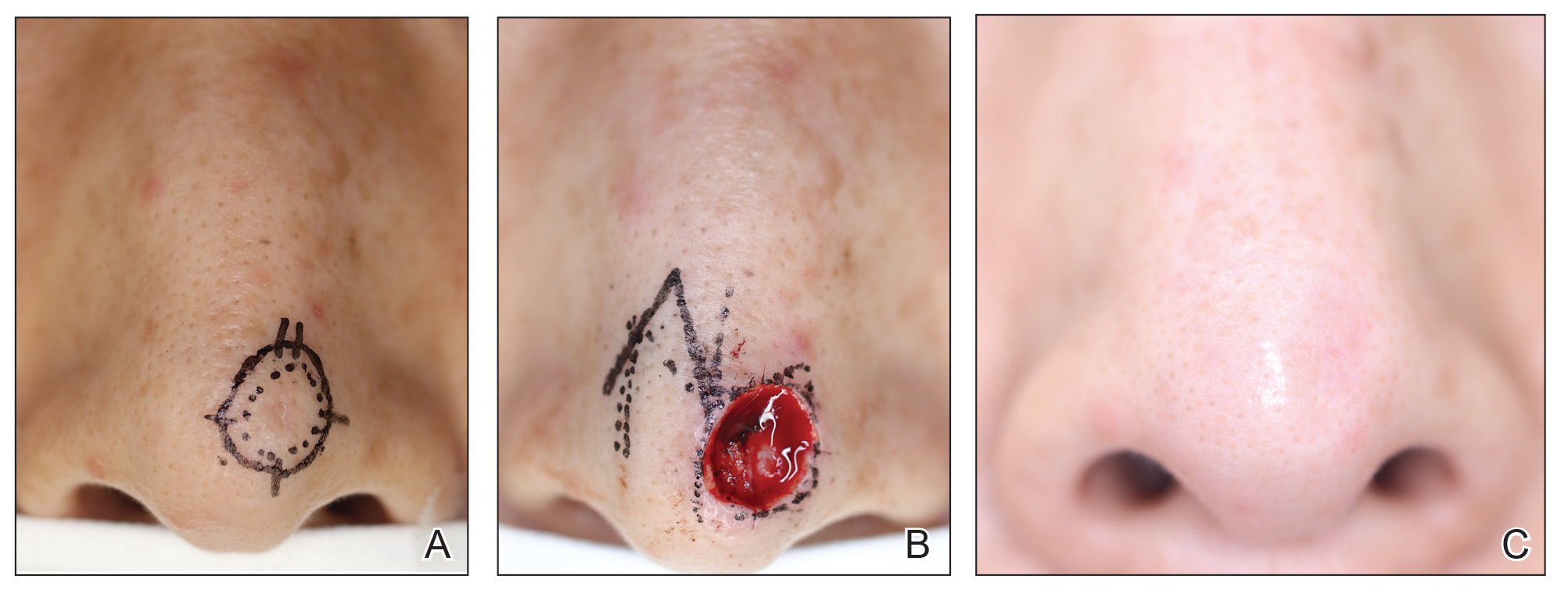
Patient 2—A 63-year-old Japanese man presented to dermatology with a brown macule on the right lower eyelid of 2 years’ duration. A biopsy of the lesion was positive for nodular BCC. After being advised to undergo WLE and extensive reconstruction with plastic surgery, the patient learned of MMS through an internet search and found our clinic. Physical examination revealed a 7×5-mm brown macule on the right lower eyelid. The patient had supplemental private insurance that covered the cost of MMS, and he provided consent for the procedure. A 1.5-mm margin was taken for the first stage, resulting in a 10×8-mm defect superficial to the orbicularis oculi muscle. The frozen section revealed residual tumor exposure in the dermis at the 9- to 10-o’clock position. A second-stage excision was performed to remove an additional 1.5 mm of skin at the 9- to 12-o’clock position with a thin layer of the orbicularis oculi muscle. The subsequent histologic examination revealed no residual BCC, and the final 13×9-mm skin defect was reconstructed with a rotation flap. There were no signs of recurrence at 2.5-year follow-up with an excellent cosmetic outcome.
Patient 3—A 73-year-old Japanese man presented to a local university dermatology clinic with a new papule on the nose. The dermatologist suggested WLE with 4-mm margins and reconstruction of the skin defect 2 weeks later by a plastic surgeon. The patient was not satisfied with the proposed surgical plan, which led him to learn about MMS on the internet; he subsequently found our clinic. Physical examination revealed a 4×3.5-mm brown papule on the tip of the nose. He understood the nature of MMS and chose to pay out-of-pocket because Japanese health insurance did not cover the procedure. We used a 2-mm margin for the first stage, which created a 7.5×7-mm skin defect. The frozen section pathology revealed no residual BCC at the cut surface. The skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Patient 4—A 45-year-old man presented to a dermatology clinic with a papule on the right side of the nose of 1 year’s duration. A biopsy revealed the lesion was a nodular BCC. The dermatologist recommended WLE at a general hospital, but the patient refused after learning about MMS. He subsequently made an appointment with our clinic. Physical examination revealed a 7×4-mm white papule on the right side of the nose. The patient had private insurance that covered the cost of MMS. The first stage was performed with 1.5-mm margins and was clear of residual tumor. A Limberg rhombic flap from the adjacent cheek was used to repair the final 10×7-mm skin defect. There were no signs of recurrence at 1 year and 9 months’ follow-up with a favorable cosmetic outcome.
Patient 5—A 76-year-old Japanese woman presented to a university hospital near Tokyo with a black papule on the left cutaneous lip of 5 years’ duration. A biopsy revealed nodular BCC, and WLE with flap reconstruction was recommended. The patient’s son learned about MMS through internet research and referred her to our clinic. Physical examination revealed a 7×5-mm black papule on the left upper lip. The patient’s private insurance covered the cost of MMS, and she consented to the procedure. We used a 2-mm initial margin, and the immediate frozen section revealed no signs of BCC at the cut surface. The 11×9-mm skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Comment
We presented 5 cases of MMS in Japanese patients with BCC. More than 7000 new cases of nonmelanoma skin cancer occur every year in Japan.3 Only 0.04% of these cases—the 5 cases presented here—were treated with MMS in Japan in 2020 and 2021, in contrast to 25% in the United States in 2006.2
MMS vs Other BCC Treatments—Mohs micrographic surgery offers 2 distinct advantages over conventional excision: an improved cure rate while achieving a smaller final defect size, generally leading to better cosmetic outcomes. Overall 5-year recurrence rates of BCC are 10% for conventional surgical excision vs 1% for MMS, while the recurrence rates for SCC are 8% and 3%, respectively.5 A study of well-demarcated BCCs smaller than 2 cm that were treated with MMS with 2-mm increments revealed that 95% of the cases were free of malignancy within a 4-mm margin of the normal-appearing skin surrounding the tumor.6 Several articles have reported a 95% cure rate or higher with conventional excision of localized BCC,7 but 4- to 5-mm excision margins are required, resulting in a greater skin defect and a lower cure rate compared to MMS.
Aggressive subtypes of BCC have a higher recurrence rate. Rowe et al8 reported the following 5-year recurrence rates: 5.6% for MMS, 17.4% for conventional surgical excision, 40.0% for curettage and electrodesiccation, and 9.8% for radiation therapy. Primary BCCs with high-risk histologic subtypes has a 10-year recurrence rate of 4.4% with MMS vs 12.2% with conventional excision.9 These findings reveal that MMS yields a better prognosis compared to traditional treatment methods for recurrent BCCs and BCCs of high-risk histologic subtypes.
The primary reason for the excellent cure rate seen in MMS is the ability to perform complete margin assessment. Peripheral and deep en face margin assessment (PDEMA) is crucial in achieving high cure rates with narrow margins. In WLE (Figure 1), vertical sectioning (also known as bread-loafing) does not achieve direct visualization of the entire surgical margin, as this technique only evaluates random sections and does not achieve PDEMA.10 The bread-loafing method is used almost exclusively in Japan and visualizes only 0.1% of the entire margin compared to 100% with MMS.11 Beyond the superior cure rate, the MMS technique often yields smaller final defects compared to WLE. All 5 of our patients achieved complete tumor removal while sparing more normal tissue compared to conventional WLE, which takes at least a 4-mm margin in all directions.
Barriers to Adopting MMS in Japan—There are many barriers to the broader adoption of MMS in Japan. A guideline of the Japanese Dermatological Association says, MMS “is complicated, requires special training for acquisition, and requires time and labor for implementation of a series of processes, and it has not gained wide acceptance in Japan because of these disadvantages.”3 There currently are no MMS training programs in Japan. We refute this statement from the Japanese Dermatological Association because, in our experience, only 1 surgeon plus a single histotechnician familiar with MMS is sufficient for a facility to offer the procedure (the lead author of this study [S.S.] acts as both the surgeon and the histotechnician). Another misconception among some physicians in Japan is that cancer on ethnically Japanese skin is uniquely suited to excision without microscopic verification of tumor clearance because the borders of the tumors are easily identified, which was based on good cure rates for the excision of well-demarcated pigmented BCCs in a Japanese cohort. This study of a Japanese cohort investigated the specimens with the conventional bread-loafing technique but not with the PDEMA.12
Eighty percent (4/5) of our patients presented with nodular BCC, and only 1 required a second stage. In comparison, we also treated 16 White patients with nodular BCC with MMS during the same period, and 31% (5/16) required more than 1 stage, with 1 patient requiring 3 stages. This cohort, however, is too small to demonstrate a statistically significant difference (S.S., unpublished data, 2020-2022).
A study in Singapore reported the postsurgical complication rate and 5-year recurrence rate for 481 tumors (92% BCC and 7.5% SCC). The median follow-up duration after MMS was 36 months, and the recurrence rate was 0.6%. The postsurgical complications included 11 (2.3%) cases with superficial tip necrosis of surgical flaps/grafts, 2 (0.4%) with mild wound dehiscence, 1 (0.2%) with minor surgical site bleeding, and 1 (0.2%) with minor wound infection.13 This study supports the notion that MMS is equally effective for Asian patients.
Awareness of MMS in Japan is lacking, and most Japanese dermatologists do not know about the technique. All 5 patients in our case series asked their dermatologists about alternative treatment options and were not offered MMS. In each case, the patients learned of the technique through internet research.
The lack of insurance reimbursement for MMS in Japan is another barrier. Because the national health insurance does not reimburse for MMS, the procedure is relatively unavailable to most Japanese citizens who cannot pay out-of-pocket for the treatment and do not have supplemental insurance. Mohs micrographic surgery may seem expensive compared to WLE followed by repair; however, in the authors’ experience, in Japan, excision without MMS may require general sedation and multiple surgeries to reconstruct larger skin defects, leading to greater morbidity and risk for the patient.
Conclusion
Mohs micrographic surgery in Japan is in its infancy, and further studies showing recurrence rates and long-term prognosis are needed. Such data should help increase awareness of MMS among Japanese physicians as an excellent treatment option for their patients. Furthermore, as Japan becomes more heterogenous as a society and the US Military increases its presence in the region, the need for MMS is likely to increase.
Acknowledgments—We appreciate the proofreading support by Mark Bivens, MBA, MSc (Tokyo, Japan), as well as the technical support from Ben Tallon, MBChB, and Robyn Mason (both in Tauranga, New Zealand) to start MMS at our clinic.
- Asgari MM, Olson J, Alam M. Needs assessment for Mohs micrographic surgery. Dermatol Clin. 2012;30:167-175. doi:10.1016/j.det.2011.08.010
- Connolly SM, Baker DR, Baker DR, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Ansai SI, Umebayashi Y, Katsumata N, et al. Japanese Dermatological Association Guidelines: outlines of guidelines for cutaneous squamous cell carcinoma 2020. J Dermatol. 2021;48:E288-E311.
- Schmults CD, Blitzblau R, Aasi SZ, et at. Basal cell skin cancer, version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21:1181-1203. doi:10.6004/jncn.2023.0056
- Snow SN, Gunkel J. Mohs surgery. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:2445-2455. doi:10.1016/b978-0-070-94171-3.00041-7
- Wolf DJ, Zitelli JA. Surgical margins for basal cell carcinoma. Arch Dermatol. 1987;123:340-344.
- Quazi SJ, Aslam N, Saleem H, et al. Surgical margin of excision in basal cell carcinoma: a systematic review of literature. Cureus. 2020;12:E9211.
- Rowe DE, Carroll RJ, Day Jus CL. Mohs surgery is the treatment of choice for recurrent (previously treated) basal cell carcinoma. J Dermatol Surg Oncol. 1989;15:424-431.
- Van Loo, Mosterd K, Krekels GA. Surgical excision versus Mohs’ micrographic surgery for basal cell carcinoma of the face. Eur J Cancer. 2014;50:3011-3020.
- Schmults CD, Blitzblau R, Aasi SZ, et al. NCCN Guidelines Insights: Squamous Cell Skin Cancer, Version 1.2022. J Natl Compr Canc Netw. 2021;19:1382-1394.
- Hui AM, Jacobson M, Markowitz O, et al. Mohs micrographic surgery for the treatment of melanoma. Dermatol Clin. 2012;30:503-515.
- Ito T, Inatomi Y, Nagae K, et al. Narrow-margin excision is a safe, reliable treatment for well-defined, primary pigmented basal cell carcinoma: an analysis of 288 lesions in Japan. J Eur Acad Dermatol Venereol. 2015;29:1828-1831.
- Ho WYB, Zhao X, Tan WPM. Mohs micrographic surgery in Singapore: a long-term follow-up review. Ann Acad Med Singap. 2021;50:922-923.
Margin-controlled surgery for squamous cell carcinoma (SCC) on the lower lip was first performed by Dr. Frederic Mohs on June 30, 1936. Since then, thousands of skin cancer surgeons have refined and adopted the technique. Due to the high cure rate and sparing of normal tissue, Mohs micrographic surgery (MMS) has become the gold standard treatment for facial and special-site nonmelanoma skin cancer worldwide. Mohs micrographic surgery is performed on more than 876,000 tumors annually in the United States.1 Among 3.5 million Americans diagnosed with nonmelanoma skin cancer in 2006, one-quarter were treated with MMS.2 In Japan, basal cell carcinoma (BCC) is the most common skin malignancy, with an incidence of 3.34 cases per 100,000 individuals; SCC is the second most common, with an incidence of 2.5 cases per 100,000 individuals.3
The essential element that makes MMS unique is the careful microscopic examination of the entire margin of the removed specimen. Tissue processing is done with careful en face orientation to ensure that circumferential and deep margins are entirely visible. The surgeon interprets the slides and proceeds to remove the additional tumor as necessary. Because the same physician performs both the surgery and the pathologic assessment throughout the procedure, a precise correlation between the microscopic and surgical findings can be made. The surgeon can begin with smaller margins, removing minimal healthy tissue while removing all the cancer cells, which results in the smallest-possible skin defect and the best prognosis for the malignancy (Figure 1).
At the only facility in Japan offering MMS, the lead author (S.S.) has treated 52 lesions with MMS in 46 patients (2020-2022). Of these patients, 40 were White, 5 were Japanese, and 1 was of African descent. In this case series, we present 5 Japanese patients who had BCC treated with MMS.
Case Series
Patient 1—A 50-year-old Japanese woman presented to dermatology with a brown papule on the nasal tip of 1.25 year’s duration (Figure 2). A biopsy revealed infiltrative BCC (Figure 3), and the patient was referred to the dermatology department at a nearby university hospital. Because the BCC was an aggressive variant, wide local excision (WLE) with subsequent flap reconstruction was recommended as well as radiation therapy. The patient learned about MMS through an internet search and refused both options, seeking MMS treatment at our clinic. Although Japanese health insurance does not cover MMS, the patient had supplemental private insurance that did cover the cost. She provided consent to undergo the procedure. Physical examination revealed a 7.5×6-mm, brown-red macule with ill-defined borders on the tip of the nose. We used a 1.5-mm margin for the first stage of MMS (Figure 4A). The frozen section revealed that the tumor had been entirely excised in the first stage, leaving only a 10.5×9-mm skin defect that was reconstructed with a Dufourmentel flap (Figure 4B). No signs of recurrence were noted at 3.5-year follow-up, and the cosmetic outcome was favorable (Figure 4C). National Comprehensive Cancer Network guidelines recommend a margin greater than 4 mm for infiltrative BCCs4; therefore, our technique reduced the total defect by at least 4 mm in a cosmetically sensitive area. The patient also did not need radiation therapy, which reduced morbidity. She continues to be recurrence free at 3.5-year follow-up.
Patient 2—A 63-year-old Japanese man presented to dermatology with a brown macule on the right lower eyelid of 2 years’ duration. A biopsy of the lesion was positive for nodular BCC. After being advised to undergo WLE and extensive reconstruction with plastic surgery, the patient learned of MMS through an internet search and found our clinic. Physical examination revealed a 7×5-mm brown macule on the right lower eyelid. The patient had supplemental private insurance that covered the cost of MMS, and he provided consent for the procedure. A 1.5-mm margin was taken for the first stage, resulting in a 10×8-mm defect superficial to the orbicularis oculi muscle. The frozen section revealed residual tumor exposure in the dermis at the 9- to 10-o’clock position. A second-stage excision was performed to remove an additional 1.5 mm of skin at the 9- to 12-o’clock position with a thin layer of the orbicularis oculi muscle. The subsequent histologic examination revealed no residual BCC, and the final 13×9-mm skin defect was reconstructed with a rotation flap. There were no signs of recurrence at 2.5-year follow-up with an excellent cosmetic outcome.
Patient 3—A 73-year-old Japanese man presented to a local university dermatology clinic with a new papule on the nose. The dermatologist suggested WLE with 4-mm margins and reconstruction of the skin defect 2 weeks later by a plastic surgeon. The patient was not satisfied with the proposed surgical plan, which led him to learn about MMS on the internet; he subsequently found our clinic. Physical examination revealed a 4×3.5-mm brown papule on the tip of the nose. He understood the nature of MMS and chose to pay out-of-pocket because Japanese health insurance did not cover the procedure. We used a 2-mm margin for the first stage, which created a 7.5×7-mm skin defect. The frozen section pathology revealed no residual BCC at the cut surface. The skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Patient 4—A 45-year-old man presented to a dermatology clinic with a papule on the right side of the nose of 1 year’s duration. A biopsy revealed the lesion was a nodular BCC. The dermatologist recommended WLE at a general hospital, but the patient refused after learning about MMS. He subsequently made an appointment with our clinic. Physical examination revealed a 7×4-mm white papule on the right side of the nose. The patient had private insurance that covered the cost of MMS. The first stage was performed with 1.5-mm margins and was clear of residual tumor. A Limberg rhombic flap from the adjacent cheek was used to repair the final 10×7-mm skin defect. There were no signs of recurrence at 1 year and 9 months’ follow-up with a favorable cosmetic outcome.
Patient 5—A 76-year-old Japanese woman presented to a university hospital near Tokyo with a black papule on the left cutaneous lip of 5 years’ duration. A biopsy revealed nodular BCC, and WLE with flap reconstruction was recommended. The patient’s son learned about MMS through internet research and referred her to our clinic. Physical examination revealed a 7×5-mm black papule on the left upper lip. The patient’s private insurance covered the cost of MMS, and she consented to the procedure. We used a 2-mm initial margin, and the immediate frozen section revealed no signs of BCC at the cut surface. The 11×9-mm skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Comment
We presented 5 cases of MMS in Japanese patients with BCC. More than 7000 new cases of nonmelanoma skin cancer occur every year in Japan.3 Only 0.04% of these cases—the 5 cases presented here—were treated with MMS in Japan in 2020 and 2021, in contrast to 25% in the United States in 2006.2
MMS vs Other BCC Treatments—Mohs micrographic surgery offers 2 distinct advantages over conventional excision: an improved cure rate while achieving a smaller final defect size, generally leading to better cosmetic outcomes. Overall 5-year recurrence rates of BCC are 10% for conventional surgical excision vs 1% for MMS, while the recurrence rates for SCC are 8% and 3%, respectively.5 A study of well-demarcated BCCs smaller than 2 cm that were treated with MMS with 2-mm increments revealed that 95% of the cases were free of malignancy within a 4-mm margin of the normal-appearing skin surrounding the tumor.6 Several articles have reported a 95% cure rate or higher with conventional excision of localized BCC,7 but 4- to 5-mm excision margins are required, resulting in a greater skin defect and a lower cure rate compared to MMS.
Aggressive subtypes of BCC have a higher recurrence rate. Rowe et al8 reported the following 5-year recurrence rates: 5.6% for MMS, 17.4% for conventional surgical excision, 40.0% for curettage and electrodesiccation, and 9.8% for radiation therapy. Primary BCCs with high-risk histologic subtypes has a 10-year recurrence rate of 4.4% with MMS vs 12.2% with conventional excision.9 These findings reveal that MMS yields a better prognosis compared to traditional treatment methods for recurrent BCCs and BCCs of high-risk histologic subtypes.
The primary reason for the excellent cure rate seen in MMS is the ability to perform complete margin assessment. Peripheral and deep en face margin assessment (PDEMA) is crucial in achieving high cure rates with narrow margins. In WLE (Figure 1), vertical sectioning (also known as bread-loafing) does not achieve direct visualization of the entire surgical margin, as this technique only evaluates random sections and does not achieve PDEMA.10 The bread-loafing method is used almost exclusively in Japan and visualizes only 0.1% of the entire margin compared to 100% with MMS.11 Beyond the superior cure rate, the MMS technique often yields smaller final defects compared to WLE. All 5 of our patients achieved complete tumor removal while sparing more normal tissue compared to conventional WLE, which takes at least a 4-mm margin in all directions.
Barriers to Adopting MMS in Japan—There are many barriers to the broader adoption of MMS in Japan. A guideline of the Japanese Dermatological Association says, MMS “is complicated, requires special training for acquisition, and requires time and labor for implementation of a series of processes, and it has not gained wide acceptance in Japan because of these disadvantages.”3 There currently are no MMS training programs in Japan. We refute this statement from the Japanese Dermatological Association because, in our experience, only 1 surgeon plus a single histotechnician familiar with MMS is sufficient for a facility to offer the procedure (the lead author of this study [S.S.] acts as both the surgeon and the histotechnician). Another misconception among some physicians in Japan is that cancer on ethnically Japanese skin is uniquely suited to excision without microscopic verification of tumor clearance because the borders of the tumors are easily identified, which was based on good cure rates for the excision of well-demarcated pigmented BCCs in a Japanese cohort. This study of a Japanese cohort investigated the specimens with the conventional bread-loafing technique but not with the PDEMA.12
Eighty percent (4/5) of our patients presented with nodular BCC, and only 1 required a second stage. In comparison, we also treated 16 White patients with nodular BCC with MMS during the same period, and 31% (5/16) required more than 1 stage, with 1 patient requiring 3 stages. This cohort, however, is too small to demonstrate a statistically significant difference (S.S., unpublished data, 2020-2022).
A study in Singapore reported the postsurgical complication rate and 5-year recurrence rate for 481 tumors (92% BCC and 7.5% SCC). The median follow-up duration after MMS was 36 months, and the recurrence rate was 0.6%. The postsurgical complications included 11 (2.3%) cases with superficial tip necrosis of surgical flaps/grafts, 2 (0.4%) with mild wound dehiscence, 1 (0.2%) with minor surgical site bleeding, and 1 (0.2%) with minor wound infection.13 This study supports the notion that MMS is equally effective for Asian patients.
Awareness of MMS in Japan is lacking, and most Japanese dermatologists do not know about the technique. All 5 patients in our case series asked their dermatologists about alternative treatment options and were not offered MMS. In each case, the patients learned of the technique through internet research.
The lack of insurance reimbursement for MMS in Japan is another barrier. Because the national health insurance does not reimburse for MMS, the procedure is relatively unavailable to most Japanese citizens who cannot pay out-of-pocket for the treatment and do not have supplemental insurance. Mohs micrographic surgery may seem expensive compared to WLE followed by repair; however, in the authors’ experience, in Japan, excision without MMS may require general sedation and multiple surgeries to reconstruct larger skin defects, leading to greater morbidity and risk for the patient.
Conclusion
Mohs micrographic surgery in Japan is in its infancy, and further studies showing recurrence rates and long-term prognosis are needed. Such data should help increase awareness of MMS among Japanese physicians as an excellent treatment option for their patients. Furthermore, as Japan becomes more heterogenous as a society and the US Military increases its presence in the region, the need for MMS is likely to increase.
Acknowledgments—We appreciate the proofreading support by Mark Bivens, MBA, MSc (Tokyo, Japan), as well as the technical support from Ben Tallon, MBChB, and Robyn Mason (both in Tauranga, New Zealand) to start MMS at our clinic.
Margin-controlled surgery for squamous cell carcinoma (SCC) on the lower lip was first performed by Dr. Frederic Mohs on June 30, 1936. Since then, thousands of skin cancer surgeons have refined and adopted the technique. Due to the high cure rate and sparing of normal tissue, Mohs micrographic surgery (MMS) has become the gold standard treatment for facial and special-site nonmelanoma skin cancer worldwide. Mohs micrographic surgery is performed on more than 876,000 tumors annually in the United States.1 Among 3.5 million Americans diagnosed with nonmelanoma skin cancer in 2006, one-quarter were treated with MMS.2 In Japan, basal cell carcinoma (BCC) is the most common skin malignancy, with an incidence of 3.34 cases per 100,000 individuals; SCC is the second most common, with an incidence of 2.5 cases per 100,000 individuals.3
The essential element that makes MMS unique is the careful microscopic examination of the entire margin of the removed specimen. Tissue processing is done with careful en face orientation to ensure that circumferential and deep margins are entirely visible. The surgeon interprets the slides and proceeds to remove the additional tumor as necessary. Because the same physician performs both the surgery and the pathologic assessment throughout the procedure, a precise correlation between the microscopic and surgical findings can be made. The surgeon can begin with smaller margins, removing minimal healthy tissue while removing all the cancer cells, which results in the smallest-possible skin defect and the best prognosis for the malignancy (Figure 1).
At the only facility in Japan offering MMS, the lead author (S.S.) has treated 52 lesions with MMS in 46 patients (2020-2022). Of these patients, 40 were White, 5 were Japanese, and 1 was of African descent. In this case series, we present 5 Japanese patients who had BCC treated with MMS.
Case Series
Patient 1—A 50-year-old Japanese woman presented to dermatology with a brown papule on the nasal tip of 1.25 year’s duration (Figure 2). A biopsy revealed infiltrative BCC (Figure 3), and the patient was referred to the dermatology department at a nearby university hospital. Because the BCC was an aggressive variant, wide local excision (WLE) with subsequent flap reconstruction was recommended as well as radiation therapy. The patient learned about MMS through an internet search and refused both options, seeking MMS treatment at our clinic. Although Japanese health insurance does not cover MMS, the patient had supplemental private insurance that did cover the cost. She provided consent to undergo the procedure. Physical examination revealed a 7.5×6-mm, brown-red macule with ill-defined borders on the tip of the nose. We used a 1.5-mm margin for the first stage of MMS (Figure 4A). The frozen section revealed that the tumor had been entirely excised in the first stage, leaving only a 10.5×9-mm skin defect that was reconstructed with a Dufourmentel flap (Figure 4B). No signs of recurrence were noted at 3.5-year follow-up, and the cosmetic outcome was favorable (Figure 4C). National Comprehensive Cancer Network guidelines recommend a margin greater than 4 mm for infiltrative BCCs4; therefore, our technique reduced the total defect by at least 4 mm in a cosmetically sensitive area. The patient also did not need radiation therapy, which reduced morbidity. She continues to be recurrence free at 3.5-year follow-up.
Patient 2—A 63-year-old Japanese man presented to dermatology with a brown macule on the right lower eyelid of 2 years’ duration. A biopsy of the lesion was positive for nodular BCC. After being advised to undergo WLE and extensive reconstruction with plastic surgery, the patient learned of MMS through an internet search and found our clinic. Physical examination revealed a 7×5-mm brown macule on the right lower eyelid. The patient had supplemental private insurance that covered the cost of MMS, and he provided consent for the procedure. A 1.5-mm margin was taken for the first stage, resulting in a 10×8-mm defect superficial to the orbicularis oculi muscle. The frozen section revealed residual tumor exposure in the dermis at the 9- to 10-o’clock position. A second-stage excision was performed to remove an additional 1.5 mm of skin at the 9- to 12-o’clock position with a thin layer of the orbicularis oculi muscle. The subsequent histologic examination revealed no residual BCC, and the final 13×9-mm skin defect was reconstructed with a rotation flap. There were no signs of recurrence at 2.5-year follow-up with an excellent cosmetic outcome.
Patient 3—A 73-year-old Japanese man presented to a local university dermatology clinic with a new papule on the nose. The dermatologist suggested WLE with 4-mm margins and reconstruction of the skin defect 2 weeks later by a plastic surgeon. The patient was not satisfied with the proposed surgical plan, which led him to learn about MMS on the internet; he subsequently found our clinic. Physical examination revealed a 4×3.5-mm brown papule on the tip of the nose. He understood the nature of MMS and chose to pay out-of-pocket because Japanese health insurance did not cover the procedure. We used a 2-mm margin for the first stage, which created a 7.5×7-mm skin defect. The frozen section pathology revealed no residual BCC at the cut surface. The skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Patient 4—A 45-year-old man presented to a dermatology clinic with a papule on the right side of the nose of 1 year’s duration. A biopsy revealed the lesion was a nodular BCC. The dermatologist recommended WLE at a general hospital, but the patient refused after learning about MMS. He subsequently made an appointment with our clinic. Physical examination revealed a 7×4-mm white papule on the right side of the nose. The patient had private insurance that covered the cost of MMS. The first stage was performed with 1.5-mm margins and was clear of residual tumor. A Limberg rhombic flap from the adjacent cheek was used to repair the final 10×7-mm skin defect. There were no signs of recurrence at 1 year and 9 months’ follow-up with a favorable cosmetic outcome.
Patient 5—A 76-year-old Japanese woman presented to a university hospital near Tokyo with a black papule on the left cutaneous lip of 5 years’ duration. A biopsy revealed nodular BCC, and WLE with flap reconstruction was recommended. The patient’s son learned about MMS through internet research and referred her to our clinic. Physical examination revealed a 7×5-mm black papule on the left upper lip. The patient’s private insurance covered the cost of MMS, and she consented to the procedure. We used a 2-mm initial margin, and the immediate frozen section revealed no signs of BCC at the cut surface. The 11×9-mm skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Comment
We presented 5 cases of MMS in Japanese patients with BCC. More than 7000 new cases of nonmelanoma skin cancer occur every year in Japan.3 Only 0.04% of these cases—the 5 cases presented here—were treated with MMS in Japan in 2020 and 2021, in contrast to 25% in the United States in 2006.2
MMS vs Other BCC Treatments—Mohs micrographic surgery offers 2 distinct advantages over conventional excision: an improved cure rate while achieving a smaller final defect size, generally leading to better cosmetic outcomes. Overall 5-year recurrence rates of BCC are 10% for conventional surgical excision vs 1% for MMS, while the recurrence rates for SCC are 8% and 3%, respectively.5 A study of well-demarcated BCCs smaller than 2 cm that were treated with MMS with 2-mm increments revealed that 95% of the cases were free of malignancy within a 4-mm margin of the normal-appearing skin surrounding the tumor.6 Several articles have reported a 95% cure rate or higher with conventional excision of localized BCC,7 but 4- to 5-mm excision margins are required, resulting in a greater skin defect and a lower cure rate compared to MMS.
Aggressive subtypes of BCC have a higher recurrence rate. Rowe et al8 reported the following 5-year recurrence rates: 5.6% for MMS, 17.4% for conventional surgical excision, 40.0% for curettage and electrodesiccation, and 9.8% for radiation therapy. Primary BCCs with high-risk histologic subtypes has a 10-year recurrence rate of 4.4% with MMS vs 12.2% with conventional excision.9 These findings reveal that MMS yields a better prognosis compared to traditional treatment methods for recurrent BCCs and BCCs of high-risk histologic subtypes.
The primary reason for the excellent cure rate seen in MMS is the ability to perform complete margin assessment. Peripheral and deep en face margin assessment (PDEMA) is crucial in achieving high cure rates with narrow margins. In WLE (Figure 1), vertical sectioning (also known as bread-loafing) does not achieve direct visualization of the entire surgical margin, as this technique only evaluates random sections and does not achieve PDEMA.10 The bread-loafing method is used almost exclusively in Japan and visualizes only 0.1% of the entire margin compared to 100% with MMS.11 Beyond the superior cure rate, the MMS technique often yields smaller final defects compared to WLE. All 5 of our patients achieved complete tumor removal while sparing more normal tissue compared to conventional WLE, which takes at least a 4-mm margin in all directions.
Barriers to Adopting MMS in Japan—There are many barriers to the broader adoption of MMS in Japan. A guideline of the Japanese Dermatological Association says, MMS “is complicated, requires special training for acquisition, and requires time and labor for implementation of a series of processes, and it has not gained wide acceptance in Japan because of these disadvantages.”3 There currently are no MMS training programs in Japan. We refute this statement from the Japanese Dermatological Association because, in our experience, only 1 surgeon plus a single histotechnician familiar with MMS is sufficient for a facility to offer the procedure (the lead author of this study [S.S.] acts as both the surgeon and the histotechnician). Another misconception among some physicians in Japan is that cancer on ethnically Japanese skin is uniquely suited to excision without microscopic verification of tumor clearance because the borders of the tumors are easily identified, which was based on good cure rates for the excision of well-demarcated pigmented BCCs in a Japanese cohort. This study of a Japanese cohort investigated the specimens with the conventional bread-loafing technique but not with the PDEMA.12
Eighty percent (4/5) of our patients presented with nodular BCC, and only 1 required a second stage. In comparison, we also treated 16 White patients with nodular BCC with MMS during the same period, and 31% (5/16) required more than 1 stage, with 1 patient requiring 3 stages. This cohort, however, is too small to demonstrate a statistically significant difference (S.S., unpublished data, 2020-2022).
A study in Singapore reported the postsurgical complication rate and 5-year recurrence rate for 481 tumors (92% BCC and 7.5% SCC). The median follow-up duration after MMS was 36 months, and the recurrence rate was 0.6%. The postsurgical complications included 11 (2.3%) cases with superficial tip necrosis of surgical flaps/grafts, 2 (0.4%) with mild wound dehiscence, 1 (0.2%) with minor surgical site bleeding, and 1 (0.2%) with minor wound infection.13 This study supports the notion that MMS is equally effective for Asian patients.
Awareness of MMS in Japan is lacking, and most Japanese dermatologists do not know about the technique. All 5 patients in our case series asked their dermatologists about alternative treatment options and were not offered MMS. In each case, the patients learned of the technique through internet research.
The lack of insurance reimbursement for MMS in Japan is another barrier. Because the national health insurance does not reimburse for MMS, the procedure is relatively unavailable to most Japanese citizens who cannot pay out-of-pocket for the treatment and do not have supplemental insurance. Mohs micrographic surgery may seem expensive compared to WLE followed by repair; however, in the authors’ experience, in Japan, excision without MMS may require general sedation and multiple surgeries to reconstruct larger skin defects, leading to greater morbidity and risk for the patient.
Conclusion
Mohs micrographic surgery in Japan is in its infancy, and further studies showing recurrence rates and long-term prognosis are needed. Such data should help increase awareness of MMS among Japanese physicians as an excellent treatment option for their patients. Furthermore, as Japan becomes more heterogenous as a society and the US Military increases its presence in the region, the need for MMS is likely to increase.
Acknowledgments—We appreciate the proofreading support by Mark Bivens, MBA, MSc (Tokyo, Japan), as well as the technical support from Ben Tallon, MBChB, and Robyn Mason (both in Tauranga, New Zealand) to start MMS at our clinic.
- Asgari MM, Olson J, Alam M. Needs assessment for Mohs micrographic surgery. Dermatol Clin. 2012;30:167-175. doi:10.1016/j.det.2011.08.010
- Connolly SM, Baker DR, Baker DR, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Ansai SI, Umebayashi Y, Katsumata N, et al. Japanese Dermatological Association Guidelines: outlines of guidelines for cutaneous squamous cell carcinoma 2020. J Dermatol. 2021;48:E288-E311.
- Schmults CD, Blitzblau R, Aasi SZ, et at. Basal cell skin cancer, version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21:1181-1203. doi:10.6004/jncn.2023.0056
- Snow SN, Gunkel J. Mohs surgery. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:2445-2455. doi:10.1016/b978-0-070-94171-3.00041-7
- Wolf DJ, Zitelli JA. Surgical margins for basal cell carcinoma. Arch Dermatol. 1987;123:340-344.
- Quazi SJ, Aslam N, Saleem H, et al. Surgical margin of excision in basal cell carcinoma: a systematic review of literature. Cureus. 2020;12:E9211.
- Rowe DE, Carroll RJ, Day Jus CL. Mohs surgery is the treatment of choice for recurrent (previously treated) basal cell carcinoma. J Dermatol Surg Oncol. 1989;15:424-431.
- Van Loo, Mosterd K, Krekels GA. Surgical excision versus Mohs’ micrographic surgery for basal cell carcinoma of the face. Eur J Cancer. 2014;50:3011-3020.
- Schmults CD, Blitzblau R, Aasi SZ, et al. NCCN Guidelines Insights: Squamous Cell Skin Cancer, Version 1.2022. J Natl Compr Canc Netw. 2021;19:1382-1394.
- Hui AM, Jacobson M, Markowitz O, et al. Mohs micrographic surgery for the treatment of melanoma. Dermatol Clin. 2012;30:503-515.
- Ito T, Inatomi Y, Nagae K, et al. Narrow-margin excision is a safe, reliable treatment for well-defined, primary pigmented basal cell carcinoma: an analysis of 288 lesions in Japan. J Eur Acad Dermatol Venereol. 2015;29:1828-1831.
- Ho WYB, Zhao X, Tan WPM. Mohs micrographic surgery in Singapore: a long-term follow-up review. Ann Acad Med Singap. 2021;50:922-923.
- Asgari MM, Olson J, Alam M. Needs assessment for Mohs micrographic surgery. Dermatol Clin. 2012;30:167-175. doi:10.1016/j.det.2011.08.010
- Connolly SM, Baker DR, Baker DR, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Ansai SI, Umebayashi Y, Katsumata N, et al. Japanese Dermatological Association Guidelines: outlines of guidelines for cutaneous squamous cell carcinoma 2020. J Dermatol. 2021;48:E288-E311.
- Schmults CD, Blitzblau R, Aasi SZ, et at. Basal cell skin cancer, version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21:1181-1203. doi:10.6004/jncn.2023.0056
- Snow SN, Gunkel J. Mohs surgery. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:2445-2455. doi:10.1016/b978-0-070-94171-3.00041-7
- Wolf DJ, Zitelli JA. Surgical margins for basal cell carcinoma. Arch Dermatol. 1987;123:340-344.
- Quazi SJ, Aslam N, Saleem H, et al. Surgical margin of excision in basal cell carcinoma: a systematic review of literature. Cureus. 2020;12:E9211.
- Rowe DE, Carroll RJ, Day Jus CL. Mohs surgery is the treatment of choice for recurrent (previously treated) basal cell carcinoma. J Dermatol Surg Oncol. 1989;15:424-431.
- Van Loo, Mosterd K, Krekels GA. Surgical excision versus Mohs’ micrographic surgery for basal cell carcinoma of the face. Eur J Cancer. 2014;50:3011-3020.
- Schmults CD, Blitzblau R, Aasi SZ, et al. NCCN Guidelines Insights: Squamous Cell Skin Cancer, Version 1.2022. J Natl Compr Canc Netw. 2021;19:1382-1394.
- Hui AM, Jacobson M, Markowitz O, et al. Mohs micrographic surgery for the treatment of melanoma. Dermatol Clin. 2012;30:503-515.
- Ito T, Inatomi Y, Nagae K, et al. Narrow-margin excision is a safe, reliable treatment for well-defined, primary pigmented basal cell carcinoma: an analysis of 288 lesions in Japan. J Eur Acad Dermatol Venereol. 2015;29:1828-1831.
- Ho WYB, Zhao X, Tan WPM. Mohs micrographic surgery in Singapore: a long-term follow-up review. Ann Acad Med Singap. 2021;50:922-923.
Practice Points
- Mohs micrographic surgery (MMS) is a safe and effective treatment method for nonmelanoma skin cancer. In some cases, this procedure is superior to standard wide local excision and repair.
- For the broader adaptation of this vital technique in Japan—where MMS is not well established—increased awareness of treatment outcomes among Japanese physicians is needed.
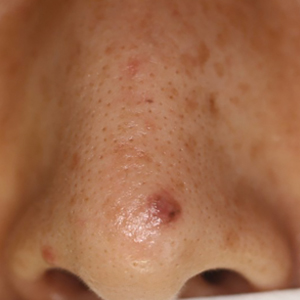
Tackling Inflammatory and Infectious Nail Disorders in Children
Nail disorders are common among pediatric patients but often are underdiagnosed or misdiagnosed because of their unique disease manifestations. These conditions may severely impact quality of life. There are few nail disease clinical trials that include children. Consequently, most treatment recommendations are based on case series and expert consensus recommendations. We review inflammatory and infectious nail disorders in pediatric patients. By describing characteristics, clinical manifestations, and management approaches for these conditions, we aim to provide guidance to dermatologists in their diagnosis and treatment.
INFLAMMATORY NAIL DISORDERS
Nail Psoriasis
Nail involvement in children with psoriasis is common, with prevalence estimates ranging from 17% to 39.2%.1 Nail matrix psoriasis may manifest with pitting (large irregular pits) and leukonychia as well as chromonychia and nail plate crumbling. Onycholysis, oil drop spots (salmon patches), and subungual hyperkeratosis can be seen in nail bed psoriasis. Nail pitting is the most frequently observed clinical finding (Figure 1).2,3 In a cross-sectional multicenter study of 313 children with cutaneous psoriasis in France, nail findings were present in 101 patients (32.3%). There were associations between nail findings and presence of psoriatic arthritis (P=.03), palmoplantar psoriasis (P<.001), and severity of psoriatic disease, defined as use of systemic treatment with phototherapy (psoralen plus UVA, UVB), traditional systemic treatment (acitretin, methotrexate, cyclosporine), or a biologic (P=.003).4
Topical steroids and vitamin D analogues may be used with or without occlusion and may be efficacious.5 Several case reports describe systemic treatments for psoriasis in children, including methotrexate, acitretin, and apremilast (approved for children 6 years and older for plaque psoriasis by the US Food and Drug Administration [FDA]).2 There are 5 biologic drugs currently approved for the treatment of pediatric psoriasis—adalimumab, etanercept, ustekinumab, secukinumab, ixekizumab—and 6 drugs currently undergoing phase 3 studies—brodalumab, guselkumab, risankizumab, tildrakizumab, certolizumab pegol, and deucravacitinib (Table 1).6-15 Adalimumab is specifically approved for moderate to severe nail psoriasis in adults 18 years and older.
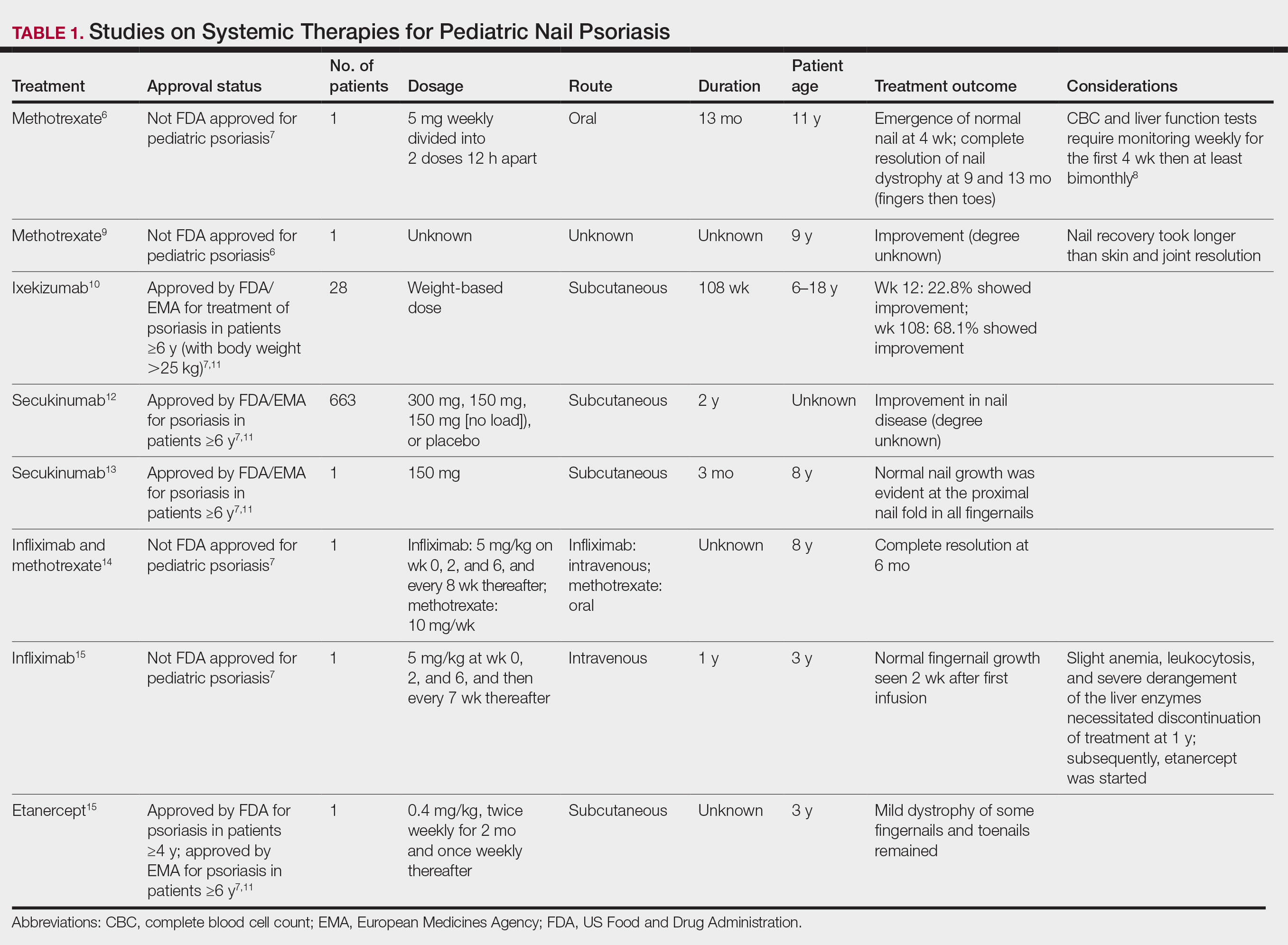
Intralesional steroid injections are sometimes useful in the management of nail matrix psoriasis; however, appropriate patient selection is critical due to the pain associated with the procedure. In a prospective study of 16 children (age range, 9–17 years) with nail psoriasis treated with intralesional triamcinolone (ILTAC) 2.5 to 5 mg/mL every 4 to 8 weeks for a minimum of 3 to 6 months, 9 patients achieved resolution and 6 had improvement of clinical findings.16 Local adverse events were mild, including injection-site pain (66%), subungual hematoma (n=1), Beau lines (n=1), proximal nail fold hypopigmentation (n=2), and proximal nail fold atrophy (n=2). Because the proximal nail fold in children is thinner than in adults, there may be an increased risk for nail fold hypopigmentation and atrophy in children. Therefore, a maximum ILTAC concentration of 2.5 mg/mL with 0.2 mL maximum volume per nail per session is recommended for children younger than 15 years.16
Nail Lichen Planus
Nail lichen planus (NLP) is uncommon in children, with few biopsy-proven cases documented in the literature.17 Common clinical findings are onychorrhexis, nail plate thinning, fissuring, splitting, and atrophy with koilonychia.5 Although pterygium development (irreversible nail matrix scarring) is uncommon in pediatric patients, NLP can be progressive and may cause irreversible destruction of the nail matrix and subsequent nail loss, warranting therapeutic intervention.18
Treatment of NLP may be difficult, as there are no options that work in all patients. Current literature supports the use of systemic corticosteroids or ILTAC for the treatment of NLP; however, recurrence rates can be high. According to an expert consensus paper on NLP treatment, ILTAC may be injected in a concentration of 2.5, 5, or 10 mg/mL according to disease severity.19 In severe or resistant cases, intramuscular (IM) triamcinolone may be considered, especially if more than 3 nails are affected. A dosage of 0.5 to 1 mg/kg/mo for at least 3 to 6 months is recommended for both children and adults, with 1 mg/kg/mo recommended in the active treatment phase (first 2–3 months).19 In a retrospective review of 5 pediatric patients with NLP treated with IM triamcinolone 0.5 mg/kg/mo, 3 patients had resolution and 2 improved with treatment.20 In a prospective study of 10 children with NLP, IM triamcinolone at a dosage of 0.5 to 1 mg/kg every 30 days for 3 to 6 months resulted in resolution of nail findings in 9 patients.17 In a prospective study of 14 pediatric patients with NLP treated with 2.5 to 5 mg/mL of ILTAC, 10 achieved resolution and 3 improved.16
Intralesional triamcinolone injections may be better suited for teenagers compared to younger children who may be more apprehensive of needles. To minimize pain, it is recommended to inject ILTAC slowly at room temperature, with use of “talkesthesia” and vibration devices, 1% lidocaine, or ethyl chloride spray.18
Trachyonychia
Trachyonychia is characterized by the presence of sandpaperlike nails. It manifests with brittle thin nails with longitudinal ridging, onychoschizia, and thickened hyperkeratotic cuticles. Trachyonychia typically involves multiple nails, with a peak age of onset between 3 and 12 years.21,22 There are 2 variants: the opaque type with rough longitudinal ridging, and the shiny variant with opalescent nails and pits that reflect light. The opaque variant is more common and is associated with psoriasis, whereas the shiny variant is less common and is associated with alopecia areata.23 Although most cases are idiopathic, some are associated with psoriasis and alopecia areata, as previously noted, as well as atopic dermatitis (AD) and lichen planus.22,24
Fortunately, trachyonychia does not lead to permanent nail damage or pterygium, making treatment primarily focused on addressing functional and cosmetic concerns.24 Spontaneous resolution occurs in approximately 50% of patients. In a prospective study of 11 patients with idiopathic trachyonychia, there was partial improvement in 5 of 9 patients treated with topical steroids, 1 with only petrolatum, and 1 with vitamin supplements. Complete resolution was reported in 1 patient treated with topical steroids.25 Because trachyonychia often is self-resolving, no treatment is required and a conservative approach is strongly recommended.26 Treatment options include topical corticosteroids, tazarotene, and 5-fluorouracil. Intralesional triamcinolone, systemic cyclosporine, retinoids, systemic corticosteroids, and tofacitinib have been described in case reports, though none of these have been shown to be 100% efficacious.24
Nail Lichen Striatus
Lichen striatus involving the nail is uncommon and is characterized by onycholysis, longitudinal ridging, splitting, and fraying, as well as what appears to be a subungual tumor. It can encompass the entire nail or may be isolated to a portion of the nail (Figure 2). Usually, a Blaschko-linear array of flesh-colored papules on the more proximal digit directly adjacent to the nail dystrophy will be seen, though nail findings can occur in isolation.27-29 The underlying pathophysiology is not clear; however, one hypothesis is that a triggering event, such as trauma, induces the expression of antigens that elicit a self-limiting immune-mediated response by CD8 T lymphocytes.30
Generally, nail lichen striatus spontaneously resolves in 1 to 2 years without treatment. In a prospective study of 5 patients with nail lichen striatus, the median time to resolution was 22.6 months (range, 10–30 months).31 Topical steroids may be used for pruritus. In one case report, a 3-year-old boy with nail lichen striatus of 4 months’ duration was treated with tacrolimus ointment 0.03% daily for 3 months.28
Nail AD
Nail changes with AD may be more common in adults than children or are underreported. In a study of 777 adults with AD, nail dystrophy was present in 124 patients (16%), whereas in a study of 250 pediatric patients with AD (aged 0-2 years), nail dystrophy was present in only 4 patients.32,33
Periungual inflammation from AD causes the nail changes.34 In a cross-sectional study of 24 pediatric patients with nail dystrophy due to AD, transverse grooves (Beau lines) were present in 25% (6/24), nail pitting in 16.7% (4/24), koilonychia in 16.7% (4/24), trachyonychia in 12.5% (3/24), leukonychia in 12.5% (3/24), brachyonychia in 8.3% (2/24), melanonychia in 8.3% (2/24), onychomadesis in 8.3% (2/24), onychoschizia in 8.3% (2/24), and onycholysis in 8.3% (2/24). There was an association between disease severity and presence of toenail dystrophy (P=.03).35
Topical steroids with or without occlusion can be used to treat nail changes. Although there is limited literature describing the treatment of nail AD in children, a 61-year-old man with nail changes associated with AD achieved resolution with 3 months of treatment with dupilumab.36 Anecdotally, most patients will improve with usual cutaneous AD management.
INFECTIOUS NAIL DISORDERS
Viral Infections
Hand, Foot, and Mouth Disease—Hand, foot, and mouth disease (HFMD) is a common childhood viral infection caused by various enteroviruses, most commonly coxsackievirus A16, with the A6 variant causing more severe disease. Fever and painful vesicles involving the oral mucosa as well as palms and soles give the disease its name. Nail changes are common. In a prospective study involving 130 patients with laboratory-confirmed coxsackievirus CA6 serotype infection, 37% developed onychomadesis vs only 5% of 145 cases with non-CA6 enterovirus infection who developed nail findings. There was an association between CA6 infection and presence of nail changes (P<.001).37
Findings ranging from transverse grooves (Beau lines) to complete nail shedding (onychomadesis)(Figure 3) may be seen.38,39 Nail findings in HFMD are due to transient inhibition of nail growth and present approximately 3 to 6 weeks after infection.40 Onychomadesis is seen in 30% to 68% of patients with HFMD.37,41,42 Nail findings in HFMD spontaneously resolve with nail growth (2–3 mm per month for fingernails and 1 mm per month for toenails) and do not require specific treatment. Although the appearance of nail changes associated with HFMD can be disturbing, dermatologists can reassure children and their parents that the nails will resolve with the next cycle of growth.
Kawasaki Disease—Kawasaki disease (KD) is a vasculitis primarily affecting children and infants. Although the specific pathogen and pathophysiology is not entirely clear, clinical observations have suggested an infectious cause, most likely a virus.43 In Japan, more than 15,000 cases of KD are documented annually, while approximately 4200 cases are seen in the United States.44 In a prospective study from 1984 to 1990, 4 of 26 (15.4%) patients with KD presented with nail manifestations during the late acute phase or early convalescent phase of disease. There were no significant associations between nail dystrophy and severity of KD, such as coronary artery aneurysm.45
Nail changes reported in children with KD include onychomadesis, onycholysis, orange-brown chromonychia, splinter hemorrhages, Beau lines, and pincer nails. In a review of nail changes associated with KD from 1980 to 2021, orange-brown transverse chromonychia, which may evolve into transverse leukonychia, was the most common nail finding reported, occurring in 17 of 31 (54.8%) patients.44 It has been hypothesized that nail changes may result from blood flow disturbance due to the underlying vasculitis.46 Nail changes appear several weeks after the onset of fever and are self-limited. Resolution occurs with nail growth, with no treatment required.
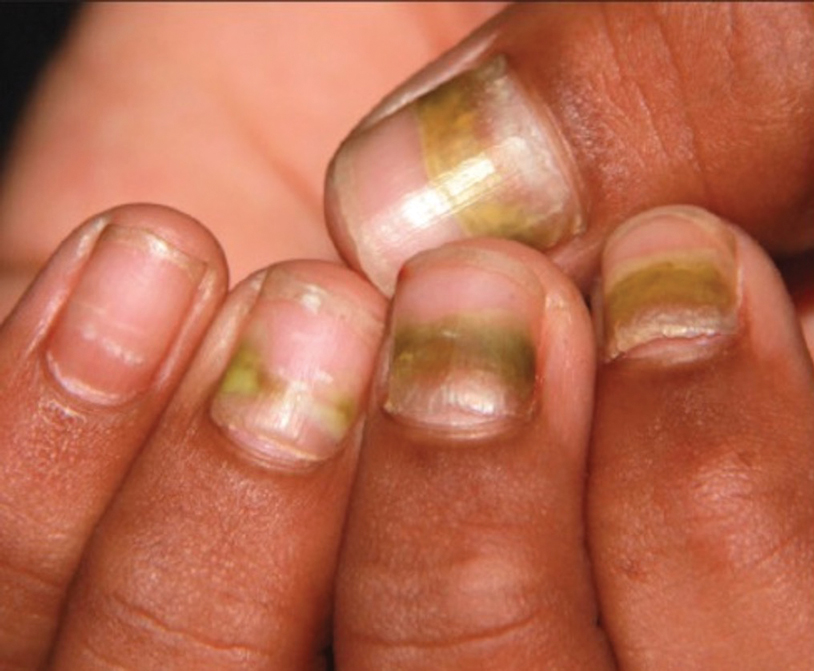
FUNGAL INFECTIONS
Onychomycosis
Onychomycosis is a fungal infection of the nails that occurs in 0.2% to 5.5% of pediatric patients, and its prevalence may be increasing, which may be due to environmental factors or increased rates of diabetes mellitus and obesity in the pediatric population.47 Onychomycosis represents 15.5% of nail dystrophies in pediatric patients.48 Some dermatologists treat presumptive onychomycosis without confirmation; however, we do not recommend that approach. Because the differential is broad and the duration of treatment is long, mycologic examination (potassium hydroxide preparation, fungal culture, polymerase chain reaction, and/or histopathology) should be obtained to confirm onychomycosis prior to initiation of antifungal management. Family members of affected individuals should be evaluated and treated, if indicated, for onychomycosis and tinea pedis, as household transmission is common.
Currently, there are 2 topical FDA-approved treatments for pediatric onychomycosis in children 6 years and older (Table 2).49,50 There is a discussion of the need for confirmatory testing for onychomycosis in children, particularly when systemic treatment is prescribed. In a retrospective review of 269 pediatric patients with onychomycosis prescribed terbinafine, 53.5% (n=144) underwent laboratory monitoring of liver function and complete blood cell counts, and 12.5% had grade 1 laboratory abnormalities either prior to (12/144 [8.3%]) or during (6/144 [4.2%]) therapy.51 Baseline transaminase monitoring is recommended, though subsequent routine laboratory monitoring in healthy children may have limited utility with associated increased costs, incidental findings, and patient discomfort and likely is not needed.51
Pediatric onychomycosis responds better to topical therapy than adult disease, and pediatric patients do not always require systemic treatment.52 Ciclopirox is not FDA approved for the treatment of pediatric onychomycosis, but in a 32-week clinical trial of ciclopirox lacquer 8% use in 40 patients, 77% (27/35) of treated patients achieved mycologic cure. Overall, 71% of treated patients (25/35) vs 22% (2/9) of controls achieved efficacy (defined as investigator global assessment score of 2 or lower).52 In an open-label, single-arm clinical trial assessing tavaborole solution 5% applied once daily for 48 weeks for the treatment of toenail onychomycosis in pediatric patients (aged 6–17 years), 36.2% (20/55) of patients achieved mycologic cure, and 8.5% (5/55) achieved complete cure at week 52 with mild or minimal adverse effects.53 In an open-label, phase 4 study of the safety and efficacy of efinaconazole solution 10% applied once daily for 48 weeks in pediatric patients (aged 6 to 16 years) (n=60), 65% (35/60) achieved mycologic cure, 42% (25/60) achieved clinical cure, and 40% (24/60) achieved complete cure at 52 weeks. The most common adverse effects of efinaconazole were local and included ingrown toenail (1/60), application-site dermatitis (1/60), application-site vesicles (1/60), and application-site pain (1/60).54
In a systematic review of systemic antifungals for onychomycosis in 151 pediatric patients, itraconazole, fluconazole, griseofulvin, and terbinafine resulted in complete cure rates similar to those of the adult population, with excellent safety profiles.55 Depending on the situation, initiation of treatment with topical medications followed by addition of systemic antifungal agents only if needed may be an appropriate course of action.
BACTERIAL INFECTIONS
Acute Paronychia
Acute paronychia is a nail-fold infection that develops after the protective nail barrier has been compromised.56 In children, thumb-sucking, nail-biting, frequent oral manipulation of the digits, and poor skin hygiene are risk factors. Acute paronychia also may develop in association with congenital malalignment of the great toenails.57
Clinical manifestations include localized pain, erythema, and nail fold edema (Figure 4). Purulent material and abscess formation may ensue. Staphylococcus aureus as well as methicillin-resistant S aureus and Streptococcus pyogenes are classically the most common causes of acute paronychia. Treatment of paronychia is based on severity. In mild cases, warm soaks with topical antibiotics are indicated. Oral antibiotics should be prescribed for more severe presentations. If there is no improvement after 48 hours, surgical drainage is required to facilitate healing.56
FINAL THOUGHTS
Inflammatory and infectious nail disorders in children are relatively common and may impact the physical and emotional well-being of young patients. By understanding the distinctive features of these nail disorders in pediatric patients, dermatologists can provide anticipatory guidance and informed treatment options to children and their parents. Further research is needed to expand our understanding of pediatric nail disorders and create targeted therapeutic interventions, particularly for NLP and psoriasis.
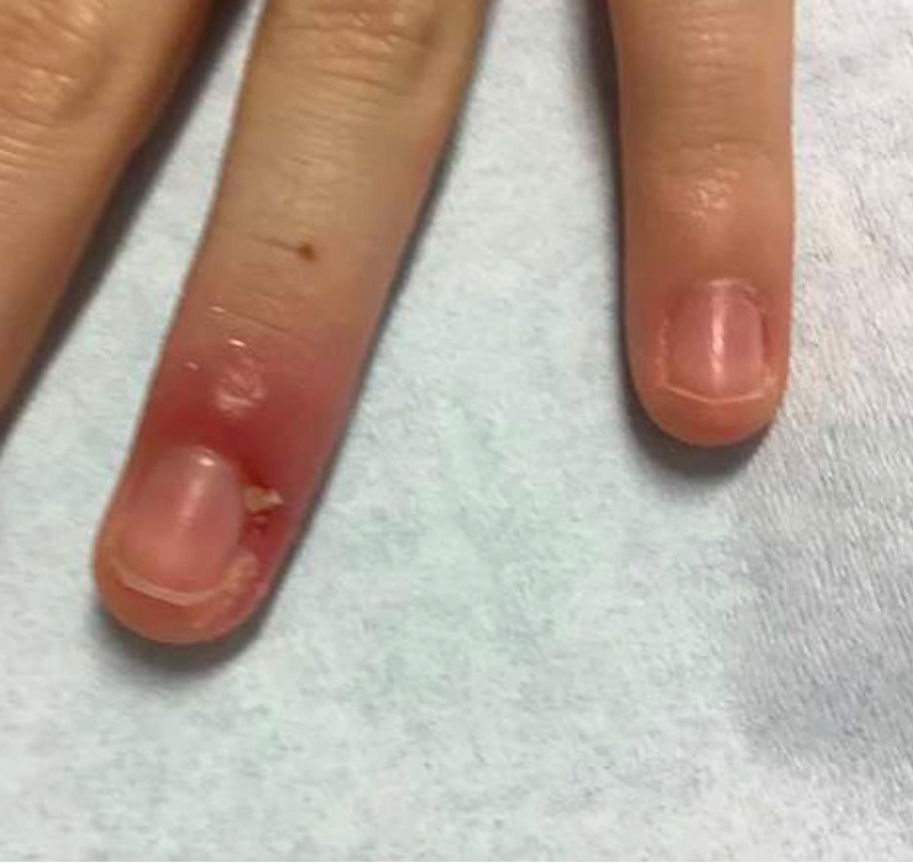
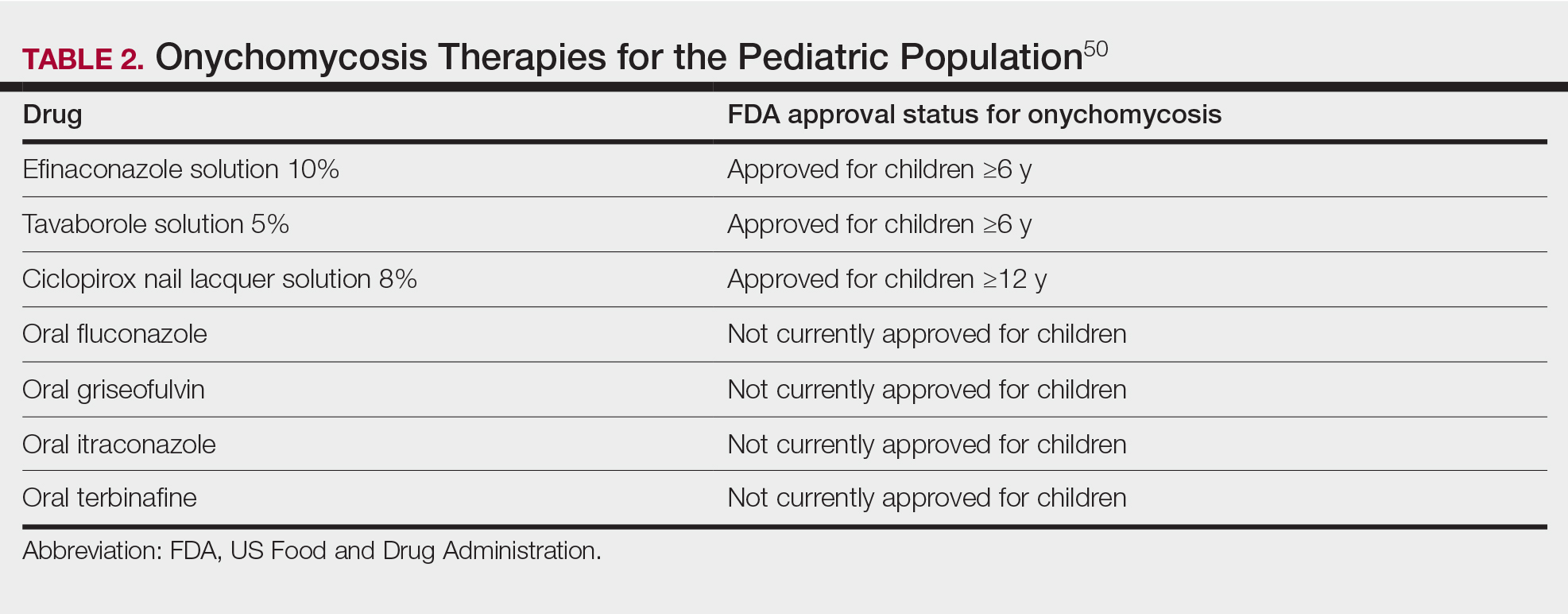
- Uber M, Carvalho VO, Abagge KT, et al. Clinical features and nail clippings in 52 children with psoriasis. Pediatr Dermatol. 2018;35:202-207. doi:10.1111/pde.13402
- Plachouri KM, Mulita F, Georgiou S. Management of pediatric nail psoriasis. Cutis. 2021;108:292-294. doi:10.12788/cutis.0386
- Smith RJ, Rubin AI. Pediatric nail disorders: a review. Curr Opin Pediatr. 2020;32:506-515. doi:10.1097/mop.0000000000000921
- Pourchot D, Bodemer C, Phan A, et al. Nail psoriasis: a systematic evaluation in 313 children with psoriasis. Pediatr Dermatol. 2017;34:58-63. doi:10.1111/pde.13028
- Richert B, André J. Nail disorders in children: diagnosis and management. Am J Clin Dermatol. 2011;12:101-112. doi:10.2165/11537110-000000000-00000
- Lee JYY. Severe 20-nail psoriasis successfully treated by low dose methotrexate. Dermatol Online J. 2009;15:8.
- Nogueira M, Paller AS, Torres T. Targeted therapy for pediatric psoriasis. Paediatr Drugs. May 2021;23:203-212. doi:10.1007/s40272-021-00443-5
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. Updated August 16, 2023. Accessed July 1, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Teran CG, Teran-Escalera CN, Balderrama C. A severe case of erythrodermic psoriasis associated with advanced nail and joint manifestations: a case report. J Med Case Rep. 2010;4:179. doi:10.1186/1752-1947-4-179
- Paller AS, Seyger MMB, Magariños GA, et al. Long-term efficacy and safety of up to 108 weeks of ixekizumab in pediatric patients with moderate to severe plaque psoriasis: the IXORA-PEDS randomized clinical trial. JAMA Dermatol. 2022;158:533-541. doi:10.1001/jamadermatol.2022.0655
- Diotallevi F, Simonetti O, Rizzetto G, et al. Biological treatments for pediatric psoriasis: state of the art and future perspectives. Int J Mol Sci. 2022;23:11128. doi:10.3390/ijms231911128
- Nash P, Mease PJ, Kirkham B, et al. Secukinumab provides sustained improvement in nail psoriasis, signs and symptoms of psoriatic arthritis and low rate of radiographic progression in patients with concomitant nail involvement: 2-year results from the Phase III FUTURE 5 study. Clin Exp Rheumatol. 2022;40:952-959. doi:10.55563/clinexprheumatol/3nuz51
- Wells LE, Evans T, Hilton R, et al. Use of secukinumab in a pediatric patient leads to significant improvement in nail psoriasis and psoriatic arthritis. Pediatr Dermatol. 2019;36:384-385. doi:10.1111/pde.13767
- Watabe D, Endoh K, Maeda F, et al. Childhood-onset psoriaticonycho-pachydermo-periostitis treated successfully with infliximab. Eur J Dermatol. 2015;25:506-508. doi:10.1684/ejd.2015.2616
- Pereira TM, Vieira AP, Fernandes JC, et al. Anti-TNF-alpha therapy in childhood pustular psoriasis. Dermatology. 2006;213:350-352. doi:10.1159/000096202
- Iorizzo M, Gioia Di Chiacchio N, Di Chiacchio N, et al. Intralesional steroid injections for inflammatory nail dystrophies in the pediatric population. Pediatr Dermatol. 2023;40:759-761. doi:10.1111/pde.15295
- Tosti A, Piraccini BM, Cambiaghi S, et al. Nail lichen planus in children: clinical features, response to treatment, and long-term follow-up. Arch Dermatol. 2001;137:1027-1032.
- Lipner SR. Nail lichen planus: a true nail emergency. J Am Acad Dermatol. 2019;80:e177-e178. doi:10.1016/j.jaad.2018.11.065
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Piraccini BM, Saccani E, Starace M, et al. Nail lichen planus: response to treatment and long term follow-up. Eur J Dermatol. 2010;20:489-496. doi:10.1684/ejd.2010.0952
- Mahajan R, Kaushik A, De D, et al. Pediatric trachyonychia- a retrospective study of 17 cases. Indian J Dermatol. 2021;66:689-690. doi:10.4103/ijd.ijd_42_21
- Leung AKC, Leong KF, Barankin B. Trachyonychia. J Pediatr. 2020;216:239-239.e1. doi:10.1016/j.jpeds.2019.08.034
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
- Jacobsen AA, Tosti A. Trachyonychia and twenty-nail dystrophy: a comprehensive review and discussion of diagnostic accuracy. Skin Appendage Disord. 2016;2:7-13. doi:10.1159/000445544
- Kumar MG, Ciliberto H, Bayliss SJ. Long-term follow-up of pediatric trachyonychia. Pediatr Dermatol. 2015;32:198-200. doi:10.1111/pde.12427
- Tosti A, Piraccini BM, Iorizzo M. Trachyonychia and related disorders: evaluation and treatment plans. Dermatolog Ther. 2002;15:121-125. doi:10.1046/j.1529-8019.2002.01511.x
- Leung AKC, Leong KF, Barankin B. Lichen striatus with nail involvement in a 6-year-old boy. Case Rep Pediatr. 2020;2020:1494760. doi:10.1155/2020/1494760
- Kim GW, Kim SH, Seo SH, et al. Lichen striatus with nail abnormality successfully treated with tacrolimus ointment. J Dermatol. 2009;36:616-617. doi:10.1111/j.1346-8138.2009.00720.x
- Iorizzo M, Rubin AI, Starace M. Nail lichen striatus: is dermoscopy useful for the diagnosis? Pediatr Dermatol. 2019;36:859-863. doi:10.1111/pde.13916
- Karp DL, Cohen BA. Onychodystrophy in lichen striatus. Pediatr Dermatol. 1993;10:359-361. doi:10.1111/j.1525-1470.1993.tb00399.x
- Tosti A, Peluso AM, Misciali C, et al. Nail lichen striatus: clinical features and long-term follow-up of five patients. J Am Acad Dermatol. 1997;36(6, pt 1):908-913. doi:10.1016/s0190-9622(97)80270-8
- Simpson EL, Thompson MM, Hanifin JM. Prevalence and morphology of hand eczema in patients with atopic dermatitis. Dermatitis. 2006;17:123-127. doi:10.2310/6620.2006.06005
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C. Nail alterations in 250 infant patients: a clinical study. J Eur Acad Dermatol Venereol. 2008;22:741-744. doi:10.1111/j.1468-3083.2008.02592.x
- Milanesi N, D’Erme AM, Gola M. Nail improvement during alitretinoin treatment: three case reports and review of the literature. Clin Exp Dermatol. 2015;40:533-536. doi:10.1111/ced.12584
- Chung BY, Choi YW, Kim HO, et al. Nail dystrophy in patients with atopic dermatitis and its association with disease severity. Ann Dermatol. 2019;31:121-126. doi:10.5021/ad.2019.31.2.121
- Navarro-Triviño FJ, Vega-Castillo JJ, Ruiz-Villaverde R. Nail changes successfully treated with dupilumab in a patient with severe atopic dermatitis. Australas J Dermatol. 2021;62:e468-e469. doi:10.1111/ajd.13633
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346. doi:10.1186/1471-2334-11-346
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283. doi:10.5021/ad.2014.26.2.280
- Verma S, Singal A. Nail changes in hand-foot-and-mouth disease (HFMD). Indian Dermatol Online J. 2021;12:656-657. doi:10.4103 /idoj.IDOJ_271_20
- Giordano LMC, de la Fuente LA, Lorca JMB, et al. Onychomadesis secondary to hand-foot-mouth disease: a frequent manifestation and cause of concern for parents. Article in Spanish. Rev Chil Pediatr. 2018;89:380-383. doi:10.4067/s0370-41062018005000203
- Justino MCA, da SMD, Souza MF, et al. Atypical hand-foot-mouth disease in Belém, Amazon region, northern Brazil, with detection of coxsackievirus A6. J Clin Virol. 2020;126:104307. doi:10.1016/j.jcv.2020.104307
- Cheng FF, Zhang BB, Cao ML, et al. Clinical characteristics of 68 children with atypical hand, foot, and mouth disease caused by coxsackievirus A6: a single-center retrospective analysis. Transl Pediatr. 2022;11:1502-1509. doi:10.21037/tp-22-352
- Nagata S. Causes of Kawasaki disease-from past to present. Front Pediatr. 2019;7:18. doi:10.3389/fped.2019.00018
- Mitsuishi T, Miyata K, Ando A, et al. Characteristic nail lesions in Kawasaki disease: case series and literature review. J Dermatol. 2022;49:232-238. doi:10.1111/1346-8138.16276
- Lindsley CB. Nail-bed lines in Kawasaki disease. Am J Dis Child. 1992;146:659-660. doi:10.1001/archpedi.1992.02160180017005
- Matsumura O, Nakagishi Y. Pincer nails upon convalescence from Kawasaki disease. J Pediatr. 2022;246:279. doi:10.1016/j.jpeds.2022.03.002
- Solís-Arias MP, García-Romero MT. Onychomycosis in children. a review. Int J Dermatol. 2017;56:123-130. doi:10.1111/ijd.13392
- Gupta AK, Mays RR, Versteeg SG, et al. Onychomycosis in children: safety and efficacy of antifungal agents. Pediatr Dermatol. 2018;35:552-559. doi:10.1111/pde.13561
- 49. Gupta AK, Venkataraman M, Shear NH, et al. Labeled use of efinaconazole topical solution 10% in treating onychomycosis in children and a review of the management of pediatric onychomycosis. Dermatol Ther. 2020;33:e13613. doi:10.1111/dth.13613
- Falotico JM, Lipner SR. Updated perspectives on the diagnosis and management of onychomycosis. Clin Cosmet Investig Dermatol. 2022;15:1933-1957. doi:10.2147/ccid.S362635
- Patel D, Castelo-Soccio LA, Rubin AI, et al. Laboratory monitoring during systemic terbinafine therapy for pediatric onychomycosis. JAMA Dermatol. 2017;153:1326-1327. doi:10.1001/jamadermatol.2017.4483
- Friedlander SF, Chan YC, Chan YH, et al. Onychomycosis does not always require systemic treatment for cure: a trial using topical therapy. Pediatr Dermatol. 2013;30:316-322. doi:10.1111/pde.12064
- Rich P, Spellman M, Purohit V, et al. Tavaborole 5% topical solution for the treatment of toenail onychomycosis in pediatric patients: results from a phase 4 open-label study. J Drugs Dermatol. 2019;18:190-195.
- Gupta AK, Venkataraman M, Abramovits W, et al. JUBLIA (efinaconazole 10% solution) in the treatment of pediatric onychomycosis. Skinmed. 2021;19:206-210.
- Gupta AK, Paquet M. Systemic antifungals to treat onychomycosis in children: a systematic review. Pediatr Dermatol. 2013;30:294-302. doi:10.1111/pde.12048
- Leggit JC. Acute and chronic paronychia. Am Fam Physician. 2017;96:44-51.
- Lipner SR, Scher RK. Congenital malalignment of the great toenails with acute paronychia. Pediatr Dermatol. 2016;33:e288-e289.doi:10.1111/pde.12924
Nail disorders are common among pediatric patients but often are underdiagnosed or misdiagnosed because of their unique disease manifestations. These conditions may severely impact quality of life. There are few nail disease clinical trials that include children. Consequently, most treatment recommendations are based on case series and expert consensus recommendations. We review inflammatory and infectious nail disorders in pediatric patients. By describing characteristics, clinical manifestations, and management approaches for these conditions, we aim to provide guidance to dermatologists in their diagnosis and treatment.
INFLAMMATORY NAIL DISORDERS
Nail Psoriasis
Nail involvement in children with psoriasis is common, with prevalence estimates ranging from 17% to 39.2%.1 Nail matrix psoriasis may manifest with pitting (large irregular pits) and leukonychia as well as chromonychia and nail plate crumbling. Onycholysis, oil drop spots (salmon patches), and subungual hyperkeratosis can be seen in nail bed psoriasis. Nail pitting is the most frequently observed clinical finding (Figure 1).2,3 In a cross-sectional multicenter study of 313 children with cutaneous psoriasis in France, nail findings were present in 101 patients (32.3%). There were associations between nail findings and presence of psoriatic arthritis (P=.03), palmoplantar psoriasis (P<.001), and severity of psoriatic disease, defined as use of systemic treatment with phototherapy (psoralen plus UVA, UVB), traditional systemic treatment (acitretin, methotrexate, cyclosporine), or a biologic (P=.003).4
Topical steroids and vitamin D analogues may be used with or without occlusion and may be efficacious.5 Several case reports describe systemic treatments for psoriasis in children, including methotrexate, acitretin, and apremilast (approved for children 6 years and older for plaque psoriasis by the US Food and Drug Administration [FDA]).2 There are 5 biologic drugs currently approved for the treatment of pediatric psoriasis—adalimumab, etanercept, ustekinumab, secukinumab, ixekizumab—and 6 drugs currently undergoing phase 3 studies—brodalumab, guselkumab, risankizumab, tildrakizumab, certolizumab pegol, and deucravacitinib (Table 1).6-15 Adalimumab is specifically approved for moderate to severe nail psoriasis in adults 18 years and older.
Intralesional steroid injections are sometimes useful in the management of nail matrix psoriasis; however, appropriate patient selection is critical due to the pain associated with the procedure. In a prospective study of 16 children (age range, 9–17 years) with nail psoriasis treated with intralesional triamcinolone (ILTAC) 2.5 to 5 mg/mL every 4 to 8 weeks for a minimum of 3 to 6 months, 9 patients achieved resolution and 6 had improvement of clinical findings.16 Local adverse events were mild, including injection-site pain (66%), subungual hematoma (n=1), Beau lines (n=1), proximal nail fold hypopigmentation (n=2), and proximal nail fold atrophy (n=2). Because the proximal nail fold in children is thinner than in adults, there may be an increased risk for nail fold hypopigmentation and atrophy in children. Therefore, a maximum ILTAC concentration of 2.5 mg/mL with 0.2 mL maximum volume per nail per session is recommended for children younger than 15 years.16
Nail Lichen Planus
Nail lichen planus (NLP) is uncommon in children, with few biopsy-proven cases documented in the literature.17 Common clinical findings are onychorrhexis, nail plate thinning, fissuring, splitting, and atrophy with koilonychia.5 Although pterygium development (irreversible nail matrix scarring) is uncommon in pediatric patients, NLP can be progressive and may cause irreversible destruction of the nail matrix and subsequent nail loss, warranting therapeutic intervention.18
Treatment of NLP may be difficult, as there are no options that work in all patients. Current literature supports the use of systemic corticosteroids or ILTAC for the treatment of NLP; however, recurrence rates can be high. According to an expert consensus paper on NLP treatment, ILTAC may be injected in a concentration of 2.5, 5, or 10 mg/mL according to disease severity.19 In severe or resistant cases, intramuscular (IM) triamcinolone may be considered, especially if more than 3 nails are affected. A dosage of 0.5 to 1 mg/kg/mo for at least 3 to 6 months is recommended for both children and adults, with 1 mg/kg/mo recommended in the active treatment phase (first 2–3 months).19 In a retrospective review of 5 pediatric patients with NLP treated with IM triamcinolone 0.5 mg/kg/mo, 3 patients had resolution and 2 improved with treatment.20 In a prospective study of 10 children with NLP, IM triamcinolone at a dosage of 0.5 to 1 mg/kg every 30 days for 3 to 6 months resulted in resolution of nail findings in 9 patients.17 In a prospective study of 14 pediatric patients with NLP treated with 2.5 to 5 mg/mL of ILTAC, 10 achieved resolution and 3 improved.16
Intralesional triamcinolone injections may be better suited for teenagers compared to younger children who may be more apprehensive of needles. To minimize pain, it is recommended to inject ILTAC slowly at room temperature, with use of “talkesthesia” and vibration devices, 1% lidocaine, or ethyl chloride spray.18
Trachyonychia
Trachyonychia is characterized by the presence of sandpaperlike nails. It manifests with brittle thin nails with longitudinal ridging, onychoschizia, and thickened hyperkeratotic cuticles. Trachyonychia typically involves multiple nails, with a peak age of onset between 3 and 12 years.21,22 There are 2 variants: the opaque type with rough longitudinal ridging, and the shiny variant with opalescent nails and pits that reflect light. The opaque variant is more common and is associated with psoriasis, whereas the shiny variant is less common and is associated with alopecia areata.23 Although most cases are idiopathic, some are associated with psoriasis and alopecia areata, as previously noted, as well as atopic dermatitis (AD) and lichen planus.22,24
Fortunately, trachyonychia does not lead to permanent nail damage or pterygium, making treatment primarily focused on addressing functional and cosmetic concerns.24 Spontaneous resolution occurs in approximately 50% of patients. In a prospective study of 11 patients with idiopathic trachyonychia, there was partial improvement in 5 of 9 patients treated with topical steroids, 1 with only petrolatum, and 1 with vitamin supplements. Complete resolution was reported in 1 patient treated with topical steroids.25 Because trachyonychia often is self-resolving, no treatment is required and a conservative approach is strongly recommended.26 Treatment options include topical corticosteroids, tazarotene, and 5-fluorouracil. Intralesional triamcinolone, systemic cyclosporine, retinoids, systemic corticosteroids, and tofacitinib have been described in case reports, though none of these have been shown to be 100% efficacious.24
Nail Lichen Striatus
Lichen striatus involving the nail is uncommon and is characterized by onycholysis, longitudinal ridging, splitting, and fraying, as well as what appears to be a subungual tumor. It can encompass the entire nail or may be isolated to a portion of the nail (Figure 2). Usually, a Blaschko-linear array of flesh-colored papules on the more proximal digit directly adjacent to the nail dystrophy will be seen, though nail findings can occur in isolation.27-29 The underlying pathophysiology is not clear; however, one hypothesis is that a triggering event, such as trauma, induces the expression of antigens that elicit a self-limiting immune-mediated response by CD8 T lymphocytes.30
Generally, nail lichen striatus spontaneously resolves in 1 to 2 years without treatment. In a prospective study of 5 patients with nail lichen striatus, the median time to resolution was 22.6 months (range, 10–30 months).31 Topical steroids may be used for pruritus. In one case report, a 3-year-old boy with nail lichen striatus of 4 months’ duration was treated with tacrolimus ointment 0.03% daily for 3 months.28
Nail AD
Nail changes with AD may be more common in adults than children or are underreported. In a study of 777 adults with AD, nail dystrophy was present in 124 patients (16%), whereas in a study of 250 pediatric patients with AD (aged 0-2 years), nail dystrophy was present in only 4 patients.32,33
Periungual inflammation from AD causes the nail changes.34 In a cross-sectional study of 24 pediatric patients with nail dystrophy due to AD, transverse grooves (Beau lines) were present in 25% (6/24), nail pitting in 16.7% (4/24), koilonychia in 16.7% (4/24), trachyonychia in 12.5% (3/24), leukonychia in 12.5% (3/24), brachyonychia in 8.3% (2/24), melanonychia in 8.3% (2/24), onychomadesis in 8.3% (2/24), onychoschizia in 8.3% (2/24), and onycholysis in 8.3% (2/24). There was an association between disease severity and presence of toenail dystrophy (P=.03).35
Topical steroids with or without occlusion can be used to treat nail changes. Although there is limited literature describing the treatment of nail AD in children, a 61-year-old man with nail changes associated with AD achieved resolution with 3 months of treatment with dupilumab.36 Anecdotally, most patients will improve with usual cutaneous AD management.
INFECTIOUS NAIL DISORDERS
Viral Infections
Hand, Foot, and Mouth Disease—Hand, foot, and mouth disease (HFMD) is a common childhood viral infection caused by various enteroviruses, most commonly coxsackievirus A16, with the A6 variant causing more severe disease. Fever and painful vesicles involving the oral mucosa as well as palms and soles give the disease its name. Nail changes are common. In a prospective study involving 130 patients with laboratory-confirmed coxsackievirus CA6 serotype infection, 37% developed onychomadesis vs only 5% of 145 cases with non-CA6 enterovirus infection who developed nail findings. There was an association between CA6 infection and presence of nail changes (P<.001).37
Findings ranging from transverse grooves (Beau lines) to complete nail shedding (onychomadesis)(Figure 3) may be seen.38,39 Nail findings in HFMD are due to transient inhibition of nail growth and present approximately 3 to 6 weeks after infection.40 Onychomadesis is seen in 30% to 68% of patients with HFMD.37,41,42 Nail findings in HFMD spontaneously resolve with nail growth (2–3 mm per month for fingernails and 1 mm per month for toenails) and do not require specific treatment. Although the appearance of nail changes associated with HFMD can be disturbing, dermatologists can reassure children and their parents that the nails will resolve with the next cycle of growth.
Kawasaki Disease—Kawasaki disease (KD) is a vasculitis primarily affecting children and infants. Although the specific pathogen and pathophysiology is not entirely clear, clinical observations have suggested an infectious cause, most likely a virus.43 In Japan, more than 15,000 cases of KD are documented annually, while approximately 4200 cases are seen in the United States.44 In a prospective study from 1984 to 1990, 4 of 26 (15.4%) patients with KD presented with nail manifestations during the late acute phase or early convalescent phase of disease. There were no significant associations between nail dystrophy and severity of KD, such as coronary artery aneurysm.45
Nail changes reported in children with KD include onychomadesis, onycholysis, orange-brown chromonychia, splinter hemorrhages, Beau lines, and pincer nails. In a review of nail changes associated with KD from 1980 to 2021, orange-brown transverse chromonychia, which may evolve into transverse leukonychia, was the most common nail finding reported, occurring in 17 of 31 (54.8%) patients.44 It has been hypothesized that nail changes may result from blood flow disturbance due to the underlying vasculitis.46 Nail changes appear several weeks after the onset of fever and are self-limited. Resolution occurs with nail growth, with no treatment required.
FUNGAL INFECTIONS
Onychomycosis
Onychomycosis is a fungal infection of the nails that occurs in 0.2% to 5.5% of pediatric patients, and its prevalence may be increasing, which may be due to environmental factors or increased rates of diabetes mellitus and obesity in the pediatric population.47 Onychomycosis represents 15.5% of nail dystrophies in pediatric patients.48 Some dermatologists treat presumptive onychomycosis without confirmation; however, we do not recommend that approach. Because the differential is broad and the duration of treatment is long, mycologic examination (potassium hydroxide preparation, fungal culture, polymerase chain reaction, and/or histopathology) should be obtained to confirm onychomycosis prior to initiation of antifungal management. Family members of affected individuals should be evaluated and treated, if indicated, for onychomycosis and tinea pedis, as household transmission is common.
Currently, there are 2 topical FDA-approved treatments for pediatric onychomycosis in children 6 years and older (Table 2).49,50 There is a discussion of the need for confirmatory testing for onychomycosis in children, particularly when systemic treatment is prescribed. In a retrospective review of 269 pediatric patients with onychomycosis prescribed terbinafine, 53.5% (n=144) underwent laboratory monitoring of liver function and complete blood cell counts, and 12.5% had grade 1 laboratory abnormalities either prior to (12/144 [8.3%]) or during (6/144 [4.2%]) therapy.51 Baseline transaminase monitoring is recommended, though subsequent routine laboratory monitoring in healthy children may have limited utility with associated increased costs, incidental findings, and patient discomfort and likely is not needed.51
Pediatric onychomycosis responds better to topical therapy than adult disease, and pediatric patients do not always require systemic treatment.52 Ciclopirox is not FDA approved for the treatment of pediatric onychomycosis, but in a 32-week clinical trial of ciclopirox lacquer 8% use in 40 patients, 77% (27/35) of treated patients achieved mycologic cure. Overall, 71% of treated patients (25/35) vs 22% (2/9) of controls achieved efficacy (defined as investigator global assessment score of 2 or lower).52 In an open-label, single-arm clinical trial assessing tavaborole solution 5% applied once daily for 48 weeks for the treatment of toenail onychomycosis in pediatric patients (aged 6–17 years), 36.2% (20/55) of patients achieved mycologic cure, and 8.5% (5/55) achieved complete cure at week 52 with mild or minimal adverse effects.53 In an open-label, phase 4 study of the safety and efficacy of efinaconazole solution 10% applied once daily for 48 weeks in pediatric patients (aged 6 to 16 years) (n=60), 65% (35/60) achieved mycologic cure, 42% (25/60) achieved clinical cure, and 40% (24/60) achieved complete cure at 52 weeks. The most common adverse effects of efinaconazole were local and included ingrown toenail (1/60), application-site dermatitis (1/60), application-site vesicles (1/60), and application-site pain (1/60).54
In a systematic review of systemic antifungals for onychomycosis in 151 pediatric patients, itraconazole, fluconazole, griseofulvin, and terbinafine resulted in complete cure rates similar to those of the adult population, with excellent safety profiles.55 Depending on the situation, initiation of treatment with topical medications followed by addition of systemic antifungal agents only if needed may be an appropriate course of action.
BACTERIAL INFECTIONS
Acute Paronychia
Acute paronychia is a nail-fold infection that develops after the protective nail barrier has been compromised.56 In children, thumb-sucking, nail-biting, frequent oral manipulation of the digits, and poor skin hygiene are risk factors. Acute paronychia also may develop in association with congenital malalignment of the great toenails.57
Clinical manifestations include localized pain, erythema, and nail fold edema (Figure 4). Purulent material and abscess formation may ensue. Staphylococcus aureus as well as methicillin-resistant S aureus and Streptococcus pyogenes are classically the most common causes of acute paronychia. Treatment of paronychia is based on severity. In mild cases, warm soaks with topical antibiotics are indicated. Oral antibiotics should be prescribed for more severe presentations. If there is no improvement after 48 hours, surgical drainage is required to facilitate healing.56
FINAL THOUGHTS
Inflammatory and infectious nail disorders in children are relatively common and may impact the physical and emotional well-being of young patients. By understanding the distinctive features of these nail disorders in pediatric patients, dermatologists can provide anticipatory guidance and informed treatment options to children and their parents. Further research is needed to expand our understanding of pediatric nail disorders and create targeted therapeutic interventions, particularly for NLP and psoriasis.
Nail disorders are common among pediatric patients but often are underdiagnosed or misdiagnosed because of their unique disease manifestations. These conditions may severely impact quality of life. There are few nail disease clinical trials that include children. Consequently, most treatment recommendations are based on case series and expert consensus recommendations. We review inflammatory and infectious nail disorders in pediatric patients. By describing characteristics, clinical manifestations, and management approaches for these conditions, we aim to provide guidance to dermatologists in their diagnosis and treatment.
INFLAMMATORY NAIL DISORDERS
Nail Psoriasis
Nail involvement in children with psoriasis is common, with prevalence estimates ranging from 17% to 39.2%.1 Nail matrix psoriasis may manifest with pitting (large irregular pits) and leukonychia as well as chromonychia and nail plate crumbling. Onycholysis, oil drop spots (salmon patches), and subungual hyperkeratosis can be seen in nail bed psoriasis. Nail pitting is the most frequently observed clinical finding (Figure 1).2,3 In a cross-sectional multicenter study of 313 children with cutaneous psoriasis in France, nail findings were present in 101 patients (32.3%). There were associations between nail findings and presence of psoriatic arthritis (P=.03), palmoplantar psoriasis (P<.001), and severity of psoriatic disease, defined as use of systemic treatment with phototherapy (psoralen plus UVA, UVB), traditional systemic treatment (acitretin, methotrexate, cyclosporine), or a biologic (P=.003).4
Topical steroids and vitamin D analogues may be used with or without occlusion and may be efficacious.5 Several case reports describe systemic treatments for psoriasis in children, including methotrexate, acitretin, and apremilast (approved for children 6 years and older for plaque psoriasis by the US Food and Drug Administration [FDA]).2 There are 5 biologic drugs currently approved for the treatment of pediatric psoriasis—adalimumab, etanercept, ustekinumab, secukinumab, ixekizumab—and 6 drugs currently undergoing phase 3 studies—brodalumab, guselkumab, risankizumab, tildrakizumab, certolizumab pegol, and deucravacitinib (Table 1).6-15 Adalimumab is specifically approved for moderate to severe nail psoriasis in adults 18 years and older.
Intralesional steroid injections are sometimes useful in the management of nail matrix psoriasis; however, appropriate patient selection is critical due to the pain associated with the procedure. In a prospective study of 16 children (age range, 9–17 years) with nail psoriasis treated with intralesional triamcinolone (ILTAC) 2.5 to 5 mg/mL every 4 to 8 weeks for a minimum of 3 to 6 months, 9 patients achieved resolution and 6 had improvement of clinical findings.16 Local adverse events were mild, including injection-site pain (66%), subungual hematoma (n=1), Beau lines (n=1), proximal nail fold hypopigmentation (n=2), and proximal nail fold atrophy (n=2). Because the proximal nail fold in children is thinner than in adults, there may be an increased risk for nail fold hypopigmentation and atrophy in children. Therefore, a maximum ILTAC concentration of 2.5 mg/mL with 0.2 mL maximum volume per nail per session is recommended for children younger than 15 years.16
Nail Lichen Planus
Nail lichen planus (NLP) is uncommon in children, with few biopsy-proven cases documented in the literature.17 Common clinical findings are onychorrhexis, nail plate thinning, fissuring, splitting, and atrophy with koilonychia.5 Although pterygium development (irreversible nail matrix scarring) is uncommon in pediatric patients, NLP can be progressive and may cause irreversible destruction of the nail matrix and subsequent nail loss, warranting therapeutic intervention.18
Treatment of NLP may be difficult, as there are no options that work in all patients. Current literature supports the use of systemic corticosteroids or ILTAC for the treatment of NLP; however, recurrence rates can be high. According to an expert consensus paper on NLP treatment, ILTAC may be injected in a concentration of 2.5, 5, or 10 mg/mL according to disease severity.19 In severe or resistant cases, intramuscular (IM) triamcinolone may be considered, especially if more than 3 nails are affected. A dosage of 0.5 to 1 mg/kg/mo for at least 3 to 6 months is recommended for both children and adults, with 1 mg/kg/mo recommended in the active treatment phase (first 2–3 months).19 In a retrospective review of 5 pediatric patients with NLP treated with IM triamcinolone 0.5 mg/kg/mo, 3 patients had resolution and 2 improved with treatment.20 In a prospective study of 10 children with NLP, IM triamcinolone at a dosage of 0.5 to 1 mg/kg every 30 days for 3 to 6 months resulted in resolution of nail findings in 9 patients.17 In a prospective study of 14 pediatric patients with NLP treated with 2.5 to 5 mg/mL of ILTAC, 10 achieved resolution and 3 improved.16
Intralesional triamcinolone injections may be better suited for teenagers compared to younger children who may be more apprehensive of needles. To minimize pain, it is recommended to inject ILTAC slowly at room temperature, with use of “talkesthesia” and vibration devices, 1% lidocaine, or ethyl chloride spray.18
Trachyonychia
Trachyonychia is characterized by the presence of sandpaperlike nails. It manifests with brittle thin nails with longitudinal ridging, onychoschizia, and thickened hyperkeratotic cuticles. Trachyonychia typically involves multiple nails, with a peak age of onset between 3 and 12 years.21,22 There are 2 variants: the opaque type with rough longitudinal ridging, and the shiny variant with opalescent nails and pits that reflect light. The opaque variant is more common and is associated with psoriasis, whereas the shiny variant is less common and is associated with alopecia areata.23 Although most cases are idiopathic, some are associated with psoriasis and alopecia areata, as previously noted, as well as atopic dermatitis (AD) and lichen planus.22,24
Fortunately, trachyonychia does not lead to permanent nail damage or pterygium, making treatment primarily focused on addressing functional and cosmetic concerns.24 Spontaneous resolution occurs in approximately 50% of patients. In a prospective study of 11 patients with idiopathic trachyonychia, there was partial improvement in 5 of 9 patients treated with topical steroids, 1 with only petrolatum, and 1 with vitamin supplements. Complete resolution was reported in 1 patient treated with topical steroids.25 Because trachyonychia often is self-resolving, no treatment is required and a conservative approach is strongly recommended.26 Treatment options include topical corticosteroids, tazarotene, and 5-fluorouracil. Intralesional triamcinolone, systemic cyclosporine, retinoids, systemic corticosteroids, and tofacitinib have been described in case reports, though none of these have been shown to be 100% efficacious.24
Nail Lichen Striatus
Lichen striatus involving the nail is uncommon and is characterized by onycholysis, longitudinal ridging, splitting, and fraying, as well as what appears to be a subungual tumor. It can encompass the entire nail or may be isolated to a portion of the nail (Figure 2). Usually, a Blaschko-linear array of flesh-colored papules on the more proximal digit directly adjacent to the nail dystrophy will be seen, though nail findings can occur in isolation.27-29 The underlying pathophysiology is not clear; however, one hypothesis is that a triggering event, such as trauma, induces the expression of antigens that elicit a self-limiting immune-mediated response by CD8 T lymphocytes.30
Generally, nail lichen striatus spontaneously resolves in 1 to 2 years without treatment. In a prospective study of 5 patients with nail lichen striatus, the median time to resolution was 22.6 months (range, 10–30 months).31 Topical steroids may be used for pruritus. In one case report, a 3-year-old boy with nail lichen striatus of 4 months’ duration was treated with tacrolimus ointment 0.03% daily for 3 months.28
Nail AD
Nail changes with AD may be more common in adults than children or are underreported. In a study of 777 adults with AD, nail dystrophy was present in 124 patients (16%), whereas in a study of 250 pediatric patients with AD (aged 0-2 years), nail dystrophy was present in only 4 patients.32,33
Periungual inflammation from AD causes the nail changes.34 In a cross-sectional study of 24 pediatric patients with nail dystrophy due to AD, transverse grooves (Beau lines) were present in 25% (6/24), nail pitting in 16.7% (4/24), koilonychia in 16.7% (4/24), trachyonychia in 12.5% (3/24), leukonychia in 12.5% (3/24), brachyonychia in 8.3% (2/24), melanonychia in 8.3% (2/24), onychomadesis in 8.3% (2/24), onychoschizia in 8.3% (2/24), and onycholysis in 8.3% (2/24). There was an association between disease severity and presence of toenail dystrophy (P=.03).35
Topical steroids with or without occlusion can be used to treat nail changes. Although there is limited literature describing the treatment of nail AD in children, a 61-year-old man with nail changes associated with AD achieved resolution with 3 months of treatment with dupilumab.36 Anecdotally, most patients will improve with usual cutaneous AD management.
INFECTIOUS NAIL DISORDERS
Viral Infections
Hand, Foot, and Mouth Disease—Hand, foot, and mouth disease (HFMD) is a common childhood viral infection caused by various enteroviruses, most commonly coxsackievirus A16, with the A6 variant causing more severe disease. Fever and painful vesicles involving the oral mucosa as well as palms and soles give the disease its name. Nail changes are common. In a prospective study involving 130 patients with laboratory-confirmed coxsackievirus CA6 serotype infection, 37% developed onychomadesis vs only 5% of 145 cases with non-CA6 enterovirus infection who developed nail findings. There was an association between CA6 infection and presence of nail changes (P<.001).37
Findings ranging from transverse grooves (Beau lines) to complete nail shedding (onychomadesis)(Figure 3) may be seen.38,39 Nail findings in HFMD are due to transient inhibition of nail growth and present approximately 3 to 6 weeks after infection.40 Onychomadesis is seen in 30% to 68% of patients with HFMD.37,41,42 Nail findings in HFMD spontaneously resolve with nail growth (2–3 mm per month for fingernails and 1 mm per month for toenails) and do not require specific treatment. Although the appearance of nail changes associated with HFMD can be disturbing, dermatologists can reassure children and their parents that the nails will resolve with the next cycle of growth.
Kawasaki Disease—Kawasaki disease (KD) is a vasculitis primarily affecting children and infants. Although the specific pathogen and pathophysiology is not entirely clear, clinical observations have suggested an infectious cause, most likely a virus.43 In Japan, more than 15,000 cases of KD are documented annually, while approximately 4200 cases are seen in the United States.44 In a prospective study from 1984 to 1990, 4 of 26 (15.4%) patients with KD presented with nail manifestations during the late acute phase or early convalescent phase of disease. There were no significant associations between nail dystrophy and severity of KD, such as coronary artery aneurysm.45
Nail changes reported in children with KD include onychomadesis, onycholysis, orange-brown chromonychia, splinter hemorrhages, Beau lines, and pincer nails. In a review of nail changes associated with KD from 1980 to 2021, orange-brown transverse chromonychia, which may evolve into transverse leukonychia, was the most common nail finding reported, occurring in 17 of 31 (54.8%) patients.44 It has been hypothesized that nail changes may result from blood flow disturbance due to the underlying vasculitis.46 Nail changes appear several weeks after the onset of fever and are self-limited. Resolution occurs with nail growth, with no treatment required.
FUNGAL INFECTIONS
Onychomycosis
Onychomycosis is a fungal infection of the nails that occurs in 0.2% to 5.5% of pediatric patients, and its prevalence may be increasing, which may be due to environmental factors or increased rates of diabetes mellitus and obesity in the pediatric population.47 Onychomycosis represents 15.5% of nail dystrophies in pediatric patients.48 Some dermatologists treat presumptive onychomycosis without confirmation; however, we do not recommend that approach. Because the differential is broad and the duration of treatment is long, mycologic examination (potassium hydroxide preparation, fungal culture, polymerase chain reaction, and/or histopathology) should be obtained to confirm onychomycosis prior to initiation of antifungal management. Family members of affected individuals should be evaluated and treated, if indicated, for onychomycosis and tinea pedis, as household transmission is common.
Currently, there are 2 topical FDA-approved treatments for pediatric onychomycosis in children 6 years and older (Table 2).49,50 There is a discussion of the need for confirmatory testing for onychomycosis in children, particularly when systemic treatment is prescribed. In a retrospective review of 269 pediatric patients with onychomycosis prescribed terbinafine, 53.5% (n=144) underwent laboratory monitoring of liver function and complete blood cell counts, and 12.5% had grade 1 laboratory abnormalities either prior to (12/144 [8.3%]) or during (6/144 [4.2%]) therapy.51 Baseline transaminase monitoring is recommended, though subsequent routine laboratory monitoring in healthy children may have limited utility with associated increased costs, incidental findings, and patient discomfort and likely is not needed.51
Pediatric onychomycosis responds better to topical therapy than adult disease, and pediatric patients do not always require systemic treatment.52 Ciclopirox is not FDA approved for the treatment of pediatric onychomycosis, but in a 32-week clinical trial of ciclopirox lacquer 8% use in 40 patients, 77% (27/35) of treated patients achieved mycologic cure. Overall, 71% of treated patients (25/35) vs 22% (2/9) of controls achieved efficacy (defined as investigator global assessment score of 2 or lower).52 In an open-label, single-arm clinical trial assessing tavaborole solution 5% applied once daily for 48 weeks for the treatment of toenail onychomycosis in pediatric patients (aged 6–17 years), 36.2% (20/55) of patients achieved mycologic cure, and 8.5% (5/55) achieved complete cure at week 52 with mild or minimal adverse effects.53 In an open-label, phase 4 study of the safety and efficacy of efinaconazole solution 10% applied once daily for 48 weeks in pediatric patients (aged 6 to 16 years) (n=60), 65% (35/60) achieved mycologic cure, 42% (25/60) achieved clinical cure, and 40% (24/60) achieved complete cure at 52 weeks. The most common adverse effects of efinaconazole were local and included ingrown toenail (1/60), application-site dermatitis (1/60), application-site vesicles (1/60), and application-site pain (1/60).54
In a systematic review of systemic antifungals for onychomycosis in 151 pediatric patients, itraconazole, fluconazole, griseofulvin, and terbinafine resulted in complete cure rates similar to those of the adult population, with excellent safety profiles.55 Depending on the situation, initiation of treatment with topical medications followed by addition of systemic antifungal agents only if needed may be an appropriate course of action.
BACTERIAL INFECTIONS
Acute Paronychia
Acute paronychia is a nail-fold infection that develops after the protective nail barrier has been compromised.56 In children, thumb-sucking, nail-biting, frequent oral manipulation of the digits, and poor skin hygiene are risk factors. Acute paronychia also may develop in association with congenital malalignment of the great toenails.57
Clinical manifestations include localized pain, erythema, and nail fold edema (Figure 4). Purulent material and abscess formation may ensue. Staphylococcus aureus as well as methicillin-resistant S aureus and Streptococcus pyogenes are classically the most common causes of acute paronychia. Treatment of paronychia is based on severity. In mild cases, warm soaks with topical antibiotics are indicated. Oral antibiotics should be prescribed for more severe presentations. If there is no improvement after 48 hours, surgical drainage is required to facilitate healing.56
FINAL THOUGHTS
Inflammatory and infectious nail disorders in children are relatively common and may impact the physical and emotional well-being of young patients. By understanding the distinctive features of these nail disorders in pediatric patients, dermatologists can provide anticipatory guidance and informed treatment options to children and their parents. Further research is needed to expand our understanding of pediatric nail disorders and create targeted therapeutic interventions, particularly for NLP and psoriasis.
- Uber M, Carvalho VO, Abagge KT, et al. Clinical features and nail clippings in 52 children with psoriasis. Pediatr Dermatol. 2018;35:202-207. doi:10.1111/pde.13402
- Plachouri KM, Mulita F, Georgiou S. Management of pediatric nail psoriasis. Cutis. 2021;108:292-294. doi:10.12788/cutis.0386
- Smith RJ, Rubin AI. Pediatric nail disorders: a review. Curr Opin Pediatr. 2020;32:506-515. doi:10.1097/mop.0000000000000921
- Pourchot D, Bodemer C, Phan A, et al. Nail psoriasis: a systematic evaluation in 313 children with psoriasis. Pediatr Dermatol. 2017;34:58-63. doi:10.1111/pde.13028
- Richert B, André J. Nail disorders in children: diagnosis and management. Am J Clin Dermatol. 2011;12:101-112. doi:10.2165/11537110-000000000-00000
- Lee JYY. Severe 20-nail psoriasis successfully treated by low dose methotrexate. Dermatol Online J. 2009;15:8.
- Nogueira M, Paller AS, Torres T. Targeted therapy for pediatric psoriasis. Paediatr Drugs. May 2021;23:203-212. doi:10.1007/s40272-021-00443-5
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. Updated August 16, 2023. Accessed July 1, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Teran CG, Teran-Escalera CN, Balderrama C. A severe case of erythrodermic psoriasis associated with advanced nail and joint manifestations: a case report. J Med Case Rep. 2010;4:179. doi:10.1186/1752-1947-4-179
- Paller AS, Seyger MMB, Magariños GA, et al. Long-term efficacy and safety of up to 108 weeks of ixekizumab in pediatric patients with moderate to severe plaque psoriasis: the IXORA-PEDS randomized clinical trial. JAMA Dermatol. 2022;158:533-541. doi:10.1001/jamadermatol.2022.0655
- Diotallevi F, Simonetti O, Rizzetto G, et al. Biological treatments for pediatric psoriasis: state of the art and future perspectives. Int J Mol Sci. 2022;23:11128. doi:10.3390/ijms231911128
- Nash P, Mease PJ, Kirkham B, et al. Secukinumab provides sustained improvement in nail psoriasis, signs and symptoms of psoriatic arthritis and low rate of radiographic progression in patients with concomitant nail involvement: 2-year results from the Phase III FUTURE 5 study. Clin Exp Rheumatol. 2022;40:952-959. doi:10.55563/clinexprheumatol/3nuz51
- Wells LE, Evans T, Hilton R, et al. Use of secukinumab in a pediatric patient leads to significant improvement in nail psoriasis and psoriatic arthritis. Pediatr Dermatol. 2019;36:384-385. doi:10.1111/pde.13767
- Watabe D, Endoh K, Maeda F, et al. Childhood-onset psoriaticonycho-pachydermo-periostitis treated successfully with infliximab. Eur J Dermatol. 2015;25:506-508. doi:10.1684/ejd.2015.2616
- Pereira TM, Vieira AP, Fernandes JC, et al. Anti-TNF-alpha therapy in childhood pustular psoriasis. Dermatology. 2006;213:350-352. doi:10.1159/000096202
- Iorizzo M, Gioia Di Chiacchio N, Di Chiacchio N, et al. Intralesional steroid injections for inflammatory nail dystrophies in the pediatric population. Pediatr Dermatol. 2023;40:759-761. doi:10.1111/pde.15295
- Tosti A, Piraccini BM, Cambiaghi S, et al. Nail lichen planus in children: clinical features, response to treatment, and long-term follow-up. Arch Dermatol. 2001;137:1027-1032.
- Lipner SR. Nail lichen planus: a true nail emergency. J Am Acad Dermatol. 2019;80:e177-e178. doi:10.1016/j.jaad.2018.11.065
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Piraccini BM, Saccani E, Starace M, et al. Nail lichen planus: response to treatment and long term follow-up. Eur J Dermatol. 2010;20:489-496. doi:10.1684/ejd.2010.0952
- Mahajan R, Kaushik A, De D, et al. Pediatric trachyonychia- a retrospective study of 17 cases. Indian J Dermatol. 2021;66:689-690. doi:10.4103/ijd.ijd_42_21
- Leung AKC, Leong KF, Barankin B. Trachyonychia. J Pediatr. 2020;216:239-239.e1. doi:10.1016/j.jpeds.2019.08.034
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
- Jacobsen AA, Tosti A. Trachyonychia and twenty-nail dystrophy: a comprehensive review and discussion of diagnostic accuracy. Skin Appendage Disord. 2016;2:7-13. doi:10.1159/000445544
- Kumar MG, Ciliberto H, Bayliss SJ. Long-term follow-up of pediatric trachyonychia. Pediatr Dermatol. 2015;32:198-200. doi:10.1111/pde.12427
- Tosti A, Piraccini BM, Iorizzo M. Trachyonychia and related disorders: evaluation and treatment plans. Dermatolog Ther. 2002;15:121-125. doi:10.1046/j.1529-8019.2002.01511.x
- Leung AKC, Leong KF, Barankin B. Lichen striatus with nail involvement in a 6-year-old boy. Case Rep Pediatr. 2020;2020:1494760. doi:10.1155/2020/1494760
- Kim GW, Kim SH, Seo SH, et al. Lichen striatus with nail abnormality successfully treated with tacrolimus ointment. J Dermatol. 2009;36:616-617. doi:10.1111/j.1346-8138.2009.00720.x
- Iorizzo M, Rubin AI, Starace M. Nail lichen striatus: is dermoscopy useful for the diagnosis? Pediatr Dermatol. 2019;36:859-863. doi:10.1111/pde.13916
- Karp DL, Cohen BA. Onychodystrophy in lichen striatus. Pediatr Dermatol. 1993;10:359-361. doi:10.1111/j.1525-1470.1993.tb00399.x
- Tosti A, Peluso AM, Misciali C, et al. Nail lichen striatus: clinical features and long-term follow-up of five patients. J Am Acad Dermatol. 1997;36(6, pt 1):908-913. doi:10.1016/s0190-9622(97)80270-8
- Simpson EL, Thompson MM, Hanifin JM. Prevalence and morphology of hand eczema in patients with atopic dermatitis. Dermatitis. 2006;17:123-127. doi:10.2310/6620.2006.06005
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C. Nail alterations in 250 infant patients: a clinical study. J Eur Acad Dermatol Venereol. 2008;22:741-744. doi:10.1111/j.1468-3083.2008.02592.x
- Milanesi N, D’Erme AM, Gola M. Nail improvement during alitretinoin treatment: three case reports and review of the literature. Clin Exp Dermatol. 2015;40:533-536. doi:10.1111/ced.12584
- Chung BY, Choi YW, Kim HO, et al. Nail dystrophy in patients with atopic dermatitis and its association with disease severity. Ann Dermatol. 2019;31:121-126. doi:10.5021/ad.2019.31.2.121
- Navarro-Triviño FJ, Vega-Castillo JJ, Ruiz-Villaverde R. Nail changes successfully treated with dupilumab in a patient with severe atopic dermatitis. Australas J Dermatol. 2021;62:e468-e469. doi:10.1111/ajd.13633
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346. doi:10.1186/1471-2334-11-346
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283. doi:10.5021/ad.2014.26.2.280
- Verma S, Singal A. Nail changes in hand-foot-and-mouth disease (HFMD). Indian Dermatol Online J. 2021;12:656-657. doi:10.4103 /idoj.IDOJ_271_20
- Giordano LMC, de la Fuente LA, Lorca JMB, et al. Onychomadesis secondary to hand-foot-mouth disease: a frequent manifestation and cause of concern for parents. Article in Spanish. Rev Chil Pediatr. 2018;89:380-383. doi:10.4067/s0370-41062018005000203
- Justino MCA, da SMD, Souza MF, et al. Atypical hand-foot-mouth disease in Belém, Amazon region, northern Brazil, with detection of coxsackievirus A6. J Clin Virol. 2020;126:104307. doi:10.1016/j.jcv.2020.104307
- Cheng FF, Zhang BB, Cao ML, et al. Clinical characteristics of 68 children with atypical hand, foot, and mouth disease caused by coxsackievirus A6: a single-center retrospective analysis. Transl Pediatr. 2022;11:1502-1509. doi:10.21037/tp-22-352
- Nagata S. Causes of Kawasaki disease-from past to present. Front Pediatr. 2019;7:18. doi:10.3389/fped.2019.00018
- Mitsuishi T, Miyata K, Ando A, et al. Characteristic nail lesions in Kawasaki disease: case series and literature review. J Dermatol. 2022;49:232-238. doi:10.1111/1346-8138.16276
- Lindsley CB. Nail-bed lines in Kawasaki disease. Am J Dis Child. 1992;146:659-660. doi:10.1001/archpedi.1992.02160180017005
- Matsumura O, Nakagishi Y. Pincer nails upon convalescence from Kawasaki disease. J Pediatr. 2022;246:279. doi:10.1016/j.jpeds.2022.03.002
- Solís-Arias MP, García-Romero MT. Onychomycosis in children. a review. Int J Dermatol. 2017;56:123-130. doi:10.1111/ijd.13392
- Gupta AK, Mays RR, Versteeg SG, et al. Onychomycosis in children: safety and efficacy of antifungal agents. Pediatr Dermatol. 2018;35:552-559. doi:10.1111/pde.13561
- 49. Gupta AK, Venkataraman M, Shear NH, et al. Labeled use of efinaconazole topical solution 10% in treating onychomycosis in children and a review of the management of pediatric onychomycosis. Dermatol Ther. 2020;33:e13613. doi:10.1111/dth.13613
- Falotico JM, Lipner SR. Updated perspectives on the diagnosis and management of onychomycosis. Clin Cosmet Investig Dermatol. 2022;15:1933-1957. doi:10.2147/ccid.S362635
- Patel D, Castelo-Soccio LA, Rubin AI, et al. Laboratory monitoring during systemic terbinafine therapy for pediatric onychomycosis. JAMA Dermatol. 2017;153:1326-1327. doi:10.1001/jamadermatol.2017.4483
- Friedlander SF, Chan YC, Chan YH, et al. Onychomycosis does not always require systemic treatment for cure: a trial using topical therapy. Pediatr Dermatol. 2013;30:316-322. doi:10.1111/pde.12064
- Rich P, Spellman M, Purohit V, et al. Tavaborole 5% topical solution for the treatment of toenail onychomycosis in pediatric patients: results from a phase 4 open-label study. J Drugs Dermatol. 2019;18:190-195.
- Gupta AK, Venkataraman M, Abramovits W, et al. JUBLIA (efinaconazole 10% solution) in the treatment of pediatric onychomycosis. Skinmed. 2021;19:206-210.
- Gupta AK, Paquet M. Systemic antifungals to treat onychomycosis in children: a systematic review. Pediatr Dermatol. 2013;30:294-302. doi:10.1111/pde.12048
- Leggit JC. Acute and chronic paronychia. Am Fam Physician. 2017;96:44-51.
- Lipner SR, Scher RK. Congenital malalignment of the great toenails with acute paronychia. Pediatr Dermatol. 2016;33:e288-e289.doi:10.1111/pde.12924
- Uber M, Carvalho VO, Abagge KT, et al. Clinical features and nail clippings in 52 children with psoriasis. Pediatr Dermatol. 2018;35:202-207. doi:10.1111/pde.13402
- Plachouri KM, Mulita F, Georgiou S. Management of pediatric nail psoriasis. Cutis. 2021;108:292-294. doi:10.12788/cutis.0386
- Smith RJ, Rubin AI. Pediatric nail disorders: a review. Curr Opin Pediatr. 2020;32:506-515. doi:10.1097/mop.0000000000000921
- Pourchot D, Bodemer C, Phan A, et al. Nail psoriasis: a systematic evaluation in 313 children with psoriasis. Pediatr Dermatol. 2017;34:58-63. doi:10.1111/pde.13028
- Richert B, André J. Nail disorders in children: diagnosis and management. Am J Clin Dermatol. 2011;12:101-112. doi:10.2165/11537110-000000000-00000
- Lee JYY. Severe 20-nail psoriasis successfully treated by low dose methotrexate. Dermatol Online J. 2009;15:8.
- Nogueira M, Paller AS, Torres T. Targeted therapy for pediatric psoriasis. Paediatr Drugs. May 2021;23:203-212. doi:10.1007/s40272-021-00443-5
- Hanoodi M, Mittal M. Methotrexate. StatPearls [Internet]. Updated August 16, 2023. Accessed July 1, 2024. https://www.ncbi.nlm.nih.gov/books/NBK556114/
- Teran CG, Teran-Escalera CN, Balderrama C. A severe case of erythrodermic psoriasis associated with advanced nail and joint manifestations: a case report. J Med Case Rep. 2010;4:179. doi:10.1186/1752-1947-4-179
- Paller AS, Seyger MMB, Magariños GA, et al. Long-term efficacy and safety of up to 108 weeks of ixekizumab in pediatric patients with moderate to severe plaque psoriasis: the IXORA-PEDS randomized clinical trial. JAMA Dermatol. 2022;158:533-541. doi:10.1001/jamadermatol.2022.0655
- Diotallevi F, Simonetti O, Rizzetto G, et al. Biological treatments for pediatric psoriasis: state of the art and future perspectives. Int J Mol Sci. 2022;23:11128. doi:10.3390/ijms231911128
- Nash P, Mease PJ, Kirkham B, et al. Secukinumab provides sustained improvement in nail psoriasis, signs and symptoms of psoriatic arthritis and low rate of radiographic progression in patients with concomitant nail involvement: 2-year results from the Phase III FUTURE 5 study. Clin Exp Rheumatol. 2022;40:952-959. doi:10.55563/clinexprheumatol/3nuz51
- Wells LE, Evans T, Hilton R, et al. Use of secukinumab in a pediatric patient leads to significant improvement in nail psoriasis and psoriatic arthritis. Pediatr Dermatol. 2019;36:384-385. doi:10.1111/pde.13767
- Watabe D, Endoh K, Maeda F, et al. Childhood-onset psoriaticonycho-pachydermo-periostitis treated successfully with infliximab. Eur J Dermatol. 2015;25:506-508. doi:10.1684/ejd.2015.2616
- Pereira TM, Vieira AP, Fernandes JC, et al. Anti-TNF-alpha therapy in childhood pustular psoriasis. Dermatology. 2006;213:350-352. doi:10.1159/000096202
- Iorizzo M, Gioia Di Chiacchio N, Di Chiacchio N, et al. Intralesional steroid injections for inflammatory nail dystrophies in the pediatric population. Pediatr Dermatol. 2023;40:759-761. doi:10.1111/pde.15295
- Tosti A, Piraccini BM, Cambiaghi S, et al. Nail lichen planus in children: clinical features, response to treatment, and long-term follow-up. Arch Dermatol. 2001;137:1027-1032.
- Lipner SR. Nail lichen planus: a true nail emergency. J Am Acad Dermatol. 2019;80:e177-e178. doi:10.1016/j.jaad.2018.11.065
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Piraccini BM, Saccani E, Starace M, et al. Nail lichen planus: response to treatment and long term follow-up. Eur J Dermatol. 2010;20:489-496. doi:10.1684/ejd.2010.0952
- Mahajan R, Kaushik A, De D, et al. Pediatric trachyonychia- a retrospective study of 17 cases. Indian J Dermatol. 2021;66:689-690. doi:10.4103/ijd.ijd_42_21
- Leung AKC, Leong KF, Barankin B. Trachyonychia. J Pediatr. 2020;216:239-239.e1. doi:10.1016/j.jpeds.2019.08.034
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
- Jacobsen AA, Tosti A. Trachyonychia and twenty-nail dystrophy: a comprehensive review and discussion of diagnostic accuracy. Skin Appendage Disord. 2016;2:7-13. doi:10.1159/000445544
- Kumar MG, Ciliberto H, Bayliss SJ. Long-term follow-up of pediatric trachyonychia. Pediatr Dermatol. 2015;32:198-200. doi:10.1111/pde.12427
- Tosti A, Piraccini BM, Iorizzo M. Trachyonychia and related disorders: evaluation and treatment plans. Dermatolog Ther. 2002;15:121-125. doi:10.1046/j.1529-8019.2002.01511.x
- Leung AKC, Leong KF, Barankin B. Lichen striatus with nail involvement in a 6-year-old boy. Case Rep Pediatr. 2020;2020:1494760. doi:10.1155/2020/1494760
- Kim GW, Kim SH, Seo SH, et al. Lichen striatus with nail abnormality successfully treated with tacrolimus ointment. J Dermatol. 2009;36:616-617. doi:10.1111/j.1346-8138.2009.00720.x
- Iorizzo M, Rubin AI, Starace M. Nail lichen striatus: is dermoscopy useful for the diagnosis? Pediatr Dermatol. 2019;36:859-863. doi:10.1111/pde.13916
- Karp DL, Cohen BA. Onychodystrophy in lichen striatus. Pediatr Dermatol. 1993;10:359-361. doi:10.1111/j.1525-1470.1993.tb00399.x
- Tosti A, Peluso AM, Misciali C, et al. Nail lichen striatus: clinical features and long-term follow-up of five patients. J Am Acad Dermatol. 1997;36(6, pt 1):908-913. doi:10.1016/s0190-9622(97)80270-8
- Simpson EL, Thompson MM, Hanifin JM. Prevalence and morphology of hand eczema in patients with atopic dermatitis. Dermatitis. 2006;17:123-127. doi:10.2310/6620.2006.06005
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C. Nail alterations in 250 infant patients: a clinical study. J Eur Acad Dermatol Venereol. 2008;22:741-744. doi:10.1111/j.1468-3083.2008.02592.x
- Milanesi N, D’Erme AM, Gola M. Nail improvement during alitretinoin treatment: three case reports and review of the literature. Clin Exp Dermatol. 2015;40:533-536. doi:10.1111/ced.12584
- Chung BY, Choi YW, Kim HO, et al. Nail dystrophy in patients with atopic dermatitis and its association with disease severity. Ann Dermatol. 2019;31:121-126. doi:10.5021/ad.2019.31.2.121
- Navarro-Triviño FJ, Vega-Castillo JJ, Ruiz-Villaverde R. Nail changes successfully treated with dupilumab in a patient with severe atopic dermatitis. Australas J Dermatol. 2021;62:e468-e469. doi:10.1111/ajd.13633
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346. doi:10.1186/1471-2334-11-346
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283. doi:10.5021/ad.2014.26.2.280
- Verma S, Singal A. Nail changes in hand-foot-and-mouth disease (HFMD). Indian Dermatol Online J. 2021;12:656-657. doi:10.4103 /idoj.IDOJ_271_20
- Giordano LMC, de la Fuente LA, Lorca JMB, et al. Onychomadesis secondary to hand-foot-mouth disease: a frequent manifestation and cause of concern for parents. Article in Spanish. Rev Chil Pediatr. 2018;89:380-383. doi:10.4067/s0370-41062018005000203
- Justino MCA, da SMD, Souza MF, et al. Atypical hand-foot-mouth disease in Belém, Amazon region, northern Brazil, with detection of coxsackievirus A6. J Clin Virol. 2020;126:104307. doi:10.1016/j.jcv.2020.104307
- Cheng FF, Zhang BB, Cao ML, et al. Clinical characteristics of 68 children with atypical hand, foot, and mouth disease caused by coxsackievirus A6: a single-center retrospective analysis. Transl Pediatr. 2022;11:1502-1509. doi:10.21037/tp-22-352
- Nagata S. Causes of Kawasaki disease-from past to present. Front Pediatr. 2019;7:18. doi:10.3389/fped.2019.00018
- Mitsuishi T, Miyata K, Ando A, et al. Characteristic nail lesions in Kawasaki disease: case series and literature review. J Dermatol. 2022;49:232-238. doi:10.1111/1346-8138.16276
- Lindsley CB. Nail-bed lines in Kawasaki disease. Am J Dis Child. 1992;146:659-660. doi:10.1001/archpedi.1992.02160180017005
- Matsumura O, Nakagishi Y. Pincer nails upon convalescence from Kawasaki disease. J Pediatr. 2022;246:279. doi:10.1016/j.jpeds.2022.03.002
- Solís-Arias MP, García-Romero MT. Onychomycosis in children. a review. Int J Dermatol. 2017;56:123-130. doi:10.1111/ijd.13392
- Gupta AK, Mays RR, Versteeg SG, et al. Onychomycosis in children: safety and efficacy of antifungal agents. Pediatr Dermatol. 2018;35:552-559. doi:10.1111/pde.13561
- 49. Gupta AK, Venkataraman M, Shear NH, et al. Labeled use of efinaconazole topical solution 10% in treating onychomycosis in children and a review of the management of pediatric onychomycosis. Dermatol Ther. 2020;33:e13613. doi:10.1111/dth.13613
- Falotico JM, Lipner SR. Updated perspectives on the diagnosis and management of onychomycosis. Clin Cosmet Investig Dermatol. 2022;15:1933-1957. doi:10.2147/ccid.S362635
- Patel D, Castelo-Soccio LA, Rubin AI, et al. Laboratory monitoring during systemic terbinafine therapy for pediatric onychomycosis. JAMA Dermatol. 2017;153:1326-1327. doi:10.1001/jamadermatol.2017.4483
- Friedlander SF, Chan YC, Chan YH, et al. Onychomycosis does not always require systemic treatment for cure: a trial using topical therapy. Pediatr Dermatol. 2013;30:316-322. doi:10.1111/pde.12064
- Rich P, Spellman M, Purohit V, et al. Tavaborole 5% topical solution for the treatment of toenail onychomycosis in pediatric patients: results from a phase 4 open-label study. J Drugs Dermatol. 2019;18:190-195.
- Gupta AK, Venkataraman M, Abramovits W, et al. JUBLIA (efinaconazole 10% solution) in the treatment of pediatric onychomycosis. Skinmed. 2021;19:206-210.
- Gupta AK, Paquet M. Systemic antifungals to treat onychomycosis in children: a systematic review. Pediatr Dermatol. 2013;30:294-302. doi:10.1111/pde.12048
- Leggit JC. Acute and chronic paronychia. Am Fam Physician. 2017;96:44-51.
- Lipner SR, Scher RK. Congenital malalignment of the great toenails with acute paronychia. Pediatr Dermatol. 2016;33:e288-e289.doi:10.1111/pde.12924
Practice Points
- Nail plate pitting is the most common clinical sign of nail psoriasis in children.
- Nail changes are common in hand, foot, and mouth disease, with the most frequent being onychomadesis.
- Because onychomycosis may resemble other nail disorders, mycologic confirmation is recommended to avoid misdiagnosis.
- Many nail conditions in children self-resolve but recognizing these manifestations is important in providing anticipatory guidance to patients and caregivers.
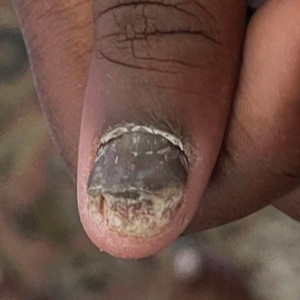
Draining Nodule of the Hand
The Diagnosis: Cutaneous Nocardiosis
The wound culture was positive for Nocardia farcinica. The patient received a 5-day course of intravenous sulfamethoxazole-trimethoprim in the hospital and was transitioned to oral sulfamethoxazoletrimethoprim (800 mg/160 mg taken as 1 tablet twice daily) for 6 months. Complete resolution of the infection was noted at 6-month follow-up (Figure).
Nocardia is a gram-positive, aerobic bacterium that typically is found in soil, water, and decaying organic matter.1 There are more than 50 species; N farcinica, Nocardia nova, and Nocardia asteroides are the leading causes of infection in humans and animals. Nocardia asteroides is the most common cause of infection in humans.1,2 Nocardiosis is an uncommon opportunistic infection that usually targets the skin, lungs, and central nervous system.3 Although it mainly affects individuals who are immunocompromised, up to 30% of infections can be seen in immunocompetent hosts who can contract cutaneous nocardiosis after experiencing traumatic injury to the skin.1
Nocardiosis is difficult to diagnose due to its diverse clinical presentations. For example, cutaneous nocardiosis can manifest similar to mycetoma, sporotrichosis, spider bites, nontuberculous mycobacteria such as Mycobacterium marinum, or methicillin-resistant Staphylococcus aureus infections, thus making cutaneous nocardiosis one of the great imitators.1 A culture is required for definitive diagnosis, as Nocardia grows well on nonselective media such as blood or Löwenstein-Jensen agar. It grows as waxy, pigmented, cerebriform colonies 3 to 5 days following incubation.3 The bacterium can be difficult to culture, and it is important to notify the microbiology laboratory if there is a high index of clinical suspicion for infection.
A history of exposure to gardening or handling animals can increase the risk for an individual contracting Nocardia.3 Although nocardiosis can be found across the world, it is native to tropical and subtropical climates such as those found in India, Africa, Latin America, and Southeast Asia.1 Infections mostly are observed in individuals aged 20 to 40 years and tend to affect men more than women. Lesions typically are seen on the lower extremities, but localized infections also can be found on the torso, neck, and upper extremities.1
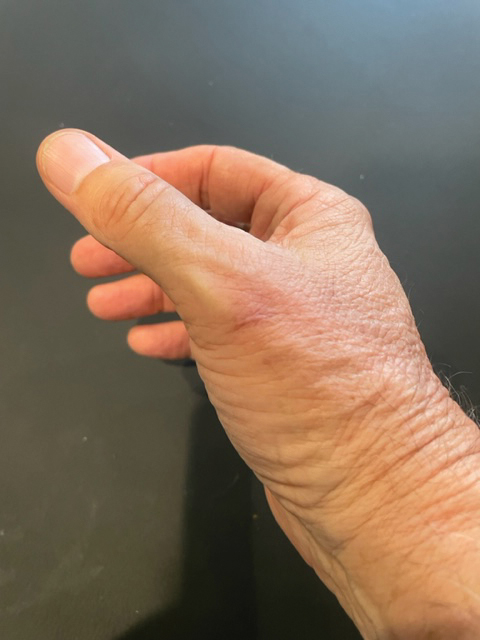
Cutaneous nocardiosis is a granulomatous infection encompassing both cutaneous and subcutaneous tissue, which ultimately can lead to injury of bone and viscera.1 Primary cutaneous nocardiosis can manifest as tumors or nodules that have a sporotrichoid pattern, in which they ascend along the lymphatics. Histopathology of infected tissue frequently shows a subcutaneous dermal infiltrate of neutrophils accompanied with abscess formation, and everlasting lesions may show signs of chronic inflammation and nonspecific granulomas.3
Treatment of nocardiosis should be guided by in vitro susceptibility tests. Sulfamethoxazole-trimethoprim 800 mg/160 mg taken as 1 tablet twice daily is the first-line option. The treatment duration is contingent on the extent, severity, and complications of infection but typically is 3 to 6 months.1
- Yu Q, Song J, Liu Y, et al. Progressive primary cutaneous nocardiosis in an immunocompetent patient. Cutis. 2023;111:E22-E25.
- Gaines RJ, Randall CJ, Ruland RT. Lymphocutaneous nocardiosis from commercially treated lumber: a case report. Cutis. 2006;78:249-251.
- Riswold KJ, Tjarks BJ, Kerkvliet AM. Cutaneous nocardiosis in an immunocompromised patient. Cutis. 2019;104:226-229.
The Diagnosis: Cutaneous Nocardiosis
The wound culture was positive for Nocardia farcinica. The patient received a 5-day course of intravenous sulfamethoxazole-trimethoprim in the hospital and was transitioned to oral sulfamethoxazoletrimethoprim (800 mg/160 mg taken as 1 tablet twice daily) for 6 months. Complete resolution of the infection was noted at 6-month follow-up (Figure).
Nocardia is a gram-positive, aerobic bacterium that typically is found in soil, water, and decaying organic matter.1 There are more than 50 species; N farcinica, Nocardia nova, and Nocardia asteroides are the leading causes of infection in humans and animals. Nocardia asteroides is the most common cause of infection in humans.1,2 Nocardiosis is an uncommon opportunistic infection that usually targets the skin, lungs, and central nervous system.3 Although it mainly affects individuals who are immunocompromised, up to 30% of infections can be seen in immunocompetent hosts who can contract cutaneous nocardiosis after experiencing traumatic injury to the skin.1
Nocardiosis is difficult to diagnose due to its diverse clinical presentations. For example, cutaneous nocardiosis can manifest similar to mycetoma, sporotrichosis, spider bites, nontuberculous mycobacteria such as Mycobacterium marinum, or methicillin-resistant Staphylococcus aureus infections, thus making cutaneous nocardiosis one of the great imitators.1 A culture is required for definitive diagnosis, as Nocardia grows well on nonselective media such as blood or Löwenstein-Jensen agar. It grows as waxy, pigmented, cerebriform colonies 3 to 5 days following incubation.3 The bacterium can be difficult to culture, and it is important to notify the microbiology laboratory if there is a high index of clinical suspicion for infection.
A history of exposure to gardening or handling animals can increase the risk for an individual contracting Nocardia.3 Although nocardiosis can be found across the world, it is native to tropical and subtropical climates such as those found in India, Africa, Latin America, and Southeast Asia.1 Infections mostly are observed in individuals aged 20 to 40 years and tend to affect men more than women. Lesions typically are seen on the lower extremities, but localized infections also can be found on the torso, neck, and upper extremities.1
Cutaneous nocardiosis is a granulomatous infection encompassing both cutaneous and subcutaneous tissue, which ultimately can lead to injury of bone and viscera.1 Primary cutaneous nocardiosis can manifest as tumors or nodules that have a sporotrichoid pattern, in which they ascend along the lymphatics. Histopathology of infected tissue frequently shows a subcutaneous dermal infiltrate of neutrophils accompanied with abscess formation, and everlasting lesions may show signs of chronic inflammation and nonspecific granulomas.3
Treatment of nocardiosis should be guided by in vitro susceptibility tests. Sulfamethoxazole-trimethoprim 800 mg/160 mg taken as 1 tablet twice daily is the first-line option. The treatment duration is contingent on the extent, severity, and complications of infection but typically is 3 to 6 months.1
The Diagnosis: Cutaneous Nocardiosis
The wound culture was positive for Nocardia farcinica. The patient received a 5-day course of intravenous sulfamethoxazole-trimethoprim in the hospital and was transitioned to oral sulfamethoxazoletrimethoprim (800 mg/160 mg taken as 1 tablet twice daily) for 6 months. Complete resolution of the infection was noted at 6-month follow-up (Figure).
Nocardia is a gram-positive, aerobic bacterium that typically is found in soil, water, and decaying organic matter.1 There are more than 50 species; N farcinica, Nocardia nova, and Nocardia asteroides are the leading causes of infection in humans and animals. Nocardia asteroides is the most common cause of infection in humans.1,2 Nocardiosis is an uncommon opportunistic infection that usually targets the skin, lungs, and central nervous system.3 Although it mainly affects individuals who are immunocompromised, up to 30% of infections can be seen in immunocompetent hosts who can contract cutaneous nocardiosis after experiencing traumatic injury to the skin.1
Nocardiosis is difficult to diagnose due to its diverse clinical presentations. For example, cutaneous nocardiosis can manifest similar to mycetoma, sporotrichosis, spider bites, nontuberculous mycobacteria such as Mycobacterium marinum, or methicillin-resistant Staphylococcus aureus infections, thus making cutaneous nocardiosis one of the great imitators.1 A culture is required for definitive diagnosis, as Nocardia grows well on nonselective media such as blood or Löwenstein-Jensen agar. It grows as waxy, pigmented, cerebriform colonies 3 to 5 days following incubation.3 The bacterium can be difficult to culture, and it is important to notify the microbiology laboratory if there is a high index of clinical suspicion for infection.
A history of exposure to gardening or handling animals can increase the risk for an individual contracting Nocardia.3 Although nocardiosis can be found across the world, it is native to tropical and subtropical climates such as those found in India, Africa, Latin America, and Southeast Asia.1 Infections mostly are observed in individuals aged 20 to 40 years and tend to affect men more than women. Lesions typically are seen on the lower extremities, but localized infections also can be found on the torso, neck, and upper extremities.1
Cutaneous nocardiosis is a granulomatous infection encompassing both cutaneous and subcutaneous tissue, which ultimately can lead to injury of bone and viscera.1 Primary cutaneous nocardiosis can manifest as tumors or nodules that have a sporotrichoid pattern, in which they ascend along the lymphatics. Histopathology of infected tissue frequently shows a subcutaneous dermal infiltrate of neutrophils accompanied with abscess formation, and everlasting lesions may show signs of chronic inflammation and nonspecific granulomas.3
Treatment of nocardiosis should be guided by in vitro susceptibility tests. Sulfamethoxazole-trimethoprim 800 mg/160 mg taken as 1 tablet twice daily is the first-line option. The treatment duration is contingent on the extent, severity, and complications of infection but typically is 3 to 6 months.1
- Yu Q, Song J, Liu Y, et al. Progressive primary cutaneous nocardiosis in an immunocompetent patient. Cutis. 2023;111:E22-E25.
- Gaines RJ, Randall CJ, Ruland RT. Lymphocutaneous nocardiosis from commercially treated lumber: a case report. Cutis. 2006;78:249-251.
- Riswold KJ, Tjarks BJ, Kerkvliet AM. Cutaneous nocardiosis in an immunocompromised patient. Cutis. 2019;104:226-229.
- Yu Q, Song J, Liu Y, et al. Progressive primary cutaneous nocardiosis in an immunocompetent patient. Cutis. 2023;111:E22-E25.
- Gaines RJ, Randall CJ, Ruland RT. Lymphocutaneous nocardiosis from commercially treated lumber: a case report. Cutis. 2006;78:249-251.
- Riswold KJ, Tjarks BJ, Kerkvliet AM. Cutaneous nocardiosis in an immunocompromised patient. Cutis. 2019;104:226-229.
A 67-year-old man presented to the emergency department with a draining nodule on the right hand of 4 days’ duration. He reported that the swelling and redness started 1 hour after handling a succulent plant. The following day, the nodule increased in size and exudated yellow pus. He presented with swelling of the thumb and hand, which resulted in a decreased range of motion. He had a history of prediabetes and denied any recent travel, allergies, or animal exposures. Physical examination revealed a draining nodule on the dorsal aspect of the right hand that measured approximately 15×15 mm with surrounding erythema and tenderness. There also was progression of ascending erythema up to the axilla. The patient was admitted to the hospital.
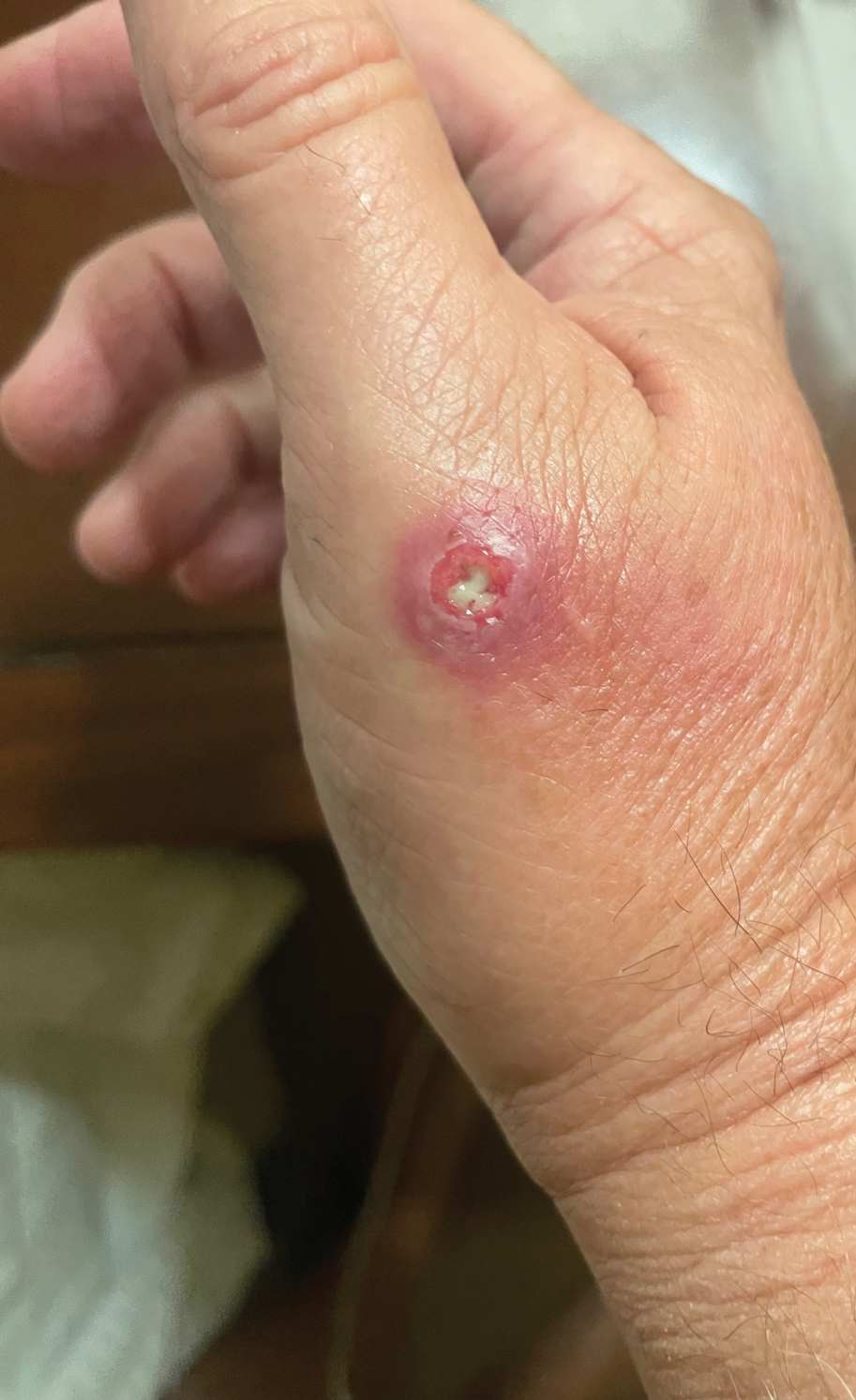
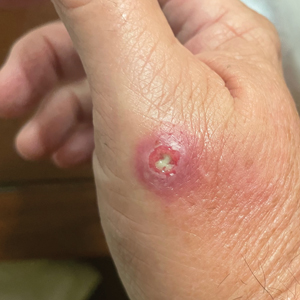
Histiocytoid Pyoderma Gangrenosum: A Challenging Case With Features of Sweet Syndrome
To the Editor:
Neutrophilic dermatoses—a group of inflammatory cutaneous conditions—include acute febrile neutrophilic dermatosis (Sweet syndrome), pyoderma gangrenosum, and neutrophilic dermatosis of the dorsal hands. Histopathology shows a dense dermal infiltrate of mature neutrophils. In 2005, the histiocytoid subtype of Sweet syndrome was introduced with histopathologic findings of a dermal infiltrate composed of immature myeloid cells that resemble histiocytes in appearance but stain strongly with neutrophil markers on immunohistochemistry.1 We present a case of histiocytoid pyoderma gangrenosum with histopathology that showed a dense dermal histiocytoid infiltrate with strong positivity for neutrophil markers on immunohistochemistry.
An 85-year-old man was seen by dermatology in the inpatient setting for a new-onset painful abdominal wound. He had a medical history of myelodysplastic syndrome (MDS), high-grade invasive papillary urothelial carcinoma of the bladder, and a recent diagnosis of low-grade invasive ascending colon adenocarcinoma. Ten days prior he underwent a right colectomy without intraoperative complications that was followed by septic shock. Workup with urinalysis and urine culture showed minimal pyuria with Pseudomonas aeruginosa. Additional studies, including blood cultures, abdominal wound cultures, computed tomography of the abdomen and pelvis, renal ultrasound, and chest radiographs, were unremarkable and showed no signs of surgical site infection, intra-abdominal or pelvic abscess formation, or pulmonary embolism. Broad-spectrum antibiotics—vancomycin and piperacillin-tazobactam—were started. Persistent fever (Tmax of 102.3 °F [39.1 °C]) and leukocytosis (45.3×109/L [4.2–10×109/L]) despite antibiotic therapy, increasing pressor requirements, and progressive painful erythema and purulence at the abdominal surgical site led to debridement of the wound by the general surgery team on day 9 following the initial surgery due to suspected necrotizing infection. Within 24 hours, dermatology was consulted for continued rapid expansion of the wound. Physical examination of the abdomen revealed a large, well-demarcated, pink-red, indurated, ulcerated plaque with clear to purulent exudate and superficial erosions with violaceous undermined borders extending centrifugally from the abdominal surgical incision line (Figure 1A). Two punch biopsies sent for histopathologic evaluation and tissue culture showed dermal edema with a dense histiocytic infiltrate with nodular foci and admixed mature neutrophils to a lesser degree (Figure 2). Special staining was negative for bacteria, fungi, and mycobacteria. Immunohistochemistry revealed positive staining of the dermal inflammatory infiltrate with CD68, myeloperoxidase, and lysozyme, as well as negative staining with CD34 (Figure 3). These findings were suggestive of a histiocytoid neutrophilic dermatosis such as Sweet syndrome or pyoderma gangrenosum. Due to the morphology of the solitary lesion and the abrupt exacerbation shortly after surgical intervention, the patient was diagnosed with histiocytoid pyoderma gangrenosum. At the same time, the patient’s septic shock was treated with intravenous hydrocortisone (100 mg 3 times daily) for 2 days and also achieved a prompt response in the cutaneous symptoms (Figure 1B).
Sweet syndrome and pyoderma gangrenosum are considered distinct neutrophilic dermatoses that rarely coexist but share several clinical and histopathologic features, which can become a diagnostic challenge.2 Both conditions can manifest clinically as abrupt-onset, tender, erythematous papules; vesiculopustular lesions; or bullae with ulcerative changes. They also exhibit pathergy; present with systemic symptoms such as pyrexia, malaise, and joint pain; are associated with underlying systemic conditions such as infections and/or malignancy; demonstrate a dense neutrophilic infiltrate in the dermis on histopathology; and respond promptly to systemic corticosteroids.2-6 Bullous Sweet syndrome, which can present as vesicles, pustules, or bullae that progress to superficial ulcerations, may represent a variant of neutrophilic dermatosis characterized by features seen in both Sweet syndrome and pyoderma gangrenosum, suggesting that these 2 conditions may be on a spectrum.5Clinical features such as erythema with a blue, gray, or purple hue; undermined and ragged borders; and healing of skin lesions with atrophic or cribriform scarring may favor pyoderma gangrenosum, whereas a dull red or plum color and resolution of lesions without scarring may support the diagnosis of Sweet syndrome.7 Although both conditions can exhibit pathergy secondary to minor skin trauma such as venipuncture and biopsies,2,3,5,8 Sweet syndrome rarely has been described to develop after surgery in a patient without a known history of the condition.9 In contrast, postsurgical pyoderma gangrenosum has been well described as secondary to the pathergy phenomenon.5
Our patient was favored to have pyoderma gangrenosum given the solitary lesion, its abrupt development after surgery, and the morphology of the lesion that exhibited a large violaceous to red ulcerative and exudative plaque with undermined borders with atrophic scarring. In patients with skin disease that cannot be distinguished with certainty as either Sweet syndrome or pyoderma gangrenosum, it is essential to recognize that, as neutrophilic dermatoses, both conditions can be managed with either the first-line treatment option of high-dose systemic steroids or one of the shared alternative first-line or second-line steroid-sparing treatments, such as dapsone and cyclosporine.2
Although the exact pathogenesis of pyoderma gangrenosum remains to be fully understood, paraneoplastic pyoderma gangrenosum is a frequently described phenomenon.10,11 Our patient’s history of multiple malignancies, both solid and hematologic, supports the likelihood of malignancy-induced pyoderma gangrenosum; however, given his history of MDS, several other conditions were ruled out prior to making the diagnosis of pyoderma gangrenosum.
Classically, neutrophilic dermatoses such as pyoderma gangrenosum have a dense dermal neutrophilic infiltrate. Concurrent myeloproliferative disorders can alter the maturation of leukocytes, subsequently leading to an atypical appearance of the inflammatory cells on histopathology. Further, in the setting of myeloproliferative disorders, conditions such as leukemia cutis, in which there can be a cutaneous infiltrate of immature or mature myeloid or lymphocytic cells, must be considered. To ensure our patient’s abdominal skin changes were not a cutaneous manifestation of hematologic malignancy, immunohistochemical staining with CD20 and CD3 was performed and showed only the rare presence of B and T lymphocytes, respectively. Staining with CD34 for lymphocytic and myeloid progenitor cells was negative in the dermal infiltrate and further reduced the likelihood of leukemia cutis. Alternatively, patients can have aleukemic cutaneous myeloid sarcoma or leukemia cutis without an underlying hematologic condition or with latent peripheral blood or bone marrow myeloproliferative disorder, but our patient’s history of MDS eliminated this possibility.12 After exclusion of cutaneous infiltration by malignant leukocytes, our patient was diagnosed with histiocytoid neutrophilic dermatosis.
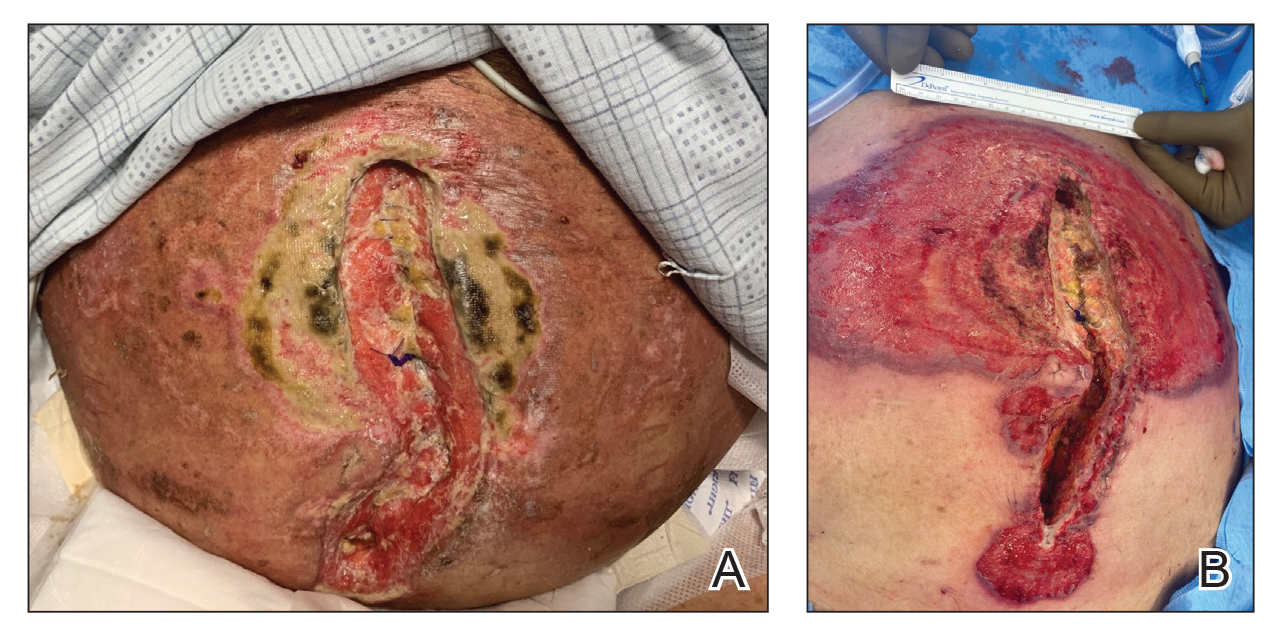
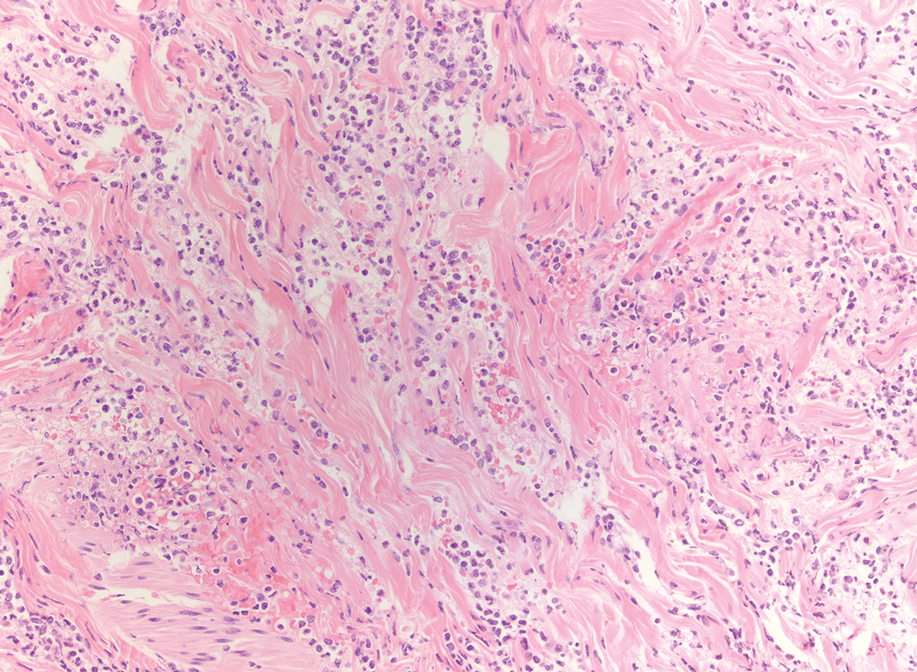
Multiple reports have described histiocytoid Sweet syndrome, in which there is a dense dermal histiocytoid infiltrate on histopathology that demonstrates myeloid lineage with immunologic staining.1,13 The typical pattern of histiocytoid Sweet syndrome includes a predominantly unaffected epidermis with papillary dermal edema, an absence of vasculitis, and a dense dermal infiltrate primarily composed of immature histiocytelike mononuclear cells with a basophilic elongated, twisted, or kidney-shaped nucleus and pale eosinophilic cytoplasm.1,13 In an analogous manner, Morin et al12 described a patient with congenital hypogammaglobulinemia who presented with lesions that clinically resembled pyoderma gangrenosum but revealed a dense dermal infiltrate mostly made of large immature histiocytoid mononuclear cells on histopathology, consistent with the histopathologic features observed in histiocytoid Sweet syndrome. The patient ultimately was diagnosed with histiocytoid pyoderma gangrenosum. Similarly, we believe that our patient also developed histiocytoid pyoderma gangrenosum. As with histiocytoid Sweet syndrome, this diagnosis is based on histopathologic and immunohistochemical findings of a dense dermal infiltrate composed of histiocyte-resembling immature neutrophils.
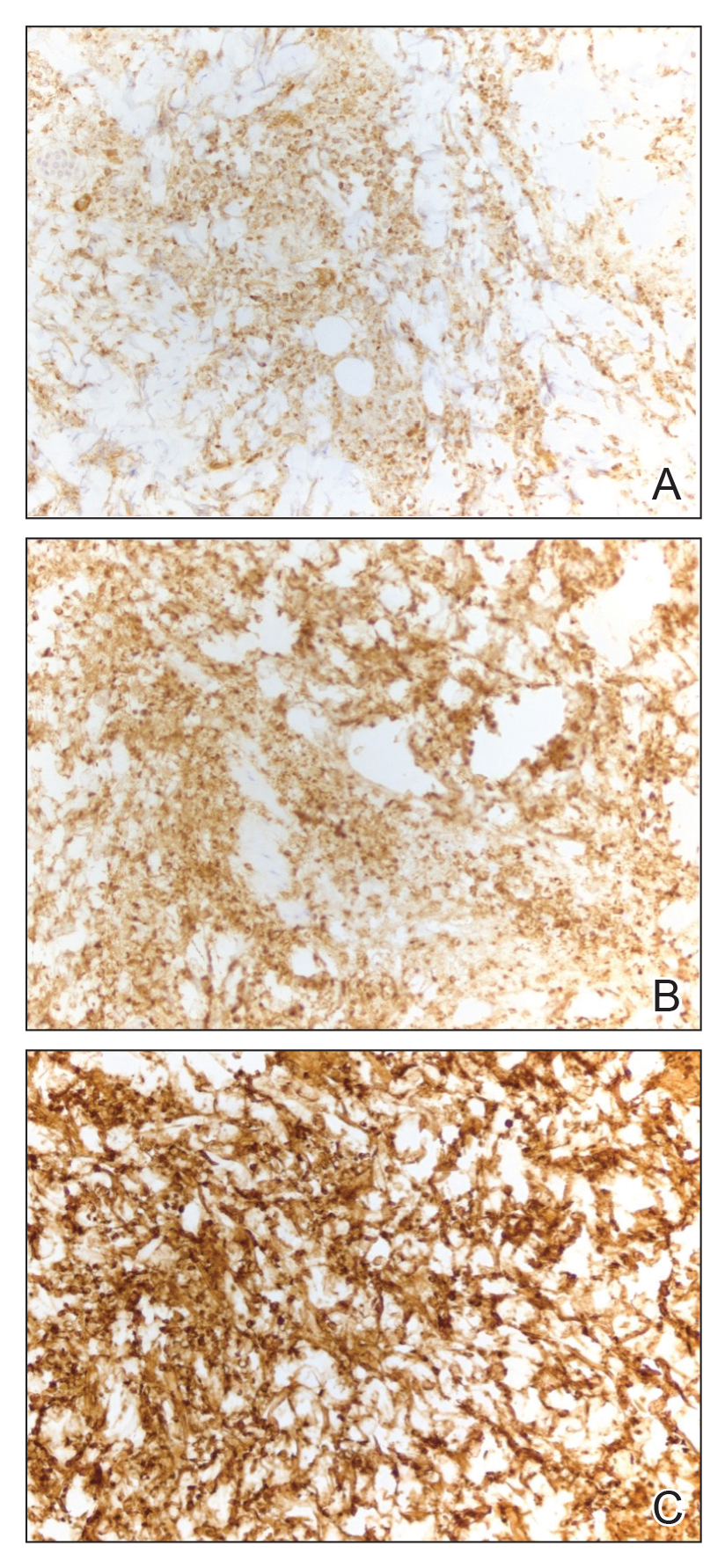
Typically, pyoderma gangrenosum responds promptly to treatment with systemic corticosteroids.4 Steroid-sparing agents such as cyclosporine, azathioprine, dapsone, and tumor necrosis factor α inhibitors also may be used.4,10 In the setting of MDS, clearance of pyoderma gangrenosum has been reported upon treatment of the underlying malignancy,14 high-dose systemic corticosteroids,11,15 cyclosporine with systemic steroids,16 thalidomide,17 combination therapy with thalidomide and interferon alfa-2a,18 and ustekinumab with vacuum-assisted closure therapy.19 Our patient’s histiocytoid pyoderma gangrenosum in the setting of solid and hematologic malignancy cleared rapidly with high-dose systemic hydrocortisone.
In the setting of malignancy, as in our patient, neutrophilic dermatoses may develop from an aberrant immune system or tumor-induced cytokine dysregulation that leads to increased neutrophil production or dysfunction.4,10,11 Although our patient’s MDS may have contributed to the atypical appearance of the dermal inflammatory infiltrate, it is unclear whether the hematologic disorder increased his risk for the histiocytoid variant of neutrophilic dermatoses. Alegría-Landa et al13 reported that histiocytoid Sweet syndrome is associated with hematologic malignancy at a similar frequency as classic Sweet syndrome. It is unknown if histiocytoid pyoderma gangrenosum would have a strong association with hematologic malignancy. Future reports may elucidate a better understanding of the histiocytoid subtype of pyoderma gangrenosum and its clinical implications.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Wallach D, Vignon-Pennamen MD. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med. 1997;73:65-68.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:E74-E76.
- Shah M, Sachdeva M, Gefri A, et al. Paraneoplastic pyoderma gangrenosum in solid organ malignancy: a literature review. Int J Dermatol. 2020;59:154-158.
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359.
- Morin CB, Côté B, Belisle A. An interesting case of pyoderma gangrenosum with immature histiocytoid neutrophils. J Cutan Pathol. 2018;45:63-66.
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Saleh MFM, Saunthararajah Y. Severe pyoderma gangrenosum caused by myelodysplastic syndrome successfully treated with decitabine administered by a noncytotoxic regimen. Clin Case Rep. 2017;5:2025-2027.
- Yamauchi R, Ishida K, Iwashima Y, et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J Infect Chemother. 2003;9:268-271.
- Ha JW, Hahm JE, Kim KS, et al. A case of pyoderma gangrenosum with myelodysplastic syndrome. Ann Dermatol. 2018;30:392-393.
- Malkan UY, Gunes G, Eliacik E, et al. Treatment of pyoderma gangrenosum with thalidomide in a myelodysplastic syndrome case. Int J Med Case Rep. 2016;9:61-64.
- Koca E, Duman AE, Cetiner D, et al. Successful treatment of myelodysplastic syndrome-induced pyoderma gangrenosum. Neth J Med. 2006;64:422-424.
- Nieto D, Sendagorta E, Rueda JM, et al. Successful treatment with ustekinumab and vacuum-assisted closure therapy in recalcitrant myelodysplastic syndrome-associated pyoderma gangrenosum: case report and literature review. Clin Exp Dermatol. 2019;44:116-119.
To the Editor:
Neutrophilic dermatoses—a group of inflammatory cutaneous conditions—include acute febrile neutrophilic dermatosis (Sweet syndrome), pyoderma gangrenosum, and neutrophilic dermatosis of the dorsal hands. Histopathology shows a dense dermal infiltrate of mature neutrophils. In 2005, the histiocytoid subtype of Sweet syndrome was introduced with histopathologic findings of a dermal infiltrate composed of immature myeloid cells that resemble histiocytes in appearance but stain strongly with neutrophil markers on immunohistochemistry.1 We present a case of histiocytoid pyoderma gangrenosum with histopathology that showed a dense dermal histiocytoid infiltrate with strong positivity for neutrophil markers on immunohistochemistry.
An 85-year-old man was seen by dermatology in the inpatient setting for a new-onset painful abdominal wound. He had a medical history of myelodysplastic syndrome (MDS), high-grade invasive papillary urothelial carcinoma of the bladder, and a recent diagnosis of low-grade invasive ascending colon adenocarcinoma. Ten days prior he underwent a right colectomy without intraoperative complications that was followed by septic shock. Workup with urinalysis and urine culture showed minimal pyuria with Pseudomonas aeruginosa. Additional studies, including blood cultures, abdominal wound cultures, computed tomography of the abdomen and pelvis, renal ultrasound, and chest radiographs, were unremarkable and showed no signs of surgical site infection, intra-abdominal or pelvic abscess formation, or pulmonary embolism. Broad-spectrum antibiotics—vancomycin and piperacillin-tazobactam—were started. Persistent fever (Tmax of 102.3 °F [39.1 °C]) and leukocytosis (45.3×109/L [4.2–10×109/L]) despite antibiotic therapy, increasing pressor requirements, and progressive painful erythema and purulence at the abdominal surgical site led to debridement of the wound by the general surgery team on day 9 following the initial surgery due to suspected necrotizing infection. Within 24 hours, dermatology was consulted for continued rapid expansion of the wound. Physical examination of the abdomen revealed a large, well-demarcated, pink-red, indurated, ulcerated plaque with clear to purulent exudate and superficial erosions with violaceous undermined borders extending centrifugally from the abdominal surgical incision line (Figure 1A). Two punch biopsies sent for histopathologic evaluation and tissue culture showed dermal edema with a dense histiocytic infiltrate with nodular foci and admixed mature neutrophils to a lesser degree (Figure 2). Special staining was negative for bacteria, fungi, and mycobacteria. Immunohistochemistry revealed positive staining of the dermal inflammatory infiltrate with CD68, myeloperoxidase, and lysozyme, as well as negative staining with CD34 (Figure 3). These findings were suggestive of a histiocytoid neutrophilic dermatosis such as Sweet syndrome or pyoderma gangrenosum. Due to the morphology of the solitary lesion and the abrupt exacerbation shortly after surgical intervention, the patient was diagnosed with histiocytoid pyoderma gangrenosum. At the same time, the patient’s septic shock was treated with intravenous hydrocortisone (100 mg 3 times daily) for 2 days and also achieved a prompt response in the cutaneous symptoms (Figure 1B).
Sweet syndrome and pyoderma gangrenosum are considered distinct neutrophilic dermatoses that rarely coexist but share several clinical and histopathologic features, which can become a diagnostic challenge.2 Both conditions can manifest clinically as abrupt-onset, tender, erythematous papules; vesiculopustular lesions; or bullae with ulcerative changes. They also exhibit pathergy; present with systemic symptoms such as pyrexia, malaise, and joint pain; are associated with underlying systemic conditions such as infections and/or malignancy; demonstrate a dense neutrophilic infiltrate in the dermis on histopathology; and respond promptly to systemic corticosteroids.2-6 Bullous Sweet syndrome, which can present as vesicles, pustules, or bullae that progress to superficial ulcerations, may represent a variant of neutrophilic dermatosis characterized by features seen in both Sweet syndrome and pyoderma gangrenosum, suggesting that these 2 conditions may be on a spectrum.5Clinical features such as erythema with a blue, gray, or purple hue; undermined and ragged borders; and healing of skin lesions with atrophic or cribriform scarring may favor pyoderma gangrenosum, whereas a dull red or plum color and resolution of lesions without scarring may support the diagnosis of Sweet syndrome.7 Although both conditions can exhibit pathergy secondary to minor skin trauma such as venipuncture and biopsies,2,3,5,8 Sweet syndrome rarely has been described to develop after surgery in a patient without a known history of the condition.9 In contrast, postsurgical pyoderma gangrenosum has been well described as secondary to the pathergy phenomenon.5
Our patient was favored to have pyoderma gangrenosum given the solitary lesion, its abrupt development after surgery, and the morphology of the lesion that exhibited a large violaceous to red ulcerative and exudative plaque with undermined borders with atrophic scarring. In patients with skin disease that cannot be distinguished with certainty as either Sweet syndrome or pyoderma gangrenosum, it is essential to recognize that, as neutrophilic dermatoses, both conditions can be managed with either the first-line treatment option of high-dose systemic steroids or one of the shared alternative first-line or second-line steroid-sparing treatments, such as dapsone and cyclosporine.2
Although the exact pathogenesis of pyoderma gangrenosum remains to be fully understood, paraneoplastic pyoderma gangrenosum is a frequently described phenomenon.10,11 Our patient’s history of multiple malignancies, both solid and hematologic, supports the likelihood of malignancy-induced pyoderma gangrenosum; however, given his history of MDS, several other conditions were ruled out prior to making the diagnosis of pyoderma gangrenosum.
Classically, neutrophilic dermatoses such as pyoderma gangrenosum have a dense dermal neutrophilic infiltrate. Concurrent myeloproliferative disorders can alter the maturation of leukocytes, subsequently leading to an atypical appearance of the inflammatory cells on histopathology. Further, in the setting of myeloproliferative disorders, conditions such as leukemia cutis, in which there can be a cutaneous infiltrate of immature or mature myeloid or lymphocytic cells, must be considered. To ensure our patient’s abdominal skin changes were not a cutaneous manifestation of hematologic malignancy, immunohistochemical staining with CD20 and CD3 was performed and showed only the rare presence of B and T lymphocytes, respectively. Staining with CD34 for lymphocytic and myeloid progenitor cells was negative in the dermal infiltrate and further reduced the likelihood of leukemia cutis. Alternatively, patients can have aleukemic cutaneous myeloid sarcoma or leukemia cutis without an underlying hematologic condition or with latent peripheral blood or bone marrow myeloproliferative disorder, but our patient’s history of MDS eliminated this possibility.12 After exclusion of cutaneous infiltration by malignant leukocytes, our patient was diagnosed with histiocytoid neutrophilic dermatosis.
Multiple reports have described histiocytoid Sweet syndrome, in which there is a dense dermal histiocytoid infiltrate on histopathology that demonstrates myeloid lineage with immunologic staining.1,13 The typical pattern of histiocytoid Sweet syndrome includes a predominantly unaffected epidermis with papillary dermal edema, an absence of vasculitis, and a dense dermal infiltrate primarily composed of immature histiocytelike mononuclear cells with a basophilic elongated, twisted, or kidney-shaped nucleus and pale eosinophilic cytoplasm.1,13 In an analogous manner, Morin et al12 described a patient with congenital hypogammaglobulinemia who presented with lesions that clinically resembled pyoderma gangrenosum but revealed a dense dermal infiltrate mostly made of large immature histiocytoid mononuclear cells on histopathology, consistent with the histopathologic features observed in histiocytoid Sweet syndrome. The patient ultimately was diagnosed with histiocytoid pyoderma gangrenosum. Similarly, we believe that our patient also developed histiocytoid pyoderma gangrenosum. As with histiocytoid Sweet syndrome, this diagnosis is based on histopathologic and immunohistochemical findings of a dense dermal infiltrate composed of histiocyte-resembling immature neutrophils.
Typically, pyoderma gangrenosum responds promptly to treatment with systemic corticosteroids.4 Steroid-sparing agents such as cyclosporine, azathioprine, dapsone, and tumor necrosis factor α inhibitors also may be used.4,10 In the setting of MDS, clearance of pyoderma gangrenosum has been reported upon treatment of the underlying malignancy,14 high-dose systemic corticosteroids,11,15 cyclosporine with systemic steroids,16 thalidomide,17 combination therapy with thalidomide and interferon alfa-2a,18 and ustekinumab with vacuum-assisted closure therapy.19 Our patient’s histiocytoid pyoderma gangrenosum in the setting of solid and hematologic malignancy cleared rapidly with high-dose systemic hydrocortisone.
In the setting of malignancy, as in our patient, neutrophilic dermatoses may develop from an aberrant immune system or tumor-induced cytokine dysregulation that leads to increased neutrophil production or dysfunction.4,10,11 Although our patient’s MDS may have contributed to the atypical appearance of the dermal inflammatory infiltrate, it is unclear whether the hematologic disorder increased his risk for the histiocytoid variant of neutrophilic dermatoses. Alegría-Landa et al13 reported that histiocytoid Sweet syndrome is associated with hematologic malignancy at a similar frequency as classic Sweet syndrome. It is unknown if histiocytoid pyoderma gangrenosum would have a strong association with hematologic malignancy. Future reports may elucidate a better understanding of the histiocytoid subtype of pyoderma gangrenosum and its clinical implications.
To the Editor:
Neutrophilic dermatoses—a group of inflammatory cutaneous conditions—include acute febrile neutrophilic dermatosis (Sweet syndrome), pyoderma gangrenosum, and neutrophilic dermatosis of the dorsal hands. Histopathology shows a dense dermal infiltrate of mature neutrophils. In 2005, the histiocytoid subtype of Sweet syndrome was introduced with histopathologic findings of a dermal infiltrate composed of immature myeloid cells that resemble histiocytes in appearance but stain strongly with neutrophil markers on immunohistochemistry.1 We present a case of histiocytoid pyoderma gangrenosum with histopathology that showed a dense dermal histiocytoid infiltrate with strong positivity for neutrophil markers on immunohistochemistry.
An 85-year-old man was seen by dermatology in the inpatient setting for a new-onset painful abdominal wound. He had a medical history of myelodysplastic syndrome (MDS), high-grade invasive papillary urothelial carcinoma of the bladder, and a recent diagnosis of low-grade invasive ascending colon adenocarcinoma. Ten days prior he underwent a right colectomy without intraoperative complications that was followed by septic shock. Workup with urinalysis and urine culture showed minimal pyuria with Pseudomonas aeruginosa. Additional studies, including blood cultures, abdominal wound cultures, computed tomography of the abdomen and pelvis, renal ultrasound, and chest radiographs, were unremarkable and showed no signs of surgical site infection, intra-abdominal or pelvic abscess formation, or pulmonary embolism. Broad-spectrum antibiotics—vancomycin and piperacillin-tazobactam—were started. Persistent fever (Tmax of 102.3 °F [39.1 °C]) and leukocytosis (45.3×109/L [4.2–10×109/L]) despite antibiotic therapy, increasing pressor requirements, and progressive painful erythema and purulence at the abdominal surgical site led to debridement of the wound by the general surgery team on day 9 following the initial surgery due to suspected necrotizing infection. Within 24 hours, dermatology was consulted for continued rapid expansion of the wound. Physical examination of the abdomen revealed a large, well-demarcated, pink-red, indurated, ulcerated plaque with clear to purulent exudate and superficial erosions with violaceous undermined borders extending centrifugally from the abdominal surgical incision line (Figure 1A). Two punch biopsies sent for histopathologic evaluation and tissue culture showed dermal edema with a dense histiocytic infiltrate with nodular foci and admixed mature neutrophils to a lesser degree (Figure 2). Special staining was negative for bacteria, fungi, and mycobacteria. Immunohistochemistry revealed positive staining of the dermal inflammatory infiltrate with CD68, myeloperoxidase, and lysozyme, as well as negative staining with CD34 (Figure 3). These findings were suggestive of a histiocytoid neutrophilic dermatosis such as Sweet syndrome or pyoderma gangrenosum. Due to the morphology of the solitary lesion and the abrupt exacerbation shortly after surgical intervention, the patient was diagnosed with histiocytoid pyoderma gangrenosum. At the same time, the patient’s septic shock was treated with intravenous hydrocortisone (100 mg 3 times daily) for 2 days and also achieved a prompt response in the cutaneous symptoms (Figure 1B).
Sweet syndrome and pyoderma gangrenosum are considered distinct neutrophilic dermatoses that rarely coexist but share several clinical and histopathologic features, which can become a diagnostic challenge.2 Both conditions can manifest clinically as abrupt-onset, tender, erythematous papules; vesiculopustular lesions; or bullae with ulcerative changes. They also exhibit pathergy; present with systemic symptoms such as pyrexia, malaise, and joint pain; are associated with underlying systemic conditions such as infections and/or malignancy; demonstrate a dense neutrophilic infiltrate in the dermis on histopathology; and respond promptly to systemic corticosteroids.2-6 Bullous Sweet syndrome, which can present as vesicles, pustules, or bullae that progress to superficial ulcerations, may represent a variant of neutrophilic dermatosis characterized by features seen in both Sweet syndrome and pyoderma gangrenosum, suggesting that these 2 conditions may be on a spectrum.5Clinical features such as erythema with a blue, gray, or purple hue; undermined and ragged borders; and healing of skin lesions with atrophic or cribriform scarring may favor pyoderma gangrenosum, whereas a dull red or plum color and resolution of lesions without scarring may support the diagnosis of Sweet syndrome.7 Although both conditions can exhibit pathergy secondary to minor skin trauma such as venipuncture and biopsies,2,3,5,8 Sweet syndrome rarely has been described to develop after surgery in a patient without a known history of the condition.9 In contrast, postsurgical pyoderma gangrenosum has been well described as secondary to the pathergy phenomenon.5
Our patient was favored to have pyoderma gangrenosum given the solitary lesion, its abrupt development after surgery, and the morphology of the lesion that exhibited a large violaceous to red ulcerative and exudative plaque with undermined borders with atrophic scarring. In patients with skin disease that cannot be distinguished with certainty as either Sweet syndrome or pyoderma gangrenosum, it is essential to recognize that, as neutrophilic dermatoses, both conditions can be managed with either the first-line treatment option of high-dose systemic steroids or one of the shared alternative first-line or second-line steroid-sparing treatments, such as dapsone and cyclosporine.2
Although the exact pathogenesis of pyoderma gangrenosum remains to be fully understood, paraneoplastic pyoderma gangrenosum is a frequently described phenomenon.10,11 Our patient’s history of multiple malignancies, both solid and hematologic, supports the likelihood of malignancy-induced pyoderma gangrenosum; however, given his history of MDS, several other conditions were ruled out prior to making the diagnosis of pyoderma gangrenosum.
Classically, neutrophilic dermatoses such as pyoderma gangrenosum have a dense dermal neutrophilic infiltrate. Concurrent myeloproliferative disorders can alter the maturation of leukocytes, subsequently leading to an atypical appearance of the inflammatory cells on histopathology. Further, in the setting of myeloproliferative disorders, conditions such as leukemia cutis, in which there can be a cutaneous infiltrate of immature or mature myeloid or lymphocytic cells, must be considered. To ensure our patient’s abdominal skin changes were not a cutaneous manifestation of hematologic malignancy, immunohistochemical staining with CD20 and CD3 was performed and showed only the rare presence of B and T lymphocytes, respectively. Staining with CD34 for lymphocytic and myeloid progenitor cells was negative in the dermal infiltrate and further reduced the likelihood of leukemia cutis. Alternatively, patients can have aleukemic cutaneous myeloid sarcoma or leukemia cutis without an underlying hematologic condition or with latent peripheral blood or bone marrow myeloproliferative disorder, but our patient’s history of MDS eliminated this possibility.12 After exclusion of cutaneous infiltration by malignant leukocytes, our patient was diagnosed with histiocytoid neutrophilic dermatosis.
Multiple reports have described histiocytoid Sweet syndrome, in which there is a dense dermal histiocytoid infiltrate on histopathology that demonstrates myeloid lineage with immunologic staining.1,13 The typical pattern of histiocytoid Sweet syndrome includes a predominantly unaffected epidermis with papillary dermal edema, an absence of vasculitis, and a dense dermal infiltrate primarily composed of immature histiocytelike mononuclear cells with a basophilic elongated, twisted, or kidney-shaped nucleus and pale eosinophilic cytoplasm.1,13 In an analogous manner, Morin et al12 described a patient with congenital hypogammaglobulinemia who presented with lesions that clinically resembled pyoderma gangrenosum but revealed a dense dermal infiltrate mostly made of large immature histiocytoid mononuclear cells on histopathology, consistent with the histopathologic features observed in histiocytoid Sweet syndrome. The patient ultimately was diagnosed with histiocytoid pyoderma gangrenosum. Similarly, we believe that our patient also developed histiocytoid pyoderma gangrenosum. As with histiocytoid Sweet syndrome, this diagnosis is based on histopathologic and immunohistochemical findings of a dense dermal infiltrate composed of histiocyte-resembling immature neutrophils.
Typically, pyoderma gangrenosum responds promptly to treatment with systemic corticosteroids.4 Steroid-sparing agents such as cyclosporine, azathioprine, dapsone, and tumor necrosis factor α inhibitors also may be used.4,10 In the setting of MDS, clearance of pyoderma gangrenosum has been reported upon treatment of the underlying malignancy,14 high-dose systemic corticosteroids,11,15 cyclosporine with systemic steroids,16 thalidomide,17 combination therapy with thalidomide and interferon alfa-2a,18 and ustekinumab with vacuum-assisted closure therapy.19 Our patient’s histiocytoid pyoderma gangrenosum in the setting of solid and hematologic malignancy cleared rapidly with high-dose systemic hydrocortisone.
In the setting of malignancy, as in our patient, neutrophilic dermatoses may develop from an aberrant immune system or tumor-induced cytokine dysregulation that leads to increased neutrophil production or dysfunction.4,10,11 Although our patient’s MDS may have contributed to the atypical appearance of the dermal inflammatory infiltrate, it is unclear whether the hematologic disorder increased his risk for the histiocytoid variant of neutrophilic dermatoses. Alegría-Landa et al13 reported that histiocytoid Sweet syndrome is associated with hematologic malignancy at a similar frequency as classic Sweet syndrome. It is unknown if histiocytoid pyoderma gangrenosum would have a strong association with hematologic malignancy. Future reports may elucidate a better understanding of the histiocytoid subtype of pyoderma gangrenosum and its clinical implications.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Wallach D, Vignon-Pennamen MD. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med. 1997;73:65-68.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:E74-E76.
- Shah M, Sachdeva M, Gefri A, et al. Paraneoplastic pyoderma gangrenosum in solid organ malignancy: a literature review. Int J Dermatol. 2020;59:154-158.
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359.
- Morin CB, Côté B, Belisle A. An interesting case of pyoderma gangrenosum with immature histiocytoid neutrophils. J Cutan Pathol. 2018;45:63-66.
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Saleh MFM, Saunthararajah Y. Severe pyoderma gangrenosum caused by myelodysplastic syndrome successfully treated with decitabine administered by a noncytotoxic regimen. Clin Case Rep. 2017;5:2025-2027.
- Yamauchi R, Ishida K, Iwashima Y, et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J Infect Chemother. 2003;9:268-271.
- Ha JW, Hahm JE, Kim KS, et al. A case of pyoderma gangrenosum with myelodysplastic syndrome. Ann Dermatol. 2018;30:392-393.
- Malkan UY, Gunes G, Eliacik E, et al. Treatment of pyoderma gangrenosum with thalidomide in a myelodysplastic syndrome case. Int J Med Case Rep. 2016;9:61-64.
- Koca E, Duman AE, Cetiner D, et al. Successful treatment of myelodysplastic syndrome-induced pyoderma gangrenosum. Neth J Med. 2006;64:422-424.
- Nieto D, Sendagorta E, Rueda JM, et al. Successful treatment with ustekinumab and vacuum-assisted closure therapy in recalcitrant myelodysplastic syndrome-associated pyoderma gangrenosum: case report and literature review. Clin Exp Dermatol. 2019;44:116-119.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Wallach D, Vignon-Pennamen MD. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med. 1997;73:65-68.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:E74-E76.
- Shah M, Sachdeva M, Gefri A, et al. Paraneoplastic pyoderma gangrenosum in solid organ malignancy: a literature review. Int J Dermatol. 2020;59:154-158.
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359.
- Morin CB, Côté B, Belisle A. An interesting case of pyoderma gangrenosum with immature histiocytoid neutrophils. J Cutan Pathol. 2018;45:63-66.
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Saleh MFM, Saunthararajah Y. Severe pyoderma gangrenosum caused by myelodysplastic syndrome successfully treated with decitabine administered by a noncytotoxic regimen. Clin Case Rep. 2017;5:2025-2027.
- Yamauchi R, Ishida K, Iwashima Y, et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J Infect Chemother. 2003;9:268-271.
- Ha JW, Hahm JE, Kim KS, et al. A case of pyoderma gangrenosum with myelodysplastic syndrome. Ann Dermatol. 2018;30:392-393.
- Malkan UY, Gunes G, Eliacik E, et al. Treatment of pyoderma gangrenosum with thalidomide in a myelodysplastic syndrome case. Int J Med Case Rep. 2016;9:61-64.
- Koca E, Duman AE, Cetiner D, et al. Successful treatment of myelodysplastic syndrome-induced pyoderma gangrenosum. Neth J Med. 2006;64:422-424.
- Nieto D, Sendagorta E, Rueda JM, et al. Successful treatment with ustekinumab and vacuum-assisted closure therapy in recalcitrant myelodysplastic syndrome-associated pyoderma gangrenosum: case report and literature review. Clin Exp Dermatol. 2019;44:116-119.
Practice Points:
- Dermatologists and dermatopathologists should be aware of the histiocytoid variant of pyoderma gangrenosum, which can clinical and histologic features that overlap with histiocytoid Sweet syndrome.
- When considering a diagnosis of histiocytoid neutrophilic dermatoses, leukemia cutis or aleukemic cutaneous myeloid sarcoma should be ruled out.
- Similar to histiocytoid Sweet syndrome and neutrophilic dermatoses in the setting of hematologic or solid organ malignancy, histiocytoid pyoderma gangrenosum may respond well to high-dose systemic corticosteroids.
Nail Alterations From Musical Instruments: Insights for Dermatologists Treating Musicians
A variety of skin problems can occur in musicians due to the repetitive movements of playing instruments.1,2 Musicians’ nails are continuously exposed to the mechanical forces and chemical substances characteristic of their instruments.3 Occupational nail alterations in musicians caused by repetitive physical trauma, allergic contact dermatitis, and/or infection may lead to disability and compromise their professional career.
We conducted a systematic review of the literature on the clinical features of musical instrument–related nail alterations to optimize the management and prevention of these conditions.
Methods
We conducted a systematic review of PubMed, Scopus, and Google Scholar databases for eligible publications on instrument-related nail alterations in musicians using the search terms musicians with nail, onychopathy, and Raynaud. No time or language criteria were applied. Reviews, editorials, and articles not related to the topic were excluded. Bibliographies/reference lists were checked to find any additional relevant publications. Relevant articles in English and French were screened by 2 independent reviewers (A.G. and N.L.), and the following data were extracted for qualitative synthesis: sex, age, musical instrument, clinical features, number of years practicing the instrument, laboratory investigations, and disease course.
Results
The literature search yielded 11 publications. Sixteen additional articles were identified by other methods (ie, references, related publications). Overall, 3 full-text articles described general nail alterations but did not describe the clinical data, and 11 publications were editorials, commentaries, reviews, or not relevant. Thirteen contributions fulfilled the inclusion criteria and were eligible for qualitative synthesis. The flow diagram illustrates the screening process (Figure 1).
Twenty-three patients were included. The instruments identified were divided into 2 groups: string instruments (ie, guitar, violin, harp) and percussion instruments (ie, drums, piano, slap bass). Nail alterations were clinically expressed as: (1) modifications of the nail surface; (2) nail bed, soft-tissue, and bone abnormalities; and (3) periungual tissue and distal pulp disorders. All cases are summarized in the Table.4-16 Three articles described occupational Raynaud phenomenon.12-14
Comment
Modifications of the Nail Surface—Onychodystrophy, such as deformity or discoloration of the nail plate, was described in 6 patients among a cohort of 295 musicians and an additional 6 patients among 199 musicians with induced skin lesions. This condition was most common in string instrument players and pianists due to injury and irritation.
One patient, who had been a professional violist for 27 years, presented with lamellar onychoschizia, which corresponds to a horizontal splitting of the nail toward its distal portion (Figure 2). The 3 fingernails of the dominant hand were involved with a V-shaped incision of the distal margin of the nail due to the repetitive friction of the nails with the strings.6
Striations of the nail plate were reported in a guitarist who played for 10 years.7 Physical examination revealed linear transverse ridges alternating with depressions on the central aspect of the nail plate of the right thumbnail, as the patient was right-handed. This condition, attributed to sustained pressure on the string applied by the thumb, also has been called habit tic deformity.7
Nail Bed, Soft-Tissue, and Bone Lesions—Purpura (or hemorrhage) of the nail bed was associated with a percussion instrument (ie, piano) in 1 patient, affecting the second, third, and fourth fingernails of the right hand.8 Especially when performing ascending glissando passages, the pianist applies pressure that may damage the finger and cause fingernail purpura. This condition improved after the patient stopping practicing glissandi.8
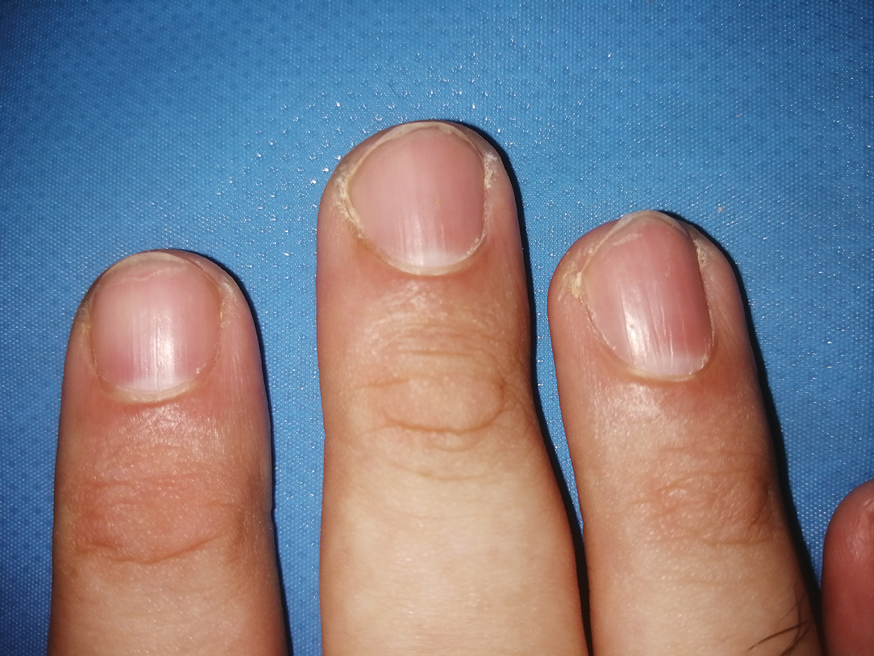
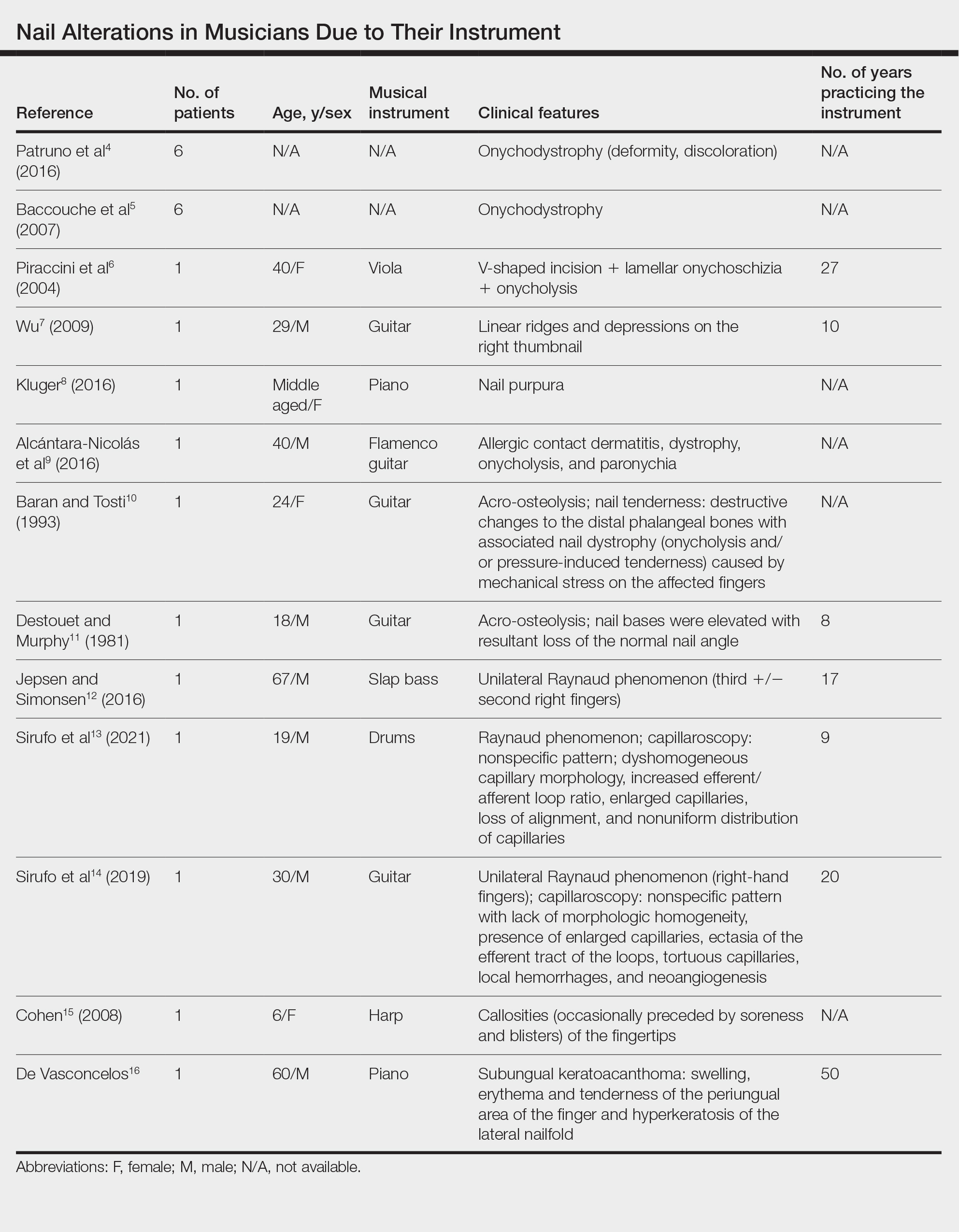
Three patients—2 guitarists and 1 violist—had onycholysis, defined by a loss of the attachment between the nail bed and the nail plate (Figure 3). It may result from repetitive trauma when strings are plucked.6,9,10
Acro-osteolysis associated with pain was reported in 2 guitarists.10,11 This condition is defined as transverse lytic bands in the distal phalanges (Figure 4). Acro-osteolysis may be secondary to multiple causes, such as vinyl chloride exposure, connective tissue diseases, thermal injuries, neuropathic diseases, hyperparathyroidism, nutritional deficiencies, psoriasis, and biomechanical stress.10 In musicians playing instruments, the mechanical stress to the guitar-playing fingers is the causative factor.17
Periungual Tissue and Distal Pulp Disorders—Paronychia is an important occupational hazard of harpists, violists, and pianists.2 It represents an inflammatory condition involving the folds of tissue surrounding fingernails. Pizzicato paronychia is related to infection in the nail fold in string players and secondary to pizzicato playing, whereby the musician plucks the instrument strings with the nails and fingertips.3
Acrylates in artificial nails frequently are used among guitarists to strengthen their nails. A case of occupational allergic contact dermatitis induced by acrylic gel nails in a flamenco guitarist was described.9 The patient developed dystrophy, onycholysis, and paronychia involving the nails of the right hand where acrylic materials were used, which resolved following the removal of the artificial nails. Patch tests were performed and were positive for 2-hydroxyethyl methacrylate, 2-hydroxyethyl acrylate, ethylene glycol dimethacrylate, and 2-hydroxypropyl methacrylate, supporting the diagnosis of allergic contact dermatitis to acrylates.9 Therefore, musicians should be aware of the sensitizing potential of acrylates and adopt preventive measures.
Unilateral Raynaud phenomenon of the dominant hand was noted in 3 cases of musicians who played string instruments due to the increased tendency to vasospasm in the digital capillaries from the direct transmission of vibrations of the strings (>100 Hz).12-14 Consequently, the disruption of the digital blood circulation leads to an abnormal reaction to cold, which is called vibration-induced white fingers or vasospastic white finger disease.19 In these 3 patients, capillaroscopy showed a nonspecific pattern with a lack of morphologic homogeneity of capillaries, the presence of enlarged capillaries, ectasia of the efferent tract of the loops, tortuous capillaries, local hemorrhages, and neoangiogenesis.13,14
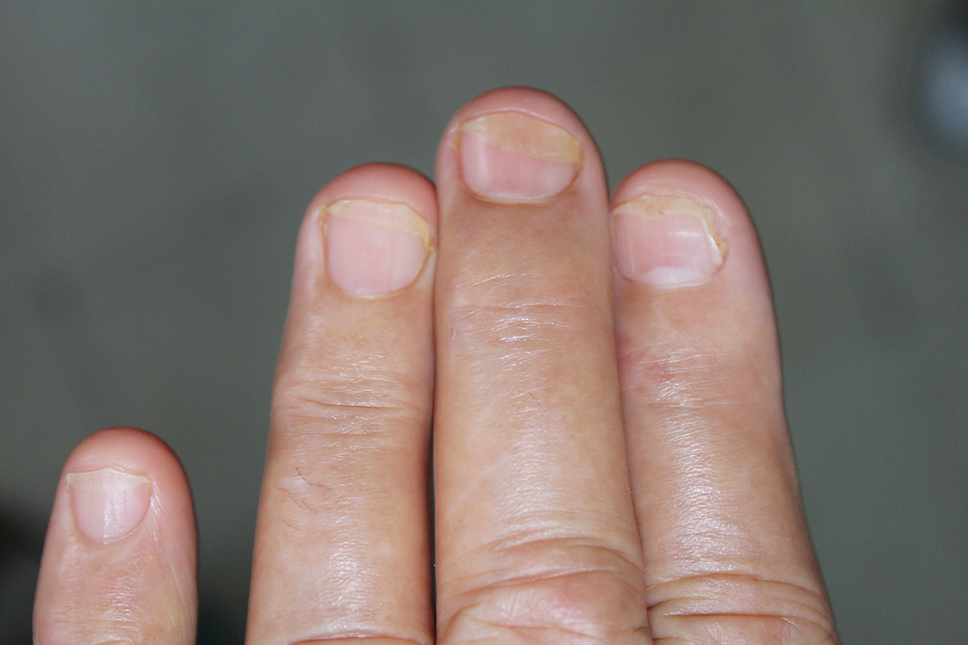
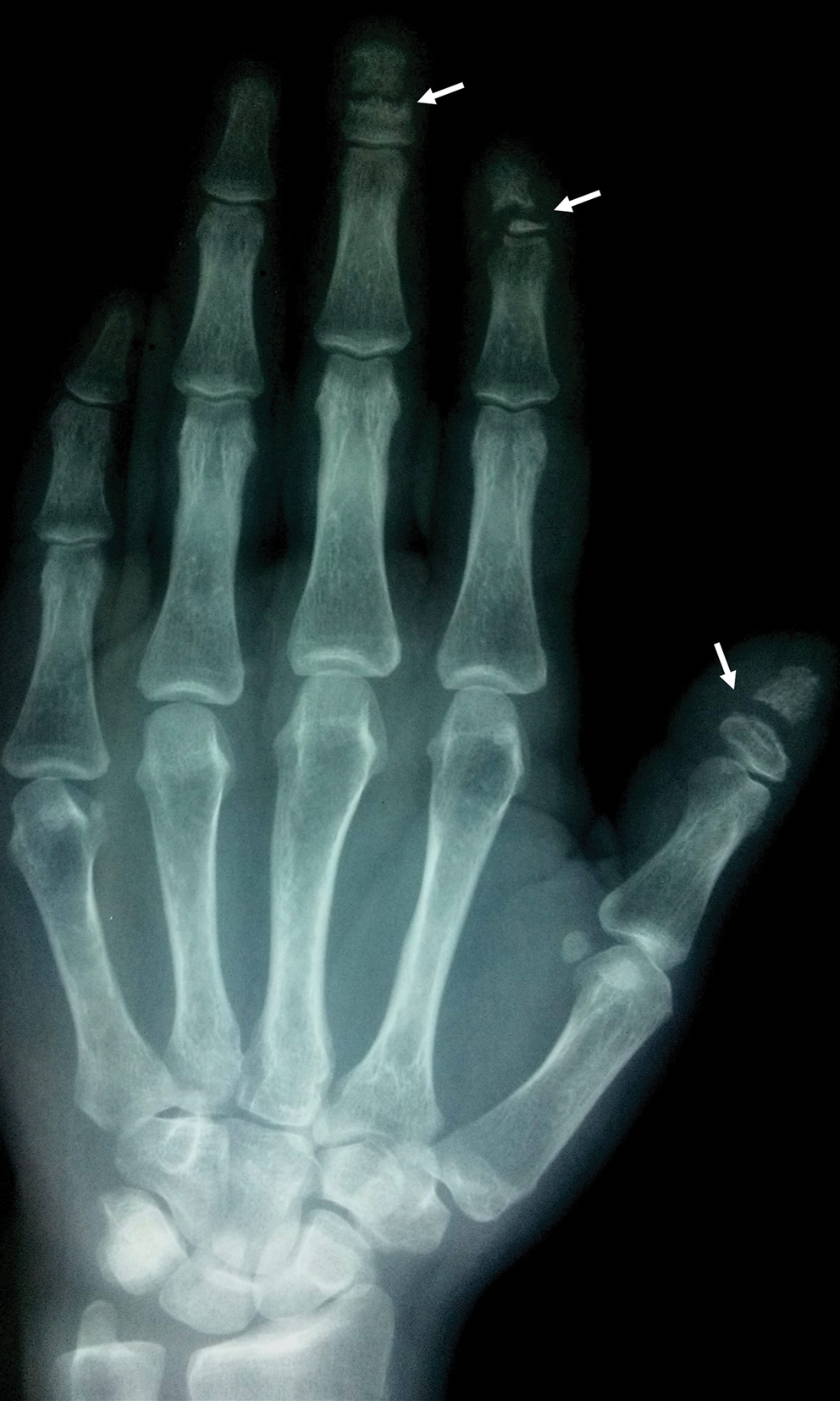
A middle-aged professional concert pianist presented with paronychia with hyperkeratosis of the lateral nail fold. Histopathology revealed a subungual keratoacanthoma eroding the distal phalanx tip, which was removed by surgical excision. The repeated fingertip trauma associated with pianistic activity was suspected to be the causative event.16
Callosities also are common on the fingertips of musicians, including 18.4% of patients in a cohort of 628 musicians, and involving fingers in 64.6% of these patients.4 These callosities are explained by the chronic mechanical forces and characterize the way musicians grasp and hold their instruments. Callosities could be preceded by soreness and blisters of the fingertips in a harpist (harpist’s finger).1,15 Calluses were located on the lateral fourth fingertip of a drummer corresponding to the friction with the drumsticks (drummer’s digit) and on the thumb of a bassoon player. Trumpet calluses generally overlie the proximal interphalangeal joint of the left index finger.
Conclusion
Healthy nails are essential for playing a musical instrument. This review highlights the occurrence of fingertip callosities, paronychia, onycholysis, and subungual hemorrhages among musicians who play instruments. Additionally, the transmission of string-vibratory movements can produce microvascular damage and occupational Raynaud phenomenon in some musicians. These occupational nail disorders are underrecognized and may be underdiagnosed. Thus, musicians and clinicians must be aware of these alterations to adopt preventive measures and to provide adequate treatment.
- Rimmer S, Spielvogel RL. Dermatologic problems of musicians. J Am Acad Dermatol. 1990;22:657-663.
- Adams RM. Skin conditions of musicians. Cutis. 2000;65:37-38.
- Vine K, DeLeo V. Dermatologic manifestations of musicians: a case report and review of skin conditions in musicians. Cutis. 2011;87:117-121.
- Patruno C, Napolitano M, La Bella S, et al. Instrument-related skin disorders in musicians. Dermatitis. 2016;27:26-29.
- Baccouche D, Mokni M, Ben Abdelaziz A, et al. Dermatological problems of musicians: a prospective study in musical students . Article in French. Ann Dermatol Venereol. 2007;134(5 Pt 1):445-449.
- Piraccini BM, Antonucci A, Iorizzo M, et al. Occupational nail fragility in a professional violist. Contact Dermatitis. 2004;51:35-36.
- Wu JJ. Habit tic deformity secondary to guitar playing. Dermatol Online J. 2009;15:16.
- Kluger N. Piano glissando purpura: another cutaneous curiosity in musicians. J Eur Acad Dermatol Venereol. 2016;30:683.
- Alcántara-Nicolás FA, Pastor-Nieto MA, Sánchez-Herreros C, et al. Allergic contact dermatitis from acrylic nails in a flamenco guitarist. Occup Med (Lond). 2016;66:751-753.
- Baran R, Tosti A. Occupational acroosteolysis in a guitar player. Acta Derm Venereol. 1993;73:64-65.
- Destouet JM, Murphy WA. Guitar player acro-osteolysis. Skeletal Radiol. 1981;6:275-277.
- Jepsen JR, Simonsen JA. Raynaud’s phenomenon in a slap bass player: a case report. Med Probl Perform Art. 2016;31:51-53.
- Sirufo MM, Catalogna A, De Pietro F, et al. Raynaud’s phenomenon in a drummer player: microvascular disorder and nailfold video capillaroscopic findings. EXCLI J. 2021;20:1526-1531.
- Sirufo MM, Ginaldi L, De Martinis M. Raynaud’s phenomenon and the nailfold capillaroscopic findings in a guitar player. QJM. 2019;112:531-533.
- Cohen PR. Harpist’s finger: case report of a trauma-induced blister in a beginner harpist and review of string instrument-associated skin problems in musicians. Cutis. 2008;82:329-334.
- De Vasconcelos P, Soares-Almeida L, Filipe P. Subungual keratoacanthoma in a pianist. G Ital Dermatol Venereol. 2016;151:455-456.
- Young RS, Bryk D, Ratner H. Selective phalangeal tuft fractures in a guitar player. Br J Radiol. 1977;50:147-148.
- Vázquez-Osorio I, Espasandín-Arias M, García-Gavín J, et al. Allergic contact dermatitis due to acrylates in acrylic gel nails: a report of 3 cases. Actas Dermosifiliogr. 2014;105:430-432.
- Atashpaz S, Ghabili K. Color triad in guitarist’s fingers: a probable case of Raynaud’s phenomenon due to string vibration phenomenon. Med Probl Perform Art. 2008;23:143.
A variety of skin problems can occur in musicians due to the repetitive movements of playing instruments.1,2 Musicians’ nails are continuously exposed to the mechanical forces and chemical substances characteristic of their instruments.3 Occupational nail alterations in musicians caused by repetitive physical trauma, allergic contact dermatitis, and/or infection may lead to disability and compromise their professional career.
We conducted a systematic review of the literature on the clinical features of musical instrument–related nail alterations to optimize the management and prevention of these conditions.
Methods
We conducted a systematic review of PubMed, Scopus, and Google Scholar databases for eligible publications on instrument-related nail alterations in musicians using the search terms musicians with nail, onychopathy, and Raynaud. No time or language criteria were applied. Reviews, editorials, and articles not related to the topic were excluded. Bibliographies/reference lists were checked to find any additional relevant publications. Relevant articles in English and French were screened by 2 independent reviewers (A.G. and N.L.), and the following data were extracted for qualitative synthesis: sex, age, musical instrument, clinical features, number of years practicing the instrument, laboratory investigations, and disease course.
Results
The literature search yielded 11 publications. Sixteen additional articles were identified by other methods (ie, references, related publications). Overall, 3 full-text articles described general nail alterations but did not describe the clinical data, and 11 publications were editorials, commentaries, reviews, or not relevant. Thirteen contributions fulfilled the inclusion criteria and were eligible for qualitative synthesis. The flow diagram illustrates the screening process (Figure 1).
Twenty-three patients were included. The instruments identified were divided into 2 groups: string instruments (ie, guitar, violin, harp) and percussion instruments (ie, drums, piano, slap bass). Nail alterations were clinically expressed as: (1) modifications of the nail surface; (2) nail bed, soft-tissue, and bone abnormalities; and (3) periungual tissue and distal pulp disorders. All cases are summarized in the Table.4-16 Three articles described occupational Raynaud phenomenon.12-14
Comment
Modifications of the Nail Surface—Onychodystrophy, such as deformity or discoloration of the nail plate, was described in 6 patients among a cohort of 295 musicians and an additional 6 patients among 199 musicians with induced skin lesions. This condition was most common in string instrument players and pianists due to injury and irritation.
One patient, who had been a professional violist for 27 years, presented with lamellar onychoschizia, which corresponds to a horizontal splitting of the nail toward its distal portion (Figure 2). The 3 fingernails of the dominant hand were involved with a V-shaped incision of the distal margin of the nail due to the repetitive friction of the nails with the strings.6
Striations of the nail plate were reported in a guitarist who played for 10 years.7 Physical examination revealed linear transverse ridges alternating with depressions on the central aspect of the nail plate of the right thumbnail, as the patient was right-handed. This condition, attributed to sustained pressure on the string applied by the thumb, also has been called habit tic deformity.7
Nail Bed, Soft-Tissue, and Bone Lesions—Purpura (or hemorrhage) of the nail bed was associated with a percussion instrument (ie, piano) in 1 patient, affecting the second, third, and fourth fingernails of the right hand.8 Especially when performing ascending glissando passages, the pianist applies pressure that may damage the finger and cause fingernail purpura. This condition improved after the patient stopping practicing glissandi.8
Three patients—2 guitarists and 1 violist—had onycholysis, defined by a loss of the attachment between the nail bed and the nail plate (Figure 3). It may result from repetitive trauma when strings are plucked.6,9,10
Acro-osteolysis associated with pain was reported in 2 guitarists.10,11 This condition is defined as transverse lytic bands in the distal phalanges (Figure 4). Acro-osteolysis may be secondary to multiple causes, such as vinyl chloride exposure, connective tissue diseases, thermal injuries, neuropathic diseases, hyperparathyroidism, nutritional deficiencies, psoriasis, and biomechanical stress.10 In musicians playing instruments, the mechanical stress to the guitar-playing fingers is the causative factor.17
Periungual Tissue and Distal Pulp Disorders—Paronychia is an important occupational hazard of harpists, violists, and pianists.2 It represents an inflammatory condition involving the folds of tissue surrounding fingernails. Pizzicato paronychia is related to infection in the nail fold in string players and secondary to pizzicato playing, whereby the musician plucks the instrument strings with the nails and fingertips.3
Acrylates in artificial nails frequently are used among guitarists to strengthen their nails. A case of occupational allergic contact dermatitis induced by acrylic gel nails in a flamenco guitarist was described.9 The patient developed dystrophy, onycholysis, and paronychia involving the nails of the right hand where acrylic materials were used, which resolved following the removal of the artificial nails. Patch tests were performed and were positive for 2-hydroxyethyl methacrylate, 2-hydroxyethyl acrylate, ethylene glycol dimethacrylate, and 2-hydroxypropyl methacrylate, supporting the diagnosis of allergic contact dermatitis to acrylates.9 Therefore, musicians should be aware of the sensitizing potential of acrylates and adopt preventive measures.
Unilateral Raynaud phenomenon of the dominant hand was noted in 3 cases of musicians who played string instruments due to the increased tendency to vasospasm in the digital capillaries from the direct transmission of vibrations of the strings (>100 Hz).12-14 Consequently, the disruption of the digital blood circulation leads to an abnormal reaction to cold, which is called vibration-induced white fingers or vasospastic white finger disease.19 In these 3 patients, capillaroscopy showed a nonspecific pattern with a lack of morphologic homogeneity of capillaries, the presence of enlarged capillaries, ectasia of the efferent tract of the loops, tortuous capillaries, local hemorrhages, and neoangiogenesis.13,14
A middle-aged professional concert pianist presented with paronychia with hyperkeratosis of the lateral nail fold. Histopathology revealed a subungual keratoacanthoma eroding the distal phalanx tip, which was removed by surgical excision. The repeated fingertip trauma associated with pianistic activity was suspected to be the causative event.16
Callosities also are common on the fingertips of musicians, including 18.4% of patients in a cohort of 628 musicians, and involving fingers in 64.6% of these patients.4 These callosities are explained by the chronic mechanical forces and characterize the way musicians grasp and hold their instruments. Callosities could be preceded by soreness and blisters of the fingertips in a harpist (harpist’s finger).1,15 Calluses were located on the lateral fourth fingertip of a drummer corresponding to the friction with the drumsticks (drummer’s digit) and on the thumb of a bassoon player. Trumpet calluses generally overlie the proximal interphalangeal joint of the left index finger.
Conclusion
Healthy nails are essential for playing a musical instrument. This review highlights the occurrence of fingertip callosities, paronychia, onycholysis, and subungual hemorrhages among musicians who play instruments. Additionally, the transmission of string-vibratory movements can produce microvascular damage and occupational Raynaud phenomenon in some musicians. These occupational nail disorders are underrecognized and may be underdiagnosed. Thus, musicians and clinicians must be aware of these alterations to adopt preventive measures and to provide adequate treatment.
A variety of skin problems can occur in musicians due to the repetitive movements of playing instruments.1,2 Musicians’ nails are continuously exposed to the mechanical forces and chemical substances characteristic of their instruments.3 Occupational nail alterations in musicians caused by repetitive physical trauma, allergic contact dermatitis, and/or infection may lead to disability and compromise their professional career.
We conducted a systematic review of the literature on the clinical features of musical instrument–related nail alterations to optimize the management and prevention of these conditions.
Methods
We conducted a systematic review of PubMed, Scopus, and Google Scholar databases for eligible publications on instrument-related nail alterations in musicians using the search terms musicians with nail, onychopathy, and Raynaud. No time or language criteria were applied. Reviews, editorials, and articles not related to the topic were excluded. Bibliographies/reference lists were checked to find any additional relevant publications. Relevant articles in English and French were screened by 2 independent reviewers (A.G. and N.L.), and the following data were extracted for qualitative synthesis: sex, age, musical instrument, clinical features, number of years practicing the instrument, laboratory investigations, and disease course.
Results
The literature search yielded 11 publications. Sixteen additional articles were identified by other methods (ie, references, related publications). Overall, 3 full-text articles described general nail alterations but did not describe the clinical data, and 11 publications were editorials, commentaries, reviews, or not relevant. Thirteen contributions fulfilled the inclusion criteria and were eligible for qualitative synthesis. The flow diagram illustrates the screening process (Figure 1).
Twenty-three patients were included. The instruments identified were divided into 2 groups: string instruments (ie, guitar, violin, harp) and percussion instruments (ie, drums, piano, slap bass). Nail alterations were clinically expressed as: (1) modifications of the nail surface; (2) nail bed, soft-tissue, and bone abnormalities; and (3) periungual tissue and distal pulp disorders. All cases are summarized in the Table.4-16 Three articles described occupational Raynaud phenomenon.12-14
Comment
Modifications of the Nail Surface—Onychodystrophy, such as deformity or discoloration of the nail plate, was described in 6 patients among a cohort of 295 musicians and an additional 6 patients among 199 musicians with induced skin lesions. This condition was most common in string instrument players and pianists due to injury and irritation.
One patient, who had been a professional violist for 27 years, presented with lamellar onychoschizia, which corresponds to a horizontal splitting of the nail toward its distal portion (Figure 2). The 3 fingernails of the dominant hand were involved with a V-shaped incision of the distal margin of the nail due to the repetitive friction of the nails with the strings.6
Striations of the nail plate were reported in a guitarist who played for 10 years.7 Physical examination revealed linear transverse ridges alternating with depressions on the central aspect of the nail plate of the right thumbnail, as the patient was right-handed. This condition, attributed to sustained pressure on the string applied by the thumb, also has been called habit tic deformity.7
Nail Bed, Soft-Tissue, and Bone Lesions—Purpura (or hemorrhage) of the nail bed was associated with a percussion instrument (ie, piano) in 1 patient, affecting the second, third, and fourth fingernails of the right hand.8 Especially when performing ascending glissando passages, the pianist applies pressure that may damage the finger and cause fingernail purpura. This condition improved after the patient stopping practicing glissandi.8
Three patients—2 guitarists and 1 violist—had onycholysis, defined by a loss of the attachment between the nail bed and the nail plate (Figure 3). It may result from repetitive trauma when strings are plucked.6,9,10
Acro-osteolysis associated with pain was reported in 2 guitarists.10,11 This condition is defined as transverse lytic bands in the distal phalanges (Figure 4). Acro-osteolysis may be secondary to multiple causes, such as vinyl chloride exposure, connective tissue diseases, thermal injuries, neuropathic diseases, hyperparathyroidism, nutritional deficiencies, psoriasis, and biomechanical stress.10 In musicians playing instruments, the mechanical stress to the guitar-playing fingers is the causative factor.17
Periungual Tissue and Distal Pulp Disorders—Paronychia is an important occupational hazard of harpists, violists, and pianists.2 It represents an inflammatory condition involving the folds of tissue surrounding fingernails. Pizzicato paronychia is related to infection in the nail fold in string players and secondary to pizzicato playing, whereby the musician plucks the instrument strings with the nails and fingertips.3
Acrylates in artificial nails frequently are used among guitarists to strengthen their nails. A case of occupational allergic contact dermatitis induced by acrylic gel nails in a flamenco guitarist was described.9 The patient developed dystrophy, onycholysis, and paronychia involving the nails of the right hand where acrylic materials were used, which resolved following the removal of the artificial nails. Patch tests were performed and were positive for 2-hydroxyethyl methacrylate, 2-hydroxyethyl acrylate, ethylene glycol dimethacrylate, and 2-hydroxypropyl methacrylate, supporting the diagnosis of allergic contact dermatitis to acrylates.9 Therefore, musicians should be aware of the sensitizing potential of acrylates and adopt preventive measures.
Unilateral Raynaud phenomenon of the dominant hand was noted in 3 cases of musicians who played string instruments due to the increased tendency to vasospasm in the digital capillaries from the direct transmission of vibrations of the strings (>100 Hz).12-14 Consequently, the disruption of the digital blood circulation leads to an abnormal reaction to cold, which is called vibration-induced white fingers or vasospastic white finger disease.19 In these 3 patients, capillaroscopy showed a nonspecific pattern with a lack of morphologic homogeneity of capillaries, the presence of enlarged capillaries, ectasia of the efferent tract of the loops, tortuous capillaries, local hemorrhages, and neoangiogenesis.13,14
A middle-aged professional concert pianist presented with paronychia with hyperkeratosis of the lateral nail fold. Histopathology revealed a subungual keratoacanthoma eroding the distal phalanx tip, which was removed by surgical excision. The repeated fingertip trauma associated with pianistic activity was suspected to be the causative event.16
Callosities also are common on the fingertips of musicians, including 18.4% of patients in a cohort of 628 musicians, and involving fingers in 64.6% of these patients.4 These callosities are explained by the chronic mechanical forces and characterize the way musicians grasp and hold their instruments. Callosities could be preceded by soreness and blisters of the fingertips in a harpist (harpist’s finger).1,15 Calluses were located on the lateral fourth fingertip of a drummer corresponding to the friction with the drumsticks (drummer’s digit) and on the thumb of a bassoon player. Trumpet calluses generally overlie the proximal interphalangeal joint of the left index finger.
Conclusion
Healthy nails are essential for playing a musical instrument. This review highlights the occurrence of fingertip callosities, paronychia, onycholysis, and subungual hemorrhages among musicians who play instruments. Additionally, the transmission of string-vibratory movements can produce microvascular damage and occupational Raynaud phenomenon in some musicians. These occupational nail disorders are underrecognized and may be underdiagnosed. Thus, musicians and clinicians must be aware of these alterations to adopt preventive measures and to provide adequate treatment.
- Rimmer S, Spielvogel RL. Dermatologic problems of musicians. J Am Acad Dermatol. 1990;22:657-663.
- Adams RM. Skin conditions of musicians. Cutis. 2000;65:37-38.
- Vine K, DeLeo V. Dermatologic manifestations of musicians: a case report and review of skin conditions in musicians. Cutis. 2011;87:117-121.
- Patruno C, Napolitano M, La Bella S, et al. Instrument-related skin disorders in musicians. Dermatitis. 2016;27:26-29.
- Baccouche D, Mokni M, Ben Abdelaziz A, et al. Dermatological problems of musicians: a prospective study in musical students . Article in French. Ann Dermatol Venereol. 2007;134(5 Pt 1):445-449.
- Piraccini BM, Antonucci A, Iorizzo M, et al. Occupational nail fragility in a professional violist. Contact Dermatitis. 2004;51:35-36.
- Wu JJ. Habit tic deformity secondary to guitar playing. Dermatol Online J. 2009;15:16.
- Kluger N. Piano glissando purpura: another cutaneous curiosity in musicians. J Eur Acad Dermatol Venereol. 2016;30:683.
- Alcántara-Nicolás FA, Pastor-Nieto MA, Sánchez-Herreros C, et al. Allergic contact dermatitis from acrylic nails in a flamenco guitarist. Occup Med (Lond). 2016;66:751-753.
- Baran R, Tosti A. Occupational acroosteolysis in a guitar player. Acta Derm Venereol. 1993;73:64-65.
- Destouet JM, Murphy WA. Guitar player acro-osteolysis. Skeletal Radiol. 1981;6:275-277.
- Jepsen JR, Simonsen JA. Raynaud’s phenomenon in a slap bass player: a case report. Med Probl Perform Art. 2016;31:51-53.
- Sirufo MM, Catalogna A, De Pietro F, et al. Raynaud’s phenomenon in a drummer player: microvascular disorder and nailfold video capillaroscopic findings. EXCLI J. 2021;20:1526-1531.
- Sirufo MM, Ginaldi L, De Martinis M. Raynaud’s phenomenon and the nailfold capillaroscopic findings in a guitar player. QJM. 2019;112:531-533.
- Cohen PR. Harpist’s finger: case report of a trauma-induced blister in a beginner harpist and review of string instrument-associated skin problems in musicians. Cutis. 2008;82:329-334.
- De Vasconcelos P, Soares-Almeida L, Filipe P. Subungual keratoacanthoma in a pianist. G Ital Dermatol Venereol. 2016;151:455-456.
- Young RS, Bryk D, Ratner H. Selective phalangeal tuft fractures in a guitar player. Br J Radiol. 1977;50:147-148.
- Vázquez-Osorio I, Espasandín-Arias M, García-Gavín J, et al. Allergic contact dermatitis due to acrylates in acrylic gel nails: a report of 3 cases. Actas Dermosifiliogr. 2014;105:430-432.
- Atashpaz S, Ghabili K. Color triad in guitarist’s fingers: a probable case of Raynaud’s phenomenon due to string vibration phenomenon. Med Probl Perform Art. 2008;23:143.
- Rimmer S, Spielvogel RL. Dermatologic problems of musicians. J Am Acad Dermatol. 1990;22:657-663.
- Adams RM. Skin conditions of musicians. Cutis. 2000;65:37-38.
- Vine K, DeLeo V. Dermatologic manifestations of musicians: a case report and review of skin conditions in musicians. Cutis. 2011;87:117-121.
- Patruno C, Napolitano M, La Bella S, et al. Instrument-related skin disorders in musicians. Dermatitis. 2016;27:26-29.
- Baccouche D, Mokni M, Ben Abdelaziz A, et al. Dermatological problems of musicians: a prospective study in musical students . Article in French. Ann Dermatol Venereol. 2007;134(5 Pt 1):445-449.
- Piraccini BM, Antonucci A, Iorizzo M, et al. Occupational nail fragility in a professional violist. Contact Dermatitis. 2004;51:35-36.
- Wu JJ. Habit tic deformity secondary to guitar playing. Dermatol Online J. 2009;15:16.
- Kluger N. Piano glissando purpura: another cutaneous curiosity in musicians. J Eur Acad Dermatol Venereol. 2016;30:683.
- Alcántara-Nicolás FA, Pastor-Nieto MA, Sánchez-Herreros C, et al. Allergic contact dermatitis from acrylic nails in a flamenco guitarist. Occup Med (Lond). 2016;66:751-753.
- Baran R, Tosti A. Occupational acroosteolysis in a guitar player. Acta Derm Venereol. 1993;73:64-65.
- Destouet JM, Murphy WA. Guitar player acro-osteolysis. Skeletal Radiol. 1981;6:275-277.
- Jepsen JR, Simonsen JA. Raynaud’s phenomenon in a slap bass player: a case report. Med Probl Perform Art. 2016;31:51-53.
- Sirufo MM, Catalogna A, De Pietro F, et al. Raynaud’s phenomenon in a drummer player: microvascular disorder and nailfold video capillaroscopic findings. EXCLI J. 2021;20:1526-1531.
- Sirufo MM, Ginaldi L, De Martinis M. Raynaud’s phenomenon and the nailfold capillaroscopic findings in a guitar player. QJM. 2019;112:531-533.
- Cohen PR. Harpist’s finger: case report of a trauma-induced blister in a beginner harpist and review of string instrument-associated skin problems in musicians. Cutis. 2008;82:329-334.
- De Vasconcelos P, Soares-Almeida L, Filipe P. Subungual keratoacanthoma in a pianist. G Ital Dermatol Venereol. 2016;151:455-456.
- Young RS, Bryk D, Ratner H. Selective phalangeal tuft fractures in a guitar player. Br J Radiol. 1977;50:147-148.
- Vázquez-Osorio I, Espasandín-Arias M, García-Gavín J, et al. Allergic contact dermatitis due to acrylates in acrylic gel nails: a report of 3 cases. Actas Dermosifiliogr. 2014;105:430-432.
- Atashpaz S, Ghabili K. Color triad in guitarist’s fingers: a probable case of Raynaud’s phenomenon due to string vibration phenomenon. Med Probl Perform Art. 2008;23:143.
Practice Points
- Long-term practice and performance with a musical instrument predispose musicians to several skin conditions and nail disorders.
- Nail alterations in musicians include onychodystrophy, callosities of the fingertips, paronychia, distal onycholysis, lamellar onychoschizia, striations, subungual hemorrhage, and occupational Raynaud phenomenon.
- Nail lesions in musicians may be caused by localized pressure, friction-induced mechanical forces, allergic or irritant contact dermatitis, or infections.
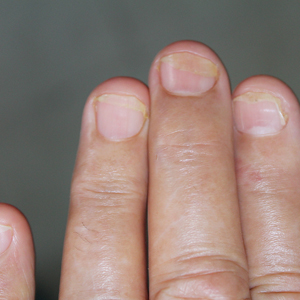
Act Fast With Traction Alopecia to Avoid Permanent Hair Loss
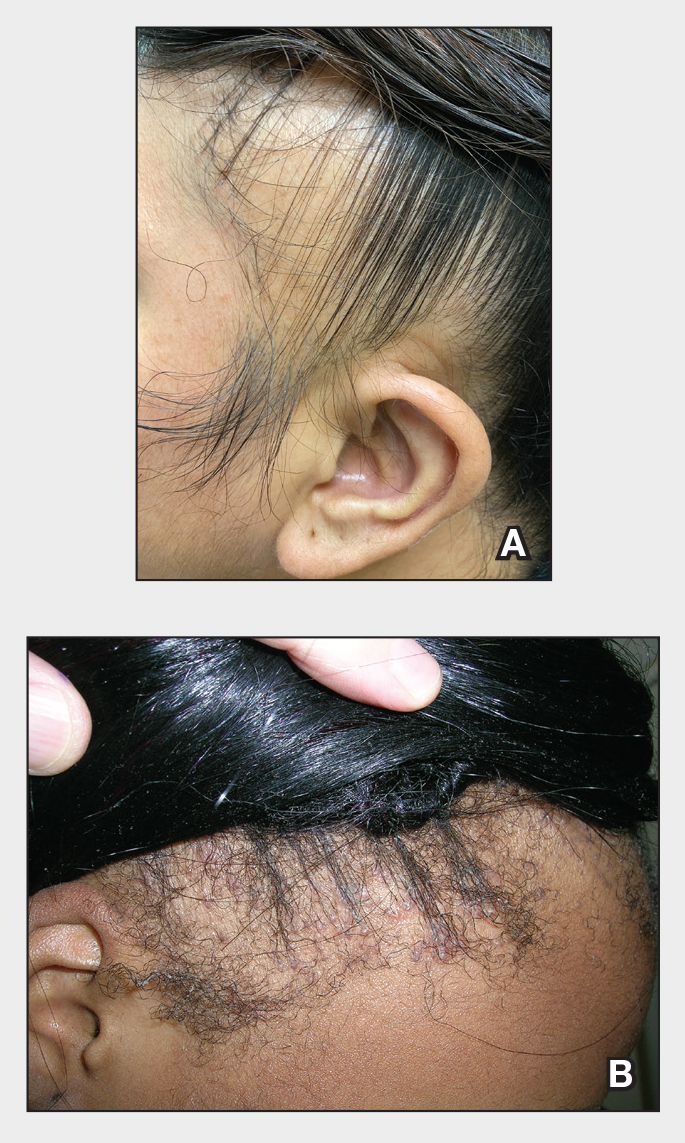
The Comparison
Traction alopecia (TA) is a common type of alopecia that ultimately can result in permanent hair loss. It often is caused or worsened by repetitive and prolonged hairstyling practices such as tight ponytails, braids, or locs, or use of wigs or weaves.1 Use of headwear, as in certain religious or ethnic groups, also can be contributory.2 Individuals participating in or training for occupations involving military service or ballet are at risk for TA due to hairstyling-specific policies. Early stages of TA are reversible with proper treatment and avoidance of exacerbating factors, emphasizing the importance of prompt recognition.3
Epidemiology
Data on the true prevalence of TA are lacking. It can occur in individuals of any race or any hair type. However, it is most common in women of African descent, affecting approximately one-third of this population.4 Other commonly affected groups include ballerinas and active-duty service members due to tight ponytails and buns, as well as the Sikh population due to the use of turbans as a part of their religious practice.2,5,6
Traction alopecia also impacts children, particularly those of African descent. A 2007 study of schoolchildren in South Africa determined that more than 17% of young African girls had evidence of TA—even some as young as 6 years of age.7
Traction alopecia can be caused or exacerbated by the use of hair clips and bobby pins that aid holding styles in place.8
Hair shaft morphology may contribute to the risk for TA, with more tightly coiled hair types being more susceptible.8 Variables such as use of chemical relaxers also increase the risk for disease, especially when combined with high-tension styling methods such as braids.9
Key clinical features
Patients with TA clinically present with hair loss and breakage in areas with tension, most commonly the marginal areas of the scalp as well as the frontal hairline and temporal scalp. Hair loss can result in a “fringe sign,” in which a patient may have preservation of a thin line of hairs at the frontal aspect of the hairline with a band of hair loss behind.10 This presentation may be used to differentiate TA from other forms of alopecia, including frontal fibrosing alopecia and female pattern hair loss. When the hair loss is not marginal, it may mimic other forms of patchy hair loss including alopecia areata and trichotillomania. Other clinical findings in TA may include broken hairs, pustules, and follicular papules.10 Patients also may describe symptoms such as scalp tenderness with specific hairstyles or headaches,11 or they may be completely asymptomatic.
Trichoscopy can be helpful in guiding diagnosis and treatment. Patients with TA often have perifollicular erythema and hair casts (cylindrical structures that encircle the proximal hair shafts) in the earlier stages of the disease, with eventual loss of follicular ostia in the later stages.10,12 Hair casts also may indicate ongoing traction.12 The flambeau sign—white tracks seen on trichoscopy in the direction the hair is pulled—resembles a lit torch.13
Worth noting
Early-stage TA can be reversed by avoiding hair tension. However, patients may not be amenable to this due to personal hairstyling preferences, job duties, or religious practices. Treatment with topical or intralesional steroids or even oral antibiotics such as doxycycline for its anti-inflammatory ability may result in regrowth of lost hair if the follicles are not permanently lost and exacerbating factors are avoided.3,14 Both topical and oral minoxidil have been used with success, with minoxidil thought to increase hair density by extending the anagen (growth) phase of hair follicles.3,15 Culturally sensitive patient counseling on the condition and potential exacerbating factors is critical.16
At later stages of the disease—after loss of follicular ostia has occurred—surgical interventions should be considered,17 such as hair transplantation, which can be successful but remains a technical challenge due to variability in hair shaft curvature.18 Additionally, the cost of the procedure can limit use, and some patients may not be optimal candidates due to the extent of their hair loss. Traction alopecia may not be the only hair loss condition present. Examining the scalp is important even if the chief area of concern is the marginal scalp.
Health disparity highlight
Prevention, early identification, and treatment initiated in a timely fashion are crucial to prevent permanent hair loss. There are added societal and cultural pressures that impact hairstyle and hair care practices, especially for those with tightly coiled hair.19 Historically, tightly coiled hair has been unfairly viewed as “unprofessional,” “unkempt,” and a challenge to “manage” by some. Thus, heat, chemical relaxers, and tight hairstyles holding hair in one position have been used to straighten the hair permanently or temporarily or to keep it maintained in a style that did not necessitate excessive manipulation—often contributing to further tension on the hair.
Military service branches have evaluated and changed some hair-related policies to reflect the diverse hair types of military personnel.20 The CROWN Act (www.thecrownact.com/about)—“Creating a Respectful and Open World for Natural Hair”—is a model law passed by 26 states that prohibits race-based hair discrimination, which is the denial of employment and educational opportunities because of hair texture. Although the law has not been passed in every state, it may help individuals with tightly coiled hair to embrace natural hairstyles. However, even hairstyles with one’s own natural curl pattern can contribute to tension and thus potential development of TA.
- Larrondo J, McMichael AJ. Traction alopecia. JAMA Dermatol. 2023;159:676. doi:10.1001/jamadermatol.2022.6298
- James J, Saladi RN, Fox JL. Traction alopecia in Sikh male patients. J Am Board Fam Med. 2007;20:497-498. doi:10.3122/jabfm.2007.05.070076
- Callender VD, McMichael AJ, Cohen GF. Medical and surgical therapies for alopecias in black women. Dermatol Ther. 2004;17:164-176.
- Loussouarn G, El Rawadi C, Genain G. Diversity of hair growth profiles. Int J Dermatol. 2005;44(suppl 1):6-9.
- Samrao AChen CZedek Det al. Traction alopecia in a ballerina: clinicopathologic features. Arch Dermatol. 2010;146:918-935. doi:10.1001/archdermatol.2010.183
- Korona-Bailey J, Banaag A, Nguyen DR, et al. Free the bun: prevalence of alopecia among active duty service women, fiscal years 2010-2019. Mil Med. 2023;188:e492-e496. doi:10.1093/milmed/usab274
- Khumalo NP, Jessop S, Gumedze F, et al. Hairdressing is associated with scalp disease in African schoolchildren. Br J Dermatol. 2007;157:106-110. doi:10.1111/j.1365-2133.2007.07987.x
- Billero V, Miteva M. Traction alopecia: the root of the problem. Clin Cosmet Investig Dermatol. 2018;11:149-159. doi:10.2147/CCID.S137296
- Haskin A, Aguh C. All hairstyles are not created equal: what the dermatologist needs to know about black hairstyling practices and the risk of traction alopecia (TA). J Am Acad Dermatol. 2016;75:606-611. doi:10.1016/j.jaad.2016.02.1162
- Samrao A, Price VH, Zedek D, et al. The “fringe sign”—a useful clinical finding in traction alopecia of the marginal hair line. Dermatol Online J. 2011;17:1.
- Kararizou E, Bougea AM, Giotopoulou D, et al. An update on the less-known group of other primary headaches—a review. Eur Neurol Rev. 2014;9:71-77. doi:10.17925/ENR.2014.09.01.71
- Tosti A, Miteva M, Torres F, et al. Hair casts are a dermoscopic clue for the diagnosis of traction alopecia. Br J Dermatol. 2010;163:1353-1355.
- Agrawal S, Daruwalla SB, Dhurat RS. The flambeau sign—a new dermoscopy finding in a case of marginal traction alopecia. Australas J Dermatol. 2020;61:49-50. doi:10. 1111/ajd.13187
- Lawson CN, Hollinger J, Sethi S, et al. Updates in the understanding and treatments of skin & hair disorders in women of color. Int J Womens Dermatol. 2017;3:S21-S37.
- Awad A, Chim I, Sharma P, et al. Low-dose oral minoxidil improves hair density in traction alopecia. J Am Acad Dermatol. 2023;89:157-159. doi:10.1016/j.jaad.2023.02.024
- Grayson C, Heath CR. Counseling about traction alopecia: a “compliment, discuss, and suggest” method. Cutis. 2021;108:20-22.
- Ozçelik D. Extensive traction alopecia attributable to ponytail hairstyle and its treatment with hair transplantation. Aesthetic Plast Surg. 2005;29:325-327. doi:10.1007/s00266-005-0004-5
- Singh MK, Avram MR. Technical considerations for follicular unit extraction in African-American hair. Dermatol Surg. 2013;39:1282-1284. doi:10.1111/dsu.12229
- Jones NL, Heath CR. Hair at the intersection of dermatology and anthropology: a conversation on race and relationships. Pediatr Dermatol. 2021;38(suppl 2):158-160.
- Franklin JMM, Wohltmann WE, Wong EB. From buns to braids and ponytails: entering a new era of female military hair-grooming standards. Cutis. 2021;108:31-35. doi:10.12788/cutis.0296
The Comparison
Traction alopecia (TA) is a common type of alopecia that ultimately can result in permanent hair loss. It often is caused or worsened by repetitive and prolonged hairstyling practices such as tight ponytails, braids, or locs, or use of wigs or weaves.1 Use of headwear, as in certain religious or ethnic groups, also can be contributory.2 Individuals participating in or training for occupations involving military service or ballet are at risk for TA due to hairstyling-specific policies. Early stages of TA are reversible with proper treatment and avoidance of exacerbating factors, emphasizing the importance of prompt recognition.3
Epidemiology
Data on the true prevalence of TA are lacking. It can occur in individuals of any race or any hair type. However, it is most common in women of African descent, affecting approximately one-third of this population.4 Other commonly affected groups include ballerinas and active-duty service members due to tight ponytails and buns, as well as the Sikh population due to the use of turbans as a part of their religious practice.2,5,6
Traction alopecia also impacts children, particularly those of African descent. A 2007 study of schoolchildren in South Africa determined that more than 17% of young African girls had evidence of TA—even some as young as 6 years of age.7
Traction alopecia can be caused or exacerbated by the use of hair clips and bobby pins that aid holding styles in place.8
Hair shaft morphology may contribute to the risk for TA, with more tightly coiled hair types being more susceptible.8 Variables such as use of chemical relaxers also increase the risk for disease, especially when combined with high-tension styling methods such as braids.9
Key clinical features
Patients with TA clinically present with hair loss and breakage in areas with tension, most commonly the marginal areas of the scalp as well as the frontal hairline and temporal scalp. Hair loss can result in a “fringe sign,” in which a patient may have preservation of a thin line of hairs at the frontal aspect of the hairline with a band of hair loss behind.10 This presentation may be used to differentiate TA from other forms of alopecia, including frontal fibrosing alopecia and female pattern hair loss. When the hair loss is not marginal, it may mimic other forms of patchy hair loss including alopecia areata and trichotillomania. Other clinical findings in TA may include broken hairs, pustules, and follicular papules.10 Patients also may describe symptoms such as scalp tenderness with specific hairstyles or headaches,11 or they may be completely asymptomatic.
Trichoscopy can be helpful in guiding diagnosis and treatment. Patients with TA often have perifollicular erythema and hair casts (cylindrical structures that encircle the proximal hair shafts) in the earlier stages of the disease, with eventual loss of follicular ostia in the later stages.10,12 Hair casts also may indicate ongoing traction.12 The flambeau sign—white tracks seen on trichoscopy in the direction the hair is pulled—resembles a lit torch.13
Worth noting
Early-stage TA can be reversed by avoiding hair tension. However, patients may not be amenable to this due to personal hairstyling preferences, job duties, or religious practices. Treatment with topical or intralesional steroids or even oral antibiotics such as doxycycline for its anti-inflammatory ability may result in regrowth of lost hair if the follicles are not permanently lost and exacerbating factors are avoided.3,14 Both topical and oral minoxidil have been used with success, with minoxidil thought to increase hair density by extending the anagen (growth) phase of hair follicles.3,15 Culturally sensitive patient counseling on the condition and potential exacerbating factors is critical.16
At later stages of the disease—after loss of follicular ostia has occurred—surgical interventions should be considered,17 such as hair transplantation, which can be successful but remains a technical challenge due to variability in hair shaft curvature.18 Additionally, the cost of the procedure can limit use, and some patients may not be optimal candidates due to the extent of their hair loss. Traction alopecia may not be the only hair loss condition present. Examining the scalp is important even if the chief area of concern is the marginal scalp.
Health disparity highlight
Prevention, early identification, and treatment initiated in a timely fashion are crucial to prevent permanent hair loss. There are added societal and cultural pressures that impact hairstyle and hair care practices, especially for those with tightly coiled hair.19 Historically, tightly coiled hair has been unfairly viewed as “unprofessional,” “unkempt,” and a challenge to “manage” by some. Thus, heat, chemical relaxers, and tight hairstyles holding hair in one position have been used to straighten the hair permanently or temporarily or to keep it maintained in a style that did not necessitate excessive manipulation—often contributing to further tension on the hair.
Military service branches have evaluated and changed some hair-related policies to reflect the diverse hair types of military personnel.20 The CROWN Act (www.thecrownact.com/about)—“Creating a Respectful and Open World for Natural Hair”—is a model law passed by 26 states that prohibits race-based hair discrimination, which is the denial of employment and educational opportunities because of hair texture. Although the law has not been passed in every state, it may help individuals with tightly coiled hair to embrace natural hairstyles. However, even hairstyles with one’s own natural curl pattern can contribute to tension and thus potential development of TA.
The Comparison
Traction alopecia (TA) is a common type of alopecia that ultimately can result in permanent hair loss. It often is caused or worsened by repetitive and prolonged hairstyling practices such as tight ponytails, braids, or locs, or use of wigs or weaves.1 Use of headwear, as in certain religious or ethnic groups, also can be contributory.2 Individuals participating in or training for occupations involving military service or ballet are at risk for TA due to hairstyling-specific policies. Early stages of TA are reversible with proper treatment and avoidance of exacerbating factors, emphasizing the importance of prompt recognition.3
Epidemiology
Data on the true prevalence of TA are lacking. It can occur in individuals of any race or any hair type. However, it is most common in women of African descent, affecting approximately one-third of this population.4 Other commonly affected groups include ballerinas and active-duty service members due to tight ponytails and buns, as well as the Sikh population due to the use of turbans as a part of their religious practice.2,5,6
Traction alopecia also impacts children, particularly those of African descent. A 2007 study of schoolchildren in South Africa determined that more than 17% of young African girls had evidence of TA—even some as young as 6 years of age.7
Traction alopecia can be caused or exacerbated by the use of hair clips and bobby pins that aid holding styles in place.8
Hair shaft morphology may contribute to the risk for TA, with more tightly coiled hair types being more susceptible.8 Variables such as use of chemical relaxers also increase the risk for disease, especially when combined with high-tension styling methods such as braids.9
Key clinical features
Patients with TA clinically present with hair loss and breakage in areas with tension, most commonly the marginal areas of the scalp as well as the frontal hairline and temporal scalp. Hair loss can result in a “fringe sign,” in which a patient may have preservation of a thin line of hairs at the frontal aspect of the hairline with a band of hair loss behind.10 This presentation may be used to differentiate TA from other forms of alopecia, including frontal fibrosing alopecia and female pattern hair loss. When the hair loss is not marginal, it may mimic other forms of patchy hair loss including alopecia areata and trichotillomania. Other clinical findings in TA may include broken hairs, pustules, and follicular papules.10 Patients also may describe symptoms such as scalp tenderness with specific hairstyles or headaches,11 or they may be completely asymptomatic.
Trichoscopy can be helpful in guiding diagnosis and treatment. Patients with TA often have perifollicular erythema and hair casts (cylindrical structures that encircle the proximal hair shafts) in the earlier stages of the disease, with eventual loss of follicular ostia in the later stages.10,12 Hair casts also may indicate ongoing traction.12 The flambeau sign—white tracks seen on trichoscopy in the direction the hair is pulled—resembles a lit torch.13
Worth noting
Early-stage TA can be reversed by avoiding hair tension. However, patients may not be amenable to this due to personal hairstyling preferences, job duties, or religious practices. Treatment with topical or intralesional steroids or even oral antibiotics such as doxycycline for its anti-inflammatory ability may result in regrowth of lost hair if the follicles are not permanently lost and exacerbating factors are avoided.3,14 Both topical and oral minoxidil have been used with success, with minoxidil thought to increase hair density by extending the anagen (growth) phase of hair follicles.3,15 Culturally sensitive patient counseling on the condition and potential exacerbating factors is critical.16
At later stages of the disease—after loss of follicular ostia has occurred—surgical interventions should be considered,17 such as hair transplantation, which can be successful but remains a technical challenge due to variability in hair shaft curvature.18 Additionally, the cost of the procedure can limit use, and some patients may not be optimal candidates due to the extent of their hair loss. Traction alopecia may not be the only hair loss condition present. Examining the scalp is important even if the chief area of concern is the marginal scalp.
Health disparity highlight
Prevention, early identification, and treatment initiated in a timely fashion are crucial to prevent permanent hair loss. There are added societal and cultural pressures that impact hairstyle and hair care practices, especially for those with tightly coiled hair.19 Historically, tightly coiled hair has been unfairly viewed as “unprofessional,” “unkempt,” and a challenge to “manage” by some. Thus, heat, chemical relaxers, and tight hairstyles holding hair in one position have been used to straighten the hair permanently or temporarily or to keep it maintained in a style that did not necessitate excessive manipulation—often contributing to further tension on the hair.
Military service branches have evaluated and changed some hair-related policies to reflect the diverse hair types of military personnel.20 The CROWN Act (www.thecrownact.com/about)—“Creating a Respectful and Open World for Natural Hair”—is a model law passed by 26 states that prohibits race-based hair discrimination, which is the denial of employment and educational opportunities because of hair texture. Although the law has not been passed in every state, it may help individuals with tightly coiled hair to embrace natural hairstyles. However, even hairstyles with one’s own natural curl pattern can contribute to tension and thus potential development of TA.
- Larrondo J, McMichael AJ. Traction alopecia. JAMA Dermatol. 2023;159:676. doi:10.1001/jamadermatol.2022.6298
- James J, Saladi RN, Fox JL. Traction alopecia in Sikh male patients. J Am Board Fam Med. 2007;20:497-498. doi:10.3122/jabfm.2007.05.070076
- Callender VD, McMichael AJ, Cohen GF. Medical and surgical therapies for alopecias in black women. Dermatol Ther. 2004;17:164-176.
- Loussouarn G, El Rawadi C, Genain G. Diversity of hair growth profiles. Int J Dermatol. 2005;44(suppl 1):6-9.
- Samrao AChen CZedek Det al. Traction alopecia in a ballerina: clinicopathologic features. Arch Dermatol. 2010;146:918-935. doi:10.1001/archdermatol.2010.183
- Korona-Bailey J, Banaag A, Nguyen DR, et al. Free the bun: prevalence of alopecia among active duty service women, fiscal years 2010-2019. Mil Med. 2023;188:e492-e496. doi:10.1093/milmed/usab274
- Khumalo NP, Jessop S, Gumedze F, et al. Hairdressing is associated with scalp disease in African schoolchildren. Br J Dermatol. 2007;157:106-110. doi:10.1111/j.1365-2133.2007.07987.x
- Billero V, Miteva M. Traction alopecia: the root of the problem. Clin Cosmet Investig Dermatol. 2018;11:149-159. doi:10.2147/CCID.S137296
- Haskin A, Aguh C. All hairstyles are not created equal: what the dermatologist needs to know about black hairstyling practices and the risk of traction alopecia (TA). J Am Acad Dermatol. 2016;75:606-611. doi:10.1016/j.jaad.2016.02.1162
- Samrao A, Price VH, Zedek D, et al. The “fringe sign”—a useful clinical finding in traction alopecia of the marginal hair line. Dermatol Online J. 2011;17:1.
- Kararizou E, Bougea AM, Giotopoulou D, et al. An update on the less-known group of other primary headaches—a review. Eur Neurol Rev. 2014;9:71-77. doi:10.17925/ENR.2014.09.01.71
- Tosti A, Miteva M, Torres F, et al. Hair casts are a dermoscopic clue for the diagnosis of traction alopecia. Br J Dermatol. 2010;163:1353-1355.
- Agrawal S, Daruwalla SB, Dhurat RS. The flambeau sign—a new dermoscopy finding in a case of marginal traction alopecia. Australas J Dermatol. 2020;61:49-50. doi:10. 1111/ajd.13187
- Lawson CN, Hollinger J, Sethi S, et al. Updates in the understanding and treatments of skin & hair disorders in women of color. Int J Womens Dermatol. 2017;3:S21-S37.
- Awad A, Chim I, Sharma P, et al. Low-dose oral minoxidil improves hair density in traction alopecia. J Am Acad Dermatol. 2023;89:157-159. doi:10.1016/j.jaad.2023.02.024
- Grayson C, Heath CR. Counseling about traction alopecia: a “compliment, discuss, and suggest” method. Cutis. 2021;108:20-22.
- Ozçelik D. Extensive traction alopecia attributable to ponytail hairstyle and its treatment with hair transplantation. Aesthetic Plast Surg. 2005;29:325-327. doi:10.1007/s00266-005-0004-5
- Singh MK, Avram MR. Technical considerations for follicular unit extraction in African-American hair. Dermatol Surg. 2013;39:1282-1284. doi:10.1111/dsu.12229
- Jones NL, Heath CR. Hair at the intersection of dermatology and anthropology: a conversation on race and relationships. Pediatr Dermatol. 2021;38(suppl 2):158-160.
- Franklin JMM, Wohltmann WE, Wong EB. From buns to braids and ponytails: entering a new era of female military hair-grooming standards. Cutis. 2021;108:31-35. doi:10.12788/cutis.0296
- Larrondo J, McMichael AJ. Traction alopecia. JAMA Dermatol. 2023;159:676. doi:10.1001/jamadermatol.2022.6298
- James J, Saladi RN, Fox JL. Traction alopecia in Sikh male patients. J Am Board Fam Med. 2007;20:497-498. doi:10.3122/jabfm.2007.05.070076
- Callender VD, McMichael AJ, Cohen GF. Medical and surgical therapies for alopecias in black women. Dermatol Ther. 2004;17:164-176.
- Loussouarn G, El Rawadi C, Genain G. Diversity of hair growth profiles. Int J Dermatol. 2005;44(suppl 1):6-9.
- Samrao AChen CZedek Det al. Traction alopecia in a ballerina: clinicopathologic features. Arch Dermatol. 2010;146:918-935. doi:10.1001/archdermatol.2010.183
- Korona-Bailey J, Banaag A, Nguyen DR, et al. Free the bun: prevalence of alopecia among active duty service women, fiscal years 2010-2019. Mil Med. 2023;188:e492-e496. doi:10.1093/milmed/usab274
- Khumalo NP, Jessop S, Gumedze F, et al. Hairdressing is associated with scalp disease in African schoolchildren. Br J Dermatol. 2007;157:106-110. doi:10.1111/j.1365-2133.2007.07987.x
- Billero V, Miteva M. Traction alopecia: the root of the problem. Clin Cosmet Investig Dermatol. 2018;11:149-159. doi:10.2147/CCID.S137296
- Haskin A, Aguh C. All hairstyles are not created equal: what the dermatologist needs to know about black hairstyling practices and the risk of traction alopecia (TA). J Am Acad Dermatol. 2016;75:606-611. doi:10.1016/j.jaad.2016.02.1162
- Samrao A, Price VH, Zedek D, et al. The “fringe sign”—a useful clinical finding in traction alopecia of the marginal hair line. Dermatol Online J. 2011;17:1.
- Kararizou E, Bougea AM, Giotopoulou D, et al. An update on the less-known group of other primary headaches—a review. Eur Neurol Rev. 2014;9:71-77. doi:10.17925/ENR.2014.09.01.71
- Tosti A, Miteva M, Torres F, et al. Hair casts are a dermoscopic clue for the diagnosis of traction alopecia. Br J Dermatol. 2010;163:1353-1355.
- Agrawal S, Daruwalla SB, Dhurat RS. The flambeau sign—a new dermoscopy finding in a case of marginal traction alopecia. Australas J Dermatol. 2020;61:49-50. doi:10. 1111/ajd.13187
- Lawson CN, Hollinger J, Sethi S, et al. Updates in the understanding and treatments of skin & hair disorders in women of color. Int J Womens Dermatol. 2017;3:S21-S37.
- Awad A, Chim I, Sharma P, et al. Low-dose oral minoxidil improves hair density in traction alopecia. J Am Acad Dermatol. 2023;89:157-159. doi:10.1016/j.jaad.2023.02.024
- Grayson C, Heath CR. Counseling about traction alopecia: a “compliment, discuss, and suggest” method. Cutis. 2021;108:20-22.
- Ozçelik D. Extensive traction alopecia attributable to ponytail hairstyle and its treatment with hair transplantation. Aesthetic Plast Surg. 2005;29:325-327. doi:10.1007/s00266-005-0004-5
- Singh MK, Avram MR. Technical considerations for follicular unit extraction in African-American hair. Dermatol Surg. 2013;39:1282-1284. doi:10.1111/dsu.12229
- Jones NL, Heath CR. Hair at the intersection of dermatology and anthropology: a conversation on race and relationships. Pediatr Dermatol. 2021;38(suppl 2):158-160.
- Franklin JMM, Wohltmann WE, Wong EB. From buns to braids and ponytails: entering a new era of female military hair-grooming standards. Cutis. 2021;108:31-35. doi:10.12788/cutis.0296
Progressive Eyelash Loss and Scale of the Right Eyelid
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
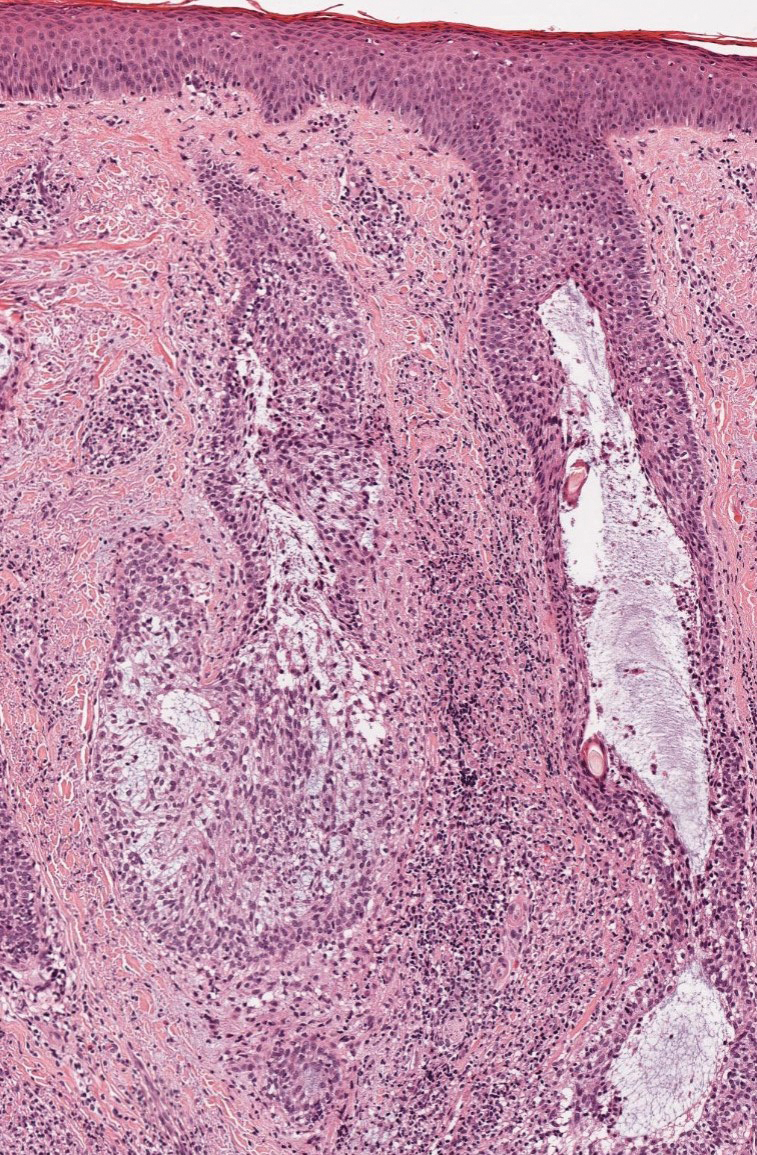
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
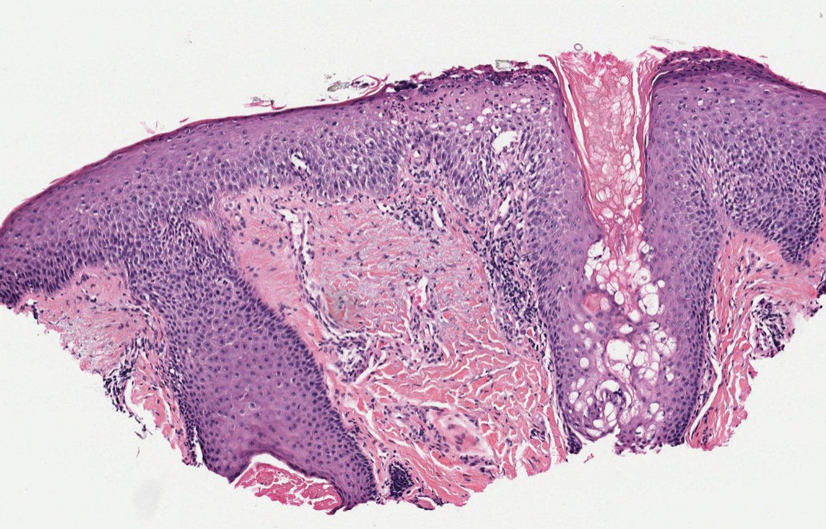
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
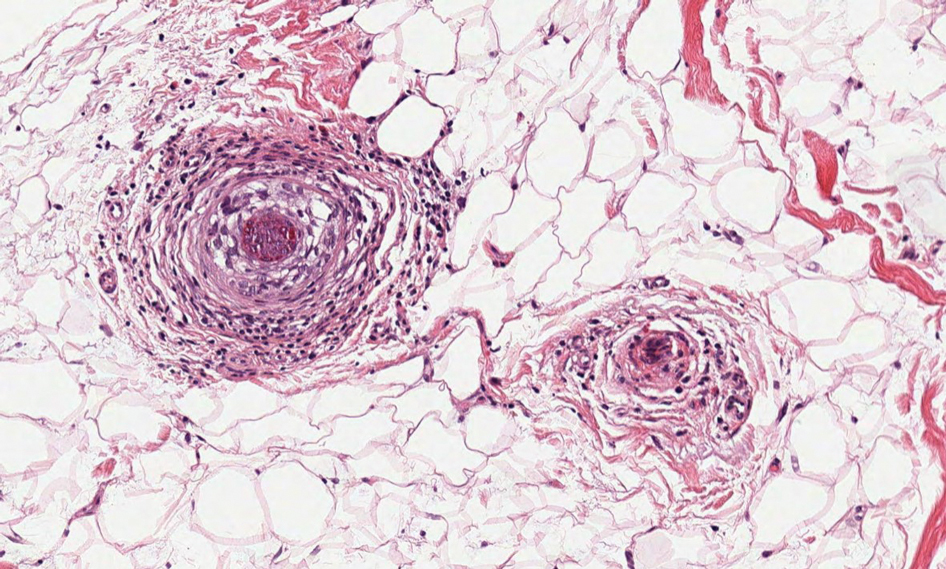
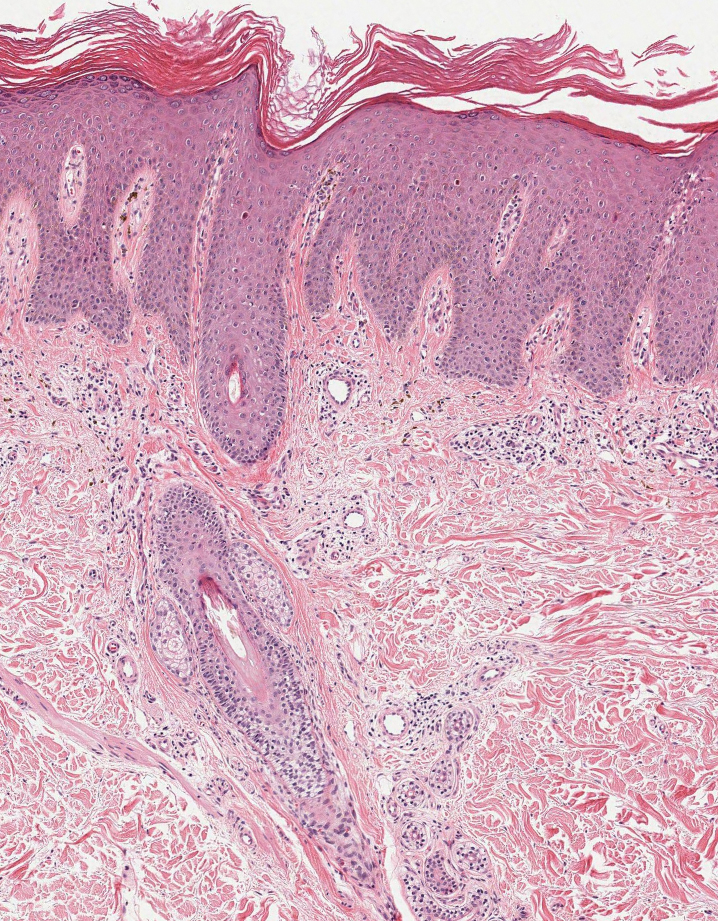
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
The Diagnosis: Folliculotropic Mycosis Fungoides
Folliculotropic mycosis fungoides (FMF) is a variant of mycosis fungoides (MF) characterized by folliculotropism and follicular-based lesions. The clinical manifestation of FMF can vary and includes patches, plaques, or tumors resembling nonfolliculotropic MF; acneform lesions including comedones and pustules; or areas of alopecia. Lesions commonly involve the head and neck but also can be seen on the trunk or extremities. Folliculotropic mycosis fungoides can be accompanied by pruritus or superimposed secondary infection.
Histologic features of FMF include follicular (perifollicular or intrafollicular) infiltration by atypical T cells showing cerebriform nuclei.1 In early lesions, there may be only mild superficial perivascular inflammation without notable lymphocyte atypia, making diagnosis challenging. 2,3 Mucinous degeneration of the follicles—termed follicular mucinosis—is a common histologic finding in FMF.1,2 Follicular mucinosis is not exclusive to FMF; it can be primary/idiopathic or secondary to underlying inflammatory or neoplastic disorders such as FMF. On immunohistochemistry, FMF most commonly demonstrates a helper T cell phenotype that is positive for CD3 and CD4 and negative for CD8, with aberrant loss of CD7 and variably CD5, which is similar to classic MF. Occasionally, larger CD30+ cells also can be present in the dermis. T-cell gene rearrangement studies will demonstrate T-cell receptor clonality in most cases.2
Many large retrospective cohort studies have suggested that patients with FMF have a worse prognosis than classic MF, with a 5-year survival rate of 62% to 87% for early-stage FMF vs more than 90% for classic patchand plaque-stage MF.4-7 However, a 2016 study suggested histologic evaluation may be able to further differentiate clinically identical cases into indolent and aggressive forms of FMF with considerably different outcomes based on the density of the perifollicular infiltrate.5 The presence of follicular mucinosis has no impact on prognosis compared to cases without follicular mucinosis.1,2
Alopecia mucinosa is characterized by infiltrating, erythematous, scaling plaques localized to the head and neck.8 It is diagnosed clinically, and histopathology shows follicular mucinosis. The terms alopecia mucinosa and follicular mucinosis often are used interchangeably. Over the past few decades, 3 variants have been categorized: primary acute, primary chronic, and secondary. The primary acute form manifests in children and young adults as solitary lesions, which often resolve spontaneously. In contrast, the primary chronic form manifests in older adults as multiple disseminated lesions with a chronic relapsing course.8,9 The secondary form can occur in the setting of other disorders, including lupus erythematosus, hypertrophic lichen planus, alopecia areata, and neoplasms such as MF or Hodgkin lymphoma.9 The histopathologic findings are similar for all types of alopecia mucinosa, with cystic pools of mucin deposition in the sebaceous glands and external root sheath of the follicles as well as associated inflammation composed of lymphocytes and eosinophils (Figure 1).9,10 The inflammatory infiltrate rarely extends into the epidermis or upper portion of the hair follicle. Although histopathology alone cannot reliably distinguish between primary and secondary forms of alopecia mucinosa, MF (including follicular MF) or another underlying cutaneous T-cell lymphoma should be considered if inflammation extends into the upper dermis, epidermis, or follicles or is in a dense bandlike distribution.11 On immunohistochemistry, lymphocytes should show positivity for CD3, CD4, and CD8. The CD4:CD8 ratio often is 1:1 in alopecia mucinosa, while in FMF it is approximately 3:1.10 CD7 commonly is negative but can be present in a small percentage of cases.12 T-cell receptor gene rearrangement studies have detected clonality in both primary and secondary alopecia mucinosa and thus cannot be used alone to distinguish between the two.10 Given the overlap in histopathologic and immunohistochemical features of primary and secondary alopecia mucinosa, definitive diagnosis cannot be made with any single modality and should be based on correlating clinical presentation, histopathology, immunohistochemistry, and molecular analyses.
Inflammatory dermatoses including seborrheic dermatitis also are in the differential diagnosis for FMF. Seborrheic dermatitis is a common chronic inflammatory skin disorder affecting 1% to 3% of the general population. 13 Patients usually present with scaly and greasy plaques and papules localized to areas with increased sebaceous glands and high sebum production such as the face, scalp, and intertriginous regions. The distribution often is symmetrical, and the severity of disease can vary substantially.13 Sebopsoriasis is an entity with overlapping features of seborrheic dermatitis and psoriasis, including thicker, more erythematous plaques that are more elevated. Histopathology of seborrheic dermatitis reveals spongiotic inflammation in the epidermis characterized by rounding of the keratinocytes, widening of the intercellular spaces, and accumulation of intracellular edema, causing the formation of clear spaces in the epidermis (Figure 2). Focal parakeratosis, usually in the follicular ostia, and mounds of scaly crust often are present. 14 A periodic acid–Schiff stain should be performed to rule out infectious dermatophytes, which can show similar clinical and histologic features. More chronic cases of seborrheic dermatitis often can take on histologic features of psoriasis, namely epidermal hyperplasia with thinning over dermal papillae, though the hyperplasia in psoriasis is more regular.
Alopecia areata is an immune-mediated disorder characterized by nonscarring hair loss; it affects approximately 0.1% to 0.2% of the general population.15 The pathogenesis involves the premature transition of hair follicles in the anagen (growth) phase to the catagen ( nonproliferative/involution) and telogen (resting) phases, resulting in sudden hair shedding and decreased regrowth. Clinically, it is characterized by asymptomatic hair loss that occurs most frequently on the scalp and other areas of the head, including eyelashes, eyebrows, and facial hair, but also can occur on the extremities. There are several variants; the most common is patchy alopecia, which features smooth circular areas of hair loss that progress over several weeks. Some patients can progress to loss of all scalp hairs (alopecia totalis) or all hairs throughout the body (alopecia universalis). 15 Patients typically will have spontaneous regrowth of hair, with up to 50% of those with limited hair loss recovering within a year.16 The disease has a chronic/ relapsing course, and patients often will have multiple episodes of hair loss. Histopathologic features can vary depending on the stage of disease. In acute cases, a peribulbar lymphocytic infiltrate preferentially involving anagen-stage hair follicles is seen, with associated necrosis, edema, and pigment incontinence (Figure 3).16 In chronic alopecia areata, the inflammation may be less brisk, and follicular miniaturization often is seen. Additionally, increased proportions of catagen- or telogen-stage follicles are present.16,17 On immunohistochemistry, lymphocytes express both CD4 and CD8, with a slightly increased CD4:CD8 ratio in active disease.18
Psoriatic alopecia describes hair loss that occurs in patients with psoriasis. Patients present with scaly, erythematous, psoriasiform plaques or patches, as well as decreased hair density, finer hairs, and increased dystrophic hair bulbs within the psoriatic plaques.19 It often is nonscarring and resolves with therapy, though scarring may occur with secondary infection. Psoriatic alopecia may occur in the setting of classic psoriasis and also may occur in psoriasiform drug eruptions, including those caused by tumor necrosis factor inhibitors.20,21 Histologic features include atrophy of sebaceous glands, epidermal changes with hypogranulosis and psoriasiform hyperplasia, decreased hair follicle density, and neutrophils in the stratum spinosum (Figure 4). There often is associated perifollicular lymphocytic inflammation with small lymphocytes that do not have notable morphologic abnormalities.
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
- Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019;133:1703-1714. doi:10.1182/blood-2018-11-881268
- Malveira MIB, Pascoal G, Gamonal SBL, et al. Folliculotropic mycosis fungoides: challenging clinical, histopathological and immunohistochemical diagnosis. An Bras Dermatol. 2017;92(5 suppl 1):73-75. doi:10.1590/abd1806-4841.20175634
- Flaig MJ, Cerroni L, Schuhmann K, et al. Follicular mycosis fungoides: a histopathologic analysis of nine cases. J Cutan Pathol. 2001;28:525- 530. doi:10.1034/j.1600-0560.2001.281006.x
- van Doorn R, Scheffer E, Willemze R. Follicular mycosis fungoides: a distinct disease entity with or without associated follicular mucinosis: a clinicopathologic and follow-up study of 51 patients. Arch Dermatol. 2002;138:191-198. doi:10.1001/archderm.138.2.191
- van Santen S, Roach REJ, van Doorn R, et al. Clinical staging and prognostic factors in folliculotropic mycosis fungoides. JAMA Dermatol. 2016;152:992-1000. doi:10.1001/jamadermatol.2016.1597
- Lehman JS, Cook-Norris RH, Weed BR, et al. Folliculotropic mycosis fungoides: single-center study and systematic review. Arch Dermatol. 2010;146:607-613. doi:10.1001/archdermatol.2010.101
- Gerami P, Rosen S, Kuzel T, et al. Folliculotropic mycosis fungoides: an aggressive variant of cutaneous T-cell lymphoma. Arch Dermatol. 2008;144:738-746. doi:10.1001/archderm.144.6.738
- Büchner SA, Meier M, Rufli TH. Follicular mucinosis associated with mycosis fungoides. Dermatology. 1991;183:66-67. doi:10.1159/000247639
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746. doi:10.7759/cureus.4746
- Rongioletti F, De Lucchi S, Meyes D, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15-19. doi:10.1111/j.1600-0560.2009.01338.x
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165. doi:10.1111/ijd.15165
- Zvulunov A, Shkalim V, Ben-Amitai D, et al. Clinical and histopathologic spectrum of alopecia mucinosa/follicular mucinosis and its natural history in children. J Am Acad Dermatol. 2012;67:1174-1181. doi:10.1016/j.jaad.2012.04.015
- Dessinioti C, Katsambas A. Seborrheic dermatitis: etiology, risk factors, and treatments: facts and controversies. Clin Dermatol. 2013;31:343-351. doi:10.1016/j.clindermatol.2013.01.001
- Gupta AK, Bluhm R. Seborrheic dermatitis. J Eur Acad Dermatol Venereol. 2004;18:13-26; quiz 19-20. doi:10.1111/j .1468-3083.2004.00693.x
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: disease characteristics, clinical evaluation, and new perspectives on pathogenesis. J Am Acad Dermatol. 2018;78:1-12. doi:10.1016/j .jaad.2017.04.1141
- Alkhalifah A, Alsantali A, Wang E, et al. Alopecia areata update: part I. clinical picture, histopathology, and pathogenesis. J Am Acad Dermatol. 2010;62:177-88, quiz 189-90. doi:10.1016/j.jaad.2009.10.032
- Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555-1559. doi:10.1001/archderm .139.12.1555
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11(2 pt 1):216-223. doi:10.1016 /s0190-9622(84)70152-6
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721. doi:10.1111/ced.12715
- Afaasiev OK, Zhang CZ, Ruhoy SM. TNF-inhibitor associated psoriatic alopecia: diagnostic utility of sebaceous lobule atrophy. J Cutan Pathol. 2017;44:563-539. doi:10.1111/cup.12932
- Silva CY, Brown KL, Kurban AK, et al. Psoriatic alopecia—fact or fiction? A clinicohistologic reappraisal. Indian J Dermatol Venereol Leprol. 2012;78:611-619. doi:10.4103/0378-6323.100574
An 88-year-old man presented with progressive eyelash loss and scale involving the right eyelids (top). Dermatopathologic examination was performed (bottom).